
The fabrication of trifunctional polyoxometalate hybrids for the cascade conversion of glycerol to lactic acid†
Meilin
Tao
,
Ningyue
Sun
,
Yiming
Li
,
Shengtian
Wang
and
Xiaohong
Wang
 *
*
Key Lab of Polyoxometalate Science of Ministry of Education, Northeast Normal University, Changchun 130024, P. R. China. E-mail: wangxh665@nenu.edu.cn; Fax: +0086 431 85099759
First published on 27th November 2019
Abstract
Lactic acid (LA) has been produced with cascade reactions under non-noble metal and base-free conditions. In this work, amino acid-functionalized polyoxometalates (POMs) LyxH3−xPMo12O40 (abbreviated as LyxH3−xPMo, x = 1–3, Ly = C6H15O2N2, lysine) with triple active sites were fabricated, which presented a 87% LA yield at 96% glycerol conversion under mild reaction conditions attributed to the combination of basic sites, Brønsted acidic sites, suitable redox ability, and special microenvironments. The mechanism study showed that with LyxH3−xPMo, glycerol was oxidized first to glyceraldehyde (GCA) and dihydroxyacetone (DHA) by the redox sites; then, the existence of basic sites led to the dehydration of DHA and GCA to pyruvaldehyde (PRA) and finally, the Brønsted acid sites catalyzed the hydration of PRA to LA. Meanwhile, the activation energies for different POMs were calculated using Arrhenius plots to confirm the effect of Ly2H1PMo for its highest efficiency. These hybrids acted as heterogeneous catalysts with long duration and high stability.
Introduction
Tandem reactions, which enable multi-step reactions in one pot, increase economic competitiveness in the production of target compounds and the greenness of the whole process by reducing the overall cost; this is effective in the minimization of waste and energy consumption and for optimizing the use of solvents.1,2 The oxidation of glycerol to lactic acid (LA) is a classic reaction; it involves the combined oxidation to dihydroxyacetone (DHA) and glyceraldehyde (GCA), dehydration of GCA or DHA, and hydration of pyruvaldehyde (PRA).3Recently, an environmental and economic assessment of lactic acid production from glycerol using cascade bio- or chemo-catalysis was reported by the collaboration of S. Papadokonstantakis and J. Pérez-Ramírez groups.4–7 LA is manufactured biotechnologically with a capacity estimated to be 300–400 kt per annum.8 The fermentative production of LA has the advantage of the very high enantiomeric purity of the product. This is of utmost importance for the polymer industry, which relies mainly on L-LA as a substrate for the production of high-quality crystalline polylactic acid (PLA).9 Nevertheless, the microbial production of LA also possesses drawbacks including the utilization of expensive feedstocks and the co-production of enormous amounts of waste. Although the utilization of various carbon-containing sources such as glucose or glycerol for fermentation is possible, most producers rely on pure and thus more expensive carbohydrate feedstocks.10 Additionally, the neutral environment is essential in the fermentation process of producing LA; therefore, calcium hydroxide or calcium carbonate is added to the broth during fermentation for the neutralization of the formed LA.10 As a result, calcium lactate is produced instead of LA. Traditionally, calcium lactate solutions are acidified with sulfuric acid to precipitate calcium sulphate, leaving LA in the aqueous phase. The need for the disposal of calcium sulfate co-produced in equal amounts with LA presents a major drawback of these biotechnological processes.
To the best of our knowledge, glycerol-based routes are more sustainable than the traditional sugar fermentation-based processes. Compared to the biocatalytic oxidation of glycerol to LA, alternatives based on chemocatalysis have been reported. Pt/ZrO2,11 Ru–Zn–CuI/HAP,12 CuO/ZrO2,13 and Au–Pt/nCeO2 (ref. 14) show high catalytic activity in the presence of a base. Based on previous works, the dehydration reaction of DHA and GCA to PRA can be catalyzed by a base better than by an acid or a neutral medium, while the hydration reaction of PRA to LA prefers acid catalysts.15 Compared to metal Lewis acid catalysts, metal-free Brønsted acids for catalyzing hydration reactions have been boosted during these years. Ponzi et al.16 used trichloroacetic acid (TCA) on different supports such as silica, titania and zirconia (TCA/SiO2, TCA/TiO2 and TCA/ZrO2·nH2O) as catalysts for α-pinene hydration. Bo Xu et al.17 used both AcOH-TfOH and AcOH-Ga(OTf)3 for the hydration of phenylacetylene and obtained a 99% yield of the product after 10 h and 8 h, respectively. These results illustrate that both Lewis and Brønsted acids can catalyze hydration reactions. The dehydrogenation–oxidation of glycerol to DHA and GCA constitutes an essential step in the production of LA from glycerol. Under this circumstance, redox–acid–base trifunctional catalysts are considerably attractive towards the target production of LA.
Polyoxometalates (POMs), especially phosphomolybdic acid (H3PMo12O40, HPMo), due to their controllable redox and acidic properties have been used as effective catalysts in all kinds of oxidation reactions.18,19 In our previous work, we found that H3PMo12O40 could act as a suitable bifunctional catalyst with both redox and acidic properties in glycerol oxidation. Through controlling the proton exchange reaction by some Lewis metal cations (L), a series of LHPMo species with redox and Lewis acidity were evaluated for glycerol conversion.20,21 By now, no triple-functional POMs have been designed and evaluated in the cascade conversion of glycerol.
To the best of our knowledge, some multifunctional catalysts based on POMs have been successfully used in other kinds of cascade reactions. Mizuno's group22 reported the design of acid–base POM catalysts obtained by incorporating rare-earth-metals into lacunary [SiW10O36]8−. These heteropolyacids (HPAs) exhibited Lewis acidity due to the rare-earth metals (RE) and basicity due to the nucleophilic surface of HPAs; the catalytic activity in the cyanosilylation reactions was enhanced by the combination of RE Lewis acids and HPA Lewis bases. Wang's group23 also reported the synthesis of Schiff base-structured acid–base catalysts [PySAIm]3PW12O40 using a Schiff base ionic liquid (IL) and Na3PW12O40 as the precursors, which led to highly efficient heterogeneous Knoevenagel condensations. Shiju's group reported mesoporous SiO2 modified with site-isolated amines and phosphotungstic acidic groups, which had tunable antagonistic acid–base functions for one-pot tandem reactions.24 Can Li's group reported the self-assembly of heteropolyacids hosted in layered double hydroxides, but the Brønsted acidity of HPAs and the basicity of the host were suppressed by each other.25 Most recently, Kim's group fabricated acid–base bifunctional hybrids using a zeolitic imidazolate framework-8 (ZIF-8) and heteropolyacids (HPAs) as precursors, which showed high efficiency in the transesterification reaction to produce biodiesel.26 It can be concluded that there are two ways to fabricate base–acid functional POMs: changing the components of POMs and loading POMs on base-containing supports. Compared to the second method, the first one shows a higher potential due to the ease of preparation and the ease of controlling the acid or base distribution only through altering the composition.
During our research on biomass conversion, we achieved the fabrication of acid–base bifunctional POMs using amino acids with double amino groups as precursors like Ly3−xHxPW,27 which showed high efficiency in the one-pot preparation of biodiesel and 5-hydroxymethylfurfural from glucose. Compared to the reported basic ionic liquids, amino acids are easily available with numerous functions for biological processes;28,29 moreover, they are inexpensive and non-toxic and can act as biomimics. Following our recent development of POMs in cascade biomass conversion reactions,3,20,21,30–32 we herein disclose amino acid-functionalized POMs LyxH3−xPMo (Ly = lysine, x = 1–3) catalyzing the glycerol transformation specifically to lactic acid. Also, a mechanism was determined to clarify the effect of isolated acidic, basic, and redox sites on each pathway. The synthetic procedure of the LyxH3−xPMo catalyst, the production of lactic acid from the by-product of biodiesel, and the reusability of the catalysts were highlighted in this investigation.
Results and discussion
FT-IR spectra of pyridine absorption were recorded to probe accessible surface acid sites; FT-IR spectroscopy is a powerful tool for identifying the nature of acid sites.33 As shown in Fig. 1, all the samples present typical bands, corresponding to strong Brønsted and Lewis acid-bound pyridines at around 1540 and 1450 cm−1.34 The calculated results for Lewis or Brønsted acidity based on the Lambert–Beer equation are listed in Table 1. Lewis acid sites emerged as a result of the proton exchange between lysine and HPMo as we know that some amino acids have a Lewis structure.35 Among them, Ly1H2PMo presented the maximum total acid amount and Lewis acid amount. Ly1H2PMo with the least lysine amount gave the highest Lewis acidity maybe owing to the strong interaction between PMo12O403+ and lysine. In comparison with the fully lysine-exchanged HPMo sample, the partially substituted HPMo catalyst had increased available acid sites due to the presence of residual protons capable of mobility, inducing new strong acid sites.36 The acid and base amounts were also tested by the conductivity titration method (Table 2), which were similar to the pyridine absorption FT-IR results.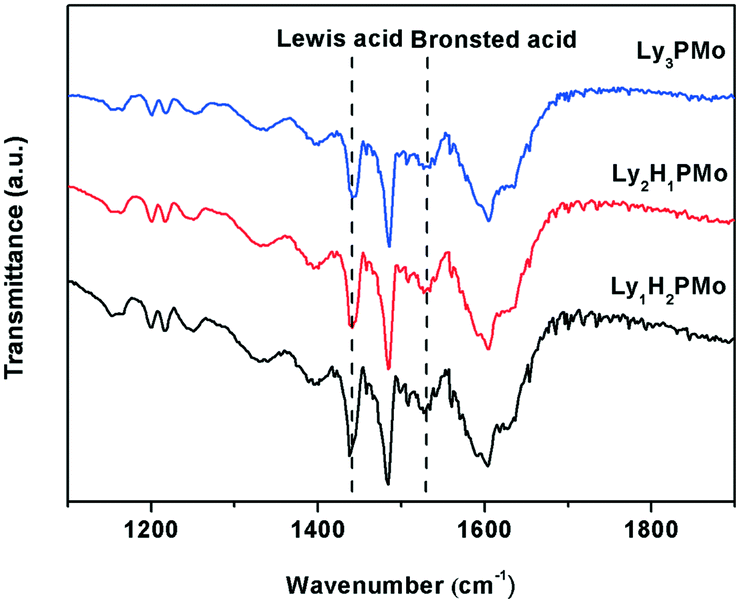 | ||
| Fig. 1 FT-IR spectra of pyridine absorption of LyxH3−xPMo. | ||
| Catalysts | Lewis acid mmol g−1 | Brønsted acid mmol g−1 | Total acid mmol g−1 |
|---|---|---|---|
| HPMo | 0.03 | 1.01 | 1.04 |
| Ly1H2PMo | 0.35 | 0.66 | 1.01 |
| Ly2H1PMo | 0.29 | 0.61 | 0.90 |
| Ly3PMo | 0.22 | 0.51 | 0.73 |
| Entry | Catalysts | Acid contents mmol g−1 | Base contents mmol g−1 | Conversion of glycerol% | Yield of DHA + GCA% | Yield of PRA% | Yield of LA% |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 5 mL, 1 M of glycerol, 4 mM of catalyst, 1 MPa O2, 80 °C, 800 rpm, 3 h. b The same reaction conditions except for the pH: the pH is the same as the Ly2H1PMo conditions. | |||||||
| 1 | Ly1H2PMo | 0.96 | 0.28 | 78 | 37 | 11 | 23 |
| 2 | Ly2H1PMo | 0.89 | 0.51 | 86 | 30 | 14 | 38 |
| 3 | Ly3PMo | 0.81 | 0.79 | 74 | 25 | 26 | 20 |
| 4 | H3PMo | 1.04 | 0 | 65 | 38 | 8 | 12 |
| 5 | Lysine | 0.27 | 0.46 | 8 | 6 | 0 | 0 |
| 6 | K1H2PMo | 0.68 | — | 56 | 24 | 19 | 10 |
| 7 | K2H1PMo | 0.61 | — | 47 | 20 | 16 | 8 |
| 8 | K3PMo | 0.55 | — | 38 | 12 | 14 | 5 |
| 9 | H3PMob | — | — | 68 | 27 | 21 | 14 |
| 10 | K2H1PMob | — | — | 70 | 23 | 30 | 16 |
| 11 | (C6H14N)1H2PMo | 0.65 | — | 52 | 42 | 3 | 5 |
| 12 | (C6H14N)2H1PMo | 0.58 | — | 45 | 37 | 1 | 2 |
| 13 | (C6H14N)3PMo | 0.52 | — | 40 | 34 | 0 | 0 |
Scheme 1 and Table 2 summarize the catalytic performances of different catalysts in the one-pot cascade reaction involving sequential redox, dehydration and hydration reactions. We found the following conclusions:
 | ||
| Scheme 1 Reaction pathways of glycerol oxidation. | ||
(1) The conversion of glycerol depended on the redox ability of the catalysts. Lysine only gave 8% conversion of glycerol due to the absence of redox sites. The oxidation of glycerol improved significantly with H3PMo and LyxH3−xPMo, showing the essential role of redox sites for glycerol oxidation. The existence of redox sites in the structure of the catalysts was also confirmed by H2-TPR (Fig. S1†). Peaks appeared between 400 and 1000 s after the process started. The consumed H2 amounts based on peak areas were 5.42 × 10−3, 4.57 × 10−3 and 3.89 × 10−3 mol g−1, corresponding to Ly1H2PMo, Ly2H1PMo, and Ly3PMo. The decrease in H2 consumption with the increase of lysine was attributed to the drop in oxidative ability, which was consistent with the cyclic voltammetry results (Fig. S2†).
(2) The conversion of glycerol for LyxH3−xPMo varied as Ly2H1PMo > Ly1H2PMo > Ly3PMo with the TOFgly values of 72, 65 and 61 h−1 (TOFgly = [Gly] × Conversion/[actual POM amount] × reaction time), respectively. Such Ly-functionalized POMs exhibited higher activity than H3PMo (TOF = 53 h−1), which might be attributed to the presence of basic sites. Control experiments were carried out to check the essential role of the basic sites. H3PMo gave enhanced conversion of glycerol in the presence of a base (Table 2, entry 9), indicating that the existence of a base might promote glycerol conversion. Nevertheless, the addition of liquid base is not economical, which requires further treatment due to the generation of lactate. Then, (C6H16N)xH3−xPMo (C6H14N is the abbreviation of 1-aminohexane) was synthesized to further determine the effect of the base. (C6H16N)xH3−xPMo is a kind of amphiphilic molecule, which can form the same assembly as LyxH3−xPMo in water, but it has no basic sites. In the aerobic oxidation of glycerol, all (C6H16N)xH3−xPMo samples presented lower activity than LyxH3−xPMo. This might be attributed to the lack of basic sites in (C6H16N)xH3−xPMo. This confirmed that the presence of the basic sites of lysine in POMs is useful for glycerol conversion.
(3) The conversion of glycerol depended on the different molar ratios between protons and amino groups, i.e., 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1.5:1 and 1:1, corresponding to LyxH3−xPMo (n = 1, 2, 3). Fig. 2 shows that Ly2H1PMo presents the highest conversion of 96% and LA yield of 87% within 4 h among all LyxH3−xPMo. This result showed that the conversion of glycerol depends on the components of POMs with the multifunctional sites of the base and Brønsted acid. Therefore, the co-existence of a base and acid in one unit might favor glycerol conversion with a suitable molar ratio for protons and amino groups of 1.5:1. The molar ratio of protons and amino groups also affects the distribution of LA with the yield order of Ly2H1PMo (87%) > Ly1H2PMo (79%) > Ly3PMo (76%). Ly2H1PMo exhibited the highest LA yield due to its perfect acidic and basic ratio, and it could catalyze the cascade reaction successfully.
:1, 1.5:1 and 1:1, corresponding to LyxH3−xPMo (n = 1, 2, 3). Fig. 2 shows that Ly2H1PMo presents the highest conversion of 96% and LA yield of 87% within 4 h among all LyxH3−xPMo. This result showed that the conversion of glycerol depends on the components of POMs with the multifunctional sites of the base and Brønsted acid. Therefore, the co-existence of a base and acid in one unit might favor glycerol conversion with a suitable molar ratio for protons and amino groups of 1.5:1. The molar ratio of protons and amino groups also affects the distribution of LA with the yield order of Ly2H1PMo (87%) > Ly1H2PMo (79%) > Ly3PMo (76%). Ly2H1PMo exhibited the highest LA yield due to its perfect acidic and basic ratio, and it could catalyze the cascade reaction successfully.
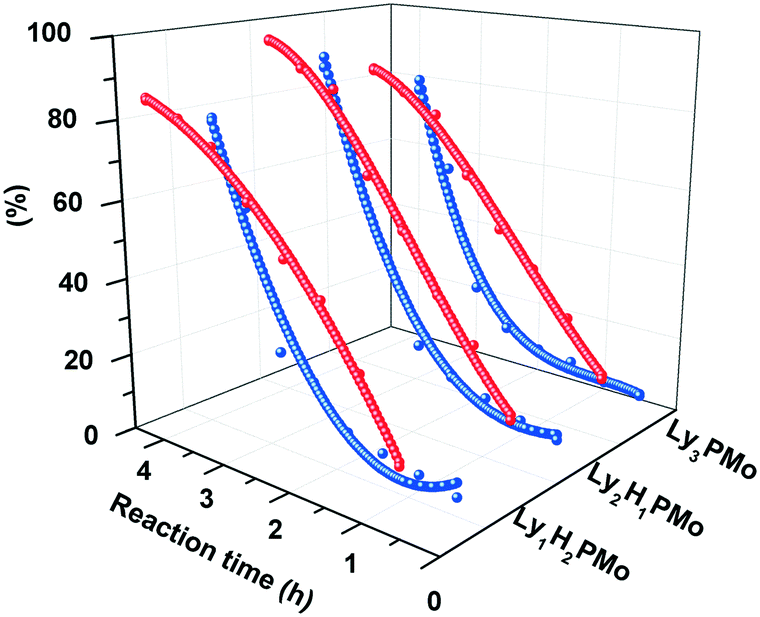 | ||
| Fig. 2 The relationship between efficiency and components of LyxH3−xPMo. Red curves: conversion of glycerol; blue curves: yield of LA. | ||
(4) The conversion of glycerol via LyxH3−xPMo was also enhanced by the self-assembly of nanostructures in water, which was determined by TEM and CMC (Fig. S3 and S4 and Table S2†). The comparison experiments using KxH3−xPMo determined this (Table 2, entries 6–8). LyxH3−xPMo is a kind of surfactant molecule, which can form micelles in water to concentrate the substrate molecules around the active sites of POMs.27 The IR spectrum of Ly2H1PMo after adsorbing glycerol further confirmed this effect (Fig. S5†). KxH3−xPMo presented lower activity compared to relative LyxH3−xPMo even at the same pH (Table 2, entries 9–10) due to the lack of nanostructure formation for KxH3−xPMo (Scheme 1 and Table 2). These observations again confirmed that except for the acid and basic sites, the activity of the catalysts was also influenced by the morphology.
(5) The existence of Ly+ also influenced the redox potentials, and the E1/2 (E1/2 = 1/2Epa + 1/2Epb) of LyxH3−xPMo was exhibited in Fig. S2.† The order of the redox potentials of LyxH3−xPMo was HPMo (183.6 mV) > Ly1H2PMo (113.8 mV) > Ly2H1PMo (92.8 mV) > Ly3PMo (74.9 mV). To our surprise, it was found for the first time that Ly+ had a significant influence on the redox potentials of POMs. Compared to the value for their parent H3PMo12O40, the redox potentials shifted by 69.8, 90.8 and 108.7 mV towards the negative direction for Ly1H2PMo, Ly2H1PMo, and Ly3PMo, respectively. This result showed that the addition of Ly+ could decrease the oxidation–reduction performance of POMs. The reason was the interaction between Ly+ and PMo12O403+, which mainly contributed to the adjustment of the electron transfer rate in oxidation. Therefore, the combination of multifunctional sites and self-assembly in these nanoreactors allowed LyxH3−xPMo to achieve high conversions of glycerol.
To further demonstrate the advantage of LyxH3−xPMo, the apparent activation energies for the three catalysts were calculated (Fig. 3). The reaction data were collected within 30 min because at the beginning, almost all glycerol was converted to DHA and GCA, which can be considered as a one-step reaction. The results indicated that the glycerol to DHA and GCA step might be a second-order reaction since the concentrations of glycerol (1/Ct) increased linearly with the reaction time in the beginning according to the reaction rate equation 1/Ct = kt + 1/C0.37 To the best of our knowledge, the rate-determining step of glycerol oxidation to LA is the dehydrogenation of glycerol.38 Accordingly, the calculated activation energies for Ly1H2PMo, Ly2H1PMo and Ly3PMo were 119, 128 and 149 kJ mol−1, respectively. Thus, the lowest activation energy for Ly1H2PMo in this cascade reaction was responsible for its highest catalytic efficiency in the first step, which was caused by the higher redox ability. The activation energies of the second and third steps were also calculated using DHA and PRA as substrates within a short time (Fig. S6 and S7†). The order of the activation energies of DHA conversion was Ly1H2PMo (36 kJ mol−1) > Ly2H1PMo (31 kJ mol−1) > Ly3PMo (27 kJ mol−1), which confirmed that the basic sites preferred to catalyse the dehydration of DHA to PRA. For PRA conversion to LA, the order was opposite, i.e., Ly1H2PMo (16 kJ mol−1) < Ly2H1PMo (17 kJ mol−1) < Ly3PMo (19 kJ mol−1), which confirmed that the basic sites were better for the rehydration of PRA to LA.
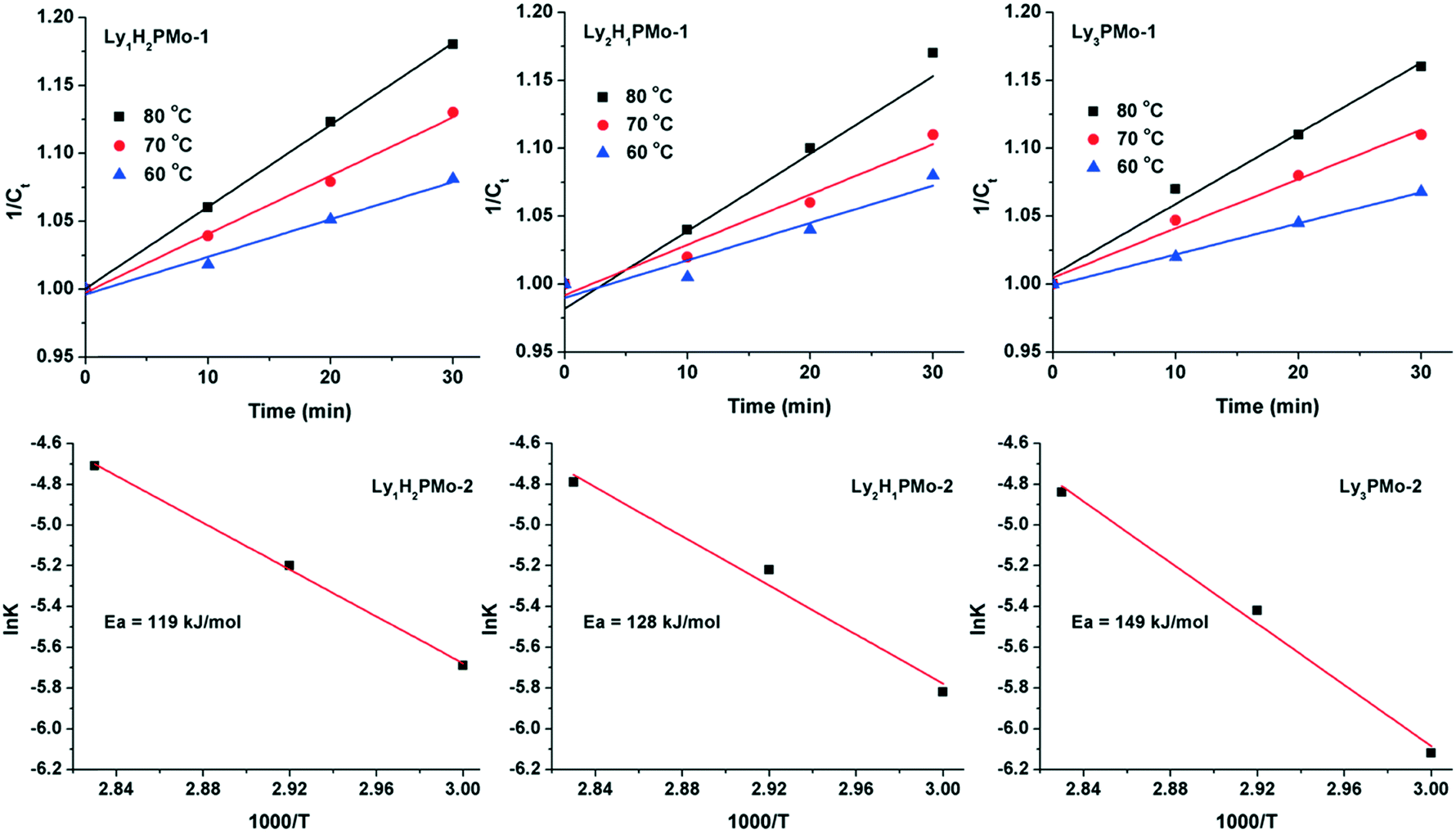 | ||
| Fig. 3 The plots of the concentration of glycerol versus reaction time (1/Ct − t curve) and the Arrhenius plots (ln(K − t)) over LyxH3−xPMo catalysts in the temperature range from 60 to 80 °C (Ct: the concentration of glycerol with the reaction time). | ||
In order to clarify the effect of multifunctional sites on glycerol transformation, different feedstocks were used as substrates including glycerol, DHA and PRA for LyxH3−xPMo (Fig. 4). When glycerol was chosen to be the substrate, for Ly1H2PMo, the generation rate of DHA and GCA was the highest and its consumption rate was the lowest, while the performances of Ly3PMo were the opposite (Fig. 4a and d). This phenomenon could be explained by the fact that the redox potential of Ly1H2PMo is the highest, but its base content is less than that of Ly3PMo, which is preferred for the dehydrogenation oxidation of glycerol but is strongly unfavourable for the dehydration reaction. A similar phenomenon also occurred in the conversion of DHA (Fig. 4b and e). Ly3PMo exhibited the best production rate of PRA but the worst consumption rate because of its highest number of basic sites and the lowest number of acidic sites. According to the LA yield beginning from PRA, Ly1H2PMo showed the best efficiency among all materials, which was attributed to its superior acidic properties (Fig. 4c and f).
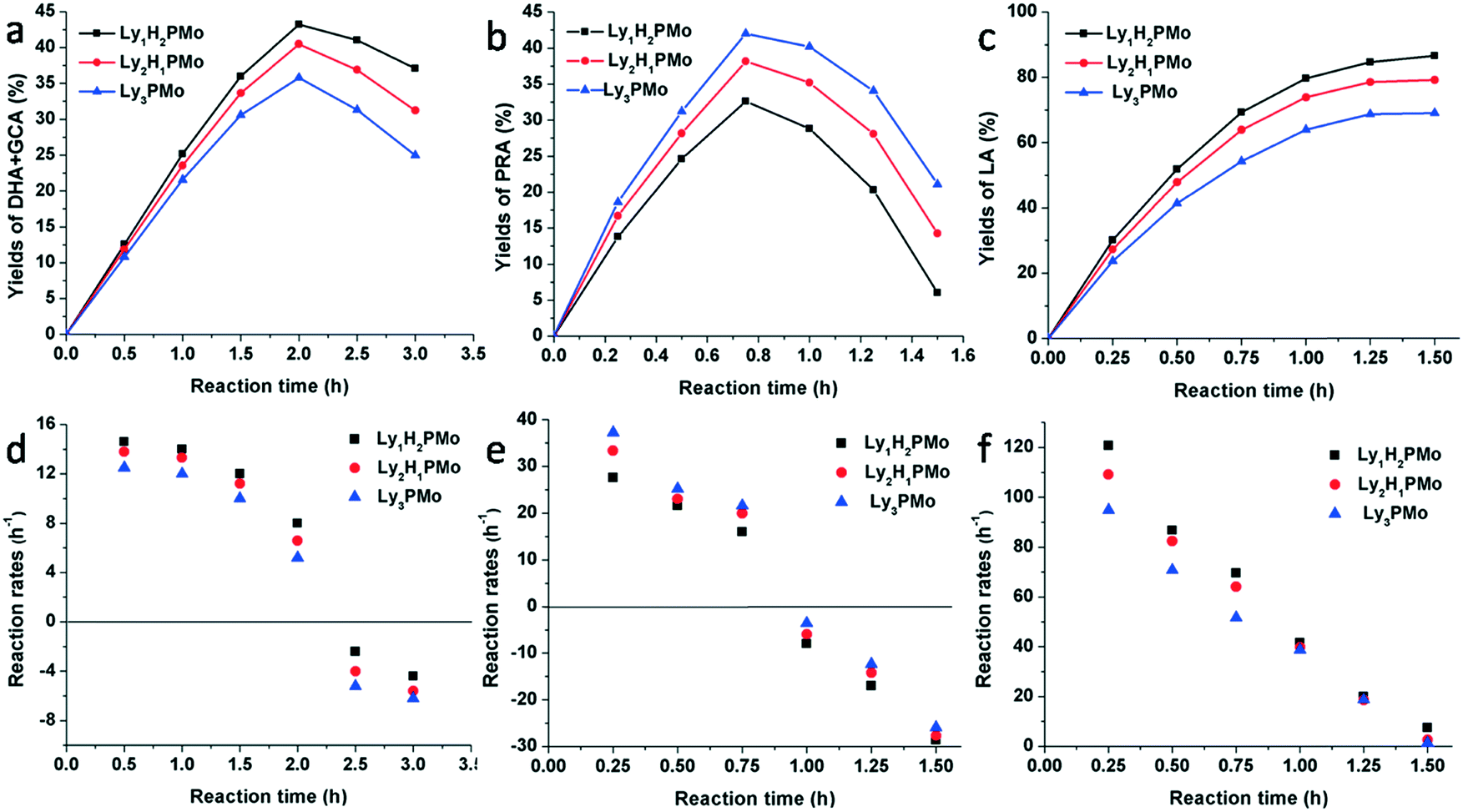 | ||
| Fig. 4 Time course of different intermediates: a) glycerol as substrate; b) DHA as substrate; c) PRA as substrate; d) reaction rate of DHA and GCA production; e) reaction rate of PRA production; f) reaction rate of LA production. Reaction condition: 5 mL, 1 M of substrates, 4 mM of catalyst, 1 MPa O2, 80 °C, 800 rpm. | ||
The reaction conditions were optimized by changing the reaction temperature, the catalyst, the concentration of glycerol and the pressure of O2 (Fig. 5). Increasing the reaction temperature (80 to 90 °C) and O2 pressure (1–1.25 MPa) resulted in a gradual decrease in the selectivity for LA owing to the over-oxidation of intermediates (Fig. 5a and b). The optimal molar ratio of glycerol to Ly2H1PMo appeared to be 250:1 to complete the reaction within 4 h. Using lower amounts of the catalyst (Fig. 5c) or more concentrated solutions of the substrate (Fig. 5d), lower selectivity for LA was observed after 4 h of reaction owing to the incomplete conversion of the intermediates. The optimum reaction conditions for glycerol oxidation to LA were 1 M of glycerol, 4 mM of catalyst, 1 MPa O2, 80 °C, and 4 h, which gave 96% of glycerol conversion and 87% of LA yield.
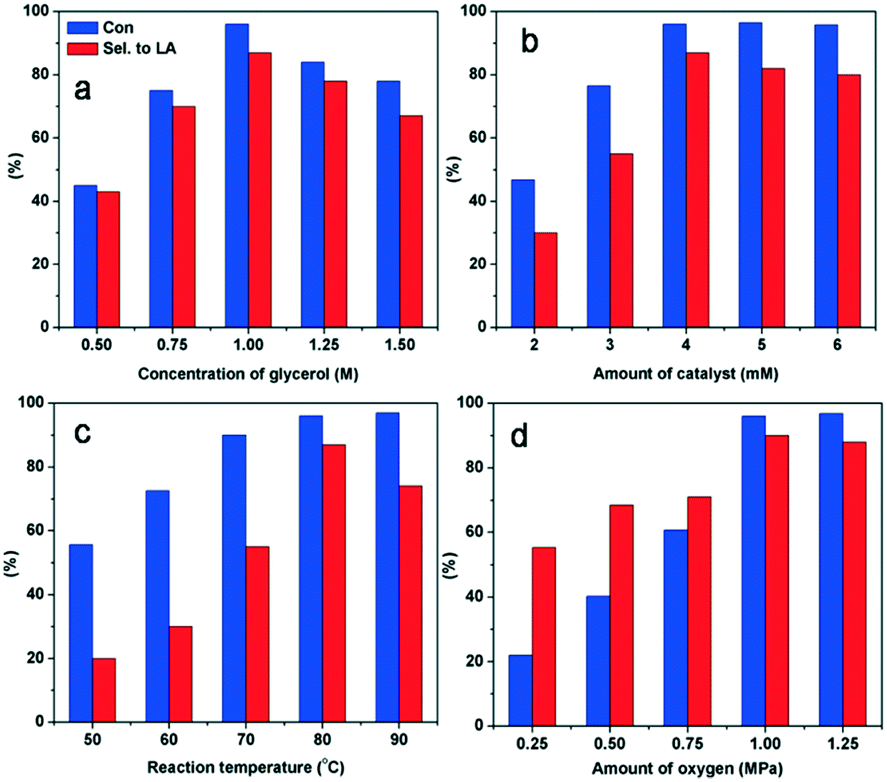 | ||
| Fig. 5 The screening process of optimum conditions in glycerol oxidation: (a) reaction temperature (4 mM Ly2H1PMo, 1 M glycerol, 4 h, 1 MPa O2); (b) amount of oxygen (4 mM of Ly2H1PMo, 1 M glycerol, 80 °C, 4 h); (c) amount of catalyst (1 M glycerol, 80 °C, 4 h, 1 MPa O2); (d) concentration of glycerol (4 mM of Ly2H1PMo, 80 °C, 4 h, 1 MPa O2). | ||
An important merit of heterogeneous catalysts is that they can be conveniently recycled and reused. As shown in Fig. 6, our LyxH3−xPMo catalysts can be used repetitively for at least 10 times without a significant decrease in the yield of LA. The mixture solutions after reaction and after removing the catalyst were tested by UV-vis spectroscopy (Fig. S8†). No signal for POM was observed, which confirmed that LyxH3−xPMo acted as a heterogeneous catalyst with little leaching during the reaction. The loss of the amount of the catalyst was due to manual errors (about 4.2%). To further determine the leaching of Ly2H1PMo, the catalyst was separated after reacting for 2 h (54% of glycerol conversion) and was allowed to react further for over 2 h under the same conditions. The result showed that the conversion of glycerol was only 55.8%, which indicated that Ly2H1PMo acted as a heterogeneous catalyst with little leaching into the reaction mixture during the reaction. The IR and 31P MAS NMR spectra of LyxH3−xPMo remained almost the same after the reaction (Fig. S9†), implying the excellent stability against leaching owing to the electrostatic interactions between Ly+ and PMo12O403−.
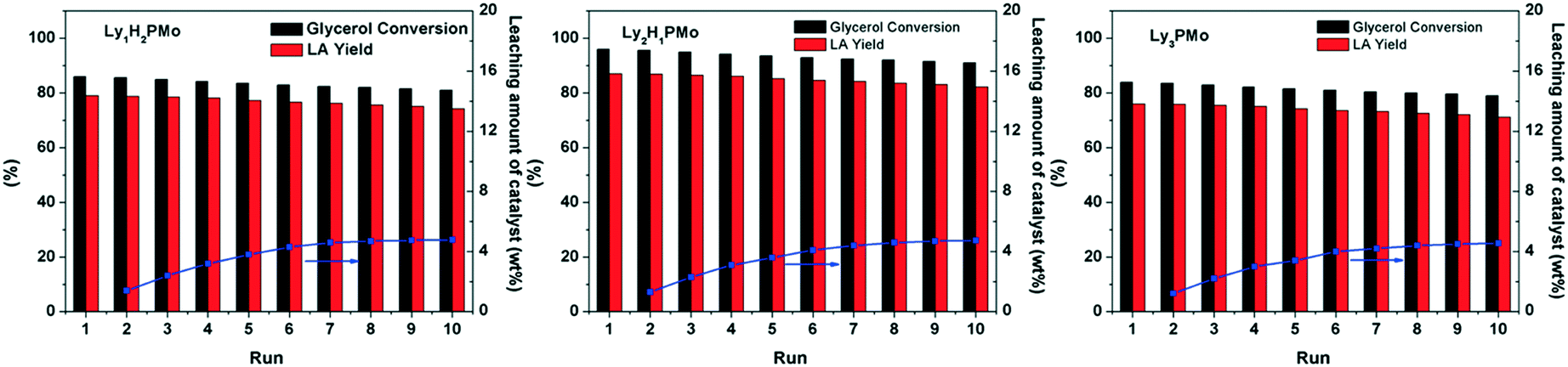 | ||
| Fig. 6 Reusability of LyxH3−xPMo in glycerol oxidation. Reaction conditions: 5 mL, 1 M of glycerol, 4 mM of catalyst, 1 MPa O2, 80 °C, 800 rpm. | ||
Experimental
The synthesis and self-assembly of LyxH3−xPMo are exhibited in Fig. 7: 6 mmol of H3PMo12O40 (HPMo) was dissolved in distilled water (30 mL) and divided into 3 parts. Different molar ratios (2, 4 and 6 mmol) of lysine were added into the HPMo solutions with vigorous stirring at room temperature. A precipitate was obtained immediately, which was continuously stirred for 30 min, followed by filtering. The yellow precipitate was dried at 30 °C under vacuum for 4 h. The resulting Ly1H2PMo, Ly2H1PMo, and Ly3PMo were obtained with a yield of 76, 82 and 85%, respectively. Multiple techniques, including ICP, FTIR, Low-angle-XRD, 31P MAS NMR and BET, were used to characterize the LyxH3−xPMo catalysts, which are discussed in detail in the ESI† (Tables S1 and S2 and Fig. S10–S12). The surface areas of the three catalysts were observed to be similar. For comparison, (C6H16N)3PMo was synthesized according to the same procedure using C6H16N instead of lysine.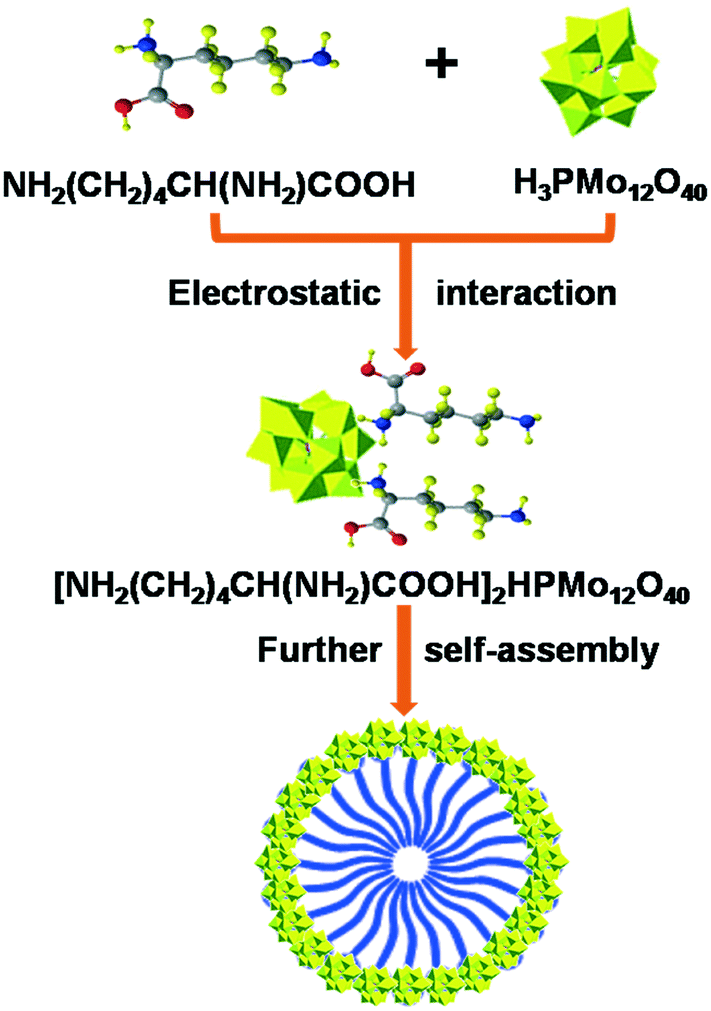 | ||
| Fig. 7 Illustration of the self-assembly of lysine and HPA to hybrid micelles. | ||
The surface acidities and the base strengths of the catalysts were determined by the conductivity titration method. For acid amounts, 0.01 g of sample was added to 50 mL of n-butylamine in acetonitrile (0.001 M) with stirring for 6 h. Then, the samples were filtrated out and the extra n-butylamine solution was titrated by 0.01 M of HCl using a conductivity meter. The same method was also used in the measurement of basicity: 0.01 g of sample was added to 50 mL of acetic acid (0.001 M) with stirring for 6 h. Then, the samples were filtrated out and the extra acetic acid was titrated by 0.01 M of KOH using a conductivity meter.
The IR spectra of adsorbed pyridine (Py-IR) helped to measure the acid content and distinguish the properties of acid sites (Lewis or Brønsted). The samples were exposed to pyridine vapor for 12 h under vacuum (10−3 Pa) at 60 °C. The quantification of acidity was calculated by the Lambert–Beer equation: A = (ε·w·c)/S.
Here, A is the absorbance (area in cm−1), ε is the extinction coefficient (m2 mol−1), w is the sample weight (kg), c is the concentration of acid (mol kg−1 or mmol g−1), and S is the sample disk area (m2). The amounts of Brønsted and Lewis acid sites were estimated from the integrated areas of the adsorption bands at 1540 and 1450 cm−1, respectively, using the extinction coefficient values based on a previous report.33
Glycerol oxidation was performed in a high-pressure batch autoclave of stainless steel with a polytetrafluoroethylene inlet (10 mL). The autoclave was equipped with a gas supply system and a magnetic stirrer. The catalysts were suspended in 5 mL aqueous glycerol, and the mixtures were heated up to 80 °C. During the reactions, oxygen pressure was maintained at 1 MPa. The products were analyzed by high performance liquid chromatography (HPLC).
Conclusions
In summary, this work developed a series of novel redox–acid–base trifunctional catalysts by connecting amine groups with POM, which exhibited high activity and selectivity in one-pot cascade reactions comprising sequential dehydrogenation oxidation, dehydration and hydration reactions. The reaction kinetics of every intermediate was investigated to confirm the functions of the three different active sites during the process. The activation energies of the three catalysts based on different substrates were also calculated using Arrhenius plots to illustrate the relationship between the three active sites and catalytic activity. All the three catalysts could be recycled at least 10 times without any significant loss. Other redox–acid–base trifunctional catalysts can be further designed in this way and can be used in various one-pot cascade reactions catalyzed by redox, acid and base catalysts, which might offer a general route for simple and green organic synthesis with high efficiencies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. 51578119]; the major projects of Jilin Provincial Science and Technology Department [20180414069GH]; and the “111” Program.Notes and references
- A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392 CrossRef CAS PubMed.
- L. L. Chang, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825 CrossRef PubMed.
- M. Tao, X. Yi, I. Delidovich, R. Palkovits, J. Shi and X. Wang, ChemSusChem, 2015, 8, 4195 CrossRef CAS PubMed.
- S. C. D'Angelo, A. Dall'Ara, C. Mondelli, J. Pérez-Ramírez and S. Papadokonstantakis, ACS Sustainable Chem. Eng., 2018, 6, 16563 CrossRef.
- G. M. Lari, C. Mondelli, S. Papadokonstantakis, M. Morales, K. Hungerbühler and J. Pérez-Ramírez, React. Chem. Eng., 2016, 1, 106 RSC.
- G. M. Lari, G. Pastore, M. Haus, Y. Ding, S. Papadokonstantakis, C. Mondelli and J. Pérez-Ramírez, Energy Environ. Sci., 2018, 1 Search PubMed.
- M. Morales, P. Y. Dapsens, I. Giovinazzo, J. Witte, C. Mondelli, S. Papadokonstantakis, K. Hungerbühler and J. Pérez-Ramírez, Energy Environ. Sci., 2015, 8, 558 RSC.
- P. F. H. Harmsen, M. M. Hackmann and H. L. Bos, Biofuels, Bioprod. Biorefin., 2014, 8, 306 CrossRef CAS.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082 CrossRef CAS PubMed.
- P. Gruber, D. E. Henton and J. Starr in Biorefineries-Industrial Processes and Products, Wiley-VCH, 2008, pp. 381–407 Search PubMed.
- J. Ftouni, N. Villandier, F. Auneau, M. Besson, L. Djakovitch and C. Pinel, Catal. Today, 2015, 257, 267 CrossRef CAS.
- Z. Jiang, Z. Zhang, T. Wu, P. Zhang, J. Song, C. Xie and B. Han, Chem. – Asian J., 2017, 12, 1598 CrossRef CAS PubMed.
- G. Y. Yang, Y. H. KeandH and F. Ren, Chem. Eng. J., 2016, 283, 759 CrossRef CAS.
- R. K. P. Purushothaman, J. Haveren, D. S. Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, Appl. Catal., B, 2014, 147, 92 CrossRef CAS.
- F. Zhang, H. Jiang, X. Li, X. Wu and H. Li, ACS Catal., 2014, 4, 394 CrossRef CAS.
- M. C. Ávila, N. A. Comelli, E. Rodríguez-Castellón, A. Jiménez-López, R. C. Flores, E. N. Ponzi and M. I. Ponzi, J. Mol. Catal. A: Chem., 2010, 322, 106 CrossRef.
- S. Liang, G. B. Hammond and B. Xu, Chem. Commun., 2015, 51, 903 RSC.
- X. Yang, J. Wang, Q. Zhang, X. Wang, L. Xu, H. Wu, X. Jiang and F. Chai, J. Nanomater., 2014, 22, 1 Search PubMed.
- A. E. S. Choi, S. Roces, N. Dugos and M. W. Wan, Sustainable Environ. Res., 2016, 26, 184 CrossRef CAS.
- M. Tao, D. Zhang, H. Guan, G. Huang and X. Wang, Sci. Rep., 2016, 6, 1 CrossRef PubMed.
- M. Tao, D. Zhang, X. Deng, X. Li, J. Shi and X. Wang, Chem. Commun., 2016, 52, 3332 RSC.
- K. Suzuki, M. Sugawa, Y. Kikukawa, K. Kamata, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2012, 51, 6953 CrossRef CAS PubMed.
- M. J. Zhang, P. P. Zhao, Y. Leng, G. J. Chen, J. Wang and J. Huang, Chem. – Eur. J., 2012, 18, 12773 CrossRef CAS PubMed.
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., 2011, 123, 9789 CrossRef.
- P. Liu, C. H. Wang and C. Li, J. Catal., 2009, 262, 159 CrossRef CAS.
- Y. Jeon, W. S. Chi, J. Hwang, D. H. Kim, J. H. Kim and Y. G. Shul, Appl. Catal., B, 2019, 242, 51 CrossRef CAS.
- Q. Zhao, Z. Sun, S. Wang, G. Huang, X. Wang and Z. Jiang, RSC Adv., 2014, 4, 63055 RSC.
- H. Qu, Y. Zhou, Y. Ma, P. Zhao, B. Gao, M. Guo and C. Feng, J. Taiwan Inst. Chem. Eng., 2018, 93, 667 CrossRef CAS.
- Q. Zhao, H. Wang, H. Zheng, Z. Sun, W. Shi, S. Wang and X. Wang, Catal. Sci. Technol., 2013, 3, 2204 RSC.
- M. Tao, N. Sun, Y. Li, T. Tong, M. Wielicako, S. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 8325 RSC.
- M. Tao, Y. Li, Y. V. Geletii, C. L. Hill and X. Wang, Appl. Catal., A, 2019, 579, 52 CrossRef CAS.
- M. Tao, Y. Li, X. Zhang, Z. Li, C. L. Hill and X. Wang, ChemSusChem, 2019, 12, 1 CrossRef.
- S. Zhu, X. Gao, F. Dong, Y. Zhu, H. Zheng and Y. Li, J. Catal., 2013, 306, 155 CrossRef CAS.
- R. Kourieh, S. Bennici, M. Marzo, A. Gervasini and A. Auroux, Catal. Commun., 2012, 19, 119 CrossRef CAS.
- I. Milosev, J. Pavlinac, M. Hodoscek and A. Lesar, J. Serb. Chem. Soc., 2013, 78, 2069 CrossRef CAS.
- K. Narasimharao, D. R. Brown, A. F. Lee, A. D. Newman, P. F. Siril, S. J. Tavener and K. Wilson, J. Catal., 2007, 248, 226 CrossRef CAS.
- S. X. Xia, R. F. Nie, X. Y. Lu, L. N. Wang, P. Chen and Z. Y. Hou, J. Catal., 2012, 296, 1 CrossRef CAS.
- T. Komanoya, A. Suzuki, K. Nakajima, M. Kitano, K. Kamata and M. Hara, ChemCatChem, 2016, 8, 1094 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01851d |
| This journal is © The Royal Society of Chemistry 2020 |