
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatization of ethylene over zeolite-based catalysts†
Evgeny A.
Uslamin
 a,
Hikaru
Saito
b,
Nikolay
Kosinov
a,
Evgeny
Pidko‡
a,
Yasushi
Sekine
*b and
Emiel J. M.
Hensen
*a
a,
Hikaru
Saito
b,
Nikolay
Kosinov
a,
Evgeny
Pidko‡
a,
Yasushi
Sekine
*b and
Emiel J. M.
Hensen
*a
aLaboratory of Inorganic Chemistry and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: ysekine@waseda.jp
First published on 6th January 2020
Abstract
Light aromatic compounds (BTX: benzene, toluene and xylenes) represent an important class of building blocks in the chemical industry. Currently, light aromatics are obtained exclusively from fossil feedstock, whose utilization is associated with serious environmental concerns. Developing new routes for a more sustainable BTX production is, therefore, of high importance. In this work, aromatization of ethylene over well-defined metal-modified HZSM-5 zeolite catalysts is examined. The results show that modification of zeolite with gallium, zinc and silver leads to a significant increase in aromatics production. Metal species are responsible for catalysing dehydrogenation pathways with Ga being the most efficient for BTX production. Increasing temperature and ethylene partial pressure facilitate ethylene aromatization. Employing a combination of isotope labelling with a thorough characterization of zeolite-entrapped species by means of IR and MAS NMR spectroscopy provides evidence for the involvement of intra-zeolite aromatic hydrocarbon species in the catalytic cycle.
1. Introduction
Aromatics constitute an important class of chemical building blocks. Especially, benzene, toluene and xylenes (BTX) are pivotal for the manufacture of many plastics and fine chemicals. Currently, BTX are mainly obtained as by-products of naphtha cracking to light olefins. The abundance of cheap natural gas feedstock, which usually contains an appreciable amount of ethane, prompts chemical producers to change the feed of crackers from naphtha to ethane, which leads to a decreased aromatics supply from the traditional naphtha cracking route.1,2 As a consequence, there is a growing need to convert light paraffins to aromatics via catalytic dehydrogenation processes.Over the last decades, zeolites have evolved as workhorses of the chemical industry, widely used in various hydrocarbon transformations.3–5 Zeolite ZSM-5 (MFI) is the preferred solid acid for the aromatization of ethane, propane and other short-chain hydrocarbons.6,7 Typically, ZSM-5-based catalysts are modified with metals to promote the alkane dehydrogenation. The most active and extensively investigated catalysts are ZSM-5 zeolites modified with zinc,8,9 gallium10 and silver,11 whose catalytic behaviour has been the subject of numerous experimental and computational studies published in the last few decades.6,12–14 Most of the works focused on unravelling the C–H activation mechanism15,16 and the associated promotion of the dehydrogenation function by metals, whereas the subsequent aromatization steps were studied to a much lesser extent. It is commonly assumed that the overall process proceeds through a bifunctional mechanism, where the transition metal species are responsible for the dehydrogenation of alkanes to produce olefins, which are then converted to aromatics over the Brønsted acid sites (BAS).17 Although experimental findings point to an important role of the transition metal cations in the secondary conversion of unsaturated hydrocarbons,18 the role in the aromatization network is much less understood. The aromatization involves a complex networks of elementary processes such as the oligomerization to higher olefins and their cracking, cyclization and further dehydrogenation to produce aromatic compounds.12 Such a bifunctional mechanism has been supported by several detailed kinetic studies on HZSM-5 (ref. 19) and Zn/HZSM-5 (ref. 20) catalysts.
Most current mechanistic proposals discuss the role of the zeolite in terms of the BAS and dispersed (cationic) metal species inside the zeolite pores. Recently, an alternative picture emphasizing the importance of confined reactive organic intermediates for different zeolite-catalyzed conversion processes has been gaining growing experimental and theoretical support.21–24 Such processes as the MTO (methanol-to-olefins),25 dehydroaromatization of methane26 and the aromatization of furanics27 are thought to proceed via a hydrocarbon pool-type mechanism.28 Similar proposals involving radical- and carbocation-type polyaromatic species as the catalytic sites have also been put forward for ethanol dehydration/aromatization reaction over HZSM-5.29–31 The formation of the hydrocarbon pool species in these processes is commonly associated with the intermediate formation and subsequent oligomerization and dehydrocyclization of unsaturated intermediates. One can therefore propose that similar mechanisms can manifest themselves during the dehydroaromatization of light alkanes over metal-exchanged zeolites.
This work reports a detailed catalytic and mechanistic analysis of ethylene aromatization by HZSM-5 zeolites modified with Ag, Ga and Zn. The influence of metal loading, ethylene partial pressure and temperature were investigated. The addition of a metal function leads to an increased aromatics selectivity, although it also causes a more rapid catalyst deactivation due to enhanced coke deposition rate. The differences in catalytic performance of promoted zeolites can be associated with changes in the rate of dehydrogenation. The dehydrogenation activity increases in the order Ga < Zn < Ag. Ga-Modified HZSM-5 catalysts were found to be preferred for ethylene aromatization. The dehydrogenation and hydrogen transfer reactions demonstrate a strong dependence on the reaction conditions such as reactant partial pressure and temperature. Temperature-programmed IR and 13C MAS NMR spectroscopy studies together with the isotopic labelling results suggest that zeolite-entrapped aromatic hydrocarbon species are involved in the catalytic cycle.
2. Experimental
2.1. Catalyst preparation
The proton form of zeolite ZSM-5 was obtained by calcining commercial NH4ZSM-5 zeolite (Si/Al = 20, Alfa Aesar) at 550 °C for 5 h. HZSM-5 supported gallium, zinc and silver catalysts were prepared via incipient wetness impregnation of the proton form of ZSM-5 zeolite with aqueous solutions of Ga(NO3)3 (Alfa Aesar, 99.9% metal based), Zn(NO3)2 (Merck, 99.9% metal based) and AgNO3 (Merck, ≥99.0%), respectively. The concentration was adjusted to achieve metal loadings of 1.4 wt% Ga, 1.3 wt% Zn and 2.2 wt% Ag. This corresponds to an atomic M/Al ratio of 0.25. Additional Ga samples were prepared with Ga/Al ratios of 0.1 and 0.5. The exact loading of the metal precursors used in the preparation are summarized in Table S1.† As-prepared samples were dried overnight at 110 °C and calcined at 550 °C for 5 h under static air. Then the zeolites were sieved to obtain a fraction with a particle size in a range of 250–500 μm.To promote solid-state ion exchange, Ga/HZSM-5 and Zn/HZSM-5 samples were subjected to a reductive treatment in hydrogen following a procedure described elsewhere.32 The sieved catalyst was loaded into a quartz tube, and then reduced in a fixed bed reactor at 550 °C for 5 h in a flow of 33 vol% H2 in He.
The resulting samples are denoted as xM/HZSM-5, where M represents the metal loaded onto the zeolite and x – the atomic M/Al ratio. For example, 0.25Ga/ZSM-5 stands for Ga-containing HZSM-5 with a Ga/Al ratio of 0.25.
2.2. Catalyst characterization
The elemental composition (Si/Al ratio and metal content) of the zeolite catalysts was determined by ICP-OES (Spectro CIROS CCD ICP optical emission spectrometer). Prior to analysis, the zeolite samples were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (by weight) mixture of HF (Merck, 40% for trace analysis), HNO3 (Merck, 65% for trace analysis) and H2O (18.2 MΩ cm).
:1:1 (by weight) mixture of HF (Merck, 40% for trace analysis), HNO3 (Merck, 65% for trace analysis) and H2O (18.2 MΩ cm).
The crystallinity of the zeolite samples was determined by powder X-ray diffraction (XRD). XRD measurements were performed on a Bruker D2 powder diffraction system (Cu Kα radiation, scan speed 0.01° s−1, 2θ range 5–60°). The relative crystallinity was normalized to the same weight of a parent HZSM-5 by comparing the integral areas of the reflections (Miller indices in parentheses) at 23.1° (051), 23.3° (501), 23.7° (511), 24.0° (033), and 24.4° (313).
1H magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were measured using a 11.7 Tesla Bruker DMX500 NMR spectrometer operating at 500 MHz. The experiments were performed using a Bruker Triple Channel 4 mm MAS probe head spinning at 10 kHz. 1H MAS NMR spectra were recorded with a Hahn-echo pulse sequence p1–τ1–p2–τ2–aq with a 90° pulse p1 = 5 μs and a 180° pulse p2 = 10 μs. The inter-scan delay was set to 120 s for quantitative measurements. Quantitative 13C MAS NMR spectra were measured with a high power proton decoupling Hahn-echo pulse sequence p1–τ1–p2–τ2–aq with a 90° pulse p1 = 5 μs and a 180° pulse p2 = 10 μs with an interscan delay of 20 s. One-dimensional 13C{1H} cross-polarization (CP) MAS and two-dimensional 1H–13C{1H} heteronuclear correlation (HETCOR) spectra were recorded with a ramped contact pulse of 3 ms and an interscan delay of 3 s. During the acquisition the 1H heteronuclear decoupling was applied using the spinal-64 pulse scheme.
Adsorption capacity and textural properties of the zeolites were evaluated by Ar porosimetry at −186 °C using a Micromeritics ASAP2020 machine. Prior to measurements, the samples were degassed at 500 °C under evacuation. The micropore volume was determined by the t-plot method in a thickness range from 3.5 Å to 4.5 Å.
The coke content and the combustion properties of the coke in used catalysts were determined using a Mettler Toledo TGA/DSC 1 instrument. Typically, an amount of 15 mg of catalyst was placed in an alumina crucible and heated to 750 °C at a rate of 10 °C min−1 in an O2/He flow. The coke content (g gcat−1) was calculated as the weight loss in the range 450–750 °C normalized by the original weight of the sample.
2.3. Catalytic activity measurements
Catalytic activity measurements were carried out in a fixed-bed quartz reactor (i.d. 3 mm) operated at atmospheric pressure. In a typical experiment, the sieved catalyst (100 mg) was charged into the reactor and heated in an O2 flow (20 vol% in He) to 550 °C at a rate of 5 °C min−1. After the pretreatment, the temperature of the reactor was set to the desired target value and the reactor was purged with He. The gases were metered into the reactor using mass flow controllers (Brooks® Instruments). For activity measurements at different ethylene partial pressures, the ethylene flow was varied whilst keeping the total gas flow constant. The reactor outlet was connected to a gas chromatograph (GC, Thermo Trace 1300) and mass spectrometer (MS, Pfeiffer Omnistar GSD 301 T3) system via a heated transfer line. The GC was equipped with two columns and detectors. A TCD with an RT-Q-Bond column (length 20 m; i.d. 0.32 mm; film thickness 10 μm) was used for the analysis of the light reaction products. Light aromatics and heavier products, including substituted naphthalenes, were analysed on a Stabilwax-DB column (length 30 m; i.d. 0.25 mm; film thickness 0.25 μm) coupled with an FID.Ethylene conversion and carbon selectivity to each product are defined as follows:
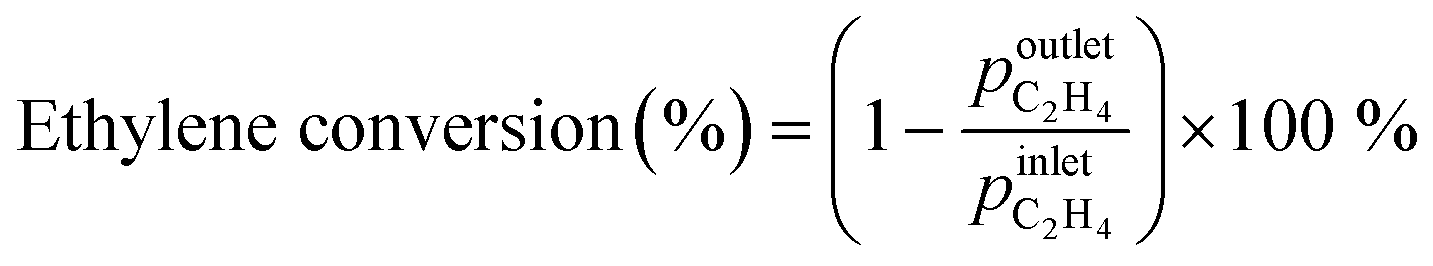
Herein, n and p denote the number of carbon atoms and partial pressure of the hydrocarbons, respectively. Analysed products include CH4, C2H4, C3, C4, C5, benzene, toluene, ethylbenzene, xylenes, naphthalene and methylnaphthalene.
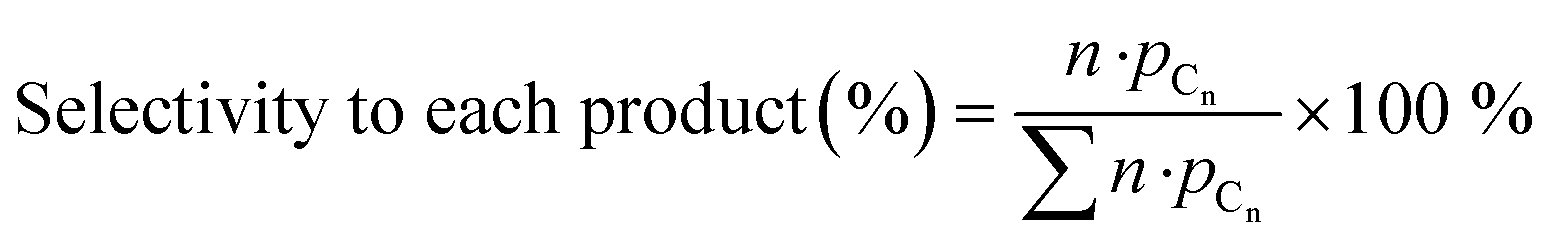
2.4. IR and TPR-IR experiments
IR spectroscopy of adsorbed species was carried out using a Bruker Vertex 70v Fourier-Transform IR spectrometer in the 4000–1000 cm−1 range. First, a zeolite sample was pressed into a self-supporting wafer (5–10 mg, diameter 13 mm), which was then placed in an environmental transmission IR cell. Samples were first pre-treated in an O2/N2 flow at 550 °C (heating rate 5 °C min−1), followed by cooling in dynamic vacuum of ca. 10−5 mbar.For IR spectroscopy of adsorbed ethylene, the sample was cooled to 50 °C. Then, an ethylene flow (10 kPa in He) was admitted to the IR cell, whilst recording IR spectra. For the in situ temperature programmed IR (TPR-IR) experiment, the sample was held in an ethylene flow for 60 min followed by heating to 550 °C at a rate of 5 °C min−1 in the closed cell. IR spectra were recorded every 2 min.
2.5. Isotope labelling experiments
13C-Ethylene (13C2H4, Eurisotop, 98%; 99% 13C) was used for isotope reaction experiments. Prior to these experiments, the zeolite catalyst was pre-calcined at 550 °C in a flow of 20 vol% O2 in He. The aromatization reaction was run for 30 min at 500 °C in 4 different modes: (i) in pure 12C-ethylene, (ii) in 12C-ethylene followed by a switch to 13C-ethylene, (iii) in 13C-ethylene followed by a switch to 12C-ethylene and (iv) in pure 13C ethylene. In modes (ii) and (iii), the switches were done after 15 min on stream. To minimize the gas-phase hold-up in the system, the reactor was filled with SiC and all connections were made of 1/16′′ stainless-steel capillaries. The dead time of the system was monitored by adding an Ar tracer (5 vol%) to the feed gas mixture. Additional isotopic labelling experiments were carried out in an equimolar mixture of 13C- and 12C-ethylene at different temperatures (300 °C, 400 °C and 500 °C) maintaining a total partial pressure of ethylene of 10 kPa. The isotopic composition of the reaction products was followed with an online MS system (Pfeiffer Omnistar GSD 301 T3) connected to the reaction outlet via a heated transfer line. To estimate the 13C/12C exchange degree between ethylene isotopologues, the corresponding m/z signals at 28, 29 and 30 were analysed and compared to the reaction runs under the same conditions with pure 12C and 13C ethylene feeds.3. Results and discussion
3.1. Catalyst characterization
To compare the effect of different metals on the catalytic performance, we prepared samples with an equal molar amount of metal (M = Ag, Ga, Zn) with respect to the Al content (M/Al 0.25). Different methods were used to introduce the metals into the zeolites. For Ag, we employed incipient wetness impregnation.11 In this way, a large fraction of BAS can be replaced by Ag+ ions. Solvated Zn2+ and Ga3+ ions cannot enter the micropores of zeolite ZSM-5 because of their large ionic radii. Therefore, incipient wetness impregnation followed by a reductive treatment in H2 was applied. Such treatment results in Zn and Ga species dispersed inside the zeolite pores.32,33 The catalyst characterization data are summarized in Table 1.| Catalyst | S total, m2 g−1 | S micro, m2 g−1 | S external, m2 g−1 | V micro, cm3 g−1 | Metal amount, μmol g−1 | M/Al molar ratio | Exchange degree,a % | Exchanged M/BAS |
|---|---|---|---|---|---|---|---|---|
| a Determined by the fractional occupation of initial BAS by metal ions as probed by 1H NMR spectroscopy (BASinitial − BAS)/BASinitial. | ||||||||
| HZSM-5 | 398 | 322 | 76 | 0.12 | 0 | 0 | 0 | 0 |
| 0.1Ga/HZSM-5 | 347 | 297 | 50 | 0.11 | 83 | 0.09 | 35 | 0.32 |
| 0.25Ga/HZSM-5 | 227 | 192 | 34 | 0.07 | 181 | 0.21 | 44 | 0.56 |
| 0.5Ga/HZSM-5 | 250 | 211 | 39 | 0.08 | 385 | 0.44 | 59 | 0.91 |
| 0.25Zn/HZSM-5 | 368 | 323 | 45 | 0.12 | 198 | 0.24 | 45 | 0.45 |
| 0.25Ag/HZSM-5 | 376 | 331 | 45 | 0.12 | 190 | 0.24 | 47 | 0.53 |
ICP elemental analysis show that the obtained metal content corresponds to the targeted loading for all samples. According to XRD, the main phase present in the modified zeolites was ZSM-5 zeolite without indication of aggregated forms of the added metals or metal oxides (XRD data are shown in Fig. S1†). Ar physisorption results show that all metal-modified zeolites remain highly microporous and exhibit only minor changes in textural properties with respect to the parent zeolite (physisorption isotherms in Fig. S2†). A decrease in the microporous volume is noted for the samples with higher Ga loading.
Zeolite samples were further characterized by solid-state 1H MAS NMR spectroscopy. Fig. 1a shows the 1H MAS NMR spectrum of the parent HZSM-5 zeolite. A signal at 1.6 ppm is attributed to silanol Si–OH groups, a weak signal at 2.5 ppm to Al–OH groups to extra-framework aluminium species, while a signal at 3.8 ppm and a broad signal at 5.4 ppm are related to bridged Si–O(H)–Al groups, which are either isolated or interact with neighbouring hydroxyl groups or framework oxygen atoms, respectively.34
 | ||
| Fig. 1 1H MAS NMR spectra of a) parent HZSM-5; b) xGa/HZSM-5 and c) 0.25Ga/HZSM-5, 0.25Zn/HZSM-5 and 0.25Ag/HZSM-5 as compared to the parent HZSM-5 zeolite. The peaks with a chemical shift of 3.8 ppm and 5.4 ppm correspond to isolated and H-bonded bridged Si–O(H)–Al respectively. The signals at 2.5 ppm and 1.6 ppm are assigned to extra-framework Al–OH and Si–OH groups, respectively. | ||
Zeolite modification with more Ga resulted in a gradual decrease in BAS concentration (1H MAS NMR spectra are shown on Fig. 1b). The ratio of the amount of Ga atom per BAS exchanged varies with Ga content and increases from 0.32 for 0.1Ga/HZSM-5 to 0.56 for 0.25Ga/HZSM-5 and 0.91 for 0.5Ga/HZSM-5. This trend can be ascribed to the formation of different cationic Ga-oxo complexes as earlier reported.32 The data show that the BAS exchange degree was found to be almost the same for all 0.25 M/HZSM-5 samples (Fig. 1c). For all three metals, about two equivalents of BAS were exchanged by one metal atom, indicative of the formation of extra-framework cationic species with a “+2” charge. This stoichiometry is typical for Zn- and Ga-modified HZSM-5. It has been reported that Zn tends to form Zn2+ species stabilized by two bridged hydroxyls, which are in close proximity. The formation of Zn2+ species is therefore favoured at relatively low Zn loading (M/Al < 0.5) and low Si/Al ratio of zeolite.35 For Ga, formation of clustered species of (Ga2O)2x2x+ stoichiometry is anticipated.32,36,37 However, the obtained exchange value is not the typically expected one for Ag, for which formation of single atom Ag+ sites is most likely.38 The latter discrepancy might be explained by the partial removal of framework Al followed by formation of extra-framework Al species as indicated by the increasing 1H NMR signal at 2.5 ppm. This could influence the observed BAS concentration. Thus, Ga-modified ZSM-5 with a loading in the Ga/Al = 0.1–0.5 range and Ag- and Zn-modified ZSM-5 with a metal loading of M/Al = 0.25 were successfully prepared. Importantly, the BAS exchange degree for different metals is similar (ca. 45%) at the M/Al ratio of 0.25, which makes a direct comparison in ethylene aromatization possible.
3.2. Influence of metal promotion on ethylene aromatization
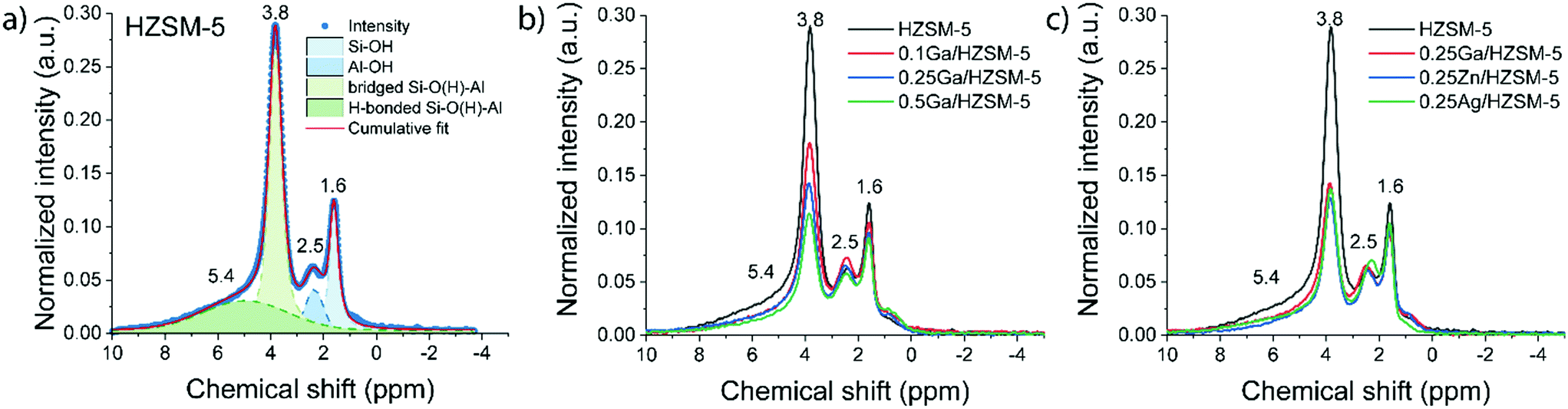
Ethylene aromatization was carried out at 500 °C and the ethylene partial pressure was set at 10 kPa (WHSV 3.75 g gcat−1 h−1). The conversion of ethylene and the product distribution profiles are shown in Fig. 2. In all cases, the catalysts deactivate due to coke deposition. For the parent HZSM-5 zeolite, the ethylene conversion, which was initially 43%, decreased nearly linearly over the course of the reaction, reaching 30% after 20 h. For Ag-, Zn- and Ga-modified zeolite, the initial ethylene conversions were 10%, 55% and 85%, respectively. Both Zn and Ga catalysts deactivate faster than HZSM-5. Although Ag-modified ZSM-5 exhibited a significantly lower activity (initially 10% ethylene conversion), it became inactive already after 2 h on stream. For all catalysts, ethylene was predominantly converted to a mixture of aromatic and aliphatic hydrocarbons. The product distribution is shown in Fig. 2b. HZSM-5 produces mostly propylene and C4–C5 hydrocarbons. In contrast, the fraction of aromatic products is significantly increased for the 0.25 M/HZSM-5 zeolites. The highest aromatic selectivity of 80% is achieved with the Ga-modified catalyst. | ||
| Fig. 2 (a) Conversion of ethylene as a function of time-on-stream; (b) the initial product distribution in the reaction of ethylene on parent and metal-modified 0.25 M/HZSM-5 zeolites measured after 15 min time-on-stream; (c) total coke content in used catalyst samples measured with TGA (reaction conditions: 500 °C, atmospheric pressure; ethylene partial pressure 10 kPa; carrier flow 50 mL min−1 He, WHSV = 3.75 h−1). | ||
The carbonaceous deposits on the used catalysts were then characterized using thermogravimetric analysis (TGA and DTG profiles shown in Fig. S3†). Despite the differences in the rates of deactivation, the coke content and its combustion properties (a single peak at 600 °C in the DTG profile) were similar for all 0.25 M/HZSM-5 catalysts. The lower coke content of used HZSM-5 is in line with the lower rate of deactivation of this sample.39 The peak at 150 °C is associated with water desorption. It is important to note that the overall coke selectivity for Zn/HZSM-5 and Ag/HZSM-5 are much higher than on Ga/HZSM-5 and HZSM-5. These results suggest that under the applied conditions coke is much faster deposited on Zn/HZSM-5 and Ag/HZSM-5 than on Ga/HZSM-5.
To further elucidate the effect of Ga modification, the influence of Ga content on the catalytic performance was investigated. The data in Fig. 3 demonstrate that the rate of deactivation increased with Ga content. Extrapolated initial deactivation rates expressed as the rate at which the conversion decreased ranged from 0.8% Closs h−1 for HZSM-5 to 7, 12 and 25% Closs h−1 for the Ga/HZSM-5 samples with increasing Ga loading. Despite this substantial impact of Ga content on the deactivation rate, the changes observed in the reaction product distribution for the catalysts are relatively small (Fig. 3b).
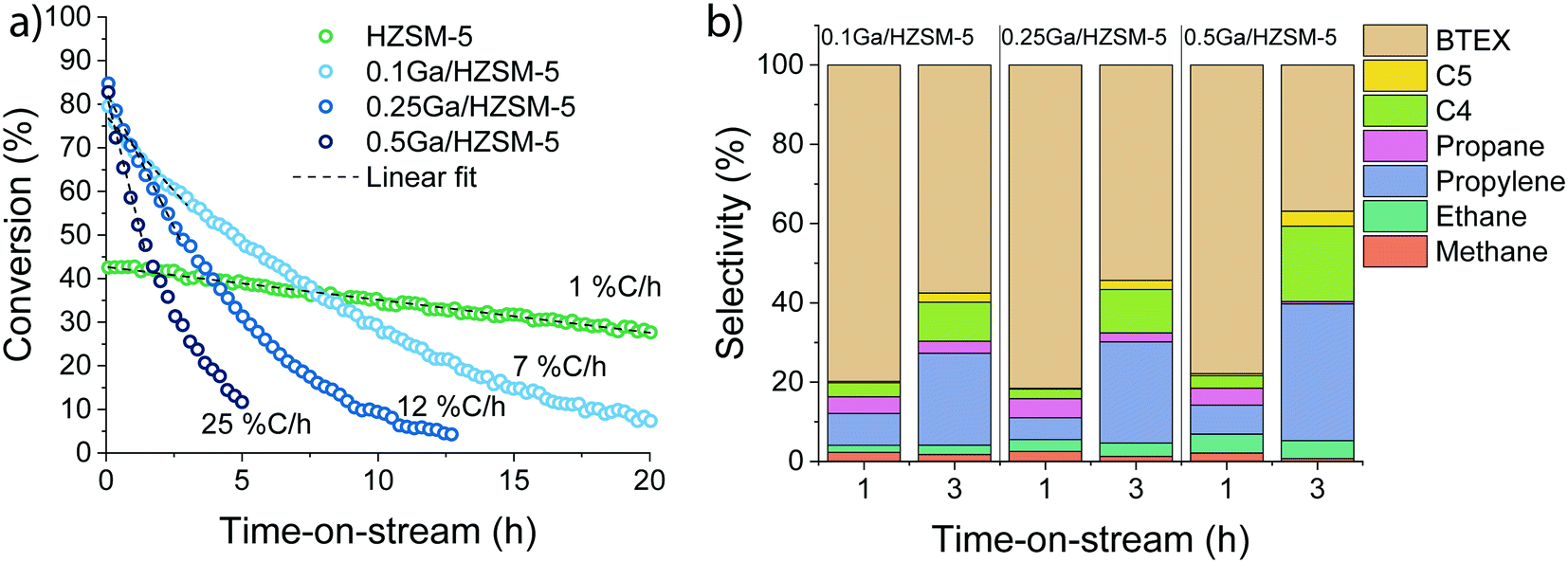 | ||
| Fig. 3 (a) Ethylene conversion as a function of time-on-stream and (b) initial product selectivity in ethylene aromatization reaction on the various Ga/HZSM-5 zeolites. The initial deactivation rates are given as % conversion loss per h (%(C) h−1) (reaction conditions: 500 °C; atmospheric pressure; ethylene partial pressure 10 kPa; carrier flow 50 mL min−1 He, WHSV = 3.75 h−1). | ||
The modification of zeolite with Ag, Zn and Ga resulted in a significant increase in the aromatic selectivity, which suggests that these species play an important role in ethylene conversion. While the formation of higher olefins can be attributed to the oligomerization of ethylene catalysed by BAS, formation of paraffins and aromatics (including coke) is related to dehydrogenation and hydrogen transfer processes.40,41 The low yield of saturated hydrocarbons indicates that the main pathway to form aromatics is dehydrogenation. Dehydrogenation can be catalysed by both BAS and Lewis acidic sites.16 However, the activity of BAS in ethylene activation is not so high (Fig. 2(a)), presumable because it involves formation of a primary carbenium cation resulting in a less stable activation state. The overall dehydrogenation activity increases in the order H < Ga < Zn < Ag, where Ga demonstrates the highest activity for the production of aromatics.
3.3. Influence of temperature
To better understand the mechanistic details of ethylene aromatization reaction, the influence of the reaction temperature (300–650 °C) on the catalytic performance of HZSM-5 and 0.25Ga/HZSM-5 was compared. Ethylene conversion and the reaction products distribution are shown in Fig. 4.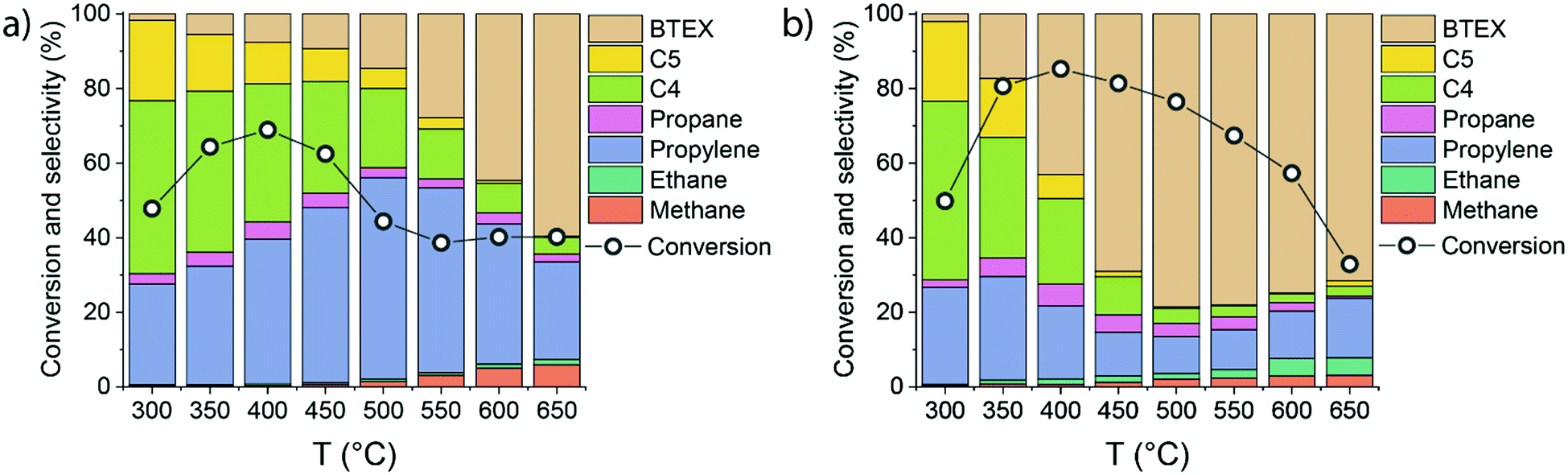 | ||
| Fig. 4 Influence of temperature on the initial ethylene conversion (measured after 15 min time-on-stream) and the product distribution during ethylene conversion over (a) HZSM-5 and (b) 0.25Ga/HZSM-5 zeolites (reaction conditions: 500 °C; atmospheric pressure; ethylene partial pressure 10 kPa; carrier flow 50 mL min−1 He, WHSV = 3.75 h−1). | ||
Ga/HZSM-5 displays substantially higher aromatics selectivity than HZSM-5 in a wide temperature range. While HZSM-5 is mostly selective to propylene and C4–C5 olefins below 550 °C, the BTX selectivity is already close to 50% at 400 °C for Ga/HZSM-5. At higher temperatures, high dehydrogenation rates are manifested in a substantial increase in aromatics formation. The reaction pathways involving hydrogen transfer are also enhanced at increased temperature as follows from an increased production of saturated products such as ethane. However, the relatively low amount of these saturated products indicates that the contribution of this pathway is limited. Nearly identical conversion rates and product distributions observed for the two catalysts at 300 °C suggest that Ga-species are not active below 350 °C. For both catalysts, the conversion increased from 300 °C to 400 °C and then gradually decreased. The decreasing conversion at higher temperature can be understood within the framework of an oligomerization/cracking mechanism. The lower pore occupancy with ethylene at higher temperature results in a decreased rate of formation of oligomers and subsequent reaction products. An alternative explanation is that the reaction involves a set of aromatic intermediates, as previously demonstrated for methanol and ethanol conversion on zeolites.21–24,31 In such a case, the apparent ethylene conversion would involve alkylation/dealkylation processes of the intermediate aromatic species.29,30,42 This hypothesis was evaluated by (i) studying the influence of ethylene partial pressure and (ii) performing isotope labelling experiments to follow the intrinsic activation of ethylene molecules.
The influence of ethylene partial pressure was studied for HZSM-5 at 500 °C for different ethylene feed compositions (pethylene = 10 kPa, 20 kPa and 30 kPa corresponding to WHSV values of 3.75 h−1, 7.5 h−1, and 11.25 h−1). The catalytic results summarized in Fig. 5 reveal a strong influence of the ethylene partial pressure on the conversion and the product distribution.
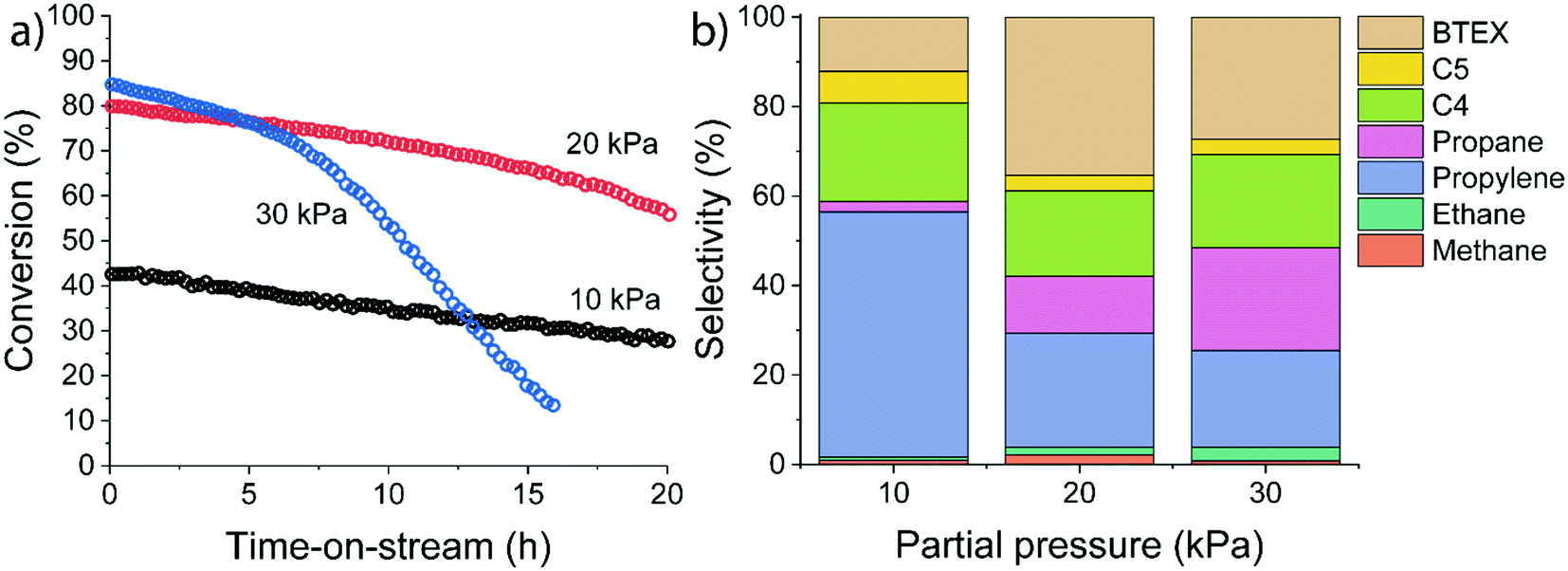 | ||
| Fig. 5 Influence of ethylene partial pressure on (a) ethylene conversion and (b) initial product distribution for ethylene conversion over HZSM-5 measured after 15 minutes time-on-stream (reaction conditions: T = 500 °C; atmospheric pressure; carrier flow 50 mL min−1 He). | ||
The initial ethylene conversion increased from 43% at 10 kPa of ethylene to 80% and 85% at 20 kPa and 30 kPa, respectively. Besides an increase in ethylene conversion, we observed a difference in the deactivation behaviour. Deactivation rates increased at higher ethylene partial pressure. Especially at longer reaction times, a strong deactivation rate was observed after ∼17 h for the 20 kPa experiment and after ∼4 h for the 30 kPa experiment. We explain these differences by a higher rate of (bimolecular) hydrogen transfer reactions at higher reactant partial pressure, as also evidenced by the substantial increase in the propane selectivity. In contrast, at lower reactant partial pressure the dominant pathway is ethylene oligomerization followed by cracking. Overall, the conversion of ethylene over HZSM-5 has a positive order with respect to ethylene. Therefore, a lower pore occupancy (due to a decrease in adsorption) of ethylene with increasing temperature can explain the decreasing conversion (Fig. 5).
Next, isotope labelling experiments were carried out, where a feed composed of an equimolar mixture of 13C- and 12C-ethylene was used. The rate of isotope exchange resulting in the formation of a mixed H213C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 12CH2 ethylene can be directly related to CC bond cleavage in ethylene and recombination. The results of these scrambling experiments show that the isotope exchange rate between 12C- and 13C-ethylene feeds significantly increased when the temperature was raised from 300 °C to 500 °C (MS data and estimated carbon isotope exchange rate shown in Fig. 6). This is a clear indication that the intrinsic activation of ethylene is enhanced at higher temperature, while the apparent conversion is lower due to a higher ethylene selectivity of hydrocarbon pool processes.
12CH2 ethylene can be directly related to CC bond cleavage in ethylene and recombination. The results of these scrambling experiments show that the isotope exchange rate between 12C- and 13C-ethylene feeds significantly increased when the temperature was raised from 300 °C to 500 °C (MS data and estimated carbon isotope exchange rate shown in Fig. 6). This is a clear indication that the intrinsic activation of ethylene is enhanced at higher temperature, while the apparent conversion is lower due to a higher ethylene selectivity of hydrocarbon pool processes.
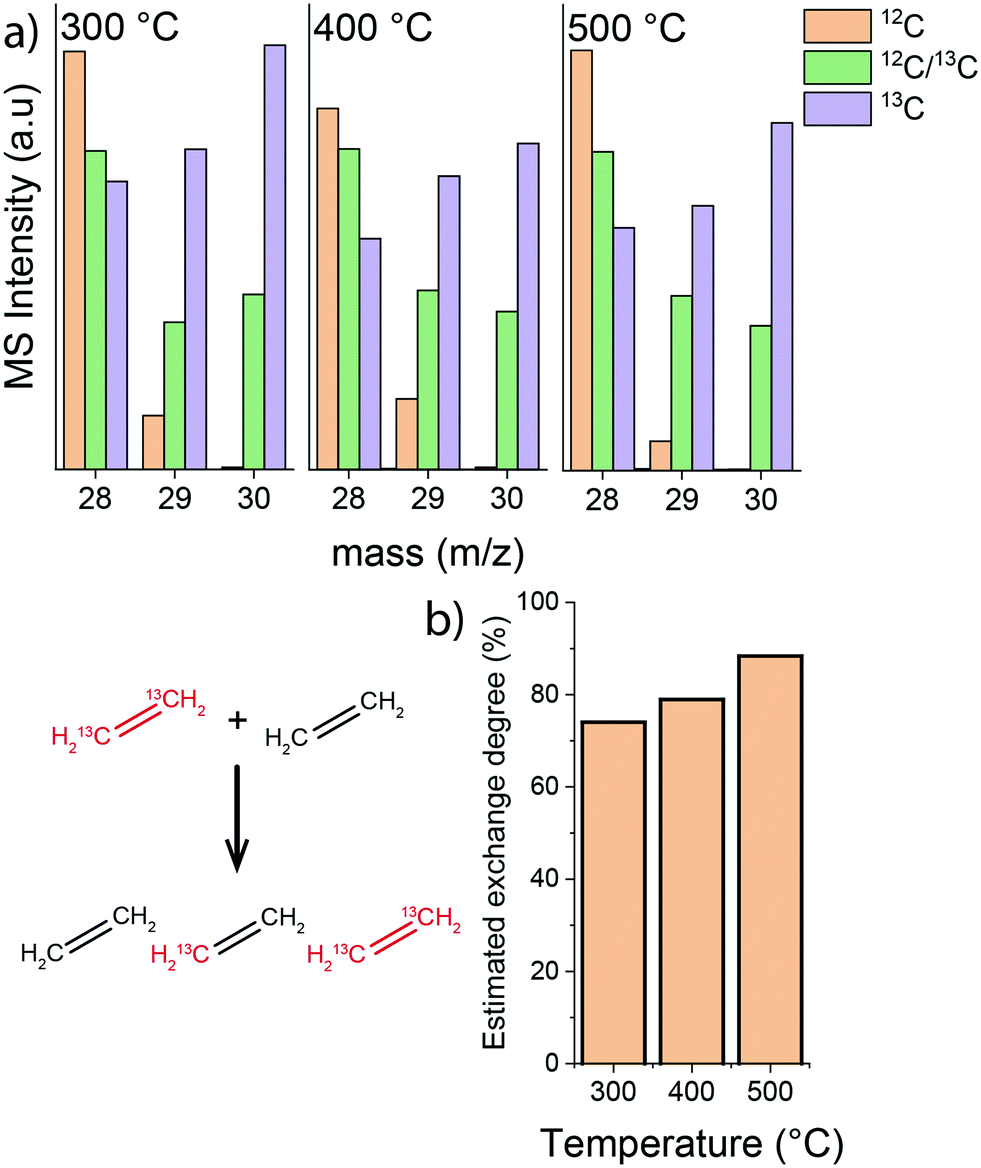 | ||
| Fig. 6 Influence of temperature on ethylene isotope exchange rate during ethylene conversion on HZSM-5: (a) MS signal at mass numbers corresponding to ethylene isotopologues (12C2H4 – m/z 28; H212C–13CH2 – m/z 29; 13C2H4 – m/z 30); (b) estimated isotope exchange degree calculated as 100 × H212C–13CH2/0.5 × (H212C–12CH2 + H212C–13CH2 + H213C–13CH2) (reaction conditions: atmospheric pressure; total ethylene partial pressure 10 kPa; 13C2H4/12C2H4 ratio 1; carrier flow 50 mL min−1 He). | ||
The results obtained suggest that hydrogen transfer reactions during ethylene conversion are catalysed exclusively by BAS and enhanced by higher temperature. The observation that the apparent conversion of ethylene decreases with temperature can be explained to a decreasing ethylene occupancy of the zeolite pores and an increasing selectivity of hydrocarbon pool processes in which intermediates are converted to ethylene, as indicated by the positive ethylene reaction order and faster rate of isotope recombination, respectively. With respect to the involvement of a pool of hydrocarbon intermediates, we investigated their formation and evolution by IR and 13C NMR spectroscopy.
3.4. Structure and role of intra-zeolite species
To determine the structure of intra-zeolitic hydrocarbon species formed during the ethylene conversion reaction, in situ TPD-IR measurements were performed. Pre-calcined HZSM-5 and 0.25Ga/HZSM-5 zeolites were exposed to 10 kPa of ethylene in He at a temperature of 50 °C. Fig. 7 shows the evolution of IR spectra in time upon the interaction of the catalysts with ethylene during the adsorption at 50 °C. Then, the IR cell was closed and heated to 500 °C at a rate of 5 °C min−1 in this atmosphere, while recording IR spectra at regular intervals. The TPD-IR profiles are shown in Fig. 8. During the adsorption step, a gradual disappearance of the band at 3610 cm−1 related to BAS can be observed for both catalysts. Interaction of the adsorbed species with BAS is manifested in the formation of a broad band of disturbed BAS at around 3300–3600 cm−1. Unfortunately, due to the high zeolite framework absorbance at ∼1100 cm−1 (due to v(Si–O) vibrations) it was not possible to directly observe the alkoxy-species. Such species would give rise to stretching v(C–O) vibrations in the 1055–1175 cm−1 range. New bands are visible in the C–H stretching (3200–2800 cm−1) and C–H bending (1800–1300 cm−1) regions. Other bands due to ethylene in the gas phase, i.e. 3104 cm−1 (vasymCH2) and 2990 cm−1 (vsymCH2), were also observed. Upon adsorption, only minor differences are observed for the HZSM-5 and 0.25Ga/HZSM-5 samples. There are small differences in the intensities in the C–H stretching region (features at 2937 cm−1 and 2935 cm−1) and C–H bending regions (1469 cm−1, 1444 cm−1 and 1419 cm−1). As the wavenumbers of these bands are not affected by the presence of Ga, these changes can be ascribed to slight differences in the structure of oligomeric species, such as the length of oligomers and the degree of branching.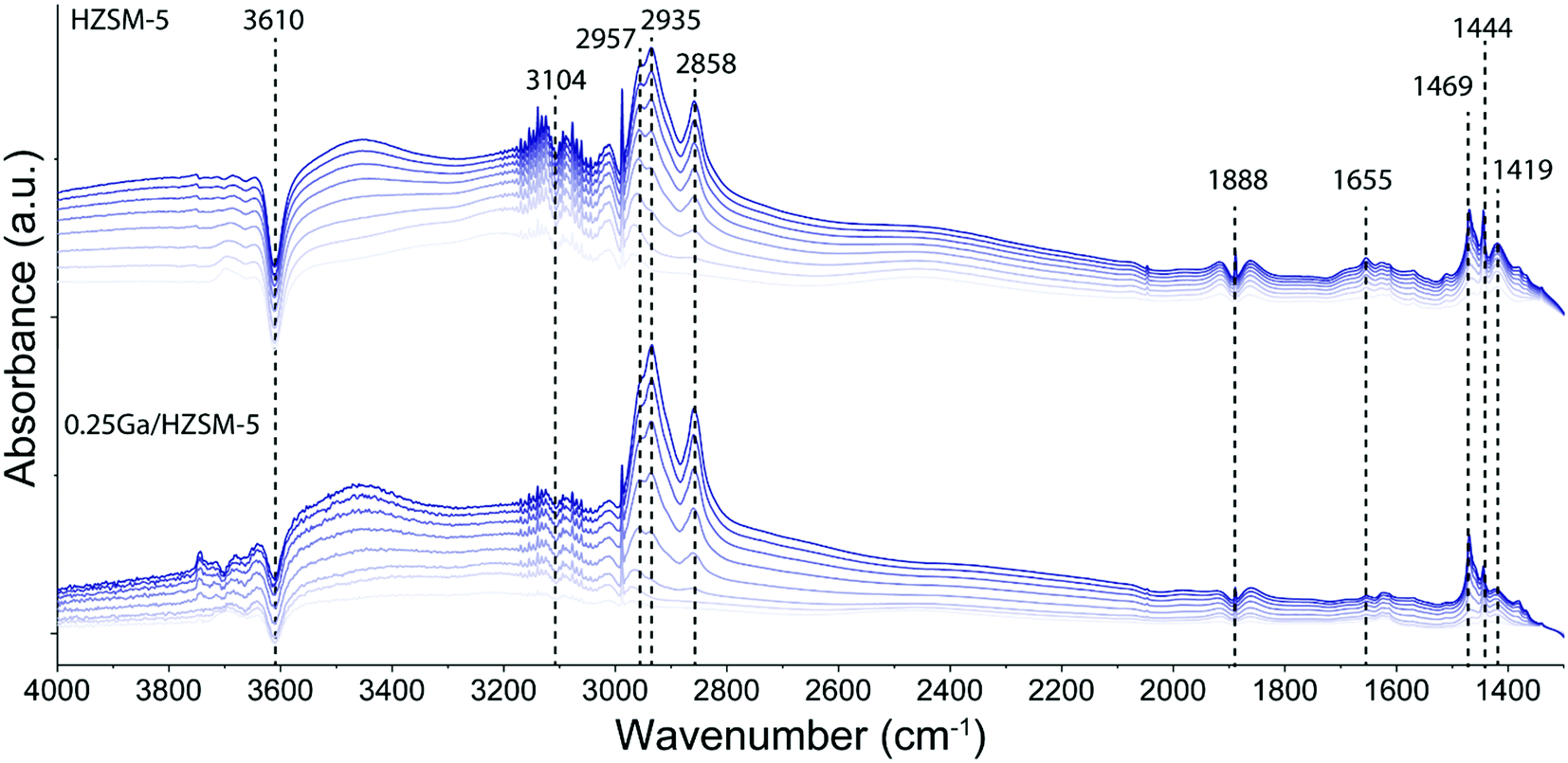 | ||
| Fig. 7 Evolution of IR spectra in time upon adsorption of ethylene on HZSM-5 and 0.25Ga/HZSM-5 zeolites at 50 °C. The spectra are taken every 2.5 min; ethylene partial pressure 10 kPa. | ||
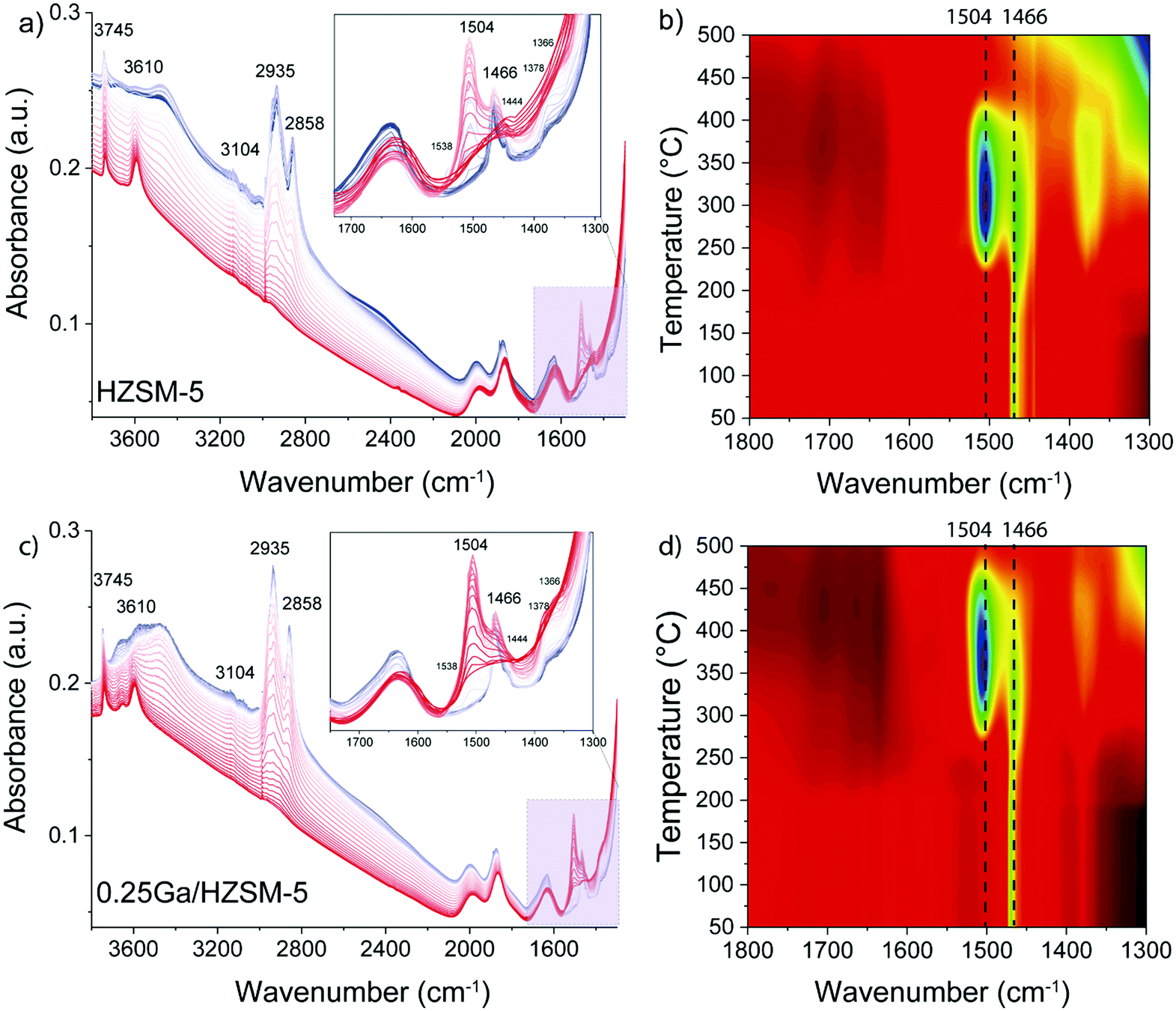 | ||
| Fig. 8 IR spectra and heat maps of low wavenumber region (1800–1300 cm−1) recorded during TPR during exposure to ethylene on (a and b) HZSM-5 and (c and d) 0.25Ga/HZSM-5 zeolites during heating from 50 °C to 500 °C (colour transition from blue to red on the graphs a and c). | ||
The decomposition of the occluded oligomeric species was followed by in situ TPD-IR experiments. The assignment of the IR bands observed is collected in Table 2. A gradual disappearance of IR bands in the 2935–2858 cm−1 range and at 1470 cm−1 followed by formation of new features at 1504 cm−1, 1466 cm−1 and 1444 cm−1 was observed. These bands can be assigned to C–H bending vibrations of hydrogen-deficient aromatic species. The formation of features at 1504 cm−1 and 1466 cm−1 is shifted to higher temperature for 0.25Ga/HZSM-5 samples as compared with HZSM-5 (Fig. 8b and d). Another distinct band at 1504 cm−1 appears below 250 °C for HZSM-5, while it is observed at ca. 300 °C for Ga/HZSM-5. However, the increase in the intensity of these two bands is more pronounced for HZSM-5. Most of these features disappear at 475 °C and 425 °C for Ga/HZSM-5 and HZSM-5, respectively. This difference in aromatics desorption suggests that the interaction of aromatic hydrocarbon intermediates was stronger in Ga/HZSM-5 (Fig. 8b and d). Although the structure of intra-zeolitic species formed at high temperature is nearly identical, there is a certain correlation between the influence of temperature on the formation of bands at 1504 cm−1 and 1466 cm−1 and the reaction performance discussed above (see temperature dependence data in Fig. 4). Indeed, the ethylene conversion was similar at 300 °C for both Ga/HZSM-5 and HZSM-5 samples, while at elevated temperatures the activity of HZSM-5 was lower than that of Ga/HZSM-5. The correlation with the IR spectra suggests that the observed adsorbate species are involved in the catalytic process.
| Wavenumber (cm−1) | Assignment |
|---|---|
| 3745 | ν(SiO–H) of silanols34,43 |
| 3610 | ν(SiO–(H)Al) of BAS34,43 |
| 3300–3600 | Disturbed ν(SiO–(H)Al)34,43 |
| 3104 | Olefinic vCC–H of the gas phase ethylene44 |
| 2957 | v asym(C–H) in –CH3 of oligomers44 |
| 2935 | v asym(C–H) in –CH2– of oligomers44 |
| 2858 | v sym(C–H) in –CH2– of oligomers/vsymC–H in –CH3 of oligomers44 |
| 1610 | δ(C–H) of hydrogen-deficient polyaromatic species45,46 |
| 1540–1538 | Substituted naphthalenes45,46 |
| 1494–1504 | Highly unsaturated (CCC) species45,46 |
| 1600, 1590, 1480–1466, 1380–1360 | Light substituted aromatics47,48 |
To further examine the structure and involvement of hydrocarbon reaction intermediates in the zeolite pores, reactions involving switches between pure 13C- and 12C-ethylene feeds were carried out. The distribution of 13C and 12C atoms in ethylene, propylene, benzene and toluene after switching from 13C- to 12C-ethylene is shown in Fig. S5.† The MS data shows that 13C-ethylene present in the gas phase and adsorbed on the zeolite was quickly removed from the reactor after the switch (∼5 s), while 13C-enriched products (benzene and toluene) were being formed for a longer time. The concentration of 13C in aromatic products slowly decreased and reached the natural abundance level after ∼60 s. These observations show that the deposited intra-zeolitic hydrocarbon species are involved in the ethylene conversion reaction.26,39
The catalyst samples after the reaction involving a switch from 13C- to 12C-ethylene were then analysed by 13C MAS NMR spectroscopy. The 13C direct excitation (DE) and 13C{1H} CP NMR spectra are shown in Fig. 9 and S6.† The sample exposed to pure 13C-ethylene exhibits two signals. A broad signal at 130 ppm corresponds to sp2 carbon and can be assigned to highly unsaturated polyaromatic species inside the zeolite.26,49 A sharp signal at 20 ppm can be attributed to aliphatic sp3 carbon species.50 When 13C-ethylene was switched to 12C-ethylene, the feature at 20 ppm disappeared, while the polyaromatic peak was retained. In contrast, when 12C-ethylene was switched to a 13C-ethylene feed, the initially absent feature at 20 ppm appeared. These changes can be explained by the fact that during the reaction both aliphatic and aromatic species are formed and retained by the zeolite. This is in good agreement with the IR data, which showed the formation of alkylated aromatic species, and also confirms the results of isotope switch experiments. An important observation is that especially the aliphatic part of the intra-zeolite species shows a dynamic response to isotopic switching of the ethylene feed, while the aromatic species retain their isotope composition.
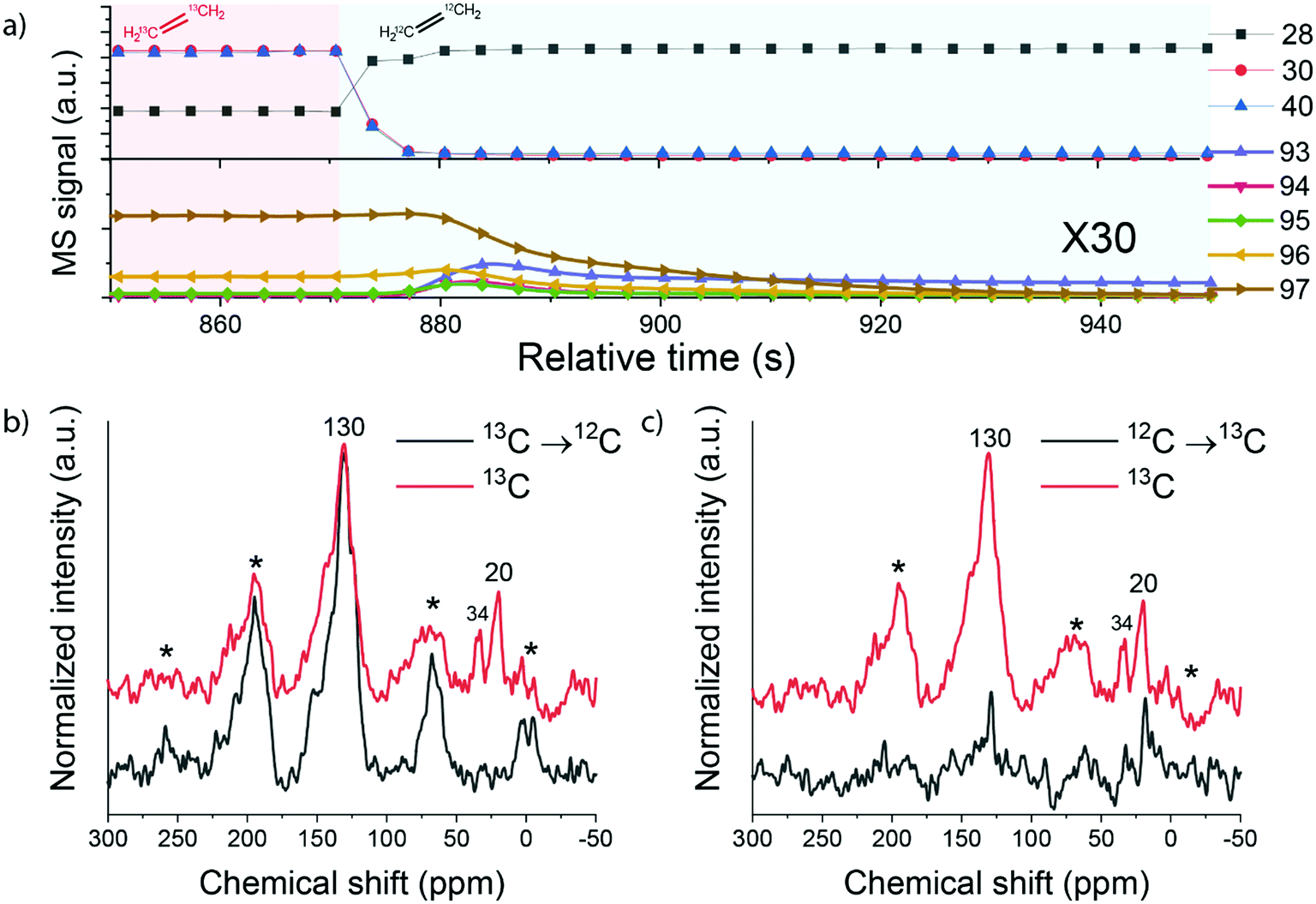 | ||
| Fig. 9 (a) MS data collected during ethylene reaction following switches between 13C-ethylene and 12C-ethylene; 13C MAS NMR of HZSM-5 subjected to ethylene aromatization reaction for 30 minutes with (b) switch from 13C-ethylene to 12C-ethylene after 15 min of reaction and (c) switch from 12C-ethylene to 13C-ethylene feed after 15 min of reaction (reaction conditions: T = 500 °C; atmospheric pressure; carrier flow 50 mL min−1 He). | ||
To summarize, the obtained results point to the catalytic role of hydrocarbon deposits occluded in the pores of transition-metal modified ZSM-5 for the transformations of ethylene to aromatic products. The spectroscopic data indicate that the presence of alkylated (poly)aromatic species occluded in the zeolite pores. Isotope labelling experiments and 13C MAS NMR characterization show that ethylene can reversibly react with aromatics to form alkylaromatics. The direct involvement of confined aromatic intermediates in the catalytic conversion of ethylene is supported by the correlation between their concentration as judged from in situ IR and the catalytic performance. We propose that the mechanism of ethylene conversion by zeolites shares similarities with the dual-cycle hydrocarbon pool mechanism previously postulated for methanol conversion.25 Conventionally, ethylene conversion is thought to proceed through oligomerization of ethylene and cracking of higher olefins, followed by dehydrogenation and cyclization on BAS of the zeolite (Scheme 1a). The oligomerization stage in this reaction scheme resembles the alkene cycle of the MTO process, which opens a pathway towards the formation of aromatic products and the arene cycle in the dual-cycle hydrocarbon pool mechanism (Scheme 1b). We show that the intermediate aromatic species retained in the zeolite micropores can contribute to the activation of ethylene via their alkylation. The alkylated aromatic compounds can then undergo a cascade of trans-alkylation, isomerization and dealkylation processes. The isotope labelling experiments demonstrated that ethylene is a product of dealkylation. For the overall ethylene conversion reaction, the formation of propylene, which is also a part of the dual-cycle hydrocarbon pool mechanism in methanol conversion, is relevant. Olefin oligomerization reactions required to obtain aromatics occur at higher rates for propylene involving secondary carbenium ions than for ethylene, which is converted via very unstable primary carbenium ions. Thus, we suggest a facile pathway from ethylene to propylene via the arene cycle as a contribution to a high overall reaction rate.
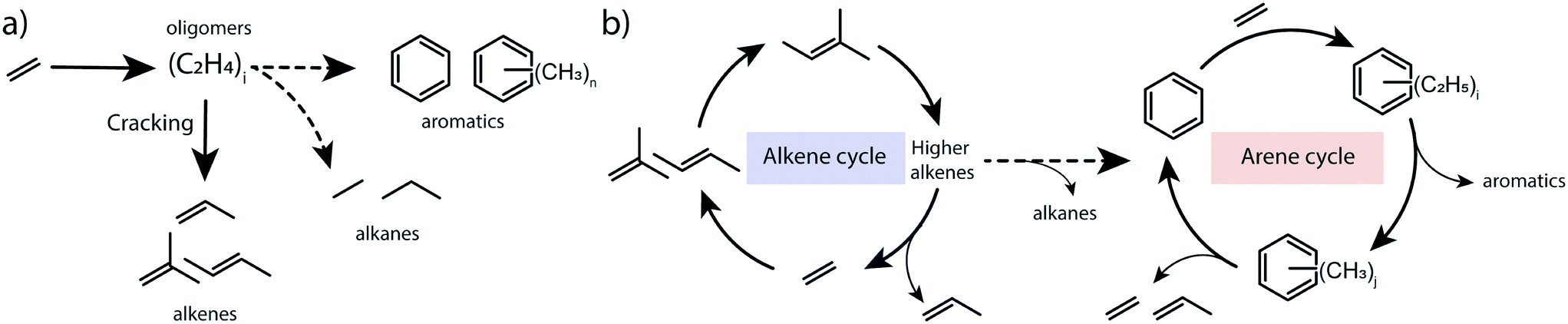 | ||
| Scheme 1 Possible reaction pathways: (a) oligomerization of ethylene followed by cracking of formed oligomers and cyclization/dehydrogenation; (b) dual-cycle hydrocarbon pool mechanism including alkene and arene cycles. Hydrogen transfer processes are indicated with dashed lines. | ||
Introduction of a metal like Ga promotes dehydrogenation, resulting amongst others in a higher rate of aromatics formation. In the concept of the dual-cycle hydrocarbon pool mechanism, this implies a shift to the arene cycle and can explain the higher overall ethylene conversion rate. Another aspect of increased hydrogen transfer rate is a higher rate of deactivation due to the deposition of more coke, usually in the form of polyaromatic deposits. On the other hand, we cannot exclude the possibility of intra-zeolitic Lewis acidic Ga+ species playing a role in ethylene dimerization as postulated before.51 Further dedicated characterization, mechanistic and theoretical studies are required to unravel the nature and role of the occluded hydrocarbon intermediates, zeolite BAS and extra-framework cations as well as their involvement in the complex reaction network underlying the catalytic conversion of ethylene by transition-metal modified zeolites.
4. Conclusions
This work presents a detailed study of ethylene aromatization catalysed by zeolites. Ag-, Zn- and Ga-modified HZSM-5 zeolites with similar concentration of remaining BAS were prepared and characterized. The catalytic results revealed a strong influence of metal promotion on the dehydrogenation activity of the catalyst. It ranges from an extreme for Ag leading to a rapid catalyst deactivation and highest coke selectivity to Zn and then to Ga, the latter one providing an optimal dehydrogenation and aromatization activity and the lowest coking selectivity. Nevertheless, the rate of deactivation for Ga/HZSM-5 is higher than that of HZSM-5. Furthermore, it was found that dehydrogenation activity and aromatics selectivity are enhanced by working at higher temperatures, while an increase in the ethylene partial pressure facilitates hydrogen transfer processes. Isotopic scrambling experiments reveal that, despite a lower apparent ethylene conversion with increasing temperature, the intrinsic rate of activation of ethylene is enhanced. Further isotope labelling experiments in combination with 13C MAS NMR points to the importance of intra-zeolite aromatic hydrocarbon species for ethylene conversion on HZSM-5. The data suggests that alkylation of aromatic intermediates provide a facile pathway to the formation of higher olefins. Further investigations are needed to verify the importance of such a mechanism for metal-promoted HZSM-5. The present findings provide strong indications that hydrocarbon pool type processes might play an important role in ethylene conversion on zeolites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation programme funded by the Ministry of Education, Culture and Science of the government of the Netherlands. H. S. acknowledges the Leading Graduate Program in Science and Engineering, Waseda University from MEXT, Japan.References
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- J. J. Siirola, AIChE J., 2014, 60, 810–819 CrossRef CAS.
- A. Martínez, M. A. Arribas and S. Moussa, in Zeolites in Catalysis: Properties and Applications, The Royal Society of Chemistry, 2017, pp. 351–408 Search PubMed.
- E. T. C. Vogt, G. T. Whiting, A. Dutta Chowdhury and B. M. Weckhuysen, in Advances in Catalysis, Elsevier, 2015, vol. 58, pp. 143–314 Search PubMed.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- M. Guisnet, N. S. Gnep and F. Alario, Appl. Catal., A, 1992, 89, 1–30 CrossRef CAS.
- A. Hagen and F. Roessner, Catal. Rev.: Sci. Eng., 2000, 42, 403–437 CrossRef CAS.
- X. Chen, M. Dong, X. Niu, K. Wang, G. Chen, W. Fan, J. Wang and Z. Qin, Chin. J. Catal., 2015, 36, 880–888 CrossRef CAS.
- H. Saito, S. Inagaki, K. Kojima, Q. Han, T. Yabe, S. Ogo, Y. Kubota and Y. Sekine, Appl. Catal., A, 2018, 549, 76–81 CrossRef CAS.
- V. R. Choudhary, P. Devadas, S. Banerjee and A. K. Kinage, Microporous Mesoporous Mater., 2001, 47, 253–267 CrossRef CAS.
- M. F. Hsieh, Y. Zhou, H. Thirumalai, L. C. Grabow and J. D. Rimer, ChemCatChem, 2017, 9, 1675–1682 CrossRef CAS.
- G. Caeiro, R. H. Carvalho, X. Wang, M. A. N. D. A. Lemos, F. Lemos, M. Guisnet and F. Ramôa Ribeiro, J. Mol. Catal. A: Chem., 2006, 255, 131–158 CrossRef CAS.
- Y. Ono, H. Kitagawa and Y. Sendoda, Sekiyu Gakkaishi, 1987, 30, 77–88 CrossRef CAS.
- L. Mahoney, L. Emdadi, A. C. Leff, D. T. Tran, W. Wu, S. Cheng, D. Liu, C. K. Nguyen and I. C. Lee, Fuel, 2019, 256, 115953 CrossRef CAS.
- M. J. Truitt, S. S. Toporek, R. Rovira-Truitt and J. L. White, J. Am. Chem. Soc., 2006, 128, 1847–1852 CrossRef CAS PubMed.
- T. F. Narbeshuber, A. Brait, K. Seshan and J. A. Lercher, J. Catal., 1997, 172, 127–136 CrossRef CAS.
- M. Guisnet, N. S. Gnep, D. Aittaleb and Y. J. Doyemet, Appl. Catal., A, 1992, 87, 255–270 CrossRef CAS.
- N. Rane, M. Kersbulck, R. A. van Santen and E. J. M. Hensen, Microporous Mesoporous Mater., 2008, 110, 279–291 CrossRef CAS.
- A. G. Gayubo, A. M. Tarrío, A. T. Aguayo, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2001, 40, 3467–3474 CrossRef CAS.
- T. Liang, H. Toghiani and Y. Xiang, Ind. Eng. Chem. Res., 2018, 57, 15301–15309 CAS.
- D. Lesthaeghe, A. Horré, M. Waroquier, G. B. Marin and V. Van Speybroeck, Chem. – Eur. J., 2009, 15, 10803–10808 CrossRef CAS PubMed.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- M. Bjorgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjrgen, P. Beato, T. V. W. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS PubMed.
- E. A. Uslamin, N. Kosinov, G. A. Filonenko, B. Mezari, E. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 8547–8554 CrossRef CAS.
- U. Olsbye, M. Bjørgen, S. Svelle, K. P. Lillerud and S. Kolboe, Catal. Today, 2005, 106, 108–111 CrossRef CAS.
- R. Johansson, S. L. Hruby, J. Rass-Hansen and C. H. Christensen, Catal. Lett., 2009, 127, 1–6 CrossRef CAS.
- A. D. Chowdhury, A. Lucini Paioni, G. T. Whiting, D. Fu, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2019, 58, 3908–3912 CrossRef CAS PubMed.
- F. F. Madeira, H. Vezin, N. S. Gnep, P. Magnoux, S. Maury and N. Cadran, ACS Catal., 2011, 1, 417–424 CrossRef CAS.
- E. A. Uslamin, B. Luna-Murillo, N. Kosinov, P. C. A. Bruijnincx, E. A. Pidko, B. M. Weckhuysen and E. J. M. Hensen, Chem. Eng. Sci., 2019, 198, 305–316 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, L. V. Pirutko, D. Freude and A. G. Stepanov, J. Phys. Chem. C, 2018, 122, 25386–25395 CrossRef CAS.
- V. B. Kazansky and A. I. Serykh, Phys. Chem. Chem. Phys., 2004, 6, 3760–3764 RSC.
- E. A. Pidko, E. J. M. Hensen and R. A. van Santen, Proc. R. Soc. A, 2012, 468, 2070–2086 CrossRef CAS.
- E. A. Pidko, R. A. van Santen and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2009, 11, 2893–2902 RSC.
- Y. Kuroda, H. Onishi, T. Mori, Y. Yoshikawa, R. Kumashiro, M. Nagao and H. Kobayashi, J. Phys. Chem. B, 2002, 106, 8976–8987 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- S. Müller, Y. Liu, F. M. Kirchberger, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 15994–16003 CrossRef PubMed.
- C. Liu, R. A. van Santen, A. Poursaeidesfahani, T. J. H. Vlugt, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2017, 7, 8613–8627 CrossRef CAS PubMed.
- Y. Ni, A. Sun, X. Wu, G. Hai, J. Hu, T. Li and G. Li, Microporous Mesoporous Mater., 2011, 143, 435–442 CrossRef CAS.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- A. G. Stepanov, M. V. Luzgin, V. N. Romannikov, V. N. Sidelnikov and E. A. Paukshtis, J. Catal., 1998, 178, 466–477 CrossRef CAS.
- H. G. Karge, W. Nießen and H. Bludau, Appl. Catal., A, 1996, 146, 339–349 CrossRef CAS.
- A. Vimont, O. Marie, J. P. Gilson, J. Saussey, F. Thibault-Starzyk and J. C. Lavalley, in Studies in Surface Science and Catalysis, 1999, vol. 126, pp. 147–154 Search PubMed.
- F. Thibault-Starzyk, A. Vimont and J. P. Gilson, Stud. Surf. Sci. Catal., 2001, 135, 133 CrossRef.
- G. Mirth and J. A. Lercher, J. Catal., 1991, 132, 244–252 CrossRef CAS.
- S. Du, D. P. Gamliel, M. V. Giotto, J. A. Valla and G. M. Bollas, Appl. Catal., A, 2016, 513, 67–81 CrossRef CAS.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed.
- E. A. Pidko, E. J. M. Hensen and R. van Santen, J. Phys. Chem. C, 2008, 112, 19604–19611 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02108f |
| ‡ Present address: Inorganic Systems Engineering group, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2020 |