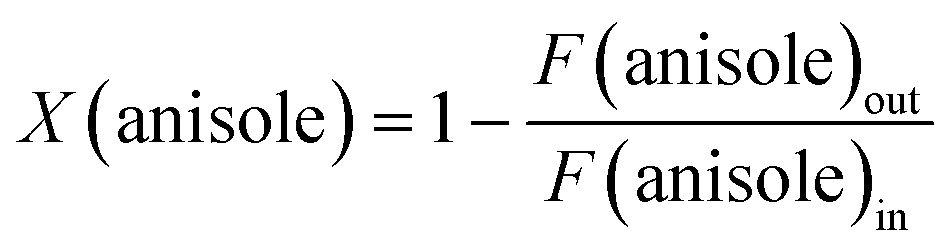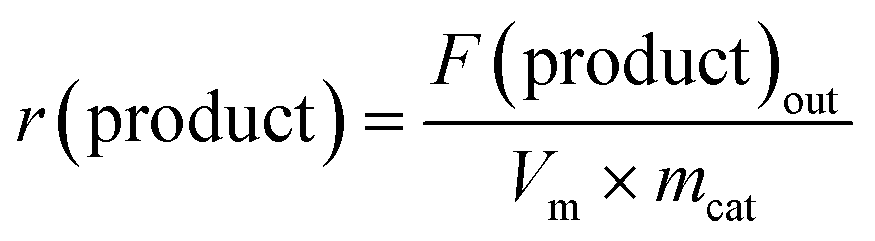Tuning the reactivity of molybdenum (oxy)carbide catalysts by the carburization degree: CO2 reduction and anisole hydrodeoxygenation†
Received
10th March 2020
, Accepted 15th May 2020
First published on 18th May 2020
Abstract
Molybdenum (oxy)carbide catalysts supported on activated carbon were prepared by a carbothermal hydrogen reduction method without passivation step. Four carburization temperatures (500 °C, 600 °C, 700 °C and 800 °C) were selected to control the catalyst carburization degree based on studies of catalyst precursor carburization process by TGA-MS and in situ XANES. Quasi in situ XRD, XAS and XPS revealed that two types of material were produced – molybdenum oxycarbide (500 °C and 600 °C) and molybdenum carbide (700 °C and 800 °C) catalysts. The oxycarbide catalysts are rich in Mo-oxide and Mo-oxycarbide species (MoO2 and MoOxCy) and the carbide catalysts rich in Mo-carbide species (α-MoC1−x and β-Mo2C) with a certain remaining oxygen atoms. The carbidic and oxophilic Mo sites in the catalysts were respectively probed by CO and N2O chemisorption. The structure–performance relationships of these catalysts in CO2 hydrogenation and anisole hydrodeoxygenation (HDO) were studied. For CO2 hydrogenation, the carbide catalysts were much active than the oxycarbide catalysts and CO was the main product in all the catalysts. In contrast, the oxycarbide and carbide catalysts displayed comparable activity towards anisole conversion and the main products shifted from a mixture of phenol and benzene to only benzene upon increasing the carburization temperature from 600 °C to 700 °C. These catalytic results demonstrate that the catalytic performance of molybdenum (oxy)carbide material can be effectively tuned by varying the carburization degree and such tuning effect depends on the nature of reactant molecules: the carbidic Mo sites in Mo-carbide species are associated with CO2 and anisole-to-benzene conversions, and the oxophilic Mo sites in Mo-oxycarbide species are related to anisole-to-phenol conversion.
1. Introduction
Transition metal carbides and molybdenum carbide in particular are efficient and earth-abundant heterogeneous catalysts.1–3 Molybdenum carbide displays promising catalytic performance in reactions such as alkane hydrogenolysis,4 hydrodenitrogenation of organonitrogen compounds,5 dry reforming of methane,6 the water–gas shift (WGS) reaction7 and CO hydrogenation.8 In recent years, the application of molybdenum carbide catalysts has also been extended to catalytic reactions relevant for the transition to a sustainable chemical industry, including CO2 hydrogenation9–14 and biomass valorization.15–19
Notwithstanding its wide catalytic applications, the nature of active site(s) and structure–performance relationship of molybdenum carbide catalysts remain rather unclear. Compared to metallic catalysts, the surface of molybdenum carbide is more complex due to the incorporation of carbon atoms. For example, it has been demonstrated that the chemical and catalytic properties of molybdenum carbides are governed by the carbon-to-metal ratio of the catalyst particles.20 Moreover, the carbidic surface is typically prone to dynamic changes under the reaction conditions. It has, for instance, been demonstrated that oxygen atoms can be incorporated into the carbidic phase during the conversion of oxygen-containing molecules, but different opinions exist regarding the influence of incorporated oxygen on the catalytic performance.21–26 Using density functional theory, Liu et al. showed that C-terminated Mo2C covered with oxygen is more active than Mo2C itself for the WGS reaction due to an optimal bonding of reaction intermediates.25 On the other hand, Choi and co-workers demonstrated that the presence of oxygen in molybdenum carbide is detrimental to benzene hydrogenation activity.22 Kumar et al. reported that increasing the oxygen content in molybdenum carbide catalysts shifts the selectivity in anisole hydrodeoxygenation (HDO) from benzene to phenol.26 The authors postulated that oxygen treatment results in MoOx/MoOxCy clusters with proper surface ensembles required for the aromatic C–O bond cleavage.
Another challenge in establishing accurate structure–performance relationship for molybdenum carbide catalysts is related to the synthesis procedure. Dispersed molybdenum carbide is a highly pyrophoric material and passivation in diluted oxygen is therefore commonly used after the preparation.27,28 Nevertheless, it has been observed that oxidation still slowly proceeds at ambient conditions even after passivation.29 Prior to evaluating its catalytic performance, the passivated samples are usually activated in hydrogen atmosphere at elevated temperature, which leads to the partial removal of O atoms but likely C atoms as well, further complicating the comparison and analysis of catalysts.29,30 A (quasi) in situ approach without passivation–activation procedure is therefore of great advantage to understand the metal carbide catalysts.31
In this study, we prepared a series of molybdenum (oxy)carbide catalysts by a carbothermal hydrogen reduction method using activated carbon support as the carbon source.32 These catalysts were tested for CO2 hydrogenation and anisole hydrodeoxygenation (HDO) to study the influence of catalyst carburization degree on these two reactions. For this purpose, we first studied the catalyst precursor carburization process by TPR-MS and in situ XANES. Based on the obtained insights, four different temperatures (500 °C, 600 °C, 700 °C and 800 °C) were selected to tune the carburization degree of the resulting catalysts. The as-prepared catalysts were directly transferred to a glovebox without passivation, characterized in detail and tested in the catalytic reactions without air exposure we established that the combination of CO chemisorption (sensitive to carbidic Mo sites) and N2O chemisorption (sensitive to all oxophilic Mo sites) is a powerful approach to probe the surface of (oxy)carbide catalysts. The catalytic results demonstrated that the CO2 conversion is govern by carbidic Mo sites, while hydrodeoxygenation of anisole is associated with both carbidic and oxophilic Mo sites.
2. Experimental section
2.1 Catalyst preparation
Activated carbon (NORIT RX-3 extra, SBET = 1219 m2 g−1) was used as-received, crushed and sieved to a 125–250 μm fraction before depositing the molybdenum precursor. The Mo/C catalyst precursor was prepared by wet impregnation using an aqueous solution of (NH4)6Mo7O24·4H2O (AHM, Merck, ≥99%). The starting Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C element weight ratio was 1:5. In a typical synthesis, 1.47 g of AHM was firstly dissolved in 40 mL of demineralized water followed by adding 4.0 g of the carbon support. The resulting dispersion was sonicated for 15 min and stirred at room temperature for 1 h. Next, the water was removed by rotatory evaporation and the obtained solid was further dried at 110 °C overnight. The carburization of Mo/C precursor was performed at a home-built catalyst preparation setup, enabling direct sample transfer to a glovebox. For each sample, 1.0 g of the dried Mo/C precursor was loaded into a quartz reactor and then carburized at selected temperatures (500 °C, 600 °C, 700 °C and 800 °C) for 6 h at a rate of 3 °C min−1 in a 10 vol% H2 in Ar flow (50 mL min−1). After carburization, the catalysts were cooled to room temperature in a flow of Ar (45 mL min−1) and directly transferred to a glovebox, where the samples were prepared for further characterization and catalytic testing. The obtained catalysts are denoted as Mo/C(T), where T stands for the carburization temperature.
:C element weight ratio was 1:5. In a typical synthesis, 1.47 g of AHM was firstly dissolved in 40 mL of demineralized water followed by adding 4.0 g of the carbon support. The resulting dispersion was sonicated for 15 min and stirred at room temperature for 1 h. Next, the water was removed by rotatory evaporation and the obtained solid was further dried at 110 °C overnight. The carburization of Mo/C precursor was performed at a home-built catalyst preparation setup, enabling direct sample transfer to a glovebox. For each sample, 1.0 g of the dried Mo/C precursor was loaded into a quartz reactor and then carburized at selected temperatures (500 °C, 600 °C, 700 °C and 800 °C) for 6 h at a rate of 3 °C min−1 in a 10 vol% H2 in Ar flow (50 mL min−1). After carburization, the catalysts were cooled to room temperature in a flow of Ar (45 mL min−1) and directly transferred to a glovebox, where the samples were prepared for further characterization and catalytic testing. The obtained catalysts are denoted as Mo/C(T), where T stands for the carburization temperature.
Another series of catalysts (passivated and non-passivated) was also prepared at a different preparation setup in order to examine the effect of passivation–activation procedure. Similar conditions were used to carburize the Mo/C precursor. After the carburization the obtained samples were passivated in a 2.5 vol% O2 in He flow (200 ml min−1) at room temperature for 3 h. The resulting passivated catalysts were further stored under ambient conditions. The non-passivated catalysts were transferred and stored in a glovebox without exposing to air.
2.2 Catalyst characterization
Inductively coupled plasma optical emission spectroscopy (ICP-OES).
The Mo loading in Mo/C precursor was determined by ICP-OES (Spectro CIROS CCD spectrometer). Prior to analysis, the sample was heated in 5 mL of concentrated nitric acid at 150 °C for 1 h, and the residual activated carbon support was removed by filtration.
Thermogravimetric analysis (TGA).
The weight loss of Mo/C precursor during carburization process was monitored by TGA using Mettler Toledo TGA/DSC 1 instrument. About 15 mg of sample was placed in an uncovered crucible. The sample was heated to 900 °C at a rate of 5 °C min−1 in a flow of 60 mL min−1 He and 6 mL min−1 H2. The gas effluent was analysed by an online mass spectrometer (Pfeiffer ThermoStar GSD 320 T). The weight loss of as-prepared and used catalysts (after anisole HDO) was also analysed using the same instrument. For this purpose, about 15 mg of catalyst was placed in an uncovered crucible, and then heated to 800 °C at a rate of 5 °C min−1 in a flow of 40 mL min−1 He and 20 mL min−1 O2.
X-ray diffraction (XRD).
The crystal structure of as-prepared catalysts was studied with a Bruker D2 Phaser diffractometer using Cu Kα radiation with a step size of 0.02° at 1.0 s per step in the 2θ range of 20–80°. The samples were grinded, loaded to a sample holder and sealed by Kapton tape in the glovebox before being transferred to the diffractometer.
X-ray photoelectron spectroscopy (XPS).
The surface composition of as-prepared catalysts was analysed using a K-Alpha XPS instrument (Thermo Scientific) with a monochromatic small-spot X-ray source and an 180° double focusing hemispherical analyser. The samples were placed on a double-sided carbon tape in the glovebox, and then transferred to the spectrometer using an air-tight transfer holder. Spectra were collected using an aluminium anode (Al Kα = 1486.68 eV) operating at 72 W and a spot size of 400 μm. Survey scans were measured at a constant pass energy of 200 eV and region scans at 50 eV. All spectra were analysed with CasaXPS software and energy calibration was done against activated carbon C 1s binding energy fixed at 284.6 eV.
CO chemisorption.
The carbidic Mo sites in the catalysts were probed by exposure the catalysts to small pulses of CO at 50 °C using a home-built plug-flow pulsing setup equipped with an online mass spectrometer (Balzers TPG 251). An appropriate amount (ca. 30 mg) of catalyst was loaded into a stainless-steel reactor and sealed by two valves in the glovebox before being transferred to the setup. After heating to 50 °C in a He flow (50 mL min−1), CO pulses were injected into the He flow by a six-way valve with a sample loop of 10 μl until no CO consumption by the catalyst was observed by the mass spectrometry.
N2O chemisorption.
The oxophilic Mo sites in the catalysts were titrated by N2O pulsing using the same setup as for CO chemisorption. N2O reacts with surface oxophilic Mo sites, leading to N2 formation (see below) in a similar way as metallic Cu sites titrated by N2O.33 The same sample preparation and pretreatment were adopted for N2O titration as for CO chemisorption. After reaching 50 °C in a He flow (50 mL min−1), 1 mL of 2 vol% N2O in He was periodically pulsed into the He flow until no N2O consumption was observed by online mass spectrometry.
X-ray absorption spectroscopy (XAS).
The oxidation state and local structure of molybdenum phases were investigated using XAS. Extended X-ray adsorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) spectra were collected at the Mo K-edge in a transmission mode on beamline BM26 (DUBBLE) at the European Synchrotron Radiation Facility (Grenoble, France). The X-ray energy was selected using a Si(111) monochromator and calibrated with molybdenum foil. Ex situ samples were prepared by pressing grinded catalysts, diluted by boron nitride, into stainless-steel sample holders in the glovebox, and sealed with Kapton tape. In situ XANES measurements during Mo/C carburization and anisole HDO reaction were carried out using a high-temperature plug-flow setup as described elsewhere.34 The Mo/C precursor was carburized in situ at 700 °C for 30 min at a rate of 10 °C min−1 in a H2 flow (30 mL min−1), and then cooled to 300 °C before starting the anisole HDO reaction. The reaction conditions were the same as these used for the catalytic measurements (see below). A separate in situ XANES experiment was performed to trace the Mo/C catalyst precursor carburization process during which the temperature was increased to 750 °C at a rate of 5 °C min−1 in a flow of hydrogen. The EXAFS and XANES spectra were background-subtracted and analysed with Athena, which is an interface of the IFRFFIT software package.35
2.3 Catalytic activity measurements
CO2 hydrogenation.
The catalytic performance in CO2 hydrogenation was evaluated in a down-flow stainless-steel reactor (ID = 4 mm) at 250 °C and 30 bar. Typically, 50 mg of catalyst was loaded in the reactor, sealed with two valves in the glovebox and transferred to the catalytic setup. After flushing the reactor with nitrogen, the reactor was pressurized with the reaction gas mixture (20 vol% CO2, 60 vol% H2 and 20 vol% N2) at a total flow of 25 NmL min−1. The reactor was then heated to the reaction temperature at a rate of 5 °C min−1. The effluent gas mixture was analysed by an online gas chromatograph (Interscience, CompactGC) equipped with Rtx-1 (FID), Rt-QBond and Molsieve 5A (TCD), and Rt-QBond (TCD) columns. The CO2 conversion and product selectivity were calculated after ca. 3 h time-on-stream using the following equations:| |  | (1) |
| |  | (2) |
where F stands for the volumetric flow rate calculated based on the internal standard (N2) using calibrated response factors. For the study of passivation–activation effect, the passivated catalysts were activated in a 10 vol% H2 in He flow (50 ml min−1) at 550 °C for 1 h before the catalytic measurements.
Anisole hydrodeoxygenation (HDO).
The catalytic performance in anisole HDO was evaluated in a down-flow quartz reactor (ID = 4 mm) at 300 °C and atmospheric pressure. For this purpose, about 50 mg catalyst was loaded in the reactor and sealed in the glovebox and transferred to the catalytic setup. After flushing with hydrogen, the reactor was switched to a hydrogen flow (30 NmL min−1) saturated with anisole vapour and heated to the reaction temperature at a rate of 5 °C min−1. The anisole vapour (0.3 kPa) was supplied by flowing hydrogen through a thermostated saturator at 19 °C. The effluent gas mixture was analysed by an online chromatograph (Trace GC 1300, Thermo, equipped with an FID detector coupled with Rxi-5Sil MS column). The anisole conversion, product selectivity and product formation rate were calculated after 18 h time-on-stream using the following equations:| |  | (3) |
| |  | (4) |
| |  | (5) |
where F stands for the volumetric flow rate calculated using calibrated response factors and Vm is the molar volume of ideal gas at normal temperature and pressure.
3. Results and discussion
3.1 Mo/C precursor carburization
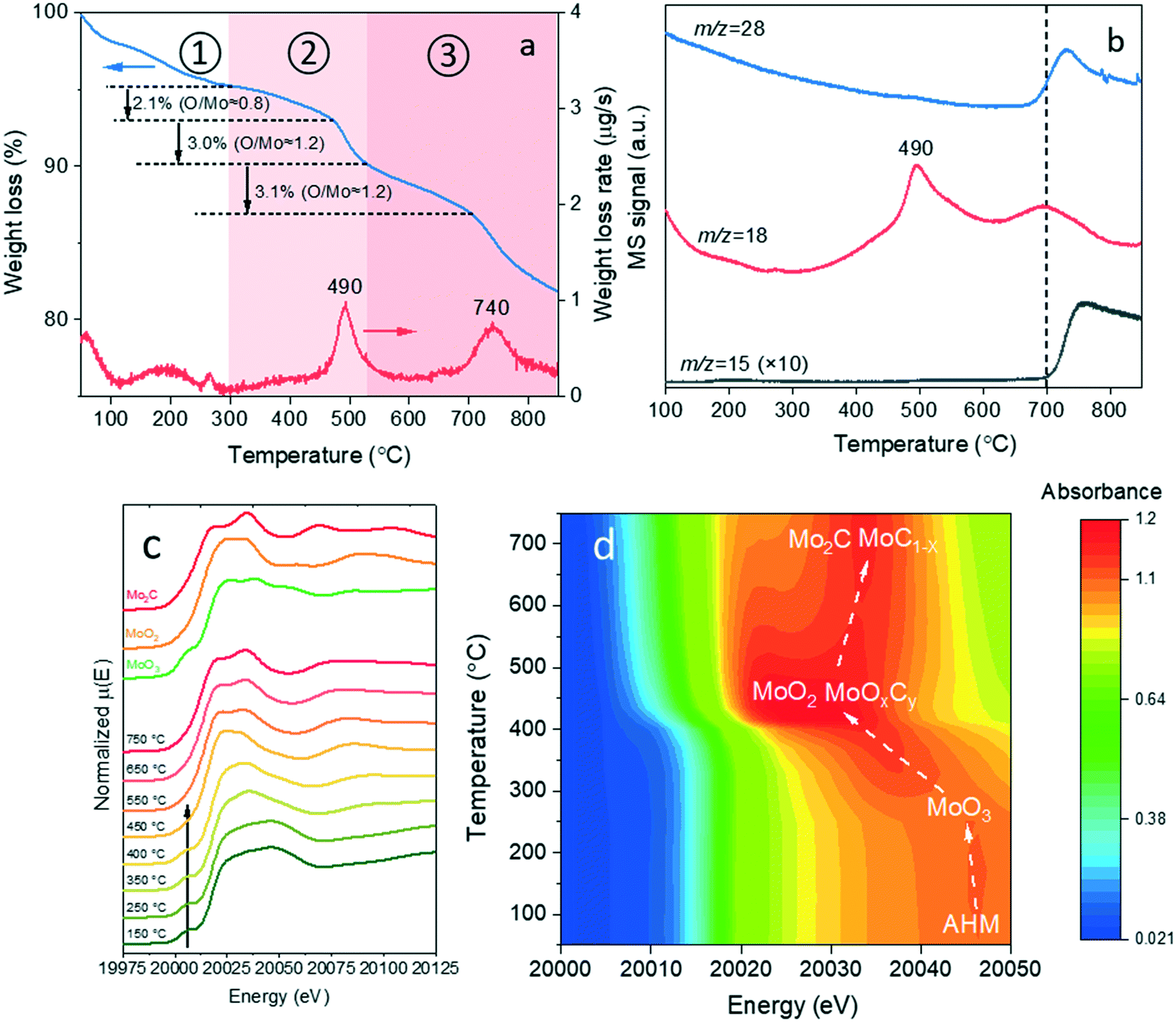
It is well known that interconversions between molybdenum oxides, oxycarbides and carbides are complicated and influenced by various parameters such as the carbon source, gas atmosphere, pretreatment, metal loading, etc.36–39 To select appropriate temperatures to obtain molybdenum (oxy)carbide catalysts, the carburization of Mo/C precursor with Mo loading of 14.9 wt% in a diluted hydrogen flow was first studied by TGA-MS (Fig. 1a and b). The weight loss below 300 °C (the first weight loss region) is due to both the desorption of physisorbed water from the support and the gradual decomposition of AHM precursor to MoO3, likely in a highly dispersed form.40 After the complete decomposition of AHM, a weight loss peak at 490 °C appeared together with water (m/z = 18) formation probed by MS, which can be related to the reduction of MoO3 to MoO2.40 Added to that, we observed the oxygen atoms were also slowly removed as water before the bulk reduction in the second weight loss region. This observation can be tentatively explained by the formation of metastable MoOxCy from MoO3, when the formed MoO3 surface vacancies are filled by carbon atoms.41 The presence of MoOxCy in addition to MoO2 is consistent with the total weight loss during the second weight loss region, showing that the average oxygen atom loss per molybdenum atom is higher than unity. In the third weight loss region, oxygen atoms were continuously removed as water until the second weight loss peak appeared (ca. 740 °C). In addition to water, CO (m/z = 28) and a small amount of CH4 (m/z = 15) were observed starting from 700 °C, pointing to the involvement of the carbon support in the molybdenum carburization process. Nevertheless, it should be noted that the molybdenum carburization was not complete even at 850 °C as indicated by the ongoing CO (m/z = 28) production.
 |
| | Fig. 1 Mo/C catalyst precursor carburization process studies. (a) TGA and DTGA profiles and (b) corresponding mass spectra during precursor carburization. (c) In situ XANES spectra and (d) magnification of white line region at Mo K-edge during precursor carburization. | |
In situ XANES (Fig. 1c and d) was further used to trace the evolution of the molybdenum phases during the carbothermal reduction synthesis. Fig. 1c shows the XANES spectra of Mo/C precursor during carburization with increasing temperature together with reference spectra of Mo2C, MoO2 and MoO3. The characteristic pre-edge feature related to the dipole-forbidden 1s → 4d electronic transition and assigned to tetrahedrally coordinated MoVI species42 disappeared, as the temperature increased from 400 °C to 450 °C. We ascribe this observation to the bulk reduction of MoO3 to MoO2.40 The MoO2-like phase was further reduced and carburized into a Mo2C-like phase as inferred from the similarity between the final spectrum at 750 °C and the Mo2C reference spectrum. Based on the TG and XANES results, three major carburization stages are tentatively suggested in Fig. 1d: (i) the AHM precursor decomposition to MoO3 (<300 °C), (ii) MoO3 reduction into a mixture of MoO2 and MoOxCy (300–450 °C) followed by (iii) further carburization at higher temperature (>450 °C) leads to the formation of molybdenum carbide(s). It should be mentioned that both α-MoC1−x and β-Mo2C phases can be formed from a mixture of MoO2 and MoOxCy.9 The nature of the formed molybdenum carbide phase(s) cannot be determined conclusively at this stage and will be studied in the following sections.
3.2 Effect of passivation and activation
To examine the effect of passivation–activation procedure on the catalytic performance, we carried out several CO2 hydrogenation tests with passivated and non-passivated molybdenum carbide catalysts. These catalysts were prepared at 800 °C. The catalytic results (Fig. S1a†) showed that, without the activation procedure, the passivated catalyst was significantly less active than the non-passivated catalyst. Moreover, even after activating at 550 °C and ambient pressure for 1 h in 10 vol% hydrogen flow (50 mL min−1), the resulting catalyst was still much less active than the non-passivated catalyst. Quasi in situ XPS analysis of these catalysts after reaction (Fig. S1b†) showed that the surface Mo oxidation degree (non-passivated < passivated–activated < passivated) was inversely correlated to the CO2 conversion (non-passivated > passivated–activated > passivated). This observation clearly points to the superiority of passivation-free approach over the passivation–activation approach for the preparation of molybdenum carbide hydrogenation catalysts. Based on these findings in the present work we will focus on the catalysts prepared by the passivation-free approach.
3.3 Mo/C(T) catalyst characterization
Based on the Mo/C carburization studies and analysis of passivated and non-passivated catalysts, we chose four temperatures (500 °C, 600 °C, 700 °C and 800 °C) to prepare molybdenum (oxy)carbide catalysts with controlled carburization degree without passivation step. We characterized these materials in detail with respect to their structural and surface properties. The as-prepared catalysts were firstly examined by XRD (Fig. 2a). The XRD patterns of the samples carburized at 500 °C and 600 °C can be assigned to MoO2 phase. The relevant diffraction peaks become broader as the carburization temperature increased, indicating the further reduction/carburization of MoO2 phase. As the reduction temperature reached 700 °C, diffraction peaks of β-Mo2C phase appeared while the peaks related to MoO2 phase disappeared. The β-Mo2C phase was crystallized further from 700 °C to 800 °C as evidenced by the narrower and more intense XRD peaks observed in the Mo/C(800) sample. A new diffraction peak at 42.4° was observed in the Mo/C(800) sample, which can be assigned to α-MoC1−x phase.9 Given the significant broadening of this new peak, we speculate that the α-MoC1−x phase may originate from a highly disordered and/or dispersed molybdenum oxycarbide phase.9
 |
| | Fig. 2 (a) XRD patterns and (b) k3-weighted R-space plots of as-prepared molybdenum (oxy)carbide catalysts. | |
We further studied the (oxy)carbide catalysts by XAS. The data was plotted in R-space with MoO2 and β-Mo2C standards to help data interpretation (Fig. 2b). In line with the XRD results, the MoO2 phase was consumed when the temperature was increased from 500 °C to 600 °C as indicated by the decreasing contribution of the Mo–Mo shell (3.2 Å) from the MoO2 phase. Moreover, the β-Mo2C phase formed at 700 °C and its contribution increased upon further raising the temperature to 800 °C as revealed by the increasing contribution of the Mo–Mo shell (2.7 Å) from the β-Mo2C phase. Additional weak Mo–Mo contributions (marked by red arrows) with a coordination distance of 2.5 Å appeared in the Mo/C(500) and Mo/C(600) samples. Based on the above Mo/C precursor carburization and XRD results, we suggest that this contribution can be related to dispersed molybdenum oxycarbide phase (i.e. MoOxCy).
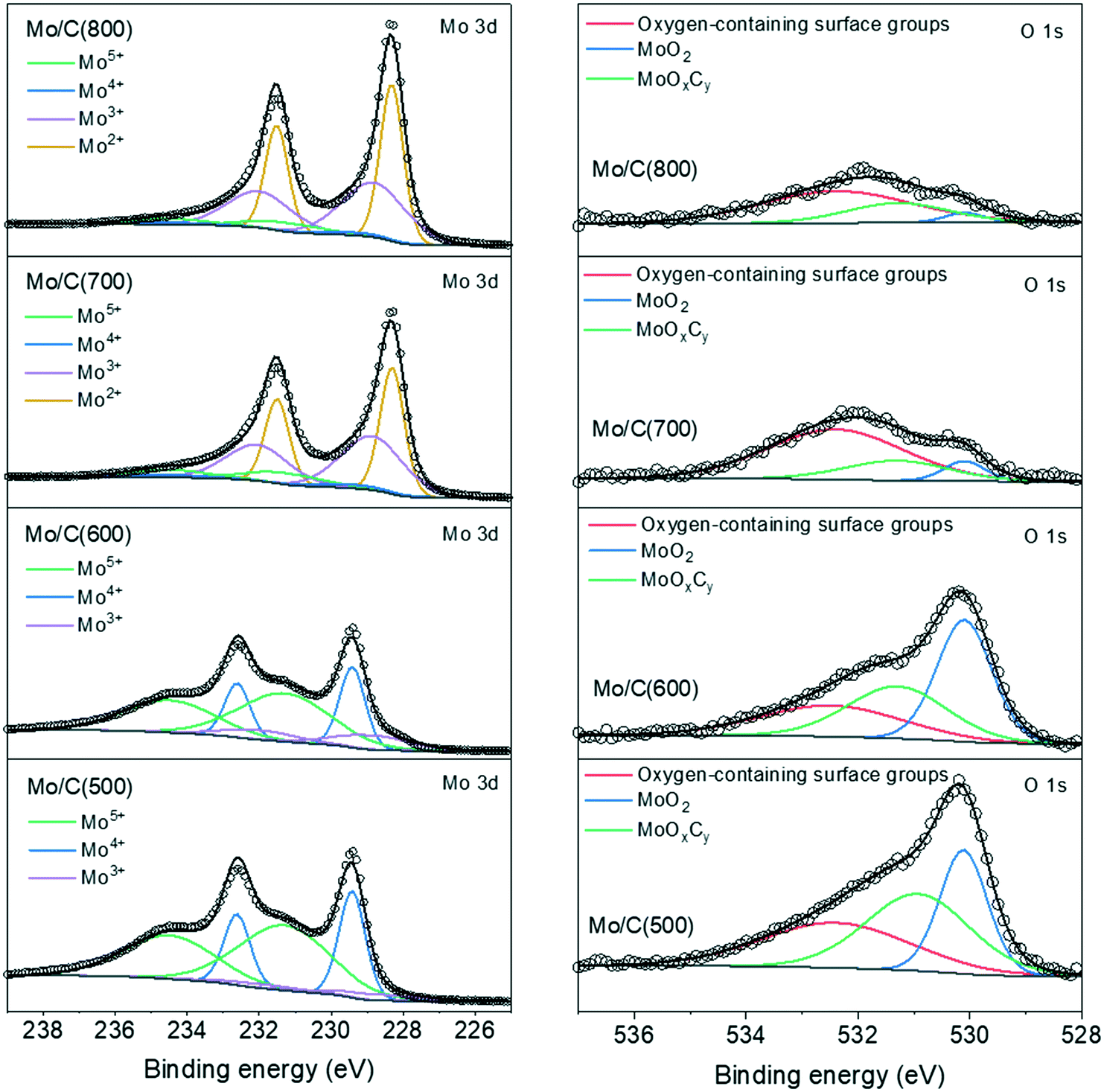
The XRD and XAS data points to the presence of MoO2 and Mo-carbide species (β-Mo2C and α-MoC1−x) in the catalysts, but the presence of MoOxCy species requires additional characterization. For this purpose, we employed XPS to study the Mo oxidation state and surface composition of the catalysts (Fig. 3). We focused on the Mo 3d and O 1s regions in this study since activated carbon was used as the support and its signal dominates the C 1s region. The Mo 3d spectra were deconvoluted into four doublet contributions (Mo5+, Mo4+, Mo3+ and Mo2+) and the deconvolution results are shown in Table 1. The Mo6+ contribution assigned to MoO3 was negligible in all the samples owing to the passivation-free approach.43 The Mo4+ contribution is assigned to MoO2 (ref. 13 and 44) and the Mo3+ and Mo2+ contributions to Mo-carbides (α-MoC1−x and β-Mo2C).13,31,45 Significant Mo5+ contributions were observed in the Mo/C(500) and Mo/C(600) catalysts, which are assigned to MoOxCy.41,46 For the O 1s spectra, the contributions with binding energies of 532.4 eV, 530.9 eV and 530.1 eV are assigned to oxygen from surface groups on carbon support, MoOxCy and MoO2, respectively.47 The oxygen contributions from MoOxCy and MoO2 species were used to calculate the overall O/Mo atomic ratio in the catalysts (Table 1).
 |
| | Fig. 3 Mo 3d and O 1s XP spectra of as-prepared molybdenum (oxy)carbide catalysts. | |
Table 1 O/Mo ratio and Mo 3d XPS analysis of as-prepared molybdenum (oxy)carbide catalysts
| Catalyst |
O/Mo atomic ratio |
Mo 3d5/2 binding energy (eV) |
Composition (mol%) |
| Mo5+ |
Mo4+ |
Mo3+ |
Mo2+ |
Mo5+ |
Mo4+ |
Mo3+ |
Mo2+ |
| Mo/C(500) |
1.43 |
231.3 |
229.4 |
228.9 |
228.3 |
64 |
30 |
6 |
0 |
| Mo/C(600) |
1.48 |
231.3 |
229.4 |
228.9 |
228.3 |
58 |
21 |
15 |
0 |
| Mo/C(700) |
0.38 |
231.3 |
229.4 |
228.9 |
228.3 |
12 |
4 |
44 |
40 |
| Mo/C(800) |
0.22 |
231.3 |
229.4 |
228.8 |
228.3 |
9 |
3 |
44 |
45 |
The XPS results showed that significant surface molybdenum carburization occurs upon increasing the temperature from 600 °C to 700 °C as indicated by the shift of the predominant oxidation states from Mo5+ and Mo4+ to Mo3+ and Mo2+, and by the significant decrease of the O/Mo atomic ratio. This observation is consistent with the previous XRD and XAS observations. Importantly, significant amounts of MoOxCy species (Mo5+) were observed by XPS which were not detected by XRD. Such difference can be understood since the formed MoOxCy surface species from partially reduced MoO3 are highly disordered and/or dispersed in nature.41 In line with TG-MS results, molybdenum carburization was not complete even at the highest carburization temperature (800 °C) as indicated by the presence of Mo5+ and Mo4+ contributions and presence of oxygen on the surface of the Mo/C(800) catalyst. Based on these observations, we conclude that Mo/C(700) and Mo/C(800) samples can be described as carbide surfaces with a certain amount of remaining oxygen atoms.22
We further used chemisorption (CO and N2O) to directly probe the surface sites in the catalysts.48 Control experiments showed that the bare carbon support did not chemisorb CO or N2O. The results in Table 2 show that the Mo/C(500) and Mo/C(600) catalysts contained almost no carbidic Mo sites, while Mo/C(700) and Mo/C(800) catalysts contained considerable amounts of these sites. This observation indicates that significant molybdenum carburization took place between 600 °C and 700 °C. Next, N2O was used as a probe molecule to measure surface oxophilic Mo sites (i.e. all oxidizable surface centres) in the catalysts. A representative N2O titration profile is shown in Fig. S2.† The high surface-sensitivity of this technique was confirmed by a quasi in situ XPS analysis (Fig. S3†), which showed that no significant oxidation took place after N2O chemisorption. Given that XPS probes top few nm of the surface, the absence of significant oxidation observed in XP spectra implies that only the top atomic layer of the catalysts is probed by N2O. In contrast to CO chemisorption, the N2O titration results show that significant amounts of oxophilic Mo sites were probed by N2O in the Mo/C(500) and Mo/C(600) catalysts. The substantial difference in the Mo sites probed by CO and N2O in the Mo/C(500) or Mo/C(600) suggests that other molybdenum-containing species contribute to the N2O consumption in addition to Mo-carbide species. Reference experiment with MoO2 standards showed that this material was completely inactive for N2O chemisorption. As such, we suggest that N2O molecule also probes the oxophilic Mo sites in the MoOxCy species (i.e., oxygen vacancies49), which can not be probed by CO. The Mo/C(700) and Mo/C(800) also contain significant amounts of oxophilic Mo sites, in line with previous oxygen chemisorption study on molybdenum carbide.48 It should be mentioned that the difference between the amounts of carbidic and oxophilic Mo sites in the Mo/C(700) and Mo/C(800) is due to the incomplete molybdenum carburization22 and the relatively high chemisorption temperature (50 °C) used in current study.50 Based on these results, we propose that CO and N2O can be used as complementary probe molecules for the quantification of carbidic and oxophilic Mo sites in the (oxy)carbide catalysts.
Table 2 Carbidic and oxophilic Mo site density in as-prepared molybdenum (oxy)carbide catalysts
| Catalyst |
Carbidic Mo sites (μmol gcat−1) |
Oxophilic Mo sites (μmol gcat−1) |
| Mo/C(500) |
0.2 |
64.0 |
| Mo/C(600) |
0.3 |
62.2 |
| Mo/C(700) |
7.2 |
129.8 |
| Mo/C(800) |
11.2 |
120.8 |
Using a combination of XRD, XAS, XPS and chemisorption (CO and N2O), we have demonstrated that molybdenum (oxy)carbide catalysts with controlled carburization degree (i.e. oxygen removal extent) can be prepared by adjusting the carburization temperature. Specifically, the Mo/C(500) and Mo/C(600) catalysts are rich in MoOxCy and MoO2 species and the Mo/C(700) and Mo/C(800) catalysts are rich in Mo-carbide species (α-MoC1−x and β-Mo2C) with a certain amount of remaining oxygen atoms. In the following sections, therefore, we refer to the Mo/C(500) and Mo/C(600) samples as oxycarbide catalysts and to the Mo/C(700) and Mo/C(800) samples as carbide catalysts.
3.4 Catalytic performance
3.4.1 CO2 hydrogenation.
The CO2 hydrogenation reaction was evaluated over the as-prepared molybdenum (oxy)carbide catalysts to study the influence on the molybdenum carburization degree (Fig. 4a). Blank catalytic test with carbon support showed that it is inactive for CO2 conversion. Fig. 4a shows that the catalysts prepared at higher carburization temperature displayed enhanced CO2 conversion and all the catalysts showed similar product selectivity profiles independent of CO2 conversion level. CO was the predominant product with a selectivity in the range of 60–75%. In addition, other products included methane (10–20%), methanol (10–20%) and small amounts of ethane and propane (<2%). The CO2 conversion and product distribution as a function of carburization temperature, together with previous characterization results, suggest that Mo-carbide species are responsible for CO2 hydronation. To gain more insight into the active sites for CO2 hydrogenation, the CO2 conversion was plotted against the amount of carbidic Mo (Fig. 4b) and oxophilic Mo sites (Fig. S6†) determined from chemisorption. Fig. 4b shows that CO2 conversion scales well with the amount of carbidic Mo sites in the catalysts, which underpins the involvement of Mo-carbide species in CO2 conversion. Such strong correlation, however, between the CO2 conversion and the amount of oxophilic Mo sites does not exist.
 |
| | Fig. 4 (a) CO2 conversion and product selectivity and (b) correlation between CO2 conversion and carbidic Mo site density measured by CO chemisorption. Reaction conditions: 250 °C, 30 bar and SV = 30 L gcat−1 h−1. | |
3.4.2 Anisole hydrodeoxygenation (HDO).
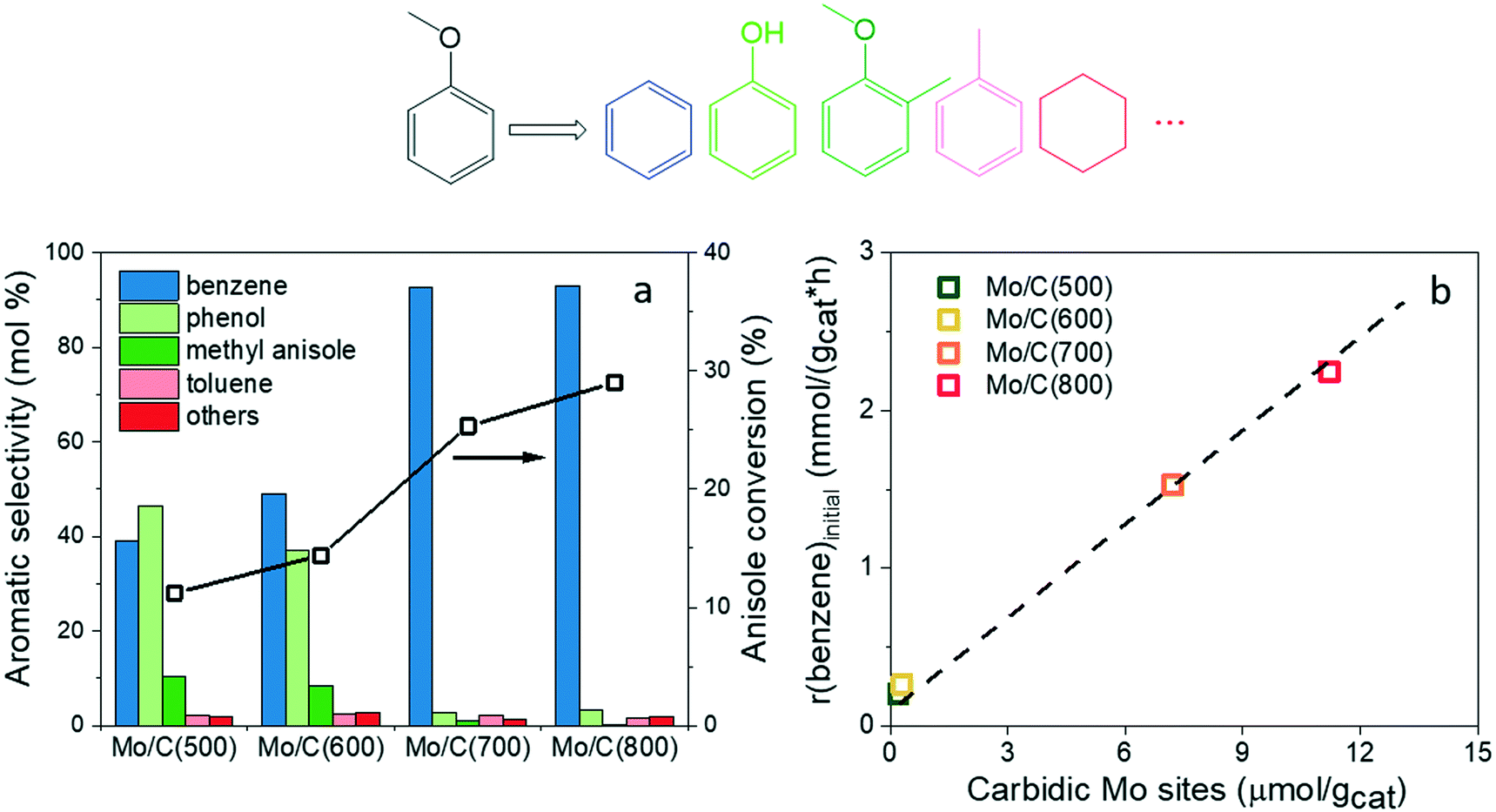
Next, anisole HDO was performed over the as-prepared molybdenum (oxy)carbide catalysts. As similar in above CO2 hydrogenation, no activity was observed over the bare carbon support. However, the conversion and product distribution in anisole HDO over the as-prepared catalysts show different dependence on the carburization temperature (Fig. 5a) as compared to CO2 conversion. For example, the conversion difference between the oxycarbide and carbide catalysts is not as significant as in CO2 hydrogenation. Moreover, different product selectivity profiles were observed between the oxycarbide and carbide catalysts at the end of the test (ca. 1 h) – the selectivity shifted from a mixture of benzene and phenol to predominantly benzene (ca. 93%) as the carburization temperature increases from 600 °C to 700 °C. Moreover, the methyl anisole selectivity over the oxycarbide catalysts was significantly higher than that over the carbide catalysts. It should be mentioned that no significant changes in selectivity were observed during the reaction (ca. 17–20 h) as showed in Fig. S8.† Clearly, active sites other than carbidic Mo sites are available in the oxycarbide catalysts for anisole activation. To gain insights into the active sites for anisole conversion, initial benzene formation rate (TOS = 30 min) was plotted against CO chemisorption data (Fig. 5b). The initial reaction rate was used here as a severe catalyst deactivation occurs at the beginning of the reaction over carbide catalysts (see below). Similar to CO2 hydrogenation, we found that the initial benzene formation rate is proportional to the amounts of carbidic Mo sites, pointing to the involvement of these sites in anisole-to-benzene conversion. Regarding anisole-to-phenol conversion, it is likely associated with the oxygen content in the oxycarbide catalysts. This hypothesis is supported by a recent report by Kumar et al.26 in which the authors showed that oxygen treatment of molybdenum carbide catalysts led to higher phenol selectivity in anisole HDO. In the current study, the phenol production from anisole over the oxycarbide catalysts is likely associated with MoOxCy species because MoO2 was proven to be completely inactive for anisole HDO under similar conditions.51 Importantly, the presence of such oxycarbide species is explicitly revealed by quasi in situ XPS analysis in this study. Moreover, the oxophilic Mo sites in MoOxCy species (e.g. oxygen vacancies49), the potential active sites for phenol formation, can be quantified by N2O chemisorption.
 |
| | Fig. 5 (a) Anisole conversion and product selectivity and (b) correlation between initial benzene formation rate and carbidic Mo site density measured by CO chemisorption. Reaction conditions: 300 °C and 1 bar. | |
As molybdenum carbide appears to be a promising catalyst for selective benzene production from anisole, we further investigated its time-on-stream behaviour. Fig. 6a shows that the benzene formation rate decreased significantly at the beginning of the reaction, and then decreased at a slower rate as the reaction continued. Similar catalyst deactivation behaviour was observed in literature31 and two main deactivation mechanisms were proposed in literature: carbide oxidation and carbonaceous deposition.52 To shed light on the deactivation mechanism over the Mo/C(700) catalyst, several (in situ) techniques were used to study the catalyst modification during anisole HDO. In situ XANES spectra (Fig. 6b) revealed that no bulk phase oxidation occurred at the beginning of reaction. Further quasi in situ XPS measurements (Fig. 6c) also showed that no significant surface oxidation took place after anisole HDO reaction, although even more surface-sensitive technique might be necessary to answer this question definitively.31 On the other hand, carbonaceous deposition after anisole HDO was evidenced by TG analysis of the used catalyst. A significant weight loss occurred in the temperature range of 100–300 °C. It should be noted that the initial weight gain in the as-prepared catalyst is due to oxidation of molybdenum carbide in air.53 The total weight loss until 600 °C was about 8 wt% higher for the used catalyst compared to the as-prepared catalyst. Further calculation indicates that the carbonaceous deposition in the used catalyst after anisole HDO was ∼25 wt%. The combined XANES, XPS and TG results suggest that carbonaceous deposition, instead of carbide phase oxidation, is likely the main reason for the quick carbide catalyst deactivation during anisole HDO. To mitigate the deactivation caused by carbonaceous deposition, one might carry out anisole HDO at higher hydrogen pressure and temperature54 or enhance the hydrogenation ability of molybdenum carbide catalyst such as by increasing phase crystallinity55 or by adding a second metal function.56
 |
| | Fig. 6 (a) Time-on-stream benzene formation rate and (b) in situ XANES spectra at Mo K-edge during anisole HDO (TOS from 0 min to 90 min) over Mo/C(700) catalyst. (c) Quasi in situ XPS spectra and (d) TGA and DTGA profiles of Mo/C(700) catalysts before and after anisole HDO. | |
3.4.3 Active site(s) of CO2 and anisole conversion.
Although the reactions of CO2 hydrogenation and anisole HDO both involve CO bond cleavage, our catalytic results show that these two reactions correlate differently to the Mo sites measured by chemisorption. Specifically, the carbidic Mo sites (probed by CO) are associated with CO2 hydrogenation and anisole-to-benzene conversion, while the oxophilic Mo sites (probed by N2O) in oxycarbide phase are inactive for CO2 conversion but associated with anisole-to-phenol conversion. The bond strength of the cleaved C–O bond may explain such difference: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2 (BDE: 532.2 kJ mol−1) is much stronger than aryl C–O (BDE: 418.8 kJ mol−1) and particularly alkyl C–O (BDE: 263.2 kJ mol−1) bonds in anisole.57 In other words, CO2 activation is more difficult than anisole activation, and it is possible that the oxygen binding energy to oxycarbide surface is not strong enough to dissociate the CO bond in CO2 as compared to the carbide surface.58 Furthermore, the selectivity shift in anisole HDO may be explained by the adsorption geometry anisole adopts over the (oxy)carbide surface. Eng et al. used HREELS to show that benzene interacts with a carbide-modified Mo(110) surface in a planar manner, and such interaction is much weaker on an oxide-modified carbide surface.59 Based on that, we postulate that anisole may adopt an on-top geometry over the carbide surface and an end-on geometry over the oxycarbide surface. Such difference in adsorption geometry leads to the selective cleavage of aryl C–O or alkyl C–O in anisole. Further kinetic and spectroscopic analysis together with theoretical calculation are necessary in order to fully understand the mechanism of CO2 and anisole conversion over (oxy)carbide surfaces.
O bond in CO2 (BDE: 532.2 kJ mol−1) is much stronger than aryl C–O (BDE: 418.8 kJ mol−1) and particularly alkyl C–O (BDE: 263.2 kJ mol−1) bonds in anisole.57 In other words, CO2 activation is more difficult than anisole activation, and it is possible that the oxygen binding energy to oxycarbide surface is not strong enough to dissociate the CO bond in CO2 as compared to the carbide surface.58 Furthermore, the selectivity shift in anisole HDO may be explained by the adsorption geometry anisole adopts over the (oxy)carbide surface. Eng et al. used HREELS to show that benzene interacts with a carbide-modified Mo(110) surface in a planar manner, and such interaction is much weaker on an oxide-modified carbide surface.59 Based on that, we postulate that anisole may adopt an on-top geometry over the carbide surface and an end-on geometry over the oxycarbide surface. Such difference in adsorption geometry leads to the selective cleavage of aryl C–O or alkyl C–O in anisole. Further kinetic and spectroscopic analysis together with theoretical calculation are necessary in order to fully understand the mechanism of CO2 and anisole conversion over (oxy)carbide surfaces.
4. Conclusions
We have demonstrated that the carburization degree of molybdenum (oxy)carbide catalyst can be tuned by varying the carbothermal synthesis temperature. In situ XANES, TGA, XRD, EXAFS, XPS and chemisorption results revealed that the catalyst was transformed from a surface rich in MoO2 and MoOxCy species to a surface rich in Mo-carbide species (α-MoC1−x and β-Mo2C) between 600 °C and 700 °C. Catalytic results showed that carbide catalysts were much more active than oxycarbide catalysts for CO2 conversion, pointing to the relevance of carbidic Mo sites in CO2 activation. In contrast, oxycarbide and carbide catalysts displayed comparable anisole conversion and different product selectivity profile during anisole HDO. The oxophilic Mo sites in MoOxCy species promote the anisole-to-phenol conversion, whereas the carbidic Mo sites in carbide species facilitate the deeper hydrogenolysis of anisole to benzene.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge financial support from The Netherlands Organization for Scientific Research (NWO) for a Vici grant. We acknowledge DUBBLE beamline (BM26A) at ESRF for allocating the time for XAS measurements.
References
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- S. T. Gyama, Catal. Today, 1992, 15, 179–200 CrossRef.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- G. S. Ranhotra, G. W. Haddix, A. T. Bell and J. A. Reimer, J. Catal., 1987, 108, 24–39 CrossRef CAS.
- J. G. Choi, J. R. Brenner and L. T. Thompson, J. Catal., 1995, 154, 33–40 CrossRef CAS.
- A. J. Brungs, A. P. E. York and M. L. H. Green, Catal. Lett., 1999, 57, 65–69 CrossRef CAS.
- J. Patt, D. Moon, C. Phillips and L. Thompson, Catal. Lett., 2000, 65, 193–195 CrossRef CAS.
- P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2003, 251, 449–455 CrossRef CAS.
- W. Xu, P. J. Ramirez, D. Stacchiola and J. A. Rodriguez, Catal. Lett., 2014, 144, 1418–1424 CrossRef CAS.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- S. F. Viñes, J. A. Rodriguez and C. Sci, Catal. Sci. Technol., 2016, 6, 6766–6777 RSC.
- C. Kunkel, F. Vines and F. Illas, Energy Environ. Sci., 2016, 1, 141–144 RSC.
- J. Gao, Y. Wu, C. Jia, Z. Zhong, F. Gao, Y. Yang and B. Liu, Catal. Commun., 2016, 84, 147–150 CrossRef CAS.
- X. Liu, C. Kunkel, P. Ram, D. Piscina and F. Vin, ACS Catal., 2017, 7, 4323–4335 CrossRef CAS.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef CAS.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed.
- W. S. Lee, Z. Wang, R. J. Wu and A. Bhan, J. Catal., 2014, 319, 44–53 CrossRef CAS.
- W. S. Lee, Z. Wang, W. Zheng, D. G. Vlachos and A. Bhan, Catal. Sci. Technol., 2014, 4, 2340–2352 RSC.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef CAS.
- S. Posada-Perez, F. Vines, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- Y. Li, Y. Fan, J. He, B. Xu, H. Yang, J. Miao and Y. Chen, Chem. Eng. J., 2004, 99, 213–218 CrossRef CAS.
- J.-S. Choi, G. Bugli and G. Djéga-Mariadassou, J. Catal., 2000, 193, 238–247 CrossRef CAS.
- C. J. Chen and A. Bhan, ACS Catal., 2017, 7, 1113–1122 CrossRef CAS.
- M. Dixit, X. Peng, M. D. Porosoff, H. D. Willauer and G. Mpourmpakis, Catal. Sci. Technol., 2017, 7, 5521–5529 RSC.
- P. Liu and J. A. Rodriguez, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- A. Kumar and A. Bhan, Chem. Eng. Sci., 2019, 197, 371–378 CrossRef CAS.
- A. Mehdad, R. E. Jentoft and F. C. Jentoft, J. Catal., 2017, 347, 89–101 CrossRef CAS.
- H.-M. Wang, X.-H. Wang, M.-H. Zhang, X.-Y. Du, W. Li and K.-Y. Tao, Chem. Mater., 2007, 19, 1801–1807 CrossRef CAS.
- K. J. Leary, J. N. Michaels and A. M. Stacy, J. Catal., 1986, 101, 301–313 CrossRef CAS.
- D. C. LaMont, A. J. Gilligan, A. R. S. Darujati, A. S. Chellappa and W. J. Thomson, Appl. Catal., A, 2003, 255, 239–253 CrossRef CAS.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- C. Liang, P. Ying and C. Li, Chem. Mater., 2002, 14, 3148–3151 CrossRef CAS.
- J. R. Jensen, T. Johannessen and H. Livbjerg, Appl. Catal., A, 2004, 266, 117–122 CrossRef CAS.
- N. Kosinov, A. Wijpkema, E. Uslamin, R. Rohling, F. Coumans, B. Mezari, A. Parastaev, A. Poryvaev, M. Fedin, E. Pidko and E. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. A. W. Hollak, R. W. Gosselink, D. S. Van Es and J. H. Bitter, ACS Catal., 2013, 3, 2837–2844 CrossRef CAS.
- R. Guil-López, E. Nieto, J. A. Botas and J. L. G. Fierro, J. Solid State Chem., 2012, 190, 285–295 CrossRef.
- M. L. Frauwallner, F. López-Linares, J. Lara-Romero, C. E. Scott, V. Ali, E. Hernández and P. Pereira-Almao, Appl. Catal., A, 2011, 394, 62–70 CrossRef CAS.
- E. Ochoa, D. Torres, R. Moreira, J. L. Pinilla and I. Suelves, Appl. Catal., B, 2018, 239, 463–474 CrossRef CAS.
- J. Wienold, R. E. Jentoft and T. Ressler, Eur. J. Inorg. Chem., 2003, 1058–1071 CrossRef CAS.
- P. Delporte, F. Meunier, C. Pham-Huu, P. Vennegues, M. J. Ledoux and J. Guille, Catal. Today, 1995, 23, 251–267 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068–7072 CrossRef CAS PubMed.
- H. Wang, S. Liu and K. J. Smith, Energy Fuels, 2016, 30, 6039–6049 CrossRef CAS.
- J. A. Schaidle, A. C. Lausche and L. T. Thompson, J. Catal., 2010, 272, 235–245 CrossRef CAS.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399–3405 RSC.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., 2014, 126, 6525–6528 CrossRef.
- J. A. Schaidle, J. Blackburn, C. A. Farberow, C. Nash, K. X. Steirer, J. Clark, D. J. Robichaud and D. A. Ruddy, ACS Catal., 2016, 6, 1181–1197 CrossRef CAS.
- T. P. St. Clair, B. Dhandapani and S. T. Oyama, Catal. Lett., 1999, 58, 169–171 CrossRef.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- J. S. Lee, K. H. Lee and J. Y. Lee, J. Phys. Chem., 1992, 96, 362–366 CrossRef CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- W. S. Lee, A. Kumar, Z. Wang and A. Bhan, ACS Catal., 2015, 5, 4104–4114 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- D. R. Stellwagen and J. H. Bitter, Green Chem., 2015, 17, 582–593 RSC.
- Q. Bkour, O. G. Marin-Flores, M. G. Norton and S. Ha, Appl. Catal., B, 2019, 245, 613–622 CrossRef CAS.
-
Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Taylor & Francis Group, 1st edn, 2007 Search PubMed.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- J. Eng, B. E. Bent, B. Frühberger and J. G. Chen, J. Phys. Chem. B, 1997, 101, 4044–4054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00484g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Evgeny A.
Uslamin
,
Evgeny A.
Uslamin