
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoated sulfated zirconia/SAPO-34 for the direct conversion of CO2 to light olefins†
Adrian
Ramirez
 a,
Abhishek
Dutta Chowdhury
a,
Mustafa
Caglayan
a,
Alberto
Rodriguez-Gomez
a,
Nimer
Wehbe
b,
Edy
Abou-Hamad
b,
Lieven
Gevers
a,
Samy
Ould-Chikh
a and
Jorge
Gascon
*a
a,
Abhishek
Dutta Chowdhury
a,
Mustafa
Caglayan
a,
Alberto
Rodriguez-Gomez
a,
Nimer
Wehbe
b,
Edy
Abou-Hamad
b,
Lieven
Gevers
a,
Samy
Ould-Chikh
a and
Jorge
Gascon
*a
aKAUST Catalysis Center (KCC), Advanced Catalytic Materials, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bCore Labs, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia
First published on 30th January 2020
Abstract
The conversion of CO2 to light olefins via bifunctional catalysts (i.e. metal oxides/zeolites) is a promising approach to tackle CO2 emissions and, at the same time, reduce fossil-fuel dependence by closing the carbon cycle. However, designing a catalyst, especially a zeolite, that can operate at mid-low temperatures (where the selective CO2 conversion is favored) is still a challenge for the scientific community. Herein, we report the synthesis of a novel coated ZrS/SAPO-34 in combination with an iron catalyst (Fe2O3@KO2) that successfully fills this temperature gap. While making use of the properties of the zeolite to maximize light olefin selectivity, the unique nature of the sulfated zircona (ZrS) layer allows the cracking of C5+ heavy hydrocarbons produced on the Fe component at temperatures where conventional zeolites fall short. In particular, total short olefin yields per pass over 20% at CO2 conversions near 50% (total C2–C4 olefin selectivity = 40–45) can be achieved at T = 375 °C, P = 30 bar, H2/CO2 = 3 and 5000 mL g−1 h−1. Light olefin space–time yields here reported (10.4 mmol gcat−1 h−1) clearly outperform conventional zeolite based bifunctional systems for CO2 conversion.
Introduction
One of the greatest challenges that our society faces nowadays is to achieve real carbon neutrality by balancing carbon emissions with carbon dioxide capture, storage and conversion.1 The CO2 feedstock is widely available, recklessly released today into the atmosphere at a pace that surpasses 35 Gt per year.2,3 Therefore, the development of technologies able to convert CO2 into high value-added products using renewable hydrogen in an economical manner (high productivity and selectivity) is of great interest.4–6Among all possible alternatives, the direct thermocatalytic conversion of CO2 using electrochemically generated H2 holds great promise.7,8 The combination of several catalytic functions into one single reactor has been shown capable of converting CO2 into valuable chemicals with high selectivities, often above the limitation imposed by the classical Anderson–Schulz–Flory distribution (ASF).9,10 In these bifunctional systems CO2 conversion can proceed through two different routes depending on the first catalytic component: i) the transformation of CO2 into methanol (MeOH) over a methanol synthesis catalyst11 followed by a classical methanol to hydrocarbons (MTH) mechanism on the zeolite component12 or, ii) the transformation of CO2 into hydrocarbons via Fischer–Tropsch (FTS) over Fe based catalyst13 followed by hydrocarbon oligomerization, cracking, isomerization and/or aromatization on the zeolite.14
Nowadays, the MeOH route is most popular among researchers, as high selectivities to olefins among the total hydrocarbons can be achieved.9,15–22 Most of these works are built on the combination of Zr-based methanol catalysts with SAPO-34. However, the overall selectivity to undesired CO generally represents more than half of the total products.9,15,17 This high CO selectivity is direct consequence of the moderated exothermicity of the methanol synthesis via hydrogenation of CO2, favored thermodynamically at low temperatures. Therefore, in order to make the MTH mechanism react in the zeolite, higher temperatures than the optimal are needed (typically above 350 °C), leading ultimately to suboptimal performance of the MeOH component. So far, the best performance in these systems has been achieved by Dang et al. with an InZr/SAPO-34 catalyst.18 The authors where able to reach a light olefins productivity of 7.1 mmol gcat−1 h−1 at 380 °C, 30 bars and 9000 ml h−1 gcat−1. Nevertheless, the CO selectivity remained as high as 64%. On the other hand, the FTS pathway has proven to be more productive and to lead to significantly lower selectivities to CO.10,14 In this line, our group recently showed14 that by combining an Fe catalyst with a MOR zeolite productivities of light olefins up to 9.2 mmol gcat−1 h−1 with a CO selectivity of 14% can be obtained at 375 °C, 30 bars and 5000 ml h−1 gcat−1 (see Table S1† for an overview of state of the art). However, in this case the product distribution is much wider,14 with an important C5+ fraction (resulting from the Fisher–Tropsch step) that is very difficult to convert into light olefins on the zeolite via cracking at the typical reaction temperatures.3,23
Catalytic cracking of medium-range hydrocarbon fractions over solid acid zeolites is a well-known process, responsible for the manufacture of over 50% of gasoline in the world.24 However, due to diffusion limitations, the typical temperature range of the reaction over zeolites ranges from 480 °C to 550 °C, way above the temperatures at which CO2 conversion is selective for the formation of hydrocarbons (∼350 °C). Although zeolites are the most common catalyst among solid acids, there is a particular group of materials with even higher acidity: solid super acids.25 Among super acids, sulfated zirconia (ZrS) has attracted much attention26–30 as it shows remarkable activity at low temperatures and in the presence of water29 for multiple acid catalyzed reactions (isomerization, cracking, alkylation, acylation, esterification, oligomerization, etc.). However, these materials lack the shape selectivity features of zeolites.
In our approach, we combine the excellent pre-cracking ability of sulfated zirconia with the remarkable shape selectivity of SAPO-34 by designing SAPO-34 core ZrS shell particles. This coated material combined with a potassium doped Fe catalyst and under reaction conditions favorable for the FTS like transformation of CO2, including high concentrations of water vapor,31 results in the selective formation of C2–C4 olefins with unrivaled productivities.
Experimental
Chemicals
Iron oxide (Fe2O3, Aldrich), potassium superoxide (KO2, Aldrich), zirconium sulfate hydrate (Zr(SO4)2·xH2O, Aldrich), SAPO-34 (ACS Materials), 1-octene (99%, Aldrich), silica (SiO2, fumed, Aldrich) were used as received. The 13CO isotopic gas mixture (25% 13CO in helium, 10 L of 13CO in lecture bottle, 100 bar) was provided by CK Isotopes Limited.Catalyst preparation
The Fe2O3@KO2 catalyst was prepared following the procedure described elsewhere31via mortar mixing of Fe2O3 and KO2 at a molar ratio of Fe/K = 2. The resultant mixture was heated up to 100 °C for 12 h prior to the catalytic measurements. The ZrS/SAPO-34 was prepared by incipient wetness impregnation of commercial SAPO-34 with an aqueous solution of 0.125 mg μl−1 of Zr(SO4)2. Afterwards, the sample was dried at 120 °C for 12 h and calcined in stagnant air at 550 °C for 2 hours. The 0.5×ZrS/SAPO-34 and the 2×ZrS/SAPO-34 samples were prepared by adding half (0.0625 mg μl−1) or double (0.250 mg μl−1) the amount of Zr(SO4)2 to the aqueous solution. The stand-alone ZrS catalyst was prepared by direct calcination of the Zr(SO4)2 precursor30 in stagnant air at 550 °C for 2 hours. Impregnation on SiO2 was done in the same way as the impregnation in ZrS/SAPO-34.CO2 hydrogenation tests
Catalytic tests were executed in a 16 channel Flowrence® from Avantium. 50 mg of the stand-alone Fe2O3@KO2 catalyst and 100 mg of composite catalyst with Fe2O3@KO2/zeolite with mass ratio 1/1 in a dual bed configuration were typically used. The mixed feed had 130 SCCM (standard cubic centimeters per minute) of a mixture of 25 vol% of CO2 and 75 vol% of H2. In addition, 8 SCCM of He were added as internal standard. The total flow is distributed among the 16 channels with a relative standard deviation of less than 2%. We aimed to have 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1 per channel in the stand-alone catalyst and 5000 mL g−1 h−1 in the composite catalyst. One of the 16th channels was always used without catalyst as blank. The reaction temperature was typically set at 375 °C. Prior to feeding the reaction mixture all samples were pretreated in situ with a pure H2 atmosphere for 4 hours at 350 °C. The tubes were then pressurized to 30 bar using a membrane based pressure controller.
000 mL g−1 h−1 per channel in the stand-alone catalyst and 5000 mL g−1 h−1 in the composite catalyst. One of the 16th channels was always used without catalyst as blank. The reaction temperature was typically set at 375 °C. Prior to feeding the reaction mixture all samples were pretreated in situ with a pure H2 atmosphere for 4 hours at 350 °C. The tubes were then pressurized to 30 bar using a membrane based pressure controller.
The GC is an Agilent 7890B GC with heated valves for switching the sample loops and for backflushing – if required – part of the sample. Three channels are available with three detectors: 2 FID's and 1 TCD. Helium is used as carrier gas for the FID channels, to be able to measure hydrocarbons. Argon is used in the TCD channel to enable detection of helium as internal standard and hydrogen as analyte. The front FID also has the possibility for CO and CO2 measurement after CO and CO2 is converted to CH4 with a methanizer. The TCD channel has a PPQ as backflush column and a Hayesep Q column for separation of CO2. A Molsieve as analytical column is used for the separation of He, H2, N2 and CH4 and CO. All other compounds (water, hydrocarbons and oxygenates) will be backflushed to waste. The FID channel is equipped with a 4 m column with a wax stationary phase to enable fast transfer of hydrocarbons C1–C14 to the analytical column. Hydrocarbons with higher carbon content will be back-flushed to vent. Separation of hydrocarbons C1–C8 is carried out on a 30 m Gaspro stationary phase. Separation of aromatics and hydrocarbons C9–C14 is carried out on a 30 m Wax stationary phase. The sample is transferred from the selector valve in the Flowrence® through the sample line to the GC. The temperature of the traced line is slightly lower or at least equal to the temperature of the valve oven of the GC.
Conversion (X, %), space time yields (STY, mmol gcat−1 h−1), hydrocarbon selectivity among hydrocarbons (SCn, %) and CO selectivity (SCO, %) are defined as follows:
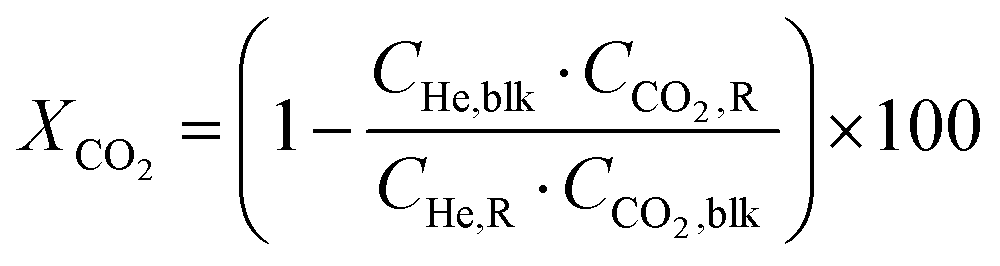
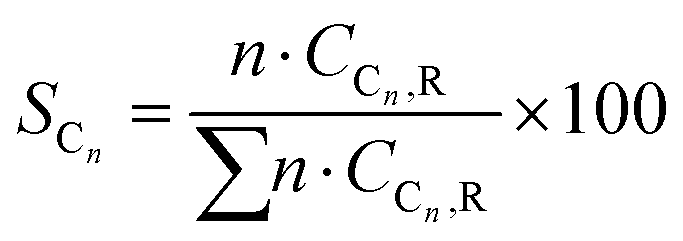
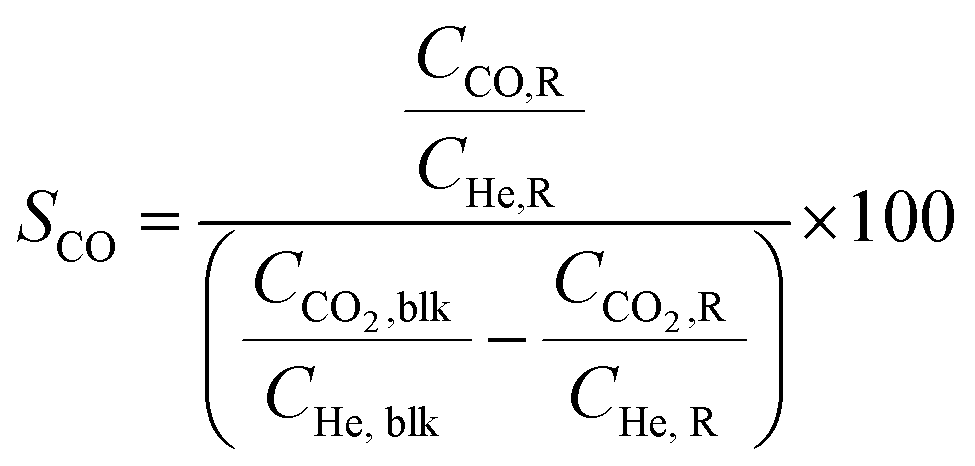
where CHe,blk, CHe,R, CCO2,blk, CCO2,R are the concentrations of He in the blank, He in the reactor effluent, CO2 in the blank, and CO2 in the reactor effluent, respectively, CCn,R is the concentration of a hydrocarbon with n carbon atoms in the reactor effluent, CCO,R is the concentration of CO in the reactor effluent, and GHSVCO2 is the CO2 gas space time in ml gcat−1 h−1. The error in carbon balance was better than 2.5% in all cases.
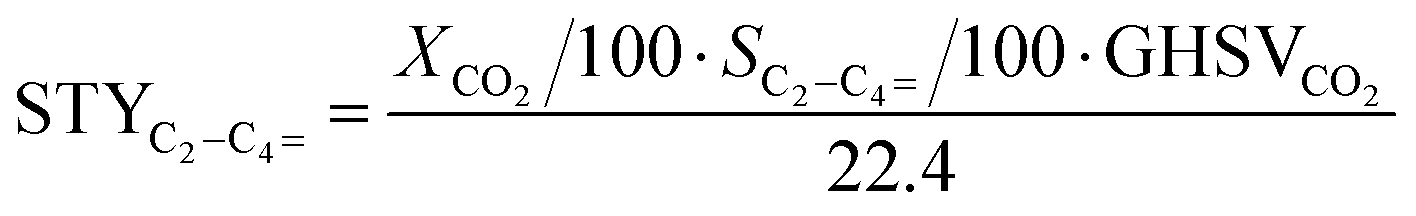
1-Octene cracking tests
The performance of the different samples in the catalytic cracking of 1-octene was carried out in a homemade tubular quartz reactor (Ø = 10 mm, length = 320 mm) using 100 mg of catalysts in fixed-bed mode at atmospheric pressure. The sample was heated-up until reaction temperature, 375 °C, through a tubular oven (304 stainless steel chassis, max. temperature 950 °C, with low thermal inertia), using 46 mL min−1 of nitrogen as carrier flow and 4 mL min−1 of He as internal standard. After temperature stabilized, a vaporized mixture of octane/water (50/50 vol), previously emulsified in an ultrasonic bath (Branson 3510) at 50 °C and pumped through two piston pumps (Gilson 305 HPLC), was introduced into the reactor using a traced line at 150 °C and a total flow of 0.1 mL min−1. Then, feedstock stream was maintained for 2 hours to evaluate the catalytic performance and stability. The gas products were analyzed on-line using a Trace 1310 gas chromatograph (Thermo Scientific) provided of a MolSieve analytical column and TCD for the analysis of He (internal standard) and H2, and a Alumina Plot column and FID for the separation an analysis of C2–C5 hydrocarbons.Nitrogen adsorption measurements
Nitrogen adsorption and desorption isotherms were recorded on a Micromeritics ASAP 2420 high throughput analysis system at 77 K. Samples were previously evacuated at 350 °C and 90 °C for 8 and 2 hours, respectively.X-ray diffraction measurements (XRD)
X-ray diffraction patterns were obtained in a Bruker D8 equipment in Bragg Bentano configuration using Cu Kα radiation. The diffractograms were scanned with step size of 0.02° in the 2θ range of 20–80°. The crystalline phase was identified by comparison data from the inorganic crystal structure database, ICSD.Temperature programed desorption (TPD) measurements
The temperature programmed desorption (TPD) experiments were carried out in an AMI-200ip Catalyst Characterization System (Altamira). In a typical experiment 55 mg were loaded into the system. The catalyst samples were first heated in argon flow at 100 °C and 550 °C sequentially. Afterwards, the samples we cooled down to 120 °C and ammonia was fed to the system. After the samples were statured in ammonia at 120 °C the temperature was increased to 750 °C linearly at a rate of 10 °C min−1. The ammonia desorption was continuously monitored by a thermal conductivity detector (TCD). For the experiments with water vapor, we introduced an additional step before the ammonia feeding. In this step, zeolites were heated at 375 °C in argon and then treated with water by flowing the argon (25 ml min−1) though a bubbler kept at room temperature. Then, the samples were purged with argon for 3 hours at 120 °C and ammonia was fed to the system.Electron microscopy and elemental mapping
Transmission electron microscopy (TEM) of the samples was performed with a Titan Themis-Z microscope from Thermo Fisher Scientific by operating it at the accelerating voltage of 300 kV and with a beam current of 0.16 nA. Dark field imaging was performed by scanning TEM (STEM) coupled to a high-angle annular dark-field (HAADF) detector. The STEM-HAADF data were acquired with a convergence angle of 29.9 mrad and a HAADF inner angle of 69 mrad. Furthermore, a X-ray energy dispersive spectrometer (FEI SuperX, ≈0.7 sR collection angle) was also utilized in conjunction with DF-STEM imaging to acquire STEM-EDS spectrum-imaging datasets (image size: 512 × 512 pixels, dwell time 5 μs). During the acquisition of these datasets, at every image-pixel, a corresponding EDS spectrum was also acquired for generating simultaneously the elemental maps of Al, Zr, S, and P atoms. It is also pertinent to note herein that spectrum-imaging datasets were acquired in so-called frame mode in which electron beam was allowed to dwell at each pixel for only a time of few microseconds in order to keep a total frame time to 6 seconds or less. Both imaging and spectroscopy datasets for each sample were both acquired as well as analyzed with a newly developed software package called Velox from Thermo Fisher Scientific. The elemental maps for Al, Zr, S and P atoms were computed using the extracted intensity of their respective Kα lines after background subtraction. The generated maps were slightly post-filtered by applying a Gaussian filter (sigma = 0.5).X-ray photoelectron spectroscopy (XPS) measurements
The XPS experiments were performed on a Kratos Axis Ultra DLD instrument equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operated at a power of 75 W and under UHV conditions in the range of ∼10–9 mbar. All spectra were recorded in hybrid mode using electrostatic and magnetic lenses and an aperture slot of 300 μm × 700 μm. The survey and high-resolution spectra were acquired at fixed analyzer pass energies of 160 eV and 20 eV, respectively. The samples were mounted in floating mode in order to avoid differential charging.Nuclear magnetic resonance (NMR) measurements
The 1H, 13C, and 31P-related magic angle spinning (MAS) solid-state nuclear magnetic resonance (ssNMR) spectroscopic experiments were performed on Bruker AVANCE III spectrometers operating at 400 MHz frequency for 1H using a conventional double-resonance 4 mm CPMAS probe (CP: cross-polarization). For 1D 1H–13C CP experiments, the following sequence was used: 90° pulse on the proton (pulse length 2.4 s), then a CP step with the contact time of typically 2 ms, and finally the acquisition of the 13C NMR signal under high-power proton decoupling. The delay between the scans was set to 4 s to allow the complete relaxation of the 1H nuclei. The number of scans (NS) was as follows: 5k (SAPO-34) and 16k (ZrS/SAPO-34). An exponential apodization function corresponding to a line broadening of 80 Hz was applied prior to Fourier transformation. For 31P MAS ssNMR experiments, 5 s recycle delay and 32 scans were applied. All of the 27Al and 29Si-related experiments were carried out on a 900 MHz Bruker AVANCE IV 21.1T spectrometers quipped with 3.2 mm CPMAS probes. For 27Al MAS ssNMR experiments, 1 s recycle delay and 128 scans were applied (except ZrS/SAPO-34, where 1024 scans were applied). The 27Al 3QMAS spectra were obtained by utilization of a three-pulse, z-filter sequence. The RF strengths of the first two hard pulses and the third soft selective 90 degrees pulse were individually optimized, and the optimized pulse lengths were typically 4.2, 1.4 (both at power level = 282 W) and 25 ms (at power level = 0.5 W) for the consecutive pulses. For 27Al 3QMAS experiments, size of fid (TD) was 1048(F2)/64 (F1) in all cases, and number of scans was 128 (for fresh and spent SAPO), 240 (for fresh ZrS/SAPO-34), 256 (for fresh and boiled ZrS/SAPO-34), and 480 (for spent ZrS/SAPO-34). For the 1H–29Si CP experiments, CP contact times of 5 ms and a 5 s recycle delay were used as well as the number of scans was 32k (for fresh/spent SAPO-34 as well as fresh and boiled ZrS/SAPO-34), 20k (for fresh ZrS/SAPO-34), and 21k (for spent ZrS/SAPO-34). 1H/13C, 31P, 27Al, and 29Si chemical shifts were externally referenced to adamantane, H3PO4, Al(NO3)3, and tetramethylsilane, respectively. In all cases, other relevant acquisition parameters were described in figure captions. All NMR spectra were processed and analyzed by Bruker TopSpin 3.6.0.Isotopic 13CO cofeeding experiments
The experiments with isotopic 13CO we carried out by feeding to the Flowrence® reactor a 13C labeled CO mixture (25% 13CO in helium) plus ethylene and hydrogen. In particular, we aimed for 5 SCCM of the 25% 13CO in helium mixture, 2.5 SCCM of H2 and 0.5 SCCM of ethylene per reactor. 50 mg of stand-alone SAPO-34 were used as catalyst. The reaction temperature was set at 375 °C and the reactor pressurized to 30 bar using a membrane based pressure controller. The mixed flow was kept at these conditions for 6 hours. Following, the spent SAPO-34 was collected and analyzed by 13C NMR.Results and discussion
The ZrS/SAPO-34 catalyst was synthesized through incipient wetness impregnation (IWI) with an aqueous solution of Zr(SO4)2 (see materials and methods). Because the zirconium precursor tends to form a bulky tetramer ([Zr4(OH)8·16H2O]8+) in solution, it cannot enter the SAPO-34 micropores during impregnation (15 Å vs. 4 Å),32 forming a thin shell layer (150 nm) over the cubic SAPO-34. Fig. 1A to 1F show scanning transmission electron microscopy (STEM) images of the ZrS/SAPO-34 material. Elemental mapping shows that Zr and S are mostly present in the shell of the structure, forming a fine coated layer alongside the cubic SAPO-34 (see Fig. 1F and S1 and S2†). This is further confirmed by intensity line profiles across the coated core–shell like structure (see Fig. S2C†). In parallel, the high acidity of the Zr(SO4)2 solution partially leaches the surface of the SAPO-34 (vide infra),33 resulting in a strongly merged structure after evaporation of the solvent. As a result, Al and P are also observed in the ZrS shell (see Fig. S2A and B†). | ||
| Fig. 1 HAADF-STEM characterization the coated ZrS/SAPO-34 material. (A) HAADF-STEM image of the ZrS/SAPO-34. (B) S elemental mapping. (C). Zr elemental mapping. (D) Al elemental mapping. (E) P elemental mapping. (F) Al + P + S + Zr combined elemental mapping. | ||
X-ray diffraction analysis (see Fig. S3†) confirms that the structure of the SAPO-34 (ref. 34) is preserved after impregnation. Albeit thin, the formation of the ZrS layer decreases the surface area of the SAPO-34 by roughly 30%, as calculated from the nitrogen adsorption–desorption isotherms (see Fig. S4 and Table S2†). We also studied the effect of the loading of Zr(SO4)2 on the final structure of the coated catalysts (0.5 and 2 times the initial loading, see materials and methods). We can observe that only the initial sample results in a homogenous coated structure: half loading is not enough to form a uniform shell of ZrS (see 0.5×ZrS/SAPO-34, Fig. S5†) while 2 times of ZrS loading partially dissolves the SAPO-34 structure (see 2×ZrS/SAPO-34, Fig. S5†). This result is a direct consequence of the extreme acidity of the highly concentrated aqueous solution employed to prepare the 20% ZrS/SAPO-34 by IWI. Additionally, this 2×ZrS/SAPO material only shows a surface area 313 m2 g−1 (see Fig. S4†), in line with the partial leaching of the SAPO-34 framework observed in Fig. S5.† Therefore, based on the above results, the initial ZrS SAPO-34 was selected as the optimal loading to obtain a uniform coated structure while keeping the SAPO-34 integrity and was further employed for the catalytic testing.
To derive further structural information in these ZrS/SAPO-34 composites we first performed temperature programmed desorption of ammonia (see Fig. 2A). Interestingly, the ammonia TPD shows a shift to lower temperatures upon ZrS incorporation, suggesting an increase of Lewis-like acid sites (vide infra for the solid-state NMR characterization), in line with the type of acidity already reported for some of the ZrS stand-alone super acids.35 This type of acidity has been reported as responsible for the direct cracking of C5+ olefins via the classical β-scission mechanism.36–38 Again, in line with the literature29 additional Brønsted-like acid sites are generated after pretreatment of the ZrS/SAPO-34 composite with water vapor as, overall, a shift in the main desorption peak to higher temperatures is observed (see yellow spectra in Fig. 2A). Furthermore, the concentration of desorbed NH3 is increased from 0.86 to 1.15 μmol m−2 after the water treatment (see Table S3†).
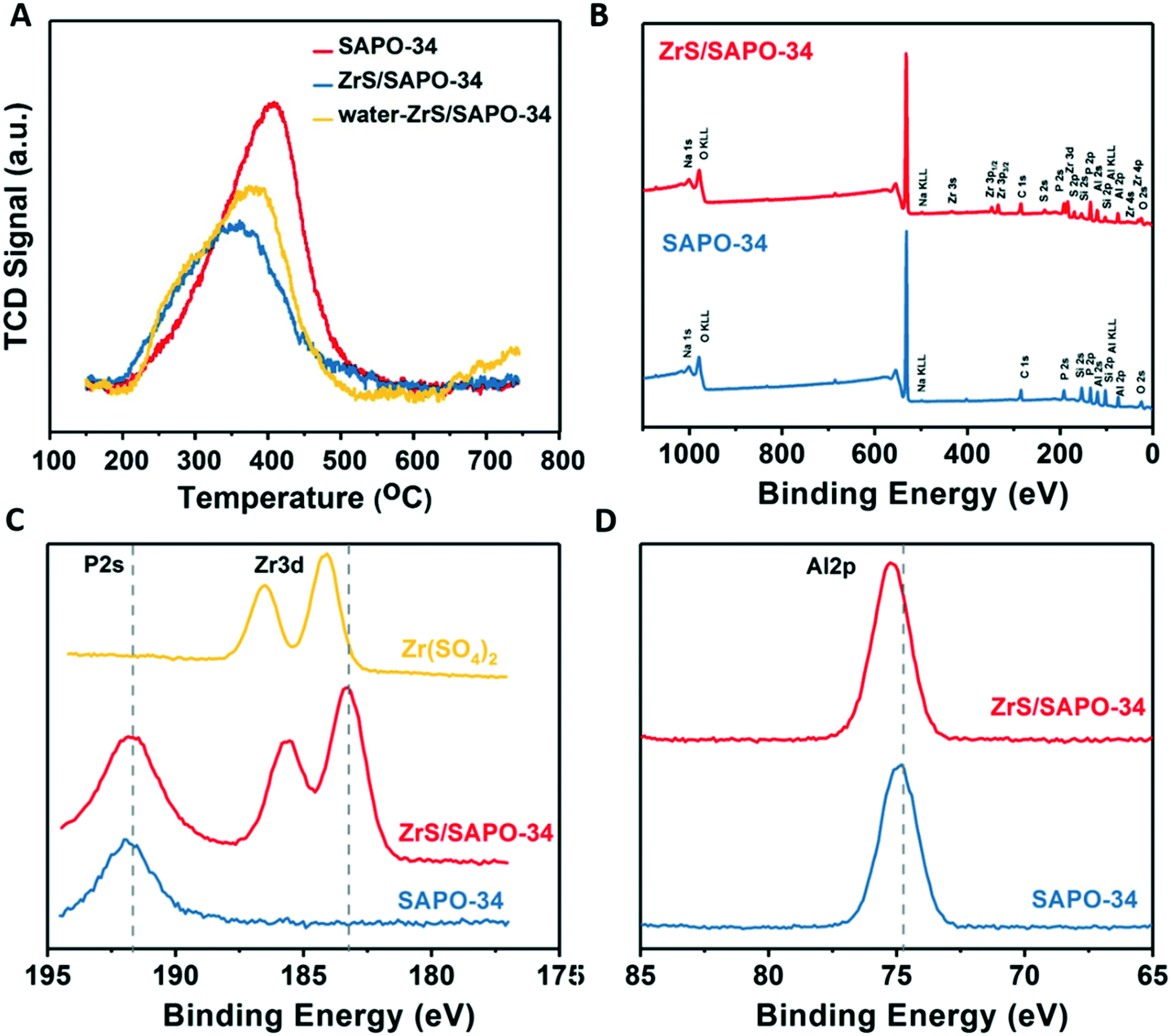 | ||
| Fig. 2 TDP and XPS characterization the ZrS/SAPO-34 structures. (A) Temperature programmed desorption of ammonia of the ZrS/SAPO-34 materials. (B) X-ray photoelectron spectroscopy (XPS) survey spectra of the ZrS/SAPO-34 materials. (C) XPS spectra at the Zr3d and P2s levels of the ZrS/SAPO-34 materials. (D) XPS spectra at the Al2p level of the ZrS/SAPO-34 materials. | ||
X-ray photoelectron spectroscopy (XPS) was next used to further unravel chemical state of the ZrS shell. The survey spectra (see Fig. 2B) clearly pinpoints the presence of Zr and S in the shell in agreement with the STEM profiles (see Fig. S2C†) while, in the Zr3d level, a shift in binding energies from 184.4 to 183.2 eV is observed (see Fig. 2C). This shift has been attributed to the chemical bonding of Zr with P (ref. 39 and 40) and suggests a different nature of the Zr in comparison to the bare Zr(SO4)2 precursor. Another shift in the Al2p level (see Fig. 2D) from shift 74.8 to 75.3 eV in comparison to the bare SAPO-34 also suggests a different nature of the Al species present in the shell after the super acid leaching.33
Following, advanced magic angle spinning solid-state NMR (ssNMR) spectroscopy was performed on the SAPO and ZrS/SAPO-34 materials. In particular, 29Si, 31P and 27Al ssNMR were performed on both fresh and post-reacted catalytic materials after 50 hours on the reaction stream. The 1D 1H–29Si CP ssNMR (see Fig. 3A) determines the local Si-environment, specifically the number of ‘next door neighbor’ Si or Al atoms. Herein, chemical shifts at around −110, −101, −98, −94, and −88 ppm were assigned to Si(OSi)4, Si(OAl)(OSi)3, Si(OAl)2(OSi)2, Si(OAl)3(OSi), and Si(OAl)4, respectively.41–44 This last signal of Si(OAl)4 is significantly higher than others in CP NMR as it is situated near a proton to compensate for the charge imbalance, resulting in the formation of Brønsted acid sites. We can also observe in Fig. 3A that both post-reacted catalysts have better signal-to-noise ratio than the fresh samples, illustrating the enhanced 1H–29Si CP transfer due to the accumulation of trapped (hydrogen-containing) organics after the reaction.
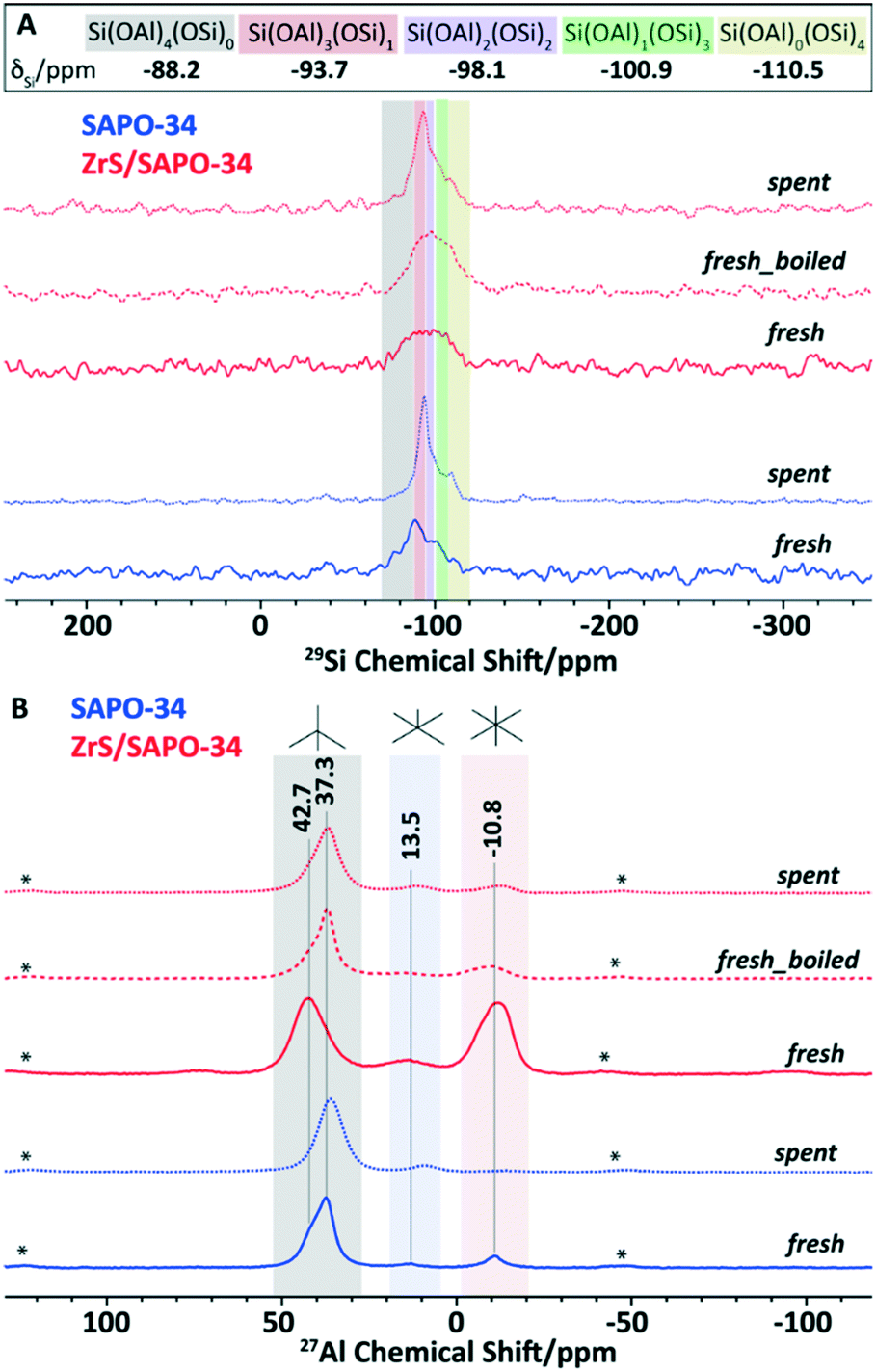 | ||
| Fig. 3 Solid-state nuclear magnetic resonance of the ZrS/SAPO-34 structures. (A) 1H–29Si cross polarization magic angle spinning (MAS, 10 kHz) solid-state nuclear magnetic resonance spectra of fresh SAPO-34, post-reacted SAPO-34, fresh ZrS/SAPO-34, fresh ZrS/SAPO-34 after boiling in water for 12 hours, and post-reacted ZrS/SAPO-34. (B) 27Al magic angle spinning (MAS, 20 kHz) solid-state nuclear magnetic resonance spectra of fresh SAPO-34, post-reacted SAPO-34, fresh ZrS/SAPO-34, fresh ZrS/SAPO-34 after boiling in water for 12 hours, and post-reacted ZrS/SAPO-34. Post-reacted materials were prepared after the hydrogenation of carbon dioxide for 50 hours. | ||
If now we compare the fresh samples, we can observe a strikingly lower signal-to-noise ratio of fresh ZrS/SAPO-34 sample that implies the lack of proton in the sample, suggesting that Brønsted acid sites were dehydroxylated to a great extent upon the incorporation of Zr, in line with our TPD observations (see Fig. 2A). Additionally, the fresh ZrS/SAPO-34 sample was boiled for 12 hours, showing now a much better signal-to-noise ratio. Therefore, it is safe to assume that ZrS/SAPO-34 material undergoes changes under hydration, again in line with our TPD observations (see Fig. 2A).
The non-identical nature of fresh ZrS/SAPO-34 with respect to the stand-alone SAPO-34 is also clear from the 31P ssNMR spectra (see Fig. S6†). Except for fresh ZrS/SAPO-34 sample, all other samples show only one peak around 30 ppm due to the AlPO-phase.45 However, in the ZrS/SAPO-34 sample, at least 2 non-identical and independent P sites have been identified at around −26 and −15 ppm. Although the assignment of −15 peak is very ambiguous in the literature,43,45 yet it could be attributed to either free chain end-groups without any attachment to Al (like pyrophosphates) or to polymerized short-chain middle phosphate groups.43 Nevertheless, the latter option is rather less probable within the AlPO-framework. Interestingly, upon boiling, the AlPO framework regained its parent-state up to a certain extent: the peak at −30 ppm was increased in the intensity at the expense of P-sites responsible for −15 ppm peak.
Under such circumstances, it is necessary to probe the local-Al environment through 1D 27Al ssNMR and 2D 27Al multiple-quantum (MQ) MAS ssNMR (Fig. 3B and S7†). Since the Brønsted acid sites originate from the bridging hydroxyl groups between Al and Si atoms, local-probing of Al in SAPO materials could intimately be correlated to the acidic properties. Remarkably, all samples demonstrated a similar spectral pattern, (again) except for ZrS/SAPO-34 sample. Upon Zr-incorporation, the content of octahedral Al-sites increased at the expense of tetrahedral Al-sites (see Fig. 3B), which indicates an increase of Lewis acidity, in line with the observed TPD data (see Fig. 2A) and also in line with the different nature of the Al species observed by XPS (see Fig. 2D). Moreover, the peak-maxima of tetrahedral-Al was shifted completely to −43 ppm, which also supports its dehydroxylated-nature, as it is also indicated in 1H–29Si CP NMR (see Fig. 3A).36–38
As expected, after boiling, the 27Al spectrum of the ZrS/SAPO-34 is consistent with the fresh SAPO-34 material itself, indicating the internal rearrangement to regain Brønsted acid sites. However, 27Al is a quadrupolar nucleus and its NMR line-shape typically suffers from residual second-order quadrupolar interactions. Therefore, 2D MQMAS experiments are obligatory to confirm the 27Al assignments (see Fig. S7†).41,42,45 In addition, the sheared 2D 27Al MQMAS spectra could demonstrate the heterogeneity with more clarity. Again, except for ZrS/SAPO-34 sample, other samples distinctly showcase at least two independent Al-sites, which can be assigned to Al in the SAPO- and residual AlPO-phases.41–43,45 However, in the ZrS/SAPO-34 sample the contribution to tetrahedral Al sites was predominantly originated from the AlPO-phase, which indicates the lack of Brønsted acidity (see Fig. S7C†) of the sample, regained upon boiling, see Fig. S5D.† Moreover, 2D 27Al MQMAS spectra illustrate that the incorporation of Zr led to the formation of an independent type of octahedral Al-sites at −5 ppm characteristically different from the residual six coordinated Al-sites in the AlPO-framework (see Fig. S7C†).
Altogether, these characterization results demonstrate the successful formation of the envisaged coated structures and the beneficial effect of water in both acidity and framework reconstruction, making the catalyst ideal to work under the very demanding conditions (in terms of water vapor concentration) of CO2 hydrogenation.
Hence, the multifunctional Fe2O3@KO2/ZrS/SAPO-34 system was assembled by combing a Fe catalyst with the coated ZrS/SAPO in a dual bed configuration (i.e. Fe2O3@KO2 on the top and ZrS/SAPO in the bottom of the tubular reactor) with a mass ratio of 1:1. The synthesis of the stand-alone Fe based catalyst was done according to our recent work.31 Note that due to the high content of K in the Fe based catalyst, mixed bed configurations result in the loss of acidity of the zeolite component.14 The catalytic performance of the multifunctional systems is summarized in Fig. 4A. Under the studied reaction conditions (T = 375 °C, P = 30 bar, H2/CO2 = 3 and 5000 mL g−1 h−1), the stand-alone Fe catalyst gives a CO2 conversion of 48% with a light olefin selectivity of 40.5% within the total hydrocarbons and a total CO selectivity of only 16%.
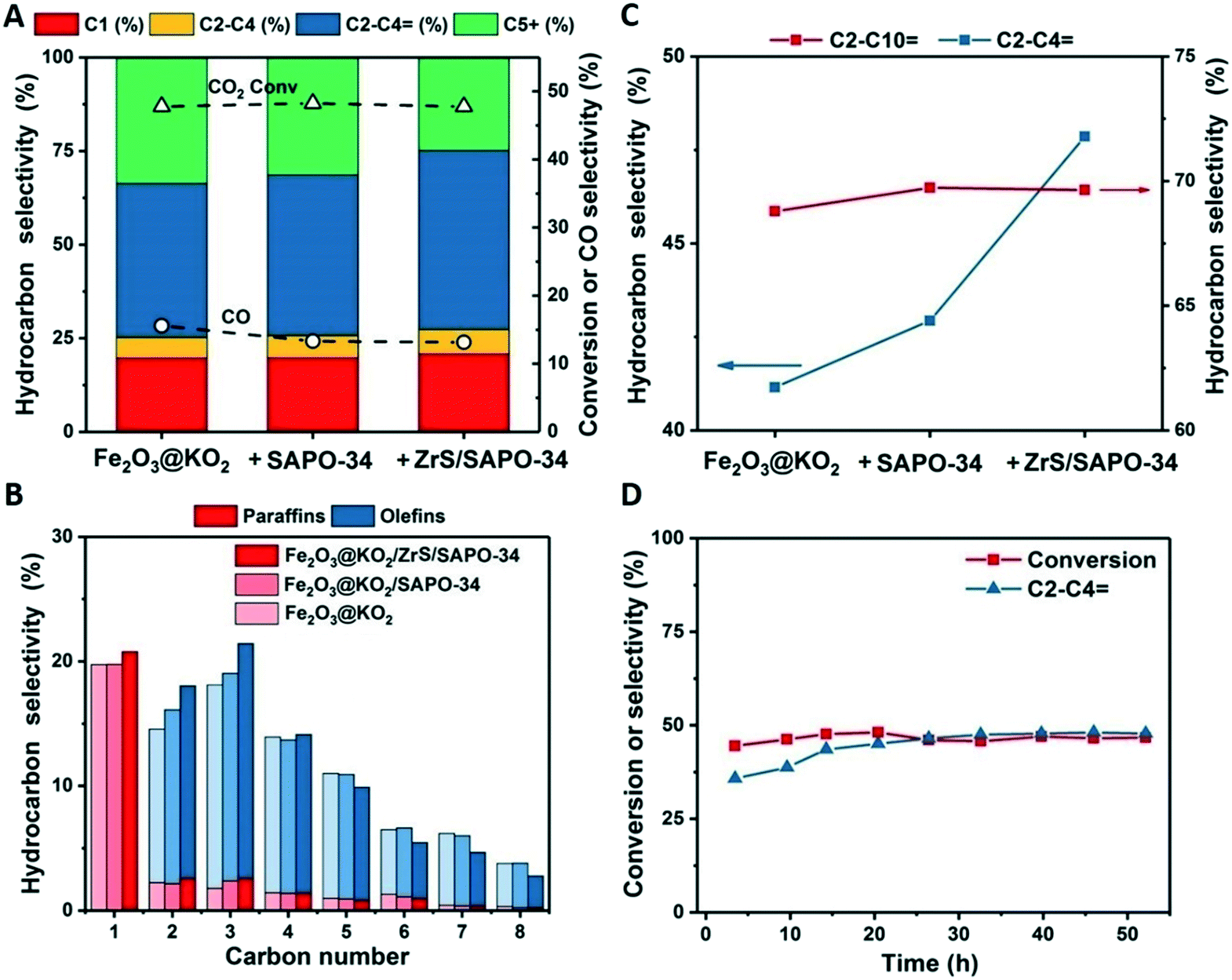 | ||
| Fig. 4 Catalytic performance of the multifunctional catalysts. (A) Catalytic performance of the multifunctional materials after a TOS of 50 h. (B) Hydrocarbon distribution of the multifunctional materials after a TOS of 50 h. (C) Olefin distribution of the multifunctional catalysts after a TOS of 50 h. (D) Stability of the Fe2O3@KO2/ZrS/SAPO-34 catalyst. Reaction conditions: 375 °C, 30 bar, H2/CO2 = 3, and 5000 mL g−1 h−1. | ||
The addition of SAPO-34 to the Fe catalyst slightly enhances the formation of ethylene and propylene, achieving a light olefin selectivity of 43% among hydrocarbons while the CO selectivity decreases to 13%. This behavior is in line with our previous observations of CO derived carbonylated reactive intermediates in the zeolite framework during the reaction.14 Now, when the coated of the light olefin fraction, adding up to 48% of the total hydrocarbon fraction. Interestingly, the effect of the SAPO-34 is not lost and the CO selectivity remains at the same low value of 13%.
Looking into detail at the hydrocarbon distribution (see Fig. 4B) we can observe that this increase of light olefins in the ZrS/SAPO-34 is accompanied by a decrease of the C5+ olefins. This C5+ fraction remains unreacted in the SAPO-34, as the reaction temperature, and probably diffusion of these species into the SAPO-34 framework, is not enough as to result in any heavy olefins cracking.24 However, by adding the super acidic functionality to the ZrS/SAPO-34, a part of this fraction is cracked, increasing more than 7 points the selectivity to light olefins at isoconversion. This effect is better observed if we compare the total olefin selectivity versus the light olefin selectivity (see Fig. 4C). Furthermore, catalytic performance is stable during the whole duration of the experiment (see Fig. 4D).
The superior performance of the ZrS/SAPO-34 material was evaluated by comparing it with the stand-alone ZrS catalyst (see Fig. S8† and materials and methods), a physical mixture of ZrS and SAPO-34 and ZrS impregnated onto silica with the same mass ratios. Our results show that the coated structure clearly outperforms these materials (see Fig. S9†). The ZrS stand-alone catalyst slightly increases the light olefin selectivity but the CO selectivity remains unchanged. Additionally, the light olefin selectivity increase is lower than in the coated material due to the bigger size of the bulk ZrS (see Fig. S8†) and the associated transport limitations. Similar results were obtained for the ZrS + SAPO-34 physical mixture and for the ZrS/SiO2 although, in the ZrS + SAPO-34 material, a decrease of the CO selectivity is also observed thanks to the SAPO-34 component.
In order to better understand the reaction mechanism we performed additional ssNMR characterization of samples after reaction, with special focus on characterizing organic species trapped in the zeolitic component.14 In the 1D 1H–13C cross-polarization (CP) spectra (Fig. 5A), two primary features were clearly visible: (i) 10–30 ppm aliphatic/methyl groups, and (ii) 125–140 ppm olefinic/aromatic moieties. Interestingly, the intensity of the aromatic region is appeared to be lower upon the incorporation of the ZrS layer (even with a higher number of scans), which could be attributed to the less efficient CP transfer and thus, a relatively more hydrogen-deficient system in ZrS/SAPO-34. This observation is consistent with the formation of more light olefins at the expense of the C5+ fraction in ZrS/SAPO-34 (see Fig. 4C).
 | ||
| Fig. 5 Solid-state nuclear magnetic resonance of post-reacted ZrS/SAPO-34 structures and state of the art comparison. (A) 1D 1H–13C cross-polarization MAS solid-state NMR spectra of post-reacted SAPO (blue, NS = 5k) and ZrS/SAPO-34 (red, NS = 16k) material trapped organic products after the hydrogenation of carbon dioxide for 50 hours (* = spinning sideband, MAS = magic angle spinning = 10 kHz, NS = number of scans). (B) Light olefin space–time yield as function of the CO + CH4 selectivity for the Fe2O3@KO2/ZrS/SAPO-34 systems compared with the state of the art for the competitive MeOH route.9,15–22 | ||
Therefore, based on all the above results, we propose a reaction pathway for the CO2 hydrogenation to light olefins on the multifunctional Fe2O3@KO2/ZrS/SAPO-34 catalyst that involves: (i) the conversion of CO2 into CO and water through the RWGS reaction31 on the Fe2O3@KO2 catalyst followed by (ii) traditional Fischer Tropsch synthesis on the same solid, (iii) CO diffusing into the pores of the SAPO-34, yielding C2–C3 light olefins (see Fig. 4A and B), presumably through carbonylated intermediates14 and (iv) the C5+ heavy hydrocarbon fraction produced on the FTS step is cracked on the water (from RWGS) boosted acid sites of the ZrS layer via β-scission,36–38 further increasing selectivity to light olefins (see Fig. 4C).
To prove this pathway we performed catalytic cracking of 1-octene in the presence of water (see materials and methods) on coated and uncoated SAPO-34 samples. After 2 hours on stream, the cracking capacity of ZrS/SAPO-34 more than doubles that of the naked zeolite (see Table S4†). Moreover, for similar experiments carried out in the absence of water the cracking capacity of both materials is drastically decreased, confirming the importance of the water generated in the first Fe2O3@KO2 bed to the overall mechanism. Additionally, to unravel the role of the SAPO-34 on the CO incorporation we have performed supplementary reaction experiments on the zeolite by feeding 13C labeled CO plus ethylene and hydrogen at the reaction conditions (i.e. 375 °C and 30 bars, see materials and methods). The selective isotope enrichment of “13CO” in the reactant feed allowed us to perform direct 13C excitation solid-state NMR spectroscopy to investigate the effect of incorporation of CO on the spent samples (see Fig. S10†). In this case, we have observed two responses, around 127 ppm (major, sp2 carbons) and 21 ppm (minor, sp3 carbons), which demonstrates that CO is primarily incorporated into unsaturated hydrocarbon moieties (both aromatic and olefinic). Since we have performed 13C direct excitation experiment for a short acquisition time, it ensures that the spectrum does not have any contribution from the naturally occurring ethylene in the reactant feed. Additionally, the product distribution clearly show the formation of a majority of C3 products (>60% selectivity, see Table S5†). Hence, these observations unequivocally supports that CO is incorporated into the product during catalysis and, therefore, the main effect of the SAPO component is to reduce the undesired CO selectivity.
Last but not least, the multifunctional Fe2O3@KO2/ZrS/SAPO-34 system here reported displays a light olefin yield of 19.4% and light olefin space time yield 10.4 of mmol gcat−1 h−1, the highest reported to date to the best of our knowledge (see Table S1†). Moreover, if we compare this productivity as function of the C1 undesired byproducts (CO + CH4) with the state of the art bifunctional catalysts for the competitive MeOH route9,15–22,46,47 we can observe that, with the Fe2O3@KO2/ZrS/SAPO-34 system, we do not only obtain the highest light olefin space time yield, but also with the lowest undesired CO + CH4 selectivity (see Fig. 5B).
Conclusions
In conclusion, we have designed and synthesized a novel ZrS/SAPO-34 structure with a super acid functionality that, when combined with an iron CO2 hydrogenation catalyst, enhances the selectivity of the C2–C4 light olefin fraction. This is due to the positive effect of H2O produced on the Fe component via RWGS that increases the acidity of the coated ZrS shell, allowing the cracking of the undesired heavy hydrocarbon fraction via a β-scission mechanism. At the same time, the SAPO-34 core incorporates CO to the reaction network, reducing the overall CO selectivity. In particular, 48% light olefins hydrocarbon selectivity was achieved at a CO2 conversion of 49% with a CO selectivity of only 13% at 375 °C, 30 bar, H2/CO2 = 3 and 5000 mL g−1 h−1. Our findings demonstrate that, through catalysis and reactor engineering, it should be possible to soon achieve economically viable routes for the valorization of carbon dioxide via thermocatalysis using renewable H2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by King Abdullah University of Science and Technology (KAUST).Notes and references
- S. Fennell, N. Shah and G. C. Maitland, The role of CO2 capture and utilization in mitigating climate change, Nat. Clim. Change, 2017, 7, 243–249 CrossRef.
- C. F. Shih, T. Zhang, L. Li and C. Bai, Powering the Future with Liquid Sunshine, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- R. Dittmeyer, M. Klumpp, P. Kant and G. Ozin, Crowd oil not crude oil, Nat. Commun., 2019, 10, 1818 CrossRef PubMed.
- E. Alper and O. Y. Orhan, CO2 utilization: Developments in conversion processes, Petroleum, 2017, 3, 109–126 CrossRef.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Catalysis for CO2 conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- A. Dokania, A. Ramirez, A. V. Bavykina and J. Gascon, Heterogeneous Catalysis for the Valorization of CO2: Role of Bifunctional Processes in the Production of Chemicals, ACS Energy Lett., 2019, 4, 167–179 CrossRef CAS.
- Z. Ma and M. D. Porosoff, Development of Tandem Catalysts for CO2 Hydrogenation to Olefins, ACS Catal., 2019, 9, 2639–2656 CrossRef CAS.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, Direct Production of Lower Olefins from CO2 Conversion via Bifunctional Catalysis, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and I. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Catalytic Carbon Dioxide Hydrogenation to Methanol: A Review of Recent Studies, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Recent Trends and Fundamental insights in the Methanol-to-Hydrocarbons Process, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- A. Ramirez, L. Gevers, A. Bavykina, S. Ould-Chikh and J. Gascon, Metal Organic Framework-Derived Iron Catalysts for the Direct Hydrogenation of CO2 to Short Chain Olefins, ACS Catal., 2018, 8, 9174–9182 CrossRef CAS.
- A. Ramirez, A. D. Chowdhury, A. Dokania, P. Cnudde, M. Caglayan, I. Yarulina, E. Abou-Hamad, L. Gevers, S. Ould-Chikh, K. De Wispelaere, V. Van Speybroeck and J. Gascon, Effect of zeolite topology and reactor configuration on the direct conversion of CO2 to light olefins and aromatics, ACS Catal., 2019, 9, 6320–6334 CrossRef CAS.
- J. Gao, C. Jia and B. Liu, Direct and Selective Hydrogenation of CO2 to Ethylene and Propene by Bifunctional Catalysts, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Selective Transformation of Carbon Dioxide into Lower Olefins with a Bifunctional Catalyst Composed of ZnGa2O4 and SAPO-34, Chem. Commun., 2018, 54, 140–143 RSC.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, Highly Selective Conversion of Carbon Dioxide to Lower Olefins, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- S. Dang, P. Gao, Z. Liu, X. Chen, C. Yang, H. Wang, L. Zhong, S. Li and Y. Sun, Role of Zirconium in Direct CO2 Hydrogenation to Lower Olefins on Oxide/Zeolite Bifunctional Catalysts, J. Catal., 2018, 364, 382–393 CrossRef CAS.
- G. Wang, L. Zeng, J. Cao, F. Liu, Q. Lin, Y. Yi and H. Pan, Highly selective conversion of CO2 to hydrocarbons over composite catalysts of ZnO-ZrO2 and SAPO-34, Microporous Mesoporous Mater., 2019, 284, 133–140 CrossRef CAS.
- C. Chen, X. Wang, D. Wu, J. Zhang, Q. Ma, X. Gao, X. Lai, H. Xia, S. Fan and T. S. Zhao, Hydrogenation of CO2 to light olefins on CuZnZr@(Zn-)SAPO-34 catalysts: Strategy for product distribution, Fuel, 2019, 239, 44–52 CrossRef.
- J. Wang, A. Zhang, X. Jiang, C. Song and X. Guo, Highly selective conversion of CO2 to lower hydrocarbons over bifunctional catalysts composed of In2O3-ZrO2 and zeolite, J. CO2 Util., 2018, 27, 81–88 CrossRef CAS.
- J. Li, T. Yu, D. Miao, X. Pan and X. Bao, Carbon dioxide hydrogenation to light olefins over ZnO-Y2O3 and SAPO-34 bifunctional catalysts, Catal. Commun., 2019, 129, 105711 CrossRef.
- N. Rahimi and R. Karimzadeh, Catalytic Cracking of Hydrocarbons over Modified ZSM-5 Zeolites to Produce Light Olefins: A Review, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- Y. V. Kissin, Chemical mechanisms of catalytic cracking over solid acidic catalysts: alkanes and alkenes, Catal. Rev.: Sci. Eng., 2001, 43, 85–146 CrossRef CAS.
- A. Corma, Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- K. Arata, Solid Superacids, Adv. Catal., 1990, 37, 165–211 CAS.
- X. Xong and A. Sayari, Sulfated Zirconia-Based Strong SolidAcid Catalysts: Recent Progress, Catal. Rev.: Sci. Eng., 1996, 38, 329–412 CrossRef.
- G. D. Yadav and J. J. Nair, Sulfated zirconia and its modified versions as promising catalysts for industrial processes, Microporous Mesoporous Mater., 1999, 33, 1–48 CrossRef CAS.
- N. Katada, J. Endo, K. Notsu, N. Yasunobu, N. Naito and M. Niwa, Superacidity and Catalytic Activity of Sulfated Zirconia, J. Phys. Chem. B, 2000, 104, 10321–10328 CrossRef CAS.
- D. Farcasiu, J. Q. Li and S. Cameron, Preparation of sulfated zirconia catalysts with improved control of sulfur content, Appl. Catal., A, 1995, 128, 97–105 CrossRef CAS.
- A. Ramirez, S. Ould-Chikh, L. Gevers, A. D. Chowdhury, E. Abou-hamad, A. Aguilar-Tapia, J. Hazemann, N. Wehbe, A. J. Al Abdulghani, S. M. Kozlov, L. Cavallo and J. Gascon, Tandem Conversion of CO2 to Valuable Hydrocarbons in Highly Concentrated Potassium Iron Catalysts, ChemCatChem, 2019, 11, 1–9 CrossRef.
- V. V. Kanazhevskii, V. P. Shmachkova, N. S. Kotsarenko, V. N. Kolomiichuk and D. I. Kochubei, Changes in the zirconium local surrounding on ligand substitution in solutions, J. Struct. Chem., 2006, 47, 860–868 CrossRef CAS.
- Y. Kameshima, K. Kageyama, Y. Mizunuma, T. Komatsu, T. Isobe, A. Nakajima and K. Okada, Preparation and solid acidity of zirconia-cluster/saponite composites, J. Ceram. Soc. Jpn., 2013, 121, 49–53 CrossRef CAS.
- K. Mirza, M. Ghadiri, M. Haghighi and A. Afghan, Hydrothermal synthesize of modified Fe, Ag and K-SAPO-34 nanostructured catalysts used in methanol conversion to light olefins, Microporous Mesoporous Mater., 2018, 260, 155–165 CrossRef CAS.
- R. Srinivasan, R. A. Keogh and B. H. Davis, Sulfated zirconia catalysts: Are Bronsted acid sites the source of the activity?, Catal. Lett., 1996, 36, 51–57 CrossRef CAS.
- A. Corma, J. Planelles, J. Sánchez-Marín and F. Tomás, The Role of Different Types of Acid Site in the Cracking of Alkanes on Zeolite Catalysts, J. Catal., 1985, 93, 30–37 CrossRef CAS.
- L. F. Lin, S. F. Zhao, D. W. Zhang, H. Fan, Y. M. Liu and M. Y. He, Acid Strength Controlled Reaction Pathways for the Catalytic Cracking of 1-Pentene to Propene over ZSM-5, ACS Catal., 2015, 57, 4048–4059 CrossRef.
- Z. Nawaz, X. Tang, J. Zhu, F. Wei and S. Naveed, Catalytic Cracking of 1-Hexene to Propylene Using SAPO-34 Catalysts with Different Bulk Topologies, Chin. J. Catal., 2019, 30, 1049–1057 CrossRef.
- A. P. Dementjev, O. P. Ivanova, L. A. Vasilyev, A. V. Naumkin, D. M. Nemirovsky and D. Y. Shalaev, Altered layer as sensitive initial chemical state indicator, J. Vac. Sci. Technol., A, 1994, 12, 423 CrossRef CAS.
- E. Paparazzo, E. Severini, A. Jimenez-Lopez, P. Maireles-Torres, P. Olivera-Pastor, E. Rodriguez-Castellon and A. A. G. Tomlinson, Surface chemistry of chromia-pillared tin and zirconium phosphate materials: an X-ray photoelectron spectroscopic study, J. Mater. Chem., 1992, 2, 1175 RSC.
- L. Zhang, J. Bates, D. Chen, H. Y. Nie and Y. Huang, Investigations of Formation of Molecular Sieve SAPO-34, J. Phys. Chem. C, 2011, 115, 22309–22319 CrossRef CAS.
- W. Shen, X. Li, Y. Wei, P. Tian, F. Deng, X. Han and X. Bao, A study of the acidity of SAPO-34 by solid-state NMR spectroscopy, Microporous Mesoporous Mater., 2012, 158, 19–25 CrossRef CAS.
- R. M. Danisi, J. E. Schmidt, A. Lucini Paioni, K. Houben, J. D. Poplawsky, M. Baldus, B. M. Weckhuysen and E. T. C. Vogt, Revealing long- and short-range structural modifications within phosphorus-treated HZSM-5 zeolites by atom probe tomography, nuclear magnetic resonance and powder X-ray diffraction, Phys. Chem. Chem. Phys., 2018, 20, 27766–27777 RSC.
- J. Schmidt, L. Peng, A. Lucini Paioni, H. L. Ehren, W. Guo, B. Mazumder, D. A. M. Winter, O. Attila, F. Fu, A. Dutta Chowdhury, K. Houben, M. Baldus, J. Poplawsky and B. Weckhuysen, Isolating Clusters of Light Elements in Molecular Sieves with Atom Probe Tomography, J. Am. Chem. Soc., 2018, 140, 9154–9158 CrossRef CAS PubMed.
- Z. Yan, B. Chen and Y. Huang, A solid-state NMR study of the formation of molecular sieve SAPO-34, Solid State Nucl. Magn. Reson., 2009, 35, 49–60 CrossRef CAS PubMed.
- S. Dang, S. Li, C. Yang, X. Chen, X. Li, L. Zhong, P. Gao and Y. Sun, Selective Transformation of CO2 and H2 into Lower Olefins over In2O3-ZnZrOx/SAPO-34 Bifunctional Catalysts, ChemSusChem, 2019, 12, 3582 CrossRef CAS PubMed.
- G. Wan, Y. Wang, J. Cao, X. Wang, Y. Yi and F. Liu, Fabrication of ZnZrO2@Al2O3@SAPO-34 tandem catalyst for CO2 conversion to hydrocarbons, Microporous Mesoporous Mater., 2020, 291, 109693 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02532d |
| This journal is © The Royal Society of Chemistry 2020 |