
Catalytic mechanisms of oxygen-containing groups over vanadium active sites in an Al-MCM-41 framework for production of 2,5-diformylfuran from 5-hydroxymethylfurfural†
Li-Juan
Liu
 a,
Zhao-Meng
Wang
a,
Ya-Jing
Lyu
b,
Jin-Feng
Zhang
a,
Zhou
Huang
a,
Ting
Qi
a,
Zhen-Bing
Si
a,
Hua-Qing
Yang
*a and
Chang-Wei
Hu
b
a,
Zhao-Meng
Wang
a,
Ya-Jing
Lyu
b,
Jin-Feng
Zhang
a,
Zhou
Huang
a,
Ting
Qi
a,
Zhen-Bing
Si
a,
Hua-Qing
Yang
*a and
Chang-Wei
Hu
b
aCollege of Chemical Engineering, Sichuan University, Chengdu, Sichuan 610065, P.R. China. E-mail: huaqingyang@scu.edu.cn; Fax: +028 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 415608; Tel: +028 85415608
415608; Tel: +028 85415608
bKey Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, P.R. China
First published on 22nd November 2019
Abstract
V-containing catalysts exhibit good catalytic performance toward the selective oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF). Here, we report our study on the catalytic mechanism of –(SiO)3V(O) ([V-0]) and –(SiO)2V(O)(OH) ([V-1]) on a V-doped Al-MCM-41 pore model (V/Al-MCM-41) for the aerobic oxidation of HMF to DFF. For the two active sites, there are three types of oxygen-containing functional groups, which are hydroxyl-oxygen ([V–OH]), lattice-oxygen ([V–O–Si]), and terminal-oxygen ([V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]). We show that the catalytic cycle involves two HMF molecules, and there are mainly two activation steps, i.e., both O–H and C–H bond cleavages of HMF, and the rate-determining step is associated with the C–H bond cleavage of the first HMF molecule. We illustrate the efficiency of the catalytic contribution as [V–OH] > [V–O–Si] > [VO], and the [V-1] active site with a hydroxyl group displays higher catalytic activity than the [V-0] active site without a hydroxyl group. The present study not only brings an in-depth understanding of the activation of both O–H and C–H bonds which has been proposed based on experimental results for biomass molecules, but also makes one step forward toward the mechanism-guided design and synthesis of efficient, environmentally-friendly, and low temperature recyclable heterogeneous catalysts.
O]). We show that the catalytic cycle involves two HMF molecules, and there are mainly two activation steps, i.e., both O–H and C–H bond cleavages of HMF, and the rate-determining step is associated with the C–H bond cleavage of the first HMF molecule. We illustrate the efficiency of the catalytic contribution as [V–OH] > [V–O–Si] > [VO], and the [V-1] active site with a hydroxyl group displays higher catalytic activity than the [V-0] active site without a hydroxyl group. The present study not only brings an in-depth understanding of the activation of both O–H and C–H bonds which has been proposed based on experimental results for biomass molecules, but also makes one step forward toward the mechanism-guided design and synthesis of efficient, environmentally-friendly, and low temperature recyclable heterogeneous catalysts.
1. Introduction
With gradually diminishing fossil resources, biomass, as a green and renewable carbon resource, has attracted widespread attention for the sustainable production of chemicals and fuels.1,2 Particularly, carbohydrates, as the major component of biomass, can be effectively converted into furans, e.g., 5-hydroxymethylfurfural (HMF).3 As a building block platform chemical bridging biomass and petrochemicals, HMF can be used to produce numerous high-value-added chemicals, such as 2,5-diformylfuran (DFF).4,5 As a partially oxidized product of HMF, DFF can be used as a versatile intermediate for the synthesis of functional polymers, pharmaceuticals, and antifungal agents.6,7For the oxidation of HMF into DFF, incipiently, a stoichiometric amount of oxidant is employed, which is high cost and generates a large amount of waste.8,9 From the view point of sustainable chemistry, the oxidation of HMF is conducted using molecular oxygen as the oxidant together with heterogeneous catalysts. Toward the aerobic oxidation of HMF, various heterogeneous catalytic systems were developed, e.g., noble metal-catalysts10–12 and transition metal-based catalysts.5,13 Compared with the use of noble metal catalysts, the use of transition metal based catalysts is much more preferable due to its low cost. Among the transition metal-based catalysts, vanadium-based catalysts have shown high catalytic activity. However, the topological structures of V species and the variations in reactions are complicated because vanadium has abundant coordination modes.14–16 Moreover, these vanadium-based catalysts also exhibit some obvious disadvantages, e.g., immobilized vanadyl–pyridine complexes with vanadium leaching,17 V2O5 supported on H-beta zeolite with fast deactivation,18 vanadium supported activated carbon in MIBK with difficulty in extracting solvents,19 immobilized vanadyl on amino modified SBA-15 with a comparatively low selectivity,8 vanadium oxide supported on copper-containing mordenite (MOR)-type zeolite with the high-boiling dimethyl sulfoxide (DMSO) solvent,20 and nanobelt-arrayed vanadium oxide hierarchical microspheres with a relatively high O2 pressure.21 Shortly, although vanadium-based catalysts have been widely used in the production of DFF from HMF, one of the most urgent problems is to develop highly stable heterogeneous vanadium-based catalysts. To achieve this end, it is necessary to gain a deep insight into the catalytic nature of V-containing active sites.
Because the instability of HMF limits pure HMF as a raw feedstock for the large scale synthesis of DFF, the direct synthesis of DFF from carbohydrates has received widespread attention, owing to the easy availability of raw materials. Currently, for the direct transformation of carbohydrates into DFF, a one-pot approach has been developed, involving the selective dehydration of carbohydrates into HMF over acid sites and the successive oxidation of HMF to DFF over redox sites,22e.g., nanobelt α-CuV2O6 with a mesoporous poly(ionic liquid),23 vanadium oxide supported high-silica MOR zeolites in the presence of hydrochloric acid,24 vanadium-containing all-silica beta-zeolites in the presence of sulfuric acid,25 and MoO3-containing protonated nitrogen doped carbon.26 The one-pot approach has achieved significant progress in the production of DFF from fructose. However, the carbohydrates were easily oxidized into humins or other undesired by-products due to the co-existence of acid and redox sites on the catalysts.27 Thus, it remains a challenge to develop a low cost, highly efficient and stable heterogeneous catalyst for isomerization, dehydration, and oxidation, which may directly convert carbohydrates into DFF.
Particularly, the mesoporous MCM-41, as one of the most common silicas, possesses a hexagonal array of roughly cylindrical, straight, and unconnected pores with an engineered diameter. It has been regarded as an environment-friendly catalyst, owning to its good thermal stability, large pore size, high surface areas, and easy preparation. Moreover, the incorporation of transition metals into the framework of all-silica MCM-41 may provide many promising heterogeneous catalysts, e.g., Ti,28 Fe and Co,29 V,30–32 Mo,33 and so on. Encouragingly, under the normal experimental temperature of about 400 K for the conversion of fructose into DFF,20,23–25 MCM-41 is supposed to be stable in the liquid phase, since the hydrothermal synthesis temperature is up to 438 K.34 Moreover, the mesoporous aluminium doped MCM-41 silica (Al-MCM-41) catalyst exhibits good catalytic performance toward the direct conversion of glucose into HMF.35 Inspired by the above studies, consolidating both acid sites over Al-MCM-41 toward the dehydration of glucose into HMF and redox sites over V-containing species toward the oxidation of HMF to DFF, one can expect that V-doped Al-MCM-41 should display good catalytic performance for the direct synthesis of DFF from glucose. Considering the reaction mechanism for the aerobic oxidation of HMF to DFF, over V2O5@Cu-MOR, Wang's group was the first to propose a possible lattice oxygen vacancy-mediated mechanistic pathway.20 Over nanobelt-arrayed vanadium oxide hierarchical microspheres, Yang's group persuasively demonstrated that the (010) facet with the highest hydrogen adsorption capability of vanadyl group (VO) sites plays a crucial role in both O–H and C–H bonds cleavages of HMF before the reduction product V–OH species is further dehydrogenated by molecular oxygen.21 Nevertheless, toward the aerobic oxidation of HMF to DFF, the detailed catalytic mechanism of V-containing active sites over the Al-MCM-41 framework is lacking. In particular, the catalytic contribution of typical oxygen-containing groups in V-containing active sites over the Al-MCM-41 framework remains unclear at the molecular level.
In this study, on the Al-MCM-41 framework, two kinds of V-doped active sites are constructed, i.e., both hydroxyl-containing –(SiO)2V(OH)(O) ([V-1]) and hydroxyl-free –(SiO)3V(O) ([V-0]) active sites. Over these active sites, the catalytic mechanisms are established toward DFF production from the aerobic oxidation of HMF, involving a deep insight into the determining intermediate (TDI) and the determining transition state (TDTS) of the turnover frequency (TOF). The goals are as follows: (a) to ascertain the structure and stability of the V-containing active sites on the Al-MCM-41 framework, (b) to determine the catalytic performance of V-containing active sites on Al-MCM-41, and (c) to correlate the relationship between catalytic activity and functional groups of the V-containing active sites on Al-MCM-41 toward the aerobic oxidation of HMF to DFF.
2. Methodology
All calculations were performed using the QMERA module implemented in the Materials Studio 7.0 software package. The QMERA module combines quantum mechanical (QM) and molecular mechanics (MM) force field calculations using the ChemShell environment.36 For the QM region (V, Si, O, C, and H atoms), DMol3 was employed,37 using the generalized gradient approximation (GGA) with the density functional of Perdew, Burke, and Ernzerhof (PBE).38,39 This PBE method has been successfully applied to vanadium-containing systems.40,41 Furthermore, the double numerical plus polarization (DNP) basis sets are used in the QM region.42 For the MM region (Si, Al, O, and H atoms), the GULP module was applied,43 using the universal force field (UFF),44 which has been successfully employed to study zeolites.45,46 For the geometric optimization, the forces imposed on each atom were converged to be less than 0.002 Hartree Å−1, the total energy was converged to be less than 1.0 × 10−5 Hartree, and the displacement convergence was less than 5 × 10−3 Å. A smearing of 0.005 Hartree to the orbital occupation was applied to achieve an accurate electronic convergence. A real-space cutoff of 5.0 Å was used, which is sufficient to accurately evaluate the energies.For all the reactants, intermediates and products, the optimized geometric structures were verified to have real frequencies. The transformation pathways and transition state structures were calculated using the complete linear synchronous transit and quadratic synchronous transit (LST/QST) methods.47 Each transition state was identified to have only one imaginary frequency and its vibration mode had the right direction connecting the reactant and product.
The turnover frequency (TOF) of the catalytic cycle determines the efficiency of the catalyst. Based on the transition state theory (TST), the TOF can be computed through the energetic span,48–53 in which both the TOF determining transition state (TDTS) and the TOF determining intermediate (TDI) are ascertained. The rate constants k(T) were calculated according to conventional TST k′(T), including tunneling correction κ(T), as mentioned in our previous study.54,55 Considering the energetic span model, the used equations are described in the ESI.†
For the mesoporous MCM-41 pore model with a 42.8 × 64.2 × 42.8 Å3 matrix of amorphous silica, containing Si1242O2746H545, the initial structure was optimized, using the MM method. Then, for saving computing time, the semi porous surface of Al-MCM-41 was slabbed for optimization using the MM method, and the model comprised Si1118Al124O2746H669. After that, for the V-doped Al-MCM-41 model, the semi porous surface of V/Al-MCM-41 was constructed using the QM/MM methods, involving Si81Al9VO231H107 and Si78Al9VO225H107, respectively. For the V/Al-MCM-41 model, the QM region is associated with the vanadium atom, its adjacent oxygen atoms, silicon atoms and hydroxyl group (−(Si7O8)(SiO)3V(O)[V-0] and –(Si6O6)(SiO)2V(O)(OH)[V-1]), while the MM region is concerned with the remainder. Besides, the reactant substrates were also involved in the QM region.
3. Results and discussion
3.1. V-Doped Al-MCM-41 model
For the mesoporous MCM-41 pore model, the construction procedure has been described in our previous study, according to the procedure proposed by Pellenq and Tielens et al.,56,57 where two kinds of silicon atoms exist on the surface, i.e., −(SiO)3Si(OH) and –(SiO)2Si(OH)2.58Next, the Al-doped MCM-41 pore model (Al-MCM-41) was created by replacing one Si atom in the –(SiO)3Si(OH) and –(SiO)2Si(OH)2 moieties on the MCM-41 pore surface with an Al atom, based on the optimal mole ratio of Si:Al = 10:1 from the experimental Al-MCM-41.35 Herein, for maintaining the +3 valence state of the Al atom, an extra hydrogen atom was added, while the resultant water molecule was deleted, leading to two kinds of Al sites, i.e., −(SiO)3Al and –(SiO)2Al(OH). As reported in the literature,59–61 the Al-containing active sites play an important role in both the isomerization of glucose to fructose and the dehydration of fructose to HMF. However, the relevant reaction mechanism for the conversion of glucose to HMF catalyzed by the Al active sites on MCM-41 is outside of the present topic scope, which will be focused on in our future work.
Subsequently, for the V-doped Al-MCM-41 pore model (V/Al-MCM-41), the V-containing active sites were generated by replacing one Si atom in the –(SiO)3Si(OH) and –(SiO)2Si(OH)2 moieties with one V atom, keeping the +5 valence state of the V atom through deleting the hydrogen atom. Thereupon, there were two kinds of V-containing active sites on the pore surface, −(SiO)3V(O) ([V-0]) and –(SiO)2V(O)(OH) ([V-1]), as shown in Fig. 1. This construction method of surface models has been successfully applied to the Mo-doped MCM-41 pore model.58
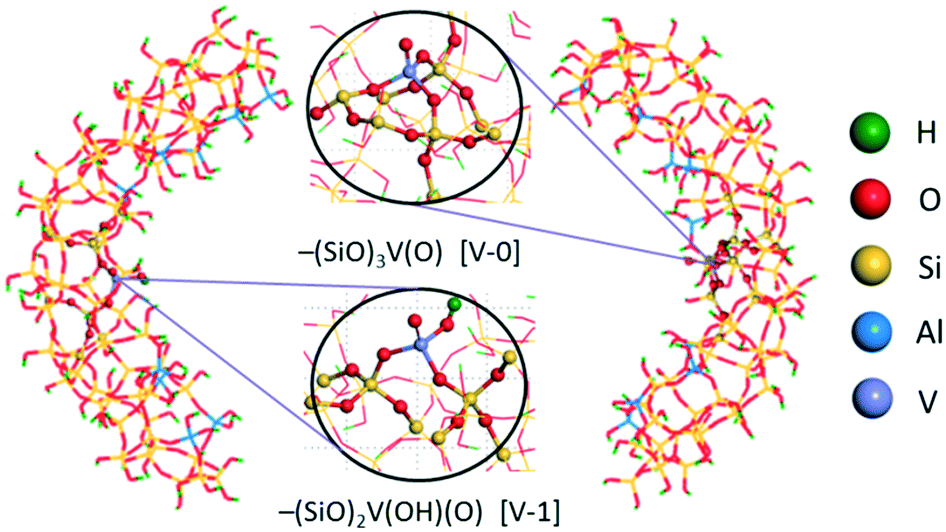 | ||
| Fig. 1 V/Al-MCM-41 QM/MM model, where the two kinds of V active sites, −(SiO)3V(O) (429 atoms, [V-0]) and –(SiO)2V(O)(OH) (420 atoms, [V-1]), are shown. Atoms shown in green, red, yellow, blue and purple represent H, O, Si, Al and V atoms, respectively. Spherical atoms are QM atoms, others are MM atoms. | ||
According to the reported method by Tielens' group,62 the stabilities of the doped two V active sites on Al-MCM-41, [V-0] and [V-1], are evaluated by the following reaction equation (eqn (1)):
| Na4V2O7 + 2[−(SiO)5(Si)2(OH)3]→ 2[−(SiO)5Si(OH)2−xV(OH)x(O)] + H2O + 2Na2SiO3 | (1) |
Then, we will examine the catalytic performances of both the [V-0] and [V-1] active sites toward the aerobic oxidation of HMF to DFF. We will mainly discuss the following reaction over the V/Al-MCM-41 catalyst:
| 2HMF + O2 → 2DFF + 2H2O | (2) |
Because the ground state of O2 molecules is the triplet state with the singlet state as the first excited state, particular attention is devoted to the possible occurrence of a two-state reactivity phenomenon. Thus, in the presence of O2, the potential energy profiles for the ground and the first excited states are investigated. The superscript prefixes “1” and “3” will be used to display the singlet and triplet states, respectively. Unless otherwise stated, the default state is referred to the ground singlet state.
3.2. Catalytic mechanism over the [V-0] active site
Based on relevant literature studies for the oxidation of HMF to DFF,58,63 over the [V-0] active site, reaction (2) can be divided into two reaction stages:| [V-0] + HMF + O2 → [V-0 + O] + DFF + H2O | (3) |
| [V-0 + O] + HMF → [V-0] + DFF + H2O | (4) |
Thus, we will discuss their thermodynamics and kinetics, vide infra.
 | ||
| Fig. 2 The geometric structures (a) and the schematic energy diagrams (b) for the reaction [V-0] + HMF + O2 → [V-0 + O] + DFF + H2O. Relative energies (kJ mol−1) for the corresponding species plus HMF + O2 relative to those for the reactants [V-0] + 2HMF + O2 are shown. For clarity, hydrogen atoms on the rings are not shown. Bond lengths are reported in Å. The blue, green, pink and yellow lines represent P3-th, P3-tb, P3-bt and P3-th2, respectively. | ||
For P3-th, at the beginning, the hydrogen atom of the –OH group in HMF interacts with the oxygen atom of –VO in the [V-0] active site through H-bonding, forming a molecular complex 0-IM1a. Next, from 0-IM1a, terminal-oxygen-mediated O–H bond cleavage takes place through a [2 + 2] addition reaction via a four-membered ring, 0-TS1a, leading to a hydroxylate 0-IM2a. After that, from 0-IM2a, hydroxyl-oxygen-mediated C–H bond cleavage occurs through a [1,4]-H shift via a five-membered ring 0-TS2a1, producing a DFF- and H2O-containing molecular complex 0-IM3a1. Then, from 0-IM3a1, there are two reaction pathways, i.e., preferential release of free H2O or DFF, resulting in 0-IM4a or [V-0+2H], denoted as P3-th1 and P3-th2, respectively. On the one hand, for P3-th1, from 0-IM4a, O2 is introduced, giving rise to an O2- and DFF-containing molecular complex 0-IM5a1. Because the singlet 0-IM5a1 lies 59.1 kJ mol−1 below the triplet 30-IM5a1, triplet–singlet spin-crossing may take place near 0-IM5a1. From 0-IM5a1, the MERP should advance on the singlet state. This may stem from the fact that the O2 in 0-IM5a1 is absorbed through a V–OO single bond, in which the Mayer bond orders of two V–OO bonds are 1.02 and 1.07, respectively. Afterward, 0-IM5a1 liberates the DFF molecule free, leaving [V-0 + O] aside. Herein, the triplet 3[V-0 + O] is located 91.3 kJ mol−1 above the singlet [V-0 + O]. This may originate from the fact that O2 is chemically adsorbed on the singlet [V-0 + O]. This can be evidenced by the 1.00 and 1.08 of the Mayer bond orders of two V–OO bonds, respectively, in the singlet [V-0 + O]. On the other hand, for P3-th2, from [V-0 + 2H], O2 is imported, forming an O2- and H2O-containing molecular complex 0-IM5a2. Because the singlet 0-IM5a2 lies 77.2 kJ mol−1 below the triplet 30-IM5a2, the triplet–singlet spin-crossing may take place near 0-IM5a2. From 0-IM5a2, the MERP should go along the singlet state. This may stem from the fact that the O2 in 0-IM5a2 is chemically absorbed through a V–OO single bond, in which the Mayer bond orders of two V–OO bonds are 1.02 and 0.97, respectively. Next, 0-IM5a2 releases the H2O molecule free, leaving a peroxide [V-0 + O] behind. It is obvious that the minimal energy reaction pathway (MERP) should go through P3-th1 other than P3-th2, because the energy height of the highest point (EHHP) in P3-th1 at 0-IM4a lies 35.1 kJ mol−1 below that in P3-th2 at [V-0 + 2H]. In the MERP, P3-th includes an EHHP of 124.4 kJ mol−1 at 0-TS2a1 and a highest energy barrier (HEB) of 95.3 kJ mol−1 at the reaction step 0-IM2a → 0-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage.
For P3-tb, from the reactants to 0-IM2a, the reaction pathway is identical to that for P3-th, which is concerned with the terminal-oxygen-mediated O–H bond cleavage. Then, from 0-IM2a, lattice-oxygen-mediated C–H bond cleavage takes place through a [1,4]-H shift via a five-membered ring 0-TS2a2, generating a DFF-containing molecular complex 0-IM3a2. After that, O2 is introduced, giving rise to an O2- and DFF-containing molecular complex 0-IM4b. Since the singlet 0-IM4b lies 41.7 kJ mol−1 below the triplet 30-IM4b, the triplet–singlet spin-crossing may occur near 0-IM4b. From 0-IM4b, the MERP should proceed along the singlet state. Afterwards, 0-IM4b sets the DFF molecule free, saving 0-IM5b aside. Next, from 0-IM5b, a [1,4]-H shift occurs via a five-membered ring 0-TS3b, leading to a peroxide 0-IM6b, in which the single-bond rotates easily to produce a more stable compound 0-IM7. With that, a [1,4]-H shift takes place again via a five-membered ring 0-TS4, yielding a H2O-containing molecular complex 0-IM8. Finally, 0-IM8 releases the H2O molecule free, leaving [V-0 + O] behind. In the MERP, P3-tb comprises an EHHP of 134.9 kJ mol−1 at 0-TS2a2 and a HEB of 105.8 kJ mol−1 at the reaction step 0-IM2a → 0-TS2a2 for the lattice-oxygen-mediated C–H bond cleavage.
For P3-bt, initially, the hydrogen atom of the –OH group in HMF interacts with the lattice-oxygen atom in the [V-0] active site through H-bonding, generating a molecular complex 0-IM1b. Next, from 0-IM1b, lattice-oxygen-mediated O–H bond cleavage occurs through a [1,3]-H shift via a four-membered ring 0-TS1b, resulting in a hydroxylate 0-IM2b. After that, from 0-IM2b, terminal-oxygen-mediated C–H bond cleavage takes place through a [1,4]-H shift via a five-membered ring 0-TS2b, yielding a DFF-containing molecular complex 0-IM3b, and then, O2 is imported, producing an O2- and DFF-containing molecular complex 0-IM4b. Afterwards, from 0-IM4b to [V-0 + O] + DFF + H2O, the reaction pathway is the same as that in P3-tb. In the MERP, P3-bt involves an EHHP of 109.8 kJ mol−1 at 0-TS2b and a HEB of 138.4 kJ mol−1 at the reaction step 0-IM2b → 0-TS2b for the terminal-oxygen-mediated C–H bond cleavage.
In view of these three reaction pathways, the EHHP of P3-bt is the lowest, while the HEB of P3-th is the lowest. That is to say, it is difficult to judge which pathway is kinetically the most favorable only from their EHHPs and HEBs.
 | ||
| Fig. 3 The geometric structures (a) and the schematic energy diagrams (b) for the reaction [V-0+O] + HMF → [V-0] + DFF + H2O. Relative energies (kJ mol−1) for the corresponding species plus DFF + 2H2O relative to those for the reactants [V-0] + 2HMF + O2 are shown. For clarity, hydrogen atoms on the rings are not shown. Bond lengths are reported in Å. The blue and pink lines represent P4-pp and P4-bp, respectively. | ||
For P4-pp, at the beginning, the hydrogen atom of hydroxyl in HMF interacts with the peroxide-oxygen atom on [V-0 + O] through H-bonding, forming a molecular complex 0-IM9a. Next, peroxide-oxygen-mediated O–H bond cleavage takes place through a [2 + 3] addition reaction via a five-membered ring 0-TS5a, resulting in a peroxide 0-IM10a. In 0-IM10a, the Mayer bond order of V–OCH2 is 1.01, indicating a single bond being basically formed. After that, from 0-IM10a, a single-bond rotates readily to form another compound 0-IM11a. Then, from 0-IM11a, peroxide-oxygen-mediated C–H bond cleavage occurs through a [1,5]-H shift via a six-membered ring 0-TS6a, leading to a DFF- and H2O-containing molecular complex 0-IM12a. Finally, 0-IM12a successively releases both DFF and H2O molecules free, regenerating the active site [V-0]. P4-pp includes an EHHP of 72.6 kJ mol−1 at 0-TS6a and a HEB of 106.2 kJ mol−1 at the reaction step 0-IM11a → 0-TS6a for the peroxide-oxygen-mediated C–H bond cleavage.
Alternatively, for P4-bp, initially, the hydrogen atom of hydroxyl in HMF interacts with the lattice-oxygen atom on [V-0 + O] through H-bonding, generating a molecular complex 0-IM9b. Then, from 0-IM9b, lattice-oxygen-mediated O–H bond cleavage occurs through a [1,3]-H shift via a four-membered ring 0-TS5b, affording a hydroxylate 0-IM10b. Afterwards, migration–extrusion takes place, yielding a more stable vacancy-containing intermediate 0-IM11b. Next, from 0-IM11b, peroxide-oxygen-mediated C–H bond cleavage occurs through a [1,4]-H shift via a six-membered chair-type 0-TS6b, leading to a DFF-containing molecular complex 0-IM12b. After that, 0-IM12b liberates the DFF molecule free, leaving 0-IM13 behind, where intramolecular rearrangement occurs near the vanadium site with the vacancy disappearing. With that, from 0-IM13, hydroxyl-oxygen mediated O–H bond cleavage takes place again through a [1,4]-H shift via a five-membered 0-TS7, producing a H2O-containing molecular complex 0-IM14. Finally, 0-IM14 releases the H2O molecule free, regenerating the active site [V-0]. P4-bp involves an EHHP of 23.2 kJ mol−1 at 0-TS6c and a HEB of 107.0 kJ mol−1 at the reaction step 0-IM11b → 0-TS6b for the peroxide-oxygen-mediated C–H bond cleavage.
Compared to P4-pp, P4-bp possesses a lower EHHP (23.2 vs. 72.6 kJ mol−1) for the C–H bond cleavage and similar HEB (107.0 vs. 106.2 kJ mol−1). Thus, P4-bp is kinetically more favorable than P4-pp. In P4-bp, after a lattice-oxygen mediates O–H bond cleavage, an oxygen vacancy is generated, which further promotes the C–H bond cleavage, combining with the peroxide-oxygen group. This result embodies the mediation effect of an oxygen vacancy toward the C–H bond cleavage, which is in good agreement with the experimental proposal for the lattice oxygen vacancy from the lattice-oxygen ([V–O–Cu]) group in the aerobic oxidation of HMF to DFF over the V2O5@Cu-MOR catalyst.20
Summarily, based on the above results, over the active site [V-0], there are two possible catalytic cycles in the MERPs, i.e.P3-bt + P4-bp ([C-0-bt]) and P3-th + P4-bp ([C-0-th]). Taking the schematic energy diagrams into account, [C-0-bt] includes the EHHP of 109.8 kJ mol−1 at 0-TS2b and HEB of 138.4 kJ mol−1 at the reaction step 0-IM2b → 0-TS2b for the terminal-oxygen-mediated C–H bond cleavage in the first HMF. Alternatively, [C-0-th] involves the EHHP of 124.4 kJ mol−1 at 0-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage in the first HMF and the HEB of 107.0 kJ mol−1 at the reaction step 0-IM11b → 0-TS6b for the peroxide-oxygen-mediated C–H bond cleavage in the second HMF. Therefore, it is also difficult to judge which catalytic cycle is kinetically more preferable only from their EHHPs and HEBs. Thus, it is necessary to investigate the global catalytic cycle using the energetic span, vide infra.
3.3. Catalytic mechanism over the [V-1] active site
Over the [V-1] active site, reaction (2) can be also divided into two reaction stages:| [V-1] + HMF + O2 → [V-1 + O] + DFF + H2O | (5) |
| [V-1 + O] + HMF → [V-1] + DFF + H2O | (6) |
Then, we will discuss their thermodynamics and kinetics, vide infra.
 | ||
| Fig. 4 The geometric structures (a) and the schematic energy diagrams (b) for the reaction [V-1] + HMF + O2 → [V-1 + O] + DFF + H2O. Relative energies (kJ mol−1) for the corresponding species plus HMF + O2 relative to those for the reactants [V-1] + 2HMF + O2 are shown. For clarity, hydrogen atoms on the rings are not shown. Bond lengths are reported in Å. The blue, green, pink and yellow lines represent P5-th, P5-tt, P5-ht and P5-th2, respectively. | ||
For P5-th, at the beginning, the hydrogen atom of hydroxyl in HMF interacts with the terminal-oxygen atom on the [V-1] active site through H-bonding, giving rise to a molecular complex 1-IM1a. Next, terminal-oxygen-mediated O–H bond cleavage occurs through a [2 + 2] addition reaction via a four-membered ring 1-TS1a, leading to a dihydroxyl complex 1-IM2a. After that, hydroxyl-oxygen-mediated C–H bond cleavage takes place through a [1,4]-H shift via a five-membered ring 1-TS2a1, resulting in a DFF- and H2O-containing molecular complex 1-IM3a1. Then, from 1-IM3a1, there are two reaction pathways, i.e., preferential release of free H2O or DFF, leading to 1-IM5 or [V-1 + 2H], denoted as P5-th1 and P5-th2, respectively. On the one hand, for P5-th1, from 1-IM5, O2 is introduced, forming an O2- and DFF-containing molecular complex 1-IM6. As the singlet 1-IM6 lies 75.7 kJ mol−1 below the triplet 31-IM6, the triplet–singlet spin-crossing may take place near 1-IM6. From 1-IM6, the MERP should proceed along the singlet state. This can be ascribed to the fact that the O2 is chemically absorbed through a V–OO single bond in 1-IM6, in which the Mayer bond orders of two V–OO bonds are 1.09 and 0.93, respectively. Afterward, 1-IM6 sets the DFF molecule free, leaving 1-IM7 behind. Finally, from 1-IM7, a [1,5]-H shift takes place via a six membered ring 1-TS4, producing a more stable complex [V-1 + O]. On the other hand, for P5-th2, from [V-1 + 2H], O2 is imported, giving rise to an O2- and H2O-containing molecular complex [V-1 + 2OH]. Since the singlet [V-1 + 2OH] lies 69.0 kJ mol−1 below the triplet 3[V-1+2OH], the triplet–singlet spin-crossing may take place near [V-1 + 2OH]. From [V-1 + 2OH], the MERP should advance on the singlet state. This may stem from the fact that the O2 in [V-1 + 2OH] is chemically absorbed through a V–OO single bond, in which the Mayer bond orders of two V–OO bonds are 1.06 and 0.93, respectively. Then, [V-1 + 2OH] liberates the H2O molecule free, saving 1-IM7 aside. In view of Fig. 4, the EHHP in P5-th1 at 1-IM4a lies 36.9 kJ mol−1 below that in P5-th2 at [V-0 + 2H], Thereupon, the MERP should go through P5-th1 other than P5-th2. In the MERP, P5-th involves an EHHP of 101.5 kJ mol−1 at 1-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage and a HEB of 95.1 kJ mol−1 at the reaction step 1-IM1a → 1-TS1a for the terminal-oxygen-mediated O–H bond cleavage.
For P5-tt, from the reactants to 1-IM2a, the reaction pathway is the same as that for P5-th, which is associated with the terminal-oxygen-mediated O–H bond cleavage. Next, from 1-IM2a, a [1,3]-H shift takes place via a four-membered ring 1-TS2a2, leading to a H2O-containing molecular complex 1-IM3a2. Then, 1-IM3a2 sets the H2O molecule free, leaving 1-IM4a behind. After that, terminal-oxygen-mediated C–H bond cleavage occurs through a [1,4]-H shift via a five-membered ring 1-TS3a, generating a DFF-containing molecular complex 1-IM5. With that, from 1-IM5 + O2 to [V-1 + O] + DFF, the reaction pathway is identical to that in P5-th. In the MERP, P5-tt includes an EHHP of 121.3 kJ mol−1 at 1-TS3a and a HEB of 140.9 kJ mol−1 at the reaction step 1-IM4a → 1-TS3a for the terminal-oxygen-mediated C–H bond cleavage.
For P5-ht, initially, the hydrogen atom of the –OH group in HMF interacts with the hydroxyl-oxygen atom in the [V-1] active site through H-bonding, forming a molecular complex 1-IM1b. Next, from 1-IM1b, hydroxyl-oxygen-mediated O–H bond cleavage takes place through a [1,3]-H shift via a four-membered ring 1-TS1b, generating a H2O-containing molecular complex 1-IM2b. Afterwards, from 1-IM2b, terminal-oxygen-mediated C–H bond cleavage occurs through a [1,4]-H shift via a five-membered ring 1-TS2b, yielding a H2O- and DFF-containing molecular complex 1-IM3a1. After that, from (1-IM3a1 + O2) to ([V-1 + O] + DFF + H2O), the reaction pathway is identical to that in P5-th. In the MERP, P5-ht comprises an EHHP of 109.8 kJ mol−1 at 1-TS2b and a HEB of 122.6 kJ mol−1 at the reaction step 1-IM2b → 1-TS2b for the terminal-oxygen-mediated C–H bond cleavage.
Among these three reaction pathways, P5-th possesses the lowest EHHP and the lowest HEB. Therefore, P5-th is kinetically the most favorable. This echoes the superiority of hydroxyl-oxygen-mediation to terminal-oxygen mediation in the vanadium-containing active site, toward the C–H bond cleavage in HMF.
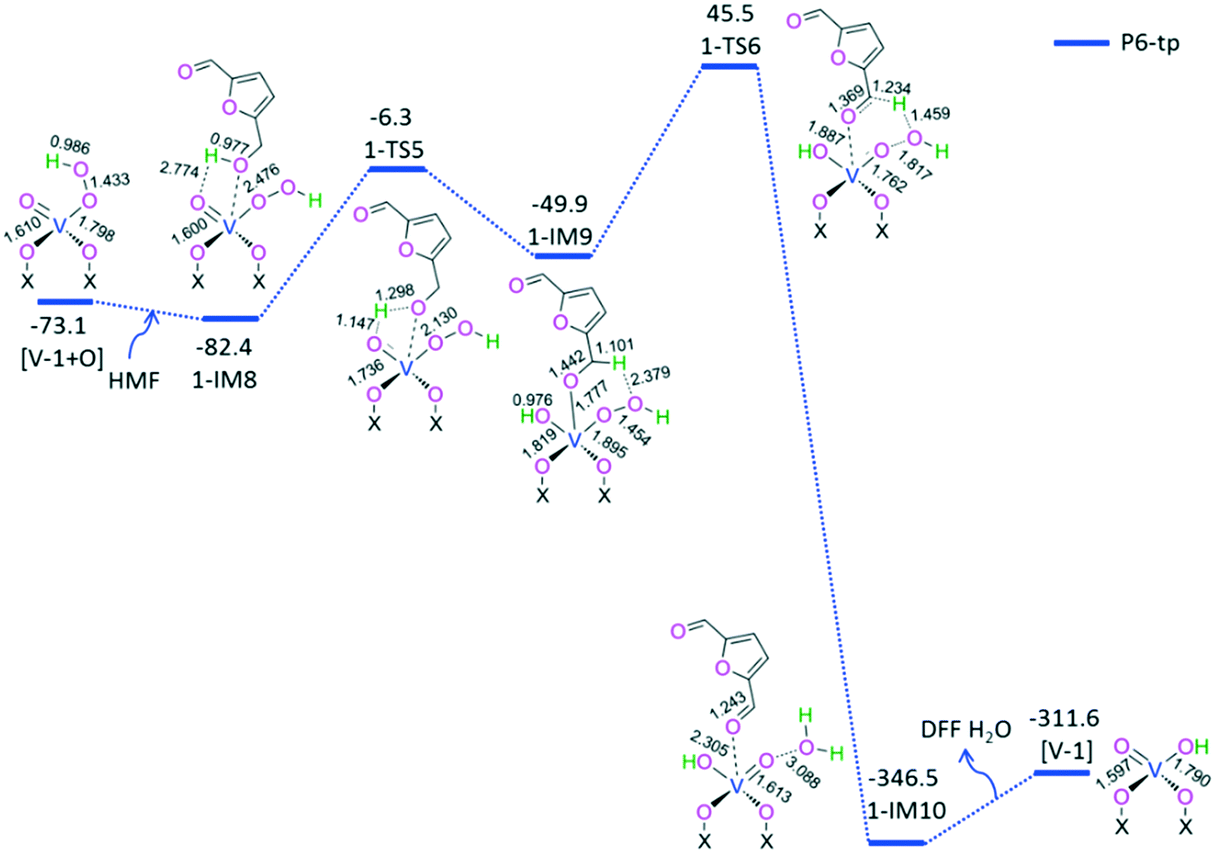 | ||
| Fig. 5 The geometric structures and the schematic energy diagrams for the reaction [V-1 + O] + HMF → [V-1] + DFF + H2O. Relative energies (kJ mol−1) for the corresponding species plus DFF + 2H2O relative to those of the reactants [V-1] + 2HMF + O2 are shown. For clarity, hydrogen atoms on the rings are not shown. Bond lengths are reported in Å. The blue line represents P6-tp. | ||
For P6-tp, initially, a HMF molecule is physically adsorbed on [V-1 + O] through H-bonding, giving rise to a molecular complex 1-IM8. Next, terminal-oxygen-mediated O–H bond cleavage takes place through a [2 + 2] addition via a four-membered 1-TS5, leading to a hydroxyl compound 1-IM9. Then, peroxide-oxygen-mediated C–H bond cleavage occurs through a [1,5]-H shift via a six-membered ring 1-TS6, affording a DFF- and H2O-containing molecular complex 1-IM10. Finally, 1-IM10 releases both DFF and H2O molecules free, regenerating the active site [V-1]. P6-tp involves an EHHP of 45.5 kJ mol−1 at 1-TS6 and a HEB of 95.4 kJ mol−1 at the reaction step 1-IM9 → 1-TS6 for the peroxide-oxygen-mediated C–H bond cleavage.
Summarily, over the active site [V-1], on account of the above results, there are two probable MERPs of catalytic cycles, i.e., P5-th + P6-tp ([C-1-th]) and P5-ht + P6-tp ([C-1-ht]). In view of the schematic energy diagrams, [C-1-th] includes the EHHP of 101.5 kJ mol−1 at 1-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage in the first HMF and the HEB of 95.4 kJ mol−1 at the reaction step 1-IM9 → 1-TS6 for the peroxide-oxygen-mediated C–H bond cleavage in the second HMF. Additionally, [C-1-ht] involves the EHHP of 109.8 kJ mol−1 at 1-TS2b and HEB of 122.6 kJ mol−1 at the reaction step 1-IM2b → 1-TS2b for the terminal-oxygen-mediated C–H bond cleavage in the first HMF. Compared to [C-1-ht], [C-1-th] is kinetically more favorable, because of its lower EHHP (101.5 vs. 109.8 kJ mol−1) and lower HEB (95.4 vs. 122.6 kJ mol−1).
3.4. Comparison of catalytic performances over the [V-1] and [V-0] active sites
As mentioned earlier, over the [V-0] active site, there are two probable catalytic cycles, i.e., [C-0-bt] and [C-0-th]. Here, we will compare their kinetics preference from the global catalytic cycle using the energetic span model. By TOF analysis, for [C-0-bt], the TDI and TDTS are computed to be 0-IM2b and 0-TS2b for the terminal-oxygen-mediated C–H bond cleavage in the first HMF, respectively. Furthermore, for [C-0-th], the TDI and TDTS are evaluated to be 0-IM1a and 0-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage in the first HMF, respectively. Herein, the rate constant kC-0-bt of 0-IM2b → 0-TS2b and the rate constant kC-0-th of 0-IM1a → 0-TS2a1 are representative of those in the gross catalytic cycles, respectively. Over the 325–425 K temperature range, the rate constants kC-0-bt (in s−1) and kC-0-th (in s−1) can be calculated by the following expressions:| kC-0-bt = 1.43 × 1013 exp (−133074/RT) | (i) |
| kC-0-th = 1.79 × 1013 exp (−122931/RT) | (ii) |
The rate constant kC-0-th is computed to be about 52–21 times higher than kC-0-bt, over the 325–425 K temperature range. That is to say, [C-0-th] is kinetically more dominant than [C-0-bt].
Over the [V-1] active site, there are also two probable catalytic cycles, i.e., [C-1-th] and [C-1-ht]. For [C-1-th], the TDI and TDTS are determined to be 1-IM1a and 1-TS2a1 for the hydroxyl-oxygen-mediated C–H bond cleavage in the first HMF, respectively. In addition, for [C-1-ht], the TDI and TDTS are found to be 1-IM1b and 1-TS2b for the terminal-oxygen-mediated C–H bond cleavage in the first HMF, respectively. Then, over the 325–425 K temperature range, the rate constants kC-1-th (s−1) and kC-1-ht (in s−1) can be calculated by the following expressions:
| kC-1-th = 4.35 × 1010 exp (−95 827/RT) | (iii) |
| kC-1-ht = 8.24 × 1011 exp (−113 744/RT) | (iv) |
The rate constant kC-1-th is calculated to be about 39–7 times higher than kC-1-ht, over the 325–425 K temperature range. In other words, [C-1-th] is kinetically more preferable than [C-1-ht].
Furthermore, over the 325–425 K temperature range, the rate constant kC-1-th is calculated to be about 54–4 times higher than kC-0-th. One can see that [V-1] displays better catalytic activity than [V-0], toward the aerobic oxidation of HMF to DFF. Next, we will discuss the nature of the catalytic activity difference between [V-0] and [V-1], vide infra.
In addition, over both the [V-0] and [V-1] active sites, the crucial reaction step is associated with the C–H bond cleavage in the first HMF. This result is in good agreement with those reported experimentally over Ru/C catalysts and theoretically over Mo-containing Keggin heteropolyacids and Mo-doped MCM-41.11,58,63 In other words, the reaction mechanism over these catalysts might be similar. Nevertheless, in a previous study, the contribution regularity of functional groups on catalytically active sites toward the O–H and C–H bond cleavages is still elusive.
On the other hand, for the activation step of O2 over the [V-0] active site, P3-bt includes the HEB of 53.6 kJ mol−1 at the reaction step 0-IM7 → 0-TS4 → 0-IM8, as shown in Fig. 2. For the activation step of O2 over the [V-1] active site, P5-th involves the HEB of 71.2 kJ mol−1 at the reaction step 1-IM7 → 1-TS4 → [V-1 + O], as shown in Fig. 4. It is indicated that the [V-0] active site displays better catalytic performance than the [V-1] active site toward the activation step of O2, because of its lower HEB (53.6 vs. 71.2 kJ mol−1).
3.5. Catalytic contribution of oxygen-containing groups over the [V-0] and [V-1] active sites
As mentioned earlier, toward the aerobic oxidation of HMF to DFF, the active site [V-1] shows better catalytic activity than [V-0]. To explore the origin of the catalytic activity difference of the active sites, the corresponding energy barriers (EBs) of oxygen-containing functional groups activating the O–H and C–H bond cleavages are analyzed, vide infra. From both the [V-0] and [V-1] active sites, there are three kinds of oxygen-containing groups, i.e., hydroxyl-oxygen ([V–OH]), lattice-oxygen ([V–O–Si]), and terminal-oxygen ([VO]). Table 1 lists the EBs for the O–H and C–H bond cleavages of HMF in the presence of oxygen-containing functional group mediation over the [V-0] and [V-1] active sites.
| Functional group | Reaction step | Energy barrier (kJ mol−1) |
|---|---|---|
| O–H bond cleavage | ||
| [V–OH] | 1-IM1b → 1-TS1b | 25.1 |
| [V–O–Si] | 0-IM1b → 0-TS1b | 56.3 |
| 0-IM9b → 0-TS5b | 47.3 | |
| [VO] |
0-IM1a → 0-TS1a | 90.6 |
| 1-IM1a → 1-TS1a | 95.1 | |
| 1-IM8 → 1-TS5 | 76.1 | |
| C–H bond cleavage | ||
| [V–OH] | 0-IM2a → 0-TS2a1 | 95.3 |
| 1-IM2a → 1-TS2a1 | 76.1 | |
| 1-IM9 → 1-TS6 | 95.4 | |
| [V–O–Si] | 0-IM2a → 0-TS2a2 | 105.8 |
| [VO] |
0-IM2b → 0-TS2b | 138.4 |
| 1-IM2b → 1-TS2b | 122.6 | |
| 1-IM4a → 1-TS3a | 140.9 | |
As shown in Table 1, toward the O–H cleavage, the EBs of functional group mediation increase in the order [V–OH] (25.1 kJ mol−1) < [V–O–Si] (47.3 and 56.3 kJ mol−1) < [VO] (76.1, 90.6, and 95.1 kJ mol−1). It is indicated that the mediation effect of functional groups decreases in the order [V–OH] > [V–O–Si] > [VO], toward the O–H cleavage. In addition, toward the C–H cleavage, the EBs of function group mediation increase in the order [V–OH] (76.1, 95.3, and 95.4 kJ mol−1) < [V–O–Si] (105.8 kJ mol−1) < [VO] (122.6, 138.4, and 140.9 kJ mol−1). It is inferred that the mediation effect of functional groups decreases in the order [V–OH] > [V–O–Si] > [VO], toward the C–H cleavage. In brief, for both O–H and C–H bond cleavages of HMF, the catalytic contribution of functional groups increases in the order [VO] < [V–O–Si] < [V–OH]. That is to say, the hydroxyl-oxygen-mediation effect is the strongest among these three functional group mediation effects, toward both O–H and C–H bond cleavages of HMF. This can be explained that [V-1] with a hydroxyl group exhibits better catalytic activity than [V-0] without a hydroxyl group. In other words, the higher catalytic activity of [V-1] stems from its hydroxyl group, compared to that of [V-0].
As mentioned earlier, the rate-determining step is concerned with the C–H bond cleavages of the first HMF, toward the aerobic oxidation of HMF to DFF over the vanadium-containing active sites. To gain a deep insight into the trend of functional groups catalyzing C–H bond cleavage, some characteristic transition states are chosen to visualize their interaction from electron density differences, i.e., 1-TS2a1, 0-TS2a2, 1-TS2b, respectively. Fig. 6 shows the electron density difference contour map of typical transition states.
 | ||
| Fig. 6 Electron density difference contour plots of 1-TS2a1 (A), 0-TS2a2 (B) and 1-TS2b (C). The red and blue colors represent increased and decreased electron density, respectively. | ||
In parts A, B, and C of Fig. 6, the contour plots of the activating C–H bond mediated by [V–OH] (1-TS2a1), [V–O–Si] (0-TS2a2), and [VO] (1-TS2b) are depicted, respectively. As shown in Fig. 6, in terms of the electron density rich region (red color) in the activating C–H bond, A (1-TS2a1) is weaker than both B (0-TS2a2) and C (1-TS2b). This visual observation is in accordance with the fact that the corresponding EB of 1-TS2a1 is lower than those of 0-TS2a2 and 1-TS2b. Moreover, in terms of the electron density rich region (red color) in the forming O–H bond, B (0-TS2a2) is stronger than C (1-TS2b). This phenomenon is in agreement with the fact that the corresponding EB of 0-TS2a2 is lower than that of 1-TS2b. These results show that the promotive contribution of oxygen-containing groups over the vanadium-containing active sites decreases in the order [V–OH] > [V–O–Si] > [VO], toward the crucial C–H bond cleavage of the HMF moiety.
Based on the above results, the lattice-oxygen ([V–O–Si]) exhibits more catalytic contribution than the terminal-oxygen ([VO]), toward both O–H and C–H bond cleavages of HMF. It is obvious that the catalytic cycle [C-0-bt] with the P3-bt and P4-bp reaction pathways truly embodies the idea of a lattice oxygen vacancy-mediated mechanistic pathway, which follows the Mars–van Krevelen mechanism by referencing previously related studies.20,64,65 Particularly, for the aerobic oxidation of HMF to DFF over V2O5@Cu-MOR, Wang et al. suggested that HMF is initially adsorbed on the catalyst surface and oxidized to generate DFF mainly by lattice oxygen (V5+–O2−–Cu), and then the resultant V4+–ϒ–Cu (ϒ denotes the lattice oxygen vacancy) transition active site is further reoxidized with O2 to regenerate [V5+–O2−–Cu] species according to the experimental results.20 That is to say, with regard to the role of lattice oxygen over the vanadium-containing active sites, the present theoretical prediction result of [V–O–Si] is similar to that in the experimental speculation of [V–O–Cu].20
Furthermore, the hydroxyl-oxygen ([V–OH]) group displays the best catalytic activity among the three kinds of oxygen-containing groups ([V–OH], [V–O–Si], and [VO]), toward both O–H and C–H bond cleavages of HMF. The prominent catalytic performance of the [V–OH] group can be verified by the experimental finding over nanobelt-arrayed vanadium oxide hierarchical microspheres by Yang's group, in which the (010) facet of vanadium oxide with the highest hydrogen adsorption capability of vanadyl group ([VO]) sites plays an important role in both O–H and C–H bond cleavages of HMF before the reduction product [V–OH] species is further dehydrogenated by molecular oxygen.21 In other words, the vanadyl group ([VO]) on the (010) facet of vanadium oxide is readily hydrogenated, resulting in the hydride, e.g., [V–OH] group, which can possess better catalytic activity than the [VO] group, toward O–H and C–H bond cleavages of HMF. In brief, with respect to the excellent catalytic function of the [V–OH] group over the vanadium-containing active site, the present theoretical result is in good accordance with the experimental findings.21
4. Conclusions
For a V-doped Al-MCM-41 pore model (V/Al-MCM-41), two kinds of vanadium active sites on the pore surface, −(SiO)3V(O) ([V-0]) and –(SiO)2V(O)(OH) ([V-1]), have been theoretically established. Over the two active sites ([V-0] and [V-1]), the catalytic mechanisms for the aerobic oxidation of HMF to DFF have been theoretically explored. The following conclusions can be drawn from the present investigation.For the overall reaction 2HMF + O2 → 2DFF + 2H2O catalyzed over both the [V-0] and [V-1] active sites, the rate-determining reaction step is concerned with the C–H bond cleavage of the –CH2 group of the first HMF molecule, where there are two main activation steps, i.e., both O–H and C–H bond cleavages of HMF. The [V-1] active site with a hydroxyl group exhibits better catalytic activity than the [V-0] active site without a hydroxyl group, toward the aerobic oxidation of HMF to DFF. Toward the activation step of O2, the [V-0] active site shows better catalytic performance than the [V-1] active site.
Over both the [V-0] and [V-1] active sites, there are some oxygen-containing functional groups, i.e., hydroxyl-oxygen, lattice-oxygen, and terminal-oxygen, which may catalyze both O–H and C–H bond cleavages of HMF. For both O–H and C–H bond cleavages of HMF, the catalytic activity of oxygen-containing functional groups increases in the order [VO] < [V–O–Si] < [V–OH]. This regularity can be visualized in the electron density depletion region for the bond cleavage and electron density rich region for the bond formation from the corresponding crucial transition states. The proposed roles of oxygen-containing groups over the V-containing active sites are in good agreement with the experimental investigation. These regular insights may engineer the rational design of catalytically active sites toward the activation of O–H and C–H bonds at low temperature, especially for some typical platform molecules of biomass, such as HMF.
Author contributions
The manuscript was written through the contributions of all the authors. L.-J. Liu is responsible for the main computations and analysis, Z.-M. Wang, Y.-J. Lyu, J.-F. Zhang, Z. Huang, T. Qi, and Z.-B. Si are responsible for some computations and analysis, H.-Q. Yang is responsible for the design, analysis, and writing, and C.-W. Hu is responsible for the design and revision. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No: 21573154) and the 111 Project (B17030).Notes and references
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS.
- A. S. Amarasekara, D. Green and L. D. Williams, Eur. Polym. J., 2009, 45, 595–598 CrossRef CAS.
- S. Zhang, W. Li, X. Zeng and L. Lin, J. Bioprocess Eng. Biorefinery, 2014, 3, 23–34 CrossRef.
- X. Liu, J. Xiao, H. Ding, W. Zhong, Q. Xu, S. Su and D. Yin, Chem. Eng. J., 2016, 283, 1315–1321 CrossRef CAS.
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, Green Chem., 2016, 18, 2302–2307 RSC.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS.
- J. Nie, J. Xie and H. Liu, J. Catal., 2013, 301, 83–91 CrossRef CAS.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS.
- H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101–3110 CrossRef CAS.
- A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101 CrossRef CAS.
- Y. Zhou, Z. Ma, J. Tang, N. Yan, Y. Du, S. Xi, K. Wang, W. Zhang, H. Wen and J. Wang, Nat. Commun., 2018, 9, 2931 CrossRef PubMed.
- O. C. Navarro, A. C. Canós and S. I. Chornet, Top. Catal., 2009, 52, 304–314 CrossRef CAS.
- I. Sádaba, Y. Y. Gorbanev, S. Kegnaes, S. S. R. Putluru, R. W. Berg and A. Riisager, ChemCatChem, 2013, 5, 284–293 CrossRef.
- C. A. Antonyraj, B. Kim, Y. Kim, S. Shin, K.-Y. Lee, I. Kim and J. K. Cho, Catal. Commun., 2014, 57, 64–68 CrossRef CAS.
- W. Zhang, J. Xie, W. Hou, Y. Liu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 23122–23132 CrossRef CAS.
- Y. Yan, K. Li, J. Zhao, W. Cai, Y. Yang and J.-M. Lee, Appl. Catal., B, 2017, 207, 358–365 CrossRef CAS.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS.
- W. Hou, Q. Wang, Z. Guo, J. Li, Y. Zhou and J. Wang, Catal. Sci. Technol., 2017, 7, 1006–1016 RSC.
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS.
- W. H. Wei Zhang, Tongsuo Meng, Wenxia Zhuang, Jingyan Xie and Y. Z. a. J. Wang, Catal. Sci. Technol., 2017, 7, 6050–6058 RSC.
- J. Zhao, A. Jayakumar, Z.-T. Hu, Y. Yan, Y. Yang and J.-M. Lee, ACS Sustainable Chem. Eng., 2017, 6, 284–291 CrossRef.
- R. Fang, R. Luque and Y. Li, Green Chem., 2017, 19, 647–655 RSC.
- S. Wang, Y. Shi, X. Ma and J. Gong, ACS Appl. Mater. Interfaces, 2011, 3, 2154–2160 CrossRef CAS.
- A. L. Cánepa, V. R. Elías, V. M. Vaschetti, E. V. Sabre, G. A. Eimer and S. G. Casuscelli, Appl. Catal., A, 2017, 545, 72–78 CrossRef.
- Y. Hu, M. Anpo and C. Wei, J. Photochem. Photobiol., A, 2013, 264, 48–55 CrossRef CAS.
- K. Wu, B. Li, C. Han and J. Liu, Appl. Catal., A, 2014, 479, 70–75 CrossRef CAS.
- X. Wang, G. Zhou, Z. Chen, W. Jiang and H. Zhou, Microporous Mesoporous Mater., 2016, 223, 261–267 CrossRef CAS.
- R. K. Rana and B. Viswanathan, Catal. Lett., 1998, 52, 25–29 CrossRef CAS.
- C.-F. Cheng, W. Zhou and J. Klinowski, Chem. Phys. Lett., 1996, 263, 247–252 CrossRef CAS.
- I. Jiménez-Morales, M. Moreno-Recio, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2015, 164, 70–76 CrossRef.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Struct., 2003, 632, 1–28 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- X. Li, H. Zhang, S. Lu, W. Li, J. Zhao, B. Johansson and L. Vitos, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 1–12 Search PubMed.
- X. Ji, K. Xu, C. Chen, B. Zhang, H. Wan, Y. Ruan, L. Miao and J. Jiang, J. Mater. Chem. A, 2015, 3, 9909–9914 RSC.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- J. D. Gale, Z. Kristallogr. - Cryst. Mater., 2005, 220, 552–554 CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. G. Iii and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- R. E. Patet, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2016, 18, 26094–26106 RSC.
- X. Nie, M. J. Janik, X. Guo, X. Liu and C. Song, Catal. Today, 2011, 165, 120–128 CrossRef CAS.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250–258 CrossRef CAS.
- S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355–3365 CrossRef CAS.
- S. Kozuch and S. Shaik, J. Phys. Chem. A, 2008, 112, 6032–6041 CrossRef CAS.
- A. Uhe, S. Kozuch and S. Shaik, J. Comput. Chem., 2011, 32, 978–985 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- S. Kozuch, ACS Catal., 2015, 5, 5242–5255 CrossRef CAS.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS.
- L.-K. Ren, L.-F. Zhu, T. Qi, J.-Q. Tang, H.-Q. Yang and C.-W. Hu, ACS Catal., 2017, 7, 2199–2212 CrossRef CAS.
- B. Xiang, Y. Wang, T. Qi, H.-Q. Yang and C.-W. Hu, J. Catal., 2017, 352, 586–598 CrossRef CAS.
- L. N. Ho, J. P. Pellitero, F. Porcheron and R. J. Pellenq, Langmuir, 2011, 27, 8187–8197 CrossRef CAS PubMed.
- M. Gierada, I. Petit, J. Handzlik and F. Tielens, Phys. Chem. Chem. Phys., 2016, 18, 32962–32972 RSC.
- Z.-M. Wang, L.-J. Liu, B. Xiang, Y. Wang, Y.-J. Lyu, T. Qi, Z.-B. Si, H.-Q. Yang and C.-W. Hu, Catal. Sci. Technol., 2019, 9, 811–821 RSC.
- T. Qi, M.-F. He, L.-F. Zhu, Y.-J. Lyu, H.-Q. Yang and C.-W. Hu, J. Phys. Chem. C, 2019, 123, 4879–4891 CrossRef CAS.
- J. Tang, X. Guo, L. Zhu and C. Hu, ACS Catal., 2015, 5, 5097–5103 CrossRef CAS.
- J. Tang, L. Zhu, X. Fu, J. Dai, X. Guo and C. Hu, ACS Catal., 2016, 7, 256–266 CrossRef.
- H. Guesmi, R. Grybos, J. Handzlik and F. Tielens, Phys. Chem. Chem. Phys., 2014, 16, 18253–18260 RSC.
- L.-K. Ren, H.-Q. Yang and C.-W. Hu, Catal. Sci. Technol., 2016, 6, 3776–3787 RSC.
- J. Nie and H. Liu, J. Catal., 2014, 316, 57–66 CrossRef CAS.
- G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293–301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic energy diagrams, energies, activation strain analysis, standard orientations, and geometric structures. See DOI: 10.1039/c9cy02130b |
| This journal is © The Royal Society of Chemistry 2020 |