
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identifying the roles of acid–base sites in formation pathways of tolualdehydes from acetaldehyde over MgO-based catalysts†
Marcella
Lusardi
 a,
Thomas
Struble
b,
Andrew R.
Teixeira
c and
Klavs F.
Jensen
*ab
a,
Thomas
Struble
b,
Andrew R.
Teixeira
c and
Klavs F.
Jensen
*ab
aMaterials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA, 02139 USA
bChemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, 02139 USA
cChemical Engineering, Worcester Polytechnic Institute, Worcester, MA, 01609 USA. E-mail: kfjensen@mit.edu
First published on 16th December 2019
Abstract
Pure and Al-substituted MgO catalysts are studied to identify the contributions of acid–base sites in the formation of two valuable xylene analogs, ortho- and para-tolualdehydes, from an ethanol derivative, acetaldehyde. The catalyst properties are characterized through XRD, 27Al MAS NMR, ICP-AES, N2 physisorption, TPD-MS, and DRIFTS experiments. Reactivity comparisons of untreated and CO2-titrated catalysts at 250 °C, coupled with CO2 DRIFTS studies on fresh and spent samples, indicate the formation of tolualdehydes from intermediates is initiated through deprotonation by a medium-strength basic site in a specific, metal–oxygen (M–O)-type coordination environment. Analyses of the catalytic surface properties and reactivity, pathways of formation, and natural bond orbital (NBO) charge distribution suggest C4 + C4 (rather than C2 + C6) mechanistic steps dominate tolualdehyde production over these catalysts under the investigated reaction conditions. Isomeric selectivity to ortho-tolualdehyde is 92 and 81 mol% over pure and Al-substituted MgO catalysts, respectively. We propose that the shift in isomeric selectivity towards para- upon introduction of a proximal Lewis acidic functionality (Al3+/MgO) to the catalyst is caused by electron redistribution in the conjugated enolate from the γ-C (forming ortho-) towards the α-C (forming para-) due to the carbonyl-O/Lewis acid coordination. This insight provides a framework for the development of next generation catalysts that give improved reactivity in cascade reactions of C2 feedstocks to aromatics.
1 Introduction
The plastics, textiles, solvents, and composite materials derived from BTX (benzene, toluene, and xylenes) are fundamental to many industries.1 As a result, global demand for these aromatic platform chemicals is high (∼85 MT y−1)1–4 and is expected to increase 5–10% over the next decade as existing industries grow and international economies develop.5–8 Currently, catalytic reforming of petroleum naphtha produces approximately 70% of BTX.8 The volatility of petroleum markets, coupled with environmental impacts of petroleum consumption, motivate the diversification of sources for BTX and other valuable petroleum derivatives.3,4,9 A major challenge in obtaining drop-in chemicals from alternative feeds (e.g., biomass) is the development of new process pathways and catalysts, given their distinct chemical makeup (oxygen content, molecular weight, etc.) compared to that of petroleum.A bottom-up approach involving the construction of aromatic rings from smaller molecules is one potential route, which could find utility for low-molecular weight, oxygenated feedstocks produced from renewably-sourced biomass or syngas conversion technologies.8,10,11 Ethanol is one such feed that could be upgraded through a complex series of reactions via its dehydrogenated intermediate, acetaldehyde.12,13 Aromatic compounds (benzene, tolualdehydes) can be produced through several different mechanisms (aldol condensation, Michael addition, dehydration, electrocyclization, etc.) involving acetaldehyde and its condensation products (Scheme 1).11,12,14,15 While the benzene mechanism is straightforward,14 the formation of tolualdehydes, which are valuable xylene analogs (Scheme S1†), is more complicated. To date, work on this reaction has focused predominantly on understanding the mechanistic routes and kinetics associated with the complex pathways.11,16,17 Moteki et al. identified the different pathways that can yield two isomers of tolualdehyde, ortho- and para-, from ethanol, acetaldehyde, and intermediates over hydroxyapatite catalysts.12 Zhang and co-workers investigated these routes as well by directly feeding the acetaldehyde dimer, 2-butenal, over MgO- and ZrO2-incorporated faujasite.18 They proposed a likely route of ortho- and para-tolualdehyde formation based on their experimental yields and a more-energetically favorable enolate intermediate calculated through DFT analysis. Recent work17 investigated the use of a dual-bed Cu/C and Co-hydroxyapatite system to catalyze ethanol dehydrogenation to acetaldehyde followed by coupling to aromatic aldehydes and alcohols, and the associated rates of each step.
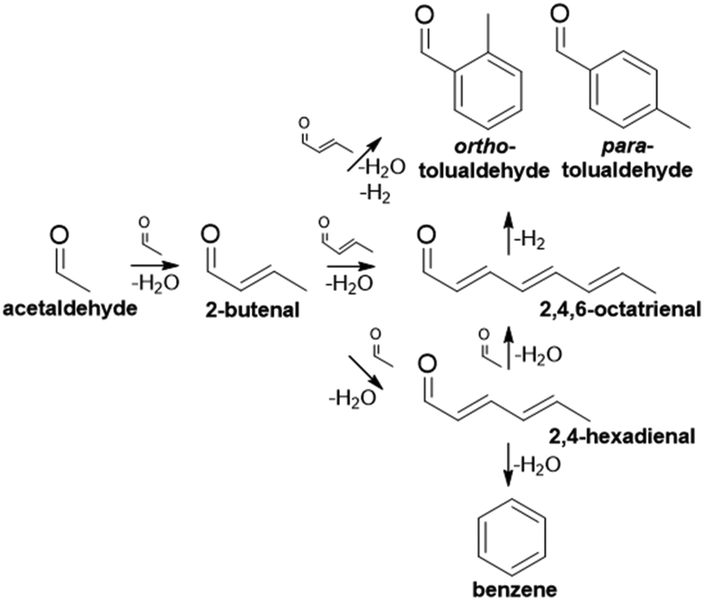 | ||
| Scheme 1 Overview of cascade reaction pathways that produce aromatics (benzene, tolualdehydes) from acetaldehyde. | ||
As mechanistic and kinetic understanding of these tolualdehyde formation routes has increased, the nature of the active sites for obtaining tolualdehydes from intermediates remains largely unexplored. Prior investigations19–23 have identified the active sites for the formation of C4 intermediates (butenal—the first step in the cascade reaction) and products (butanol, butadiene) from C2 compounds over a range of different catalysts. Recently, the active coordination environment for the formation of C6+ products from ethanol over hydroxyapatite was identified.24 These products were strictly aliphatic and formed through a direct ethanol coupling mechanism, not via an acetaldehyde intermediate that can form tolualdehydes. To the best of our knowledge, no work has studied the specific nature of the active basic site(s) responsible for initiating the tolualdehyde formation mechanism via proton abstraction from intermediates. Thus, the primary focus and contribution of this work is the characterization of active sites in the second step of the cascade reaction to form C8 aromatics as the terminal products through C2 + C6 cross-condensation and/or C4 + C4 self-condensation routes. In parallel, we determine the more likely of those two routes for the catalysts and reaction conditions used in this study.
MgO-based catalysts were chosen as the candidates to study the nature of the active site(s) in this reaction. MgO-based catalysts exhibit the high basic character necessary to promote condensation pathways over other potential acetaldehyde routes (e.g., coking and cracking, reductive coupling)25–29 and have shown multiple condensation activity (i.e., condensations that yield trimers, tetramers, etc.) in previous studies.15,18,30–32 Different coordination environments on MgO surfaces give rise to a wide distribution of basic site types and strengths, which enables the screening of such sites for their contribution to the formation of aromatic products from C2 feeds.33 To probe the impact of acidity, which can contribute in aldol condensation-type mechanisms through acid–base cooperation,30,34–38 varying amounts of Al3+ were introduced into the MgO lattice. Partial isomorphic substitution of Al3+ for Mg2+ in the octahedral sites of the face-centered cubic lattice generates Lewis acidity through Al3+ cations exposed on the surface.39 In addition to introducing an acidic functionality, Al incorporation can affect the basic properties of the mixed oxide by creating both relatively weaker and very strong basic centers in the mixed oxide compared to that of the pure periclase (MgO) phase.19,40,41 Thus, varying the amount of Al incorporated can have a significant impact on the acid–base character of the Mg–Al oxide.
To understand the impact of the surface chemistry on the formation of aromatics from an ethanol derivative, with the overall goal of identifying the nature of the active basic site(s) and potential contributions from catalyst acidity, we prepared catalysts with various degrees of Al content (molar Al/(Mg + Al) = 0, 0.25, and 0.33). In particular, we studied the formation of ortho- and para-tolualdehydes from acetaldehyde over these catalysts, and analyzed the catalytic reactivities in the contexts of their surface properties and possible mechanistic routes. From these studies, we characterize the roles of different site types and propose likely pathways of tolualdehydes formation over the MgO-based catalysts studied here. The insight obtained from these studies is critical to direct next generation catalyst design for improved reactivity in the formation of valuable aromatics from C2 feedstocks.
2 Materials and methods
2.1 Catalyst synthesis
2.2 Catalyst characterization
Diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) experiments were run to further characterize the nature of the basic sites present on the oxide catalysts using CO2 as a probe molecule.42 DRIFTS measurements were taken on a Bruker Vortex 70 benchtop FTIR equipped with a Harrick Praying Mantis diffuse reflectance cell and a liquid N2-cooled mercury-cadmium-telluride (MCT) detector. Atmosphere and temperature control were achieved with a high-temperature chamber built with ZnSe windows. DRIFTS samples were prepared by diluting the catalyst to 20 wt% with KBr. After pretreatment in He at 450 °C for 1 h, the sample was cooled to 50 °C in 50 °C increments, and a reference sample was collected at each temperature. The sample was then saturated at 50 °C with the probe molecule (10% CO2, 90% N2) and flushed with He to remove physisorbed species. Subsequently, a sample was collected after 15 min evacuation at each temperature to evaluate the CO2 interaction with the oxide surface. The results are plotted in Kubelka Munk (f(R∞)) units at each temperature, where R∞ is the CO2-saturated sample referenced to the cleaned sample at the same temperature.43 CO2 DRIFTS analysis using the same procedure was also conducted on spent MgO after a given time on steam.
2.3 Catalytic reactivity
The activity of the catalysts was evaluated in a vapor phase flow system at ambient pressure. 10 mg of catalyst sized between 75 and 125 μm was packed onto a bed of stainless steel beads in a 2.5′′ L × 0.049′′ ID micro packed bed reactor. After in situ pretreatment of the catalyst in 10 sccm of N2 for 0.5 h at 350 °C, the acetaldehyde vapor (0.1%/99.9% N2, Airgas) was introduced at 10 sccm, which corresponds to a ∼10 ms residence time. The reactor temperature was 250 °C in all runs. Transfer lines downstream of the reactor were heated to prevent condensation of heavier intermediates and products. Samples were analyzed with an Agilent G1888 head space sampler retrofitted for online gas-phase analysis and an Agilent 7890A gas chromatograph (GC) equipped with a flame ionization detector (FID) for species quantification. The carbon balance for all reactions (inclusive of carbonaceous deposits quantification by oxidative thermogravimetric analysis, TGA) was >90%. Yields are reported on a mole carbon basis. Experiments were duplicated and the variation between the two runs is shown by error bars. For the mechanical mixture test, we gently mixed together weights of MgO and γ-Al2O3 that gave 10 mg total oxide with a molar Mg/Al = 2.In situ CO2 titration studies were conducted to distinguish the activity contribution by strong basic sites (i.e., sites whose temperature of CO2 desorption is higher than the reaction temperature of 250 °C). In these experiments, the catalyst was subjected to an additional pretreatment step. After cleaning and drying the catalyst at 350 °C in N2, the packed bed was cooled to 50 °C, and a 10 sccm flowrate of 10% CO2/90% N2 was introduced to saturate the catalyst surface. Subsequently, the bed temperature was increased to the reaction temperature (250 °C) in the diluted CO2 mixture. The reaction proceeded as outlined above, except that CO2 was co-fed with the 0.1% acetaldehyde vapor (0.1% acetaldehyde/10% CO2/89.9% N2, Airgas).
2.4 Computational methods
DFT calculations were performed using Gaussian 16 (ref. 44) in order to carry out the NBO analysis (NBO version 3.1 (ref. 45)). Geometry optimizations were done using the B3LYP46/6-311+g(d,p) functional. All optimized structures were verified by frequency computations as minima (zero imaginary frequencies).3 Results and discussion
3.1 Catalyst characterization
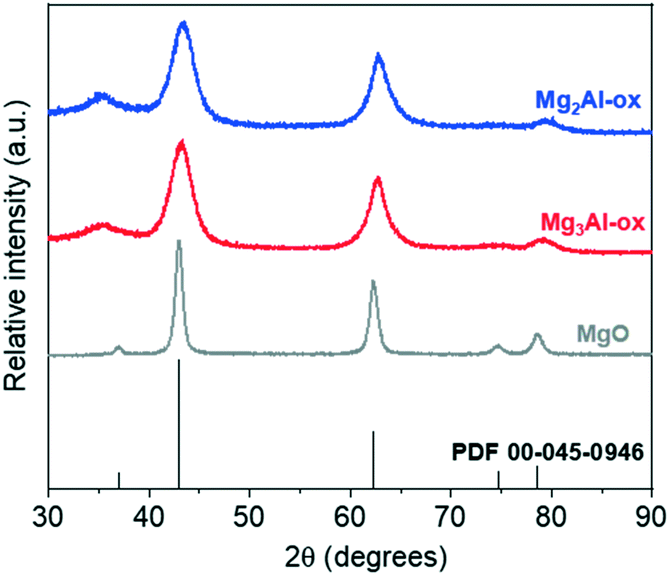 | ||
| Fig. 1 XRD patterns for MgxAl-ox and MgO, and the reference pattern for periclase. | ||
| Sample | Lattice parameter aa (Å) | Mg/Alb (mol/mol) | BET surface areac (m2 g−1) | Total pore volumec (cm3 g−1) | n B (μmol m−2) | n A (μmol m−2) |
|---|---|---|---|---|---|---|
| a Calculated from (200) diffraction. b Measured by ICP-AES. c Calculated from N2 adsorption–desorption isotherms (77 K). d Measured by CO2 (nB) and NH3 (nA) TPD-MS. | ||||||
| Mg2Al-ox | 4.155 | 2.07 | 249 | 0.783 | 1.25 | 1.46 |
| Mg3Al-ox | 4.172 | 3.09 | 268 | 0.598 | 1.36 | 0.87 |
| MgO | 4.208 | — | 140 | 0.318 | 2.59 | 0.07 |
27Al MAS NMR spectra (Fig. S3†) show two resonances corresponding to two major Al coordination environments: octahedral (AlO, 10–14 ppm) and tetrahedral (AlT, 72 ppm). The absence of peak splitting to reflect the presence of octahedral and tetrahedral Al3+ in periclase and spinel-type phases indicates that the MgxAl-ox samples are more disordered in nature. Thus, a wide distribution of coordination environments and corresponding acid–base sites is possible. ICP-AES results summarized in Table 1 show that the actual Mg/Al values obtained are in good agreement with the expected values based on the initial metal salts concentrations prepared during the co-precipitation syntheses. No residual Na content from the syntheses was detected by ICP or XPS (Fig. S4†).
From the N2 adsorption–desorption measurements at 77 K, we report BET surface areas and total pore volumes in Table 1. The raw isotherms for the MgxAl-ox and MgO catalysts are presented in Fig. S5.† The smallest pore diameters (pore size distributions, Fig. S6†) measured in MgxAl-ox and MgO are much larger (>2×) than the pores that give rise to aromatics shape and/or size selectivity due to confinement (e.g., ZSM-5 with dpore ∼ 6 Å).51 Thus, differences in isomeric selectivity amongst these catalysts can be attributed to variations in surface chemistry, rather than to pore confinement effects.
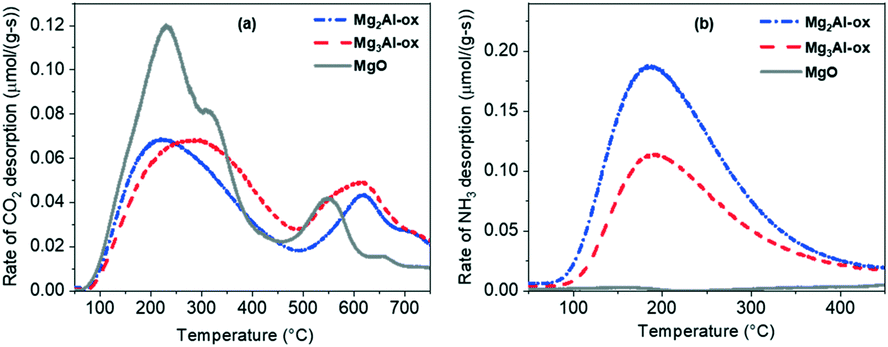 | ||
| Fig. 2 (a) CO2 and (b) NH3 TPD-MS profiles for MgxAl-ox and MgO for basic and acid site quantification, respectively. | ||
CO2 DRIFTS studies were run to further understand the nature of basic sites present on the MgxAl-ox and MgO catalysts. CO2 DRIFTS profiles in the full temperature range (50 to 350 °C) are reported in Fig. S7† and a comparison of profiles for MgxAl-ox and MgO at 50 °C and 200 °C is shown in Fig. 3a and b, respectively. Given the difficulty of linearizing DRIFTS signals with adsorbate concentrations,43,54 the analysis here is limited to strictly qualitative comparisons.
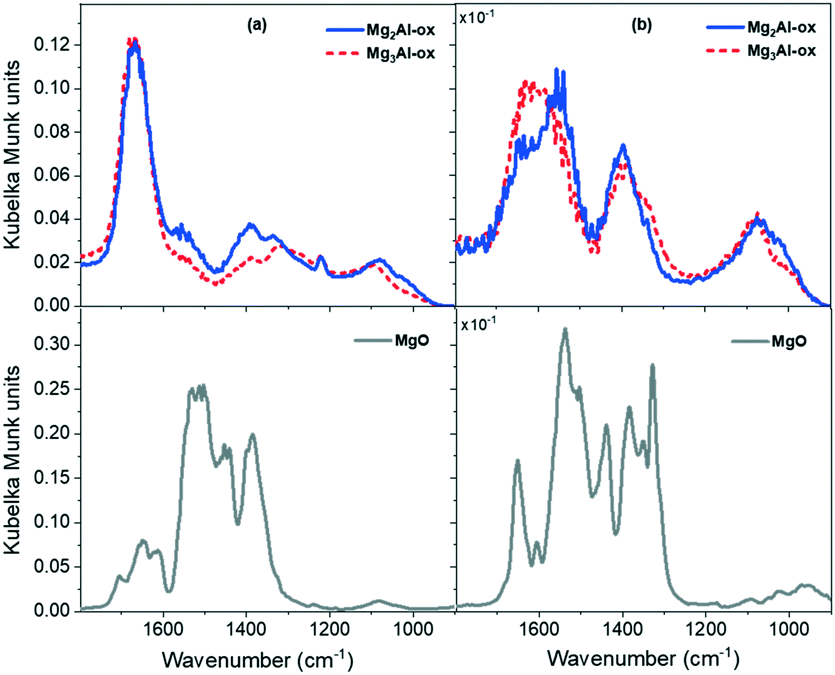 | ||
| Fig. 3 DRIFTS spectra of CO2 adsorbed on MgxAl-ox and MgO at (a) 50 °C and (b) 200 °C. | ||
The spectra for all samples (Fig. 3a) show features attributed to bicarbonate, bidentate, and monodentate structures, which correspond to CO2 adsorption on weak hydroxyl groups, medium-strength metal–oxygen (M–O) pairs, and strong low-coordinated oxygen sites, respectively.19,33,39 The MgxAl-ox spectra exhibit quite similar features, indicative of similar basic site coordination environments on these Al-substituted samples regardless of Al content (25 and 33 mol% Al). Over MgxAl-ox, a strong peak appears around 1660–1700 cm−1 which corresponds to the asymmetric stretching mode of O–C–O–H in the bicarbonate structure.19,42,55 The symmetric stretching mode appears as an indistinct feature around 1450 cm−1.19,40 The C–OH bending vibration, δ(C–OH), appears clearly at 1220 cm−1 for the MgxAl-ox samples.19,40,42 In the MgO spectrum (Fig. 3a), the asymmetric stretching mode of the bicarbonate manifests from 1660–1710 cm−1. The symmetric stretching and bending modes for bicarbonate over MgO appear at 1460–1470 and 1235 cm−1, respectively.
Bidentate carbonate features in the MgxAl-ox spectra appear at 1590–1650 cm−1 for the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode; and at 1315–1330 cm−1 and ∼1075 cm−1 for the ν(O–C–O) asymmetric and symmetric stretching modes, respectively.19,42,55,56 For MgO, bidentate modes present as distinct peaks at 1600–1650 cm−1, a shoulder at 1310–1350 cm−1, and a broad feature at 1085 cm−1. Fig. 3b shows the spectra taken at 200 °C for MgxAl-ox and MgO. At this temperature, bicarbonate structures are no longer present; and bidentate structures, which correspond to CO2 adsorbed on medium-strength M–O pairs, are stable on MgO materials and disappear by 300 °C.33 Two bidentate ν(CO) modes appear at frequencies ≥1600 cm−1 over MgO. While these modes exhibit comparable temperature stabilities (disappear by 250 °C), the difference in their frequency values (45 cm−1) suggests different site strengths and/or local geometries due to varied M–O coordination environments, both of which can impact catalytic activity.57–59 The breadth of the MgxAl-ox spectra prevents distinguishing different bidentate structures corresponding to varied M–O coordination types that might be present, but a shoulder is visible at 1620–1650 cm−1 that corresponds to at least one ν(CO) mode on the Al-substituted samples (Fig. 3b).
O) mode; and at 1315–1330 cm−1 and ∼1075 cm−1 for the ν(O–C–O) asymmetric and symmetric stretching modes, respectively.19,42,55,56 For MgO, bidentate modes present as distinct peaks at 1600–1650 cm−1, a shoulder at 1310–1350 cm−1, and a broad feature at 1085 cm−1. Fig. 3b shows the spectra taken at 200 °C for MgxAl-ox and MgO. At this temperature, bicarbonate structures are no longer present; and bidentate structures, which correspond to CO2 adsorbed on medium-strength M–O pairs, are stable on MgO materials and disappear by 300 °C.33 Two bidentate ν(CO) modes appear at frequencies ≥1600 cm−1 over MgO. While these modes exhibit comparable temperature stabilities (disappear by 250 °C), the difference in their frequency values (45 cm−1) suggests different site strengths and/or local geometries due to varied M–O coordination environments, both of which can impact catalytic activity.57–59 The breadth of the MgxAl-ox spectra prevents distinguishing different bidentate structures corresponding to varied M–O coordination types that might be present, but a shoulder is visible at 1620–1650 cm−1 that corresponds to at least one ν(CO) mode on the Al-substituted samples (Fig. 3b).
Lastly, these materials exhibit features attributed to monodentate carbonates, which form from adsorption of CO2 on low-coordinated O2− proximal to defects and are stable at high temperature (≥300 °C).33,55 The asymmetric and symmetric ν(O–C–O) modes for the MgxAl-ox appear at 1500–1575 cm−1 and 1335–1425 cm−1, respectively; a broad feature corresponding to ν(C–O) weakly presents at ∼1030 cm−1 (Fig. 3a).19,33,42,55,60 Over MgO, the asymmetric and symmetric ν(O–C–O) modes appear in the ranges 1495–1545 cm−1 and 1385–1440 cm−1, respectively, while the ν(C–O) appears below 1000 cm−1. At higher temperature (Fig. 3b), a more complex spectrum manifests quite distinct site types corresponding to carbonate formation on medium-strength and strong basic sites. Some of these features for MgO (e.g., the sharp peak at 1330 cm−1) have frequencies outside the expected range for monodenate modes. Given their persistence at high temperature (350 °C), and the magnitude of the split, Δν3, between symmetric and asymmetric modes, we assign them to polydentate structures attributed to a strong, basic O2− center.61,62
As a general conclusion, the CO2 DRIFTS studies show that varying the amount of Al incorporated (in the range of molar Mg/Al = 2 and 3) does not significantly impact the nature or type of basic sites present, while CO2 TPD-MS experiments illustrate that Al content does affect total basic site density and the densities of various site types (e.g., lower temperature peak maximum for Mg3Al-ox is shifted +100 °C relative to maximum for Mg2Al-ox). In the absence of isomorphic Al substitution (i.e., pure MgO), the resulting oxide has a narrower distribution of sites and the strongest basic character.
3.2 Aromatics production from acetaldehyde over MgxAl-ox and MgO catalysts
Acetaldehyde and its products can undergo a complex reaction network (Scheme 1) to yield the target aromatic species. The yields for the products 2-butenal, benzene, and tolualdehydes are reported in Fig. 4(a)–(c). Conversion and full product distributions are plotted in Fig. S8 and S9,† respectively. Given the extremely low yields of benzene, this discussion will focus on the other aromatic product of interest, tolualdehydes. The benzene results are summarized in the ESI† (Fig. S10 and Scheme S2).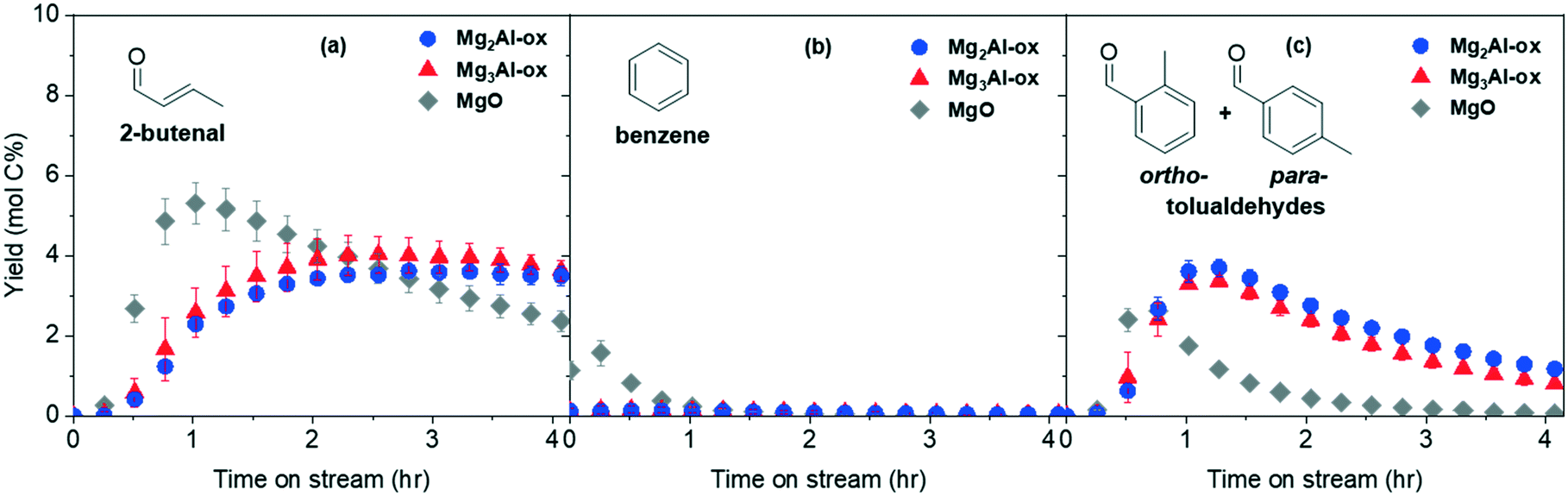 | ||
| Fig. 4 Yields of products (a) 2-butenal, (b) benzene, and (c) tolualdehydes formed from acetaldehyde over MgxAl-ox and MgO catalysts (0.1% acetaldehyde/99.9% N2, 10 sccm total flowrate, 10 mg catalyst, 250 °C, 1 bar). | ||
2-Butenal forms through the self-aldol condensation of acetaldehyde. This mechanism is initiated by base deprotonation at the α-carbon (α-C) of acetaldehyde, generating an enolate intermediate. The enolate then executes a nucleophilic attack onto the carbonyl-C of the second acetaldehyde, forming the C4 aldol which dehydrates to 2-butenal.63–65 Previous studies of the vapor phase aldol condensation of acetaldehyde have reported the active 2-butenal-forming site to be a medium-strength, M–O-type site.19,23,65,66
Tolualdehydes (ortho- and para-) can form through 2 main routes.12 The first route involves a cascade reaction: acetaldehyde self-condenses to its dimer, 2-butenal. Next, acetaldehyde undergoes a cross-condensation with 2-butenal to form the linear trimer, 2,4-hexadienal. Finally, acetaldehyde undergoes a second cross-condensation with the trimer to form the linear tetramer, 2,4,6-octatrienal (route 1, C2 + C6). The second route (route 2, C4 + C4) occurs via a bimolecular reaction between two molecules of the acetaldehyde dimer, 2-butenal. These routes form C8 intermediates, which can undergo further reaction (deprotonation, electrocyclization, etc.) to form tolualdehydes. We did not detect the formation of any linear (2,4-hexadienal, 2,4,6-octatrienal) enals, which is not surprising given the unstable nature of polyenals, particularly at high temperatures (Treaction = 250 °C).11,67
Yields of tolualdehyde reach a maximum at time on stream (TOS) ≤ 1.25 h over all catalysts prior to decaying (Fig. 4c). Regardless of which routes are contributing (C2 + C6, C4 + C4), 2-butenal is an intermediate in tolualdehydes formation. Over the MgxAl-ox catalysts, the 2-butenal and tolualdehyde yields are comparable regardless of Al content. If the availability of the intermediate (2-butenal) is the same for these catalysts (given by comparable 2-butenal yields and similar desorption quantities after flushing the reactor at high temperature), this suggests that the density of the active basic site(s) is approximately constant over these materials. The tolualdehyde-formation activity loss over time could be due to acetaldehyde and 2-butenal consumption in other pathways and/or site deactivation. We observe a decrease in side product formation (e.g., ethyl acetate) from acetaldehyde after 1 h on stream and detect no other major products that consume 2-butenal (<1 mol% of the total carbon). This indicates deactivation of the tolualdehyde-forming site(s) is the primary cause of the decrease in tolualdehyde yields over time. Deactivation is consistent with the decay in acetaldehyde conversion over all samples (Fig. S8†), the color change of the spent catalysts to brown from white (e.g., spent MgO in Fig. S8 inset†), and the weight loss between 400 and 500 °C in the TGA profiles of the spent samples that is absent in those of the fresh samples (not shown). The active site for 2-butenal is also likely deactivating, albeit slowly, because we do not measure a commensurate increase in the intermediate (2-butenal, Fig. 4a) yields as the consumption in tolualdehyde pathways decreases.
MgO exhibits similar trends for both 2-butenal and tolualdehydes, except that activity occurs at earlier TOS and decays more quickly. The strong basic character of pure MgO contributes to extensive formation of multiple condensation products that are nonvolatile at reaction conditions, particularly at early TOS prior to deactivating.15,26,30 Even if the active basic site(s) are similar over MgO and MgxAl-ox, the deactivation mechanisms available over purely basic MgO differ from those over acid–base bifunctional MgxAl-ox, creating disparate profiles over time.68
MgxAl-ox and MgO show tolualdehyde formation activity that does not directly correlate with the total basic site density (Table 1). This suggests that, assuming comparable availabilities of relevant intermediates (e.g., similar yield profiles for 2-butenal at 1.5 < TOS < 3.5 h, and no significant surface coverages evidenced by post-reaction flushes) not all basic site types contribute to the formation of tolualdehydes. Within each main tolualdehyde-forming route, there are subroutes that differ based on the position of the enolate (nucleophile) and the site of the nucleophilic attack (electrophile). The relative contributions of these subroutes dictate the observed selectivity towards each isomer (ortho- or para-). The isomeric selectivity is constant throughout TOS (section 3.4), despite the concurrent activity loss for tolualdehydes production. This strongly suggests that there is one dominant basic site and corresponding subroutes (i.e., subroutes initiated by the same basic site deprotonation) responsible for the formation of these isomers; otherwise, we would expect to see a change in the isomeric selectivity as deactivation occurs.11,69 It is possible that multiple basic sites and subroutes initiated by distinct deprotonation steps are contributing and exhibit identical rates of deactivation, but this is less likely.
3.3 Nature of the active basic site for tolualdehyde formation from intermediates
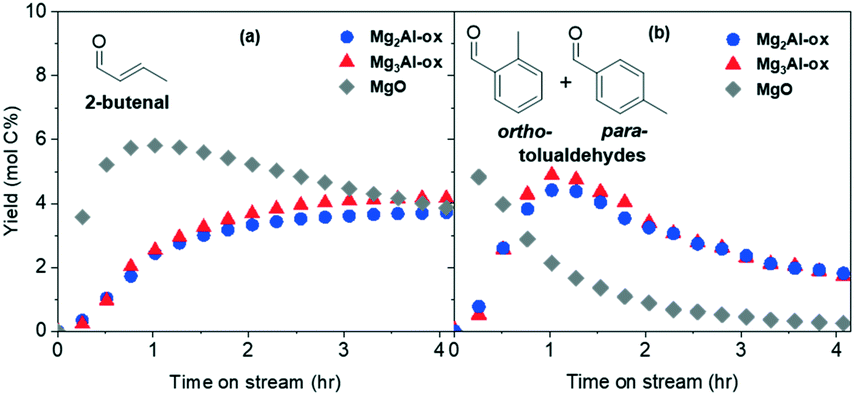 | ||
| Fig. 5 Yields of products (a) 2-butenal and (b) tolualdehydes formed from acetaldehyde over in situ CO2-titrated MgxAl-ox and MgO catalysts (0.1% acetaldehyde/10% CO2/89.9% N2, 10 sccm total flowrate, 10 mg catalyst, 250 °C, 1 bar). | ||
The tolualdehyde yields are maintained for all four catalysts, indicating that strong sites are not responsible for tolualdehydes (or 2-butenal) production over Al-substituted or pure MgOs at these reaction conditions. The slight increase in tolualdehyde yields over CO2-titrated MgxAl-ox can be attributed to the fact that, upon selective deactivation of strong sites, relatively more acetaldehyde and intermediates are available to interact with the medium-strength or weak basic site responsible for tolualdehydes production. Over CO2-titrated MgO, the maximum yield of tolualdehydes is greater (4.8 mol C%) and also occurs at earlier TOS (0.25 h) compared to the untreated sample (2.4 mol C%, 0.5 h). This can also be attributed to the elimination of activity from strong sites, which consume acetaldehyde and/or intermediates in nonvolatile product-forming routes at early TOS prior to deactivating.30 Similarly, because early TOS activity is not dominated by strong sites, yields of 2-butenal are also detected earlier (0.25 h, rather than 0.5 h) over CO2-titrated MgO. Since 2-butenal is an intermediate involved in the production of tolualdehyde, the increased production of 2-butenal likely contributes to improved tolualdehyde formation kinetics. Despite the slight increases in tolualdehyde yields over all catalysts, the profile shapes (after the initial activity shift over MgO) are essentially unchanged across untreated and CO2-titrated samples, which shows that strong, low-coordinated O2− basic sites present in MgO-based catalysts do not participate significantly in the tolualdehydes formation mechanism.
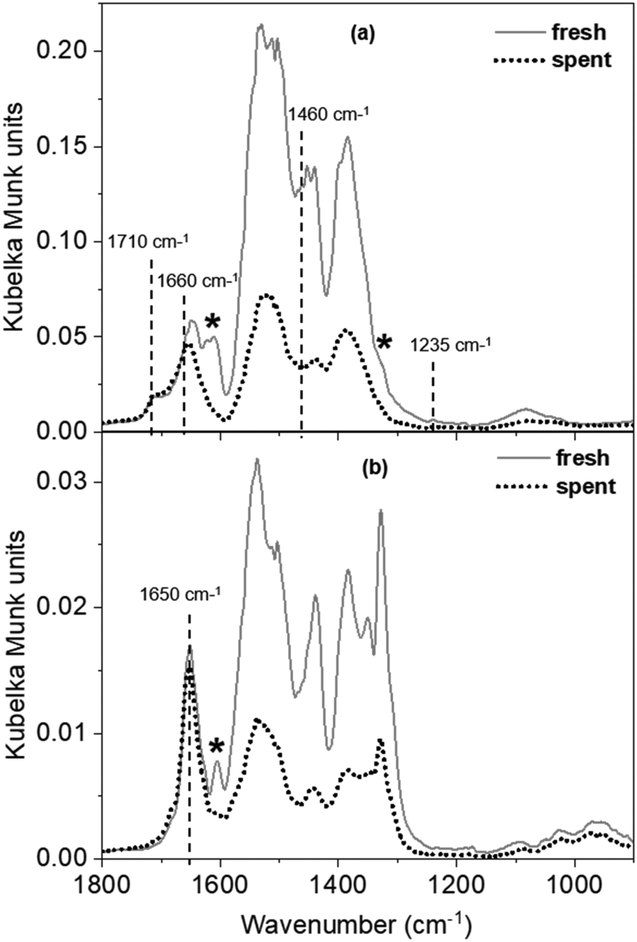 | ||
| Fig. 6 CO2 DRIFTS spectra over fresh and spent MgO at (a) 100 °C and (b) 200 °C. Frequency values marked with dashed lines indicate the features that are present in both spectra for the fresh and spent samples. Asterisks (*) denote the carbonate modes that are absent in the spent MgO spectra due to deactivation of the sites that give rise to these modes. | ||
All three carbonate structures (bicarbonate, bidentate, and monodentate-type) are present on the fresh MgO sample at 100 °C. The spectrum for the spent sample at this temperature (Fig. 6a) shows retainment of bicarbonate features corresponding to the distinct modes at 1710 and 1660 cm−1 (asymmetric), and those with relatively weaker intensities at 1460 cm−1 (symmetric) and 1235 cm−1 (bending; Fig. S12b† shows the y axis decreased to 0.02 Kubelka Munk units to see this small feature more clearly). Weakly basic hydroxyl groups, which are responsible for the formation of bicarbonate structures on MgO catalysts, are still available for CO2 uptake on the spent sample. Because no substantial activity is observed (<0.001 mol C% tolualdehydes) despite the availability of the intermediate (2-butenal) and the weak basic sites to adsorb CO2, these sites are not the tolualdehydes-forming active basic site. These results, in conjunction with the analysis from the strong site titration experiments, leave the remaining active site candidate to be a medium-strength, M–O-type site. The absence of bidentate carbonate structures that arise from CO2 adsorption on these M–O-type sites at ∼1610 cm−1 and ∼1325 cm−1 (ν(CO) and asymmetric ν(O–C–O), respectively, denoted by asterisks in Fig. 6a) supports this result. The loss of these carbonate structures is consistent with the loss of tolualdehyde activity, both due to site deactivation.
Many M–O-type surface coordination environments can give rise to medium-strength (between 120 and 150 kJ mol−1) basic centers, depending on the oxygen site's nearest neighbors (≤5 bonds with Mn+, where Mn+ is Mg2+ for pure MgO, and Mg2+ or Al3+ for MgxAl-ox) and proximity to defects (e.g., edges, vacancies). However, not all medium-strength sites deactivated after 5 h on stream, which suggests that a site in specific M–O coordination is responsible for the formation of tolualdehydes over these materials. This is apparent in the CO2 DRIFTS spectra at 200 °C (Fig. 6b), where the loss of bicarbonate structures due to increased temperature facilitates visualization of different bidentate ν(CO) modes (1600–1650 cm−1) that likely correspond to CO2 adsorbed on distinctly-coordinated M–O-type sites.70 Features at lower frequencies are more difficult to differentiate because of the strong intensities of monodentate/polydentate modes that persist at this temperature. The M–O-type site that gives rise to the mode at 1605 cm−1 deactivates, as this feature is absent in the spent spectrum (Fig. 6b, denoted by an asterisk), in contrast to the site that produces the persistent peak at 1650 cm−1. This suggests that this former (lower frequency ν(CO) mode) site is the active site for tolualdehydes formation. At this point, the exact coordination environment of this M–O site, and whether it is a unique site from that which is active for 2-butenal production, remain unclear.
The breadth of the CO2 DRIFTS spectra for MgxAl-ox caused by the wider distribution of basic sites on these relatively more disordered structures makes distinguishing specific features more difficult. However, we expect the active basic site that initiates the formation of tolualdehyde through deprotonation to be in the same coordination sphere in MgxAl-ox (M–O-type) as in MgO because they are all the same phase (periclase). The small spinel domains do not appear to contribute to tolualdehyde activity since the tolualdehyde yields do not scale with spinel content (Fig. S2† and 4c). Similar to pure MgO, the active site is specifically-coordinated; otherwise, MgxAl-ox with different densities of different medium-strength sites (Fig. 2, CO2 TPD profile for Mg2Al-ox compared to that for Mg3Al-ox at 200 < T < 450 °C) would give rise to dissimilar yields of tolualdehydes for comparable 2-butenal and side product activity. Yet, these yields are very similar for both Al-substituted samples (Fig. 4a and c).
3.4 Tolualdehydes formation pathways and isomeric selectivity
The isomeric fraction (defined by the molar ratio of ortho- to total (ortho- + para-) tolualdehydes produced) is plotted in Fig. 7. The regions corresponding to the first 0.5 h on stream over all catalysts and at ≥3 h over MgO are not quantitatively reliable because the yields are so low (see Fig. 4c). An ASPEN simulation (Table S1†) determined that the equilibrium distribution of ortho- and para-tolualdehydes favors para- by over a factor of 2 (Fig. 7). Given that the ortho-isomeric fraction is >0.5 over all catalysts in the present study, ortho-tolualdehyde is the kinetic product. At TOS > 0.5 h, a constant isomeric fraction of 0.81 and 0.92 is measured over MgxAl-ox and MgO, respectively.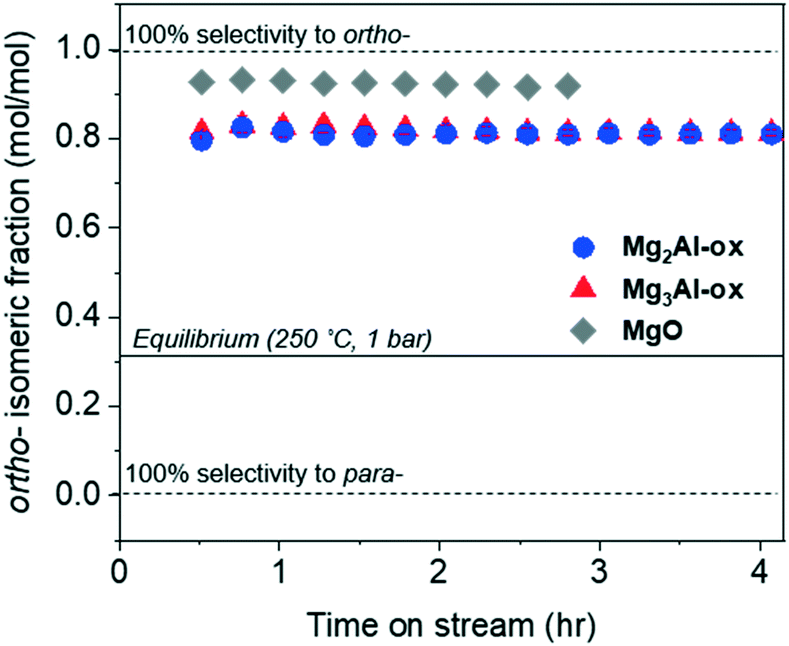 | ||
| Fig. 7 The ortho-tolualdehyde isomeric fraction measured over TOS for MgxAl-ox and MgO catalysts. | ||
Within route 1 (C2: acetaldehyde + C6: 2,4-hexadienal), we considered six different subroutes including the most probable enolates, electrophiles, and dehydration positions involved in an aldol-type condensation based on a natural bond orbital (NBO) charge distribution analysis (Table 2). Computational details for the NBO charge density analysis are contained in the ESI† (Tables S2–S4†). The C2 α-enolate can add to the C6-carbonyl, -β, and -δ carbons; and the C6 α-, γ-, and ε- enolates can add to the C2-carbonyl carbon (Scheme S3†). Of these subroutes, only three yield C8 enals capable of electrocyclization to tolualdehydes: 1A, 1B, and 1C. Routes 1A, 1B2, and 1C all yield ortho-tolualdehyde, while 1B1 produces para-tolualdehyde. 1B1 and 1B2 both involve the C6 α-enolate adding to the C2 carbonyl-C, but the formation mechanism of the enolate is different. In 1B1, the C6 α-enolate is formed by direct deprotonation of the sp2 α-C, while in 1B2, it forms as a resonance structure from deprotonation at the sp3 ε-C. Direct deprotonation at the sp2 α-C is considerably less favorable, given the inability to undergo π-delocalization of the negative charge, unlike for the ε-C deprotonation, which results in a highly conjugated anion.71,72
|
|
|||
|---|---|---|---|
| Carbon position | Acetaldehyde (C2) | 2-butenal (C4) | 2,4-hexadienal (C6) |
| Carbonyl | 0.438 | 0.391 | 0.381 |
| α | −0.683 | −0.309 | −0.288 |
| β | — | −0.087 | −0.131 |
| γ | — | −0.618 | −0.247 |
| δ | — | — | −0.104 |
| ε | — | — | −0.613 |
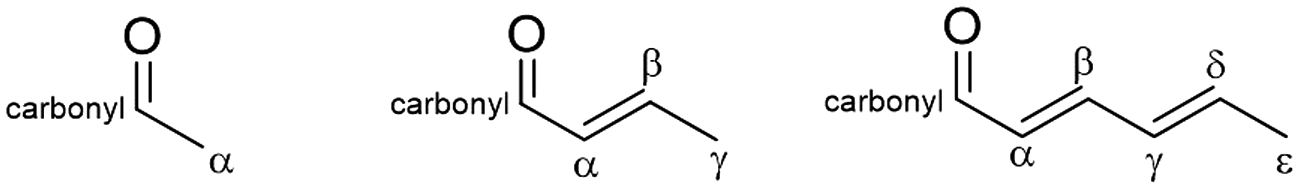
In the route 1 mechanisms outlined in Scheme S3,† no single deprotonation step initiates the formation of both ortho- and para-tolualdehyde; the formation of both isomers, which we experimentally observe, would require C6-ε or C2-α and C6-α deprotonation. Given the active basic site and deactivation analysis that suggests a single site and deprotonation step are responsible for the formation of both isomers, in addition to the high kinetic cost of deprotonation of an sp2 carbon (C6-α), it is improbable that route 1 (C2 + C6) is occurring to any significant extent in the formation of tolualdehydes over these MgxAl-ox and MgO catalysts. This is further supported by the fact that benzene activity is very low over these materials (Fig. 4b, <0.5 mol C% over MgxAl-ox, 2 mol C% at TOS < 1 h before decay to <0.5 mol C% over MgO). At this reaction temperature (250 °C), cyclodehydration of the C6 linear trimer (2,4-hexadienal) to benzene is likely more facile than the aldol condensation (C2 + C6).14,67 Thus, if route 1 mechanisms were contributing significantly to tolualdehydes production, we would expect greater benzene activity over these catalysts.
Within route 2 (C4: 2-butenal + C4: 2-butenal), C4 enolates (α- and γ-) can add to C4 electrophiles (carbonyl- and β- carbons), giving rise to 4 subroutes, 2A–2D.12 In this analysis, we consider only the C4 α-enolate formed as the resonance structure achieved by deprotonation at the C4 γ-C (sp3), not the C4 α-enolate that could theoretically form by direct C4 α-C (sp2) deprotonation (Fig. S13†). While we discuss these two enolates (C4 α- and γ-) as distinct intermediates to differentiate the positions of the nucleophilic attack, it is a single intermediate with a delocalized charge. Scheme 2 summarizes the four subroutes initiated by base deprotonation of the C4 γ-C. Routes 2A, 2C2, and 2D result in ortho-tolualdehyde, while 2B and 2C1 form the para- isomer.
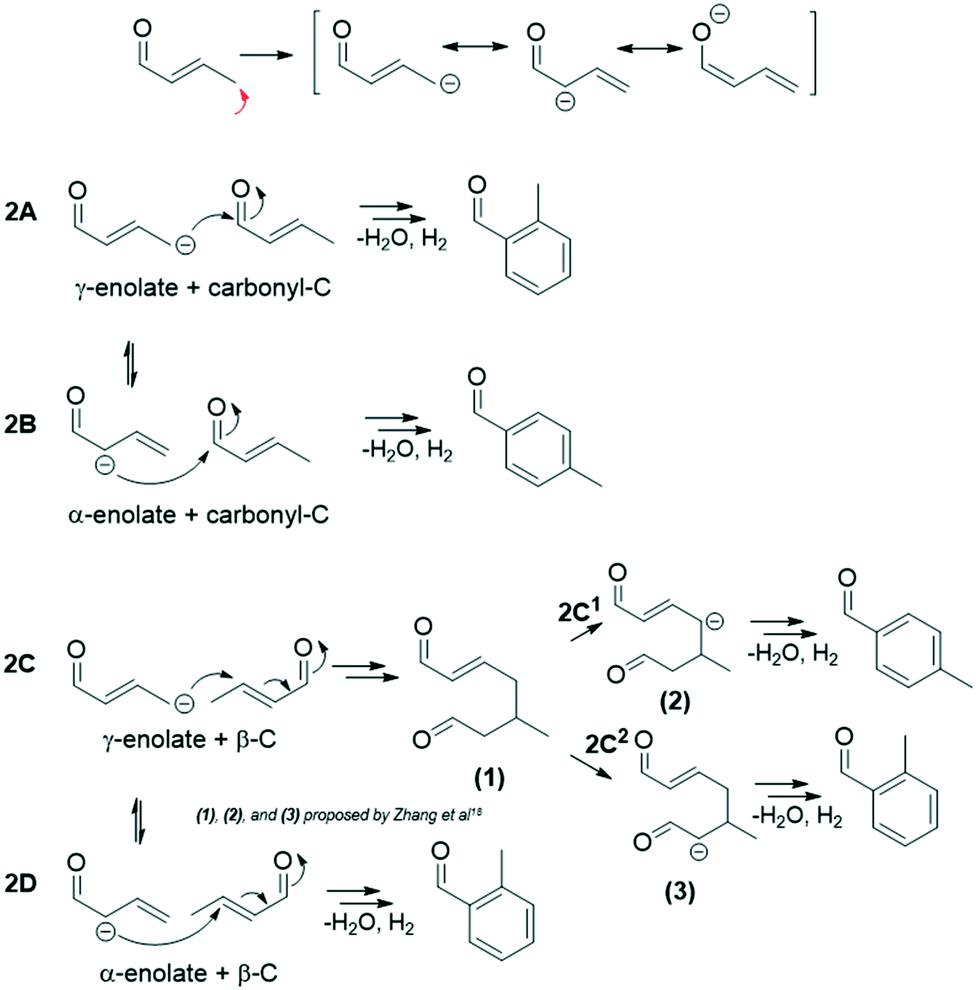 | ||
| Scheme 2 The four subroutes of route 2 (C4: 2-butenal + C4: 2-butenal) initiated by γ-C deprotonation that yield tolualdehydes.12,18 | ||
Previous kinetic studies12 of 2-butenal and 2-butenal surrogates (2-butenol, 3-methyl-2-butenal) over hydroxyapatite catalysts at similar temperatures investigated here revealed that routes 2A and 2B dominate tolualdehydes production. We postulate that routes 2A and 2B are primarily responsible for the yields of ortho- and para-tolualdehydes, respectively, in this work as well, given that the carbonyl-C position is significantly more electrophilic (0.391) than the β-C (−0.087), and that routes 2C and 2D require additional deprotonation steps.12,18 While we cannot conclusively rule out routes 2C and 2D, these factors make them less kinetically favorable a priori. In-depth mechanistic understanding through a complete DFT study of intermediate and transition state barriers for these complex pathways would facilitate further understanding of feasible tolualdehyde formation routes.
We measure a significantly higher selectivity towards the ortho- isomer (>80 mol C%) across all catalysts (Fig. 7). Wang and co-workers17 measured similar isomeric selectivity (∼85 mol C%) over Co-hydroxyapatite catalysts at 325 °C and a factor of 10 larger acetaldehyde concentration (diluted in N2) than the concentration investigated here. Steric and electronic effects could be contributing to this, among other factors (e.g., transition state stability).73 Sterically, attack from the terminal γ-C might be less hindered than that from the mid-chain α-C, which would favor higher ortho- selectivity. Electronic considerations involve the negative charge delocalization throughout the C4–O chain due to conjugation after the γ-C is deprotonated by the base site.71,72 The NBO charge distribution calculation of the γ-deprotonated enolate intermediate (Table S5†) shows that the electronegative oxygen atom in the carbonyl group bears the most negative charge (−0.749); yet the γ-C still has a large negative charge density (−0.607), more so than the α-C (−0.474). The excess negative charge in the enolate intermediate is more concentrated at the γ-C position compared to the α-C, which might suggest that the γ-enolate is a more represented nucleophile compared to the α-enolate. The higher density of the γ-C nucleophile would result in more contributions by route 2A (γ-enolate + carbonyl-C) compared to 2B (α-enolate + carbonyl-C), giving rise to a high ortho- selectivity as measured over these catalysts.
3.5 Role of catalyst acidity
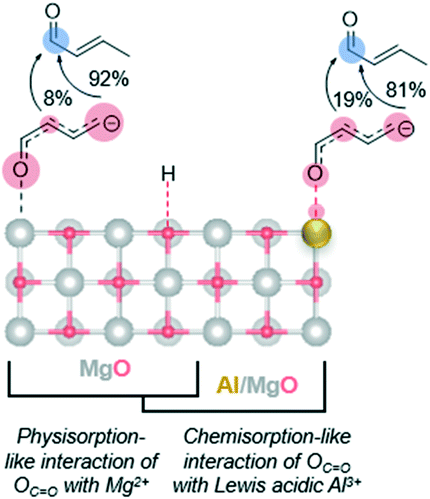 | ||
| Fig. 8 A schematic of the proposed negative charge distribution (indicated by the size of red bubbles) in the conjugated C4 enolate over the {100} surface of pure MgO (minimal acidity) and MgxAl-ox (Lewis acidity via Al3+). Red and blue denote electron-rich and electron-poor atoms, respectively. Mg: silver, O: red, Al: gold. | ||
As an electron acceptor, the Lewis acid site draws the negative charge and stabilizes the enolate intermediate, which is feasible because the conjugation of the molecule is preserved.77 We hypothesize that this electron flow towards the carbonyl-O coordinated to the Lewis acid shifts the negative charge density in the carbon chain originally localized on the γ-C towards the α-C, thereby making the α-enolate a more represented nucleophile. This allows route 2B to contribute more appreciably. As a result, we detect a selectivity shift towards para-tolualdehyde for Al-substituted MgO. The isomeric selectivity is not increasingly affected by an increased density of Lewis acid Al3+ sites in MgxAl-ox with increasing Al content (Table 1), even for a sample with Mg/Al = 1 and an even higher acid site density (1.74 μmol m−2; not shown). This indicates two primary findings. First, the charge redistribution by Al3+/carbonyl-O reduces the ortho- selectivity to a minimum value of 81%. This could be due to reduced, but still large, negative charge density on the γ-C, as well as other persistent steric or transition state stability effects that are not surmounted by the presence of more Lewis acidic Al3+ sites. Second, the density of acid centers needed to impact isomeric selectivity appears to far exceed that of the density of enolates formed by base deprotonation of the γ-carbon of 2-butenal. Even for the lower Al content of the MgxAl-ox (Mg/Al = 3), Al3+ effects on the enolate stability are saturated; otherwise, we expect the isomeric selectivity to be measured as a weighted fraction given by the relative amounts of sites with and without Al3+ effects and this value would change as Al content increases from 25 to 33 mol%.
Conclusions
In this work, we investigated the impact of MgO catalyst surface chemistry on the formation of valuable aromatic platform chemicals from acetaldehyde, an ethanol derivative. In particular, we studied the nature of the active basic site for the formation of tolualdehydes, the isomers of which are oxidized intermediates in the synthesis of high-volume chemicals (phthalic anhydride, terephthalic acid) from xylenes. Through selective poisoning of strong basic sites, we showed that low-coordinated O2− sites do not measurably contribute to the formation of tolualdehydes. This, in conjunction with the CO2 DRIFTS experiments, identifies the active basic site over MgO-based catalysts to be a medium-strength site in a specific M–O coordination environment. The wide distribution of sites obtained on samples synthesized through the co-precipitation technique, coupled with the inherent disorder (e.g., spinel nuclei) of calcined LDHs, make elucidating the precise coordination environment of this active site difficult on these catalysts. Yet, the finding that the active coordination sphere is M–O-type enables targeted syntheses of materials with well-defined M–O surface environments for continued investigation of potential catalysts.While tolualdehydes can be produced from the cross-condensation of acetaldehyde with its linear trimer aldol product, 2,4-hexadienal (route 1, C2 + C6), the high likelihood of a single active basic site initiating the formation of both isomers, the large kinetic costs of sp2-C deprotonation, and the minimal benzene yields rule out significant contributions from this pathway. Ortho- and para-tolualdehydes are more probably forming through the condensation of the acetaldehyde self-condensation product, 2-butenal (route 2, C4 + C4), via nucleophilic attack by the C4 γ- and α-enolate, respectively, on the carbonyl-C of 2-butenal. We attribute the isomeric selectivity shift towards para- observed over the MgxAl-ox to a charge distribution effect that occurs when the carbonyl-O of the enolate coordinates with a nearby Lewis acid Al3+ center: the negative charge in the intermediate is drawn to and stabilized by the electron acceptor, redistributing the charge density down the carbon chain from the initial locus of negative charge (γ-C) towards the acid site. This results in a higher charge density on the α-C than in the pure MgO case (minimal acid character), which increases the relative number of nucleophilic attacks occurring from the α-C to the carbonyl-C (route 2B) and shifts the isomeric selectivity towards para-. Even with this Lewis acid-mediated charge distribution effect that improves para- selectivity, ortho-tolualdehyde remains the dominant product. Tailoring of the catalyst surface chemistry, along with other catalyst and reaction parameters, can be explored to further probe active site requirements for enhanced tolualdehyde yields and selectivity to the significantly more valuable para-isomer.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by BP through the MIT Energy Initiative Advanced Conversion Research Program. The authors gratefully acknowledge Professor Yuriy Román and his group members for sharing instrumentation that allowed us to run the N2 adsorption–desorption and DRIFTS experiments. The authors thank Lagnajit Pattanaik and Professor Bill Green for performing the DFT calculations on their machines, and Professor Haomiao Zhang for his help with ASPEN calculations. The authors also greatly appreciate the insight of collaborators at BP, in particular Casey Hetrick, John Shabaker, and Eric Doskocil.Notes and references
-
J. L. Pellegrino, Energy and Environmental Profile of the U.S. Chemical Industry, 2000 Search PubMed
.
- B. Thompson, M. Machas and D. R. Nielsen, Curr. Opin. Biotechnol., 2015, 36, 1–7 CrossRef CAS PubMed
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed
- A. M. Niziolek, O. Onel, Y. A. Guzman and C. A. Floudas, Energy Fuels, 2016, 30, 4970–4998 CrossRef CAS
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed
- C. L. Williams, K. P. Vinter, R. E. Patet, C. C. Chang, N. Nikbin, S. Feng, M. R. Wiatrowski, S. Caratzoulas, W. Fan, D. G. Vlachos and P. J. Dauenhauer, ACS Catal., 2016, 6, 2076–2088 CrossRef CAS
-
B. Dudley and S. Dale, BP Statistical Review of World Energy, 2017 Search PubMed
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS
- R. A. Sheldon, J. Mol. Catal. A: Chem., 2016, 422, 3–12 CrossRef CAS
- T. Q. Hoang, X. Zhu, T. Sooknoi, D. E. Resasco and R. G. Mallinson, J. Catal., 2010, 271, 201–208 CrossRef CAS
- T. Moteki, A. T. Rowley and D. W. Flaherty, ACS Catal., 2016, 6, 7278–7282 CrossRef CAS
- T. Moteki, A. T. Rowley, D. T. Bregante and D. W. Flaherty, ChemCatChem, 2017, 9, 1921–1929 CrossRef CAS
- S. Hanukovich, A. Dang and P. Christopher, ACS Catal., 2019, 9, 3537–3550 CrossRef CAS
- Y.-C. Chang and A.-N. Ko, Appl. Catal., A, 2000, 190, 149–155 CrossRef CAS
- H. Kurokawa, M. Yanai, M. A. Ohshima and H. Miura, React. Kinet., Mech. Catal., 2012, 105, 401–412 CrossRef CAS
- T. Moteki and D. W. Flaherty, ACS Catal., 2016, 6, 4170–4183 CrossRef CAS
- Q.-N. Wang, X.-F. Weng, B.-C. Zhou, S.-P. Lv, S. Miao, D. Zhang, Y. Han, S. L. Scott, F. Schüth, A.-H. Lu, P. F. Schüth and A.-H. Lu, ACS Catal., 2019, 9, 7204–7216 CrossRef CAS
- L. Zhang, T. N. Pham, J. Faria, D. Santhanaraj, T. Sooknoi, Q. Tan, Z. Zhao and D. E. Resasco, ChemSusChem, 2016, 9, 736–748 CrossRef CAS PubMed
- J. I. Di Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef CAS
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. a. Bruijnincx, Catal. Sci. Technol., 2015, 5, 2869–2879 RSC
- S. Ordóñez, E. Díaz, M. León and L. Faba, Catal. Today, 2011, 167, 71–76 CrossRef
- M. León, E. Díaz and S. Ordóñez, Catal. Today, 2011, 164, 436–442 CrossRef
- C. R. Ho, S. Shylesh and A. T. Bell, ACS Catal., 2016, 6, 939–948 CrossRef CAS
- Q.-N. Wang, B.-C. Zhou, X.-F. Weng, S.-P. Lv, F. Schüth and A.-H. Lu, Chem. Commun., 2019, 1, 2–5 Search PubMed
- S. Luo and J. L. Falconer, J. Catal., 1999, 185, 393–407 CrossRef CAS
- E. Garrone, D. Bartalini, S. Coluccia, G. Martra, D. Tichit and F. Figueras, Stud. Surf. Sci. Catal., 1994, 90, 183–193 CrossRef CAS
- Y. Tang, G. Chen and Y. Lu, Res. Chem. Intermed., 2012, 38, 937–946 CrossRef CAS
- H. Idriss and M. A. Barteau, Catal. Lett., 1996, 40, 147–153 CrossRef CAS
- H. Idriss, C. Diagne, J. P. Hindermann, A. Kiennemann and M. A. Barteau, J. Catal., 1995, 155, 219–237 CrossRef CAS
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93 CrossRef CAS
- J. Quesada, L. Faba, E. Díaz, S. Bennici, A. Auroux and S. Ordóñez, J. Catal., 2015, 329, 1–9 CrossRef CAS
- G. S. Salvapati, K. V. Ramanamurty and M. Janardanarao, J. Mol. Catal., 1989, 54, 9–30 CrossRef CAS
- J. I. Di Cosimo, V. K. Díez, C. Ferretti and C. R. Apesteguía, Catalysis, 2014, 26, 1–28 CAS
- R. W. Snell, E. Combs and B. H. Shanks, Top. Catal., 2010, 53, 1248–1253 CrossRef CAS
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Mol. Catal. A: Chem., 2002, 182–183, 327–342 CrossRef CAS
- G. D. Yadav and P. Aduri, J. Mol. Catal. A: Chem., 2012, 355, 142–154 CrossRef CAS
- J. Tai and R. J. Davis, Catal. Today, 2007, 123, 42–49 CrossRef CAS
- E. Dumitriu, V. Hulea, C. Chelaru, C. Catrinescu, D. Tichit and R. Durand, Appl. Catal., A, 1999, 178, 145–157 CrossRef CAS
- J. Shen, J. M. Kobe, Y. Chen and J. A. Dumesic, Langmuir, 1994, 10, 3902–3908 CrossRef CAS
- J. Shen, M. Tu and C. Hu, J. Solid State Chem., 1998, 137, 295–301 CrossRef CAS
- P. Kustrowski, L. Chmielarz, E. Boz, P. Kus, M. Sawalha and F. Roessner, Mater. Res. Bull., 2004, 39, 263–281 CrossRef CAS
- I. M. Hill, S. Hanspal, Z. D. Young and R. J. Davis, J. Phys. Chem. C, 2015, 119, 9186–9197 CrossRef CAS
- J. Sirita, S. Phanichphant and F. C. Meunier, Anal. Chem., 2007, 79, 3912–3918 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS
- D. O. Scanlon, A. Walsh, B. J. Morgan, M. Nolan, J. Fearon and G. W. Watson, J. Phys. Chem. C, 2007, 111, 7971–7979 CrossRef CAS
- S. V. Cherepanova, N. N. Leont'eva, A. B. Arbuzov, V. a. Drozdov, O. B. Belskaya and N. V. Antonicheva, J. Solid State Chem., 2015, 225, 417–426 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef
- T. Sato, K. Kato, T. Endo and M. Shimada, React. Solids, 1986, 2, 253–260 CrossRef CAS
- J. Zhang, W. Qian, C. Kong and F. Wei, ACS Catal., 2015, 5, 2982–2988 CrossRef CAS
- D. Meloni, R. Monaci, V. Solinas, A. Auroux and E. Dumitriu, Appl. Catal., A, 2008, 350, 86–95 CrossRef CAS
- J. A. Lercher, React. Kinet. Catal. Lett., 1982, 20, 409–413 CrossRef CAS
- T. Armaroli, T. Bécue and S. Gautier, Oil Gas Sci. Technol., 2004, 59, 215–237 CrossRef CAS
- M. A. Aramendia, V. Borau, C. Jimenez, J. M. Marinas, A. Porras and F. J. Urbano, J. Mater. Chem., 1996, 6, 1943–1949 RSC
- F. Prinetto, G. Ghiotti, V. P. Giuria, R. Durand and D. Tichit, J. Phys. Chem. B, 2000, 104, 11117–11126 CrossRef CAS
- J. A. Lercher, C. Colombier and H. Noller, J. Chem. Soc., Faraday Trans., 1984, 80, 949–959 RSC
- J. C. Lavalley, Catal. Today, 1996, 27, 377–401 CrossRef CAS
- A. Travert, A. Vimont, A. Sahibed-Dine, M. Daturi and J. C. Lavalley, Appl. Catal., A, 2006, 307, 98–107 CrossRef CAS
- F. Prinetto, G. Ghiotti, P. Graffin and D. Tichit, Microporous Mesoporous Mater., 2000, 39, 229–247 CrossRef CAS
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS
- G. N. Vayssilov, M. Mihaylov, P. S. Petkov, K. I. Hadjiivanov and K. M. Neyman, J. Phys. Chem. C, 2011, 115, 23435–23454 CrossRef CAS
- M. Singh, N. Zhou, D. K. Paul and K. J. Klabunde, J. Catal., 2008, 260, 371–379 CrossRef CAS
- D. Fan, X. Dong, Y. Yu and M. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 25671–25682 RSC
- H. Zhang, M. Y. S. Ibrahim and D. W. Flaherty, J. Catal., 2018, 361, 290–302 CrossRef CAS
- B. I. Stefanov, Z. Topalian, C. G. Granqvist and L. Österlund, J. Mol. Catal. A: Chem., 2014, 381, 77–88 CrossRef CAS
- M. Ros and E. J. J. Groenen, J. Chem. Phys., 1991, 94, 7640–7648 CrossRef CAS
- V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, Lat. Am. Appl. Res., 2003, 33, 79–86 Search PubMed
- J. E. Rekoske and M. A. Barteau, Ind. Eng. Chem. Res., 2011, 50, 41–51 CrossRef CAS
- G. Martra, Appl. Catal., A, 2000, 200, 275–285 CrossRef CAS
- J. E. Bartmess and J. P. Kiplinger, J. Org. Chem., 1986, 51, 2173–2176 CrossRef CAS
- I. Iriarte, O. Olaizola, S. Vera, I. Gamboa, M. Oiarbide and C. Palomo, Angew. Chem., Int. Ed., 2017, 56, 8860–8864 CrossRef CAS PubMed
- E. M. P. Silva and A. M. S. Silva, Synthesis, 2012, 44, 3109–3128 CrossRef CAS
- M. del Arco, C. Martin, I. Martin, V. Rives and R. Trujillano, Spectrochim. Acta, Part A, 1993, 49, 1575–1582 CrossRef
- Y. Roman-Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566–1580 CrossRef CAS
- J. D. Lewis, S. Van de Vyver and Y. Roman-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef CAS PubMed
- S. Herrmann and E. Iglesia, J. Catal., 2017, 346, 134–153 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01927h |
| This journal is © The Royal Society of Chemistry 2020 |