
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand electronic fine-tuning and its repercussion on the photocatalytic activity and mechanistic pathways of the copper-photocatalysed aza-Henry reaction†
Chenfei
Li
 a,
Robert
Dickson
a,
Nils
Rockstroh
b,
Jabor
Rabeah
b,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Paul
Hünemörder
b,
Anke
Spannenberg
b,
Michael
Bühl
a,
Esteban
Mejía
*b,
Eli
Zysman-Colman
*a and
Paul C. J.
Kamer
*b
a,
Robert
Dickson
a,
Nils
Rockstroh
b,
Jabor
Rabeah
b,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Paul
Hünemörder
b,
Anke
Spannenberg
b,
Michael
Bühl
a,
Esteban
Mejía
*b,
Eli
Zysman-Colman
*a and
Paul C. J.
Kamer
*b
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, North Haugh, KY16 9ST St Andrews, UK
bLeibniz Institute for Catalysis, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 18th September 2020
Abstract
A family of six structurally related heteroleptic copper(I) complexes of the form of [Cu(N^N)(P^P)]+ bearing a 2,9-dimethyl-1,10-phenanthroline diimine (N^N) ligand and a series of electronically tunable xantphos (P^P) ligands have been synthesized and their optoelectronic properties characterized. The reactivity of these complexes in the copper-photocatalyzed aza-Henry reaction of N-phenyltetrahydroisoquinoline was evaluated, while the related excited state kinetics were comprehensively studied. By subtlety changing the electron-donating properties of the P^P ligands with negligible structural differences, we could tailor the photoredox properties and relate them to the reactivity. Moreover, depending on the exited-state redox potential of the catalysts, the preferred mechanism can shift between reductive quenching, energy transfer and oxidative quenching pathways. A combined study of the structural modulation of copper(I) photocatalysts, optoelectronic properties and photocatalytic reactivity resulted in a clearer understanding of both the rational design of the photocatalyst and the complexity of competing photoinduced electron and energy transfer mechanisms.
Introduction
Photoredox catalysis has become an increasingly employed tool in synthesis as many organic transformations can be promoted under mild conditions with often excellent chemoselectivity and high yields.1–3 The key to the vast majority of photoredox catalysis reactions is the use of a visible-light photoactive compound that, when photoexcited, can initiate a single electron transfer (SET) reaction with an organic substrate.4 The most popular photocatalysts5 used are those based on ruthenium(II) and iridium(III) complexes as their optoelectronic properties have been widely studied and their excited state redox potentials enable a wide range of chemical transformations. For instance, potent photooxidants such as [Ru(bpz)3]2+ ( = 1.45 V vs. SCE in MeCN) and [Ir(dF(CF3)ppy)2(dtBubpy)]+ (
= 1.45 V vs. SCE in MeCN) and [Ir(dF(CF3)ppy)2(dtBubpy)]+ ( = 1.21 V vs. SCE in MeCN) have been used for the photocatalytic oxidation of benzylic alcohols to aldehydes,6 while powerful photoreductants such as fac-Ir(ppy)3 (
= 1.21 V vs. SCE in MeCN) have been used for the photocatalytic oxidation of benzylic alcohols to aldehydes,6 while powerful photoreductants such as fac-Ir(ppy)3 (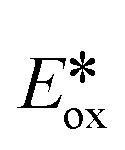 of −1.73 V vs. SCE in MeCN) have enabled the reduction of unactivated alkyl, alkenyl and aryl iodides.7 Recently, non-toxic, Earth-abundant and inexpensive copper(I) complexes have been investigated as alternative photocatalysts with, in certain cases, superior excited-state redox properties.8 For instance, [Cu(XyBnta)(dppb)]+ is a very strong photoreductant (
of −1.73 V vs. SCE in MeCN) have enabled the reduction of unactivated alkyl, alkenyl and aryl iodides.7 Recently, non-toxic, Earth-abundant and inexpensive copper(I) complexes have been investigated as alternative photocatalysts with, in certain cases, superior excited-state redox properties.8 For instance, [Cu(XyBnta)(dppb)]+ is a very strong photoreductant (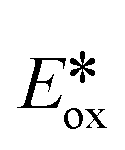 = −2.40 V vs. SCE in THF)9 while [Cu(bath)(ThioPOP)]+ is a strong photooxidant (
= −2.40 V vs. SCE in THF)9 while [Cu(bath)(ThioPOP)]+ is a strong photooxidant ( = 1.56 V vs. SCE in MeCN).10 Copper(I) photocatalysts have been successfully employed in a diverse set of transformations such as photo-catalytic hydrogen production, cyclisations, atom transfer radical additions (ATRAs) and C–H activation chemistry.10–15 In terms of their chemical structure, the majority of copper(I) photocatalysts can be divided into one of two families: homoleptic complexes of the form [Cu(N^N)2]+ and heteroleptic complexes of the form [Cu(N^N)(P^P)]+, where N^N and P^P are diimine and bis(phosphine) ligands, respectively. Unlike homoleptic copper(I) complexes, where the exited state energies are usually small (E(0,0) = ∼2 eV)16 and the optoelectronic properties difficult to modulate, the modular structure of heteroleptic complexes provides great opportunity to fine-tune their photophysical, structural and electrochemical properties.13,17,18 Collins et al. studied a library of 50 heteroleptic copper(I) complexes with different and mostly commercial bis(phosphine) and diimine ligands in a series of photoredox, proton-coupled electron transfer (PCET) and energy transfer reactions and demonstrated how the yields could be optimized through the tuning of the optoelectronic properties of the photocatalyst.19 However, to date this strategy has been underexplored in the context of photoredox catalysis. Indeed, the development of [Cu(N^N)(P^P)]+-based photocatalysts has relied on a combination of commercially available bis(phosphine) ligands and electronically tuned N^N ligands to modulate their photophysical and/or photocatalytic performance. For instance, Costa et al. demonstrated how the solid-state photophysical and electroluminescent properties of [Cu(4,4′-R2bpy)(xantphos)]+ complexes could be systematically tuned as a function of the nature of the R group on the bipyridine ligand.17 More recently, Housecroft et al. studied the effect of phosphine tuning in heteroleptic [Cu(N^N)(P^P)]+ complexes as emitters for light-emitting electrochemical cells (LEECs) and demonstrated that the enhanced performance of the LEECs could be attributed to steric modification of the backbone of the xantphos ligand with tert-butyl groups.20 To date, electronic tuning of the P^P ligand and its impact on the optoelectronic and photocatalytic properties of heteroleptic copper(I) complexes remains comparatively unexplored, likely due to the challenging and air-sensitive syntheses required.21 Among the different combinations of (P^P) and (N^N) ligands, complexes bearing xantphos and phenanthroline derivatives have shown the most promising results as they have been successfully employed in the production of hydrogen from water,10–12,22–24 reduction of CO2 to CO,25 C–C bond formation in the synthesis of carbazoles,26,27 cyclisations,28 polymerizations.29 and oxygen sensing.30
= 1.56 V vs. SCE in MeCN).10 Copper(I) photocatalysts have been successfully employed in a diverse set of transformations such as photo-catalytic hydrogen production, cyclisations, atom transfer radical additions (ATRAs) and C–H activation chemistry.10–15 In terms of their chemical structure, the majority of copper(I) photocatalysts can be divided into one of two families: homoleptic complexes of the form [Cu(N^N)2]+ and heteroleptic complexes of the form [Cu(N^N)(P^P)]+, where N^N and P^P are diimine and bis(phosphine) ligands, respectively. Unlike homoleptic copper(I) complexes, where the exited state energies are usually small (E(0,0) = ∼2 eV)16 and the optoelectronic properties difficult to modulate, the modular structure of heteroleptic complexes provides great opportunity to fine-tune their photophysical, structural and electrochemical properties.13,17,18 Collins et al. studied a library of 50 heteroleptic copper(I) complexes with different and mostly commercial bis(phosphine) and diimine ligands in a series of photoredox, proton-coupled electron transfer (PCET) and energy transfer reactions and demonstrated how the yields could be optimized through the tuning of the optoelectronic properties of the photocatalyst.19 However, to date this strategy has been underexplored in the context of photoredox catalysis. Indeed, the development of [Cu(N^N)(P^P)]+-based photocatalysts has relied on a combination of commercially available bis(phosphine) ligands and electronically tuned N^N ligands to modulate their photophysical and/or photocatalytic performance. For instance, Costa et al. demonstrated how the solid-state photophysical and electroluminescent properties of [Cu(4,4′-R2bpy)(xantphos)]+ complexes could be systematically tuned as a function of the nature of the R group on the bipyridine ligand.17 More recently, Housecroft et al. studied the effect of phosphine tuning in heteroleptic [Cu(N^N)(P^P)]+ complexes as emitters for light-emitting electrochemical cells (LEECs) and demonstrated that the enhanced performance of the LEECs could be attributed to steric modification of the backbone of the xantphos ligand with tert-butyl groups.20 To date, electronic tuning of the P^P ligand and its impact on the optoelectronic and photocatalytic properties of heteroleptic copper(I) complexes remains comparatively unexplored, likely due to the challenging and air-sensitive syntheses required.21 Among the different combinations of (P^P) and (N^N) ligands, complexes bearing xantphos and phenanthroline derivatives have shown the most promising results as they have been successfully employed in the production of hydrogen from water,10–12,22–24 reduction of CO2 to CO,25 C–C bond formation in the synthesis of carbazoles,26,27 cyclisations,28 polymerizations.29 and oxygen sensing.30
The parent complex, 3 [Cu(dmphen)(xantphos)]+, has been previously reported as a photocatalyst and sensor in polymerisations, oxidative-cyclisations of triarylamines to make N-substituted carbazoles and stilbenoids to make helicenes.26,28,29 More widely, xantphos-type ligands have been widely used in homogeneous catalysis31,32 and are particularly attractive as they possess rigid backbones and enforce large bite-angles, which at the same time stabilize the tetrahedral geometry and disfavor the square planar geometry. The latter feature has proven to be a powerful tool in catalytic transformations.33–35 Xantphos was originally designed to stabilize the bisequatorial coordination mode in trigonal bipyramidal rhodium hydroformylation catalysis leading to extremely high selectivity for the linear aldehyde.36 An additional advantage of these wide bite-angle ligands is the facile electronic fine-tuning leading to further optimization of catalyst activity and selectivity.37 The combination of steric constraints to enforce a preferred coordination mode with subtle fine-tuning of electronic properties can lead to unprecedented performance in homogeneous catalysis. A striking example is the Ni catalyzed hydrocyanation of alkenes, where traditional phosphines had been inactive, but xantphos-type ligands induced high activity and selectivity.38,39 We anticipated that we could also observe these structure–activity relationships in the context of photocatalytic transformations.
Herein, we report a Hammett series of five well-defined [Cu(dmphen)(p-R xantphos)]PF6 (dmphen = neocuproine; R = CF3, F, H, Me, OMe) complexes (1–5, Fig. 1) where the optoelectronic properties of the complexes are systematically tuned as a function of the substituent located on the arylphosphine moiety of the xantphos ligand. We studied the reactivity of these complexes in the aerobic photoinduced cross-dehydrogenative coupling of tetrahydroisoquinoline with nitromethane, which is one of the most well-studied photocatalytic transformations. Previous mechanistic studies have shown that this reaction can proceed through one or several different pathways simultaneously.40–43 However, the effect of the intrinsic photoredox properties of the catalyst on their preferred reaction mechanism are not well understood. In this study, we have systematically tuned the electronic properties of a series of Cu(I) photocatalysts. We have investigated how excited state kinetics and (photo)stability as a function of the structure of the photocatalyst can impact reaction yield and working mechanism in this model reaction. By applying the gained photophysical and mechanistic knowledge, a more conscious catalysts and process design can be anticipated.
 | ||
| Fig. 1 Hammett series of [Cu (dmphen)(P^P)]+ complexes in this study. | ||
Results and discussion
The [Cu(dmphen)(P^P)]PF6 complexes, 1–5, were obtained in good to excellent yield in two steps: stoichiometric reaction of [Cu(NCMe)4]PF6 with xantphos type ligands substituted by 4-CF3, 4-F, 4-H, 4-Me and 4-OMe (ref. 36, 37 and 44) followed by addition of dmphen in dichloromethane (Fig. 2). | ||
| Fig. 2 Synthesis of [Cu(dmphen)(P^P)]PF6 complexes. | ||
Crystals suitable for X-ray analysis of 1 and 5 were obtained by slow diffusion of diethyl ether into a saturated dichloromethane solution at room temperature, confirming a distorted tetrahedral coordination sphere of the copper center. Ellipsoid representations and the complete crystallographic information can be found in the ESI.†
Solution-state stability studies
Dynamic ligand exchange in solution has been observed for certain [Cu(dmphen)(P^P)]+ complexes. As described by Armaroli et al. the ratio of heteroleptic [Cu(dmphen)(P^P)]+ to homoleptic [Cu(dmphen)2]+ and [Cu(P^P)2]+ is highly dependent on the nature of the P^P ligand and its impact on the relative thermodynamic stabilities of the complexes.45 However, the homoleptic complex [Cu(xantphos)2][PF6], was identified unequivocally by single crystal X-ray diffraction by Yuasa et al., corroborating the stabilization of the tetrahedral coordination mode of xantphos.46 This complex was found to be poorly soluble in most organic solvents and thus the potential precipitation of this product from the reaction mixture can take the system out of equilibrium and affect the catalytic species in solution.We next studied the stability of 1–5 in MeCN under inert atmosphere upon photoirradiation with a 420 nm blue LED light (see Fig. S4 in ESI† for selected examples).
Complex 3, was found to be stable over the 4-hour experiment as were complexes 4 and 5 possessing electron-donating substituents on the P^P ligand. By contrast, complexes bearing electron-withdrawing substituents on the P^P ligand (1 and 2) showed decomposition with the apparent formation of [Cu(dmphen)2]+ as evidenced by the emergence of a new MLCT band at ca. 480 nm, results consistent with those of Beller et al.10 and Walton et al.47
Redox properties
The electrochemistry of 1–5 was studied by cyclic voltammetry (CV) in order to discern the redox behaviour of the complexes.48 Measurements were carried out in acetonitrile (MeCN) under a nitrogen atmosphere. The CV traces are shown in Fig. 3 and the data are collected in Table 1. All five complexes show irreversible oxidation and quasi-reversible reduction waves. | ||
| Fig. 3 CV traces of 1–5 measured in degassed MeCN with 0.1 M TBAPF6 as the supporting electrolyte using a glassy carbon working electrode, a platinum wire counter electrode, a Ag/Ag+ pseudo-reference electrode and referenced vs. SCE using Fc/Fc+ as an internal standard (0.38 V in MeCN).49 Scan rate: 20 mV s−1, scanning only from the first reduction to first oxidation. | ||
| Complex | E ox | E red | ΔEredoxa | E (0,0) |
|
|
E HOMO | E LUMO | ΔEH–L |
|---|---|---|---|---|---|---|---|---|---|
| [V] | [V] | [V] | [V] | [V] | [V] | [eV] | [eV] | [eV] | |
a Electrochemical measurements carried out in a degassed HPLC grade MeCN with glassy carbon working electrode, Ag/Ag+ reference electrode and a platinum wire counter electrode. Fc/Fc+ was used as the internal standard and the data reported versus SCE (0.38 V vs. SCE in MeCN).49
b Optical gap inferred from the intersection points of the normalized absorption spectra and the tangent of the onset of the normalized emission spectra in MeCN, defined as the energy at 10% relative intensity of the maximum on the low energy tail.
c Excited state redox potentials calculated with equation 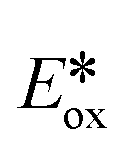 = Eox − E(0,0), = Eox − E(0,0),  = Ered + E(0,0).50
d
E
HOMO = −(E[onset,ox vs. Fc/Fc+] + 4.8 eV); ELUMO = −(E[onset,red vs. Fc/Fc+] + 4.8 eV).51 = Ered + E(0,0).50
d
E
HOMO = −(E[onset,ox vs. Fc/Fc+] + 4.8 eV); ELUMO = −(E[onset,red vs. Fc/Fc+] + 4.8 eV).51
|
|||||||||
| 1 | 1.51 | −1.63 | 3.14 | 2.79 | −1.28 | 1.16 | −5.82 | −2.78 | 3.03 |
| 2 | 1.20 | −1.73 | 2.93 | 2.66 | −1.46 | 0.93 | −5.54 | −2.69 | 2.85 |
| 3 | 1.21 | −1.74 | 2.95 | 2.61 | −1.40 | 0.87 | −5.54 | −2.70 | 2.84 |
| 4 | 1.19 | −1.72 | 2.91 | 2.58 | −1.39 | 0.86 | −5.49 | −2.71 | 2.78 |
| 5 | 1.17 | −1.75 | 2.92 | 2.53 | −1.36 | 0.78 | −5.49 | −2.69 | 2.80 |

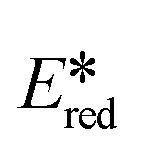
Density functional theory (DFT) calculations indicate that the lowest unoccupied molecular orbital (LUMO) of 1–5 is located on the dmphen ligand while the highest occupied molecular orbital (HOMO) consists of copper d-orbitals with contributions from the P^P ligands (Fig. 4). There is a strong correlation between the nature of the substituent on the P^P ligand on both the experimentally inferred and calculated HOMO and LUMO energies, as evidenced by the Hammett plots in Fig. 5.
 | ||
| Fig. 4 DFT calculations for 1–5 (PBE0/ECP1/PCM(DCM)//PBE0-D3/ECP1 level). Kohn–Sham energy diagram with electron density distribution of HOMO and LUMO shown. Red bars represent Cu orbitals, green bars represent orbitals on the N^N ligand and the blue bars represent orbitals on the P^P ligand. | ||
 | ||
| Fig. 5 a) Hammett plot of the optical band gap; b) Hammett plot of the HOMO and LUMO energies determined from electrochemistry. | ||
The oxidation wave is shifted to less positive potentials in the presence of electron-donating substituents on the P^P ligand, leading to a shallower HOMO while the oxidation wave is shifted anodically, leading to a more stabilized HOMO in the presence of electron-withdrawing substituents on the P^P ligand. There is a strong Hammett correlation with respect to Eox (R2 = 0.93, Fig. 5a).
A Hammett analysis of the MeCN Epaox data shows a correlation with an R2 of 0.89 (Fig. S12a†). The reduction potentials of complexes 1–5 showed a moderate Hammett correlation (Fig. S12b,†R2 = 0.83), which can be rationalized by the small influence of the electronics of the P^P ligand on the copper metal, which is also implicated, albeit to a very small extent, in the reduction along with on the dmphen ligand. It has been reported in the literature that [Cu(bathocuproine)(xantphos)]+ has an Eox of 1.31 V vs. SCE in DCM, which is more positive than that of complex 3 (Eox = 1.18 V in DCM) despite possessing the same P^P ligand, while the corresponding Ered of −1.67 V is also cathodically shifted by 0.36 V compared to that of 3 in DCM, which is due to the extended π-conjugation of the N^N ligand.10 A second literature complex [Cu(dmphen)(DPEphos)]BF4 has a reversible 1st reduction at Ered of −1.72 V vs. SCE in MeCN that is anodically shifted by 20 mV compared to that of 3 but is close to the reduction potential of 2 (DPEPhos = bis(2-diphenylphosphino)phenyl ether).29 Coppo et al.52 reported the electrochemistry of the structurally related [Cu(dmbpy)(xantphos)]+ with Eox = 1.27 V and Ered = −1.10 V (dmbpy = 6,6′-dimethyl-2,2′-bipyridine). Their reported oxidation potential is moderately more positive than that of 3.53
The energies of HOMO and LUMO were estimated from the onset of the redox waves (Table 1).51 The trend in HOMO energies follows a Hammett trend where complexes 4 and 5 have the next highest HOMO (EHOMO = −5.49 eV) while complexes with electron-withdrawing substituents (1 and 2) show the lowest-lying HOMO (1, EHOMO = −5.82 eV). There is a moderate Hammett correlation for the LUMO energies (R2 = 0.72, Fig. 5b); however, this correlation is expectedly not as strong, as the electronics of the P^P ligand influence the HOMO more strongly than the LUMO. The trends in the experimentally determined redox gaps are consistent with those in the DFT-calculated HOMO–LUMO gaps (Table 1, Fig. 4).
Further analysis of Mulliken electronic contributions showed that Cu metal ion contributes less (P^P ligand contributes more) to the HOMO with more electron-donating ligands, while there was no strong trend found for contributions to the LUMO as the this is localized on the dmphen ligand.
In order to assess the viability of this family of complexes as photoredox catalysts, the excited state redox potentials were calculated from the ground state electrochemistry and the optical gap (Table 1).50 Given that the trends in redox behaviour and absorption spectroscopy complement each other, there is therefore no Hammett correlation observed for the excited state redox potentials (Fig. S12c and d†). The excited state oxidation potentials of 1–5 vary from −1.46 eV for 2 to −1.28 eV for 1. These values are close to the well-studied photocatalyst fac-Ir(ppy)3 ( = −1.73 V vs. SCE in MeCN), which is one of the most powerful photoreductants, and are higher than that of [Ru(bpy)3]2+ (
= −1.73 V vs. SCE in MeCN), which is one of the most powerful photoreductants, and are higher than that of [Ru(bpy)3]2+ ( = −0.81 V vs. SCE in MeCN), which is widely used in photoredox catalysis reactions following oxidative quenching pathways.54 Comparing 1–5 with [Cu(dap)2]Cl (dap = 2,9-anisylphenanthroline) (
= −0.81 V vs. SCE in MeCN), which is widely used in photoredox catalysis reactions following oxidative quenching pathways.54 Comparing 1–5 with [Cu(dap)2]Cl (dap = 2,9-anisylphenanthroline) (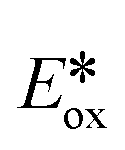 = −1.43 V), a well-studied copper-based photoreductant,55 complex 2 was found to be stronger photoreductants (
= −1.43 V), a well-studied copper-based photoreductant,55 complex 2 was found to be stronger photoreductants ( = −1.46 V).
= −1.46 V).
In the literature, the redox potentials for 3 were likewise measured in degassed acetonitrile (Eox = 1.20 V; Ered = −1.73 V;  = −1.44 V;
= −1.44 V;  = 0.91 V);56 the discrepancy is primarily due to the small difference in the extrapolated Eopt (Eopt = 2.64 eV). On the other hand, these copper complexes also have the potential of being photooxidants. The excited state reduction potentials of 1–5 vary from 0.78 V (for 5) to 1.16 V (for 1). The
= 0.91 V);56 the discrepancy is primarily due to the small difference in the extrapolated Eopt (Eopt = 2.64 eV). On the other hand, these copper complexes also have the potential of being photooxidants. The excited state reduction potentials of 1–5 vary from 0.78 V (for 5) to 1.16 V (for 1). The  for 1 is close to that of workhorse photocatalyst [Ir(dF(CF3)ppy)2(dtBubpy)]+ (
for 1 is close to that of workhorse photocatalyst [Ir(dF(CF3)ppy)2(dtBubpy)]+ (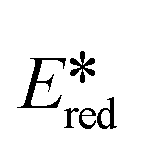 = 1.21 V),57 while all six complexes have larger
= 1.21 V),57 while all six complexes have larger 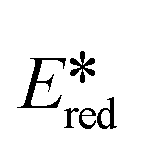 than [Ru(bpy)3]2+ (
than [Ru(bpy)3]2+ (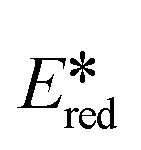 = 0.77 V)54 and all complexes are more photooxidizing than the commonly used fluorescent organic photocatalyst Eosin Y (
= 0.77 V)54 and all complexes are more photooxidizing than the commonly used fluorescent organic photocatalyst Eosin Y (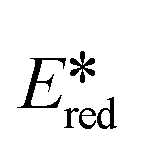 = 0.79 V).58
= 0.79 V).58
UV-visible absorption
The absorption spectra for 1–5 are shown in Fig. 6 and the data compiled in Table 2.48 The absorption spectra are dominated by two features: an intense (ε ≈ 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) high energy (250–300 nm), ligand-centred (1LC) π–π* transition and a low energy (350–450 nm), hypochromic (ε ≈ 2000 M−1 cm−1) mixed charge-transfer transition. These absorption profiles are in agreement with literature reports of [Cu(dmphen)(P^P)]+ complexes1,3,59 where the high-energy bands are usually assigned as LC transitions of both the dmphen and P^P ligands, while the broad low-energy bands are assigned as mixed charge transfer states containing
000 M−1 cm−1) high energy (250–300 nm), ligand-centred (1LC) π–π* transition and a low energy (350–450 nm), hypochromic (ε ≈ 2000 M−1 cm−1) mixed charge-transfer transition. These absorption profiles are in agreement with literature reports of [Cu(dmphen)(P^P)]+ complexes1,3,59 where the high-energy bands are usually assigned as LC transitions of both the dmphen and P^P ligands, while the broad low-energy bands are assigned as mixed charge transfer states containing 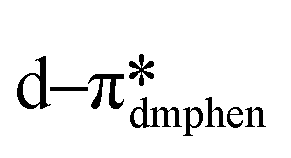 metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) from the P^P ligand to dmphen. The experimental spectra are consistent with the predicted electronic transitions from TD-DFT (Table S4†).
metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) from the P^P ligand to dmphen. The experimental spectra are consistent with the predicted electronic transitions from TD-DFT (Table S4†).
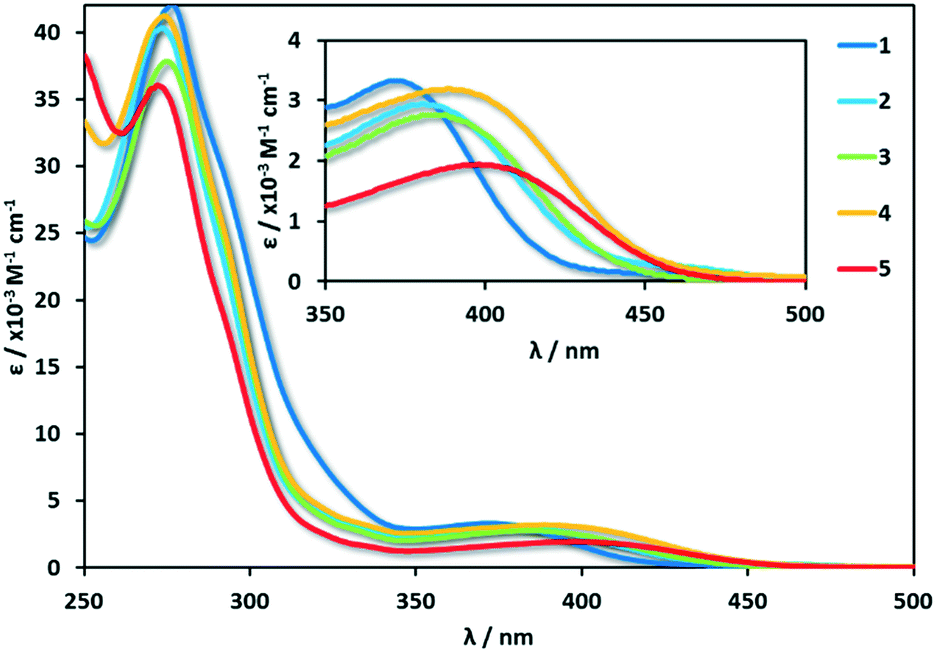 | ||
| Fig. 6 Absorption spectra of 1–5. Measurements were carried out at 298 K in degassed HPLC grade DCM. | ||
| Complexa | λ/nm (ε × 10−3/M−1 cm−1)b | Calculated λ/nm (f)c | Contributionsc | Naturec | |
|---|---|---|---|---|---|
| LC | CT | ||||
| a [BF4] was used as counteranion. b In HPLC grade DCM at 298 K. c TD-DFT calculated HOMO–LUMO transitions (oscillator strength), PBE0/ECP1/PCM(DCM) level. | |||||
| 1 | 278 (41) | 376 (3.3) | 375 (0.084) | HOMO–LUMO (97%) | MLCT&ILCT |
| 2 | 275 (40) | 385 (2.8) | 390 (0.106) | HOMO–LUMO (93%) | MLCT&ILCT |
| 3 | 276 (38) | 393 (2.7) | 391 (0.110) | HOMO–LUMO (98%) | MLCT&ILCT |
| 4 | 276 (41) | 392 (3.2) | 398 (0.080) | HOMO–LUMO (96%) | MLCT&ILCT |
| 5 | 274 (36) | 403 (1.9) | 413 (0.087) | HOMO–LUMO (95%) | MLCT&ILCT |
Across the series, there is no discernible trend in the energies of the π–π* bands. This is understandable as the substituents on the P^P ligands affect to a similar extent both occupied and unoccupied orbitals on the bis(phosphine) and thus the energy of this transition is not expected to change significantly. By contrast, the energy of the CT bands and the E0,0 transition (Eopt) extracted from the intersection points of the normalized emission and absorption spectra, both correlate strongly with the Hammett σp parameter of the substituent on the bis(phosphine) (Fig. 7).60 This is due to the HOMO-directing influence of the P^P, which indirectly modulates the Cu-based HOMO level; the LUMO is mainly localized on the dmphen ligand. When the P^P ligand is substituted with electron-withdrawing groups both the Eopt and the 1CT band move to higher energy while when the P^P ligand contains electron-donating groups, these transitions move to lower energy. This trend is reflected in the TD-DFT calculated HOMO–LUMO transitions, though the computations systematically predict slightly lower energies for the 1CT transition. The extinction coefficients of the CT states for 1–5 are also consistent with calculated oscillator strengths, f, where complex 5 (ε = 1900 @ 403 nm, f = 0.132 @ 434 nm) shows the weakest CT states.
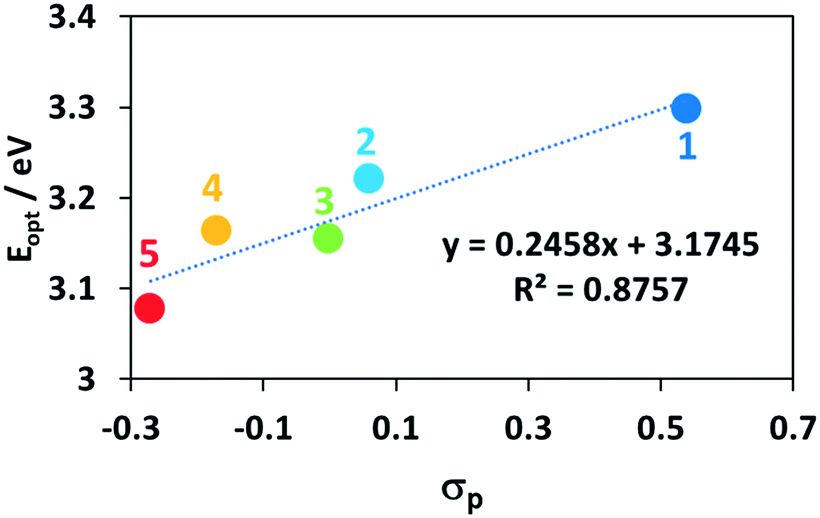 | ||
| Fig. 7 Hammett plot of the energies of CT states for 1–5. | ||
Solution-state photophysics
The photophysical properties of 1–5 were studied in degassed DCM at 298 K.48 This solvent was chosen as solution-state photophysics is most commonly reported in DCM for this class of complexes.61 Their emission profiles are shown in Fig. 8, and the photophysical data are given in Table 3. All five complexes show broad and unstructured CT emission profiles and luminesce over a narrow range in the green-yellow with λPL spanning from 520 for 1 to 558 nm for 5. The emission energies strongly fit a Hammett trend (Fig. S9a†), indicating that the excited state is influenced in a similar manner to the ground state and demonstrating that modulation of the electronics of the P^P ligand can tune the emission energy of the copper complex in a controlled manner. | ||
| Fig. 8 Normalised emission spectra for 1–5 in deaerated DCM solution. λexc = 360 nm. | ||
| Complexb | λ PL /nm | λ PL /nm | τ PL /μs | τ PL , /μs | Φ PL /% | k r × 10−4/s−1 | k nr × 10−4/s−1 |
|---|---|---|---|---|---|---|---|
| a Measurements in deaerated DCM at 298 K. b [BF4] was used as counteranion. c λ exc = 360 nm. d Quinine sulfate used as the reference (ΦPL = 54.6% in 0.5 M H2SO4 at 298 K).65 e λ exc = 369 nm. f Measurements in deaerated MeCN. | |||||||
| 1 | 520 | 535 | 3.09 | 0.03 | 8 | 2.60 | 29.77 |
| 2 | 537 | 560 | 3.37 | 0.10 | 12 | 3.50 | 29.67 |
| 3 | 550 | 580 | 3.00 | 0.17 | 13 | 4.17 | 33.33 |
| 4 | 550 | 580 | 6.17 | 0.56 | 9 | 1.51 | 16.21 |
| 5 | 558 | 585 | 6.83 | 0.98 | 15 | 2.12 | 14.64 |
A second order Jahn–Teller distortion is commonly observed for copper(I) complexes, which as a result are typically not very emissive in solution.62,63 For example, [Cu(pypz)(DPEphos)]BF4 (pypz = 1-(2-pyridyl)pyrazole) is very emissive in the solid state (ΦPL = 56% for the crystalline powder) yet is almost non-emissive in DCM (ΦPL = 2.1%). Thanks to the large degree of steric hindrance about the metal centre conferred by both the dmphen and the bulky P^P ligands, photoluminescence quantum yields, ΦPL, of ca. 10% were observed for 1–5. The excited state lifetimes, τPL, of 1–5 range from 3.00 to 6.83 μs, indicating that the triplet excited state is involved in the emission. Notably, though complex 3 has been employed as a photocatalyst in a number of papers,26,30,64 its solution-state photophysics data has as of yet not been reported. Interestingly, the values of both the radiative and non-radiative decay rate constants (kr, and knr) across the series are smaller for complexes bearing electron-donating substituents on the P^P ligand (4 and 5).
In the context of photoredox catalysis, an analysis of the excited state redox behaviour of 1–5 reveals the attractive potential of 5 as it has the highest excited state oxidation potential, highest solution state ΦPL and longest τPL.
The solid state photophysics and a discussion of the TADF properties can be found in ESI.†
Photocatalysis
In order to assess the impact of the electronic properties of the P^P ligands on the photocatalytic activity of copper-based photocatalysts, these were tested in the model aerobic photocatalyzed cross-dehydrogenative coupling (CDC) aza-Henry reaction between N-phenyl-tetrahydroisoquinoline (6) and nitromethane (Fig. 9). This model reaction can be easily monitored by kinetic profiling enabling comparison of the reactivity among our series of structurally related photocatalysts (see below). | ||
| Fig. 9 General scheme for the aerobic photocatalyzed cross-dehydrogenative coupling (CDC) aza-Henry reaction between N-phenyl-tetrahydroisoquinoline (7) and nitromethane catalyzed by [Cu(dmphen)(P^P)]PF6 complexes 1–5. | ||
This reaction has previously been studied using a wide range of different photocatalysts, including homogeneous iridium(III),41 and chromium(III)66 complexes as well as heterogeneous metal oxides (TiO2, ZnO).67 Relevant to the current study, Che et al. used zwitterionic copper(I) complexes bearing phenanthroline-based N^N and nido-carborane-diphosphine ligands.42
Photocatalytic reactions with both pre-made PC 3 and in situ generated PC 3 were performed and the use of both showed similar yields of 78% and 85%, respectively (Table S6,† entries 1 and 2). Blank experiments showed that the substrate 6 itself can be directly photoactivated providing access to the coupling product 7 in a less efficient background reaction (Table S6,† entry 9), while control experiments demonstrated that light was an essential component and that the reaction could not be thermally activated (Table S6,† entries 4 and 5). These results are consistent with experiments previously reported by Stephenson et al.41 and Gschwind et al.40 The concentration of oxygen greatly influences the reaction efficiency. Without O2 bubbling, only a modest 45% yield was achieved (Table S6,† entry 3). The absence of [Cu(NCMe)4][PF6], neocuproine or xantphos led to somewhat lower product yield after 18 hours of reaction (Table S6,† entries 6–8). In order to obtain more detailed insight on reaction progression, reaction progress kinetic analysis was followed by gas chromatography (GC) and yields as a function of time are shown in Fig. 10.
 | ||
| Fig. 10 Reaction progress (yield of 7) of the aerobic photocatalyzed aza-Henry reaction between 6 and nitromethane followed by GC (λexc = 450), a) the first 108 min; b) the entire 960 min. Data shown represent the average and standard deviations of three reactions. | ||
For all of the five tested photocatalysts (under the conditions outlined in Table S6,† entry 1) high yields (68–94%) were observed after 16 hours. During the first two hours of the reaction, rapid conversion of the starting material to the product was observed for all photocatalysts (Fig. 10a). The photocatalysts with more electron-donating substituents on the P^P ligand showed higher conversion rates, and generally followed a Hammett relationship (Fig. 11a), except for complex 5, which showed a higher conversion rate than complex 1 and 2, but lower than 3 and 4. These findings point to a different or additional mechanistic pathway operational when PC 5 is employed. At the late stages of the reaction (2–16 hours), photodegradation of the product was observed (Fig. 10b), with greater degradation observed for photocatalysts with more electron-donating substituents (Fig. 11b); again the behaviour of PC 5 is an outlier, which showed a larger degradation of 7 than with PC 1 and 2, but smaller than with 3 and 4. The degradation of the product was especially apparent for PC 4, where the GC yield was 89% yield after 108 min but this decreased to 68% after 16 hours.
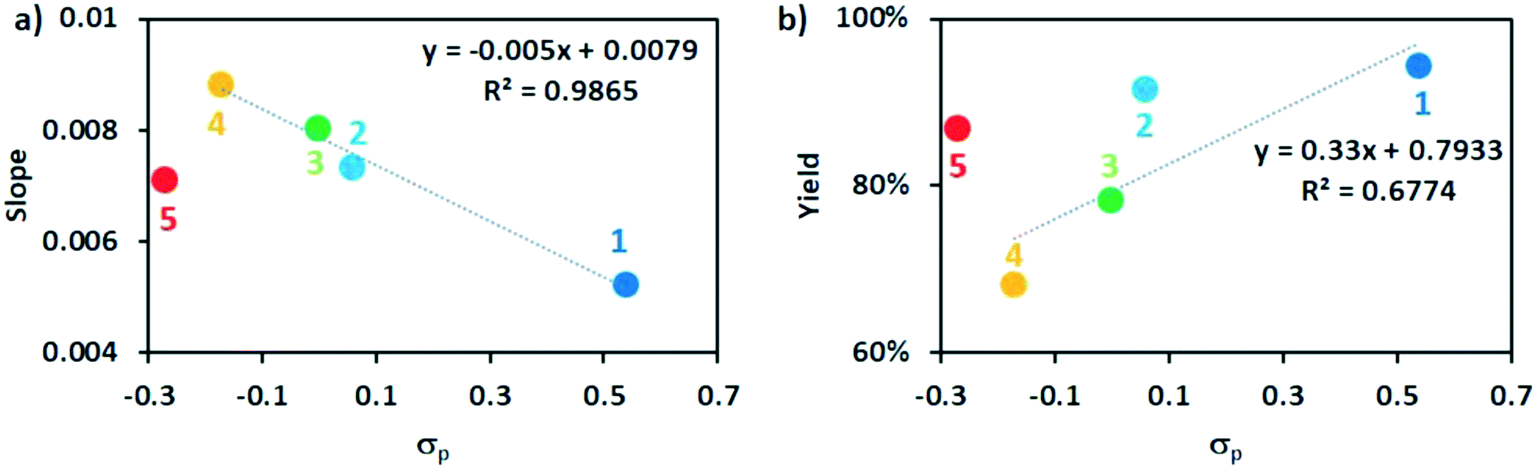 | ||
| Fig. 11 Hammett plots (complex 5 was excluded from the linear fitting, see text) of a) slope of kinetic profile at the early stage of reactions (0–2 hours); b) final yield of the reaction at 16 hours. | ||
Based on the reaction profiles obtained from the kinetic studies of the photocatalyzed aza-Henry reaction, we performed additional mechanistic studies in order to assess the nature of the unusual behavior of photocatalyst 5. Gschwind et al. had previously studied the mechanism of the photocatalyzed aza-Henry reaction of N-aryltetrahydroisoquinolines, and proposed three possible pathways with their corresponding intermediates.40 However, the role of the photocatalyst, the excited state kinetics during the reaction and the impact of the photocatalysts' optoelectronic properties on the reaction activity were not explored. Che et al. had proposed that product formation proceeded via a reductive quenching cycle based on the EPR detection of O2˙− using nitrone spin traps.42 However, this observation cannot exclude the possibility of an oxidative quenching pathway since O2 can also be directly reduced by the excited copper(I) complexes (Ered = 0.99 V <  of PCs 1–5 or Ered of PCs 1–5)68 while 6 can likewise be oxidized by the oxidized copper(II) species (Eox > 1.1 V for PCs 1–5). More recently, Detty et al. reported that singlet oxygen can activate the quinoline substrate 6 while using a chalcogenorosamine photocatalyst.69 Complicating the analysis even further, oxygen can be activated to singlet oxygen by photo-induced energy transfer (PET) from the Cu(I) photocatalysts, and 1O2 can then oxidize 6.43 Thus, there are three possible operative quenching pathways of the photocatalyst in this reaction: reductive (A) or oxidative (B) quenching via single-electron transfer and oxygen photosensitization (C) via energy transfer (Fig. 12).
of PCs 1–5 or Ered of PCs 1–5)68 while 6 can likewise be oxidized by the oxidized copper(II) species (Eox > 1.1 V for PCs 1–5). More recently, Detty et al. reported that singlet oxygen can activate the quinoline substrate 6 while using a chalcogenorosamine photocatalyst.69 Complicating the analysis even further, oxygen can be activated to singlet oxygen by photo-induced energy transfer (PET) from the Cu(I) photocatalysts, and 1O2 can then oxidize 6.43 Thus, there are three possible operative quenching pathways of the photocatalyst in this reaction: reductive (A) or oxidative (B) quenching via single-electron transfer and oxygen photosensitization (C) via energy transfer (Fig. 12).
 | ||
| Fig. 12 Proposed mechanism for the aerobic photocatalyzed cross-dehydrogenative coupling between 6 and nitromethane performed by [Cu(dmphen)(P^P)]+ complexes, [Cu]+. Path A: quenched via reductive quenching pathway; path B: quenched via oxidative quenching pathway; path C: quenched by oxygen via energy transfer. | ||
In order to evaluate the relative contributions of each one of the possible mechanisms, stoichiometric solutions in nitromethane of each photocatalyst 1–5 were irradiated at 420 nm using an LED array under air and monitored by UV-vis spectroscopy (Fig. S17†). For complexes 1–3, a new absorption band at ∼455 nm was observed, which is consistent with the MLCT absorption bands of the corresponding homoleptic copper species, as previously observed via spectroelectrochemistry studies after electrochemical oxidation of 3.56 For complexes 4 and 5, another band evolved over time at ca. 420 nm, which is reminiscent to the absorption spectrum of an analogous reduced copper complex reported by Dietzek and co-workers.70 However, these small changes in the UV spectra do not provide irrefutable evidence of a contribution from a reductive quenching pathway.
Furthermore, in the photostability studies (Fig. S4†), we observed the appearance of additional absorption bands for complexes 1 and 2. Hence, we cannot exclude direct photodecomposition of the complex. Therefore, we next investigated the photocatalyst reactivity with oxygen and 6 by monitoring changes in the UV-visible absorption spectra using MeCN solutions of complexes 3 and 4 (Fig. S18†). The results show that 3 and 4 likely cannot be directly oxidized by oxygen (Fig. S19a and d†), suggesting that the reaction is unlikely to proceed via the oxidative quenching pathway (B), while the more electron-rich photoredox catalyst 5 can be directly oxidized by oxygen (Fig. S18e†). On the other hand, with 6, new absorption bands were observed, which are consistent with reduced 3, based on the cross comparison with the spectroelectrochemistry (Fig. S18b†).70
Noting that 6 is itself photoactive under these conditions, we next investigated NEt3 as a non-chromophore surrogate of 6, which possesses a similar Eox to 6 (Eox = 0.83 for NEt3,71Ered = 0.82 V for 6). When NEt3 is used as the quencher, only small absorption changes were found at ∼455 nm (Fig. S18c†). Thus, for the most electron-rich photoredox catalyst, 5, all three paths contribute together to yield the radical cation 8, while for the other analogues, only path A and path C remain possible.
We next proceeded to quantify the magnitude of the quenching by performing Stern–Volmer experiments from which we were able to obtain quenching rates of oxygen and triethylamine (the latter under a nitrogen atmosphere, Fig. S17 and S18†). Quenching rates could not be ascertained using 6 as the quencher due to the overlap in the absorption of 6 and 3 at the excitation wavelength. The pseudo first-order quenching rates as well as quenching quantum yields are summarized in Table 4 (see also ESI†).
| k q/s−1 | Φ | |||||
|---|---|---|---|---|---|---|
| O2a | NEt3b | k 0 | O2a | NEt3b | k 0 | |
| a Bubbled with pure O2 gas, O2 atmosphere (1 bar), same as the reaction conditions. b 66 equiv. (1.5 mol% catalyst loading) quenching rates calculated from the Stern–Volmer quenching experiment results. c k 0 represent the rate of radiative and non-radiative decay, which shows the rate of the intrinsic deactivation of the excited state of copper(I) complexes. | ||||||
| 1 | 1.57 × 108 | 0.46 × 108 | 0.32 × 108 | 0.67 | 0.20 | 0.13 |
| 2 | 1.21 × 108 | 0.18 × 108 | 0.10 × 108 | 0.81 | 0.12 | 0.06 |
| 3 | 1.10 × 108 | 0.06 × 108 | 0.06 × 108 | 0.90 | 0.05 | 0.05 |
| 4 | 0.66 × 108 | 0.06 × 108 | 0.02 × 108 | 0.89 | 0.08 | 0.02 |
| 5 | 0.74 × 108 | 0.03 × 108 | 0.01 × 108 | 0.95 | 0.04 | 0.01 |
Among the five photocatalysts, 1 shows the highest oxygen quenching rate of 0.157 ns−1, which is more than two times faster than that of 4, which has the lowest kq of 0.066 ns−1. There is a correlation between the oxygen quenching rate shown and the energy of the T1 state of the complexes. Thus, 1 and 2, with electron-withdrawing substituents showed the highest quenching rates. Due to their higher excited state reduction potentials, these two complexes also showed the largest quenching rates with triethylamine (0.046 ns−1 for 1 and 0.018 ns−1 for 2). On the other hand, due to their longer τPL, the rates of intrinsic deactivation of the excited states of PCs 4 and 5 are much slower than those of PCs 1 and 2. For all complexes, the quenching rate by oxygen is significantly faster than that by NEt3, suggesting that path C dominates over path A. As a result, the sum of ΦO2 and ΦNEt3 for 3–5 (0.95–0.99) are higher than those of 1 and 2 (0.87 for 1 and 0.93 for 2), thereby pointing to fewer photons lost via radiative or other non-productive non-radiative decay pathways. The quenching kinetic studies showed that 6 can be oxidized to 8 by both 1O2 (path C) and reductive quenching of the photocatalyst (path A), while the reaction is likely to proceed via an oxidative quenching mechanism (path B) only when 5 is used. In order to assess the formation of singlet oxygen in the reaction via energy transfer (ET) from the photoexcited sensitizer, we performed scavenging experiments using TEMP (2,2,6,6-tetramethylpiperidine). This compound is commonly employed as a singlet oxygen probe, which upon oxidation with 1O2 produces the stable radical TEMPO that can be easily detected by EPR.43 After some minutes of irradiation, we could detect the characteristic EPR signal of TEMPO as it accumulated in solution (see Fig. S5†). To exclude an electron transfer from the photosensitizer to TEMP and a subsequent radical pathway for TEMPO formation with 3O2 we also performed emission quenching experiments with TEMP in the absence of oxygen.72 No-to-negligible quenching was observed in the presence of a more than 200 fold excess of TEMP, thus proving the presence of a singlet oxygen sensitizing pathway (see Fig. S24†). The results are consistent with our proposed mechanism for photocatalysts 1–4 that path C dominates, but that path A must also contribute to the overall reaction yield.
After the formation of the radical cation 8, hydrogen atom abstraction from the benzylic position of 8 by O2˙− forms radical 9 along with the hydroperoxyl radical HOO˙. Reaction of these two species, either by direct recombination or single electron oxidation, leads to an equilibrating mixture of hydroperoxide 10 and iminium ion 11.73–75 Finally, trapping of 11 with nitromethane leads to the formation of the CDC product 7.
Complicating the analysis, we observed significant photodegradation of the product commencing after the first 2 hours of the reaction, where photosensitizers containing more electron-donating ligands caused more significant degradation. We therefore hypothesized that the product can be reduced by the copper complexes. UV-vis absorption spectroscopy of 7 in MeCN (Fig. S23†), showed a slightly red-shifted absorption spectrum compared to that of 6 with an onset of 505 nm. The oxidation and reduction potentials of 7 are 1.02 V and −1.60 V, respectively, while those of 6 are 0.89 V and −2.78 V, respectively (Fig. S21†). Thus, photoactive product 7 can potentially both oxidize and reduce the excited photocatalyst. Considering the trend observed in the kinetic reaction profile, reductive degradation seems to be the more likely process.
From this study, it becomes apparent that in order to improve the photocatalytic activity, photocatalysts with higher E(0,0) and longer τPL would promote the energy transfer pathway, and would therefore lead to a higher initial conversion rate at early reaction times. These features could be achieved by employing P^P ligands with more σ-donating substituents. However, special care should be taken to limit the σ-donating power of the P^P ligand as exemplified by the reduced rate of conversion with complex 5, which behaves differently from the other photocatalysts studied. The design of a photocatalyst with P^P ligands that are even more σ-donating than that found in 5 would likewise turn on the oxidative quenching pathway, but this may not necessarily lead to faster rates or higher conversions. On the other hand, once all of the starting material is consumed, there would be nothing left to turn the oxidative quenching cycle over and thus, 5 eventually photodecomposed in the presence of oxygen and could therefore not participate in the photodegradation of the product 7.
Conclusions
We studied a family of five structurally related heteroleptic copper(I) complexes of the form of [Cu(N^N)(P^P)]+ bearing a 2,9-dimethyl-1,10-phenanthroline diimine (N^N) ligand and a series of electronically tuned xantphos (P^P) ligands. The complexes showed green to yellow emission in DCM with moderate ΦPLs of 8 to 15% and microsecond emission lifetimes. In the spin-coated neat films, complexes 2–5 showed bright yellowish green emission with longer τPLs ranging from 11–33 μs while complex 1 is very poorly emissive at solid states (ΦPL < 1%). The MLCT nature of the emission is reflected in the very small ΔEST values, which ranged from 6–43 meV. An efficient reverse intersystem crossing from T1 to S1 state as well as thermally activated delayed fluorescence (TADF) was observed for complexes 2 to 5. The electrochemical and photophysical studies showed strong correlations between the para-Hammett parameters and the optoelectronic properties of Cu(I) complexes. The photostability of these species is dictated also by the electronics of the P^P ligand that contributes to the strength of the π-back bonding from the metal ion to the dmphen ligand. Indeed, use of the electron-rich photocatalyst 4 resulted in the highest conversion rate over the first two hours. When the reactions progressed over longer time courses, a photodegradation of the product was observed, itself most prominent with electron-rich complexes were used, and the more electron-poor photocatalyst 1 resulted in the highest final yield of 7 (94%). In order to interpret the correlation between the photocatalytic activity and optoelectronic properties, we interrogated the mechanism via a series of quenching studies. We found that the reaction follows mainly two pathways: singlet oxygen sensitization and reductive quenching. Notably, when the most electron-rich catalyst 5 was used an additional oxidative quenching process became accessible and led to a lower conversion rate at early reaction times. However, we also discovered that electron-poor photocatalysts, though generating product faster, also contributed more readily to photodegradation of the product, resulting in lower yields at long reaction times. Therefore, in order to improve the photocatalytic activity, photocatalysts with higher E(0,0) and longer τPL should be selected. While higher and E(0,0) could be obtained by destabilizing the N^N-based LUMO or stabilizing the metal-based HOMO, longer τPL could be obtained through the use of electron-rich P^P ligands. A caveat is that employing P^P ligands that are sufficiently electron-rich opens up an oxidative quenching pathway that correlates with reduced reaction rates. In short, the electron-donating properties of the P^P ligands can be systematically changed without altering the structural properties. Thus, we could tailor the photoredox properties of the catalysts and modulate their activity. Remarkably, the preferred mechanistic pathway can shift between reductive quenching, energy transfer and oxidative quenching depending on the inherent redox potential of the photo-excited catalysts. Importantly, when the product of a photocatalysis reaction is itself photoactive, it is essential to probe its reactivity under the photocatalysis conditions. As exemplified here, the combination of photocatalyst and product contributed to the latter's undesired decomposition.
and E(0,0) could be obtained by destabilizing the N^N-based LUMO or stabilizing the metal-based HOMO, longer τPL could be obtained through the use of electron-rich P^P ligands. A caveat is that employing P^P ligands that are sufficiently electron-rich opens up an oxidative quenching pathway that correlates with reduced reaction rates. In short, the electron-donating properties of the P^P ligands can be systematically changed without altering the structural properties. Thus, we could tailor the photoredox properties of the catalysts and modulate their activity. Remarkably, the preferred mechanistic pathway can shift between reductive quenching, energy transfer and oxidative quenching depending on the inherent redox potential of the photo-excited catalysts. Importantly, when the product of a photocatalysis reaction is itself photoactive, it is essential to probe its reactivity under the photocatalysis conditions. As exemplified here, the combination of photocatalyst and product contributed to the latter's undesired decomposition.
Acronyms
| bpz | 2,2′-Bipyrazine |
| bpy | 2,2′-Bipyridine |
| SCE | Saturated calomel electrode |
| MeCN | Acetonitrile |
| dF(CF3)ppy | 2-(2,4-Difluorophenyl)-5-(trifluoromethyl)pyridine |
| dtBubpy | 4,4′-Di-tert-butyl-2,2′-bipyridine |
| ppy | 2-phenylpyridinato |
| XyBnta | N-(2-(1-Benzyl-1H-1,2,3-triazol-4-yl)phenyl)-2,6-dimethylaniline |
| dppb | 1,2-Bis(diphenylphosphanyl)benzene |
| THF | Tetrahydrofuran |
| Bath | 2,9-Dimethyl-4,7-diphenyl-1,10-phenanthroline |
| ThioPOP | 10,10′-(2,8-Dimethylphenoxathiine-4,6-diyl)bis(2,8-dimethyl-10H-phenoxaphosphinine) |
| PCET | Proton-coupled electron transfer |
| NMR | Nuclear magnetic resonance |
| DCM | Dichloromethane |
| dmphen | 2,9-Dimethyl-1,10-phenanthroline |
| DPEphos | (Oxybis(2,1-phenylene))bis(diphenylphosphane) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Li thanks the Prof. & Mrs Purdie Bequests Scholarship and AstraZeneca for a PhD Studentship. We acknowledge P. Forero for the initial synthesis of the BF4 salts of some of the complexes.Notes and references
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- The terms “photocatalyst” and “photosensitizer” will be considered as equivalents and henceforth used exchangeablythroughout the manuscript. Photochemical sensitization and photocatalysis are closely related processes with very similar definitions, their preferred use depending on the scientific community using them. See for reference: C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046–12055 CrossRef CAS.
- H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517–5520 CrossRef CAS.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS.
- B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC.
- G. F. Manbeck, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2011, 50, 3431–3441 CrossRef CAS.
- E. Mejia, S.-P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A.-E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chem. – Eur. J., 2013, 19, 15972–15978 CrossRef CAS.
- M. Karnahl, E. Mejia, N. Rockstroh, S. Tschierlei, S.-P. Luo, K. Grabow, A. Kruth, V. Brueser, H. Junge, S. Lochbrunner and M. Beller, ChemCatChem, 2014, 6, 82–86 CrossRef CAS.
- S.-P. Luo, E. Mejia, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419–423 CrossRef CAS.
- A. C. Hernandez-Perez and S. K. Collins, Acc. Chem. Res., 2016, 49, 1557–1565 CrossRef CAS.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039–2058 CrossRef CAS.
- M. M. Cetin, R. T. Hodson, C. R. Hart, D. B. Cordes, M. Findlater, D. J. Casadonte Jr, A. F. Cozzolino and M. F. Mayer, Dalton Trans., 2017, 46, 6553–6569 RSC.
- M. D. Weber, M. Viciano-Chumillas, D. Armentano, J. Cano and R. D. Costa, Dalton Trans., 2017, 46, 6312–6323 RSC.
- H. Junge, N. Rockstroh, S. Fischer, A. Brückner, R. Ludwig, S. Lochbrunner, O. Kühn and M. Beller, Inorganics, 2017, 5, 14 CrossRef.
- C. Minozzi, A. Caron, J.-C. Grenier-Petel, J. Santandrea and S. K. Collins, Angew. Chem., Int. Ed., 2018, 57, 5477–5481 CrossRef CAS.
- F. Brunner, A. Babaei, A. Pertegás, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, Dalton Trans., 2019, 48, 446–460 RSC.
- N. I. Rinehart, A. J. Kendall and D. R. Tyler, Organometallics, 2018, 37, 182–190 CrossRef CAS.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S.-P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2016, 22, 1233–1238 CrossRef CAS.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brueckner, ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
- Y.-Y. Sun, H. Wang, N.-Y. Chen, A. J. J. Lennox, A. Friedrich, L.-M. Xia, S. Lochbrunner, H. Junge, M. Beller, S. Zhou and S.-P. Luo, ChemCatChem, 2016, 8, 2340–2344 CrossRef CAS.
- A. Rosas-Hernandez, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- A. C. Hernandez-Perez and S. K. Collins, Angew. Chem., Int. Ed., 2013, 52, 12696–12700 CrossRef CAS.
- A. C. Hernandez-Perez, A. Caron and S. K. Collins, Chem. – Eur. J., 2015, 21, 16673–16678 CrossRef CAS.
- A. C. Hernandez-Perez, A. Vlassova and S. K. Collins, Org. Lett., 2012, 14, 2988–2991 CrossRef CAS.
- P. Xiao, F. Dumur, J. Zhang, J. P. Fouassier, D. Gigmes and J. Lalevee, Macromolecules, 2014, 47, 3837–3844 CrossRef CAS.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079–14085 CrossRef CAS.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC.
- G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- J. A. Gillespie, D. L. Dodds and P. C. J. Kamer, Dalton Trans., 2010, 39, 2751–2764 RSC.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- L. A. Van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. Van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616–11626 CrossRef CAS.
- W. Goertz, W. Keim, D. Vogt, U. Englert, M. D. K. Boele, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1998, 2981–2988 RSC.
- M. Kranenburg, P. C. J. Kamer, P. W. N. M. van Leeuwen, D. Vogt and W. Keim, J. Chem. Soc., Chem. Commun., 1995, 2177–2178, 10.1039/c39950002177.
- H. Bartling, A. Eisenhofer, B. Koenig and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860–11871 CrossRef CAS.
- A. G. Condie, J. C. Gonzalez-Gomez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS.
- B. Wang, D. P. Shelar, X.-Z. Han, T.-T. Li, X. Guan, W. Lu, K. Liu, Y. Chen, W.-F. Fu and C.-M. Che, Chem. – Eur. J., 2015, 21, 1184–1190 CrossRef CAS.
- X.-S. Ke, Y. Ning, J. Tang, J.-Y. Hu, H.-Y. Yin, G.-X. Wang, Z.-S. Yang, J. Jie, K. Liu, Z.-S. Meng, Z. Zhang, H. Su, C. Shu and J.-L. Zhang, Chem. – Eur. J., 2016, 22, 9676–9686 CrossRef CAS.
- L. Shaw, D. M. U. K. Somisara, R. C. How, N. J. Westwood, P. C. A. Bruijnincx, B. M. Weckhuysen and P. C. J. Kamer, Catal. Sci. Technol., 2017, 7, 619–626 RSC.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J.-J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J.-F. Nierengarten, Inorg. Chem., 2013, 52, 12140–12151 CrossRef CAS.
- J. Yuasa, M. Dan and T. Kawai, Dalton Trans., 2013, 42, 16096–16101 RSC.
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS.
- In order to avoid trace impurities of the complex's precursors, [BF4] was used as counteranion for these studies.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- S. E. Braslavsky, A. U. Acuna, W. Adam, F. Amat, D. Armesto, T. D. Z. Atvars, A. Bard, E. Bill, L. O. Bjoern, C. Bohne, J. Bolton, R. Bonneau, H. Bouas-Laurent, A. M. Braun, R. Dale, K. Dill, D. Doepp, H. Duerr, M. A. Fox, T. Gandolfi, Z. R. Grabowski, A. Griesbeck, A. Kutateladze, M. Litter, J. Lorimer, J. Mattay, J. Michl, R. J. D. Miller, L. Moggi, S. Monti, S. Nonell, P. Ogilby, G. Olbrich, E. Oliveros, M. Olivucci, G. Orellana, V. Prokorenko, K. R. Naqvi, W. Rettig, A. Rizzi, R. A. Rossi, E. San Roman, F. Scandola, S. Schneider, E. W. Thulstrup, B. Valeur, J. Verhoeven, J. Warman, R. Weiss, J. Wirz and K. Zachariasse, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS.
- I. Andres-Tome, J. Fyson, F. Baiao Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669–8674 RSC.
- The reduction potential is, however, significantly different to those of 1–6 (e.g., for 4Ered = −1.74 V in MeCN), which, taking into account their reported ΔEredox (2.37 V) and EPL (2.34 eV) values, we believe is due to the mis-assignment of the reduction wave to that of an electrochemically produced by-product, a function of the poor electrochemical stability.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336–7340 CrossRef CAS.
- Y. Zhang, M. Heberle, M. Waechtler, M. Karnahl and B. Dietzek, RSC Adv., 2016, 6, 105801–105805 RSC.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, , Jr., G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- M. Neumann, S. Fueldner, B. Koenig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS.
- L. Qin, Q. Zhang, W. Sun, J. Wang, C. Lu, Y. Cheng and L. Wang, Dalton Trans., 2009, 9388–9391, 10.1039/b916782j.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2007, 129, 5248–5256 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS.
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem. – Eur. J., 2012, 18, 3478–3481 CrossRef CAS.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS.
- J. L. Clark, J. E. Hill, I. D. Rettig, J. J. Beres, R. Ziniuk, T. Y. Ohulchanskyy, T. M. McCormick and M. R. Detty, Organometallics, 2019, 38, 2431–2442 CrossRef CAS.
- Y. Zhang, P. Traber, L. Zedler, S. Kupfer, S. Graefe, M. Schulz, W. Frey, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2018, 20, 24843–24857 RSC.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- G. Nardi, I. Manet, S. Monti, M. A. Miranda and V. Lhiaubet-Vallet, Free Radical Biol. Med., 2014, 77, 64–70 CrossRef CAS.
- E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Fares and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106–8109 CrossRef CAS.
- E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317–5325 CrossRef CAS.
- M. O. Ratnikov and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 1549–1557 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1950442 and 1979865. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy01221a |
| This journal is © The Royal Society of Chemistry 2020 |