
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalysis, kinetics and mechanisms of organo-iridium enantioselective hydrogenation-reduction
Joseph M.
Mwansa
 and
Michael I.
Page
*
and
Michael I.
Page
*
School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: mike.page@ed.ac.uk
First published on 11th December 2019
Abstract
The synthesis of chiral molecules is of great importance to the pharmaceutical, agrochemical, flavour and fragrance industries. The use of organo-iridium complexes has gained a reputation for its great utility in key enantioselective synthetic procedures. Prime examples include the catalytic reduction of carbonyls and imines; iridium-catalysed allylic substitution and catalysed enantioselective hydrogenation of unsaturated carboxylic acids. Important aspects in these processes are the reaction conditions such as the catalyst loading, metal-ion ligands, the substrate, solvent and the reaction times-all of which can affect the degree of enantioselectivity. Understanding the mechanisms of these hydrogenation/reduction reactions through kinetic and other related studies makes a vital contribution to improving catalytic efficiency.
1. Introduction
Iridium is an extremely rare element, estimated to be 1 ppb of the Earth's crust and found mainly in Alaska and South Africa as two naturally occurring isotopes. It is the densest element. Its electronic configuration is [Xe]4f145d76s2 and, as a result of a large crystal field stabilisation energy associated with the t2g6 configuration, its most important oxidation state is +3 (d6). However, the +1 state (d8), particularly with π-acceptor ligands, and the +4 state (d5) also occur to a significant extent. Most d6 complexes are low spin and octahedral with an effective ionic radius for Ir3+ of 68 pm. The number of ligand atoms bonded to the iridium is indicated by η followed by a superscript indicating that number of ligand atoms. For example, if the cyclopentadienyl anion ligand bonds through all five atoms, they are designated η5-C5H5. The three most important catalyst classes for enantioselective reactions are neutral iridium(I), cationic iridium(I) and iridium(III).Many organo-iridium complexes obey the 18-electron rule and are typically “exchange inert”. In general ligand exchange occurs by dissociative substitution mechanisms, wherein the rate of reaction is determined by the rate of dissociation of a ligand. Conversely, 18-electron compounds can be highly reactive towards electrophiles such as protons and such reactions are associative in mechanism. Complexes that undergo dissociative substitution are often co-ordinatively saturated and have octahedral molecular geometry. The entropy of activation is characteristically positive for these reactions, which indicates that the molecularity of the reacting system increases on going from the initial to the transition state (TS). Analogous to SN1 mechanisms, these dissociative pathways are characterized by a rate determining step that involves release of a ligand from the coordination sphere of the metal undergoing substitution. Consequently, the concentration of the substituting nucleophile has no influence on the rate and an intermediate of reduced coordination number can sometimes be detected. Although the 18-electron rule would suggest non-reactivity in either a stoichiometric or catalytic sense, there are many exceptions. Complexes with fewer than 18 valence electrons tend to show enhanced reactivity.
The composition of enantiomerically enriched products is traditionally expressed as the percent enantiomeric excess ee, defined as:
| % ee = ((R − S) × 100/(R + S)) |
| %R = 50 + ee/2 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10).1 Enantioselective synthesis is usually achieved under conditions of kinetic control so that a prochiral substrate, S, yielding enantiomeric products, PR and PS, is determined by the relative rates of formation of the two products, with rate constants kR and kS, respectively (eqn 1). The product ratio (PR/PS) is given by (eqn (2)) where ΔΔG‡ is the difference in free energies of activation for the formation of PR and PS whereas the relationship between %ee and kR/kS is given in eqn (3).
:10).1 Enantioselective synthesis is usually achieved under conditions of kinetic control so that a prochiral substrate, S, yielding enantiomeric products, PR and PS, is determined by the relative rates of formation of the two products, with rate constants kR and kS, respectively (eqn 1). The product ratio (PR/PS) is given by (eqn (2)) where ΔΔG‡ is the difference in free energies of activation for the formation of PR and PS whereas the relationship between %ee and kR/kS is given in eqn (3).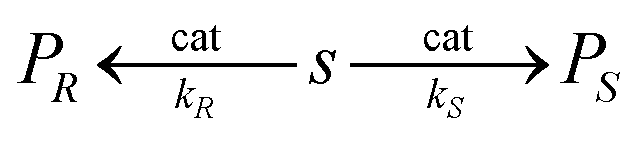 | (1) |
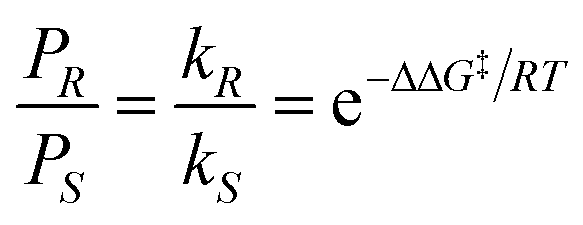 | (2) |
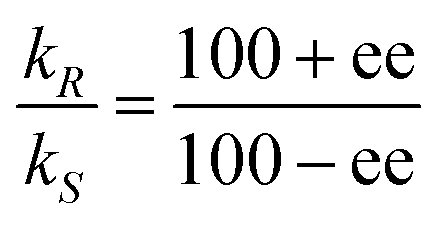 | (3) |
:1 and an ee of 82% (Table 1).
| k R /kS | %ee | ΔΔG‡ (kJ mol−1) |
|---|---|---|
| 2 | 33.3 | 1.7 |
| 5 | 66.6 | 4.0 |
| 10 | 81.8 | 5.7 |
| 25 | 92.3 | 8.0 |
| 50 | 96.1 | 9.7 |
| 100 | 98.0 | 11.4 |
| 1000 | 99.8 | 17.1 |
The changes in concentration of the reactant substrate [A] and enantiomeric products [R] and [S] (eqn (1)) with time are given by eqn (4)–(6).
| [A] = [Ao]e−(kR+kS)t | (4) |
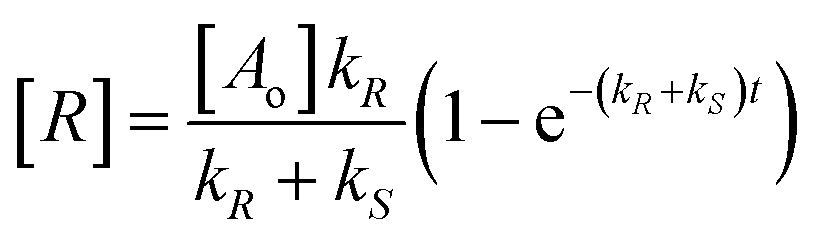 | (5) |
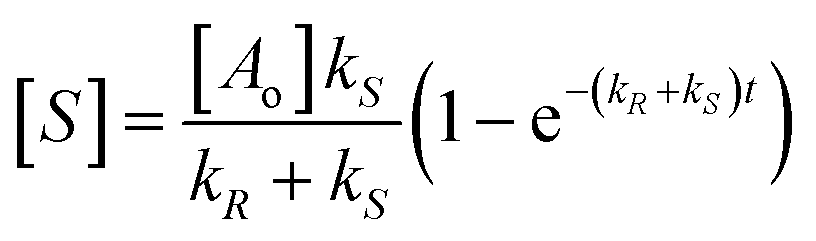 | (6) |
In general, an activated Ir catalyst (Ircat) can react with a substrate S through initial complex formation to give intermediate species after chemical reaction and eventually to generate bound product before product release and catalyst regeneration (Scheme 1).
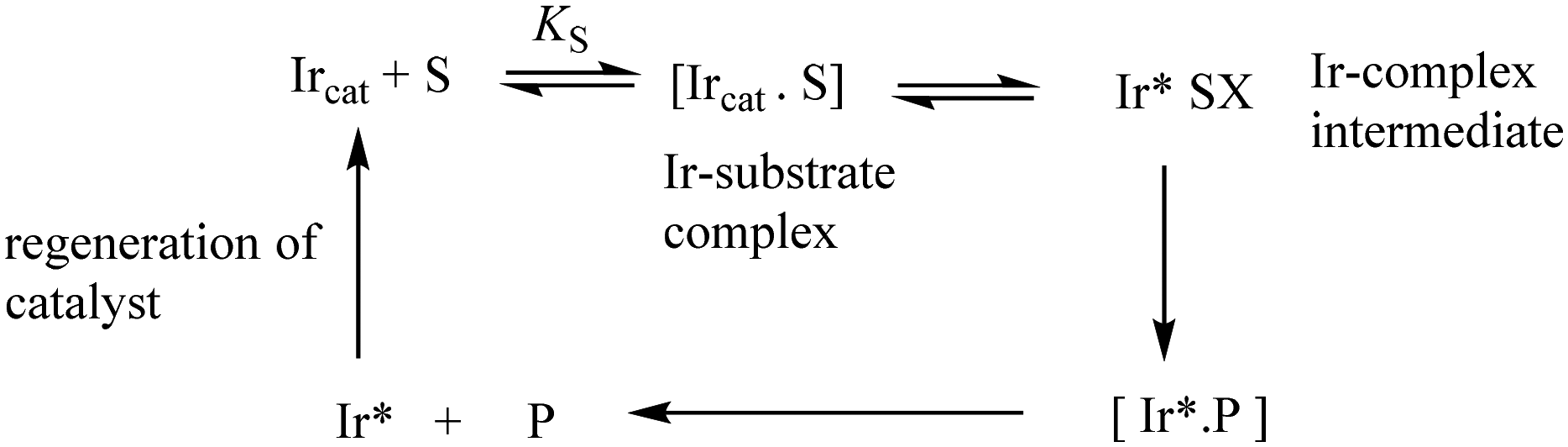 | ||
| Scheme 1 Possible pathway for the iridium catalysed reactions. | ||
The binding of S and intermediates can be very weak or so strong that they accumulate and represent the major Ir species present under the reaction conditions. Consequently, the rate of reaction can show saturation kinetics i.e. zero-order in substrate. A rate independent of substrate concentration can be indicative of a rate-limiting step involving the catalyst bound to substrate, intermediate or product or regeneration of the catalytic species. If coordination is strong and an apparent binding constant can be measured it is important to remember that this constant is the overall binding constant of all Ir-bound species and not necessarily KS (Scheme 1), e.g. the Ir*SX intermediate may accumulate and represent the major Ir species present under steady-state conditions.
2. External factors affecting enantioselectivity
Later sections will emphasise the importance of the catalytic iridium structure, particularly the ligands and oxidation state of the metal-ion, and the nature of the substrate that influence reactivity and selectivity, but here other external factors are briefly discussed.In general, for a catalysed conversion of an achiral substrate into a chiral product, any enantioselectivity results from the differences in the free energies of the transition states of the two enantiomers being formed (eqn (2)). Changing the solvent does not change this conclusion, although relative reactivities in the rates of formation of the products in the two solvents can sometimes result from changes in the free energies of the achiral reactants and catalyst in the two solvents as well as transition state effects.2 For example, good reactivities and selectivities in the Ir(I) catalysed reduction of imines with hydrogen are often favoured in dichloromethane and toluene.3 Solvents may interact directly with the catalyst, substrate and product which can therefore increase or decrease reactivity and selectivity.4 There are many solvent properties which can influence these parameters and include polarity, hydrogen-bond donating and accepting ability, basicity and nucleophilicity.5 Interaction or coordination of the solvent with the catalyst can influence catalytic stability, activity and selectivity. The degree of ion-pairing between a charged catalyst and counter-ion charged substrate can be greatly affected by the nature of the solvent and hence catalytic activity. Similarly, a solvent poor at solvating ions can have an impact on ion-pairing between a positively charged catalyst and its counter-anion and so influence catalytic activity. This can be adverse if the counter-ion blocks the active site of the catalyst by blocking the vacant coordination site where the substrate may bind.6 If there is an increase in polarisation – charge density – of the transition state compared with the reactant state, including the catalyst, then using a more polar solvent will increase the rate of reaction. Conversely, a less polar solvent will stabilise the transition state if its polarisation is less than that of the reactants, thus increasing the rate of reaction.7 A solvent that preferentially stabilizes one of the enantiomeric transition states controlling selectivity should enhance the enantioselectivity of the product i.e. changes the ratio kR/kS.
Several reactions using iridium catalysts involve reduction with gaseous hydrogen and the mass transfer of gases can sometimes be affected by the solvent viscosity leading to a reduction in the rate of reaction or even changes in selectivity. For example, the rate of hydrogenation of imines by a cyclometallated iridium complex containing an imino ligand increases linearly with hydrogen pressure but is zero-order in imine concentration compatible with Ir-hydride formation rather than hydride transfer to the imine.8 Highly viscous solvents generally have low rates of mass transfer of gases which can affect both rate and selectivity.9
Changing the temperature generally has only a small effect on enantioselectivity.10,11 A difference in activation energies for formation of two enantiomeric products of 12 kJ mol−1 requires a temperature change of 50 °C to give a difference in their rates of formation, kR/kS, of 2.
The role of various additives has been explored including changing the anion associated with positively charged catalysts. Other common additives include bases, acids and metal-ions. Many iridium catalyzed reactions involve nucleophilic attack on an unsaturated centre and the addition of electrophiles such as a proton or metal-ion may facilitate this by coordination to the unsaturated substrate (Scheme 2). The proton is more effective than even multiply charged metal-ions. The reactivity and selectivity of the hydrogenation of the imine N-(1-phenylethylidene)benzylamine using the chiral Ir catalyst, [Cp*IrCl{(S,S)-Tscydn}], is increased by the presence of silver salts. An excess of AgSbF6 increased enantioselectivity by up to 72% ee which is attributed to coordination of Ag+ to the imine nitrogen facilitating hydride transfer from Ir–H.12
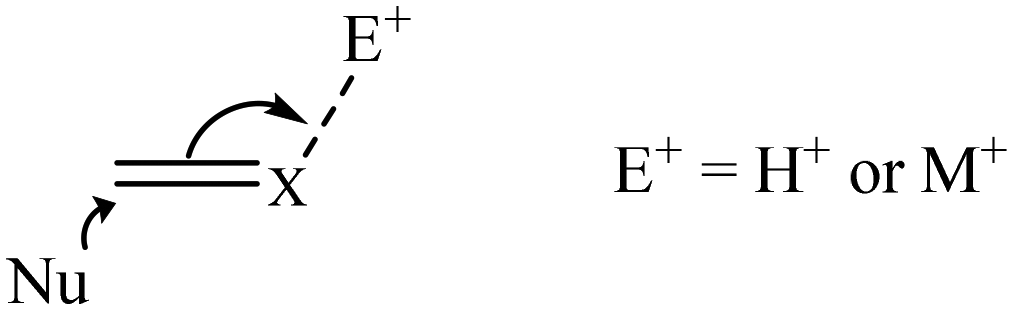 | ||
| Scheme 2 Nucleophilic attack on an activated prochiral substrate. | ||
3. Historical background
Homogeneous iridium catalysis had a slow start mainly because it apparently displayed no special properties compared with other platinum metals. Vaska13 prepared several iridium species (Scheme 3) in which co-ordinatively unsaturated Ir(I) 1a reacted with a variety of A–B molecules to give the corresponding oxidative addition octahedral Ir(III) products 1b. This formally represents a two electron transfer from Ir to A–B and a change in oxidation state of the metal-ion and so is referred to as oxidative addition. The importance of these new complexes centres on the addition of small ligands and serves as the platform for homogeneous catalysis.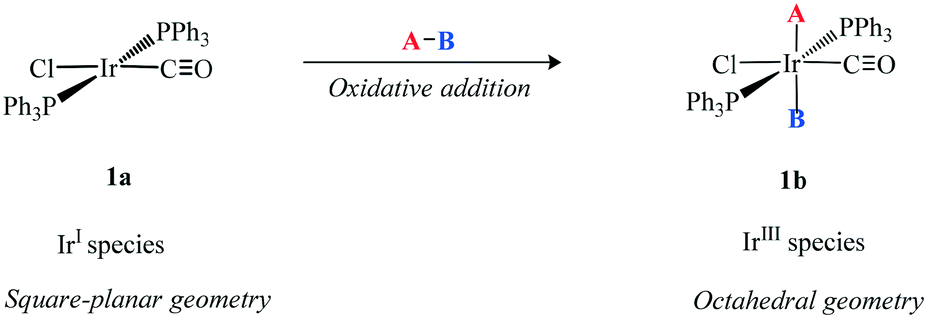 | ||
| Scheme 3 Oxidative addition of A–B to Ir(I) (1a) species to generate the octahedral Ir(III) species (1b). | ||
The ability of a transition metal compound to activate H–H bonds at ambient temperature was fundamental to later discoveries of catalysts for the homogeneous hydrogenation of unsaturated organic substrates.14 The isolable oxidative product Ir(III)H2Cl(PPh3)3 is extremely stable whereas relatively unstable intermediates are required for any successful catalytic protocol. The dissociation of a ligand to generate a vacant coordination site for the substrate or reactant to bind is often a pre-requisite for a catalytic cycle. The main reason for the lack of dissociation of the phosphine ligand in the iridium complex compared with rhodium is due to the increase in bond strength on going from the second to third row of the transition metals. This factor has been one of the disadvantages of using iridium in catalytic processes.
Due to the isoelectronic nature of iridium and rhodium advances in either area have often been transferred and tested on the respective counterpart. The discovery by Schrock and Osborn that 2 could be converted in situ to 3 demonstrated that dissociation of the ligand was not restricted to phosphine ligands.15 This discovery not only paved the way for Crabtree's catalyst 4 but also offered the possibility of incorporating other chelating ligands including chiral ones (Scheme 4).16
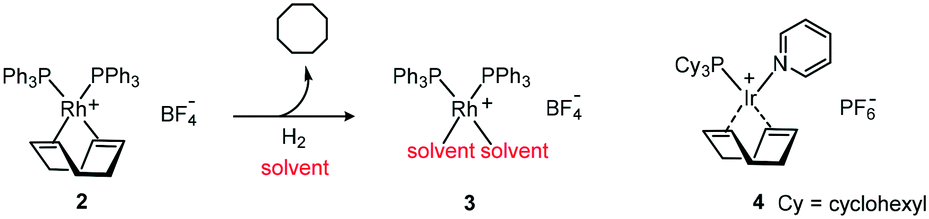 | ||
| Scheme 4 COD displacement by solvent molecules. | ||
After the discovery that coordinating solvents such as acetone, ethanol and THF resulted in complexes [M(cod)L2] ClO4 (cod = 1,5 cyclooctadiene) which reacted with hydrogen to obtain isolable complexes, Crabtree reported enhanced catalytic activity of a series of [M(cod)L(py)]PF6 (M = Rh(I), Ir(I); py = pyridine) complexes which dissolve readily in dichloromethane. The new complex 4 exhibited enhanced activity in the hydrogenation of even tri- and tetra-substituted alkenes. Whereas the coordinating solvent stabilises the iridium complexes,17 it was demonstrated that only the alkene can stabilise the catalyst by coordination with the rhodium analogue. The higher catalytic activity indicated the critical role of the solvent in homogeneous catalysis.18
The hydrogenation of steroids such as 5 with [Ir(cod)L(py)]PF64 (Scheme 5) gave yields ranging from 70–100% and the best rates were obtained by bubbling hydrogen through the solution rather than by working under one atmosphere of hydrogen. However, steroids bearing an alcohol functionality deactivated the catalyst and so protection of this moiety was required.19
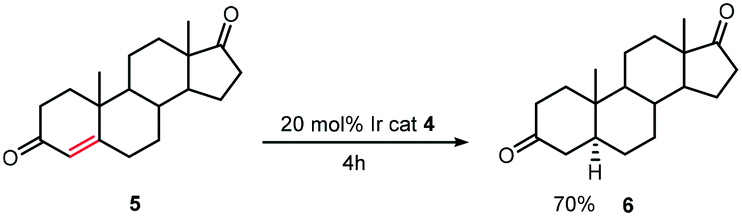 | ||
| Scheme 5 Hydrogenation of steroid 5 with [Ir(cod)L(py)]PF6. | ||
The coordination of the hydroxyl group can exert a stereocontrolling effect in Ir catalyzed reactions.20 The hydrogenation of indenones with a β-OH or carbonyl gave exclusively cis-indanone, whereas the α-derivative in DCM gave the trans-indanone (trans/cis 96:4).
The ability of the alcohol to control the stereogenicity of hydrogenation21 reaches an impressive 1000:1 ratio for the isomeric product alkanes from the reduction of terpinen-4-ol 7 (Scheme 6). The proposed intermediate complex was not observed but the more stable, but poorer catalytic, derivative Ir(I)(PPh3)2+, binds to the hindered endo-5-norbornen-2-ol 10 as shown by NMR due to the sensitivity of the Ir–H chemical shift to the nature of the trans ligand (Scheme 7).22
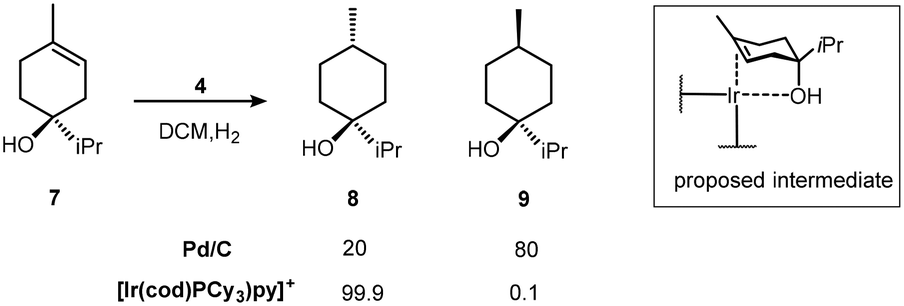 | ||
| Scheme 6 Terpinen-4-ol hydrogenation with Ir catalyst 4. | ||
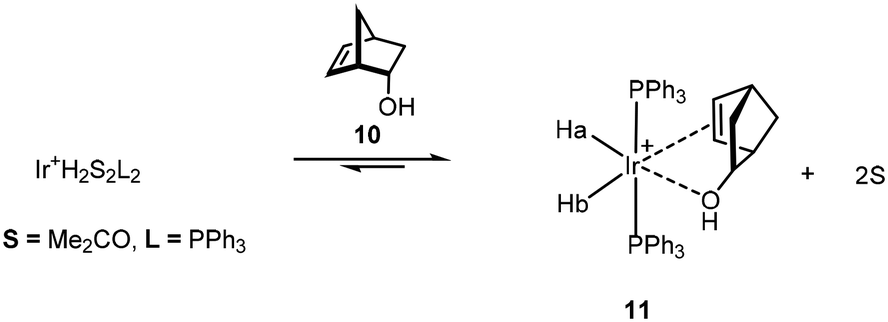 | ||
| Scheme 7 Proposed structure for the stable intermediate 11. | ||
Application of this new coordination possibility to increase selectivity was used in the synthesis of (+)-pumiliotoxin C. One of the key transformations during this synthesis (Scheme 8) was the transformation of 12, the intermediate product 13 was obtained in quantitative yield with high selectivity.23 A ligating group, particularly OH, in alkenes can thus bind to the catalyst and so direct addition of H2 from that face of the molecule which contains the ligating group. Furthermore, the OH also protects the catalyst from deactivation.
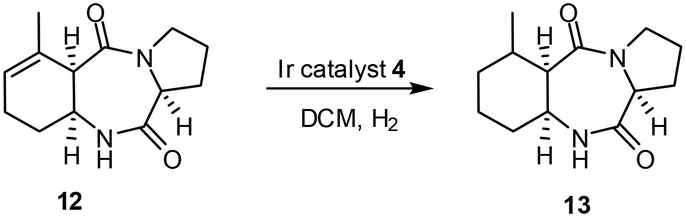 | ||
| Scheme 8 Hydrogenation of 12, a key intermediate in the synthesis of (+)-pumiliotoxin C. | ||
4. Ligand development
In general iridium catalytic species can be divided into cationic(I), neutral(I) and Iridium(III) clusters, each displaying different mechanisms in their ability to transform prochiral substrates into their respective chiral counterparts.One of the prominent examples in ligand development was the introduction of a novel BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) ligand 14 with a ruthenium complex (Scheme 9) which exhibited high reactivity and excellent selectivity for allylic and homoallylic alcohols and showed the dependency of these on the relative position of the hydroxyl group and alkene. Further developments of this catalytic protocol went on to earn Noyori the Nobel prize in 2001.24,25
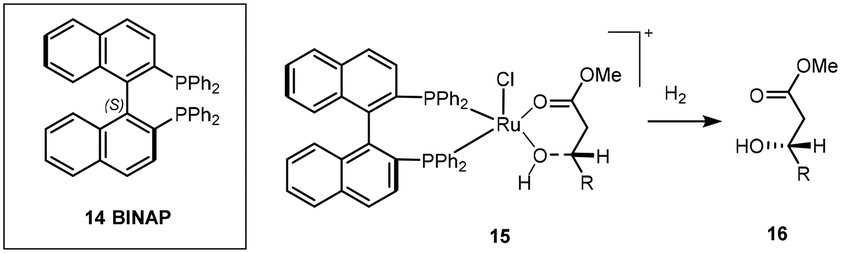 | ||
| Scheme 9 Stereo-directing binding effect of BINAP for Ru dihydride reduction to afford chiral product 16. | ||
The discovery of the chloroacetanilide (S)-metolachlor, one of the most important herbicides for use in maize production initiated the quest to find a viable enantioselective catalyst as 95% of the herbicidal activity of metolachlor resides in the two (1′S)-diastereoisomers. Although rhodium was initially recruited as the metal, the maximum ee of 69% for this process was not viable on an industrial scale and so attention turned to using iridium with a BDPP ligand giving 84% ee. A disadvantage was that the catalyst suffered from active complex dimerization 19, an irreversible process (Scheme 10).26
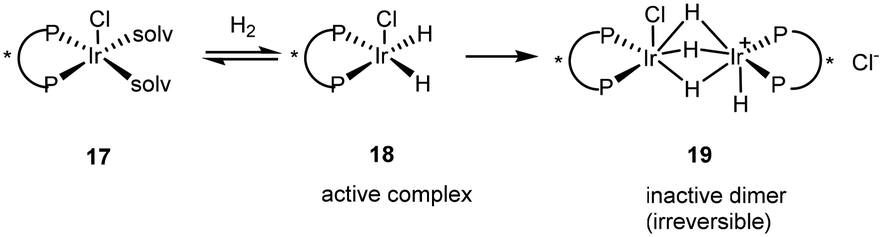 | ||
| Scheme 10 Catalyst deactivation via active complex 18 dimerisation. | ||
The enantioselective hydrogenation of 2-methyl-1,2,3,4-tetrahydroquinoxaline (MeQ) using the ortho-metallated dihydride complex fac-exo-(R) 22 as the catalyst precursor increased ee using methanol as the solvent rather than usual DCM.27 The two imines are hydrogenated at similar rates. The sixteen electron intermediate 23 (Scheme 11), is thought to be the active catalytic species, coordinating the substrate imine, allowing hydride transfer to give an anionic amine species, followed by hydrogen oxidative addition and reductive elimination of the product amine to complete the cycle with regeneration of the catalyst.
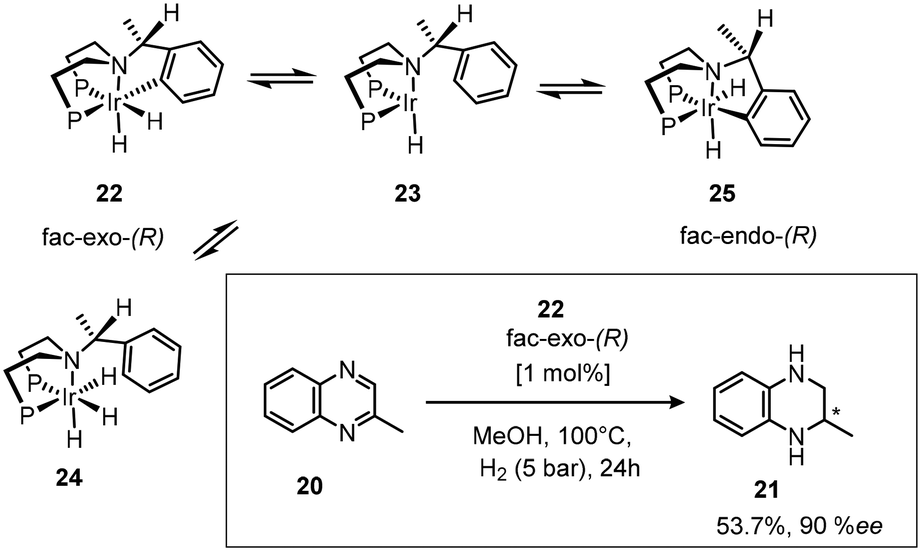 | ||
| Scheme 11 ortho-Metallated dihydride complex catalytic species. | ||
The introduction of a chiral phosphane ligand, (R,R)-f-binaphane 39, modified by a ferrocene backbone, altered the electronic and steric properties of the resultant catalyst (Scheme 12).28 The low rotation barrier of the ferrocene backbone offers flexibility which can facilitate binding of sterically demanding imines to the Ir centre, and more importantly the strong electron back-donating ability from an Ir complex to an imine substrate is increased by the electron-donating f-binaphane ligand with a large P–Ir–P bite angle. A significant change in the enantioselectivities was observed in the presence of I2.
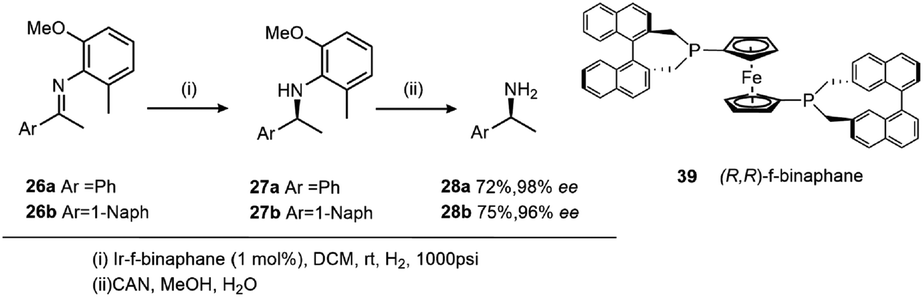 | ||
| Scheme 12 Asymmetric hydrogenation of 26 using 39. | ||
4.1 Ligands for neutral iridium(I) catalysts
The following section will provide a brief coverage of neutral Ir(I) complexes as one of the key catalytic species in transfer hydrogenation of various substrates. Bis(1,5-cyclooctadiene) (COD) diiridium(I) dichloride 30 serves as the main precursor and the ancillary labile COD ligands allow for complex architecture to be introduced around the metal centre by various ligands (Scheme 13, 31–48); providing rich chiral environments for hydrogenation processes.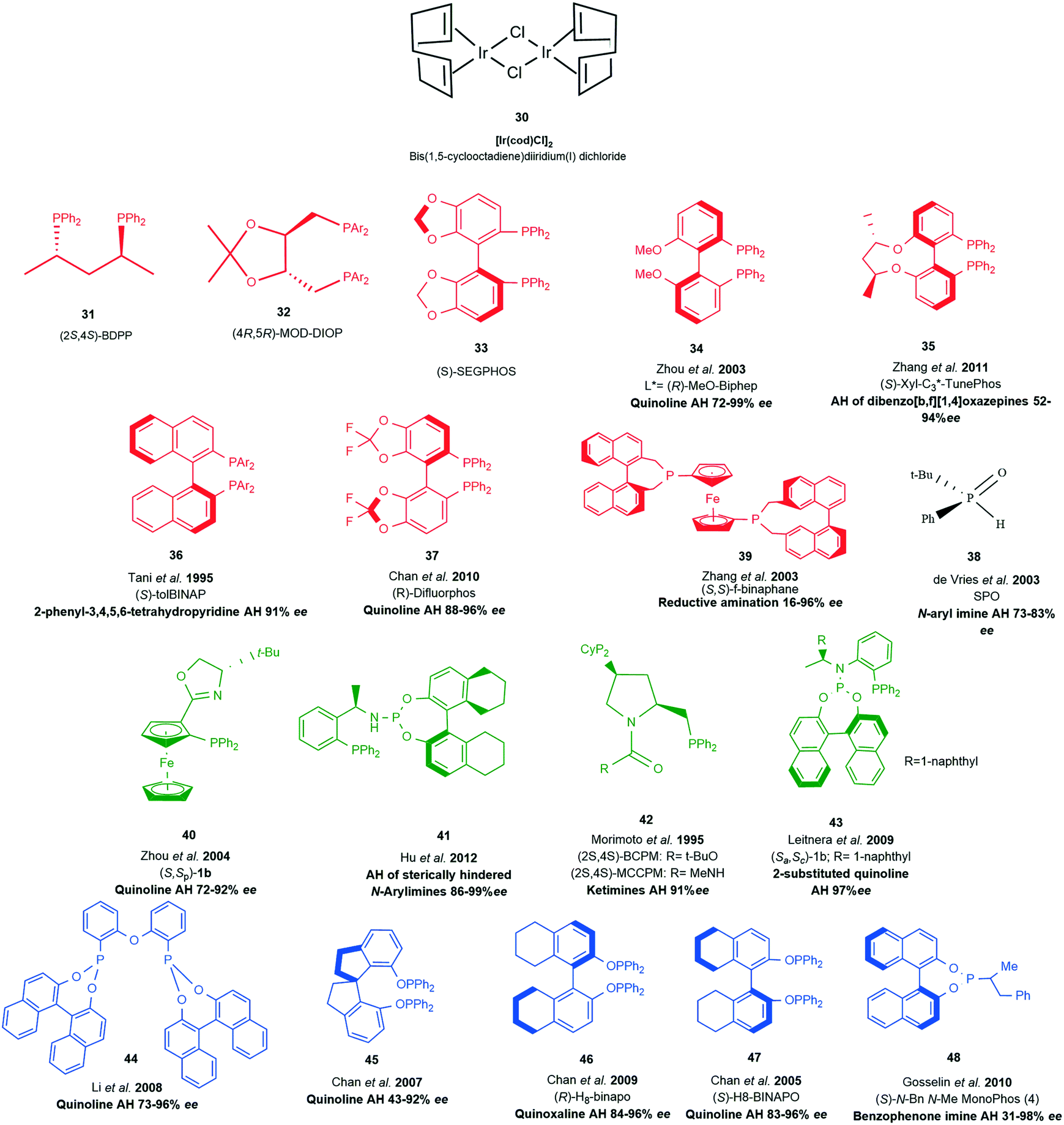 | ||
| Scheme 13 Ligand examples with [Ir(cod)Cl2]2 serving as the precursor. RED (diphosphino ligands), BLACK (phosphino oxide) GREEN (phosphine phosphoramidite P–N ligands), BLUE (diphosphonite ligands). | ||
The introduction of chiral phosphine ligands had a tremendous impact on iridium catalysed reactions of industrial interest. A series of diphosphinoiridium complexes26 for the enantioselective hydrogenation of N-aryl ketimines were synthesised and among this crop of ligands BDPP (2S,4S)-2,4-bis(diphenylphosphino)pentane 31 (Scheme 13) gave rise to highly reactive complexes capable of good activity and enantioselectivities due to the conformationally flexible six- or seven-membered metallacycle (Scheme 14).
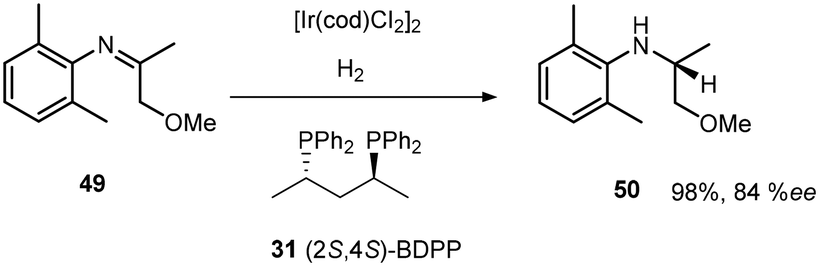 | ||
| Scheme 14 Chiral diphosphine ligand utility in the synthesis of 50. | ||
The asymmetric hydrogenation of ketimines by a neutral Ir(I) complex of (4R,5R)-MOD-DIOP((4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-diyl) bis(methylene)bis(bis(3,5-dimethylphenyl)phosphine) 32, a derivative of the original DIOP ligand produced an efficient reduction of 2,3,3-trimethylindolenine 51 (Scheme 15). The introduction of p-methoxy and m′m-dimethyl groups into the phenyl groups of biphosphine ligands improved the enantioselectivity of this Ir(I) complex to 81.4% ee.29
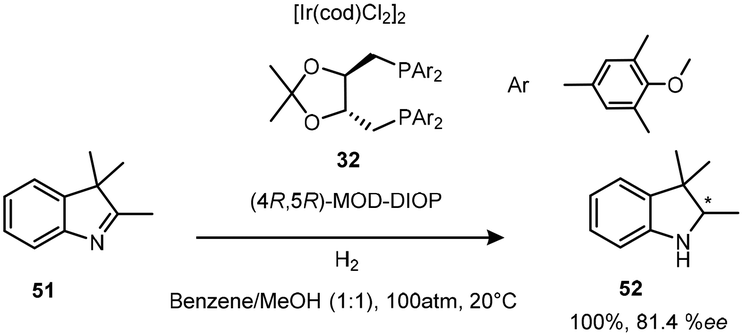 | ||
| Scheme 15 Hydrogenation of 2,3,3-trimethylindolenine with the newly developed ligand 32. | ||
The development of Josiphos, extremely modular and tunable chiral ferrocenyl diphosphine ligands, provided very efficient ligands for several commercial applications.30,31 Xyliphos 53, (Scheme 16) was capable of full conversion of the substrate 49 in 4 h with 79% ee. This hydrogenation not only represents the largest enantioselective catalytic process in use in industry, but also presented one of the fastest homogeneous systems known, second only to certain Ziegler–Natta polymerization catalysts.32
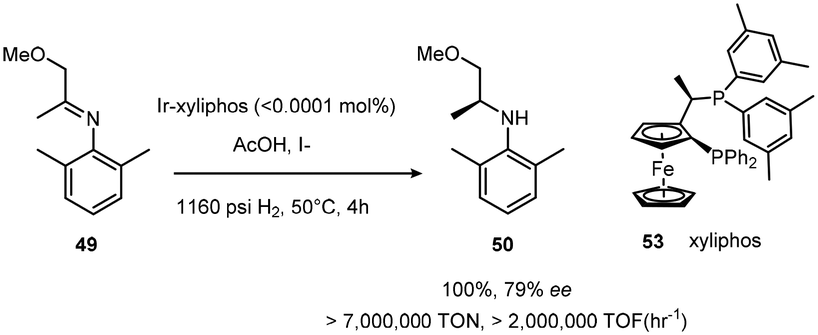 | ||
| Scheme 16 Hydrogenation of 49 with 53 resulting in enhanced conversion capability. | ||
Some heteroaromatic compounds with resonance stability that retards reduction and also contain nitrogen and sulfur atoms capable of poisoning catalytic species are a challenge to asymmetric transfer hydrogenation (ATH). Employing choloroformate as an activating agent partially disrupts aromaticity by the formation of quinolinium and isoquinolinium salts so that the ATH of quinolines and isoquinolines could be achieved with 83 and 90% ee's respectively.33 Ir(I) complexes with BCPM and MCCPM 42 (Scheme 13) in the presence of tetrabutylammonium iodide or bismuth(III) iodide were applied in the reduction of 2,3,3-trimethylindolenine and other cyclic imines with impressive enantioselectivities up to 91% ee and conversions of around 92%.34 New phosphine–phosphoramidite ligands containing two elements of chirality were among a new crop of ligands introduced, 43 proved to be very efficient for the ATH of 2-substituted quinolones with 99% yields and 97% ee.
In an effort to improve access to sterically hindered chiral amines a family of unsymmetrical hybrid phosphine phosphoramidite ligands 41 where developed.35 Modification of the binaphthyl backbone to further improve the overall catalytic performance by increasing steric hindrance on the amino moiety and chiral carbon centre of this ligand resulted in high turnover numbers (up to 100000) and good enantioselectivities (up to 99% ee) for the hydrogenation of a variety of sterically hindered N-arylimines. These results indicated that the presence of an N–H proton on the amino moiety and a H8-binaphthyl moiety on this ligand is crucial for achieving high catalytic activity and enantioselectivity. The utility of this catalytic system was demonstrated by the synthesis of the chiral herbicide (1S)-metolachlor at a catalyst loading of 0.001 mol%.
(R)-MeO-Biphep 34 represented the first example of a highly enantioselective iridium/phosphine/I2 catalytic system for the hydrogenation of quinolines; an efficient methodology granting access to a variety of optically active tetrahydroquinolines with up to 96% ee. This methodology was successfully applied to asymmetric synthesis of tetrahydroquinoline alkaloids providing examples of access to naturally occurring tetrahydroquinoline alkaloids angustureine, galipinine and cuspareine in high yields.36
Following the highly enantioselective catalytic asymmetric hydrogenation of quinolines with MeO-Biphep/I2 as the catalyst system, ferrocene derived phosphino-oxazoline ligands 40 gave up to 92% ee's. The chiral N,P ligands of ferrocenyl–oxazolines derived from different amino alcohols show varied effectiveness for the enantioselectivity with absolute configuration of the product being mainly controlled by the central chirality of the oxazoline ring.37
A highly modular and inexpensive chiral phosphoramidite ligand 48 provided a protection-free approach to asymmetric synthesis of diarylmethylamines.38 This environmentally sound, and atom-economical approach performing well under acidic reaction conditions yielded 18 examples with ee ranging from 31–98%. The necessity for ortho substitution of benzophenone N–H imines was noted as being a key feature for obtaining high enantioselectivity. It was also reported that the enantioselective hydrogenation of seven-membered substituted dibenzo[b,f][1,4]oxazepines provided an efficient route to optically active 11-substituted-10,11-dihydrodibenzo[b,f][1,4]oxazepines. The sterically hindered (S)-Xyl-C3*-TunePhos 35 gave the highest enantioselectivity, providing 13 examples with ee's ranging from 52–94%.39
An Ir(I)-(S)-tolBINAP-protic amine catalytic system 36 for the asymmetric hydrogenation of the cyclic imine, 2-phenyl-3,4,5,6-tetrahydropyridine achieved an enantioselectivity of 90%. Enhanced catalytic activity was observed with the addition of sub-stoichiometric addition of MeOH or EtOH without impairing the enantioselectivity.40
Chiral diphosphonate ligands, derived from BINOL with an achiral diphenyl ether backbone, are excellent for the Ir-catalyzed asymmetric hydrogenation of quinolines. The ligand 44 comprises a diphosphonite derived from BINOL and an achiral backbone originating from diphenyl ether.
An Ir-f-binaphane complex 39 allows for the complete conversions and high enantioselectivities (up to 96% ee) in the asymmetric reductive amination of aryl ketones in the presence of Ti(OiPr)4 and I2. Formation of the imine substrate appears to be the limiting step, however the approach to avoid isolation and purification of this substrate allows for the asymmetric reductive amination of ketones or aldehydes with amines to be undertaken in a simple and practical method for the preparation of chiral amines.41
By employing an easily accessible H8-BINAPO chiral phosphinite ligand 47 for the ATH of quinolines derivatives it was demonstrated that the ligand provided better enantioselectivities in the less polar biphasic system poly (ethylene glycol) dimethyl ether (DMPEG, MW1/4500)/hexane reaction medium than in THF; with up to 11% increase of enantioselectivity being observed compared with those obtained in THF.42 An iridium catalyst containing a new chiral phosphinite ligand Spiropo 45 derived from (R)-1,19-spirobiindane-7,79-diol exhibits high catalytic activity (substrate/catalyst ratio up to 5000 with 91% conversion) and excellent enantioselectivity (up to 94% ee) in the asymmetric hydrogenation of quinolines. The catalytic species was noted to be stable in THF; a feat that was maintained even after two months under an inert atmosphere with activity and enantioselectivity still unchanged. If the hydrogenation was carried out in a DMPEG/hexane biphasic system this resulted in efficient separation and recycling of the catalyst. Although the conversion dropped to 40% after four runs, the enantioselectivities remained high.43
The use of Ir/diphosphinite 46 catalyst in the asymmetric hydrogenation of quinoxalines gives one of the highest ee values attained so far in the catalytic asymmetric hydrogenation of 2-methylquinoxaline.44
A high-performance catalytic system with readily available DifluorPhos 37, a biaryl bisphosphine ligand, to enantioselectively hydrogenate a series of quinolines at high (S/C) ratios, afforded up to 96% ee with up to 43000 TON and up to 3510 h−1. The substrate scope was also extended to asymmetric hydrogenation of tri-substituted pyridines showing excellent reactivity and enantioselectivity in the hydrogenation with nearly quantitative yields and up to 98% ee.45
A series of secondary phosphine oxide (SPO) monodentate ligands 38 (ref. 46) have been developed and although the phosphine oxide is the most stable form at room temperature, the tautomeric phosphinite is capable of complexing with the iridium precursor. The acidic proton of SPOs was noted as having an accelerating effect upon the transition metal-catalysed hydrogenation of imines. Higher rates were obtained with SPO/Ir ratios of 1, but this decreased enantioselectivity. An increment to a ratio of 2 (SPO/Ir) resulted in increased enantioselectivity. Additives such as n-Bu4NI, K2CO3, t-BuOK, Na2CO3, primary amines, phthalimide, CF3CO2Ag and AgBF4 were found to have a negative effect on hydrogenation.
4.2 Ligands for cationic iridium(I) catalysts
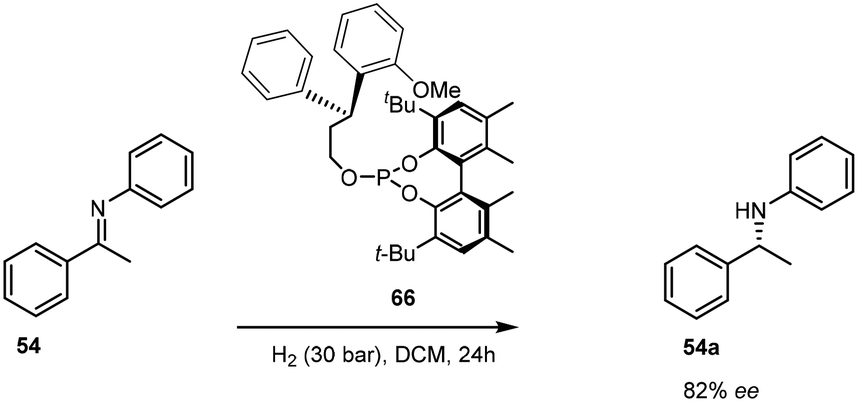
Of significance in the development of cationic Ir(I) complexes was the introduction of diphosphine ligands including the synthesis of phosphinooxazoline (PHOX) derivatives.47 The phosphanodihydrooxazole had been used in Pd-catalysed allylic substitution,48 allylic amination49,50 and the Heck reaction.51 The new catalyst paved the way for one of the most important transformations in organic chemistry – the asymmetric transfer hydrogenation of imines to afford amines (Scheme 17). This air stable complex is able to convert imines to amines with an ee of 89%.52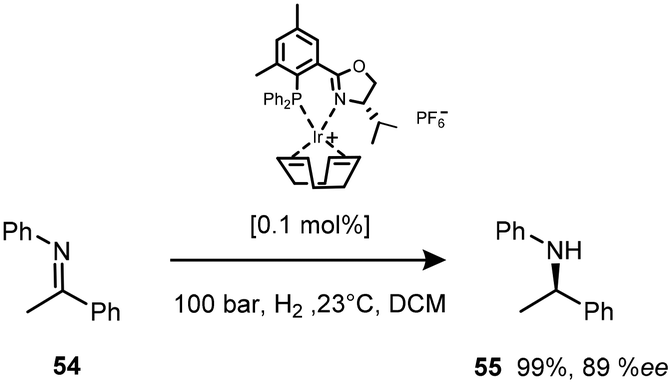 | ||
| Scheme 17 ATH of the imine 54. | ||
Only the catalytic asymmetric hydrogenation of unfunctionalised tri-substituted alkenes had been reported using the chiral C2-symmetric titanocene catalyst which gave yields ranging from 70–94% with ee's of 83–99%.53 However, the high catalyst loading required for this protocol (>5 mol%) limits its applications and so the introduction of novel chiral ligands further enhanced the potential of iridium complexes as catalysts.54
The functionalisation of 2-azanorbornane-oxazoline with phosphine led to novel PHOX ligands.55 A decrease in reaction rate and enantioselectivity was observed for substrates bearing an o-methyl substituent. Imines with electron withdrawing groups were reduced with high yields and %ee (Scheme 18). The analogous 62 however did not display any activity in the reduction of imines but it did show high activity in the hydrogenation of acyclic and cyclic tri-substituted alkenes.
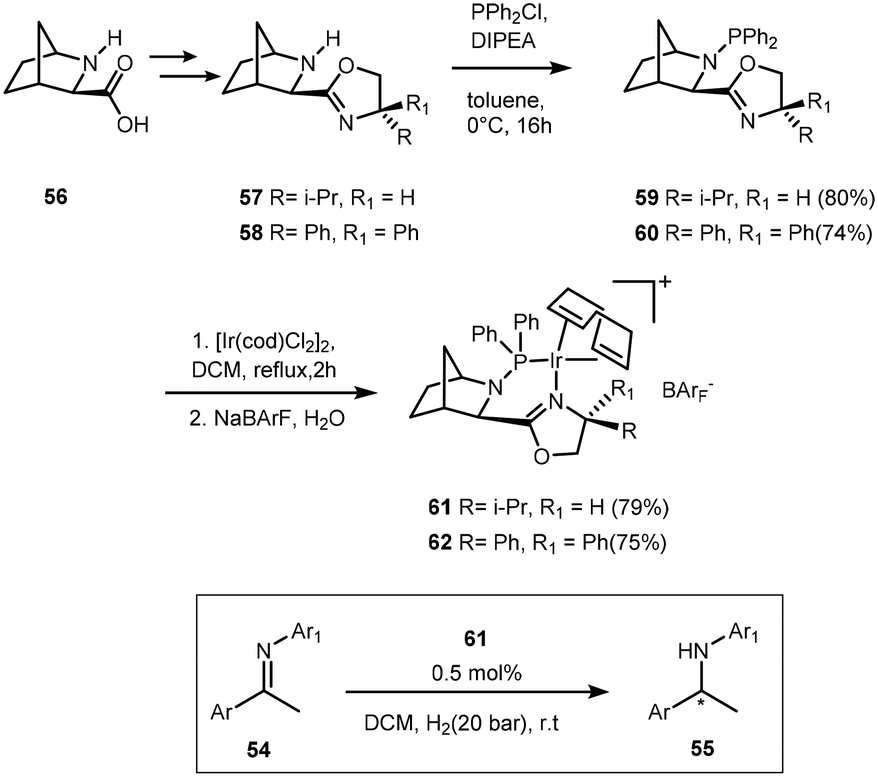 | ||
| Scheme 18 Phosphine–oxazoline synthesis and utilisation in the hydrogenation of imines 54. | ||
The utility of the chiral PHOX iridium catalysts was expanded using a combination of ionic liquids (ILs) and super-critical carbon dioxide (scCO2). The ionic environment provides beneficial molecular interactions with the cationic organometallic intermediates (Scheme 19).56 The CO2 in the biphasic system allows for efficient hydrogenation in ILs, dissolving the catalyst in the IL leads to activation by anion exchange and the process allows for easy removal of the product from the catalyst solution by CO2 extraction without cross-contamination of IL or catalyst. The highest activity was achieved in the presence of scCO2 at 40 °C in 1-methyl-3-pentylimidazolium hexafluorophosphate [PMIM][PF6] 63 with ee varying from 53–78%.57
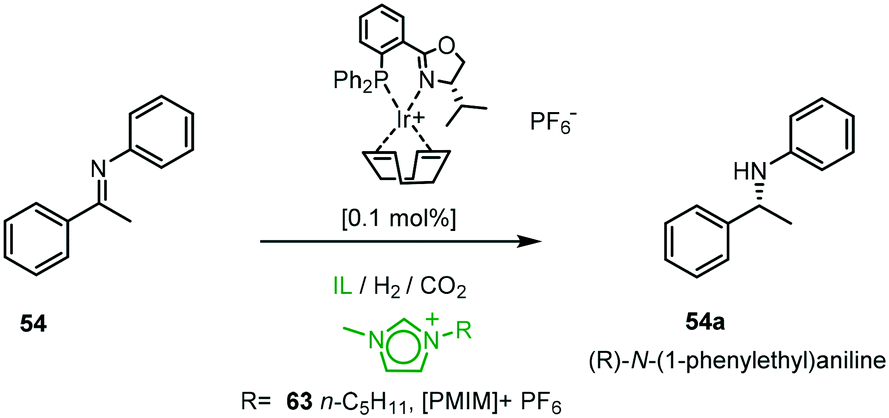 | ||
| Scheme 19 Hydrogenation of 54 in various ionic liquids (IL). | ||
Cationic Ir(I) complexes bearing aminophosphine-oxazolines (Scheme 20) were used for the hydrogenation of imines providing chiral amines up to 90% ee.53
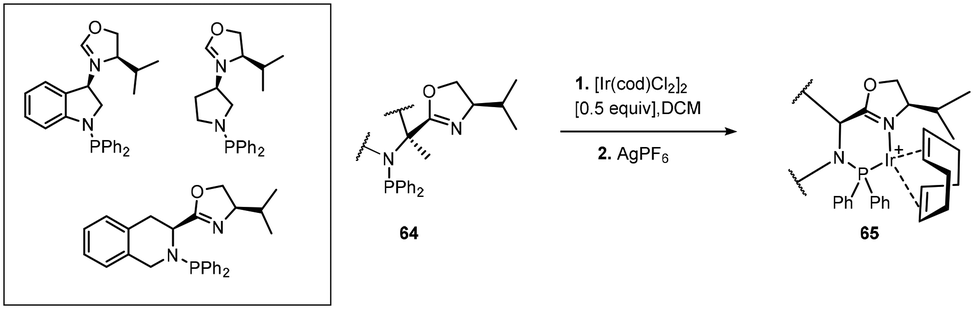 | ||
| Scheme 20 Aminophosphine-oxazoline based iridium complexes. | ||
Iridium complexes with phosphine–phosphite ligands demonstrate the importance of backbone flexibility on the enantioselectivity of the reaction and showing unusual better asymmetric induction with a more flexible ligand (Scheme 21).51,58
 | ||
| Scheme 21 Phosphine–phosphite ligands. | ||
Examples of cationic Ir(I) and Ir(III) complexes used for asymmetric hydrogenation are shown in Scheme 22. Using an in situ prepared iridium catalyst based on the cheap chiral monodentate ligand (S)-PipPhos 68 a new low pressure hydrogenation method of a range of acyclic N-aryl imines was developed with excellent enantioselectivities.59
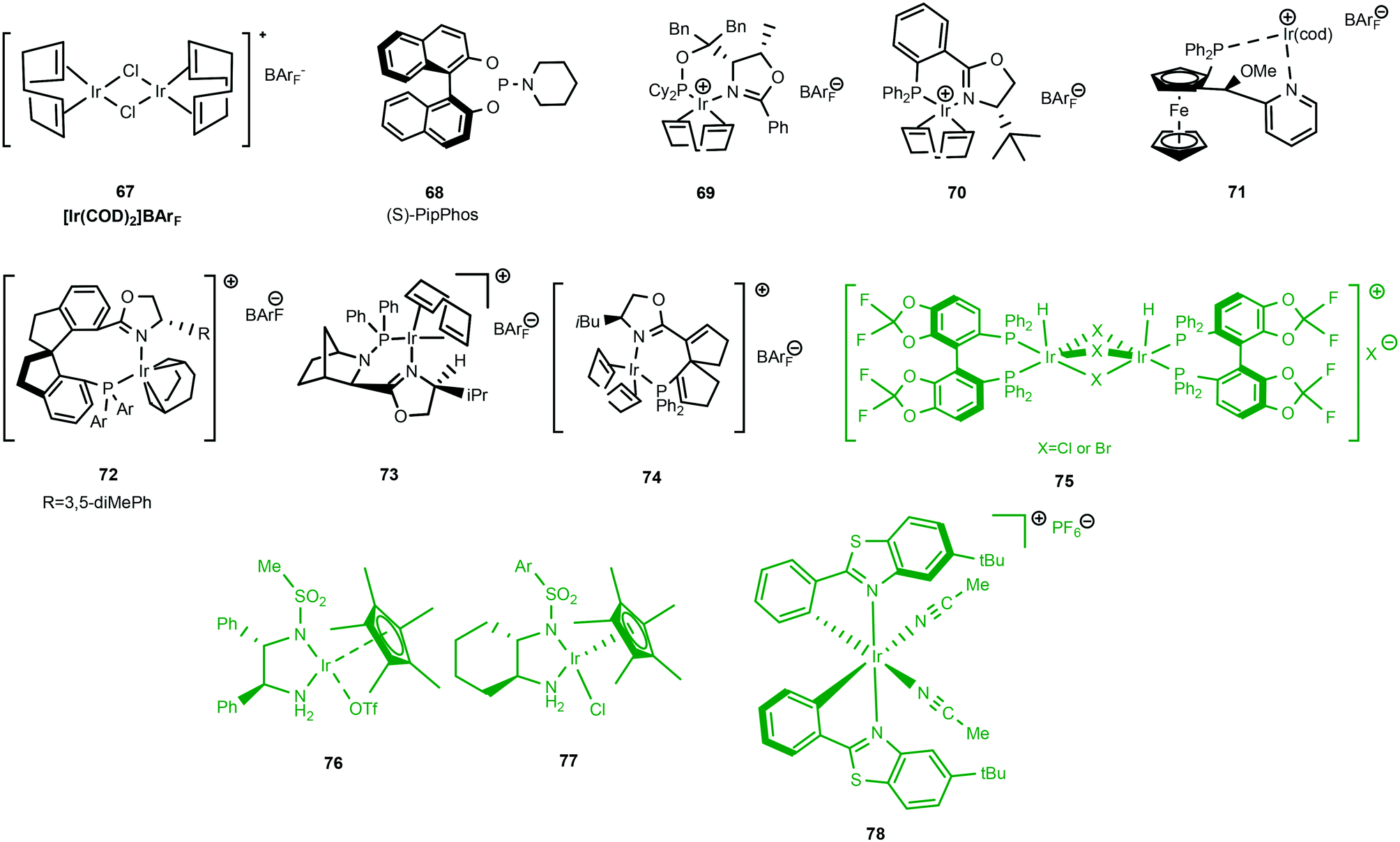 | ||
| Scheme 22 Cationic Ir(I) in BLACK and Ir(III) complexes in GREEN for asymmetric hydrogenation. | ||
Unfunctionalised enamines present challenging substrates in asymmetric hydrogenation but cationic Ir(I) complexes with chiral oxazoline or pyridine-based N,P ligands are active catalysts for their reduction (Scheme 23). Complexes 69 and 70 exhibited excellent activity giving full conversion with low catalyst loading (0.5–1 mol%). High ee's were attributed to substitution at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and the nitrogen atom with N-aryl or N-benzyl groups providing beneficial effects on the enantioselectivity.60
C and the nitrogen atom with N-aryl or N-benzyl groups providing beneficial effects on the enantioselectivity.60
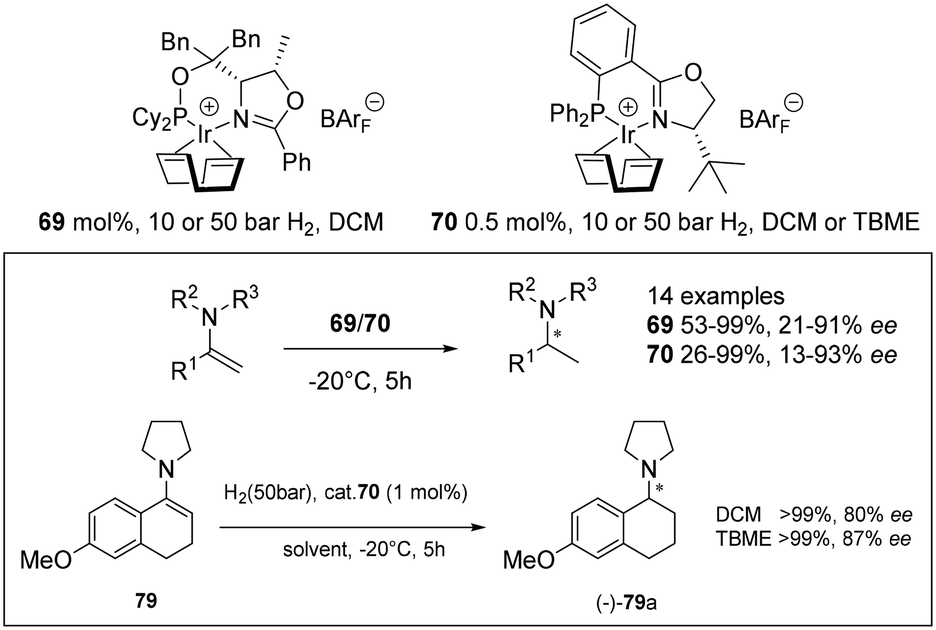 | ||
| Scheme 23 Chiral oxazoline cationic Ir(I) complexes 69 and 70 for hydrogenation of enamines. | ||
Preparation of γ- and δ-lactams in 92–97% ee was achieved with novel class of chiral P,N-ferrocenyl ligands 71; the cationic iridium catalyst also showing great utility in the enantioselective reduction of imines (up to 99% ee).61 The spirobiindane scaffold is extremely rigid and bulky, a feature which was exploited in the synthesis of chiral phosphine–oxazoline ligands, providing well-defined cationic iridium complexes. A particular disadvantage of cationic iridium catalysts is that they easily form inactive trimers under a hydrogen atmosphere something that SIPHOX 72 was reported to be resistant to when exposed to a high pressure of hydrogen. SIPHOX created a crowded but efficient chiral environment around the metal centre. The catalytic system was indeed efficient in the hydrogenation of acyclic N-aryl ketimines, providing chiral amines in excellent ee's (>97%).62
Extending the chiral phosphine–oxazoline library, the iridium-catalysed hydrogenation of acyclic N-arylimines with 73, induced high enantioselectivities (66–90% ee) of acyclic aromatic N-arylimines.55
Spiro-[4,4]-1,6-nonadiene offers convenient functionalization of the backbone because the sp2 carbon centres are well suited for further anchoring of the chelating ligation moieties during chiral-ligand construction. As well as providing excellent enantioselectivities of challenging ketimines (>98%) the cationic iridium complexes featuring chiral phosphine–oxazoline spiro-[4,4]-1,6-nonadiene backbone (SpinPHOX 74) were subsequently employed in the asymmetric synthesis of sertraline ((+)-cis-(1S,4S)-1-methylamino-4-(3,4-dichlorophenyl)tetralin, (Scheme 24) (S,S)-81), an antidepressant chiral drug, affording the chiral amine isomers cis-81 and trans-81 with excellent enantioselectivities (89 and 98% ee, respectively) in a ratio of approximately 1:1.63
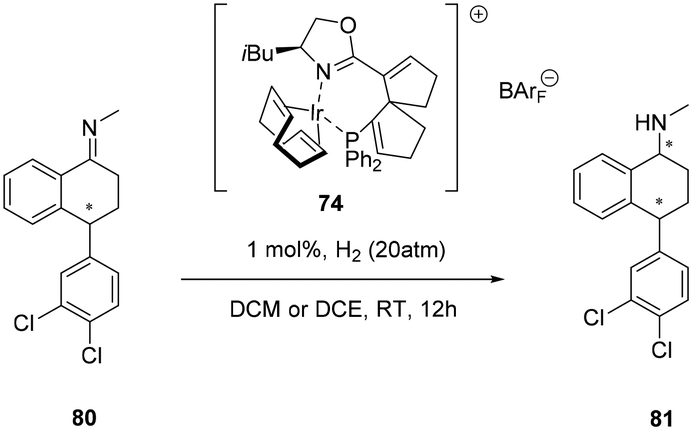 | ||
| Scheme 24 Spiro-[4,4]-1,6-nonadiene phosphino-oxazoline cationic Ir(I) complex in the synthesis of sertraline 81. | ||
4.3 Ligands for iridium(III) catalysts
Of the three clusters encompassing iridium catalytic species, Ir(III) complexes (Scheme 22) [green] have found limited application in the reduction of imines. One of the representative examples in this class is the exceptional cationic dinuclear Ir(III) complex 75.64,65 The substrate scope features excellent hydrogenation of 2-aryl substituted quinolines and 2-alkyl and 2-aryl quinoxalines. Additionally, complex 75 has also proved to be formidable in the reduction of 1- or 3-substituted isoquinolinium salts.Ir(III) complexes featuring chiral monosulfonylated diamines have indeed provided some of the highest ee's in the reduction of 2-alkyl and functionalized 2 alkyl quinolines with low catalyst loading (S/C = 500); the air stable 76 gave yields of 95% with high ee's (94–99%).66
Similar half-sandwich Ir(III) complexes 77 have also been reported;12 in this work the utilisation of a cyclohexan-1,2-diamine based monosulfonylated diamine was active in the reduction of aryl methyl N-benzyl imines, achieving full conversion albeit with moderate enantioselectivity (39–78% ee). The efficient enantioselective ATH of ketones was achieved using bis-cyclometalated Ir(III) complex 78 with catalyst loadings as low as to 0.002 mol% (96.6% ee).67 The rigidity of the complex limits the degree of conformational flexibility of intermediates in the catalytic cycle, an important factor that provides an entropic advantage during catalysis. The marked increase in both catalysis and enantioselectivity is also attributed to the metal–ligand cooperativity (bifunctional catalysis) due in part to the presence of the pyrazole co-ligands in the catalytic system.
5. Mechanisms of transfer hydrogenation
There are several questions to be addressed to elucidate the mechanism of iridium complex catalysed enantioselective synthesis. What is the structure of the active catalyst? Is there significant binding between the substrate and iridium complex? If so, is the binding intermolecular or does it involve significant bonding? If the latter what is the site of bonding? Are any intermediates formed? Is the reaction mechanism homo- or hetero-lytic? What is the rate-limiting step of catalysis? How is the catalyst regenerated? The previous section has catalogued an extensive library of applications of iridium catalysts in enantioselective transformations, this next section is aimed at giving a brief overview of the current understanding of mechanistic pathways encompassing this field of organometallic chemistry.5.1 Hydrogen activation
The most common reduction catalysed by iridium complexes involves the formal hydrogen addition to unsaturated bonds generally in the form of proton and hydride transfers (Scheme 25a). The source of hydrogen can be molecular hydrogen or hydride donors such as formate anions or iso-propanol (Scheme 25). Most involve iridium hydride as an active species, probably best described as a polarized covalent bond rather than as a pure ionic interaction. A particular concern in hydrogenation mechanisms is the source of the proton for reductions with molecular hydrogen in aprotic solvents and with no obvious acidic groups available. Although the formal oxidative addition of H2 to Ir(I) to form dihydrides of Ir(III) could, in principle, provide a simple solution via a concerted addition to the unsaturated centre.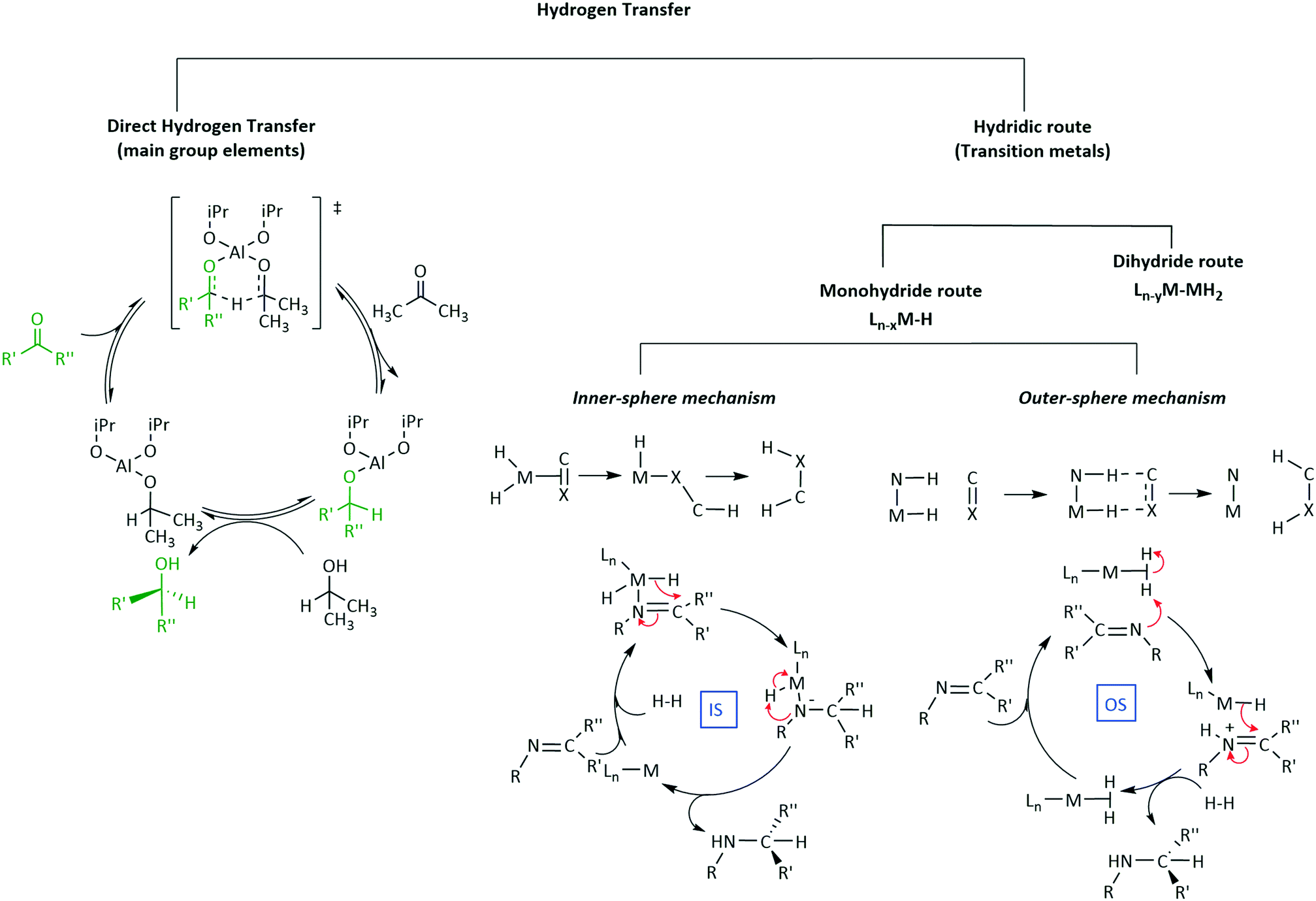 | ||
| Scheme 25 Transfer hydrogenation mode using different hydrogen sources. | ||
Generation of hydridic species from hydrogen can occur through heterolytic hydrogen cleavage [Scheme 26(a)] usually through oxidative addition, in which formally there are two electron transfers from Ir(I) to hydrogen to form two metal hydrides and a change of the oxidation state of the metal to Ir(III).68 This of course is not an easy transformation given the H–H bond dissociation energy is 436 kJ mol−1; however, transition metal complexes achieve this feat under mild conditions.
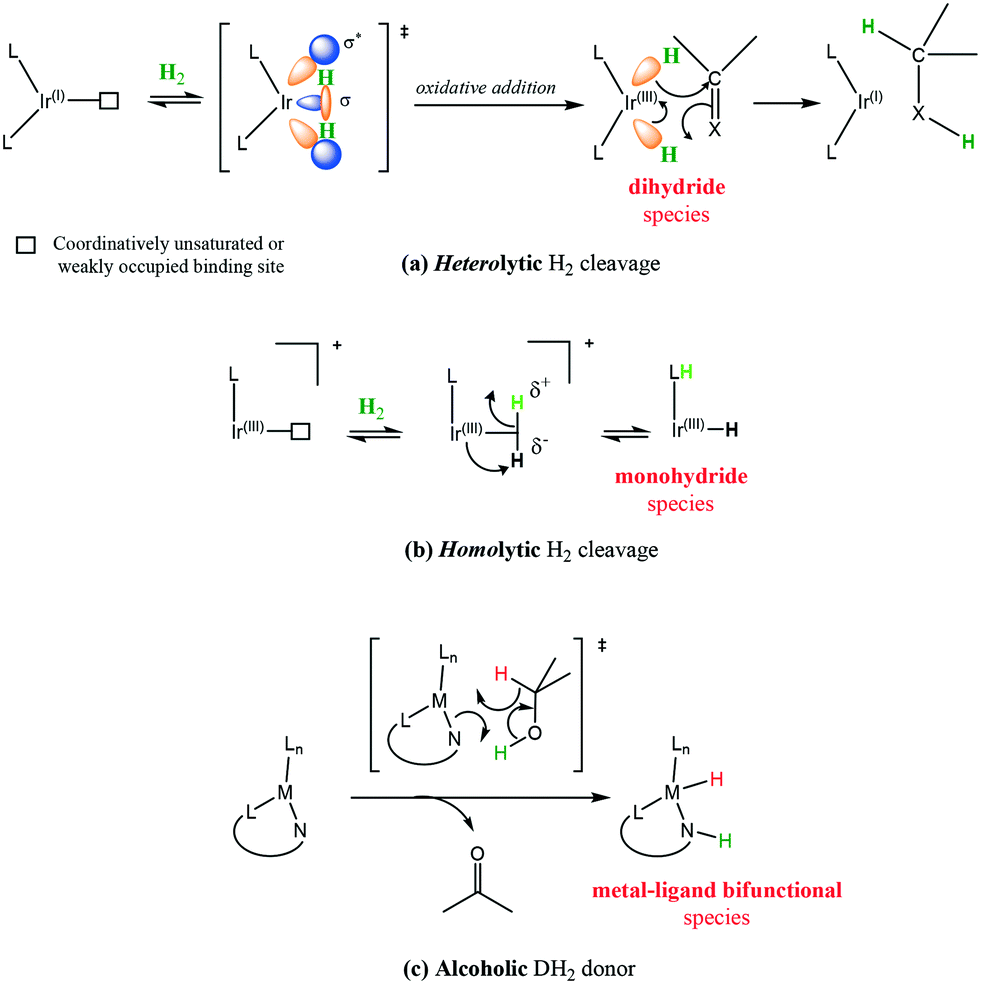 | ||
| Scheme 26 Transfer hydrogenation mode using different hydrogen sources. | ||
Homolytic cleavage of hydrogen [Scheme 26(b)] involves hydride transfer to Ir and proton abstraction to give a mono-hydride without formal change of oxidation state.69 This process can be further split into intramolecular or intermolecular processes (mediated by an external base). Ir(III) activation by hydride donors generally yields the mono-hydride again with no formal change of oxidation state.70
Employing a sacrificial DH2 donor [Scheme 26(c)] represents another alternative for the source of hydrogen; with the donor species transferring two hydrogen molecules to an acceptor species.
5.2 Substrate considerations
The mechanistic question about the formal hydrogen addition to unsaturated bonds in the form of proton and hydride transfers (Scheme 27) is are the steps concerted or stepwise and, if the latter, which comes first? The two prochiral substrates considered here are CO and CN centres. CO substrates are well suited to undergo reduction as they are highly polarized; exhibiting weak binding to the metal. Indeed, if binding of CO to the metal centre was strong this would retard H2 activation which is a weak ligand. On the other hand, imines are not very polarized but the lone pair residing on the nitrogen is capable of metal coordination which can suppress catalysis; prior N-protonation thus serves to increase its affinity for the nucleophilic addition of a hydride ion (H−).71
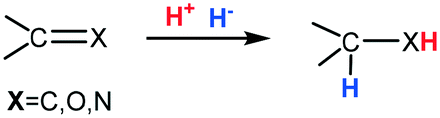 | ||
| Scheme 27 Addition of H− and H+ across a CX bond. | ||
It is sometimes suggested that proton transfer can occur from an amine NH in which this involves the formation of a very unstable nitrogen anion.72 Similarly, hydride addition to a neutral imine generates a nitrogen anion. The pKa of amines, involving proton loss from NH, in water is ∼30 and is even higher in non-aqueous solvents such as DCM or acetonitrile and so the generation of nitrogen anions under the common conditions used in the reduction of unsaturated systems would require enormous stabilisation by other factors and seems generally unlikely.
The substrate directs classification of the reaction pathway based on the role it plays in the coordination sphere. If the substrate directly coordinates to the metal it is termed inner-sphere (IS) (Scheme 25b) whilst that with no coordination is outer-sphere (OS) (Scheme 25c). Those reactions involving sacrificial H donors can also be classified within these two families. Typically, in an IS mechanism (Scheme 25b), sometimes referred to as ionic hydrogenation, the substrate binds directly to the metal-ion, followed by subsequent insertion into the M–H bond, elimination of the product and oxidative H2 addition steps to regenerate the catalyst and complete the cycle. This mechanism is common for homogenous catalysis involving iridium.73 OS mechanisms (Scheme 25c) on the other hand do not involve direct binding of the substrate to the metal-ion but rather proceed via initial intermolecular interactions followed by hydride and proton transfers. The transfer of H+ and H− can either proceed concertedly or in a stepwise fashion to the unbound substrate. This addition of H+ and H− offers a sub-level classification in OS mechanisms as the processes can be considered as bifunctional or ionic mechanisms.
Mechanistic considerations in transfer hydrogenation have been extensively studied and while all catalysts discussed below exhibit similar features, unfortunately, some of the proposals rely more on conjecture than hard experimental evidence. This section is divided into parts, some of which involve a degree of overlap.
5.3 Outer sphere mechanisms of hydrogenation
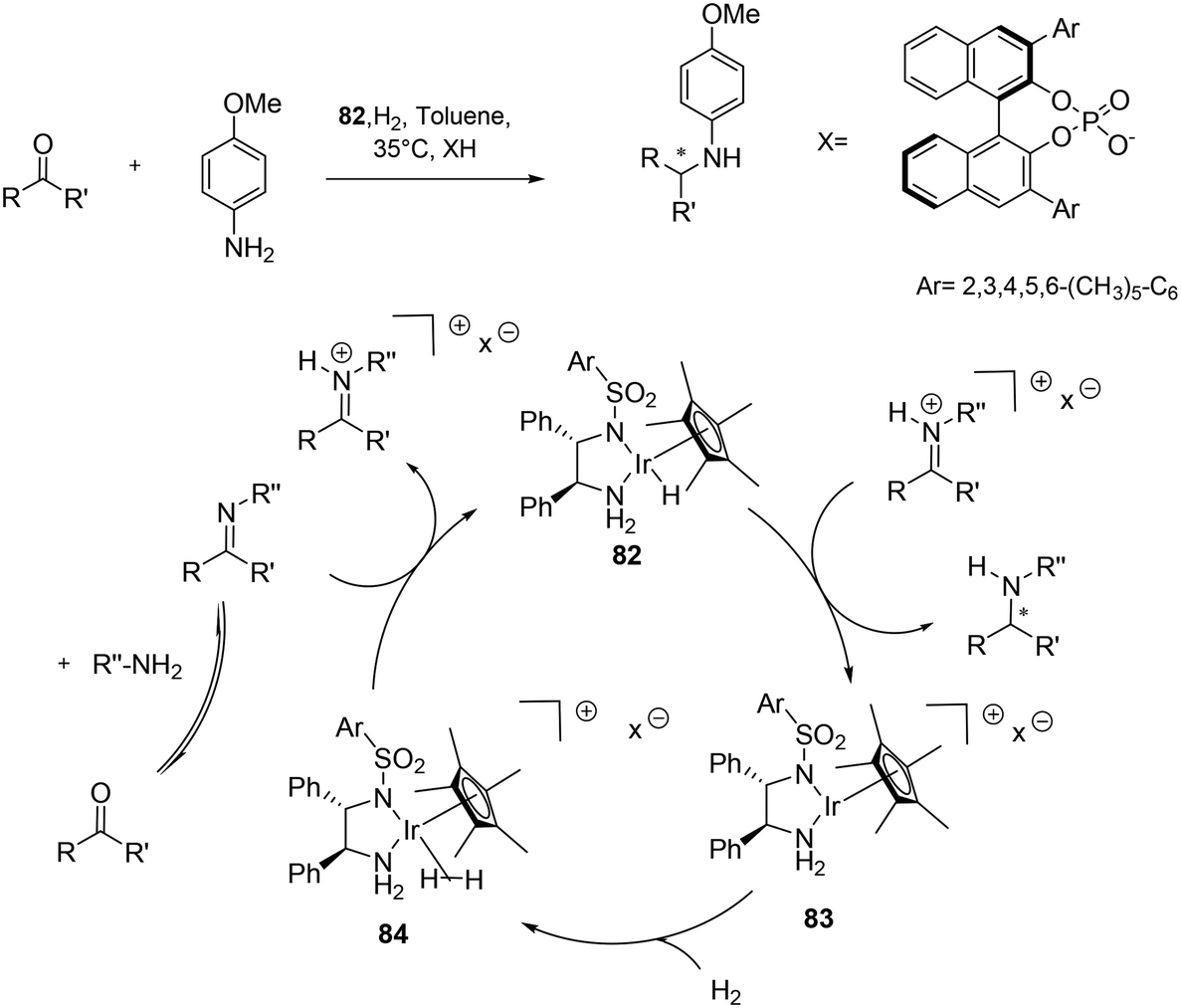 | ||
| Scheme 28 Direct asymmetric reduction amination (DARA) of ketones via metal-counter anion cooperative catalysis. | ||
The addition of AgSbF6 in asymmetric hydrogenation of acyclic imines enhances the reactivity of the imine with diamine half-sandwich Ir(III) species, attributed to coordination of the Ag+ to the imine nitrogen thus facilitating hydride transfer (Scheme 29). The SbF6− anion can be envisaged as forming an ion-pair.11 In this outer sphere proposal, a base mediated heterolytic H2 cleavage generates the active 18 electron monohydride species 88 which delivers a hydride to the Ag+ coordinated imine. Protonation of the bound product results in amine formation. Whereas proton transfer precedes hydride transfer in the general OS mechanism, in this proposal, Ag+ coordination to the imine causes a reversal, thus hydride transfer occurs first, followed by protonation, although again it involves the formation of an unstable nitrogen anion.
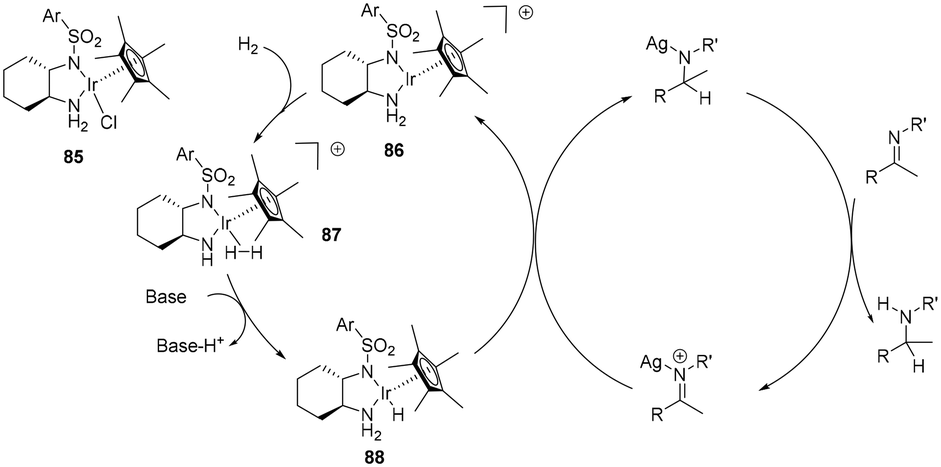 | ||
| Scheme 29 Half-sandwich Ir(III) complex 85 in the Ag+ assisted hydrogenation of acyclic imines. | ||
Computational mechanistic investigations are currently leading the charge to unravel key transformations in catalytic organic processes. In a theoretical study of the PHOX system80 an outer sphere mechanism in the asymmetric hydrogenation of acyclic imines was proposed (Scheme 30). COD hydrogenation results in the formation of a cis-dihydride complex 90 and an additional ligand, possibly a solvent molecule, occupies the vacant coordination site in the apical position. Though arbitrary, the solvent molecule is assumed to be stable throughout the cycle. An incoming H2 is then assumed to coordinate in an η2 fashion trans to the phosphorus ligand 91. Formation of the active catalytic species is thus dependent on displacement of the imine by H2. Heterolytic cleavage of the bound H2(η2) by the imine results in formation of an iminium species. In an outer sphere mechanism, the iridium species 92 then delivers hydride in a stereoselective fashion to the protonated species, thus affording the amine product which is assumed to temporarily occupy a coordination site that in the next step is vacated in favour of either an imine or H2. It is interesting to note that throughout the cycle, several inert hydride ligands are present, a justification for maintaining the +3 oxidation state.
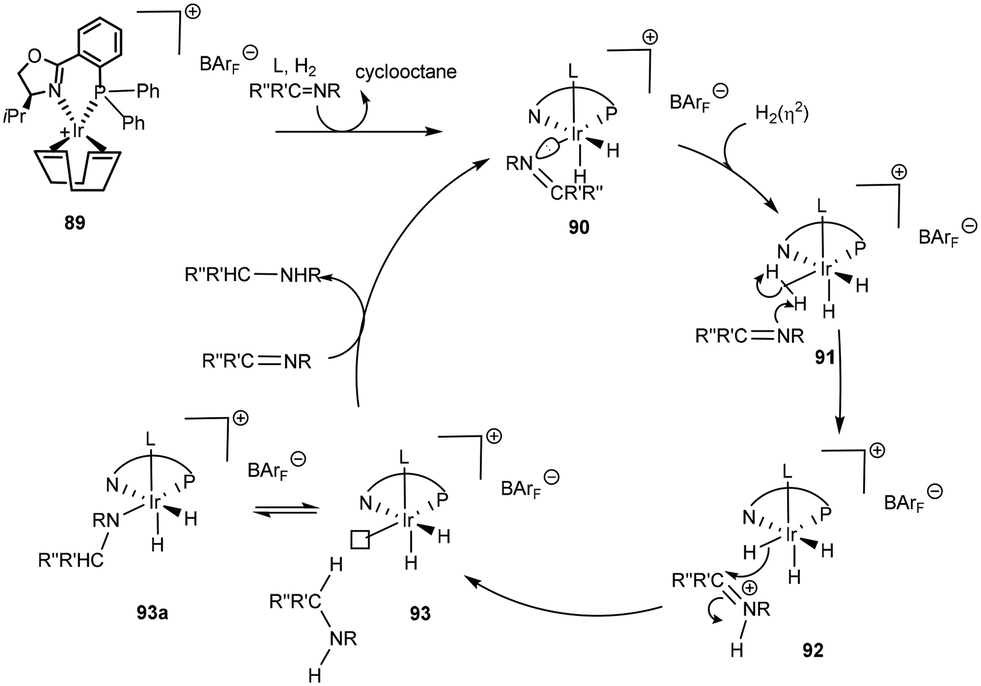 | ||
| Scheme 30 Proposed inner sphere mechanism with homolytic H2 cleavage with 89 based on a computational study.86 | ||
In an unorthodox outer sphere mechanism, a ligand assisted outer sphere hydrogenation of acyclic imines (Scheme 31) 94 was proposed.81 Fundamental to this proposal is the replacement of benzene (η6) by an amine, exhibiting both η6 (94) and η4 (96) binding modes. A free amine abstracts a proton from the Ir(III) species 95 which allows for subsequent oxidative addition of dihydrogen that eventually forces the proton to migrate to the η4 bound amine ligand, reducing the metal to Ir(I) as evidenced by H/D scrambling experiments. The rate law would agree with either a slow dissociative amine-by-imine exchange or a rate-determining bifunctional hydrogenation step. The long induction periods observed in the different solvents employed could reflect the amine-assisted dihydrogen oxidative addition step becoming the rate-determining step at low amine concentration – a feature that could be enhanced by competition of the imine for the hydrogen-bonding sites.81 A trihydride Ir(III) complex 97 is generated by the oxidative addition of H2 which subsequently proceeds to deliver a hydride to the carbon and a proton from the amine ligand in a bifunctional catalytic process 98, thus affording the product amine.
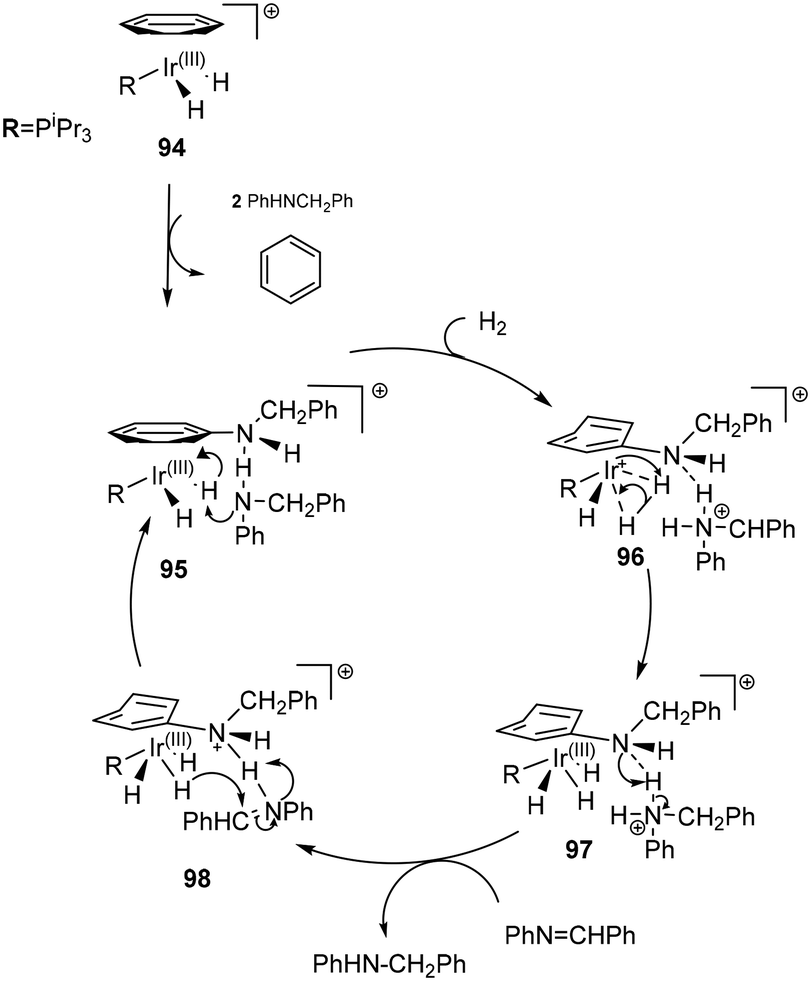 | ||
| Scheme 31 Proposed outer sphere mechanism for complex 94. | ||
N-Heterocyclic carbene (NHC) complexes represent a growing area of research and have been established as highly stereo-directing ligands in transfer hydrogenations.82–87 The strong σ-donor properties exhibited by NHCs provide beneficial reactivity of the metal hydride intermediates formed during the catalysis, but also stabilize the configuration of the catalyst.88 An outer-sphere mechanism for the 99 ATH of cyclic N-sulfonylimines was proposed leading to 24 examples of chiral sultams with high enantioselectivities (94–98% ee) (Scheme 32). The addition of ammonium formate is crucial in generating the catalytic species and serving as the hydrogen source. The expulsion of CO2 and ammonia generates the active species 102. Though not conclusive it is reasoned that generation of the 16-electron species is preceded by dissociation of MeCN or NH3, followed by fast hydride transfer to generate a highly reactive intermediate 102. The catalytic species then proceeds to transfer H+ and H− in a concerted fashion, with a hydrogen bond holding the substrate in place, subsequent product release leads to regeneration of the catalytic species. Of chief significance in the stereoselectivity is the reduced steric hindrance in the TS in addition to the hydrogen-bonding network. The outer-sphere mechanism is aided by the presence of bulky tert-butyl groups in the complex that restrict potential amine/imine coordination.89
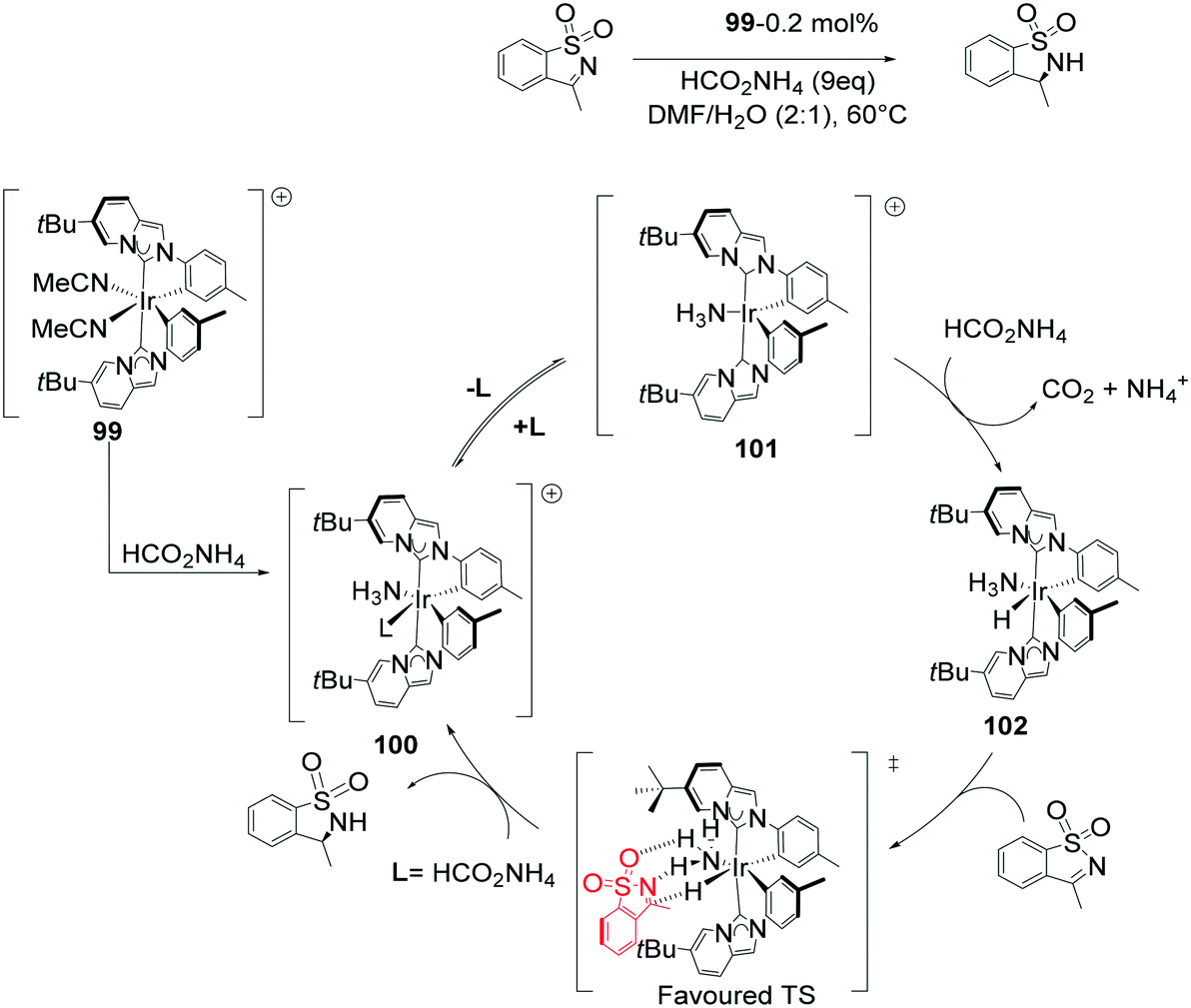 | ||
| Scheme 32 ATH of N-sulfonylimines with a NHC furnished Ir complex 99. | ||
Other outer sphere mechanisms of cyclic substrates have been proposed following a thorough investigation in the enantioselective reduction of 2-aryl-pyridinium salts with the Ir-MP (ref. 2)-SEGPHOS catalyst (Scheme 33). A series of deuteration experiments confirmed a multi-step hydrogenation cascade that involves multiple tautomerisations between the intermediate enaminium and iminium ions followed by an enantioselective addition via an outer-sphere mechanism. One of the challenges in hydrogenating pyridine based substrates is their strong coordination ability, a feature that can be detrimental to the catalyst. The rate of reduction of the tetra-substituted iminium was slower than that of tautomerization. Therefore, the second tautomerization had longer time to undergo a reversible process of protonation and deprotonation. A 1,4-addition was thought to be the first reduction step in the hydrogenation process with selectivity heavily relying on the geometrical fit between the catalyst and substrate. Hydride transfer and subsequent chiral reduction takes place through another outer-sphere pathway to release the final chiral piperidine to complete the catalytic cycle.76
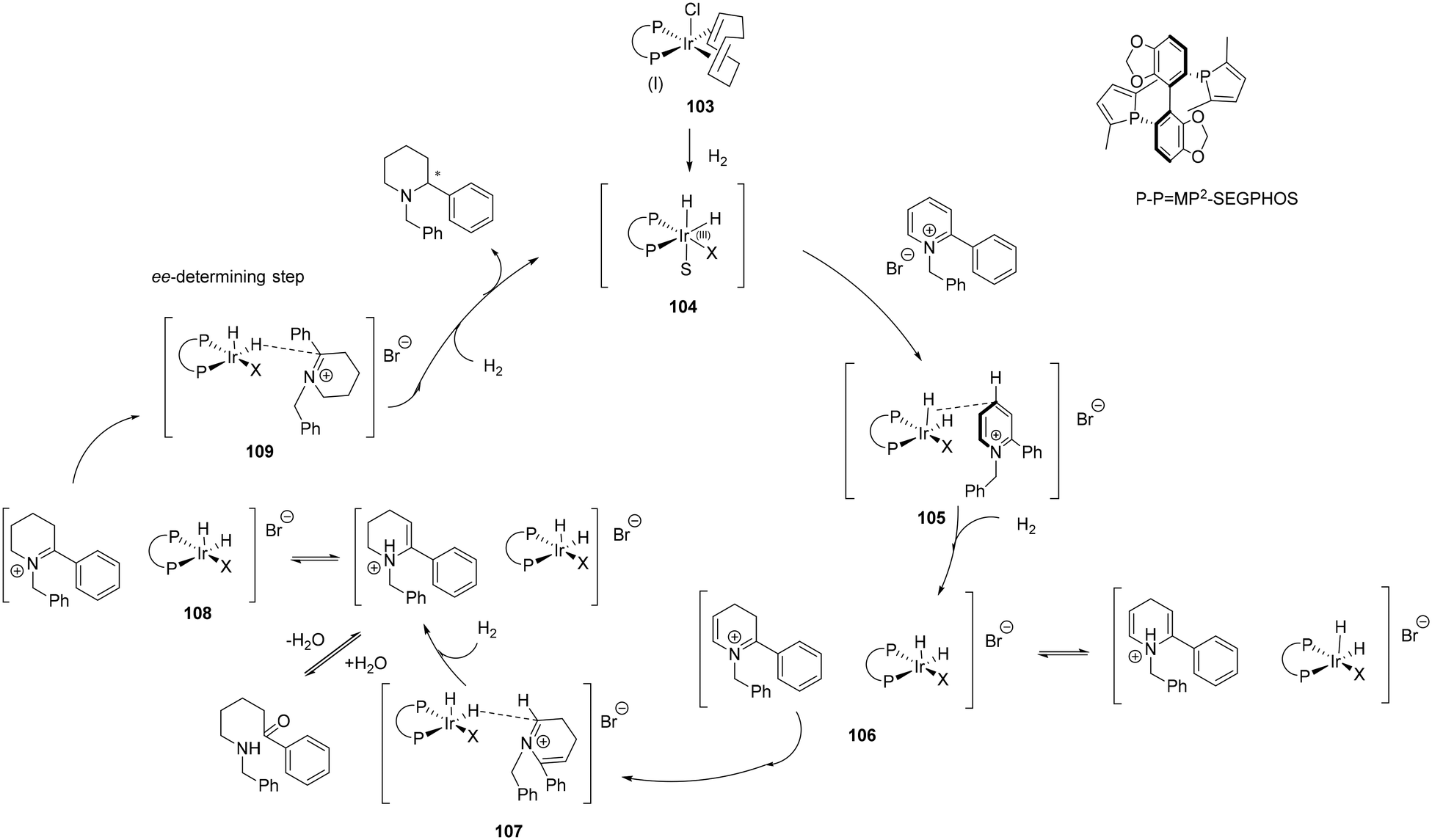 | ||
| Scheme 33 Outer-sphere mechanism in the hydrogenation 2-aryl-pyridinium salts with 103. | ||
The reduction of multiple bonds as in quinolines has also been proposed to occur through an outer sphere mechanism in a stepwise fashion (Scheme 34). Similar to Zhang's76 cycle the reduction encompasses two simultaneous cycles. Initially, a 1,4-addition in the first catalytic cycle proceeds via step-wise proton transfer and hydride transfer to the fourth position 111. The reduction is followed by enamine–imine isomerisation. The imine is protonated in the second cycle, leading to a final 1,2-addition, that results in the amine product.90,94
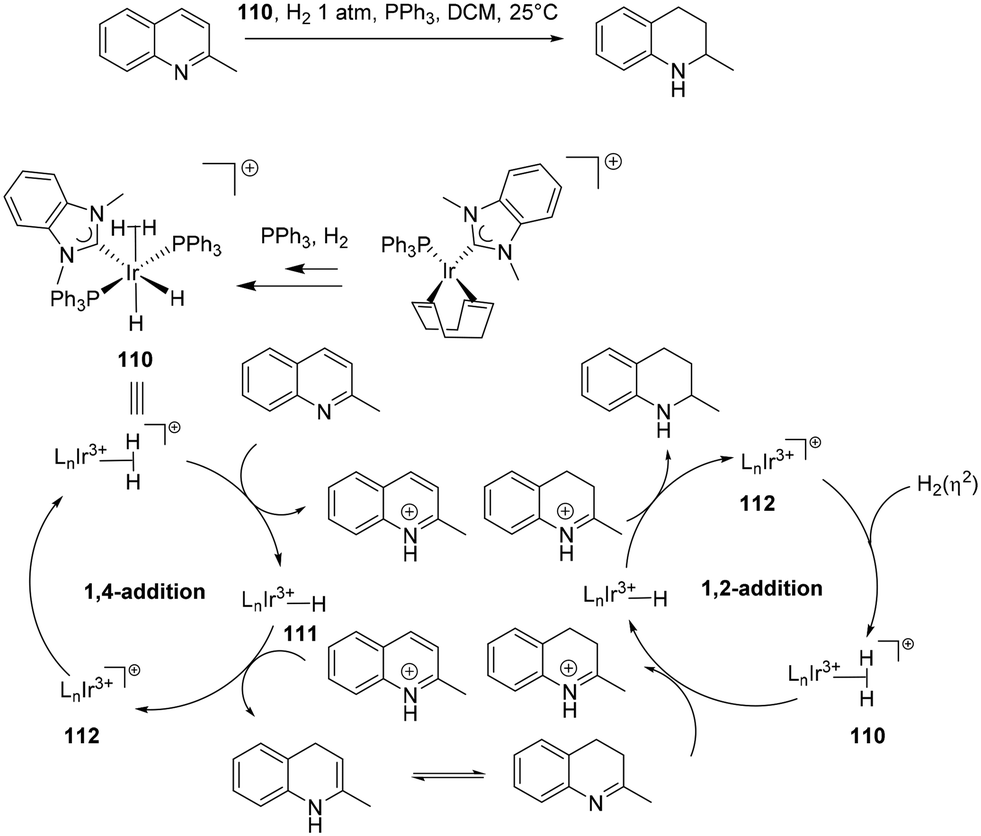 | ||
| Scheme 34 The proposed mechanism for the double hydrogenation of quinolines via a 1,4 and 1,2-addition with 110. | ||
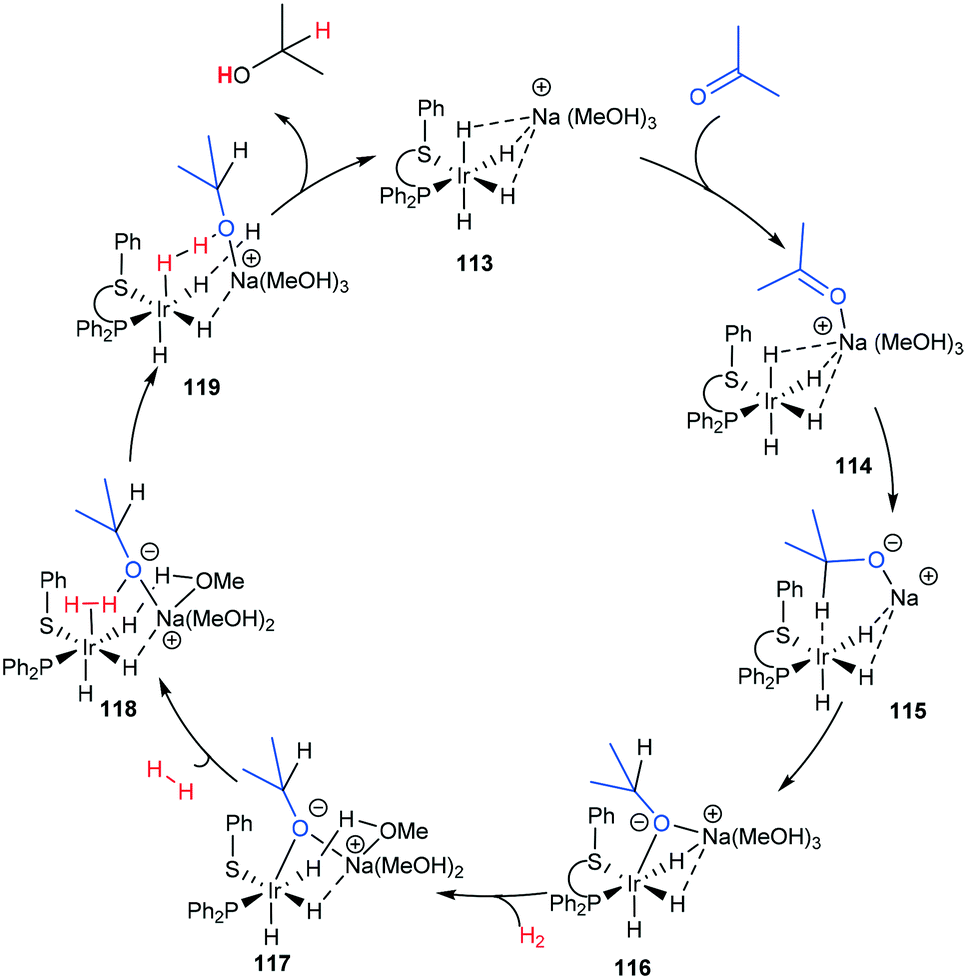 | ||
| Scheme 35 Ketone outer-sphere proposal based on a DFT calculation.97 | ||
5.4 Inner sphere mechanisms of hydrogenation
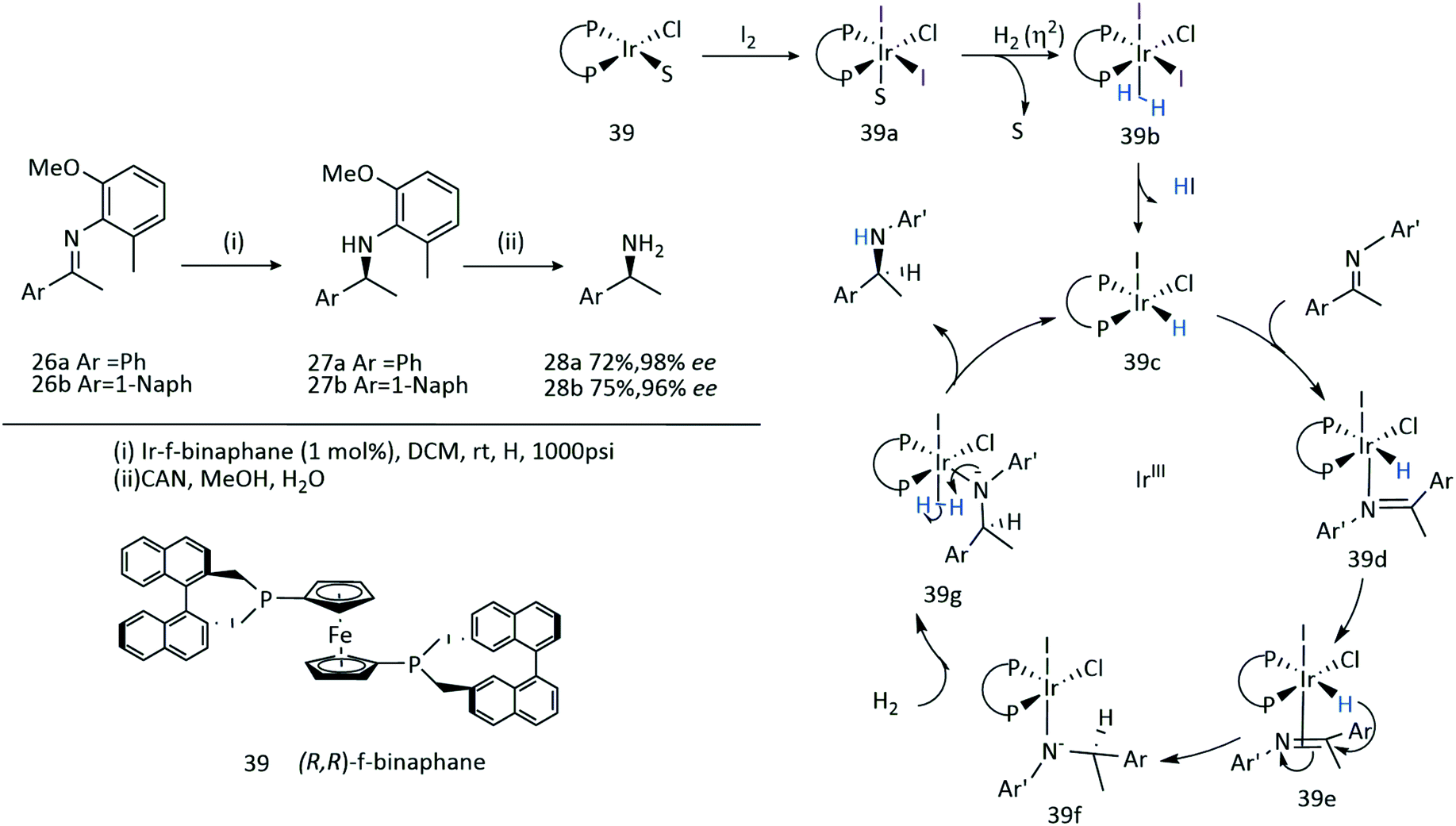 | ||
| Scheme 36 Proposed catalytic cycle for hydrogenation of 26a–b with Ir-f-binaphane 39. | ||
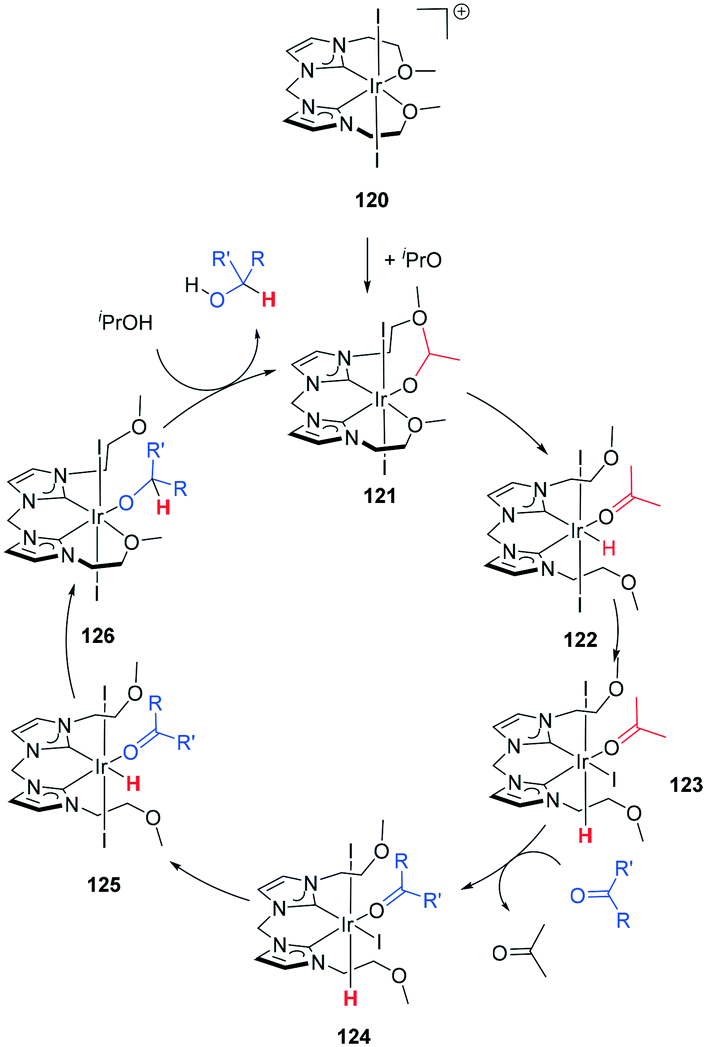 | ||
| Scheme 37 Ketone hydrogenation preceding via an inner-sphere mechanism with 120. | ||
6. Hydrogen transfer steps and other mechanistic considerations
6.1 Asymmetric transfer hydrogenation
Three main pathways have been proposed (Scheme 38) for the transfer hydrogenation of ketones using iridium catalysts to give corresponding alcohol products. Pathway A involves a direct hydrogen transfer proceeding by means of simultaneous interaction of the substrate and hydrogen donor with the metal centre – the close proximity of these two entities results in a concerted delivery of a formal hydride from the alcohol donor to the acceptor ketone.98,99 This pathway does not involve a metal hydride as the hydride is provided by the donor alcohol. The metal's purpose is to aid enhancement of the electrophilic nature of the carbonyl, making it more receptive to the hydride attack and providing a highly organised transition state where the correct orientations for bond making and breaking are met. This direct transfer of hydrogen has been extensively studied computationally and has given support to this mechanism for some iridium catalysts.94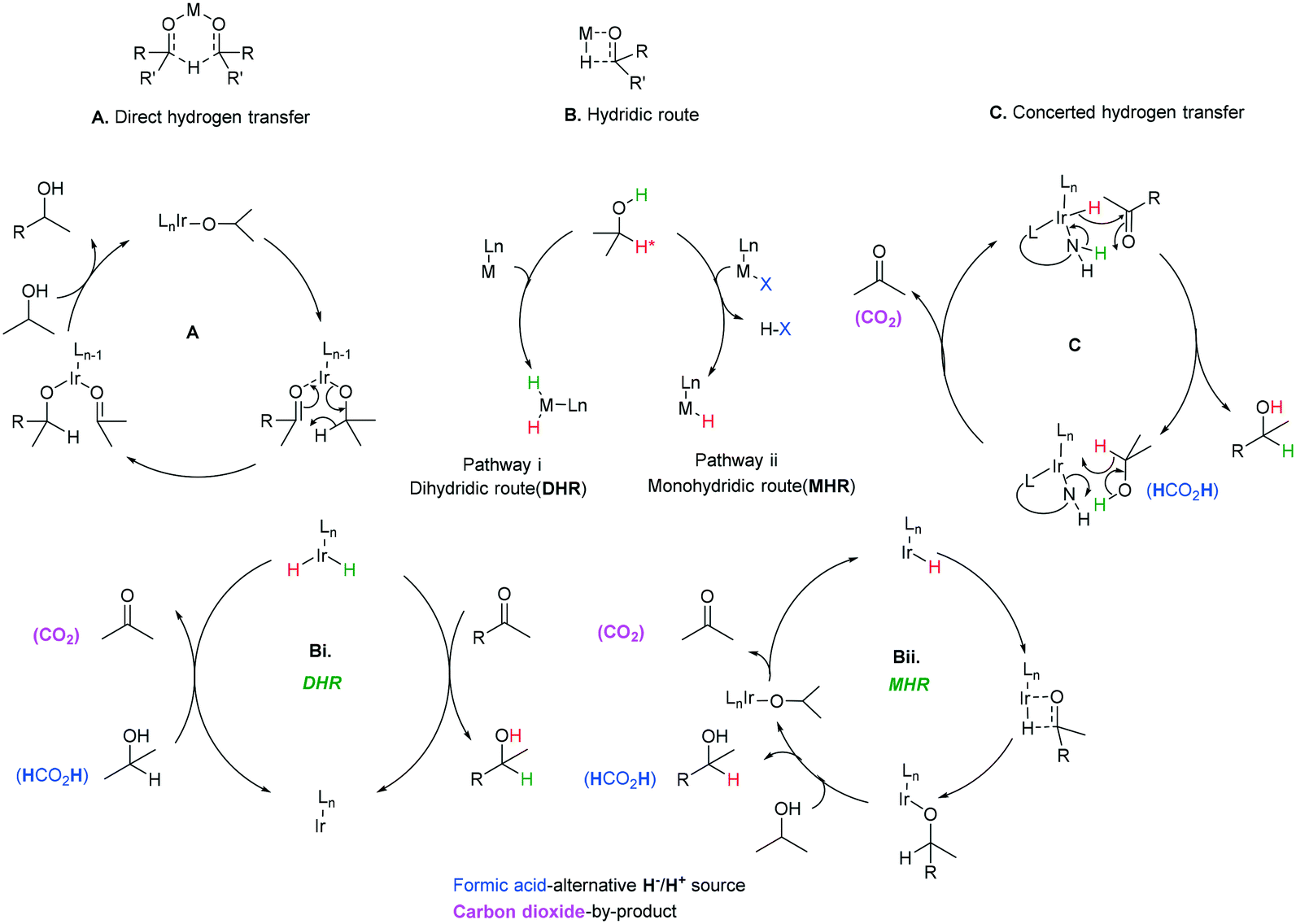 | ||
| Scheme 38 Mechanisms for the ATH of ketones. | ||
Pathway B (Scheme 38B) involves metal hydride complexes and deuterium labelling experiments have been used to probe whether hydrogen transfer proceeds through monohydride or dihydride mechanisms. The monohydride pathway (Scheme 38Bii) arises from the α-C–H of the donor and the dihydride results from abstraction of both the O–H and α-C–H.
In the case of Ir(I) complexes, the monohydride mechanism (Bii) is favoured. Activation of the H-donor occurs upon coordination of 2-propanol to the metal, providing the alkoxy complex, which subsequently undergoes an intramolecular β-hydrogen extraction to produce the metal hydride and acetone. The hydride is then delivered to the carbonyl group according to two different limiting mechanisms: (i) displacement of the coordinated acetone by acetophenone giving the alkoxide ion, followed by migratory insertion of the ketone into the metal–hydrogen bond to provide the new alkoxy derivative (inner sphere mechanism) and (ii) displacement of the alkoxy ligand from the coordination sphere of the metal by a second 2-propanol to deliver the reduced product and restarts the catalytic cycle.95
Delivery of hydrogen in a concerted fashion is described by pathway (C) (Scheme 38C) – metal–ligand bifunctional catalysis. These systems are unorthodox in that substrate reduction proceeds via concerted hydridic H delivery from the metal centre to the electrophilic CO carbon, while the ligand amine NH ligand delivers a H proton to the oxygen simultaneously. The presence of an available ionisable hydrogen on the ligand is imperative and its absence results in no catalytic activity.96,97 Catalyst regeneration then follows in a reverse process using the donor alcohol. This in turn led to the development of a repertoire of related catalysts containing an N–H functionality including iridium-based complexes (Scheme 39).98,99
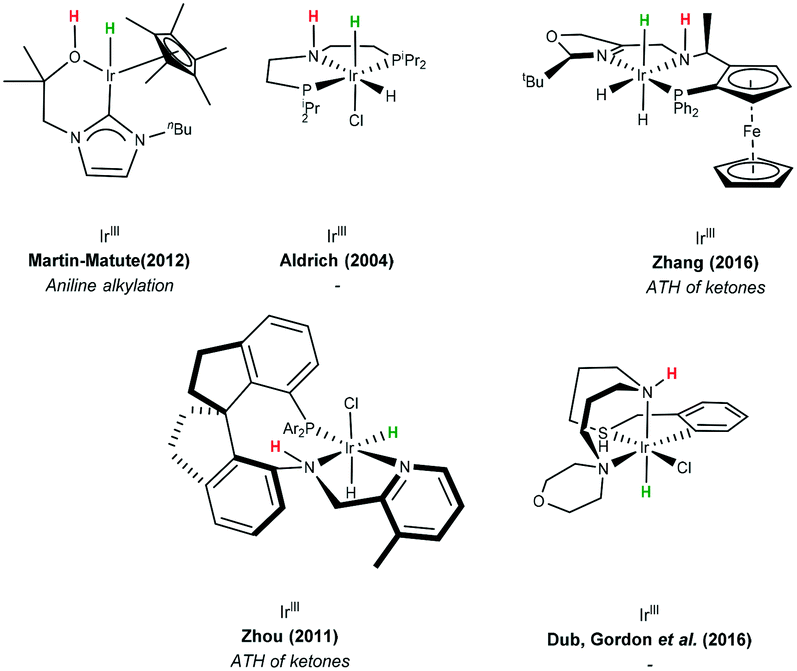 | ||
| Scheme 39 Examples of metal–ligand bifunctional catalysts. | ||
All three reaction pathways are feasible and several factors such as the catalyst, substrate and solvent can influence which pathway is favoured by an iridium catalyst.
6.2 ATH of imines
The significant difference between imines, alkenes and ketones is the basicity of the substrate due to the nitrogen lone pair, which in turn impacts mechanistic pathways adopted. In terms of reactive catalytic species formed in any hydrogenation protocol, one of the most important intermediates formed is the hydride species and subsequently whether hydride or proton transfer occurs first. The reaction conditions are obviously important and for Ir(III) catalysis where formic acid or iso-propanol are the source of the hydride, then for imines it is reasonable to assume that protonation of the substrate occurs first. Initial protonation significantly activates the functionality by increasing electrophilicity of the carbon hence facilitating hydride transfer which occurs subsequently.Even with Ir(I) catalysed reduction of imines with molecular hydrogen, it seems likely that the reaction involves rapid, reversible protonation of the substrate, followed by hydride transfer from the metal. A kinetic study on the hydrogenation of CN substrates with piano-stool ruthenium hydride complexes demonstrated that hydride transfer from the transition metal to the pre-formed iminium ion is the enantioselectivity-determining step with transfer of hydride being the rate-limiting step of the reaction.73,100,101 This ionic hydrogenation reaction mechanism allows selective reduction of polar CX double bonds over CC ones, with selectivity and rate all appearing to depend on H− transfer as the key step.73,102
Due to differences in basicity and susceptibility to nucleophilic attack as well as binding mode, imine hydrogenation may follow different pathways from ketonic reduction, although they do share some similarities. The control of stereochemistry in ketones is due to a favourable π/CH interaction between the hydrogen atom on the η6-arene ligand and the aromatic ring of the substrate. The origin of enantioselection with respect to the transfer hydrogenation of imines using Noyori/CATHy catalysts is unclear. The transfer hydrogenation of imines is rather unusual as it leads to chiral amines with the opposite stereochemistry from that expected applying a similar rational. Various transition states have been proposed that could result in the correct product with the correct configuration103,104 (Scheme 40). The suggestions are based on interactions that lead to the substrate adopting configurations that allow for transfer hydrogenation to occur yielding the enantiomeric product.
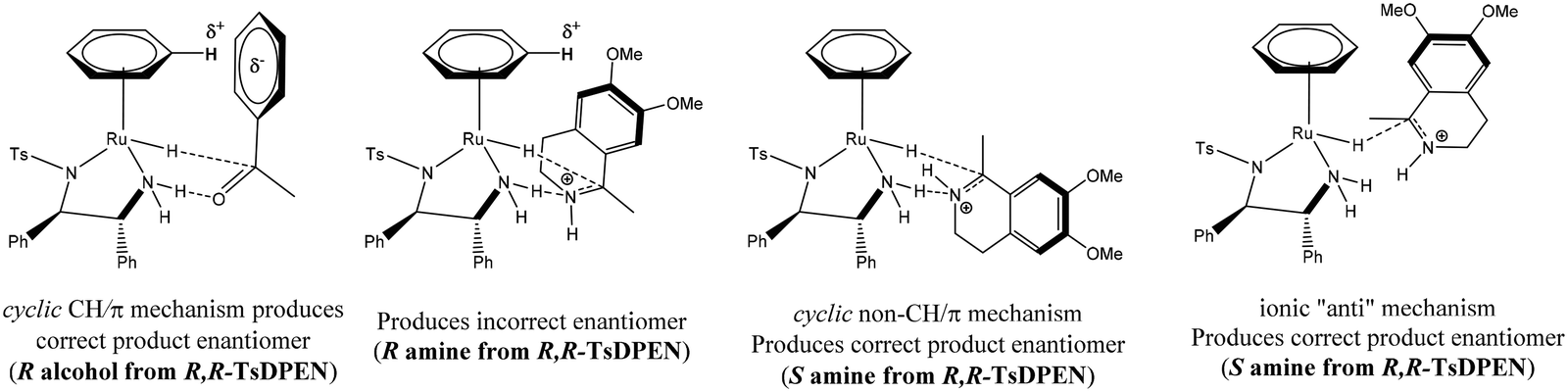 | ||
| Scheme 40 Consideration of interactions in the enantioselectivity of imine and ketone reduction to afford the correct product. | ||
Though not conclusive, the study did confirm that the stereo-controlling effects observed in ketonic transfer hydrogenation cannot simply be superimposed onto their imine counterparts as this results in the ‘wrong’ product.
An outer-sphere mechanism for the iridacycle mediated hydrogenation of simple imines (Scheme 41) is initiated by chloride dissociation affording the catalytic species. The next step is hydride abstraction from the coordinated formate, releasing CO2 and generating the hydridic species, subsequently transferring hydride-ion to the protonated imine substrate. Kinetic measurements showed that the hydrogenation rate is second order in HCO2H concentration, first order in catalyst and zero order in imine. Using DCO2D in place of HCO2H yielded a kH/kD of 1.9, supporting Ir-hydride formation being the turnover-limiting step. Hydrogen bonding of MeOH to the formate anion stabilises the ion pair.105
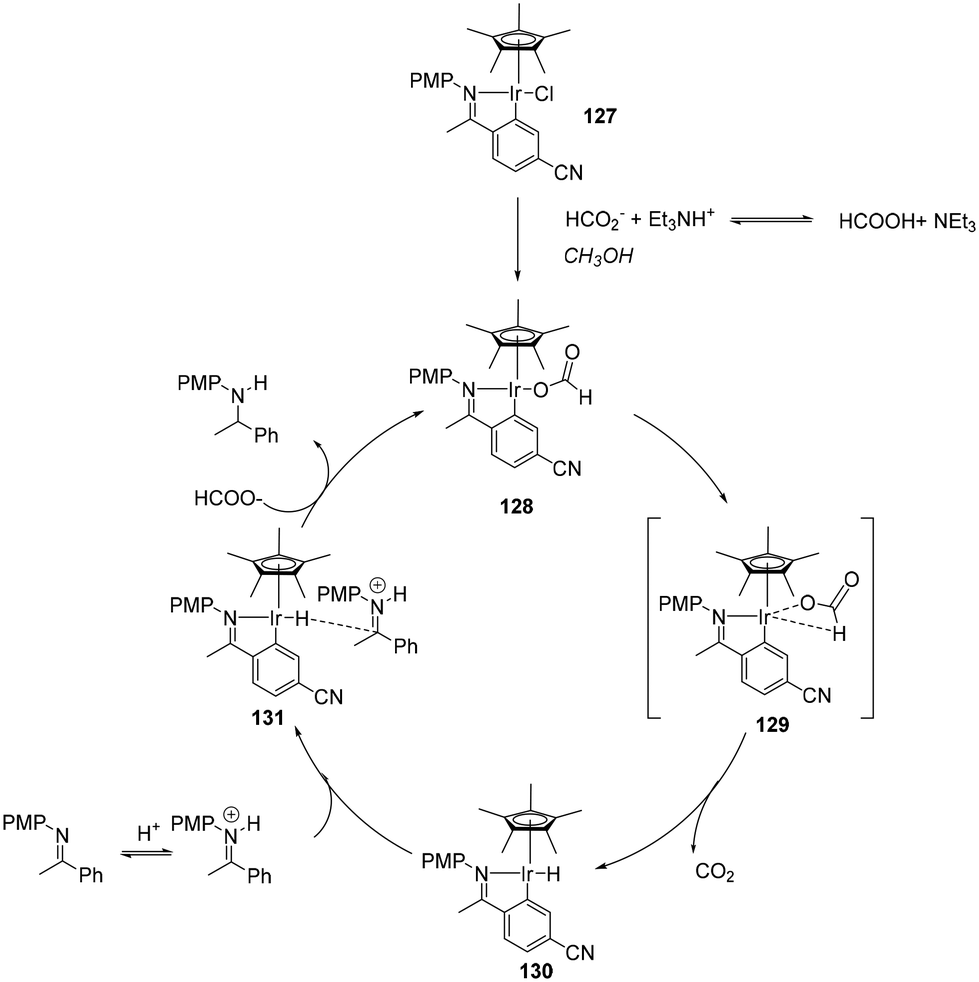 | ||
| Scheme 41 ATH of acyclic imines with cyclometalated 127. | ||
The Ir(III)-catalysed asymmetric transfer hydrogenation of 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline 132 (Scheme 42a) uncovered some unusual enantiomeric excess profiles for formation of the respective product amines 133 using the Ir(III) catalyst IrCp*I (R,R TsDPEN) in acetonitrile or DCM (Scheme 42) under acidic conditions.106 In general, enantioselectivity arises from differences in transition state stabilities for the two enantiomers with common kinetic orders in their rate laws. However, if the formation of the two enantiomeric products follow different rate laws then enantioselectivity can arise simply from different rates as a function of concentration of substrates.
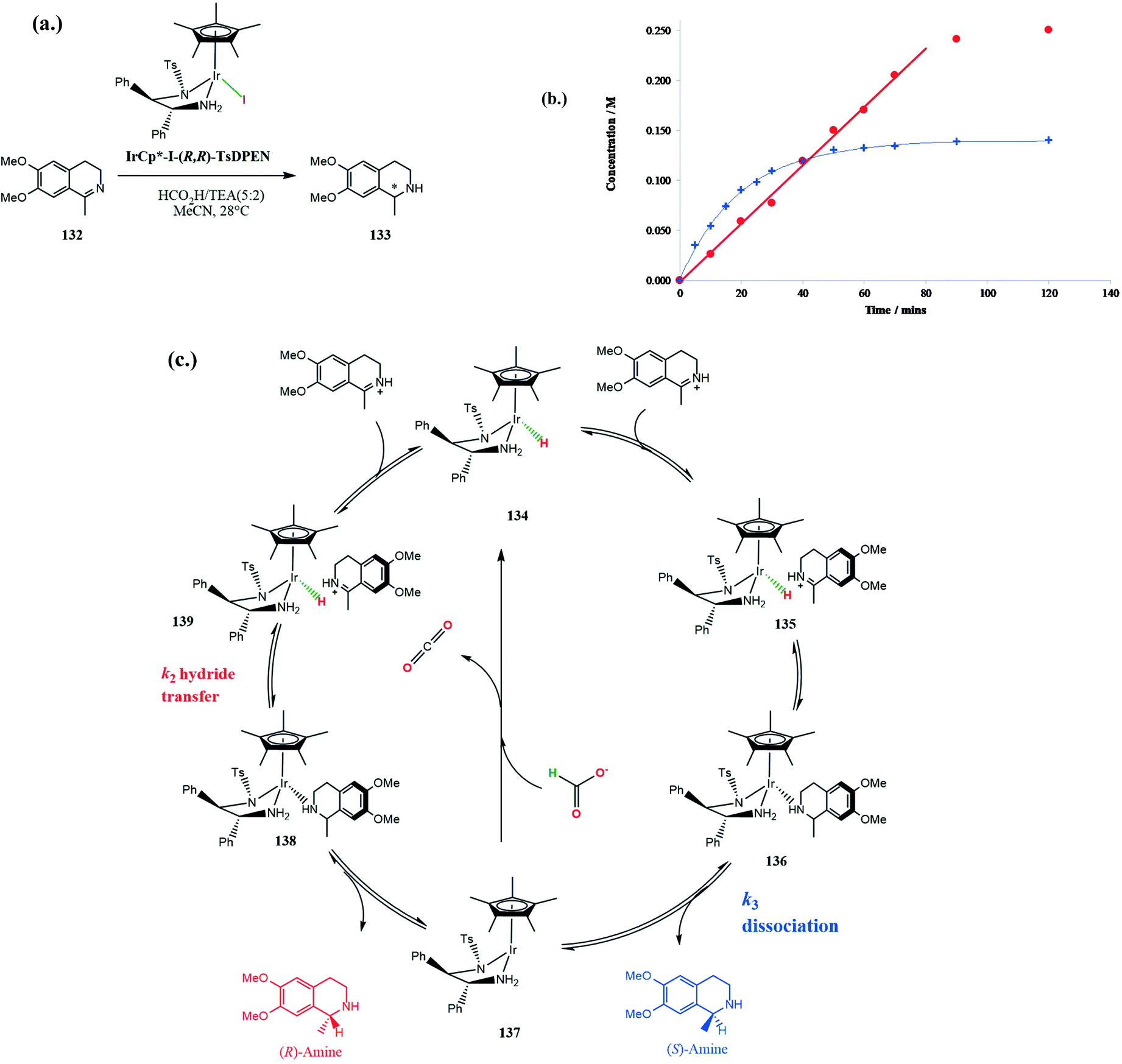 | ||
| Scheme 42 Kinetic and mechanistic considerations for the formation of (R) and (S) amine. | ||
Initially the reaction gives predominantly the (R)-enantiomer which then decreases significantly during further reaction. The enantioselectivity of the reduction is solvent dependent, with faster rates in dichloromethane but greater enantioselectivity in acetonitrile. The changes in ee as the reaction proceeds is a result of the rate of formation of the (R)-enantiomer following observed pseudo first order kinetics whereas that for the (S)-enantiomer is pseudo zero order. Consequently, the rate of formation of the (R)-enantiomer decreases as the concentration of imine decreases whereas that for (S)-enantiomer remains constant and so the relative rates of formation of the two enantiomers changes with time as the reaction proceeds (Scheme 42b).
Formation of the (S)-amine product involves rate-limiting dissociation from the catalyst 136, step k3 (Scheme 42c) giving rise to zero-order kinetics. The rate-limiting step for formation of the (R)-amine is hydride transfer 139, supported by a H/D kinetic isotope effect, and so exhibiting normal first-order kinetics. The rate of formation of the (S)-amine is independent of imine concentration. Alternatively, they may be two distinct catalytic species, each responsible for the formation of the respective enantiomers.106 Using a series of 1-fluorinated methyl-3,4-dihydroisoquinolines as substrates changes the rate-limiting step of dissociation of the (S)-amine product from Ir(III) k3, so that both enantiomers are formed with first-order kinetics.107 This results in almost complete removal of the enantioselectivity of the reduction.
It has been suggested that reduction of imines using transition metal complexes occurs through the neutral imine rather than the more reactive iminium-ion.108 However, α-substituted imines with electron-withdrawing fluorinated groups make protonation more difficult but on the other hand enhance the electrophilicity of the imine carbon so facilitating nucleophilic attack. The α-difluoromethyl (CHF2) 3,4-dihydroisoquinoline has a much reduced pKa so there is little protonated species present under the reaction conditions and it is 10-fold less reactive than the α-monofluoromethyl imine in the cyclopentadienyl Ir(III) complex catalysed reduction in acetonitrile under the acidic conditions of a 5:2 ratio of formic acid:triethylamine. Despite a more electrophilic imine carbon the α-trifluoromethyl derivative is completely unreactive towards the cyclopentadienyl Ir(III) complex catalysed reduction, showing that the iminium ion is the reactive species.107 It appears in this case that selectivity is almost exclusively due to the different kinetic orders for the rates of formation of the two enantiomers. Changing the ligand to S,S-TsDPEN rather than R,R inverts the kinetic orders of the rates of formation of the two enantiomeric product amines.106,107
6.3 ATH of ketones
A lithium mediated hydrogenation of ketones with a novel class of air-stable tridentate ferrocene-based amino-phosphine sulfonamide (f-amphamide) ligands displays excellent activity in the iridium-catalyzed asymmetric hydrogenation of aryl ketones (Scheme 43). Lithium oxide successfully promoted the hydrogenation of the carbonyl group. The catalytic cycle involves enantio-determining hydride transfer from Ir centre to the keto carbon and the H2 activation process to yield the chiral product and regenerate the catalyst. TS1(S) leading to (S)-alcohol 142a was found to be more stable than (R)-alcohol TS 1 (R) by 2.8 kcal mol−1142b, leading to 98% ee. The origin of enantioselectivity is attributed to greater steric repulsion between Ph and P of the ligand and the substrate Ph of TS1(R) and TS1 (S). This repulsion results in different bond lengths (Ir–H and C–H) in the two TS's with TS1(R) 142b having longer distances than its counterpart. The repulsion leads to better overlap between the d-orbitals of the metal centre and the TS1(S) than TS1(R).109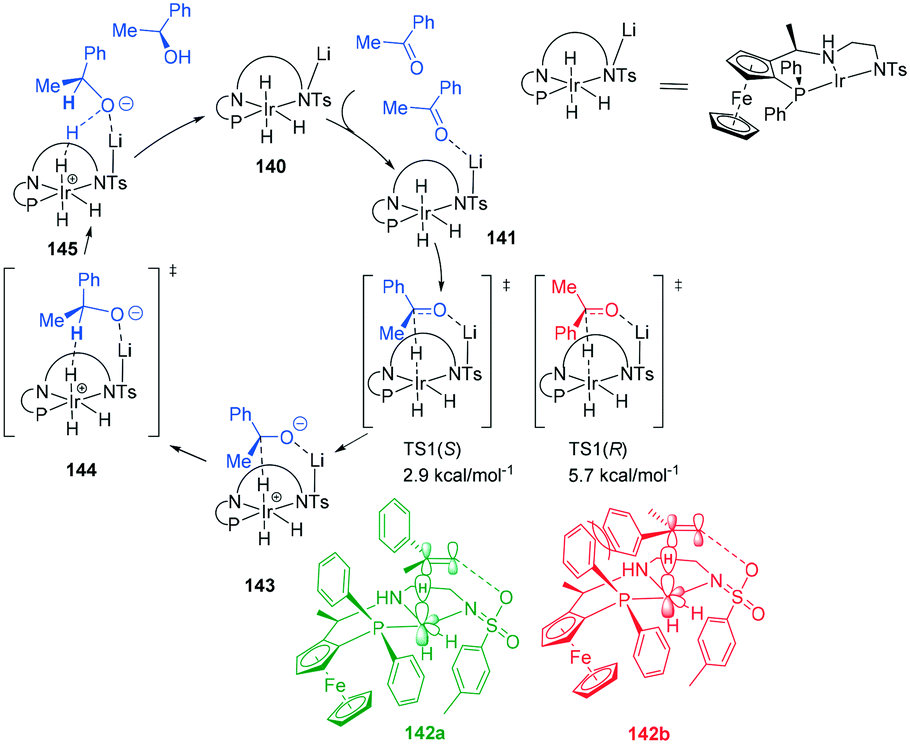 | ||
| Scheme 43 Lithium assisted ATH of ketones, showing the favoured TS1(S) leading to 98% ee. | ||
Metal-bifunctional complexes provide a different approach in hydrogenation of ketones. The presence of a proton source on a ligand provides insights into the reduction process. Ferrocene-based amino-phosphine-alcohol ligands (f-amphol ligand) are efficient in the ATH of various ketones affording excellent enantioselectivities (up to 99.9% ee) and high activities (TON up to 200000) (Scheme 44).110 DFT calculations suggest that the hydroxyl group of f-amphol ligands plays a key role in obtaining high reactivities and excellent enantioselectivities. The f-amphol ligand contains both NH and OH labile protons. Calculated TS1 details the approach of the ketone on the Re-face, generating the (R)-alcohol whereas approach from the Si-face as in TS2 generates the (S)-alcohol. The higher free energy of TS2 of 3.3 kcal mol−1 highlights the kinetically unfavourable pathway. ATH at the amino side was also considered with the Re-face approach occurring through TS4 and Si-face approach occurring through TS5. The free energy of TS4 is higher than that of TS1, indicating the preferential ATH of acetophenone at the hydroxyl site. This study confirmed that the generation of (R)-alcohol was favoured in this catalytic protocol.110
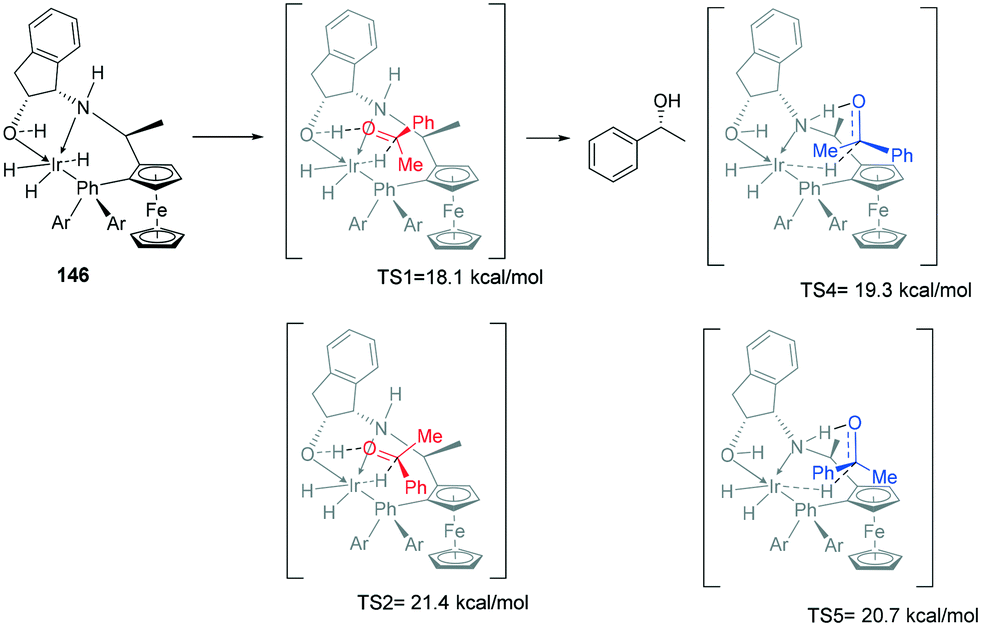 | ||
| Scheme 44 Metal bifunctional complexes TS, showing the four proposed TS's with the DFT calculations predicting TS1 as being the most stable, leading to 99.9% ee. | ||
7. Outlook
The use of iridium in catalysis, both homogeneous and heterogeneous, remains an exciting topic with many new developments, not only for hydrogenation/reduction reactions but for many other reactions.111–113 The observation that enantioselectivity of some reactions can result from enantiomers being formed by different kinetic orders106,107 highlights the importance and requirement for careful kinetic studies. The field is ripe for the application of physical organic techniques including linear free-energy relationships and kinetic isotope effects.Conflicts of interest
The authors declare no conflict of interest.References
- R. E. Gawley, J. Org. Chem., 2006, 71, 2411–2416 CrossRef CAS PubMed.
- R. P. Bell, J. E. Critchlow and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1974, 66–70 RSC.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2010, 16, 4003–4009 CrossRef CAS PubMed.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, John Wiley & Sons, 4th edn, 2011 Search PubMed.
- P. S. Pregosin, Pure Appl. Chem., 2009, 81, 615–633 CAS.
- M. I. Page and A. Williams, Organic and Bio-organic Mechanisms, Pearson Education Limited, Harlow, 1997, vol. 1 Search PubMed.
- B. Villa-Marcos, W. Tang, X. Wu and J. Xiao, Org. Biomol. Chem., 2013, 11, 6934–6939 RSC.
- P. G. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2666–2694 CrossRef CAS.
- Y. Schramm, F. Barrios-Landeros and A. Pfaltz, Chem. Sci., 2013, 4, 2760–2766 RSC.
- C. Réthoré, F. Riobé, M. Fourmigué, N. Avarvari, I. Suisse and F. Agbossou-Niedercorn, Tetrahedron: Asymmetry, 2007, 18, 1877–1882 CrossRef.
- S. Y. Shirai, H. Nara, Y. Kayaki and T. Ikariya, Organometallics, 2009, 28, 802–809 CrossRef CAS.
- L. Vaska and J. W. DiLuzio, J. Am. Chem. Soc., 1961, 83, 2784–2785 CrossRef CAS.
- R. U. Kirss, Bull. Hist. Chem., 2013, 38, 52–60 CAS.
- A. Cadu and P. G. Andersson, Dalton Trans., 2013, 42, 14345–14356 RSC.
- P. A. Fryzuk and M. D. MacNeil, Organometallics, 1983, 2, 682–684 CrossRef.
- J. A. Schrock and R. R. Osborn, J. Am. Chem. Soc., 1976, 98, 2143–2147 CrossRef.
- R. H. Crabtree and G. E. Morris, J. Organomet. Chem., 1977, 135, 395–403 CrossRef CAS.
- J. W. Suggs, S. D. Cox, R. H. Crabtree and J. M. Quirk, Tetrahedron Lett., 1981, 22, 303–306 CrossRef CAS.
- G. Stork and D. E. Kahne, J. Am. Chem. Soc., 1983, 105, 1072–1073 CrossRef CAS.
- R. H. Crabtree and M. W. Davis, Organometallics, 1983, 2, 681–682 CrossRef CAS.
- M. W. Davis and R. H. Crabtree, J. Org. Chem., 1986, 51, 2655–2661 CrossRef.
- A. G. Schultz, P. J. McCloskey and J. J. Court, J. Am. Chem. Soc., 1987, 109, 6493–6502 CrossRef CAS.
- H. Takaya, T. Ohta, N. Sayo, H. Kumobayashi, S. Akutagawa, S. Inoue, I. Kasahara and R. Noyori, J. Am. Chem. Soc., 1987, 109, 1596–1597 CrossRef CAS.
- R. Sablong and J. A. Osborn, Tetrahedron Lett., 1996, 37, 4937–4940 CrossRef CAS.
- F. Spindler, B. Pugin and H.-U. Blaser, Angew. Chem., Int. Ed. Engl., 1990, 29, 558–559 CrossRef.
- C. Bianchini, P. Barbaro, G. Scapacci, E. Farnetti and M. Graziani, Organometallics, 1998, 17, 3308–3310 CrossRef CAS.
- D. Xiao and X. Zhang, Angew. Chem., Int. Ed., 2001, 40, 3425–3428 CrossRef CAS PubMed.
- K. Morimoto, T. Nakajima and N. Achiwa, Chem. Pharm. Bull., 1994, 42, 1951–1953 CrossRef.
- H. U. Blaser, B. Pugin, F. Spindler, E. Mejía and A. Togni, Josiphos Ligands: From Discovery to Technical Applications, Top. Catal., 2002, 19, 3, DOI:10.1023/A:1013832630565.
- R. Dorta, D. Broggini, R. Stoop, H. Rüegger, F. Spindler and A. Togni, Chem. – Eur. J., 2004, 10, 267–278 CrossRef CAS PubMed.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 426–426 CrossRef CAS.
- S. M. Lu, Y. Q. Wang, X. W. Han and Y. G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260–2263 CrossRef CAS PubMed.
- T. Morimoto, N. Nakajima and K. Achiwa, Synlett, 1995, 7, 748–750 CrossRef.
- C. J. Hou, Y. H. Wang, Z. Zheng, J. Xu and X. P. Hu, Org. Lett., 2012, 14, 3554–3557 CrossRef CAS PubMed.
- W. B. Wang, S. M. Lu, P. Y. Yang, X. W. Han and Y. G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536–10537 CrossRef CAS PubMed.
- S. M. Lu, X. W. Han and Y. G. Zhou, Adv. Synth. Catal., 2004, 346, 909–912 CrossRef CAS.
- G. Hou, R. Tao, Y. Sun, X. Zhang and F. Gosselin, J. Am. Chem. Soc., 2010, 132, 2124–2125 CrossRef CAS PubMed.
- K. Gao, C. Bin Yu, W. Li, Y. G. Zhou and X. Zhang, Chem. Commun., 2011, 47, 7845–7847 RSC.
- T. Kazuhide, O. Jun-ichiro, Y. Tsuneaki and K. Yasutaka, Chem. Lett., 1995, 955–956 Search PubMed.
- Y. Chi, Y. G. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120–4122 CrossRef CAS PubMed.
- H. L. Kim, L. Xu, L. Feng, Q. H. Fan, L. L. Fuk, W. H. Lo and A. S. C. Chan, Adv. Synth. Catal., 2005, 347, 1755–1758 CrossRef.
- W. J. Tang, S. F. Zhu, L. J. Xu, Q. L. Zhou, Q. H. Fan, H. F. Zhou, K. Lam and A. S. C. Chan, Chem. Commun., 2007, 613–615 RSC.
- W. Tang, L. Xu, Q. H. Fan, J. Wang, B. Fan, Z. Zhou, K. H. Lam and A. S. C. Chan, Angew. Chem., Int. Ed., 2009, 48, 9135–9138 CrossRef CAS PubMed.
- W. Tang, Y. Sun, X. Lijin, T. Wang, Q. Fan, K. H. Lam and A. S. C. Chan, Org. Biomol. Chem., 2010, 8, 3464–3471 RSC.
- X. Bin Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. F. Boogers and J. G. De Vries, Org. Lett., 2003, 5, 1503–1506 CrossRef PubMed.
- A. Pfaltz, Acta Chem. Scand., 1996, 50, 189–194 CrossRef CAS.
- P. von Matt and A. Pfaltz, Angew. Chem., Int. Ed. Engl., 1993, 32, 566–568 CrossRef.
- P. von Matt, O. Loiseleur, G. Koch, A. Pfaltz, C. Lefeber, T. Feucht and G. Helmchen, Tetrahedron: Asymmetry, 1994, 5, 573–584 CrossRef CAS.
- D. Müller, G. Umbricht, B. Weber and A. Pfaltz, Helv. Chim. Acta, 1991, 74, 232–240 CrossRef.
- S. Vargas, M. Rubio, A. Suárez and A. Pizzano, Tetrahedron Lett., 2005, 46, 2049–2052 CrossRef CAS.
- P. Schnider, G. Koch, R. Prétôt, G. Wang, F. M. Bohnen, C. Krüger and A. Pfaltz, Chem. – Eur. J., 1997, 3, 887–892 CrossRef CAS.
- C. Blanc, F. Agbossou-Niedercorn and G. Nowogrocki, Tetrahedron: Asymmetry, 2004, 15, 2159–2163 CrossRef CAS.
- A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 CrossRef CAS.
- A. Triforiova, J. S. Diesen, C. J. Chapman and P. G. Andersson, Org. Lett., 2004, 6, 3825–3827 CrossRef PubMed.
- S. Kainz, A. Brinkmann, W. Leitner and A. Pfaltz, J. Am. Chem. Soc., 1999, 121, 6421–6429 CrossRef CAS.
- M. Solinas, A. Pfaltz, P. G. Cozzi and W. Leitner, J. Am. Chem. Soc., 2004, 126, 16142–16147 CrossRef CAS PubMed.
- S. Vargas, M. Rubio, A. Suárez, D. Del Río, E. Álvarez and A. Pizzano, Organometallics, 2006, 25, 961–973 CrossRef CAS.
- N. Mršić, A. J. Minnaard, B. L. Feringa and J. G. De Vries, J. Am. Chem. Soc., 2009, 131, 8358–8359 CrossRef PubMed.
- A. Baeza and A. Pfaltz, Chem. – Eur. J., 2009, 15, 2266–2269 CrossRef CAS PubMed.
- M. N. Cheemala and P. Knochel, Org. Lett., 2007, 9, 3089–3092 CrossRef CAS PubMed.
- S. F. Zhu, J. B. Xie, Y. Z. Zhang, S. Li and Q. L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886–12891 CrossRef CAS PubMed.
- Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345–5349 CrossRef CAS PubMed.
- A. Iimuro, K. Yamaji, S. Kandula, T. Nagano, Y. Kita and K. Mashima, Angew. Chem., Int. Ed., 2013, 52, 2046–2050 CrossRef CAS.
- H. Tadaoka, D. Cartigny, T. Nagano, T. Gosavi, T. Ayad, J. P. Genêt, T. Ohshima, V. Ratovelomanana-Vidal and K. Mashima, Chem. – Eur. J., 2009, 15, 9990–9994 CrossRef CAS PubMed.
- Z. W. Li, T. L. Wang, Y. M. He, Z. J. Wang, Q. H. Fan, J. Pan and L. J. Xu, Org. Lett., 2008, 10, 5265–5268 CrossRef CAS PubMed.
- C. Tian, L. Gong and E. Meggers, Chem. Commun., 2016, 52, 4207–4210 RSC.
- G. J. Kubas, J. Organomet. Chem., 2014, 751, 33–49 CrossRef CAS.
- M. Ito and T. Ikariya, Chem. Commun., 2007, 5134–5142 RSC.
- M. Peruzzini and R. Poli, Recent Advances in Hydride Chemistry, Elsevier SA, Amsterdam, 2001 Search PubMed.
- O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21–27 RSC.
- K. H. Hopmann and A. Bayer, Organometallics, 2011, 30, 2483–2497 CrossRef CAS.
- R. M. Bullock, Chem. – Eur. J., 2004, 10, 2366–2374 CrossRef CAS PubMed.
- N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS.
- J. B. Johnson and J. E. Bäckvall, J. Org. Chem., 2003, 68, 7681–7684 CrossRef CAS PubMed.
- Y. Huang, S. Liu, Y. Liu, Y. Chen, M. Weisel, R. T. Williamson, I. W. Davies and X. Zhang, Tetrahedron, 2018, 74, 2182–2190 CrossRef CAS.
- J. S. M. Samec, A. H. Éll and J. E. Bäckvall, Chem. Commun., 2004, 2748–2749 RSC.
- A. Comas-Vives, G. Ujaque and A. Lledós, Inner- and Outer-Sphere Hydrogenation Mechanisms: A Computational Perspective, Elsevier Inc., 2010, vol. 62 Search PubMed.
- B. Villa-Marcos, C. Li, K. R. Mulholland, P. J. Hogan and J. Xiao, Molecules, 2010, 15, 2453–2472 CrossRef CAS PubMed.
- G. Koch, G. C. Lloyd-Jones, O. Loiseleur, A. Pfaltz, R. Prétôt, S. Schaffner, P. Schnider and P. von Matt, Recl. Trav. Chim. Pays-Bas, 1995, 114, 206–210 CrossRef CAS.
- M. Martín, E. Sola, S. Tejero, J. A. López and L. A. Oro, Chem. – Eur. J., 2006, 12, 4043–4056 CrossRef PubMed.
- D. Zhao, L. Candish, D. Paul and F. Glorius, ACS Catal., 2016, 6, 5978–5988 CrossRef CAS.
- S. Gülcemal, A. G. Gökçe and B. Çetinkaya, Inorg. Chem., 2013, 52, 10601–10609 CrossRef PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- C. Diez and U. Nagel, Appl. Organomet. Chem., 2010, 24, 509–516 CrossRef CAS.
- F. Aznarez, M. Iglesias, A. Hepp, B. Veit, P. J. Sanz Miguel, L. A. Oro, G. X. Jin and F. E. Hahn, Eur. J. Inorg. Chem., 2016, 2016, 4598–4603 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- H. Chiyojima and S. Sakaguchi, Tetrahedron Lett., 2011, 52, 6788–6791 CrossRef CAS.
- Y. Li, M. Lei, W. Yuan, E. Meggers and L. Gong, Chem. Commun., 2017, 53, 8089–8092 RSC.
- G. E. Dobereiner, A. Nova, N. D. Schley, N. Hazari, S. J. Miller, O. Eisenstein, R. H. Crabtree and I. C. Gerhardt, J. Am. Chem. Soc., 2011, 133(19), 7547–7562 CrossRef CAS PubMed.
- J. M. Hayes, E. Deydier, G. Ujaque, A. Lledós, R. Malacea-Kabbara, E. Manoury, S. Vincendeau and R. Poli, ACS Catal., 2015, 5, 4368–4376 CrossRef CAS.
- M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC.
- N. García, E. A. Jaseer, J. Munarriz, P. J. Sanz Miguel, V. Polo, M. Iglesias and L. A. Oro, Eur. J. Inorg. Chem., 2015, 2015, 4388–4395 CrossRef.
- J. W. Handgraaf, J. N. H. Reek and E. J. Meijer, Organometallics, 2003, 22, 3150–3157 CrossRef CAS.
- O. Pàmies and J. E. Bäckvall, Chem. – Eur. J., 2001, 7, 5052–5058 CrossRef.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- R. Soni, F. K. Cheung, G. C. Clarkson, J. E. D. Martins, M. A. Graham and M. Wills, Org. Biomol. Chem., 2011, 9, 3290–3294 RSC.
- C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS.
- W. Wu, S. Liu, M. Duan, X. Tan, C. Chen, Y. Xie, Y. Lan, X. Q. Dong and X. Zhang, Org. Lett., 2016, 18, 2938–2941 CrossRef CAS PubMed.
- N. Onishi, M. Z. Ertem, S. Xu, A. Tsurusaki, Y. Manaka, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2016, 6, 988–992 RSC.
- V. Herrera, B. Muñoz, V. Landaeta and N. Canudas, J. Mol. Catal. A: Chem., 2001, 174, 141–149 CrossRef CAS.
- M. P. Magee and J. R. Norton, J. Am. Chem. Soc., 2001, 123, 1778–1779 CrossRef CAS PubMed.
- J. E. D. Martins, G. J. Clarkson and M. Wills, Org. Lett., 2009, 11, 847–850 CrossRef CAS PubMed.
- R. Soni, K. E. Jolley, S. Gosiewska, G. J. Clarkson, Z. Fang, T. H. Hall, B. N. Treloar, R. C. Knighton and M. Wills, Organometallics, 2018, 37, 48–64 CrossRef CAS.
- C. Wang and J. Xiao, Chem. Commun., 2017, 53, 3399–3411 RSC.
- M. J. Stirling, G. Sweeney, K. Macrory, A. J. Blacker and M. I. Page, Org. Biomol. Chem., 2016, 14, 3614–3622 RSC.
- J. M. Mwansa, M. J. Stirling and M. I. Page, Pure Appl. Chem. DOI:10.1515/pac-2019-0222.
- D. G. Blackmond, M. Ropic and M. Stefinovic, Org. Process Res. Dev., 2006, 10, 457–463 CrossRef CAS.
- Z. Liang, T. Yang, G. Gu, L. Dang and X. Zhang, Chin. J. Chem., 2018, 36, 851–856 CrossRef CAS.
- J. Yu, M. Duan, W. Wu, X. Qi, P. Xue, Y. Lan, X. Q. Dong and X. Zhang, Chem. – Eur. J., 2017, 23, 970–975 CrossRef CAS PubMed.
- Q. Cheng, H. F. Tu, C. Zheng, J. P. Qu, G. Helmchen and S. L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- M. M. Zhang, Y. N. Wang, B. C. Wang, X. W. Chen, L. Q. Lu and W. J. Xiao, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- A. G. Schafer and S. B. Blakey, Chem. Soc. Rev., 2015, 44, 5969–5980 RSC.
| This journal is © The Royal Society of Chemistry 2020 |