
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The direct synthesis of hydrogen peroxide using a combination of a hydrophobic solvent and water†
Adeeba
Akram
a,
Greg
Shaw
a,
Richard J.
Lewis
 a,
Marco
Piccinini
a,
David J.
Morgan
a,
Thomas E.
Davies
a,
Simon J.
Freakley
b,
Jennifer K.
Edwards
a,
Jacob A.
Moulijn
a and
Graham J.
Hutchings
*a
a,
Marco
Piccinini
a,
David J.
Morgan
a,
Thomas E.
Davies
a,
Simon J.
Freakley
b,
Jennifer K.
Edwards
a,
Jacob A.
Moulijn
a and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 21st October 2020
Abstract
The direct synthesis of hydrogen peroxide (H2O2) has been studied using a solvent system comprising a hydrophobic alcohol (decan-1-ol) and water. It is demonstrated that, with the optimum combination of solvent and catalyst the contribution of H2O2 degradation pathways can be minimised to achieve industrially acceptable H2O2 concentrations under moderate conditions. This is achieved through the use of a catalyst that is retained by the organic component and the extraction of synthesised H2O2 into the aqueous phase, consequently limiting contact between the synthesised H2O2, catalyst and reactant gases, resulting in an improved selectivity towards H2O2. Investigation of the reaction parameters provides an insight into the proposed solvent system, and optimised conditions to produce H2O2 from molecular H2 and O2 have been identified. Through this optimisation H2O2 concentrations up to 1.9 wt% have been achieved via sequential gas replacement experiments.
Introduction
The direct synthesis of hydrogen peroxide (H2O2) from molecular hydrogen and oxygen offers an attractive alternative to the current means of production, the anthraquinone oxidation or in-direct process. The direct route to H2O2 has been carried out in a wide range of reaction media, including water.1–9 For many applications the most convenient solvent for the reaction would be water as it is readily available, non-toxic, non-flammable and completely miscible with H2O2. Indeed a large number of industrial processes utilise aqueous H2O2, in particular for application as a bleaching agent in the pulp and textiles sector (accounting for nearly 50% of annual usage10) H2O2 diluted in water is favoured.11 However, a major drawback of using H2O as a solvent is the low solubility of reagent gases H2 and O2, (1.62 mg L−1 and 40 mg L−1 respectively, at room temperature)12 which has been shown to limit H2O2 synthesis rates, compared to that observed when using alcohol/water co-solvent systems.1,2 To overcome these limitations solvents other than water have been investigated including supercritical CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13–16 and halogenated solvents17–19 as well as short chain alcohols,9,20,21 with Paunovic et al.22 providing a comprehensive study on the role of the co-solvent in the direct synthesis reaction. It has been reported that the rate of H2O2 production in short chain alcohols is much higher than that in aqueous media due to higher gas solubility.20,23 The solubility of H2 in short chain alcohols, in particular methanol, has been reported to be 4–5 times higher than in water whereas that of O2 may increase up to eightfold.24,25 Furthermore, additional studies have reported that using a methanol co-solvent can lead to the suppression of H2O2 decomposition activity, likely due to the ability of methanol to act as a hydroxyl radical scavenger.26
13–16 and halogenated solvents17–19 as well as short chain alcohols,9,20,21 with Paunovic et al.22 providing a comprehensive study on the role of the co-solvent in the direct synthesis reaction. It has been reported that the rate of H2O2 production in short chain alcohols is much higher than that in aqueous media due to higher gas solubility.20,23 The solubility of H2 in short chain alcohols, in particular methanol, has been reported to be 4–5 times higher than in water whereas that of O2 may increase up to eightfold.24,25 Furthermore, additional studies have reported that using a methanol co-solvent can lead to the suppression of H2O2 decomposition activity, likely due to the ability of methanol to act as a hydroxyl radical scavenger.26
Despite this approach, the presence of subsequent reactions in the H2O2 direct synthesis process (Scheme 1) suggests that it will remain challenging to achieve high H2O2 concentrations, due to the thermodynamically favoured H2O2 degradation pathways (hydrogenation and decomposition). Typically, these unwanted reaction pathways have been inhibited through the use of a combination of sub-ambient reaction temperatures,27 acidic promoters,28–30 halide stabilizing agents31,32 (with obvious drawbacks associated with reactor corrosion and the need for their down-stream removal) or complicated catalyst design, such as pre-treatment of catalyst supports prior to metal immobilisation33 or sequential heat treatment cycles.34 While a growing number of catalysts have been reported that are capable of achieving high H2O2 selectivity,33–35 in the absence of stabilizing agents, the consecutive reaction pathways still prohibit the production of H2O2 concentrations on an industrial scale, with H2O2 concentrations between 0.6–1.8 wt% typically produced in initial stages of the industrial process, prior to concentration and shipping.36
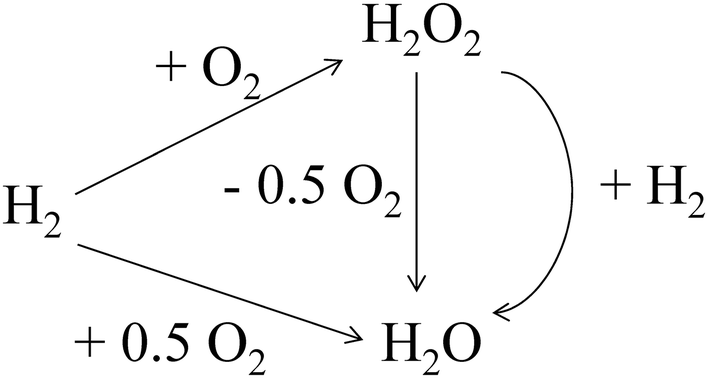 | ||
| Scheme 1 Reaction pathways associated with the direct synthesis of H2O2 from H2 and O2. | ||
With the first order dependence of H2O2 synthesis on H2 partial pressure,37 increasing H2 concentration at the catalyst surface through application of a short chain alcohol as solvent or increasing reaction pressure (which is not attractive for a commercial process) can inherently be related to a proportional increase in the rate of H2O2 production. A bi-phasic solvent system consisting of long chain alcohols and water could provide both high solubility of gases and the ability to extract H2O2 into the aqueous phase under moderate conditions. Ideally in this system, the catalyst and gases will be held in the organic phase where H2O2 will be produced and subsequently extracted in situ into the aqueous phase due to the higher solubility of H2O2 in water than the organic solvent. The use of a bi-phasic solvent system has the potential to lead to enhanced selectivity towards H2O2, through minimising contact time between H2O2, the catalyst and reactant gases, which in turn leads to a reduction in the subsequent H2O2 degradation reactions and, thus, an increase in H2O2 concentration. Indeed similar approaches have been recently been reported for the production of H2O2via photocatalytic processes38–40 as well as in the biomass conversion literature, where bi-phasic solvent systems often lead to higher product yields than with mono-phasic aqueous systems due to the higher solubility of products in the organic phase.41 Furthermore, the use of multi-phasic systems has been well established in processes that utilise homogeneous catalysts.42,43 In the case of H2O2 synthesis, when selecting a suitable candidate for the organic phase two key features are required, an ability to produce H2O2 in the solvent, and a low water solubility. In order to narrow down the search for a suitable organic solvent only primary alcohols were considered in this study. Water was selected as the co-solvent due to it being low cost, readily available and its full miscibility with H2O2.
The choice of the catalyst support is also an important parameter to consider, with the ability of the catalyst to be retained in the hydrophobic solvent, during the course of the reaction, crucial to inhibiting H2O2 degradation. In this optimum scenario (Scheme 2a) the contact between catalyst and reagent gases would be maximized, while also isolating the formed H2O2 and inhibiting degradation pathways. On the other hand, the use of a catalyst that is retained primarily in the aqueous phase (Scheme 2b), or indeed the use of a single phase solvent system, such as water/ethanol44 or water/methanol,35,45,46 which are both commonly studied in the literature, (Scheme 2c) would not limit contact between synthesised H2O2, catalyst and reagent gases, leading to increased rates of H2O2 degradation. With the application of stabilizing agents, to inhibit catalytic degradation of H2O2, typical in these miscible solvent systems.
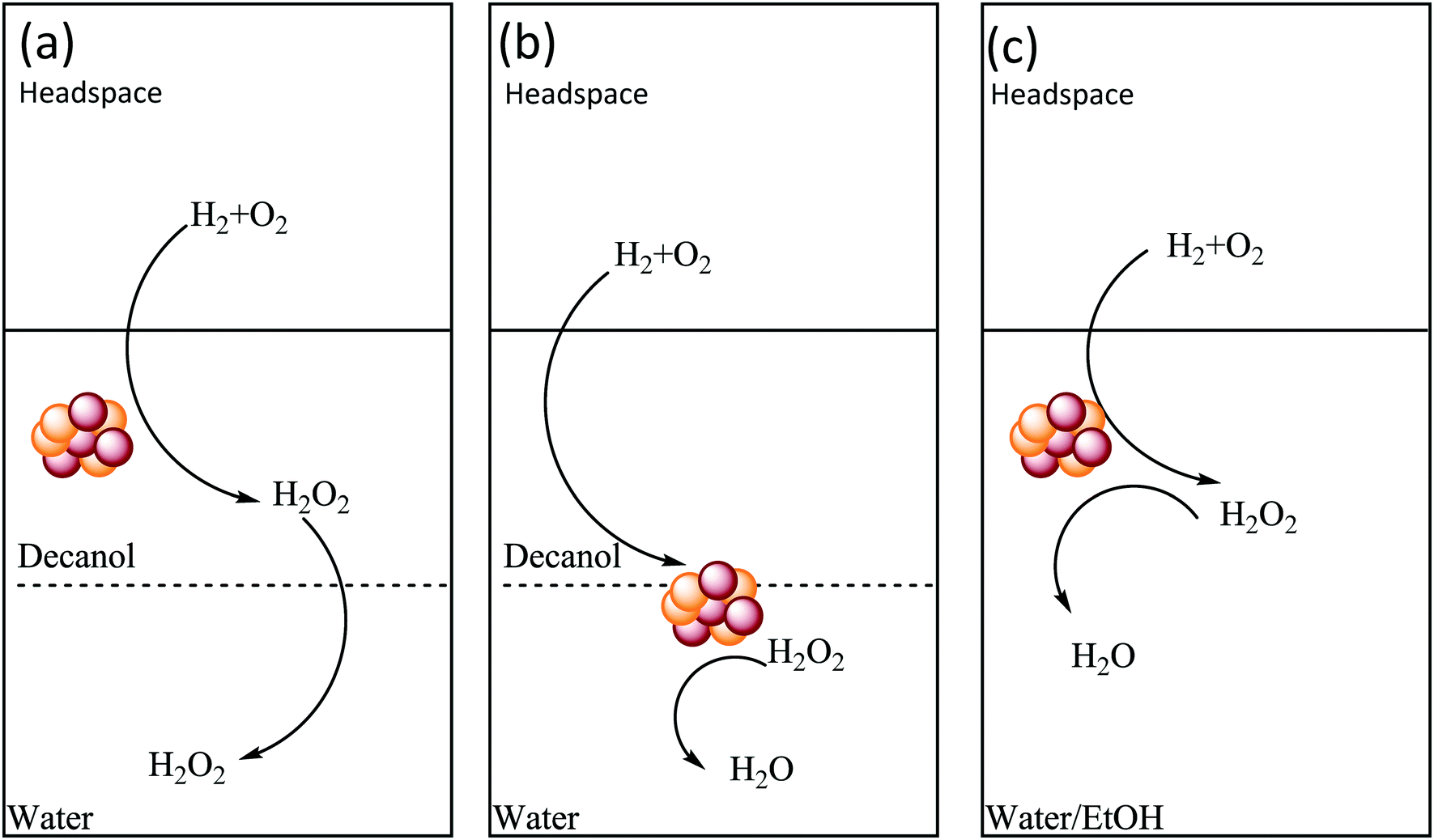 | ||
| Scheme 2 The effect of solvent and catalyst support on promoting H2O2 selectivity in (a) a non-miscible solvent system where the catalyst is retained in the organic component of the solvent, (b) a non-miscible solvent system where the catalyst is retained in the aqueous component of the solvent and (c) a completely miscible solvent system. | ||
This paper outlines the investigation of the direct synthesis of H2O2 based on the described hydrophobic alcohol and water solvent system. Reaction parameters have been varied, namely water concentration, catalyst support, solvent mass and reaction time to deduce the feasibility of the outlined solvent system.
Experimental
Preparation of AuPd supported catalysts by wet impregnation
AuPd bimetallic catalysts were prepared via a wet co-impregnation onto a range of supports; Darco G60 carbon (C), Aeroxide TiO2 (Degussa, Aeroxide, P25), SiO2 and CeO2. The standard preparation of 2.5 wt% Au–2.5 wt% Pd supported catalyst is described as follows (all quantities stated are per 1 g of catalyst). PdCl2 (0.0417 g, Sigma Aldrich) was added to HAuCl4·3H2O (2.04 mL, 12.25 mg mL−1, Strem Chemicals). The solution was stirred and heated (80 °C) until the PdCl2 dissolved completely to form a homogeneous solution. The appropriate support (0.95 g) was added to the solution and stirred until a paste was formed. The resultant material was dried in an oven (110 °C, 16 h) before being ground and calcined in static air (400 °C, 3 h, 20 °C min−1).Characterisation
The volumetric Karl Fischer method was performed using a TitroLine® 7500 KF volumetric Karl Fischer titrator where the titrating agent was accurately added through a piston burette. The standard procedure for each titration was as follows. The burette was filled with the titrating agent (Hydranal®-Composite 5). The working medium (Hydranal®-CompoSolver E) was added to the titration vessel, which was titrated to dryness with the titrating agent. The organic solvent sample was added (between 0.1–1.0 g depending on expected water concentration) and the sample was titrated with the titrating agent to determine the water concentration.1H NMR spectra were recorded on a Bruker Ultrashield 500 MHz spectrometer, using a H2O solvent suppression program. Filtered solvent (0.7 mL) was added to an NMR tube containing D2O (0.1 mL).
Investigation of the bulk structure of the crystalline materials was carried out using a (θ–θ) PANalytical X'pert Pro powder diffractometer using a Cu Kα radiation source, operating at 40 KeV and 40 mA. Standard analysis was carried out using a 40 min run with a back filled sample, between 2θ values of 10–80°. Phase identification was carried out using the International Centre for Diffraction Data (ICDD).
X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C (1s) binding energy of adventitious carbon contamination that was taken to be 284.7 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans while 40 eV was employed for detailed regional scans. The intensities of the Au (4f) and Pd (3d) features were used to derive the Au/Pd surface ratios.
To allow for quantification of total metal loading catalysts were digested via an aqua-regia assisted, microwave digestion method using a Milestone Connect Ethos UP microwave with an SK15 sample rotor. Digested samples were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All samples were diluted by a factor of 10 using HPLC grade H2O (1% HNO3 and 0.5% HCl matrix). All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent.
Direct synthesis of H2O2
Catalyst testing was performed using a Parr Instruments stainless steel autoclave which had a nominal volume of 100 mL and a maximum working pressure of 14 MPa. The autoclave was equipped with an overhead stirrer (0–2000 rpm) and provision for measurement of temperature and pressure. To test the direct synthesis of H2O2, the autoclave was typically charged with catalyst (0.01 g) and saturated decan-1-ol (0.39 g H2O and 8.11 g decan-1-ol). The charged autoclave was purged three times with 5% H2/CO2 (100 psi) and then filled with 5% H2/CO2 (420 psi) and 25% O2/CO2 (160 psi) to give a H2:O2 ratio of 0.5 and a total working pressure of 580 psi, with no continual introduction of gas. The reaction mixture was allowed to stabilise at the desired temperature (25 °C) after which stirring commenced (1200 rpm) and experiments were carried out for 30 minutes, unless stated otherwise. H2O2 yield was determined by titrating all of the final solution (unless stated otherwise) with acidified Ce(SO4)2. The Ce(SO4)2 solutions were standardised against (NH4)2Fe(SO4)2·6H2O using ferroin as an indicator. These reaction conditions were systematically varied in this study.
In this paper, results have been primarily obtained with saturated (with water) decan-1-ol, with the amount of hydrogen peroxide discussed in terms of the theoretical H2O2 concentration in water (wt%). The H2O2 concentration is expressed assuming facile separation of decan-1-ol and water can be achieved, with no H2O2 retained in the organic solvent.
Time-on-line analysis for the direct synthesis of H2O2
An identical procedure to that outlined above for the direct synthesis of H2O2 is followed for the desired reaction time. It should be noted that individual experiments are carried out and the reaction mixture is not sampled on-line.Gas replacement experiments for the direct synthesis of H2O2
An identical procedure to that outlined above for the direct synthesis of H2O2 is followed for a reaction time of 0.5 h. After this, stirring is stopped and the reactant gas mixture is vented prior to replacement with the standard pressures of 5% H2/CO2 (420 psi) and 25% O2/CO2 (160 psi). The reaction is then stirred (1200 rpm) for a further 0.5 h.Degradation of H2O2
Catalytic activity towards H2O2 degradation was determined in a similar manner to the direct synthesis activity of a catalyst. The autoclave was typically charged with catalyst (0.01 g) and reaction solution (0.39 g 4 wt% H2O2 and 8.11 g organic solvent). The charged autoclave was purged three times with 5% H2/CO2 (100 psi) and then filled with 5% H2/CO2 (420 psi). The reaction mixture was allowed to stabilise at the desired temperature (25 °C) after which stirring commenced (1200 rpm) and experiments were carried out for 30 minutes. H2O2 degraded was determined by titrating all of the final solution with acidified Ce(SO4)2. The Ce(SO4)2 solutions were standardised against (NH4)2Fe(SO4)2·6H2O using ferroin as an indicator. Catalytic activity towards H2O2 degradation is reported herein in terms of rate (molH2O2 kgcat−1 h−1) to better allow for comparison to the literature and accounts for hydrogenation and decomposition pathways.Note: within this work reactant gases have been diluted with CO2 to ensure that at no time do mixtures of H2 and O2 enter the explosive region (4–94 mol%).
Results and discussion
Within this study we evaluate the catalytic performance of a range of previously studied supported AuPd catalysts prepared by a wet co-impregnation methodology, (5% AuPd/C,33 5% AuPd/TiO2,6 5% AuPd/CeO2 (ref. 47) and 5% AuPd/SiO2 (ref. 27)) (actual metal loading, as determined by aqua-regia assisted microwave digestion reported in Table S.1†) towards H2O2 synthesis, using a solvent system consisting of a hydrophobic alcohol and water. We aim to address H2O2 selectivity through minimizing contact between catalyst and the synthesised H2O2, with the choice of solvent ensuring the former is preferentially retained in the hydrophobic alcohol while the latter extracted into the aqueous phase.We have previously demonstrated that catalytic selectivity of the materials studied within this work correlates strongly with the surface charge of the support, with catalysts synthesised on supports such as C and SiO2, which have low isoelectric points, offering the greatest rates of H2O2 synthesis.48 Although other parameters such as nanoparticle dispersion,49 the degree of alloying50 and Pd oxidation state51,52 are clearly also strongly related to catalytic performance.
With this in mind we initially investigated the as-prepared catalysts via XRD (Fig. S.1†), with reflections observed at 38, 44, 66 and 78° corresponding to Au (111), (200) (220) and (311) planes in all samples, in addition to reflections at 65 and 79° corresponding to the Pd (220) and (311) planes observed in the AuPd/SiO2 catalyst. This is indicative of the supported nanoparticles generally being large or poorly dispersed. This is perhaps unsurprising, with catalysts prepared via a wet-impregnation procedure well known to display a bimodal distribution of nanoparticle size.53 Indeed detailed STEM-XEDS analysis of oxide supported AuPd nanoparticles has previously revealed a distinct relationship between particle size and elemental composition, with larger nanoparticles found to be Au-rich, while smaller nanoparticles are Pd-rich.54 In a similar manner we have demonstrated that analogously prepared carbon supported catalysts also adopt a size dependent nanoparticle-composition. However, unlike with catalysts prepared on oxide supports, which adopt a Au-core Pd-shell morphology after exposure to an oxidative heat treatment, analogous catalysts prepared on carbon have been found to maintain the random alloy morphology observed prior to calcination.55
We55 and others56,57 have extensively studied the synergistic effect achieved through the incorporation of Au into supported Pd catalysts with the reduction in contiguous Pd ensembles often attributed as the cause for the enhanced catalytic performance observed compared to monometallic analogues. Further studies have highlighted the role Au plays in electronically modifying Pd, with Au incorporation supressing O–O bond cleavage, inhibiting the formation of H2O, as well as promoting H2O2 desorption through weakening the interaction between the as-synthesised H2O2 and the catalyst surface.58–60 In particular the formation of Au-core Pd-shell nanoparticle morphology, commonly adopted upon the calcination of AuPd nanoparticles supported on oxide supports, has been widely reported to result in enhanced catalytic performance. Analysis of the various AuPd catalyst (Table S.2 and Fig. S.2†) via XPS reveals that the observed Pd:Au surface atomic ratios for the oxide supported catalysts are markedly higher than the nominal compositions, with these observations consistent with a Au-core Pd-shell nanoparticle morphology. By comparison, the Au:Pd surface ratio for the carbon supported catalyst is more in keeping with the formation of a homogeneous alloy, again with this aligning well with our previous studies.54,55
The as-prepared catalysts were investigated for their affinity to be retained in a hydrophobic solvent. Samples were added to a solvent with two distinct phases, decan-1-ol and water. After vigorous shaking it was observed (Fig. 1) that only the 5% AuPd/C remained in the alcohol phase, whereas the analogous TiO2, CeO2 and SiO2 supported catalysts were predominately present in the aqueous phase. These results are not unexpected as TiO2, CeO2 and SiO2 are more polar than C.
 | ||
| Fig. 1 Images of 5% AuPd/supported catalysts (carbon, SiO2, TiO2 and CeO2) in a two-phase solvent system consisting of decan-1-ol (top layer) and water (bottom layer). | ||
We have previously investigated the ability of these catalysts to synthesise H2O2 under conditions optimised to promote H2O2 stability, namely the use of a methanol–water solvent system and sub-ambient temperature1 (see Table S.3† for H2O2 synthesis and degradation under these conditions). Subsequently we have investigated the catalytic activity of these bi-metallic catalysts towards H2O2 degradation using a water/decan-1-ol solvent system, (Table S.4†) with the 5% AuPd/C catalyst demonstrating significantly lower rates of H2O2 degradation compared to oxide supported analogues, presumably due to the greater retention of the catalyst in the organic phase and decreased contact between catalyst and synthesised H2O2.
We have previously reported that the 5% AuPd/C catalyst, offers a reasonably high activity towards H2O2 synthesis using a methanol–water co-solvent system under reaction conditions that are similar to those reported herein (98 molH2O2 kgcat−1 h−1).3
We next investigated the efficacy of the 5% AuPd/C catalyst towards the direct synthesis of H2O2 in a range of straight chain alcohols (containing no aqueous co-solvent) as solvent (Table 1). Previous studies investigating the direct synthesis of H2O2 in short chain alcohol solvents have typically utilised acidic or halide promoters (or a combination thereof) to achieve high selectivity towards H2O2.20,61 Under our reaction conditions, where no halide or acidic promoters are utilised, low concentrations of H2O2 were produced in all alcohols studied with H2O2 concentration decreasing with increasing carbon chain length, with this possibly resulting from a number of factors including (i) decreased O2 solubility as a function of alcohol carbon-chain length,62 or (ii) increasing H2O2 degradation due to improved H2 solubility.24 The ability to produce H2O2 therefore strongly depends on the overall rate of mass transfer of gaseous reactants to the catalyst surface. Thus, the decreasing concentration of observed H2O2 is likely to be due to a mixture of both chemical and physical properties of the solvent such as higher viscosity and surface tension as the alcohol carbon chain length increases. A larger pressure drop of the reagent gases was also observed during reaction (Table S.5†), indicating increased solubility of reactant gases, although the possibility for increased solubility of the CO2 diluent should not be ruled out, in keeping with observations by Francesconi et al.24 and Wainwright et al.25 who have shown that H2 solubility in the C1–4 primary alcohol series increases with carbon chain length and therefore it is reasonable to expect H2 to dissolve better in decan-1-ol rather than methanol.
| Solvent | H2O2 productivity (molH2O2 kgcat−1 h−1) | [H2O2] (wt%) | H2O2 produced (μmol) | H2O solubilitya (gH2O kgsolvent−1) |
|---|---|---|---|---|
| H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent, 25 °C, 30 min (note: no H2O added). a As measured by Karl Fisher titration. | ||||
| Water | 10 | 0.016 | 50 | — |
| Methanol | 50 | 0.094 | 250 | — |
| Propan-1-ol | 32 | 0.063 | 160 | — |
| Butan-1-ol | 26 | 0.030 | 130 | — |
| Hexan-1-ol | 4.4 | 0.008 | 22 | 94 |
| Octan-1-ol | 1.4 | 0.003 | 7 | 53 |
| Decan-1-ol | 0.8 | 0.002 | 4.2 | 46 |
Another important consideration when selecting the organic solvent was the requirement to have low water solubility in the alcohol phase in order to create a bi-phasic solvent system, which would allow for ease of separation of H2O2. Table 1 shows that the use of a methanol-only solvent results in the highest H2O2 concentration (250 μmol). However, due to the complete miscibility of methanol with water, it is not a suitable solvent choice for this investigation. Higher carbon chain alcohols (>C6) have lower water solubility and the addition of excess water can create a bi-phasic solvent system.63 The saturation point of the alcohols in water has been determined experimentally by Karl Fischer titration (Table 2). As decan-1-ol was determined to have the lowest water solubility (46 gH2O kgdecan-1-ol−1), while also allowing for the formation of H2O2, it was chosen as a suitable solvent for the hydrophobic solvent layer of the proposed two-phase system. Furthermore, decan-1-ol has been shown to be stable under reaction conditions by NMR analysis (Fig. S.3†).
| Entry | Total pressure/psi | [H2O2]/wt% | H2O2 productivity/molH2O2 kgcat−1 h−1 |
|---|---|---|---|
| H2O2 synthesis reaction conditions: 5% AuPd/C (0.02 g), total pressure (580–700 psi), H2/O2 (0.5), 1200 rpm, 4 g total solvent (39 gH2O kgdecan-1-ol−1), 25 °C, 30 min. a Non-optimised reaction conditions. 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent (39 gH2O kgdecan-1-ol−1), 25 °C, 30 min. | |||
| 1a | 580 | 0.17 | 3.3 |
| 2 | 580 | 0.45 | 2.0 |
| 3 | 700 | 0.81 | 3.7 |
The effect of solvent composition on the direct synthesis of H2O2, by varying the water concentration in decan-1-ol, from 0 to 590 gH2O kgdecan-1-ol−1, was investigated (Fig. 2), while maintaining all other reaction variables, including total solvent mass which was maintained at 8.5 g. Increasing water content is observed to lead to an increase in H2O2 concentration, with the maximum H2O2 concentration (0.17 wt%) being observed at the saturation point of decan-1-ol; 46 gH2O kgdecan-1-ol−1 (0.39 g of H2O and 8.11 g of decan-1-ol). This is ascribed to the inhibition of H2O2 degradation (via hydrogenation and decomposition pathways) through the separation of H2O2 from the catalyst and the in situ formation of carbonic acid in the aqueous phase, through solvation of the CO2 gaseous diluent, with the improved stability of H2O2 under acidic conditions well known.64,65 Indeed similar improvements in H2O2 concentrations have been observed through the introduction of water into methanol solvent systems.1–3 The sharp increase in the H2O2 concentration up to 46 gH2O kgdecan-1-ol−1 demonstrates that the presence of water has a significant effect on the concentrations of H2O2 that can be achieved over the 5% AuPd/C catalyst. On further increasing the water content, to form a bi-phasic solvent, there was little effect on the H2O2 concentration up to 120 gH2O kgdecan-1-ol−1. Increasing H2O content beyond 150 gH2O kgdecan-1-ol−1 lead to a significant decrease in H2O2 concentration, with this attributed to a combination of decreased H2 availability, with increasing water content, and the inability of the decan-1-ol phase to retain the totality of the catalyst, leading to increased contact between catalyst and synthesised H2O2 and in turn increased H2O2 degradation.
 | ||
| Fig. 2 The effect of H2O content in a H2O/decan-1-ol solvent system on the direct synthesis of H2O2. H2O2 concentration (black squares). H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent (decan-1-ol + water), 25 °C, 30 min. (---) H2O saturation point of decan-1-ol. | ||
With water concentrations above 120 gH2O kgdecan-1-ol−1 (1 g H2O and 7.5 g decan-1-ol) it was possible to separate the water and decan-1-ol in order to directly calculate the H2O2 concentration in the aqueous phase. Comparing these extracted H2O2 concentrations with the results obtained from titrating the entire decan-1-ol and water solvent are shown in Fig. 3. When 120 gH2O kgdecan-1-ol−1 (1 g H2O and 7.5 g decan-1-ol) and 235 gH2O kgdecan-1-ol−1 (2 g H2O and 6.5 g decan-1-ol) were added the difference between the results obtained through analysis of the total reaction solution or the aqueous phase only is 16–17%, suggesting that at low H2O extraction of H2O2 is incomplete. The difference between the two measures of H2O2 content was subsequently decreased to 6% when the water phase was more easily separated from the decan-1-ol at 590 gH2O kgdecan-1-ol−1 (5 g H2O and 3.5 g decan-1-ol). This indicates that the efficiency of separating the two solvents will be a critical step to maintaining the formed H2O2 concentrations. All preceding results were obtained from titrating the whole solvent mixture to allow for accurate determination of H2O2 concentrations.
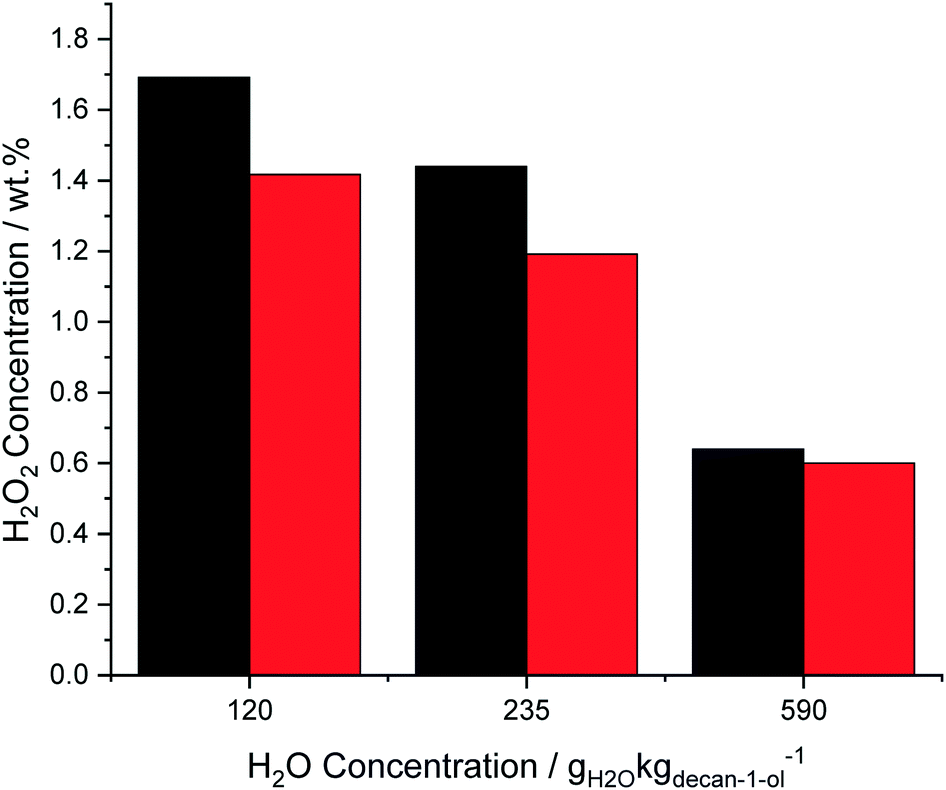 | ||
| Fig. 3 Calculation of the H2O2 concentration in a mixture of decan-1-ol and water (black bars) and the separated water phase (red bars). H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent (decan-1-ol + H2O), 25 °C, 30 min. | ||
The solvent system was next investigated for its ability to suppress H2O2 degradation, under an atmosphere of 5% H2/CO2 (Fig. 4). Reactions were carried out with increasing amounts of pre-formed 4 wt% aqueous H2O2 added to decan-1-ol; from a saturated decan-1-ol solution (0.39 g) to a truly bi-phasic solvent system (3.0 g), keeping the total solvent mass constant (8.5 g). When 0.39 g of 4 wt% H2O2, was added to decan-1-ol (8.11 g), to achieve a saturated decan-1-ol solution, a degradation activity of 29 molH2O2 kgcat−1 h−1 was observed. Catalytic activity towards H2O2 degradation was seen to increase to a value of 113 molH2O2 kgcat−1 h−1 when using 1 g of 4 wt% H2O2, before plateauing when a higher volume, 3.0 g of 4 wt% H2O2 was added to the decan-1-ol solvent. With this plateau ascribed to the ability of H2O2 to catalyse the oxidation of Pd, as previously reported by Choudhary et al.,64 with Pd2+ species known to be far less active towards H2O2 degradation than Pd0.66,67 The observed degradation activity, even at low amounts of H2O2 suggests that there is still some contact between catalyst and H2O2, which may be expected given the shear forces that result from high stirring speed used in this study it is promising that the amount of H2O2 degraded is relatively low. Indeed, it should be noted that the degradation activity observed using the proposed solvent system is far less than that previously reported over the same catalyst when using a miscible, H2O/methanol solvent system, under identical reaction conditions (352 molH2O2 kgcat−1 h−1), although there are clear differences in H2 solubility between H2O/methanol solvent systems and those used within this study.3
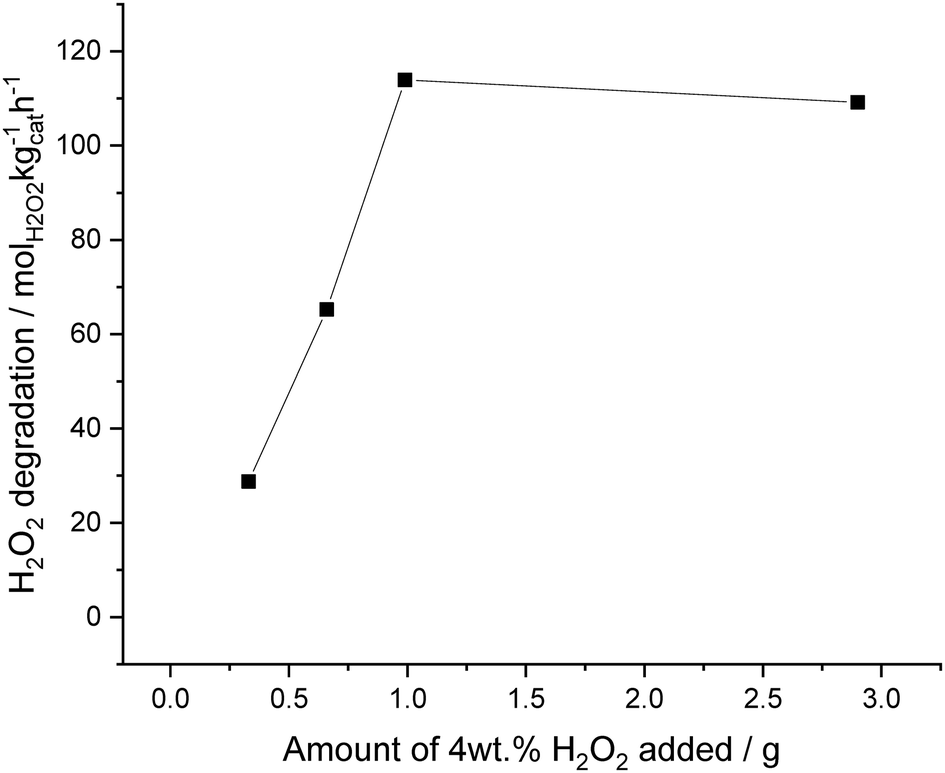 | ||
| Fig. 4 Catalytic activity of a 5% AuPd/C catalyst towards H2O2 degradation with increasing amounts of aqueous 4 wt% H2O2. H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent decan-1-ol + X g 4 wt% H2O2, 25 °C, 30 min. | ||
The effect of solvent mass, while maintaining the mass of the 5% AuPd/C catalyst at 0.01 g, was next investigated (Fig. 5). As expected these results demonstrate that it is possible to enhance the concentration of H2O2 by decreasing the total solvent mass, this is mainly due to a dilution effect, producing the same amount of H2O2 in a smaller volume and increasing H2 partial pressure relative to the solvent, due to increased reactor head space.
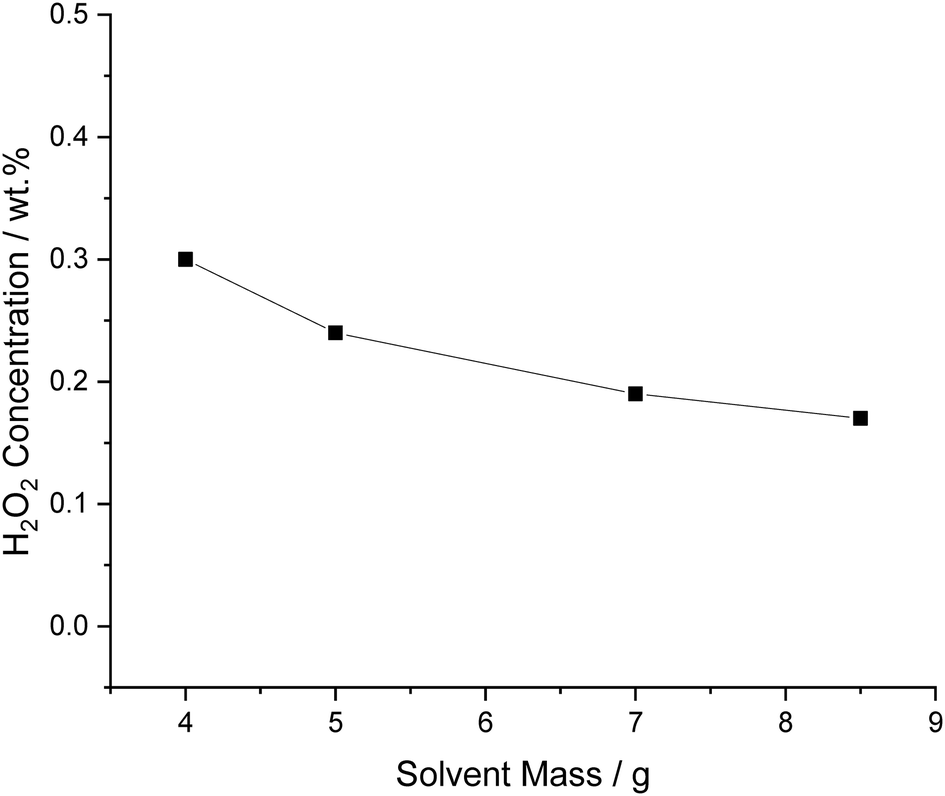 | ||
| Fig. 5 The effect of total solvent mass on catalytic activity towards H2O2 synthesis. H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, X g total solvent (46 gH2O kgdecan-1-ol−1), 25 °C, 30 min. | ||
The effect of varying catalyst mass from 0.005 to 0.04 g, whilst keeping the solvent mass constant at 8.5 g, was next investigated (Fig. 6). It can be seen that with increasing mass of catalyst, H2O2 concentration increases in a non-linear manner, to a value of 0.25 wt% when using 0.02 g of catalyst, beyond which the increase in H2O2 concentration is far less pronounced. It is likely that the observed plateau in H2O2 concentration can be attributed to a combination of factors; (i) limitations associated with diffusion of reactant gas; (ii) the inability of the decan-1-ol to retain the catalyst forcing a higher concentration of catalyst into the aqueous phase, thus further increasing contribution of H2O2 degradation.
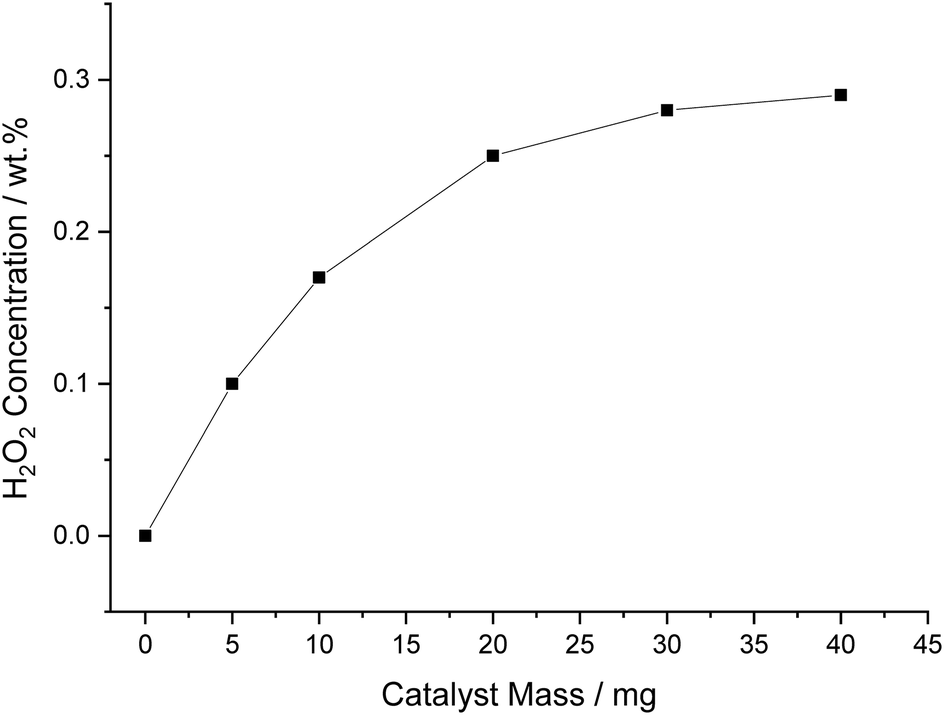 | ||
| Fig. 6 The effect of catalyst mass on catalytic activity towards the direct synthesis of H2O2. H2O2 synthesis reaction conditions: 5% AuPd/C (X mg), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent (46 gH2O kgdecan-1-ol−1), 25 °C, 30 min. | ||
Time-on-line analysis was next carried out, with the vast majority of H2O2 seen to be produced over 30 minutes (0.17 wt%), although a steady rise in H2O2 concentration is observed over 60 minutes (0.19 wt%) (Fig. 7). Beyond 60 minutes there is a minor decrease in H2O2 concentration, presumably, this is due to a depletion in reagent gas availability, in particular, H2 and the rate of H2O2 degradation exceeding that of H2O2 synthesis, with similar observations previously reported by Crole et al. in a range of solvents.2 However, it is promising that H2O2 concentrations can be generally retained, even at extended reaction times.
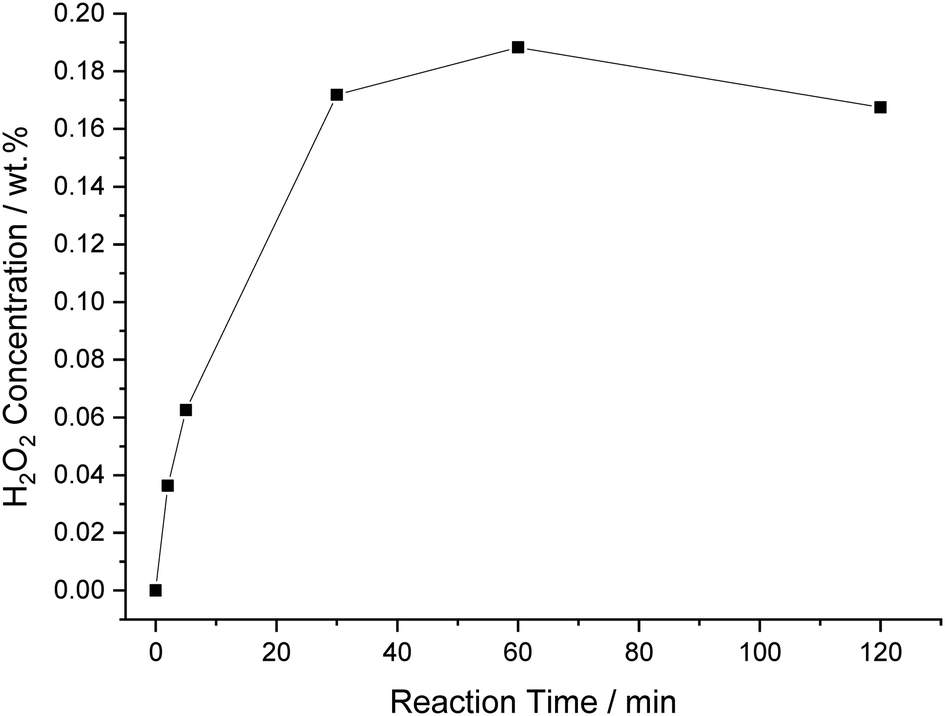 | ||
| Fig. 7 The effect of reaction time on catalytic activity towards the direct synthesis of H2O2. H2O2 synthesis reaction conditions: 5% AuPd/C (0.01 g), total pressure (580 psi), H2/O2 (0.5), 1200 rpm, 8.5 g total solvent (46 gH2O kgdecan-1-ol−1), 25 °C, X min. | ||
In an attempt to further enhance H2O2 concentration, optimised conditions were combined from the previous experiments carried out above (Fig. 2–7); 0.02 g catalyst, 4 g total solvent with 46 gH2O kgdecan-1-ol−1 and a reaction time of 30 minutes. It can be seen (Table 2, entry 2) that when the direct synthesis reaction was carried out with a total reactant gas pressure of 580 psi (H2:O2 = 0.5) it resulted in an H2O2 concentration of 0.45 wt%, over 2.5 times greater than that observed over non-optimised conditions (0.17 wt%). Further increasing total reaction pressure to 700 psi, while maintaining optimised reaction conditions (Table 2, entry 3) is observed to result in a 4-fold increase in H2O2 concentration (0.81 wt%) over standard conditions.
Finally, with the plateau in H2O2 concentration, previously observed (Fig. 7) we carried out a series of sequential H2O2 direct synthesis reactions under our optimised reaction conditions (Fig. 8). We observe a steady increase in H2O2 concentration up to 1.9 wt% after 5 sequential reactions. Although a limited volume of H2O2 is reported (0.16 ml of 1.9 wt% H2O2) at low productivities, the concentration achieved highlights the advantages of using a bi-phasic solvent for the direct synthesis of H2O2. If facile separation of the organic and aqueous phases can be achieved, the H2O2 concentration is comparable to that achieved in the initial stages of the current indirect method of industrial H2O2 production, prior to the use of multiple distillation steps to raise H2O2 concentrations to exceed ∼70 wt%.36
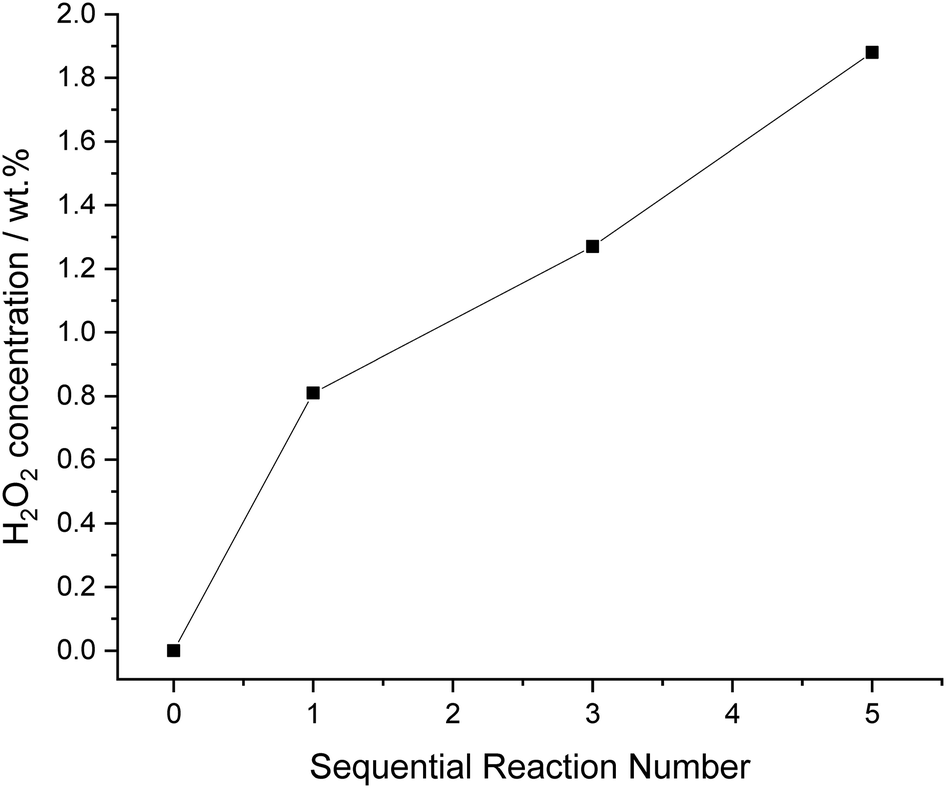 | ||
| Fig. 8 Sequential H2O2 synthesis reactions, under optimised reaction conditions using a 5% AuPd/C catalyst. H2O2 synthesis reaction conditions: 5% AuPd/C (0.02 g), total pressure (700 psi), H2/O2 (0.5), 1200 rpm, 4 g total solvent (46 gH2O kgdecan-1-ol−1), 25 °C, 30 min. | ||
Conclusion
In this paper the direct synthesis of H2O2 from H2 and O2 over a 5% AuPd/C catalyst has been investigated using a solvent system containing decan-1-ol and water. As a result of the preference for the catalyst to be retained in the hydrophobic, organic component, thus limiting contact between the synthesised H2O2 and catalyst a significant improvement in catalytic efficacy is observed, compared to the use of a miscible solvent mixture. Through optimisation of the reaction conditions, and assuming facile separation of the organic and aqueous solvent, we demonstrate that it is possible to reach concentrations of H2O2 comparable to that produced in the initial stages of H2O2 production, on an industrial scale, with a H2O2 concentration of 1.9 wt% reported. Perhaps more importantly we demonstrate the feasibility of a bi-phasic solvent system in allowing simultaneous reaction and extraction, minimising H2O2 degradation rates, resulting in increased concentrations of H2O2. We consider that this work represents a promising basis for further exploration of a wider set of solvents and catalysts for use in the direct synthesis of H2O2. In particular we believe that, given the high desirability for a continuous production of H2O2, this approach would lend itself well to a flow regime.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors wish to acknowledge the financial support of and research discussion with Solvay S. A.References
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- D. A. Crole, S. J. Freakley, J. K. Edwards and G. J. Hutchings, Proc. R. Soc. London, Ser. A, 2016, 472, 20160156 Search PubMed.
- S. J. Freakley, R. J. Lewis, D. J. Morgan, J. K. Edwards and G. J. Hutchings, Catal. Today, 2015, 248, 10–17 CrossRef CAS.
- T. Deguchi and M. Iwamoto, J. Catal., 2011, 280, 239–246 CrossRef CAS.
- E. N. Ntainjua, M. Piccinini, S. J. Freakley, J. C. Pritchard, J. K. Edwards, A. F. Carley and G. J. Hutchings, Green Chem., 2012, 14, 170 RSC.
- J. Edwards, B. Solsona, P. Landon, A. Carley, A. Herzing, C. Kiely and G. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- M. Piccinini, E. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488–2492 RSC.
- V. V. Krishnan, A. G. Dokoutchaev and M. E. Thompson, J. Catal., 2000, 196, 366–374 CrossRef CAS.
- R. Burch and P. R. Ellis, Appl. Catal., B, 2003, 42, 203–211 CrossRef CAS.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- R. E. Brooks and S. B. Moore, Cellulose, 2000, 7, 263–286 CrossRef CAS.
- C. Samanta, Appl. Catal., A, 2008, 350, 133–149 CrossRef CAS.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917–1923 RSC.
- C. M. Piqueras, J. García-Serna and M. J. Cocero, J. Supercrit. Fluids, 2011, 56, 33–40 CrossRef CAS.
- T. Moreno, J. García-Serna and M. J. Cocero, Green Chem., 2010, 12, 282–289 RSC.
- Q. Chen and E. J. Beckman, Green Chem., 2007, 9, 802–808 RSC.
- F. Moseley and P. N. Dyer, US Pat., 4336240, 1982 Search PubMed.
- I. T. Horváth, Acc. Chem. Res., 1998, 31, 641–650 CrossRef.
- M. Kawakami, Y. Ishiuchi, H. Nagashima, T. Tomita and Y. Hiramatsu, US Pat., 5399334, 1995 Search PubMed.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Appl. Catal., A, 2008, 339, 130–136 CrossRef CAS.
- S. Melada, F. Pinna, G. Strukul, S. Perathoner and G. Centi, J. Catal., 2006, 237, 213–219 CrossRef CAS.
- V. Paunovic, V. V. Ordomsky, V. L. Sushkevich, J. C. Schouten and T. A. Nijhuis, ChemCatChem, 2015, 7, 1161–1176 CrossRef CAS.
- E. J. Beckman and J. Supercrit, Fluids, 2004, 28, 121–191 CAS.
- J. V. H. d'Angelo and A. Z. Francesconi, J. Chem. Eng. Data, 2001, 46, 671–674 CrossRef.
- M. S. Wainwright, T. Ahn, D. L. Trimm and N. W. Cant, J. Chem. Eng. Data, 1987, 32, 22–24 CrossRef CAS.
- Z. Laughrey, E. Bear, R. Jones and M. A. Tarr, Ultrason. Sonochem., 2001, 8, 353–357 CrossRef CAS.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- T. Pospelova and N. Kobozev, Russ. J. Phys. Chem., 1961, 35, 584–587 Search PubMed.
- E. N. Ntainjua, M. Piccinini, J. C. Pritchard, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, ChemSusChem, 2009, 2, 575–580 CrossRef.
- S. Abate, P. Lanzafame, S. Perathoner and G. Centi, Catal. Today, 2011, 169, 167–174 CrossRef CAS.
- S. Melada, F. Pinna, G. Strukul, S. Perathoner and G. Centi, J. Catal., 2005, 235, 241–248 CrossRef CAS.
- E. N. Ntainjua, M. Piccinini, J. C. Pritchard, Q. He, J. K. Edwards, A. F. Carley, J. A. Moulijn, C. J. Kiely and G. J. Hutchings, ChemCatChem, 2009, 1, 479–484 CrossRef.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS.
- N. M. Wilson, J. Schröder, P. Priyadarshini, D. T. Bregante, S. Kunz and D. W. Flaherty, J. Catal., 2018, 368, 261–274 CrossRef CAS.
- H. Li, B. Zheng, Z. Pan, B. Zong and M. Qiao, Front. Chem. Sci. Eng., 2018, 12, 124–131 CrossRef CAS.
- Q. Liu and J. H. Lunsford, Appl. Catal., A, 2006, 314, 94–100 CrossRef CAS.
- Y. Kawase, Y. Isaka, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2019, 55, 6743–6746 RSC.
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS.
- X. Chen, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, J. Mater. Chem. A, 2020, 8, 1904–1910 RSC.
- J. E. Romo, N. V. Bollar, C. J. Zimmermann and S. G. Wettstein, ChemCatChem, 2018, 10, 4805–4816 CrossRef.
- V. M. Blasucci, Z. A. Husain, A. Z. Fadhel, M. E. Donaldson, E. Vyhmeister, P. Pollet, C. L. Liotta and C. A. Eckert, J. Phys. Chem. A, 2010, 114, 3932–3938 CrossRef CAS.
- X. Jin, J. Feng, H. Song, J. Yao, Q. Ma, M. Zhang, C. Yu, S. Li and S. Yu, Green Chem., 2019, 21, 3583–3596 RSC.
- G.-H. Han, X. Xiao, J. Hong, K.-J. Lee, S. Park, J.-P. Ahn, K.-Y. Lee and T. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 6328–6335 CrossRef CAS.
- Z. Cheng, R. Lippi, C. E. Li, Y. Yang, L. Tang, S. Huang, W. J. Lee, S. Lim, X. Ma and J. Patel, Ind. Eng. Chem. Res., 2019, 58, 20573–20584 CrossRef CAS.
- R. J. Lewis, J. K. Edwards, S. J. Freakley and G. J. Hutchings, Ind. Eng. Chem. Res., 2017, 56, 13287–13293 CrossRef CAS.
- E. N. Ntainjua, M. Piccinini, J. C. Pritchard, J. K. Edwards, A. F. Carley, C. J. Kiely and G. J. Hutchings, Catal. Today, 2011, 178, 47–50 CrossRef CAS.
- E. N. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 1162 RSC.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y.-F. Han, J. Catal., 2017, 349, 30–40 CrossRef CAS.
- A. Cybula, J. B. Priebe, M.-M. Pohl, J. W. Sobczak, M. Schneider, A. Zielińska-Jurek, A. Brückner and A. Zaleska, Appl. Catal., B, 2014, 152–153, 202–211 CrossRef CAS.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- L. Ouyang, P.-F. Tian, G.-J. Da, X.-C. Xu, C. Ao, T.-Y. Chen, R. Si, J. Xu and Y.-F. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, D. Kovačič, D. J. Morgan, T. E. Davies, J. K. Edwards, M. S. Skjøth-Rasmussen and G. J. Hutchings, Catal. Lett., 2020 DOI:10.1007/s10562-020-03281-1.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225 RSC.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388 RSC.
- S. Kanungo, V. Paunovic, J. C. Schouten and M. F. Neira D'Angelo, Nano Lett., 2017, 17, 6481–6486 CrossRef CAS.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. Neira d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- A. V. Beletskaya, D. A. Pichugina, A. F. Shestakov and N. E. Kuz'menko, J. Phys. Chem. A, 2013, 117, 6817–6826 CrossRef CAS.
- F. Menegazzo, P. Burti, M. Signoretto, M. Manzoli, S. Vankova, F. Boccuzzi, F. Pinna and G. Strukul, J. Catal., 2008, 257, 369–381 CrossRef CAS.
- S. Bo, R. Battino and E. Wilhelm, J. Chem. Eng. Data, 1996, 41, 644 CrossRef CAS.
- N. Šegatin and C. Klofutar, Monatsh. Chem., 2004, 135, 241–248 CrossRef.
- V. R. Choudhary, C. Samanta and T. V. Choudhary, J. Mol. Catal. A: Chem., 2006, 260, 115–120 CrossRef CAS.
- V. R. Choudhary, C. Samanta and P. Jana, Appl. Catal., A, 2007, 317, 234–243 CrossRef CAS.
- A. G. Gaikwad, S. D. Sansare and V. R. Choudhary, J. Mol. Catal. A: Chem., 2002, 181, 143–149 CrossRef CAS.
- G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. G. Fierro, Chem. Commun., 2004, 1184–1185 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01163k |
| This journal is © The Royal Society of Chemistry 2020 |