
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The direct synthesis of hydrogen peroxide from H2 and O2 using Pd–Ga and Pd–In catalysts†
Sheng
Wang
ab,
Richard J.
Lewis
 c,
Dmitry E.
Doronkin
ad,
David J.
Morgan
c,
Jan-Dierk
Grunwaldt
ad,
Graham J.
Hutchings
*c and
Silke
Behrens
*ab
c,
Dmitry E.
Doronkin
ad,
David J.
Morgan
c,
Jan-Dierk
Grunwaldt
ad,
Graham J.
Hutchings
*c and
Silke
Behrens
*ab
aInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: Silke.Behrens@kit.eu
bInstitute of Inorganic Chemistry, Ruprecht-Karls University Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: Hutch@Cardiff.ac.uk
dInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of technology (KIT), Engesserstr. 20, 76131 Karlsruhe, Germany
First published on 25th February 2020
Abstract
The direct synthesis of hydrogen peroxide is investigated using PdGa/TiO2 and PdIn/TiO2 catalysts prepared by an acid-washed sol-immobilisation procedure, which allows for good control of particle size. The introduction of both Ga and In into a supported Pd catalyst leads to significantly reduced rates of H2O2 degradation in comparison to the monometallic counterpart. Detailed characterisation reveals that this enhancement is a result of selectively tuning the ratio of Pd oxidation states.
Introduction
Hydrogen peroxide (H2O2) is a powerful, green oxidant that finds use primarily in the textile/paper and pulp industry and in the chemical synthesis sector.1 This is in part attributed to the growing demand for propylene oxide which is used primarily in the production of polyurethane plastics, produced via the integrated HPPO process. In addition, H2O2 finds application in the treatment of waste streams, where it is superseding chlorine containing oxidants, primarily due to increasing environmental legislation.1 In recent years, global H2O2 production has exceeded 3.3 million tons per annum2 and is predicted to rise annually at a rate of approximately 4% with demand forecast to reach 5.2 million tons per annum by 2020.3 This increase in demand in part can be related to the growing application of H2O2 in the chemical synthesis sector; in particular the rise in demand for H2O2 can be related to its use in the production of both propylene oxide, via the integrated HPPO process, and cyclohexanone oxime, which are key intermediates in the production of polyurethane and nylon-6 respectively. Other significant applications of H2O2 are found in, but not limited to, alkene epoxidation,4,5 organo-sulphur oxidation6,7 and ketone oxidation.8,9Currently, the production of H2O2 on an industrial scale is met via the well-established anthraquinone oxidation (AO) process. Although highly efficient the underlying chemistry has changed little since first developed by BASF, utilising an anthraquinone carrier molecule, H2 and O2 with the former reduced over a Pd-based catalyst producing a diol which is oxidised to produce H2O2 and regenerate the anthraquinone. Initially relatively low concentrations of H2O2 (0.8–1.9 wt%) are produced, with this raised through numerous extraction and purification and distillation steps to yield H2O2 solutions in excess of 70 wt% prior to shipping to the end user. As such the AO process is only economically viable on a large scale.10 It should be noted that the typical on-site application of H2O2 requires concentrations in the range of 1–10 wt% with dilution prior to use required. Furthermore, the low stability of H2O2, even at relatively mild temperatures, requires the use of acidic stabilizing agents to prevent its decomposition to water. Although effective in enhancing H2O2 stability the use of such stabilizing agents adds additional costs associated with decreased reactor lifetime and their removal from product streams.1,10
The on-site production of H2O2via the direct combination of H2 and O2 would offer an attractive alternative to the AO process, allowing for production of H2O2 at appropriate concentrations and alleviating the additional costs associated with the shipping and storage of H2O2. The direct synthesis of H2O2 represents a greener, more atom efficient process for H2O2 production that can potentially be adopted at point of use. The use of Pd-based catalysts has received the greatest academic focus, with the first patent filed by Henkel and Weber in 1914.11 However, Pd-based catalysts often suffer from poor selectivity and require the use of acidic or halide stabilizing agents to limit the production of H2O via decomposition and hydrogenation pathways. Building on the work of Landon et al.12 and Haruta and co-workers,13 who simultaneously demonstrated the activity of Au-based catalysts for the direct synthesis of H2O2, numerous studies have since focused on AuPd nanoparticles, which have been demonstrated to offer excellent selectivities towards H2O2 in the absence of acidic or halide stabilizing agents. Indeed, Edwards et al.14 have previously demonstrated that through acidic pre-treatment of a carbon support prior to immobilization of Au and Pd it is possible to reach H2O2 selectivities in excess of 95%, with a similar enhancement subsequently reported when utilizing oxide supports.15,16 More recently, Freakley et al.17 have reported that it is possible to replace Au with cheaper, more abundant base-metals such as Sn, Ni and Co, while maintaining excellent selectivity towards H2O2. Further research has since been placed on the modification of Pd with a range of secondary metals including Sn,18 Ag,19 Zn,20 Ni,21 Sb22 and Te23 in an attempt to enhance catalytic selectivity towards H2O2.
The use of sol-immobilisation procedures to prepare bimetallic catalysts has been extensively studied in the literature and is known to offer excellent control of elemental composition and mean particle size in comparison to more widely used catalyst preparation techniques such as wet-impregnation.24,25 The formation of bimetallic alloyed nanoparticles is well known to offer distinct differences in catalytic performance with electronic and chemical properties that are typically very different from those of the monometallic analogues and often display enhanced selectivity, activity, and stability.26 In particular, the choice of ligand is well known to play a key role in influencing catalytic activity and selectivity.27,28 Therefore, obtaining “clean” particles, while also controlling particle size and composition has become a major challenge in the case of supported catalysts prepared via colloidal methods.29 Instead of removing ligands to cause sinter, here we prepared nanoparticles with same method, same type and amount of ligands to limit the effect on catalytic properties.
In this work, we investigate the catalytic activity of colloidal PdGa and PdIn nanoparticles immobilized on a TiO2 support for the direct synthesis of H2O2 from molecular H2 and O2.
Experimental
Materials
Palladium acetylacetonate (Pd(acac)2; 99%) and indium(III) acetylacetonate (In(acac)3; 98%) were purchased from Alfa Aesar. Gallium(III) acetylacetonate (Ga(acac)3; 99.99%) was obtained from ABCR. Oleylamine (70%, OLAM) and trioctylphosphine (97%, TOP) were purchased from Sigma-Aldrich. TiO2 (P25) was received from Evonik (Degussa). Sulfuric acid (95–97%) was from Merck. Ethanol and chloroform were from Fisher Chemical. All of the solvents were of reagent grade and all reagents were used as-received.Synthesis of unsupported Pd, Ga, In and bimetallic Pd–Ga and Pd–In nanoparticles
Unsupported Pd–Ga and Pd–In nanoparticles, of varying ratio were prepared with the ratio of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M (M = Ga, In) indicated by the nomenclature used, so that the Pd2Ga has a theoretical molar Pd:Ga ratio of 2:1.
:M (M = Ga, In) indicated by the nomenclature used, so that the Pd2Ga has a theoretical molar Pd:Ga ratio of 2:1.
All syntheses were performed using standard Schlenk techniques. In a typical synthesis, Pd(acac)2 and M(acac)3 (M = Ga, In) were dissolved in OLAM (40 mL) in a four-neck flask. The synthesis parameter within this work can be seen in (Table S1†). Pd(acac)3 (0.6 mmol) and M(acac)3 (0.3 mmol) were employed to prepare Pd2M. Pd(acac)3 (0.45 mmol) and M(acac)3 (0.45 mmol) were mixed to prepare Pd1M. Pd(acac)3 (0.3 mmol) and M(acac)3 (0.6 mmol) were used to prepare Pd0.5M. The mixture was flushed with argon, heated from room temperature to 60 °C over a period of 5 min and stirred for 30 min. After adding 2 mL TOP, the mixture was heated to 200 °C (heating rate 8 °C min−1) for 30 min with stirring at 400 rpm. Next the temperature was further increased to 300 °C (heating rate 8 °C min−1) for 30 min. After this the solution was cooled to room temperature, the bimetallic Pd–M nanoparticles were precipitated by addition of ethanol and thoroughly purified by dissolution in CHCl3, precipitation with ethanol, and centrifugation steps.
Pd nanoparticles were prepared using a similar procedure. Pd(acac)2 (0.90 mmol) was dissolved in OLAM (40 mL) in a four-neck flask. The mixture was flushed with argon, then heated to 60 °C quickly and stirred for 30 min. After adding 2 mL TOP, and the mixture was heated to 200 °C (heating rate 9 °C min−1) which was kept for 30 min. After cooling to room temperature, the Pd nanoparticles were purified as outlined above.
Ga nanoparticles were synthesized in a similar procedure as well. Ga(acac)3 (0.90 mmol) was dissolved in OLAM (40 mL) in a four-neck flask. The mixture was flushed with argon, then heated to 60 °C and kept for 30 min. After adding 2 mL TOP, heat the mixture to 330 °C at the rate 9 °C min−1 and stirred for 50 min. After cooling to room temperature, collect the samples by washing with CHCl3 and ethanol.
In nanoparticles were obtained with the typical method. In(acac)3 (0.90 mmol) was dissolved in OLAM (40 mL) in a four-neck flask. The mixture was flushed with argon, then heated to 60 °C and kept for 30 min. After adding 2 mL TOP, the mixture was heated to 200 °C (heating rate 8 °C min−1) which was kept for another 30 min while stirring. Afterwards, the temperature was further increased to 300 °C (heating rate 8 °C min−1) and kept at 300 °C for additional 30 min. After cooling to room temperature, the In nanoparticles were purified as outlined above.
Acid pretreatment of TiO2 support
The treatment of the support prior to metal particle immobilization is based upon our previous investigations into the role of acid washing both oxide15,16 and carbon supports prior to immobilization of precious metals.14 In a typical procedure, TiO2 support (10 g) was stirred in an aqueous solution of H2SO4 (2 wt%, 100 mL) for 3 hours, then filtered and washed with H2SO4 (2 wt%) solution and dried under vacuum, 16 h at 30 °C.Supported catalyst preparation
Metal nanoparticles were immobilized on the pre-treated TiO2 support from the colloidal solution by adding TiO2 (0.4 g) to the appropriate amount of metal nanoparticles in chloroform and stirring for 3 h at 800 rpm. For most catalysts, the colourless supernatant indicated the complete immobilisation of metal nanoparticles. In some cases, where some nanoparticles remained in solution (as indicated by a black coloured supernatant), ethanol (3 mL) was added and the suspension stirred for another 3 h at 800 rpm. The catalysts were recovered by centrifugation, followed by washing with chloroform and ethanol. The final catalysts were dried (30 °C, 3 hours) under vacuum and ground to a fine powder. Table S2† summarizes the total metal loading of the Pd–Ga and Pd–In supported catalysts.Supported catalysts of varying Pd:Ga and Pd:In ratio have been prepared with this ratio indicated by the nomenclature used, so that the Pd2Ga/TiO2 catalyst has a theoretical Pd:Ga molar ratio of 2:1.
Catalyst characterization
Powder X-ray diffraction (XRD) patterns were recorded on a PANalytical X'Pert Pro X-ray diffractometer employing Bragg–Brentano geometry with Cu Kα radiation and a Ni filter. The range between 5 and 120° was measured lasting 16 h. The reflections were compared to reference data reported in the Joint Committee of Powder Diffraction Standards (JCPDS) data base. Pd and Pd–M (M = Ga, In) nanoparticles were precipitated by ethanol from colloidal chloroform solution with centrifugation (7850 rpm, 10 min) and dried at ambient temperature under vacuum. The resulting material was then deposited onto a XRD sample holder under air.Total metal loading and Pd:M (M = Ga, In) ratio were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 725 ICP-OES Spectrometer). PdM nanoparticles were dissolved in aqua regia. To measure the metal loading of supported catalyst, HF and aqua regia (volume ratio of HF:aqua regia = 2:1) were used to dissolve the catalysts.
X-ray photoelectron spectroscopy (XPS) analyses were made on a Kratos Axis Ultra DLD spectrometer. Samples were mounted using double-sided adhesive tape and binding energies were referenced to the C (1s) binding energy of adventitious carbon contamination taken to be 284.8 eV. Monochromatic AlKα radiation was used for all measurements; an analyser pass energy of 160 eV was used for survey scans while 40 eV was employed for detailed regional scans. The intensities of the Pd (3d) and Ga (2p) features were used to derive the Pd(0)/Pd(II) and Ga/Pd surface ratios. The intensities of the Pd (3d) and In (3d) features were used to derive the Pd(0)/Pd(II) and In/Pd surface ratios.
Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. TEM images of metal nanoparticles and high angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDS analysis) of supported metal nanoparticles were carried out with a FEI Tecnai F20 ST TEM (operating voltage 200 kV) which was equipped with a field emission gun and an EDAX EDS X-ray spectrometer (Si (Li) detecting unit, super ultra-thin window, active area 30 mm2, resolution 135 eV at 5.9 keV). For TEM/EDS analysis, a small droplet of the colloidal nanoparticle solution in chloroform or the catalyst powder, accordingly, were deposited on amorphous carbon-coated, 400 mesh Cu grids and air dried.
X-ray absorption spectroscopy (XAS) measurements at Pd K absorption edge were performed at the CAT-ACT beamline30 (CAT experimental station, using Si (311) double crystal monochromator) of the KIT synchrotron (Karlsruhe, Germany) and the P64 beamline (using Si(111) channel-cut QEXAFS monochromator) of the PETRA III at DESY (Hamburg, Germany) in transmission mode using ionization chambers as detectors. Catalyst samples were measured ex situ as powders packed in 3 mm o.d. quartz capillaries (0.02 mm wall thickness). The spectra were normalized and the extended X-ray absorption fine structure spectra (EXAFS) background subtracted using the ATHENA program (IFFEFIT).31 The k1-, k2-, and k3-weighted EXAFS functions were Fourier transformed in the k range of 2.5–12.5 Å−1 and multiplied by a Hanning window with sill size of 1 Å−1. The structural model was based on a Pd metal core (ICSD collection code 52251) and a PdO shell (ICSD collection code 24692). For the Pd2Ga/TiO2 sample, addition of a Ga shell from Pd2Ga (ICSD collection code 409939) model structure improved the fit. The structure refinement was performed using ARTEMIS (IFFEFIT)31 using theoretical backscattering amplitudes and phases calculated by FEFF 6.0.32 The theoretical data were then adjusted to the experimental spectra by a least square method in R-space between 1.0 and 3.2 Å−1. First, the amplitude reduction factors (S02 = 0.78 for CAT-ACT and 0.88 for P64) were calculated using the Pd foil reference spectrum and then the coordination numbers, interatomic distances, energy shift (δE0) and mean square deviation of interatomic distances (σ2) were refined. The absolute misfit between theory and experiment was expressed by ρ.31
Metal leaching during the direct synthesis reaction was quantified using an Agilent 7900 ICP-MS equipped with an I-AS autosampler using a 5-point calibration using certified reference materials from Perkin Elmer and certified internal standard from Agilent. All calibrants were matrix matched.
Catalyst evaluation
Direct synthesis of H2O2
Hydrogen peroxide synthesis activity was evaluated using a Parr Instruments stainless steel autoclave, equipped with PTFE liner, with a nominal volume of 100 ml and a maximum working pressure of 14 MPa. To test each catalyst for H2O2 synthesis, the autoclave was charged with catalyst (0.01 g) and solvent (5.6 g MeOH and 2.9 g H2O). The charged autoclave was then purged three times with 5% H2/CO2 (0.7 MPa) before filling with 5% H2/CO2 to a pressure of 2.9 MPa, followed by the addition of 25% O2/CO2 (1.1 MPa). The temperature was then decreased to 2 °C using a HAAKE K50 bath/circulator using an appropriate coolant (note: during cooling to 2 °C no H2O2 formation or H2 conversion is observed). Once at 2 °C the reaction mixture is stirred (1200 rpm) for 0.5 h. The above reaction parameters represent the optimum conditions we have previously used for the synthesis of H2O2.35 H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.01 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1.Catalytic conversion of H2 and selectivity towards H2O2 were determined using a Varian 3800 GC fitted with TCD and equipped with a Porapak Q column.
H2 conversion (eqn (1)) and H2O2 selectivity (eqn (2)) are defined as follows:
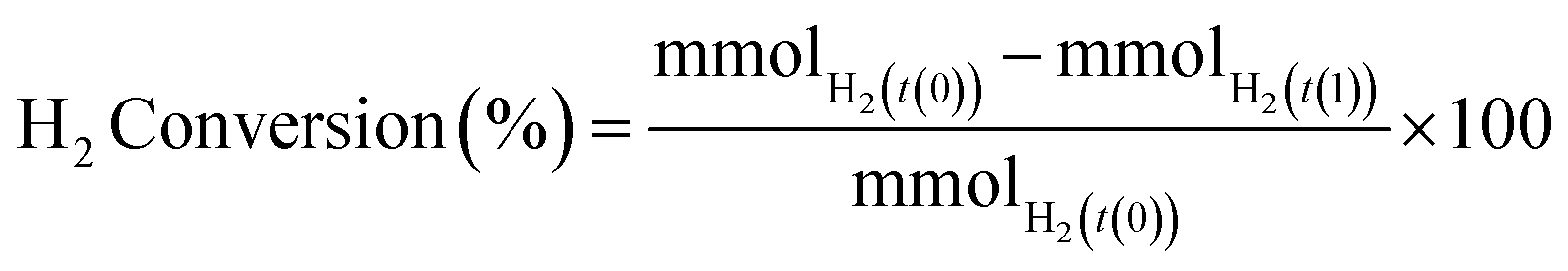 | (1) |
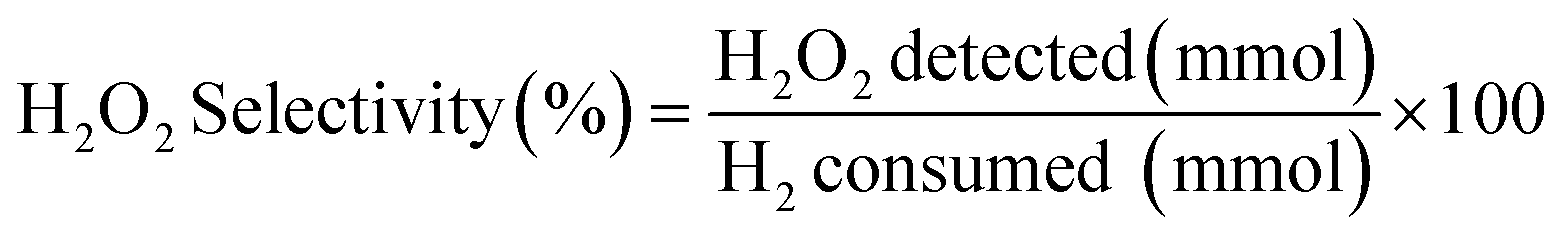 | (2) |
Degradation of H2O2
Catalytic activity towards H2O2 was determined in a manner similar to the direct synthesis activity reaction. The autoclave was charged with MeOH (5.6 g), H2O2 (50 wt% 0.69 g) HPLC standard H2O (2.21 g) and catalyst (0.01 g), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the solution 2 aliquots of 0.05 g were removed and titrated with acidified Ce(SO4)2 solution using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. The autoclave was pressurised with 2.9 MPa 5% H2/CO2, cooled to 2 °C and the reaction mixture was stirred at 1200 rpm for 0.5 h (note: during cooling to 2 °C no degradation of H2O2 is observed). After the reaction was complete, the catalyst was removed from the reaction solvents and as previously two aliquots of 0.05 g were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.Results and discussion
We initially investigated the unsupported monometallic Pd, Ga, In and bimetallic Pd–Ga, Pd–In nanoparticles via XRD (Fig. 1). The XRD diffractograms revealed very broad reflections of low intensity, indicative of small nanoparticles. In particular, a broad, low intensity reflection centred at 33.8° can be observed for the monometallic Ga nanoparticles, with the diffraction pattern in keeping with that of the rhombohedral phase of Ga2O3 (JCPDS 00-043-1013). Further analysis of the monometallic In nanoparticles revealed reflections associated with the In2O3 cubic phase (JCPDS 01-089-4595) and tetragonal In (JCPDS 01-085-1409). Analysis of the alloyed Pd–Ga and Pd–In nanoparticles revealed a single, broad reflection at approximately 40° (2θ) which is consistent with the (111) reflection of the face centred cubic Pd phase, also present in the XRD diffractograms of the analogous monometallic Pd sample. Analysis of the Pd–Ga and Pd–In nanoparticles did not show any reflections characteristic for an ordered intermetallic phase, suggesting the random elemental distribution of these bimetallic nanoparticles. Analysis of the TiO2 supported monometallic Ga and bimetallic Pd–Ga and Pd–In catalysts via XRD (Fig. S1†) does not reveal the presence of the reflections associated with the supported metals, with only reflections associated with the anatase (JCPDS 01-073-1764) and rutile (JCPDS 01-078-1510) phases present in the TiO2 support, indicative of a high dispersion of metal nanoparticles on the support. By comparison reflections associated with In2O3 (JCPDS 00-001-0929) can be observed for the In/TiO2 catalyst, possibly indicating the poor dispersion of nanoparticles on the support in this case. Further analysis of the unsupported Pd and Pd–M (M = Ga, In) nanoparticles via microscopic techniques (Table S3† and Fig. 2) demonstrated a very narrow size distribution and spherical shape of the as prepared metal nanoparticles.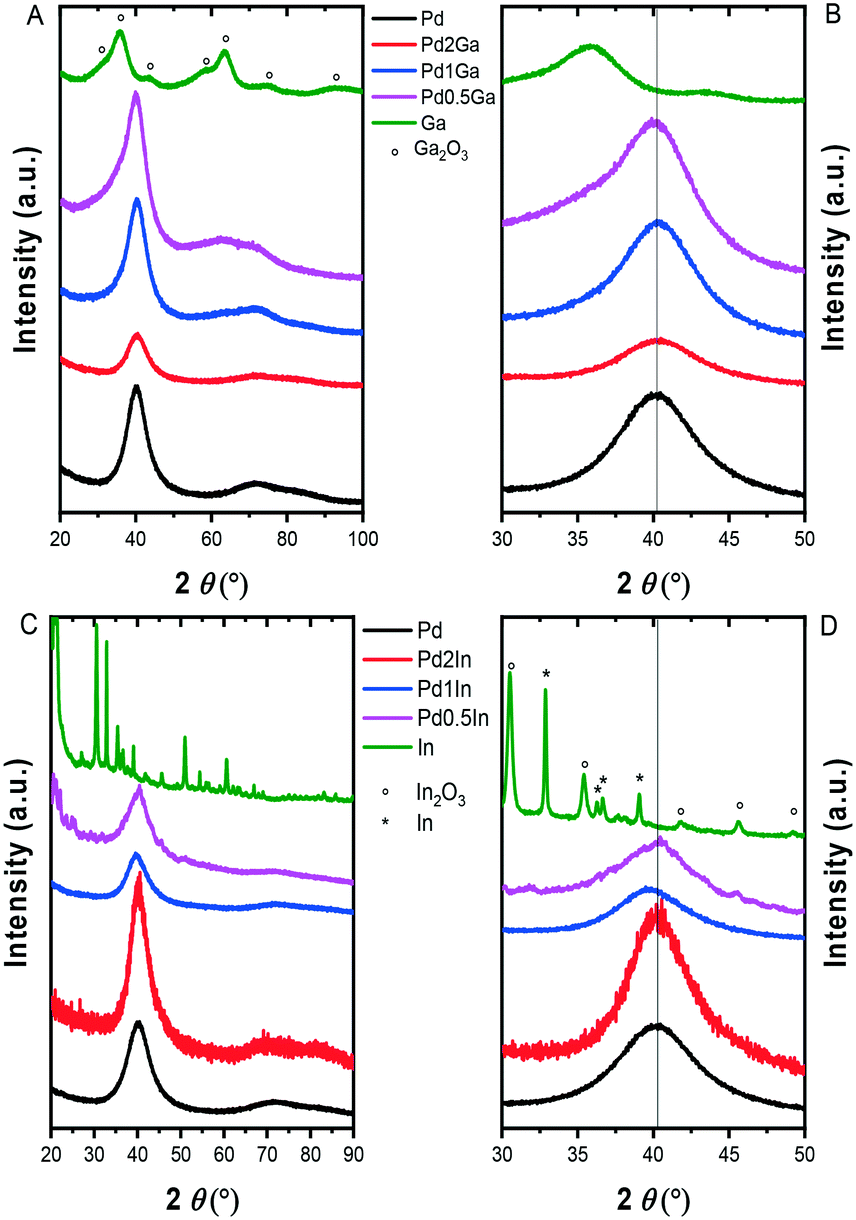 | ||
| Fig. 1 XRD diffractograms of A) unsupported Pd, Pd–Ga and Ga nanoparticles; B) magnified between 30 and 50°; C) unsupported Pd, Pd–In and In nanoparticles; D) magnified diffractograms between 30 and 50°. | ||
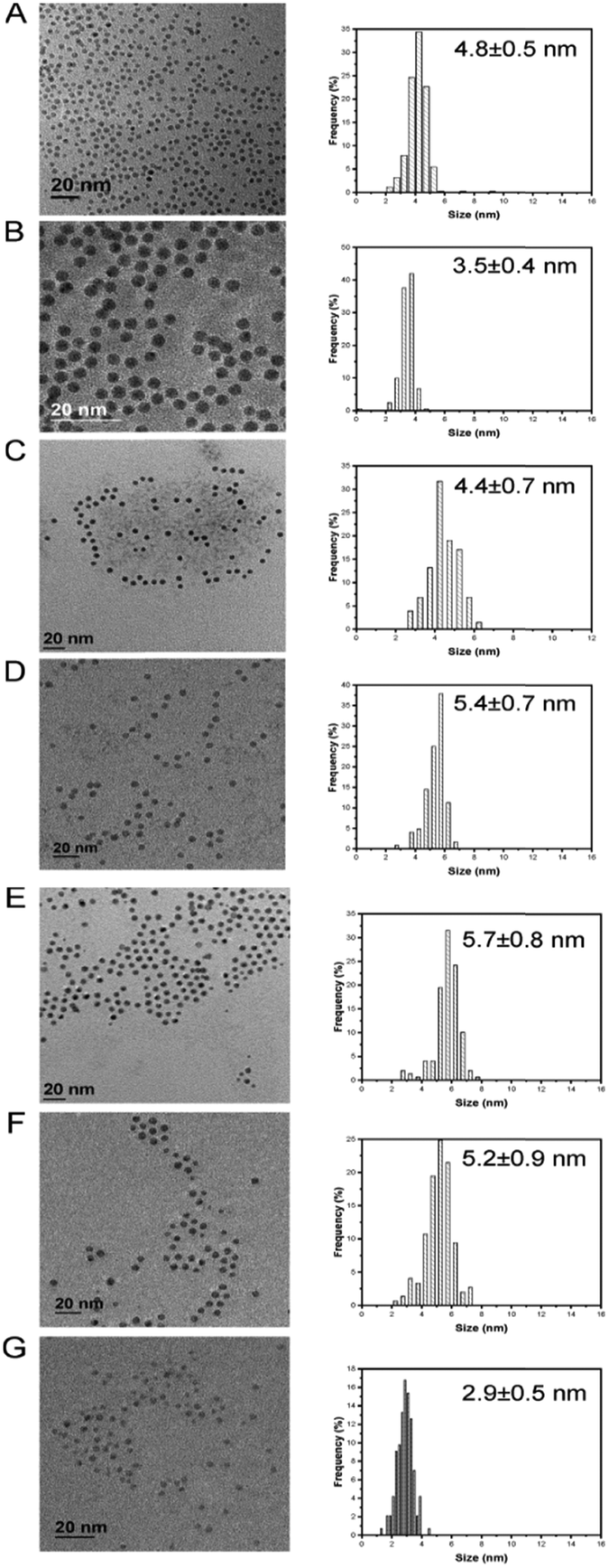 | ||
| Fig. 2 TEM micrographs and particle size distribution: A) Pd; B) Pd2Ga; C) Pd1Ga; D) Pd0.5Ga; E) Pd2In; F) Pd1In; G) Pd0.5In. | ||
Upon immobilization of Pd–M nanoparticles onto the acid-washed TiO2 support, mean particle size of the as-prepared alloyed particles, determined by transmission electron microscopy (TEM), remained generally well controlled, with mean particle size in the range of 1–4 nm (Table S3† and Fig. 3). However, upon immobilisation a relatively minor decrease in mean particle size is observed. Although, in the case of the Pd-only and PdGa/TiO2 catalysts this is not considered to be statistically relevant; in the case of the PdIn/TiO2 series it is possible that this decrease is a result of restructuring of the nanoparticles upon immobilisation. In an attempt to elucidate the extent of restructuring/segregation, if any, EDX analysis of the supported PdIn metal nanoparticles was carried out (Fig. S2–S8†). Unfortunately, the low signal intensities of the metals mean it is not possible to clearly and distinctly observe the extent of surface segregation. As such a relationship between nanoparticle restructuring and enhancement in the catalytic performance cannot be ruled out.
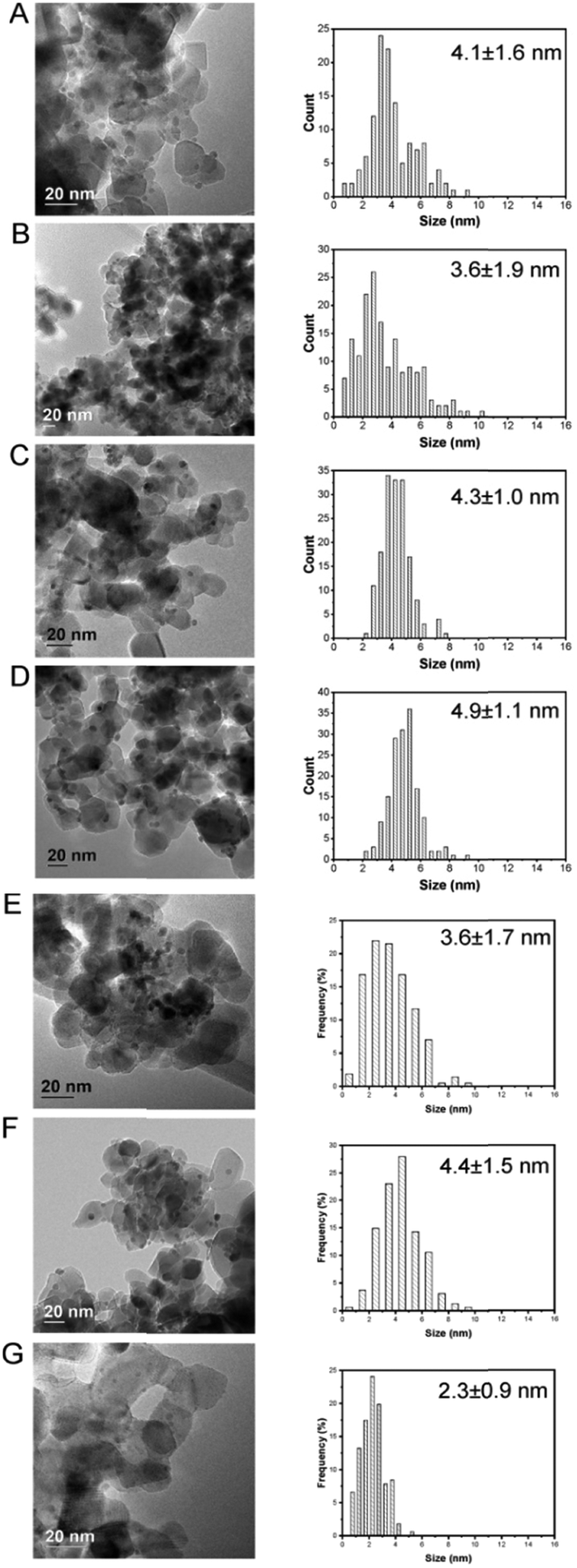 | ||
| Fig. 3 TEM micrographs and particle size distribution: A) Pd/TiO2; B) Pd2Ga/TiO2; C) Pd1Ga/TiO2; D) Pd0.5Ga/TiO2; E) Pd2In/TiO2; F) Pd1In/TiO2; G) Pd0.5In/TiO2. | ||
While ICP-OES (Tables S1 and S2†) was carried out in order to confirm total metal loading and Pd:Ga and Pd:In ratios of nanoparticles and the supported catalysts. The structure and composition of the supported catalysts was further investigated by X-ray absorption spectroscopy (XAS). XANES analysis (Fig. 4) show mixed features attributed to the presence of both PdO (high intensity just above the absorption edge at ca. 24367 eV, the so-called “white line”) and metallic Pd (e.g. a valley at 24367 eV and a peak at approx. 24390 eV). This has previously been observed by Centomo et al.33 and Selinsek et al.34 for Pd-based catalysts investigated for the direct synthesis of H2O2 and attributed to surface oxidation of Pd.34 Analysis of EXAFS spectra (Fig. S9 and Table S4†) reveal the dominance of backscattering from transition metal (mostly Pd) neighbours similar to the spectra of the Pd foil. Hence, our EXAFS analysis indicates the formation of metallic nanoparticles with a structure similar to monometallic Pd. Further analysis confirms the presence of Pd–O neighbours (i.e. partial oxidation of Pd). Average Pd–Pd distances can be used to evaluate structure distortion in the metal nanoparticles. Since all nanoparticles are of similar size and were measured under identical conditions, changes of Pd–Pd distance relative to that in Pd–M could be attributed to the doping of Pd nanoparticles with a second metal and the formation of alloyed nanoparticles. Pd–Pd (In) bond distances of all PdIn/TiO2 catalysts are seen to be slightly higher than that of the monometallic Pd/TiO2 analogue, indicative of the presence of alloyed nanoparticles. Similarly, for Pd2Ga/TiO2 the Pd–Pd (Ga) bond distance is slightly lower than Pd, hence it is likely that nanoparticles exist as mixed metal alloys. It should be noted that the changes in Pd–Pd (Ga) bond length observed for the Pd1Ga/TiO2 and Pd0.5Ga/TiO2 catalysts are not statistically significant.
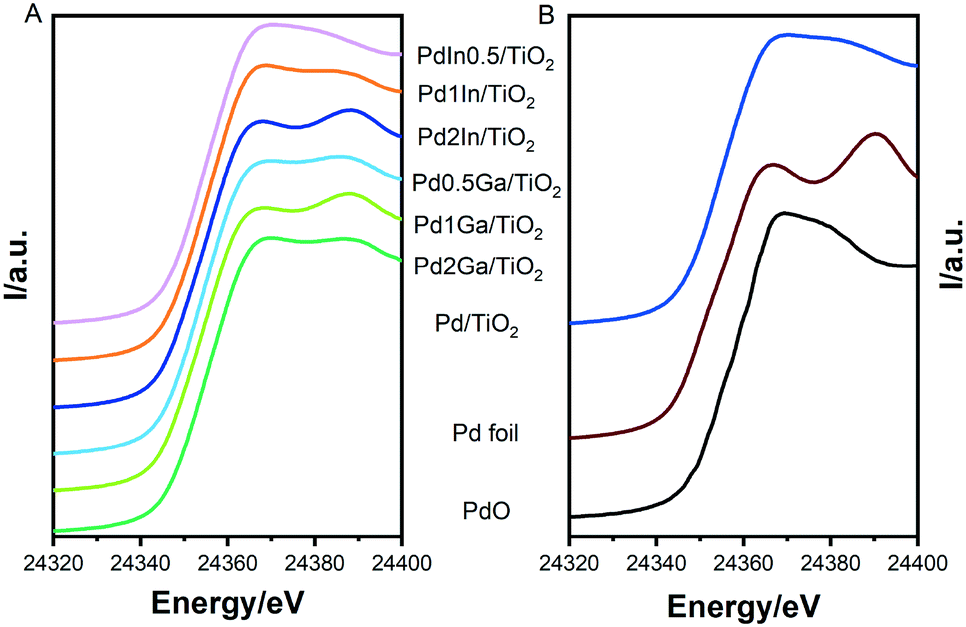 | ||
| Fig. 4 Pd–K edge XANES spectra of A) Pd2Ga/TiO2, Pd1Ga/TiO2, Pd0.5Ga/TiO2, Pd2In/TiO2, Pd1In/TiO2 and Pd0.5In/TiO2; B) Pd/TiO2, metallic Pd, and PdO reference spectra. | ||
Catalyst evaluation for the direct synthesis of H2O2
Our previous work has demonstrated the beneficial effect that a range of secondary metals including Ga and In introduction into a supported Pd catalyst can have on catalytic selectivity towards H2O2 formation.17 Building on these previous studies we have now investigated the Pd–Ga and Pd–In supported catalysts prepared via an acid-washed, sol-immobilisation technique for the direct synthesis of H2O2 and its subsequent degradation (Fig. 5 and Table S5†). It is observed that introducing a small amount of Ga into supported Pd catalysts results in a significant reduction in rates of H2O2 degradation, with a decrease of approximately 35% observed for the Pd2Ga/TiO2 catalyst (695 molH2O2 kgcat−1 h−1) compared to that for the Pd/TiO2 catalyst (1056 molH2O2 kgcat−1 h−1), with a minor increase in H2O2 synthesis rate, from 103 to 111 molH2O2 kgcat−1 h−1, also observed. However, further addition of Ga is observed to significantly decrease catalytic activity towards H2O2 synthesis to 86 molH2O2 kgcat−1 h−1 for the Pd0.5Ga/TiO2 catalyst, with minimal change in H2O2 degradation rates.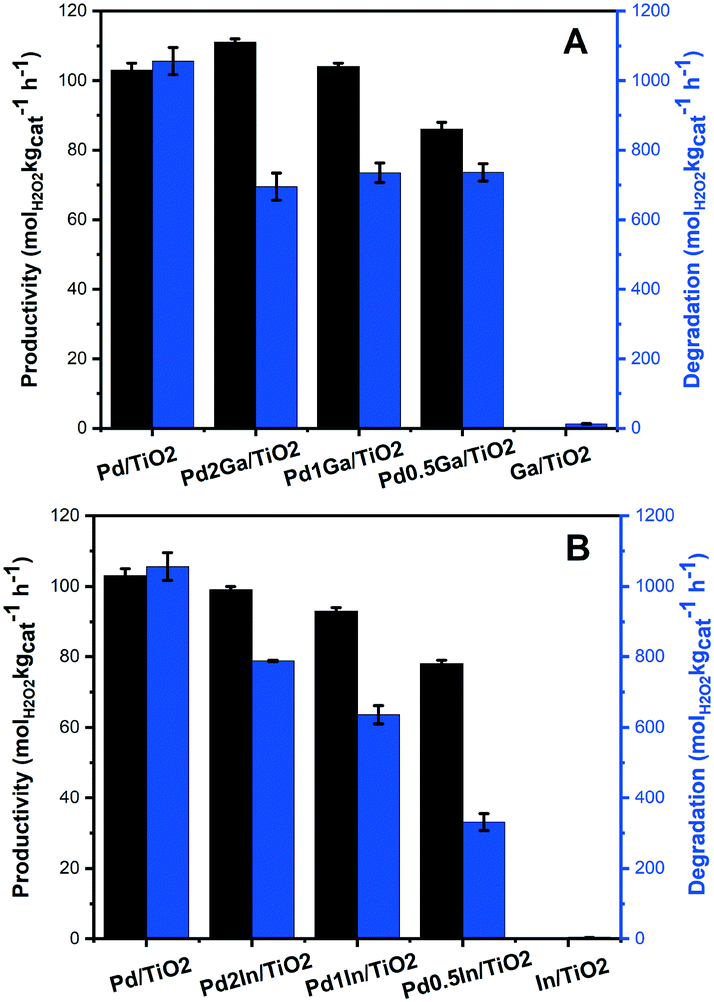 | ||
| Fig. 5 Catalytic activity of TiO2 supported (A) Pd–Ga and (B) Pd–In catalysts towards the direct synthesis and subsequent degradation of H2O2. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. | ||
By comparison incorporation of In into a supported Pd/TiO2 catalyst is seen to result in an inhibition of catalytic activity towards both H2O2 formation and degradation. However, it should be noted that even with significant In incorporation the rate of H2O2 synthesis is still particularly high, with the synthesis activity of the Pd0.5In/TiO2 (78 molH2O2 kgcat−1 h−1) catalyst comparable to that of the analogous Pd0.5Ga/TiO2 catalyst, while displaying significantly lower rates of H2O2 degradation (331 molH2O2 kgcat−1 h−1).
Evaluation of catalytic selectivity towards H2O2 can be seen in Table 1 (full details of reaction parameters shown in Table S6†). The modification of Pd via the addition of a range of secondary, non-precious metals has previously been demonstrated to enhance catalytic selectivity, with a reduction in the amount of contiguous Pd ensemble sites and an enhancement in the number of isolated Pd sites often reported as the cause for the improvement in catalytic selectivity towards H2O2.35 In keeping with these studies, we now report the addition of both Ga and In to a supported Pd/TiO2 catalyst results in an relatively minor enhancement in catalytic selectivity towards H2O2.
For any catalyst operating in a three-phase system the possibility of leaching of the active phase is of great concern, with the low stability of sol-immobilized catalysts well known.36 Indeed previous work by Dissanayake and Lunsford has demonstrated the high activity of homogeneous Pd species in the direct synthesis of H2O2.37 To this end, post reaction solutions were analysed by ICP-AES (Table S7†) with negligible levels of all metals observed.
To further investigate the effect of Ga and In addition to supported Pd catalysts analysis by X-ray photoelectron spectroscopy (XPS) was carried out (Table 2 and Fig. S10†). From our analysis it is clear to see that Pd predominantly exits as a metallic species, which can in part be related to the high rates of H2O2 degradation observed over these catalysts. With the catalytic selectivity of Pd-based catalysts known to be highly dependent on the oxidation state of Pd, with metallic Pd species typically more active towards both H2O2 synthesis and its subsequent degradation than an analogous PdO catalyst38–40 or those of mixed oxidation state.41 Upon introduction of either Ga or In a general rise in PdO content is observed, which in turn can be related to the inhibition of H2O2 degradation activity. This enhancement in PdO content is far more pronounced through.
:Ga and Pd:In ratio
| Catalyst | Pd2+:Pd0 |
Pd:M (M = Ga, In) |
|---|---|---|
|
a The high Pd:Ga ratio observed is ascribed to a combination of the increased Pd content compared to the other PdGa catalysts and the smaller mean nanoparticle size, resulting in greater penetration of the XPS beam.
|
||
| Pd/TiO2 | 0.03 | — |
| Pd2Ga/TiO2 | 0.09 | 14.38a |
| Pd1Ga/TiO2 | 0.17 | 2.38 |
| Pd0.5Ga/TiO2 | 0.03 | 1.18 |
| Pd2In/TiO2 | 0.28 | 2.24 |
| Pd1In/TiO2 | 0.15 | 1.85 |
| Pd0.5In/TiO2 | 0.18 | 1.63 |
In incorporation and may explain the lower activity of the Pd–In catalysts towards both H2O2 synthesis and its subsequent degradation.
Conclusions
In conclusion, we have demonstrated an effective means of producing supported bimetallic Pd–Ga and Pd–In catalysts, with effective control for the particle size. The materials resulting from these preparations were found to offer enhanced selectivity towards H2O2 compared to the analogous Pd catalyst. With the introduction of small quantities of Ga in particular shown to markedly inhibit H2O2 degradation and enhance catalytic selectivity, through modification of Pd oxidation states.We consider that these catalysts represent a promising basis for further exploration for the direct synthesis of H2O2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge Cardiff University electron microscope facility for the transmission electron microscopy. XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’), operated by Cardiff University and UCL, under contract No. PR16195. S. W. acknowledges financial support of the Karlsruhe House of Young Scientists (KHYS) for her research visit to Cardiff University, KIT.Notes and references
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- M.-g. Seo, H. J. Kim, S. S. Han and K.-Y. Lee, Catal. Surv. Asia, 2017, 21, 1–12 CrossRef CAS.
- C. Peng, X.-H. Lu, X.-T. Ma, Y. Shen, C.-C. Wei, J. He, D. Zhou and Q.-H. Xia, J. Mol. Catal. A: Chem., 2016, 423, 393–399 CrossRef CAS.
- R. Fareghi-Alamdari, S. M. Hafshejani, H. Taghiyar, B. Yadollahi and M. R. Farsani, Catal. Commun., 2016, 78, 64–67 CrossRef CAS.
- F. Gregori, I. Nobili, F. Bigi, R. Maggi, G. Predieri and G. Sartori, J. Mol. Catal. A: Chem., 2008, 286, 124–127 CrossRef CAS.
- M. Kirihara, J. Yamamoto, T. Noguchi and Y. Hirai, Tetrahedron Lett., 2009, 50, 1180–1183 CrossRef CAS.
- Z. Cui, H. Chen, M. Zhao and F. J. DiSalvo, Nano Lett., 2016, 16, 2560–2566 CrossRef CAS PubMed.
- X. Li, R. Cao and Q. Lin, Catal. Commun., 2015, 63, 79–83 CrossRef CAS.
- N. M. Wilson, D. T. Bregante, P. Priyadarshini and D. W. Flaherty, Catalysis, 2017, 29, 122–212 CAS.
- H. Henkel and W. Weber, US1108752 A, Henkel AG and Co KGaA, 1914.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- M. Okumura, Y. Kitagawa, K. Yamagcuhi, T. Akita, S. Tsubota and M. Haruta, Chem. Lett., 2003, 32, 822–823 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- J. K. Edwards, S. F. Parker, J. Pritchard, M. Piccinini, S. J. Freakley, Q. He, A. F. Carley, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 812–818 RSC.
- J. K. Edwards, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512–8515 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley and A. Y. Borisevich, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- F. Li, Q. Shao, M. Hu, Y. Chen and X. Huang, ACS Catal., 2018, 8, 3418–3423 CrossRef CAS.
- J. Gu, S. Wang, Z. He, Y. Han and J. Zhang, Catal. Sci. Technol., 2016, 6, 809–817 RSC.
- S. Wang, K. Gao, W. Li and J. Zhang, Appl. Catal., A, 2017, 531, 89–95 CrossRef CAS.
- S. Maity and M. Eswaramoorthy, J. Mater. Chem. A, 2016, 4, 3233–3237 RSC.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y.-F. Han, Chin. J. Catal., 2018, 39, 673–681 CrossRef CAS.
- P. Tian, X. Xu, C. Ao, D. Ding, W. Li, R. Si, W. Tu, J. Xu and Y.-F. Han, ChemSusChem, 2017, 10, 3342–3346 CrossRef CAS PubMed.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards and C. J. Kiely, Langmuir, 2010, 26, 16568–16577 CrossRef CAS PubMed.
- D. I. Sharapa, D. E. Doronkin, F. Studt, J.-D. Grunwaldt and S. Behrens, Adv. Mater., 2019, 31, 1807381 CrossRef PubMed.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- G. M. Lari, B. Puertolas, M. Shahrokhi, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2017, 56, 1775–1779 CrossRef CAS PubMed.
- L. Abis, S. J. Freakley, G. Dodekatos, D. J. Morgan, M. Sankar, N. Dimitratos, Q. He, C. J. Kiely and G. J. Hutchings, ChemCatChem, 2017, 9, 2914–2918 CrossRef CAS.
- Y. Zhao, J. A. Baeza, N. Koteswara Rao, L. Calvo, M. A. Gilarranz, Y. D. Li and L. Lefferts, J. Catal., 2014, 318, 162–169 CrossRef CAS.
- A. Zimina, K. Dardenne, M. Denecke, J. Grunwaldt, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe and R. Steininger, J. Phys.: Conf. Ser., 2016, 712, 012–019 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621 CrossRef CAS.
- P. Centomo, C. Meneghini, S. Sterchele, A. Trapananti, G. Aquilanti and M. Zecca, Catal. Today, 2015, 248, 138–141 CrossRef CAS.
- M. Selinsek, B. J. Deschner, D. E. Doronkin, T. L. Sheppard, J.-D. Grunwaldt and R. Dittmeyer, ACS Catal., 2018, 8, 2546–2557 CrossRef CAS.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 308–317 RSC.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2003, 214, 113–120 CrossRef CAS.
- V. R. Choudhary, A. G. Gaikwad and S. D. Sansare, Catal. Lett., 2002, 83, 235–239 CrossRef CAS.
- A. Gaikwad, S. Sansare and V. Choudhary, J. Mol. Catal. A: Chem., 2002, 181, 143–149 CrossRef CAS.
- G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. Fierro, Chem. Commun., 2004, 1184–1185 RSC.
- L. Ouyang, P.-f. Tian, G.-j. Da, X.-C. Xu, C. Ao, T.-y. Chen, R. Si, J. Xu and Y.-F. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Composition, particle size, catalytic performance, XRD pattern, XAeS, XPS. See DOI: 10.1039/c9cy02210d |
| This journal is © The Royal Society of Chemistry 2020 |