
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCpCo(I) precatalysts for [2 + 2 + 2] cycloaddition reactions: synthesis and reactivity†‡
Fabian
Fischer
a,
Tobias
Pientka
a,
Haijun
Jiao
 a,
Anke
Spannenberg
a and
Marko
Hapke
*ab
a,
Anke
Spannenberg
a and
Marko
Hapke
*ab
aLeibniz-Institut für Katalyse e.V. (LIKAT Rostock), Albert-Einstein-Strasse 29a, D-18059 Rostock, Germany
bJohannes Kepler Universität Linz, Institut für Katalyse (INCA), Altenberger Strasse 69, A-4040 Linz, Austria. E-mail: marko.hapke@jku.at
First published on 2nd October 2020
Abstract
The efficient synthesis and structural characterisation of a series of novel CpCo(I)–olefin–phosphite/phosphoramidite complexes and their evaluation in catalytic cyclotrimerisation reactions are reported. The protocol for precatalyst synthesis is widely applicable to different P-containing ligands, especially phosphites and phosphoramidites, as well as acyclic and cyclic olefins. A selection of the prepared complexes was investigated towards their catalytic performance in [2 + 2 + 2] cycloaddition reactions of diynes and nitriles, as well as triynes. While revealing significant differences in reactivity, the most reactive precatalysts work even already at 75 °C. One of these precatalysts also proved its potential in exemplary (co)cyclotrimerisations towards functionalised pyridines and benzenes. The energetics of complex formation and exemplary ligand exchange with a substrate diyne were elucidated by theoretical calculations and compared with the catalytic reactivity.
Introduction
The application of transition metal complexes as catalysts for the execution of [2 + 2 + 2] cycloaddition reactions with a multitude of unsaturated functionalities like alkynes, olefins, nitriles, isocyanates etc. has virtually become routine for many organic chemists concerned with synthesising aromatic and heteroaromatic compounds.1,2 In certain cases cyclotrimerisations can also be performed without applying transition metal complexes, when e.g. Brønsted acids together with appropriate substrates were combined or rather drastic conditions were applied.3 However, here also the allocation of specifically designed substrates such as functionalised alkynes, diynes, triynes and oligoynes, plays a distinguished role for the successful transformation. This becomes equally important in cases, when complex natural product syntheses utilise the [2 + 2 + 2] cycloaddition reaction as a key transformation in a late step of the overall sequence.4 The arsenal of catalysts available for [2 + 2 + 2] cyclotrimerisation reactions is now covering basically the whole range of transition metals in the periodic table, expanding the availability of a suitable catalyst to principally every substrate imaginable.2d,5Since the reawakening of the cyclotrimerisation reaction by Vollhardt and co-workers, cobalt complexes have played a vital role in performing carbo- and heterocyclic ring-forming reactions, including applications in natural product assembly.6,7 Significant contributions and detailed studies on the reactivity of differently substituted cyclopentadienyl (Cp)–Co(I)-complexes in cyclotrimerisations including alkynes and nitriles were reported by Bönnemann.8 The experimental results have been interpreted and explained by a series of theoretical work and calculations on such cyclisations at the CpCo(I)-fragment, including alkynes and nitriles.7,9 Especially commercially available CpCo(CO)2 has become the work horse in many synthetic applications to date.7,10 However, CpCo(I)–olefin complexes as precatalysts of high reactivity were first synthesised by Jonas and co-workers and the sensitive Jonas' complex CpCo(H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)2 was applied in a number of reactions.11 In general, the chemistry of the CpCo(I)-fragment with respect to coordination and reactivity with neutral ligands is quite extensive,12 however, only very few complexes were investigated in more detail for their applicability as precatalysts for the [2 + 2 + 2] cycloaddition reaction.13 Interestingly, olefin complexes of the higher congener fragments CpM (M = Rh, Ir) are much less reactive compared to the CpCo-analog in cyclotrimerisation reactions, requiring considerably more energy for activation.14 A significant advance towards more stable precatalysts was published by Gandon and co-workers through the synthesis of the first CpCo(CO)(alkene) complexes, being employed as precatalysts for cyclotrimerisations (Scheme 1, left).15 The combination of CO and an electron-deficient olefin as neutral ligands stabilises the CpCo(I)-fragment, furnishing air-stable precatalysts. However, rather high temperatures are required for activation in cyclotrimerisation reactions.
CH2)2 was applied in a number of reactions.11 In general, the chemistry of the CpCo(I)-fragment with respect to coordination and reactivity with neutral ligands is quite extensive,12 however, only very few complexes were investigated in more detail for their applicability as precatalysts for the [2 + 2 + 2] cycloaddition reaction.13 Interestingly, olefin complexes of the higher congener fragments CpM (M = Rh, Ir) are much less reactive compared to the CpCo-analog in cyclotrimerisation reactions, requiring considerably more energy for activation.14 A significant advance towards more stable precatalysts was published by Gandon and co-workers through the synthesis of the first CpCo(CO)(alkene) complexes, being employed as precatalysts for cyclotrimerisations (Scheme 1, left).15 The combination of CO and an electron-deficient olefin as neutral ligands stabilises the CpCo(I)-fragment, furnishing air-stable precatalysts. However, rather high temperatures are required for activation in cyclotrimerisation reactions.
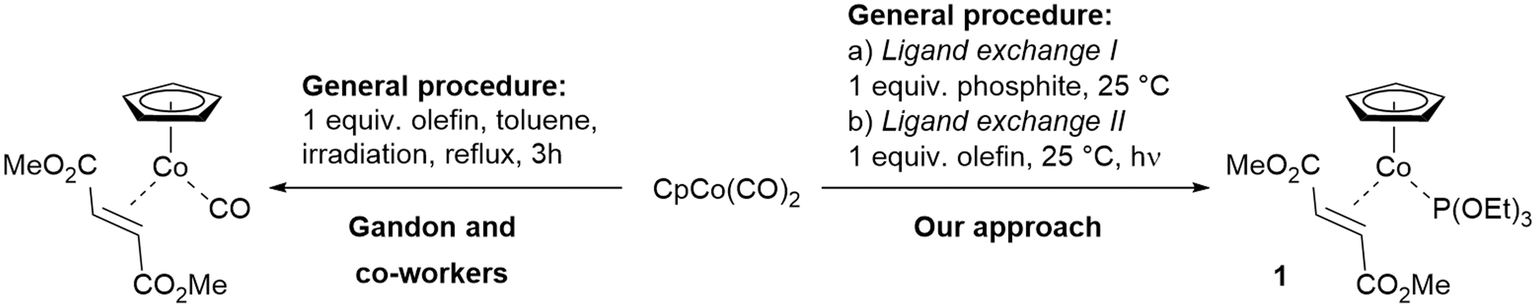 | ||
| Scheme 1 Synthetic approach for the preparation of CpCo(I)–olefin–phosphite complexes by step-wise ligand substitution from CpCo(CO)2. | ||
From our recent work emerged a unique class of CpCo(I) precatalysts containing an olefin/phosphite combination, which turned out to be significantly more reactive compared to the known complexes.16 The approach of introducing electron-deficient olefins and different phosphites as unique ligand combination furnished air-stable and recyclable CpCo(I)-precatalyst, which can be used for carbocyclisation of alkynes, as well as cross-cyclisations of alkynes and nitriles.17 The complex CpCo[P(OEt)3](dimethyl fumarate) (1) turned out to be a versatile precatalyst, which could be successfully applied under conventional thermal, photochemical and microwave-assisted reaction conditions for the assembly of carbo- and heterocycles.18 The two-step synthetic approach also started from commercial CpCo(CO)2 (route outlined in Scheme 1, right), including a unique photochemical ligand exchange step, allowing introduction of the second ligand under very mild conditions.
The influence of the nature of the olefin has become clear from the previous investigations, with electron-deficient olefins leading to significantly higher stability of the precatalyst while affording higher reaction temperatures for activation due to better stabilisation of the CpCo-fragment by raised π back-bonding capability of the electron-deficient olefin. However, the complex stability should be depending on the properties of both neutral ligands, to a larger extent from the olefin, however.17a Therefore, we were highly interested to exploit and generalise the procedure and to prove its generality for the synthesis of an array of diversified complexes with structurally different phosphorus and olefin components. Furthermore, calculating the ligand substitution energetics of the synthesis starting from CpCo(CO)2 should provide clues on the stability of the precatalysts as guidance to the activation energy required. Finally, the reactivity of selected complexes in cyclotrimerisations to synthesise pyridines and benzenes should be investigated.
Results and discussion
Synthesis of CpCo(I) complexes
Based on the synthetic two-step pathway exploited for the synthesis of the first CpCo(I)–olefin–phosphite complex like 1 from CpCo(CO)2, we envisioned to explore the broadness and generality of this approach by using commercially available or easy accessible phosphite/phosphoramidite and olefins in various ligand combinations first (Scheme 2).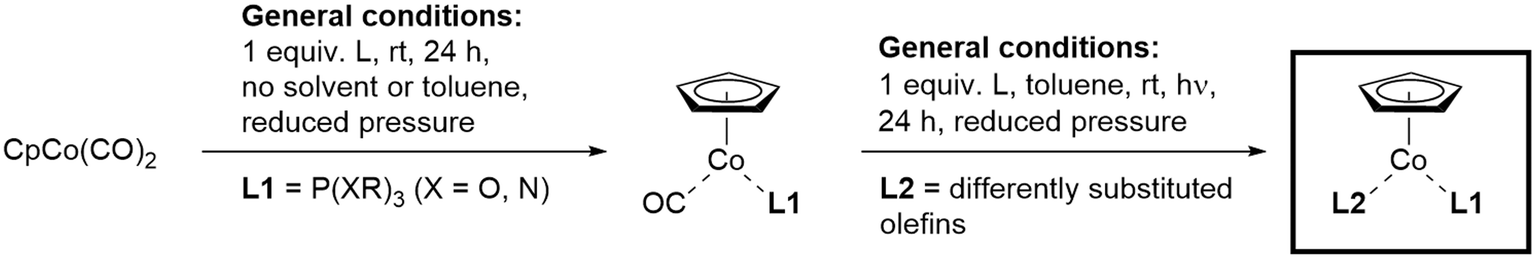 | ||
| Scheme 2 General synthetic scheme for the preparation of CpCo–olefin–phosphite complexes. | ||
The initial step is the exchange of the first CO ligand from CpCo(CO)2 for a phosphite ligand, usually proceeding smoothly at slightly reduced pressure to facilitate CO expulsion and substitution.19 We investigated the scope of different phosphites in this reaction and the results are shown in Scheme 3. Phosphites with electron-rich (L1a, L1c, L1d, L1e), as well as electron-deficient alkyl and aryl groups (L1b), and especially those with different sterically encumbered groups (e.g.L1c, L1f, L1g) were identified to be suitable replacements for the first CO group (Scheme 3, top). The reactions usually occur in good to excellent yield (79–99%) at room temperature, except for P(OtBu)3 (L1c), corresponding complex 2c of which could only be isolated in lower yield (24%). For the sterically large phosphite P(O-2,4-bis-tBu-phenyl)3 (L1g) the product was obtained after irradiation of 1 for 24 h in the presence of the phosphite, however, with excellent yield of 2g. The complex 2f containing the phosphoramidite L1f required a larger reaction time, when it was formed with practically quantitative yield. The 31P NMR spectra of the CpCo(CO)–phosphite complexes in general showed large downfield shifts (>25 ppm at least), except for 2c, showing only 2.6 ppm difference compared to the resonance for the free ligand P(Ot-Bu)3 (L1c).20 This might correspond to a weak coordination, which would explain the obtained low yield of 2c. The physical appearance of the complexes differs in that the complexes 2b, 2c, 2d, 2f and 2g form solids, while the complexes 2a and 2e are of oily consistence. The molecular structure of complex 2b and 2c were elucidated by X-ray structure analysis.21 These complexes turned out to be stable under inert conditions, with no significant catalytic reactivity under moderate conditions.
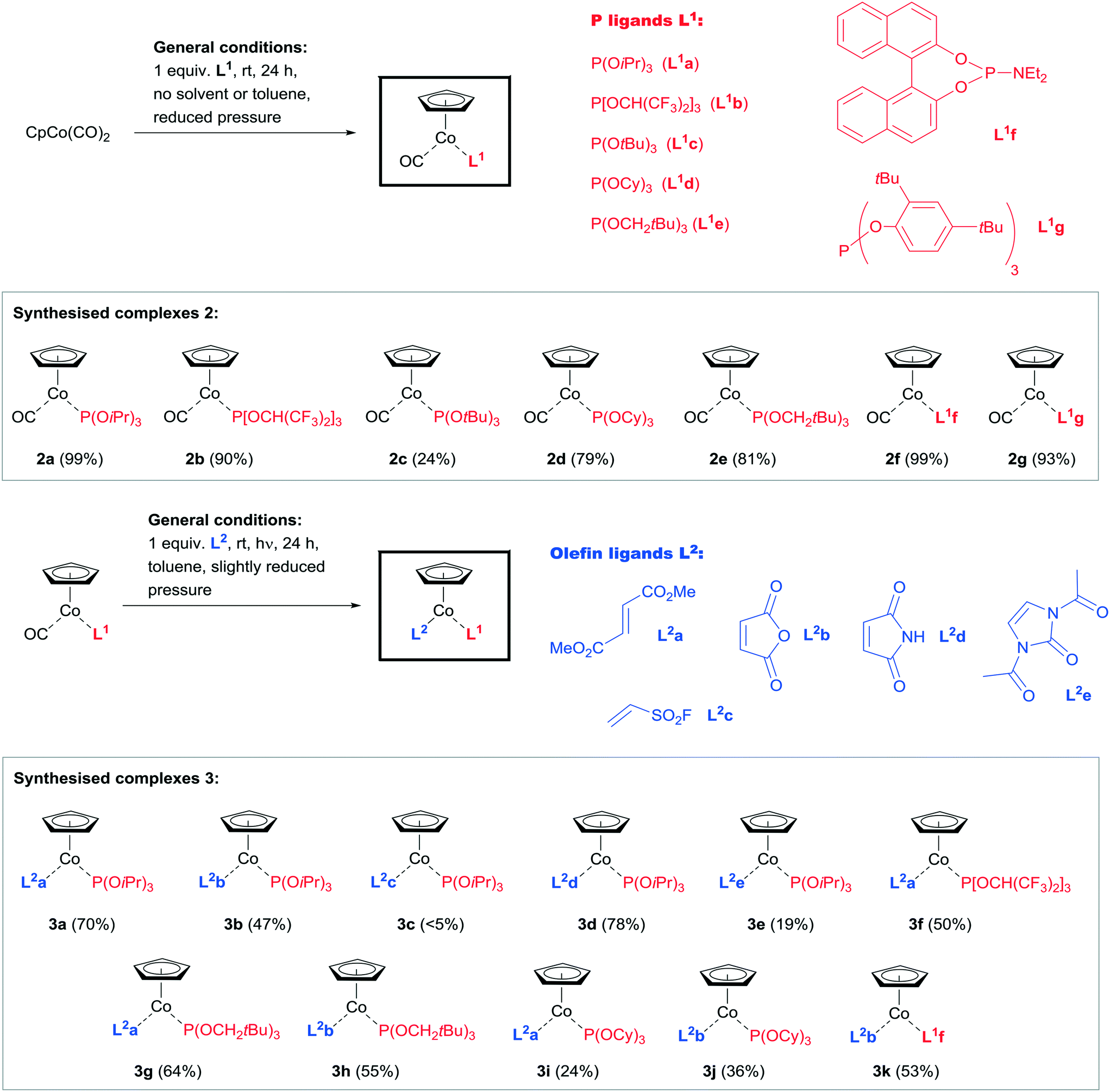 | ||
| Scheme 3 Syntheses of complex classes 2 and 3 following the two-step ligand substitution protocol. | ||
In the next step, the second CO ligand in complexes 2 was exchanged for an olefin derivative.22 We have found that the nature of the olefin plays an essential role for the stability/reactivity of the precatalyst.17 While electron-rich olefins usually lead to very reactive and therefore less stable complexes,16 olefins containing electron-withdrawing substituents stabilise the CpCo(phosphite) fragment, leading to a significant increase in stability. However, also the configuration of the double bond plays an extremely important role, exemplified by the successful formation of complexes applying fumaric acid ester (trans-1,2-ethylenedicarboxylic acid ester, L2a), while the complexation using maleic acid ester (cis-1,2-ethylenedicarboxylic acid ester) did not furnish isolable complexes. From the literature, however, mononuclear cobalt complexes containing maleic acid anhydride (L2b) are known to exist, which motivated us to investigate this compound for the photochemical substitution step, comprising also the cyclic maleic acid imide (L2d).23 We therefore selected the different olefins L2a–L2e for our examination of olefin complexation, aiming at controlling the stability/reactivity of the intended CpCo–olefin–phosphite complexes by different substitution of the olefinic double bond. Selected results for the photochemical ligand exchange experiments in step 2 of the synthesis sequence are given in Scheme 3 (below, complexes 3a–k).
The results for the synthesis of a variety of complexes 3 were highly promising. It turned out that either dimethyl fumarate (L2a) or maleic acid anhydride (L2b) were the most suitable ligands in the exchange of the second CO ligand, accepting a variety of phosphites present in the starting complexes 2. It must be noticed that the complexes 2c, 2f and 2g (containing the ligands P(OtBu)3 (L1c), L1f and L1g) did not yield any of the expected products in the second CO ligand exchange step with dimethyl fumarate (L2a). Performing the photochemical ligand exchange with 2c and 2f in the presence of dimethyl fumarate (L1a) even at 40 °C reaction temperature for 48 h did not yield the expected complexes. In general, complexes containing P(OiPr3) (L1a) were used in most cases due to the convenient access to complex 2a. It was found that the CO exchange here occurs with good yields for several olefins (L2a, L2b and L2d, complexes 3a, 3b and 3d). Even for such unusual olefins like ethenesulfonyl fluoride (L2c, complex 3c) a complexation was observed, however, with a very low yield of isolated product. The olefin L2e was synthesised according to a known procedure and can be regarded as an protected 1,3-dihydro-2H-imidazol-2-one derivative, and being more electron-rich even with acetylated nitrogen substituents compared to L2a or L2b.24 The exchange reaction using complex 2a gave the expected product 3e with 19% isolated yield, however, the yield was significantly lower compared to the other cyclic olefins L2b and L2d. The olefin L2a is furthermore very conveniently applied in complexes containing neopentyl-substituted electron-rich (3g) and partially fluorinated electron-deficient (3f) phosphites with 50 and 64% yield. Complexes of L2a and cyclic L2b with tricyclohexylphosphite (3i and 3j) were furnished with lower yields than most other compounds 3 (24 and 35% yield). The second ligand exchange in 2f was as described above not possible for dimethyl fumarate (L2a), however, using L2b allowed access to 3k with favourable 53% yield. This can serve as an interesting example for the importance of the double bond configuration in alkene ligand exchange reactions.
The complexes 3 are all solids, which can except compound 3e be possible to be handled at least for short periods of time in air.25 If stored under an inert atmosphere and with exclusion of light, they are long-life stable compounds.
We were able to characterise several of the maleic acid anhydride (L2b) complexes, namely 3h, 3j and, 3k and also complex 3e, containing the 1,3-dihydro-2H-imidazol-2-one core in ligand L2e, by X-ray structure analysis (Chart 1).26 The structural features of the complexes are expectedly very similar, with small variations depending on the coordinated neutral molecules. The distances Co–P are slightly longer for P(OCy)3 (L1d, 2.1308(5) Å) and the phosphoramidite L1f (2.1253(6) Å) compared to P(OiPr)3 (L1a, 2.1118(5) Å) and P(OCH2tBu)3 (L1e, 2.1181(3) Å). The bond lengths Co–Colefin are in the range of 1.9926(14) to 2.013(2) Å. The double bond of the coordinated olefins is elongated compared to the uncoordinated olefin as expected. These bond lengths are in the range of 1.43 Å and do not show any significant difference.
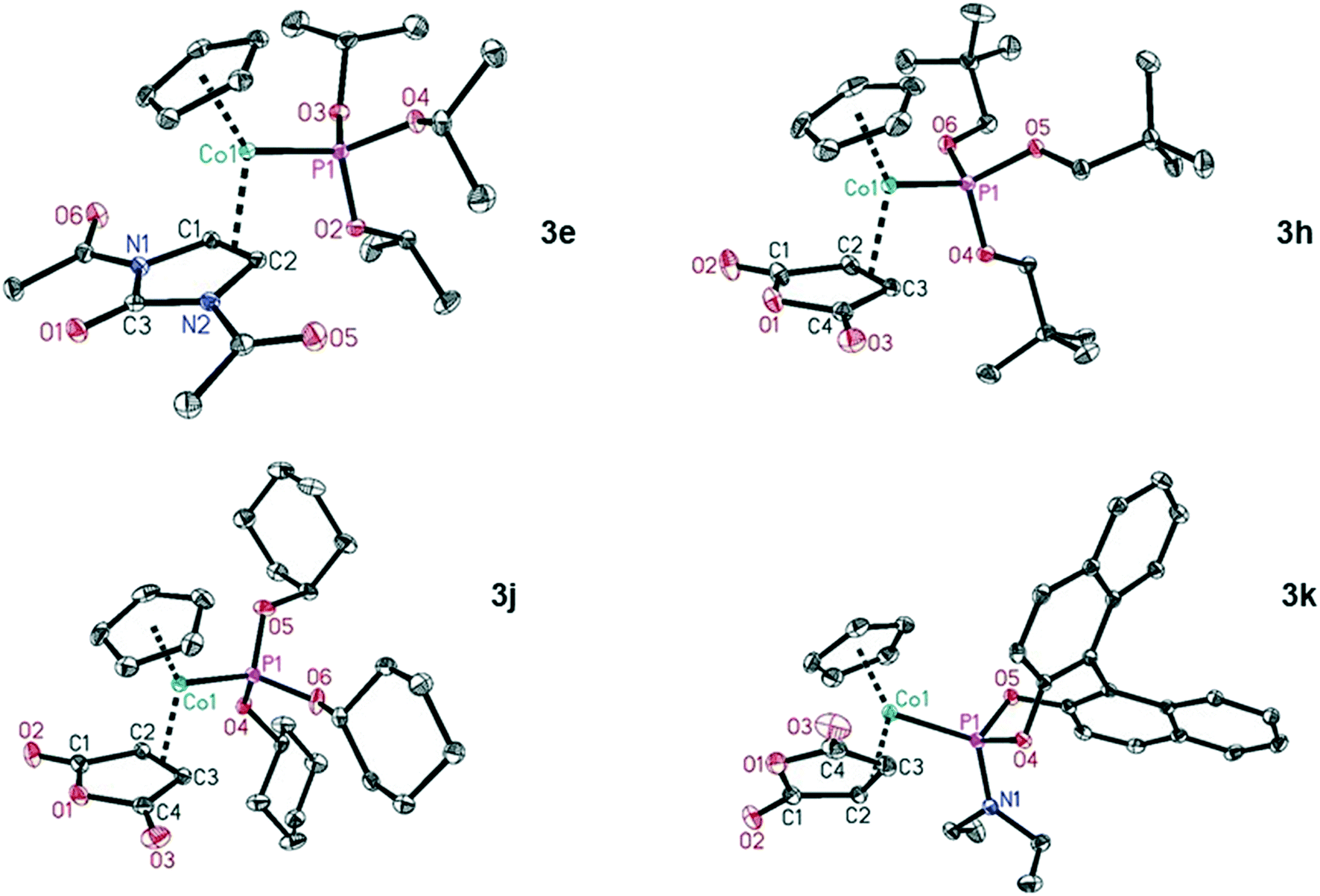 | ||
| Chart 1 Molecular structures of the CpCo–olefin–phosphite complexes 3e, 3h, 3j and 3k. Displacement ellipsoids correspond to 30% probability. Hydrogen atoms are omitted for clarity. | ||
In summary, the structural features of these complexes do not allow to draw any conclusion onto which reactivity can be expected in the cyclisation reactions.
Screening on catalytic activity of the CpCo(I) precatalysts
After having systematically synthesised a number of precatalysts 3, we set out to evaluate their reactivity in cyclotrimerisation reactions. The reactivity of such complexes varied largely with the electronic properties of the olefin ligand, as the very initial work showed, however, electron-rich olefins led to complexes prone to decomposition and quite unhandy to work with.16,17a Why not using the CpCo(CO)-phosphite complexes directly as precatalysts? It was demonstrated that they were in principle able to mediate cyclotrimerisations, but only at rather high temperatures, which are also needed for CpCo(CO)2 as precatalyst itself and without achieving superior yields. In addition, complexes 2 are not very convenient to experiment with due to their consistency in most cases. We expected the moderation of the reactivity by both, phosphite and olefin, and the interplay of different ligands to provide stable yet highly reactive complexes under comparably mild temperatures. For this reason, we investigated selected complexes 3 in benchmark cycloaddition reactions of 1,6-heptadiyne and benzonitrile, as well as triyne, 4 under thermal conditions. Results of the initial investigations on the cylisation of triyne 4 at 75 °C are presented in Chart 2.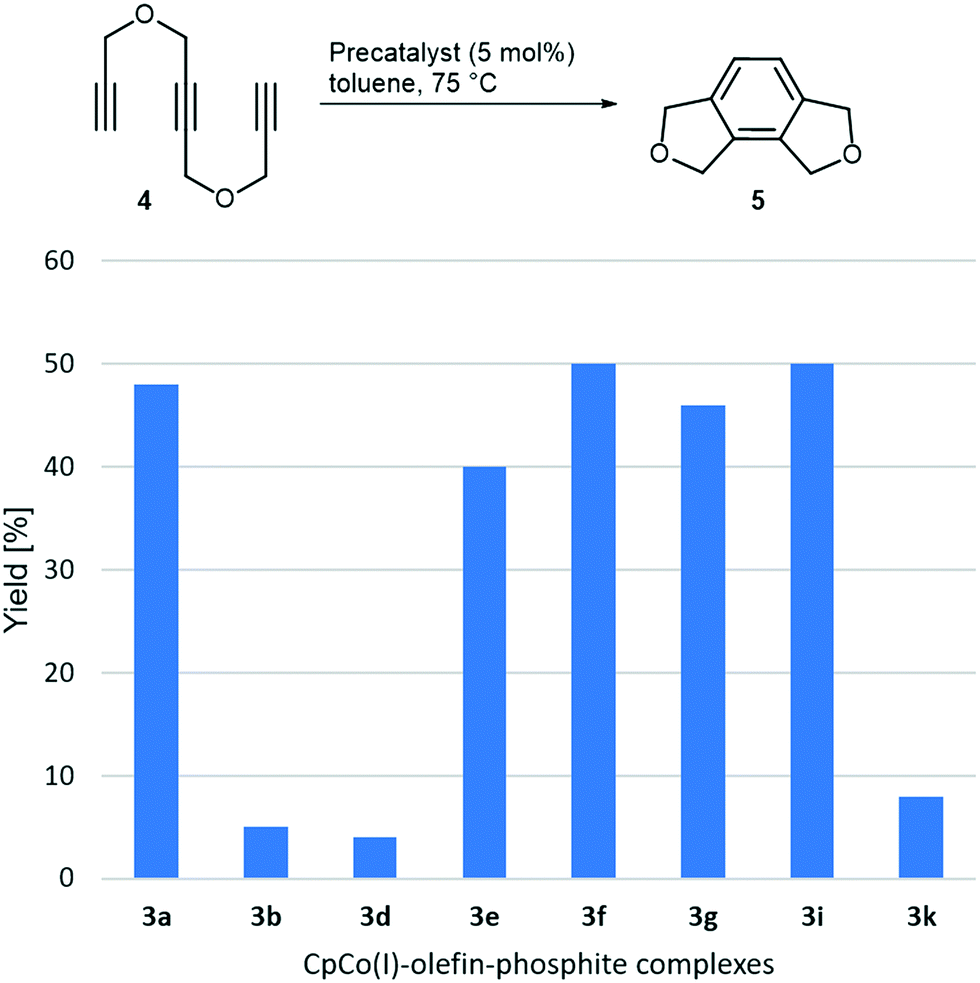 | ||
| Chart 2 Screening of catalytic activity for triyne 4 cyclisation by selected novel complexes 3. | ||
Significant differences in reactivity can be recognised. The most reactive complexes 3a and 3e–3i afforded the annelated benzene derivative 5 with yields between 40 and 50% at the moderate reaction temperature. Interestingly, the (re)activity of the complexes 3a and 3f, differing in the fluorinated methyl groups of the phosphite ligand of complex 3f, are basically identical. Therefore, the presence of a less electron-rich phosphite alkyl groups obviously does not play a significant role here, neither does the role of the amide substituted olefin in 3e to a major extent. Precatalysts containing cyclic olefins like in 3b, 3d and 3k appear to be less reactive at this reaction temperature. Increasing the catalyst loading from 5 mol% to 10 mol% was tested for 3g and 3i and did just give minor higher yields (up to 3%).
For the assembly of pyridine derivative, 6 the initial results including complexes 3a, 3b, 3d–3i and 3k showed broad variations in their performance under identical conditions at 75 °C. However, rapid conversions of the diyne were encountered for the complexes 3a and 3e, while for the other complexes conversions in the range of 50–80% were encountered only after 24 h by GC analysis.27 The data were corroborated by obtaining isolated yields for the respective catalysts as shown in the overview in Chart 3. In this case of co-cyclotrimerisation, complex 3e and 3i showed significant higher activity, while all other complexes are significantly less active. Precatalysts 3a, 3f and 3g provided some activity, however, remarkable is the difference for complexes CpCo[P(OiPr)3](dimethyl fumarate) (3a) and CpCo[P(OiPr)3](maleic anhydride) (3b) both containing triisopropylphosphite as ligand and olefins, which are electron-deficient substituted donors, differentiated by the configuration of the double bond. The comparably electron-rich cyclic olefin L1e, however, is the olefin component of the most reactive precatalyst 3e, active in both investigated types of cyclisation. Precatalyst 3i containing P(OCy)3 (L1d) and the dimethyl fumarate (L2a) ligand shows a similar performance to 3e and is the most active of the complexes containing L2a. Tricyclohexyl phosphite (L1d) is one of the most electron-rich phosphites applied in the study; unfortunately the related complex containing the even more electron-rich tri-tert-butyl phosphite (L1c) and L2a was synthetically not accessible as described above. The activity/reactivity of the precatalysts seems to be not obviously dominated by either a single phosphite component or olefin component, however, electron-deficient (E)-alkene ligands (like L2a) provides significantly higher precatalyst reactivities compared to electron-deficient cyclic olefins (like L2b and L2d). Comparing benzene ring formation and pyridine ring formation (Charts 2 and 3), for the latter case a smaller number of precatalysts performed well. Further interpretation of the screening data will be given below in connection to the calculated energetics of the complex assembly process.
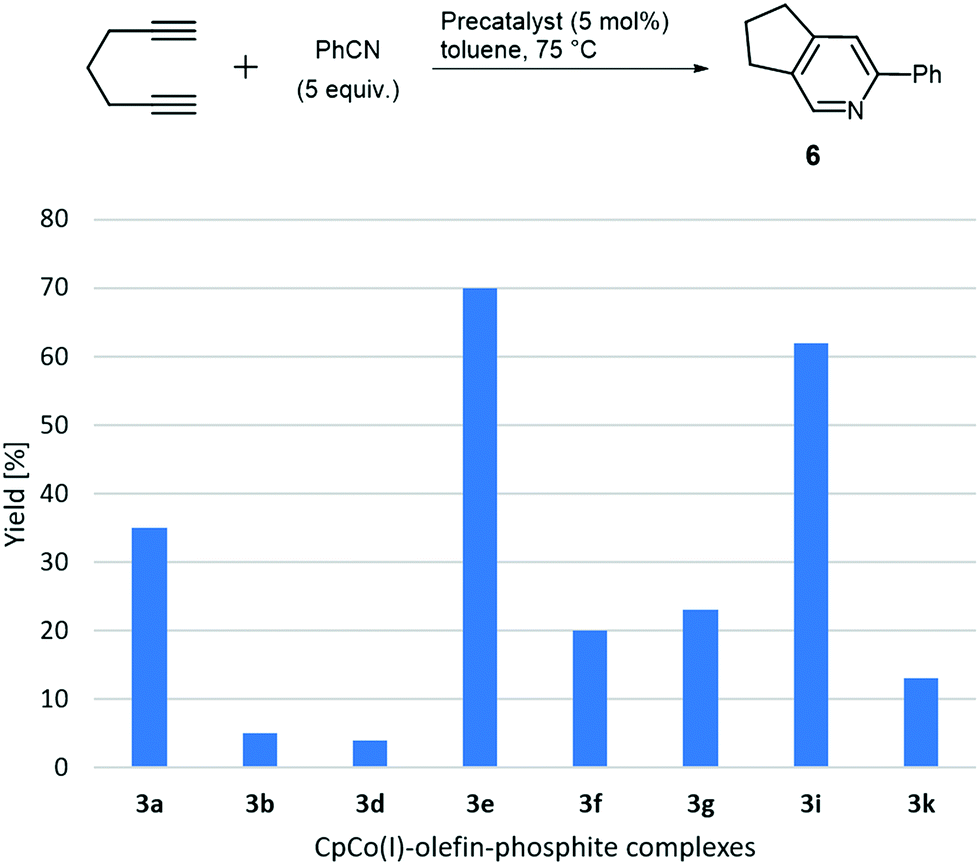 | ||
| Chart 3 Screening of catalytic activity in the cyclisation of 1,6-heptadiyne and benzonitrile with selected novel complexes 3. | ||
For the most active complex CpCo[P(O-iPr)3](N,N′-diacetyl imidazol-2-one) (3e), the reaction scope was investigated in some more detail, to elucidate the reactivity at such rather low cyclisation temperatures for this CpCo(I) precatalyst (Scheme 4). Reaction of diyne 7 as often used substrate for heterobiaryl synthesis in the presence of PhCN furnished product 8 with 56% in only 2 h of reaction time at 75 °C. Transformation of the 1,6-heptadiyne derivative 9 with piperidine-1-carbonitrile yielded the expected biaryl product 10 with nearly quantitative yield. Also, very good yields were obtained with diethylamino carbonitrile, furnishing biaryl 11 and with cyclopropanecarbonitrile, yielding cyclopropyl-substituted biaryl pyridine 12.
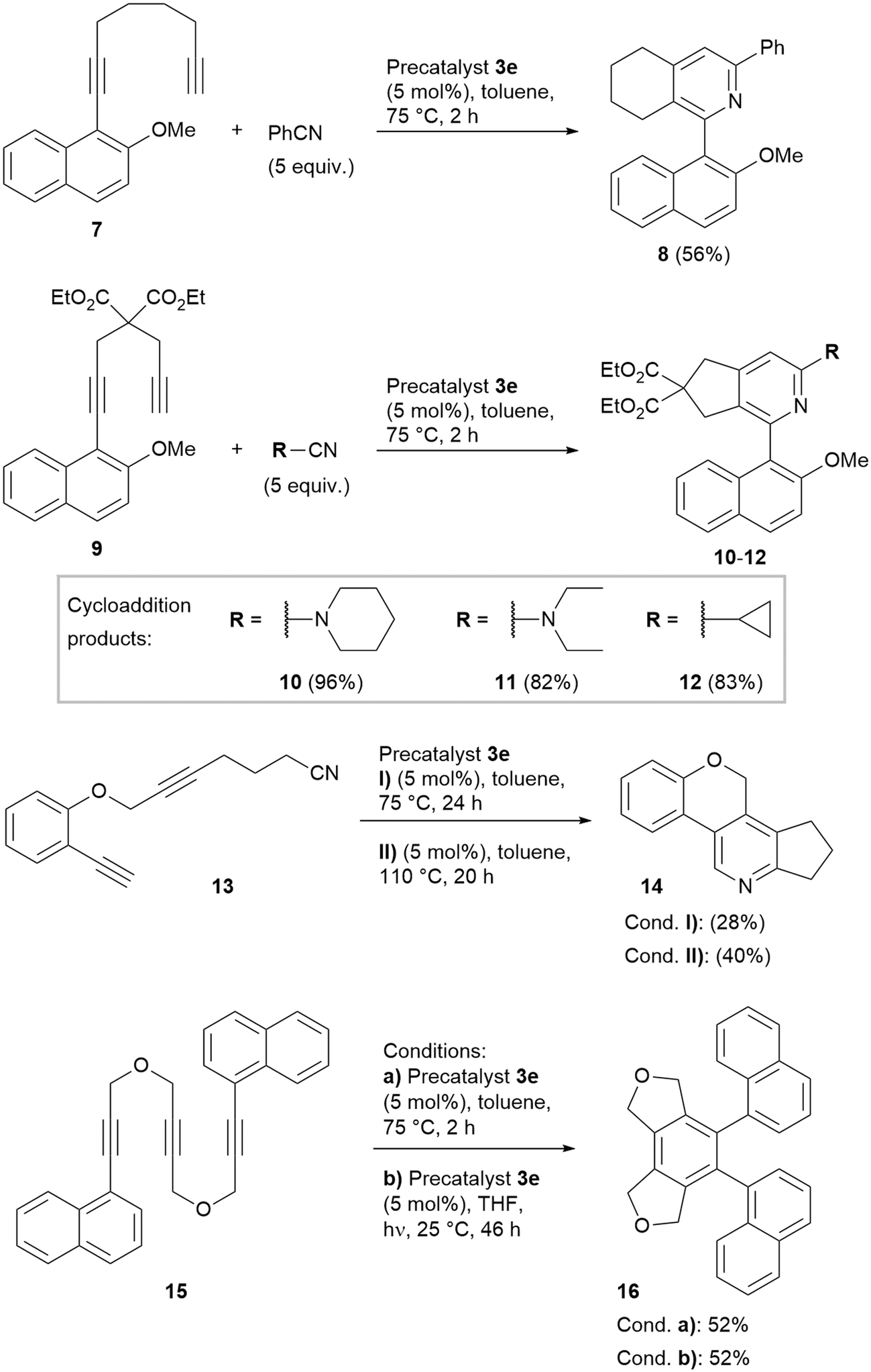 | ||
| Scheme 4 Cyclisations using precatalyst 3e. | ||
Intramolecular cyclisation of cyanodiyne 13 proceeded also at 75 °C reaction temperature, however, the isolated yield of the annelated pyridine product 14 was 28%. Increase of the reaction temperature to 110 °C gave 40% yield of 14. Finally, the intramolecular cyclotrimerisation of triyne 15 yielded the triaryl 16 with acceptable 52% yield in only 2 h reaction time at 75 °C. For comparison, applying catalyst 1 after 19 h reaction time at 100 °C only 44% yield were obtained.18 Repetition of this experiment under photochemical conditions gave the identical yield at 25 °C over 46 h irradiation time.
Calculations on the stability of the CpCo(I) complexes and initial catalytic steps, modification of complex 3e
We have calculated the energetics of both ligand-exchange reactions to arrive at an estimate of the stability of the different precatalysts and to gain insight into the potential stability/activation parameters of the synthesised complexes. The Gibbs free energies of the endergonic two-step substitution processes described in Scheme 3 have been calculated, as it has been shown that they can provide a good lead on the stability and also reactivity of the CpCo complexes.28Chart 4 shows the energy values for the complexes 3a–k. The exchange of the first CO ligand from CpCo(CO)2 for the different phosphites and phosphoramidites is of little difference despite the electronically and sterically different nature of the P-based ligands (red bars in Chart 4). The endergonic exchange of the second CO for the olefin ligands is energetically significantly more different compared to the first process (blue bars in Chart 4). In the complexes 3a–e containing P(OiPr)3 as phosphite, remarkable differences were observed between some of the ligands, with the 1,3-dihydro-2H-imidazol-2-one ligand L2e giving the highest ΔG value for this second substitution and therefore also the overall energy over all complexes. Consistingly, complex 3e proved to be the most reactive complex during the cyclisation experiments described above and, macroscopically, the complex was identified to be the most unstable compound against air as evidenced by significant decomposition after short time. Nearly identical data were calculated for the fluorinated phosphite complex 3f, which was quite active in the cyclisation of triyne 4. In general, the differences of substitution for the dimethyl fumarate (L2a) and the maleic acid anhydride (L2d) were particularly striking, as was demonstrated with P(OiPr)3 (L1a) as phosphite ligand in complexes 3a und 3d. Their activity in cyclisation reactions differs remarkably as could be seen in Charts 2 and 3. Precatalyst 3i containing the electron-rich P(OCy)3 (L1d) and dimethyl fumarate (L2a) features a large ΔG for the second CO substitution and overall gave a similar performance to 3e, providing evidence that this might act as an indicator for the precatalyst reactivity. Moreover, 3i performed well in both benchmark cyclisations, while 3f (even larger ΔG for the second CO substitution) did perform much less favourable in the pyridine formation.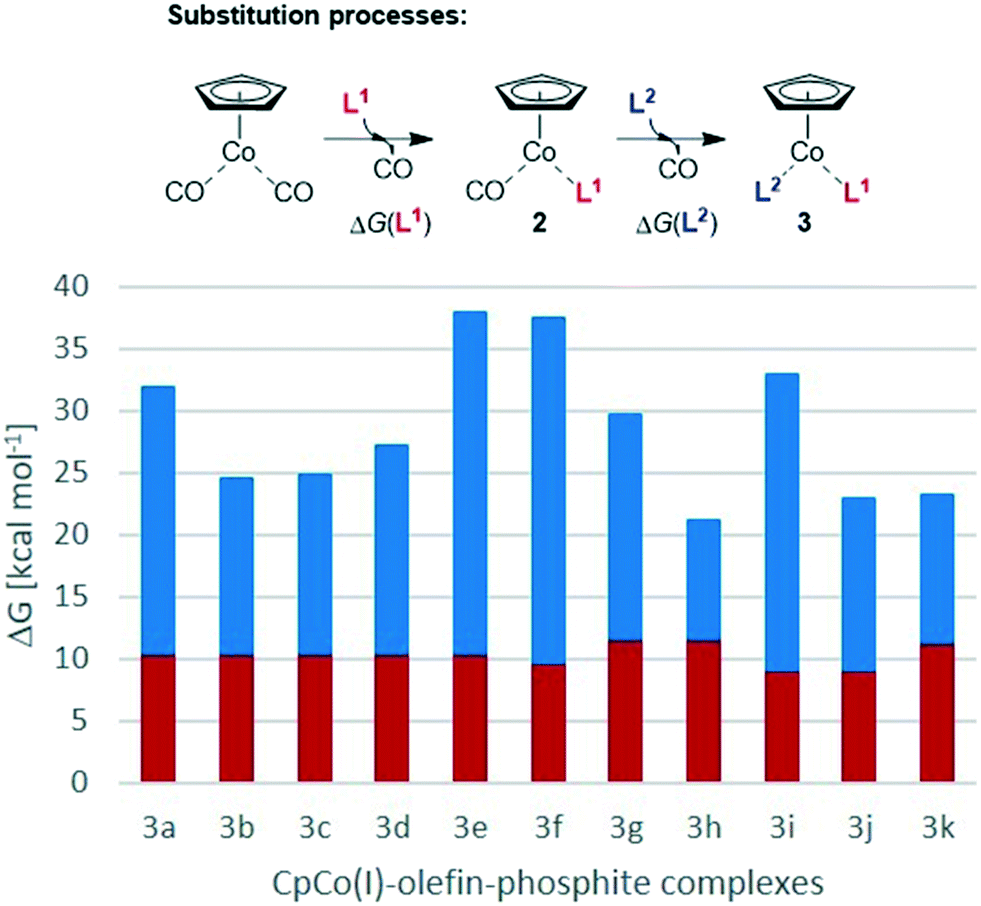 | ||
| Chart 4 BP86/SVP computed substitution Gibbs free energy of CO by phosphite (L1, in red) from CpCo(CO)2 to form CpCo(CO)(L1) (ΔG(L1), 2) as well as CO by olefin (L2, in blue) from CpCo(CO)(L1) (2) to form CpCo(L1)(L2) (ΔG(L2), 3). | ||
Finally, calculation of the Gibbs free energies for the ligand exchange of cyclisation substrate 1,6-heptadiyne in precatalysts CpCo(CO)2, 3e and 3f for the assumed first step of a cyclotrimerisation reaction, are particularly revealing (Scheme 5). The exchange against the two CO molecules from CpCo(CO)2 is largely endergonic by nearly 29 kcal mol−1. Exchange of one CO ligand for P(OiPr)3 (L1a) in 2a leads to a decrease in activation energy, however, still over 18 kcal mol−1 are required in the exchange process. On the other hand, the exchange of 1,6-heptadiyne for the olefin/phosphite ligands is exergonic by roughly 9 kcal per mole. Therefore, the significant higher reactivity of the precatalysts 3 can be explained from the thermodynamics of this initial step of the cyclotrimerisation catalytic cycle, entering the catalytic cycle by exergonic release of the neutral ligands for the alkyne π bond donors.
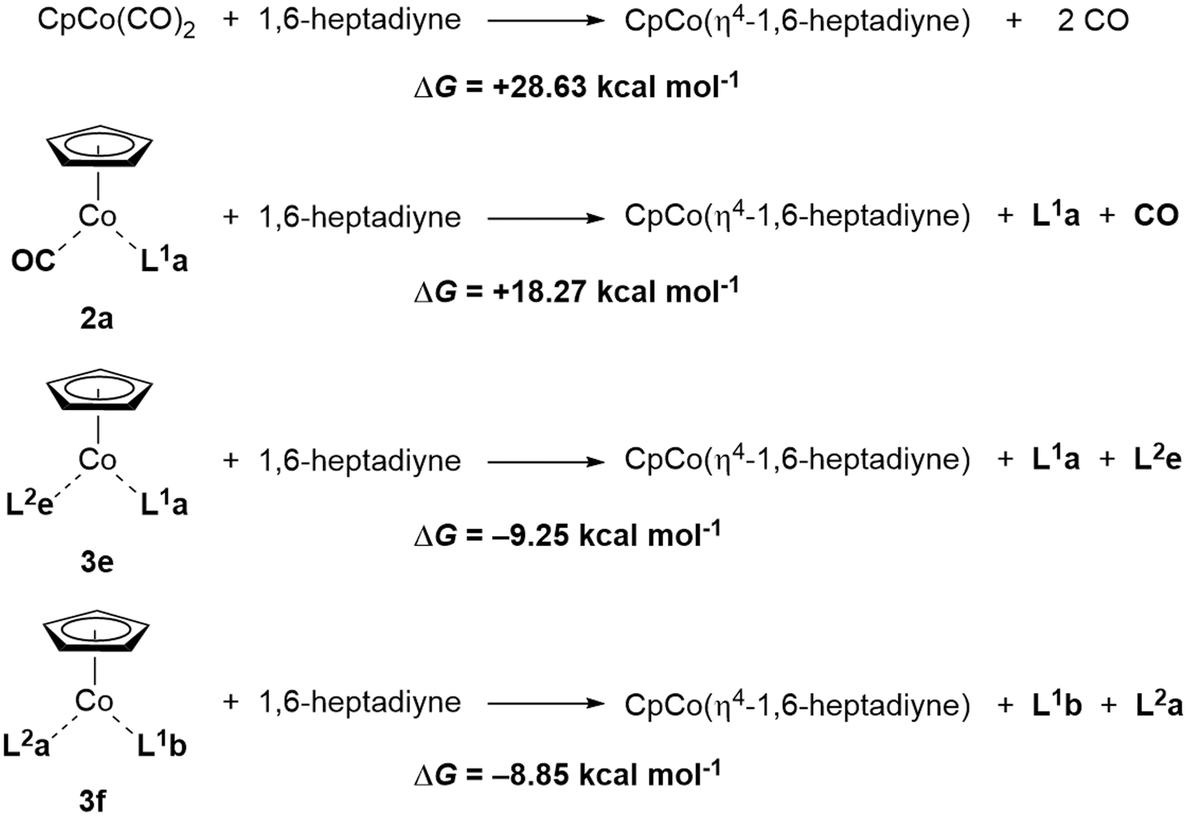 | ||
| Scheme 5 Calculation on the ligand exchange in CpCo(CO)2, 3e and 3f with 1,6-heptadiyne. | ||
The synthesis of complex 3e was found to be comparably challenging in the second substitution step, as only rather low yields of the complex were observed. Substituting the phosphite by the corresponding triisopropylphosphine (PiPr3) was expected to possibly enhance the complex stability and would also be interesting from the viewpoint of reactivity comparison. As it turned out, the first CO substitution in CpCo(CO)2 was smooth (nearly quantitative yield of CpCo(CO)(PiPr)3, 2hPhosphine), however, the second substitution under our conditions to afford complex 3lPhosphine analogous to 3e was not feasible, leaving the starting materials unreacted, therefore contrasting the comparably successful synthesis of 3e (Scheme 6). Estimations on the stability of the complexes showed that CpCo[P(OiPr)3](N,N′-diacetyl imidazol-2-one) (3e) was more stable compared to CpCo(PiPr3)(N,N′-diacetyl imidazol-2-one) (3lPhosphine).29 In addition, comparison of IR for the two complexes data showed a shift from 1921 cm−1 to 1899 cm−1 for the CO stretching band, thus indicating a higher degree of backbonding for the more electron-rich phosphine, which might also hamper a smooth substitution of the second CO for the alkene.30
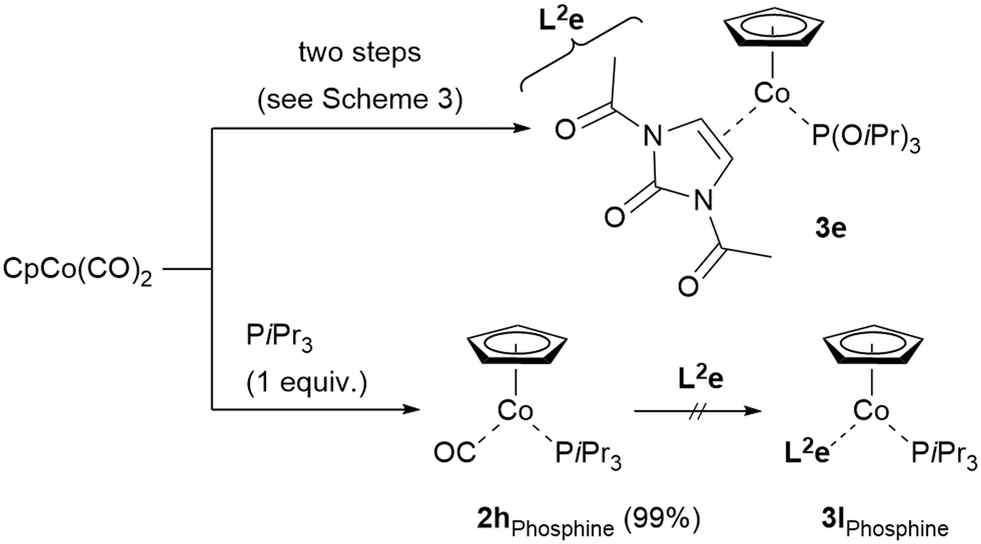 | ||
| Scheme 6 Synthesis of 3evs. attempted synthesis of the triisopropyl phosphine analog 3lPhosphine. | ||
However, the complexes except 3e are stable enough to be isolated and stored under inert atmosphere at room temperature and even survive (short) expositions to air if not being stable under air. As they can be activated by irradiation, long-term storage under exclusion of light is recommended. The possibility of applying such a large range of structurally diverse phosphites and olefins as ligand combo should allow and promote the incorporation of such CpCo(I) complexes in many applications, such as solid-phase hosted catalysts or as convenient source for the CpCo fragment not only in catalysis.31
Conclusion
The presented study described the possibilities for systematically synthesising a large array of CpCo(CO)–phosphite (2) and subsequently CpCo–olefin–phosphite (3) complexes from CpCo(CO)2 by a two-step substitution process involving one photochemical CO/olefin exchange process. The synthetic approach allows a wide variety of structurally and electronically different phosphites and phosphoramidite to be involved, as well as mostly electron-deficient acyclic and cyclic olefins and one rather electron-rich olefin. Coordination of dimethyl fumarate as (E)-olefin in combination with sterically large P ligands can be problematic as the second CO substitution does not work. Reactivity screening in [2 + 2 + 2] cycloadditions of triyne and diyne/nitrile substrates demonstrated at reaction temperatures as low as 75 °C significant differences in performance. In general, precatalysts with electron-deficient cyclic olefins (L2b, L2d) are much less reactive compared to complexes with electron-deficient (E)-configurated olefins like investigated dimethyl fumarate (L2a). The complexes CpCo[P(OiPr)3](N,N′-diacetyl imidazol-2-one) (3e) and CpCo[P(OCy)3](dimethyl fumarate) (3i) turned out to be the most active precatalysts for both processes, and subsequent cyclotrimerisations with precatalyst 3e with a variety of substrates including photochemical precatalyst activation further corroborated the screening results at this significantly lower reaction temperature than usually applied in cyclotrimerisations with CpCo(I) complexes. Calculations on the energetics of the ligand exchange in the synthesis of the CpCo–olefin–phosphite complexes showed that the complexes requiring most energy for the substitution process are indeed the most reactive precatalysts, but also revealed that the involved ΔG differences are strongly depending on the olefin component. In the combination with an electron-deficient olefin like L2a, electron-rich phosphites like P(OCy)3 (L1a) showed the best performance as was demonstrated with precatalyst 3i. The exothermicity of the neutral ligand exchange from complexes 3 for 1,6-heptadiyne as substrate shows favourable reactivity of the developed precatalysts. The presented combination of olefin/phosphite ligands therefore allows access to a more reactive alternative class of precatalysts for cyclisation reactions with a tunable reactivity profile.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Leibniz-Gemeinschaft for financial support and Prof. Dr. Uwe Rosenthal for helpful discussions.Notes and references
- (a) Transition-Metal-Mediated Aromatic Ring Construction, ed. K. Tanaka, John Wiley&Sons, Hoboken, 2013 Search PubMed; (b) A. Roglans, A. Pla-Quintana and M. Solà, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00062.
- (a) Selected reviews: K. Tanaka, Heterocycles, 2012, 85, 1017–1043 CrossRef CAS; (b) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS; (c) M. Amatore and C. Aubert, Eur. J. Org. Chem., 2015, 265–286 CrossRef CAS; (d) K. Yamamoto, H. Nagae, H. Tsurugi and K. Mashima, Dalton Trans., 2016, 45, 17072–17081 RSC; (e) Y. Yamamoto, Tetrahedron Lett., 2017, 58, 3787–3794 CrossRef CAS; (f) M. Babazadeh, S. Soleimani-Amiri, E. Vessally, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 43716–43736 RSC; (g) S. Kotha, K. Lahiri and G. Sreevani, Synlett, 2018, 29, 2342–2361 CrossRef CAS; (h) I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144–158 CrossRef.
- (a) Recent overview: M. Hapke, Tetrahedron Lett., 2016, 57, 5719–5729 CrossRef CAS; (b) recent example: M. Sasaki, P. J. Hamzik, H. Ikemoto, S. G. Bartko and R. L. Danheiser, Org. Lett., 2018, 20, 6244–6249 CrossRef CAS.
- (a) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 44, 2639–2672 CrossRef CAS; (b) B. Witulski and J. Grand, Application to the Synthesis of Natural Products in Transition-Metal-Mediated Aromatic Ring Construction, ed. K. Tanaka, John Wiley&Sons, Hoboken, 2013, pp. 207–242 Search PubMed.
- M. Hapke, N. Weding and K. Kral, Cyclotrimerization Reactions of Alkynes, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH, Weinheim, 2018, vol. 1, pp. 375–409 Search PubMed.
- (a) K. P. C. Vollhardt, Angew. Chem., Int. Ed. Engl., 1984, 23, 539–556 CrossRef; (b) Recent review: J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef CAS.
- T. Gläsel and M. Hapke, Cobalt-Catalysed [2+2+2] Cycloadditions, in Cobalt Catalysis in Organic Synthesis, ed. M. Hapke and G. Hilt, Wiley-VCH, Weinheim, 2020, ch. 9, pp. 287–335 Search PubMed.
- (a) H. Bönnemann, W. Brijoux, R. Brinkmann, W. Meurers, R. Mynott, W. von Philipsborn and T. Egolf, J. Organomet. Chem., 1984, 272, 231–249 CrossRef; (b) H. Bönnemann, Angew. Chem., Int. Ed. Engl., 1985, 24, 248–262 CrossRef.
- (a) J. H. Hardesty, J. B. Koerner, T. A. Albright and G.-Y. Lee, J. Am. Chem. Soc., 1999, 121, 6055–6067 CrossRef CAS; (b) A. A. Dahy, C. H. Suresh and N. Koga, Bull. Chem. Soc. Jpn., 2005, 78, 792–803 CrossRef CAS; (c) A. A. Dahy and N. Koga, Bull. Chem. Soc. Jpn., 2005, 78, 781–791 CrossRef CAS; (d) N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2007, 129, 8860–8871 CrossRef CAS; (e) A. A. Dahy, K. Yamada and N. Koga, Organometallics, 2009, 28, 3636–3649 CrossRef CAS; (f) A. A. Dahy and N. Koga, J. Organomet. Chem., 2010, 695, 2240–2250 CrossRef CAS.
- V. Gandon, C. Aubert and M. Malacria, Chem. Commun., 2006, 2209–2217 RSC.
- (a) K. Jonas, E. Deffense and D. Habermann, Angew. Chem., Int. Ed. Engl., 1983, 22, 716–717 CrossRef; K. Jonas, E. Deffense and D. Habermann, Angew. Chem. Suppl., 1983, 1005–1016 Search PubMed; (b) Applications: P. Sehnal, Z. Krausová, F. Teplý, I. G. Stará, I. Starý, L. Rulisek, D. Šaman and I. Císařová, J. Org. Chem., 2008, 73, 2074–2082 CrossRef CAS; (c) S. Amslinger, C. Aubert, V. Gandon, M. Malacria, E. Paredes and K. P. C. Vollhardt, Synlett, 2008, 2056–2060 CAS; (d) T. Aechtner, D. A. Barry, E. David, C. Ghellamallah, D. F. Harvey, A. de la Houpliere, M. Knopp, M. J. Malaska, D. Pérez, K. A. Schärer, B. A. Siesel, K. P. C. Vollhardt and R. Zitterbart, Synthesis, 2018, 50, 1053–1089 CrossRef CAS.
- Actual overview: D. A. Loginov, L. S. Shul'pina, D. V. Muratov and G. B. Shul'pin, Coord. Chem. Rev., 2019, 387, 1–31 CrossRef CAS.
- I. Thiel and M. Hapke, Rev. Inorg. Chem., 2014, 34, 217–245 CAS.
- N. Weding, R. Jackstell, H. Jiao, A. Spannenberg and M. Hapke, Adv. Synth. Catal., 2011, 353, 3423–3433 CrossRef CAS.
- A. Geny, N. Agenet, L. Iannazzo, M. Malacria, C. Aubert and V. Gandon, Angew. Chem., Int. Ed., 2009, 48, 1810–1813 CrossRef CAS.
- I. Thiel, H. Jiao, A. Spannenberg and M. Hapke, Chem. – Eur. J., 2013, 19, 2548–2554 CrossRef CAS.
- (a) I. Thiel, A. Spannenberg and M. Hapke, ChemCatChem, 2013, 5, 2865–2868 CrossRef CAS; (b) I. Thiel and M. Hapke, J. Mol. Catal. A: Chem., 2014, 383–384, 153–158 CrossRef CAS.
- F. Fischer and M. Hapke, Eur. J. Org. Chem., 2018, 3193–3201 CrossRef CAS.
- (a) H. G. Schuster-Woldan and F. Basolo, J. Am. Chem. Soc., 1966, 88, 1657–1663 CrossRef CAS; (b) A. Spencer and H. Werner, J. Organomet. Chem., 1979, 171, 219–228 CrossRef CAS; (c) M. E. Rerek and F. Basolo, Organometallics, 1983, 2, 372–376 CrossRef CAS.
- See the ESI,† section 8.
- The bond between Co and P is slightly shorter in 2b, while the Co–CO bond is slightly longer compared to 2c, which is consistent with the less electron-donating character of the partially fluorinated phosphite of 2b. For details see the ESI† and ref. 26.
- H. Werner and B. Juthani, J. Organomet. Chem., 1981, 209, 211–218 CrossRef CAS.
- (a) Examples: K. Leonhard and H. Werner, Angew. Chem., Int. Ed. Engl., 1977, 16, 649–650 CrossRef; (b) P. Hong, Y. Yamamoto and H. Yamazaki, J. Organomet. Chem., 1982, 232, 71–79 CrossRef CAS; (c) H. Werner and J. Mahr, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 1364–1376 CAS; (d) G. Schmid, B. Kilanowski, R. Boese and D. Bläser, Chem. Ber., 1993, 126, 899–906 CrossRef CAS.
- For the synthesis of L2e, see: S. Han and S. Z. Zard, Org. Lett., 2014, 16, 5386–5389 CrossRef CAS.
- Storage of complexes 3a, 3f, 3g and 3i for at least 24 h under air at room temperature (air-conditioned laboratory) and examination by 1H and 31P NMR did not show any sign of decomposition of the complexes.
- CCDC 1957193 (2b) and CCDC 1957192 (2c) for the CpCo–CO–phosphite complexes as well as CCDC 1957196 (3e), CCDC 1957195 (3h), CCDC 1957194 (3j) and CCDC 1957197 (3k) for the CpCo–olefin–phosphite complexes contain the ESI† crystallographic data for Chart 1.
- For details see the ESI†.
- M. Hapke, N. Weding and A. Spannenberg, Organometallics, 2010, 29, 4298–4304 CrossRef CAS.
- The calculations show that complex 3l phosphine is overall less stable compared to 3e by 7.5 kcal mol−1. While the main energetical difference exists in the first step (+5.4 kcal mol−1 for 3l phosphine) the second step is more endothermic for 3e. Possibly also the sterical hindrance around the Co atom is higher for PiPr3 (2h phosphine).
- L. Hofmann and H. Werner, J. Organomet. Chem., 1985, 289, 141–155 CrossRef CAS.
- Example for application of CpCo(olefin)(phosphite) complexes: C. Georgi, M. Hapke, I. Thiel, A. Hildebrandt, T. Waechtler, S. E. Schulz and H. Lang, Thin Solid Films, 2015, 578, 180–184 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr. Rüdiger Beckhaus on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthesis of ligands and complexes, reaction conditions for reactivity screening, as well as substrate scope cyclisation conditions and characterisation of cyclisation products, are described in the ESI. The IR and NMR spectra of the prepared complexes and compounds (where applicable) are also given. CCDC 1957192–1957197. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy00876a |
| This journal is © The Royal Society of Chemistry 2020 |