
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd–Au bimetallic catalysts supported on ZnO for selective 1,3-butadiene hydrogenation†
Belén
Bachiller-Baeza
 *a,
Ana
Iglesias-Juez
a,
Giovanni
Agostini
b and
Eva
Castillejos-López
c
*a,
Ana
Iglesias-Juez
a,
Giovanni
Agostini
b and
Eva
Castillejos-López
c
aInstituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie No. 2, Cantoblanco, 28049 Madrid, Spain. E-mail: b.bachiller@icp.csic.es
bESRF–The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
cDpto. Química Inorgánica y Técnica, Fac. de Ciencias, UNED, C/Senda del Rey No. 9, 28040, Madrid, Spain
First published on 5th March 2020
Abstract
Bimetallic Pd–Au catalysts were synthesized using two differently shaped ZnO supports, ZnO-t (tetrapods) and ZnO-n (needles). These catalysts were tested in the partial hydrogenation of 1,3-butadiene and the results were compared to those for the corresponding monometallic catalysts of Pd. The structural and electronic properties of the catalysts were analysed by transmission electron microscopy and scanning transmission electron microscopy (TEM-STEM), Fourier transform infrared spectroscopy analysis of adsorbed CO (CO-FTIR), X-ray photoelectron spectroscopy (XPS) and in situ X-ray absorption spectroscopy (XAS) measurements combined with in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). On both catalysts, gold diffusion on the Pd(111) faces and formation of small Pd-rich particles and big Au-rich core/Pd-rich shell like structures were confirmed. Besides, both Pd–Au bimetallic samples gave similar conversion and selectivity values in the hydrogenation reaction. The product distribution resembled that for monometalic Pd on ZnO-t, except for the selectivity to 1-butene among the butenes which was slightly higher. The best catalyst in terms of selectivity to the butenes is Pd–ZnO-n. The differences found in the catalytic performance were analysed based on electronic and ensemble effects. In contrast to that found for the monometallic catalysts, the support morphology does not seem to be a determinant for the properties in the case of the Pd–Au bimetallic samples.
Introduction
Bimetallic gold–palladium catalysts have attracted considerable interest due to their superior performance in terms of activity, selectivity and stability compared to pure Au and Pd catalysts in a wide variety of reactions such as selective oxidation of alcohols or polyols, oxidation of hydrocarbons, low temperature oxidation of CO, and direct synthesis of hydrogen peroxide from H2–O2 mixtures.1 However, there have been relatively few studies on the effects of Pd/Au nanoparticles in hydrogenation reactions. Some of those studies have focused on the selective hydrogenation of p-chloronitrobenzene,2 1,3-butadiene3 or cyclohexene.4 The overall enhancement of the catalytic activity of these systems is attributable to a synergistic effect between both noble metals that can be explained by ligand and ensemble effects. These effects modify the local electronic and geometric structures of nanoparticles and are often intertwined; therefore, it is complicated to separate and understand the contribution of each factor to the alloy performance.The supports generally used for PdAu bimetallic catalysts are non-reducible supports, such as silica, alumina and carbon, and reducible supports like titania or iron oxide.1,5–8 Apart from stabilizing highly dispersed nanoparticles, the choice of an appropriate support can affect the catalyst properties by promoting metal–support interaction. In this sense, ZnO is an interesting support due to its unique properties, such as excellent thermal and chemical stability and easily modified electrical conductivity. These properties are strongly dependent on its nanostructures, including morphology, polar/nonpolar exposed facet ratio, and size, orientation, and density of crystal.9,10 Accordingly, apart from its efficient role in various fields (semiconductors, gas sensors, photocatalysts, etc.),11–14 ZnO has also been employed as a carrier of different metal nanoparticles, Pd and Au among them. Pd–ZnO catalysts have exhibited high performance in terms of selectivity and stability in different reactions including steam reforming of methanol, water–gas shift and hydrogenation of alkenes.15–18 On the other hand, Au/ZnO catalysts have also been reported as efficient catalysts for: methanol steam reforming for hydrogen and CO2 production;19 carbon monoxide hydrogenation into methanol; CO oxidation and CO oxidation in an excess of H2 (PROX reaction);20,21 photodegradation of dyes in the liquid phase and benzene in the gas phase acting as a photocatalyst under UV and/or visible irradiation.22
Depending on the thermal treatments and gas atmospheres, the formation of α- and/or β-PdZn and AuZn alloys and intermetallic structures, as well as the presence of strong-metal support interaction (SMSI) states, have been reported.16–18,23–26 Zn incorporation seems to change the number of ensemble active atoms as well as the Pd or Au electronic properties due to charge transfer between Zn and the metal, consequently affecting the catalytic behaviour. In addition, the catalytic performance of PdZn and AuZn phases seem to be influenced by the exposed ZnO facets. In this sense, differences in catalytic performance have been described between Pd/ZnO (0001) and Pd/ZnO (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0)27 or between Pd supported on ZnO with predominant non-polar facets and that on commercial ZnO without any dominant facets.28 We have also previously observed that the exposed planar surface of the ZnO support affected the Zn content, morphology and chemical properties of the formed PdZn nanostructures towards the hydrogenation of 1,3-butadiene.29 Similarly, Au catalysts prepared on ZnO materials with hierarchical architectures30 or with distinct morphologies31,32 have shown different CO sensing responses or photocatalytic activities in several applications. Besides, in some cases, epitaxial growth of metal particles has been claimed to explain their particular behaviour in catalytic reactions.21,33,34
0)27 or between Pd supported on ZnO with predominant non-polar facets and that on commercial ZnO without any dominant facets.28 We have also previously observed that the exposed planar surface of the ZnO support affected the Zn content, morphology and chemical properties of the formed PdZn nanostructures towards the hydrogenation of 1,3-butadiene.29 Similarly, Au catalysts prepared on ZnO materials with hierarchical architectures30 or with distinct morphologies31,32 have shown different CO sensing responses or photocatalytic activities in several applications. Besides, in some cases, epitaxial growth of metal particles has been claimed to explain their particular behaviour in catalytic reactions.21,33,34
However, very few studies of bimetallic PdAu catalysts on ZnO can be found.35 Thus, motivated by our own studies21,29,33 and the above-mentioned results showing the sometimes unusual behavior of both Pd and Au monometallic catalysts supported on different ZnO supports, herein, we present a comparative study on the performance of bimetallic Pd–Au catalysts supported on two shaped ZnO supports with different polar/nonpolar exposed facet ratios: needles (ZnO-n) and tetrapods (ZnO-t), preferentially exposing polar (0001) and nonpolar (100) facets, respectively. The aim is to understand how the morphology and exposed facets of the ZnO support influence the formation of bimetallic PdAu nanoparticles, also bearing in mind the initial Pd– and Au–support interactions that, as can be foreseen by the exposition above, will be conditioned by the ZnO support structure. In order to unveil the structural and electronic properties, a combination of characterization techniques were applied (TEM-STEM, XPS, XAS, CO-FTIR), and the selective hydrogenation of 1,3-butadiene was selected to evaluate the catalytic properties.
Experimental
Preparation of catalysts
Two ZnO supports with different shapes and different exposed faces were used: needles (ZnO-n) and tetrapods (ZnO-t). Briefly, material ZnO-n was prepared using a microemulsion method,14 and high purity tetrapods of ZnO (ZnO-t) were prepared by gas phase oxidation of zinc in the presence of air at 1073 K.36 More details about material preparation can be consulted in the corresponding references.Pd supported on ZnO, with a 2 wt% Pd loading, was prepared by incipient wetness impregnation using water solutions of Pd(NO3)2. The bimetallic PdAu catalysts were prepared by wet impregnation using a water/methanol (15/1) solution of Pd(NO3)2 and HAuCl4·3H2O precursors. The mixture was treated with a water solution of Na2CO3 (0.1 M) until pH = 10. Then, the support was added to the flask with this solution and kept at room temperature and under stirring with a magnetic stirrer until evaporation. The corresponding catalysts, with a palladium and gold nominal loading of 1 wt%, which corresponds to an Au/Pd atomic ratio of 0.53 (35 at% Au), were labelled PdAu–ZnO-t and AuPd–ZnO-n. Monometallic Au catalysts (2 wt%) were prepared for comparison by the deposition–precipitation method with sodium carbonate as a precipitating agent.33 All samples were dried overnight in air at 373 K.
Instrumentation and catalyst characterization
Transmission electron microscopy (TEM) and annular dark-field scanning transmission electron microscopy (ADF-STEM) images of the catalysts were measured using a JEOL JEM-2100 field-emission gun electron microscope operated at 200 kV. The samples were dispersed in ethanol and sonicated for 5 min. Then, a small amount of the solution was dropped onto a holey carbon film supported by a copper grid and allowed to dry at room temperature. The average particle diameter (d) was calculated on the basis of a minimum of 100 measured particles, using the following expression, where ni is the number of particles with diameter di: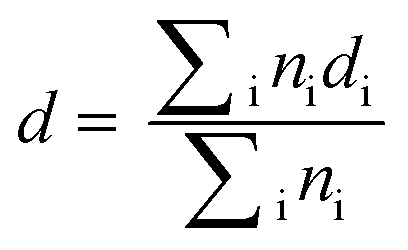
Scanning transmission electron microscopy (STEM) mode imaging was done using a spot size of 1 nm. X-ray energy dispersive spectroscopic (EDS) mapping analysis was carried out using the Oxford Instruments Inca software package. Quantification of the experimental EDS spectra was performed by the Cliff–Lorimer method based on relative intensities of the Pd-Lα (2.839 keV) and Au-Mα (2.123 keV) peaks using k-factors provided by the EDS system manufacturer.
The infrared spectra of adsorbed CO were recorded on a Varian 670 Fourier transform IR spectrophotometer at a resolution of 4 cm−1 using a liquid N2 cooled MCT detector. Self-supporting wafers of the samples were placed in a glass cell that can be coupled to a conventional vacuum gas handling line, reduced in a H2 flow of 30 ml min−1 at 523 K for 1 h and then outgassed at the same temperature for 1 h. After cooling to room temperature, CO (80 Torr) was introduced and the IR spectra were recorded after removing the gas phase. The infrared spectra were background corrected and subtracted from the reference spectrum of the clean sample obtained prior to the addition of CO.
X-ray photoelectron spectroscopy (XPS) analyses were performed on an ESCA-PROBE P (Omicron) spectrometer and spectra were recorded using non-monochromatized Mg-Kα (1253.6 eV) or Al-Kα radiation (1486.7 eV). Powder samples were pressed into pellets, placed in the sample holder and then transferred to the sample treatment chamber where they were reduced at 523 K under a flow of a mixture of 10% H2 in Ar (50 ml min−1). Then, after cooling down to room temperature, the sample was transferred without air contact to the preparation chamber, degassed for 6–8 h and finally transferred to the ultra-high vacuum analysis chamber. XPS data analysis was performed using CASA XPS software. The C 1s peak at 284.6 eV was used as the energy reference. For the fitting, the relative intensity ratio and the energy separation between the spin–orbit components Pd 3d5/2 and Pd 3d3/2 were fixed, whereas the other parameters such as intensity, FWHM and peak position were allowed to vary within a reasonable range. A tail exponent factor has been introduced in the fitting of the metallic palladium to match the asymmetry of the metallic state. The FWHM values found were in the range 1.8–2.0 eV for the oxidized species and in the range 1.5–1.7 eV for the reduced species. The relative concentrations and atomic ratios were estimated from the integrated intensities of photoelectron lines corrected with the corresponding atomic sensitivity factor.
Synchrotron in situ XANES-DRIFTS combined experiments
Pd K edge (24.350 eV) X-ray absorption measurements combined simultaneously with diffuse reflectance infrared spectroscopy (DRIFTS) and mass spectrometry (MS) were carried out at beamline BM23 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France).37 The samples, ca. 30–40 mg, were loaded into a cell specifically designed to work in combined DRIFTS and XAS measurements.38 XAS data were collected using a Si(111) crystal monochromator and ionization chambers in transmission mode. Pd foil XANES spectra were simultaneously acquired to ensure appropriate energy calibration. DRIFTS measurements were made employing a Varian 670 (mid-range) IR spectrometer coupled with a MCT detector and specially designed DRIFTS optics for this application. Evolved gas analysis was carried out using a quadrupole mass spectrometer (Pfeiffer Omnistar). The flow of reaction gas feed to the reactor was constant at 60 mL min−1, with compositions of 15% H2 in helium during temperature programmed reduction experiments and 15% H2 + 3% 1,3-butadiene (Bd) in helium under hydrogenation reaction conditions. The temperature was raised by 3 K min−1 up to 523 K during the TPR. XANES spectra were collected in continuous acquisition mode (q-EXAFS) every 1.5 min during the heating and cooling ramps. After cooling to the reaction temperature, EXAFS spectra were acquired (up to 11 Å−1, with an integration time of 2 s for all spectra with Δk = 0.03 Å−1 constant step in the EXAFS region) and then XANES-DRIFTS time resolved experiments (0.5 s) with H2/H2 + Bd/H2 alternating exposures at constant temperature were performed with an exposure time of 30 min for each atmosphere. The exhaust products from the reactor cell were analysed using MS.Catalytic measurements: hydrogenation of 1,3-butadiene
1,3-Butadiene (Bd) hydrogenation reaction was carried out in a continuous flow fixed-bed reactor at atmospheric pressure. Prior to the reaction test, the catalyst was reduced in flowing H2 at 523 K for 1 h. Then, a H2 (10% vol), Bd (2% vol) and N2 (balance) gas mixture flowing at 60 mL min−1 was passed through the catalyst bed. The reaction temperature was varied between 298 and 313 K. The effluents of the reactor were online analysed using a gas chromatograph (Varian 3400) with a flame ionization detector (FID) and thermal conductivity detector (TDC), and with a 20% BMEA Chromosorb P80/100 column. The conversion of Bd and selectivities to 1-butene, cis-2-butene, trans-2-butene and butane were calculated.Results and discussion
Catalyst characterization
Representative TEM images and particle size distributions of the nanoparticles for the mono- and bimetallic catalysts prepared on the ZnO materials are shown in Fig. 1 and S1 to S4 (see the ESI†). As previously reported, the different structures of the two ZnO supports studied are clearly reflected in the TEM images.29 While ZnO-t presents rods that are elongated along the c crystallographic orientation and a larger contribution of nonpolar (100) planes, ZnO-n shows sharp needle-like structures that grow in the a direction perpendicular to the c-axis and, therefore, has a larger proportion of polar faces. As far as the metal phase is concerned, the addition of Au to Pd catalysts leads to a significant increase in the mean particle size when supported on ZnO-t, 6 vs. 3 nm for PdAu–ZnO-t and Pd–ZnO-t, respectively, as well as leading to a wider particle size distribution, (Fig. S1 and S2†). An apparent fraction of NPs with a larger diameter (>15 nm) can be also observed in different regions of the sample. The opposite situation was observed for the samples supported on ZnO-n, (Fig. S3 and S4†), i.e. smaller NPs with a diameter of ca. 3 nm were found to be mainly present on the surface of PdAu–ZnO-n resulting in a narrower distribution than that for its monometallic counterpart. A small proportion but definitely a minor phase of bigger particles with diameters of around 20 nm was also observed.
 | ||
| Fig. 1 TEM images of the (A) Pd–ZnO-t, (B) Pd–ZnO-n, (C) PdAu–ZnO-t and (D) PdAu–ZnO-n catalysts. | ||
EDS elemental maps for both bimetallic catalysts (Fig. 2 and 3) showed that the population of alloyed particles in areas of higher dispersion can be described as Pd-rich nanoparticles and that the larger particles (>15 nm in size) are also bimetallic particles but with higher Au concentration (between 50 and 70 at% compared to the theoretical bulk Au composition of 35 at%). Differences in the atomic ratios as a function of particle size and the existence of Au-rich PdAu alloyed particles have also been reported in the literature on PdAu bimetallic catalysts prepared by impregnation methods.39 Our results also agree with calculations according to Maeland and Flanagan's theory40 that showed that the solubility limit of Au in Pd was about 12 at% and that Pd was more soluble in Au with a maximum of 31 at%. On the other hand, the Pd-Lα signal seems to originate from a bigger spatial area compared to the Au-Mα signal, which could indicate Pd surface enrichment or a core–shell like structure. In general, the Pd/Au composition of the bulk and of the catalytically active surface can differ markedly due to the differences in the surface free energies of Pd and Au, 2.043 J m−2 and 1.626 J m−2, respectively. The PdcoreAushell arrangement is theoretically favored due to the lowest surface energy of Au and the minimization of the lattice mismatch, about 4%, between Au and Pd (Au, 4.079 Å; Pd 3.8898 Å).41 Notably, our big PdAu nanoparticles would have the opposite configuration (AucorePdshell like structure), as it has been also reported for other PdAu catalysts.1,8
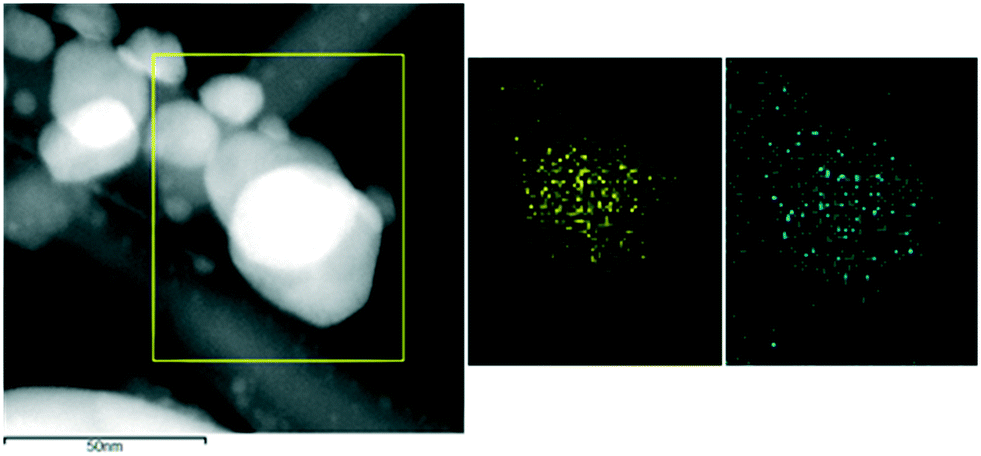 | ||
| Fig. 2 ADF-STEM image and corresponding EDS elemental maps of the highlighted area showing the chemical distribution of Au (yellow) and Pd (blue) for the PdAu–ZnO-t catalyst. | ||
 | ||
| Fig. 3 ADF-STEM image and corresponding EDS elemental maps of the highlighted area showing the chemical distribution of Au (yellow) and Pd (blue) of the PdAu–ZnO-n catalyst. | ||
Alloy formation and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370 eV) becomes decreased (as shown in Fig. S8† for sample PdAu–ZnO-n as an example), leading to a spectrum similar to that of Pd(0). The reduction of the Pd initial species during the H2-TPR experiment was performed following the shifting of the adsorption edge towards lower energies of the set of XANES spectra obtained. The results are presented in Fig. 4. The onset temperature of reduction varies and follows the trend Pd–ZnO-n < PdAu–ZnO-t ≤ PdAu–ZnO-n < Pd–ZnO-t. Notably, while the addition of Au retards the reduction of Pd when supported on ZnO-n, it promotes the reduction when supported on ZnO-t. In addition, while the reduction seems more gradual for PdAu–ZnO-n, for the bimetallic PdAu–ZnO-t, it is as quick as that for both monometallic catalysts. During the reduction process, surface diffusion of the two deposited metal precursors is needed to yield the bimetallic Pd–Au aggregates. But, apart from the Pd–Au bond strength, the relative strengths of the Pd–Zn and Au–Zn bonds can influence the degree of segregation/mixing.42 Then, the trends and shifts observed at the temperature of reduction of the Pd–Au alloy during the TPR would be a consequence of these primary Pd–support interactions. We have observed for the Pd monometallic samples that the Pd reduction led to the formation of a PdZn alloy, and its temperature of formation depended on the type of support where the stronger the interaction, the higher the temperatures and the lower the alloying degree.29 Previous in situ XRD results also showed the formation of a PdZn intermetallic phase by diffusion of Zn into the Pd bulk, which agree with results reported for studies on model surfaces under vacuum43 and is in contrast with the formation of an intermediate PdH phase.44 On the other hand, different metal–support interactions depending on the two shaped ZnO can also be assumed for Au. In this sense, gold nanoparticle epitaxial growth on a ZnO-t support has been previously reported.21,33 Additionally, the TEM of the reference monometallic gold catalysts (Fig. S5 and S6†) would confirm this fact since both particle size and morphology are different, i.e. bigger and spherical on ZnO-n. Therefore, the addition of Au seems to inhibit the formation of PdZn by possibly limiting the interaction between the metal and the support. A similar suppression of the SMSI effect has been proposed for the addition of Cu to Pd/Nb2O5 catalysts.45 On PdAu–ZnO-t, we observed that Pd is even partially reduced under vacuum (see XPS results below), and therefore, the addition of gold facilitates the reduction of Pd forming the PdAu alloy. For the PdAu–ZnO-n catalyst, the lack of intensity reduction for the CO-FTIR signals in the spectra (see below) also revealed the limited formation of PdZn alloy that was mainly present on the monometallic Pd–ZnO-n catalyst. So, although the TEM and STEM-EDS maps have reflected the limited (partial) solubility between Au and Pd metals in the alloy crystals with the presence of small Pd-rich and big Au-rich particles the absence (or minor presence) of PdZn alloy on any of the bimetallic catalysts can be assessed. Besides, the different particle size distributions observed by TEM for the bimetallic samples are also a consequence of the different reducibility. Reduction of Pd is easier for PdAu–ZnO-t, which would imply that it is less resistant to sintering than PdAu–ZnO-n with the increase in the temperature of reduction, and the higher average particle size and wider distribution after reduction at 523 K seem to corroborate this.
370 eV) becomes decreased (as shown in Fig. S8† for sample PdAu–ZnO-n as an example), leading to a spectrum similar to that of Pd(0). The reduction of the Pd initial species during the H2-TPR experiment was performed following the shifting of the adsorption edge towards lower energies of the set of XANES spectra obtained. The results are presented in Fig. 4. The onset temperature of reduction varies and follows the trend Pd–ZnO-n < PdAu–ZnO-t ≤ PdAu–ZnO-n < Pd–ZnO-t. Notably, while the addition of Au retards the reduction of Pd when supported on ZnO-n, it promotes the reduction when supported on ZnO-t. In addition, while the reduction seems more gradual for PdAu–ZnO-n, for the bimetallic PdAu–ZnO-t, it is as quick as that for both monometallic catalysts. During the reduction process, surface diffusion of the two deposited metal precursors is needed to yield the bimetallic Pd–Au aggregates. But, apart from the Pd–Au bond strength, the relative strengths of the Pd–Zn and Au–Zn bonds can influence the degree of segregation/mixing.42 Then, the trends and shifts observed at the temperature of reduction of the Pd–Au alloy during the TPR would be a consequence of these primary Pd–support interactions. We have observed for the Pd monometallic samples that the Pd reduction led to the formation of a PdZn alloy, and its temperature of formation depended on the type of support where the stronger the interaction, the higher the temperatures and the lower the alloying degree.29 Previous in situ XRD results also showed the formation of a PdZn intermetallic phase by diffusion of Zn into the Pd bulk, which agree with results reported for studies on model surfaces under vacuum43 and is in contrast with the formation of an intermediate PdH phase.44 On the other hand, different metal–support interactions depending on the two shaped ZnO can also be assumed for Au. In this sense, gold nanoparticle epitaxial growth on a ZnO-t support has been previously reported.21,33 Additionally, the TEM of the reference monometallic gold catalysts (Fig. S5 and S6†) would confirm this fact since both particle size and morphology are different, i.e. bigger and spherical on ZnO-n. Therefore, the addition of Au seems to inhibit the formation of PdZn by possibly limiting the interaction between the metal and the support. A similar suppression of the SMSI effect has been proposed for the addition of Cu to Pd/Nb2O5 catalysts.45 On PdAu–ZnO-t, we observed that Pd is even partially reduced under vacuum (see XPS results below), and therefore, the addition of gold facilitates the reduction of Pd forming the PdAu alloy. For the PdAu–ZnO-n catalyst, the lack of intensity reduction for the CO-FTIR signals in the spectra (see below) also revealed the limited formation of PdZn alloy that was mainly present on the monometallic Pd–ZnO-n catalyst. So, although the TEM and STEM-EDS maps have reflected the limited (partial) solubility between Au and Pd metals in the alloy crystals with the presence of small Pd-rich and big Au-rich particles the absence (or minor presence) of PdZn alloy on any of the bimetallic catalysts can be assessed. Besides, the different particle size distributions observed by TEM for the bimetallic samples are also a consequence of the different reducibility. Reduction of Pd is easier for PdAu–ZnO-t, which would imply that it is less resistant to sintering than PdAu–ZnO-n with the increase in the temperature of reduction, and the higher average particle size and wider distribution after reduction at 523 K seem to corroborate this.
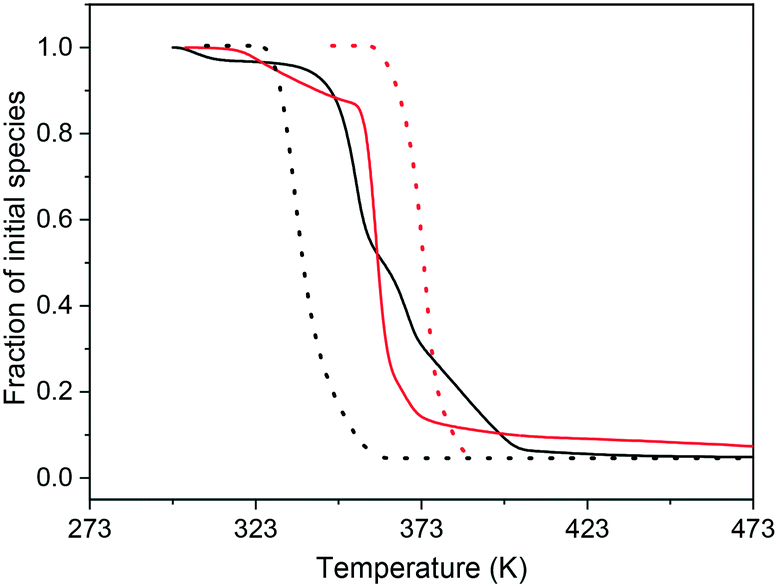 | ||
Fig. 4 Fraction of initial species during TPR in H2: ( ) Pd–ZnO-t; ( ) Pd–ZnO-t; ( ) Pd–AuZnO-t; ( ) Pd–AuZnO-t; ( ) PdZnO-n; (—) PdAu–ZnO-n. ) PdZnO-n; (—) PdAu–ZnO-n. | ||
Despite some differences in terms of the energy position of the edge and the CR characteristics with respect to the typical spectrum for pure Pd metal, the final XANES spectra can be assignable in all cases to Pd(0) species as shown in Fig. 5. For the monometallic catalysts, we already explained that the lower values of the Pd K-edge energy position compared to that for the Pd-foil reference were due to the higher electronic density of Pd entities.29 Besides, the differences found between the two monometallic samples, i.e. the slight shift observed between the spectra, suggested different Pd environments depending on the ZnO morphology. For both bimetallic catalysts, the addition of Au seems to lead to similar final spectra, and the Pd K-edge values are higher than that obtained for Pd–ZnO-n and closer to the edge of the Pd foil, suggesting a null or small electronic perturbation of Pd atoms due to Au.
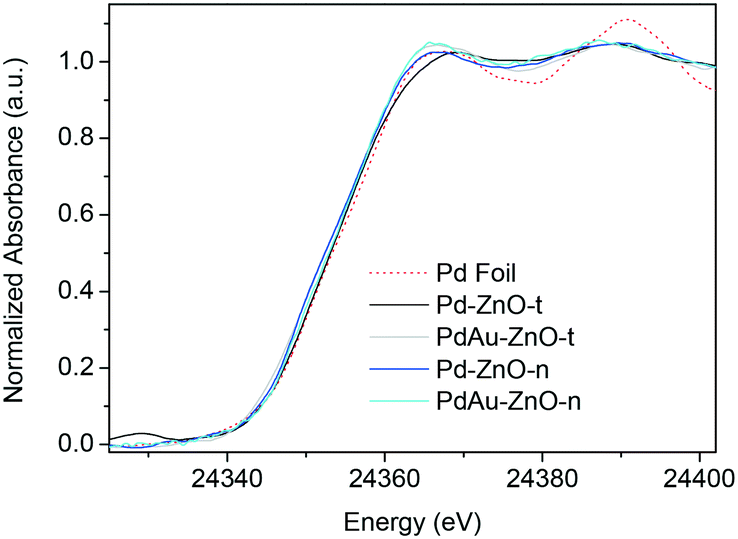 | ||
| Fig. 5 Pd K-edge XANES spectra of the final species after TPR in H2 for Pd–ZnO-t, PdAu–ZnO-t, Pd–ZnO-n, PdAu–ZnO-n, and the Pd foil reference. | ||
The local structure around the Pd atoms after the reduction treatment was analysed by acquiring the EXAFS spectra at the Pd K edge for all the samples. The EXAFS spectra of the Pd monometallic systems were fitted with Pd–Pd and Pd–Zn scattering paths while, for the PdAu bimetallic catalysts, Pd–Pd and Pd–Au scattering paths were used for the curve fitting. The obtained fitting parameters and the Fourier transforms of the Pd k3-weighted spectra are shown in Table 1 and Fig. S9,† respectively (complementary information in Table S1 in the ESI†). A thorough analysis to elucidate the extent of alloying or intra-particle composition of the PdAu structures could not be performed since, due to time constraints, the Au LIII edge data were not collected. Furthermore, the TEM results show that the clusters possessed different compositional distributions, which could lead to an inaccurate analysis. However, some inferences can be obtained. As previously reported for the monometallic catalyst, the presence of the Pd–Zn coordination shell and the atom distance modification confirmed the formation of a PdZn alloy. Furthermore, comparison of the coordination numbers for the Pd–Zn and Pd–Pd shells indicated higher Zn content in the alloy phase for the Pd–ZnO-n sample. The fits obtained for the PdAu bimetallic catalysts also confirmed the change in the Pd coordination environment, with a Pd–Pd atomic distance in the first coordination shell of 2.58–2.59 Å and a new Pd–Au coordination shell with an atomic distance of 2.73–2.77 Å. The reduced Pd–Pd distance compared to that in the bulk Pd metal will be consistent with a reduction in particle size or the presence of pseudomorphic overlayers, suggesting some level of segregation within the PdAu NPs. The sum of the coordination numbers of Pd–Pd and Pd–Au bonds (CN = NPd–Pd + NPd–Au) obtained from the EXAFS analysis provide an indication of the Pd phase particle size. We can note that these values, 11.4 and 7.5, are smaller than that of the Pd foil (CN = 12), which indicates that the nanoparticles are well dispersed on the support (as an average measurement). The higher value for PdAu–ZnO-t would be in agreement with the higher particle size measured by TEM. Both NPd–Au/NPd–Pd ratios are higher than the theoretical bulk Au/Pd atomic ratio introduced in these catalysts pointing again to some segregation of phases, as surface atoms have a lower number of neighbours. The higher NPd–Au/NPd–Pd ratio for the PdAu–ZnO-n sample, 1.3 vs. 0.7 for PdAu–ZnO-t, could be due in part to the different levels of phase segregation, and also to the different particle sizes, lower in the case of PdAu–ZnO-n.
| Catalyst | 1st shell Pd–Zn | 2nd shell Pd–Pd | ||
|---|---|---|---|---|
| R | N 1 | R | N 2 | |
| Pd–ZnO-t | 2.54 | 0.6 | 2.90 | 2.1 |
| Pd–ZnO-n | 2.51 | 1.4 | 2.88 | 1.4 |
| 1st shell Pd–Pd | 2nd shell Pd–Au | |||
| PdAu–ZnO-t | 2.61 | 6.7 | 2.77 | 4.7 |
| PdAu–ZnO-n | 2.57 | 3.3 | 2.73 | 4.2 |
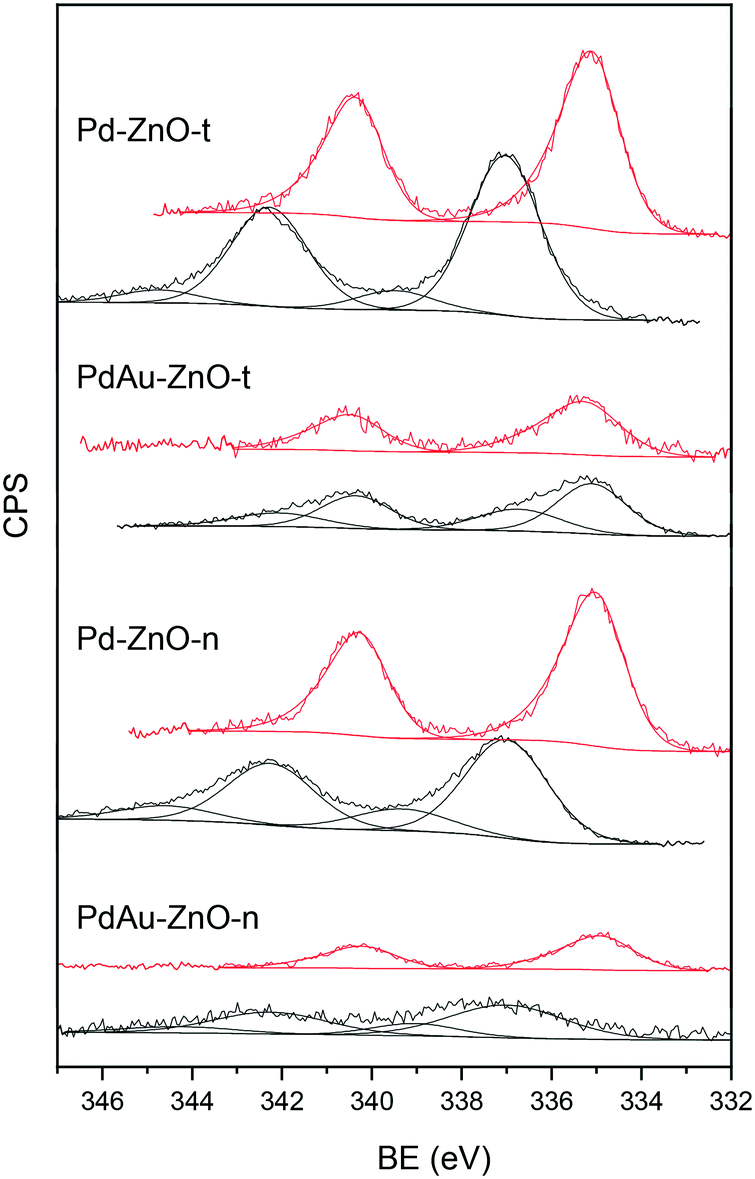 | ||
| Fig. 6 XPS spectra of the Pd 3d core level for Pd–ZnO-n, PdAu–ZnO-n, Pd–ZnO-t, and PdAu–ZnO-t. Black lines: fresh samples; red lines: after H2 treatment at 523 K. | ||
Analysis of the Au 4f spectra is complicated by the severe overlap of the Zn 3p3/2 and Zn 3p1/2 peaks (at 88.3 and 91.3 eV, respectively) and the low Au content (1 wt%). Only for the reduced bimetallic catalysts, a small peak at around 84.4 eV, the Au 4f7/2 component, could be seen more clearly (Fig. S10†). This can be assigned to reduced phase of gold or to large Au-rich nanoparticles since the observed binding energy values are close to that of pure Au foil, 84 eV.7,49 These values are similar to those obtained for the monometallic Au–ZnO-t and Au–ZnO-n catalysts prepared for comparison (included in Fig. S10†).
The atomic Au/Pd ratios calculated for the reduced bimetallic catalysts are between 0.3 and 0.4. Although they have to be taken with care due to the difficulties in the area determination, if compared with the bulk Au/Pd ratio of 0.53, a slight surface enrichment of Pd is implied. This would agree with the microscopy results that showed the presence of both small Pd-rich particles and big core–shell like particles with a higher proportion of Pd on the surface and Au-rich inside. Similar conclusions regarding Pd surface enrichment based on XPS measurements were reached in previous studies of Au–Pd catalysts.8,39 The bimetallic character of the formed nanoparticles, i.e. formation of intermetallic PdZn and/or PdAu phases, cannot be definitely concluded by XPS due to the low amounts of gold and the overlapping of Zn signals.
 | ||
| Fig. 7 FTIR spectra of CO adsorption for Pd–ZnO-t, PdAu–ZnO-t, Pd–ZnO-n and PdAu–ZnO-n after H2 treatment at 523 K. | ||
The spectra of both bimetallic samples were very similar and highlighted an important point: the band for CO adsorbed linearly on low coordinated Au atoms, which should lie at ca. 2100 cm−1, was not detected. Another remarkable point is that the overall spectrum intensity that was greatly reduced for the monometallic Pd–ZnO-n (compared to the pure Pd phase) is now recovered for the bimetallic samples. The spectra showed the peaks at 2084, 2066, 1970 and 1890 cm−1. The higher linear-to-bridging intensity ratios compared to those for the monometallic catalysts and the near absence of the peak around 1925 cm−1 (bridge-bonded sites associated to closed (111)-type surfaces) could be ascribed to ensemble effects due to incorporation of Au into the Pd particles. More specifically, preferential Au diffusion onto the closed Pd(111)-type planes forming a PdAu alloy can be suggested.53
Therefore, while a different morphology of PdZn was suggested for the monometallic samples depending on the support shape, exposing PdZn(111) or (100) planes for ZnO-t and ZnO-n, respectively, the same type of PdAu alloy in terms of morphology seems to be formed in both bimetallic samples, exposing PdAu(111) independently of the ZnO support or exposed faces (polar for ZnO-n and non-polar for ZnO-t) and, consequently, of the initial Pd–support or Au–support interaction. A density functional study of low Pd concentrations on Au clusters has shown that Pd atoms tend to occupy the (111) facets leaving the Au(001) facets free.54 In addition, the fact that CO can still adsorb on 3-fold Pd sites in the bimetallic catalysts confirmed that the surface presented vicinal (contiguous) Pd atoms. These results agree with those obtained by TEM-STEM-mapping where both samples presented small Pd-rich particles and big particles where Pd was concentrated along the perimeter, i.e. Au-rich core/Pd-rich shell like structures or Pd segregated domains.
| Catalyst | d (nm) | Redn. temp. 523 K | Redn. temp. 623 K | |
|---|---|---|---|---|
| 298 K | 313 K | 313 K | ||
| Pd–ZnO-t | 3 | 98(42) | 100(25) | 100(48) |
| PdAu–ZnO-t | 6 | 60(54) | 99(32) | 98(42) |
| Pd–ZnO-n | 4 | 86(55) | 97(60) | 97(80) |
| PdAu–ZnO-n | 3 | 59(48) | 92(23) | 100(48) |
The surface analysis has revealed the relative difficulty of Au to dissolve in Pd that resulted in Pd-rich small particles and bigger Au core–Pd-rich shell type particles for both bimetallic catalysts. Moreover, as we have shown, the Au monometallic catalysts became active only at high temperatures and none of the low coordinated Au sites, which are generally accepted as the active species on gold catalysts, were detected in the CO-FTIR spectra of our bimetallic samples. Therefore, the activity must be related to the Pd sites.
As far as the selectivities are concerned, the selectivities to butenes are in the range 42–55% for all the catalysts at RT. Besides, while the selectivity to butenes slightly increased to 60% at a higher reaction temperature for the monometallic Pd–ZnO-n, it decreased to values of 23–32% for the rest of the catalysts. Notably, this trend, although with higher values (80% and between 40 and 50% for Pd–ZnO-n and the rest of the catalysts, respectively), is also followed after reduction at 623 K. Moreover, some differences in the proportion of butenes could be also observed (Fig. 8). Monometallic Pd–ZnO-n and both PdAu bimetallic catalysts showed higher selectivity to 1-butene that slightly decreased at higher reaction temperature. Therefore, alloying Pd with Au does not appear to affect greatly the activity but does have some effect on the selectivity.55
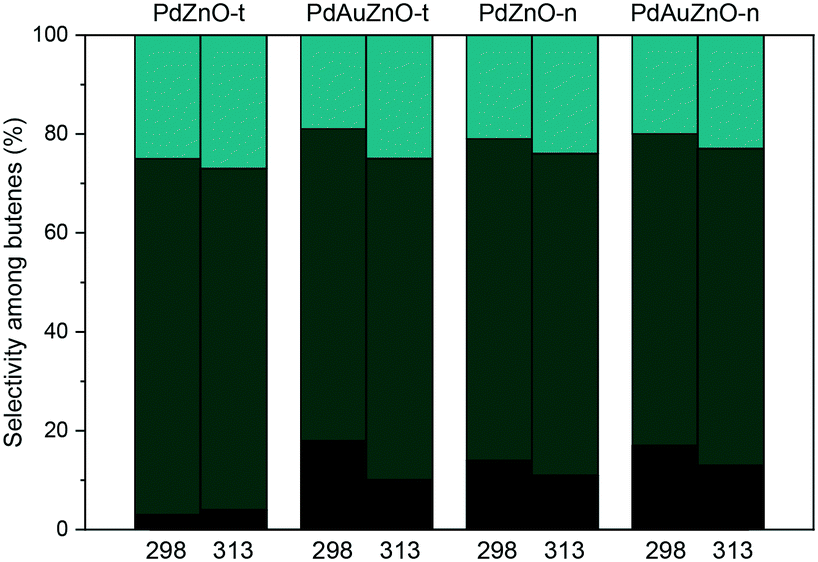 | ||
| Fig. 8 Selectivity among butenes: 1-butene (black), trans-2-butene (dark green) and cis-2-butene (light green). | ||
Both ensemble and electronic effects are generally invoked to interpret the catalytic behaviour of alloys. Then, the observed variations of the selectivity values could be associated to electronic modifications that would affect the relative concentrations of surface adsorbed species and, consequently, the relative rates of competitive surface reactions. So, the interaction of 1,3-butadiene and products was investigated by simultaneous XANES-DRIFTS experiments performed during H2/Bd + H2/H2 alternating exposures. The high stability of the PdZn and PdAu entities formed on the mono- and bimetallic catalysts, respectively, under reaction conditions was confirmed using the XANES spectra (not shown) that were unaltered during the whole cycle. Although all samples showed similar DRIFTS spectra, the general lower intensity observed for the bimetallic samples (Fig. S11†) reflects a lower concentration of adsorbed species as a result of the different Pd loading. As previously indicated, the unambiguous identification of the relative coverages of unsaturated and saturated species could not be accomplished due to overlapping of bands in the spectral region at 2900–3025 cm−1.29 However, a rough approximation can be obtained from the subtle changes in the 1400 and 1450 cm−1 peaks (assigned to butane), in the appearance of additional peaks between 3080 and 3015 cm−1, and the shifting in the position of the maximum of the wide band that is determined by the relative intensity ratio for peaks at 2976 (shoulder) and 2966 cm−1, characteristic of 1-butene and 2-butenes, and butane respectively. In this case, the effect is small since, as the catalyst characterization has shown, both bimetallic samples were very similar. Also, examination of the time evolution of the absolute intensity of the wide band at around 2966 cm−1 (related to the formation of hydrogenation products combining butenes and butane) under Bd + H2/H2 cycling conditions (Fig. S12†) is helpful. For the PdAu bimetallic catalysts, surface saturation was faster than that for the monometallic catalysts and, reached a lower value, approximately half of that for the monometallic catalysts. The saturation values are consistent if we consider the different Pd loadings and assume that the particle sizes are roughly the same for all the samples. In that case, the number of surface active sites would be also the same, which added to the faster saturation, implying that the activity per Pd active site would be higher on the PdAu samples. Comparing with previously reported data for a Pd–TiO2 catalyst,29 the initial catalytic activities of the PdAu catalysts would be similar, suggesting again that no electronic effect between Pd and Au takes place on these catalysts. In short, the results clearly unveil that the obtained PdAu and PdZn bimetallic phases present different adsorption capacities of the reactant and products leading to different reactivity.
The catalyst with the best yield to butenes at both temperatures of reaction and at the two temperatures of reduction was the monometallic Pd–ZnO-n sample that presented the β-PdZn alloy and exposed PdZn(100) facets. The XAS results showed a slight increase of the electron density of palladium and greater elongation of the lattice bonds for PdZnO-n compared to Pd–ZnO-t.29 This electronic modification improves the selectivity by decreasing the stability of adsorbed butenes that leads to a decrease in the butane production.56 For the PdAu bimetallic catalysts, this electronic effect was negligible and both catalysts behaved in a similar way, which agrees with the characterization results that confirm the little or no effect of the ZnO support on the properties of these PdAu formed phases. All this could explain the similar activity profile to that of the pure Pd catalysts. Furthermore, FTIR-CO has shown that both PdAu bimetallic samples exposed similar active sites on a Pd-rich surface and preferential Au diffusion on the Pd(111) planes. This evidence contrasts with the theoretical and experimental studies suggesting a ligand effect of the gold atoms surrounding the Pd monomers that lowers the adsorption energies of different molecules.57 In addition, this electronic effect is claimed to depend on the surface, being more pronounced for dense PdAu(111) surfaces.
On the other hand, for Pd catalysts, it is well known that dissolved hydrogen and formation of PdH hydrides, α- and β-phases, influence the product distribution obtained in reactions of hydrogenation.58 We can rule out the formation of these hydride phases under the reaction conditions based on the XAS measurements, which agrees with the XRD results previously obtained over the monometallic catalysts.29 Therefore, it is likely that the formation of the PdZn alloy on the monometallic catalysts and addition of Au on the bimetallic catalysts suppressed the formation of the palladium hydride phases due to dilution of Pd and reduction of the ensemble size. Suppression of the formation of the palladium hydride phases has also been reported for other Pd-based bimetallic systems.48 Despite the absence of the hydride phase in all the catalysts, differences in the concentration of surface hydrogen species or in the diffusion rate from the nanoparticle surface into the subsurface cannot be ruled out to explain the different selectivity results of the catalysts.59,60 Then, a higher surface hydrogen coverage or rate of diffusion favouring total hydrogenation to butane could be inferred for the Pd–ZnO-t and the PdAu bimetallic samples. Notably, the isomerization of 1-butene to cis- and trans-2 butene for both PdAu bimetallic catalysts is similar to that for Pd–ZnO-n and lower than that for Pd–ZnO-t, which reflects that the presence of gold does affect in some way the mechanism of reaction. Slight differences in the butene desorption in preference to its isomerization or in the H diffusion to the sub-surface, limiting the hydrogen available and avoiding reaching a critical value needed to lead to 1-butene isomerization, are again likely.
Conclusions
In summary, we have prepared Pd and PdAu bimetallic catalysts on two morphologically different ZnO supports (ZnO-t and ZnO-n). Independent of the exposed faces of the ZnO support and of the different initial metal–support interactions, both PdAu bimetallic systems showed small Pd-rich particles and big Au-rich core/Pd-rich shell like structures and similar physicochemical properties, as demonstrated by TEM, XPS, XANES and FTIR-CO. These catalysts also behaved similarly in the hydrogenation of 1,3-butadiene. Although the scenario is complicated, the catalytic results observed for Pd–ZnO-t and both PdAu bimetallic catalysts seem to be mainly attributable to a geometric effect due to dilution of Pd atoms at the surface of the particles without great changes in their intrinsic properties, while for Pd–ZnO-n, where a PdZn alloy was formed, apart from the geometric effect, the Pd electronic properties seem to be modified resulting in a better performance in terms of selectivity.It remains to be seen whether exploring different methods of preparation, metal loadings and Pd:Au ratios could bring further insights into how the morphology/facet of the ZnO support affects the formation mechanism as well as the structural and electronic properties of the resultant nanostructures in bimetallic PdAu/ZnO systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ESRF for access to facilities and granting beamtime at BM23. A. I.-J. thanks the CSIC for the PIM-2014-201480I012 project and Fundación Ramón Areces for the supporting funding through the project 20172353 (OTR02666). We thank R. R. Bacsa for providing the ZnO-t support.Notes and references
- G. J. Hutchings, Chem. Commun., 2008, 1148 RSC.
- F. Cardenas-Lizana, S. Gomez-Quero, A. Hugon, L. Delannoy, C. Louis and M. A. Keane, J. Catal., 2009, 262, 235 CrossRef CAS.
- A. Hugon, L. Delannoy, J.-M. Krafft and C. Louis, J. Phys. Chem. C, 2010, 114, 10823 CrossRef CAS.
- T. Ward, L. Delannoy, R. Hahn, S. Kendell, C. J. Pursell, C. Louis and B. D. Chandler, ACS Catal., 2013, 3, 2644 CrossRef CAS.
- G. J. Hutchings and C. J. Kiely, Acc. Chem. Res., 2013, 46, 1759 CrossRef CAS PubMed.
- E. Ntainjua N, J. K. Edwards, A. F. Carley, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 1162 RSC.
- C. M. Olmos, L. E. Chinchilla, E. G. Rodrigues, J. J. Delgado, A. B. Hungría, G. Blanco, M. F. R. Pereira, J. J. M. Órfão, J. J. Calvino and X. Chen, Appl. Catal., B, 2016, 197, 222 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS PubMed.
- C. Wöll, Prog. Surf. Sci., 2007, 82, 55 CrossRef.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- C. G. Van de Walle and A. Janotti, Rep. Prog. Phys., 2009, 72, 126501 CrossRef.
- C. S. Rout, A. R. Raju, A. Govindaraj and C. N. R. Rao, Solid State Commun., 2006, 138, 136–138 CrossRef CAS.
- A. Mclaren, T. Valdes-Solis, G. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540 CrossRef CAS PubMed.
- A. Iglesias-Juez, F. Viñes, O. Lamiel-García, M. Fernández-García and F. Illas, J. Mater. Chem. A, 2015, 3, 8782 RSC.
- T. Conant, A. M. Karim, V. Lebarbier, Y. Wang, F. Girgsdies, R. Schlögl and A. J. Datye, J. Catal., 2008, 257, 64 CrossRef CAS.
- M. Armbrüster, M. Behrens, K. Föttinger, M. Friedrich, É. Gaudry, S. K. Matam and H. R. Sharma, Catal. Rev.: Sci. Eng., 2013, 55, 289 CrossRef.
- M. W. Tew, H. Emerich and J. A. van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457 CrossRef CAS.
- K. Föttinger, Catalysis, 2013, 25, 77 Search PubMed.
- M. B. Boucher, N. Yi, F. Gittleson, B. Zugic, H. Saltsburg and M. Flytzani-Stephanopoulos, J. Phys. Chem. C, 2011, 115, 1261 CrossRef CAS.
- S. A. C. Carabineiro, B. F. Machado, R. R. Bacsa, P. Serp, G. Dražić, J. L. Faria and J. L. Figueiredo, J. Catal., 2010, 273, 191 CrossRef CAS.
- E. Castillejos, R. Bacsa, A. Guerrero-Ruiz, I. Rodríguez-Ramos, L. Datas and P. Serp, Nanoscale, 2011, 3, 929 RSC.
- H. Yu, H. Ming, H. Zhang, H. Li, K. Pan, Y. Liu, F. Wang, J. Gong and Z. Kang, Mater. Chem. Phys., 2012, 137, 113 CrossRef CAS.
- S. Derrouiche, C. La Fontaine, G. Thrimurtulu, S. Casale, L. Delannoy, H. Lauron-Pernota and C. Louis, Catal. Sci. Technol., 2016, 6, 6794 RSC.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054 CrossRef CAS.
- X. Liu, M. Liu, Y. Luo, C. Mou, S. D. Lin, H. Cheng, J. Chen, J. Lee and T. Lin, J. Am. Chem. Soc., 2012, 134, 10251 CrossRef CAS PubMed.
- P. Kast, M. Friedrich, F. Girgsdies, J. Kröhnert, D. Teschner, T. Lunkenbein, M. Behrens and R. Schlögl, Catal. Today, 2016, 260, 21 CrossRef CAS.
- M. P. Hyman, V. M. Lebarbier, Y. Wang, A. K. Datye and J. M. J. Vohs, J. Phys. Chem. C, 2009, 113, 7251 CrossRef CAS.
- H. Zhang, J. Sun, V. L. Dagle, B. Halevi, A. K. Datye and Y. Wang, ACS Catal., 2014, 4, 2379 CrossRef CAS.
- E. Castillejos-López, G. Agostini, M. Di Michel, A. Iglesias-Juez and B. Bachiller-Baeza, ACS Catal., 2017, 7, 796 CrossRef.
- S. Arunkumar, T. Hou, Y.-B. Kim, B. Choi, S. H. Park, S. Jung and D.-W. Lee, Sens. Actuators, B, 2017, 243, 990 CrossRef CAS.
- C. G. Silva, M. J. Sampaio, S. A. C. Carabineiro, J. W. L. Oliveira, D. L. Baptista, R. Bacsa, B. F. Machado, P. Serp, J. L. Figueiredo, A. M. T. Silva and J. L. Faria, J. Catal., 2014, 316, 182 CrossRef CAS.
- Y. Chen, D. Zeng, K. Zhang, A. Lu, L. Wand and D.-L. Peng, Nanoscale, 2014, 6, 874 RSC.
- E. Castillejos, E. Gallegos-Suarez, B. Bachiller Baeza, R. Bacsa, P. Serp, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Catal. Commun., 2012, 22, 79 CrossRef CAS.
- J. Liu, B. Qiao, Y. Song, H. Tang, Y. Huang and J. Liu, J. Energy Chem., 2016, 25, 361 CrossRef.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem. – Eur. J., 2011, 17, 6524 CrossRef CAS PubMed.
- R. R. Bacsa, J. Dexpert-Ghys, M. Verelst, A. Falqui, B. Machado, W. S. Bacsa, P. Chen, S. M. Zakeeruddin, M. Graetzel and P. Serp, Adv. Funct. Mater., 2009, 19, 875 CrossRef CAS.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet and S. Gatla, et al. , J. Synchrotron Radiat., 2015, 22, 1548 CrossRef CAS PubMed.
- G. Agostini, D. Meira, M. Monte, H. Vitoux, A. Iglesias-Juez, M. Fernadez-Garcia, O. Mathon, F. Meunier, G. Berruyer, F. Perrin, S. Pasternak, T. Mairs, S. Pascarelli and B. Gorges, J. Synchrotron Radiat., 2018, 25, 1745 CrossRef CAS PubMed.
- A. A. Herzing, M. Watanabe, J. K. Edwards, M. Conte, Z.-R. Tang, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2008, 138, 337 RSC.
- A. Maeland and T. B. Flanagan, Can. J. Phys., 1964, 42, 2364 CrossRef CAS.
- D. L. Weissman-Wenocur, P. M. Stefan, B. B. Pate, M. L. Shek, I. Lindau and W. E. Spicer, Phys. Rev. B, 1983, 27, 3308 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 3846 CrossRef PubMed.
- W. Stadlmayr, C. Rameshan, C. Weilach, H. Lorenz, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, D. Zemlyanov, S. Penner, R. Schlögl, G. Rupprechter, B. Klötzer and N. Memmel, J. Phys. Chem. C, 2010, 114, 10850 CrossRef CAS.
- Y. Niu, X. Liu, Y. Wang, S. Zhou, Z. Lv, L. Zhang, W. Shi, Y. Li, W. Zhang, D. S. Su and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 1 CrossRef.
- M. M. Pereira, F. B. Noronha and M. Schmal, Catal. Today, 1993, 16, 407 CrossRef CAS.
- P. S. Wehner, G. C. Tustin and B. L. Gustafson, J. Catal., 1984, 88, 246 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, in Handbook of X-Ray Photoelectron Spectroscopy, ed. G. E. Muilenberg, Perkin-Elmer, Eden Prairie, MN, 1979, p. 110 Search PubMed.
- B. Coq and F. Figueras, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, in Handbook of X-Ray Photoelectron Spectroscopy, ed. G. E. Muilenberg, Perkin-Elmer, Eden Prairie, MN, 1979, p. 154 Search PubMed.
- C. R. Henry, Surf. Sci. Rep., 1998, 31, 231 CrossRef CAS.
- E. Ozensoy and D. W. Goodman, Phys. Chem. Chem. Phys., 2004, 6, 3765 RSC.
- E. Groppo, S. Bertarione, F. Rotunno, G. Agostini, D. Scarano, R. Pellegrini, G. Leofanti, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2007, 111, 7021 CrossRef CAS.
- D. J. Childers, N. M. Schweitzer, S. M. K. Shahri, R. M. Rioux, J. T. Miller and R. J. Meyer, Catal. Sci. Technol., 2014, 4, 4366 RSC.
- D. W. Yuan, X. G. Gong and R. Q. Wu, Phys. Rev. B, 2008, 78, 035441 CrossRef.
- M. Neurock and D. Mei, Top. Catal., 2002, 20, 5 CrossRef CAS.
- A. Sarkany, Z. Zsoldos, B. Furlong, J. W. Hightower and L. Guczi, J. Catal., 1993, 141, 566 CrossRef CAS.
- I. Rivalta, G. Mazzone, N. Russo and E. Sicilia, J. Chem. Theory Comput., 2009, 5, 1350 CrossRef CAS PubMed.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS PubMed.
- A. Cooper, B. Bachiller-Baeza, J. A. Anderson, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Catal. Sci. Technol., 2014, 4, 1446 RSC.
- W. Ludwig, A. Savara, R. J. Madix, S. Schauermann and H.-J. Freund, J. Phys. Chem. C, 2012, 116, 3539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI:10.1039/c9cy02395j |
| This journal is © The Royal Society of Chemistry 2020 |