
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adipic acid formation from cyclohexanediol using platinum and vanadium catalysts: elucidating the role of homogeneous vanadium species†
Owen
Rogers
a,
Samuel
Pattisson
 a,
Rebecca V.
Engel
a,
Robert L.
Jenkins
a,
Keith
Whiston
b,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
a,
Rebecca V.
Engel
a,
Robert L.
Jenkins
a,
Keith
Whiston
b,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cf.ac.uk; Tel: +44 (0)2920 874059
bINVISTA Performance Technologies, The Wilton Centre, Wilton, Redcar, TS10 4RF, UK
First published on 2nd June 2020
Abstract
Vanadium compounds have shown great potential alongside Pt/C for the oxidation of cyclohexanediol to adipic acid. However, the low stability of these materials often leads to ambiguity when considering the homogeneous or heterogeneous nature of the active species. In this article we describe our attempts to synthesise stable vanadium catalysts through the utilisation of vanadium bronze structures. By the addition of sodium, copper or silver into these structures, leaching could be decreased to 5% for AgVO3, compared to 88.4% with V2O5. These reactions were run in aqueous conditions under 3 bar O2. However, despite significant stabilisation of vanadium in the bronze structures, we show that as little as 7.6 ppm of a homogeneous vanadium species in the reaction solution can cause the selective oxidation of 2-hydroxycyclohexanone to adipic acid. Analysis of the speciation by 51V NMR and UV-vis has revealed the active species to be in the +5 oxidation state in the form of a decavanadate compound with the presence of small amounts of monovanadate.
Introduction
The production of chemicals using the principles of green chemistry is becoming increasingly important.1 Drives to cut emissions are at the forefront of government policy and as such the incentives for industry to follow suit are much greater. Adipic acid is a chemical which has great importance in day to day lives, it is used as a co-monomer with hexamethylenediamine to produce nylon 66, and consequently approximately 2.5 million tonnes is produced annually.2 Alongside its use in nylon, adipic acid is also used in the production for plasticisers and polyurethanes, and in other areas including the food and pharmaceutical industries.3Currently, adipic acid is produced predominantly from a cyclohexane-based feedstock.3–5 In this process the use of nitric acid as the oxidant results in the release of N2O, which has a global warming potential of 300 relative to CO2.6 The environmental impact of this adipic acid process has been diminished due to recent catalytic and thermal abatement of the N2O emissions, resulting in significant reduction of associated N2O release. However the prospect of completely eliminating N2O from the process would be beneficial.7–9 This is gradually becoming more feasible due to the possibility of using cyclohexene as a substrate instead of cyclohexane. Cyclohexene is more easily oxidised and therefore, greener oxidants such as oxygen and hydrogen peroxide can be used, which can offer better atom economy.1,10–12
Oxidation starting from cyclohexene may need to be run in two steps. The first step would be an oxidation of cyclohexene to cyclohexanediol, which can be achieved using O2, albeit with a maximum selectivity of 50%.13 It is for this reason that a process which uses H2O2 in a first step, would be the most economically viable. However, this would likely only be economic at commercial scale if a stoichiometric amount of oxidant relative to substrate could be used.14 This could then be followed by a second step to convert cyclohexanediol to adipic acid, which can be achieved using O2.
The oxidative cleavage of vicinal diols is an important process in organic synthetic chemistry and is mostly achieved using expensive noble metal-based homogeneous catalysts. However, these catalysts suffer from a poor substrate range and do not offer reusability.15–17 The Malaprade reaction18 and the Criegee oxidation19 offer classical examples of the cleavage of 1,2-diols, however, they require the use of stoichiometric oxidants, such as high-valent iodine or lead, resulting in large amounts of toxic waste.20,21 Heterogeneous catalysts have also been used for vicinal diol cleavage, particularly, noble metals such as Pt, Ru, and Au have been reported but these usually suffer from low activity.22–24 It is rare that a non-noble metal shows activity for this reaction. However, Escande et al. found that a sodium–manganese oxide was active at 100 °C, albeit with a minimal substrate scope and, in particular, being inactive for the oxidation of cyclohexanediol.25
Brégeault and co-workers demonstrated the ability of various vanadium compounds to perform the aerobic oxidative cleavage of 2-hydroxycyclohexanone (2-HCO) to form adipic acid.26 Rozhko et al. subsequently studied the ability of Keggin type P/Mo/V polyoxometalates for the oxidation of cyclohexanediol under acidic conditions. However, it was suggested that despite high initial selectivity, the adipic acid formed underwent further reaction with cyclohexane diol to form the corresponding ester, resulting in low overall yields of the acid.27 More recently, Obara et al.24 demonstrated the potential for a system utilising Pt/C and V2O5 that can selectively cleave the vicinal diol to adipic acid (Scheme 1). Using O2 as oxidant and H2O as solvent, a yield of over 90% can be reached when both catalysts were combined in a one-pot reaction over 48 h. Cyclohexanediol was converted to 2-HCO over Pt/C alone, whereas this reaction did not proceed with only V2O5. By using a V2O5 co-catalyst, the 2-HCO was then converted to cyclohexanediol. The main problem with such a system is the solubility of the V2O5 catalyst. The researchers highlighted that over each re-use approximately half of the vanadium leached into solution, although it was suggested that the remaining solid was responsible and sufficient for the observed catalysis. Nevertheless, a truly heterogeneous vanadium catalyst would mark a major advance in this field.
 | ||
| Scheme 1 Reaction scheme for oxidation of cyclohexanediol to adipic acid as proposed by Obara et al. | ||
Supported vanadium oxides have been extensively used in oxidation reactions, notably in the selective oxidation of alkenes and alkanes and the oxidation of H2S.28–30 However, heterogeneous vanadium catalysts are well-known to leach in aqueous solutions. This can be alleviated to some extent by changing the support and reaction conditions as shown in work by Masumoto et al.31 However, even the most successful attempt at mitigating leaching, a V/Al2O3 catalyst, still showed 37.6% leaching of vanadium into the solvent. The effect of leaching of vanadium catalysts in heterogeneous liquid phase reactions is also stated in studies by Ziolek et al., whereby V/MCM-41 leaches 71 wt% of vanadium into the reaction solution.32 Interestingly, work by Tiwari et al. on polyoxometalate structures, specifically H5[PV2Mo10O40], have shown that the dissolution of two reactive pervanadyl ions by H2SO4 is a reversible process, which can be controlled by the pH.33
Vanadium bronzes, which when combined with other metals such as sodium, copper or silver, can show stability in aqueous media and have various applications in areas such as, photocatalysts and in electrochemical energy storage, however elemental analysis of aqueous media is not always reported.34,35 In this report we show the synthesis of several vanadium bronzes with the aim of achieving stabilisation in aqueous media. However, we also demonstrate that even small amounts of leaching from these catalysts can have a great effect on reactivity. The filtrate of these reactions is also analysed to determine the likely active vanadium speciation.
Experimental
Chemicals
The following chemicals were used in this investigation without further purification. Vanadium oxide (V2O5, 99.95%), sodium sulfate (NaSO4, 99.99%), copper(I) acetate (CuOAc, 97%), hydrogen peroxide (H2O2, 30 wt%), ammonium metavanadate (NH4VO3, 99.9%), vanadium carbide (99%), urea (99%), trans-1,2-cyclohexanediol (98%), 5 wt% Pt/C, 2-hydroxycyclohexanone dimer (97%) were all sourced from Sigma-Aldrich.Catalyst preparation
Catalytic testing
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) was loaded into a low-pressure glass reactor. When required, Pt/C (10 mg) and the vanadium co-catalyst (2 mg) were then added to the vessel. The reactor was purged with O2 and then charged with 3 bar O2. The reactor was then stirred at 80 °C for 4 h. Samples were centrifuged and filtered using PTFE syringe filters (0.45 μm) before HPLC analysis.
000 ppm) was loaded into a low-pressure glass reactor. When required, Pt/C (10 mg) and the vanadium co-catalyst (2 mg) were then added to the vessel. The reactor was purged with O2 and then charged with 3 bar O2. The reactor was then stirred at 80 °C for 4 h. Samples were centrifuged and filtered using PTFE syringe filters (0.45 μm) before HPLC analysis.
| Conversion (%) = [(mol of consumed substrate)/(mol of initial substrate)] × 100 |
| Selectivity (%) = [(mol of product)/(mol of consumed substrate)] × 100 |
| Mass balance (%) = [(mol of product final + mol of final substrate)/(mol of initial substrate)] × 100 |
Catalyst characterisation
Results and discussion
Obara et al.24 have demonstrated the potential for Pt/C and vanadium to operate catalytically in tandem offering a highly selective route to adipic acid from cyclohexanediol. Firstly, the results (Table 1 – entry 1) show that the blank reaction displayed no activity. The use of V2O5 (Table 1 – entry 2) as the sole catalyst resulted in little activity, with only a small amount of conversion observed. This is followed by Pt/C (Table 1 – entry 3), which showed a conversion of 39.4% and a selectivity of 59.4% towards 2-HCO. This is accompanied by low selectivity to other oxidation products, such as glutaric acid and succinic acid at 2.7% and 1.4%, respectively. When a vanadium catalyst is added to the Pt/C catalyst, selectivity towards adipic acid was observed, as shown in entry 4. In this system selectivity to adipic acid increases to 76.1% and conversion remains at a similar level of 49.5%. This reaction is likely a largely homogeneous reaction, as most of the vanadium oxide catalyst has been measured by MP-AES to be dissolved in the reaction media. Obara et al. suggested that despite losing approximately half the solid vanadium on each use, the remaining catalyst is responsible for the activity observed.24 In either case, this leached homogeneous vanadium makes the system far less industrially viable due to the poor stability, and therefore lifetime, of the vanadium oxide under reaction conditions in addition to the possible contamination of the product with toxic vanadium species. If a stable heterogeneous vanadium catalyst could be prepared, then this would be more practical industrially. For this purpose, we therefore prepared a range of vanadium bronzes incorporating Na, Cu or Ag to increase stability, as they have commonly been used previously as heterogeneous catalysts in aqueous reactions, albeit with V leaching not being reported.34–37000 ppm cyclohexanediol in water (5 ml), 10 mg 5% Pt/C, 2 mg V2O5
| Entry | Catalyst | Conversion/% | Selectivity/% | Vanadium leaching/% | Vanadium concentration/ppm | Mass balance/% | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Adipic acid | 2-HCO | Glutaric acid | Succinic acid | Unknowns | ||||||
| 1 | Blank | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | — | — | 100 |
| 2 | V2O5 | 3.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 97.5 | ||
| 3 | Pt/C | 39.4 | 0.0 | 59.4 | 2.1 | 1.3 | 9.2 | — | — | 90.4 |
| 4 | Pt/C + V2O5 | 49.5 | 76.1 | 1.2 | 4.5 | 1.4 | 5.4 | 88.4 | 168 | 96.2 |
All bronze catalysts in this study were synthesised using a hydrothermal technique and the XRD patterns are shown in Fig. 1. The Na2V6O16 bronze was synthesised according to a previous method described by Xu et al.,38 using V2O5 and NaSO4 as precursors. The XRD pattern (black) can be referenced to the monoclinic Na2V6O16 phase with a P21/m space group with reflections at 2θ = 11.70, 25.93, 28.35, 29.77, 40.06, 46.26 and 50.83° all indicative of this phase.39,40 This is accompanied by a high degree of crystallinity. Included in Fig. 1 is the XRD pattern for Cu0.5V2O6 (red), which shows reflections that would be indicative of a CuV2O6 phase with peaks at 2θ = 20.7, 26.7, 33.6, 42.1, 42.2 and 44.8°. However, there also appear to be reflections which can be identified as related to the monoclinic β-Cu0.261V2O5 phase, which is characterised by reflections at 2θ = 29.16, 12.15, 26.39, 31.16, 33.09, 37.25, 39.73, 40.93 and 46.23°.29,36 The XRD pattern for AgVO3 (blue) shows strong similarities to that of the β-AgVO3 phase with the monoclinic structure and I2/m space group.35,41 There were no other detectable reflections in the sample suggesting the sample was mainly composed of the β-AgVO3 phase.
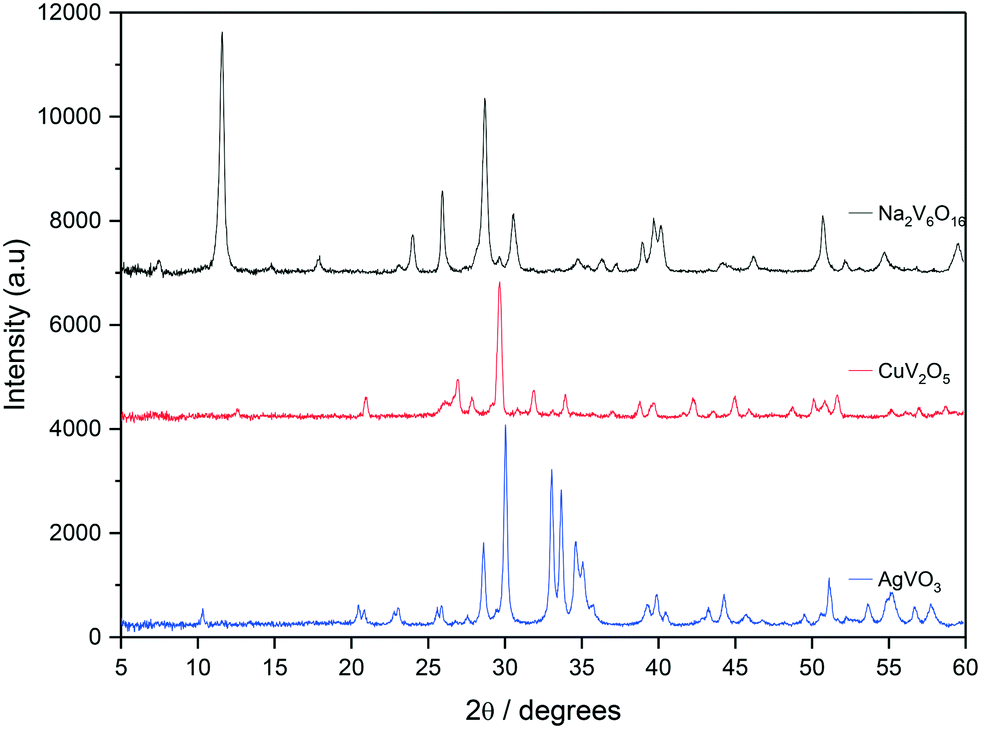 | ||
| Fig. 1 XRD patterns for synthesised bronzes: NaV6O15 (black), CuV2O5 (red), AgVO3 (blue). | ||
When used for the oxidation of cyclohexanediol, the Na2V6O16 catalyst (Table 2 – entry 1) demonstrates a similar conversion and adipic acid selectivity to the Pt/C + V2O5 system, achieving 36.7% and 70.1% respectively. However, the main drawback of this catalyst was the amount of leaching observed. MP-AES analysis of the vanadium bronze post reaction solution revealed a total of 67 ppm V which amounts to 41.8% of the overall vanadium present in the bronze. This level of leaching was still distinctly lower than that for a Pt/C + V2O5 catalytic reaction which leached 88.4%, however it is still an unsustainable amount. This level of leaching was consistent even after catalysts were washed with 1 L boiling water prior to the catalytic testing and also after multiple reuse experiments. These results show that sodium is not a suitable metal substitute to stabilise the leaching of vanadium species in the mixed metal oxide.
000 ppm cyclohexanediol in water (5 ml), 10 mg 5% Pt/C, 2 mg vanadium catalyst
| Entry | Catalyst | Conversion/% | Selectivity/% | Vanadium leaching/% | Vanadium concentration/ppm | Mass balance/% | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Adipic acid | 2-HCO | Glutaric acid | Succinic acid | Unknowns | ||||||
| 1 | Pt/C + Na2V6O16 | 36.7 | 70.1 | 4.0 | 6.5 | 1.6 | 1.5 | 41.8 | 67 | 96.0 |
| 2 | Pt/C + Cu0.25V2O6 | 32.0 | 64.0 | 4.9 | 0.0 | 0.0 | 4.3 | 16.6 | 35 | 91.2 |
| 3 | Pt/C + Cu0.33V2O6 | 27.2 | 60.4 | 9.0 | 0.0 | 0.0 | 0.0 | 5.8 | 11 | 91.2 |
| 4 | Pt/C + Cu0.5V2O6 | 25.1 | 63.7 | 9.5 | 0.0 | 2.2 | 5.2 | 19.8 | 33 | 95.5 |
As an attempt to further decrease the vanadium leaching, the effect of using copper or silver as stabilising metals in the vanadium bronze structure was investigated. Metals such as Cu and Ag are commonly substituted in these materials, due to the resulting enhanced electrochemical properties and increased activity in the oxidation of H2S.29,41–43 For the copper bronzes, shown in Table 2 entries 2–4, different molar ratios of copper to vanadium were prepared to investigate any effect that copper had on the stability of the vanadium bronze. The molar ratios of copper to vanadium studied were 0.25, 0.33, and 0.5. From Table 2 it can also be seen that vanadium leaching is reduced dramatically on inclusion of copper to 5.8% for Cu0.33V2O6. This contrasted with Cu0.25V2O6 and Cu0.5V2O6, which showed vanadium leaching of 16.6% and 19.8% respectively. This is a drastically lower leaching level than that observed with the Na2V6O16 bronzes, demonstrating that the copper successfully increases the stability of the bronze. As seen in Table 2, the copper bronzes achieve lower conversions than observed with Na2V6O16. However, this level of conversion is mostly influenced by Pt/C and therefore it is more useful to compare selectivity to adipic acid. It is evident from the data that CuxV2O6 showed lower selectivity to adipic acid. This may be due to copper atoms occupying active sites at the surface of the catalyst, or due to the additional copper affecting the electronic structure of vanadium at the surface, making it more stable but potentially less reactive. Another explanation for this change in selectivity may also be a result of less leaching in the reaction solution highlighting the role of homogeneous vanadium. These bronzes show similar selectivities towards adipic acid, despite there being a lower vanadium concentration present in the Cu0.33V2O6 reaction solution. However, this may be due to only low concentrations of homogeneous vanadium being required to have this marked effect on selectivity.
The final bronze of the series of vanadium catalysts was AgVO3, which is shown in Table 3 – entry 1. This bronze catalyst shows the highest stability with regards to vanadium leaching with only 5.0% V leached into solution. This corresponds to a total of 9 ppm V present in the reaction solution, the lowest levels of vanadium leaching from the bonzes. Unfortunately, this catalyst also showed a noticeably lower cyclohexanediol conversion than Na2V6O16 and CuxV2O6. It can be observed (Table 3 entry 1) that there is still 21.3% of 2-HCO in the solution so this had not been completely converted to adipic acid. This may be due to less active vanadium sites or also it could be attributed to the lower concentration of homogeneous vanadium in solution.
000 ppm cyclohexanediol in water (5 ml), 10 mg 5% Pt/C, 2 mg vanadium catalyst
| Entry | Catalyst | Conversion/% | Selectivity/% | Vanadium leaching/% | Vanadium concentration/ppm | Mass balance/% | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Adipic acid | 2-HCO | Glutaric acid | Succinic acid | Unknowns | ||||||
| a Reaction conditions: 80 °C, 3 bar O2, 4 h, 4000 ppm 2-hydroxycyclohexanone in water (5 ml), 5 mg vanadium carbide after washing with 5 L water. | ||||||||||
| 1 | AgVO3 | 18.4 | 41.6 | 21.3 | 0.0 | 2.1 | 0.0 | 5.0 | 9 | 92.8 |
| 2 | V–C3N4 | 24.7 | 59.3 | 5.3 | 0.0 | 1.7 | 33.7 | — | 33 | 91.4 |
| 3 | VCa | 19.4 | 55.0 | — | 0.0 | 0.0 | 16.0 | 1.2 | 17 | 91.3 |
Efforts to probe the heterogeneity of this reaction extended to synthesising a support that had a strong interaction with vanadium, to minimise leached vanadium in solution. In previous studies Ding et al.44 had shown that vanadium supported on carbon nitride can be a promising catalyst in liquid phase reactions for the synthesis of phenol from benzene. The purpose of carbon nitride as a support to stabilise vanadium was predominantly due to basic NH and NH2 groups on the surface which help to bind to the acidic vanadium species. These acid–base interactions have also been shown to decrease leaching to a negligible amount in vanadium-substituted molybdophosphoric acid, due to the strong interactions between the heteropolyacid and amine groups on SBA-15.45 The V–C3N4 material was successfully synthesised and tested in the oxidation reaction (Table 3 – entry 2). However, after a hot wash prior to the catalytic testing, 32.6 ppm V was still leached into the reaction solution. An accurate measure of vanadium leaching from V–C3N4 was not obtained, due to the difficulty of dissolving graphite and carbon nitride in aqua regia. In addition to the synthesised vanadium catalysts, a commercial vanadium carbide, which is insoluble in aqueous solutions,46 was tested (Table 3 – entry 3). However, under reaction conditions leaching was observed for this material as well. The leaching continued even after a pre-wash of the catalyst with 5 L of boiling water. However, after this treatment only 1.2% of total vanadium leached from the catalyst, giving a total concentration of 17.0 ppm vanadium in the reaction mixture. To observe whether this level of vanadium leaching was significant for cyclohexanediol oxidation a further study using solutions with known quantities of homogeneous vanadium was conducted.
Effect of homogeneous vanadium on the oxidation of cyclohexanediol
To elucidate the role of homogenous vanadium, a series of reactions using low concentrations of vanadium in solution were conducted. A time online study was also undertaken to assess the effect of conversion on selectivity, as conversion is largely controlled by Pt/C. As there is a slight correlation between conversion and selectivity (Fig. 2a), 2-HCO was used as the starting material for these reactions, and therefore the addition of Pt/C was unnecessary.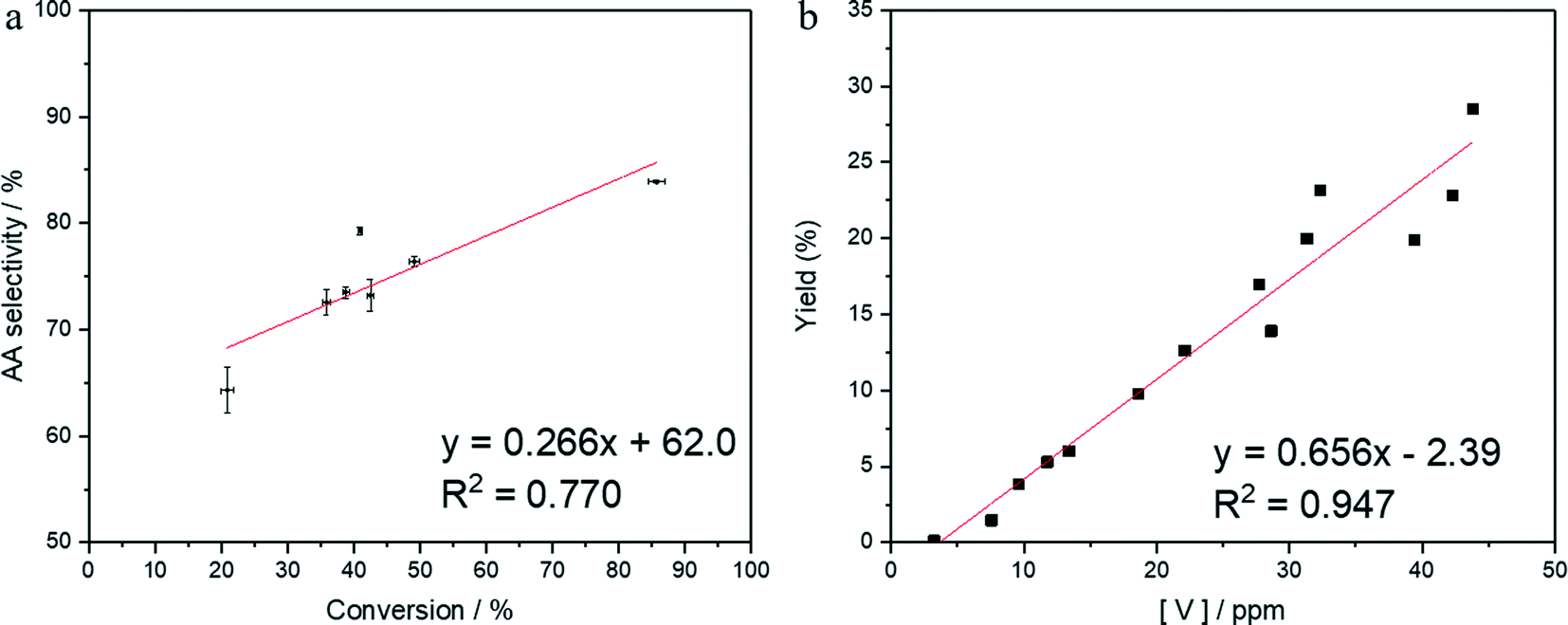 | ||
| Fig. 2 a) Effect of conversion on selectivity taken over reactions from 2 h to 22 h. Reaction conditions: 80 °C, 3 bar O2, 5 ml 10000 ppm cyclohexanediol in water, 10 mg Pt/C, 2 mg V2O5 b) oxidation of 2-HCO to adipic acid using solutions of vanadium between 0 and 45 ppm. Reaction conditions: 80 °C, 3 bar O2, 4 h, 4000 ppm 2-hydroxycyclohexanone in water (5 ml), vanadium added as a stock solution of 200 ppm V2O5 in water. Vanadium concentrations are obtained using MP-AES analysis which are run in triplicate and averaged. | ||
To study the effect of homogeneous vanadium, a solution of 4000 ppm 2-HCO in water was used as the starting solution. A stock solution of V2O5 in water was prepared with a concentration of 200 ppm. This was then inserted into the reaction as required to obtain vanadium concentrations within the range of 0 to 45 ppm. The final concentration post reaction was then analysed by MP-AES. The results can be seen in Fig. 2b. These solutions contained only soluble vanadium species so any reaction observed would be completely homogeneous. Fig. 2b demonstrates that at these low concentrations, a linear trend can be observed between vanadium in solution and yield to adipic acid. Interestingly, it is only very minimal amounts of vanadium that are required to promote conversion of 2-HCO to adipic acid. A concentration of 7.6 ppm is sufficient to observe an effect on the reaction and yield 1.5% adipic acid. This suggests that even a small amount of leaching from the above vanadium materials would likely result in catalytic activity. These vanadium species can also be determined to be acting catalytically and not as stoichiometric oxidants (SI). Higher concentrations above 45 ppm proved difficult to prepare under these reaction conditions, despite seeing concentrations above 150 ppm previously when using the bronzes as catalysts. High concentrations above 50 ppm seems to be achievable only under certain reaction conditions, which were not present in the study of 4000 ppm 2-HCO. As such, a study was undertaken to probe which conditions would induce leaching of vanadium in reaction solution and the results are illustrated in Fig. 3.
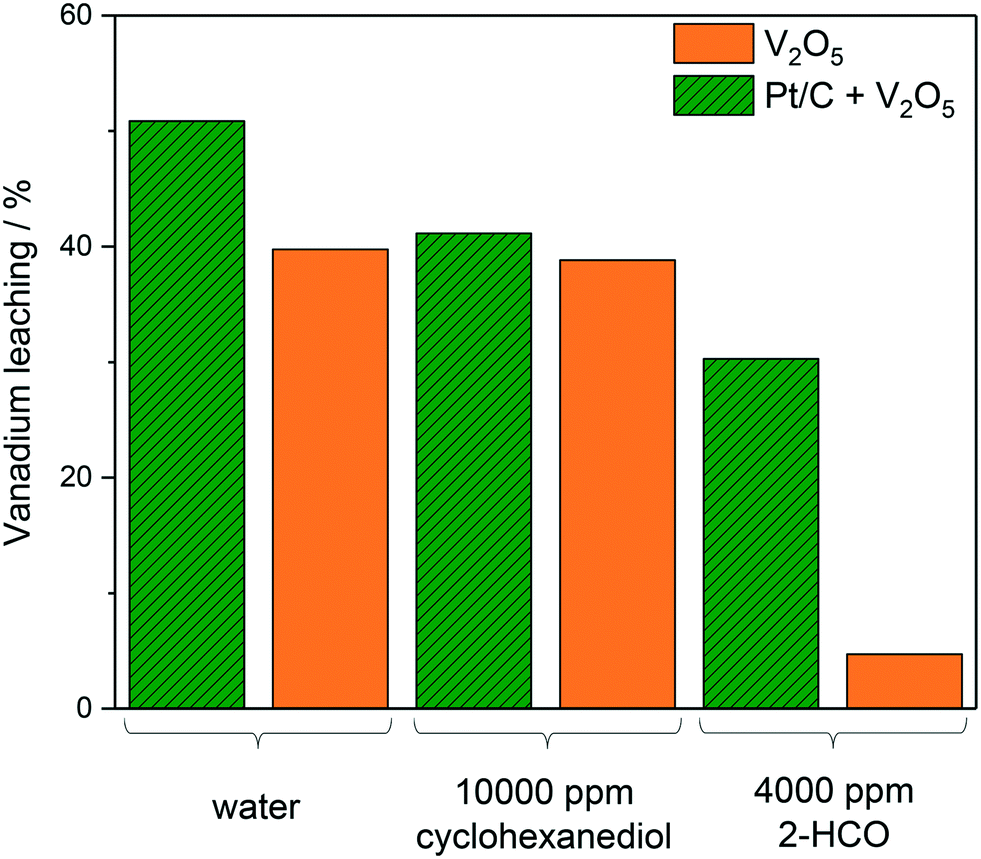 | ||
| Fig. 3 Leaching of vanadium induced by differing sets of reaction conditions. Reaction conditions: 80 °C, 3 bar O2, 4 h, 5 ml reaction solution (as specified), 10 mg Pt/C (when required), 5 mg V2O5 (when required). | ||
Fig. 3 shows the effect of reaction conditions on vanadium leaching, specifically the effect of the presence of Pt/C and the effect of different reaction mixtures. The reactions containing both Pt/C and V2O5 showed consistently more leaching than their counterpart which only included V2O5. The effect of the Pt/C on the leaching may have been due to the Pt activating the oxygen causing it to destabilise the vanadium structure and promote leaching of the V2O5 more readily. The most drastic decrease in leaching of V2O5 is observed in 4000 ppm 2-HCO where only 4.7% was homogeneous. This is compared to 38.8% in 10000 ppm cyclohexanediol. The main reason for this could be the polarity of the different substrates affecting the solubility of vanadium.
Since we have shown that adipic acid selectivity was mainly reliant on homogeneous vanadium, UV-vis and 51V NMR studies were undertaken to determine the speciation of vanadium present in the reactions. The UV-vis is shown in Fig. 4, and shows the reaction filtrate from Na2V6O16 (green) and the V2O5 reaction mixture (blue) as well as the V2O5 stock solution (pink) used for the vanadium homogeneous reactions (Fig. 2). From the UV-vis spectra of these mixtures it can be concluded that the vanadium oxidation state is +5 with no other oxidation states present.47 From the UV-vis spectra we do not see V(III) or V(IV) so the catalytic V(V) must have a closed catalytic cycle. The spectra for 51V NMR studies are shown in Fig. 5. By comparing the NMR spectrum with data published by Andersson et al.48 the vanadium species can be identified as a vanadium decavanadate, which has 3 chemical environments as shown by the spectra. These shifts can be observed at −424 ppm, −508 ppm, −526 ppm for Vc, Vb and Va respectively. An additional signal, which can be attributed to the monovanadate species, can also be seen in the NMR spectrum.49 To confirm that the spectrum observed for the reaction mixture in Fig. 5a was as a result of a V10 species (Fig. 5c), a sample of the decavanadate was prepared from an acidified solution of sodium orthovanadate.49–51 As can be seen in the spectrum the signals overlay perfectly, with the only exception of the vanadium environment signified by Va. For this environment there is a small shift from −526 ppm to −529 ppm for the reaction filtrate and the acidified sodium orthovanadate V10 species, respectively. This shift may result from a binding of Va with reaction products. There also appears to be a stronger monovanadate peak in the 51V NMR spectrum of V10, which may be a consequence of the lower pH of this sample.
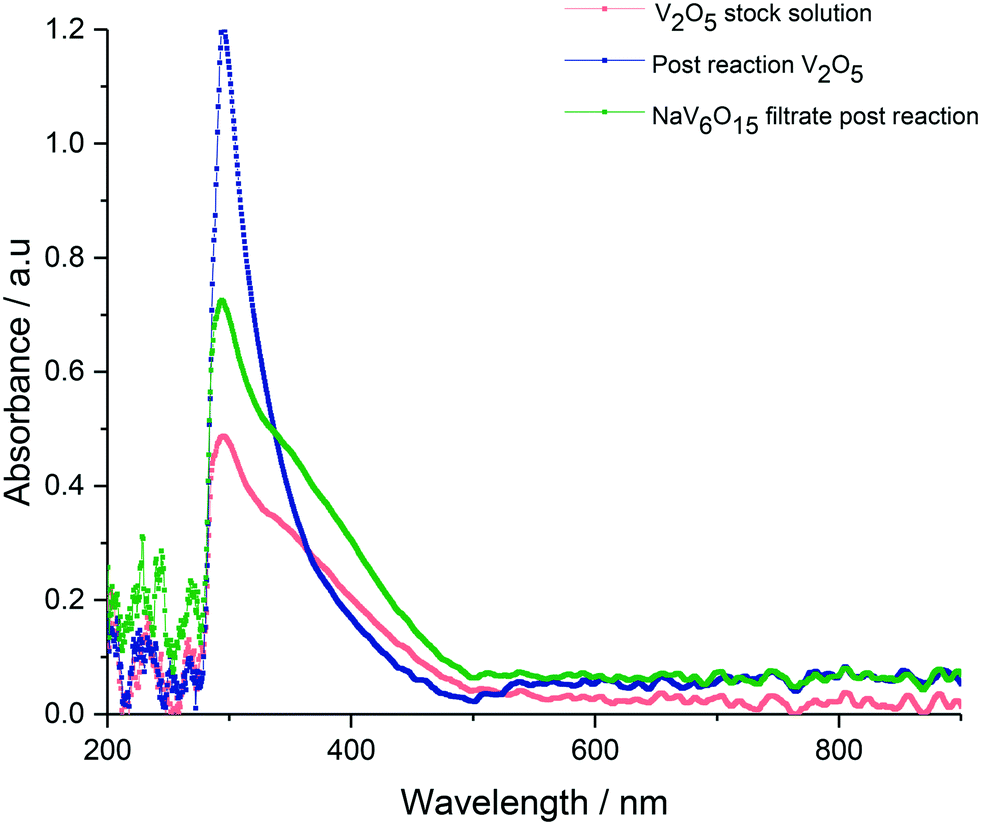 | ||
| Fig. 4 UV-vis spectra of reaction filtrate from bronze reactions showing presence of only vanadium +5. V2O5 stock solution (pink), post reaction mixture of V2O5 (blue), post reaction filtrate of NaV6O15 (green). | ||
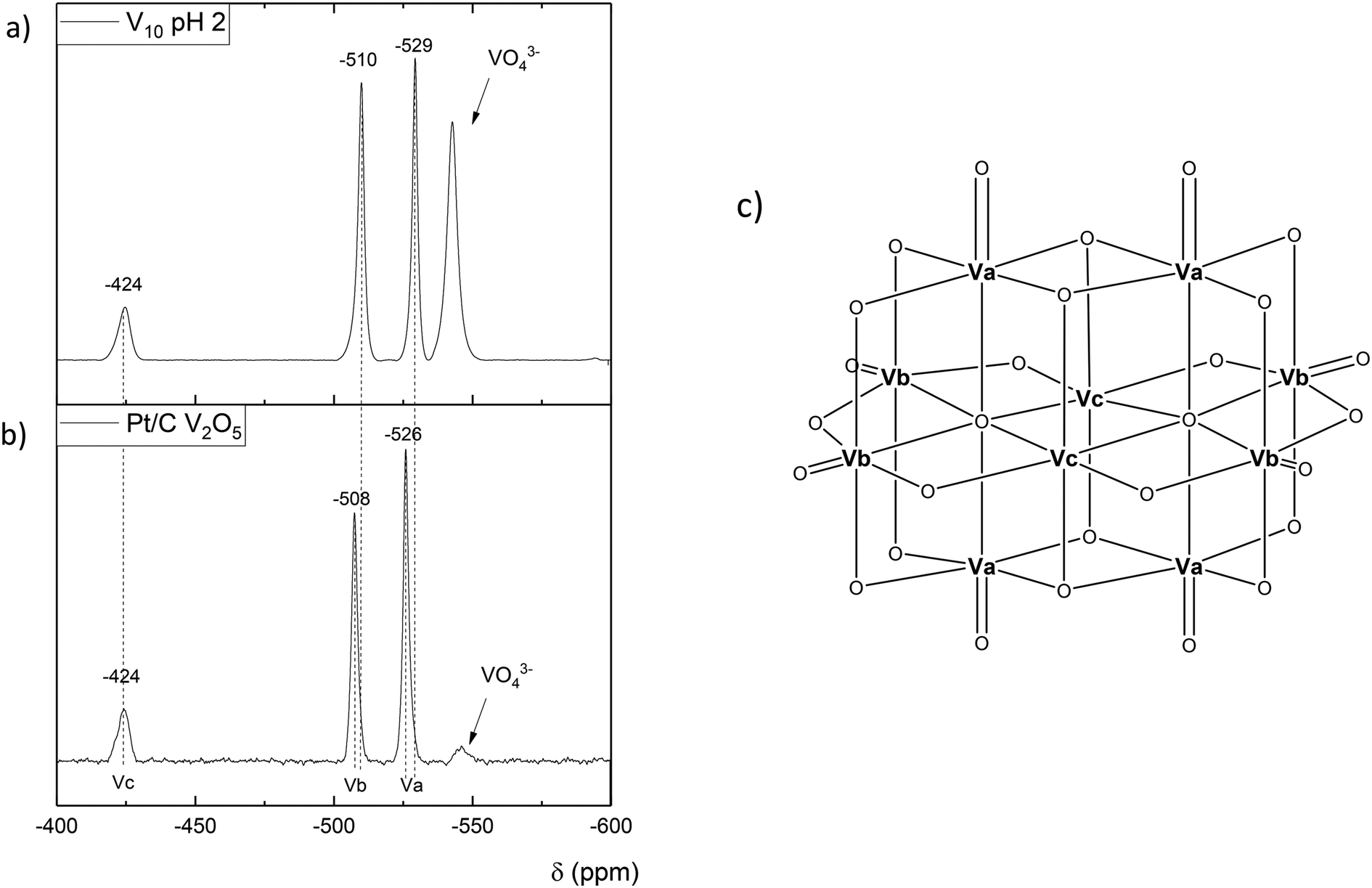 | ||
| Fig. 5 51V NMR of a) vanadium decavanadate which is present at low pH with presence of monovanadate also b) reaction sample of Pt/C and V2O5 with 4000 ppm 2-HCO solution c) V10 compound with labelled vanadium sites corresponding to the shifts shown in the NMR spectra. | ||
Conclusions
A range of vanadium bronzes were assessed for the oxidation of cyclohexanediol to adipic acid. Initial sodium containing bronzes demonstrate promising activities, however this is accompanied by high degrees of leaching. This leaching can be significantly mitigated by the incorporation of Cu or Ag. However, even the low levels of leaching, observed with the Cu0.33V2O6 and AgVO3, enable the conversion of 2-HCO to adipic acid. In addition, efforts to reduce leaching using a carbon nitride as a support proved unsuccessful despite strong acid–base interactions being reported as stabilising the vanadium onto the surface of the support. In subsequent studies it was shown that even trace levels of vanadium leached from these materials was enough to catalyse the reaction homogeneously. 51V NMR studies have revealed that the speciation in these homogeneous reactions to be in the form of a decavanadate.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge INVISTA and the EPSRC Centre for Doctoral Training in Catalysis for funding, project reference EP/L016443/1. RVE acknowledges funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 663830. Information on the data underpinning the results presented here can be found in the Cardiff University data catalogue at: http://doi.org/10.17035/d.2020.0108567396.References
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- S. Van De Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- A. Castellan, J. C. J. Bart and S. Cavallaro, Catal. Today, 1991, 9, 237–254 CrossRef CAS.
- A. F. Lindsay, Chem. Eng. Sci., 1954, 3, 78–93 CrossRef CAS.
- J. R. L. Smith, C. B. Thomas and M. Whittaker, J. Chem. Soc., Perkin Trans. 2, 1993, 2191–2194 RSC.
- R. A. Reimer, C. S. Slaten, M. Seapan, M. W. Lower and P. E. Tomlinson, Environ. Prog., 1994, 13, 134–137 CrossRef CAS.
- J. Li, G. Luo, Y. Chu and F. Wei, Chem. Eng. J., 2012, 184, 168–175 CrossRef CAS.
- R. E. Dickinson and R. J. Cicerone, Nature, 1986, 319, 109–115 CrossRef CAS.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1648 CrossRef CAS PubMed.
- R. Sheldon, Nature, 1999, 399, 33 CrossRef.
- D. S. Ovoshchnikov, B. G. Donoeva, B. E. Williamson and V. B. Golovko, Catal. Sci. Technol., 2014, 4, 752 RSC.
- P. Blach, Z. Böstrom, S. Franceschi-Messant, A. Lattes, E. Perez and I. Rico-Lattes, Tetrahedron, 2010, 66, 7124–7128 CrossRef CAS.
- Z. Z. Zhou, M. Liu, L. Lv and C. J. Li, Angew. Chem., Int. Ed., 2018, 57, 2616–2620 CrossRef CAS PubMed.
- A. Wang and H. Jiang, J. Org. Chem., 2010, 75, 2321–2326 CrossRef CAS PubMed.
- E. Takezawa, S. Sakaguchi and Y. Ishii, Org. Lett., 1999, 1, 713–715 CrossRef CAS PubMed.
- L. Malaprade, Bull. Soc. Chim. Fr., 1934, 3, 833–852 Search PubMed.
- R. Criegee, Ber. Dtsch. Chem. Ges., 1930, 64, 260–266 CrossRef.
- F. J. Wolf and J. Weijlard, Org. Synth., 1955, 35, 18 CrossRef CAS.
- M. Frigerio and M. Santagostino, Tetrahedron Lett., 1994, 35, 8019–8022 CrossRef CAS.
- T. R. Felthouse, P. B. Fraundorf, R. M. Friedman and C. L. Schosser, J. Catal., 1991, 127, 393–420 CrossRef CAS.
- S. Solmi, E. Rozhko, A. Malmusi, T. Tabanelli, S. Albonetti, F. Basile, S. Agnoli and F. Cavani, Appl. Catal., A, 2018, 557, 89–98 CrossRef CAS.
- N. Obara, S. Hirasawa, M. Tamura and Y. Nakagawa, ChemCatChem, 2016, 8, 1732–1738 CrossRef CAS.
- V. Escande, C. H. Lam, P. Coish and P. T. Anastas, Angew. Chem., Int. Ed., 2017, 56, 9561–9565 CrossRef CAS PubMed.
- M. Vennat, P. Herson, J.-M. Brégeault and G. B. Shul, Eur. J. Inorg. Chem., 2003, 908–917 CrossRef CAS.
- E. Rozhko, K. Raabova, F. Macchia, A. Malmusi, P. Righi, P. Accorinti, S. Alini, P. Babini, G. Cerrato, M. Manzoli and F. Cavani, ChemCatChem, 2013, 5, 1998–2008 CrossRef CAS.
- K. Li and T. Chien, Catal. Lett., 1999, 57, 77–80 CrossRef CAS.
- L. Ruiz-Rodríguez, T. Blasco, E. Rodríguez-Castellón and J. M. L. Nieto, Catal. Today, 2018, 333, 237–244 CrossRef.
- J. M. López Nieto, P. Concepción, A. Dejoz, H. Knözinger, F. Melo and M. I. Vázquez, J. Catal., 2000, 189, 147–157 CrossRef.
- Y.-K. Masumoto, R. Hamada, K. Yokota, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2002, 184, 215–222 CrossRef CAS.
- M. Ziolek, Catal. Today, 2004, 90, 145–150 CrossRef CAS.
- C. K. Tiwari, M. Baranov, A. Neyman, R. Neumann and I. A. Weinstock, ACS Appl. Mater. Interfaces DOI:10.1021/acs.inorgchem.9b03747.
- C. Xia, J. Guo, P. Li, X. Zhang and H. N. Alshareef, Angew. Chem., 2018, 130, 4007–4012 CrossRef.
- R. Konta, H. Kato, H. Kobayashi and A. Kudo, Phys. Chem. Chem. Phys., 2003, 5, 3061–3065 RSC.
- W. Guo, W. D. Chemelewski, O. Mabayoje, P. Xiao, Y. Zhang and C. B. Mullins, J. Phys. Chem. C, 2015, 119, 27220–27227 CrossRef CAS.
- Z. Xie, J. Lai, X. Zhu and Y. Wang, ACS Appl. Energy Mater., 2018, 1, 6401–6408 CrossRef.
- Y. Xu, X. Han, L. Zheng, W. Yan and Y. Xie, J. Mater. Chem., 2011, 21, 14466–14472 RSC.
- G. T. Zhou, X. Wang and J. C. Yu, Cryst. Growth Des., 2005, 5, 969–974 CrossRef CAS.
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang, L. Zhou, Z. Zhou and L. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- L. Mai, L. Xu, Q. Gao, C. Han, B. Hu and Y. Pi, Nano Lett., 2010, 10, 2604–2608 CrossRef CAS PubMed.
- J. Zhang, N. Wang, C. Zhao, J. Li and T. Zhang, Chin. J. Mater. Res., 2018, 32, 555–560 Search PubMed.
- Y. Xu, X. Han, L. Zheng, W. Yan and Y. Xie, J. Mater. Chem., 2011, 21, 14466–14472 RSC.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- A. N. Kharat, S. Moosavikia, B. T. Jahromi, A. Badiei, A. Nemati Kharat, S. Moosavikia, B. Tamaddoni Jahromi, A. Badiei, A. N. Kharat, S. Moosavikia, B. T. Jahromi, A. Badiei, A. Nemati Kharat, S. Moosavikia, B. Tamaddoni Jahromi and A. Badiei, J. Mol. Catal. A: Chem., 2011, 348, 14–19 CrossRef CAS.
- M. Woolery, in Kirk-Othmer Encyclopedia of Chemical Technology, American Cancer Society, 2005 Search PubMed.
- C. Choi, S. Kim, R. Kim, Y. Choi, S. Kim, H. Y. Jung, J. H. Yang and H. T. Kim, Renewable Sustainable Energy Rev., 2017, 69, 263–274 CrossRef CAS.
- I. Andersson, S. Angus-Dunne, O. Howarth and L. Pettersson, J. Inorg. Biochem., 2000, 80, 51–58 CrossRef CAS PubMed.
- D. Rehder, T. Polenova and M. Bühl, Annu. Rep. NMR Spectrosc., 2007, 62, 49–114 CrossRef CAS.
- M. Adam and J. E. Hileman, Can. J. Chem., 1980, 58, 2255–2261 CrossRef.
- N. Samart, Z. Arhouma, S. Kumar, H. A. Murakami, D. C. Crick and D. C. Crans, Front. Chem., 2018, 6, 1–16 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00914h |
| This journal is © The Royal Society of Chemistry 2020 |