
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the conversion of CO2 to value added products over composite PdZn and H-ZSM-5 catalysts: excess Zn over Pd, a compromise or a penalty?†
Christian
Ahoba-Sam
 a,
Elisa
Borfecchia
b,
Andrea
Lazzarini
ac,
Aram
Bugaev
de,
Abdulrahman Adamu
Isah
f,
Mostafa
Taoufik
f,
Silvia
Bordiga
ab and
Unni
Olsbye
*a
a,
Elisa
Borfecchia
b,
Andrea
Lazzarini
ac,
Aram
Bugaev
de,
Abdulrahman Adamu
Isah
f,
Mostafa
Taoufik
f,
Silvia
Bordiga
ab and
Unni
Olsbye
*a
aSMN Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, N-0315 Oslo, Norway. E-mail: unni.olsbye@kjemi.uio.no
bDepartment of Chemistry, NIS Center and INSTM Reference Center, University of Turin, 10125, Turin, Italy
cDepartment of Physical and Chemical Sciences, University of l'Aquila, Via Vetoio 42 (COPPITO 2), 67100, l'Aquila, Italy
dThe Smart Materials Research Institute, Southern Federal University, Sladkova 178/24, 344090, Rostov-on-Don, Russia
eSouthern Scientific Centre, Russian Academy of Sciences, Chekhova 41, 344006 Rostov-on-Don, Russia
fUniversité Lyon 1, Institut de Chimie Lyon, CPE Lyon CNRS, UMR 5265 C2P2, LCOMS, 43 Bd du 11 November 1918, 69616 Villeurbanne Cedex, France
First published on 16th June 2020
Abstract
A challenge in converting CO2 into hydrocarbons (HC) via methanol (MeOH) is the gap between the optimal temperature for each step (i.e. ≤250 °C for MeOH and ≥350 °C for HC). The focus of this study is to elucidate the cause of the limitations associated to oxygenate and hydrocarbon formation in combined PdZn and H-ZSM-5 catalysts at 250 to 350 °C. Starting with two different chemical states of Pd and Zn from two preparation approaches (physical mixture and surface organometallic chemistry grafting), operando X-ray absorption spectroscopy (XAS) and powder X-ray diffraction (PXRD) studies revealed similar PdZn alloy active phase formed during pretreatment in flowing H2/Inert at 400 °C. The physical mixture performed better than the grafted analogue, with up to 8.8% yield to oxygenates (MeOH and dimethyl ether (DME); MeOH+) at 300 °C, close to the estimated thermodynamic yield (9.0%). The space–time yield (STY) of oxygenates increased from 250 to 300 °C, reaching 8.5 mol(MeOH+) kg(PdZn)−1 h −1. The catalyst performance surpassed other reported yields in similar systems, which activity declined with temperature even below 300 °C. Operando XAS and PXRD experiments further showed that the PdZn phase active for MeOH formation was maintained during testing in the 250–350 °C range. InfraRed (FT-IR) and XAS experiments revealed the poisoning of Brønsted acid sites in H-ZSM-5 by Zn(II) exchange, thereby rendering it inactive for hydrocarbon formation. Overall, to avoid biasing the hybrid catalyst performance, a careful and balanced choice of the compositional characteristics will be crucial in designing an improved multi-functional catalytic system for CO2 valorisation.
1. Introduction
Chemical conversion of CO2 to value-added chemicals and fuels potentially presents an important substitute for the rapidly dwindling and non-renewable fossil fuels that are currently the major source of chemicals and energy in the world.1–3 Some key reactions in CO2 hydrogenation are illustrated in eqn (1)–(4). As illustrated in Fig. 1, the highly exothermic methanol (MeOH), dimethyl ether (DME) or hydrocarbons formation are thermodynamically favoured at low temperature while the contrary is true for the endothermic reverse water shift reaction (RWGS, eqn (2)). Hence, MeOH synthesis is often carried out at relatively low temperature and high pressure.4 Currently, MeOH is commercially produced from synthesis gas (mainly CO and H2) over ICI developed Cu/ZnO/Al2O3 catalyst at 50–100 bar and 200–300 °C.5 However, when the feed consists of CO2 and H2, the RWGS reaction is highly enhanced over the Cu/ZnO/Al2O3 catalyst, leading to substantial selectivity loss.6 Recently, several metals and metal oxides have been reported to be active for CO2 hydrogenation to MeOH, including Pd-based,6–16 In-based,17–22 and Zn/ZrO2-based23–29 catalysts.| Reaction |
|
| Eqn |
|---|---|---|---|
| CO2 + 3H2 ⇌ CH3OH + H2O | −49.2 | 3.7 | (1) |
| CO2 + H2 ⇌ CO + H2O | 41.2 | 28.8 | (2) |
| CO2 + 3H2 ⇌ 0.5CH3OCH3 + 1.5H2O | −61.2 | −5.7 | (3) |
| CO2+3H2 ⇌ 0.5CH2CH2 + 2H2O | −63.9 | −29.2 | (4) |


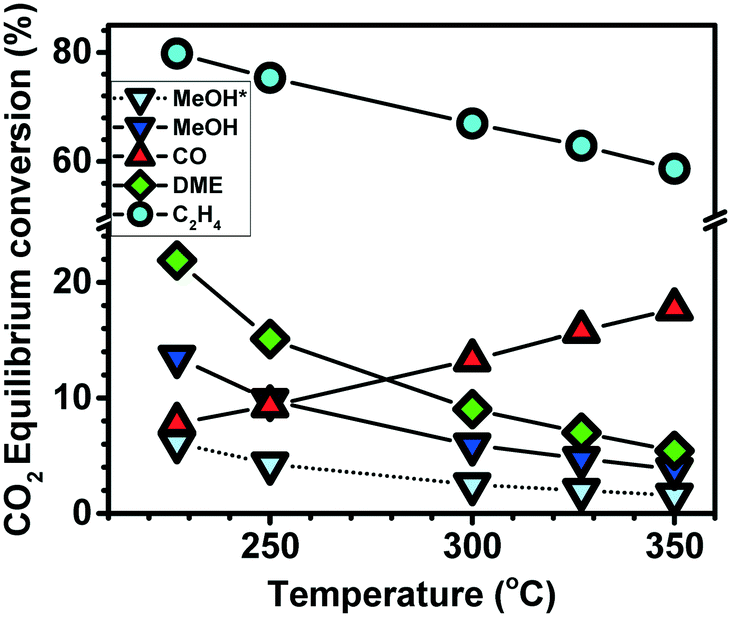 | ||
| Fig. 1 CO2 equilibrium conversion at 20 bar (*8 bar) versus temperature, following eqn (1)–(4). | ||
Among them, PdZn-based catalysts have shown promising MeOH yield especially at medium temperatures (210–270 °C).6–9,11,30 By applying different catalyst preparation methods, Bahruji et al.7,8 reported that increasing the Zn/Pd ratio led to increased Zn0 incorporation in Pd lattice, enhancing surface PdZn alloy formation and increasing MeOH production. However, when only Pd without Zn was deposited on TiO2 or Al2O3, CO was predominantly produced suggesting that pure Pd sites promote RWGS reaction while the presence of Zn is crucial to MeOH production.6,10,30 By introducing a CdSe shell around a Pd–Zn core, a study by Liao et al.6,30 indicated that deeper reduction of ZnII to Zn0 could be attained at milder temperatures (280 °C) which subsequently enhanced the number of neighbouring Zn0 around the Pd0 atoms leading to 80–90% MeOH selectivity at 270 °C. A mechanistic study using DRIFT and DFT calculations suggested that, possible intermediates in CO2 hydrogenation over PdZn catalysts could be either (i) HCOO which directly hydrogenates to MeOH or (ii) trans-COOH, which isomerize to cis-COOH for RWGS route or MeOH.6,9trans-COOH formation was found to be energetically favoured on pure Pd (111) while it was highly unstable on Pd1Zn1 (111). The authors concluded that, the PdZn site on the catalyst surface suppresses trans-COOH formation, thereby suppressing the RWGS reaction. Hence, increasing amounts of Zn minimized isolated Pd, thereby suppressing RWGS reaction and promoting MeOH formation.
Beyond the possible improvements of PdZn-based catalysts, MeOH production is still limited by thermodynamic equilibrium, especially at increasing temperatures (>200 °C). Here, instead, CO formation is thermodynamically favoured, leading to decreasing MeOH selectivity with increasing temperature. Literature examples of this effect are shown in Fig. 2 for PdZn-based catalysts. In addition to the decreasing MeOH selectivity, MeOH productivity numbers were also reported to decrease with increasing temperature for the Pd:Zn/TiO2 catalyst, already between 210–250 °C. For the other catalyst, Pd:ZnO, MeOH production was reported to decrease at temperatures beyond 250 °C.7,8
 | ||
| Fig. 2 CO2 conversion (box), MeOH selectivity (circle) and MeOH space–time yield (STY, bars) versus temperature over two Pd:Zn catalysts, Pd:Zn/TiO2 (left) and Pd:ZnO (right). Adapted from7,8 | ||
The dehydration of formed MeOH to dimethyl ether (DME) presents one means by which the reaction selectivity can be shifted away from CO at temperatures beyond 250 °C. Beside being a thermodynamic driving force (see Fig. 1), DME can be used directly as an environmentally friendly fuel or as a key feedstock in the methanol-to-hydrocarbons (MTH) reaction over zeolites.31–33 To convert CO2 to DME, there is a need for a bi-functional catalyst composed of MeOH catalyst and an acidic catalyst for dehydration. Reported, solid acid catalysts used for such reactions in combination with MeOH synthesis catalysts include sulphated ZrO2,34 alumina35,36 and zeolites.36–38 Among them, combination of MeOH based catalyst and zeolite has shown promising DME production within 220–330 °C.36,38 One drawback is that adsorbed water during the reaction can induce sintering of MeOH synthesis catalyst.38 This implies that, since water is a major product in the CO2 to DME reaction, a hydrophobic catalyst with optimum acidity is preferred. Increasing the Si/Al ratio has been reported to decrease the hydrophilicity of H-ZSM-5 zeolite significantly.39 Aside the acid sites density, the strength of the acid sites, the pore size, the micro and meso porosity of the solid acid also play an important role in MeOH dehydration.40 Even though both Lewis acid and Brønsted acid sites are known to aid in dehydration, Brønsted acid sites are more favourable.41 However, the stronger the acid strength, the more they promote coking to deactivate the catalyst.40
The method for mixing the two catalytic functions is important to obtain an optimal interaction between the active sites for MeOH synthesis and dehydration, respectively, avoiding their mutual poisoning or restricting MeOH diffusion. Prior studies have shown that composite physical mixture of PdZn alloy and H-ZSM-5 produces higher amount of oxygenates across 220 to 300 °C, compared to a chemical vapour impregnated H-ZSM-5 with PdZn.36 N2 physisorption and NH3 TPD of the H-ZSM-5 before and after impregnation showed a decrease in the accessible surface area and a clear decrease in the number of strong acid sites while the weak acid sites were preserved after impregnation. This result was attributed to blocking of the zeolite micropores by PdZn nanoparticles which restricted MeOH access to such sites. They further implied that, while the two catalysts need to be in close proximity to each other, there was no apparent MeOH diffusion restriction from the MeOH catalyst to the zeolite surface for the physically mixed hybrid catalyst.
The conversion of CO2 and H2 to hydrocarbons via MeOH would enable substantially higher per pass yields than the conversion to MeOH or DME (see Fig. 1). A general challenge of this process is the gap between the optimal temperature for each step. As described above,1,42 CO2 hydrogenation to MeOH or DME is favoured by low temperature and high pressure while MeOH or DME to hydrocarbons – a reaction that is catalysed by Brønsted acidic zeolites - is favoured by temperatures above 350 °C.43,44
In the present study, our aim is to elucidate the cause of the previously reported limitations associated to oxygenate and hydrocarbon formation in the combined PdZn and H-ZSM-5 catalyst system at 270 to 350 °C. To this aim, we have studied PdZn@ZnO alone and physically mixed with H-ZSM-5 as well as PdZn chemically grafted on H-ZSM-5 through surface organometallic chemistry (SOMC), for the CO2 hydrogenation reaction at 250 to 350 °C. The latter methodology (SOMC) offers a controlled environment of the bi-metallic nanoparticles.45 Cardinal in the current work was the use of combined operando X-ray absorption spectroscopy (XAS) and powder X-ray diffraction (PXRD) to study the hybrid catalyst stability at relatively high pressures conditions (8 bar). This study also addresses the penalty involved in having excess Zn in the PdZn mixed with H-ZSM-5 zeolite as a bi-functional catalyst in one bed.
2. Experimental
2.1 Catalyst preparation
Commercial ZSM-5 CBV-5524 G (Si/Al = 25) and CBV 8014 (Si/Al = 40) were purchased from Zeolyst International. The parent ZSM-5 was ion exchanged with 1 M NH4NO3 solution twice for 2 h each at 70 °C over water bath. The resulting mixture was centrifuged, oven dried and then calcined at 550 °C for 2 h to obtain H-ZSM-5. PdZn @ZSM-5 was prepared by grafting Pd(acac)2 in dry toluene on meso-H-ZSM-5 (obtained after desilication described elsewhere46) using SOMC (surface organometallic chemistry) on oxide followed by reduction under H2 at 500 °C (4 h) to achieve supported Pd metal particles. This procedure was followed by a second grafting, using SOMC diethylzinc in dry toluene on metal (Pd particles) and then further reduction in H2 at 500 °C (4 h) to give the bimetallic PdZn on H-ZSM-5 named PdZn@H-ZSM-5.45 PdZn@ZnO was prepared by incipient impregnation of ZnO powder with Pd(NO3)2 (both purchased from Sigma-Aldrich) following similar route as reported by Tew et al.47 0.8 g Pd(NO3)2·6H2O was dissolved in 1 ml water and thoroughly mixed with 3 g of ZnO powder to form a thick paste. The paste was dried at 80 °C overnight, and then calcined at 500 °C under static air for 3 h. The resulting material was reduced under H2 at 400 °C for 1 h to form the PdZn@ZnO sample. The PdZn@ZnO was physically mixed with H-ZSM-5 for the CO2 hydrogenation reaction.2.2 Catalyst characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H-ZSM-5 = 1:3 by weight) were carefully ground and sieved with 250 to 177 μm sieves. For Pd K-edge measurements, ca. 10 mg of powder were packed in a 2.5 mm-diameter capillary, while for Zn K-edge data collection, ca. 1 mg of catalyst was packed in a 0.7 mm-diameter capillary. The capillary reactor was then connected to an appropriate gas-flow setup for the CO2 hydrogenation reaction. Temperature at the measurement position was controlled by a heat gun. A total flow rate of 12 ml min−1 was employed during all the experiments, consisting of i) activation from ambient temperature to 400 °C at 1 bar in 10H2/2He and kept for 60 min at 400 °C, ii) cooling down to 250 °C in H2 and flushing with He, iii) pressurizing in He to 8 bar iv) CO2 hydrogenation reaction with 2.5CO2/7.5H2/2He at 250, 300, 350 °C at 60 min reaction time for each temperature.
:H-ZSM-5 = 1:3 by weight) were carefully ground and sieved with 250 to 177 μm sieves. For Pd K-edge measurements, ca. 10 mg of powder were packed in a 2.5 mm-diameter capillary, while for Zn K-edge data collection, ca. 1 mg of catalyst was packed in a 0.7 mm-diameter capillary. The capillary reactor was then connected to an appropriate gas-flow setup for the CO2 hydrogenation reaction. Temperature at the measurement position was controlled by a heat gun. A total flow rate of 12 ml min−1 was employed during all the experiments, consisting of i) activation from ambient temperature to 400 °C at 1 bar in 10H2/2He and kept for 60 min at 400 °C, ii) cooling down to 250 °C in H2 and flushing with He, iii) pressurizing in He to 8 bar iv) CO2 hydrogenation reaction with 2.5CO2/7.5H2/2He at 250, 300, 350 °C at 60 min reaction time for each temperature.
Pd K-edge and Zn K-edge XAS spectra were collected in transmission mode, using a water-cooled flat Si(111) double-crystal monochromator. To measure the incident (I0) and transmitted (I1) X-ray intensity, 30 cm-length ionization chambers filled with a mixture of He and Ar were used. We characterized static states for each catalyst collecting two consecutive XAS scans, of ca. 10 min each, in the energy ranges 24100–25300 eV and 9400–10500 eV, for Pd K-edge and Zn K-edge, respectively, with constant energy step of 0.4 eV in the continuous scanning mode. Shorter XAS scans, of ca. 2 min per scan were employed to monitor the evolution of the spectroscopic features during the activation ramp in H2. Pd and Zn foils were measured simultaneously using a third ionization chamber for energy calibration.
XAS spectra were normalized to unity edge jump using the Athena software from the Demeter package.51 Extraction of the χ(k) EXAFS functions and Fourier transforms were also performed using Athena program.
PXRD data were collected quasi-simultaneously with XAS measurements. The patterns were recorded by CMOS-Dexela 2D detector. The sample-to-detector distance l = 419.91(8) mm and photon wavelength λ = 0.50544(2) Å, selected by a Si(111) channel-cut monochromator, were calibrated using reference LaB6 and Si materials. The above geometry resulted in 2θ range 1.2–35.0 (dmin = 0.85 Å). For a given experimental condition, 5 consecutive acquisition by 10 s each were acquired and averaged. For dark field correction, dark images (with closed X-ray shutter) were collected with the same statistics and subtracted from the data. Azimuthal integration of 2D patterns was done in PyFAI code52 to obtain I(2θ) data using integration parameters initially determined in Fit2D program.53
2.3 Catalytic tests
Catalytic tests were performed in a fixed bed continuous flow stainless steel reactor (I.D. 8.3 mm). 150–300 mg pelleted catalyst in the range of 240–420 μm grain size was loaded in the reactor and pre-treated in situ at 400 °C for 1 h at 1 atm in a flow of H2 gas (10 ml min−1) before cooling to reaction temperatures. All quoted flows are at STP. The total pressure was then increased in He to 20 bar controlled by a back-pressure regulator (Bronkhorst P-702CV). The reactant gas mixture (1CO2:3H2:1(10% Ne in He)) was then introduced in the reactor system. A standard total gas flow rate was kept at 30 ml/min with mainly GHSV of 6000 ml gcat−1 h−1. In the case of testing only PdZn@ZnO without zeolite, 50% SiO2 was added. The post-reactor lines were heated at about 200 °C to avoid product condensation. The effluent was analysed using an online Agilent 7890B GC and Agilent 3000 micro-GC connected to FID and TCD respectively. CO2 conversion and product selectivity were calculated using the formulas below:

Since the PdZn alloy is generally accepted to be the active site for the formation of MeOH,6,30 the mass of the active component for MeOH was estimated based on the Pd and Zn mass.

3. Results
3.1 BET and electron microscopy
The Si/Al ratio of the parent H-ZSM-5 used in the study were 25 and 40, hereafter the samples are named “H-ZSM-5 (25)” and “H-ZSM-5 (40)” respectively. Despite the difference in the Si/Al of the H-ZSM-5 zeolites, their specific surface area and particle sizes were very similar (see Table 1 and Fig. 3). The BET surface areas were 444 and 420 m2 g−1 while the micropore volumes were 0.196 and 0.176 cm3 g−1 for the H-ZSM-5 (40) and H-ZSM-5 (25), respectively (see also ESI,† Fig. S1 for the adsorption isotherms). The H-ZSM-5 (25) was used for the PdZn grafting while the H-ZSM-5 (40) was used for the physical mixing with PdZn@ZnO. Upon grafting the PdZn on H-ZSM-5 (25), the BET surface area was 348 m2 g−1 while micropore volume also reduced to 0.121 cm3 g−1. The PdZn grafted on H-ZSM-5 (25) was named PdZn@H-ZSM-5 (25), and contained 1.5 wt% Pd. The TEM imaging (of the as-prepared PdZn@H-ZSM-5 (25), Fig. 3c) showed spherical metal or alloy (PdZn) nanoparticles (NPs) of predominantly 9 ± 3 nm (see ESI† Fig. S3 for particle distribution curve), with the smaller NPs grafted within the zeolite while larger NPs were observed at the edges. The Pd impregnated ZnO was named PdZn@ZnO, as H2 reduction pre-treatment is expected to give PdZn alloy, over ZnO support (more details will follow in section 3.3). The Pd loading in the PdZn@ZnO was 3.3 wt%. TEM imaging of the H2 reduced (pre-treated) impregnated sample (Fig. 3d) also showed spherical but larger metal/alloy particles of about 13 ± 7 nm size on top of ZnO particles (identified from EDS spot analysis, see ESI† Fig. S2).| Sample | Elemental composition | Textural properties | ||
|---|---|---|---|---|
| Si/Al | Zn/Pd | BET area (m2 g−1) | Micropore volume (cm3 g−1) | |
| H-ZSM-5 (25) | 25 | 420 | 0.176 | |
| PdZn@H-ZSM-5 (25) | 25 | 5 | 348 | 0.121 |
| H-ZSM-5 (40) | 40 | 444 | 0.196 | |
| PdZn@ZnO | 16 | |||
 | ||
| Fig. 3 SEM (a) and (b) and TEM (c) and (d) of the catalyst samples investigated. The PdZn@H-ZSM-5 sample was reduced in H2 at 500 °C for 4 h, and the PdZn@ZnO sample was pretreated in H2 at 400 °C for 1 h, prior to the investigation. The PdxZny phases were determined using d-spacing values from the HRTEM see details in the ESI† Fig. S2. | ||
3.2 Pyridine adsorption FT-IR spectroscopy
Basic probe molecules coupled with FT-IR spectroscopy are one of the easiest and most used tools for acid sites characterization in solid state samples. Among all the possible alternatives, pyridine allows to perform a quantitative analysis of the different acid sites distinguishing Brønsted from Lewis sites, with methods well described in the specialized literature.48,49 The spectral region of interest is the one between 1600 and 1400 cm−1, where we observe two specific bands unequivocally ascribable to the pyridinium ion, originated through the interaction of pyridine with strong Brønsted acidic sites (1544 cm−1) and to strongly adsorbed pyridine on Lewis sites, associated to extra-lattice Al3+ and Zn2+ (1455 cm−1).Part (a) of Fig. 4 illustrates the data associated to the sample H-ZSM-5 (25), while part (b) refers to H-ZSM-5 (40). From the two set of data it is evident that the preparation method of the two samples affects substantially the number and the type of the acidic sites. Black curves in part (a) and (b) show that the starting points (protonic parent zeolites) are very similar, presenting a majority of Brønsted acidic sites and a minor number of Lewis species ascribable to few extra-lattice Al3+ species. In case of the sample in which PdZn nanoparticles were obtained by direct deposition on the parent zeolite (orange curve in part (a) of Fig. 4), major changes occur already in the “as-prepared” sample, as evidenced by the major decrease of the band associated to the pyridinium and the substantial growth of the band ascribed to pyridine adsorbed on Lewis sites (most probably Zn2+). Conversely, the spectrum obtained after catalyst use (brown curve in part (a) of Fig. 4) does not show major changes in the number of Brønsted and Lewis acid sites apart from a small decrease (about 10%) of the latter that can be interpreted by considering that some of them are not accessible anymore to the probe. Moving to the sample obtained by physical mixture of PdZn@ZnO and H-ZSM-5 (40), (orange curve in part (b) of Fig. 4), it is clear that no significant changes are observed in the “as-prepared” sample, with respect to the respective abundance of Brønsted and Lewis acidic sites. Conversely, once the sample has been treated in H2 at 400 °C for 1 h (red curve in part (b) of Fig. 4), major decrease of the band associated to the pyridinium and the substantial growth of the band ascribed to pyridine adsorbed on Lewis sites (most probably Zn2+) occur, testifying a migration of metal ions that substitute acidic protons in the zeolitic framework. Conversely, the spectrum obtained after the catalyst use (brown curve in part (b) of Fig. 4) does not show major changes in the number of Brønsted and Lewis acidic sites. The inset of Fig. 4(a), illustrates the zeolite spectra in the ν(OH) region showing the substantial decrease of the band at 3610 cm−1, occurring upon the PdZn functionalization. The quantification of Brønsted and Lewis sites are reported in Table 2. Thanks to the full set of data, it is clear that zinc has a high affinity towards the acidic zeolites easily replacing the protons, implying a substantial decrease of the Brønsted acidity in favour of the introduction of Lewis sites.
 | ||
| Fig. 4 FT-IR spectra highlighting the bands of pyridinium originated from the strong Brønsted (B) acid sites and of pyridine adsorbed on Lewis (L) sites. Part (a): Black curve represents the parent H-ZSM-5(25), orange curve represents the PdZn@H-ZSM-5(25) as-prepared sample, brown curve represents the PdZn@H-ZSM-5(25) tested sample. Inset shows a magnification of the ν(OH) region for the same samples prior to pyridine dosing. Part (b): Black curve represents the parent H-ZSM-5(40), orange curve represents the PdZn@ZnO + H-ZSM-5(40) as-prepared sample, red represents PdZn@ZnO + H-ZSM-5(40) sample treated with H2 at 400 °C for 1 h; brown curve represents the PdZn@ZnO + H-ZSM-5(40) tested sample. | ||
| Sample | Step | Brønsted sites (mmol g−1) | Lewis sites (mmol g−1) |
|---|---|---|---|
| Fresh H-ZSM-5 | Si/Al = 25 | 0.21 | 0.07 |
| Si/Al = 40 | 0.15 | 0.07 | |
| PdZn@H-ZSM-5 (25) | As prepared | 0.02 | 0.57 |
| Tested | 0.02 | 0.46 | |
| PdZn@ZnO + H-ZSM-5 (40) | As prepared | 0.11 | 0.05 |
| Activated | 0.04 | 0.25 | |
| Tested | 0.03 | 0.32 |
3.3 Operando XAS and PXRD studies
To obtain detailed information on the electronic and structural properties of the investigated PdZn-zeolite bifunctional catalysts, we combined XAS and PXRD measurements at the BM31 beamline of the ESRF. We firstly considered the activation process, carried out in H2 from RT to 400 °C, comparing the time-dependent evolution of Pd and Zn K-edge XAS data for both PdZn@H-ZSM-5 (25) and PdZn@ZnO + H-ZSM-5 (40). Fig. 5 reports an overview of the results, together with reference spectra for selected model compounds at both absorption edges, to guide the data interpretation. | ||
| Fig. 5 Overview of operando XAS data collected during the activation of the investigated catalysts, carried out in H2 gas flow, from 50 °C to 400 °C. (a and c) Pd K-edge XANES (main panel) and FT-EXAFS (bottom inset) spectra for (a) PdZn@H-ZSM-5 (25) and (c) PdZn@ZnO + H-ZSM-5 (40). The top insets report the first derivative of the XANES spectra. (b and d) As parts (a and c) but reporting Zn K-edge XAS data for the two catalysts. (e) Pd K-edge XAS data on selected Pd-containing model compounds, namely Pd metal and PdO. (f) Zn K-edge XAS data on selected Zn-containing model compounds, namely Zn metal, ZnO, Zn-exchanged H-ZSM-5 zeolite (1% wt Zn; Si/Al = 40, pretreated in air at 400 °C, from ref. 48). For both catalysts, the reported Pd K-edge FT-EXAFS spectra were obtained by transforming the k3χ(k) functions in the 3.0–9.5 Å−1 range, while Zn K-edge FT-EXAFS spectra were obtained by transforming the k2χ(k) functions in the 2.4–9.5 Å−1 range. | ||
Considering Pd K-edge spectra in Fig. 5a and c, we note how the initial state at 50 °C in He, prior to activation in H2, differs for the two catalysts. PdZn@H-ZSM-5 (25) already shows the characteristic features of Pd0. Conversely, for the PdZn@ZnO + H-ZSM-5 (40) physical mixture, a substantial fraction of Pd is still in the PdII oxidation state, with an energy position of the edge compatible with that of the PdO model compound. Switching the gas feed to pure H2, Pd efficiently undergoes reduction to Pd0 also in the PdZn@ZnO + H-ZSM-5 (40) catalyst, as clearly evidenced by the shift of the edge position to lower energy values. Comparison with the XANES and FT-EXAFS spectra of bulk Pd metal in Fig. 5e points to the presence of Pd metal nanoparticles in both the materials. As temperature increases during activation, the position of the edge remains unchanged, supporting a stable Pd0 oxidation state. Nonetheless, from ca. 300 °C upward, the spectral features further evolve showing very similar modifications for the two investigated catalysts, albeit occurring at slightly lower temperature for PdZn@H-ZSM-5 (25) with respect to PdZn@ZnO + H-ZSM-5 (40). In particular, the XANES post-edge oscillations characteristic of metal Pd become progressively flatter and, at the end of the activation, a XANES line-shape significantly different from the one of metal Pd is observed. In parallel, the FT-EXAFS restructures towards a broad single maximum, peaking at ca. 2.15 Å in the (phase-uncorrected) spectra of both catalysts. Based on the previous literature47,54,55 and in agreement with the PXRD results presented below, these XAS fingerprints are diagnostic for the formation of a PdZn nano-alloy, representing the active phase for CO2 hydrogenation over both bi-functional catalysts. According to previous reports, the broad peak in the FT-EXAFS stems from the combination in partial antiphase of Pd–Zn and Pd–Pd scattering contributions, expected at 2.64 Å and 2.90 Å, respectively, for the tetragonal L10 structure of the PdZn alloy.47,55 According to further analysis of FT-EXAFS analysis (See ESI,† Fig. S5 and S6), only minor fraction of pure Pd nanoparticles remains after the activation, which further converts completely to a Pd–Zn alloy under reaction conditions.
In parallel, Zn K-edge XAS measurements allowed us to characterize the average properties of Zn-species in the bi-functional catalysts, keeping in mind that Zn is present in excess with respect to Pd in both cases (see Table 1). In contrast to Pd K-edge results, pointing to a very similar local structure around Pd sites in both the activated materials, Zn K-edge XAS shows markedly different trends for PdZn@H-ZSM-5 (25) and PdZn@ZnO + H-ZSM-5 (40). In particular, for PdZn@H-ZSM-5 (25) (see Fig. 5b), the evolution during activation closely resembles what has been previously reported for Zn-exchanged zeolites.48 Throughout the whole activation process, Zn-speciation appears dominated by isolated ZnII ions, without detectable contributions from ZnO. The decrease in the XANES white line peak, as well as in the first-shell maximum observed at ca. 1.5 Å in the phase-uncorrected FT-EXAFS spectra suggest a progressive coordination decrease in the first coordination sphere of ZnII ions, possibly connected with the progressive dehydration of ZnII species and their coordination to framework oxygen atoms at zeolite exchange sites. Consistently, the Zn K-edge XANES and EXAFS spectra of PdZn@H-ZSM-5 (25) at the end of the activation in H2 resembles the ones of a representative dehydrated Zn-exchanged H-ZSM-5, reported in Fig. 5f. All these evidences consistently indicate that a substantial fraction of Zn ions has exchanged into the zeolite component of the bi-functional catalysts, already prior to the activation. Notably, from Zn K-edge XANES of PdZn@H-ZSM-5 (25), a minor contribution from metallic Zn is also observed, in line with the Zn:Pd ratio of ∼5 in the catalyst. In particular, looking at the first derivative XANES spectra in the top inset of Fig. 5b, a feature at ca. 9960 eV is noted to progressively develop as temperature increases during the activation, which matches the edge energy position of metallic Zn (Fig. 5f, top inset). This minor, but still detectable contribution from Zn0 is expected to come from the Zn atoms participating into the PdZn alloy.
Zn K-edge XAS data obtained for PdZn@ZnO + H-ZSM-5 (40) during the activation (Fig. 5d) clearly show that, in this catalyst, ZnO oxide dominates. This is not surprising, since in this case ZnO represents the support for the PdZn alloy formed upon activation, with an overall Zn:Pd ratio of ~ 16. Due to the largely dominant ZnO contribution and the bulk averaging nature of the XAS techniques, Zn K-edge XAS in this case is not informative on possible ion-exchange phenomena of Zn ions inside the zeolite component upon activation in H2. Also, it is not possible to identify the tiny fraction of Zn atoms participating in the PdZn alloy (evident from Pd K-edge XAS results) solely looking at the XANES spectra or even at their first derivatives. However, the differential XANES spectra obtained by calculating the difference between the XANES of PdZn@ZnO + H-ZSM-5 (40) after and before the activation shows a signal structure very similar to the reference differential XANES obtained by subtracting the spectrum of metal Zn from the one of ZnO (Fig. S7†). This observation supports reduction to Zn0 and alloying with Pd of a very minor fraction of the total Zn atoms in the system, in line with the compositional characteristics of the catalyst.
After having investigated by XAS the activation process and the final state reached at 400 °C in H2, we also monitored the two catalysts under reaction conditions at 8 bar, the maximum pressure safely achievable with our gas flow setup. Under these conditions, we have obtained Pd K-edge XAS data, since this absorption edge enables direct structural/electronic sensitivity to the PdZn alloy. The obtained results for the two investigated catalysts monitored during CO2 hydrogenation at the three temperatures evaluated during the catalytic tests, namely 250, 300 and 350 °C; the spectra collected on the activated catalysts just prior to the reaction at 250 °C in He are also shown for comparison (see ESI,† Fig. S8). Overall, XAS indicates that the formed PdZn alloy remains stable under the reaction feed in both the studied catalysts, with no evidences of important structural modifications or formation of extra-phases. While the XANES is substantially unchanged as a function of the temperature, not surprisingly the EXAFS spectra present a progressive intensity as temperature increases, due to larger thermal contributions to the Debye-Waller factors. It is however interesting to note that, for each probed temperature, the main peak amplitude in the FT-EXAFS of PdZn@H-ZSM-5 (25) is slightly lower than in PdZn@ZnO + H-ZSM-5 (40), suggesting a higher level of structural disorder and/or an average smaller particle size in the former catalyst in agreement with the TEM results.
Fig. 6 reports an overview of the PXRD results at the various stages of the catalyst treatment. In particular, right after H2 pre-treatment at 400 °C, we observed new peaks around 41.3 and 44.2 °C, compatible with tetragonal β1 PdZn phase.47,56 The peak intensities slightly increased and their positions slightly shifted with temperature in both catalysts. The PdZn@H-ZSM-5 (25) showed more disordered reflections compared to the PdZn@ZnO + H-ZSM-5 (40) probably due to smaller crystallite sizes as the TEM and XAS measurements also revealed similar trend in particle size distribution.
 | ||
| Fig. 6 PXRD patterns collected, quasi simultaneously to XAS spectra, at key conditions for (a) PdZn@H-ZSM-5 (25) and (b) PdZn@ZnO + H-ZSM-5 (40), reported in the 2θ range where the characteristic reflections of the PdZn alloy occur. The PXRD pattern of pure H-ZSM-5 zeolite is also reported for comparison. The patterns are vertically translated for clarity. | ||
Rietveld refinement of PXRD data obtained for PdZn@ZnO + H-ZSM-5 (40) showed that the initial structure of the PdZn@ZnO + H-ZSM-5 contained orthorhombic ZSM-5 phase (ca. 90%) and hexagonal ZnO (ca. 10%). The atomic parameters of ZSM-5 were fixed to those reported by Schmidt et al.57 which already gave good agreement with experimental pattern. For ZnO phase, isotropic displacement parameters (Uiso) of both Zn and O atoms were refined while atomic positions and occupancies were kept fixed. For both structures, unit cell parameters were refined together with profile shape parameters of the Gaussian and Lorentzian components (GW and LY) of the pseudo-Voigt function. Both profile shape parameters were found very similar for the two phases, which indicates that the dominant contribution to the peak broadening is given by the experimental factors (mainly by 2.5 mm thickness of the capillary). Therefore, the profile parameters of the two phases were constrained to be equal in the final refinement. The background was fitted by 5-parameter Legendre polynomial. The final refinement with two phases is shown in the ESI,† Fig. S9, and the obtained structural parameters are reported in Table 3.
| Phase | Refined parameter | Initial | After activation | Spent |
|---|---|---|---|---|
| H-ZSM-5 | Fraction, % | 89.9(7) | 89.4(9) | 90.2(8) |
| a, Å | 20.101(1) | 20.075(1) | 20.087(1) | |
| b, Å | 19.915(1) | 19.920(1) | 19.914(1) | |
| c, Å | 13.4002(9) | 13.3939(9) | 13.3984(9) | |
| ZnO | Fraction, % | 10.1(7) | 9.8(7) | 8.9(6) |
| a, Å | 3.25076(8) | 3.25940(9) | 3.2510(1) | |
| b, Å | 5.2075(2) | 5.2166(2) | 5.2079(2) | |
| c, Å | 0.0100(4) | 0.0184(4) | 0.0096(4) | |
| PdZn | Fraction, % | — | 0.79(2) | 0.85(2) |
| a, Å | — | 4.1262(5) | 4.1005(3) | |
| c, Å | — | 3.3463(7) | 3.3373(4) | |
| U iso(Pd), Å2 | — | 0.036(5) | 0.023(3) |
Based on the peaks at 41.3° and 44.2°, we then refined the unit cell parameters of the tetragonal PdZn phase and Uiso parameter of Pd atom, fixing Uiso of Zn to 0, and keeping the atomic coordinates and occupancies fixed. LY and GW profile parameters were fixed to those of H-ZSM-5 and ZnO phases, while additional LX contribution was added to account for the size effects. The crystallite size was then calculated as  . This number is in the upper limit of the values found by TEM investigations in section 3.1 (and ESI† Fig. S3). This can be explained by (i) the stronger contribution of bigger particles to PXRD signal (see green bars in part b of Fig. S3 I†), (ii) the possible overestimation of experimental contribution (GW and LY) and (iii) the presence of both Pd and Pd–Zn particles in the TEM images, while the PXRD only considered the tetragonal PdZn phase. The results of the refinement and the obtained structural parameters are reported in Table 3. Particularly, the fraction of ZnO phase decreases, indicating that Zn atoms may be transferred either to Zn–Pd alloy nanoparticles or to the zeolite framework.
. This number is in the upper limit of the values found by TEM investigations in section 3.1 (and ESI† Fig. S3). This can be explained by (i) the stronger contribution of bigger particles to PXRD signal (see green bars in part b of Fig. S3 I†), (ii) the possible overestimation of experimental contribution (GW and LY) and (iii) the presence of both Pd and Pd–Zn particles in the TEM images, while the PXRD only considered the tetragonal PdZn phase. The results of the refinement and the obtained structural parameters are reported in Table 3. Particularly, the fraction of ZnO phase decreases, indicating that Zn atoms may be transferred either to Zn–Pd alloy nanoparticles or to the zeolite framework.
3.4 Hydrogenation of CO2
Fig. 7(a)–(c) shows the CO2 catalytic hydrogenation versus TOS over the various catalysts at 300 °C and 20 bar. Focusing first on PdZn@ZnO alone (without H-ZSM-5), it initially yielded 17% CO2 conversion, that decreased to 11.6% over 20 h (Fig. 7(a)). Selectivity to MeOH sharply increased from 0 to 32% with corresponding decrease in CO selectivity from 92% to 65% during the first 6 h, and then the MeOH and CO selectivitives gradually merged at appx. 50% selectivity by 20 h TOS, possibly due to delay in the formation of the active site for MeOH. Fig. 7(d) and Fig. 8(a) show that the space time yield (STY) of MeOH over PdZn@ZnO increased with temperature from 250 to 300 °C, and that the yield of MeOH (5.5%) approached the thermodynamic equilibrium yield of 5.9% after 20 h on stream at 300 °C. This result is superior to a previous report devoted to PdZn on ZnO support: In that study, the MeOH yield decreased from 6.4% at 250 °C to 4.2% at 300 °C, with a space time yield of 1.6 mol kg−1 h−1 at 300 °C (see Fig. 2, It should be noted that the sample was made by sol-immobilization method, and tested at 3600 ml/g h WHSV in a 0.5 × 50 cm2 stainless tube reactor).7 A further temperature increase to 350 °C led to dramatically lower MeOH STY and selectivity over the PdZn@ZnO catalyst (Fig. 7). Comparison to the thermodynamic equilibrium value (Fig. 8) showed that the system was farther from equilibrium at this temperature than at 300 °C, suggesting that the system deactivated further when reaching the higher temperature. | ||
| Fig. 7 Conversion and selectivity versus time on stream at 300 °C over (a) PdZn@ZnO + SiO2, (b) PdZn@ZnO + H-ZSM-5, (c) PdZn@H-ZSM-5. Space time yield (STY) comparison of the catalysts versus temperature for the formation of (d) MeOH+ (sum of MeOH, DME and C2+ hydrocarbons), (e) DME, (f) CO, the catalysts were pretreated in H2 flow at 400 °C and tested at 20 bar with H2/CO2 = 3, and WHSV = 6000 ml g−1 h−1. | ||
 | ||
| Fig. 8 Comparison of oxygenate ((a) MeOH and (b) DME) yields from the CO2 hydrogenation reaction at 20 bar with theoretical equilibrium yields versus temperature. | ||
The second case examined in our tests was the physically mixed H-ZSM-5 (40) and PdZn@ZnO in 1:1 ratio (Fig. 7b). Here, the CO2 conversion was 20% and slightly decreased to 17% after 11 h TOS at 300 °C. Aside the CO and MeOH main products, high amounts of DME were observed with minor amounts of methane and C2+ hydrocarbons (accounting for <2% overall). The selectivity to oxygenates (DME and MeOH) gradually increased from 35 to 52% over 11 h TOS at the expense of CO. Notably, beyond 11 h, conversion and selectivity reached a steady state that was maintained up to the 45 h TOS studied (only the first 30 h is shown here for brevity). Generally, the addition of H-ZSM-5 to the PdZn@ZnO catalysts shifted the oxygenates yield beyond MeOH thermodynamic equilibrium yield, in agreement with literature reports,36 since further conversion of MeOH to DME or hydrocarbons is more thermodynamically favourable than ending the reaction at MeOH as illustrated in Fig. 1. Overall, we observed 8.8% yield to the oxygenates at 300 °C, very close to the 9% estimated DME equilibrium yield (see Fig. 8), and surpassing the previously reported 4.2% oxygenates yield at the same temperature and pressure.36 Increasing the temperature to 350 °C, a substantial decrease was observed in MeOH and DME STY and yields, the oxygenate yield now being further away from equilibrium than at 300 °C. This result is in agreement with that observed over PdZn@ZnO alone, and suggests that similar deactivation of the MeOH forming active sites takes place with and without the physical mixture with H-ZSM-5. Turning next our attention to the CO space time yield over these two catalysts (Fig. 7(f)), it increases exponentially with temperature, and is the same for both catalysts. This result suggests that the CO forming active sites are also not influenced by mixing the two catalysts.
The third case consisting of PdZn grafted on H-ZSM-5 is shown in Fig. 7c. CO2 conversion remained at 13%, with 81 and 17% selectivity to CO and DME respectively, at 300 °C. Meanwhile, selectivities to MeOH and C2+ accounted for less than 2% over the 33 h TOS studied. Here, the conversions and selectivities were relatively stable compared to that of PdZn@ZnO + H-ZSM-5 (40) at least within the first 10 h TOS. However, clearly the physical mixture outperformed the grafted catalysts both in terms of yield and selectivity to the oxygenates at 300 °C. Next, we turn to compare the space time yield (STY) for the formation of DME, MeOH+ (sum of MeOH, DME and C2+ hydrocarbons) and CO at 250–350 °C over the three catalysts (Fig. 7(d)–(f)). As reported above, PdZn@ZnO showed no DME formation at all temperatures, however, the inclusion of H-ZSM-5 either by grafting or physical mixing induced high activity towards DME formation. STY for DME and MeOH+ increased up to 300 °C and then declined with temperature. Maxima of 6.7 and 4.5 mol(DME) kg(PdZn)−1 h−1 DME STY were observed at 300 °C for the PdZn@ZnO + H-ZSM-5 (40) and PdZn@H-ZSM-5 (25) respectively. Similarly, for MeOH+, the optimal STY measured at 300 °C were 5.4, 8.5 and 4.9 mol(MeOH+) kg(PdZn)−1 h−1 for the PdZn@ZnO, PdZn@ZnO + H-ZSM-5 (40) and PdZn@H-ZSM-5 (25) respectively. STY for CO increased with temperature over all catalysts, but more rapidly for the PdZn@H-ZSM-5 compared to that of the PdZn@ZnO and PdZn@ZnO + H-ZSM-5.
4. Discussion
The characterization data from XAS, PXRD and TEM, of the investigated hybrid PdZn/zeolite catalysts for CO2 conversion, consistently showed that upon H2 pre-treatment, Pd-Zn alloy was gradually formed from ca. 300 °C upward. The XAS data of the as-prepared PdZn@H-ZSM-5 already showed the Pd0 characteristic features, whilst the PdZn@ZnO + H-ZSM-5 still had substantial amount of PdII in its as-prepared state. Upon H2 treatment with temperature, the Pd K-edge XAS showed the characteristic features of PdZn alloy in both hybrid materials. In parallel, PXRD also showed the emergence of new peaks at 41.3° and 44.2° denoting the formation of tetragonal β1 PdZn alloy nanoparticles. PdZn@H-ZSM-5 exhibited a much higher level of structural disorder compared to the PdZn@ZnO + H-ZSM-5, as evidenced by very weak and broadened peaks in its PXRD patterns, which hampered Rietveld refinement of these data. The TEM showed the presence of the PdZn alloy with different particle sizes, smaller in the case of the PdZn@ZSM-5 compared to the PdZn@ZnO+H-ZSM-5, consistent with indications from PXRD.Regarding catalytic testing results, conversions were generally stable or slightly decreased over time. However, there was always some delay in attaining steady selectivity to products despite pre-treatment in H2 at 400 °C for all the catalysts prior to testing. The PdZn@H-ZSM-5 catalyst attained a stable selectivity much earlier than the PdZn@ZnO + SiO2 or PdZn@ZnO + H-ZSM-5 samples. On a similar line, the XAS data showed that the PdZn@H-ZSM-5 catalyst developed the PdZn alloy characteristic feature earlier than PdZn@ZnO + H-ZSM-5 during thermal activation in H2. Even though in the final state after activation, the two catalysts showed similar XAS characteristic of Pd0 sites in bimetallic PdZn, it is possible that attaining full formation of the Pd–Zn alloy took longer time in the macroscale catalytic testing setup compared to the capillary reactor cell used for the operando XAS, due to the smaller amount of sample used in the latter case. Thus, the delay in the attaining steady selectivity can be related to longer time in forming the active PdZn site.
The activity in the different combinations of catalyst differed. Firstly, comparing the PdZn@ZnO with and without H-ZSM-5, the presence of H-ZSM-5 led to higher activity towards oxygenates. Here, there is a clear promotion of MeOH+ especially at higher temperatures (>300 °C), where the conversion of MeOH to DME or hydrocarbons becomes important.43,44 Interestingly, the STYs of CO were similar over both catalysts at the different temperatures. It has been reported30 that COOH intermediate which decompose to CO (and H2O) is better stabilized on Pd(111) surface rather than HCOO intermediate which hydrogenates to MeOH on PdZn surface. Therefore, this report is in agreement with our observations and suggest that given the same reaction conditions, MeOH and CO formations occur at distinct active sites, such that the presence of H-ZSM-5 only increased oxygenates formation without any influence on CO formation.
Beyond this, the catalyst produced by SOMC, PdZn@H-ZSM-5 (25) showed rather high yield of CO compared to the PdZn@ZnO + H-ZSM-5 (40). Nonetheless, the PdZn active sites were relatively similar in both catalysts except for the particle size, which was even smaller in the PdZn@H-ZSM-5 (25). Even though there were differences in the Si/Al ratio of the H-ZSM-5, this is not expected to influence CO2 conversion to MeOH. One main difference however on the side of CO2 to MeOH catalyst, is the Zn/Pd ratio in the two hybrid systems. It has already been reported that having neighbouring Zn0 around Pd atom significantly suppresses trans-COOH which leads to RWGS reaction.6 Therefore, the enhanced oxygenates formation in the PdZn@ZnO + H-ZSM-5 can be attributed to the presence of a higher amount of Zn neighbours around Pd atom.
Despite the requirement of adequate supply of neighbouring Zn atoms around Pd atom, it appears it comes at a cost. In the hybrid catalyst systems, considerable amount of DME were observed rather than the expected hydrocarbons.58 DME formation is known to precede hydrocarbon formation in MTH reactions via MeOH dehydration over Brønsted acid sites.59,60 MeOH dehydration is mechanistically suggested to occur over the acid sites via either associative route, which involves protonated MeOH dimer formation, or dissociative route involving methoxy reacting with MeOH. Higher MeOH pressures favours dimer formation whilst dissociation of proton from MeOH is less favoured. Furthermore MTH reaction is known to involve an induction period due to initial C–C bond formation, hydrocarbon pool and autocatalytic reactions,61 more pronounced at lower temperatures.62 The induction period significantly reduces with increasing temperature, as it has been reported for example that the induction period reduced from 120 to 10 min at 260 and 280 °C respectively with about 25 to 37% MeOH conversion to hydrocarbons over H-ZSM-5 with 4 gMeOH gH-ZMS-5−1 h−1 WHSVs.62 Hence, considering the rather low WHSV (i.e. 0.27 and 0.21 gMeOH gH-ZMS-5−1 h−1, based on amount of oxygenates produced over PdZn@ZnO + H-ZSM-5 (40) in this study respectively) at 300 and 350 °C, the reaction was expected to proceed beyond DME to hydrocarbons.
One important factor in the MTH mechanism is the presence of Brønsted acid sites.63 The XAS data showed that the Zn K-edge spectrum of PdZn@H-ZSM-5 was dominated by ZnII ions which progressively decrease in coordination during pre-treatment (activation) and closely resembled dehydrated Zn-exchanged H-ZSM-5.48 Even though in the Zn K-edge XAS spectrum of PdZn@ZnO + H-ZSM-5 (40), ZnII ion migration evidence was overshadowed by the dominant response from ZnO support, we can infer from the PdZn@H-ZSM-5 (40) results that a substantial fraction of Zn ions has exchanged into the zeolite component of the bi-functional catalysts. The FT-IR data indicated a substantial decrease in the Brønsted acid site for the PdZn@H-ZSM-5 and PdZn@ZnO + H-ZSM-5 prior to the reaction feed as well as the spent hybrid catalyst whilst the amount of Lewis acid sites increased. This suggested that the ZnII ion-exchanged with the protons in the zeolite to poison their Brønsted acid sites. As a result, the Brønsted acid required to initiate hydrocarbon formation from MeOH or DME was inadequate, which explains why the MeOH conversion ended in DME formation.
As a final note, the observation of concurrent ZnO reduction to form PdZn alloy nanoparticles, and Zn2+ diffusion followed by ion exchange with H+ in the zeolite, both during pretreatment in a H2 flow, may seem contradictory. To this end, it is important to note that the Gibbs free energy of Zn oxidation (eqn (5)) is −358 kJ mol−1 at 400 °C, corresponding to an equilibrium constant of 6.5 × 1027. Hence, minor traces of O2 (or H2O) in the gas will stabilize zinc in the Zn2+ state, as experimentally demonstrated by Lew et al.64
| Zn + 0.5O2 ⇌ ZnO | (5) |
Considering next the formation of PdZn alloy phase, Kou and Chang reported high stability of intermetallic phases formed between Zn and group VIII transition metals such as Pd.65 They ascribed the stabilization to transfer of electrons from Zn to the d-band of Pd, thus suggesting that Pd co-presence impacts ZnO reduction.
5. Conclusion
CO2 hydrogenation to DME was performed over PdZn alloy mixed with H-ZSM-5 catalyst. Although two different methods (SOMC grafting and physical mixture) were employed to introduce the Pd–Zn on the zeolite, operando PXRD and XAS showed that similar active sites (PdZn alloy) were generated in both cases prior to CO2 hydrogenation reaction feed conditions. Furthermore, PdZn alloy which is the active site for MeOH synthesis remained stable under reaction conditions within the 250–350 °C temperature range with no important structural modifications or extra-phases formed except crystallite size growth. Physical mixture of PdZn@ZnO with H-ZSM-5 showed an enhanced oxygenates yield of 8.8% at 300 °C, which approached thermodynamic equilibrium yield of DME but beyond that of MeOH. With ZnO as support, PdZn@ZnO + H-ZSM-5 performed better than PdZn grafted on H-ZSM-5 by SOMC, attributed to higher amount of Zn which suppressed isolated Pd atoms. Instead of hydrocarbons, DME was rather observed in the hybrid catalysts. Clearly, this was due to Zn ion migration into the zeolite thereby poisoning the Brønsted acid sites responsible for hydrocarbon formation. Overall, even though increased number of neighbouring Zn0 is required to optimize the PdZn alloy formation, excess Zn have the tendency to diffuse into the zeolite to replace acid protons rendering it less effective for hydrocarbon formation.Cascade reactions over hybrid catalysts represent a promising approach in designing more effective routes to convert CO2 into value-added products. However, the presented results for PdZn/zeolite hybrid catalysts exemplify how the different functionalities could undertake unexpected interactions, ultimately biasing the hybrid catalyst performance. In this context, a careful and balanced choice of the compositional characteristics, together with detailed characterization under reaction-relevant conditions, are crucial to design improved multi-functional catalytic systems for CO2 valorisation.
Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
CAS, UO: acknowledge the Research Council of Norway for funding through grant number 288331 (CO2LO). EB: acknowledges the CASCADE-X MSCA-IF project; this project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 793423. ALB: acknowledges Russian Science Foundation project No. 18-73-00189 for funding the research (participation in the synchrotron experiment and data analysis). EB, SB, MT, UO: acknowledge the COZMOS project; This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 837733. We thank Alina Skorynina and Elizaveta Kamyshova for their help during XAS and PXRD measurements. We are indebted to Wouter van Beek and Dragos Stoian for their friendly and competent assistance during the measurements at BM31. We acknowledge that part of the work was carried out at NorTEM (the Norwegian Centre for Transmission Electron Microscopy) in Oslo, and we thank Dr. Phuong Dan Nguyen for her support.References
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- G. A. Florides and P. Christodoulides, Environ. Int., 2009, 35, 390–401 CrossRef CAS PubMed.
- World Energy agency, Overview and key findings.
- W. J. Thomas and S. Portalski, Ind. Eng. Chem., 1958, 50, 967–970 CrossRef CAS.
- J. B. Hansen and P. E. Højlund Nielsen, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp, H. G. Karge and H. Pfeifer, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008, pp. 2920–2949 Search PubMed.
- F. Liao, X.-P. Wu, J. Zheng, M. M.-J. Li, A. Kroner, Z. Zeng, X. Hong, Y. Yuan, X.-Q. Gong and S. C. E. Tsang, Green Chem., 2017, 19, 270–280 RSC.
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS.
- H. Bahruji, M. Bowker, W. Jones, J. Hayward, J. Ruiz Esquius, D. J. Morgan and G. J. Hutchings, Faraday Discuss., 2017, 197, 309–324 RSC.
- J. Xu, X. Su, X. Liu, X. Pan, G. Pei, Y. Huang, X. Wang, T. Zhang and H. Geng, Appl. Catal., A, 2016, 514, 51–59 CrossRef CAS.
- C.-H. Kim, J. S. Lee and D. L. Trimm, Top. Catal., 2003, 22, 319–324 CrossRef CAS.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Appl. Catal., A, 2018, 560, 42–53 CrossRef CAS.
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. Navarro Yerga and M. Behrens, J. Catal., 2012, 293, 27–38 CrossRef CAS.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- A. García-Trenco, E. R. White, A. Regoutz, D. J. Payne, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2017, 7, 1186–1196 CrossRef.
- S. E. Collins, J. J. Delgado, C. Mira, J. J. Calvino, S. Bernal, D. L. Chiavassa, M. A. Baltanás and A. L. Bonivardi, J. Catal., 2012, 292, 90–98 CrossRef CAS.
- E. M. Fiordaliso, I. Sharafutdinov, H. W. P. Carvalho, J.-D. Grunwaldt, T. W. Hansen, I. Chorkendorff, J. B. Wagner and C. D. Damsgaard, ACS Catal., 2015, 5, 5827–5836 CrossRef CAS.
- K. Sun, Z. Fan, J. Ye, J. Yan, Q. Ge, Y. Li, W. He, W. Yang and C.-j. Liu, J. CO2 Util., 2015, 12, 1–6 CrossRef CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019 CrossRef CAS PubMed.
- J. Gao, C. Jia and B. Liu, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- J. L. Snider, V. Streibel, M. A. Hubert, T. S. Choksi, E. Valle, D. C. Upham, J. Schumann, M. S. Duyar, A. Gallo, F. Abild-Pedersen and T. F. Jaramillo, ACS Catal., 2019, 9, 3399–3412 CrossRef CAS.
- N. Akkharaphatthawon, N. Chanlek, C. K. Cheng, M. Chareonpanich, J. Limtrakul and T. Witoon, Appl. Surf. Sci., 2019, 489, 278–286 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- R. Raudaskoski, M. V. Niemelä and R. L. Keiski, Top. Catal., 2007, 45, 57–60 CrossRef CAS.
- J. Liu, J. Shi, D. He, Q. Zhang, X. Wu, Y. Liang and Q. Zhu, Appl. Catal., A, 2001, 218, 113–119 CrossRef CAS.
- X.-M. Liu, G. Q. Lu and Z.-F. Yan, Appl. Catal., A, 2005, 279, 241–245 CrossRef CAS.
- J. Słoczyński, R. Grabowski, A. Kozłowska, P. Olszewski, J. Stoch, J. Skrzypek and M. Lachowska, Appl. Catal., A, 2004, 278, 11–23 CrossRef.
- J. Słoczyński, R. Grabowski, P. Olszewski, A. Kozłowska, J. Stoch, M. Lachowska and J. Skrzypek, Appl. Catal., A, 2006, 310, 127–137 CrossRef.
- T. Phongamwong, U. Chantaprasertporn, T. Witoon, T. Numpilai, Y. Poo-arporn, W. Limphirat, W. Donphai, P. Dittanet, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2017, 316, 692–703 CrossRef CAS.
- F. Liao, X.-P. Wu, J. Zheng, M.-J. Li, Z. Zeng, X. Hong, A. Kroner, Y. Yuan, X.-Q. Gong and S. C. E. Tsang, Catal. Sci. Technol., 2016, 6, 7698–7702 RSC.
- T. A. Semelsberger, R. L. Borup and H. L. Greene, J. Power Sources, 2006, 156, 497–511 CrossRef CAS.
- G. J. Hutchings and R. Hunter, Catal. Today, 1990, 6, 279–306 CrossRef CAS.
- J. S. Martinez-Espin, M. Mortén, T. V. W. Janssens, S. Svelle, P. Beato and U. Olsbye, Catal. Sci. Technol., 2017, 7, 2700–2716 RSC.
- T. Witoon, T. Permsirivanich, N. Kanjanasoontorn, C. Akkaraphataworn, A. Seubsai, K. Faungnawakij, C. Warakulwit, M. Chareonpanich and J. Limtrakul, Catal. Sci. Technol., 2015, 5, 2347–2357 RSC.
- Y. Suwannapichat, T. Numpilai, N. Chanlek, K. Faungnawakij, M. Chareonpanich, J. Limtrakul and T. Witoon, Energy Convers. Manage., 2018, 159, 20–29 CrossRef CAS.
- H. Bahruji, R. D. Armstrong, J. Ruiz Esquius, W. Jones, M. Bowker and G. J. Hutchings, Ind. Eng. Chem. Res., 2018, 57, 6821–6829 CrossRef CAS.
- G. Bonura, M. Cordaro, L. Spadaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., A, 2013, 140-141, 16–24 CrossRef CAS.
- G. Bonura, M. Migliori, L. Frusteri, C. Cannilla, E. Catizzone, G. Giordano and F. Frusteri, J. CO2 Util., 2018, 24, 398–406 CrossRef CAS.
- H. Nakamoto and H. Takahashi, Zeolites, 1982, 2, 67–68 CrossRef CAS.
- E. Catizzone, A. Aloise, M. Migliori and G. Giordano, Appl. Catal., A, 2015, 502, 215–220 CrossRef CAS.
- S. Jiang, J.-S. Hwang, T.-H. Jin, T. Cai, W. Cho, Y.-S. Baek and S.-E. Park, Bull. Korean Chem. Soc., 2004, 25, 185–189 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle, K.-P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- H. Schulz, Z. Siwei and H. Kusterer, in Studies in Surface Science and Catalysis, ed. T. Inui, S. Namba and T. Tatsumi, Elsevier, 1991, vol. 60, pp. 281–290 Search PubMed.
- P. Rouge, A. Garron, S. Norsic, C. Larabi, N. Merle, L. Delevoye, R. M. Gauvin, K. C. Szeto and M. Taoufik, Mol. Catal., 2019, 471, 21–26 CrossRef.
- K. C. Szeto, A. Gallo, S. Hernández-Morejudo, U. Olsbye, A. De Mallmann, F. Lefebvre, R. M. Gauvin, L. Delevoye, S. L. Scott and M. Taoufik, J. Phys. Chem. C, 2015, 119, 26611–26619 CrossRef CAS.
- M. W. Tew, H. Emerich and J. A. van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465 CrossRef CAS.
- I. Pinilla-Herrero, E. Borfecchia, J. Holzinger, U. V. Mentzel, F. Joensen, K. A. Lomachenko, S. Bordiga, C. Lamberti, G. Berlier, U. Olsbye, S. Svelle, J. Skibsted and P. Beato, J. Catal., 2018, 362, 146–163 CrossRef CAS.
- E. Selli and L. Forni, Microporous Mesoporous Mater., 1999, 31, 129–140 CrossRef CAS.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Kieffer and J. P. Wright, Powder Diffr., 2013, 28, S339–S350 CrossRef CAS.
- A. Hammersley, J. Appl. Crystallogr., 2016, 49, 646–652 CrossRef CAS.
- M. Gentzen, D. E. Doronkin, T. L. Sheppard, A. Zimina, H. Li, J. Jelic, F. Studt, J.-D. Grunwaldt, J. Sauer and S. Behrens, Angew. Chem., Int. Ed., 2019, 58, 15655–15659 CrossRef CAS PubMed.
- J. R. Gallagher, D. J. Childers, H. Zhao, R. E. Winans, R. J. Meyer and J. T. Miller, Phys. Chem. Chem. Phys., 2015, 17, 28144–28153 RSC.
- E. J. Peterson, B. Halevi, B. Kiefer, M. N. Spilde, A. K. Datye, J. Peterson, L. Daemen, A. Llobet and H. Nakotte, J. Alloys Compd., 2011, 509, 1463–1470 CrossRef CAS.
- W. Schmidt, U. Wilczok, C. Weidenthaler, O. Medenbach, R. Goddard, G. Buth and A. Cepak, J. Phys. Chem. B, 2007, 111, 13538–13543 CrossRef CAS PubMed.
- T. V. W. Janssens, J. Catal., 2009, 264, 130–137 CrossRef CAS.
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53, 12177–12181 CrossRef CAS PubMed.
- A. Ghorbanpour, J. D. Rimer and L. C. Grabow, ACS Catal., 2016, 6, 2287–2298 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- L. Qi, Y. Wei, L. Xu and Z. Liu, ACS Catal., 2015, 5, 3973–3982 CrossRef CAS.
- L. Qi, J. Li, Y. Wei, Y. He, L. Xu and Z. Liu, RSC Adv., 2016, 6, 52284–52291 RSC.
- S. Lew, A. F. Sarofim and M. Flytzani-Stephanopoulos, Chem. Eng. Sci., 1992, 47, 1421–1431 CrossRef CAS.
- S. Kou and Y. A. Chang, Acta Metall., 1975, 23, 1185–1190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: N2 adsorption, TEM, XAS, PXRD, test results. See DOI: 10.1039/d0cy00440e |
| This journal is © The Royal Society of Chemistry 2020 |