
Base-controlled product switch in the ruthenium-catalyzed protodecarbonylation of phthalimides: a mechanistic study†
Massimo Christian
D'Alterio
 ab,
Yu-Chao
Yuan
c,
Christian
Bruneau
c,
Giovanni
Talarico
b,
Rafael
Gramage-Doria
*c and
Albert
Poater
*a
ab,
Yu-Chao
Yuan
c,
Christian
Bruneau
c,
Giovanni
Talarico
b,
Rafael
Gramage-Doria
*c and
Albert
Poater
*a
aInstitut de Quimica Computacional i Catalisi and, Departament de Quimica, Universitat de Girona, c/ Ma Aurelia Capmany 69, 17003 Girona, Catalunya, Spain. E-mail: albert.poater@udg.edu
bDipartimento di Scienze Chimiche, Universita di Napoli Federico II, Via Cintia, I-80126 Napoli, Italy
cUniv Rennes, CNRS, ISCR-UMR6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
First published on 21st November 2019
Abstract
The whole reaction mechanism of the ruthenium-catalyzed protodecarbonylation of N-substituted phthalimides into secondary amides was unravelled by a combined experimental and theoretical study. The chemoselectivity of the reaction, which is catalyzed by para-cymene coordinated Ru(II) species all over the catalytic cycle, is exclusively controlled by the unique roles of the bases. Meanwhile, in the presence of K2CO3 or KOH at high temperatures, the same product (benzamide) is mainly formed, whereas at low temperatures, KOH led to an unexpected side-product (phthalamic acid) and no reactivity was observed with K2CO3. The non-covalent interactions between the potassium cations and the different carbonyl groups in the molecules are key to providing a thermodynamically favourable pathway with energetically accessible transition states. The unexpected formation of carbon dioxide (CO2) in the course of the reaction originates from the phthalimide substrate and the base K2CO3 in two different elementary steps, respectively.
Introduction
Over the past decades, homogeneous catalysis has played a pivotal role to cover the demands of our society.1 For instance, the synthesis of many daily life chemicals (drugs, pesticides, OLED materials, commodities, polymers, etc.) comprises at least one step involving transition metal catalysed transformation and many of these reactions nowadays are being conducted on a multiton scale (i.e. the rhodium-catalyzed hydroformylation of alkenes).2 Consequently, the fundamental understanding of metal reactivity and the discovery of new chemical reactions are extremely appealing in view of generating sustainable and environmentally benign processes in academic and industrial laboratories.Well-defined transition metal catalysts derived from platinum, rhodium, iridium and palladium are, by far, the most widely used for fine chemical synthesis due to their versatility and ability to reach multiple oxidation states and multiple coordination geometries that can affect the reactivity of given substrates.3 However, these metals are extremely expensive, very scarce on the Earth's crust and their related complexes are, in general, unstable in air and moisture.3 It seems convenient then to turn the attention to more abundant, affordable and stable metal complexes. As such, ruthenium seems promising since it exhibits a large number of oxidation states (from +8 to −2) and coordination geometries comparable to most precious metals, which makes this metal interesting to replace the less abundant ones.4 In addition, the reactivity of ruthenium complexes for fine chemical synthesis has been well-established during the last decades.4 The most relevant breakthroughs in this context are (1) the asymmetric hydrogenation of ketones into chiral alcohols with Ru–BINAP–diamine complexes by Noyori,5 (2) the olefin metathesis reaction with RuCl2(PCy3)2(CHPh) as a pre-catalyst by Grubbs,6 and (3) the C–H bond functionalization of aryl moieties with RuH2(CO)(PPh3)3 as a pre-catalyst by Murai using ketones as directing groups.7
Aiming at discovering new chemical reactivity for ruthenium, some us have recently reported a general and highly functional group tolerant protodecarbonylation of N-substituted phthalimides into amides using [RuCl2(p-cymene)]2 as the pre-catalyst in the presence of K2CO3 and water at high temperatures (Scheme 1).8 Herein, we show detailed control experiments together with theoretical calculations at the DFT level to identify the mechanism operating in this ruthenium-catalyzed reaction, in particular, regarding the five-membered ring opening step.9 The key to the success of the reaction is the various roles played by the base. Most notably, the non-covalent interactions between potassium cations and carbonyl groups as well as an unexpected intramolecular nucleophilic attack prior to the release of CO2 have been identified. An interesting switch of product selectivity is observed when using KOH instead of K2CO3 as the base. It is relevant to note that the formal opposite reaction, namely the CO-carbonylation of amides into phthalimides catalysed by ruthenium and rhodium, has been reported by Chatani;10 and Rovis, respectively.11 Recently, Goossen, Hartwig and Zhao, Hong, Baidya, and Shi, independently, have also reported appealing ruthenium-catalyzed decarboxylative reactions with structurally-related benzoic acid derivatives,12 apart from the homologous studies with nickel13 and palladium.14
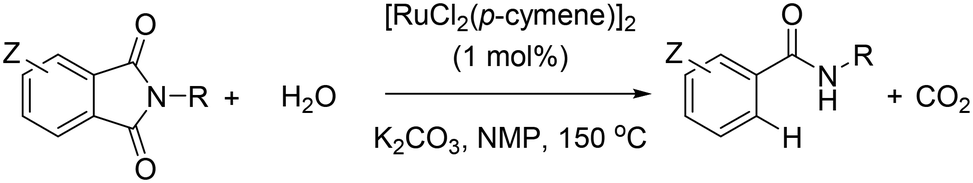 | ||
| Scheme 1 Ruthenium-catalyzed protodecarbonylation of phthalimides whose mechanism is studied in this study [NMP = N-methyl-2-pyrrolidone; R = alkyl, benzyl, and aryl; Z = F, Cl, NO2, and Me; tolerant to –CN, –OMe, –CF3, –Br, –I, –C(O)Me, –CO2Et, pyridine, quinoline, and thiophene]. | ||
Results and discussion
Catalytic experiments
The optimal reaction conditions previously developed consisted of [RuCl2(p-cymene)]2 (1 mol%), 1.5 equivalents of water and 3 equivalents of K2CO3 at 150 °C for 6 hours.8 Under these reaction conditions, N-methylphthalimide (1) has fully reacted, leading to N-methylbenzamide (2) in an almost quantitative yield (Table 1, entry 1). Previously, preliminary mechanistic investigations indicated that CO2 was formed in the course of the reaction and water served as the source of protons according to GC gas phase analysis and deuteration experiments, respectively.8 A classical hydrogenation pathway was unlikely as the reaction did not occur in the presence of H2 gas, and a slight excess of K2CO3 and water was required for the success of the transformation.8 In addition, radical trapping experiments and mercury tests suggested homogeneous pathways involving no formation of radicals.8|
|
||||
|---|---|---|---|---|
| Entry | [Ru] (× mol%) | Base | T (°C) |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3b :2:3b |
| a Reaction conditions: 1 (0.4 mmol), H2O (0.6 mmol), base (1.2 mmol), NMP (2 mL), argon. b Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard and by GC-MS analysis. c Unknown by-products are formed. | ||||
| 1 | [RuCl2(p-cymene)]2 (1) | K2CO3 | 150 | 0:100:0 |
| 2 | [Ru(p-cymene)(MesCO2)2] (2) | K2CO3 | 150 | 0:100:0 |
| 3 | [Ru(NCtBu)6(BF4)2] (2) | K2CO3 | 150 | 70:0:0c |
| 4 | [RuCl2(p-cymene)]2 (1) | KOH | 150 | 0:72:28 |
| 5 | [RuCl2(p-cymene)]2 (1) | KOH | 100 | 0:10:90 |
| 6 | [RuCl2(p-cymene)]2 (1) | KOH | 25 | 0:0:100 |
| 7 | [RuCl2(p-cymene)]2 (1) | K2CO3 | 25 | 100:0:0 |
| 8 | [RuCl2(p-cymene)]2 (1) | — | 25 | 100:0:0 |
| 9 | [RuCl2(p-cymene)]2 (1) | — | 100 | 100:0:0 |
With this background, we performed a number of experiments with different ruthenium complexes as pre-catalysts in view of understanding which ligands lead to catalytically productive ruthenium species. K2CO3 and KOH bases were evaluated at different temperatures as shown in Table 1. The same catalytic outcome was observed with 2 mol% [Ru(p-cymene)(MesCO2)2] (Mes = 2-mesityl) as the pre-catalyst (Table 1, entry 2),15 whereas a poor conversion of 30% was observed with the 2 mol% para-cymene-free [Ru(NCtBu)6(BF4)2] complex and no formation of the expected product 2 was observed (Table 1, entry 3).16 These data strongly suggested that the catalytically active metal complex involved chloride-free, monomeric para-cymene-coordinated ruthenium(II) species.
For comparison purposes, we performed a reaction with KOH as the base instead of K2CO3 as we reasoned that the anion of the base (hydroxyl versus carbonate) could influence the catalysis to some extent. At 150 °C with 1 mol% [RuCl2(p-cymene)]2, full conversion of 1 was observed with the unexpected side-formation of N-methylphthalamic acid (3) together with the benzamide 2 as the products (Table 1, entry 4). Interestingly, 3 became the major product of the reaction when the temperature was decreased to 100 °C (Table 1, entry 5) and at room temperature, it was exclusively formed with the full conversion of 1 (Table 1, entry 6). It is important to note that metal-free, base-mediated five-membered ring opening of N-substituted phthalimides is known to occur only at high temperatures,17 which further supports that the formation of 3 at room temperature using KOH is indeed catalysed by ruthenium species. In stark contrast, using K2CO3 as the base at room temperature led to no conversion of starting material 1 (Table 1, entry 7). The starting material was recovered unreacted when the reaction was performed without a base regardless of the temperature (Table 1, entries 8–9).8
DFT calculations
Taking into account all the previous experimental data and the difficulty to isolate any potential ruthenium intermediates during the catalysis,8 theoretical calculations at the DFT level were carried out to gain a better understanding of the origin of the chemoselectivity observed by swapping the base in the ruthenium-catalyzed protodecarbonylation of phthalimides (Fig. 1).18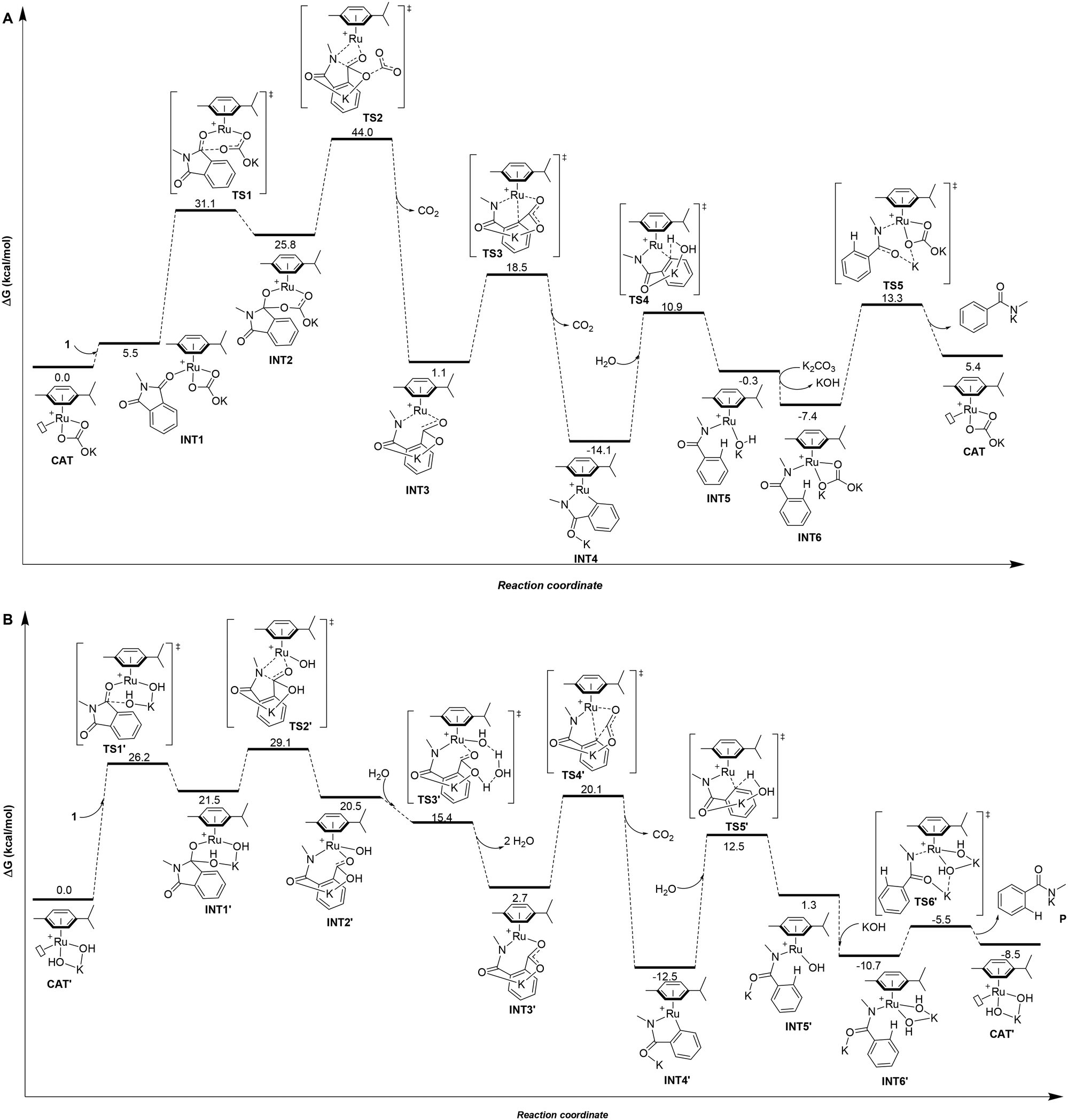 | ||
| Fig. 1 (A) Computed minimum free energy reaction profile (in kcal mol−1) of the ruthenium-catalyzed protodecarbonylation of 1 assisted by K2CO3 and (B) computed minimum free energy reaction profile (in kcal mol−1) of the ruthenium-catalyzed protodecarbonylation of 1 assisted by KOH. | ||
First, the catalytic cycle was studied for the case where K2CO3 is employed as the base, which selectively leads to benzamide 2 at 150 °C (Fig. 1A). The initial catalytically active species for the calculations was rationalized to be a cationic 16 electron Ru(II) complex with one μ6-coordinated para-cymene ligand and one κ2-coordinated carbonate ligand with a vacant site at ruthenium (CAT, Fig. 1A). Such hypothesis is further supported by the findings from Demerseman and co-workers who reported that reacting [Ru(p-cymene)Cl2(PR3)] (R = Me, Cy, and Ph) with K2CO3 in a polar solvent (i.e. acetone) led to [Ru(p-cymene)(κ2-O2CO)(PR3)] species.19 In addition, we verified that the reaction of [RuCl2(p-cymene)]2 with K2CO3, releasing KCl and forming [Ru(p-cymene)(κ2-O2COK)]+ (CAT), is highly exergonic with ΔG = −40.1 kcal mol−1 (see ESI† for details).
In the intermediate INT2, the hybridization of one carbon atom switches from sp2 in INT1 to sp3 in INT2 with a subsequent elongation of the C–N bond by 0.06 Å (dC–N = 1.39 Å in INT1versus dC–N = 1.45 Å in INT2). Next, the simultaneous five-membered ring opening of 1 and the decarboxylation from the carbonate fragment occur viaTS2 (Fig. 2A) giving rise to intermediate INT3 (Fig. 1A).
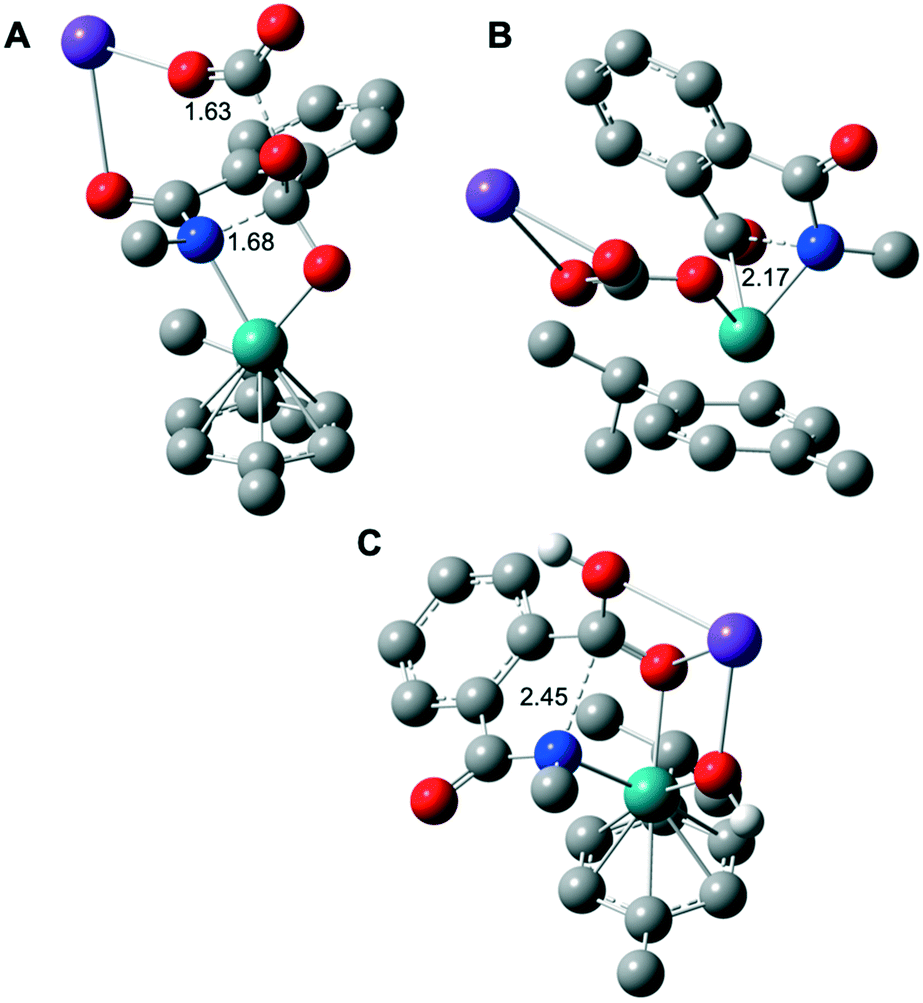 | ||
| Fig. 2 Computed molecular structure of the five-membered ring opening rate determining step TS2 in the case of K2CO3 as the base for the assisted mechanism of Fig. 1 (A); without considering the intramolecular nucleophilic attack from INT1 to INT2 (B); (C) the five-membered ring opening TS2′ of Fig. 3 for the KOH assisted mechanism. H atoms are omitted for clarity (except the protic ones). Selected distances are displayed in Å. Color code: Ru in green, K in purple, O in red, N in blue and C in gray. | ||
TS2 appears to be the rate determining step of the catalytic cycle with an energetic cost of 44.0 kcal mol−1 with respect to CAT (Fig. 1A). The calculated energetic barrier is in full agreement with the experimental reaction temperature of 150 °C required for the success of the catalysis. Despite the latter energetically-demanding step viaTS2, the previous intramolecular nucleophilic attack defined by TS1 is crucial in kinetic terms as well, since the weakening of the C–N bond in INT2 (dC–N = 1.45 Å, vide supra) makes the former substrate ready for the ring opening (Fig. 1A). Indeed, for comparison purposes, we modelled the transition state (TS) of the five-membered ring opening of 1 without the previous nucleophilic attack. In this case, the substrate 1 coordinates to ruthenium via the nitrogen atom and a carbon atom belonging to the carbonyl group (Fig. 2B) with an energetic barrier that goes up to 75.1 kcal mol−1 with respect to CAT in order to form the intermediate INT3. From intermediate INT3, decarboxylation occurs within the substrate viaTS3 with an energy barrier of 17.4 kcal mol−1 with respect to INT3, affording the very stable five-membered ruthenacycle INT4 (Fig. 1A). The high stability of INT4 is largely ascribed to the formation of the new Ru–C(aryl) bond. The next concerted TS4, with an energy barrier of 25.0 kcal mol−1, requires the participation of one molecule of H2O (Fig. 1A). More precisely, the Ru–C(aryl) bond is broken via proton transfer from H2O to the carbon atom and the hydroxyl group from H2O migrates to the potassium cation, formally releasing KOH. Thus, a new K2CO3 molecule enters in the catalytic cycle serving as a ligand to ruthenium leading to intermediate INT6, in which the benzamide product is already coordinated to the catalyst. The theoretical evidence of the second additional K2CO3 molecule is in agreement with the experimental requirements of an excess of base to reach good yields of the benzamide product. The elementary step (INT4 → INT5) is well supported by the experimental evidence that 1.5 equivalents of water are enough to complete the catalysis (Table 1). The next step is supported by the excess K2CO3, together with the thermodynamic stability of INT6 (7.1 kcal mol−1). Finally, P is released viaTS5 with an energy barrier of 20.7 kcal mol−1, but 27.4 kcal mol−1 from INT4 (Fig. 1A). Experimentally, 2 is obtained from P by an acidic work-up.
In the case of the product selectivity controlled by KOH, we reasoned that the catalytically active ruthenium species might be the mononuclear [Ru(OH)2(p-cymene)K]+ complex (CAT′, Fig. 1B) similar to the experimental findings from Stephenson and co-workers.20 For that, the calculations performed for the reaction of [RuCl2(p-cymene)]2 with KOH forming CAT′ and KCl led to a highly favourable exergonic process with ΔG = −55.6 kcal mol−1 (see ESI† for details). In this case, we considered that four water molecules form a hydration cluster with KOH, as it was already noted elsewhere.21,22
Differently, from the case of K2CO3, the substrate 1 cannot coordinate to catalyst CAT′ according to the calculations (Fig. 1B). In fact, a carbonyl group of 1 directly undergoes nucleophilic attack by an hydroxyl group coordinated to ruthenium viaTS1′ with an energy barrier of 26.2 kcal mol−1 (Fig. 1B). The resulting intermediate INT1′ presents a carbon atom that has switched its hybridization from sp2 to sp3 resulting in an elongation of the C–N bond by 0.17 Å (from 1.39 Å in 1 to 1.56 Å in INT1′). Importantly, the nucleophilic attack of the KOH base and the subsequent C–N bond elongation in 1 both together promote the five-membered ring opening of substrate 1.
The rate determining step for the OH− assisted mechanism is the five-membered ring opening of 1 (TS2′ of Fig. 1B) analogous to the KCO3− assisted mechanism (TS2 of Fig. 1A); however, we calculated a lower energy barrier for TS2′ (29.1 kcal mol−1) compared with 44.0 kcal mol−1 for TS2. Such observation is a direct consequence of the longer distance of the C–N bond observed in INT1′ (ΔdC–N = 0.17 Å, Fig. 1B) compared to that of INT2 (ΔdC–N = 0.06 Å, Fig. 1A). This finding suggests the higher reactivity of the [Ru(OH)2(p-cymene)K]+ complex (CAT′) than that of the [Ru(p-cymene)(κ2-O2COK)]+ complex (CAT) even at low temperatures. However, experimentally, CAT′ does not yield any secondary amide 2 as a product, but yields phthalamic acid derivative 3 at low temperatures (Table 1, entry 6). Indeed, the intermediate INT2′ can be considered as the product 3 coordinated to the [Ru(OH)(p-cymene)]+ complex (Fig. 1B).
Following an alternative mechanistic pathway (Fig. 3), the addition of a KOH molecule easily releases product P′, the K+ salt of 3 and the regeneration of the catalyst via the transition state TS3′′ with an overall energetic barrier of 13.0 kcal mol−1 with respect to CAT′ (Fig. 3). On the other hand, the pathway leading to the formation of product 2 (Fig. 1B) requires overcoming two energetic barriers higher than 13.0 kcal mol−1, which are (1) the decarboxylation of the substrate viaTS4′ with an energetic barrier of 20.1 kcal mol−1 and (2) the protonation of the ruthenium-coordinated C(aryl) atom by a water molecule viaTS5′ with an energetic barrier of 25.0 kcal mol−1.
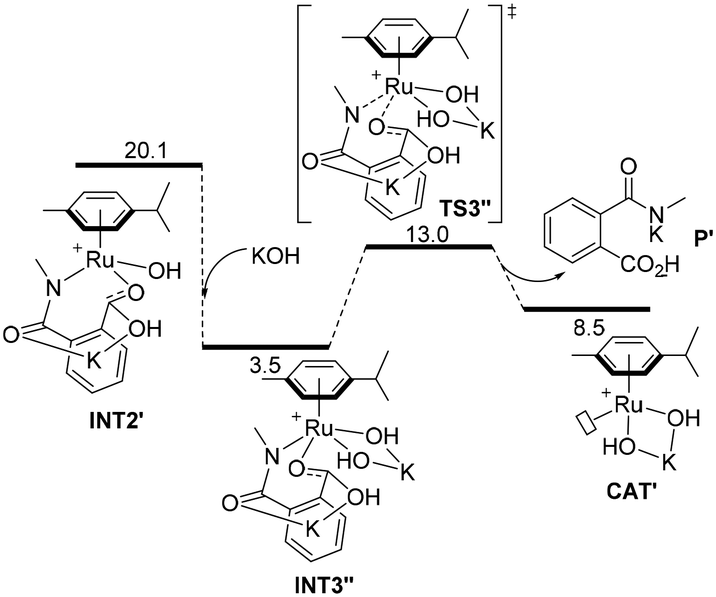 | ||
| Fig. 3 Computed minimum free energy reaction profile (in kcal mol−1) of the formation of phthalamic acid derivative P′ from the ruthenium intermediate INT2′ assisted by KOH. | ||
The results of these two competitive paths explain why at low temperature, the Ru-catalysed process leads preferentially to product 3, while by increasing the temperature, all the barriers of the catalytic process are overcome, affording selectivity toward product 2 at 150 °C with a selectivity of 72% (Table 1, entry 4).
Conclusions
In summary, by means of experimental and thorough theoretical calculations, we have shown that the ruthenium-catalyzed protodecarbonylation of phthalimide derivatives into secondary amides proceeds under a mechanism involving the release of two CO2 molecules: one from the substrate and one from K2CO3 used as a reagent. Interestingly, it was found that the ruthenium center exhibits an oxidation state of +2 through the whole catalytic cycle and the potassium cations are relevant to stabilise all the intermediates via weak interactions with the carbonyl groups present either in the substrate and/or in the carbonate ligands. The catalysis performed in the presence of KOH as the base at high temperature led to the formation of a secondary amide product; however, at low temperature, the formation of a phthalamic acid derivative was exclusively observed, indicating a switch in the reaction mechanism that has been rationalized by theoretical calculations as well. In both cases (KOH and K2CO3), the rate determining step is the same, namely the five-membered ring opening of phthalimide. However, the presence of KOH in the reaction mixture facilitates an unexpected side-reaction towards phthalamic acid, which is not the case for K2CO3. Consequently, this work shows the non-trivial, multiple roles that the bases can play in reactions catalysed by ruthenium complexes and it provides insights into a new mechanistic hypothesis that might be considered for related transition metal-based catalytic reactions.Experimental section
General methods
All reagents were obtained from commercial sources and used as supplied. All reactions were carried out in flame-dried glassware under an argon atmosphere unless otherwise noted. Catalytic experiments were performed in Schlenk-type flasks under an argon atmosphere unless otherwise noted. Organic solutions were concentrated under reduced pressure using a rotary evaporator. Thin-layer chromatography (TLC) was carried out on a 0.25 mm Merck silica gel (60-F254). Flash column chromatography was performed using a silica gel (Silica 60 M, 0.04–0.063 mm). N-Methyl-2-pyrrolidone (NMP) was distilled under reduced pressure and stored under molecular sieves and argon atmosphere. Technical grade petroleum ether (40–60) and ethyl acetate were used for column chromatography. CDCl3 was stored under nitrogen over molecular sieves. NMR spectra were recorded on an AVANCE III 400 spectrometer. 1H NMR spectra were referenced to a residual protiated solvent (δ = 7.26 ppm for CDCl3) and 13C chemical shifts were reported relative to deuterated solvents (δ = 77.0 ppm for CDCl3). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet, and br. for broad. GC-MS analyses were performed with a GCMS-QP2010S (Shimadzu) instrument with a GC-2010 equipped with a 30 m capillary column (Supelco, SLB™-5ms, fused silica capillary column, 30 m × 0.25 mm × 0.25 mm film thickness), which was used with helium as the vector gas. The following GC conditions were used: an initial temperature of 80 °C for 2 minutes, then a rate of 20 °C min−1 until 280 °C and 280 °C for 28 minutes. HRMS was carried out on a Waters Q-Tof 2 mass spectrometer at the corresponding facilities of the CRMPO, Centre Régional de Mesures Physiques de l'Ouest, Université de Rennes 1.Synthesis and characterization of substrate 1
Phthalimide (7 mmol, 1.03 g, 1 equiv.), potassium carbonate (14 mmol, 2.59 g, 2 equiv.) and iodomethane (14 mmol, 2 equiv.) were heated at 40 °C in N,N-dimethylformamide (6 mL) for 18 hours. After solvent evaporation under vacuum, water was added to the reaction mixture followed by extraction with DCM. The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The desired phthalimide 1 was purified by silica gel column chromatography with a mixture of petroleum ether and ethyl acetate as an eluent in 88% isolated yield. N-Methylphthalimide (1): 1H NMR (400 MHz, CDCl3): δ = 7.78 (dd, J = 5.6 Hz, 3.2 Hz, 2H), 7.66 (dd, J = 5.6 Hz, 3.2 Hz, 2H), 3.13 (s, 3H) ppm. The spectral data match those previously reported.23General procedure for the catalysis and characterization of 2–3
[RuCl2(p-cymene)]2 (0.004 mmol, 2.5 mg, 0.01 equiv.), base (1.2 mmol, 165.8 mg, 3 equiv.), distilled water (0.6 mmol, 10.8 mg, 10.8 μL, 1.5 equiv.), substrate 1 (0.4 mmol, 1 equiv.) and N-methyl-2-pyrrolidone (2.0 mL) were introduced in a flame-dried Schlenk tube under an argon atmosphere. The reaction mixture was stirred at 150 °C for 6 hours. Then, the reaction mixture was cooled down to room temperature and diluted with water (20 mL). Then, HCl (1 M) was added until the pH reached ca. 7. The aqueous phase was extracted with ethyl acetate (3 × 20 mL) and the combined organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. After solvent evaporation, the crude product 2 was obtained [note: in order to detect product 3, a small change in the work-up was performed using an excess of HCl in order to reach pH ca. 1].:1 to 2:1, v/v) in 93% yield (50.3 mg) as a colourless solid. 1H NMR (400 MHz, CDCl3): δ = 7.77–7.74 (m, 2H), 7.49–7.45 (m, 1H), 7.42–7.38 (m, 2H), 6.42 (br. s, 1H), 2.98 (d, J = 4.8 Hz, 3H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 168.3, 134.5, 131.2, 128.3, 126.8, 26.7 ppm. GC: tR = 8.7 min; MS (EI): m/z = 134 (M+, 48), 105 (100), 77 (91), 51 (34). The spectral data match those previously reported.24
Computational details
All the DFT static calculations were performed with the Gaussian09 set of programs,25 using the BP86 functional of Becke and Perdew,26–28 together with the Grimme D3 correction term to the electronic energy.29 The electronic configuration of the molecular systems was described with the double-ζ basis set with the polarization of Ahlrichs for main-group atoms (SVP keyword in Gaussian),30 whereas for ruthenium, the small-core quasi-relativistic Stuttgart/Dresden effective core potential, with an associated valence basis set (standard SDD keywords in Gaussian09) were employed.31–33 The geometry optimizations were performed without symmetry constraints, with analytical frequency calculations for the characterization of the located stationary points. These frequencies were used to calculate unscaled zero-point energies (ZPEs) as well as thermal corrections and entropy effects at 298.15 K. Energies were obtained by single-point calculations on the optimized geometries with the M06 functional34 and the cc-pVTZ basis set.35 The reported free energies in this work include energies obtained at the M06/cc-pVTZ∼sdd level of theory corrected with zero-point energies, thermal corrections and entropy effects evaluated at 298.15 K, achieved at the BP86-D3/SVP∼sdd level plus solvent contributions evaluated by means of the PCM (N,N-dimethylacetamide).36Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. is a Serra Húnter Fellow and thanks the Ministerio de Ciencia e Innovación (MICINN) of Spain for the project PGC2018-097722-B-I00 and EU for the FEDER fund (UNGI08-4E-003). Y.-C. Y., C. B. and R. G.-D. acknowledge the CNRS, the Universite de Rennes 1, the Rennes Metropole, the China Scholarship Council (PhD grant to Y.-C. Y.), the Agence Nationale de la Recherche (ANR-JCJC) and the COST Action CA15106 (CHAOS) for the financial support.Notes and references
- (a) R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS.
- (a) K. Sanderson, Nature, 2011, 469, 18–20 CrossRef CAS; (b) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC; (c) G. Rothenberg, Catalysis: Concepts and Green Applications, Wiley-VCH, Weinheim, 2008 CrossRef; (d) M. Beller, Chem. Soc. Rev., 2011, 40, 4891–4892 RSC; R. Franke, D. Selen and A. Borner, Chem. Rev., 2012, 112, 5675–5732 Search PubMed.
- (a) S. E. Livingstone, The Chemistry of Ruthenium, Rhodium, Palladium, Osmium, Iridium and Platinum, Pergamon Texts in Inorganic Chemistry, Elsevier, 1973 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2009 Search PubMed; (c) M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2004 CrossRef; (d) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 CrossRef; (e) A. de Meijere, S. Braese and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, 2014 CrossRef.
- (a) E. A. Seddon and K. R. Seddon, The Chemistry of Ruthenium, Elsevier Science, 1984 Search PubMed; (b) S.-I. Murahashi, Ruthenium in Organic Synthesis, Wiley-VCH, Weinheim, Germany 2004 CrossRef; (c) C. Bruneau and P. H. Dixneuf, Ruthenium in Catalysis, Top. Organomet. Chem., Springer-Verlag, Berlin-Heidelberg, Germany, 2014, vol. 48 Search PubMed.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (b) R. Noyori and T. Okhuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed; (b) R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS; (c) R. Credendino, A. Poater, F. Ragone and L. Cavallo, Catal. Sci. Technol., 2011, 1, 1287–1297 RSC; (d) A. Poater and L. Cavallo, Beilstein J. Org. Chem., 2015, 11, 1767–1780 CrossRef CAS.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- Y.-C. Yuan, R. Kamaraj, C. Bruneau, T. Labasque, T. Roisnel and R. Gramage-Doria, Org. Lett., 2017, 19, 6404–6407 CrossRef CAS.
- (a) Y.-C. Yuan, C. Bruneau, V. Dorcet, T. Roisnel and R. Gramage-Doria, J. Org. Chem., 2019, 84, 1898–1907 CrossRef CAS; (b) A. Poater, S. V. C. Vummaleti and L. Cavallo, Organometallics, 2013, 32, 6330–6336 CrossRef CAS.
- (a) S. Inoue, H. Shiota, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898–6899 CrossRef CAS; (b) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–8073 CrossRef CAS.
- Y. Du, T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 12074–12076 RSC.
- (a) L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Goossen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef CAS; (b) J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef CAS; (c) J.-L. Yu, S.-Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef CAS; (d) A. Mandal, H. Sahoo, S. Dana and M. Baidya, Org. Lett., 2017, 19, 4138–4141 CrossRef CAS PubMed; (e) F. Pu, Z.-W. Liu, L.-Y. Zhang, J. Fan and X.-Y. Shi, ChemCatChem, 2019, 11, 4116–4122 CrossRef CAS.
- T. Shiba, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 13636–13639 CrossRef CAS.
- (a) P. K. Samanta and P. Biswas, J. Org. Chem., 2019, 84, 3968–3976 CrossRef CAS; (b) T.-L. Zhou, G.-C. Li, S. P. Nolan and M. Szostak, Org. Lett., 2019, 21, 3304–3309 CrossRef CAS; (c) S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087–4101 RSC.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5883 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- M. Simonetti, G. J. P. Perry, X. C. Cambeiro, F. Julia-Hernandez, J. N. Arokianathar and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 3596–3606 CrossRef CAS.
- (a) S. K. Hasan and S. A. Abbas, Can. J. Chem., 1975, 53, 2450–2453 CrossRef CAS; (b) P. P. Kumar, B. R. Devi, P. K. Dubey and S. M. G. Mohiuddin, Green Chem. Lett. Rev., 2011, 4, 341–348 CrossRef CAS.
- Y.-Y. Meng, X.-J. Si, Y.-Y. Song, H.-M. Zhou and F. Xu, Chem. Commun., 2019, 55, 9507–9510 RSC.
- B. Demerseman, M. D. Mbaye, D. Sémeril, L. Toupet, C. Bruneau and P. H. Dixneuf, Eur. J. Inorg. Chem., 2006, 1174–1181 CrossRef CAS.
- T. Arthur, D. R. Robertson, D. A. Tocher and T. A. Stephenson, J. Organomet. Chem., 1981, 208, 389–400 CrossRef CAS.
- A. Kumar, M. Park, J. Y. Huh, H. M. Lee and K. S. Kim, J. Phys. Chem. A, 2006, 110, 12484–12493 CrossRef CAS PubMed.
- J. Mahler and I. Persson, Inorg. Chem., 2012, 51, 425–438 CrossRef PubMed.
- S. Takebayashi, J. M. John and S. H. Bergens, J. Am. Chem. Soc., 2010, 132, 12832–12834 CrossRef CAS.
- A. B. Charette, M. Grenon, A. Lemire, M. Pourashraf and J. Martel, J. Am. Chem. Soc., 2001, 123, 11829–11830 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406–7406 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- U. Haeusermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger and R. M. Pitzer, Mol. Phys., 1993, 78, 1211–1224 CrossRef.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data, all Cartesian XYZ coordinates, and energies of all species. See DOI: 10.1039/c9cy02047k |
| This journal is © The Royal Society of Chemistry 2020 |