
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold–palladium colloids as catalysts for hydrogen peroxide synthesis, degradation and methane oxidation: effect of the PVP stabiliser†
Simon J.
Freakley
 *a,
Nishtha
Agarwal
b,
Rebecca U.
McVicker
b,
Sultan
Althahban
cd,
Richard J.
Lewis
b,
David J.
Morgan
b,
Nikolaos
Dimitratos
e,
Christopher J.
Kiely
bc and
Graham J.
Hutchings
*b
*a,
Nishtha
Agarwal
b,
Rebecca U.
McVicker
b,
Sultan
Althahban
cd,
Richard J.
Lewis
b,
David J.
Morgan
b,
Nikolaos
Dimitratos
e,
Christopher J.
Kiely
bc and
Graham J.
Hutchings
*b
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: s.freakley@bath.ac.uk
bCardiff Catalysis Institute and School of Chemistry, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
cDepartment of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015, USA
dDepartment of Mechanical Engineering, Jazan University, Jazan 82822, Saudi Arabia
eDepartment of Industrial Chemistry, Alma Mater Studiorum-University of Bologna, Viale Risorgimento, 40136, Bologna, Italy
First published on 7th August 2020
Abstract
The reactivity of AuPd nanoparticle catalysts prepared by sol immobilisation is often explained by a structure activity relationship based solely on particle size or composition. In this contribution, we compare colloidal AuPd nanoparticles stabilised with polyvinylpyrrolidone (PVP) with the same AuPd nanoparticles supported on TiO2 for the direct synthesis of hydrogen peroxide and methane oxidation to methanol. We show that while the particles have similar rates of H2O2 synthesis, supporting the particles can affect the rates of H2O2 decomposition and hence the effectiveness of the catalyst for reactions which rely on H2O2 as an initiator or oxidant. We demonstrate that the absence of PVP results in high rates of H2O2 decomposition in methane oxidation experiments but this can be minimised by the addition of PVP to the reactor. These results also show that for AuPd alloys, both polymer stabiliser and support effects need to be taken into account when describing the activity of the nanoparticles and the active sites should in fact be thought of as a metal–support–polymer interface with many degrees of freedom.
Introduction
The preparation of supported precious metal catalysts by colloidal nanoparticle synthesis methods can result in highly active materials for a range of important chemical processes.1,2 The advantage of solution phase synthesis of metal nanoparticles is the high degree of control over the particle size, shape and composition that can be achieved before immobilisation onto high surface area supports.3 Typically, colloidal methods involve fast chemical reduction of metal precursors and steric stabilisation of the growing nanoparticles by polymer additives.4 In many cases these catalysts are used without high temperature oxidative heat treatments or washing protocols specifically designed to remove the strongly bound polymer.5–7 Therefore, in reality the reported performance of the catalyst material not only depends on the nature of the metal particle (size, shape, oxidation state) but also the nature of the composite particle–polymer surface.Reaction rates, product selectivity and enantioselectivity can all be altered by the interaction of the polymer ligand or organic modifier with the metal surface.8 For instance, the introduction of chiral modifiers such as cinchonidine derivatives or napthyl-ethylamine to Pt catalysts have been shown to impart increased enantioselectivity in activated ketone hydrogenation.9–11 Polymer additives can also be used to tune the oxidation states of metal surfaces through binding of electron donating or withdrawing groups which in turn effects reactant binding strengths.12 Steric effects can also be observed in many cases with changes in reaction selectivity observed in the presence of polymer additives.13 Despite these potential advantageous degrees of control available by polymer addition, detrimental effects such as site blocking by strong adsorption can reduce activity – especially in gas phase reactions.14–16 This requires careful removal of polymer additives prior to reaction without significantly changing the nanostructure of the catalyst material which has been carefully controlled in the colloidal synthesis of the nanoparticles.
Polyvinylpyrrolidone (PVP) is a common polymer additive used in the preparation of precious metal catalysts for reactions such as glycerol oxidation using Au and Pd based catalysts supported on TiO2.17–20 We recently demonstrated that while effective catalysts could be prepared in the absence of additives, the presence of PVP and polyvinylalcohol (PVA) has significant effects on product selectivity which is often not considered in structure activity relationships.21,22 Similar catalyst structures based on Au–Pd particles have been shown to be highly active for reactions such as the direct synthesis of hydrogen peroxide (H2O2) from molecular hydrogen and oxygen which would represent an attractive alternative to the current indirect anthraquinone process for on-site H2O2 production.23–27 Catalysts with varying alloy compositions and nanostructure have been extensively studied for this reaction and have been shown to be highly active for H2O2 synthesis.27 Titania supported Au–Pd catalysts prepared by colloidal methods with small particle sizes tend to have significant activity for H2O2 degradation by over hydrogenation and decomposition.28 To date, the challenge of minimising the subsequent degradation of H2O2 (via hydrogenation and decomposition pathways) has only been achieved with a few specific catalyst systems in the absence of acid and halide additives.29–31
Unsupported colloidal nanoparticles can be active for a number of reactions including glucose oxidation,32 aromatic alcohol oxidation33 and CO2 reduction.34 Nomura et al. have previously reported that unsupported AuPd colloidal particles can be active for the direct synthesis of H2O2 from H2 and O2.35 Recent studies by Deguchi et al. have shown that a range of bimetallic colloidal particles stabilised with PVP, including Pd–Ir and Pd–Pt, can be active catalysts – however the role of the polymer additive in all these cases has not been explicitly addressed.36 Recently we demonstrated that using colloidal AuPd nanoparticles stabilised with PVP, it was possible to achieve selective oxidation of methane to methanol at 25–50 °C with O2 incorporation using H2O2 as an initiator.37 A significant result in this study showed that the background H2O2 decomposition rate was minimal for unsupported colloidal AuPd–PVP particles at 50 °C, allowing the radical reaction process to propagate rather than terminate; however the H2O2 decomposition rate increased significantly when then the same particles were supported on TiO2 making this an ineffective catalyst.37
This result clearly demonstrated that colloidal particles stabilised by polymers can have significantly different behaviour in reactions involving H2O2 than analogous ‘bare’ particles supported on TiO2 and while colloidal catalyst systems could have limitations in terms of long term stability it is possible to use them to propose structure–activity relations. In this study, we aim to elucidate if the PVP stabilisers present on AuPd particles effect the H2O2 synthesis and methane oxidation reaction networks and investigate if the reactivity in these systems should be considered as not only a result of interactions with the surface atoms on the metal nanoparticle, but rather with an entity more akin to a metal core–polymer shell composite.
Results and discussion
Recent studies by Giorgianni et al. reported that the addition of PVA to Pd catalysts results in enhanced H2O2 yields due to hindered H2O2 back diffusion to the metal surface via hydrophobic interactions in addition to greater observed catalyst stability when PVA is present.38 In this current study on H2O2 synthesis and methane oxidation, we focus on PVP as an additive because in methane oxidation reactions traces of MeOH have been previously observed as a result of PVA contamination or oxidative degradation under our reaction conditions. A series Au–Pd colloidal nanoparticle solutions were synthesised according to previously described procedures.37 The colloidal solutions of Au–Pd nanoparticles (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 1:1 by moles) were synthesised via NaBH4 reduction of metal salts and stabilised by PVP. A portion of the AuPd–PVP colloidal nanoparticles were immobilised onto TiO2 (1 wt% metal loading) and dried at 110 °C to provide comparative supported samples. Both the supported and unsupported AuPd–PVP materials were then tested for the direct synthesis of H2O2 and methane oxidation, using experimental conditions which have both been previously described in the literature.28,37
:Pd = 1:1 by moles) were synthesised via NaBH4 reduction of metal salts and stabilised by PVP. A portion of the AuPd–PVP colloidal nanoparticles were immobilised onto TiO2 (1 wt% metal loading) and dried at 110 °C to provide comparative supported samples. Both the supported and unsupported AuPd–PVP materials were then tested for the direct synthesis of H2O2 and methane oxidation, using experimental conditions which have both been previously described in the literature.28,37
The colloidal metal nanoparticles and the supported colloidal nanoparticles have been previously characterised by X-ray photoelectron spectroscopy.37 XPS analysis of the colloidal AuPd–PVP nanoparticle solution in the Au(4f) and Pd(3d)/Au(4d) regions, after drop casting onto a solvent cleaned silicon wafer, showed that both elements were present in predominantly the metallic state (Au(4f) ∼ 83–84 eV, Pd(3d) ∼ 334.5–335.5 eV, the range in energies varying depending on particle size and support interactions) as could be expected from the presence of the strong reducing agent at the high molar ratio used in the preparation. For the unsupported colloid, a Au signal at 85.5 eV and a Pd signal (ca. 388 eV) were also detected and attributed to residual or leached metal chlorides. XPS analysis of the immobilised nanoparticles exhibit both Au and Pd binding energies ca. 1 eV lower compared to the unsupported colloid. Such low Pd binding energies may be attributed to a particle–support interaction making the zero-valent supported Pd more electron rich. The corresponding Au(4f7/2) signal exhibits a binding energy of 83.1 eV and lower than the characteristic 84 eV for bulk Au, although such binding energies have been widely reported, and are typically attributed to small, low coordination atoms, charge transfer from PVP to Au, and charge transfer between Pd and Au, increasing the s-state occupancy of Au indicating alloy formation.39,40
TEM analysis (Fig. 1a and b) showed that the mean size of the bimetallic particles in the colloidal solution was 3.0 ± 2.0 nm, which increased slightly to 4.1 ± 1.3 nm when the particles were immobilised onto the support. This is in good agreement with our previous studies on these materials where the apparent increase in size occurs as a result of the AuPd nanoparticle flattening slightly and faceting to form an interface with the TiO2 support (Fig. 1c and d).28
 | ||
| Fig. 1 BF-TEM images and corresponding particle size distributions of (a) colloidal and (b) TiO2 supported Au–Pd nanoparticles prepared by sol-immobilisation using PVP as the stabiliser. (c) HAADF image of Au–Pd–PVP colloidal particles drop cast onto a C TEM gird. (d) HR-TEM phase contrast lattice image of a typical AuPd–PVP particle supported on TiO2. | ||
Previous studies have demonstrated that colloidal Pd and AuPd–PVP nanoparticles, in the presence of strong acid and halide additives, can produce H2O2via the direct reaction of H2 and O2.41 These un-supported particles were stabilised by PVP and were shown to exhibit appreciable rates for direct H2O2 synthesis suggesting that the presence of a support is not essential for high activity in this reaction. We carried out comparative tests between our colloidal AuPd–PVP materials in the unsupported and TiO2 supported state ensuring that each reaction had the equivalent moles of metal (0.66 μmol). The results, shown in Table 1, entries 1–3, demonstrate that over a 30 min reaction period the unsupported colloidal Au–Pd–PVP nanoparticles were capable of producing 0.10 wt% H2O2 compared to 0.07 wt% H2O2 when using TiO2 supported counterparts; their corresponding apparent turnover frequencies (TOFs) based on total moles of metal were 8.0 × 102 and 5.2 × 102 molH2O2 molmetal−1 h1 respectively. The TiO2 support alone showed no background activity towards H2O2 synthesis or H2O2 degradation (via over-hydrogenation or decomposition). Next, the amount of AuPd–PVP colloidal nanoparticle solution used was varied to identify the kinetic regime in which the reaction was operating. Fig. 2 shows that increasing the amount of Au–Pd colloidal nanoparticle solution added to the reaction results in a linear increase in the amount of H2O2 produced, reaching 0.29 wt% after 30 min reaction, which was the point where the 2.9 mL H2O used as solvent was completely replaced by the aqueous AuPd–PVP colloidal solution. This result indicated that no external gas–liquid diffusion limitations were occurring when increasing amounts of the colloidal catalyst were used in the reaction. This finding is in contrast to our previous studies using TiO2 supported AuPd catalysts where a plateau is observed in H2O2 yield with increasing catalyst mass due to mass transfer effects.42
| Entry | Catalyst | PVP: M | H2O2a | Apparent reaction rate at 30 min | H2O2 degradationb | H2O2 decomp.c | |
|---|---|---|---|---|---|---|---|
| (wt%) | (molH2O2 molmetal−1 h−1) | (molH2O2 molsurface−1 h−1)d | (%) | (%) | |||
| a H2O2 synthesis conditions: 5% H2/CO2 (29 bar) and 25% O2/CO2 (11 bar), 8.5 g solvent (2.9 g HPLC 5.6 g MeOH), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. b H2O2 degradation conditions: 5% H2/CO2 (29 bar), 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g 50% H2O2), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. c H2O2 decomposition conditions: 25% O2/CO2 (29 bar), 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g 50% H2O2), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. d As determined from measured TEM particle size distributions. | |||||||
| 1 | AuPd colloid | 1.2 | 0.10 | 8.0 × 102 | 2.7 × 104 | 13 | 1 |
| 2 | 1% AuPd/TiO2 | 1.2 | 0.07 | 5.2 × 102 | 3.2 × 104 | 11 | 7 |
| 3 | TiO2 | 0 | 0.00 | — | — | 0 | 0 |
| 4 | 1% AuPd/C | 1.2 | 0.13 | 10 × 103 | — | 9 | 1 |
| 5 | AuPd colloid | 0.005 | 0.08 | 6.2 × 102 | 2.8 × 104 | 9 | 0 |
| 6 | AuPd colloid | 0.1 | 0.11 | 8.5 × 102 | 2.6 × 104 | 11 | 1 |
| 7 | AuPd colloid | 20 | 0.12 | 9.1 × 102 | 2.7 × 104 | 13 | 1 |
| 8 | AuPd colloid | 0 | 0.07 | 5.2 × 102 | — | 11 | 7 |
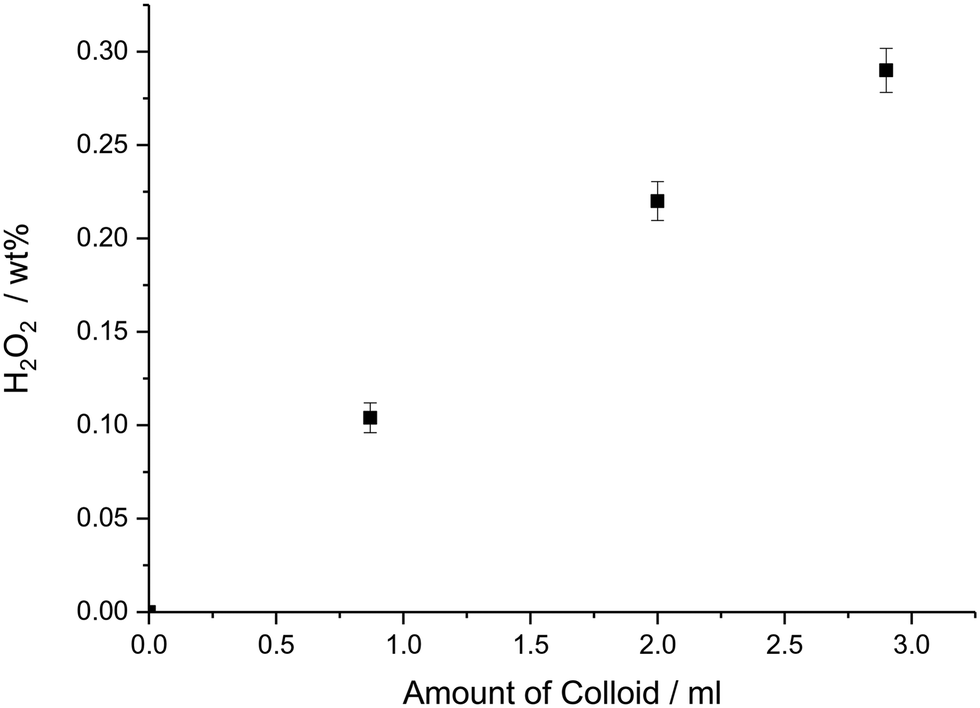 | ||
| Fig. 2 H2O2 produced with increasing amounts of unsupported Au–Pd colloid added to the reaction solution up to complete replacement of the H2O in the solvent system (2.9 ml). H2O2 synthesis conditions: 5% H2/CO2 (29 bar) and 25% O2/CO2 (11 bar), 8.5 g solvent (2.9 g HPLC 5.6 g MeOH), colloidal solution concentration [0.66 mMmetal], reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. | ||
Over a 30 min reaction time, significant H2O2 degradation is likely when using Pd containing nanoparticles in the 2–5 nm size range, with halide and acid stabilisers typically used to minimize degradation pathways, suggesting that the reported TOFs after 30 min of reaction are under-estimates of the actual TOFs, because some fraction of the H2O2 produced will have been destroyed by subsequent over-hydrogenation or decomposition reactions.43Fig. 3 shows a comparison in terms of wt% H2O2 produced of the colloidal metal nanoparticle solution and supported AuPd catalyst as a function of time for this reaction. It can be seen that at very short reaction times, (∼2 min) where the contribution from H2O2 degradation pathways can be assumed to be negligible, that the colloidal AuPd–PVP nanoparticle solution has a higher initial rate of H2O2 synthesis than the corresponding supported AuPd nanoparticles. A turnover frequency of 4630 molH2O2 molmetal−1 h1 was determined for the unsupported colloid as compared to 2140 molH2O2 molmetal−1 h1 for the TiO2 supported AuPd nanoparticles based on the total moles of metal present in this regime where subsequent reactions are minimised.
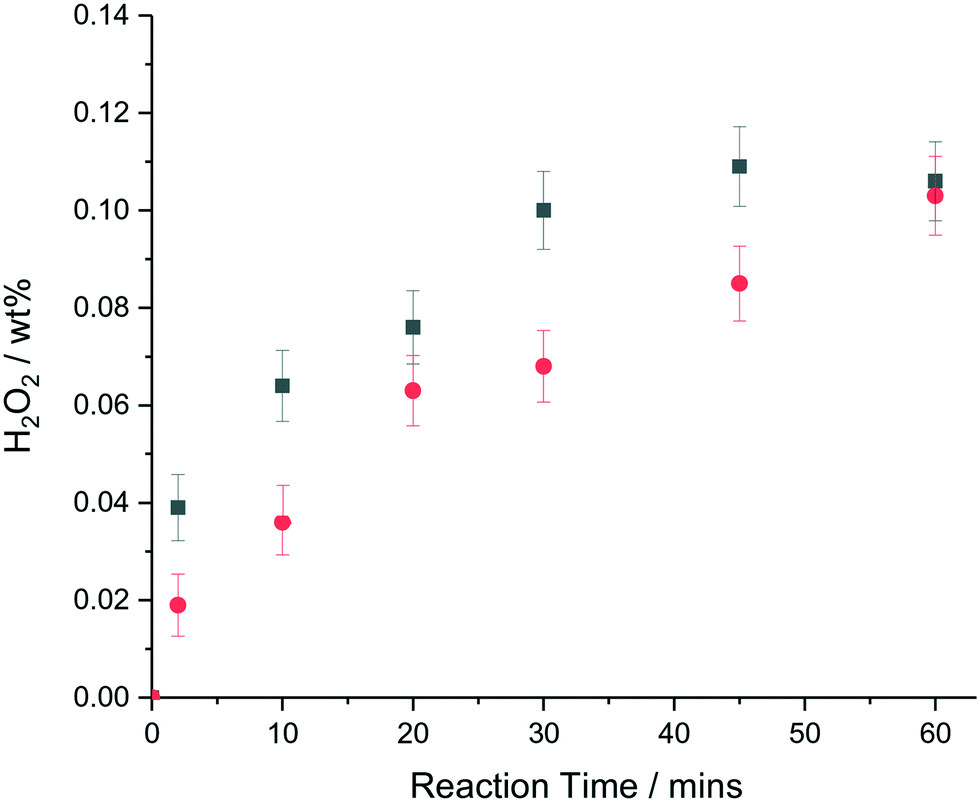 | ||
| Fig. 3 H2O2 produced (wt%) as a function of reaction time by unsupported and TiO2 supported AuPd–PVP colloid nanoparticles. wt% H2O2 produced – filled symbols; colloidal particles (■), supported particles (●). H2O2 synthesis conditions: 5% H2/CO2 (29 bar) and 25% O2/CO2 (11 bar), 8.5 g solvent (2.9 g HPLC 5.6 g MeOH), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm;, reaction time = 30 min. | ||
Due to the measured difference in particle size distribution of the two catalytic systems, the total number of surface atoms available in each reaction was estimated using the particle size distributions obtained by TEM and applying the same model for atom packing to each system. This approximation assumes activity is related to the total number of exposed surface atoms and is not correlated with specific surface sites such as edges or corner atoms. In addition it assumes that the polymer coverage of the metal surface is consistent between both samples. This analysis gave 1.2 × 1017 surface atoms for the colloidal system assuming spherical particles versus 5.7 × 1016 for the TiO2 supported catalyst assuming hemispherical supported particles meaning that the colloidal samples have ∼2.1 times the exposed metal surface. Based on the amount of H2O2 produced after 2 min of reaction, where the contribution from subsequent reactions (hydrogenation and decomposition) is minimal, we find that the moles of H2O2 produced at each exposed surface site is comparable between the colloidal metal nanoparticle solution (1.7 × 10−21 molH2O2 metal site−1 h−1) and the supported AuPd nanoparticles (1.5 × 10−21 molH2O2 per metal site per h). Considering the assumptions made during this comparison this suggests that H2O2 formation occurs on the surface of the Au–Pd nanoparticles at similar rates in both catalytic systems when minimal H2O2 degradation pathways are operating. Due to the large difference in catalyst mass between colloidal and supported nanoparticle catalysts rates normalised to both total moles of metal and moles of metal surface for 30 min reactions are reported in Table 1 assuming no significant change in the catalyst structure takes place.
Experiments were also carried out to investigate the effect that supporting the colloidal AuPd–PVP nanoparticles on TiO2 had on the degradation of a 4 wt% H2O2 solution, which included contributions from both H2O2 hydrogenation and decomposition pathways. Comparing the unsupported colloidal AuPd–PVP solution to the corresponding TiO2 supported catalyst, the total degradation after 30 min (Table 1, entries 1–2) was similar for both situations. However, when normalising to the difference in surface sites between the colloidal and supported nanoparticles, it is clear to see that the supported particles have significant higher rates per metal surface site by a factor of ∼2.5. By comparing the rates of H2O2 decomposition under a 25% O2/CO2 atmosphere to remove over-hydrogenation from the possible reaction pathways, it was observed that the colloidal AuPd–PVP solution decomposed only 1% of the H2O2 present compared to the supported particles which decomposed 7% of the initial H2O2 suggesting a significant difference in how the two samples interact with H2O2.
The decomposition of H2O2 occurs much faster when the Au–Pd particles are supported on TiO2, despite the supported particles having larger mean particle size and therefore less exposed surface available (by roughly a factor of 2) meaning an increase in observed decomposition rate of about 14 times for the supported particles as compared to the unsupported colloidal particles. Assuming that the H2O2 hydrogenation and decomposition pathways are independent of each other, this suggests that either it is the act of supporting the particles which generates a metal–metal oxide support interface or the removal of PVP on washing the solid which changes the predominant H2O2 degradation pathway. Our previous studies have shown that supporting Au colloids on crystalline metal oxides such as TiO2 results in a more highly faceted particle structure than when they are supported on amorphous carbon structures.14,15,44 We prepared an analogous sample by supporting the same colloidal AuPd nanoparticle solution on an activated carbon. This sample showed higher H2O2 production, but also no H2O2 decomposition activity, supporting the notion that immobilisation of the AuPd colloid onto TiO2 P25 surface results in increased H2O2 decomposition rates possibly through the formation of highly faceted surfaces as the particle restructures to minimise the energy needed to interact with the underlying support lattice and form an extended intimate support/metal interface. This highlights the possibility that the underlying support material can possibly indirectly effect the decomposition pathways of H2O2 on the surfaces of the alloy nanoparticle attached to it.
Numerous observations that supported catalysts prepared from PVP-stabilised colloids show low activity in gas phase reactions, such as CO oxidation, due to the presence of residual polymer suggest that the polymer is still present to some extent after the preparation of the supported catalyst material.15,16 Further studies by Han et al. report the stability of the PVP polymer under H2O2 synthesis conditions similar to those used within this work supporting the hypothesis that PVP is stable to the reaction conditions used in this study.6 To further investigate if the presence of PVP contributes to these effects colloidal Au–Pd nanoparticles were prepared using a variety of PVP:metal ratios between 20 and 0.005 in order to study the influence of PVP concentration and particle size on the reaction, (Table 1 entries 5–7, Fig. 4). The mean particle size of the colloidal particles ranged from 6 ± 1.9 nm for a PVP/metal ratio of 0.05 to 1.9 ± 1.3 nm for a PVP/metal ratio of 20. This decrease in mean size correlates well with the increasing amount of polymer allowing the formation of a higher number of smaller nuclei during the preparation. Fig. 4 shows that as the average particle size of the AuPd nanoparticles decreases, the net amount of H2O2 produced after 30 min of reaction increases from 0.08 wt% for 6 nm particles to 0.12 wt% for 1.9 nm particles. In all cases, the colloidal AuPd–PVP particles showed minimal H2O2 decomposition (Table 1, entry 5–7), even when the solution contained smaller particles (i.e., a greater number of exposed metal surface atoms) producing higher amounts of H2O2 over the 30 min reaction. Fig. 4b reports the normalised rate per surface atom as approximated by the TEM derived particle size distributions for a range of catalysts prepared with various PVP to metal ratios for H2O2 synthesis, degradation and decomposition. No significant change in apparent TOF per surface atom was observed over the mean particle size range 1.9 to 6.0 nm, however it should be noted that these reaction rates were determined after 30 min so do not represent initial rates which are independent of subsequent reaction processes. This suggests, due to the minimised H2O2 decomposition in all colloidal samples compared to the supported sample, that it is the effect of supporting the particles and their interaction with the TiO2 lattice that initiates H2O2 decomposition in this system and that the main loss of H2O2 selectivity using colloidal particles results from over hydrogenation.
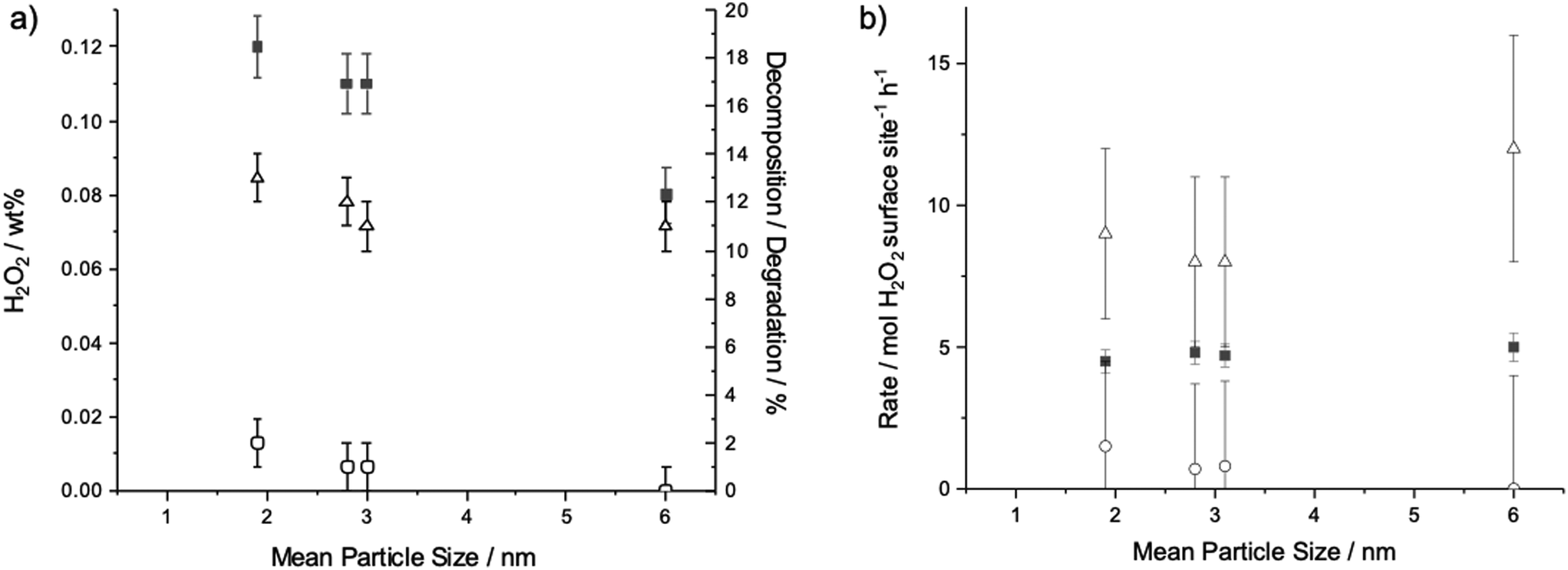 | ||
| Fig. 4 a) Catalytic activity results for unsupported AuPd–PVP colloidal nanoparticle solutions prepared with various amounts of PVP giving rise to different mean nanoparticle sizes. b) Rate normalised to available metal surface as determined by TEM particle size distributions. Key to symbols; wt% H2O2 produced (■), H2O2 degradation (△) and H2O2 decomposition (○). H2O2 synthesis conditions: 5% H2/CO2 (29 bar) and 25% O2/CO2 (11 bar), 8.5 g solvent (2.9 g HPLC 5.6 g MeOH), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. H2O2 degradation conditions: 5% H2/CO2 (29 bar), 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g 50% H2O2), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. H2O2 decomposition conditions: 25% O2/CO2 (29 bar), 8.5 g solvent (5.6 g MeOH, 2.22 g H2O and 0.68 g 50% H2O2), 0.66 μmol metal, reaction temperature = 2 °C, stirring rate = 1200 rpm, reaction time = 30 min. | ||
To support our conclusions, we investigated the decomposition of H2O2 by simply stirring both supported and unsupported catalytic systems in glass vials containing 4 wt% H2O2 under air at ambient conditions, rather than at 2 °C and under a 29 bar pressure of 5% H2/CO2 as in the regular H2O2 synthesis experiments. Fig. 5 shows that when TiO2 and the AuPd–PVP sol were stirred separately with the starting H2O2 solution, approximately 4–5% of the H2O2 was decomposed over the 30 min reaction period. In contrast, when the supported 1wt% AuPd/TiO2 material was tested, containing the same amount of metal, bubbles of gas generated from the decomposition of H2O2 were clearly visible originating from the catalyst. Over the same 30 min time period, approximately 80% of the H2O2 was decomposed. Furthermore, addition of varying extra amounts of PVP to the solution did not suppress the rapid gas generation.
 | ||
| Fig. 5 H2O2 decomposition results as a function of time at ambient conditions. Key to symbols: blank (□), TiO2 only (○), unsupported colloidal AuPd–PVP particles (◊) and TiO2 supported AuPd particles (△). H2O2 decomposition conditions: 4 wt% H2O2 in H2O (10 mL, HPLC grade), in each case 0.66 μmol metal, 9 mg of TiO2 added under magnetic stirring at ambient temperature and pressure. | ||
Astruc and co-workers have demonstrated that it is possible to prepare colloidal Au catalysts in the absence of polymer stabilisers relying solely on electrostatic stabilisation of the metal particles by residual salts in the colloidal solution.45 We recently showed that it is possible to prepare supported AuPd catalysts in a similar manner with a slightly broader particle size distribution compared to conventional immobilisation of polymer stabilised colloidal solutions.22 This colloidal nanoparticle solution prepared without polymer addition (denoted SF-stabiliser free) was also tested for direct H2O2 synthesis (Table 1, entry 8). Interestingly they showed activity towards H2O2 production and also increased rates of H2O2 decomposition suggesting that the greater accessibility to the surface in the absence of PVP results in higher H2O2 decomposition rates in these colloidal systems. Similar tests under ambient conditions using this stabiliser free AuPd nanoparticle solution clearly showed the evolution of bubbles as a result of H2O2 decomposition, which could be supressed by the addition of PVP to the reaction mixture. This is in direct contrast to the situation found when using the TiO2 supported AuPd catalyst, where there seemed to be an interplay between the presence of PVP and the effect of supporting the nanoparticles on inducing H2O2 decomposition. The addition of N-methyl pyrrolidone as an analogue of the PVP monomer unit was not able to suppress the decomposition in the same way as adding PVP to the stabiliser free colloid, implying that the presence of the hydrophobic alkyl chain backbone could be crucial in controlling the rates of H2O2 decomposition in colloidal nanoparticle solutions.
This difference in the H2O2 decomposition rates in the absence of pressurised CO2 could have significant implications in reactions involving aqueous solutions of H2O2 as an oxidant or initiator in the absence of acid stabilisers due to the reactivity of support nanoparticles being significantly different to colloidal nanoparticles in terms of H2O2 decomposition. Our previous studies on methane oxidation using unsupported and TiO2 supported colloidal AuPd–PVP nanoparticles have shown that unsupported colloidal catalysts are capable of oxidising methane in the presence of H2O2 to produce methanol under mild conditions in which gas phase O2 incorporation occurred with high efficiency with respect to H2O2.37,40 It was also demonstrated that the TiO2 supported AuPd colloidal nanoparticles showed low activity in this particular reaction due to high levels of H2O2 decomposition.
To extend our previous study and elucidate the role of PVP in this reaction we conducted further methane oxidation experiments with H2O2 only as an oxidant using the 1 wt% AuPd/TiO2 catalyst with extra PVP added to the reaction to identify if this unwanted H2O2 decomposition activity, initiated by supporting the AuPd–PVP particles, could be suppressed. Table 2 shows a summary of the catalytic testing results for a colloidal AuPd catalyst prepared with PVP, the TiO2 supported AuPd–PVP nanoparticles, and the same supported nanoparticles with the addition of extra PVP (giving a metal to PVP ratio 1.2 by weight in the reaction). The results clearly show the superior efficiency of the unsupported AuPd–PVP colloidal system with respect to H2O2 efficiency in producing oxygenates. On adding extra PVP to the supported catalyst an improvement was noted with respect to H2O2 efficiency and oxygenate production from a H2O2: product ratio from 570 to 376, however, the activity of unsupported colloidal system still remained over an order of magnitude better. This demonstrated that the support induced H2O2 decomposition could not be fully negated by the addition of further PVP stabiliser.
| Catalyst | Product amount (μmol) | Oxygenate selectivity/(%) | H2O2/products | |||
|---|---|---|---|---|---|---|
| CH3OH | CH3OOH | HCOOH | CO2 | |||
| Reaction conditions; 1000 μmol H2O2, reaction temperature = 50 °C, total volume = 10 mL, 30 bar CH4, reaction time = 30 min, stirring rate = 1500 rpm, 6.6 μmol metal used per reaction. | ||||||
| AuPd–PVP colloid | 3.19 | 9.76 | 7.04 | 3.09 | 86 | 36 |
| AuPd/TiO2 | 0.43 | 0.00 | 0.00 | 1.23 | 26 | 575 |
| AuPd/TiO2 + PVP | 0.86 | 0.43 | 0.00 | 1.51 | 46 | 326 |
Further experiments were carried out to investigate if the presence of polymer stabiliser was crucial in achieving the high efficiency of methane oxidation with H2O2 and O2 using colloidal AuPd–PVP nanoparticles. Fig. 6 shows a stark comparison of the catalytic performance of the colloidal metal nanoparticle system synthesised with and without PVP polymer. Without the PVP polymer being present, H2O2 consumption during the 30 min reaction period at 50 °C was over 95%, producing minimal oxygenated products. This compares to only 32% H2O2 consumption when the colloid incorporated the PVP ligand. The lower H2O2 consumption in this latter case resulted in significantly higher oxygenated product formation showing that the polymer is crucial in achieving high reactivity, presumably– by controlling the rate of H2O2 decomposition. We then took the stabiliser free colloidal solution prepared in the absence of PVP and added PVP to the methane oxidation reaction. The catalytic performance was significantly improved and indeed, the activity approached that of the colloidal solution prepared with PVP. The H2O2 consumption was 48% with significant oxygenate production demonstrating that it is in fact the polymer additive significantly effects the rate of H2O2 decomposition on the metal surface and therefore the overall efficiency of methane oxidation. The PVP ligand shell presumably controls the rate of H2O2 diffusion to the metal surface and therefore the production of radicals that can interact with the solubilised methane, whose concentration may also be enhanced by the hydrophobic alkyl chains in the organic layer around the metal surface.
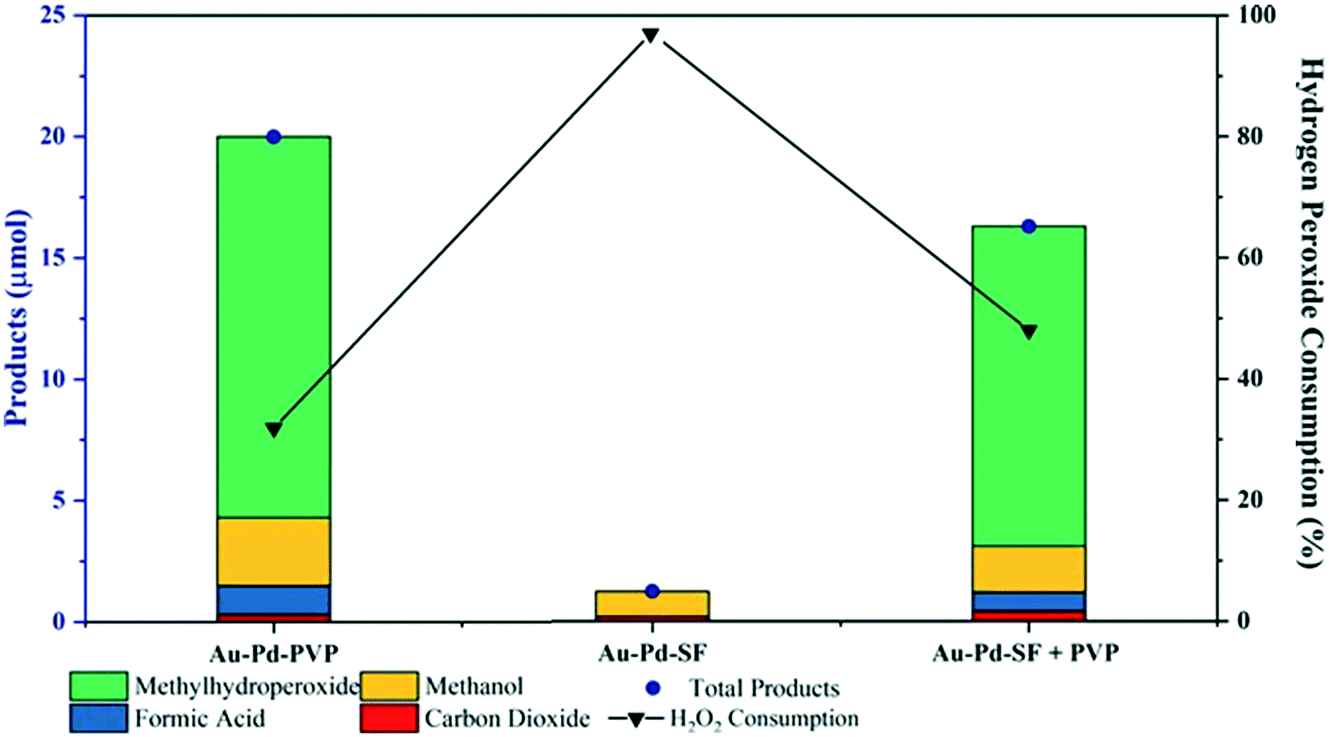 | ||
| Fig. 6 Comparative methane oxidation tests for AuPd–PVP colloidal nanoparticle sample prepared with PVP (Au–Pd–PVP), stabiliser free (Au–Pd–SF) and stabiliser free with the addition of PVP (Au–Pd–SF + PVP) equivalent to metal/PVP ratio of 1.2. Reaction conditions: pressure (CH4) = 30 bar, pressure (O2) = 5 bar, amount of catalyst: 10 mL colloid – 6.6 μmol of metal, 50 μmol H2O2, reaction temperature – 50 °C (with a ramp rate of 2.25 °C min−1), stirring rate – 1500 rpm, reaction time – 30 min. | ||
In conclusion, we have investigated the role of the PVP ligand shell and TiO2 support in controlling the reactions of H2O2 on AuPd–PVP colloidal particles. These particles display significant activity towards H2O2 direct synthesis, hydrogenation and decomposition as well as methane oxidation using H2O2 and O2. We have shown that supporting the PVP stabilised AuPd nanoparticles on TiO2 increases activity towards H2O2 decomposition which cannot effectively be suppressed by the addition of extra PVP under our H2O2 synthesis or methane oxidation reaction conditions. This could be a result of the formation of more highly faceted alloy particles and an extended metal–support interface – which is absent in the unsupported colloidal AuPd–PVP nanoparticle catalyst. The unsupported particles, when stabilised with PVP, show limited H2O2 decomposition activity while being able to produce H2O2 at similar rates to supported nanoparticles. However, the use of colloidal particles in extended reactions under flow conditions remains a challenge to translation of this activity to industrial application. Using methane oxidation as a test reaction we demonstrated that this high H2O2 decomposition rate is detrimental to the overall efficacy in this case. We also have shown that the colloidal systems require the presence of polymer stabilisers to control the rate of H2O2 decomposition. These results clearly demonstrate that for AuPd alloys prepared by colloidal methods, both polymer stabiliser and support effects need to be taken into account when describing the activity of the nanoparticles for a range of reactions involving H2O2 such as selective oxidations or reactions that use H2O2 or other peroxides as an initiator for oxidation.
Experimental details
Catalyst preparation
Bimetallic Au–Pd nanoparticles were prepared by standard colloidal methods. An aqueous solution of HAuCl4 precursor (Strem Chemicals) and acidic solution of PdCl2 (Sigma Aldrich) precursor (in 0.58 M HCl) were dissolved in 800 mL of de-ionized water (Au:Pd = 1:1 by moles) to give a total metal concentration of 0.16 mmol L−1. Polyvinylpyrrolidone (PVP, average molecular weight 1300000, Sigma Aldrich) was added as a stabilizer to give the required metal-to-PVP weight ratio (typically 1:1.2 (wt/wt)). After 2–3 min of stirring, freshly prepared 0.1 M sodium borohydride (NaBH4, Sigma Aldrich) solution was added such that the molar ratio of NaBH4-to-metal was 5:1 (mol/mol). This produced a dark brown colloid which was then left stirring for 30 minutes to ensure that all the metal precursors were reduced to the metallic form. The colloid was concentrated using a roto-evaporator to give a nominal metal loading of 0.66 mmol L−1. The colloid was stored in glass media bottles prior to use. For supported catalysts, the sol prepared as described above was immobilized onto a TiO2 (P25, Degussa, 1.98 g) or activated carbon (Darco G60) (1.98 g) in the following manner. A sufficient amount of support material was added to ensure a 1 wt% metal loading and the solution was acidified to pH 1 using sulphuric acid to enhance and achieve more homogeneous deposition of nanoparticles. The supernatant solution became clear over a 1 h period after support addition, indicating the deposition process was complete. The sol-immobilized catalyst was then filtered, washed thoroughly with distilled water and then left to dry in an oven at 110 °C for 16 h.
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 mL and a maximum working pressure of 14 MPa according to our previous optimisation studies.46 To test each catalyst for H2O2 synthesis, the autoclave was charged with catalyst (in each case 0.66 μmol), and solvent (5.6 g MeOH and 2.9 g H2O). The charged autoclave was then purged three times with 5% H2/CO2 (7 bar) before filling with 5% H2/CO2 (29 bar), followed by the addition of 25% O2/CO2 (11 bar). The temperature was then decreased to 2 °C followed by stirring (1200 rpm) of the reaction mixture for 0.5 h. H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator. Duplicate reactions gave H2O2 productivities that were typically consistent to ±2% based on multiple titration results.Degradation of H2O2
The autoclave was charged with MeOH (5.6 g), H2O2 (50 wt%, 0.69 g), HPLC grade H2O (2.21 g) and catalyst (0.66 μmol), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution, 2 aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The autoclave was pressurised with 5% H2/CO2 (29 bar) and cooled to 2 °C. Upon reaching 2 °C the reaction mixture was stirred at 1200 rpm for 0.5 h. After the reaction was complete the catalyst was removed from the reaction solvents and as previously described, i.e., two aliquots (approximately 0.05 g) were titrated against an acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as the percentage of the initial 4 wt% degraded. Duplicate reactions gave degradation values that were typically consistent to ±1% H2O2 decomposed based on multiple titration results.H2O2 degradation conducted in the presence of 5% H2/CO2 (29 bar) represents the sum of both hydrogenation and decomposition pathways, while the use of 25% O2/CO2 (29 bar) allows for catalytic activity towards H2O2 decomposition alone to be determined. All reaction conditions were as described above, apart from the gas atmosphere for the H2O2 decomposition studies.
Methane oxidation reaction conditions
Methane oxidation was carried out in a 50 mL glass-lined stainless steel Parr autoclave reactor. The reactor was charged with either colloidal or supported catalyst (0.66 μmol metal per reaction) and H2O2 (amount defined in figure captions, Sigma Aldrich, 50 wt% in water). The charged autoclave was sealed and purged three times with methane (10 bar 99.999%, Air Products). It was then pressurized with methane (30 bar) and in some cases oxygen (5 bar, BOC). The mixture was stirred at 1500 rpm and heated to 50 °C at a ramp rate of 2.25 °C min−1 and maintained at the reaction temperature for 30 min. At the end of the reaction, the autoclave was cooled in ice to a temperature below 10 °C in order to minimize the loss of volatile products. The reaction gas was removed for analysis in a gas sampling bag.Product analysis for the methane oxidation reaction
Liquid phase product analysis was carried out using 1H-NMR on a Bruker 500 MHz instrument equipped with a solvent suppression system. Tetramethylsilane (TMS) in CDCl3 was used as an internal standard. The H2O2 concentration was determined using a titanium oxalate spectrophotometric method (Agilent, Cary 60). In this procedure, 0.05 to 1.0 mL of reaction sample was acidified using dilute H2SO4 before adding potassium titanium oxalate solution (0.5 wt% in water, Sigma Aldrich) to form the yellow pertitanic acid complex with a characteristic absorption peak at 390 nm. Gaseous products were quantified using a Varian 450-GC fitted with a CP-Sil 5CB capillary column (50 m length, 0.32 mm diameter, carrier gas = He), a methaniser unit and both FID and TCD detectors.Catalyst characterization
kV equipped with an Oxford Instruments X-ray energy dispersive (XEDS) spectrometer system.
Explanation of determination of number of surface atoms is reported in the ESI† material. The same approach was used for each catalyst material for comparative purposes assuming spherical particles for colloidal nanoparticles and hemispherical particles for supported nanoparticles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Cardiff University for funding as part of the MaxNet for Chemical Energy Conversion.References
- C. J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457–2487 RSC.
- J. Quinson, S. Neumann, T. Wannmacher, L. Kacenauskaite, M. Inaba, J. Bucher, F. Bizzotto, S. B. Simonsen, L. Theil Kuhn, D. Bujak, A. Zana, M. Arenz and S. Kunz, Angew. Chem., Int. Ed., 2018, 57, 12338–12341 CrossRef CAS PubMed.
- L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855–863 CrossRef CAS PubMed.
- P. Sonström and M. Bäumer, Phys. Chem. Chem. Phys., 2011, 13, 19270–19284 RSC.
- S. Maity and M. Eswaramoorthy, J. Mater. Chem. A, 2016, 4, 3233–3237 RSC.
- G. H. Han, S. H. Lee, M. G. Seo and K. Y. Lee, RSC Adv., 2020, 10, 19952–19960 RSC.
- J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 308–317 RSC.
- L. F. de L. e Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 5202–5207 CrossRef.
- M. Studer, S. Burkhardt and H. U. Blaser, Chem. Commun., 1999, 1727–1728 RSC.
- H. U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed.
- B. Minder, M. Schürch, T. Mallat, A. Baiker, T. Heinz and A. Pfaltz, J. Catal., 1996, 160, 261–268 CrossRef CAS.
- S. Haesuwannakij, T. Kimura, Y. Furutani, K. Okumura, K. Kokubo, T. Sakata, H. Yasuda, Y. Yakiyama and H. Sakurai, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- B. Donoeva and P. E. de Jongh, ChemCatChem, 2018, 10, 989–997 CrossRef CAS PubMed.
- G. M. Lari, E. Nowicka, D. J. Morgan, S. A. Kondrat and G. J. Hutchings, Phys. Chem. Chem. Phys., 2015, 17, 23236–23244 RSC.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- J. Xu, H. Zhang, Y. Zhao, B. Yu, S. Chen, Y. Li, L. Hao and Z. Liu, Green Chem., 2013, 15, 1520–1525 RSC.
- A. Villa, C. Campione and L. Prati, Catal. Lett., 2007, 115, 133–136 CrossRef CAS.
- W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 264–273 CrossRef CAS.
- L. Abis, N. Dimitritatos, M. Sankar, S. J. Freakley and G. J. Hutchings, Top. Catal., 2019, 1–9 Search PubMed.
- L. Abis, S. J. Freakley, G. Dodekatos, D. J. Morgan, M. Sankar, N. Dimitratos, Q. He, C. J. Kiely and G. J. Hutchings, ChemCatChem, 2017, 9, 2914–2918 CrossRef CAS.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. Neira d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- S. Kanungo, V. Paunovic, J. C. Schouten and M. F. Neira D'Angelo, Nano Lett., 2017, 17, 6481–6486 CrossRef CAS PubMed.
- F. Menegazzo, M. Manzoli, M. Signoretto, F. Pinna and G. Strukul, Catal. Today, 2015, 248, 18–27 CrossRef CAS.
- E. Ghedini, F. Menegazzo, M. Signoretto, M. Manzoli, F. Pinna and G. Strukul, J. Catal., 2010, 273, 266–273 CrossRef CAS.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS PubMed.
- G. M. Lari, B. Puértolas, M. Shahrokhi, N. López and J. Pérez-Ramírez, Angew. Chem., 2017, 129, 1801–1805 CrossRef.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- J. K. Edwards, B. Solsona, E. Ntainjua, N. A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS.
- J. Zhao, W. Y. Hernández, W. Zhou, Y. Yang, E. I. Vovk, M. Capron and V. Ordomsky, ChemCatChem, 2020, 12, 238–247 CrossRef CAS.
- A. García-Trenco, E. R. White, A. Regoutz, D. J. Payne, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2017, 7, 1186–1196 CrossRef.
- Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619–621 CrossRef CAS PubMed.
- T. Deguchi, H. Yamano, S. Takenouchi and M. Iwamoto, Catal. Sci. Technol., 2018, 8, 1002–1015 RSC.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- G. Giorgianni, S. Abate, G. Centi and S. Perathoner, ChemCatChem, 2019, 11, 550–559 CrossRef CAS.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
- R. McVicker, N. Agarwal, S. J. Freakley, Q. He, S. Althahban, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Catal. Today, 2020, 342, 32–38 CrossRef CAS.
- T. Ishihara, R. Nakashima, Y. Ooishi, H. Hagiwara, M. Matsuka and S. Ida, Catal. Today, 2015, 248, 35–39 CrossRef CAS.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- Y. F. Han and J. H. Lunsford, J. Catal., 2005, 230, 313–316 CrossRef CAS.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63–86 RSC.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- M. Piccinini, E. Ntainjua, N. J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488–2492 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00915f |
| This journal is © The Royal Society of Chemistry 2020 |