
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of hydrogen peroxide without the use of acids or halide promoters in dissolution†
G.
Blanco-Brieva
a,
F.
Desmedt
b,
P.
Miquel‡
b,
J. M.
Campos-Martin
 *a and
J. L. G.
Fierro§
a
*a and
J. L. G.
Fierro§
a
aSustainable Energy and Chemistry Group, Instituto de Catálisis y Petroleoquímica, CSIC, Marie Curie, 2, Cantoblanco, 28049 Madrid, Spain. E-mail: jm.campos@csic.es; Web: http://www.icp.csic.es/eqsgroup/
bSolvay, Rue de Ransbeek, 310, B-1120 Brussels, Belgium
First published on 17th March 2020
Abstract
We show the first catalyst family able to produce hydrogen peroxide at concentrations higher than 5 wt% in the absence of any promoter or stabilizing agent in dissolution. The catalysts are composed of palladium supported on bifunctionalized silica or resin with acidic groups and bromide.
H2O2 is commonly recognized as a green oxidant and a major chemical commodity produced at an industrial scale by sequential hydrogenation and oxidation of an alkyl anthraquinone. However, this process requires high-volume reactors, large amounts of solvents and costly separation steps, thus making the process economical only for large units;1 furthermore, the above process is definitely not green. An attractive alternative is a direct reaction between H2 and O2 in the presence of catalysts.2,3 The direct synthesis seems to be a simple reaction, but there is a complex reaction network that makes controlling selectivity quite difficult; in addition to the main reaction, other reactions occur on the same catalysts: a) direct formation of water, b) decomposition, and hydrogenation of H2O2, all of which reduce the selectivity to hydrogen peroxide.4–6 To solve the problem of these sequential reactions with palladium catalysts, stabilizers are required.7–9 These are typically mineral acids and halide ions. Unfortunately, the presence of stabilizers in the reaction medium is a serious problem because they have to be removed from the product after the reaction, especially when the H2O2 is to be used without refinement (e.g., epoxidation of propylene). Perhaps, the most negative effects are the dissolution of the active components of the catalyst and the need to work with special equipment to avoid corrosion.10 These drawbacks can be eliminated by using supports that contain acidic groups. This type of catalytic system has been successfully tested by employing sulfonic acid functionalized supports, such as acidic resins or functionalized silica, as reported by our group7,11–14 and others.15–21 These catalyst types can produce hydrogen peroxide solutions by the direct reaction between H2 and O2 in methanol with high selectivity, at concentrations >10 wt%; moreover, the above solutions are non-corrosive.
In this work, we report catalytic systems in non-acidic solutions or other additives. As such, the use of acidic or bromide-containing stabilizers is avoided. Our alternative fits well in the principles of green chemistry The catalytic systems employed consist of supported palladium incorporated on commercial resin or silica functionalized with acidic groups and bromide groups to produce H2O2 by direct synthesis between H2 and O2 (outside the explosive limits of H2/O2 mixtures) in non-acidic solutions. Bifunctionalized silica with aryl sulfonic groups (Si–C6H4–SO3H) and aryl bromide groups was purchased from Silicycle Inc. (labeled Sil-Br). However, the resin was modified starting from a resin styrene–divinylbenzene copolymer functionalized with sulfonic groups (LK2621 kindly provided by Lewatit). The introduction of bromide groups was performed using two different methods: a) bromination method based on H2O2/HBr and b) bromination with Br2.
The bromination method based on H2O2/HBr consists of the oxidation of hydrobromic acid by H2O2 without the use of any transition metal catalyst (see the ESI†), labeled resin-Br1. Bromination with Br2 consists of the direct addition of Br2 catalyzed by a Lewis acid (see the ESI†), labeled resin-Br2.
Using these supports, three catalysts were prepared to obtain a Br/Pd weight ratio near 0.64 in the final catalysts (see the ESI†), no reduction treatment was employed in the preparation of the catalysts. Such a ratio was selected on the basis of earlier results,22 which demonstrated that this ratio is optimal for reaching a high hydrogen peroxide yield using acidic resin catalysts with added bromobenzene in solution as a promoter.
The concentration of S, Br and Pd of the catalysts was determined by XRF. The Br concentration in the catalysts was quite similar to that in the support (result not shown here), which indicated that no modification occurred during the Pd incorporation. However, the amount of Br depended on the matrix used (silica or resin) and the method of Br incorporation that followed. The bromination method based on H2O2/HBr yielded a higher extent of Br incorporation than the Br2 method (Table 1). For this reason, the amount of palladium added to the support was different to reach the target Br/Pd ratio near 0.64; thus, the palladium content in each catalyst was different (Table 1).
| Sample | S (wt%) | Br (wt%) | Pd (wt%) | Br/Pd weight |
|---|---|---|---|---|
| Pd/resin | 10.78 | — | 2.20 | — |
| Pd/resin-Br1 | 10.78 | 1.60 | 2.20 | 0.73 |
| Pd/resin-Br2 | 10.75 | 0.65 | 0.94 | 0.69 |
| Pd/Sil-Br | 10.52 | 1.33 | 1.73 | 0.77 |
These catalysts were tested in the direct synthesis of H2O2, introducing the same amount of palladium in the reactor modifying the amount of catalyst present in the reactor. The catalysts were employed as prepared without any reduction treatment. In all cases, H2O2 formation was detected, and the highest peroxide concentration was obtained using the catalyst prepared with silica, Pd/Sil–SO3H–Br, where the concentration of H2O2 was approximately 4 wt% and the selectivity to hydrogen peroxide was 27%, both figures were clearly lower than those previously reported by our group using a bromobenzene additive in solution.22 Based on these non-optimized results and taking into account that the interaction of palladium–bromine would be less efficient in a catalyst with tethered Br species, with respect to an additive in solution, we decided to prepare samples with a higher Br/Pd ratio, increasing from 0.6 to 1.2. These new prepared catalysts were shown in Table 2.
| Sample | S (wt%) | Br (wt%) | Pd (wt%) | Br/Pd weight |
|---|---|---|---|---|
| Pd/resin-Br1b | 10.78 | 1.60 | 1.32 | 1.21 |
| Pd/resin-Br2b | 10.75 | 0.65 | 0.55 | 1.18 |
| Pd/Sil-Brb | 10.52 | 1.33 | 1.11 | 1.20 |
This second lot of catalysts were used in the direct synthesis of hydrogen peroxide at 5 MPa and 40 °C (see the ESI†), the catalysts were employed as prepared without any reduction treatment. The hydrogen peroxide concentration and selectivity profiles, shown in Fig. 1, displayed a behavior completely different with respect to their counterparts with a low Br/Pd ratio (Table 1vs.Table 2), and were quite similar to those using a reference catalyst with the addition of a bromide promoter in dissolution (Pd/resin).22 Furthermore, the new catalytic systems had clearly better profiles from a sustainability point of view (absence of additives in dissolution). The concentration of hydrogen peroxide increases over time, reaching values higher than 6 wt% for all the catalysts after 3 h of reaction. The highest concentration of hydrogen peroxide is reached with the catalyst Pd/resin-Br2b.
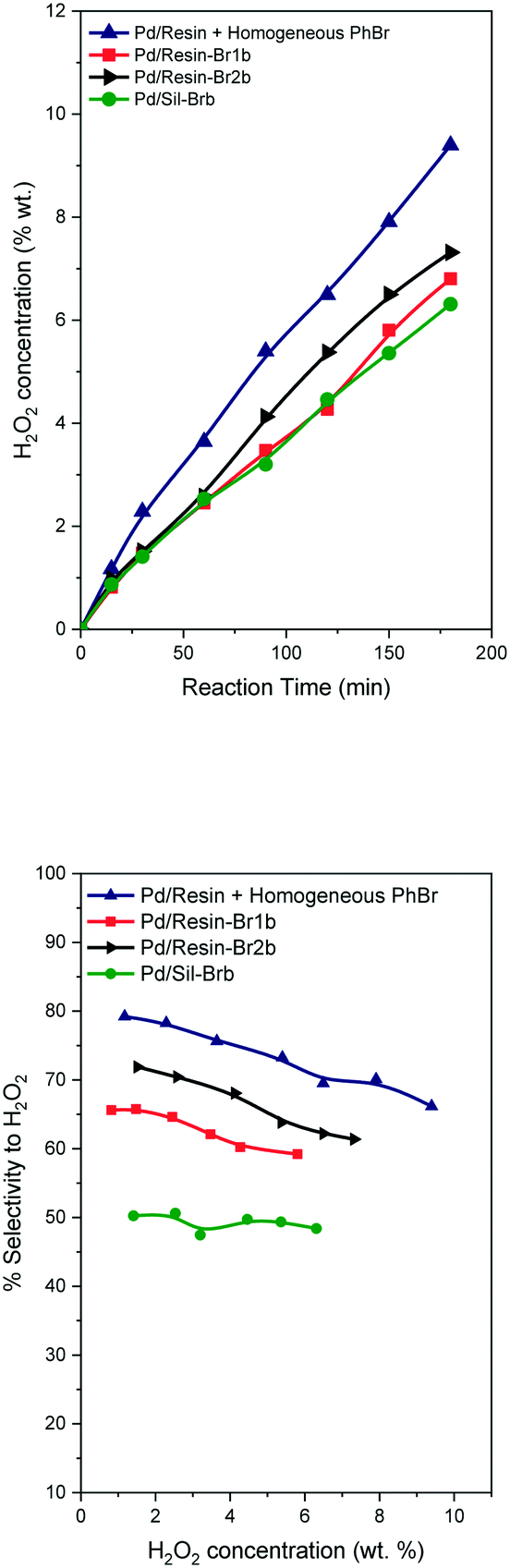 | ||
| Fig. 1 H2O2 concentration profiles versus time and H2O2 selectivity profiles versus H2O2 concentration for the direct synthesis at 40 °C and 5.0 MPa. | ||
The selectivity vs. hydrogen peroxide concentration shows a slight decrease when the H2O2 concentration increases. However, all the catalysts with bromide incorporated into the solid have lower selectivity than the reference sample with a promoter in dissolution. Taking into account that this procedure is greener, this is a good starting point. The order of selectivity values follows the same trend as the hydrogen peroxide concentration. The liquid samples at the end of the reaction were analyzed by ICP-OES and by ionic chromatography. No palladium was detected by ICP-OES (<0.1 Pd ppm) and no bromide ions in solution were detected (<0.5 ppb). The absence of palladium leaching was previously observed in our previous work where the bromide promoter was in dissolution.22 This behavior is different to that in previous work where the bromide promoter was added to the solid catalyst as NaBr, and H3PO4,21 and the authors found partial leaching of palladium. But in this work we do not use free acid and ionic bromide species in solution, and in consequence the palladium leaching is clearly suppressed.
This second group of fresh catalysts was characterized by XPS. The chemical state of Pd was determined from the binding energy of the Pd 3d5/2 peak of the XPS spectra (Table 3). Most of the samples showed the presence of two palladium species, a minor component at 336.9 eV typical of palladium oxide supported species and a main component at a higher binding energy (338.0 eV). This component could be attributed to oxidized Pd species interacting with –SO3H groups.7 No signal of reduced palladium species was detected, as expected because the samples were analyzed without any reduction treatment. The proportions of both Pd species were different among the prepared catalysts. The main component was a high binding energy species in all cases. The catalysts with a high proportion of palladium species with a binding energy of 338.0 eV had the highest selectivity for hydrogen peroxide. This observation was in agreement with previous experimental work where catalysts with more Pd(II) species interacting with the acidic groups (high BE) gave higher selectivity to H2O27,11 and in agreement with recent studies where oxidized species favored the selectivity to hydrogen peroxide.23
| Sample | BE Pd 3d5/2 (eV) |
|---|---|
| Pd/resin | 337.9 (100) |
| Pd/resin-Br1b | 336.9 (10) |
| 337.8 (90) | |
| Pd/resin-Br2b | 337.9 (100) |
| Pd/Sil-Brb | 336.9 (30) |
| 338.0 (70) |
We have made some XPS measurements of used catalysts, and the palladium signal has several components detecting the presence of a mixture of metallic palladium and oxidized species. There are several players in the reaction system, an excess of oxygen (around 46.4%) and methanol both have an opposite effect on the palladium oxidation state for a reaction at 40 °C. However, the treatment of the samples after use in the reaction, washing and drying for instance, makes it very difficult to obtain an accurate ratio of oxidized/reduced species which are really in the catalysts in the reaction media.
Finally, we decided to check the effect of the interaction of palladium and bromine species on the decomposition of hydrogen peroxide. For this purpose, the catalysts were tested with 10 wt% hydrogen peroxide at 40 °C (Fig. 2) (see the ESI†). In addition, we included the use of two reference catalysts, a Pd/resin catalyst and Pd/resin + homogeneous PhBr as a promoter.22 The change in the hydrogen peroxide concentration profiles was different depending on the catalysts employed. When a Pd/resin catalyst without the presence of a bromine promoter was used, the hydrogen peroxide concentration fell quickly to zero in approximately 90 min. However, in the presence of bromine promoters, the concentration of hydrogen peroxide was kept quite high, decreasing by less than 10% after 3 h. However, different behavior was detected among the samples. A low decrease in hydrogen peroxide was observed for the Pd/resin + homogeneous PhBr, while the other catalysts followed the order Pd/resin-Br2b < Pd/resin-Br1b < Pd/SilBrb.
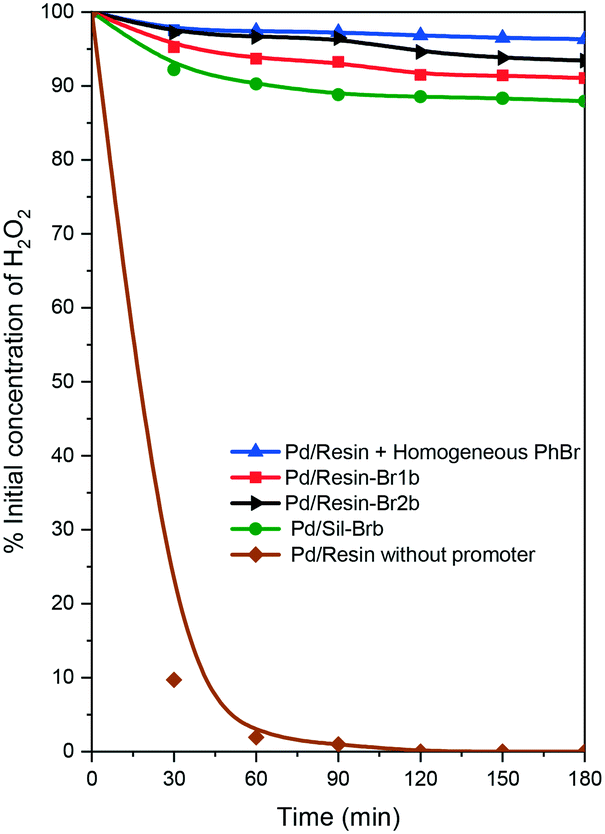 | ||
| Fig. 2 Hydrogen peroxide decomposition test at 40 °C for the resin supported catalysts with different Br/Pd ratios. | ||
These results clearly showed that there is an interaction of bromine and palladium that affects the ability to decompose the hydrogen peroxide. In addition, the catalysts with a high ability to decompose hydrogen peroxide reached low selectivity to hydrogen peroxide in direct synthesis (Fig. 1 and 2). We do not indicate that there is a direct relationship between hydrogen peroxide decomposition and selectivity, because other important reactions must be taken into account in the reaction mechanism (for instance, the hydrogenation of H2O2). But the measurement of the hydrogen decomposition curves can be a simple and quick indication of the presence or not of the interaction between palladium and bromine species.
This kind of catalyst is a very interesting starting point for direct synthesis on acidic/bromide supports and opens new ways to approach the difficult application of direct hydrogen peroxide synthesis at an industrial scale.
Conclusions
Herein, we show the possibility of producing hydrogen peroxide at high concentrations (>5 wt%) by direct synthesis in the absence of acid and halide promoters in dissolution, which is a greener option with respect to the present state of the art. For this purpose, we prepared catalysts based on Pd on supports in which both acidic and bromide groups were incorporated. This methodology was employed in two different kinds of supports, silica and resins. All prepared catalysts produced H2O2, but there was a tremendous effect on the catalytic behavior based on the Br/Pd ratio and not the concentration of Br. The more selective catalysts had a high proportion of Pd species interacting with the sulfonic groups (BE 338.2 eV).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from Solvay (Brussels, Belgium). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- C. Samanta, Appl. Catal., A, 2008, 350, 133–149 CrossRef CAS.
- A. I. Olivos-Suarez, A. Szecsenyi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- P. Biasi, J. Garcia-Serna, A. Bittante and T. Salmi, Green Chem., 2013, 15, 2502–2513 RSC.
- G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. G. Fierro, Chem. Commun., 2004, 1184–1185, 10.1039/B402530J.
- C. Samanta and V. R. Choudhary, Appl. Catal., A, 2007, 326, 28–36 CrossRef CAS.
- S. Melada, R. Rioda, F. Menegazzo, F. Pinna and G. Strukul, J. Catal., 2006, 239, 422–430 CrossRef CAS.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2002, 206, 173–176 CrossRef CAS.
- G. Blanco-Brieva, M. C. Capel-Sanchez, M. P. de Frutos, A. Padilla-Polo, J. M. Campos-Martin and J. L. G. Fierro, Ind. Eng. Chem. Res., 2008, 47, 8011–8015 CrossRef CAS.
- G. Blanco-Brieva, J. M. Campos-Martin, M. P. De Frutos and J. L. G. Fierro, Catal. Today, 2010, 158, 97–102 CrossRef.
- G. Blanco-Brieva, M. P. De Frutos Escrig, J. M. Campos-Martin and J. L. G. Fierro, Green Chem., 2010, 12, 1163–1166 RSC.
- G. Blanco-Brieva, M. Montiel-Argaiz, F. Desmedt, P. Miquel, J. M. Campos-Martin and J. L. G. Fierro, RSC Adv., 2016, 6, 99291–99296 RSC.
- Y. M. Chung, Y. T. Kwon, T. J. Kim, S. H. Oh and C. S. Lee, Chem. Commun., 2011, 47, 5705–5707 RSC.
- J. Kim, Y.-M. Chung, S.-M. Kang, C.-H. Choi, B.-Y. Kim, Y.-T. Kwon, T. J. Kim, S.-H. Oh and C.-S. Lee, ACS Catal., 2012, 2, 1042–1048 CrossRef CAS.
- S. Sterchele, P. Biasi, P. Centomo, S. Campestrini, A. Shchukarev, A.-R. Rautio, J.-P. Mikkola, T. Salmi and M. Zecca, Catal. Today, 2015, 248, 40–47 CrossRef CAS.
- C. Burato, S. Campestrini, Y. F. Han, P. Canton, P. Centomo, P. Canu and B. Corain, Appl. Catal., A, 2009, 358, 224–231 CrossRef CAS.
- C. Burato, P. Centomo, M. Rizzoli, A. Biffis, S. Campestrini and B. Corain, Adv. Synth. Catal., 2006, 348, 255–259 CrossRef CAS.
- S. Park, J. H. Choi, T. J. Kim, Y.-M. Chung, S.-H. Oh and I. K. Song, J. Mol. Catal. A: Chem., 2012, 353–354, 37–43 CrossRef CAS.
- P. Biasi, J. P. Mikkola, S. Sterchele, T. Salmi, N. Gemo, A. Shchukarev, P. Centomo, M. Zecca, P. Canu, A. R. Rautio and K. Kordas, AIChE J., 2017, 63, 32–42 CrossRef CAS.
- G. Blanco-Brieva, M. Montiel-Argaiz, F. Desmedt, P. Miquel, J. M. Campos-Martin and J. L. G. Fierro, RSC Adv., 2016, 6, 99291–99296 RSC.
- F. Wang, C. G. Xia, S. P. de Visser and Y. Wang, J. Am. Chem. Soc., 2019, 141, 901–910 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00416b |
| ‡ Deceased (1982−2016). |
| § Deceased (1948−2020). |
| This journal is © The Royal Society of Chemistry 2020 |