DOI:
10.1039/D0CY00654H
(Perspective)
Catal. Sci. Technol., 2020,
10, 3805-3824
Reduction of sugar derivatives to valuable chemicals: utilization of asymmetric carbons
Received
2nd April 2020
, Accepted 12th May 2020
First published on 12th May 2020
Abstract
Biomass is a promising renewable resource substitute for petroleum, and transformation of biomass-derived materials to valuable chemicals is an urgent issue. To date, transformation methods of biomass-based materials via furfural and 5-hydroxymethyl furfural have been intensively developed, giving some important chemicals. However, considering the versatility of chemicals from petroleum, development of other transformation methods of biomass-derived materials is essential; in particular, transformation methods of biomass-derived materials which can make the most use of the unique structures is preferable. This perspective focuses on non-furfural routes of sugar derivatives, which are the main scaffolds of biomass-derived cellulose and hemicellulose, and it summarizes recent studies on catalytic transformations to valuable chemicals. These transformation routes are based on key reactions such as hydrogenolysis, reduction with silane reducing agents using borane catalyst, deoxydehydration (DODH) (+hydrogenation (HG)) and combination of dehydration and hydrogenation (and/or hydrogenolysis), and some of these transformation methods enable the synthesis of chiral products derived from the original sugars.
Introduction
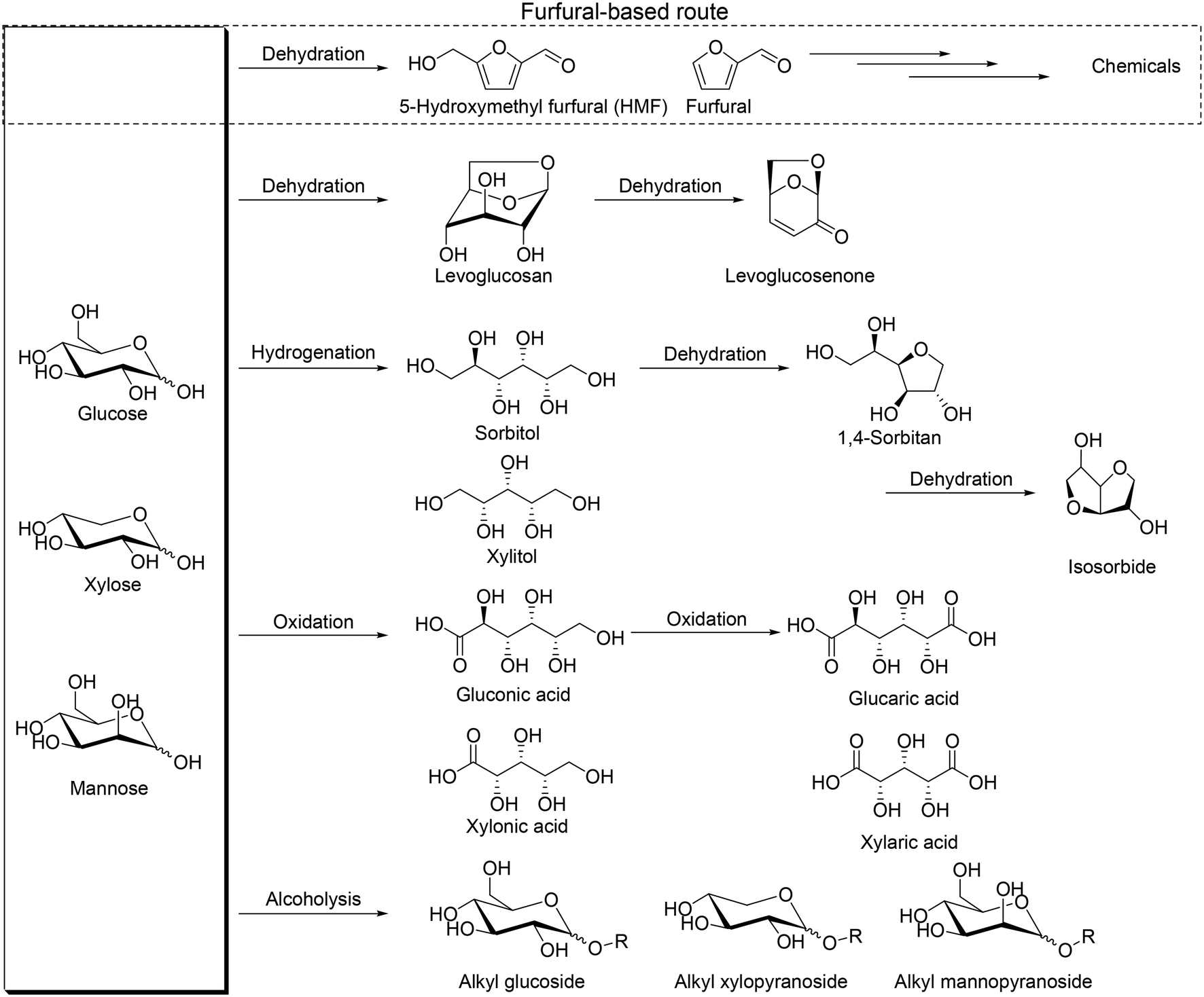
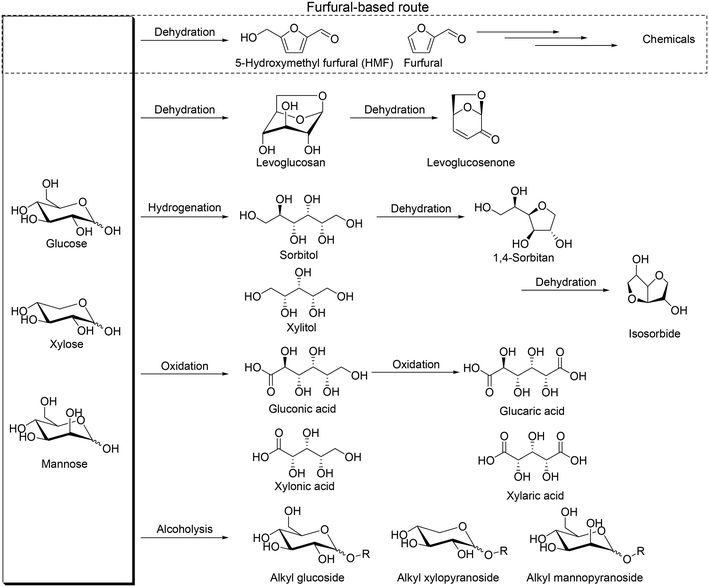
Biomass is one of the important resources for the production of chemicals in place of petroleum because biomass is the only organic compound among various renewable resources. Therefore, the development of effective transformation methods of biomass-derived materials, particularly inedible biomasses, to valuable chemicals is highly required. Woody and herbaceous biomasses are well known as inedible ones, and they are composed of cellulose, hemicellulose and lignin. Cellulose is the largest component in lignocellulose (about 30–50%), and hemicellulose and lignin have a similar composition ratio (20–40%).1 Polysaccharides are the main framework of cellulose and hemicellulose, and they are composed of monosaccharides, namely simple sugars, which are useful chemicals for various applications.2 Simple sugars have unique stereostructures, and hence, methodologies for effective conversion of poly- and monosaccharides to useful chemicals while maintaining the original stereostructure are essential to make the most use of biomass.
So far, various transformation methods of saccharides have been developed such as pyrolysis, biological transformation, and chemical transformation.3 Biological transformation such as enzymatic transformation can provide chemicals while maintaining the original stereostructure; in contrast, pyrolysis destroys the stereostructure of saccharides, and it is generally thought that the chemical approach is not good at transforming with retention of the original stereostructure. Among chemical transformation routes of saccharides, the most popular one is transformation of saccharides via formation of furfural and 5-hydroxymethyl furfural, denoted as the furfural-based route, by multiple dehydration reactions (Scheme 1), which provides various useful chemicals.4 However, generally, handling of furfural and 5-hydroxymethyl furfural is difficult due to the low stability to heat, and they are often decomposed or polymerized to form humin. At the same time, the intrinsic stereostructure of sugars is lost by the route via formation of furfurals, and therefore, development of new transformation routes and methods, particularly while maintaining the original stereostructure, is necessary for the production of various chemicals and making the most use of sugars.5 Dehydration to levoglucosan and levoglucosenone, hydrogenation to sugar alcohols, oxidation to sugar carboxylic acids and alcoholysis to alkyl glycosides can be regarded as possible alternative routes (Scheme 1). Levoglucosan, levoglucosenone, sugar alcohols, sugar carboxylic acids and alkyl glycosides are important platform chemicals from these transformation routes and can maintain some of the original stereostructures without C–C bond dissociation. The largest difference between the furfural-based route and the other transformation routes is the stereostructure of the platform chemicals, and the platform chemicals by non-furfural routes can have chiral centers derived from the mother sugars. Chiral products from sugars and sugar derivatives include deoxy sugars and chiral polyols. Deoxy sugars can be found in natural products as the scaffold, and dideoxy sugars can be used for the synthesis of rare sugars, chiral ligands, glycolipids, and medicines, for example (+)-asperlin, (−)-isofagomine, daumone, digitoxin, methymycins and 2,3-dihydroxy-epilupinine.6 Chiral polyols such as tri- and tetraols have high potential for polymers such as polyesters, polyurethanes, and alkyd resins7 and can be further transformed to polymer precursors such as tetrahydrofurandimethanol, 1,6-hexanediol and 1,5-pentanediol.8 Effective catalyst systems for the synthesis of valuable chemicals via these transformation routes are desirable to be developed from now on.
 |
| | Scheme 1 Platform chemicals from sugars derived from cellulose and hemicellulose without dissociation of C–C bonds. | |
There are many excellent reviews on the transformation of lignocellulosic biomass to saccharides,9 saccharides to furfurals,10 and furfurals to valuable chemicals.4 In this perspective, we focus on the transformation routes of C4–C6 sugar derivatives, not via formation of furfurals, particularly on reactions without dissociation of C–C bonds in sugar derivatives because C–C bonds and the stereostructure are important in organic synthesis. The routes mainly include transformation of levoglucosenone, sugar alcohols, isosorbide, sugar carboxylic acids and methyl glycosides as platform chemicals with catalysts (Scheme 1). We categorized recent catalyst systems based on the key reactions such as hydrogenolysis, reduction with borane catalyst + silane reducing agents, deoxydehydration (DODH) (+hydrogenation (HG)), and combination of dehydration and hydrogenation (and/or hydrogenolysis); simple ring formation from sugar derivatives by dehydration was excluded from this perspective.
Hydrogenolysis
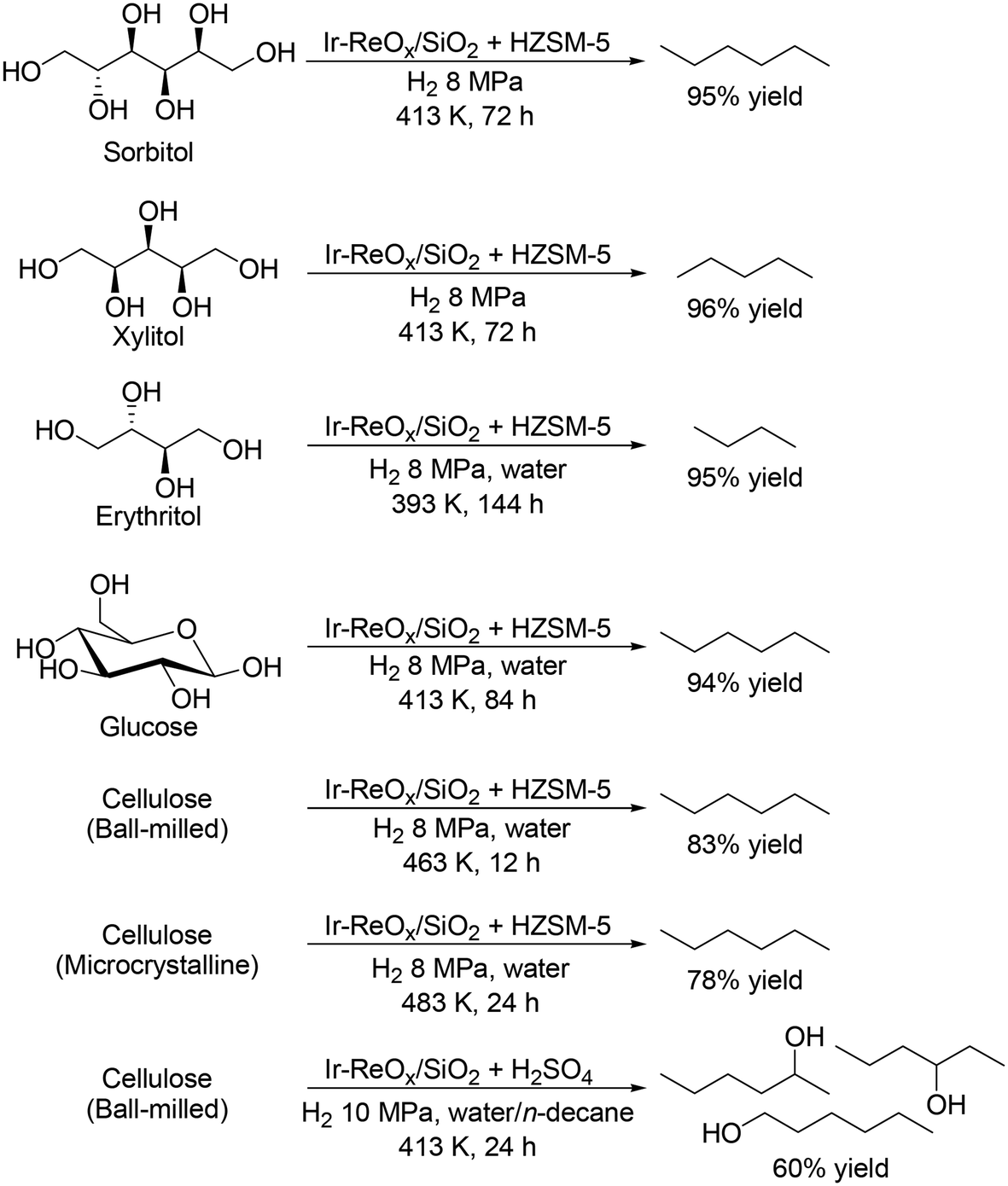
The most common and conventional method for removal of OH groups in sugar derivatives is C–O hydrogenolysis. To date, various effective heterogeneous catalysts have been developed for the hydrogenolysis of polyols such as glycerol and erythritol,11 and many important reviews on the hydrogenolysis of polyols have been already published;12 hence, the details are not introduced in this perspective. Focusing on the hydrogenolysis of polyols such as C4, C5 and C6 sugar alcohols without C–C bond dissociation, selective formation of single target products is difficult due to the multiple OH groups and similar properties of OH groups. Ir–ReOx/SiO2 combined with acid catalysts such as zeolites has been reported to be effective for total hydrogenolysis and partial hydrogenolysis of sugar alcohols at a comparatively low reaction temperature (393–413 K) to provide high yields of alkanes and mono alcohols, respectively, without dissociation of C–C bonds (Scheme 2).13 The high yield is due to the low activity to C–C bond dissociation and high activity to C–O bond dissociation. The other catalyst systems required high temperatures (typically 473 K) and provided comparatively low yields (<90%).14 Moreover, the catalyst system of Ir–ReOx/SiO2 and acid catalysts was applicable to the transformation of carbohydrates such as glucose and cellulose by combination with acid-catalyzed hydrolysis, providing the corresponding alkanes in high yields (Scheme 2).13 However, selective synthesis of diols and triols while maintaining the original stereostructure from C4, C5 and C6 sugar alcohols is quite difficult due to the high activity of the hydrogenolysis-active metal species for epimerization of OH groups at comparatively high temperature (≥423 K). Therefore, control of the selectivity and stereostructure in sugar alcohols by hydrogenolysis will be a future task. The reusability of the Ir–ReOx-based catalysts in the hydrogenolysis of polyols is fundamentally high, which is related to the high stability of the catalysts and comparatively low reaction temperature (typically below 473 K).15 Kinetic studies such as the effect of H2 pressure and substrate concentration were also investigated with Ir–ReOx-based catalysts.15 The reaction order with respect to substrate concentration is typically zero, suggesting that the interaction between polyols and Ir–ReOx-based catalysts is strong. The typical reaction order with respect to H2 pressure is +1 for C–O hydrogenolysis, suggesting that attack of the hydride species, which is formed by heterolytic dissociation of H2 at the interface between Ir metal and ReOx species, to the carbon atom of the C–O bond in polyols, leads to the high activity. However, the kinetics for multiple hydrogenolysis or combination of hydrogenolysis and hydration of polyols are complicated, and precise investigation of the kinetics will be necessary.
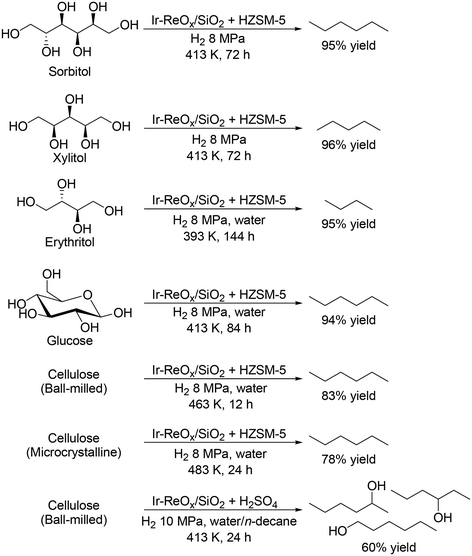 |
| | Scheme 2 Transformation of sugar alcohols and cellulose to alkanes and mono-alcohols with Ir–ReOx/SiO2 and acid catalysts.13 | |
Partial reduction with borane catalyst and silane reducing agents
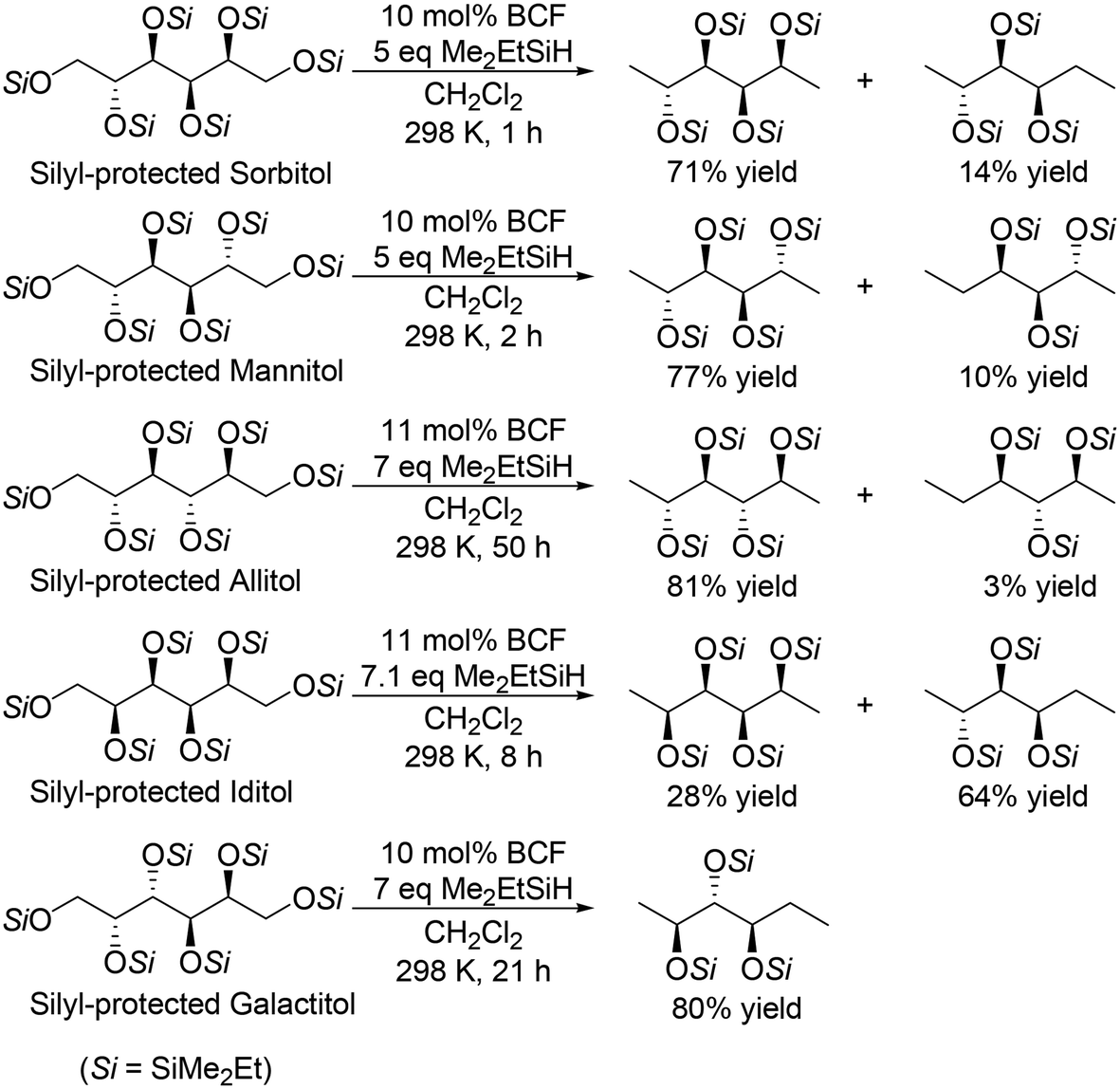
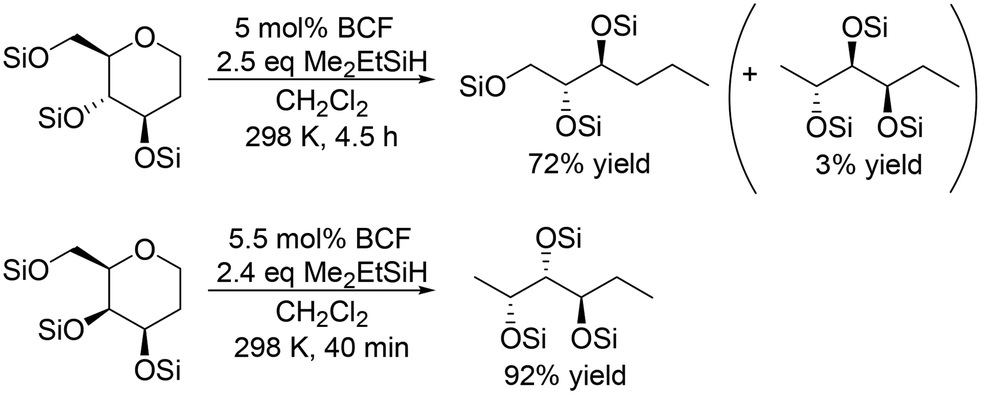
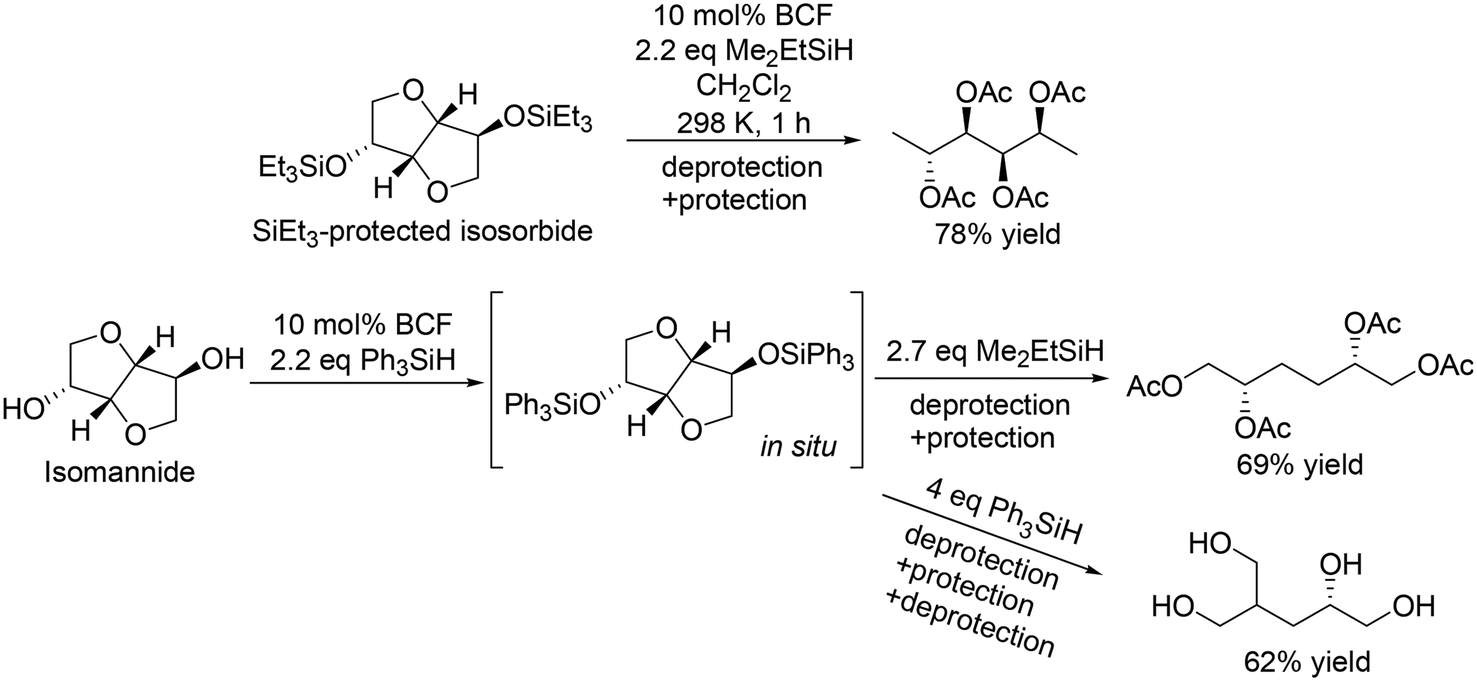
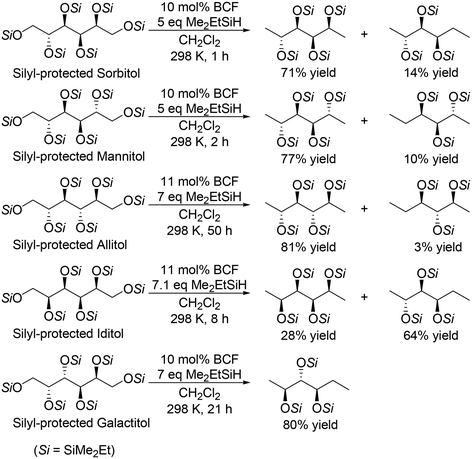
For the transformation of sugar derivatives while taking advantage of the original stereostructure, the combination of tris(pentafluorophenyl)borane (BCF) as a catalyst and tertiary silane, in particular Me2EtSiH, as a reducing agent was reported by Gagné and co-workers to be an effective catalyst system for chemoselective transformation of sugar alcohols (Scheme 3), 1,2-deoxy sugars (Scheme 4) and doubly dehydrated sugars (isosorbide and isomannide) (Scheme 5) to chiral compounds.16 In these reports, the silyl-protected sugar derivatives were basically used as the starting materials, while unprotected sugars seem to be applicable to the catalyst systems by using excess amount of silanes.
 |
| | Scheme 3 Selective transformation of SiMe2Et-protected sugar alcohols with a combination catalyst of BCF and Me2EtSiH.16 | |
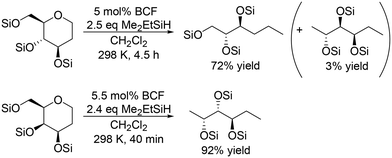 |
| | Scheme 4 Selective transformation of 1,2-dideoxy sugars with a combination catalyst of BCF and Me2EtSiH.16 | |
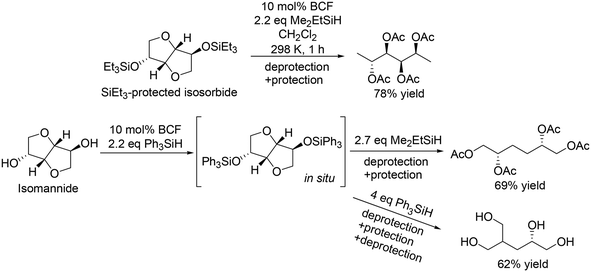 |
| | Scheme 5 Selective transformation of doubly dehydrated sugars with BCF and silanes.16 | |
In the transformation of sugar alcohols with BCF and Me2EtSiH, sorbitol, mannitol and allitol were converted mainly to the 1,6-reduced tetraols (71–81% yield) with a small amount of the triols (3–14% yield) and the original stereostructure of all the residual OH groups was maintained in these products. On the other hand, iditol and galactitol were converted mainly to the triols, and the triols had an inverted stereostructure of the OSi group at the C5 position. The difference of the results was explained by the difference of the reactivity of sugars: iditol and galactitol are reactive and the other sugars are less reactive. The inversion of the stereostructure was explained by the nucleophilic attack of H–B− (C6F5)3 on the formed 5-membered ring cyclic silyl oxonium ion as the intermediate at the sterically non-congested side.
The catalyst system was applicable to the selective reduction of 1,2-dideoxy glucose and 1,2-dideoxy galactose (Scheme 4) and the partial reduction of doubly dehydrated sugars, isosorbide and isomannide (Scheme 5). The selective reduction of 1,2-dideoxy glucose and 1,2-dideoxy galactose provided 1,2,3-trideoxy isomer in high yield (72%) and 1,2,6-trideoxy isomer in high yield (92%), respectively, with retention of the stereostructure. The difference of the reduction position could also be explained by the steric hindrance of the nucleophilic attack of H–B− (C6F5)3 on the 6-membered ring cyclic silyl oxonium ion in the intermediate. As for the partial reduction of doubly dehydrated sugars, isosorbide was converted to the tetraol, which is the same product from sorbitol. Isomannide was transformed to 3,4-dideoxy mannitol in 69% yield by using Me2EtSiH as a reducing agent and was further transformed to tetraol via skeletal rearrangement (probably an alkyl shift in the silyl oxonium intermediate + reduction of the resulting oxocarbenium ion) by using the bulky silyl-protecting group Ph3SiH.
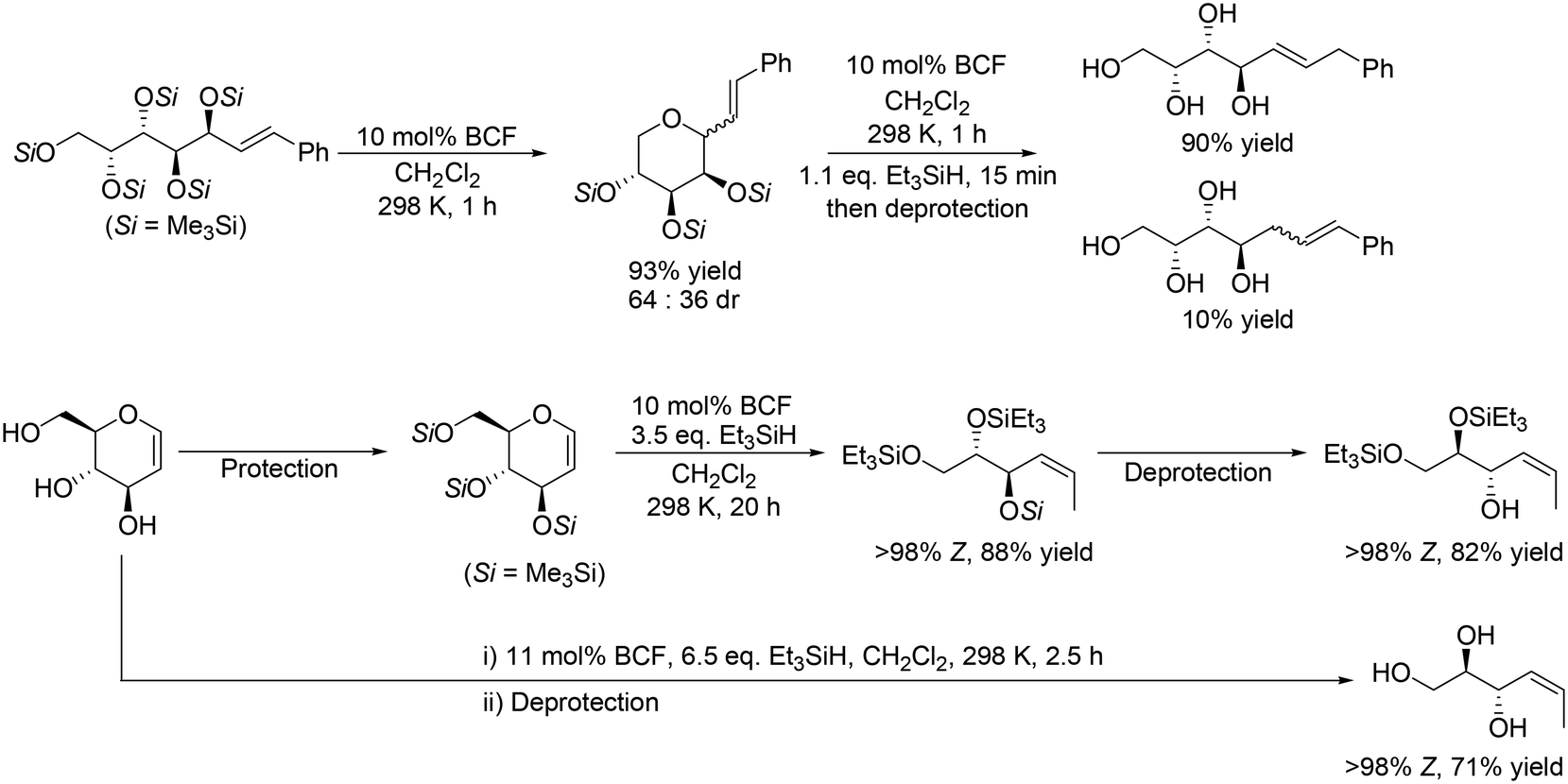
In addition, Gagné and co-workers expanded the method to the reactions of unsaturated carbohydrate derivatives such as Me3Si-protected styryl sugars and glycals, which was based on their previous results on the reductive carbocyclization of unsaturated carbohydrates.17 Some examples are shown in Scheme 6. Me3Si-protected styryl galactol was converted to the corresponding tetrahydropyran derivative by cyclization, and the target tetraol was produced in a high yield of 88% by SN2′ reduction of the tetrahydropyran derivative (Scheme 6, top) with retention of the original stereostructure. Other Me3Si-protected styryl sugar alcohols (gluco-, manno-, fuco-, ribo-, xylo- and lyxo- derivatives) were also transformed to the corresponding polyols in high yields (72–91%). Me3Si-protected glucal was also converted to anti-(Z)-Si3-triol with one SiMe3 and two SiEt3 groups in a high yield of 88%, which could be furthermore converted to the partially deprotected product with 82% yield. In addition, glucal was directly transformed to the corresponding unsaturated triol in a high yield of 71% by reduction with BCF and Et3SiH and deprotection treatment (Scheme 6, bottom).18
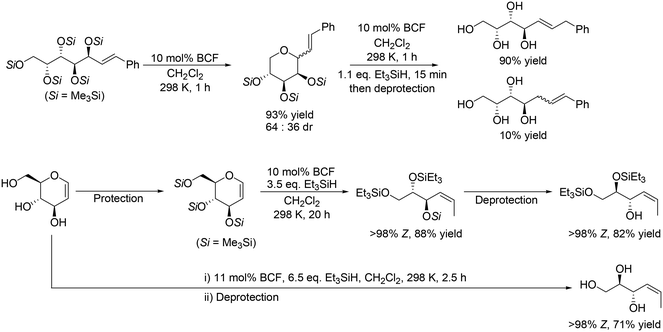 |
| | Scheme 6 Selective reduction of Me3Si-protected styryl galactol and glucal with BCF and Et3SiH.17,18 | |
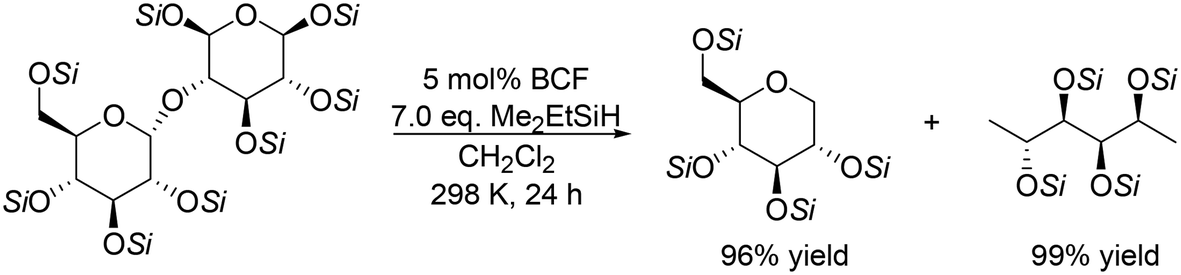
Silyl-protected disaccharides also underwent reduction with the same catalyst system (Scheme 7). Me2EtSiH-protected β-maltose was converted to 1-deoxyglucose and 1,6-dideoxysorbitol in high yields (96% and 99%, respectively).19 Other silyl-protected disaccharides such as cellobiose, trehalose and isomaltose also reacted to give the corresponding polyols with the retention of the original stereostructure.
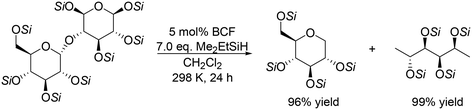 |
| | Scheme 7 Reductive transformation of Me2EtSiH-protected β-maltose to 1-deoxyglucose and 1,6-deoxysorbitol with BCF and Me2EtSiH.19 | |
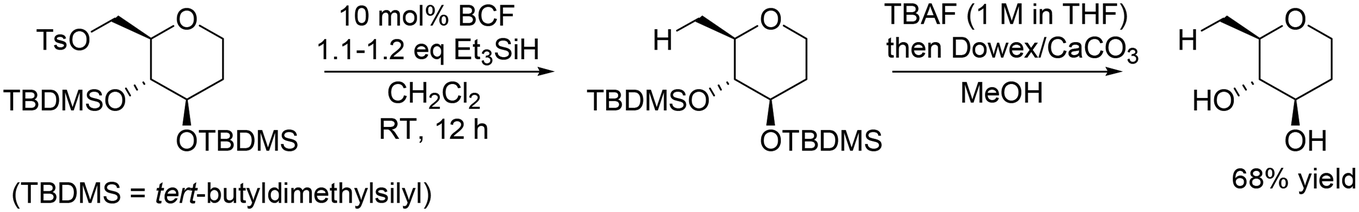
Other research groups also reported similar catalyst systems for transformation of sugar derivatives. Oestreich and co-workers applied the same catalyst method with BCF and Et3SiH to chemoselective defunctionalization of primary OH group-tosylated silyl-protected 1,2-dideoxy-D-glucose (Scheme 8).20 In combination with deprotection treatment, the target unprotected tetrahydropyran was obtained in a high yield of 68%.
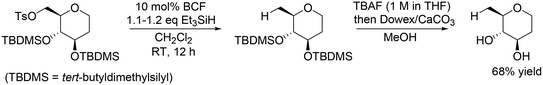 |
| | Scheme 8 Selective reduction of the tosyl group in primary OH group-tosylated silyl-protected 1,2-dideoxy-D-glucose with BCF and Et3SiH.20 | |
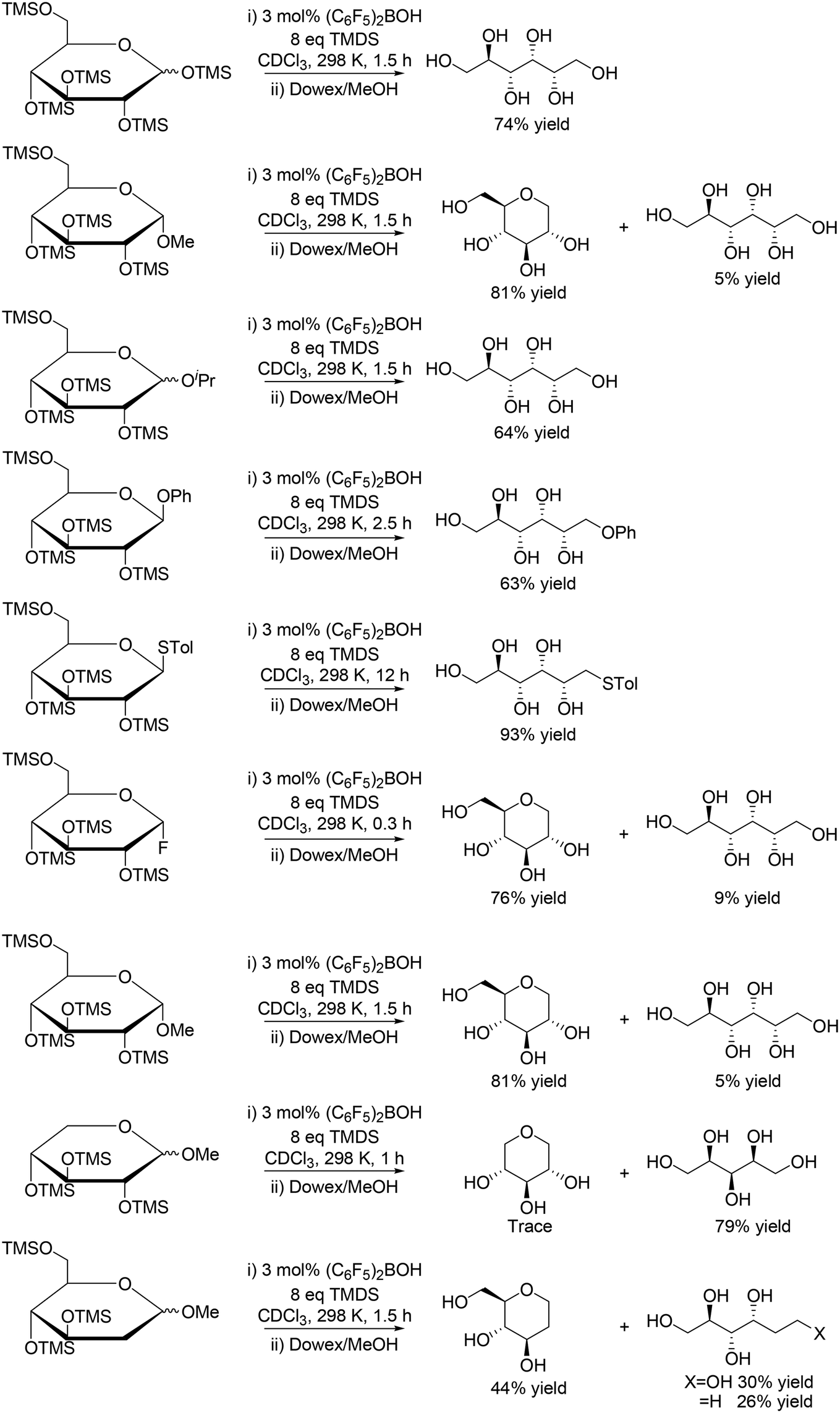
A similar catalyst system using (C6F5)2BOH and 1,1,3,3-tetramethyldisiloxane (TMDS) was reported by S. Park, S. Chang and co-workers to be also effective for reduction of silyl-protected monosaccharides (Scheme 9) and disaccharides (Scheme 10), where reactive (C6F5)2BH (Piers' borane) catalyst was in situ generated.21 The reactivity of the protected monosaccharides seems to be changed by the direction of the substituent at the anomeric carbon, and β-anomeric glucoses provided linear polyols via ring-opening reduction with the retention of the original stereostructure, and α-anomeric glucoses gave mainly 1-deoxy glucose. However, the reactivity was also influenced by the presence/absence of the other substituents, and silyl-protected methyl glucoside without the C5 silyoxy group afforded linear polyols, which is completely different from the result of the silyl-protected methyl glucoside. Silyl-protected 2-deoxy methyl glucoside gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cyclic and linear polyol products. The explanation of the reactivity tendency will be difficult and the reason for that was not clearly discussed in the report.
:1 mixture of cyclic and linear polyol products. The explanation of the reactivity tendency will be difficult and the reason for that was not clearly discussed in the report.
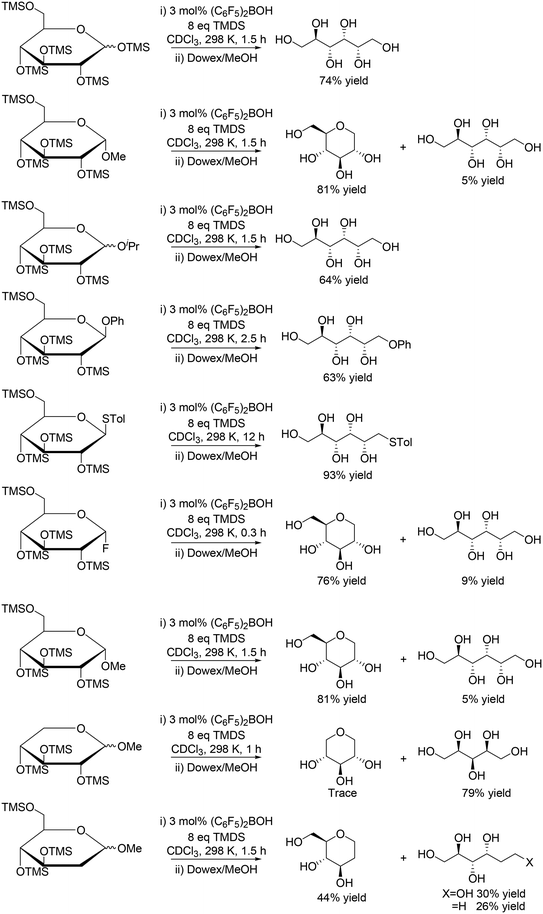 |
| | Scheme 9 Selective reduction of protected monosaccharides with (C6F5)2BOH and TMDS.21 | |
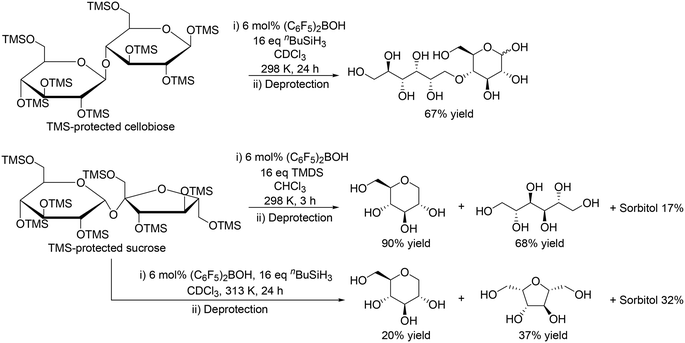 |
| | Scheme 10 Application of (C6F5)2BOH and silanes to the reduction of disaccharides.21 | |
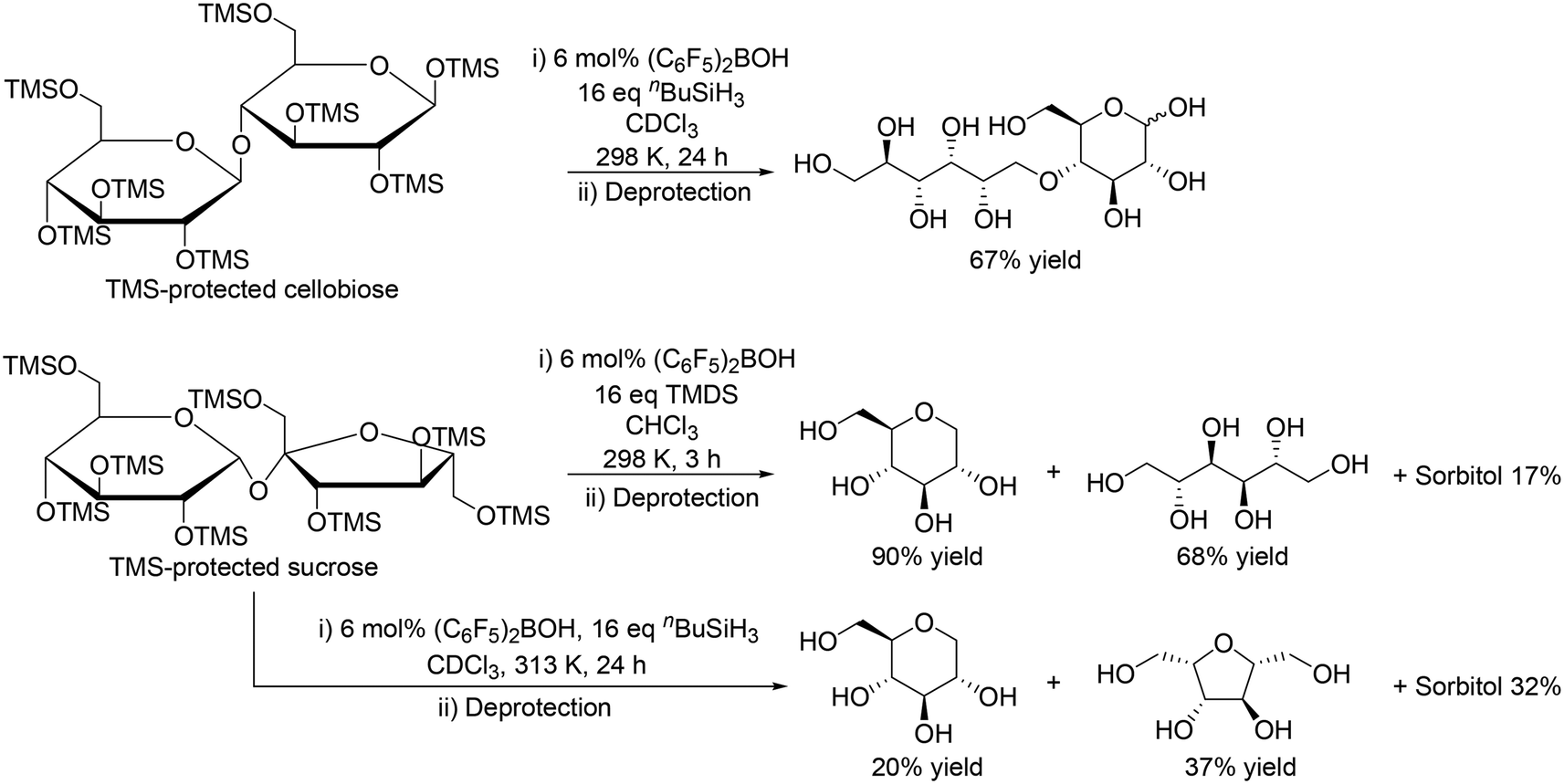
Moreover, the catalyst system was applicable to the reduction of disaccharides such as silyl-protected cellobiose and sucrose (Scheme 10).21 In the case of TMS-protected cellobiose, one ring-opened product was obtained in moderate yield (67%) by using (C6F5)2BOH and nBuSiH3. On the other hand, TMS-protected sucrose was converted to 1-deoxy glucose and mannitol in 90% and 68% yield, respectively, with TMDS at 298 K; however, with (C6F5)2BOH and nBuSiH3 at 313 K, the selectivity to the target polyols was very low.
Deoxydehydration (DODH) (+hydrogenation (HG))
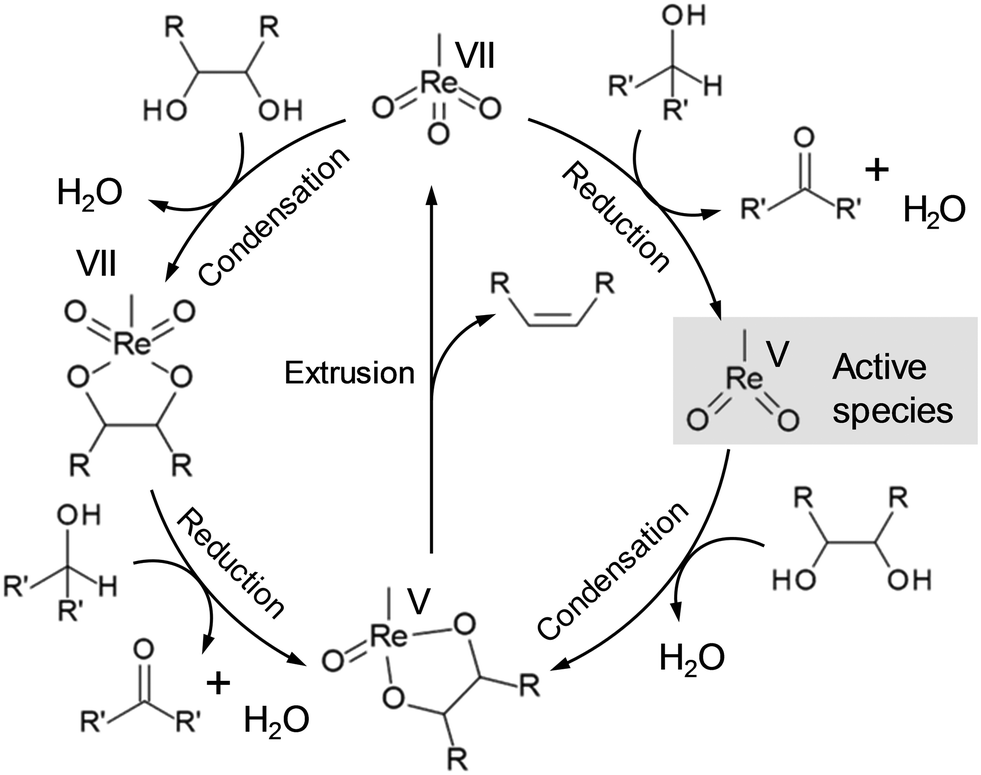
Deoxydehydration (DODH) is a powerful tool for reduction of OH groups in sugar derivatives, where vicinal OH groups in polyols are simultaneously removed to form the corresponding olefin group by high-valent metal catalysts and reducing agents without dissociation of C–C bonds.22 The most common catalyst for DODH reaction is oxo-rhenium complexes, and the proposed DODH reaction mechanism is shown in Scheme 11, where the reaction proceeds via the redox reaction between Re(VII) and Re(V). There are two main routes for the DODH reaction with oxo-rhenium complexes: (i) condensation of diols after the reduction of oxo-rhenium species and (ii) condensation of diols before the reduction of oxo-rhenium species. The route will depend on the reducing agents such as PPh3, alcohols, H2 and so on. CH3ReO(OH)2 and Re(VII)diolate were also proposed to be the active species for the DODH reaction except for the methyldioxorhenium in Scheme 11, and the presence of CH3ReO(OH)2 was strongly supported by DFT calculations.23 The details of the kinetics and the reaction mechanism of DODH by homogeneous oxo-rhenium catalysts were highlighted in previous reviews.24
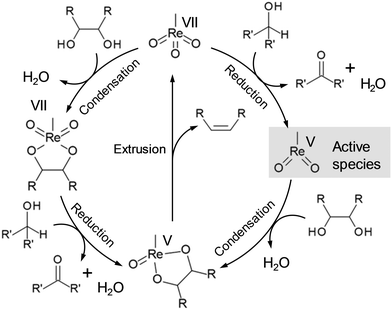 |
| | Scheme 11 Proposed reaction mechanism of DODH with oxo-rhenium complexes.22a | |
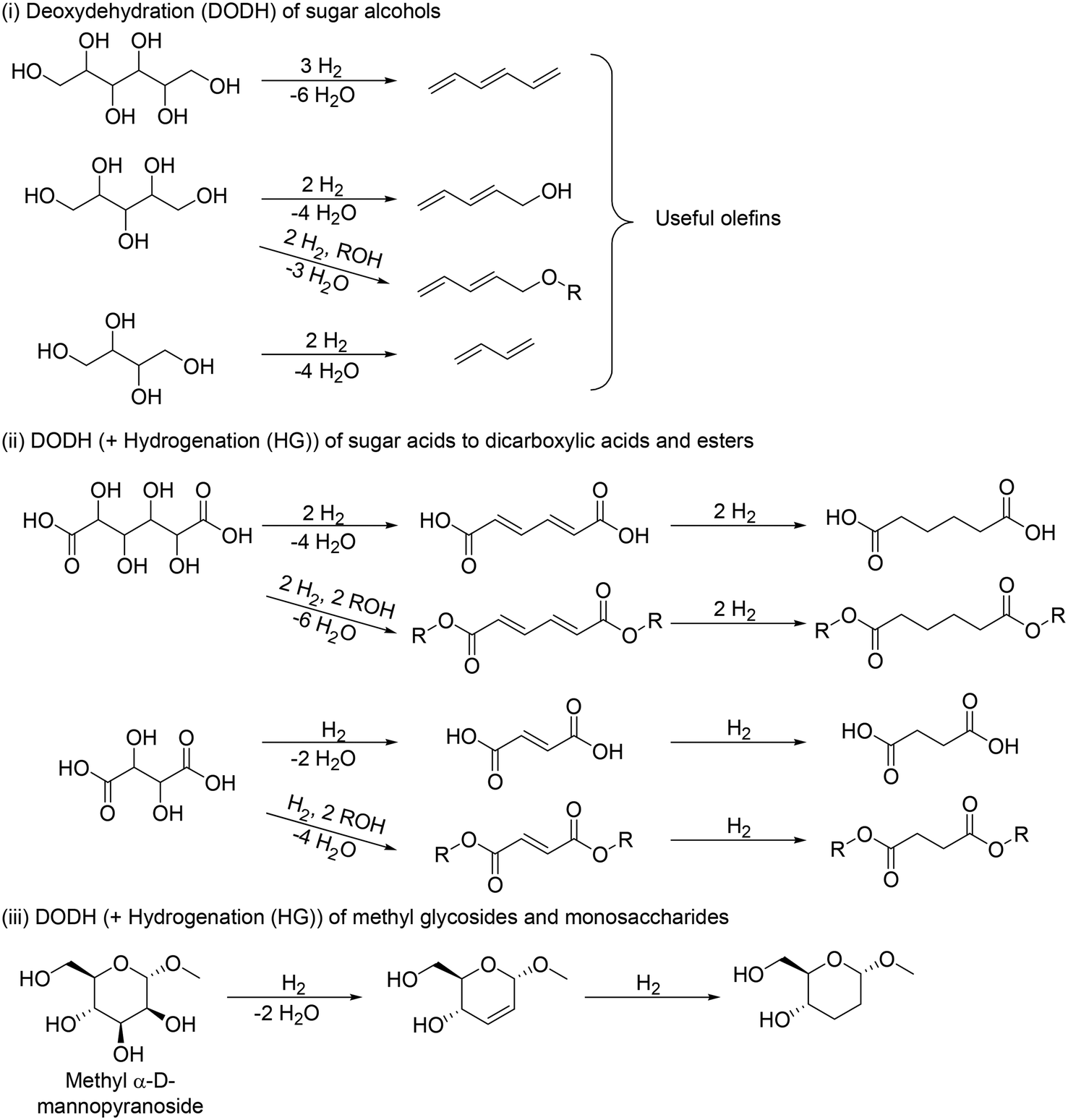
The application of DODH to the transformation of sugar derivatives is divided mainly into three cases in terms of starting materials (Scheme 12): (i) DODH of sugar alcohols to olefins, (ii) DODH (+HG) of sugar acids to dicarboxylic acids and esters, and (iii) DODH (+HG) of methyl glycosides to dideoxy methyl glycosides.
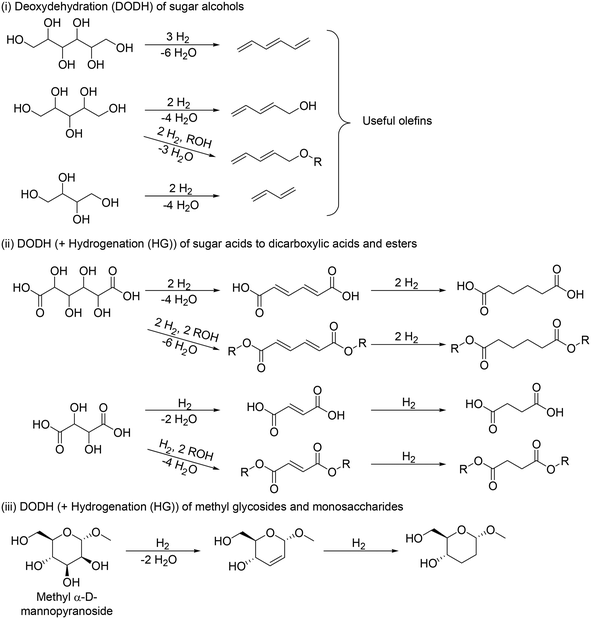 |
| | Scheme 12 DODH reaction for transformation of sugar derivatives (typical examples). | |
(i) DODH of sugar alcohols and cyclic polyols to olefins and/or aromatic compounds
In this section, we focus on the DODH of linear polyols having more than three OH groups such as C4, C5, and C6 sugar alcohols and cyclic polyols such as inositols and aldoses.
In 1996, M. A. Andrews and G. K. Cook first reported that Cp*ReO3 was an effective catalyst for the DODH of erythritol with PPh3 as a reducing agent at 408 K in chlorobenzene solvent, giving 1,3-butadiene in about 80% yield, and the catalyst system seemed to be applicable to the DODH of xylitol to 2,4-pentadiene-1-ol, although the yield was not shown.25 Moreover, J. A. Ellman, R. G. Bergman and co-worker reported that the combination of Re2(CO)10 as a catalyst and 3-octanol as a reducing agent with an organic acid of TsOH was effective for the DODH of erythritol at 433 K, and 55% yield of 2,5-dihydrofuran was obtained by the consecutive reactions of dehydration of erythritol to 1,4-anhydroerythritol and DODH of 1,4-anhydroerythritol.26
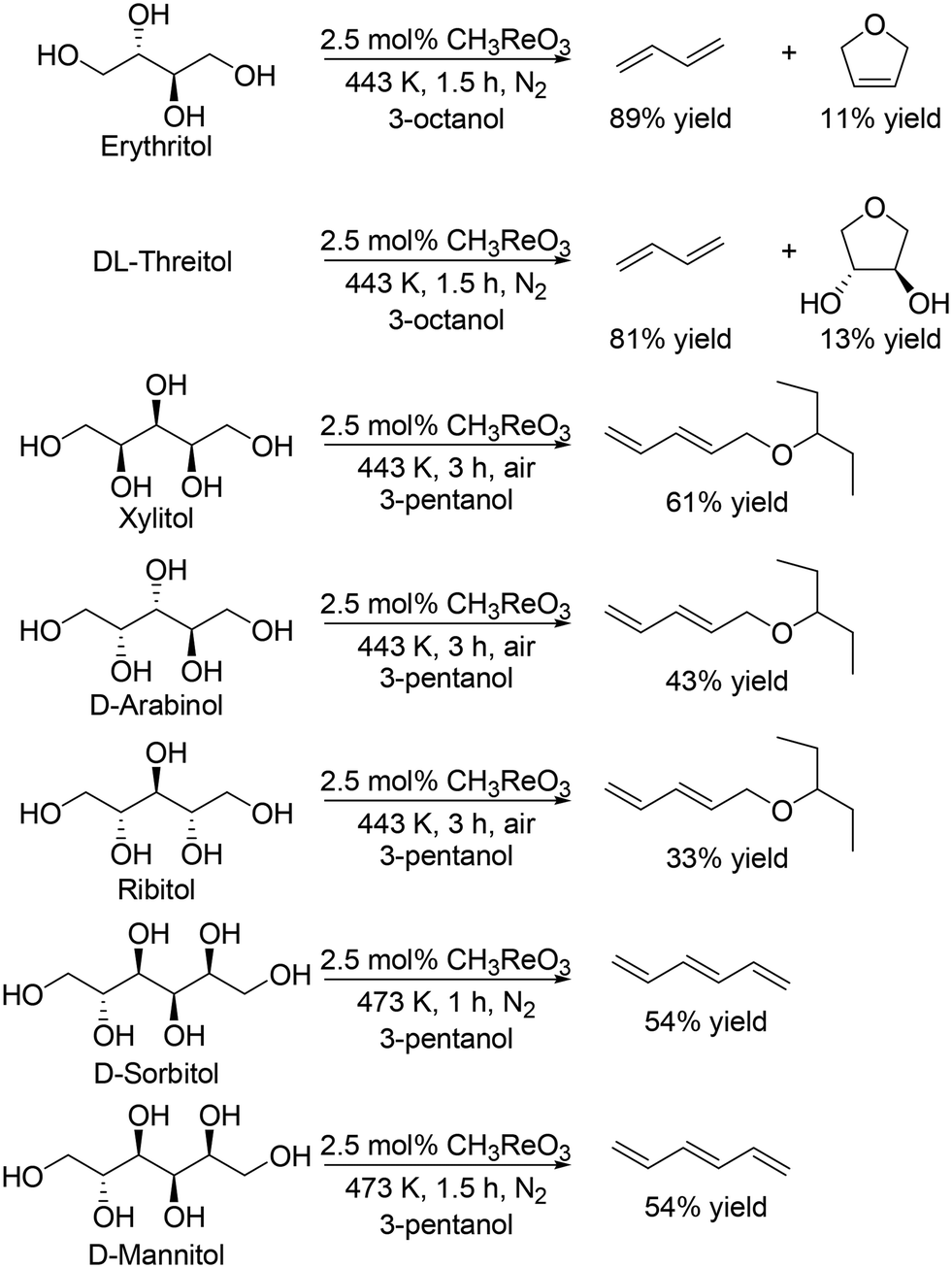
Toste and Shiramizu comprehensively studied the DODH of C4–C6 sugar derivatives.27 They found that the combination of CH3ReO3 as a catalyst and secondary mono alcohols as a reducing agent was an effective catalyst system for transformation of sugar alcohols including C4, C5 and C6 sugar alcohols, and various allylic alcohols and olefins were obtained in moderate to high yields (Scheme 13).27 In the case of C5 and C6 sugar alcohols, low concentration (0.3 M for C4 alcohols, and 0.05 M for C5 and C6 sugar alcohols) is necessary to obtain a high yield of the target DODH products.
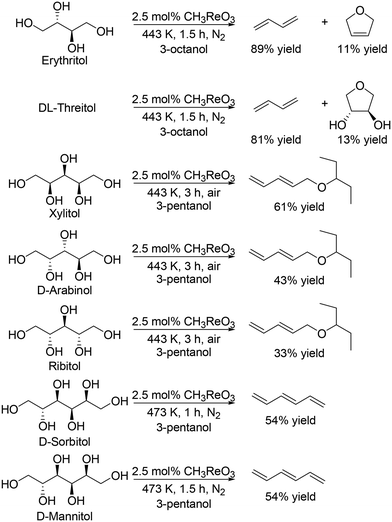 |
| | Scheme 13 Transformation of C4–C6 sugar alcohols to the corresponding olefins and allylic alcohols with CH3ReO3 catalyst and secondary alcohol.27 | |
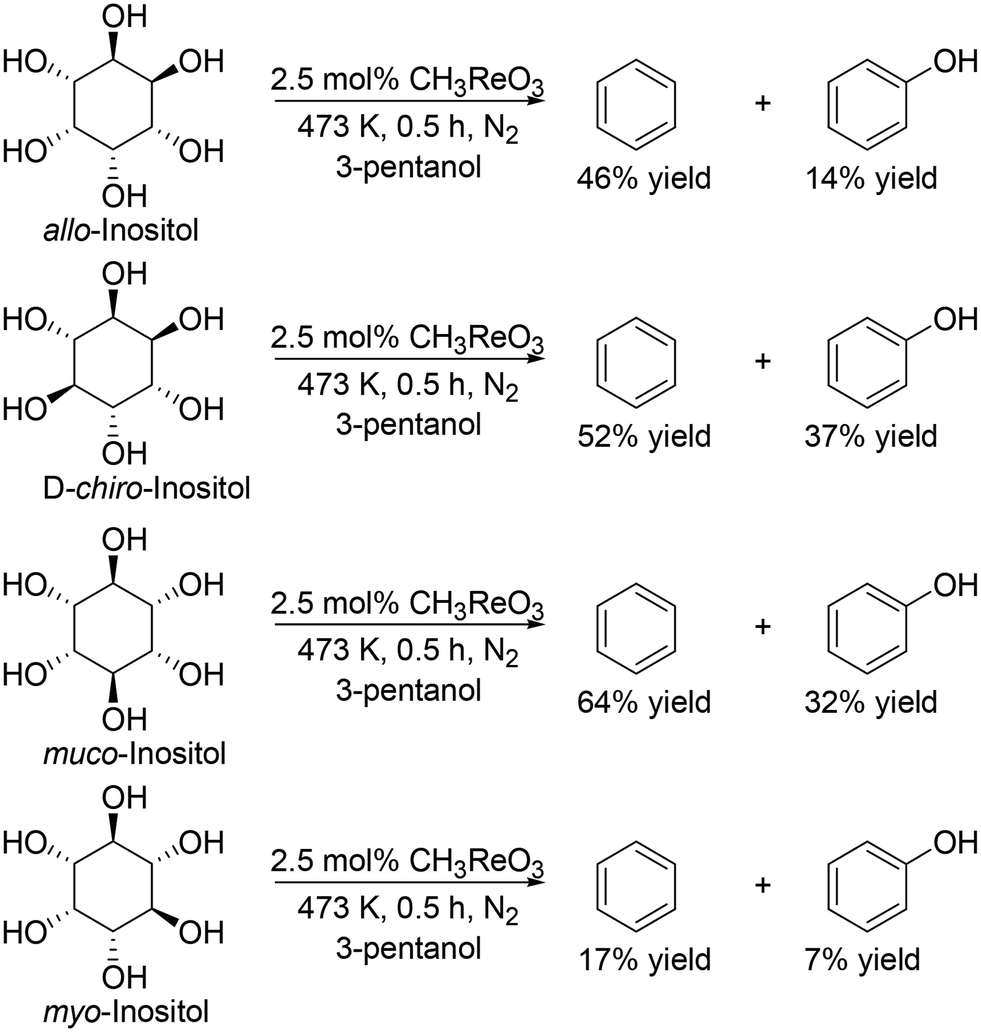
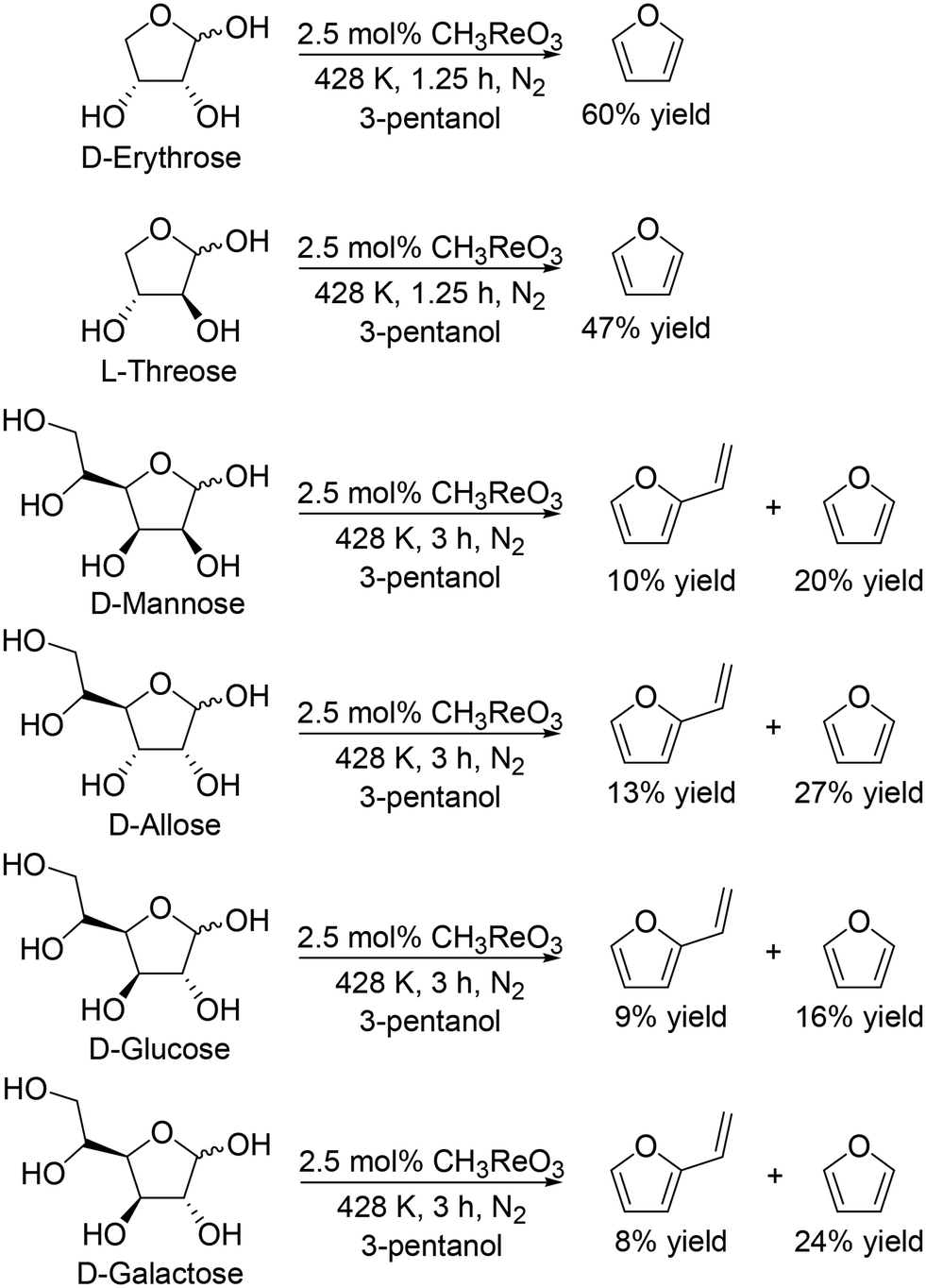
They also applied the catalyst system to inositols (Scheme 14)27 and aldoses such as D-erythrose, L-threose and hexoses (Scheme 15).27 Benzene and phenol were obtained in moderate to high yields from inositols having more than one cis-vicinal OH groups; however, the reactivity of myo-inositol which has only one cis-vicinal OH group is low, which is related to the higher reactivity of cis-vicinal OH groups than trans-vicinal OH groups. Moreover, the catalyst system was also applicable to the reactions of aldoses such as tetroses (D-erythrose and L-threose) and hexoses (D-mannose, D-allose, D-glucose and D-galactose) (Scheme 15). The tetroses were transformed to furan in moderate yields by DODH and subsequent dehydration, and the hexoses were transformed to 2-vinylfuran by DODH and dehydration with formation of furan, which can be produced via a retro-aldol reaction. The authors also tried the reaction of pentoses with the same catalyst system; however, the yield of the target product 2-(alkoxymethyl)furan seemed to be low.
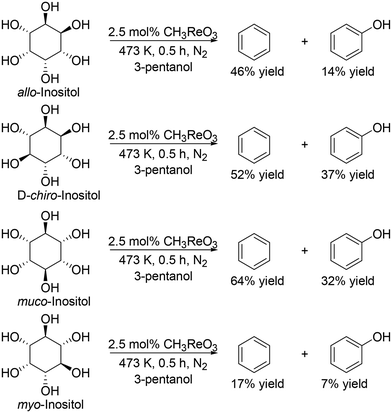 |
| | Scheme 14 DODH of inositols to aromatic compounds with CH3ReO3 catalyst and 3-pentanol.27 | |
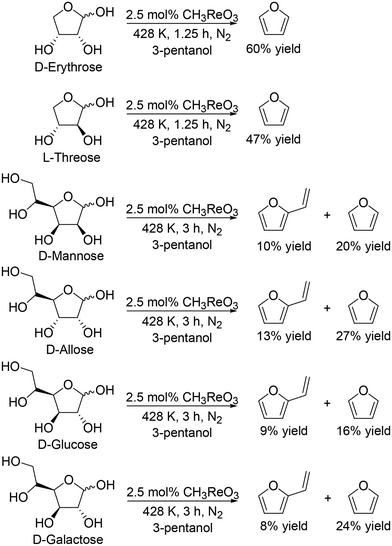 |
| | Scheme 15 DODH of aldoses to furan derivatives with CH3ReO3 catalyst and 3-pentanol.27 | |
K. M. Nicholas and co-workers found that indoline was an effective reducing agent with an oxo-rhenium complex for DODH reactions by screening various hydroaromatic compounds.28 Erythritol and xylitol were converted to 1,3-butadiene and buta-1,3-dien-1-ol, respectively, in moderate yields (43% and 56%, respectively) with the corresponding amount of indole.
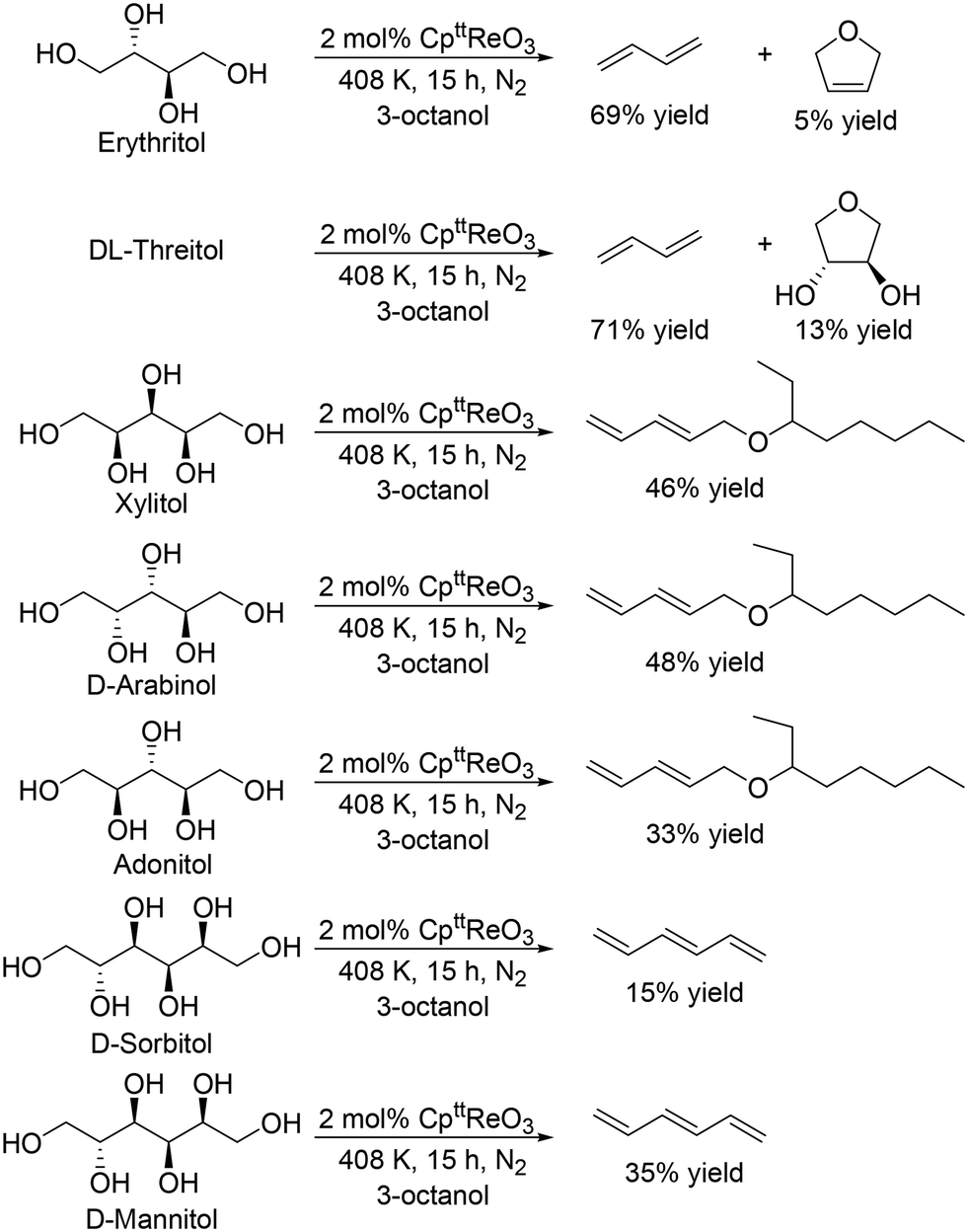
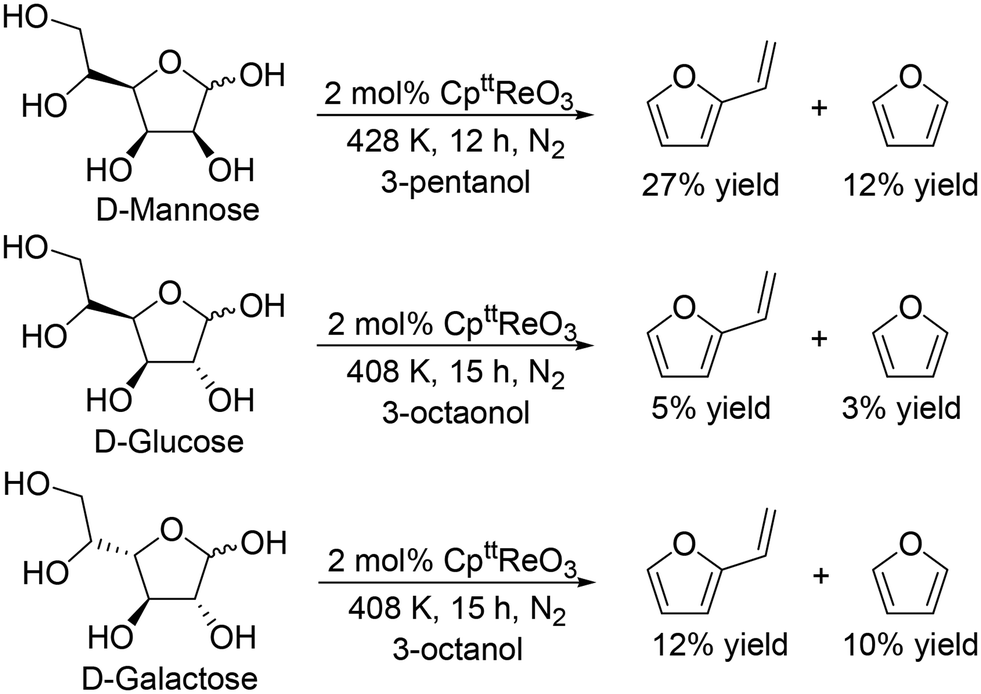
J. M. Klein Gebbink and co-workers applied 1,3-di-tert-butylcyclopentadienyl trioxorhenium (CpttReO3) and 3-octanol to the DODH of C4 sugar alcohols (erythritol and DL-threitol), C5 sugar alcohols (xylitol, D-arabinitol and adonitol), C6 sugar alcohols (D-mannitol and D-sorbitol) (Scheme 16) and various furanoses (D-glucose, D-galactose, and D-mannose) (Scheme 17).29 From C4 sugar alcohols, 1,3-butadiene was obtained in moderate yields (∼70%). From C5 sugar alcohols, the corresponding diene ether was obtained in moderate yields (42–46%); however, the yield of 1,3,5-hexatriene was low compared with the case of the CH3ReO3 and 3-pentanol catalyst system as described above. Compared with C5 and C6 sugar alcohols, the yields of the target product, vinylfuran, from furanoses were low, a tendency that is similar to the case of the CH3ReO3 and 3-pentanol catalyst system.
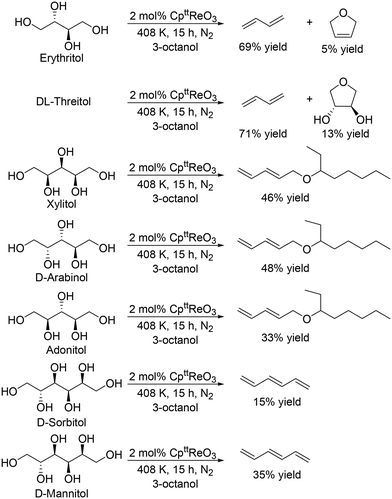 |
| | Scheme 16 DODH of C4–C6 sugar alcohols to olefins and allylic alcohols with CpttReO3 catalyst and 3-octanol.29 | |
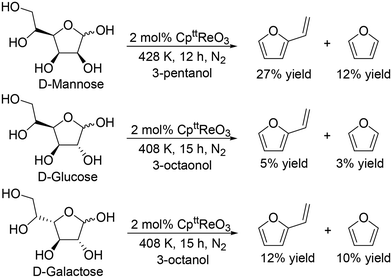 |
| | Scheme 17 DODH of furanoses to furans with CpttReO3 catalyst and secondary alcohols.29 | |
(ii) DODH (+HG) of sugar acids to dicarboxylic acids and esters
Dicarboxylic acids are important chemicals in industry for the synthesis of various valuable chemicals such as polymers and medicines. To date, various approaches for the synthesis of dicarboxylic acids and esters by DODH were reported including homogeneous and heterogeneous catalysts, and the most common catalysts for the reaction are oxorhenium complexes. In general, the acidity of the oxorhenium complex catalysts often brings about esterification of the acids with alcohols which are used as reducing agents. Therefore, the synthesis of free carboxylic acids is generally difficult due to the Lewis acidity of the catalysts. Representative examples are introduced below.
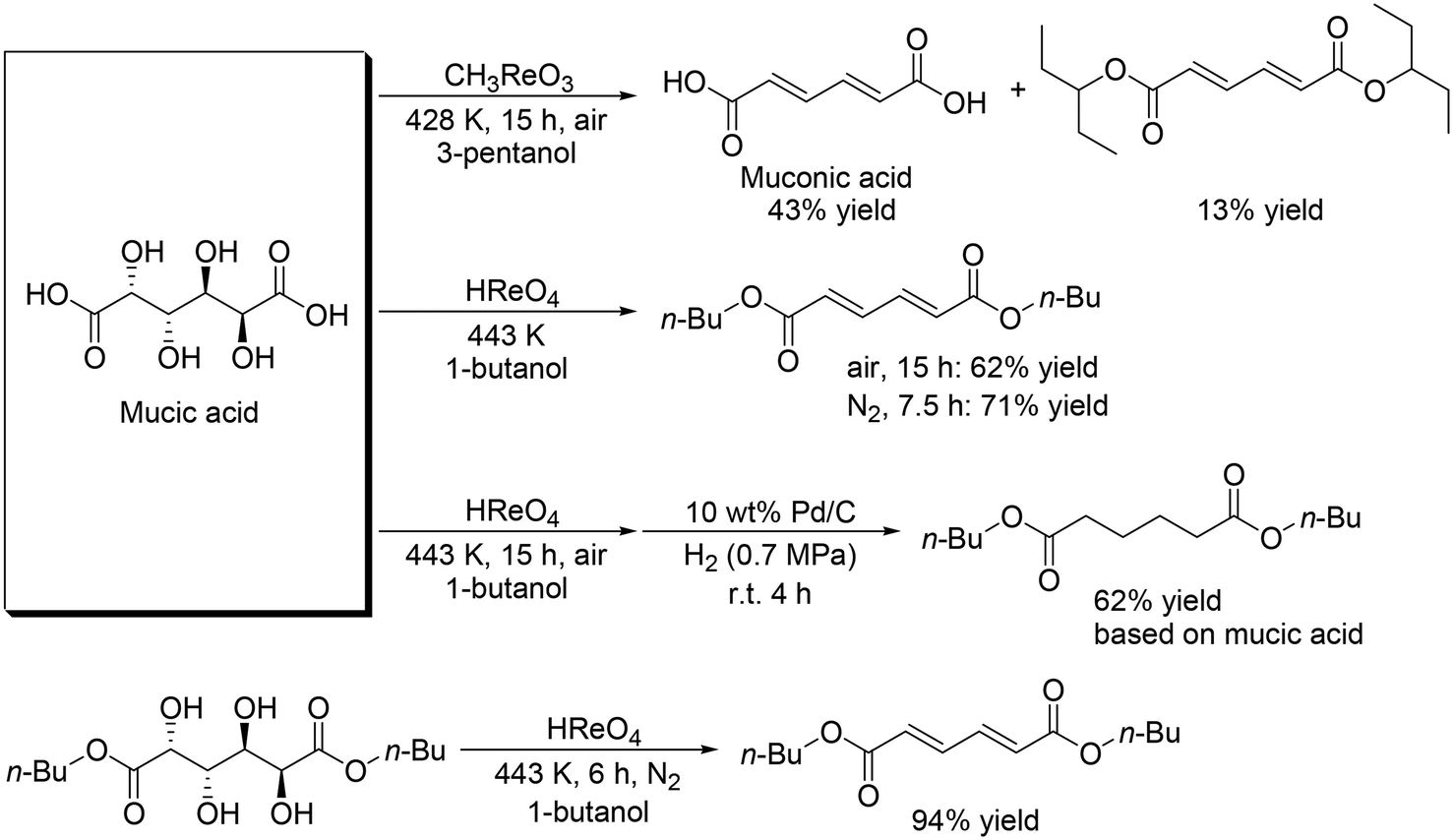
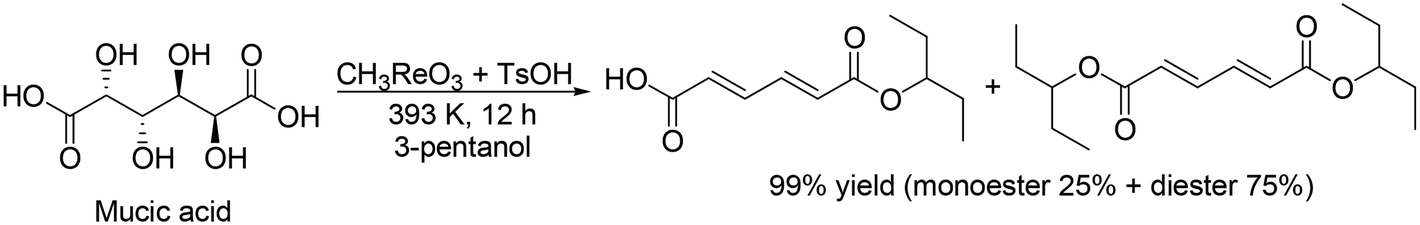
Toste and Shiramizu presented the first example for the transformation of aldaric acids, and they demonstrated that CH3ReO3 was an effective catalyst for the conversion of mucic acid, which can be obtained by the oxidation of galactose, to muconic acid and its diester (Scheme 18) with 3-pentanol as a reducing agent.30 43% yield of muconic acid and 13% yield of its ester were obtained by using CH3ReO3 catalyst at 428 K under air conditions. By changing the reaction conditions (CH3ReO3 → HReO4, 428 K → 443 K, 3-pentanol → 1-butanol, air → N2), a high yield of the target ester (71%) was obtained. Moreover, one-pot synthesis of dibutyl adipate was also achieved by the combination of DODH with a HReO4 catalyst system and hydrogenation with Pd/C and H2. They also showed that HReO4 was effective for DODH of mucic acid dibutyl ester to provide the corresponding DODH product in a high yield of 94%.
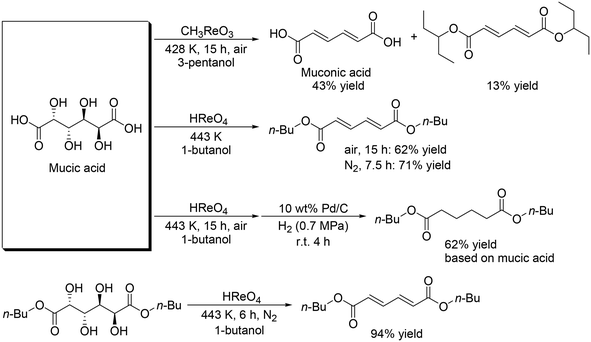 |
| | Scheme 18 DODH + (hydrogenation (HG)) of mucic acid and its ester.30 | |
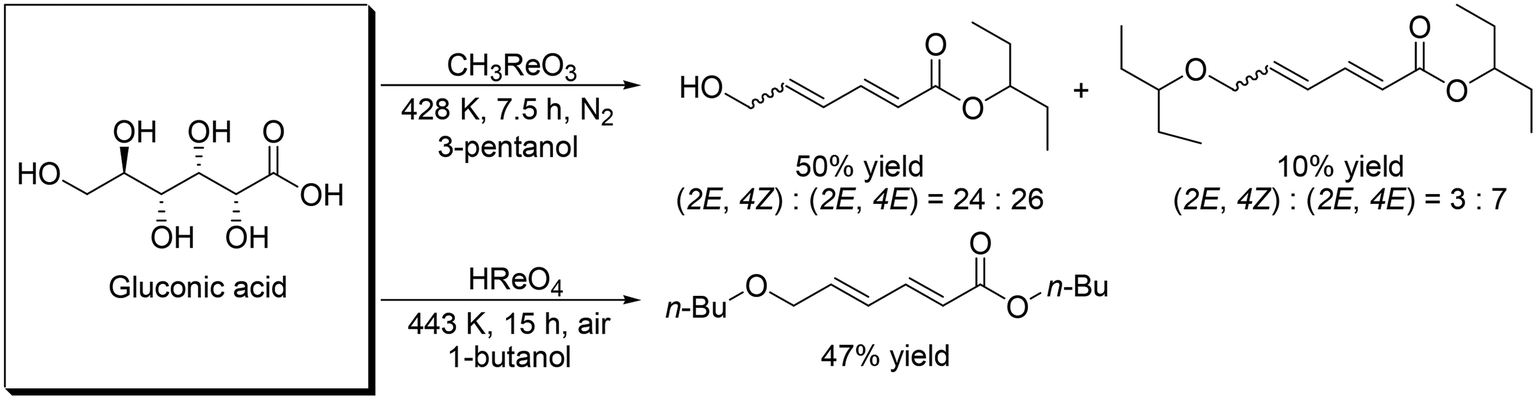
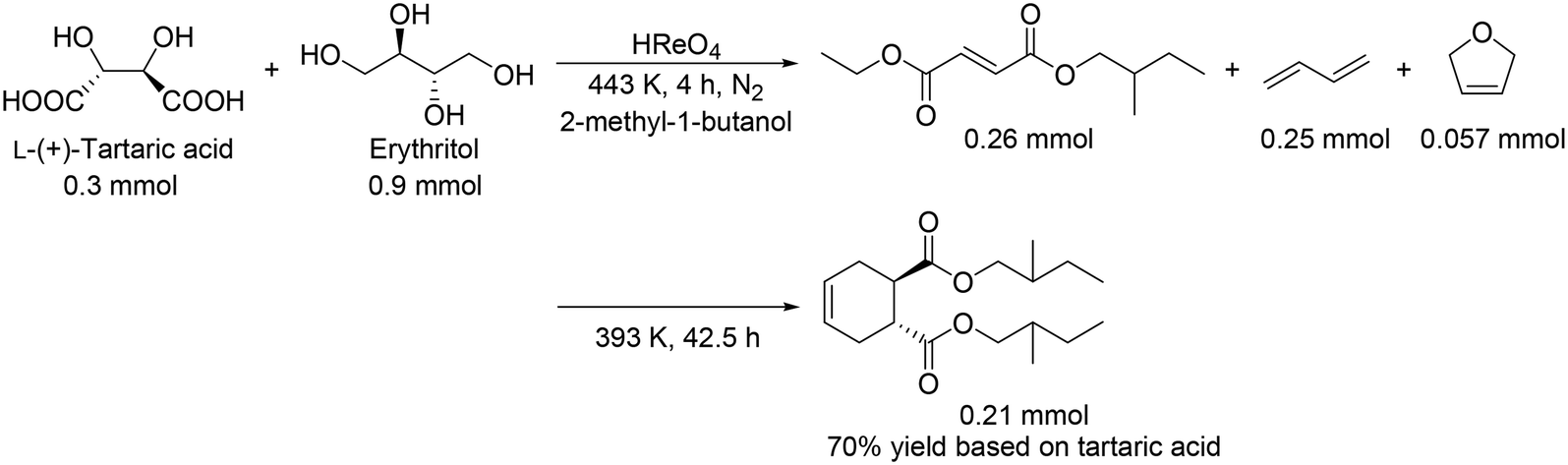
These catalyst systems were applied to the DODH of D-gluconic acid, one of the aldonic acids (Scheme 19).30 In the case of the CH3ReO3 catalyst, the DODH product was obtained in 50% yield ((2E, 4Z):(2E, 4E) = 24:26) with the corresponding ester in 10% yield ((2E, 4Z):(2E, 4E) = 3:7). In the case of the HReO4 catalyst, only (2E, 4E)-DODH product was obtained in 47% yield. Moreover, they demonstrated the tandem synthesis of 4-cyclohexene-1,2-dicarboxylic acid diester from L-(+)-tartaric acid and erythritol via DODH of L-(+)-tartaric acid and erythritol, and Diels–Alder reaction of the DODH products (Scheme 20) in one pot. L-(+)-Tartaric acid and erythritol were transformed to the corresponding DODH diester and butadiene, respectively, by DODH reaction with HReO4, and then the diester and butadiene were condensed to provide 4-cyclohexene-1,2-dicarboxylic acid in a high yield of 70% based on L-(+)-tartaric acid.
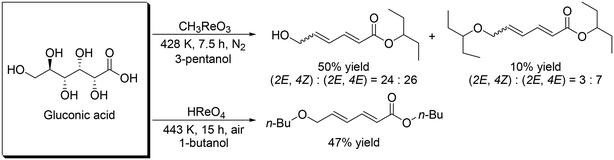 |
| | Scheme 19 DODH of gluconic acid.30 | |
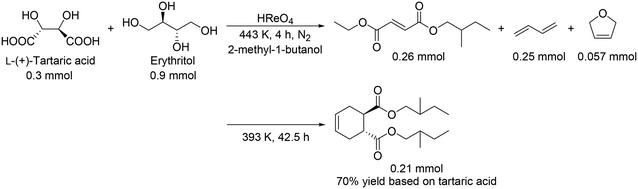 |
| | Scheme 20 One-pot synthesis of 4-cyclohexene-1,2-dicarboxylic acid diester by the combination of DODH reaction and Diels–Alder reaction.30 | |
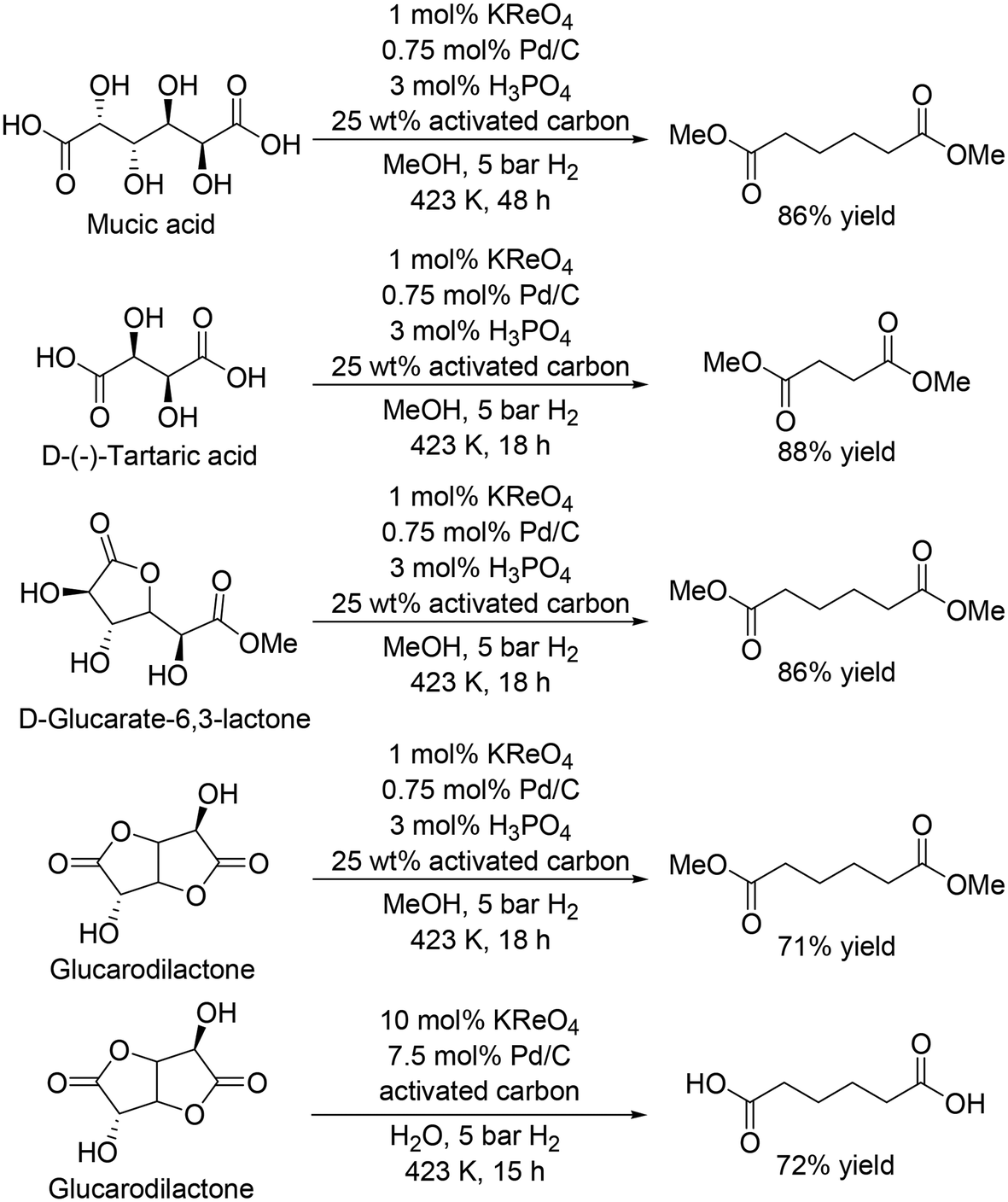
The same group recently reported that a combination catalyst of KReO4 and Pd/C with hydrogen gas as a reductant was effective for the synthesis of dicarboxylic acid methyl esters from biomass-derived dicarboxylic acids and esters, and also found that addition of H3PO4 and activated carbon improved the activity of the catalyst system (Scheme 21).31 Mucic acid and D-(−)-tartaric acid were converted to the dimethyl adipate and dimethyl succinate in 86% and 88% yield, respectively. D-Glucarate-6,3-lactone was also converted to dimethyl adipate in 86% yield. The catalyst system was applicable to the DODH and hydrogenation of glucarodilactone, affording the dimethyl adipate in 72% yield under similar conditions. Moreover, the authors used water as a solvent for the reaction and obtained adipic acid directly in 71% yield, while a large amount of KReO4 and Pd/C was necessary. The low stability of CH3ReO3 and low solubility of KReO4 limited the usage of water as a solvent, which will lead to difficulty in direct synthesis of free carboxylic acids.
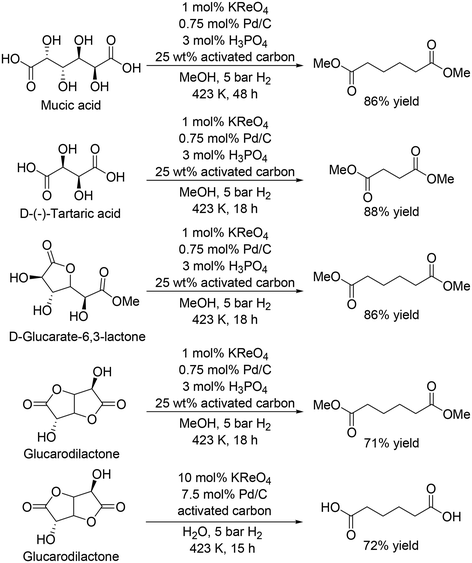 |
| | Scheme 21 DODH and hydrogenation of sugar acid and esters with KReO4 and Pd/C catalyst.31 | |
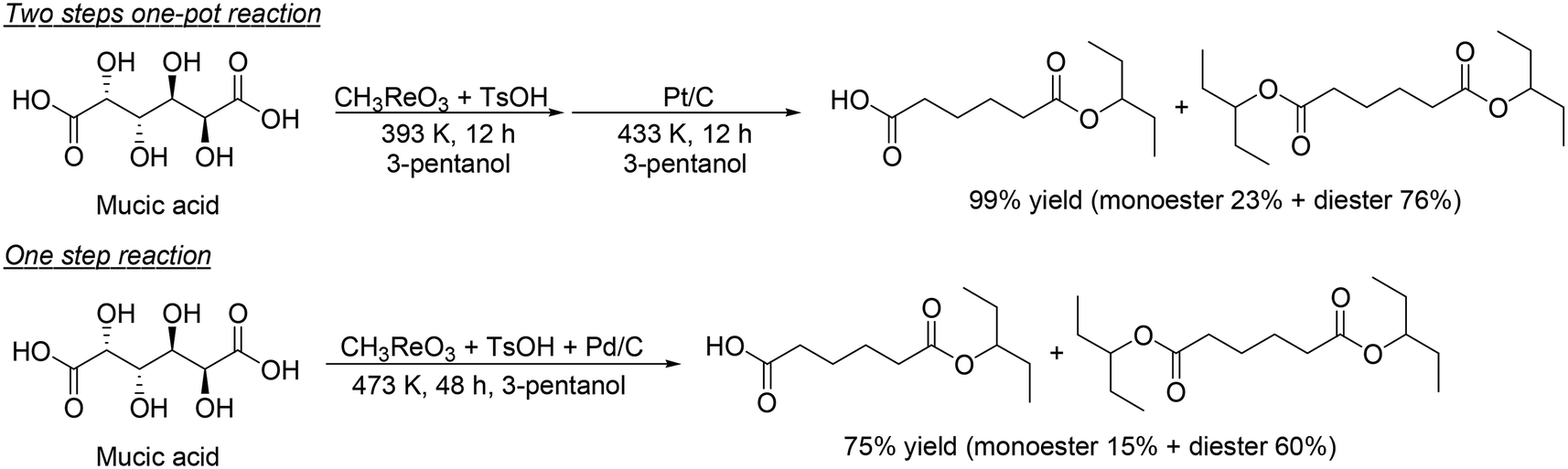
H. Su and Y. Zhang and co-workers developed an optimized process for two-step one-pot transformation of mucic acid to adipic acid esters by CH3ReO3-catalyzed DODH reaction and consecutive Pt/C-catalyzed hydrogenation (HG) reaction.32 They optimized the reactions separately; 393 K, a comparatively low reaction temperature, and combination of TsOH as an acid co-catalyst with CH3ReO3 were effective for the DODH reaction of mucic acid (Scheme 22) and they also found that Pt/C catalyst and 473 K, a comparatively high temperature, were efficient for the hydrogenation of produced DODH esters. A two-step one-pot reaction of DODH and HG of mucic acid composed of DODH reaction with MTO and TsOH catalysts and HG reaction with Pt/C catalyst quantitatively provided the target adipic diesters (99%) (Scheme 23, top). Moreover, they demonstrated a one-step reaction from mucic acid to adipic esters with MTO, TsOH and Pt/C at 473 K with 3-pentanol, giving the target product in 75% yield (Scheme 23, bottom).
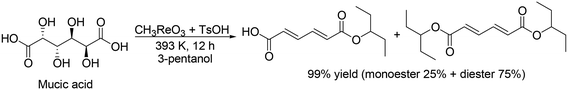 |
| | Scheme 22 DODH of mucic acid to the target esters with CH3ReO3 and TsOH catalysts.32 | |
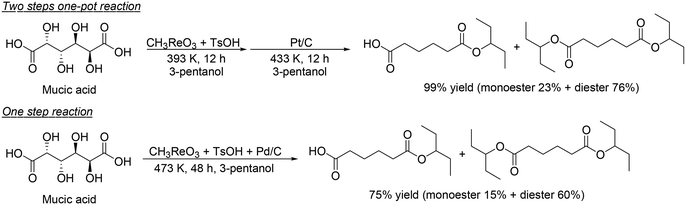 |
| | Scheme 23 Two-step one-pot reaction and one-step reaction for transformation of mucic acid to adipic esters.32 | |
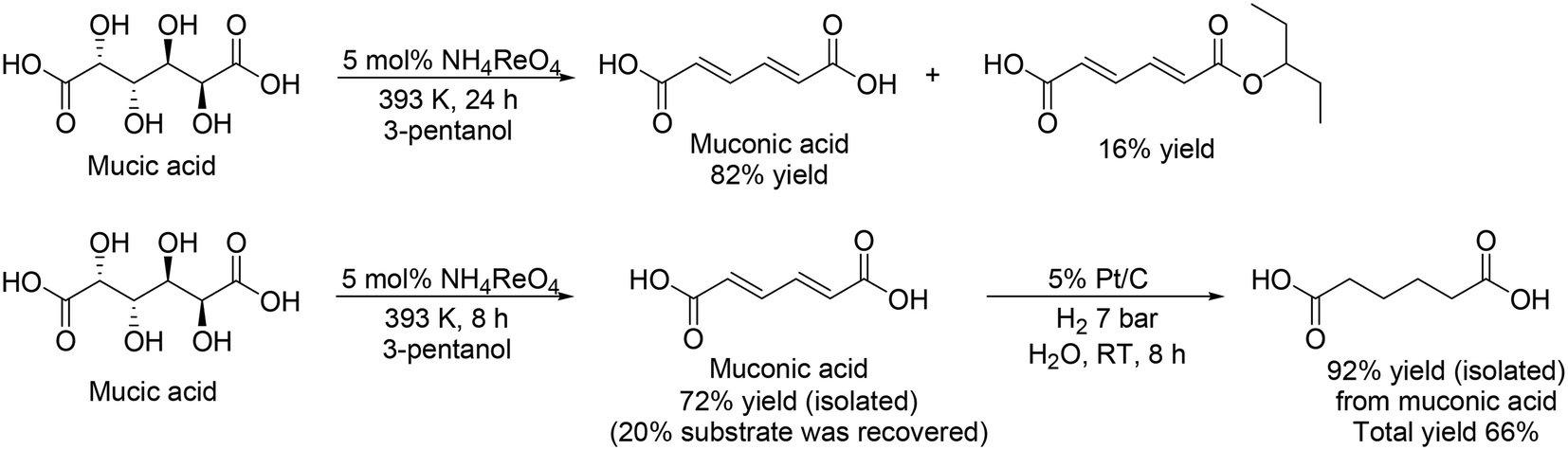
The same group and H. Zhao's group investigated Re catalysts for the DODH of mucic acid without the formation of methyl ester because the target product, adipic acid, is in the carboxylic form.33 They found that NH4ReO4 was an effective catalyst for the selective formation of muconic acid among various Re complexes, providing 82% yield of muconic acid with 16% monoester (Scheme 24, top), which was explained by the low Lewis acidity of NH4ReO4 compared with other Re complexes. From the time-course, they also found that muconic acid was obtained in quite high selectivity (∼98%) below a moderate conversion level (∼75%), and demonstrated the synthesis of adipic acid from mucic acid by the combination of DODH with NH4ReO4 catalyst and hydrogenation with Pt/C catalyst (Scheme 24, bottom). Moreover, they also demonstrated the synthesis of adipic acid from sugar beet residue by biological transformation of sugar beet residue to mucic acid with the enzyme Viscozyme® L and biocatalyst of engineered Escherichia coli strain, and chemical transformation of mucic acid to adipic acid with NH4ReO4 and Pt/C catalysts, which provided 8.4% yield of adipic acid based on the initial sugar beet residue.
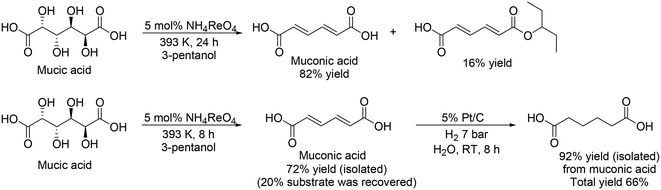 |
| | Scheme 24 DODH and DODH + HG of mucic acid with NH4ReO4 and NH4ReO4 + Pt/C catalysts.33 | |
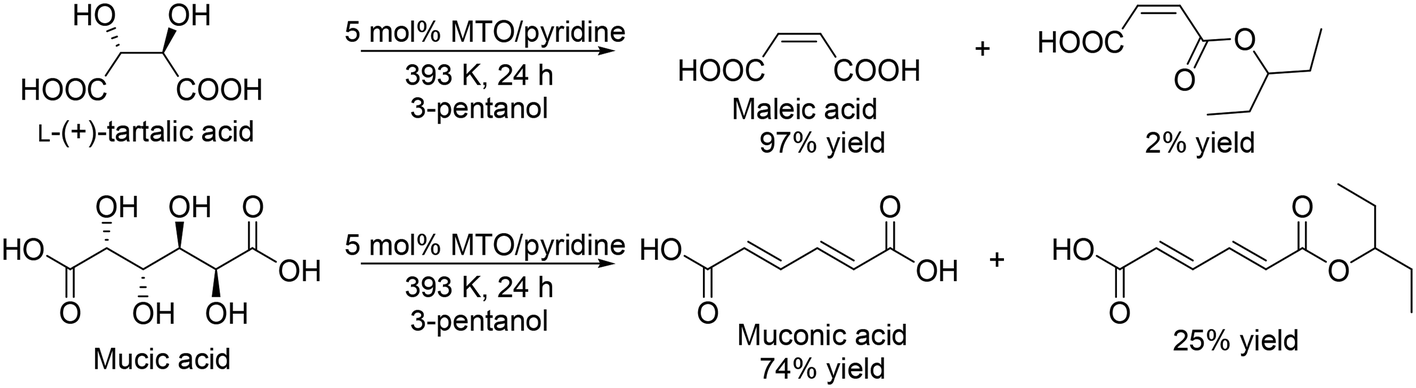
Y. Zhang's group developed a Re catalyst system for the synthesis of free carboxylic acids from sugar carboxylic acids.34 They investigated the addition of base modifiers to MTO in the DODH of L-(+)-tartaric acid and found that pyridine was the most effective additive for the selective formation of maleic acid, although the activity seems to be decreased by the addition of pyridine. The yield of maleic acid was drastically improved from 78% (no pyridine additive) to 97% (Scheme 25, top). The performance seems to be similar to that with NH4ReO4 catalyst without additives as mentioned above (96% yield at 24 h under the same reaction conditions). A combination of MTO and pyridine was applied to the DODH of mucic acid, giving muconic acid in a high yield of 74% with 25% of the monoester (Scheme 25, bottom).
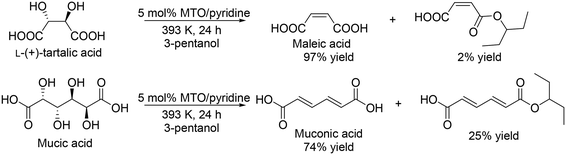 |
| | Scheme 25 DODH of L-(+)-tartaric acid and mucic acid with CH3ReO3 and pyridine.34 | |
Moreover, they studied the immobilization of CH3ReO3 and HReO4 on N-containing polymers such as PVP (poly(4-vinylpyridine)), P-Bn (poly-benzylamine) and PMF (poly(melamine-formaldehyde)) and found that 48%HReO4/P-Bn was the most effective for DODH of L-(+)-tartaric acid and 90% yield of free maleic acid with 96% total yield of maleic acid and the monoester. The problem of the immobilized HReO4/P-Bn catalyst is reusability, and the conversion was drastically decreased, which is due to leaching of Re species into the solution.
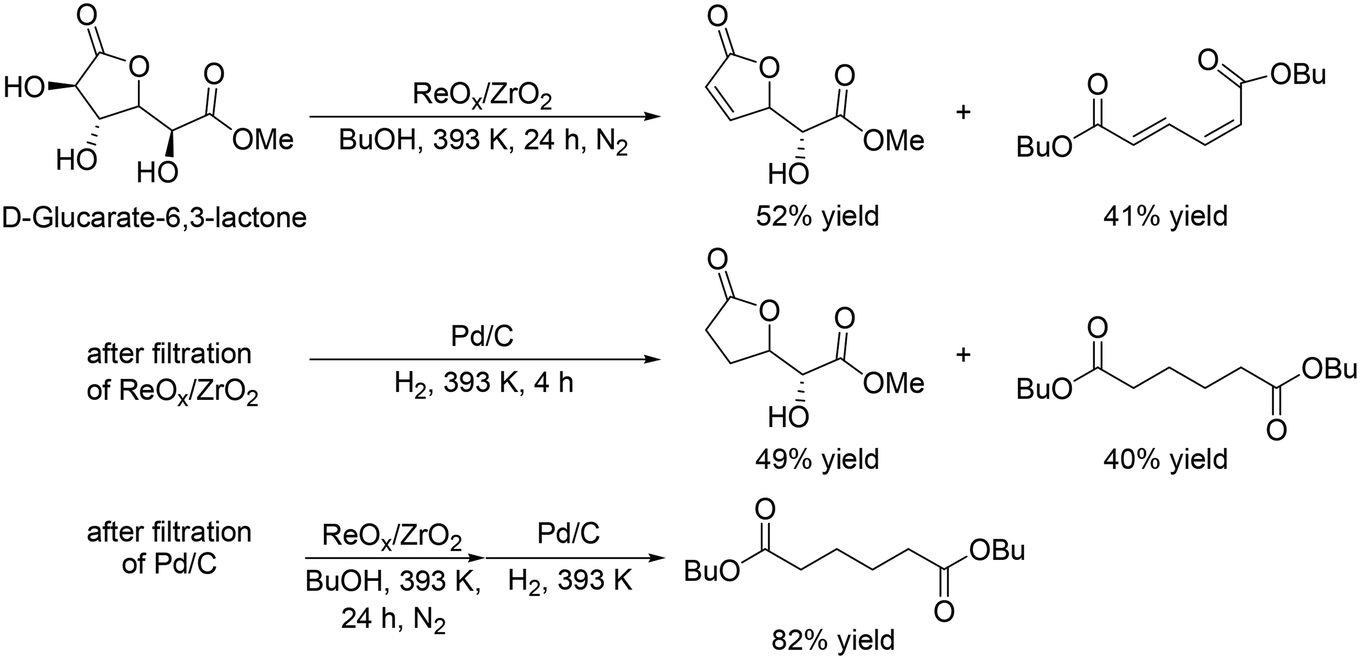
Y. Wang and co-workers reported that ReOx/ZrO2 was an effective heterogeneous catalyst for the DODH of D-glucaric acid-1,4-lactone,35 providing a high total yield of DODH products (93% at 24 h, Scheme 26, top); however, a high yield of the dibutyl hexa-2,4-dienedioate was not obtained even when the reaction time was prolonged (74%, 48 h), which is due to side reactions such as polymerization of the products and reaction of the products with 1-butanol. They demonstrated high yield synthesis of dibutyl adipate with a combination of ReOx/ZrO2 and Pd/C catalysts, and DODH and hydrogenation reactions were conducted two times (Scheme 26). The final yield of dibutyl adipate was 82%.
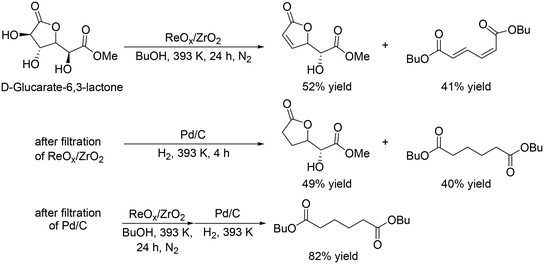 |
| | Scheme 26 DODH of D-glucarate-6,3-lactone with ReOx/ZrO2 catalyst and stepwise synthesis of dibutyl adipate from D-glucarate-6,3-lactone ReOx/ZrO2 and Pd/C catalysts.35 | |
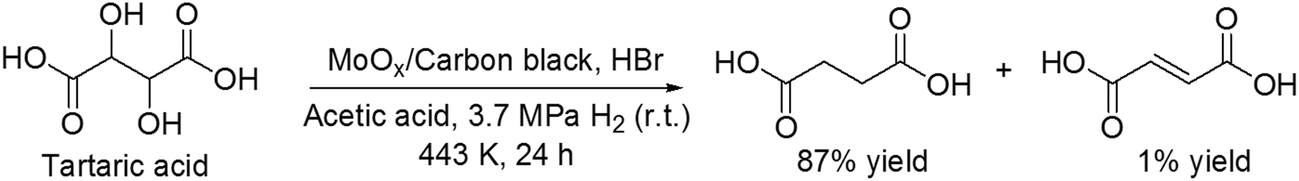
Recently, Vlachos and co-workers reported another the DODH reaction of tartaric acid to succinic acid with MoOx/carbon black catalyst and HBr in acetic acid as the solvent at 3.7 MPa H2 pressure (at r.t.) and 443 K, giving succinic acid in a high yield of 87% (Scheme 27).36 The reaction path was proposed to be composed of stepwise dissociation of C–O bonds (C–OH or C–OAc bond) to form fumaric acid and hydrogenation of fumaric acid to succinic acid. The valence of the MoOx species was estimated to be +4 to 0 based on the catalyst characterization (TPR, XRD, XPS and XAS), which played important roles in dissociation of C–O bonds and hydrogenation of fumaric acid.
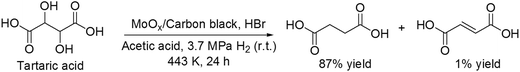 |
| | Scheme 27 Transformation of tartaric acid with MoOx/carbon black and HBr.36 | |
(iii) DODH (+HG) of methyl glycosides to dideoxy methyl glycosides
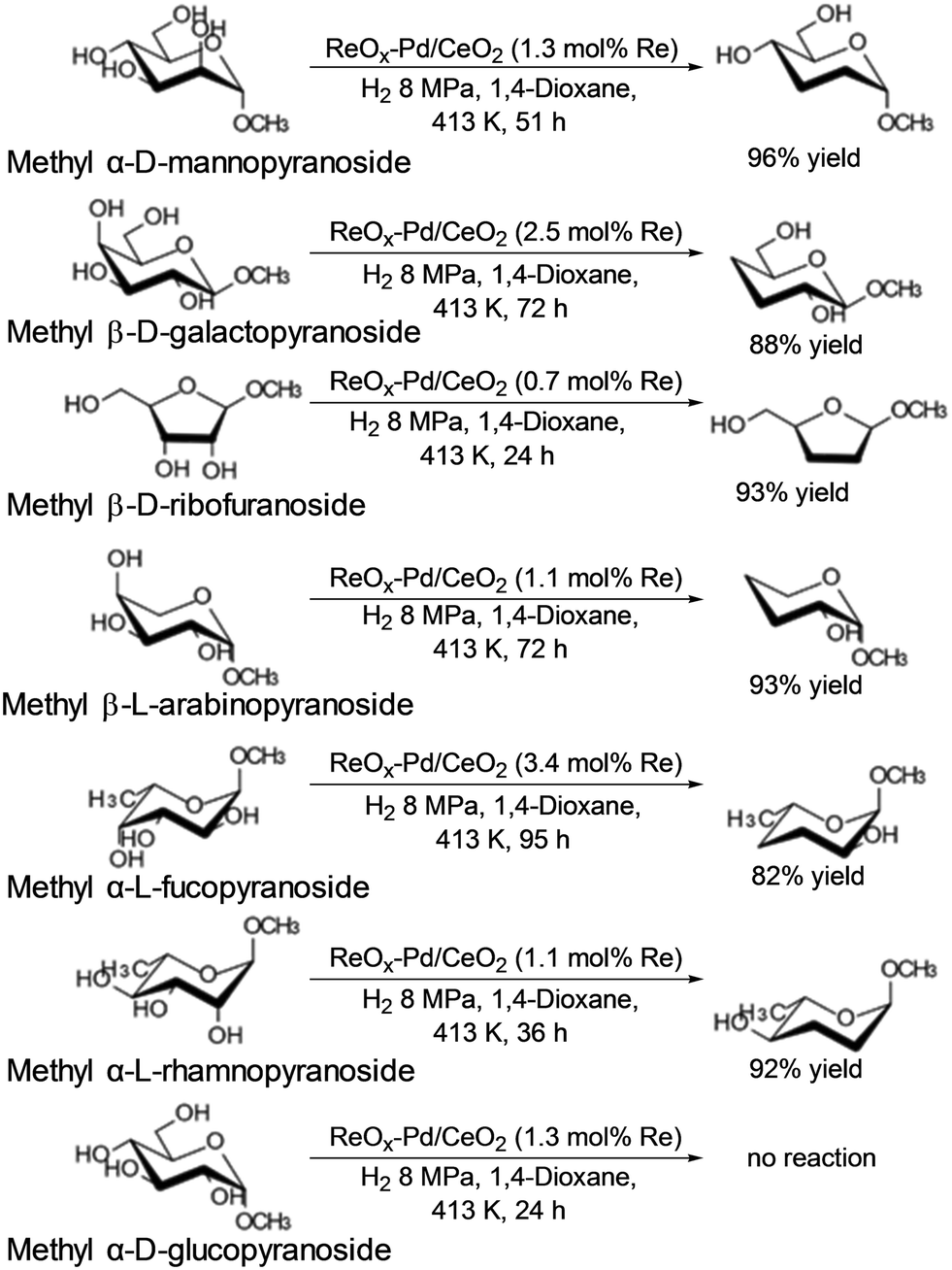
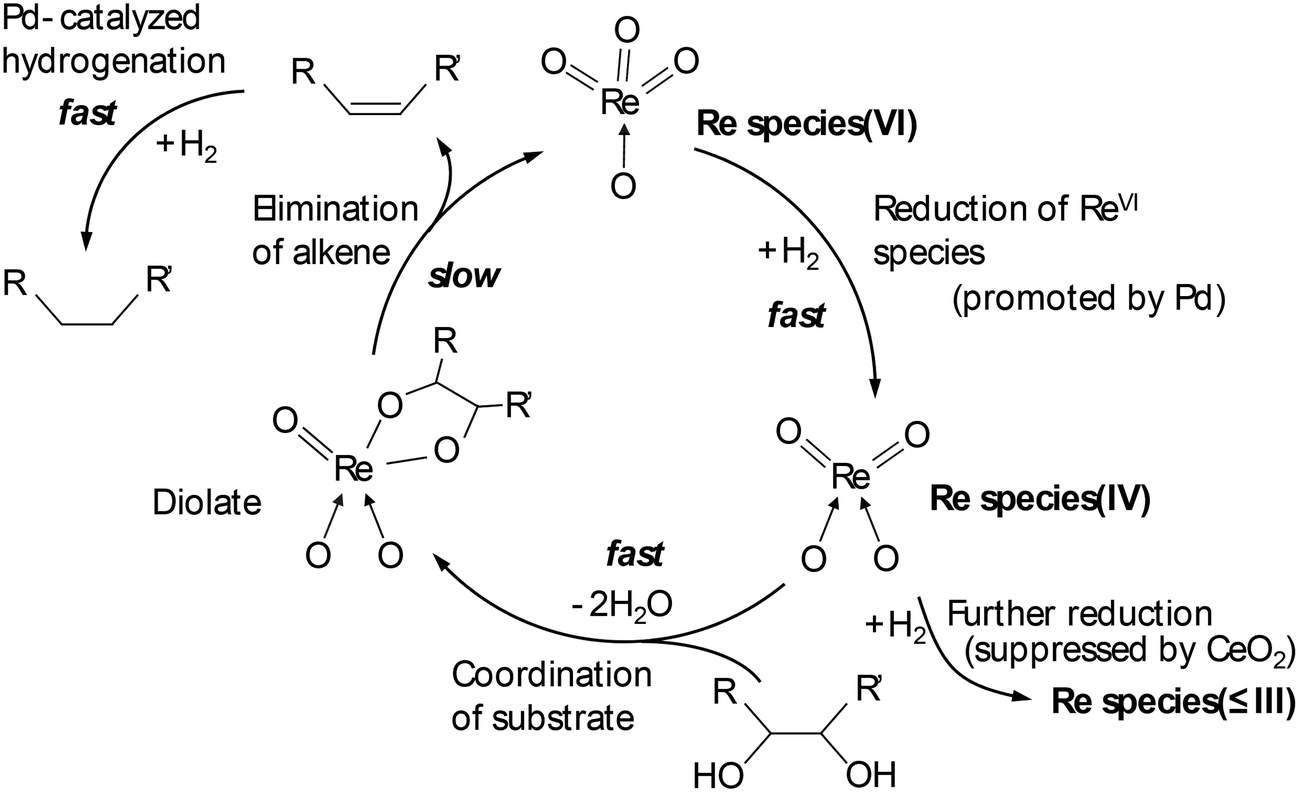
Tomishige and co-workers reported that ReOx–Pd/CeO2 was an effective heterogeneous catalyst for DODH + HG of methyl glycosides having cis-vicinal OH groups, providing the corresponding dideoxy methyl glycosides in high yields (82–96%) by selective removal of the cis-vicinal OH groups.37 The results were based on the finding that the ReOx–Pd/CeO2 catalyst was effective for the DODH + HG of vicinal OH groups in polyols such as 1,4-anhydroerythritol, glycerol and so on.38 Various methyl glycosides having cis-vicinal OH groups were converted to the dideoxy methyl glycosides; however, methyl α-D-glucoside, which has only trans-vicinal OH groups, did not react (Scheme 28). The reactivity difference between cis-vicinal OH groups and trans-vicinal OH groups can account for the high selectivity and yield for the target products. Based on kinetic studies and catalyst characterization (XPS, EXAFS, XRD, TEM, and so on), the active species of the ReOx–Pd/CeO2 catalyst is proposed to be an isolated and reduced Re4+, which is formed through the reduction of Re7+ species to Re4+ by Pd-activated H2 species. The DODH reaction proceeded by assistance of the redox between Re6+ and Re4+, and the DODH product (unsaturated dideoxy product) is produced. The produced DODH product is hydrogenated by Pd metal species, giving the target dideoxy product. The catalytic cycle is shown in Scheme 29. The difference between homogeneous Re complex catalysts and ReOx–Pd/CeO2 catalyst is the valences in the catalytic cycle (homogeneous Re complex catalysts: typically Re7+ and Re5+, ReOx–Pd/CeO2 catalyst: Re6+ and Re4+), and overreduction of Re4+ species to Re0 species seems to be suppressed by the CeO2 support.38b
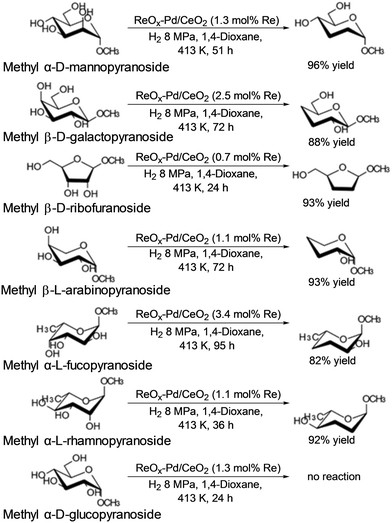 |
| | Scheme 28 DODH and HG of methyl glycosides with ReOx–Pd/CeO2 catalyst.37 | |
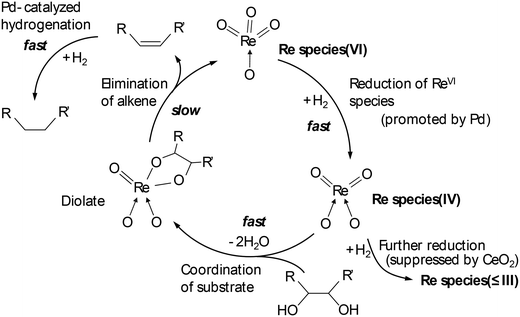 |
| | Scheme 29 Catalytic cycle of DODH and HG of polyols with ReOx–Pd/CeO2 catalyst.38 | |
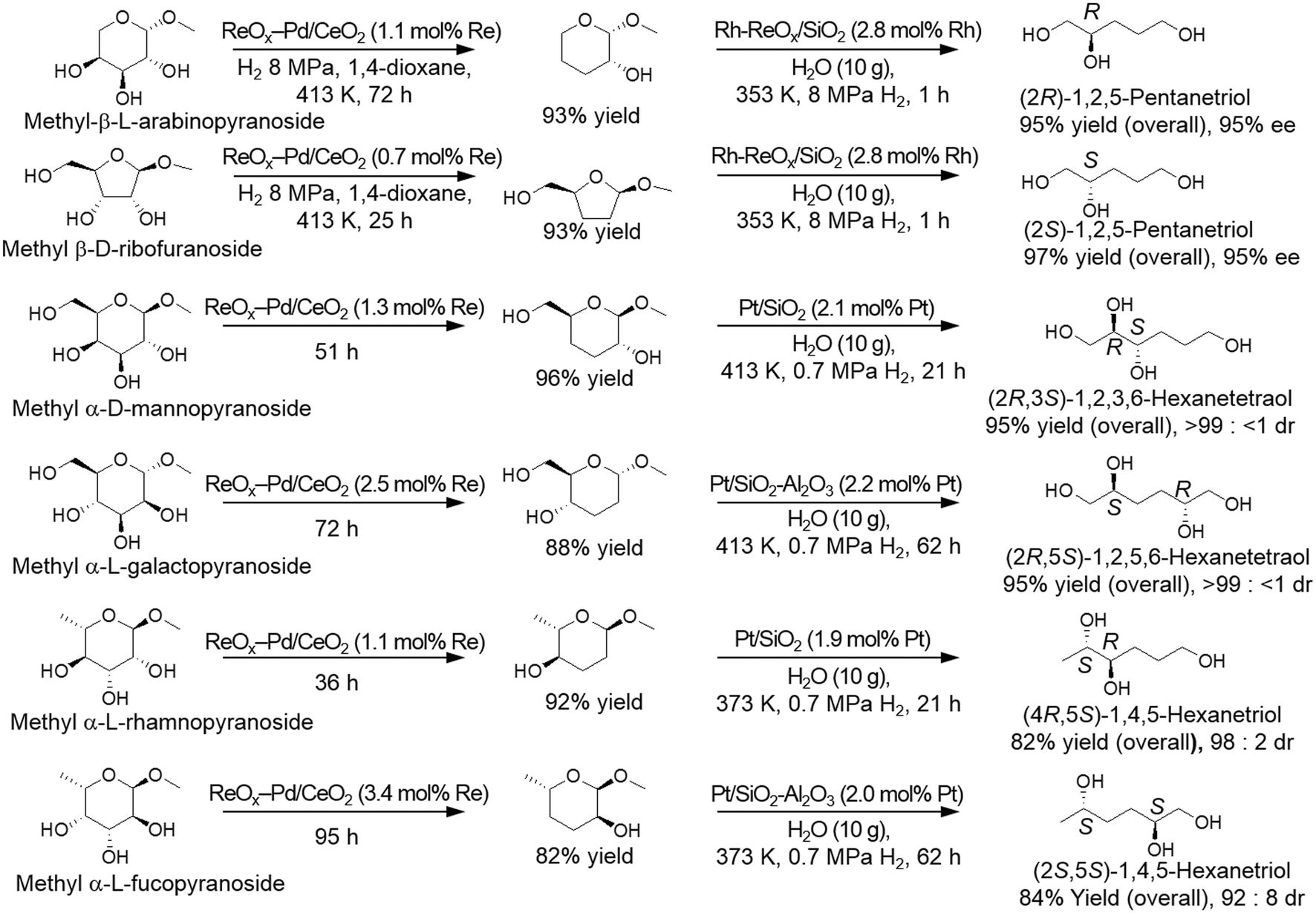
The produced dideoxy methyl glycosides were furthermore transformed to the chiral polyols by a combination of hydrolysis and hydrogenation reactions with hydrogenation catalysts such as Rh–ReOx/SiO2 catalyst and Pt-supported catalysts (Scheme 30). It was demonstrated that high yields of the target chiral polyols (82–97%) were obtained while maintaining the original chiral structures (92 ≥ 99% ee).37,39
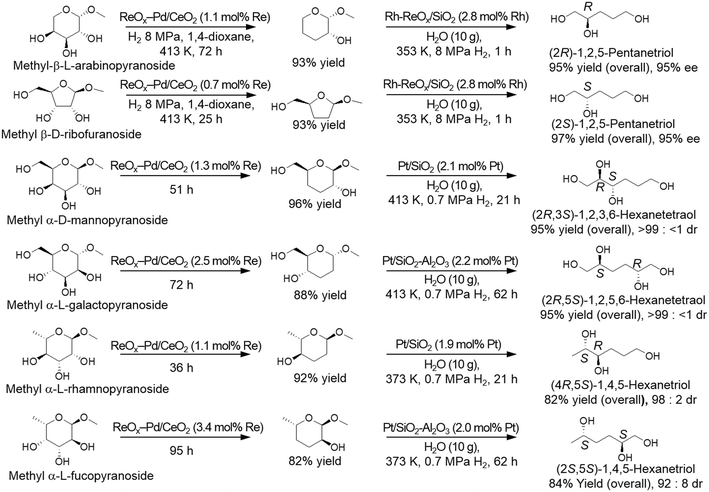 |
| | Scheme 30 Transformation of methyl glycosides to the corresponding chiral polyols by DODH, HG, hydrolysis and HG with ReOx–Pd/CeO2 and hydrogenation catalysts.37,39 | |
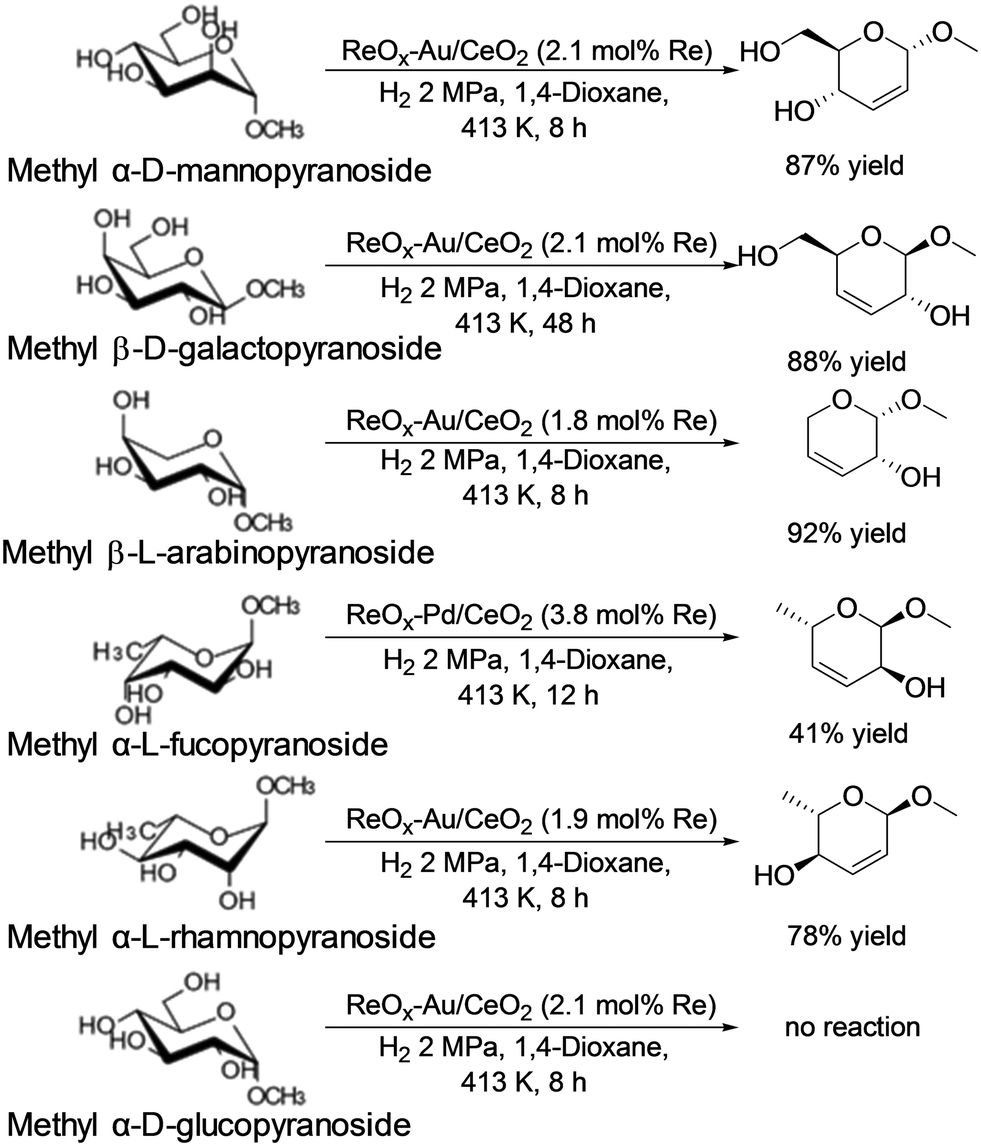
In addition, they also reported that ReOx–Au/CeO2 acted as an effective heterogeneous catalyst for the DODH of methyl glycosides having cis-vicinal OH groups to the corresponding unsaturated dideoxy methyl glycosides (Scheme 31).40 The catalyst system was based on the reports on DODH of vicinal OH groups in polyols to the corresponding olefins with the ReOx–Au/CeO2 catalyst41 and was furthermore applied to the transformation of 1,4-anhydroerythritol to 1,4-butanediol.42 High yields of the DODH products were obtained; however, the reaction of methyl β-D-ribofuranoside is difficult, which will be due to the low stability of the produced unsaturated DODH product from methyl β-D-ribofuranoside. Like the above case, methyl α-D-glucoside showed almost no conversion for the reaction. The active species of the ReOx–Au/CeO2 catalyst for the DODH reaction is the same as that of the above case; isolated and reduced Re4+, and low activity of Au species for hydrogenation of olefins resulted in high yield synthesis of DODH products. Also, the reactivity tendency of methyl glycosides is similar to the above case. Therefore, the active species will be the same as that of the ReOx–Pd/CeO2 catalyst.
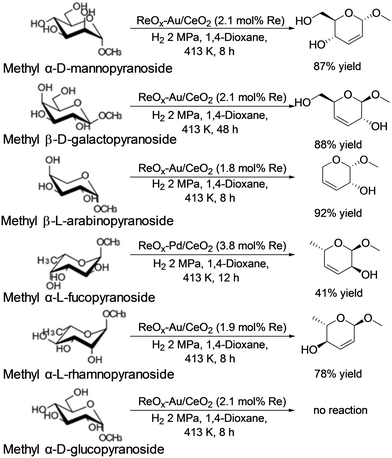 |
| | Scheme 31 DODH reaction of methyl glycosides with the ReOx–Au/CeO2 catalyst.40 | |
The important points of heterogeneous ReOx/CeO2-based catalysts (ReOx–Pd/CeO2 and ReOx–Au/CeO2 catalysts) are low-temperature transformation (typically 413 K) compared with the case of Re complex catalysts and use of H2 gas as a reducing agent. Generally, sugar derivatives decompose or polymerize at high temperature (≥423 K), which is the large hurdle for low-activity catalysts such as Re complex catalysts. In contrast, heterogeneous ReOx/CeO2-based catalysts had much higher activity per Re atom than homogeneous Re catalysts and made reactions at low temperature (∼413 K) possible, which substantiated the transformation of sugar derivatives without decomposition. Moreover, the use of H2 as a reducing agent instead of secondary alcohols decreased the side reactions of secondary alcohols with substrates or products.
In the DODH reaction with ReOx–Pd/CeO2 and ReOx–Au/CeO2 catalysts, the high catalyst stability and reusability were confirmed,37,40 and they were also confirmed in the case of the ReOx/ZrO2 catalyst.35 However, the reusability of the MoOx/carbon black and HBr catalyst system was low due to the agglomeration of MoOx species.36 Detailed kinetic studies on the effect of reaction parameters such as H2 pressure and substrate concentration on the DODH of sugar derivatives over ReOx–Pd/CeO2 and ReOx–Au/CeO2 catalysts have not been investigated, although kinetic studies on the DODH of polyols such as diols and triols were already reported.38,41 The DODH reaction mechanism of sugar derivatives will be complicated because of the multiple functional groups and the variety of configurations, and hence more detailed kinetic studies and DFT calculations are required. As discussed in the reaction mechanism, the active species, Re4+ species, over ReOx/CeO2-based catalysts is different from that of homogeneous oxo-rhenium catalysts; however, the details for the formation of the Re4+ species over ReOx/CeO2 catalysts is unclear, and further investigation such as DFT calculations is necessary.
Combination of dehydration and hydrogenation (and/or hydrogenation) via formation of levoglucosenone
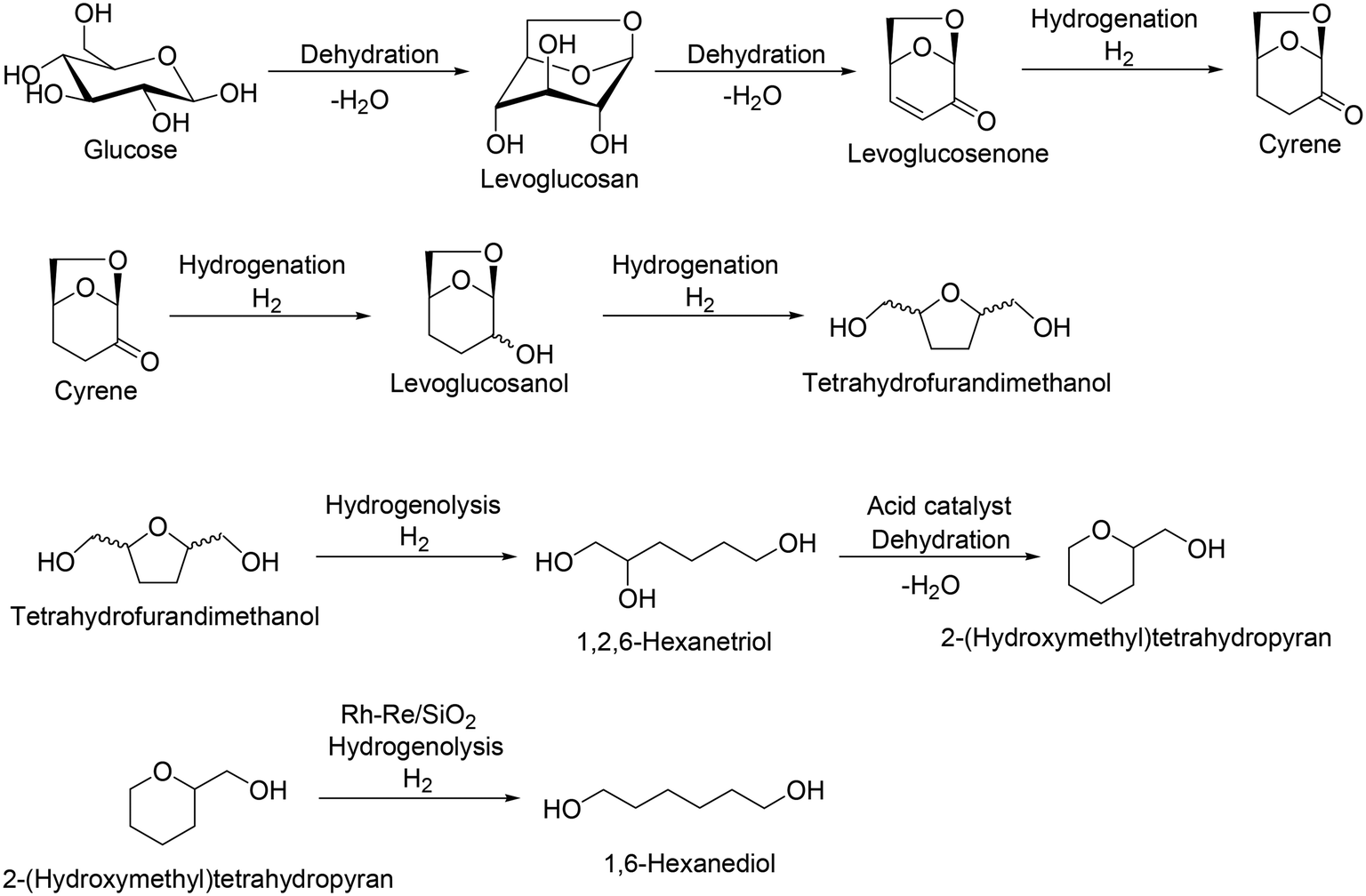
Levoglucosan and levoglucosenone are partially dehydrated products of sugars, which can be obtained by pyrolysis of cellulose with acid catalysts.43 Levoglucosenone is a promising platform chemical from cellulose, and various transformation routes from levoglucosenone to valuable chemicals such as pharmaceuticals and Cyrene® have been developed.44 Recently, the synthesis of tetrahydrofurandimethanol from levoglucosenone in water solvent with Pt/C catalyst was reported by DuPont (Scheme 32).45 Also, the transformation of tetrahydrofurandimethanol to 1,6-hexanediol using a combination of Rh–Re/SiO2 and Nafion SAC-13 catalysts was also reported by H. J. Heeres, J. G. de Vries and co-workers.46 The selective hydrogenolysis over Rh–Re/SiO2 catalyst is based on the result that Rh–ReOx and Rh–MoOx-supported catalysts were effective heterogeneous catalysts for the hydrogenolysis of the C–O ether bond in tetrahydrofurfuryl alcohol and tetrahydrofurandimethanol.47 Considering these results, combination reactions of levoglucosenone to tetrahydrofurandimethanol and tetrahydrofurandimethanol to 1,6-hexanediol can be regarded as a new route for 1,6-hexanediol synthesis.
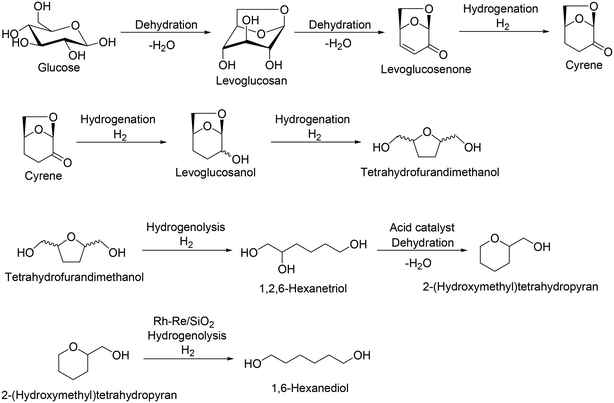 |
| | Scheme 32 Transformation route from glucose to tetrahydrofurandimethanol via formation of levoglucosenone and from tetrahydrofurandimethanol to 1,6-hexanediol.43–46 | |
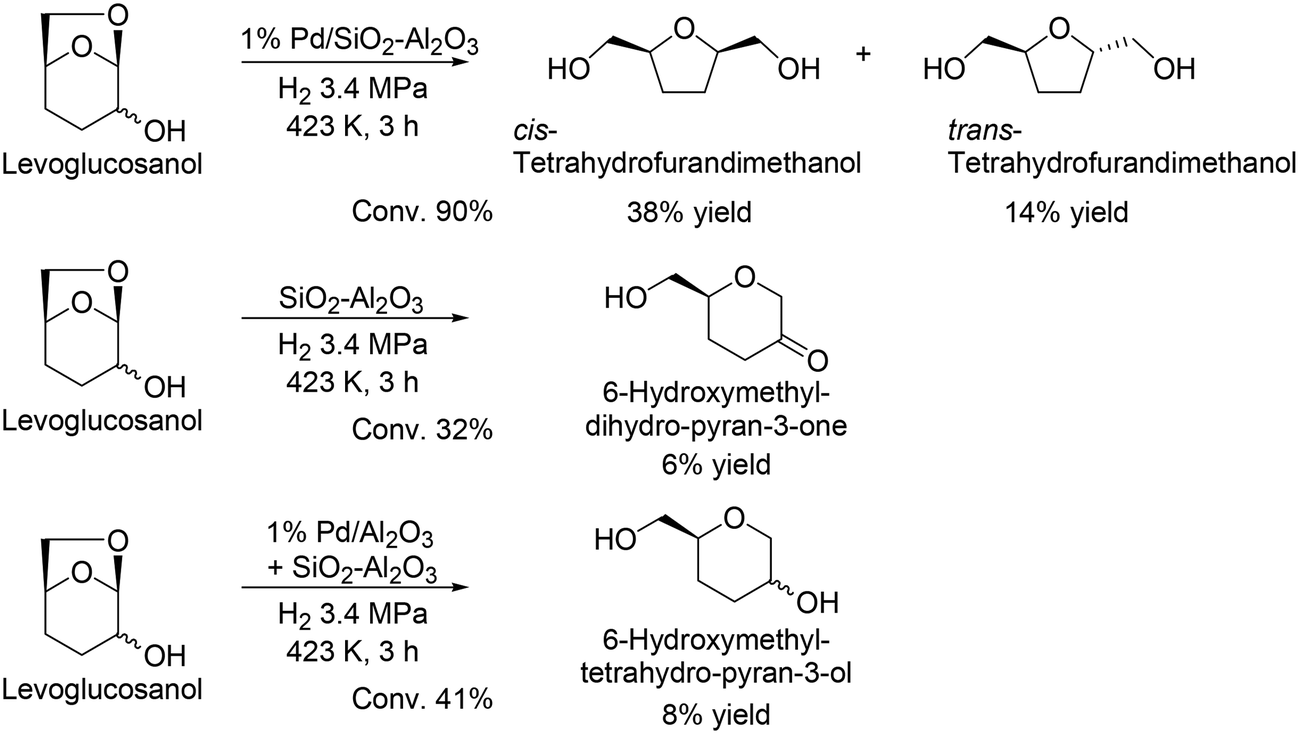
G. W. Huber and co-workers reported another route for the production of chiral compounds from levoglucosenone, and cis-tetrahydrofurandimethanol and trans-tetrahydrofurandimethanol were obtained in moderate yields (38% and 14%, respectively) by using Pd/SiO2–Al2O3 catalyst (Scheme 33).48 Moreover, they demonstrated the formation of 6-hydroxymethyl-dihydro-pyran-3-one from levoglucosanol by hydration and dehydration with the acid catalyst SiO2–Al2O3, and furthermore reacted levoglucosanol with the combination catalyst of Pd/Al2O3 and SiO2–Al2O3 to provide 6-hydroxymethyltetrahydro-pyran-3-ol. The reusability of the catalyst and detailed kinetics were not discussed in the literature, and further investigation is needed for improvement of the reaction route, particularly yield and selectivity.
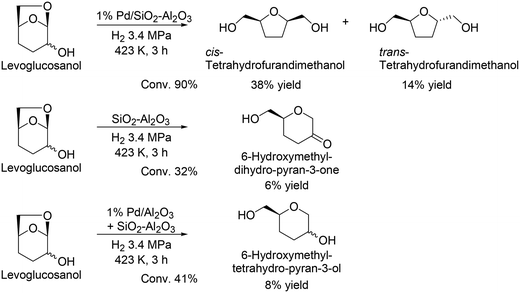 |
| | Scheme 33 Transformation of levoglucosanol to chiral products.48 | |
Summary and outlook
In this perspective, we introduced recent developments for the catalytic transformation of sugar derivatives to valuable chemicals, particularly focusing on non-furfural routes. These transformation routes were based on various key reactions such as hydrogenolysis, reduction with borane catalyst + silane reducing agents, deoxydehydration (+hydrogenation (HG)), and a combination of dehydration and hydrogenation (and/or hydrogenolysis). These routes have high potential for production of chiral chemicals from sugar derivatives; however, each route seems to have many hurdles such as many reaction steps, large amount of reagents, low yields and narrow scope and so on for industrialization. The points to be improved in hydrogenolysis include severe reaction conditions for the transformation of cellulose and selectivity to partial hydrogenolysis. Milder reaction conditions such as temperature below 473 K and pressure below 1 MPa H2 will be desirable for industrialization, and establishment of catalyst systems for selective removal of OH groups is required to expand the application. The problems of the partial reduction with borane catalyst and silane reducing agents are low yields (low selectivities) of the target products and protection of OH groups with silyl groups. Development of catalyst systems without protection of OH groups in sugar derivatives as well as improvement of product yields is highly anticipated in terms of economic and environmental viewpoints. The future improvement points in DODH (+HG) are yield for the target products, particularly olefins, scope of substrates and replacement of expensive Re metal with affordable metals. Considering the low stability of olefins, highly active catalysts which can act at lower reaction temperature are desired to be developed. In addition, DODH catalysts such as Re-based catalysts can selectively transform the cis-vicinal OH groups in cyclic sugar derivatives; however, they cannot transform the trans-vicinal OH groups, leading to the narrow scope. Development of alternative effective high-valent metal oxide catalysts for ReOx such as MoOx, VOx, WOx, and so on is essential. The improvement points for combination of dehydration and hydrogenation (and/or hydrogenolysis) via formation of levoglucosenone include multi-step reactions and low yields for target products. The development of one-pot reaction systems with multi-functional catalysts or combination of some catalysts will be necessary to improve the yield and efficiency. In the future, development of effective catalysts, particularly effective heterogeneous catalysts for each route based on new catalyst designs is essential and sophisticated control of reaction conditions is also required. Early establishment of reaction routes for the synthesis of various chemicals including chiral chemicals from biomass-derived chemicals, in particular making the most use of the original structure of sugars, is desirable. Moreover, application of these catalyst systems to flow reactors will be essential for industrialization, and there are some reports on hydrogenolysis of sugar alcohols using a fixed-bed reactor;14a,b,49 however, there seems to be no report on the other reaction systems. Development of suitable catalysts for the flow reactors and the kinetic study are required in the future.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) A. Tursi, Biofuel Res. J., 2019, 22, 962–979 CrossRef;
(b) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. M. K. Boysen, Chem. – Eur. J., 2007, 13, 8648–8659 CrossRef CAS PubMed.
-
(a) M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef PubMed;
(b) Y. Li, Y. Liao, X. Cao, T. Wang, L. Ma, J. Long, Q. Liu and Y. Xua, Biomass Bioenergy, 2015, 74, 148–161 CrossRef CAS;
(c) L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS;
(d) D. M. Alonso, S. W. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC;
(e) L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Appl. Catal., B, 2015, 174–175, 225–243 CrossRef CAS;
(f) G. W. Huber, S. Ibora and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 2, 7393–7420 Search PubMed;
(g) J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC;
(h) H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC;
(i)
K. Tomishige, Y. Nakagawa and M. Tamura, Biofuels Biorefineries, 2017, vol. 7, pp. 343–373 Search PubMed;
(j) Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520–2530 CrossRef CAS PubMed.
-
(a) M. J. Climent, A. Corma and S. Ibora, Green Chem., 2014, 16, 516–547 RSC;
(b) G. G. Millán, S. Hellsten, J. Llorca, R. Luque, H. Sixta and A. M. Balu, ChemCatChem, 2019, 11, 2022–2042 CrossRef;
(c) Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS;
(d) P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318–346 CrossRef CAS;
(e) Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC;
(f) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed;
(g) X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS;
(h) R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- K. Tomishige, ChemSusChem, 2019, 12, 1115–1117 CrossRef CAS PubMed.
-
(a)
R. M. De Lederkremer and C. Marino, Deoxy sugars: occurrence and synthesis, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, Elsevier, Amsterdam, 2007, vol. 61, pp. 143–216 Search PubMed;
(b) A. M. Gomez, F. Lobo, S. Miranda and J. C. Lopez, Molecules, 2015, 20, 8357–8394 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
-
(a)
K. Strensrud and C.-C. Ma, US2017/0044123A1, 2017;
(b) C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 CrossRef CAS;
(c) J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS;
(d) T. Buntara, I. Melián-Cabrera, Q. Tan, J. L. G. Fierro, M. Neurock, J. G. de Vries and H. J. Heeres, Catal. Today, 2013, 210, 106–116 CrossRef CAS.
-
(a) R. Rinaldi and F. Schuth, ChemCatChem, 2009, 2, 1096–1107 CAS;
(b) R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 RSC;
(c) M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed;
(d) P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52–68 CrossRef CAS;
(e) S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed;
(f) I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 RSC;
(g) E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
-
(a) L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed;
(b) P. Bhaumik and P. L. Dhepe, Catal. Rev.: Sci. Eng., 2016, 58, 36–112 CrossRef CAS;
(c) B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed;
(d) P. Zhou and Z. Zhang, Catal. Sci. Technol., 2016, 6, 3694–3712 RSC;
(e) S. Dutta, S. De, B. Saha and M. D. I. Alam, Catal. Sci. Technol., 2012, 2, 2025–2036 RSC.
-
(a) R. Arundhathi, T. Mizugaki, T. Mitsudome, K. Jitsukawa and K. Kaneda, ChemSusChem, 2013, 6, 1345–1347 CrossRef CAS;
(b) X. Zhao, J. Wang, M. Yang, N. Lei, L. Li, B. Hou, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2017, 10, 819–824 CrossRef CAS PubMed;
(c) Y. Fan, S. Cheng, H. Wang, J. Tian, S. Xie, Y. Pei, M. Qiao and B. Zong, Appl. Catal., B, 2017, 217, 331–341 CrossRef CAS;
(d) Y. Fan, S. Cheng, H. Wang, D. Ye, S. Xie, Y. Pei, H. Hu, W. Hua, Z. Li, M. Qiao and B. Zong, Green Chem., 2017, 19, 2174–2183 RSC;
(e) L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375–2380 RSC.
-
(a) Y. Nakagawa, M. Tamura and K. Tomishige, J. Mater. Chem. A, 2014, 2, 6688–6702 RSC;
(b) K. Tomishige, Y. Nakagawa and M. Tamura, Top. Curr. Chem., 2014, 353, 127–162 CrossRef CAS PubMed;
(c)
Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem. Sustain. Technol., 2016, pp. 203–225 Search PubMed;
(d) K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC;
(e) Y. Nakagawa, M. Tamura and K. Tomishige, Res. Chem. Intermed., 2018, 44, 3879–3903 CrossRef CAS;
(f) K. Tomishige, Y. Nakagawa and M. Tamura, Curr. Opin. Green Sustain. Chem., 2020, 22, 13–21 CrossRef;
(g) K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 CrossRef CAS;
(h) D. Sun, Y. Yamada, S. Sato and W. Ueda, Appl. Catal., B, 2016, 193, 75–92 CrossRef CAS;
(i) Y. Wang, J. Zhou and X. Guo, RSC Adv., 2015, 5, 74611–74628 RSC.
-
(a) K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa and K. Tomishige, ChemSusChem, 2013, 6, 613–621 CrossRef CAS PubMed;
(b) S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS;
(c) S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS PubMed;
(d) S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC;
(e) Y. Nakagawa, S. Liu, M. Tamura and K. Tomishige, ChemSusChem, 2015, 8, 1114–1132 CrossRef CAS PubMed;
(f) Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
-
(a) G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS;
(b) R. M. West, M. H. Tucker, D. J. Braden and J. A. Dumesic, Catal. Commun., 2009, 10, 1743–1746 CrossRef CAS;
(c) N. Li, G. A. Tompsett and G. W. Huber, ChemSusChem, 2010, 3, 1154–1157 CrossRef CAS PubMed;
(d) L. Vilcocq, A. Cabiac, C. Especel, S. Lacombe and D. Duprez, J. Catal., 2014, 320, 16–25 CrossRef CAS;
(e) Q. Zhang, T. Wang, B. Li, T. Jiang, L. Ma, X. Zhang and Q. Liu, Appl. Energy, 2012, 97, 509–513 CrossRef CAS;
(f) K. Murata, I. Takahara and M. Inaba, React. Kinet. Catal. Lett., 2008, 93, 59–66 CrossRef CAS;
(g) D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS;
(h) Q. Zhang, T. Jiang, B. Li, T. Wang, X. Zhang, Q. Zhang and L. Ma, ChemCatChem, 2012, 4, 1084–1087 CrossRef CAS;
(i) Q. Zhang, T. Wang, Y. Xu, Q. Zhang and L. Ma, Energy Convers. Manage., 2014, 77, 262–268 CrossRef CAS.
-
(a) Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS;
(b) Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117–127 CrossRef CAS;
(c) Y. Nakagawa, X. Ning, Y. Amada and K. Tomishige, Appl. Catal., A, 2012, 433–434, 128–134 CrossRef CAS;
(d) Y. Amada, H. Watanabe, Y. Hirai, Y. Kajikawa, Y. Nakagawa and K. Tomishige, ChemSusChem, 2012, 5, 1991–1999 CrossRef CAS PubMed;
(e) M. Tamura, Y. Amada, S. Liu, Z. Yuan, Y. Nakagawa and K. Tomishige, J. Mol. Catal. A: Chem., 2014, 388–389, 177–187 CrossRef CAS;
(f) L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., B, 2019, 256, 117775 CrossRef;
(g) L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576–581 CrossRef CAS PubMed.
- T. A. Bender, J. A. Darowski, H. Zhong and M. R. Gagné, Org. Lett., 2016, 18, 4120–4123 CrossRef CAS PubMed.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, ACS Catal., 2016, 6, 8339–8403 Search PubMed.
- Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 81–85 CrossRef CAS.
- I. Chatterjee, D. Porwal and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3389–3391 CrossRef CAS.
- J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757–13761 CrossRef CAS.
-
(a) J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed;
(b) A. R. Peterson and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef PubMed;
(c) L. J. Donnelly, S. P. Thomas and J. B. Love, Chem. – Asian J., 2019, 14, 3782–3790 CrossRef CAS.
-
(a) S. Qu, Y. Deng, M. Wen and Z. X. Wang, Chem. – Eur. J., 2013, 19, 3827–3832 CrossRef CAS PubMed;
(b) X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed.
-
(a) S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS;
(b) K. A. DeNike and S. M. Kilyanek, R. Soc. Open Sci., 2019, 6, 191165 CrossRef.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS.
- M. Shiramizu and D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- C. Boucher-Jacobs and K. M. Nicholas, Organometallics, 2015, 34, 1985–1990 CrossRef CAS.
- J. Li, M. Lutz, M. Otte and R. J. M. K. Gebbink, ChemCatChem, 2018, 10, 4755–4760 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed.
- R. T. Larson, A. Samant, J. Chen, W. Lee, M. A. Bohn, D. M. Ohlmann, S. J. Zuend and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 14001–14004 CrossRef CAS PubMed.
- X. Li, D. Wu, T. Liu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed.
- H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500–1506 CrossRef CAS.
- X. Li and Y. Zhang, ChemSusChem, 2016, 9, 2774–2778 CrossRef CAS PubMed.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC.
- J. Fu, E. S. Vasiliadou, K. A. Goulas, B. Saha and D. G. Vlachos, Catal. Sci. Technol., 2017, 7, 4944–4954 RSC.
- M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa and K. Tomishige, Angew. Chem., Int. Ed., 2018, 57, 8058–8062 CrossRef CAS PubMed.
-
(a) N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed;
(b) N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- S. H. Krishna, J. Cao, M. Tamura, Y. Nakagawa, M. De Bruyn, G. S. Jacobson, B. M. Weckhuysen, J. A. Dumesic, K. Tomishige and G. W. Huber, ACS Sustainable Chem. Eng., 2020, 8, 800–805 CrossRef CAS.
- J. Cao, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Catal., 2019, 9, 3725–3729 CrossRef CAS.
-
(a) S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS;
(b) Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
-
(a) T. Wang, S. Liu, M. Tamura, Y. Nakagawa, N. Hiyoshi and K. Tomishige, Green Chem., 2018, 20, 2547–2557 RSC;
(b) T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS PubMed.
-
(a) S. Kudo, N. Goto, J. Sperry, K. Norinaga and J.-I. Hayashi, ACS Sustainable Chem. Eng., 2017, 5, 1132–1140 CrossRef CAS;
(b) F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
-
(a) M. Ostermeier and R. Schobert, J. Org. Chem., 2014, 79, 4038–4042 CrossRef CAS PubMed;
(b) C. Muller, M. Frau, D. Ballinari, S. Colombo, A. Bitto, E. Martegani, C. Airoldi, A. S. van Neuren, M. Stein, J. Weiser, C. Battistini and F. Peri, ChemMedChem, 2009, 4, 524–528 CrossRef PubMed;
(c) J. Sherwood, M. De Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
-
A. M. Allgeier, W. I. N. De Silva, E. Korovessi, C. A. Menning, J. C. Ritter, S. K. Sengupta and C. S. Stauffer, US Pat., 8865940B2, 2014 Search PubMed.
-
(a) T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed;
(b) T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Top. Catal., 2012, 55, 612–619 CrossRef CAS.
-
(a) S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035–2037 RSC;
(b) S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89–92 CrossRef CAS;
(c) K. Chen, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, ChemCatChem, 2010, 2, 547–555 CrossRef CAS;
(d) S. Koso, Y. Nakagawa and K. Tomishige, J. Catal., 2010, 280, 221–229 CrossRef;
(e) S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2012, 111–112, 27–37 CrossRef CAS.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- P. U. Karanjkar, S. P. Burt, X. Chen, K. J. Barnett, M. R. Ball, M. D. Kumbhalkar, X. Wang, J. B. Miller, I. Hermans, J. A. Dumesic and G. W. Huber, Catal. Sci. Technol., 2016, 6, 7841–7851 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Yoshinao
Nakagawa
*,
Yoshinao
Nakagawa