
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of the ligand and activator in selective Cr–PNP ethene tri- and tetramerization catalysts – a spectroscopic study†
Bas
Venderbosch
a,
Lukas A.
Wolzak
a,
Jean-Pierre H.
Oudsen
a,
Bas
de Bruin
 b,
Ties J.
Korstanje
a and
Moniek
Tromp
*c
b,
Ties J.
Korstanje
a and
Moniek
Tromp
*c
aSustainable Materials Characterization, Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
bHomogeneous, Supramolecular and Bio-Inspired Catalysis, Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cMaterials Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: Moniek.tromp@rug.nl
First published on 11th August 2020
Abstract
The reaction of the ethene tetramerization catalyst, ((C6H5)2P)2NiPrCrCl3(THF) (complex 1), and ethene trimerization catalyst, ((o-C6H4OMe)2P)2NMeCrCl3 (complex 2), with alkylaluminum reagents (AlMe3 and MMAO) was investigated using spectroscopic techniques (Cr K-edge XAS, X-band EPR and UV-vis) and catalytic studies. In all cases the majority of chromium was reduced to the divalent oxidation state and only a minor fraction of chromium was reduced further to the monovalent oxidation state. It is demonstrated that MMAO and the ligand (through a pendant ether donor) can facilitate ion pair formation for these divalent Cr complexes, providing insights into the role of the ligand and activator in the activation process. Via the use of dienes, we succeeded in characterizing a monocationic CrII alkene complex, providing evidence that catalysis could proceed via cationic CrII/CrIV intermediates. This is supported by DFT calculations, where it is shown that a mechanism proceeding via dicationic CrII/CrIV intermediates explains the observed product selectivity.
Introduction
Linear α-olefins (LAOs) are an important class of compounds that show application in the production of linear low-density polyethylene (C4–C8), lubricants (C10–C12), detergents (C10–C16), surfactants (C16–C18) and waxes (C20+).1 The majority of LAOs are produced using ethene oligomerization catalysts that operate via a Cossee–Arlman mechanism, and consequently yield a statistical distribution of LAOs.2 One well-known example of an ethene oligomerization catalyst is the nickel-based SHOP catalyst.3,4 Due to a growing demand for shorter LAOs (C4–C8), petrochemical companies have successfully developed organochromium complexes that are capable of forming 1-hexene or 1-octene in high selectivity.5 In these systems, the selectivity arises from metallacyclic intermediates that are involved in the catalytic cycle.6–9Bis(diarylphosphino)amine ligands can facilitate the chromium-catalyzed selective oligomerization of ethene. Mixing of the ligand, a trivalent chromium precursor and an excess of an alkylaluminum compound, leads to the formation of the active species. The use of bis(diarylphosphino)amine ligands in the chromium-catalyzed selective ethene oligomerization was first described in 2002 by Wass and coworkers.10 In their study, the most active ligand contained an ortho-methoxy within the backbone of the ligand (Scheme 1a). In 2004, Bollmann et al. prepared bis(diarylphosphino)amine ligands with reduced steric bulk (e.g.Scheme 1b).11 The ligands reported in this study were the first example of ligands that could facilitate the selective tetramerization of ethene.
 | ||
| Scheme 1 Depiction of ligands used for a) selective ethene trimerization and b) selective ethene tetramerization. | ||
The (electronic) structure of the active species in these selective oligomerization systems has been the subject of numerous spectroscopic studies. For example, Brückner and coworkers investigated the activation of the Cr(acac)3/((C6H5)2P)2NiPr/modified methylaluminoxane (MMAO) system using electron paramagnetic resonance (EPR) and X-ray absorption spectroscopy (XAS) and suggested the formation of the neutral ((C6H5)2P)2NiPrCrMe2 complex.12,13 In a more recent study, Brückner and coworkers also used different activators (e.g. AlMe3 and AlOct3) to study their influence on the activation process.14 Here, they found that the ligand and the alkyl groups of the activator compete with one another for coordination to the metal center. Findings similar to the ones reported by Brückner and coworkers were obtained in our group. In a freeze-quench XAS study, we observed the formation of ((C6H5)2P)2NiPrCrMeCl in the reaction between ((C6H5)2P)2NiPr)CrCl3(THF) and AlMe3.14,15 In another study, Bercaw and coworkers performed an EPR and UV-vis study of a CrCl3 complex containing the PNP ligand shown in Scheme 1a, functionalized with solubilizing groups.16 Upon reaction of the complex with MMAO, a variety of CrI, CrIII and EPR-silent complexes (dinuclear CrI, or CrII) were identified. The authors attempted to correlate the concentration of these complexes to catalytic activity. No clear correlation was found and the authors proposed that an unidentified minority complex is responsible for catalysis.
These spectroscopic studies have provided insights into the reaction between the activator and the chromium precursor. These studies have however not provided insights into: i) the nature of the active species (e.g. neutral or cationic) and ii) the role of the ligand in the formation of the active species.
The present study aims to shed more light on these two aspects of the selective ethene oligomerization catalysts. This was investigated by studying the reaction between chromium complexes containing the earlier discussed bis(diarylphosphino)amine ligands (Scheme 2) and AlMe3 or MMAO-12 (from now on denoted as MMAO). The catalytic activity was studied and the (electronic) structure of the activated metal center was investigated (using X-band EPR, Cr K-edge XAS and UV-vis). Additionally, the reactivity of the activated complexes towards dienes was studied. The results were interpreted by comparison to DFT-D3 calculations of plausible structures. Finally, plausible structures for the active species are proposed. These proposals are validated through DFT-D3 calculations of the corresponding mechanisms.
 | ||
| Scheme 2 Structure of the complexes employed in this study. Depicted is a) ((C6H5)2P)2NiPrCrCl3(THF) (complex 1) and (b) ((C6H4OMe)2P)2NMeCrCl3 (complex 2). | ||
Results and discussion
((C6H5)2P)2NiPr and ((o-C6H4OMe)2P)2NMe (Scheme 1) were prepared following earlier reported literature procedures.17,18 ((C6H5)2P)2NiPrCrCl3(THF) (complex 1, Scheme 2a) and ((o-C6H4OMe)2P)2NMeCrCl3 (complex 2, Scheme 2b) were also prepared following literatures procedures.7,15Catalytic performance of complex 1 and 2
The two complexes were tested for their activity in the selective oligomerization of ethene in toluene at ethene pressures of 1 bar and 45 bar (Table 1). AlMe3 (40 eq.) and MMAO (400 eq.) were employed as activators.| Entry | Complex | Activatorb | T (°C)c | [Cr]d (mM) | C2H4 (bar) | 1-C6 (mg) | 1-C8 (mg) | TOF 1-C6e,f (h−1) | TOF 1-C8e,g (h−1) | PEh (mg) |
|---|---|---|---|---|---|---|---|---|---|---|
| a General conditions: reaction performed in toluene. A volume of either 12 mL (for 1 bar of C2H4) or 150 mL (for 45 bar of C2H4) was employed. Reaction times of either 60 minutes (for 1 bar of C2H4) or 30 minutes (for 45 bar of C2H4) were employed. Mesitylene (∼3 mg mL−1) was employed as an internal standard for quantification. b Activation done with either AlMe3 (40 eq.) or MMAO (400 eq.). c RT. is used to denote room temperature. d The difference in Cr concentration at 1 bar and 45 bar might also influence catalytic performance. e TOF is defined as: (mole product)·(mole per Cr) h−1. f 1-C6 is used to denote 1-hexene. g 1-C8 is used to denote 1-octene. h PE is used to denote the amount of polyethylene isolated. | ||||||||||
| 1 | 1 | MMAO | RT. | 0.42 | 1 | None | None | None | None | 261.1 |
| 2 | 1 | MMAO | 50 | 0.42 | 1 | None | None | None | None | 4.1 |
| 3 | 1 | MMAO | 45 | 0.20 | 45 | 3212 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 969 969 |
2545 | 6515 | 224.6 |
| 4 | 1 | AlMe3 | RT. | 0.42 | 1 | 9 | None | 22 | None | None |
| 5 | 1 | AlMe3 | 50 | 0.42 | 1 | 3 | None | 7 | None | None |
| 6 | 1 | AlMe3 | 45 | 0.20 | 45 | 1955 | 109 | 1549 | 64.5 | 168.0 |
| 7 | 2 | MMAO | RT. | 0.42 | 1 | 93 | 0.36 | 221 | 0.6 | 23.2 |
| 8 | 2 | MMAO | 50 | 0.42 | 1 | 69 | 0.22 | 163 | 0.4 | None |
| 9 | 2 | MMAO | 45 | 0.20 | 45 | 5005 | 715 | 3965 | 245 | 787.2 |
| 10 | 2 | AlMe3 | RT. | 0.42 | 1 | 100 | 0.98 | 237 | 1.7 | None |
| 11 | 2 | AlMe3 | 50 | 0.42 | 1 | 8 | None | 20 | None | None |
| 12 | 2 | AlMe3 | 45 | 0.20 | 45 | 1270 | 178 | 1006 | 106 | None |
Pressure and the choice of activator have a large influence on the catalytic performance of complex 1. The system shows very little activity when catalytic experiments are performed at 1 bar of ethene pressure (entry 1, 2, 4 and 5). If the pressure is increased to 45 bars of ethene pressure (entry 3 and 6), the system does become active. The choice of activator is important for selectivity. If AlMe3 is employed as an activator (entry 6), the amount of 1-hexene exceeds the amount of 1-octene formed (1-C6/1-C8 = 24.0). If MMAO is used as an activator (entry 3), the amount of 1-octene formed exceeds the amount of 1-hexene formed (1-C6/1-C8 = 0.39). The effect of the activator on the selectivity of the catalyst was not investigated by Bollmann et al. in their original report.11 In a follow-up study by McGuinness et al. it was demonstrated that the choice of activator can affect the product selectivity: the use of activators that coordinate more strongly to the metal is detrimental for the 1-octene selectivity.19
In contrast to complex 1, complex 2 is already active for the formation of 1-hexene at room temperature and at 1 bar of ethene pressure (entry 7 and 10). Increasing the temperature from room temperature to 50 °C leads to a diminished performance of the catalytic system (entry 8 and 11). At these temperatures, catalyst deactivation likely plays a more prominent role. When the pressure is increased to 45 bars of ethene pressure, the activity of the catalytic system increases further (entry 9 and 12). Interestingly, when AlMe3 is used as an activator, no PE formation is observed.
X-band EPR investigation of the activation of complex 1 and 2
Next, we investigated the activation of complex 1 and 2 in toluene using spectroscopic techniques (UV-vis, Cr K-edge XAS and X-band EPR). For complex 1 and 2, we investigated the reaction with both AlMe3 (40 eq.) and MMAO (400 eq.). Complex 2 is insoluble in toluene, but the activated complex is soluble and could still be studied spectroscopically. However, the reaction between complex 2 and MMAO is slow and could not be followed by UV-vis or Cr K-edge EXAFS due to low concentration of Cr in solution. All spectroscopic results are reported in detail in the ESI† (section 2).X-band EPR spectra for complex 1 and 2 were acquired in 1:1 mixtures of dichloromethane and toluene at cryogenic temperatures (20 K). Resonances typical for high-spin CrIII complexes are observed (Fig. S4 and Fig. S29†).20
Activation of complex 1 and 2 in toluene with either AlMe3 or MMAO leads to disappearance of their respective resonances (Fig. S14, S24, S39 and S43†) and appearance of novel CrI resonances. For the reaction of complex 1 and 2 with AlMe3 we have attempted to determine the concentration of these CrI complexes by double integration of the observed resonances at room temperature (Fig. S13 and S38†). However, the concentration of these complexes was too low to be accurately determined (≪1% of total chromium content).
The CrI concentration for the reaction between complex 1 and MMAO was larger and two CrI complexes were found to contribute to the spectrum (Fig. S24†). The first component of the spectrum has an axial symmetry (gx,y = 1.979 and gz = 2.005) and a relative contribution of 60% to the spectrum. The second component has a rhombic symmetry (gx = 1.961, gy = 1.996 and gz = 2.032) and a relative contribution of 40% to the spectrum.
The symmetry and g-factors of the first component are in the expected range for bis(toluene)CrI complexes.12,13 Evidence for this assignment is found in an EPR spectrum acquired at room temperature (Fig. 1). Here, superhyperfine interaction with 10 neighboring hydrogen atoms is observed. For the reaction between complex 2 and MMAO, a similar EPR spectrum is acquired at room temperature and similar fitting parameters were obtained (Fig. S42†).
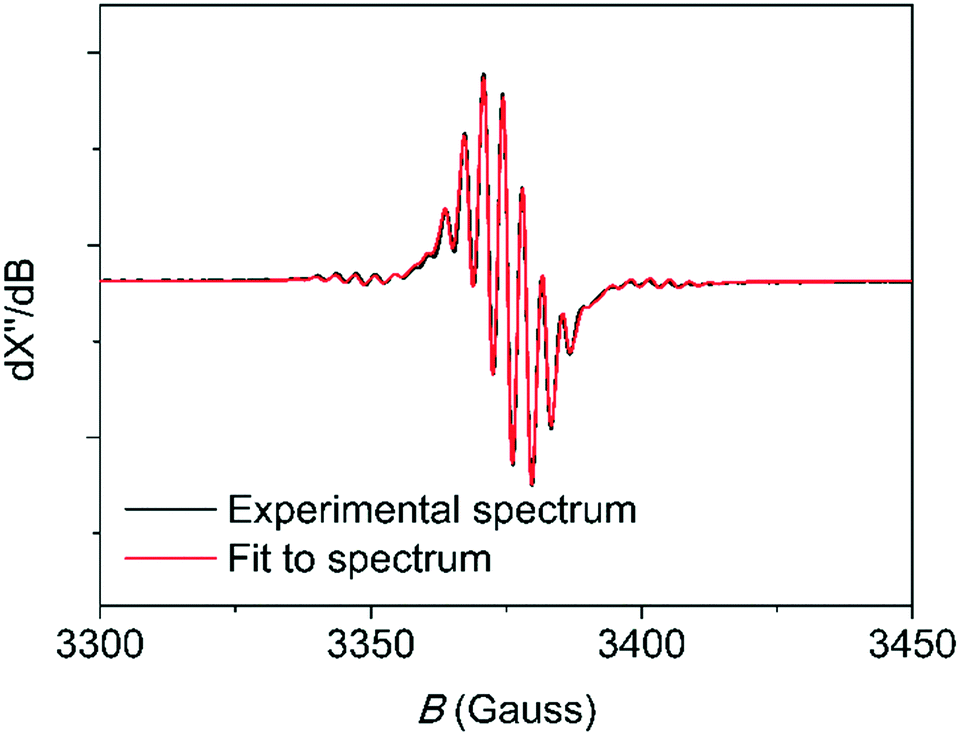 | ||
| Fig. 1 Room temperature X-band EPR spectrum (9.4 GHz) for the reaction of complex 1 with MMAO (400 eq.), acquired after 45 minutes, in toluene. The simulation was performed using giso = 1.987, by taking into account the (super)hyperfine interaction with 10 neighboring hydrogen atoms (AH = 9.95 MHz) and 1 chromium atom (ACr = 50.52 MHz) and by applying Lorentzian broadening (0.58 MHz). The spectrometer was operating at a frequency of 9.4 GHz. | ||
The origin of the second component is less clear. This complex is likely unobservable at room temperature (Fig. 1), as the spectrum could be simulated using a single component. Brückner and coworkers also observed a resonance with rhombic symmetry in a previous study.12 The g-factors reported by Brückner and coworkers (gx = 1.970, gy = 2.010 and gz = 2.040) are similar to the ones reported in the current study.12 They interpret this resonance to arise from a CrI complex ligated by a PNP ligand.
For the activation of complex 1 with MMAO, we quantified the amount of bis(toluene)CrI formed; this was found not to exceed 2% of the total chromium content after an hour (Fig. S23†). These CrI quantities are in stark contrast to the spectroscopic study performed on the Cr(acac)3/((C6H5)2P)2NiPr/MMAO system by Brückner and coworkers.13 In their study, quantitative formation of bis(toluene)CrI was observed in toluene after activation. Comparison of the results described in their study and the results obtained here show that the choice of chromium source can strongly affect the CrI concentration.
Cr K-edge XANES analysis of the activation of complex 1 and 2
The X-band EPR results thus show the formation of a large excess of EPR-silent complexes (dinuclear CrI or mononuclear CrII) after activation of complex 1 and 2. Only a minor amount of the complexes is present in the EPR-detectable monovalent oxidation state (<5% after 1 hour). To probe the (electronic) structure of these EPR-silent complexes, we have performed freeze-quench Cr K-edge XAS experiments.21 Activation experiments were performed in toluene and aliquots of these solutions were taken after 2 minutes, 10 minutes and 60 minutes and frozen in liquid nitrogen.Fig. 2a shows the Cr K-edge XANES region for the reaction between complex 1 and AlMe3 (40 eq.) or MMAO (400 eq.) after a reaction time of 10 minutes. An edge shift towards lower energies is observed when complex 1 (edge position: 5998.3 eV) is activated with either AlMe3 (edge position: 5996.5 eV) or MMAO (edge position: 5996.2 eV). In our previous study of the PNP system, we had also observed an edge shift towards lower energies and had shown that this was due to a reduction to the divalent oxidation state. Combined with the X-band EPR data, these results thus confirm that the metal is reduced in the reaction with AlMe3 and MMAO.
 | ||
| Fig. 2 Cr K-edge XANES region for the reaction between a) complex 1 and AlMe3 (40 eq.) or MMAO (400 eq.), and b) complex 2 and AlMe3 (40 eq.) or MMAO (400 eq.). Reactions were performed in toluene and frozen after 10 minutes. | ||
Notable differences are observed in the pre-edge region between the AlMe3- and MMAO-activated complex. Both complexes show two pre-edge features (5989.0 eV and 5991.1 eV). The intensity of the first pre-edge feature is roughly similar. For the MMAO-activated complex however, the second pre-edge feature gains significant intensity. These results suggest that the choice of activator affects the structure and/or geometry of the formed complex.
The Cr K-edge XANES data for the reaction between complex 1 and AlMe3 or MMAO, after reaction times of 2, 10 and 60 minutes (Fig. S6 and S16†) show only minor differences. This indicates that after the initial reduction has occurred no further changes to the (electronic) structure of the metal occur.
Fig. 2b shows the Cr K-edge XANES region for the reaction between complex 2 and AlMe3 (40 eq.) or MMAO (400 eq.) after a reaction time of 10 minutes. Again, an edge shift towards lower energies is observed when complex 2 (edge position: 6000.7 eV) is reacted with either AlMe3 (edge position: 5996.5 eV) or MMAO (edge position: 5996.5 eV). The AlMe3- and MMAO-activated complex have two pre-edge features (5989.1 eV and 5991.5 eV) with roughly similar intensity, hinting at a similar coordination environment and geometry. Similar to complex 1, only minor changes are observed in the Cr K-edge XANES region for the reaction of complex 2 and AlMe3 after reaction times of 2, 10 and 60 minutes (Fig. S31†).
Cr K-edge EXAFS analysis of the activation of complex 1 and 2
To probe the structure of the complexes formed, Cr K-edge EXAFS analysis was performed. The Cr K-edge EXAFS analysis results are summarized in Table 2 and an example of the obtained data quality is presented in Fig. 3. Cr K-edge EXAFS analysis for the reaction between complex 2 and MMAO is not reported due to earlier mentioned low concentration of chromium in solution. Additionally, while X-band EPR does show the formation of CrI during the activation of complex 1 and 2, the concentration of these CrI was shown to be low (<5%). The presence of these complexes was thus not taken into consideration in the EXAFS analysis as these complexes will likely not have a significant contribution to the observed EXAFS oscillations.| Entry | Conditions | Reaction time | Coordination shell | σ 2 (Å−2) | d (Cr–X) (Å) experimental | d (Cr–X) (Å) DFT/XRDa |
|---|---|---|---|---|---|---|
| a In entry 8, the experimental data is compared to a crystal structure provided in ref. 7. In the other entries, the experimental data is compared to a DFT-D3 geometry optimized at the BP86/TZ2P level of theory. In all cases, an average of the Cr–Cl and Cr–P distance is reported. b Samples were measured as pellets mixed with boron nitride. c Similar parameters used for the Debye–Waller factor and distance of the Cr–Cl and Cr–P shell. d Parameters within the Cr–O shell were kept fixed. | ||||||
| 1 | Complex 1b | Not applicable | 1 Cr–O | 0.002(5) | 2.15(3) | 2.134 |
| 3 Cr–Clb | 0.011(2) | 2.32(2) | 2.374 | |||
| 2 Cr–Pb | 0.011(2) | 2.32(2) | 2.374 | |||
| 2 | Complex 1 + AlMe3 | 2 minutes | 1.4(5) Cr–C | 0.002(4) | 2.03(2) | 2.080 |
| 1 Cr–Clc | 0.0060(7) | 2.43(1) | 2.386 | |||
| 2 Cr–Pc | 0.0060(7) | 2.43(1) | 2.386 | |||
| 3 | 10 minutes | 1.0(2) Cr–C | 0.000(2) | 2.046(8) | ||
| 1 Cr–Clc | 0.0059(3) | 2.443(4) | ||||
| 2 Cr–Pc | 0.0059(3) | 2.443(4) | ||||
| 4 | 60 minutes | 1.1(3) Cr–C | 0.002(3) | 2.03(1) | ||
| 1 Cr–Clc | 0.0056(4) | 2.440(5) | ||||
| 2 Cr–Pc | 0.0056(4) | 2.440(5) | ||||
| 5 | Complex 1 + MMAO | 2 minutes | 1 Cr–C | 0.003(3) | 2.14(5) | 2.032 |
| 2 Cr–P | 0.0030(7) | 2.41(1) | 2.438 | |||
| 6 | 10 minutes | 1 Cr–C | 0.003(4) | 2.08(5) | ||
| 2 Cr–P | 0.0018(7) | 2.39(1) | ||||
| 7 | 60 minutes | 1 Cr–C | 0.002(2) | 2.08(3) | ||
| 2 Cr–P | 0.0023(5) | 2.384(9) | ||||
| 8 | Complex 2b | Not applicable | 1 Cr–Od | 0.0030 | 2.16 | 2.156 |
| 3 Cr–Cl | 0.003(2) | 2.24(2) | 2.297 | |||
| 2 Cr–P | 0.002(3) | 2.43(3) | 2.448 | |||
| 9 | Complex 2 + AlMe3 | 2 minutes | 2 Cr–C/Cr–O | 0.002(2) | 2.07(4) | 2.117 |
| 2 Cr–P | 0.010(3) | 2.48(3) | 2.424 | |||
| 10 | 10 minutes | 2.3(4) Cr–C/Cr–O | 0.003(1) | 2.07(2) | ||
| 0.007(1) | 2.46(1) | |||||
| 2 Cr–P | ||||||
| 11 | 60 minutes | 1.7(4) Cr–C/Cr–O | 0.003(3) | 2.13(2) | ||
| 0.0058(9) | 2.51(1) | |||||
| 2 Cr–P | ||||||
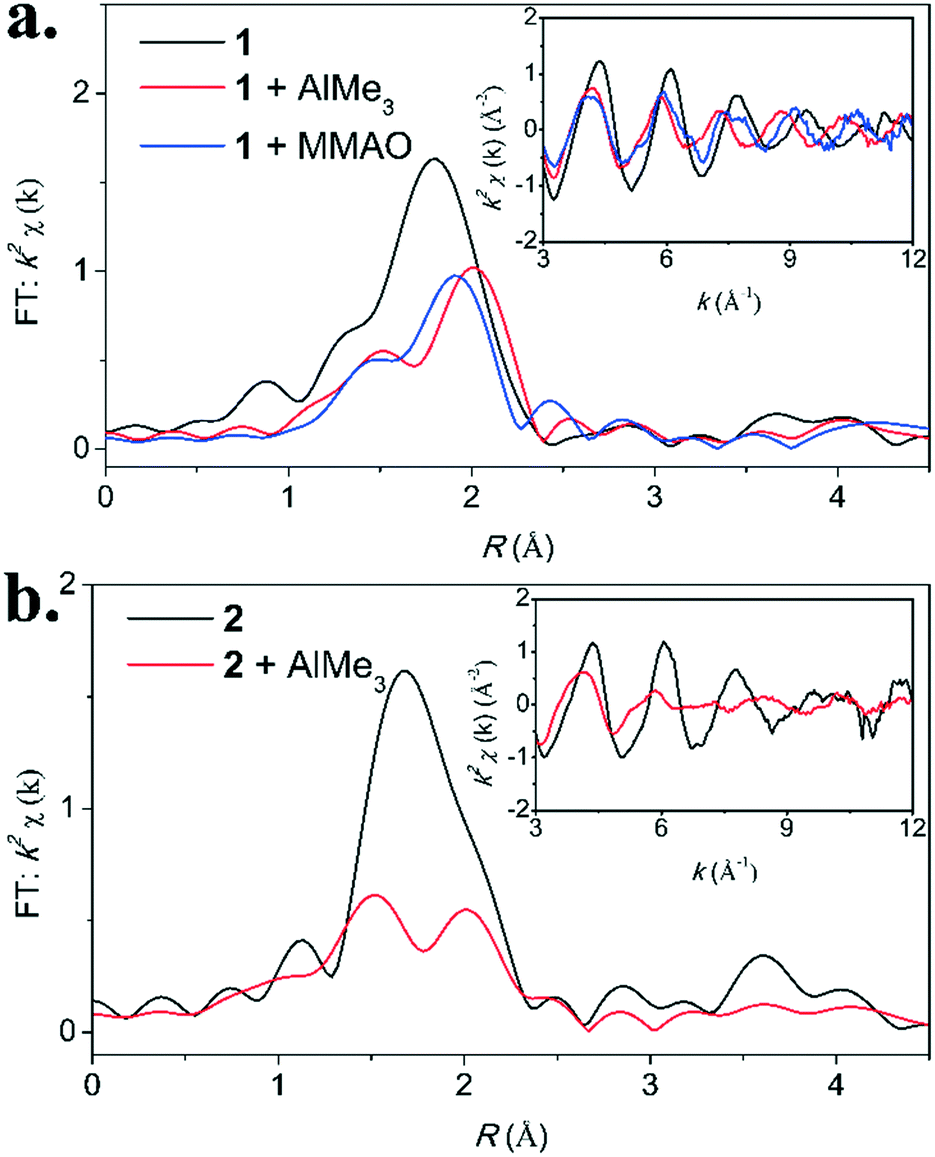 | ||
| Fig. 3 Cr K-edge EXAFS data (inset) and the corresponding Fourier transform for the reaction between a) complex 1 and AlMe3 (40 eq.) or MMAO (400 eq.), and b) complex 2 and AlMe3 (40 eq.). Reactions were performed in toluene and frozen after 10 minutes. | ||
A fit for complex 1 (entry 1) was obtained by including a Cr–O shell containing 1 atom at a distance of 2.15(3) Å from the metal center and including a mixed Cr–Cl/Cr–P shell at a distance of 2.32(2) Å from the metal center. The Cr–Cl shell contains 3 atoms and the Cr–P contains 2 atoms.
A crystal structure of ((C6H5)2P)2NiPrCrCl3(THF) has not been reported; only the dinuclear complex (((C6H5)2P)2NiPrCrCl3)2 has been isolated.22 We therefore performed DFT-D3 calculations at the BP86/TZ2P level of theory to assess the most favorable Isomer of ((C6H5)2P)2NiPrCrCl3(THF) (Table S14†). The meridional isomer was found to be most favored, and the quartet spin state was favored over the doublet spin state (ΔG° = +17.9 kcal mol−1). The calculated Cr–O (experimental: 2.15(3) Å, calculated: 2.13 Å), Cr–Cl/Cr–P (experimental: 2.32(2) Å, calculated: 2.37 Å) distances matched closely with those determined experimentally.
Reaction of complex 1 with AlMe3 (entry 2–4) leads to clear changes in the coordination environment of the metal, as is evident from the EXAFS analysis. Within error, the EXAFS analysis after a reaction time of 2, 10 and 60 minutes are identical. These findings are in line with the observations made in the Cr K-edge XANES region (Fig. S6†). The results obtained after a reaction time of 10 minutes will be discussed here.
A fit was obtained by introducing a Cr–C shell containing 1 atom at a distance 2.046(8) Å from the metal center. Additionally, a Cr–Cl containing 1 atom and a Cr–P shell containing 2 atoms were introduced at a distance of 2.443(4) Å from the metal center. These fitting parameters are similar to the ones we previously reported for the reaction between complex 1 and AlMe3 and are in line with the formation of ((C6H5)2P)2NiPrCrClMe (Scheme 3).15 In addition, based on thermochemical calculations, it is likely that AlMe3 coordinates to the methide and chloride moiety (ESI† section 3.3).
 | ||
| Scheme 3 Proposed activation pathway for the reaction between complex 1 and AlMe3 or MMAO. | ||
The Cr K-edge EXAFS analysis for the reaction between complex 1 and excess MMAO (entry 5–7) gives different results compared to the reaction of complex 1 with AlMe3. Again, we have investigated the reaction after reaction times of 2, 10 and 60 minutes and within error similar fitting parameters were obtained for all three reaction times. The results obtained after a reaction time of 10 minutes are discussed here (entry 6).
A fit was obtained by introducing a Cr–C shell center containing 1 atom at a distance of 2.08(5) Å) from the metal center. An additional Cr–P shell containing 2 atoms was introduced at a distance of 2.39(1) Å. However, we did not succeed in including an additional Cr–Cl shell, indicating that the halide is lost in the presence of MMAO.
We considered two hypotheses regarding the loss of the halide upon introduction of MMAO: i) further reduction to the monovalent oxidation state occurs or ii) upon introduction of MMAO, the halide is abstracted from the metal center and ion pair formation occurs (Scheme 3).
To assess whether reduction to the monovalent oxidation state is favorable, we performed thermochemical calculations (ESI† section 3.2). The calculations show that reduction of complex 1 to the divalent oxidation state is favorable; the formation of ((C6H5)2P)2NiPrCrClMe (ΔG° = −20.2 kcal mol−1) and ((C6H5)2P)2NiPrCrCl2 (ΔG° = −22.9 kcal mol−1) is thermodynamically almost equally favored. Further reduction of ((C6H5)2P)2NiPrCrClMe to form ((C6H5)2P)2NiPrCrCl (ΔG° = +4.5 kcal mol−1) or ((C6H5)2P)2NiPrCrMe (ΔG° = +13.9 kcal mol−1) is expected to be disfavored. These calculations suggest that the divalent oxidation state is favored over the monovalent oxidation state.
This is in line with the previously discussed EPR measurements, where it was shown that the majority of the solution consists of EPR-silent complexes and only small amounts of CrI can be detected. Further confirmation is found in the Cr K-edge XANES region (Fig. 2a): a similar edge position is observed for complex 1 activated with AlMe3 or MMAO, suggesting that the two complexes have a similar oxidation state. Based on these observations, we propose that a cationic CrII complex is formed with the structure [((C6H5)2P)2NiPrCrMe][ClMMAO].
These results thus show that the use of MMAO can facilitate ion-pair formation during the activation process. Ion-pair formation may be facilitated by Lewis acid sites contained within MMAO; these sites are absent in AlMe3.23–25 Recently, a self-activating cationic CrIII(PNP) ethene tetramerization complex has been prepared by Hirscher et al.26 These findings suggest that ion-pair formation is important for the generation of the active species.
Our findings are in contrast to the XAS study performed by Brückner and coworkers, where the neutral complex ((C6H5)2P)2NiPrCrMe2 was detected in the reaction between Cr(acac)3/((C6H5)2P)2NiPr/MAO.13 Possibly, the choice of a chloride-containing chromium source facilitates ion pair formation.
Next, we investigated the reaction of complex 2 with AlMe3. EXAFS analysis of complex 2 reveals a Cr–P shell containing 2 atoms at a distance of 2.43(3) Å (crystal structure: 2.45 Å) and a Cr–Cl shell containing 3 atoms at a distance of 2.24(2) Å (crystal structure: 2.30 Å) had to be included. Additionally, inclusion of a Cr–O shell at a distance of 2.16 Å significantly improved the fit. These parameters are in close agreement with an earlier reported crystal structure of complex 2.7
Upon reaction of complex 2 with AlMe3, marked changes are apparent in the EXAFS region (Fig. 3b). The Cr K-edge EXAFS analysis after a reaction time of 2, 10 and 60 minutes, within experimental error, are similar to one another (entry 9–11). The results obtained for the EXAFS analysis after a reaction time of 10 minutes will be discussed here; a similar interpretation applies to data obtained at other reaction times.
A fit was obtained by including a Cr–C/Cr–O shell containing 2.3(4) atoms at a distance of 2.07(2) Å and a Cr–P shell containing 2 atoms at a distance of 2.46(1) Å. Major structural differences between complex 2 and complex 2 activated with AlMe3 thus include i) loss of all chlorides and ii) alkylation of the metal center.
In line with these EXAFS results, we hypothesized two plausible structures for the complex formed: ((o-C6H4OMe)2P)2NMeCrMe2 or [((o-C6H4OMe)2P)2NMeCrMe][AlMe3Cl]. In the latter complex, the pendant ether donor coordinates to the metal, effectively forming a tridentate complex (Scheme 4).
 | ||
| Scheme 4 Proposed activation pathway for the reaction between complex 2 and AlMe3. | ||
To assess whether formation of ((o-C6H4OMe)2P)2NMeCrMe2 is feasible, we have performed thermochemical calculations (ESI† section 3.3). Based on these calculations however, the formation of ((o-C6H4OMe)2P)2NMeCrMe2 (ΔG° = −6.0 kcal mol−1) is disfavored over the formation of ((o-C6H4OMe)2P)2NMeCrMeCl (ΔG° = −15.8 kcal mol−1) or ((o-MeC6H4)2P)2NMeCrCl2 (ΔG° = −15.6 kcal mol−1) from complex 2. These calculations thus suggest that the formation of ((o-C6H4OMe)2P)2NMeCrMe2 is unlikely.
Additionally, close agreement is found between the experimentally determined and computationally (BP86+D3/TZ2P) determined Cr–C/Cr–O (experimental: 2.07(2) Å, calculated: 2.12) and Cr–P (experimental: 2.46(1) Å, calculated: 2.42) distances for [((o-C6H4OMe)2P)2NMeCrMe]+. We therefore propose the formation of the square-planar complex [((o-C6H4OMe)2P)2NMeCrMe][AlMe3Cl] in the reaction between complex 2 and AlMe3. Similar to the activation of complex 1 with MMAO, ion-pair formation is thus observed. Where ion-pair formation was facilitated by the activator for complex 1, ion-pair formation is facilitated by the ligand for complex 2.
Coordination of substrates to the metal
To obtain information on the oxidation state of the metal during the catalytic cycle, we performed activation experiments in the presence of substrates. In a previous study, we have shown for the [(R-SN(H)S-R)CrCl3] trimerization system, that coordination of the first ethene molecule is endergonic under 1 bar of ethene pressure.27 Coordination of substrates could be achieved by using more electron-rich substrates (e.g. alkynes). In the present study, to employ substrates more closely related to ethene, we have studied the reaction with dienes. It was demonstrated by Bowen et al. that both complex 1 and 2 are active for the trimerization of dienes.28 As a diene, we have employed isoprene. The advantage over butadiene is that isoprene is a liquid, and high concentrations in solution can be easily achieved.Adding isoprene (40 eq.) to an AlMe3-activated solution of complex 2 lead to no changes in the UV-vis spectrum (Fig. S44†) and the Cr K-edge XANES region (Fig. S45†). When isoprene is added to a MMAO-activated solution of complex 1 instead, clear changes in the UV-vis spectrum (Fig. S46†) are observed.
To exclude a change in oxidation state after introduction of isoprene, we have performed X-band EPR measurements at cryogenic temperatures (Fig. S47†). Major contributors the observed spectrum include an S = ½ complex with axial symmetry (gx,y = 1.979 and gz = 2.000) with a relative concentration of 36% and another S = ½ complex with rhombic symmetry (gz = 2.029, gy = 1.993 and gx = 1.960) with a relative concentration of 58%. These complexes had also been observed in the absence of isoprene (Fig. S24†). The simulation could be improved by including another S = ½ complex with axial symmetry (gx,y = 2.016 and gz = 1.987) with a relative concentration of 5%. From these measurements it can thus be concluded that the introduction of isoprene does not lead to major changes in the X-band EPR spectrum.
Next, we studied the reaction using Cr K-edge XAS. Fig. 4 shows a comparison of the Cr K-edge XANES region. The position of the edge is unaffected by the introduction of isoprene and the shape is only slightly affected. This is suggestive of no changes in the oxidation state of chromium after introduction of isoprene. The two pre-edge features shift slightly towards lower energies (5988.7 eV and 5990.8 eV compared to 5989.1 eV and 5991.5 eV) and the relative intensity of these features change. These changes are suggestive of a changing coordination environment of chromium after introduction of isoprene.
 | ||
| Fig. 4 Cr K-edge XANES region for the reaction between complex 1 and MMAO (400 eq.) in the absence and presence of isoprene (50 eq.). Reaction times of 60 minutes were employed. For the reaction in presence of isoprene, isoprene was injected after 10 minutes and the reaction was continued for another 50 minutes. | ||
To study the exact structure of the reaction product, we have investigated the reaction using Cr K-edge EXAFS. The Cr K-edge EXAFS region is presented in Fig. 5 and the EXAFS analysis for the reaction in the presence of isoprene is presented in Table 3. A successful fit was obtained by including a Cr–C shell containing 5.1(5) atoms at a distance of 2.28(5) Å and by including a Cr–P shell containing 2 atoms at a distance of 2.31(5) Å. In the presence of isoprene, the coordination number of the Cr–C shell is thus increased by 4 atoms (Scheme 5).
 | ||
| Fig. 5 Cr K-edge EXAFS data (inset) and the corresponding Fourier transform for the reaction between complex 1 and MMAO (400 eq.) in the presence of isoprene (40 eq.). | ||
| Coordination shell | σ 2 (Å−2) | d (Cr–X) (Å) experimental |
|---|---|---|
| 5.1(5) Cr–C | 0.006(3) | 2.28(5) |
| 2 Cr–P | 0.008(5) | 2.31(5) |
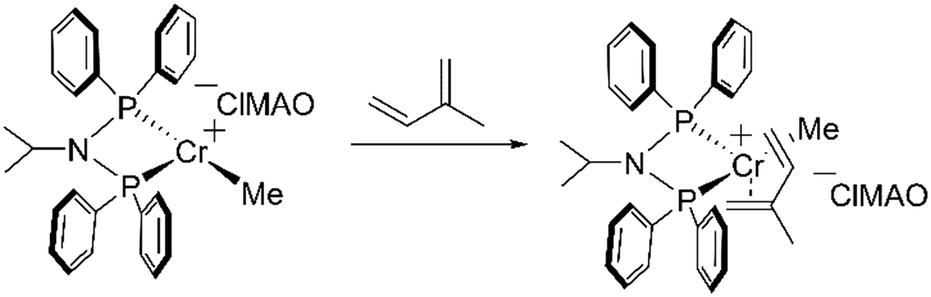 | ||
| Scheme 5 Proposed pathway for the reaction between the cationic CrII complex and isoprene. | ||
Proposed catalytic cycle
The chromium-catalyzed tri- and tetramerization of ethene, catalyzed by bis(diarylphosphino)amine ligands, has been the subject of a number of DFT studies performed by Britovsek and McGuinness and coworkers.29–32 In these DFT studies they propose that mono- and bis-ethene insertion events can occur during the catalytic cycle. 1-Hexene is predominantly formed through a pathway proceeding via mono-ethene insertion events and 1-octene is formed through a pathway proceeding via both mono- and bis-ethene insertion events. This model of bis-ethene insertion has provided a mechanistic understanding for the experimentally observed second-order ethene dependence on 1-octene formation.33 This proposal is in line with recent deuterium labeling studies performed by Hirscher et al., where it is shown that 1-hexene and 1-octene are formed through a common chromacycloheptane intermediate.9In one DFT study, they have deemed a CrII/CrIV redox couple unlikely, as the formation of a CrII–olefin complex is thermodynamically less preferred compared to the formation of a CrI–olefin complex. In addition, the barrier for 1-hexene formation is lower for a CrI/CrIII redox couple.30 These calculations have been performed using a simplified structure of the ligand ((CH3)2P)2NMe. In follow-up studies, where the full structure of the ligand (i.e. ((C6H5)2P)2NiPr and ((o-C6H4OMe)2P)2NMe) was used in the performed calculations, a CrII/CrIV redox couple was not taken into consideration.29,31,32
The present study has shown that the majority of chromium is reduced to the divalent oxidation state and that the divalent oxidation state is capable of binding olefins. To investigate whether CrII/CrIV intermediates could also explain the observed product selectivity, we have therefore performed DFT calculations at the BP86/TZP level of theory using a full structure of the two ligands. Catalysis could proceed either via monocationic or dicationic intermediates (formed through abstraction of a methyl group facilitated by AlMe3 or MMAO). For this reason, we have compared a monocationic mechanism (Scheme S15†) to a dicationic mechanism (Scheme 6) for the ((C6H5)2P)2NiPr ligand framework.
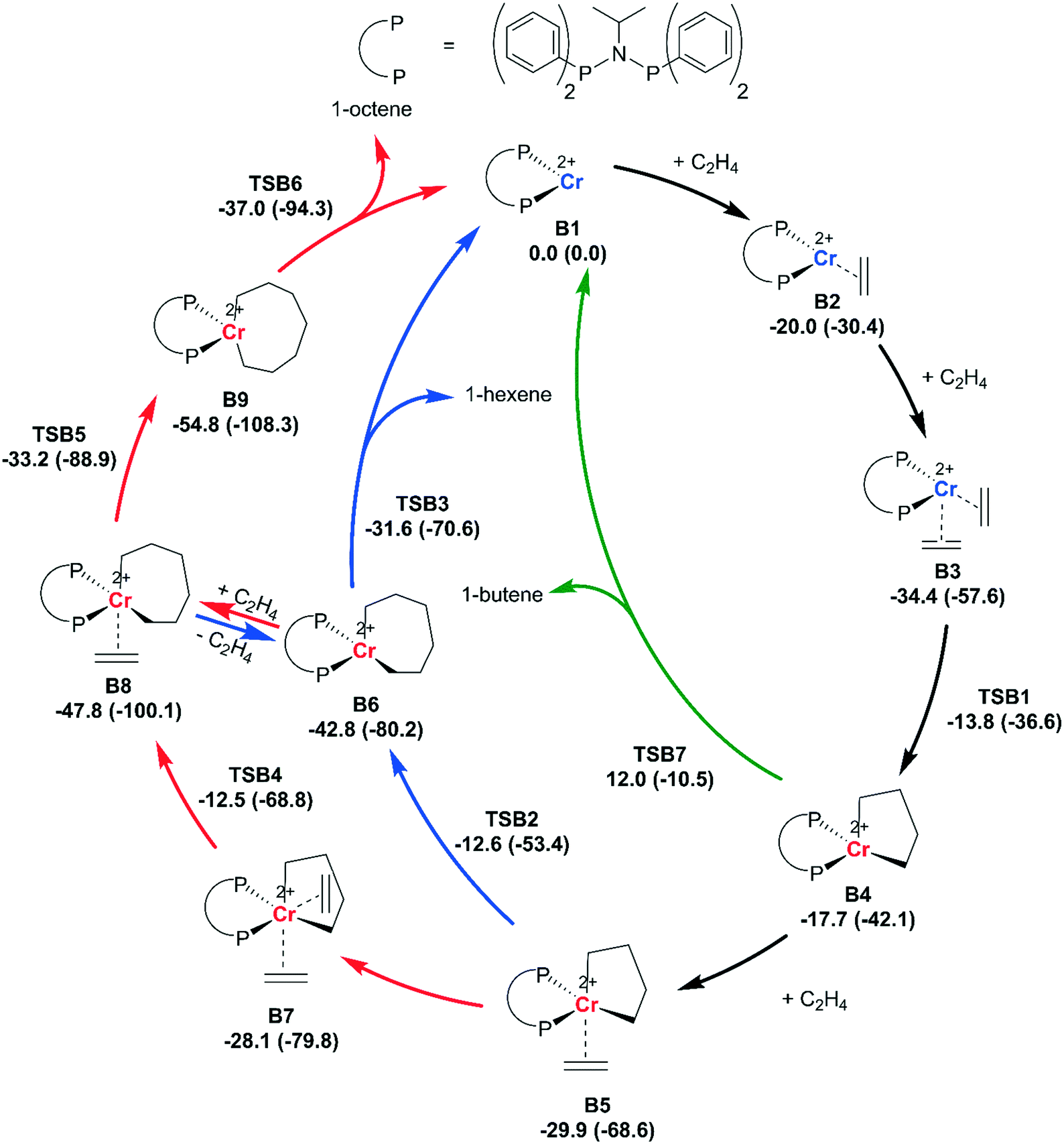 | ||
| Scheme 6 Calculated catalytic cycle for the tri- and tetramerization of ethene. Calculations were performed at the BP86/TZP level of theory in the gas phase (1 atm.). The singlet (not depicted), triplet (red) and quintet (blue) spin states were taken into consideration. Reported are the Gibbs free energies (298.15 K), and the electronic energies in parentheses, in kcal mol−1. Depicted are the catalytic cycles for the formation of 1-butene (green), 1-hexene (blue) and 1-octene (red). Parts of the ligand are removed for clarity of the image. | ||
The mechanism proceeding via monocationic intermediates is reported in the ESI† (section 5.1). This mechanism cannot explain the experimentally observed product selectivity. Firstly, the fourth ethene molecule cannot coordinate to the metal center. This would hamper the formation of 1-octene. Secondly, the barrier for the formation of 1-butene is lower compared to the formation of 1-hexene and this would make the complex active for ethene dimerization.
A mechanism proceeding via dicationic intermediates does correctly predict the observed product selectivity (Scheme 6). The starting point of the calculations is the dicationic CrII complex, B1. Coordination of the first (ΔG° = −20.0 kcal mol−1) and second (ΔG° = −34.4 kcal mol−1) ethene molecule to B1 is exergonic. Oxidative coupling of the two ethene molecules to yield chromacyclopentane B4 (ΔΔG = +16.6 kcal mol−1)) occurs with a moderate barrier (TSB1, ΔΔG‡ = +20.6 kcal mol−1). The oxidative coupling is accompanied by a spin crossover from the quintet to the triplet spin state. Subsequent elimination of 1-butene from chromocyclopentane B4 is deemed unlikely due to the high barrier (TSB7, ΔΔG‡ = +29.7 kcal mol−1).
Subsequent coordination of ethene to B4 to form B5 is exergonic (ΔΔG = −12.2 kcal mol−1). Either, this ethene molecule can insert into the metallacycle (TSB2, ΔΔG‡ = +17.3 kcal mol−1) to form chromacycloheptane intermediate B6 (ΔΔG = −12.9 kcal mol−1). Or a second ethene molecule can coordinate to B5 (ΔΔG = +1.8 kcal mol−1) and subsequently insert into the metallacycle (TSB4, ΔΔG‡ = +17.4 kcal mol−1) to form the chromacycloheptane B8 (ΔΔG = −19.7 kcal mol−1). Interestingly, the mono- and bis-ethene insertion pathway have very similar barriers (difference TSB4 and TSB2, ΔΔG = +0.1 kcal mol−1). Likely the bis-ethene insertion pathway (TSB4) becomes dominant at elevated ethene pressures.
1-hexene is formed through a concerted 3,7-H shift from B6 (TSB3, ΔΔG‡ = +11.2 kcal mol−1). 1-octene is formed through insertion of ethene into the chromacycloheptane intermediate B8 (TSB5, ΔΔG‡ = +14.6 kcal mol−1) to form chromacyclononane B9. Chromacyclononane intermediate B9 can subsequently undergo a 3,7-H shift to form 1-octene (TSB6, ΔΔG‡ = +17.8 kcal mol−1). This DFT model correctly explains that the formation of 1-octene is favored over 1-hexene (difference TSB3 and TSB5, ΔΔG = +1.6 kcal mol−1).
The increased selectivity for 1-hexene when AlMe3 is used as an activator (Table 1) might arise from the coordinating ability of the generated anions ([AlMe3Cl]− or [AlMe4]−). These anions are expected to coordinate more strongly compared to the anions generated in the presence of MMAO and will likely disfavor the formation of intermediate B8 and thus raise the barrier for 1-octene formation.19,31
We also performed calculations for the ((o-C6H4OMe)2P)2NMe ligand framework (Scheme S16†). Here, we had only considered a mechanism proceeding via dicationic CrII/CrIV intermediates. Our findings are reported in detail in the ESI† (section 5.3). Key features of the mechanism include that the bis-ethene insertion pathway is disfavored over the mono-ethene insertion pathway due to the coordination of the pendant ether donor. Additionally, liberation of 1-hexene is favored over further metallacycle growth, thus explaining the experimentally observed product selectivity.
The three DFT models have thus demonstrated that a mechanism proceeding via cationic CrII/CrIV intermediates can also reproduce the experimentally observed product selectivity. Nevertheless, a CrI/CrIII redox couple cannot be conclusively excluded. While Cr K-edge XAS shows that the bulk remains in the divalent oxidation state, a minority unobservable CrI complex might still be responsible for the catalytic activity. In this regard, Bercaw and coworkers have suggested that an unobservable minority complex is responsible for the catalytic activity in the ((o-C6H4OMe)2P)2NMeCrCl3 trimerization system.16 Additionally, Britovsek and McGuinness have demonstrated that a mechanism proceeding via CrI/CrIII intermediates also correctly predicts the observed product selectivity. Future Cr K-edge XAS and EPR experiments at elevated ethene pressures are required to conclusively assign an oxidation state to the active species.
Conclusions
In this study, we have investigated the reactivity of an ethene tri- and tetramerization with alkylaluminum reagents. During the activation process, the majority of chromium is reduced to the divalent oxidation state and only a minority is further reduced to the monovalent oxidation state. Both the choice of activator and choice of ligand were found to have an influence on the activation process. A major finding in this study includes the observation of ion pair formation. This is facilitated either through the use of MMAO or through the use of a ligand containing a pendant ether donor.Experiments in the presence of isoprene have allowed for the characterization of a monocationic CrII alkene complex, demonstrating that the divalent oxidation state can bind olefins. Additionally, DFT-D3 calculations for a mechanism proceeding via dicationic CrII/CrIV intermediates have demonstrated that a mechanism proceeding via CrII/CrIV intermediates can correctly predict the observed product selectivity.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank NWO for funding (VIDI grant 723.014.010 (to M. T. for B. V., L. W., J. P. O.), TOP-Grant 716.015.001 (to B. d. B) and VENI grant 722.016.012 (to T. J. K.). The authors thank the staff of the beamlines SuperXAS, Swiss Light Source (proposal number 20160674) in Villigen, Switzerland and B18, Diamond Light Source (proposal number SP22331) in Didcot, UK for support and access to their facilities. The authors thank Michelle Hammerton for support during synchrotron measurements. The authors thank Andreas Ehlers and Ed Zuidinga for NMR spectroscopy and mass spectrometry support.References
- G. R. Lappin and J. D. Sauer, Alpha Olefins Applications Handbook, M. Dekker, 1989 Search PubMed.
- J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS.
- E. F. Lutz, J. Chem. Educ., 1986, 63, 202 CrossRef CAS.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef CAS PubMed.
- O. L. Sydora, Organometallics, 2019, 38, 997–1010 CrossRef CAS.
- J. R. Briggs, J. Chem. Soc., Chem. Commun., 1989, 674–675 RSC.
- T. Agapie, S. J. Schofr, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 1304–1305 CrossRef CAS PubMed.
- M. J. Overett, K. Blan, A. Bollmann, J. T. Dixon, D. Haasbroek, E. Killian, H. Maumela, D. S. McGuinness and D. H. Morgan, J. Am. Chem. Soc., 2005, 127, 10723–10730 CrossRef CAS PubMed.
- N. A. Hirscher, J. A. Labinger and T. Agapie, Dalton Trans., 2019, 48, 40–44 RSC.
- A. Carter, S. A. Cohen, N. A. Cooley, A. Murphy, J. Scutt and D. F. Wass, Chem. Commun., 2002, 858–859 RSC.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed.
- A. Brückner, J. K. Jabor, A. E. C. McConnell and P. B. Webb, Organometallics, 2008, 27, 3849–3856 CrossRef.
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, ACS Catal., 2013, 3, 95–102 CrossRef CAS.
- R. Grauke, R. Schepper, J. Rabeah, R. Schoch, U. Bentrup, M. Bauer and A. Brückner, ChemCatChem, 2019, 12, 1025–1035 CrossRef.
- S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, Catal. Sci. Technol., 2016, 6, 6237–6246 RSC.
- L. H. Do, J. A. Labinger and J. E. Bercaw, ACS Catal., 2013, 3, 2582–2585 CrossRef CAS PubMed.
- S. C. Eady, T. Breault, L. Thompson and N. Lehnert, Dalton Trans., 2016, 45, 1138–1151 RSC.
- E. J. M. De Boer, H. van der Heijden, Q. A. On, J. P. Smit and A. van Zon, Shell International Research, Catalytic oligomerization of olefinic monomers, WO patent WO2007057455A1, 2007.
- D. S. McGuinness, A. J. Rucklidge, R. P. Tooze and A. M. Z. Slawin, Organometallics, 2007, 26, 2561–2569 CrossRef CAS.
- N. Shaham, H. Cohen, D. Meyerstein and E. Bill, J. Chem. Soc., Dalton Trans., 2000, 3082–3085 RSC.
- S. A. Bartlett, P. P. Wells, M. Nachtegaal, A. J. Dent, G. Cibin, G. Reid, J. Evans and M. Tromp, J. Catal., 2011, 284, 247–258 CrossRef CAS.
- A. M. Lifschitz, N. A. Hirscher, H. B. Lee, J. A. Buss and T. Agapie, Organometallics, 2017, 36, 1640–1648 CrossRef CAS.
- S. Collins, M. Linnolahti, M. G. Zamora, H. S. Zijlstra, M. T. Rodríguez Hernández and O. Perez-Camacho, Macromolecules, 2017, 50, 8871–8884 CrossRef CAS.
- M. E. Z. Velthoen, A. Muñoz-Murillo, A. Bouhmadi, M. Cecius, S. Diefenbach and B. M. Weckhuysen, Macromolecules, 2018, 51, 343–355 CrossRef CAS PubMed.
- E. P. Talsi, N. V. Semikolenova, V. N. Panchenko, A. P. Sobolev, D. E. Babushkin, A. A. Shubin and V. A. Zakharov, J. Mol. Catal. A: Chem., 1999, 139, 131–137 CrossRef CAS.
- N. A. Hirscher, D. Perez Sierra and T. Agapie, J. Am. Chem. Soc., 2019, 141, 6022–6029 CrossRef CAS PubMed.
- B. Venderbosch, J. P. H. Oudsen, D. J. Martin, B. de Bruin, T. J. Korstanje and M. Tromp, ChemCatChem, 2020, 12, 881–892 CrossRef CAS.
- L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 39, 560–567 RSC.
- G. J. P. Britovsek and D. S. McGuinness, Chem. – Eur. J., 2016, 22, 16891–16896 CrossRef CAS PubMed.
- D. S. Mcguinness, B. Chan, G. J. P. Britovsek and B. F. Yates, Aust. J. Chem., 2014, 67, 1481–1490 CrossRef CAS.
- G. J. P. Britovsek, D. S. McGuinness and A. K. Tomov, Catal. Sci. Technol., 2016, 6, 8234–8241 RSC.
- G. J. P. Britovsek, D. S. McGuinness, T. S. Wierenga and C. T. Young, ACS Catal., 2015, 5, 4152–4166 CrossRef CAS.
- S. Kühlmann, Selective Tri- and Tetramerization of Ethylene – from Ligand Design to Mini- Plant Operation, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01168a |
| This journal is © The Royal Society of Chemistry 2020 |