
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile catalyst recycling by thermomorphic behaviour avoiding organic solvents: a reactive ionic liquid in the homogeneous Pd-catalysed telomerisation of the renewable β-myrcene†
Michael
Terhorst
 a,
Anna
Kampwerth
a,
Amelie
Marschand
a,
Dieter
Vogt
a,
Andreas J.
Vorholt
b and
Thomas
Seidensticker
*a
a,
Anna
Kampwerth
a,
Amelie
Marschand
a,
Dieter
Vogt
a,
Andreas J.
Vorholt
b and
Thomas
Seidensticker
*a
aTU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Tel: +49 231 755 2310
bMax-Planck-Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45740 Mülheim an der Ruhr, Germany
First published on 3rd February 2020
Abstract
For the first time, the reactive ionic liquid N,N-dimethyl ammonium N,N-dimethyl carbamate (dimcarb) was used as a polar solvent in the palladium-catalyzed telomerisation of the renewable monoterpene β-myrcene. By doing so, it was possible to avoid any volatile organic solvents. Moreover, the special decomposition behaviour of the reactive ionic liquid dimcarb could be exploited to the extent that dimethyl amine was used as a reagent in the reaction. Accordingly, non-symmetrical C20-chain dimethyl amines were produced from β-myrcene and dimethylamine liberated by dimcarb, achieving selectivities higher than 80%. Detailed investigations revealed the thermomorphic behaviour of the reaction mixture consisting only of dimcarb, β-myrcene and the catalytic system. The advantageous temperature-dependent phase behaviour resulted in one liquid phase occurring at the reaction temperature and two liquid phases upon cooling. Consequently, a virtually pure organic product phase could be separated from the polar catalyst-containing dimcarb phase. Immobilization of the catalyst in the polar reactive ionic liquid phase was ensured by the use of sulfonated triphenylphosphine derivatives. Subsequently, the separated catalyst-containing dimcarb phase was successfully reused in 14 consecutive recycling runs with only minimal leaching of both the catalyst and dimcarb, reaching a total turnover number of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
Introduction
In homogenous catalysis, the efficient separation of a transition metal catalyst from the product mixture is a critical issue. However, due to a lack of efficient strategies to overcome this issue, the advantages of homogeneous catalysts are frequently weakened. Hence, the majority of catalysed syntheses in industries are heterogeneously catalysed since the separation of the product and catalyst is much easier.1 Nevertheless, homogeneous catalysis enables sustainable and atom-economic ways to produce valuable products with significantly lower amounts of energy used and waste produced.1 In particular, the formation of long-chain amines is still bound to waste production by non-atom-economic procedures or by implementing activated and functionalized starting materials.2 Amino-telomerisation is an alternative to the known industrial amine syntheses in this respect, featuring high atom economy and mild reaction conditions.The dimerisation of two 1,3-diene molecules combined with the attack of a Brønsted-acidic nucleophile (Scheme 1), generally referred to as telomerisation, is a long-known reaction and part of recent reviews on the topic of telomerisation and functionalization of 1,3-dienes.3–5 Mechanistic insights were given by Jolly, who investigated the telomerisation of butadiene with methanol as a nucleophile.6 It is assumed that amines act similarly as nucleophiles in this reaction.7 The potential of this reaction is illustrated by the fact that there are already large-scale industrial applications, for example, in the Kuraray process for the production of 1-octanol8 from 1,3-butadiene and water (Scheme 1, R = H and Nu = –OH) via the intermediate 2,7-octadienol and in the Dow process for the production of 1-octene from 1,3-butadiene and methanol (Scheme 1, R = H and Nu = –OMe) via the intermediate 2,7-octadienyl methyl ether.9 Also, many telomerisation examples with amines are known, ranging from ammonia to secondary amines (Scheme 1, Nu = –NH2, –NHR, and –NR2).7,10–12 Likewise, in different parts of the literature, the recovery of the valuable homogeneous transition metal catalyst has already been considered. Various recycling concepts for telomerisation, such as aqueous two-phase systems,13 thermomorphic multicomponent systems (TMS)14 and utilization of ionic liquids15 have been developed. However, they have some critical drawbacks, such as mass transfer limitations and the requirement of specific, expensive and/or hazardous solvents. Currently, research is carried out in many areas of organometallic catalysis on the use of novel solvent systems for the immobilization of catalysts, such as deep eutectic solvents, supported ionic liquid phases and other novel biphasic systems based on more sustainable solvents.16–18
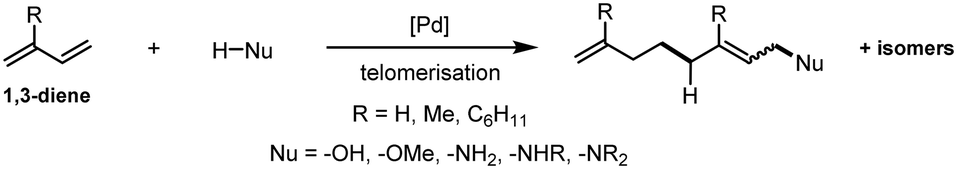 | ||
| Scheme 1 General telomerisation of 1,3-dienes with various nucleophiles. | ||
Our group recently developed a method to combine the introduction of a dimethylamine moiety by telomerisation with the recovery of the catalyst in an ionic liquid phase.19N,N-Dimethyl-ammonium-N,N-dimethyl carbamate (dimcarb) was used as a reactive ionic liquid (RIL) to immobilize the homogeneous transition metal catalyst and simultaneously react as a dimethylamine precursor. Dimcarb is a combination of two moles of dimethylamine with one mole of carbon dioxide (Scheme 2).20 It is both liquid and ionic at room temperature, but has specific properties that are untypical for ionic liquids, such as a low decomposition temperature of 60 °C.21 Thus, dimcarb reacts like dimethylamine at temperatures above that. However, below 60 °C, it is stable and can be safely handled and used as a dimethylamine source in contrast to gaseous dimethylamine, which has to be handled under strict safety conditions. Thus, dimcarb combines crucial advantages as follows:
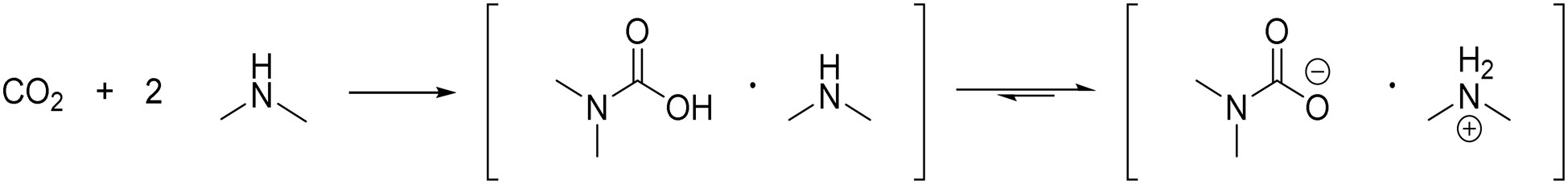 | ||
| Scheme 2 Formation of N,N-dimethyl-ammonium-N,N-dimethylcarbamate (dimcarb) and the equilibrium between its ionic and non-ionic form. | ||
▶ Highly polar solvent.
▶ Ability to react as dimethyl amine due to its decomposition above 60 °C.
▶ Liquid, safe to handle dimethyl amine source.
▶ Purification by ‘distillation’ possible.
Butadiene has already been used successfully in telomerisation with dimcarb; however, the catalyst has to be recycled by extraction of the product with cyclohexane. The active nucleophile is assumed to be the dimethyl amine that is released by the decomposition of dimcarb. In our continuous endeavour in developing sustainable amine syntheses, we envisaged employing higher molecular 1,3-dienes in the telomerisation with dimcarb. Consequently, products with even lower polarity are formed, which we thought would make their separation from the catalyst containing a reactive ionic liquid phase possible without the utilization of hazardous solvents. Hence, sustainability is improved, and additional separation steps are avoided.
For instance, terpenes exhibit a 1,3-diene unit, which is needed for telomerisation, like in the monoterpene β-myrcene. β-Myrcene as a renewable raw material22,23 has been investigated in telomerisation.24,25 Additionally, it has been used in several amination reactions, such as hydroaminomethylation26 and hydroamination,27–29 and in other homogeneously catalysed functionalization reactions such as hydroformylation.30,31 However, its telomerisation was never carried out with dimethylamine and recycling of the homogeneous palladium/phosphorus catalyst was considered but never executed.24,25 By application of β-myrcene in the telomerisation with dimcarb, highly branched, tertiary dimethylamines with a third highly branched alkyl chain (C20) at the nitrogen atom can be generated. Related molecules are industrially important for consumer products since surfactants can be formed via alkylation and N-oxidation. These long chain products should enable facile recycling, which is necessary for sustainable synthesis. No additional solvents and a straightforward decantation are expected, similarly to the hydroamination of β-farnesene (C15).19
Herein, we present our results on the generation of long-chain, tertiary dimethyl-alkyl amines in an atom-efficient way by utilizing renewable resources and the reactive ionic liquid dimcarb via homogeneously catalysed telomerisation. Furthermore, we show that the recycling of the homogeneous catalyst is possible only using reactants during the reaction and phase separation.
Results and discussion
The starting point for our reaction development was the past discoveries by our group (Behr et al. and Vorholt et al.) on the homogeneously palladium-catalysed telomerisation of β-myrcene. In the initial experiment, dimcarb was applied as a precursor for dimethylamine under the conditions previously published by Behr et al. in 2010.24 As a typical catalyst system, a combination of [Pd(MeCN)4](BF4)2 with triphenylphosphine as the ligand was used. However, the yield of the target C20-dimethylamines was only 8%. The two main side-products identified were the dimerisation product of β-myrcene and hydroamination product of β-myrcene and dimethylamine. Additional information on these preliminary results is presented in the ESI.†Scheme 3 presents an overview of the reaction system with the respective stoichiometric proportions.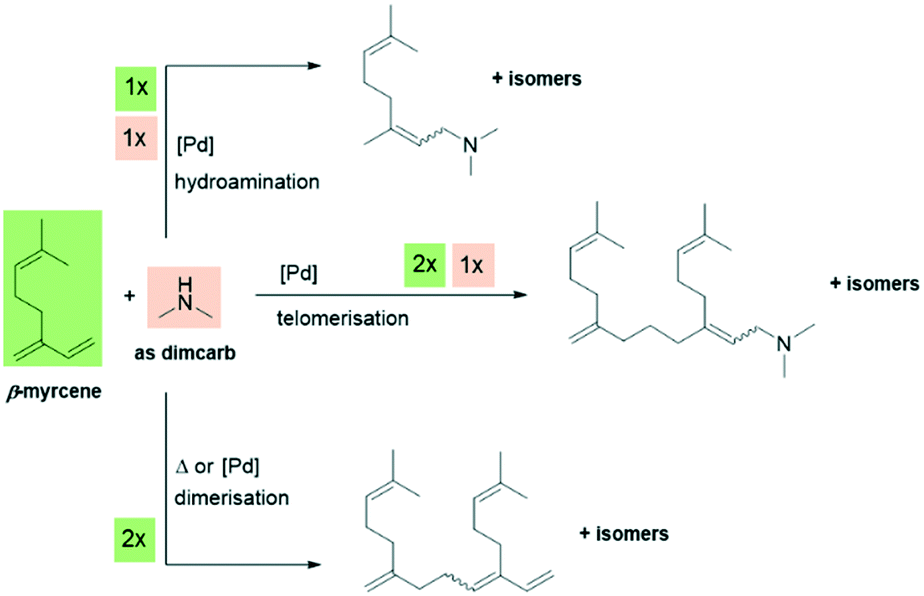 | ||
| Scheme 3 Telomerisation of β-myrcene with dimcarb and dimerisation/hydroamination as typical side reactions. | ||
The unexpected low yield of 8% compared to 91% yield reported in the literature indicates that dimcarb reacts considerably different under the chosen reaction conditions. Thus, the reaction conditions were optimized. We were able to increase the yield of the desired telomers to 46% by changing the solvent from DMF to methanol, which was used in the telomerisation of butadiene with amines before.10 To improve the sustainability by avoiding hazardous or volatile solvents, the amount of methanol was reduced, and to our delight it became apparent that the execution of the reaction without the use of any volatile organic solvent is also possible with even higher yields of the telomers of up to 83%. Detailed information on these results is presented in the ESI.† Notably, under these conditions, only the catalyst and the two substrates, dimcarb and β-myrcene, were present, and no additional solvents and auxiliaries were needed.
With these very promising initial findings in hand and the reduction of the reaction system to the two substrates only in a “neat” environment (dimcarb and β-myrcene), we focused on the recyclability of the catalytic system because the recovery of the catalyst is crucial so that the products are not contaminated and higher productivities can be achieved. In combination with β-farnesene, it was assumed by Vorholt et al. that dimcarb follows the principles of a thermomorphic multiphase system (TMS) with alkenes.32 This system is defined by the fact that two liquid phases form a monophasic system at a specific critical temperature. If this mixture falls below or exceeds the critical temperature again, spontaneous phase separation occurs. This behaviour is extremely beneficial for our reaction system due to the minimization of mass transport limitations because of a single phase at the reaction temperature and good phase separation at room temperature due to the formation of two phases. Therefore, it was necessary to investigate the phase behaviour of dimcarb in combination with β-myrcene in detail, especially after a biphasic mixture was observed after the reaction with a higher dimcarb excess (cf. ESI†). Accordingly, different dimcarb/β-myrcene ratios were investigated, and the temperature was determined at which no second phase or turbidity in the mixture was seen (Fig. 1, blue squares). The highest detected temperature of the mixtures was called the critical solution temperature (CST). The precise execution of these experiments is described in the ESI.†
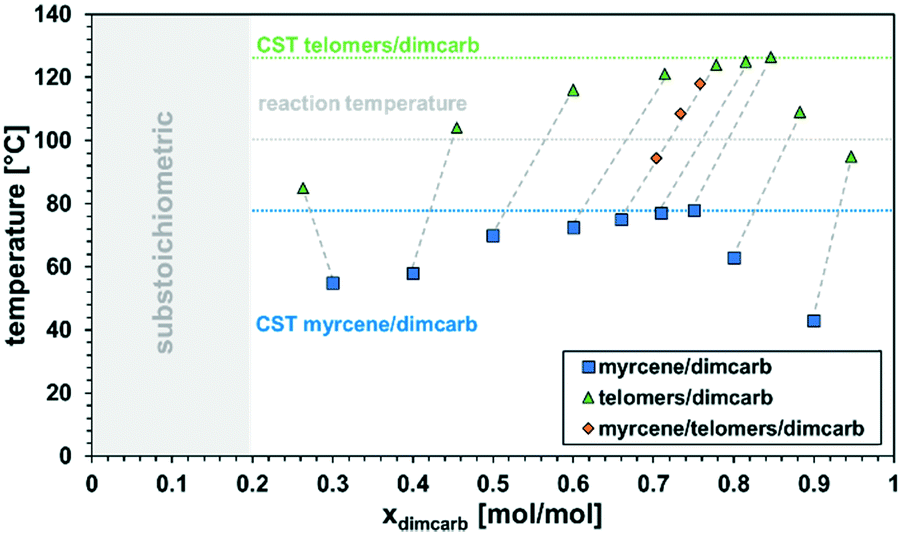 | ||
| Fig. 1 Critical solution temperatures of dimcarb/β-myrcene (blue squares) and dimcarb/telomer (green triangles) mixtures. The dashed line indicates the change in the composition during the reaction, assuming a full conversion with 100% selectivity towards telomer products. Orange diamonds represent temperatures at which model compositions with 25%, 50% and 75% conversion with a starting dimcarb molar fraction of xdimcarb = 0.66 became monophasic. | ||
It was observed that mixtures of dimcarb and β-myrcene are monophasic at temperatures >80 °C over the whole range of ratios, with a minimum CST of 43 °C at xdimcarb = 0.9. Hence, at the typical reaction temperature of 100 °C, the potential reaction mixture will be monophasic. However, this is only true for the model binary mixtures of dimcarb and β-myrcene without conversion (i.e., Xβ-myrcene = 0%), and thus no yield (Ytelomers = 0%). For catalyst recycling, the temperature-dependent phase behaviour of the mixture after the reaction also has to be considered. It was expected that the temperatures at which the reaction mixtures modelling full conversion become monophasic are generally higher due to the lower polarity of the product (C22 formed from C10). Thus, the investigation was repeated, and the critical solution temperatures of different ratios of dimcarb in combination with prior synthesized telomer products were determined (Fig. 1, green triangles). Additional information on the synthesis is presented in the ESI.† Here, the dashed lines indicate the reaction progress with the blue squares as the starting point and the green triangles representing the composition at the end of the reaction (assuming 100% selectivity towards the telomer).
Indeed, the respective critical solution temperatures shifted to higher values compared to that of binary mixtures of dimcarb and β-myrcene, but generally showed the same trends. However, at the typical reaction temperature of 100 °C, mixtures of dimcarb and the telomer-products between xdimcarb = 0.4 and 0.8 are biphasic. Hence, in the course of the reaction in such mixtures, the product will spontaneously separate from the reaction mixture. The critical conversion for this to occur was investigated at a constant xdimcarb of 0.66 at the start of the reaction with different ratios of substrate/product, modelling different degrees of conversion (Fig. 1, orange diamonds). At conversions of X > 50%, the mixture had a critical solution temperature of >100 °C, and the product formed a second phase. Hence, the concentration of the telomer product in the “active dimcarb/β-myrcene/catalyst phase” remained low and could not hinder the reaction. Assuming that generally higher critical solution temperatures indicate a lower cross-solubility of the two phases, this should lead to even better retention of the catalyst in the dimcarb phase at any desired separation temperature. A reaction temperature of 100 °C is in retrospect a compromise between high activity of the catalyst and highly beneficial phase behaviour during the reaction process since at lower temperatures the reaction is significantly slower and at higher temperatures dimerisation of β-myrcene increases, which results in decreased selectivity, as revealed in other experiments (cf. ESI†).
Here, it needs to be emphasized again that the total reaction mixture only contained reactants, products, and the catalyst system, and no additional solvents and other auxiliaries that enable thermomorphic phase behaviour or act as phase transfer agents were needed. Also, no subsequent extraction or distillation was required. The product spontaneously separated from the catalyst-containing phase and could be used without any additional labour-intensive operations. The main issue is that the catalyst should remain in the polar dimcarb phase, and in terms of homogeneous catalysis, the choice of ligand is crucial for efficient catalyst separation, but simultaneously the ligand will influence the reactivity. The reactions before were carried out with triphenylphosphine (TPP) as the ligand. The sulfonation of ligands is a well-known approach to increase their solubility in polar solvents, and for dimcarb this was previously shown to be appropriate.19 Two sulfonated analogues of triphenylphosphine are commonly used, namely triphenylphosphine monosulfonate (TPPMS) and triphenylphosphine trisulfonate (TPPTS). The former was previously used for the telomerisation of β-myrcene and the latter for the telomerisation of butadiene with dimcarb.19,25 We assumed that the solubility of the ligand in dimcarb indicates the retention of the active catalyst, where the higher the solubility, the higher the retention. It was observed (Fig. 2) that TPPMS has the highest solubility among the three ligands in dimcarb at temperatures of 25 °C and 0 °C. Surprisingly, TPPTS showed the lowest solubility, which was unexpected due to its highest polarity and also highest solubility in water.33 Accordingly, we chose TPPMS as the ligand for further optimization.
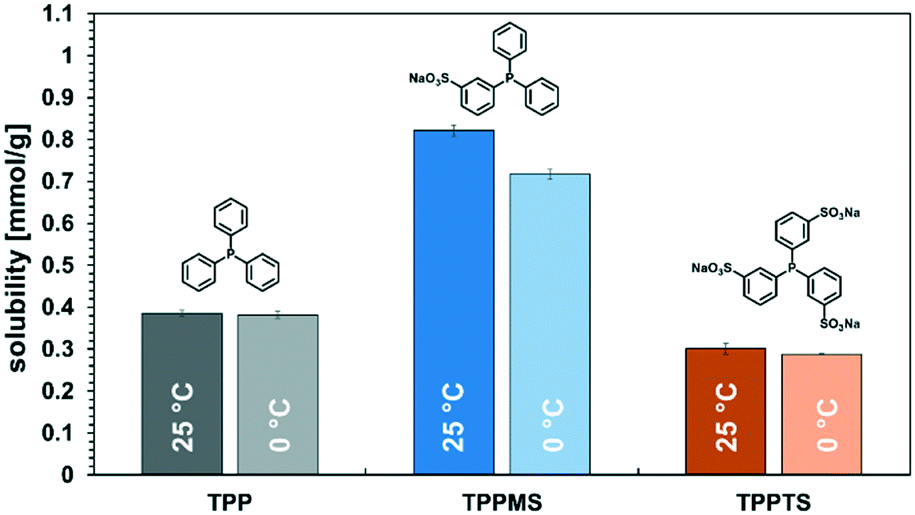 | ||
| Fig. 2 Solubility of TPP, TPPMS, and TPPTS in dimcarb at 0 °C and 25 °C. | ||
Based on these results, optimization of the reaction was performed with TPPMS as the ligand and Pd(acac)2 as the precursor. Details of these investigations can be found in the ESI.† It was found that a high excess of dimcarb (>3 eq. meaning xdimcarb >0.75 molar fraction) les to significantly lower selectivity due to the formation of hydroamination products. At low concentrations of dimcarb (<1 eq. meaning 0.5 molar fraction), an intense colouring of the product phase was observed, which indicated poor retention of the catalyst in the polar dimcarb phase. Thus, a dimcarb excess of 2 eq. (0.66 molar fraction of dimcarb) was chosen. However, the main by-products were still hydroamination products due to the excess of amine. Nevertheless, this represents the best compromise between the possibility of catalyst separation and selectivity of the reaction. Additionally, it became clear that the utilization of TPPMS and a switch of the precursor to Pd(acac)2, which is more active, selective and less hygroscopic, and therefore more preferable to work with, led to high activities for the reaction (cf. ESI†). Therefore only 0.035 mol% and a metal:ligand ratio of 1:3 were used in further investigations. The reaction profile was recorded, and after 4 h, the reaction reached a conversion of β-myrcene of 96% with a selectivity towards the telomers of 80%, resulting in a TON of 2200 toward the telomers and a TOF20 of nearly 900 h−1 at 20% conversion (cf. ESI†).
With all these data in hand, finally, the catalyst was recycled under the optimized conditions. Detailed information on the process is presented in the ESI.† A glass autoclave was used to observe the two-phase system and its thermomorphic behaviour and to efficiently separate the organic product phase from the catalyst-containing dimcarb phase after the reaction using a long cannula (Fig. 3). It was notable that only reactants were present in this reaction and that the top phase consisted mostly of the desired telomers, small amounts of by-products and non-converted β-myrcene. The bottom dimcarb phase dissolved only 4% of the product, representing a partition coefficient of 24.
 | ||
| Fig. 3 Reaction development and recycling of the telomerisation of β-myrcene and dimcarb in a glass autoclave. | ||
The catalyst-containing dimcarb phase was repeatedly used in consecutive recycling runs. After each reaction, the reactor was degassed, ensuring that surplus CO2 formed due to the decomposition of dimcarb and the reaction of dimethyl amine was removed. Subsequently, the upper product phase was withdrawn from the reactor and fresh β-myrcene and the amount of dimcarb that was lost due to the reaction of the dimethyl amine, were added (see ESI† for details). The results of this first recycle utilizing TPPMS as the ligand are shown in Fig. 4 (blue columns).
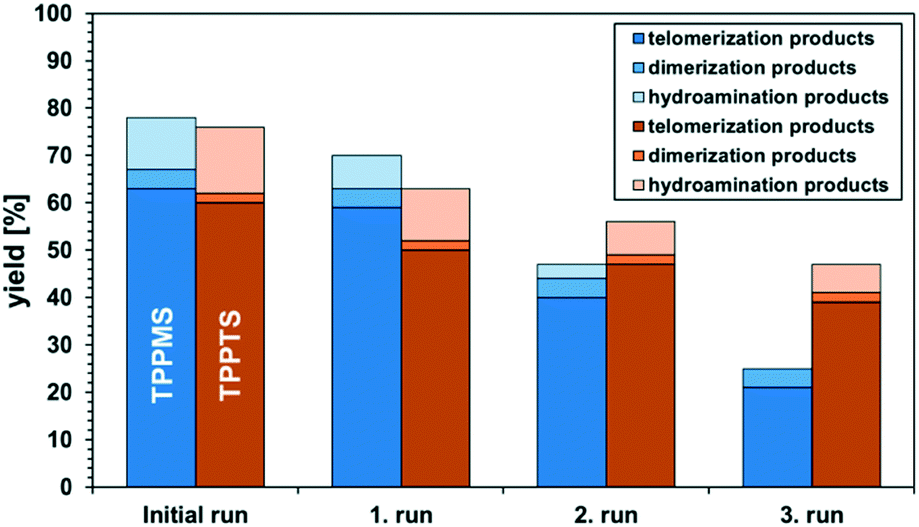 | ||
| Fig. 4 First recycling experiments of neat telomerisation of β-myrcene and dimcarb with a Pd(acac)2/TPPMS and Pd(acac)2/TPPTS system under the optimized conditions. Conditions: 35 mmol (2.9 g, 3.6 mL) β-myrcene, 70 mmol (5.4 g, 5.1 mL) dimcarb, 0.035 mol% (0.8 mmol L−1) Pd(acac)2, nPd:nligand = 1:3, T = 100 °C, 4 h, rpm = 500, and pAr = 0.01 MPa. After 1 h phase separation, 35 mmol β-myrcene and 13 mmol of dimcarb were added. Yields determined via GC-FID. | ||
The recovery of the palladium catalyst was successful by using dimcarb both as the substrate and as polar phase, showing activity in three consecutive runs. The yields were not as high as in the autoclaves that were used before, but this may be attributed to the reduced heat transfer in the glass autoclave or the scale-up. These circumstances are acceptable due to the ability to perform precise phase separation. Nevertheless, by using TPPMS as the ligand (blue columns), the activity decreased instantly, which suggests that the amount of active catalyst is diminished either by decomposition of the active catalyst or by leaching of the catalyst into the product phase. ICP-OES measurements of the product phase were carried out to verify this assumption, and it became apparent that Pd-leaching is responsible due to the loss of approximately 19% of the palladium per recycling run. Hence, the solubility of the ligand in dimcarb is not the critical parameter for efficient recyclability of the catalyst, as assumed previously. Thus, we investigated the solubility of the ligands in the telomer products (Fig. 5, left). The solubility of TPPMS in the telomer products is nearly twenty times lower than in dimcarb, which is preferable during the recovery of the catalyst. Nonetheless TPPTS shows an even lower solubility in the telomer products. Therefore, we applied the TPPTS ligand in the recycling to determine if the lower solubility in the product phase has a positive effect on the catalyst recycling (Fig. 4, orange columns).
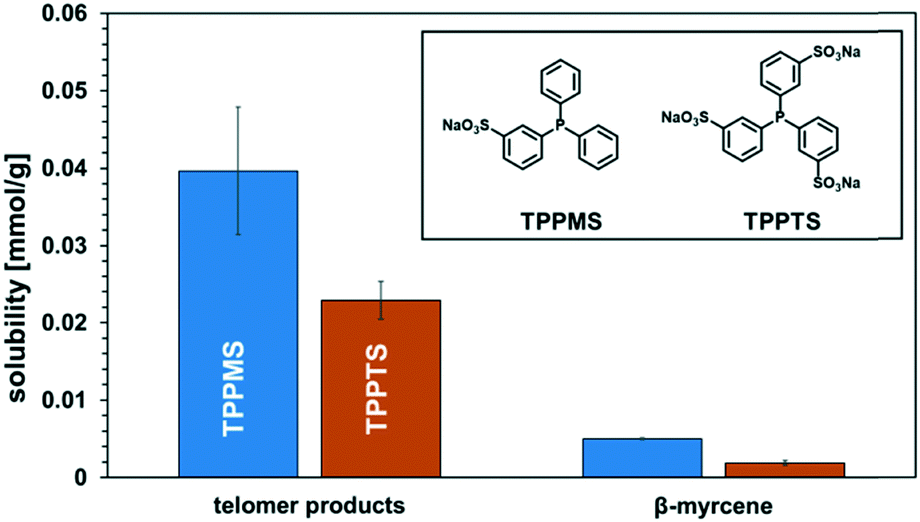 | ||
| Fig. 5 Solubility of TPPMS and TPPTS in the telomer products and β-myrcene at 25 °C. | ||
By application of TPPTS, similar activities and selectivities were observed, although a higher amount of hydroamination products was formed. In addition, higher stability of the catalytic activity was observed due to the slower declining yield of the telomer products with stable selectivity. Thus, we carried out ICP-OES measurements and observed Pd-leaching of only 6% in total over the four reactions. By application of TPPTS, which exhibited lower solubility in the product phase compared to TPPMS, the leaching was reduced by two thirds. Thus, according to these results, it can be concluded that low solubility of the ligand in the apolar product phase is more critical for efficient catalyst separation than the high solubility of the ligand in the polar catalyst-containing phase.
Nonetheless, the activity of the Pd/TPPTS catalyst decreased, and during the recycling, black Pd-particles were observed. Consequently, the loss of activity is not solely due to the leaching of the catalyst into the product phase, but also deactivation during the recycling progress. Thus, in the case of decreasing activity, two factors are responsible. Although leaching was determined by ICP-OES measurements of the product phase, the deactivation observed by the formation of black Pd-particles could not be determined. However, at the end of the recycling, it was possible to isolate the black particles. After drying for 24 h at 200 °C and 5 mbar, the residue was analysed by EDX- and ICP-OES analysis, confirming that they consist of palladium.
Thus, to minimize the Pd leaching, two alterations to the protocol were conducted. Firstly, an increase in the ligand concentration led to higher retention, although more coordinated species of palladium and TPPTS were present during the phase separation. Higher retention of Pd in the dimcarb phase was accomplished considering the fact that a higher ligand excess leads to slower reaction rates (cf. ESI†). The second alteration was that the reaction was stopped at a lower conversion as before since the ligand is less soluble in β-myrcene, and it has been previously observed that solubility may be an indication of the retention of the catalyst.
In a final recycling attempt, we applied a Pd:ligand ratio of 1:8 and carried out the reaction for 4 h (Fig. 6). By application of a higher ligand excess, a conversion of 52% in the initial run was observed, which represents a 25% lower yield than in the previous recycling, thus both mentioned alterations could be evaluated during this recycling.
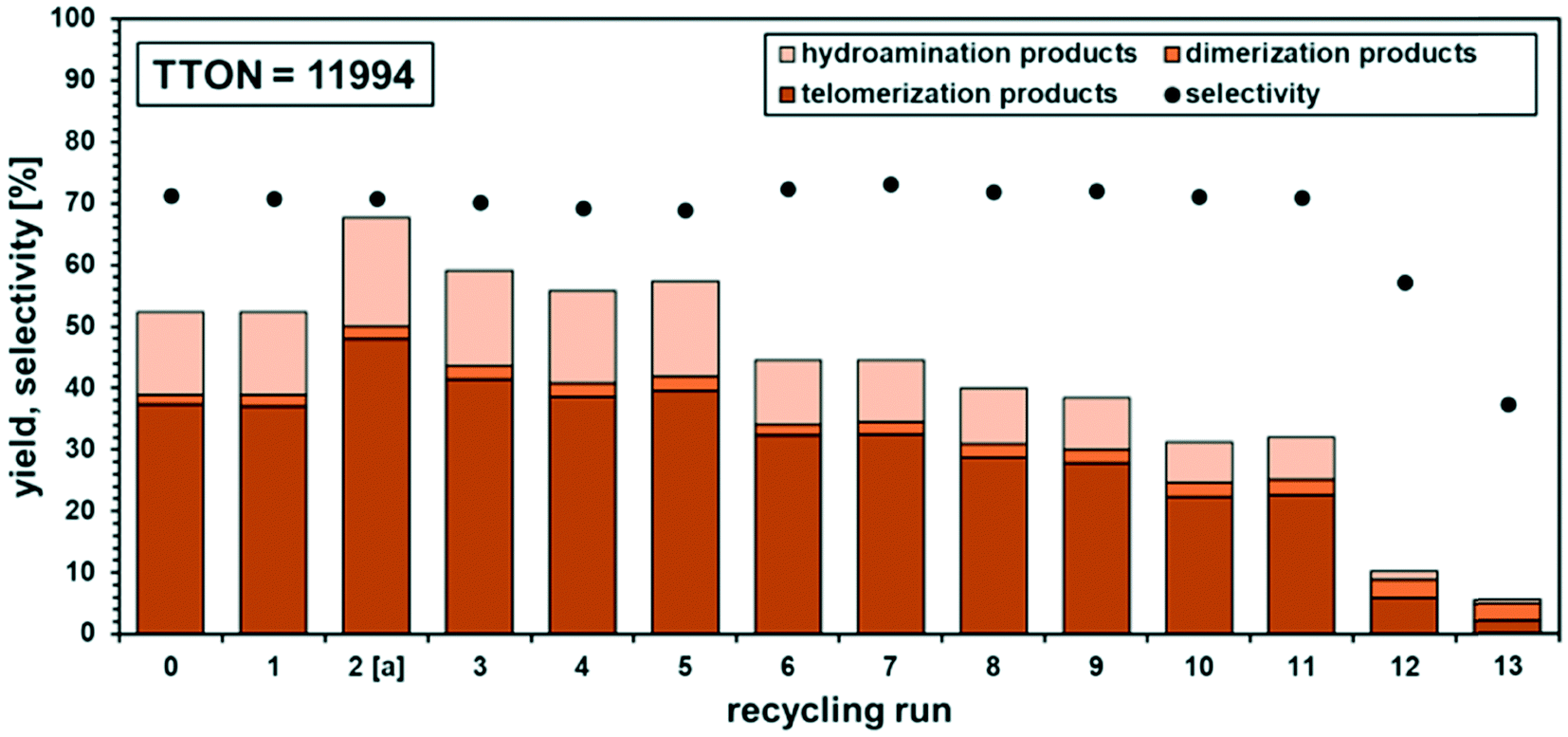 | ||
| Fig. 6 Recycling experiments with Pd(acac)2/TPPTS system with an increased Pd:L ratio of 1:8. Conditions: 35 mmol (2.9 g, 3.6 mL) β-myrcene, 70 mmol (5.4 g, 5.1 mL) dimcarb, 0.035 mol% (0.8 mmol L−1) Pd(acac)2, nPd:nTPPTS = 1:8, T = 100 °C, 4 h, rpm = 500, and pAr = 0.01 MPa. After 1 h phase separation, 35 mmol β-myrcene and 13 mmol of dimcarb were added. Yield determined via GC-FID. [a]: t = 4.5 h. | ||
During the recycling progress, it became obvious that the higher ligand excess and limitation of the conversion favoured the stability of the catalyst. The activity decreased after recycling run 5, which was accompanied by a colour change in the polar catalyst phase from light yellow to light brown. In comparison, recycling run 2 showed a better yield because the reaction was carried out for 30 min longer. Nevertheless, stable chemoselectivity of around 70% towards the telomers was observed over 11 runs. A low Pd-concentration of 0.035 mol% was applied, and a leaching of only 0.8% Pd and 0.5% P per run was observed, which is a significant improvement and supports the hypothesis of the beneficial higher ligand excess (Pd:L-ratio of 1:8) and the phase separation at a limited conversion of 50–60%, leading to less Pd leaching. However, leaching alone is not responsible for the loss of nearly half of the activity. Oxidation of the ligand is not an issue, as revealed by 31P-NMR since constant selectivity was previously confirmed. We observed two peaks in the 31P-NMR spectrum, which were identified as non-oxidized TPPTS and coordinated TPPTS (for additional info, see the ESI†). Nevertheless, the question arises why the catalyst lost its activity. Again, after recycling run 10, a formation of black particles was visible. After isolation of these particles and measurements by ICP-OES and EDX, it was found again that they were composed of palladium. Thus, it is possible that the Pd0 particles coagulate in the system and become catalytically inactive, which is consistent the observations that the activity loss is not due to the formation of a different catalytically active species since the selectivity remained the same. The particles were not part of the top product phase, and therefore not analysed as part of the ICP-OES measurements to determine the leaching. Minimizing the formation of these particles is a challenge in our ongoing research but should not outshine the good results of possible recycling of the molecular catalyst in a neat environment.
The alterations helped to minimize the leaching significantly. The higher ligand excess resulted in the formation of more stable palladium–phosphorus species. This resulted in a decrease in the selectivity also due to the formation of palladium–phosphorus species with more than one bound ligand. Therefore, hydroamination is the main side reaction in this recycling. On the other hand the ligand excess led to improved long-term stability and better retention of the catalyst in the polar phase.
Separating the phases at a conversion of 50–60% also proved to be efficient because the solubility of the ligand was lowered, and thus the palladium in the apolar product phase. By application of these two alterations, a total turnover number of almost 12000 was reached in a neat environment, only consisting of dimcarb as the polar catalyst-containing phase and dimethylamine precursor, and β-myrcene as the 1,3-diene and apolar compound.
Conclusions
In this study, it was possible to establish a system free of any volatile organic solvent in the recycling of a homogeneous palladium catalyst for the telomerisation of β-myrcene. The reaction was carried out in the reactive ionic liquid dimcarb, which simultaneously acts as a polar solvent and substrate. The liquid decomposes above 60 °C to dimethyl amine and carbon dioxide. Thus, the amine liberated was used to generate long chain, non-symmetrical tertiary dimethyl amines. One of the factors for the success was the exploitation of the thermomorphic behaviour of the dimcarb/β-myrcene-mixture. For the first time, detailed investigations of the phase behaviour were carried out and the separation of a pure product phase by decantation after the reaction was enabled. Based on the determinations of the ligand solubilities, the palladium leaching in the organic product was drastically reduced from 20% to only 0.8%. Utilization of TPPTS as the ligand, a ligand excess and executing the phase separation at 50–60% conversion of β-myrcene finally led to the recycling of the catalyst over 14 runs and a TON of 12000.
It is remarkable that in this context, no additional solvents were needed for the extraction of the products, renewable resources were used for the synthesis of long-chain dimethyl alkyl amines, and a successful recycling concept in this environment was established to show the high potential of this atom-economic reaction.
Experimental
Preparation of chemicals
All substrates used and the catalysts, ligands and solvents were commercially available and purchased from ABCR, Acros Organics, TCI, Sigma-Aldrich, VWR or UMICORE. Dimcarb was freshly distilled each week for purification. β-Myrcene was purified over an aluminium oxide column and stored under argon. For the reactions under strictly inert conditions, the substrates (dimcarb, β-myrcene) were transferred to a heated Schlenk vessel and then the oxygen was removed from the substrates using the freeze–pump–thaw method. The vessel in which the substrate was placed was sealed and connected to a Schlenk line by a hose. The valve of the swivel vessel was closed, and the substrate was frozen with liquid nitrogen. Vacuum was then drawn for approx. 20 min. Particularly, with dimcarb care must be taken that the substrate is completely frozen before the tap is opened to vacuum, otherwise the substrate would be removed. Afterwards, the valve of the Schlenk vessel was closed again, and it was removed from the liquid nitrogen and placed in a warm water bath to melt the substrate again. The development of gas bubbles was observed. After the substrate was completely melted again, it was frozen once again with liquid nitrogen and finally vacuumed again. This process was performed 4 times for each substrate.Screening experiments in 25 mL stainless steel autoclaves
Screening experiments were carried out in 25 mL stainless steel autoclaves (cf. ESI†). In an argon atmosphere, the palladium catalyst and the ligand were first introduced into the reactors and then mixed with dimcarb under inert conditions. The autoclaves were closed and gassed with 5 bars argon. After 1 h of preforming at 100 °C and 500 rpm, the reactors were cooled with an ice bath and degassed. Under inert conditions, β-myrcene was added and then stirred at 100 °C for 2 h at 500 rpm. After the reaction, the autoclaves were cooled again, the reaction solution was drawn up in syringes and the phases separated from each other. The respective phases were transferred into separate tubes, weighed and examined by gas chromatography.Recycling experiments in 40 mL glass autoclave
In an argon atmosphere, the catalyst and the ligand were first placed in a previously heated and inert Schlenk vessel. Subsequently, oxygen-free dimcarb was added under an argon atmosphere. The solution was homogenized in an ultrasonic bath. The glass autoclave (cf. ESI†) was inerted by drawing vacuum and then flushing it with argon. In the argon counter-flow, the catalyst phase was introduced into the reactor and then heated to 100 °C with an oil bath and stirred at 500 rpm for 1 h. The reactor was then cooled in an ice bath. The oxygen-free β-myrcene was added in the argon counter-flow and then stirred at 100 °C and 500 rpm for 4 h. After the reaction, the reactor was cooled again. In the argon counter-flow, the upper phase was removed with a cannula and β-myrcene and dimcarb were added again. The product phase was transferred to a glass tube, weighed and examined by gas chromatography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – TRR 63 “Integrierte chemische Prozesse in flüssigen Mehrphasensystemen” (Teilprojekt A11) – 56091768. We thankfully acknowledge Umicore AG & Co. KG for donation of the palladium precursors and OXEA GmbH for the donation of Na-TPPTS solution. We thank the workgroup of Prof. Tiller from the TU Dortmund University for carrying out the EDX-analysis under the lead of Volker Brandt.Notes and references
- A. Behr and P. Neubert, Applied Homogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015, pp. 1–55 Search PubMed.
- N. Herrmann, D. Vogelsang, A. Behr and T. Seidensticker, ChemCatChem, 2018, 10, 5342–5365 CrossRef CAS.
- T. A. Faßbach, A. J. Vorholt and W. Leitner, ChemCatChem, 2019, 1153–1166 CrossRef.
- A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598–3614 CrossRef CAS PubMed.
- P. W. Jolly, Angew. Chem., Int. Ed., 1985, 24, 283–295 CrossRef.
- A. M. Lazutkin and A. I. Lazutkina, React. Kinet. Catal. Lett., 1978, 8, 263–268 CrossRef CAS.
- N. Yoshimura, in Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, Second edn, 2004, pp. 540–550 Search PubMed.
- G. B. Jacobsen, H. L. Pelt and B. J. Schaart, Continuous process for the telomerization of conjugated dienes, WO1991009822A1, 1991.
- M. Beller, D. J. Nielsen, A. Grotevendt, L. A. Oro, A. Spannenberg, K. J. Cavell, R. Jackstell and M. Bartolome, Tetrahedron Lett., 2007, 48, 9203–9207 CrossRef.
- J. Beger and F. Meier, J. Prakt. Chem., 2004, 322, 69–80 CrossRef.
- T. Prinz, W. Keim and B. Driessen-Hölscher, Angew. Chem., Int. Ed. Engl., 1996, 35, 1708–1710 CrossRef CAS.
- A. Behr and J. Leschinski, Green Chem., 2009, 11, 609–613 RSC.
- A. Behr and R. Roll, Chem. Ing. Tech., 2005, 77, 748–752 CrossRef CAS.
- L. Völkl, D. Geburtig, S. Kiermaier, P. Wasserscheid and M. Haumann, Chem. Eng. Process., 2016, 99, 107–114 CrossRef.
- J. Garcia-Álvarez, Eur. J. Inorg. Chem., 2015, 5147–5157 CrossRef.
- N. Ajellal, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- T. Rösler, T. A. Faßbach, M. Schrimpf, A. J. Vorholt and W. Leitner, Ind. Eng. Chem. Res., 2019, 58, 2421–2436 CrossRef.
- T. A. Faßbach, R. Kirchmann, A. Behr and A. J. Vorholt, Green Chem., 2017, 19, 5243–5249 RSC.
- W. Schroth, H. Schädler and J. Andersch, Z. Chem., 1989, 29, 129–135 CrossRef CAS.
- A. E. Rosamilia, C. R. Strauss and J. L. Scott, Pure Appl. Chem., 2007, 79, 1869–1877 CAS.
- M. Eggersdorfer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, vol. 36, pp. 29–45 Search PubMed.
- T. R. Savich and L. A. Goldblatt, Process for producing myrcene from beta-pinene, US2507546A, 1950.
- A. Behr, L. Johnen and A. J. Vorholt, ChemCatChem, 2010, 2, 1271–1277 CrossRef CAS.
- T. A. Faßbach, F. O. Sommer, A. Behr, S. Romanski, D. Leinweber and A. J. Vorholt, Catal. Sci. Technol., 2017, 7, 1650–1653 RSC.
- T. A. Faßbach, T. Gaide, M. Terhorst, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 1359–1362 CrossRef.
- A. Behr, L. Johnen and N. Rentmeister, Adv. Synth. Catal., 2010, 352, 2062–2072 CrossRef CAS.
- D. Vogelsang, J. M. Dreimann, D. Wenzel, L. Peeva, J. Burgal, A. G. Livingston, A. Behr and A. J. Vorholt, Ind. Eng. Chem. Res., 2017, 56, 13634–13641 CrossRef CAS.
- T. Färber, O. Riechert, T. Zeiner, G. Sadowski, A. Behr and A. J. Vorholt, Chem. Eng. Res. Des., 2016, 112, 263–273 CrossRef.
- C. M. Foca, H. J. V. Barros, E. N. dos Santos, E. V. Gusevskaya and J. Carles Bayón, New J. Chem., 2003, 27, 533 RSC.
- H. J. V. Barros, B. E. Hanson, E. V. Gusevskaya and E. N. dos Santos, Appl. Catal., A, 2004, 278, 57–63 CrossRef CAS.
- J. Bianga, K. U. Künnemann, T. Gaide, A. J. Vorholt, T. Seidensticker, J. M. Dreimann and D. Vogt, Chem. – Eur. J., 2019, 25, 11586–11608 CrossRef CAS PubMed.
- N. Pinault and D. W. Bruce, Coord. Chem. Rev., 2003, 241, 1–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02569c |
| This journal is © The Royal Society of Chemistry 2020 |