
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN,N,O-Coordinated tricarbonylrhenium precatalysts for the aerobic deoxydehydration of diols and polyols†
Jing
Li
 a,
Martin
Lutz
b and
Robertus J. M.
Klein Gebbink
*a
a,
Martin
Lutz
b and
Robertus J. M.
Klein Gebbink
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584CG, Utrecht, The Netherlands. E-mail: R.J.M.KleinGebbink@uu.nl
bCrystal and Structural Chemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Padualaan 8, 3584 CH, Utrecht, The Netherlands
First published on 18th May 2020
Abstract
Rhenium complexes are well known catalysts for the deoxydehydration (DODH) of vicinal diols (glycols). In this work, we report on the DODH of diols and biomass-derived polyols using L4Re(CO)3 as precatalyst (L4Re(CO)3 = tricarbonylrhenium 2,4-di-tert-butyl-6-(((2-(dimethylamino)ethyl)(methyl)amino)methyl)phenolate). The DODH reaction was optimized using 2 mol% of L4Re(CO)3 as precatalyst and 3-octanol as both reductant and solvent under aerobic conditions, generating the active high-valent rhenium species in situ. Both diol and biomass-based polyol substrates could be applied in this system to form the corresponding olefins with moderate to high yield. Typical features of this aerobic DODH system include a low tendency for the isomerization of aliphatic external olefin products to internal olefins, a high butadiene selectivity in the DODH of erythritol, the preferential formation of 2-vinylfuran from sugar substrates, and an overall low precatalyst loading. Several of these features indicate the formation of an active species that is different from the species formed in DODH by rhenium-trioxo catalysts. Overall, the bench-top stable and synthetically easily accessible, low-valent NNO–rhenium complex L4Re(CO)3 represents an interesting alternative to high-valent rhenium catalysts in DODH chemistry.
Introduction
Due to the increased consumption of fossil hydrocarbon resources, the development of alternative and sustainable feedstocks is highly recommended.1,2 Biomass-derived feedstocks are considered as attractive and renewable resources for a sustainable chemical industry.3,4 One of the many challenges in biomass conversion and valorization is the defunctionalization of oxygen-rich molecular components obtained after depolymerization of cellulosic biomass.5 Deoxydehydration (DODH) reactions, which formally combine deoxygenation6,7 and dehydration,8 can efficiently remove vicinal hydroxyl groups to form the corresponding olefin. The successful application of DODH reactions on oxygen-rich biomass components would provide access to a variety of low-volume, high-value chemicals.9–11High-valent rhenium complexes are known as catalysts for the DODH of diols and polyols,9 while low-valent rhenium complexes, e.g., Re2(CO)10 and BrRe(CO)5, can also initiate DODH reactions in the presence of oxygen.12 Among the high-valent rhenium complexes, Cp-based trioxorhenium complexes (Cp′ReO3, Cp′ = substituted cyclopentadienyl) show high catalytic activity and DODH product selectivity. A drawback in the development of this type of Cp′Re complexes is that the overall yield of the synthesis of the final Cp′ReO3 complexes is rather low (4% to 49% starting from Cp′H/Re), mainly because the trioxo-rhenium species decomposes under the oxidizing conditions of its synthesis from Cp′Re(CO)3.13 A possible way to solve this problem is to use Cp′Re(CO)3 as a precatalyst to form the corresponding Cp′ReO3 species in situ during catalysis in the presence of oxygen. Yet, on the basis of the work of Ellman and Bergman, Cp*Re(CO)3 is not active towards the deoxydehydration of diols under aerobic conditions.12
For this reason, we were interested in other low-valent Re-complexes that would allow for an oxidative activation to provide in situ access to high-valent Re-trioxo complexes for DODH catalysis. Accordingly, we have started to investigate Re-complexes derived from anionic NNO-type ligands. Such ligands provide an anionic 6e− platform that is isoelectronic to Cp′-type ligands, and potentially coordinate to Re in a facial fashion similar to Cp′ ligands. Here, we report on the synthesis and characterization of novel low-valent (NNO)Re(I) tricarbonyl complexes, in which NNO represents an open chain bis-amino-phenol ligand (NNO = 2-(((2-(dimethylamino)ethyl)(methyl)amino)methyl)phenol), and on the use of these complexes as precatalysts in the aerobic DODH of vicinal diols and polyols.
Results and discussion
Synthesis and characterization
The NNO-type ligands (HL) were synthesized via direct reductive amination of N,N,N′-trimethylethylenediamine with a phenolic aldehyde using NaBH(OAc)3 (1.5 equiv.) in dichloroethane (Scheme 1).14 The preligand HL was then reflux with BrRe(CO)5 and an excess amount of NaOAc in toluene to form the corresponding LRe(CO)3 complexes.15 After recrystallization, all LRe(CO)3 complexes were isolated as needle-like crystals in >70% Re-based yield. The complexes synthesized in this way are summarized in Fig. 1.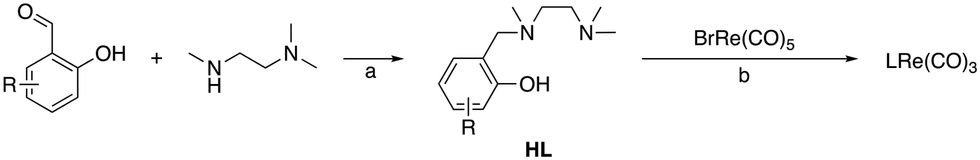 | ||
| Scheme 1 Synthesis of NNO-based tricarbonylrhenium complexes: a. NaBH(OAc)3 (1.5 equiv.), dichloroethane, r.t., overnight, N2; b. NaOAc (10 equiv.), toluene, reflux, 3 h, N2. | ||
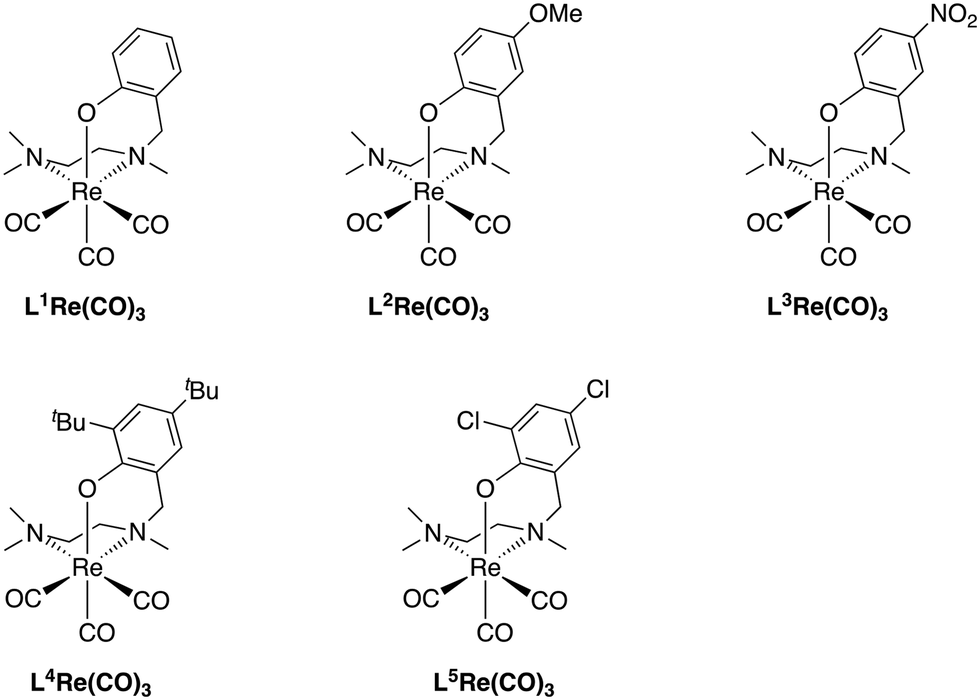 | ||
| Fig. 1 (NNO)tricarbonylrhenium complexes reported in this work. | ||
The structures of L1Re(CO)3, L3Re(CO)3, and L4Re(CO)3 were established by X-ray crystal structure determination (Fig. 2). Selected geometrical parameters are given in Table 1. Diffraction-quality crystals of L1Re(CO)3 and L3Re(CO)3 were obtained by diffusion of diethyl ether into acetonitrile solutions of the complexes at room temperature. Diffraction-quality crystals of L4Re(CO)3 were obtained by recrystallization from Et2O/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) at −30 °C. In all three complexes nitrogen atom N1 is chiral, but the compounds crystallize as racemates in centrosymmetric space groups, respectively. They showcase the overall octahedral geometry of the complexes discussed here with the tripodal N,N,O-ligand coordinated to rhenium in a facial manner. Due to the chelating bonding mode the bond angles around Re deviate from the perfect 90° and range from 79.66(14)–96.98(17)° in L1Re(CO)3, 79.51(10)–96.66(13)° in L3Re(CO)3, and 80.03(12)–96.17(15)° in L4Re(CO)3. The five-membered chelate rings in L1Re(CO)3 and L3Re(CO)3 are best described as twist conformations, while in L4Re(CO)3 the ring adopts an envelope conformation. The six-membered chelate rings in L1Re(CO)3 and L3Re(CO)3 are in an envelope conformation, in L4Re(CO)3 it is intermediate between a boat and twist conformation as indicated by the deviation of N1 and Re1 from the least-squares plane of the phenyl ring of the phenolate donor (Table 2). Further characterization of the complexes included 1H NMR, 13C NMR, IR, ESI-MS, and elemental analysis (see ESI†).
:2) at −30 °C. In all three complexes nitrogen atom N1 is chiral, but the compounds crystallize as racemates in centrosymmetric space groups, respectively. They showcase the overall octahedral geometry of the complexes discussed here with the tripodal N,N,O-ligand coordinated to rhenium in a facial manner. Due to the chelating bonding mode the bond angles around Re deviate from the perfect 90° and range from 79.66(14)–96.98(17)° in L1Re(CO)3, 79.51(10)–96.66(13)° in L3Re(CO)3, and 80.03(12)–96.17(15)° in L4Re(CO)3. The five-membered chelate rings in L1Re(CO)3 and L3Re(CO)3 are best described as twist conformations, while in L4Re(CO)3 the ring adopts an envelope conformation. The six-membered chelate rings in L1Re(CO)3 and L3Re(CO)3 are in an envelope conformation, in L4Re(CO)3 it is intermediate between a boat and twist conformation as indicated by the deviation of N1 and Re1 from the least-squares plane of the phenyl ring of the phenolate donor (Table 2). Further characterization of the complexes included 1H NMR, 13C NMR, IR, ESI-MS, and elemental analysis (see ESI†).
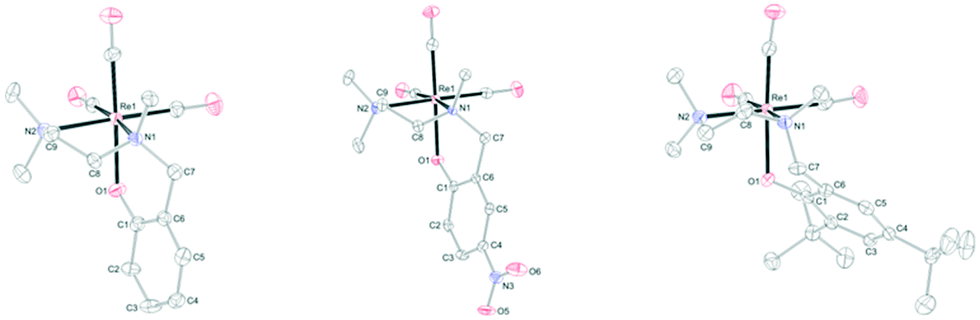 | ||
| Fig. 2 Molecular structures of L1Re(CO)3, L3Re(CO)3, and L4Re(CO)3 in the crystal. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
| L1Re(CO)3 | L3Re(CO)3 | L4Re(CO)3 | |
|---|---|---|---|
| Re1–O1 | 2.111(3) | 2.122(2) | 2.144(3) |
| Re1–N1 | 2.235(4) | 2.238(3) | 2.247(3) |
| Re1–N2 | 2.278(4) | 2.275(3) | 2.262(3) |
| C1–O1 | 1.330(5) | 1.309(4) | 1.354(4) |
| C7–N1–C8 | 109.4(3) | 110.1(3) | 108.7(3) |
| C1–C6–C7–N1 | 39.9(6) | 43.3(5) | −65.1(5) |
| N1–C8–C9–N2 | 57.5(5) | 58.8(4) | −60.0(4) |
| L1Re(CO)3 | L3Re(CO)3 | L4Re(CO)3 | |
|---|---|---|---|
| N1 | 0.819(4) | 0.872(3) | 1.017(3) |
| Re1 | 0.0427(2) | 0.1875(4) | 1.7545(4) |
In the IR spectrum of BrRe(CO)3, two absorption signals at 1956 and 2023 cm−1 were observed (Table 3). In case of the NNO-ligand coordinated tricarbonylrhenium complexes and CptttRe(CO)3, three absorption bands at 1851–1889, 1873–1921, and 2007–2016 cm−1 were observed. These absorption bands are attributed to the symmetric and asymmetric ν(CO) stretches. The similarity of IR spectra indicates that the NNO-ligand coordinated tricarbonylrhenium complexes have a similar structure in the solid state, that is, a octahedral geometry structure with the tripodal N,N,O-ligand coordinated to rhenium in a facial manner.
| Entry | [Re] | Infrared ν(CO) (cm−1) |
|---|---|---|
| 1 | BrRe(CO)5 | 1956, 2023 |
| 2 | L1Re(CO)3 | 1877, 1918, 2014 |
| 3 | L2Re(CO)3 | 1859, 1886, 2007 |
| 4 | L3Re(CO)3 | 1884, 1921, 2016 |
| 5 | L4Re(CO)3 | 1851, 1894, 2010 |
| 6 | L5Re(CO)3 | 1855, 1873, 2007 |
| 7 | CptttRe(CO)3 | 1889, 1911, 2007 |
Besides, a trend in the energies of the IR absorptions was observed in relation to the electronic properties of the substituents on the phenol ring. Fig. 3 shows the wavenumbers of the IR absorptions plotted against the Hammett constants (σp) of the para-substituents.16 Although electronic effects of the ortho-substituents may also be involved for L4Re(CO)3 and L5Re(CO)3, the overall trend clearly shows that the more electron-donating para-substituents give rise to lower energies of the CO vibrations. In other words, the CO ligands are more strongly coordinated to the more electron-rich rhenium centers.
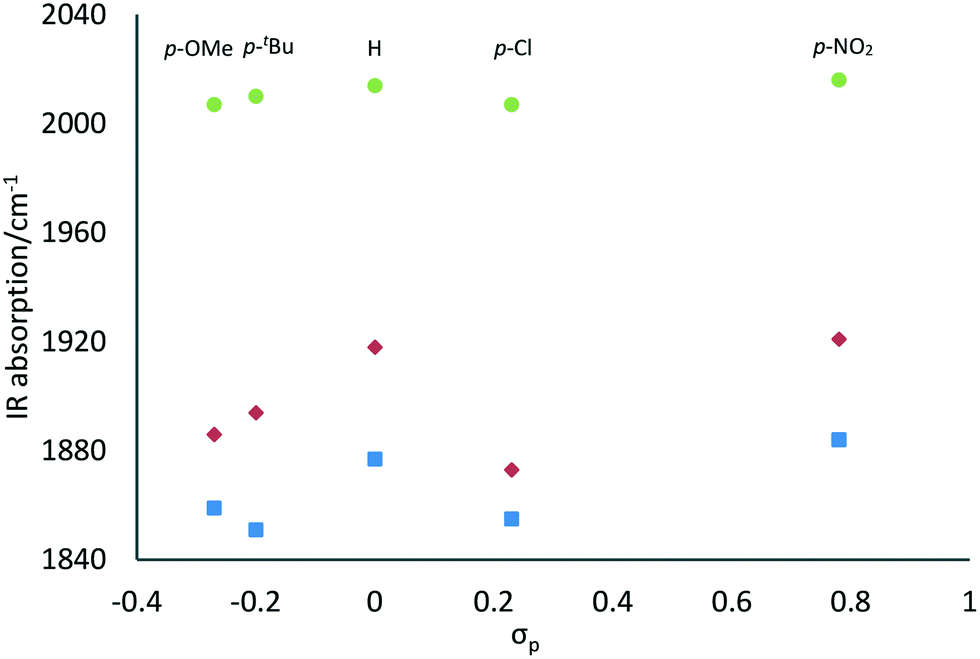 | ||
| Fig. 3 Relation between the Hammett parameter (σp) of the phenolate para-substituent and the IR wavenumbers of the CO vibrations for LRe(CO)3 complexes. | ||
Catalytic evaluation
With the new NNO–Re complexes in hand, we started our catalytic investigation by applying the parent L1Re(CO)3 complex (2 mol%) to the deoxydehydration of 1,2-octanediol using triphenylphosphine (PPh3) as reductant and chlorobenzene as solvent. Running the reaction under oxygen-free conditions gave no 1-octene formation, while 12% of 1-octene was formed under aerobic conditions. This result indicates that an active DODH species, likely a high-valent rhenium species, can be formed starting from L1Re(CO)3 in the present of oxygen. Since secondary alcohols have been reported as both reductants and solvents for Re-catalyzed DODH reactions,12 we also investigated the L1Re(CO)3 initiated DODH of 1,2-octanediol using 3-octanol as both reductant and solvent: 0.5 mmol of 1,2-octanediol was dissolved in 5 mL of 3-octanol, 2 mol% of L1Re(CO)3 was used as precatalyst (Table 4). The mixture was then heated at 180 °C in a closed pressure tube. The amount of O2 present in the reaction mixture was approximately 18 equiv. on the basis of L1Re(CO)3. A sample was taken for 1H NMR analysis after the mixture was heated for 3 h. 71% of 1-octene had formed, as well as dehydration products (2-/3-octene) derived from 3-octanol (the latter products are not derived from isomerization of 1-octene, vide infra). The ratio of 1-octene and 2-/3-octene was 3.23:1. In order to reach full conversion of 1,2-octanediol, the mixture was heated for another 3 h, after which 96% of 1-octene had formed. The ratio of 1-octene and 2-/3-octene after 3 h was 1.71:1, which can be explained on the basis of the continued dehydration of the 3-octanol solvent/reductant while diol DODH reaches close to full conversion (Table 4, entry 2). When applying BrRe(CO)5 as the precatalyst under these conditions for 3 h, 1,2-octanediol was fully converted forming 94% of 1-octene, as well as a significant amount of 2-/3-octene (ratio 0.97:1; entry 1). BrRe(CO)5 therefore seems to promote both the deoxydehydration of 1,2-octandiol and the dehydration of 3-octanol without a particular preference. The tendency of L1Re(CO)3 to promote the dehydration of 3-octanol is significantly lower as compared to BrRe(CO)5.
| Entry | [Re] | t | Yieldb |
n(1-):n(2-/3-)c |
|---|---|---|---|---|
| a Reaction conditions: 1,2-octanediol (0.5 mmol), rhenium pre-catalyst (0.01 mmol, 2 mol%), 3-octanol (5 mL), 180 °C (temperature of oil bath), air. b 1-Octene yield determined by 1H NMR using mesitylene (0.5 mmol) as an internal standard. c Ratio of 1-octene and 2-/3-octene product isomers. d The reaction mixture was heated in a closed pressure tube with air for 3 h, then the reaction was cooled to room temperature, a sample was taken from the reaction mixture. After sampling the reaction mixture, the pressure tube was closed again and heated for another 3 h, in which case more air entered the reaction system. e Instead of 5 mL, 1 mL of 3-octanol was used. Conversion of 1,2-octanediol was 56%. | ||||
| 1 | BrRe(CO)5 | 3 h | 94% | 0.97:1 |
| 2 | L1Re(CO)3 | 3 h | 71% | 3.23:1 |
| 6 hd | 96% | 1.71:1 |
||
| 3 | L2Re(CO)3 | 3 h | 83% | 2.11:1 |
| 4 | L2Re(CO)3 | 6 hd | 95% | 1.5:1 |
| 5 | L3Re(CO)3 | 3 h | 13% | 0.89:1 |
| 6 hd | 56% | 2.77:1 |
||
| 6 | L3Re(CO)3 | 6 h | 13% | 0.74:1 |
| 7 | L4Re(CO)3 | 3 h | 92% | 2.52:1 |
| 8 | L5Re(CO)3 | 3 h | 21% | 1.23:1 |
| 6 hd | 65% | 2.71:1 |
||
| 9 | CptttRe(CO)3 | 3 h | 8% | 0.62:1 |
| 6 hd | 8% | 0.62:1 |
||
| 10e | L4Re(CO)3 | 3 h | 56% | 14:1 |
After this initial study, the other NNO-based tricarbonylrhenium complexes were also investigated using the same DODH conditions (Table 4). With an electron-donating methoxy group at the para-position of the phenol moiety (complex L2Re(CO)3), a slightly higher 1-octene yield (83%) was obtained after 3 h (entry 3). Upon full 1,2-octanediol conversion after 6 h, the 1-octene yield was 95% (entry 4). With an electron-withdrawing nitro substituent at the para-position of the phenol (complex L3Re(CO)3), a 1-octene yield of only 13% was achieved after 3 h (entry 5). After having taken a sample for analysis, and exposing the reaction mixture to more air, the 1-octene yield increased to 56% after closing the reaction tube again and heating it for another 3 h. When this same reaction was carried for 6 h without opening the reaction tube (entry 6), the 1-octene yield was only 13%, i.e. the same as heating the mixture for 3 h. This result indicates that the activation of L3Re(CO)3 was not complete after heating for 3 h. After exposing the reaction mixture to more oxygen/air, L3Re(CO)3 was further activated, resulting in an enhanced formation of 1-octene, as well as an increased 1-octene to 2-/3-octene product ratio (change from 0.89:1 to 2.77:1). In case of the bis(tert-butyl)-substituted complex L4Re(CO)3, full conversion of 1,2-octanediol was achieved after 3 h, achieving a 1-octene yield of 92% at a 2.52:1 ratio of 1-octene and 2-/3-octene (entry 7). For the o,p-dichlorinated complex L5Re(CO)3, only 21% of 1-octene was formed after heating for 3 h, and 65% after heating the mixture for another 3 h after exposure to air (entry 8). As a comparison, CptttRe(CO)3 (Cpttt = 1,2,4-tri-tert-butylcyclopentadienyl) was also investigated under these reaction conditions, providing only 8% of 1-octene with or without additional exposure to air and extending the reaction time. Besides, the 1-octene to 2-/3-octene product ratio was 0.62:1.
Interestingly, this series of experiments shows the preference of the NNO-based tricarbonylrhenium complexes, upon exposure to air, to engage in diol deoxydehydration over secondary alcohol dehydration. The concentration of 1,2-octanediol in these experiments was much lower than that of 3-octanol. However, more deoxydehydration product from 1,2-octanediol was formed than dehydration product from 3-octanol. A higher concentration of 1,2-octanediol was tested (entry 10), by decreasing the amount of 3-octanol from 5 to 1 mL. In this case, 56% of 1-octene was formed at 56% of 1,2-octanediol conversion, i.e. full selectivity for olefin formation. The ratio of 1-octene and 2-/3-octene in this experiment was 14:1. This result strengthens the notion that DODH is kinetically favoured for all LRe(CO)3 precatalysts, although dehydration of the secondary alcohol reductant is always observed to a significant extent.
These initial catalysis experiments also show the beneficial effect of electron-donating substituents at the phenol moiety on both the overall DODH product formation and the DODH vs. dehydration product ratio. A likely explanation for this observation is the stabilization of the high-valent ReVII center in the anticipated trioxo-complexes by the more electron-rich donor ligands, even though a more electron-rich Re-center would also lead to more firmly bound CO-ligands which could hamper oxidation of the low-valent carbonyl complexes. The catalysis data shown in Table 4 are in line with the trend in CO vibrational energies depicted in Fig. 3. Based on these observations, the di(tert-butyl) substituted complex L4Re(CO)3 was selected as the precatalyst for further investigation and optimization of DODH reactions of diols and polyols.
As the next step in the protocol optimization, different alcohol reductants, including primary and secondary alcohols, were investigated (Table 5). No 1-octene was detected when 1-butanol and 2-butanol were used as reductant and solvent (entries 1 and 2). Surprisingly, with 3-pentanol and 2,4-dimethyl-3-pentanol, which have been reported as good reductants in high-valent rhenium-catalyzed DODH reactions,12,17–21 only 14% and trace amounts of 1-octene were formed, respectively (entry 3 and 4). When using 3-pentanol, a small amount (4%) of 2-pentene was observed. Other secondary alcohols, such as 2-octanol and 4-octanol, were also investigated. When using 2-octanol as reductant and solvent, 88% of 1-octene was formed from 1,2-octanediol, and 49% (= n2-octene/n1,2-octanediol) of 2-octene was observed (entry 5). When 4-octanol was used, only 20% of 1-octene was formed, together with 4% (= n3-/4-octene/n1,2-octanediol) of 3-/4-octene (entry 7). Besides, 1,2-octanediol itself could also be used as reductant; in this case 32% of 1-octene was formed (entry 8). Based on these results, it was decided to stick with 3-octanol as the reductant and solvent in further investigations.
| Entry | Secondary alcohol | Yieldb | b.p. of solventd |
|---|---|---|---|
| a Reaction conditions: 1,2-octanediol (0.5 mmol), L4Re(CO)3 (0.01 mmol, 2 mol%), alcohol reductant/solvent (5 mL), 180 °C (temperature of oil bath), 3 h, air. b 1-Octene yield, determined by 1H NMR using mesitylene (0.5 mmol) as an internal standard. c n.d. = no 1-octene was detected. d All solvents were purchased from Sigma-Aldrich; boiling points are reported on the basis of the corresponding Safety Data Sheet. | |||
| 1 | 1-Butanol | n.d.c | 116–118 °C |
| 2 | 2-Butanol | n.d.c | 98 °C |
| 3 | 3-Pentanol | 14% | 114–115 °C |
| 4 | 2,4-Dimethyl-3-pentanol | Trace | 139–140 °C |
| 5 | 2-Octanol | 88% | 177–182 °C |
| 6 | 3-Octanol | 92% | 174–176 °C |
| 7 | 4-Octanol | 20% | 174–176 °C |
| 8 | None | 32% | |
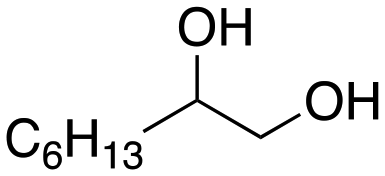
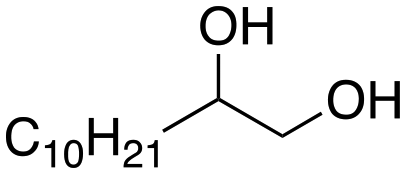
Following these optimization studies, the substrate scope using a variety of vicinal diols and polyols was investigated under optimized conditions, i.e., using 0.5 mmol of substrate, 2 mol% of L4Re(CO)3, 5 mL of 3-octanol, 180 °C, under air in a closed pressure tube (Table 6). The aliphatic vicinal diol 1,2-dodecanediol gave 93% of 1-dodecene. No olefin isomers of the 1-dodecene product were detected, lending further credit to the notion that the formation of 2-octene and 3-octene results from alcohol dehydration, instead of 1-octene isomerization when using 3-octanol as the reductant. For the previously reported Cp′Re trioxo catalyzed DODH reactions, the formation of internal olefin isomers derived from the terminal olefin product were detected at reaction temperatures ≥135 °C, typically in 2–15% yield at 135 °C, and 4–29% yield at 180 °C.13 Accordingly, in situ activation of precatalyst L4Re(CO)3 does not provide access to a species active in olefin isomerization.
| Entry | Substrate | Product | Yieldb |
|---|---|---|---|
| a Reaction conditions: substrate (0.5 mmol), L4Re(CO)3 (0.01 mmol, 2 mol%), 3-octanol (5 mL), 180 °C (temperature of oil bath), 3 h, air. Unless otherwise noted, the conversion of substrate was >98%. b Determined by 1H NMR using mesitylene (0.5 mmol) as an internal standard. c Reaction time: 1.5 h. d BrRe(CO)5 (2 mol%) was used as precatalyst. e The product was a mixture of fumaric acid and fumarates. f Reaction time: 6 h. g The product was a mixture of muconates. | |||
| 1 |
|
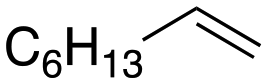
|
92% |
| 2 |
|
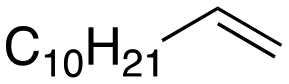
|
93% |
| 3c |
|
|
83% |
| 4 |
|
|
62% |
| 5 |
|
|
97% |
| 6 |
|
|
80% |
| 7 |
|
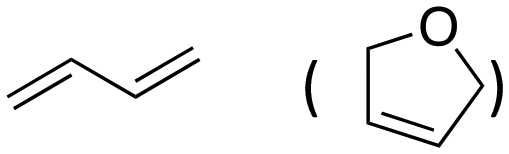
|
78% (2%) |
| 67% (6%)d | |||
| 8 |
|
|
64% (6%) |
| 9e |

|
|
81% |
| 10f,g |
|
|
46% |
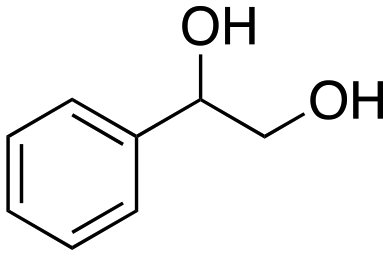
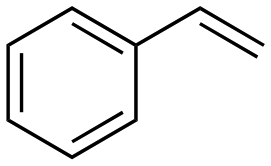
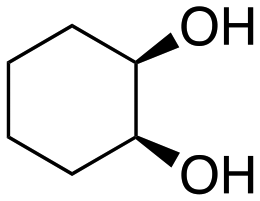
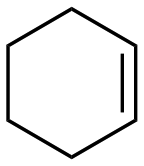
The aromatic vicinal diol 1-phenyl-1,2-ethanediol gave 83% yield of styrene at full conversion (entry 3). Interestingly, cis-1,2-cyclohexanediol gave 62% yield of 1-cyclohexene under these reaction conditions. This cyclic vicinal diol is known as a quite challenging substrate for rhenium-catalyzed DODH reactions. For example, when using the CptttRe(CO)3 catalyst only 10% of 1-cyclohexene forms at 21% substrate conversion.22
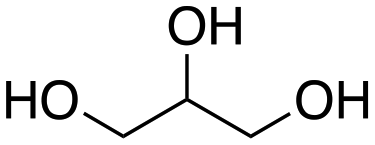
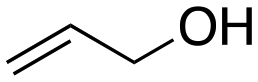
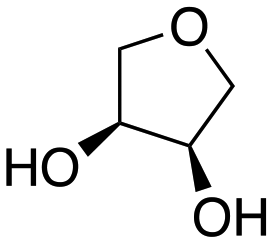
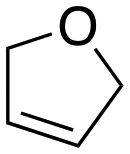
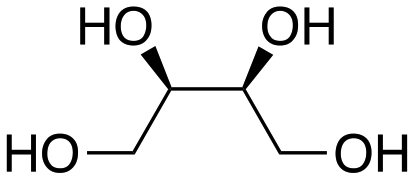
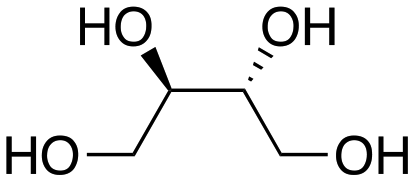
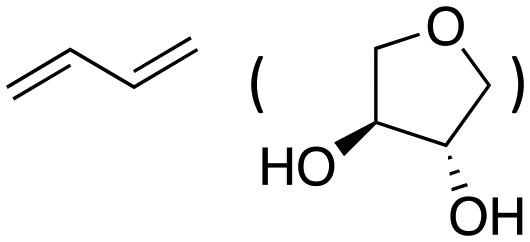
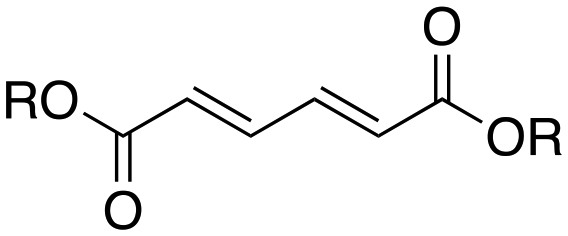
Inspired by these results, we moved to biomass-based substrates, such as 1,4-anhydroerythritol, glycerol, erythritol, and DL-threitol.23,24 When glycerol was used as substrate, 97% of allyl alcohol was formed (entry 5). The DODH activity of precatalyst L4Re(CO)3 for this substrate is competitive with respect to the activities that can be attained with high-valent rhenium trioxo catalysts.21 When erythritol and DL-threitol were used as substrate, 78% and 64% of 1,3-butadiene was formed, respectively (entry 7 and 8). On the other hand, 1,4-anhydroerythritol gave 80% of 2,5-dihydrofuran (entry 6). The latter observations suggest that the DODH of C4 tetraols using L4Re(CO)3 does not proceed via dehydrative cyclisation to form a cyclised diol as intermediate.
Given the interest in the catalytic formation of 1,3-butadiene from biomass, the activity and selectivity of precatalyst L4Re(CO)3 was compared to that of other catalysts reported in the field (Fig. 4). The butadiene yield achieved with L4Re(CO)3 competes with the highest reported yields for this reaction using either CpttReO3 or MTO as catalyst, i.e. compared to catalysts in which Re is already in a high-oxidation trioxo form from the start of the reaction.18,21,22 For catalysts included in this comparison that operate starting from a low oxidation state under oxidative conditions, L4Re(CO)3 gave a higher total yield compared to BrRe(CO)5. Among all these Re-systems, L4Re(CO)3 gave the highest 1,3-butadiene selectivity, i.e. 78% of 1,3-butadiene yield and only 2% of 2,5-dihydrofuran byproduct, though the total yield of volatile products was not as high as for the CpttReO3 and MTO catalysts.
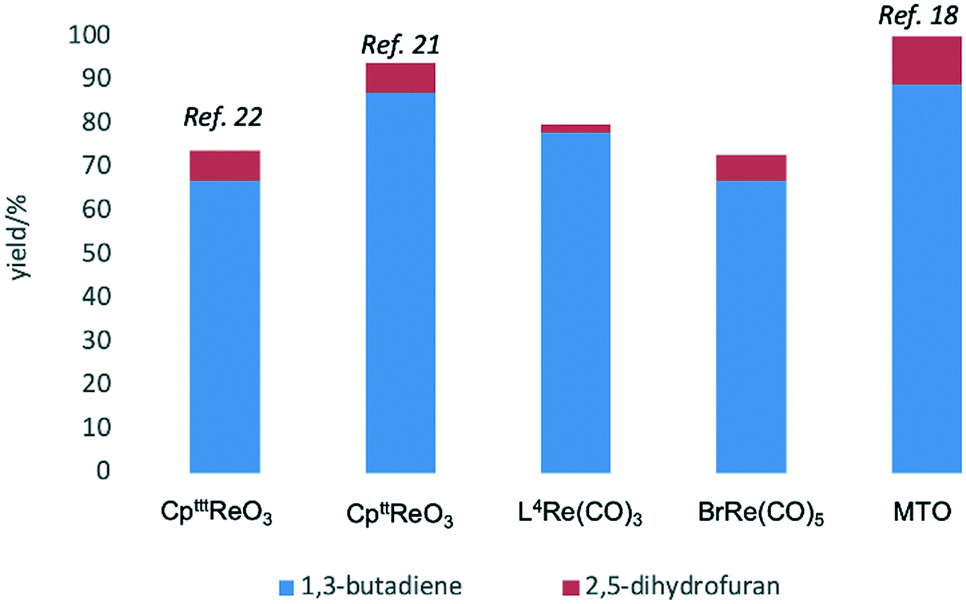 | ||
| Fig. 4 Comparison of DODH reactions of erythritol catalyzed/initiated by different Re-complexes.18,21,22 | ||
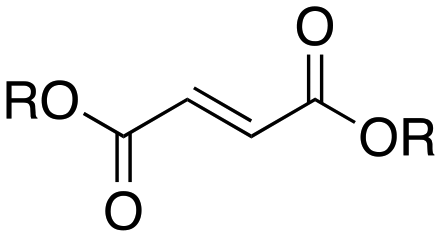
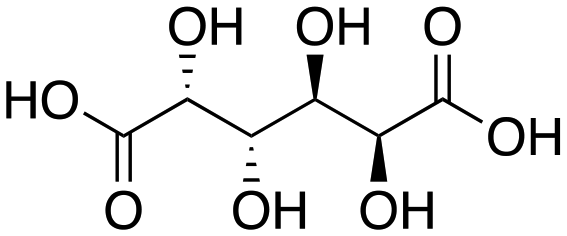
Next, two biomass-based acids, L-(+)-tartaric acid25 and mucic acid,26–28 were investigated using L4Re(CO)3. Since 3-octanol was also used as solvent, the deoxydehydration product of these two substrates was a mixture of acid and mono/di-octyl ester. L-(+)-Tartaric acid gave 81% of fumaric acid and fumarates (entry 9), while mucic acid gave 46% of muconates. Remarkably, when using Re2(CO)10 as pre-catalyst, no muconates were formed from mucic acid.29 Su and Zhang claimed that this is probably due to the poor tolerance of Re2(CO)10 to the carboxylic acid groups. Our results clearly indicate that once ligated by an N,N,O-type ligand the low-valent Re center does not suffer from the presence of these functions groups. Although 3-pentanol was not a very efficiency reductant for the deoxydehydration of 1,2-octanediol, it provided a good overall activity in case of the deoxydehydration of L-(+)-tartaric acid; 83% of a mixture of fumaric acid and fumarates was obtained at 155 °C for 12 h (Fig. 5). In previous work, L-(+)-tartaric acid was converted to 87% of fumarates while using 12 mol% of HReO4 as catalyst, and 97% of fumarates were formed while using 5 mol% of MTO as catalyst (Fig. 5).20,30 In our system, the yield of fumaric acid and fumarates is slightly lower, albeit that a lower precatalyst loading of 2 mol% is used.
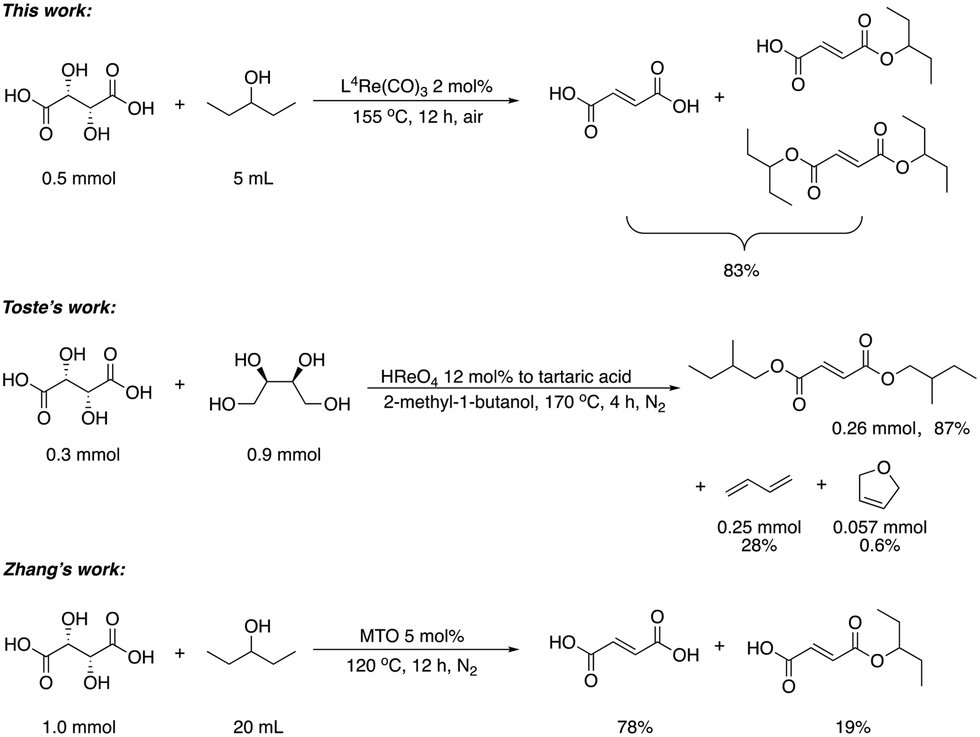 | ||
| Fig. 5 DODH of L-(+)-tartaric acid.20,30 | ||
On basis of the results above, we have discovered a rhenium system which can be applied to the deoxydehydration of diols and polyols with quite good olefin yields. Sugars, which in a way can be labelled as complicated polyol substrates, are quite challenging substrates for Re-catalyzed DODH reactions. MTO has been reportedly used for the deoxydehydration of sugars, e.g., D-mannose, D-allose, D-glucose, and D-galactose. 2-Vinylfuran (2-VF) and furan were the main products in these reactions, with furan being the major product in most cases.18 For the CpttReO3-catalyzed deoxydehydration of sugars, 2-vinylfuran and furan were also found to be the main products, but the product distribution was not the same as for MTO,21 which indicates that these systems might operate through a different active species. In a very recent report, Marrone, d'Alessandro and co-workers explored the deoxydehydration of glycerol using a series of rhenium sources (such as MTO, ReO3, Re2O7, IReO2(PPh3)2,ReCl5, and ReI3) as catalyst and secondary alcohols as reductant.31 For the latter reactions, an induction period was observed and a black solid separated from the reaction mixtures. When used in a next catalytic run, this black solid was found to be active in DODH without an induction period. The authors proposed that the black solid is the active species in these DODH reactions. Besides, they specifically analyse the MTO-catalyzed DODH and claim that MTO releases methane to form a ReVII alkoxide species with the help of a secondary alcohol at high temperature, and that the ReVII alkoxide species is the actual catalyst. Interestingly, the same authors also reported that Re2(CO)10 is not active for the deoxydehydration of glycerol at 140 °C in air with 2,4-dimethyl-3-pentanol as reductant and solvent. Since in our current investigations, a secondary alcohol was also used as reductant, we were curious if similar Re-alkoxide active species would be formed and would lead to related product profiles in the DODH of sugar substrates. D-Mannose, D-glucose, and D-galactose were then investigated using L4Re(CO)3 under optimized conditions (Table 7). Comparing the performance of L4Re(CO)3 to MTO and CpttReO3 clearly shows that for each sugar substrate the major product with L4Re(CO)3 is 2-vinylfuran instead of furan. This observation may be taken as an indication that L4Re(CO)3 operates through a different active species compared to the Re-trioxo catalysts. In addition, no black solid was observed for the deoxydehydration of simple vicinal diols in our system. The highest total yield in furans for L4Re(CO)3 was achieved using D-mannose as substrate (32%), which is quite similar to the MTO catalyst but somewhat lower than the CpttReO3 catalyst. For the other two sugars, the yields achieved with L4Re(CO)3 do not compete with those obtained with MTO.
| Sugar | [Re] (x mol%) | Total yield (2-VF:furan) |
Conditions |
|---|---|---|---|
| D-Glucose | MTO (2.5)18 | 25% (1:1.8) |
3-Pentanol (0.05 M) 155 °C, N2 |
| CpttReO3 (2.0)21 | Trace | 3-Pentanol (0.05 M) 155 °C, N2 | |
| L4Re(CO)3 (2.0) | 6% (1.8:1) |
3-Pentanol (0.1 M) 155 °C, air | |
| D-Galactose | MTO (2.5)18 | 32% (1:3.0) |
3-Pentanol (0.05 M) 155 °C, N2 |
| CpttReO3 (2.0)21 | 14% (no 2-VF) | 3-Pentanol (0.05 M) 155 °C, N2 | |
| L4Re(CO)3 (2.0) | 11% (2.0:1) |
3-Pentanol (0.1 M) 155 °C, air | |
| D-Mannose | MTO (2.5)18 | 30% (1:2.3) |
3-Pentanol (0.05 M) 155 °C, N2 |
| CpttReO3 (2.0)21 | 39% (2.3:1) |
3-Pentanol (0.05 M) 155 °C, N2 | |
| L4Re(CO)3 (2.0) | 32% (1.5:1) |
3-Pentanol (0.1 M) 155 °C, air |
Conclusions
A series of LRe(CO)3 complexes (LH = substituted 2-(((2-(dimethylamino)ethyl)(methyl)amino)methyl)phenol) were investigated as precatalyst for the deoxydehydration of diols and polyols under aerobic conditions and using a secondary alcohol as reductant and solvent. The 2,4-di-tert-butyl-substituted complex L4Re(CO)3 was selected as the pre-catalyst for further investigation after initial catalytic screening of the series. Both aliphatic and aromatic vicinal diols, as well as biomass-derived glycerol and polyols can be converted using L4Re(CO)3 to the corresponding olefins with good yields. The application of sugar substrates to these reaction conditions leads to the preferential formation of 2-vinylfuran. This unusual product distribution indicates that the active species generated from L4Re(CO)3 could be different from the ones generated by Re-trioxo systems.Compared to the Cp-based trioxo rhenium complexes previously reported by us, the new NNO-based Re(CO)3 precatalysts are much more easy to handle. Not only are the NNO-ligands easy to prepare and modify, the low-yielding oxidation from Re(I) to Re(VII) during catalyst synthesis is avoided and the (NNO)Re(CO)3 systems are air stable at room temperature while the Cp′ReO3 systems need to be stored at low temperature or under N2. In view of these considerations, the new NNO-based Re-tricarbonyl precatalysts are interesting alternatives for high-valent Re-trioxo systems in DODH chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JL acknowledges financial support through the Chinese Scholarship Council and Utrecht University. The X-ray diffractometer has been financed by the Netherlands Organization for Scientific Research (NWO).Notes and references
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562 RSC.
- E. P. Ahern, P. Deane, T. Persson and O. Brian, Renewable Energy, 2015, 78, 648 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef PubMed.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Rev. Chem., 2018, 2, 35–46 CrossRef CAS.
- M. Schlaf, Dalton Trans., 2006, 4645 RSC.
- L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., 2014, 126, 1672 ( Angew. Chem., Int. Ed. , 2014 , 53 , 1646 ) CrossRef.
- T. J. Korstanje, E. F. De Waard, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, ACS Catal., 2012, 2, 2173 CrossRef CAS.
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281 CrossRef CAS.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767 CrossRef CAS PubMed.
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem. – Asian J., 2019, 14, 3782 CrossRef CAS PubMed.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408 CAS.
- S. Raju, C. A. M. R. Van Slagmaat, J. Li, M. Lutz, J. T. B. H. Jastrzebski, M.-E. Moret and R. J. M. Klein Gebbink, Organometallics, 2016, 35, 2178 CrossRef CAS.
- D. G. Lonnon, S. B. Colbran and D. C. Craig, Eur. J. Inorg. Chem., 2006, 2006, 1190 Search PubMed.
- M. Toganoh, N. Harada and H. Furuta, Polyhedron, 2013, 52, 1153 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. Yi, S. Liu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 1401 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., 2012, 124, 8206 ( Angew. Chem., Int. Ed. , 2012 , 51 , 8082 ) CrossRef.
- C. Boucher-Jacobs and K. M. Nicholas, ChemSusChem, 2013, 6, 597 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., 2012, 125, 13143 ( Angew. Chem., Int. Ed. , 2013 , 52 , 12905 ) CrossRef.
- J. Li, M. Lutz, M. Otte and R. J. M. Klein Gebbink, ChemCatChem, 2018, 10, 4755 CrossRef CAS PubMed.
- S. Raju, J. T. B. H. Jastrzebski, M. Lutz and R. J. M. Klein Gebbink, ChemSusChem, 2013, 6, 1673 CrossRef CAS PubMed.
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520 CrossRef CAS PubMed.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393 CrossRef CAS.
- J. Fu, E. S. Vasiliadou, K. A. Goulas, B. Saha and D. G. Vlachos, Catal. Sci. Technol., 2017, 7, 4944 RSC.
- H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500 CrossRef CAS.
- R. T. Larson, A. Samant, J. Chen, W. Lee, M. A. Bohn, D. M. Ohlmann, S. J. Zuend and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 14001 CrossRef CAS PubMed.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017 RSC.
- X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., 2014, 126, 4284 ( Angew. Chem., Int. Ed. , 2014 , 53 , 4200 ) CrossRef.
- X. Li and Y. Zhang, ChemSusChem, 2016, 9, 2774 CrossRef CAS PubMed.
- M. Lupacchini, A. Mascitti, V. Canale, L. Tonucci, E. Colacino, M. Passacantando, A. Marrone and N. d'Alessandro, Catal. Sci. Technol., 2019, 9, 3005 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of LRe(CO)3 complexes and general catalysis procedures are included. CCDC 1978458–1978460. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy00618a |
| This journal is © The Royal Society of Chemistry 2020 |