
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced catalyst selectivity in the direct synthesis of H2O2 through Pt incorporation into TiO2 supported AuPd catalysts†
Xiaoxiao
Gong‡
ab,
Richard J.
Lewis‡
 a,
Song
Zhou
cde,
David J.
Morgan
a,
Thomas E.
Davies
a,
Xi
Liu
cf,
Christopher J.
Kiely
ag,
Baoning
Zong
*b and
Graham J.
Hutchings
*a
a,
Song
Zhou
cde,
David J.
Morgan
a,
Thomas E.
Davies
a,
Xi
Liu
cf,
Christopher J.
Kiely
ag,
Baoning
Zong
*b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: Hutch@cardiff.ac.uk
bLaboratory of Catalytic Materials and Chemical Engineering, Research Institute of Petroleum Processing, SINOPEC, Beijing, 100083, P.R. China. E-mail: Zongbn.ripp@sinopec.com
cSynCat@Beijing, Synfuels China Technology Co. Ltd., Beijing, 101407, P.R. China
dState Key Laboratory of Coal Convers, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, P.R. China
eSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing, 100049, P.R. China
fSchool of Chemistry and Chemical Engineering, In-situ Centre for Physical Science, Shanghai Jiao Tong University, 200240, Shanghai, P. R. China
gDepartment of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA
First published on 24th June 2020
Abstract
The introduction of small quantities of Pt into supported AuPd nanoparticles is found to result in enhanced catalytic efficiency in the direct synthesis of H2O2. This is attributed to a combination of superior H2O2 synthesis rates, as determined through calculation of initial rates of reaction, and an inhibition of H2O2 degradation pathways, achieved through the modification of Pd oxidation states. Through gas replacement experiments we demonstrate that it is possible to reach concentrations of H2O2 approaching those produced during initial stages of the current industrial means of H2O2 production.
Introduction
Hydrogen peroxide (H2O2) is a versatile, environmentally friendly oxidant that finds applications as a bleaching agent in the pulp and textile industry,1 the treatment of waste streams2,3 and is finding growing use in the production of both commodity and fine chemicals. With the demand from the chemical sector in particular driven by the growing need for both propylene oxide, via the integrated HPPO process,4,5 and cyclohexanone oxime, a key intermediate in the production of Nylon-6.6 In recent years, global H2O2 production has exceeded 3 million tons per annum7 and is predicted to continue to grow at a rate of 4% per year to exceed 4 million tons per annum by 2020.8Currently the global demand for H2O2 is met by the highly efficient anthraquinone oxidation (AO) or indirect synthesis process, first developed by BASF in 1939.9 The AO process has undergone numerous improvements since, but the underlying chemistry has changed little, utilising H2, O2 and an anthraquinone derivative, where the anthraquinone molecule undergoes sequential hydrogenation and oxidation steps to generate H2O2, while avoiding the risk of combining H2 and O2 directly. This process is able to initially yield H2O2 concentrations of 1–2 wt%, which through further distillation and purification steps can be raised to exceed 70 wt%; a concentration which can then be shipped and stored prior to dilution at point of use.
Despite the AO process being highly efficient there are some concerns regarding its carbon efficiency, with the over-hydrogenation of the anthraquinone carrier-molecule necessitating its replacement periodically. This coupled with the high infrastructure costs and complexity of the process has often prevented the large-scale generation of H2O2 at point of use. In addition, the instability of H2O2, undergoing rapid decomposition to H2O at relatively mild temperatures or in the presence of weak bases requires the use of acidic stabilising agents, which result in additional purification steps to prevent contamination of product streams and decreased reactor lifetime due to corrosion, raising costs to the end user.
The catalysed direct synthesis of H2O2 from molecular H2 and O2 offers an attractive alternative to the current means of H2O2 production on an industrial scale and would allow for H2O2 production to be adopted at point of use. Since 191410 Pd-based catalysts have received significant attention within both the academic11,12 and patent13,14 literature. However, issues around catalytic selectivity have prevented commercialisation of the direct synthesis process despite over 100 years of academic pursuit. The issue of catalyst selectivity is easy to understand given that that formation of H2O is thermodynamically favoured compared to H2O2, as summarised in Scheme 1.
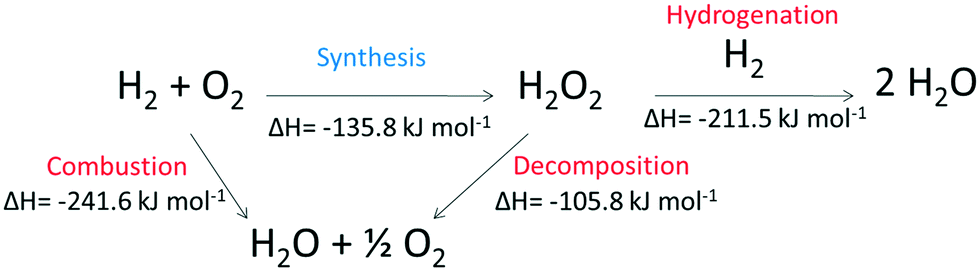 | ||
| Scheme 1 Reaction pathways associated with the direct synthesis of H2O2 from H2 and O2. | ||
In order to overcome limitations around selectivity, halide salts (e.g. NaBr)15–17 or mineral acids (e.g. HCl, HNO3)18,19 have often been employed, with Pospelova et al.20 first demonstrating increased yields of H2O2 through their application alongside a supported Pd catalyst. Although it is clear that the use of halide additives can greatly enhance catalytic selectivity, the means by which this effect is achieved is still ambiguous. Nevertheless, the use of halide and acid additives offers significant drawbacks to the user, akin to those associated with H2O2 generated via the anthraquinone process and significant attention has been placed on enhancing catalytic selectivity through catalytic design. With the incorporation of Au into Pd,21–25 a particularly well-studied catalytic system for the production of H2O2, the need for acid or halide stabilising agents is removed. The means by which the incorporation of Au into Pd-based catalysts enhances catalytic activity is still of some debate, with electronic, structural and isolation effects all being cited as potential causes. However, conclusive evidence on the nature of catalytic enhancement is still lacking and it is likely that a combination of these factors are responsible for the observed synergy. More recently Freakley et al.26 have demonstrated that it is possible to exchange Au with a range of secondary base metals to reach selectivity levels towards H2O2 in excess of 95% and this has prompted the further investigation of Pd modification with a range of non-precious metals.27–30
Further studies have demonstrated that the incorporation of low concentrations of Pt into supported Pd or AuPd catalysts can greatly enhance catalytic activity towards the direct synthesis of H2O2. Indeed, a comprehensive study by Deguchi et al.31 revealed that the incorporation of Pt into a Pd-polyvinylpyrrolidone colloid resulted in a significant increase in catalytic activity, which was attributed to the ability of Pt to readily adsorb dissociated H2. However, this rise in catalytic activity, with H2O2 formation rates doubling upon incorporation of 0.5 at% Pt, came at the expense of catalytic selectivity. By comparison we have previously demonstrated that the addition of Pt into AuPd catalysts in small concentrations enhances catalytic performance, through inhibition of H2O2 degradation pathways and leads to improved selectivity towards H2O2.32,33 However, our previous studies have focussed on catalysts prepared by a conventional wet co-impregnation procedure, primarily due to the simplicity and industrial applicability of this methodology. It is important to note that a limitation of catalysts prepared via this procedure is the considerable variation in elemental composition with nanoparticle size, with larger particles generally being Au-rich, while smaller nanoparticles are predominantly Pd-rich. As such it has been difficult to determine the key parameter responsible for the enhancement in catalytic performance upon Pt incorporation into supported AuPd nanoparticles, with the modification of Pd oxidation state and changes in mean particle size both possible causes for the observed improvement. By comparison to catalysts prepared by wet impregnation, those produced via a sol-immobilisation methodology offer better control of particle size and elemental composition.34 As such, this study now focusses on the efficacy of Pt introduction into AuPd catalysts prepared via a sol-immobilisation methodology.
Experimental methods
Catalyst preparation
Mono-, bi- and tri-metallic 1% AuPdPt/TiO2 (total metal loading of 1 wt%) catalysts have been prepared (on a molar basis) by a sol-immobilisation procedure, based on methodology previously reported in the literature, which has been shown to result in enhanced precious metal dispersion by limiting particle growth and agglomeration.34 The procedure to produce 1% Au1Pd1Pt1/TiO2 (1 g) is outlined below (where the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd:Pt molar ratio is fixed at 1:1:1) with a similar methodology utilised for mono- and bi-metallic catalysts.
:Pd:Pt molar ratio is fixed at 1:1:1) with a similar methodology utilised for mono- and bi-metallic catalysts.
Aqueous solutions of HAuCl4·3H2O (0.322 mL, 12.25 mg mL−1, Strem Chemicals), PdCl2 (0.356 mL, 6 mg mL−1, Sigma Aldrich) and H2PtCl6·6H2O (0.285 mL, 13.76 mg mL−1, Sigma Aldrich) were added to deionised water (400 mL) under vigorous stirring conditions at room temperature. The resulting solution was allowed to stir for 2 minutes prior to the addition of polyvinylalcohol (PVA) (1.30 mL, 1 wt% MW = 9000–10000 g mol−1, 80% hydrolysed, Sigma Aldrich) such that the weight ratio of metal:PVA was 1:1.3. The resulting solution was stirred for 2 minutes prior to the addition of a freshly prepared solution of NaBH4 (4.015 mL, 0.1 M), such that the molar ratio of NaBH4:(Au + Pd) was 5:1 and the molar ratio of NaBH4:Pt was 10:1. Upon the addition of NaBH4 the mixture turned dark brown and was stirred vigorously for an additional 30 min followed by the addition of TiO2 (0.99 g, Degussa P25). The solution was acidified to pH 1 via the addition of H2SO4 (>95%) and stirred for 1 h. Following this, the suspension was filtered under vacuum, washed thoroughly with distilled water, then dried (110 °C, 16 h) and calcined (400 °C, 3 h, 10 °C min−1, static air).
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 mL, equipped with a PTFE liner so that total liquid volume is reduced to 66 mL, and a maximum working pressure of 14 MPa. To test each catalyst for H2O2 synthesis, the autoclave liner was charged with catalyst (0.01 g) and solvent (5.6 g methanol and 2.9 g H2O). The charged autoclave was then purged three times with 5% H2/CO2 (0.7 MPa) before filling with 5% H2/CO2 to a pressure of 2.9 MPa, followed by the addition of 25% O2/CO2 (1.1 MPa). A pressure of 5% H2/CO2 and 25% O2/CO2 are given as gauge pressures. The reaction was conducted at a temperature of 2 °C for 0.5 h with stirring (1200 rpm). The above reaction parameters are based on optimum conditions we have previously used for the synthesis of H2O2.35 The H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.0085 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.The catalytic conversion of H2 and selectivity towards H2O2 were determined using a Varian 3800 GC fitted with TCD and equipped with a Porapak Q column.
H2 conversion (eqn (1)) and H2O2 selectivity (eqn (2)) are defined as follows:
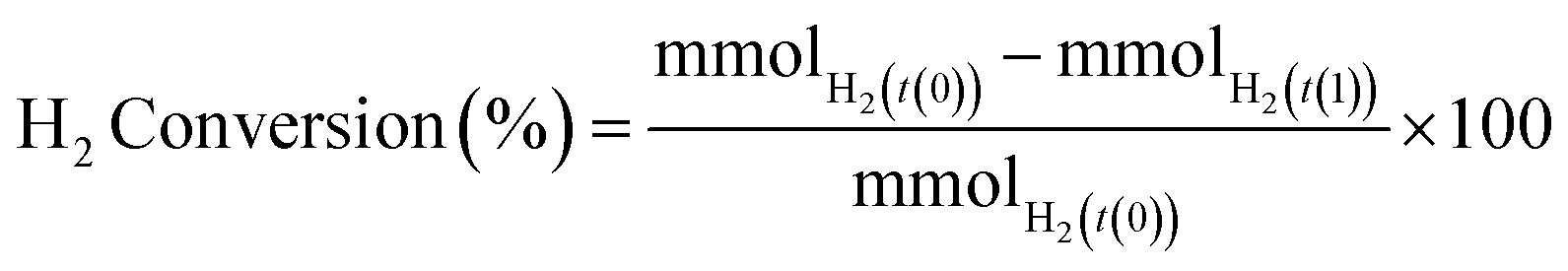 | (1) |
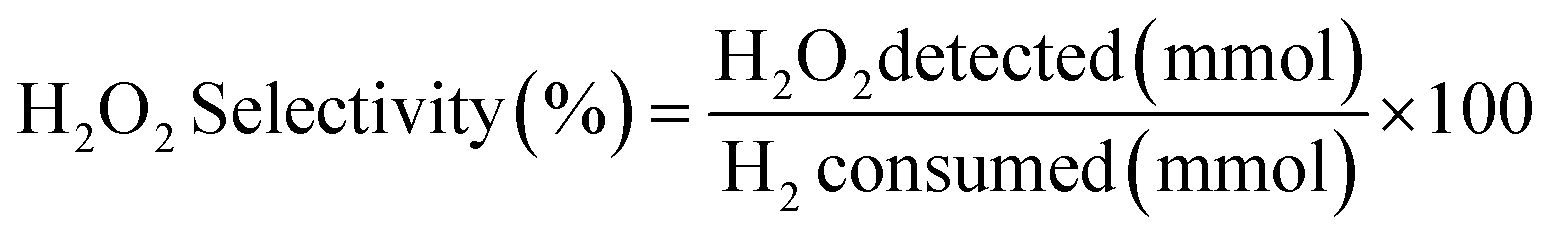 | (2) |
Degradation of H2O2
Catalytic activity towards H2O2 degradation was determined in a similar manner to the direct synthesis activity of a catalyst. The autoclave liner was charged with methanol (5.6 g), H2O2 (50 wt% 0.69 g), HPLC standard H2O (2.21 g) and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution, two 0.05 g aliquots were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The autoclave was pressurised with 2.9 MPa 5% H2/CO2 (gauge pressure). The reaction was conducted at a temperature of 2 °C, for 0.5 h with stirring (1200 rpm). After the reaction was complete the catalyst was removed from the reaction mixture and two 0.05 g aliquots were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.The reactor temperature was controlled using a HAAKE K50 bath/circulator using an appropriate coolant.
Catalyst reusability in the direct synthesis and degradation of H2O2
In order to determine catalyst reusability, a similar procedure to that outlined above for the direct synthesis of H2O2 is followed utilising 0.05 g of catalyst. Following the initial test, the catalyst was recovered by filtration and dried (30 °C, 17 h, under vacuum); from the recovered catalyst sample 0.01 g was used to conduct a standard H2O2 synthesis or degradation test.Catalyst characterisation
The as-prepared aqueous sols, contained in a quartz cuvette, were optically characterised using a UV-vis spectrometer (V-570, JASCO) operating over the 200 to 800 nm wavelength range.X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C(1s) binding energy of adventitious carbon contamination that was taken to be 284.8 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans, while 40 eV was employed for more detailed regional scans. The intensities of the Au(4f), Pt(4f) and Pd(3d) features were used to derive the Pd/Pt and Au/Pt surface composition ratios.
Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dispersion in ethanol by sonication and deposited on 300 mesh copper grids coated with holey carbon film. Energy dispersive X-ray spectroscopy (XEDS) was performed using an Oxford Instruments X-MaxN 80 detector and the data analysed using Aztec software. Aberration corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected Hitachi HF5000 S/TEM, operating at 200 kV. The instrument was equipped with bright field (BF), high angle annular dark field (HAADF) and secondary electron (SE) detectors for high spatial resolution STEM imaging experiments. This microscope was also equipped with a secondary electron detector and dual Oxford Instruments XEDS detectors (2 × 100 mm2) having a total collection angle of 2.02 sr.
Total metal leaching from the supported catalyst was quantified via inductively coupled plasma mass spectrometry (ICP-MS). Post-reaction solutions were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All samples were diluted by a factor of 10 using HPLC grade H2O (1% HNO3 and 0.5% HCl matrix). All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent.
DRIFTS measurements were taken on a Bruker Tensor 27 spectrometer fitted with a mercury cadmium telluride (MCT) detector. A sample was loaded into the Praying Mantis high temperature (HVC-DRP-4) in situ cell before exposure to N2 and then 1% CO/N2 at a flow rate of 50 cm3 min−1. A background spectrum was obtained using KBr, and measurements were recorded every 1 min at room temperature. Once the CO adsorption bands in the DRIFT spectra ceased to increase in size, the gas feed was changed back to N2 and measurements were repeated until no change in subsequent spectra was observed.
Results and discussion
Prior to immobilisation the as-synthesised Au–Pd–Pt colloids were analysed by UV-vis spectrometry (Fig. S1†) with no characteristic plasmon resonance band for Au being observed in the bi- and tri-metallic colloids, suggesting the formation of alloyed nanoparticles. Our initial studies, under conditions previously optimised for H2O2 synthesis, investigated the efficacy of supported monometallic (Au, Pd, Pt) and bi-metallic (AuPd, AuPt and PtPd) catalysts supported on TiO2 for the direct synthesis of H2O2 and its subsequent degradation, via hydrogenation and decomposition pathways, as shown in Table 1. As previously reported, the activity of the immobilised Au-only catalyst towards H2O2 synthesis is limited (4 molH2O2 kgcat−1 h−1). By comparison, the 1 wt% Pd/TiO2 catalyst was observed to offer a marginally higher activity towards both H2O2 production, with a higher synthesis rate (11 molH2O2 kgcat−1 h−1) and subsequent degradation (59 molH2O2 kgcat−1 h−1). In keeping with numerous previous studies, the co-immobilisation of Au and Pd is seen to result in an enhancement in catalytic activity towards H2O2 synthesis35 (81 molH2O2 kgcat−1 h−1), far greater than the activity observed over a physical mixture of the two mono-metallic catalysts (7 molH2O2 kgcat−1 h−1). It should be noted that the H2O2 synthesis activity of the 1% AuPd/TiO2 catalyst, prepared via the sol-immobilisation procedure is comparable to that observed for an analogous catalyst prepared via modified impregnation, where relatively high concentrations of HCl are utilized to enhance metal dispersion, (80 molH2O2 kgcat−1 h−1)35 and somewhat greater than that for the analogous catalyst prepared by conventional wet-impregnation (64 molH2O2 kgcat−1 h−1). While these latter methodologies may be more attractive for catalyst synthesis on an industrial scale, they typically result in a wider variation in particle size and elemental composition than catalysts produced via a sol-immobilisation technique.34 As such the sol-immobilisation procedure has clear advantage in producing model systems, where tight control of catalytic parameters are necessary.| Catalyst | Productivitya molH2O2 kgcat−1 h−1 | Degradationb molH2O2 kgcat−1 h−1 |
|---|---|---|
| a H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. b H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. c Reaction conditions identical to those outlined above, using 0.005 g of each catalyst. | ||
| 1% Au/TiO2 | 4 | 27 |
| 1% Pd/TiO2 | 11 | 59 |
| 1% Pt/TiO2 | 9 | 340 |
| 1% Au1Pd1/TiO2 | 81 | 257 |
| 1% Au1Pt1/TiO2 | 30 | 243 |
| 1% Pd1Pt1/TiO2 | 18 | 316 |
| 0.5% Au/TiO2 + 0.5% Pd/TiO2c | 7 | 41 |
| TiO2 | 0 | 0 |
We have previously reported that an improvement in catalytic selectivity towards H2O2 can be achieved through the introduction of small quantities of Pt into AuPd nanoparticles, prepared by a conventional wet co-impregnation methodology, dispersed on a range of supports.32,36 Additional studies have reported a similar enhancement in catalytic efficacy for a range of selective oxidation reactions, using supported AuPdPt catalysts prepared by a sol-immobilisation methodology.37,38 Building on our initial findings, we next investigated the effect of Pt addition on the catalytic activity of 1% Au1Pd1/TiO2 towards H2O2 synthesis (Fig. 1). In keeping with our previous studies, the addition of a small quantity of Pt (approx. 0.006 wt%) significantly enhances H2O2 synthesis rates, from 81 molH2O2 kgcat−1 h−1 for the 1% Au1Pd1/TiO2 catalyst to 112 molH2O2 kgcat−1 h−1 for the 1% Au1Pd1Pt0.01/TiO2 catalyst. However, further addition of Pt is observed to lead to a decrease in catalytic activity towards H2O2 synthesis, with this metric decreasing to a value of 30 molH2O2 kgcat−1 h−1 for the 1% Au1Pd1Pt1/TiO2 catalyst.
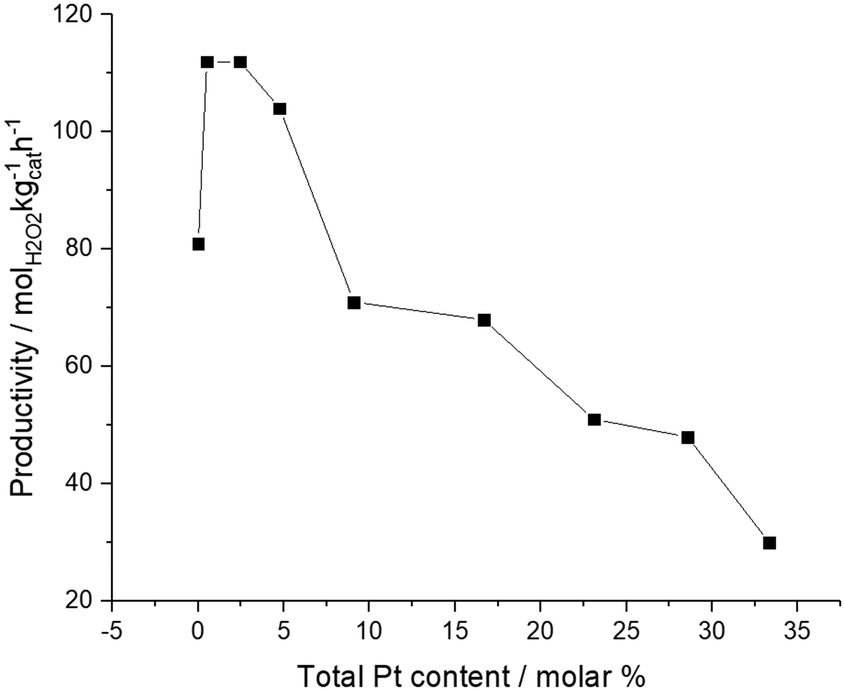 | ||
| Fig. 1 The effect of Pt incorporation into 1% AuPd/TiO2 on catalytic activity towards the direct synthesis of H2O2. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 ° C, 1200 rpm. | ||
The observation of a strong dependency between catalytic activity towards H2O2 synthesis and Pt content in the sol-immobilised materials, motivated us to further investigate the structure–activity relationships existing over the 1% Au1Pd1/TiO2, 1% Au1Pd1Pt0.01/TiO2 and 1% Au1Pd1Pt1/TiO2 catalysts.
An assessment of catalytic selectivity towards H2O2 and H2 conversion of the systematic set of 1% AuPdPt/TiO2 catalysts is presented in Table 2. In keeping with the lower rates of H2O2 degradation and higher yield of H2O2, the 1% Au1Pd1Pt0.01/TiO2 catalyst displayed a selectivity towards H2O2 (37%) which was greater than that of the 1% Au1Pd1/TiO2 (31%) or 1% Au1Pd1Pt1/TiO2 (15%) catalysts, while all catalysts displayed similar rates of H2 conversion.
| Catalyst | H2 conversion/% | H2O2 selectivity/% | Productivity/molH2O2 kgcat−1 h−1 | H2O2 concentration/wt% | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C 1200 rpm. | |||||
| 1% Au1Pd1/TiO2 | 39 | 31 | 81 | 0.16 | 257 |
| 1% Au1Pd1Pt0.01/TiO2 | 43 | 37 | 112 | 0.22 | 245 |
| 1% Au1Pd1Pt1/TiO2 | 44 | 15 | 30 | 0.10 | 271 |
| 1% Pt/TiO2 | 20 | 8 | 11 | 0.02 | 340 |
Evaluation of the as prepared Pt incorporated 1% AuPd/TiO2 catalysts by XPS can be seen in Table 3 (corresponding spectra in Fig. S.2†). Upon introduction of low quantities of Pt (approx. 0.006 wt%) the surface Pd:Au ratio remains unchanged, with further addition resulting in a minor decrease of the Pd:Au ratio. This can be attributed to a combination of significant decrease in mean particle size, as determined by TEM (Table 4) and the surface migration of Pt and disruption of the Pd-rich surface, which is a similar effect to that previously observed by Kondrat et al.38 Perhaps more interesting is the significant decrease in the Pd2+:Pd0 ratio upon Pt incorporation, with this value decreasing from a value of 1.3 for the bimetallic 1% Au1Pd1/TiO2 catalyst to 0.9 for the 1% Au1Pd1Pt0.01/TiO2 catalyst (coinciding with an enhancement in catalytic selectivity towards H2O2) with Pd0 content continuing to increase upon further Pt incorporation. This may be surprising given the low selectivity of Pd0 species towards H2O2 that has been well reported in the literature.39,40 However, Ouyang et al.41 have recently reported the enhanced selectivity and activity of supported Pd catalysts containing Pd0–Pd2+ ensembles in comparison to those catalysts with a predominance of Pd in either oxidation state. This improvement can be ascribed to the propensity of H2 to dissociate on Pd0 and the enhanced stability of O2 on Pd2+ surfaces, with the maintenance of the O–O bond required for the formation of H2O2 over H2O. It is of note to highlight the similarity in Pd2+:Pd0 oxidation ratio between the 1% PdPt/TiO2 (1.1) and 1% AuPd/TiO2 (1.3) catalysts, despite the significant differences observed in H2O2 synthesis activity, 18 and 81 molH2O2 kgcat−1 h−1 respectively for these catalysts. This clearly highlights the importance of Au incorporation into precious metal catalyst, as well reported in the literature.42–44
| Catalyst | Au:Pt |
Pd:Au |
Pd2+:Pd0 |
|---|---|---|---|
| All catalysts calcined, 400 °C, 3 h, 10 °C min−1 in static air. | |||
| 1% Au1Pt1/TiO2 | 0.6 | — | — |
| 1% Pd1Pt1/TiO2 | — | — | 1.1 |
| 1% Au1Pd1/TiO2 | — | 1.9 | 1.3 |
| 1% Au1Pd1Pt0.01/TiO2 | 0.8 | 1.9 | 0.9 |
| 1% Au1Pd1Pt1/TiO2 | 0.5 | 1.9 | 0.6 |
| Catalyst | Mean particle size/nm (standard deviation) | Productivity/molH2O2 kgcat−1 h−1 (H2O2 wt%) |
|---|---|---|
| All catalysts calcined, 400 °C, 3 h, 10 °C min−1 in static air. | ||
| 1% Au1Pd1/TiO2 | 4.2 (0.98) | 81 (0.16) |
| 1% Au1Pd1Pt0.01/TiO2 | 3.7 (0.55) | 112 (0.22) |
| 1% Au1Pd1Pt1/TiO2 | 1.8 (0.56) | 30 (0.10) |
It is therefore possible to relate the enhanced catalytic performance of the 1% Au1Pd1Pt0.01/TiO2 catalyst, compared to either the 1% AuPd/TiO2 catalyst or Pt-rich analogue to the development of these Pd0–Pd2+ domains. It can be inferred that the increased degradation rates observed over the Pt-rich 1% Au1Pd1Pt1/TiO2 catalyst results from an increase in Pd0 content, at the expense of Pd2+.
The CO-DRIFTS spectra of the as prepared 1% AuPdPt/TiO2 catalysts can be seen in Fig. 2. In the case of the 1% Au1Pd1/TiO2 catalyst, the DRIFTS spectra are typically dominated by Pd–CO bands. The peak observed at 2090 cm−1 represents linearly bonded CO to Pd atoms of low coordination (i.e., edge or corner sites) – denoted (Pd–CO) – while the broad feature that begins at 1950 cm−1 represents the 2- and 3-fold adsorption of CO on Pd.45 Upon the introduction of small quantities of Pt into AuPd, a small red-shift of the band related to the linearly bonded CO on Pd sites is observed, from 2090 to 2087 cm−1. This shift is possibly a result of the charge-transfer to Pd d-orbitals, resulting in enhanced back donation to 2π CO molecular orbitals. In keeping with our observations, Ouyang et al.46 have previously reported a similar transfer of electron density upon the alloying of Au and Pd with an associated suppression of O–O bond scission and enhancement in catalytic selectivity towards H2O2 synthesis.
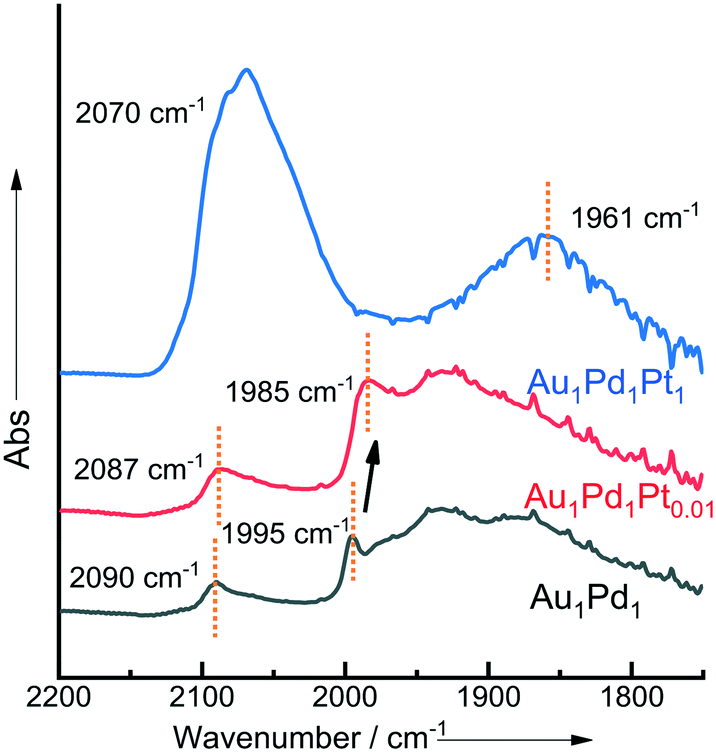 | ||
| Fig. 2 CO-DRIFTS spectra for selected sol-immobilised 1% AuPdPt/TiO2 catalysts in the as-prepared state. | ||
Catalytic activity towards H2O2 synthesis, in particular that of monometallic Pd catalysts, is widely reported to be dependent on particle size.47,48 Indeed Tian et al.49 have recently reported that an optimal particle size in the sub-nanometer range is desirable for achieving high activity and selectivity towards H2O2, with monodisperse atoms demonstrating an extremely low activity towards H2O2 formation, while larger nanoparticles offer greater activity towards the subsequent degradation pathways. Unlike with other means of catalyst preparation, such as wet-impregnation, the sol-immobilisation synthesis procedure allows for good control of mean particle size.34 Measurements of mean particle size for the various 1% AuPdPt/TiO2 catalysts (as determined from the bright field transmission electron micrographs presented in Fig. S3†) are shown in Table 4. This data reveals that the mean particle size is quite similar for the 1% Au1Pd1/TiO2 (4.2 nm) and 1% Au1Pd1Pt0.01/TiO2 (3.7 nm) catalysts despite their distinctly different catalytic performances. Given the comparable particle size, it is therefore reasonable to propose that the enhancement in catalytic activity cannot be associated with metal dispersion and instead is related to the electronic modification of Pd, as indicated by XPS and CO-DRIFTS.
Further comparison of the as-prepared 1% AuPdPt/TiO2 catalysts using complementary BF-, HAADF- and SE- AC-STEM imaging was also carried out (Fig. S4†) to illustrate the good size control and uniform dispersion of the alloy particles on the TiO2 support, with EDX analysis demonstrating the presence of precious metals in keeping with nominal ratios (Table S1†). In addition, X-ray energy dispersive spectroscopy (X-EDS) spectrum imaging and point analyses of individual particles was performed as shown in Fig. 3 to demonstrate in all cases that intimate alloying of the constituent metallic elements has occurred. Previous studies of AuPdPt nanoparticles supported on TS-1 prepared by a conventional wet-impregnation methodology have demonstrated a strong correlation between nanoparticle size and elemental composition, with larger particles (>20 nm) typically being Au-rich.32 It is expected that the sol-immobilisation preparation methodology utilised here should facilitate better control over nanoparticle elemental composition.50 In keeping with previous reports by Dimitratos et al.,51 we did not observe the development of any Au-core/Pd-shell morphologies in our sol-immobilised samples, that are typically found for AuPd nanoparticles prepared on oxide supports by impregnation methods in this case, the Au–Pd particles are a homogeneous random alloy as indicated by the STEM-XEDS elemental mapping (1% Au1Pd1/TiO2, Fig. 3a). The Pt was not detectable by X-EDS mapping at very low concentrations (1% Au1Pd1Pt0.01/TiO2, Fig. 3b) but is clearly discernible at higher concentrations (1% Au1Pd1Pt1/TiO2, Fig. 3c) where it was found to be uniformly dispersed throughout the AuPd nanoparticles.
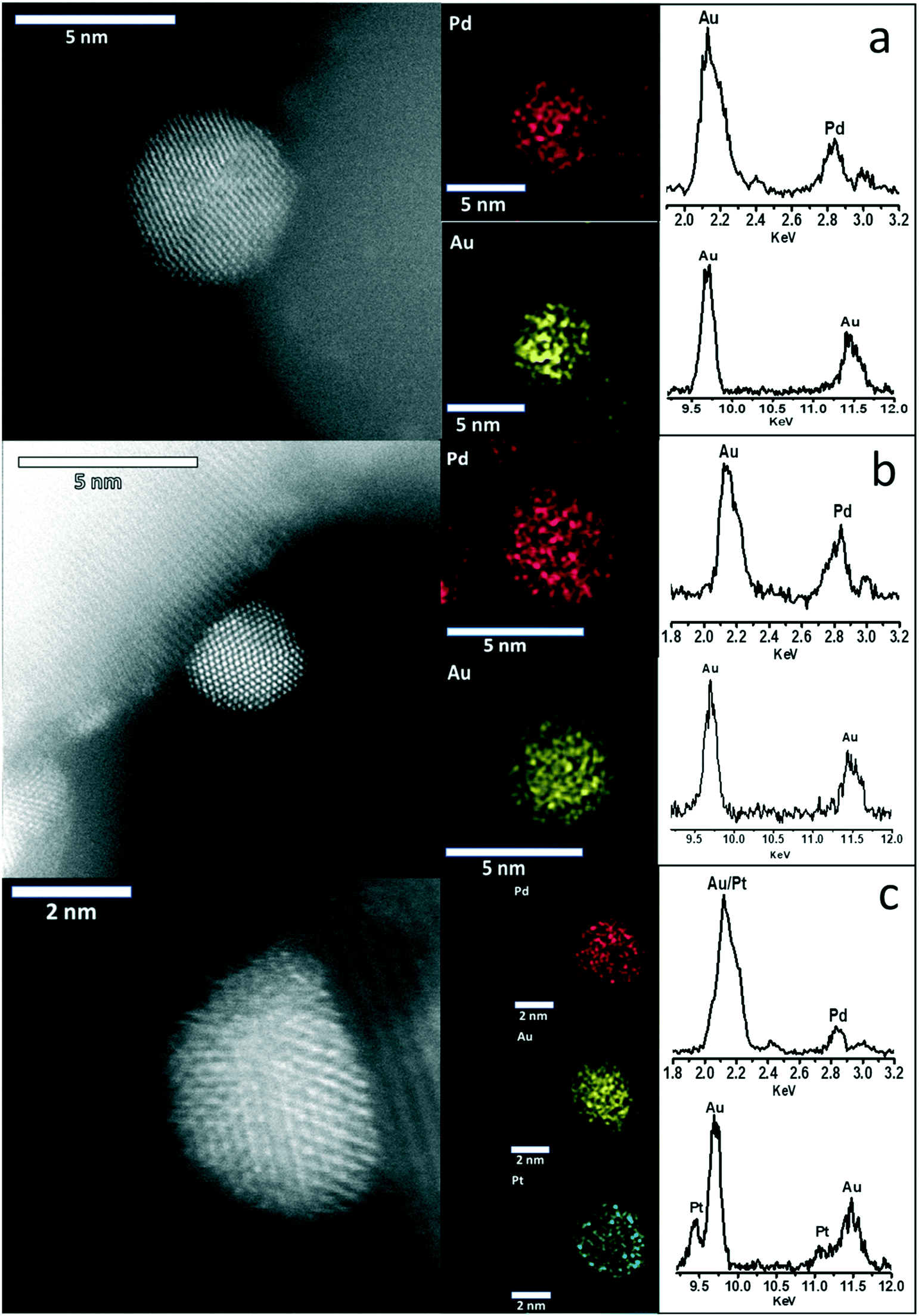 | ||
| Fig. 3 Representative STEM-ADF micrographs and complementary XEDS elemental maps and integrated point spectra of individual alloy particles in (a) 1% AuPd/TiO2, (b) 1% Au1Pd1Pt0.01/TiO2 and (c) 1% Au1Pd1Pt1/TiO2 catalysts. | ||
Time-on-line studies comparing H2O2 synthesis rates over the bi-metallic 1% AuPd/TiO2 and tri-metallic 1% AuPdPt/TiO2 catalysts can be seen in Fig. 4a, with a stark difference in catalytic activity being observed between the bi- and tri-metallic variants. The greater catalytic activity of the 1% Au1Pd1Pt0.01/TiO2 catalyst is clear, with a H2O2 concentration of 0.22 wt% being reached over a time period of 0.5 h, which is significantly greater than that achieved over either the 1% Au1Pd1/TiO2 (0.16 wt% concentration of H2O2) or 1% Au1Pd1Pt1/TiO2 (0.09 wt% concentration of H2O2) catalysts over the same reaction time. The enhanced activity of the 1% Au1Pd1Pt0.01/TiO2 catalyst is also highlighted through comparison of calculated reaction rates (Table S2†) at reaction times where there is assumed to be no contribution from subsequent degradation reactions. The rate of H2O2 synthesis over the 1% Au1Pd1Pt0.01/TiO2 catalyst was found to be over double that observed for the 1% Au1Pd1Pt1/TiO2 catalyst and 30% greater than the analogous bi-metallic 1% AuPd/TiO2 catalyst. Furthermore, the H2O2 yield achieved over the 1% Au1Pd1Pt0.01/TiO2 catalyst remained stable at extended reaction times, reaching a H2O2 concentration of 0.25 wt% at a reaction time of 1.5 h, whereas this metric was significantly lower for both the 1% Au1Pd1/TiO2 and 1% Au1Pd1Pt1/TiO2 catalysts, indicative of the comparatively higher H2O2 selectivity of the 1% Au1Pd1Pt0.01/TiO2 catalyst.
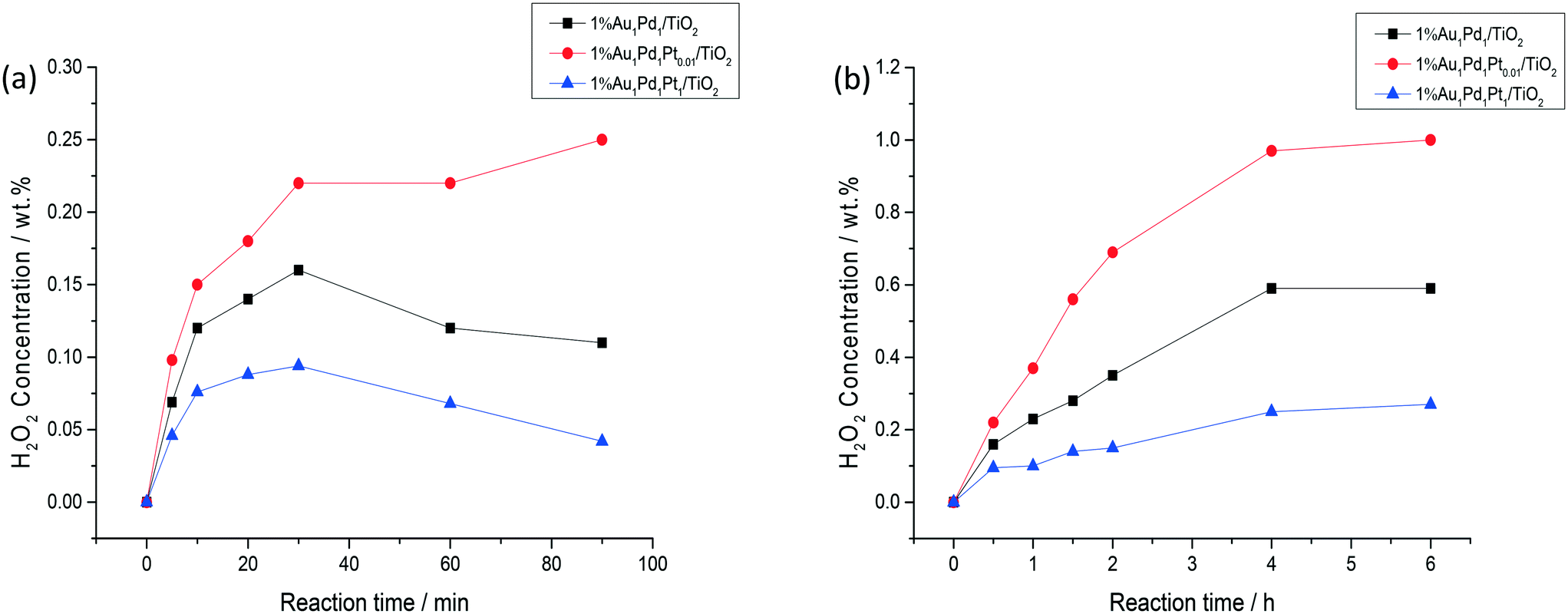 | ||
| Fig. 4 Comparison of the catalytic activity as (a) a function of reaction time and (b) over sequential H2O2 synthesis reactions. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. | ||
Evaluation of catalytic activity over multiple sequential H2O2 synthesis tests can be seen in Fig. 4b, with a marked enhancement in H2O2 concentration being observed for the 1% Au1Pd1Pt0.01/TiO2 catalyst compared to either the 1% Au1Pd1/TiO2 or 1% Au1Pd1Pt1/TiO2 catalysts. After running the reaction eight consecutive times, the H2O2 concentration increased to a value of 0.97 wt%, over the 1% Au1Pd1Pt0.01/TiO2 material which is far superior to the yields of H2O2 achieved over the 1% Au1Pd1/TiO2 (0.59 wt% H2O2) or 1% Au1Pd1Pt1/TiO2 (0.27 wt% H2O2) catalysts. Indeed the concentration of H2O2 achieved over the 1% Au1Pd1Pt0.01/TiO2 catalyst is comparable to that achieved in the initial stages of the current indirect method of industrial H2O2 production, prior to the use of multiple distillation steps to raise H2O2 concentrations to exceed ∼70 wt%.52
With the requirement to re-use a catalyst successfully at the heart of green chemistry and the activity of homogeneous species towards H2O2 synthesis well known,11 we next evaluated catalytic activity towards H2O2 synthesis and H2O2 degradation pathways (hydrogenation and decomposition) upon re-use. It can be seen that for all three catalysts evaluated the catalytic activity increased upon re-use compared to first use, under standard reaction parameters (Table 5), with a similar improvement in reaction rate at short reaction times, where the contribution from competitive degradation reactions are assumed to be negligible (Table S2†). We ascribe this to the rise in Pd0 content, at the expense of Pd2+ species, as determined by XPS (Table S3,† corresponding spectra in Fig. S2†). Numerous prior studies have reported an enhanced activity of Pd0-rich catalysts, towards both H2O2 synthesis and its subsequent degradation, compared to Pd2+ analogues.53–55 As such, balancing the ratio of Pd species (Pd0:Pd2+) is crucial to achieving a optimal catalytic performance. Analysis of the H2O2 synthesis reaction solution by ICP-MS (Table S4†) revealed the high structural stability of the supported 1% AuPdPt/TiO2 catalysts during the H2O2 synthesis reaction. It should also be noted that a minor increase in mean particle size for all catalysts tested was observed after use in the direct synthesis of H2O2 (Table S5,† as determined from the bright field transmission electron micrographs presented in Fig. S4†).
| Catalyst | Productivity molH2O2 kgcat−1 h−1 | Degradation molH2O2 kgcat−1 h−1 | Hydrogenation molH2O2 kgcat−1 h−1 | Decomposition molH2O2 kgcat−1 h−1 | ||||
|---|---|---|---|---|---|---|---|---|
| Fresh | Used | Fresh | Used | Fresh | Used | Fresh | Used | |
| H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C 1200 rpm. | ||||||||
| 1% Au1Pd1/TiO2 | 81 | 117 | 257 | 355 | 165 | 206 | 92 | 149 |
| 1% Au1Pd1Pt0.01/TiO2 | 112 | 141 | 245 | 363 | 137 | 107 | 108 | 256 |
| 1% Au1Pd1Pt1/TiO2 | 30 | 37 | 271 | 429 | 48 | 119 | 223 | 310 |
Conclusions
The addition of low quantities of Pt into AuPd nanoparticles results in a significant enhancement in catalytic selectivity and activity in the direct synthesis of H2O2 compared to AuPd or more Pt-rich AuPdPt analogues. This is attributed to a modification of Pd oxidation states and the formation of mixed Pd2+–Pd0 domains, which are well known to offer enhanced selectivity towards H2O2 compared to Pd0 or Pd2+ rich analogues. With increasing Pt addition to AuPd, the Pd0 content rises significantly with a corresponding loss of catalytic selectivity. The role of Pt in enhancing catalytic activity of supported AuPd nanoparticles can therefore be related to the electronic modification of Pd.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The China Scholarship Council (CSC) is gratefully acknowledged for the financial support given to X. G. The authors also acknowledge the National Key R&D Program of China (2016YFB0301600). We also wish to thank the Cardiff University electron microscope facility for use their instruments. The technical support from Mr Hiroaki Matsumoto and Mr Chaobin Zeng, Hitachi High-Technologies (Shanghai) Co. Ltd, for HR-STEM characterization is also greatly appreciated. This work is supported by National Natural Science Foundation of China (21673273, 21872163).References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- R. Underhill, R. J. Lewis, S. J. Freakley, M. Douthwaite, P. J. Miedziak, J. K. Edwards, O. Akdim and G. J. Hutchings, Johnson Matthey Technol. Rev., 2018, 62, 417–425 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, M. Alvaro and H. Garcia, ChemSusChem, 2012, 5, 46–64 CrossRef CAS PubMed.
- M. Lin, C. Xia, B. Zhu, H. Li and X. Shu, Chem. Eng. J., 2016, 295, 370–375 CrossRef CAS.
- Y. Wang, H. Li, W. Liu, Y. Lin, X. Han and Z. Wang, Trans. Tianjin Univ., 2018, 24, 25–31 CrossRef CAS.
- G. Liu, J. Wu and H. a. Luo, Chin. J. Chem. Eng., 2012, 20, 889–894 CrossRef CAS.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- M. Seo, H. J. Kim, S. S. Han and K. Lee, Catal. Surv. Asia, 2017, 21, 1–12 CrossRef CAS.
- H. J. Rledl and G. Pfleiderer, US2158525A, I. G. Farbenindustrie AG, 1939.
- H. Henkel and W. Weber, US1108752A, Henkel AG and Co KGaA, 1914.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2002, 206, 173–176 CrossRef CAS.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Angew. Chem., Int. Ed., 2008, 47, 6221–6224 CrossRef CAS PubMed.
- L. W. Gosser, US Pat., No. 4681751, Du Pont, 1987 Search PubMed.
- L. Kim and G. W. Schoenthal, US Pat. No. 4007256, Shell Oil, 1977 Search PubMed.
- E. N. Ntainjua, M. Piccinini, J. C. Pritchard, Q. He, J. K. Edwards, A. F. Carley, J. A. Moulijn, C. J. Kiely and G. J. Hutchings, ChemCatChem, 2009, 1, 479–484 CrossRef.
- P. Centomo, C. Meneghini, S. Sterchele, A. Trapananti, G. Aquilanti and M. Zecca, Catal. Today, 2015, 248, 138–141 CrossRef CAS.
- G. Gallina, J. García-Serna, T. O. Salmi, P. Canu and P. Biasi, Ind. Eng. Chem. Res., 2017, 56, 13367–13378 CrossRef CAS.
- Y. Han and J. Lunsford, J. Catal., 2005, 230, 313–316 CrossRef CAS.
- N. M. Wilson and D. W. Flaherty, J. Am. Chem. Soc., 2016, 138, 574–586 CrossRef CAS PubMed.
- T. Pospelova and N. Kobozev, Russ. J. Phys. Chem., 1961, 35, 584–587 Search PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. Neira d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- A. Villa, S. J. Freakley, M. Schiavoni, J. K. Edwards, C. Hammond, G. M. Veith, W. Wang, D. Wang, L. Prati, N. Dimitratos and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 694–697 RSC.
- A. Staykov, T. Kamachi, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2008, 112, 19501–19505 CrossRef CAS.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- J. Gu, S. Wang, Z. He, Y. Han and J. Zhang, Catal. Sci. Technol., 2016, 6, 809–817 RSC.
- S. Wang, K. Gao, W. Li and J. Zhang, Appl. Catal., A, 2017, 531, 89–95 CrossRef CAS.
- S. Maity and M. Eswaramoorthy, J. Mater. Chem. A, 2016, 4, 3233–3237 RSC.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y. Han, Chin. J. Catal., 2018, 39, 673–681 CrossRef CAS.
- T. Deguchi, H. Yamano, S. Takenouchi and M. Iwamoto, Catal. Sci. Technol., 2018, 8, 1002–1015 RSC.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- J. K. Edwards, J. Pritchard, L. Lu, M. Piccinini, G. Shaw, A. F. Carley, D. J. Morgan, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2014, 53, 2381–2384 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. N. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- J. K. Edwards, J. Pritchard, P. J. Miedziak, M. Piccinini, A. F. Carley, Q. He, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 3244–3250 RSC.
- Q. He, P. J. Miedziak, L. Kesavan, N. Dimitratos, M. Sankar, J. A. Lopez-Sanchez, M. M. Forde, J. K. Edwards, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2013, 162, 365–378 RSC.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. G. Fierro, Chem. Commun., 2004, 1184–1185 RSC.
- A. G. Gaikwad, S. D. Sansare and V. R. Choudhary, J. Mol. Catal. A: Chem., 2002, 181, 143–149 CrossRef CAS.
- L. Ouyang, P. Tian, G.-j. Da, X. Xu, C. Ao, T. Chen, R. Si, J. Xu and Y. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115(51), 25359–25367 CrossRef CAS.
- A. V. Beletskaya, D. A. Pichugina, A. F. Shestakov and N. E. Kuz'menko, J. Phys. Chem. C, 2013, 117(31), 6817–6826 CrossRef CAS PubMed.
- J. Kim, H. Kim, S. Kim, I. Kim, T. Tu, G. Han, K. Lee, J. Lee and J. Ahn, ACS Nano, 2019, 13(4), 4761–4770 CrossRef CAS PubMed.
- K. Duan, Z. Liu, J. Li, L. Yuan, H. Hu and S. I. Woo, Catal. Commun., 2014, 57, 19–22 CrossRef CAS.
- L. Ouyang, G. Da, P. Tian, T. Chen, G. Liang, J. Xu and Y. Han, J. Catal., 2014, 311, 129–136 CrossRef CAS.
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Angew. Chem., Int. Ed., 2008, 47, 6221–6224 CrossRef CAS PubMed.
- S. Kim, D. Lee, K. Lee and E. A. Cho, Catal. Lett., 2014, 144, 905–911 CrossRef CAS.
- P. Tian, D. Ding, Y. Sun, F. Xuan, X. Xu, J. Xu and Y. Han, J. Catal., 2019, 369, 95–104 CrossRef CAS.
- M. Khawaji and D. Chadwick, Catal. Sci. Technol., 2018, 8, 2529–2539 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142–5153 RSC.
- H. Li, B. Zheng, Z. Pan, B. Zong and M. Qiao, Front. Chem. Sci. Eng., 2018, 12, 124–131 CrossRef CAS.
- V. R. Choudhary, C. Samanta and T. V. Choudhary, Appl. Catal., A, 2006, 308, 128–133 CrossRef CAS.
- R. Burch and P. R. Ellis, Appl. Catal., B, 2003, 42, 203–211 CrossRef CAS.
- Q. Liu, K. K. Gath, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Catal. Lett., 2009, 132, 342 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01079k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |