Dual catalyst system for selective vinyl chloride production via ethene oxychlorination†
Received
5th September 2019
, Accepted 7th December 2019
First published on 11th December 2019
Abstract
A dual system comprising two catalytic reactors connected in series was developed for the direct conversion of ethene to vinyl chloride monomer (VCM). The first reactor uses ZrO2-supported ceria (CeO2/ZrO2) to perform ethene oxychlorination to 1,2-dichloroethene (EDC) that is dehydrochlorinated to VCM in the second reactor over calcium-promoted γ-Al2O3. The choice of carrier for ceria is of critical importance to maximize the EDC production by reducing combustion products. While MgO, SiO2, SiC, TiO2, ZSM-5, and γ-Al2O3 carriers induced higher overoxidation compared to bulk ceria, ZrO2 was the only carrier that suppressed COx formation. Moreover, the latter carrier led to the highest oxychlorination activity. The unique performance of the CeO2/ZrO2 catalyst was rationalized by its ability to promote chlorine evolution and to suppress the combustion of chlorinated products, as inferred from the activity evaluation in HCl and VCM oxidation, respectively. The outstanding redox properties, enabling operation at low temperature and thus high selectivity, are associated with the formation of defective CeO2 nanoparticles, contrasting the low activity over Ce–Zr mixed oxide. In order to subsequently form VCM, an efficient EDC dehydrochlorination catalyst was designed by moderating the acidity of γ-Al2O3via calcium doping and used in a reactor after CeO2/ZrO2. This dual catalyst system displayed 100% selectivity to VCM at 25% ethene conversion, surpassing the space time yield of the best ethene-to-VCM catalyst EuOCl by a factor of four, where the first step is operated at an elevated temperature of about 100–150 K with respect to cupric chloride benchmarks. In addition, the catalytic dehydrochlorination, operated at a lower temperature of 100 K than the current non-catalytic process, showed minimized coke formation. This developed system rendered stable after slight initial deactivation, offering promising potential to intensify VCM production.
Introduction
Ethene oxychlorination, i.e., the reaction of a former alkene with HCl and oxygen, constitutes an important step of the so-called balanced technology for the production of vinyl chloride monomer (VCM) that is the key precursor in the manufacture of polyvinyl chloride (PVC).1,2 Since its commercialization in the late 1950s,1–3 the oxychlorination process has been industrially practiced over catalysts comprising an active cupric chloride (CuCl2) phase supported on γ-alumina carriers and promoted by (oxy)chlorides of alkali or rare-earth metals as these materials provide high activity and selectivity to 1,2-dichloroethane (EDC, C2H4Cl2),1,2,4–11 which is subsequently dehydrochlorinated to VCM in a separate non-catalytic step.1,2 Nonetheless, cupric chloride species display an inherently low melting point.1,2,6 In combination with the high exothermicity of the oxychlorination reaction, this factor can cause particle stickiness in fluidized bed reactors, or gradual loss of the active phase due to volatilization, linked to hot-spot formation in fixed-bed reactors. Efforts to remediate both these effects by the addition of promoters were only partially effective. This limited stability of CuCl2-based catalysts triggered a search for alternative active phases that could overcome this challenge while still exhibiting exceptional activity and selectivity for ethene oxychlorination. The quest was additionally motivated by the recent progress in the development of stable non-copper catalytic systems involving RuO2- and CeO2-based catalysts that are able to preserve the structural integrity under the similarly hash reaction environment of HCl oxidation (Deacon reaction).12,13 Notably, the evaluation of bulk RuO2 and CeO2 in ethene oxychlorination revealed that in contrast to the marginal productivity of EDC and VCM over RuO2 due to preferential overoxidation into carbon oxides (COx), CeO2 displays a substantial selectivity to these products (80–90%) and activity levels that are comparable to those of benchmark CuCl2-based catalysts.14 Moreover, in addition to its stable catalytic performance, CeO2 exhibited one key feature – the ability to directly convert significant amounts of ethene to VCM. The latter catalytic property of CeO2 offers attractive potential to advance the current VCM production technology as it could eliminate the need for the second EDC-to-VCM step that is conducted at high temperatures and therefore represents one of the most energy-intensive unit operations in the process. In fact, a decrease of every 10 K of the operating temperature of EDC dehydrochlorination leads to a heat saving of 0.095 GJ tVCM−1.15 In a typical PVC plant, reducing the operating temperature by 100 K would thus amount to attractive annual savings of about 4.5 million USD. Nonetheless, the application of bulk CeO2 as an ethene oxychlorination catalyst is hampered by the (i) still significant production of COx and (ii) limited EDC-to-VCM conversion capacity. In addition, detailed mechanistic studies on CeO2 revealed intrinsic material properties that cause the formation of undesired by-products, such as dichloroethane (DCE).16 Recently, we have shown that another lanthanide-based catalyst, EuOCl, could overcome these barriers.17 However, the three orders of magnitude higher costs of europium compared to those of ceria diminish the chances for its potential commercial success.
Supporting of CeO2 on appropriate carriers is an attractive,18 yet unexplored strategy to obtain the desired product distribution in ethene oxychlorination. As demonstrated in previous studies, carriers can modify the redox properties of CeO2via geometric and electronic effects, thus offering the possibility to curb combustion.18 In addition, similar to the beneficial effects that ceria provides when used as a carrier,19 appropriate carriers for CeO2 could promote the EDC-to-VCM conversion step by introducing acid functionalities that are required for this reaction.14,16,17,20–25 In fact, as reported in the literature, several acid materials, such as Al2O3 or Pt–Cu/SiO2, are active in EDC dehydrochlorination.20,26 If applied as active carriers or co-catalysts for CeO2, these materials can allow integration of oxychlorination and dehydrochlorination reactions in a single catalytic system. Nonetheless, existing dehydrochlorination catalysts exhibit limited VCM selectivity at high EDC conversion levels and a prominent propensity for deactivation due to coking.20,26
This study aims at developing CeO2-based catalytic systems for highly selective direct VCM production via ethene oxychlorination. In the first step, CeO2 was nanostructured over a broad range of carriers in order to suppress COx formation and to determine the potential of different carriers to promote EDC dehydrohalogenation. Hereby, it is shown that overoxidation can be completely avoided only by supporting CeO2 over a ZrO2 carrier that, however, leads to a marginal EDC-to-VCM conversion. To foster the latter step, a calcium doped-γ-Al2O3 co-catalyst that exhibits a combination of high activity and unprecedented selectivity to VCM in EDC dehydrochlorination with a low propensity for coking was developed. Finally, a dual-bed reactor system that comprises CeO2/ZrO2 and calcium doped-γ-Al2O3 catalysts was designed enabling direct ethene to VCM conversion via oxychlorination with absolute selectivity to this polymer precursor, outperforming the best reported catalyst to date, EuOCl, by a factor of four in terms of space time yield.
Experimental
Catalyst preparation
Bulk CeO2 (Sigma-Aldrich, nanopowder, 99.5%) and carriers, MgO (∼20 nm, Strem Chemicals, ≥99%), SiO2 (20–60 μm, Evonik, AEROPERL 300/30, ≥99.0%), SiC (<100 nm, Sigma-Aldrich, ≥99%), TiO2 (≤70 μm, Sigma-Aldrich, ≥99%), ZrO2 (≤60 μm, Alfa-Aesar, 99%), γ-Al2O3 (5–100 μm, Sasol, PURALOX SCFa 140, ≥98%), ZSM-5-15 (Zeolyst, CBV3024E), ZSM-5-40 (Zeolyst, CBV8014), ZSM-5-140 (Zeolyst, CBV28014), ZSM-5-1040 (Tosoh, HSZ-890HOA), FER-29 (Zeochem, CP905), Y-40 (Zeolyst, CBV780), and MOR-110 (Tosoh, HSZ-690HOA), were calcined prior to their use in catalytic tests or in catalyst preparation. Dry impregnation was used as a standard method for catalyst synthesis. Herein, an appropriate amount of Ce(NO3)3·6H2O (Aldrich, 99.9%) was dissolved in a volume of deionized water equal to the carrier pore volume to achieve the desired wt% loading of Ce. The precursor solutions were then added dropwise to the carrier, which was stored under vacuum (50 mbar) at 373 K. The wetted materials were periodically mixed at room temperature for 1 h, then dried and finally calcined. The resulting catalysts are denoted as CeO2-x/C-SA-T, where x denotes the Ce loading, C the carrier, SA the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratio if C is a zeolite, and T the calcination temperature. The standard loading of 10 wt% and calcination temperature (T = 873 K) are omitted for clarity only specified in the case of deviations, where 5 wt% and 20 wt% of Ce are indicated by letters l (low) and h (high), respectively. Promoted alumina catalysts were prepared following the same dry impregnation protocol as described for the preparation of supported CeO2 catalysts, using Ce(NO3)3·6H2O, KNO3·6H2O (Aldrich, 99.9%), La(NO3)3·6H2O (Aldrich, 99.9%), and Ca(NO3)2·6H2O (Aldrich, 99.9%) as precursors. Drying was performed under vacuum (50 mbar) at 373 K for 2 h, while calcination was performed in static air at 873 K for 5 h using a heating rate of 5 K min−1. The resulting catalysts are denoted as M-x/Al2O3, where M denotes the metal promoter and x its loading. The standard promoter loading was 2 wt% and is omitted in the catalyst naming, while the reduced promoter loading of 0.5 wt% is indicated by letter l (low).
:Al ratio if C is a zeolite, and T the calcination temperature. The standard loading of 10 wt% and calcination temperature (T = 873 K) are omitted for clarity only specified in the case of deviations, where 5 wt% and 20 wt% of Ce are indicated by letters l (low) and h (high), respectively. Promoted alumina catalysts were prepared following the same dry impregnation protocol as described for the preparation of supported CeO2 catalysts, using Ce(NO3)3·6H2O, KNO3·6H2O (Aldrich, 99.9%), La(NO3)3·6H2O (Aldrich, 99.9%), and Ca(NO3)2·6H2O (Aldrich, 99.9%) as precursors. Drying was performed under vacuum (50 mbar) at 373 K for 2 h, while calcination was performed in static air at 873 K for 5 h using a heating rate of 5 K min−1. The resulting catalysts are denoted as M-x/Al2O3, where M denotes the metal promoter and x its loading. The standard promoter loading was 2 wt% and is omitted in the catalyst naming, while the reduced promoter loading of 0.5 wt% is indicated by letter l (low).
Catalyst characterisation
Powder X-ray diffraction (XRD) was measured using a PANalytical X'Pert PRO-MPD diffractometer with the Bragg–Brentano geometry by applying Cu Kα radiation (λ = 1.54060 Å). The data were recorded in the 10–70° 2θ range with an angular step size of 0.017° and a counting time of 2.04 s per step. The average size of CeO2 crystallites was estimated from the broadening of the (111) reflection of CeO2 by using the Scherrer equation with a dimensionless shape factor of K = 0.9. High-resolution transmission electron microscopy (HRTEM), high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), and energy dispersive X-ray (EDX) spectroscopy were conducted on an FEI Talos microscope operated at 200 kV. All the samples were dispersed as dry powders onto lacey carbon coated nickel grids. Over 200 CeO2 nanoparticles were examined to estimate their average size, using the ImageJ software. Raman spectroscopy was performed on a WITec CRM200 confocal Raman system using a 532 nm laser with a 20 mW power, a 100× objective lens with a numerical aperture NA = 0.9 (Nikon Plan), and a fiber-coupled grating spectrometer (2400 lines per mm), giving a spectral sampling resolution of 0.7 cm−1. N2 sorption at 77 K was measured using a Micromeritics TriStar analyser. Prior to the measurements, the samples were degassed to 50 mbar at 573 K for 12 h. The Brunauer–Emmett–Teller (BET) method was applied to calculate the total surface area, SBET, in m2 g−1. Temperature-programmed desorption of ammonia (NH3-TPD) and temperature-programmed reduction with hydrogen (H2-TPR) were performed using a Micromeritics Autochem II 2920 unit equipped with a thermal conductivity detector coupled to a Pfeiffer Vacuum OmniStar mass spectrometer. The powder sample (0.1 g) was loaded into a U-shaped quartz micro-reactor, pre-treated in He (20 cm3 STP min−1) at 573 K for 3 h, and cooled to 373 K in He. For NH3-TPD experiments, ammonia was chemisorbed at 473 K in three consecutive cycles of saturation with 5 vol.% NH3/He (20 cm3 STP min−1) for 30 min followed by purging with He (20 cm3 STP min−1) at the same temperature for 30 min. Desorption of NH3 was monitored in the range of 473–1273 K using a heating rate of 20 K min−1 and a He flow of 20 cm3 STP min−1. For H2-TPR experiments, the sample was pre-treated in He (20 cm3 STP min−1) at 423 K for 1 h, and cooled to room temperature followed by ramping the temperature at 10 K min−1 up to 1273 K in 5 vol% H2 (20 cm3 STP min−1). The resulting profiles of NH3-TPD and H2-TPR were normalised to the sample weight. Thermogravimetric analysis (TGA) to determine the amount of coking was performed on a Linseis STA-PT-1600 apparatus. The sample (30 mg) was loaded on an alumina pan and pretreated in Ar (100 cm3 STP min−1) at 373 K for 1 h, then the temperature was ramped up to 1173 K with a heating rate of 10 K min−1 under a flow of synthetic air (100 cm3 STP min−1). X-ray photoelectron spectroscopy (XPS) measurements were performed on a Physical Electronics Quantum 2000 XPS using monochromatic Al-Kα radiation generated from an electron beam operated at 15 kV, and equipped with a hemispherical capacitor electron-energy analyser. The powdered sample was firmly pressed onto indium foil. The area analysed was 150 μm in diameter and the electron take-off angle was 45°. The pass energy used for the detailed spectra of the Ce 3d core levels was 46.95 eV to yield a total analyser energy resolution of 0.95 eV. The spectrometer energy scale was calibrated for the Au 4f electrons to be at 84.0 ± 0.1 eV. Partial compensation of surface charging during spectra acquisition was obtained by the simultaneous operation of electron and argon ion neutralizers. Extended X-ray absorption fine structure spectroscopy (EXAFS) and X-ray absorption near-edge structure spectroscopy (XANES) measurements were performed at the Ce L3-edge on the XAFCA beamline at the Singapore Synchrotron Light Source (SSLS).27 The storage ring of the SSLS was operated at 0.7 GeV with a maximum current of 200 mA. The data were collected in transmission mode (to kmax = 10.5) using a Si(111) double crystal monochromator and ion chamber detectors. To ascertain the reproducibility of the experimental data, at least three scan sets were collected. EXAFS and XANES data processing was performed using the Demeter software package (Athena and Artemis).28 The amplitude reduction factor, S02 was derived from the EXAFS data analysis of the commercial CeO2 standard.
Catalyst testing
Steady-state oxychlorination of ethene was investigated at ambient pressure in a continuous-flow fixed-bed reactor (Scheme S1†). The set-up consists of (i) digital mass flow controllers (Bronkhorst®) to dose C2H4 (PanGas, 3.5), HCl (Air Liquide, 2.8, anhydrous), O2 (Messer, 19.96% in He), He (PanGas, 5.0) as a carrier gas, and Ar (PanGas, 5.0) as an internal standard at a total volumetric flow, FT, of 100 cm3 STP min−1, (ii) a syringe pump (Nexus 6000, Chemyx) to feed EDC (Fluka, 99.5%), (iii) a vaporiser operated at 403 K, accommodating a quartz T-connector filled with glass beads to vaporise EDC, (iv) an electrically heated oven hosting a quartz micro-reactor equipped with a K-type thermocouple whose tip reaches the centre of the catalyst bed, (v) downstream heat-tracing to avoid any condensation of the reactants and products, and (vi) a gas chromatograph coupled to a mass spectrometer (GC-MS) for on-line analysis. The effluent stream was neutralised by passing it through an impinging bottle containing an aqueous NaOH solution (1 M). The catalyst (Wcat = 0.5 g, particle size dp = 0.4–0.6 mm) was loaded in the micro-reactor (10 mm inner diameter) and pre-treated in He at 473 K for 30 min. Prior to the analysis of the reaction mixtures, the catalysts were equilibrated for at least 1 h under each condition. Carbon-containing compounds in the effluent gas stream at the reactor outlet (C2H4, C2H3Cl, C2H4Cl2, CO, and CO2) and Ar were quantified using an on-line gas-chromatograph, equipped with a GS-Carbon PLOT column, which was coupled to a mass spectrometer (Agilent GC 7890B, Agilent MSD 5977A) with a triple-axis detector and an electron multiplier. The conversion of reactant i, Xi, (i: C2H4, C2H3Cl, and C2H4Cl2) was calculated using eqn (1),| | 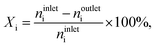 | (1) |
where ninleti and noutleti are the molar flows of reactant i at the inlet and outlet of the reactor, respectively. The selectivity to gaseous products, Sj, and yield, Yj, of product j (j: C2H4, C2H4Cl2, C2H3Cl, CO, and CO2) were determined according to eqn (2) and (3),| | 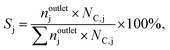 | (2) |
| | 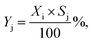 | (3) |
where noutletj is the molar flow of product j at the reactor outlet. NC,j is the number of carbon atoms in compound j. The error of the carbon balance, εC, was determined using eqn (4),| | 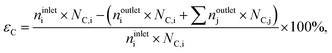 | (4) |
where NC,i/j is the number of carbon atoms in compound i or j, which was less than 5% in all experiments, i.e., the carbon mass balance reached ≥95%. Coke was not considered a product, and therefore not included in the standard selectivity calculation. However, the amount of coking that a catalyst experiences is expressed as selectivity as the ratio of the rate of coke formation in the catalyst to the rate of ethene feed. In the HCl oxidation tests, Cl2 production was quantified using a Mettler Toledo G20 compact titrator by off-line iodometric titration of triiodide, which is formed by purging a Cl2-containing reactor outlet through a 0.10 M aqueous KI solution (Sigma-Aldrich, 99.5%), with a 0.01 M sodium thiosulfate solution (Sigma-Aldrich, 99.99%). The conversion of HCl in HCl oxidation experiments, X(HCl), was calculated using eqn (5):| | 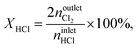 | (5) |
where nintletHCl and 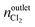 denote the molar flow of HCl and Cl2 at the reactor inlet and outlet, respectively. Calculation of the Weisz–Prater and Mears criteria indicated that all catalytic tests were performed in the absence of heat and mass transport limitations. After the tests, the reactor was quenched to room temperature in a He flow.
denote the molar flow of HCl and Cl2 at the reactor inlet and outlet, respectively. Calculation of the Weisz–Prater and Mears criteria indicated that all catalytic tests were performed in the absence of heat and mass transport limitations. After the tests, the reactor was quenched to room temperature in a He flow.
Results and discussion
Supported ceria catalysts for selective oxychlorination
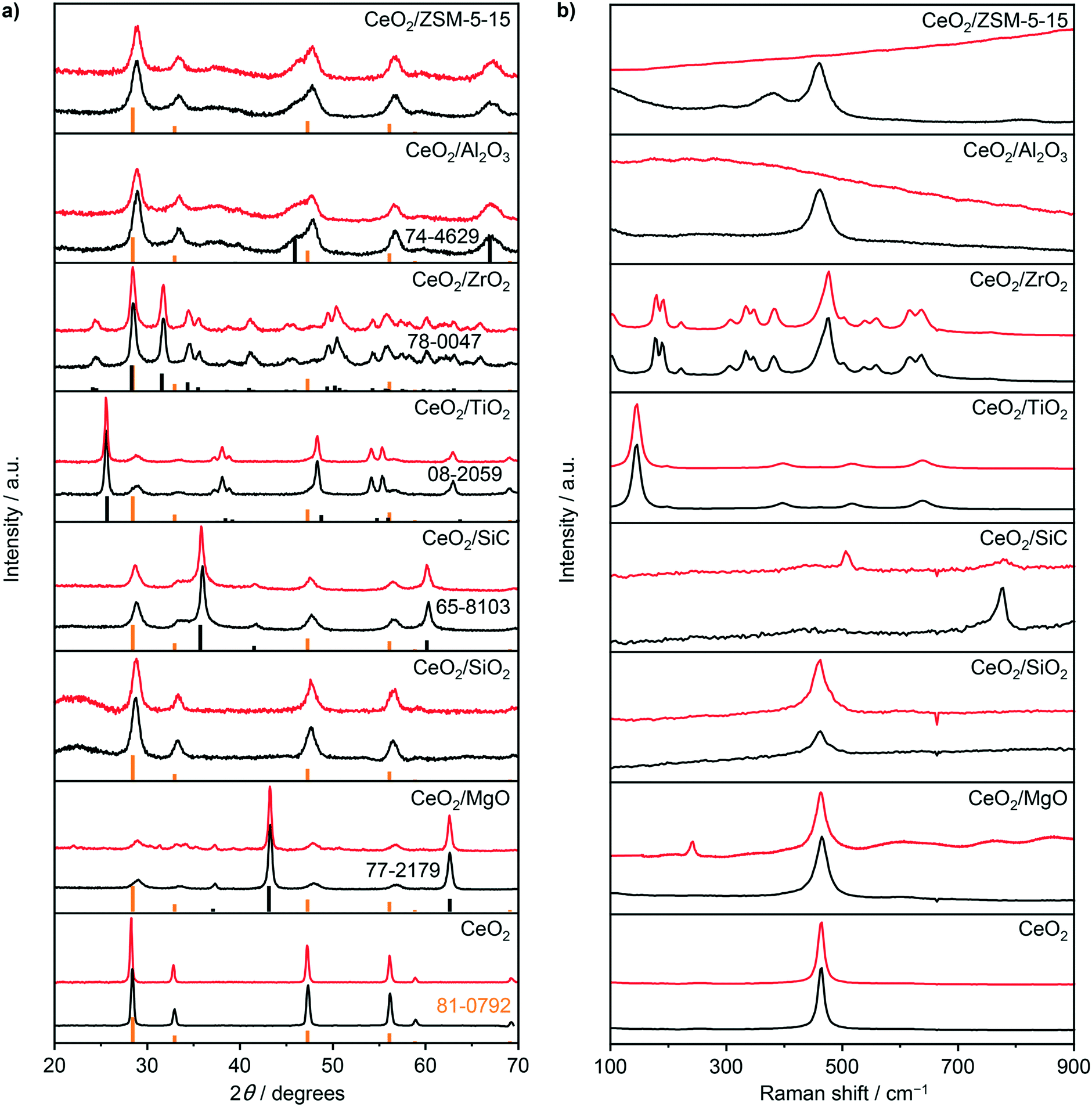
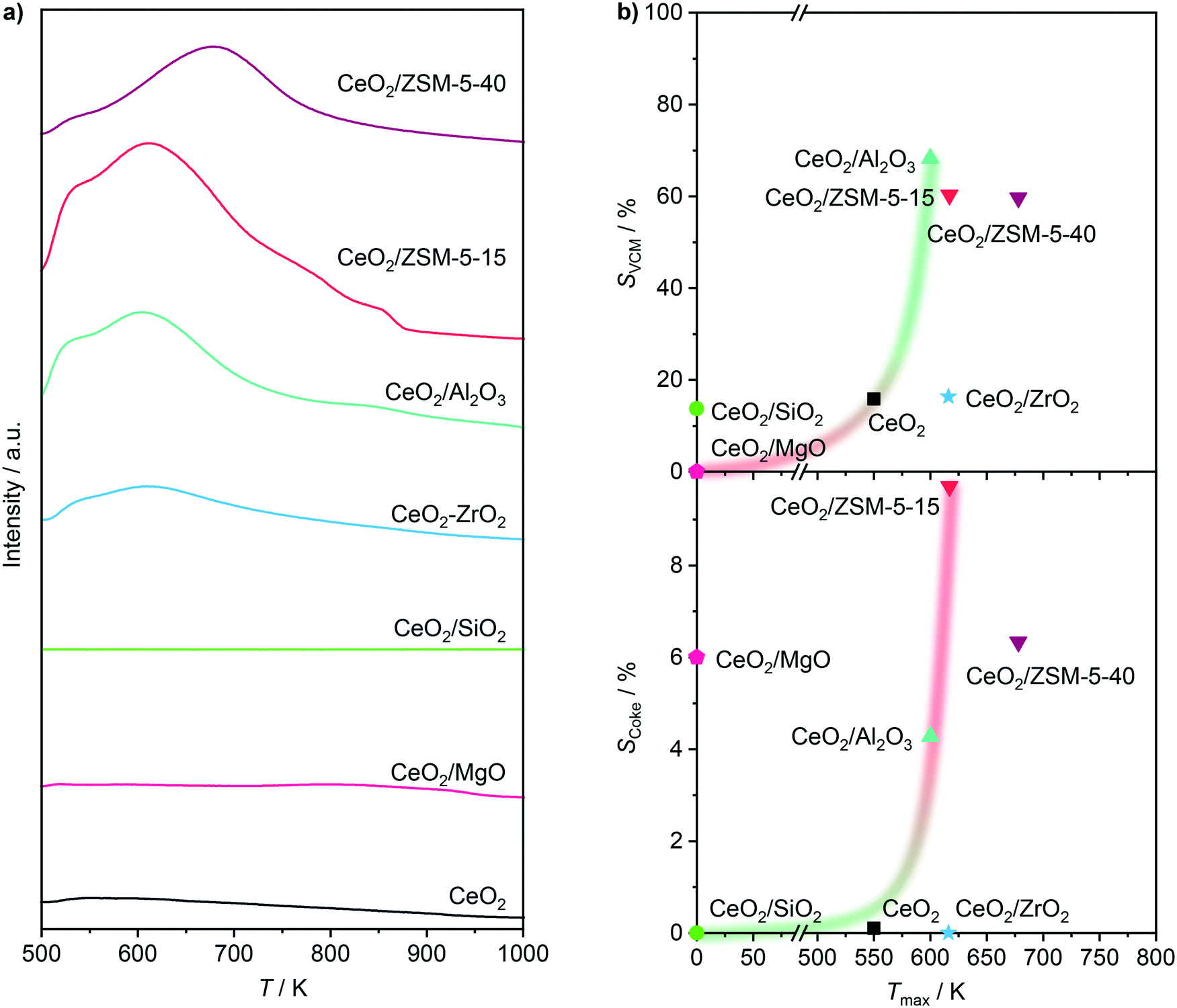
To provide a comprehensive assessment of the carrier impact on the oxychlorination performance of ceria, a range of supported catalysts with a nominal cerium loading of 10 wt% was prepared including CeO2/MgO, CeO2/SiO2, CeO2/SiC, CeO2/TiO2, CeO2/ZrO2, CeO2/Al2O3, and CeO2/ZSM-5-15. X-ray diffraction (XRD) analysis of the catalysts evidenced the ceria dispersion in the form of nanoparticles (Fig. 1a), exhibiting a size range of 5–16 nm (Table 1). The only exception was CeO2/ZrO2 which displayed no characteristic diffraction lines of the CeO2 phase. Consistent with the XRD patterns, the Raman spectra of fresh CeO2, CeO2/MgO, CeO2/SiO2, CeO2/Al2O3, and CeO2/ZSM-5-15 (Fig. 1b) exhibited a pronounced peak at 461–464 cm−1, which corresponds to the F2g mode of the fluorite CeO2,29–32 while the spectra of CeO2/ZrO2 exhibited only contributions that are ascribed to monoclinic ZrO2,33 further corroborating high Ce dispersion. Notably, the spectra of fresh CeO2/SiC and CeO2/TiO2 show only the peaks that can be ascribed to the Si–C bonds (ca. 796 cm−1) and TiO2-anatase phase,34,35 with no observable CeO2 contribution. Considering the prominent diffraction lines of CeO2 that were observed in these two samples, the absence of the characteristic active phase vibrations in the spectra of these samples can be rationalised by their significantly weaker Raman intensity as compared to their respective carriers.
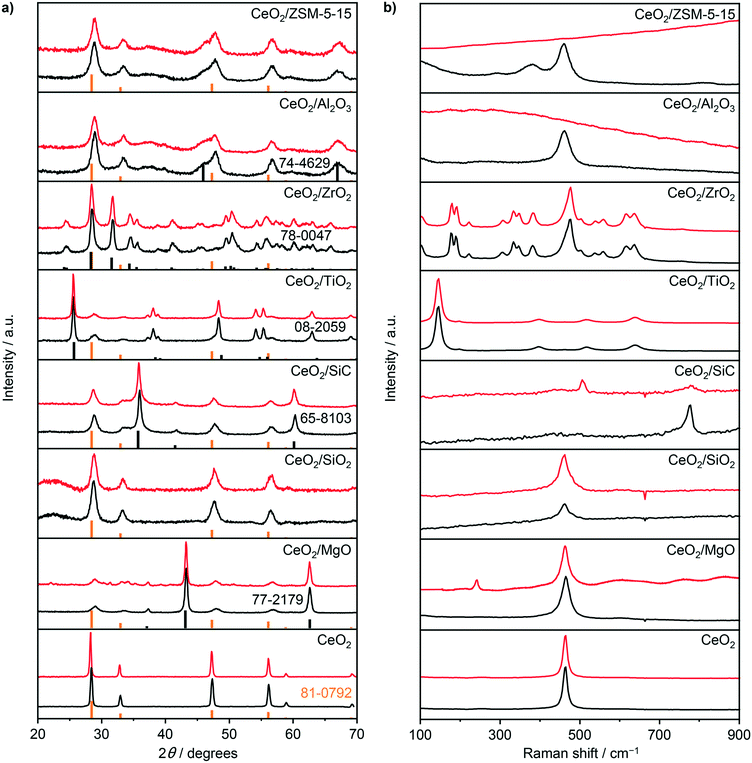 |
| | Fig. 1 a) XRD patterns and b) Raman spectra of bulk and supported CeO2 catalysts prior to (black) and after use (red). Reference patterns in a) are shown below the measured diffractograms in orange (CeO2, ICDD-PDF 81-0792) and black (supports, ICDD-PDF numbers are shown correspondingly). | |
Table 1 Particle size, surface area, and pore volume of fresh and used supported catalysts with 10 wt% Ce loading and bulk ceria as the reference material
| Catalyst |
Particle sizea/nm |
S
BET
/m2 g−1 |
V
pore
/cm3 g−1 |
| Fresh |
Used |
Fresh |
Used |
Fresh |
Used |
|
As determined by the Scherrer equation and for the selected materials, by microscopy on a particle sample size of 200 (in brackets).
As determined by N2 sorption.
|
| CeO2 |
|
|
42 |
39 |
0.08 |
0.11 |
| CeO2/MgO |
5 |
7 (5) |
44 |
14 |
0.16 |
0.06 |
| CeO2/SiO2 |
8 |
8 (7) |
185 |
187 |
0.68 |
0.68 |
| CeO2/SiC |
10 |
10 |
26 |
24 |
0.13 |
0.12 |
| CeO2/TiO2 |
5 |
6 (5) |
27 |
27 |
0.13 |
0.11 |
| CeO2/ZrO2 |
— |
— |
42 |
39 |
0.20 |
0.18 |
| CeO2/Al2O3 |
8 |
10 (12) |
207 |
225 |
0.71 |
0.69 |
| CeO2/ZSM-5-15 |
6 |
8 (16) |
291 |
179 |
0.12 |
0.09 |
To confirm the structural integrity of the investigated materials under the oxychlorination environment, all the catalyst samples were equilibrated in ethene oxychlorination at 673 K and FT = 100 cm3 STP min−1 gcat−1 for 4 h using an inlet feed composition as detailed in Table 2. Most of the equilibrated catalysts displayed similar textural and crystallographic properties as the corresponding fresh materials, indicating the absence of significant structural changes such as sintering or extensive chlorination of the ceria phase (Fig. 1, Table 1). Nonetheless, more prominent changes with respect to the fresh catalysts were observed in the Raman spectra of the used CeO2/SiC and CeO2/MgO catalysts, which displayed new peaks at around 512 and 240 cm−1, respectively (Fig. 1b). The former band can be ascribed to Mg–Cl bond vibrations, and the latter can be associated with the Si–Si bond vibration, suggesting the partial restructuring of these carriers.36,37 Moreover, the spectra of used CeO2/Al2O3, and CeO2/ZSM-5-15 exhibited strong fluorescence which is consistent with the pronounced propensity of these catalysts towards coking (vide infra). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to examine the differences in ceria dispersion between the equilibrated catalysts more closely (Fig. 2). The surface-averaged size of CeO2 nanoparticles showed a good agreement with those estimated by the Scherrer equation from the respective diffraction lines. However, in contrast to the low-surface area catalysts, CeO2/MgO (5 nm) and CeO2/TiO2 (5 nm), that comprise spherical-like ceria nanoparticles with uniform size and spatial distribution, higher-surface area materials, such as CeO2/SiO2 (7 nm), CeO2/Al2O3 (12 nm), and CeO2/ZSM-5-15 (16 nm), exhibited a broader range of particle sizes, which are distributed in the form of small clusters and larger agglomerates leading to a significant fraction of uncovered carrier areas. In addition, CeO2/ZSM-5-15 displays elongated nanoparticles, which is likely the reason for the more significant deviation between the particle size estimates between XRD and microscopy observed only for this sample. This broader particle size distribution can be attributed to the initial high dispersion of the cerium precursor (i.e. small particles) after impregnation. Depending on the affinity of CeO2 for the support and its mobility on the surface upon calcination, sintering of contacting particles leads to larger clusters or agglomerates, where smaller particles sinter more easily than larger particles,38,39 which results in a broad particle size distribution on the high surface area materials.
Table 2 Reactions and conditions used in this work
| Reaction |
T/K |
Concentrationa/vol% |
| C2H4 |
EDC |
VCM |
HCl |
O2 |
Arb |
Hec |
|
F
T/Wcat−1 = 200 cm3 STP min−1 gcat−1 unless indicated otherwise.
Internal standard.
Carrier gas.
|
| Oxychlorination |
623–723 |
3 |
0 |
0 |
4.8 |
3 |
3 |
86.2 |
| HCl oxidation |
523–723 |
0 |
0 |
0 |
3 |
3 |
3 |
91 |
| VCM oxidation |
523–723 |
0 |
0 |
1.5 |
0 |
3 |
3 |
92.5 |
| Dehydrochlorination |
523–723 |
0 |
1.5 |
0 |
4.8 |
3 |
3 |
87.7 |
 |
| | Fig. 2 HAADF-STEM micrographs of a) CeO2/MgO, b) CeO2/SiO2, c) CeO2/TiO2, d) CeO2/ZrO2, e) CeO2/Al2O3, and f) CeO2/ZSM-5-15 after use in ethene oxychlorination. The insets depict the corresponding particle size distribution of CeO2. | |
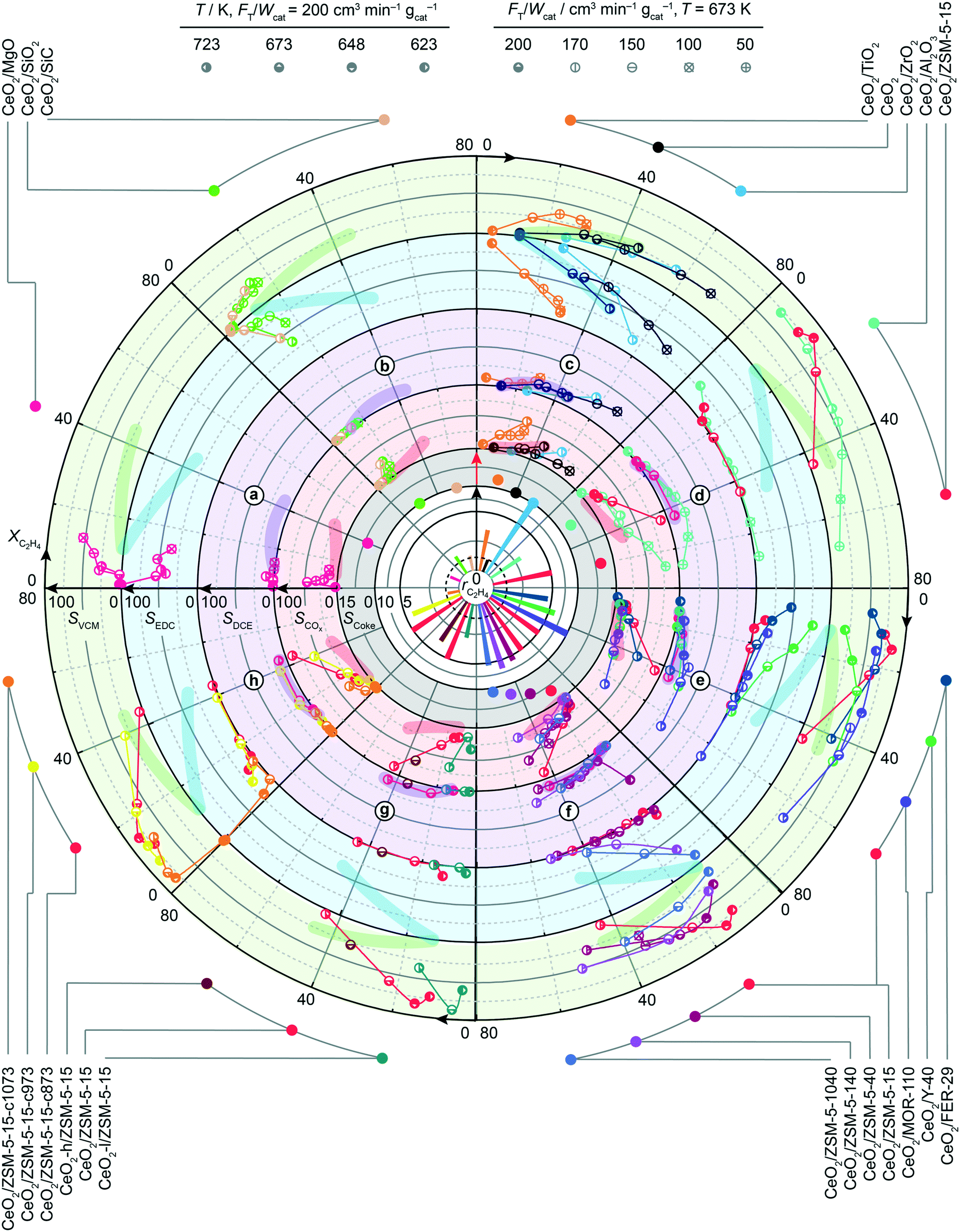
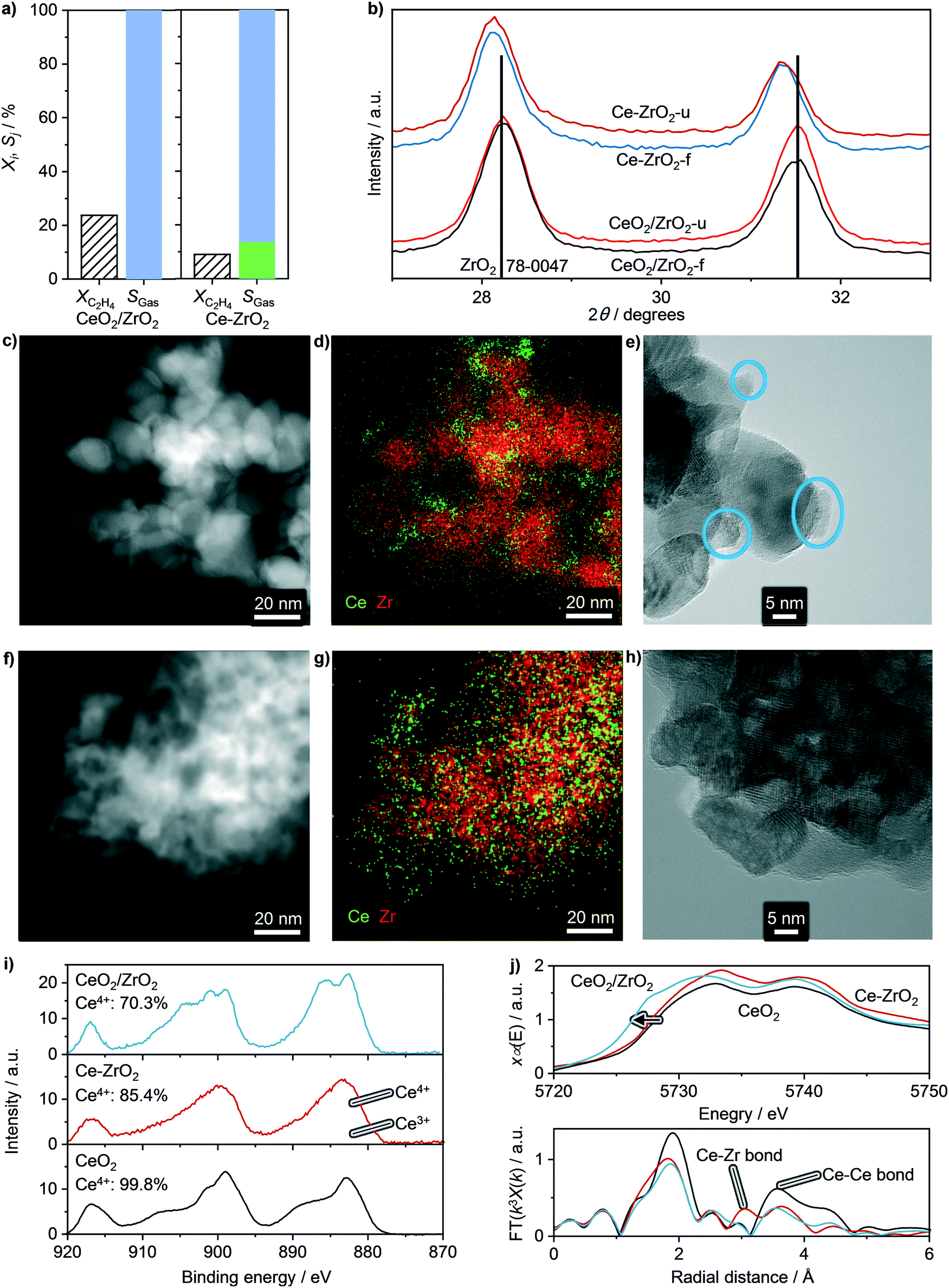
After assessing the structural stability of supported ceria catalysts, their oxychlorination performance was investigated in broader ranges of space velocities (50–200 cm3 STP min−1 gcat−1) and temperatures (623–723 K) that closely match the typical operating window of the state-of-the-art copper chloride-based materials.14 The absence of deactivation-induced measurement bias was ascertained by measuring the catalytic performance under fixed standard conditions (T = 673 K and FT = 200 cm3 STP min−1 gcat−1, feed composition as shown in Table 2) before and after the variations of temperature and space velocity, which showed identical ethene conversion levels and product selectivities. Examination of the recorded activity and selectivity patterns in the form of a circular plot allows unambiguous benchmarking with respect to the bulk ceria (highlighted regions) and simple comparison between the different classes of catalysts. Herein, the central white part of the plot presents the specific rates of ethene oxychlorination measured at constant oxychlorination feed composition (Table 2), temperature (673 K), and space velocity (200 cm3 STP min−1 gcat−1), while the grey region indicates the corresponding selectivity to coke, determined after 4 h on stream under standard conditions (vide supra). The four outer rings provide the information on the selectivity (radial direction) to different products, i.e., VCM, EDC, DCE, and COx as a function of ethene conversion that increases in a clockwise direction. Within the top half of the plot, the CeO2/MgO, CeO2/SiO2, CeO2/SiC, CeO2/TiO2, CeO2/ZrO2, CeO2/Al2O3, and CeO2/ZSM-5-15 catalysts are divided into four sections (a–d) based on their performance. The first catalyst section (a) is represented by CeO2/MgO, showing a lower activity than bulk ceria and high COx selectivity. The former could be attributed to the high degree of chlorination, or formation of chloride species, as evidenced by Raman spectroscopy. Furthermore, MgO can cover parts of small CeO2 nanoparticles, further reducing the active phase area,18 while coking is pronounced, likely linked to the basicity of MgO. In section (b), exhibiting slightly higher ethene oxychlorination rates, CeO2/SiC and in particular CeO2/SiO2 supported catalysts selectively yield EDC at conversion levels of up to 8%, while promoting combustion and VCM formation. The higher activity of CeO2/SiO2 could be linked to a difference in the surface area and CeO2 particle structure, even though the CeO2 particle size differs only marginally. However, in contrast to the MgO supported catalyst, CeO2/SiO2 exhibited no coking, as also evidenced by fully preserved pore volume after the reaction. In section (c), catalysts that mirror the performance of bulk CeO2, are shown along with this benchmark. The performance patterns of CeO2/TiO2 are shifted towards lower conversion levels, yet indicate higher VCM selectivity, at the cost of stronger combustion compared to bulk ceria. CeO2/ZrO2, on the other hand, exhibits no coking, decreases COx and DCE formation, and exceeds any other catalyst formulation in terms of activity, although the selectivity to VCM does not exceed 25%. The increased activity of CeO2/ZrO2 (Fig. 3 and 4a) however, allows operation at lower temperature, resulting in fully selective EDC formation at 25% ethene conversion. This might be attributed to a substantial CeO2–ZrO2 interaction, as shown in Fig. 1, where no CeO2 reflections could be distinguished. Considering the well-known propensity of CeO2 to form solid solutions with ZrO2, high resolution, XRD was performed on CeO2/ZrO2 and co-precipitated Ce–Zr mixed oxide (Ce–ZrO2) with 10 wt% nominal cerium loading to shed light on ceria dispersion in this sample (Fig. 4b). While the most prominent reflection of Ce–ZrO2 displays a clear shift with respect to the ZrO2 reference, the most prominent reflection of CeO2/ZrO2 coincides with the reference. This indicates the absence of substantial cerium inclusion in the ZrO2 lattice in the CeO2/ZrO2 catalyst. Consistently, in contrast to the model solid solution material Ce–ZrO2, EDX and HRTEM analyses revealed the presence of CeO2 particles in CeO2/ZrO2 (Fig. 4c–h). To determine the descriptors for the outstanding activity of CeO2/ZrO2 in comparison to the mixed oxide and bulk ceria, XPS analysis revealed that bulk CeO2 exhibits the highest Ce4+ fraction, followed by Ce–ZrO2, and CeO2/ZrO2 (Fig. 4i), which allows two considerations: first, under ultra-high vacuum conditions, the material that exhibits a lower Ce4+ fraction is more easily reducible, which explains why CeO2/ZrO2 is much more reactive than Ce–ZrO2 and bulk CeO2. Second, this good reducibility of CeO2/ZrO2 in comparison with Ce–ZrO2 could arise from the fact that nanoparticle systems exhibit a considerable number of defects, in comparison to the Ce–Zr mixed oxide. The latter hypothesis was further supported by XANES and EXAFS analyses (Fig. 4j), where XANES evidenced a red-shift of the Ce L3 edge of CeO2/ZrO2 confirming more oxygen vacancies or defects in the surface, while EXAFS showed distinct characteristics of the Ce–O paths. Even though the errors are not negligible, the Ce–ZrO2 mixed oxide exhibits the highest number of neighbouring atoms, followed by bulk CeO2 and CeO2/ZrO2 (Table 3), which indicates that in particular, CeO2/ZrO2 exhibits strongly defective CeO2 nanoparticles, in contrast to the mixed oxide, which shows an even higher number of neighbouring atoms than the reference value (8). In addition, CeO2/ZrO2 exhibits a slightly decreased Ce–O distance (2.31 Å) in comparison to the used bulk CeO2 (2.34 Å) due to the strongly defective surface, while the mixed oxide shows even lower Ce–O distances (2.27 Å), most likely related to the accommodation of additional atoms in the lattice. This incorporation of Ce into the ZrO2 matrix is indicated by a peak in the EXAFS spectra, ascribed to the Ce–Zr scattering path. In addition, the average Zr–O bond length was found shorter than that of Ce–O, while in general, the metal–oxygen bond renders much shorter in the mixed oxide than in CeO2 or CeO2/ZrO2, suggesting again that CeO2/ZrO2 predominantly features CeO2 nanoparticles.
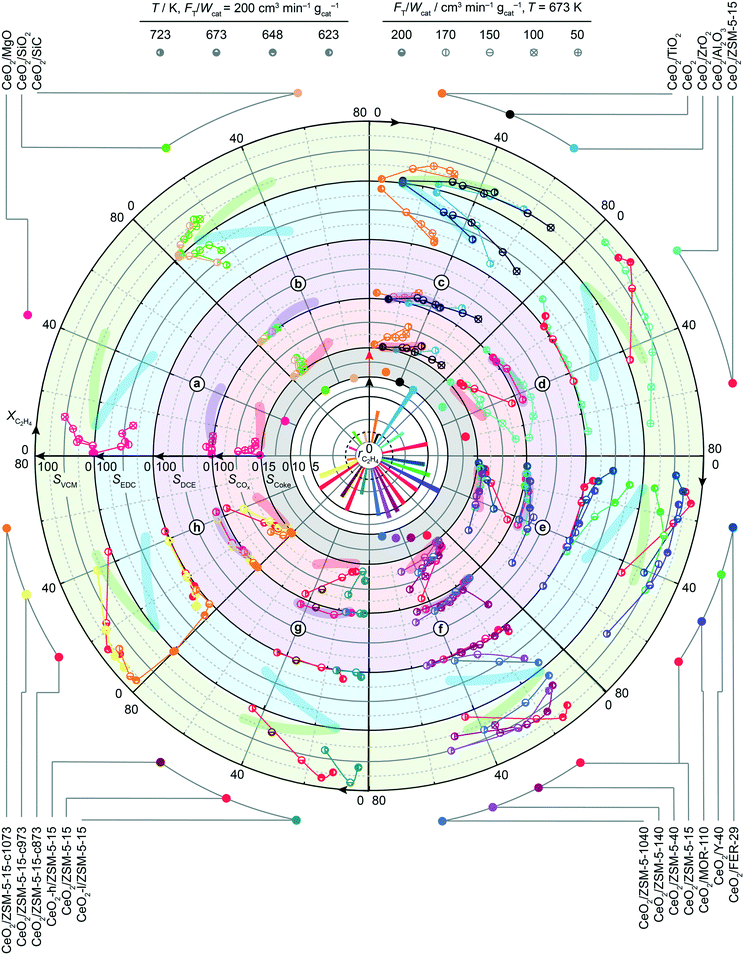 |
| | Fig. 3 Reaction rates (in mol h−1 molCe−1) and coke selectivity (in %) at 673 K, and product selectivities (in %) recorded as a function of ethene conversion (in %) for ceria catalysts supported over a)–d) different carriers, and e)–h) zeolite supports, with various framework types, Si:Al ratios, Ce-loading, and calcination temperatures, respectively. The catalysts are identified by a colour code, indicated on the outside of the circle, while catalytic data points were recorded using the conditions that are specified by the symbol interior, as indicated at the top. The performance of bulk CeO2 is used as a benchmark, indicated by the dashed circle and highlighted regions. Conditions are detailed in Table 2. | |
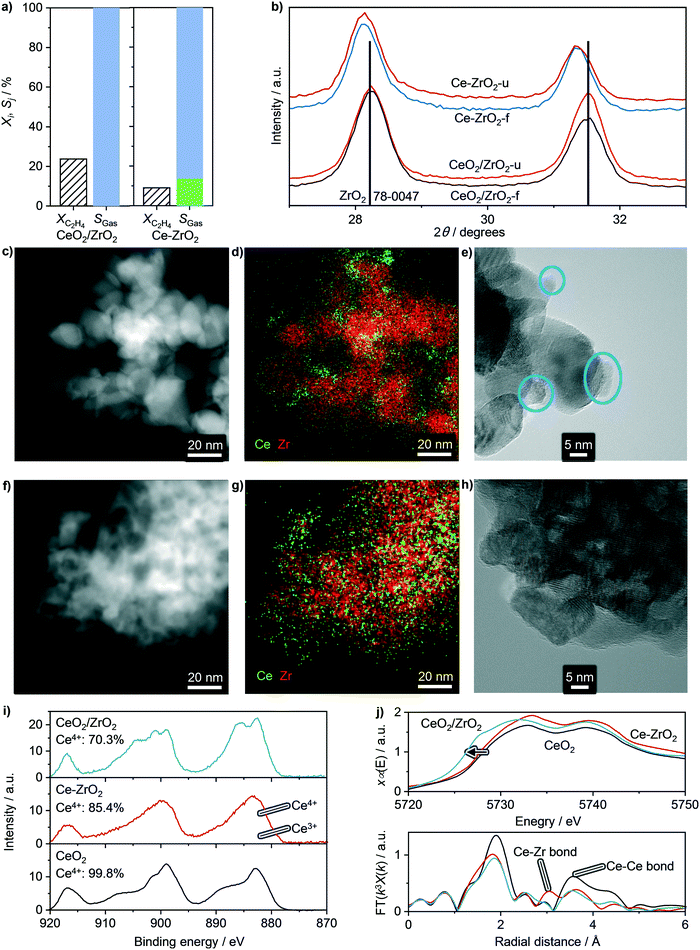 |
| | Fig. 4 a) Conversion and selectivity under standard reaction conditions and 623 K, b) XRD reflections, and c–h) HAADF-STEM, EDX mapping, and HRTEM results of CeO2/ZrO2 (c and d) and Ce–ZrO2 (f–h). i) XPS spectra (solid lines), spectral Ce4+ and Ce3+ fitting (shaded), and Ce4+ fraction and j) XANES (top, arrow indicates red-shift) and EXAFS (bottom) spectra of bulk CeO2, Ce–ZrO2, and CeO2/ZrO2. Conditions are detailed in Table 2. | |
Table 3 Number of neighbouring atoms (N), disorder in the neighbour distance (σ2), distance to neighbouring atoms (R), and R-factor, determined by EXAFS for the Ce–O path. The corresponding errors are reported in brackets
| Catalyst |
N/− |
σ
2/Å2 |
R/Å |
R-factor/− |
| CeO2 |
7.4 (1.6) |
0.0033 (0.0008) |
2.34 (0.02) |
0.018 |
| Ce–ZrO2 |
8.3 (2.1) |
0.0097 (0.0024) |
2.27 (0.02) |
0.020 |
| CeO2/ZrO2 |
5.4 (1.2) |
0.0041 (0.0008) |
2.31 (0.02) |
0.025 |
The fourth section (d) of the supported ceria catalysts in Fig. 3 is represented by CeO2/Al2O3, and CeO2/ZSM-5-15, which both exhibit an encouraging performance with respect to VCM selectivity (>75%, COx and EDC) at 10–20% conversion, while differences between the two materials arise outside this region. In contrast to the Al2O3 supported sample, the higher activity of CeO2/ZSM-5-15 is impaired by strongly reduced VCM selectivity at higher conversions. In addition, both materials form combustion products under all circumstances, in increased amounts at high temperature or low flow rates, and coking of the catalysts is considerable, especially for CeO2/ZSM5–15. However, the activity and selectivity patterns show no clear correlation with particle size, total surface area, pore volume (Table 1), or particle surface area (Fig. S1†). By aiming to better understand the observed activity and selectivity differences, we evaluated three distinct catalyst key descriptors: (i) activity in ethene oxychlorination, likely linked to the redox properties of the material, (ii) VCM selectivity, linked to the dehydrochlorination of EDC and material acidity, and (iii) propensity towards combustion, again linked to the redox properties (Fig. 5).
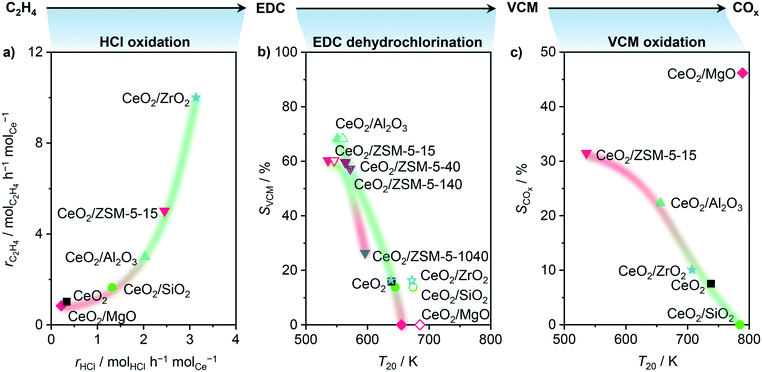 |
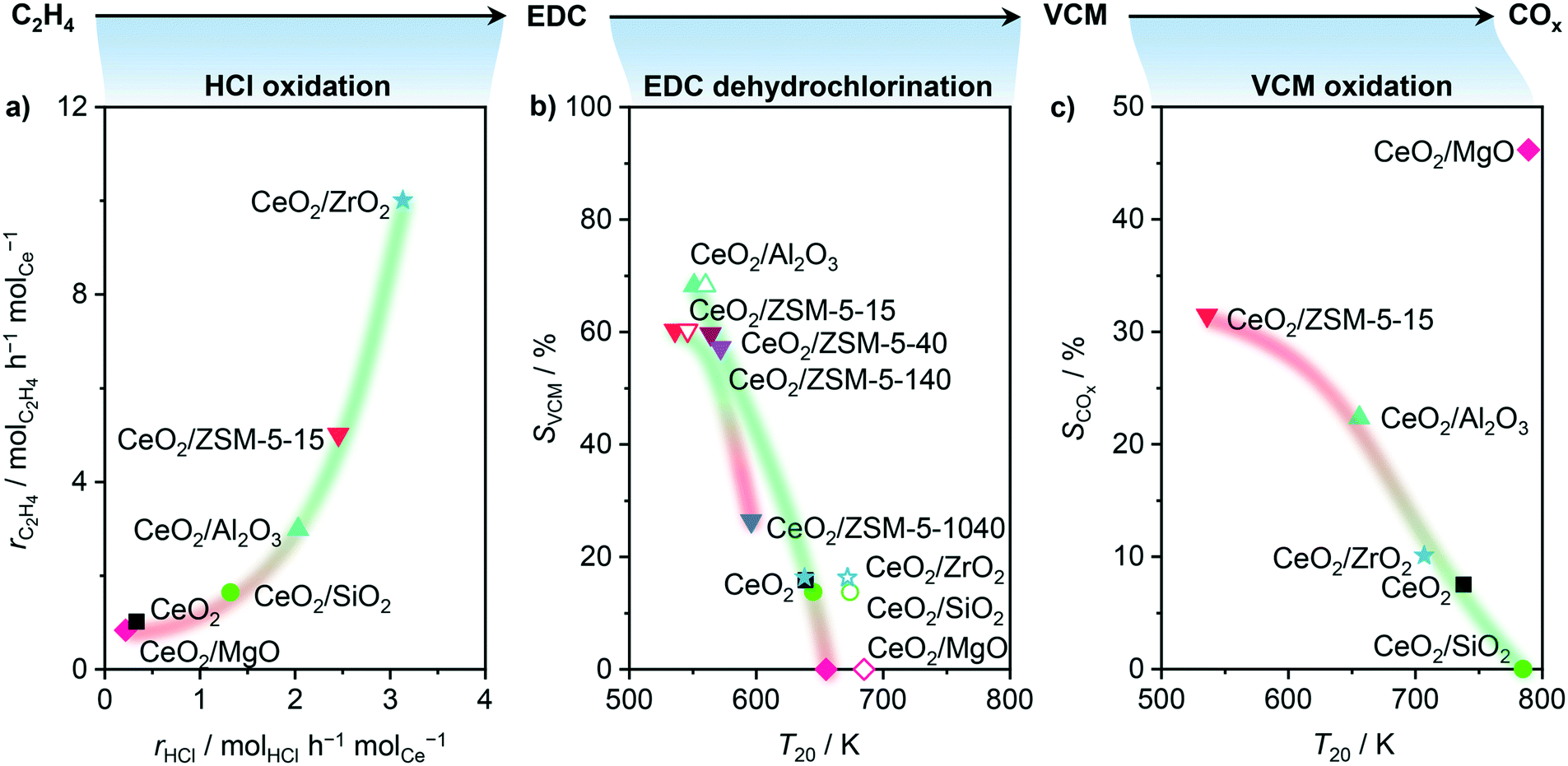
| | Fig. 5 Probe reactions: HCl oxidation, EDC dehydrochlorination, and VCM oxidation, rationalizing dedicated parts of the oxychlorination reaction, in particular activity, VCM selectivity, and propensity towards combustion, respectively. a) Rate of ethene consumption as a function of the rate of HCl oxidation at 673 K, and b) VCM selectivity in ethene oxychlorination at 673 K as a function of the temperature of 20% conversion in EDC dehydrochlorination over catalysts (solid symbols) and bulk supports (open symbols), where the corresponding VCM selectivity of the catalyst was taken into account. c) COx selectivity for ethene oxychlorination at 673 K as a function of the temperature of 20% conversion in VCM oxidation. Conditions are detailed in Table 2. | |
In order to address the first point, HCl oxidation was adopted as a suitable probe reaction to characterise the redox properties of a material for ethene oxychlorination, due to its similarities in reaction conditions and the requirement for Cl evolution in both reactions. In contrast, the typical application of H2-TPR to assess the redox properties of the used catalysts led to no convincing correlation (Fig. S2†), which is ascribed to the chlorination of the materials under reaction conditions.40,41Fig. 5a depicts the rate of ethene consumption in oxychlorination as a function of the rate of HCl oxidation over pre-equilibrated catalysts, evidencing a clear positive correlation between the two rates. This indicates that the activity of a catalyst in ethene oxychlorination for VCM formation is primarily determined by its ability to activate ethene and HCl, which requires two sites to form EDC, in line with previous theoretical studies.16 Thus, the activity of a catalyst in ethene oxychlorination is mainly determined by its ability to activate HCl, in line with the light-off curves in HCl oxidation (Fig. 5a, S3a†),13 the availability of chlorinated vacancies in ethene activation,16 and the ease of chlorine or EDC evolution from such vacancies.16 Yet, considering that all carriers were inactive in ethene oxychlorination, promotional effects of the support that go beyond the determination of the previously described CeO2 particle shape and speciation with respect to the Ce4+ fraction are unlikely. Secondly, rationalising the VCM selectivity, primarily influenced by the material acidity,24 was performed by using the EDC dehydrochlorination activity as an acidity descriptor that can be evaluated in an environment similar to ethene oxychlorination. This resulted in a clear positive trend between high VCM selectivity in ethene oxychlorination and low temperature where 20% conversion is reached in EDC dehydrochlorination (Fig. 5b). In addition to rationalising the VCM selectivity by the EDC dehydrochlorination performance, acidity characterisation by temperature programmed desorption of ammonia (NH3-TPD, Fig. 6) corroborated the found relationship, where only CeO2/ZrO2 renders as an outlier, likely due to the tight Ce–Zr integration, which is likely influenced by the presence of chlorine, and thus not correctly probed by ammonia. On the other hand, when considering the amount of acid sites (Fig. S4†) instead of the average strength (Fig. 5b), CeO2/MgO, which exhibits strong coking, contains the lowest number. Nevertheless, both correlations lead to the same conclusion that acid features of the material enhance VCM selectivity in accordance with EDC dehydrochlorination tests (Fig. 4b). However, the increase in acidity also promotes coking. Thus, optimal performance necessitates moderated acid properties. Furthermore, pure supports were tested and compared with the catalysts in EDC dehydrochlorination, evidencing only slight shifts of T20 to a higher temperature for materials with high VCM selectivity and shifts of 50 K for materials with low VCM selectivity (Fig. 5b). This indicates that indeed EDC dehydrochlorination is promoted by the presence of CeO2 in the catalyst formulation, but the major part of VCM formation seems to happen on the carrier, implying the existence of true bifunctional, two-site catalysts. Considering the ceria nanoparticle distribution as shown in Fig. 2, big particles or clusters of nanoparticles of ceria with large uncovered carrier surfaces in between therefore seem to facilitate the two-step reaction oxychlorination–dehydrochlorination on a hybrid surface. Nevertheless, the pronounced coking of CeO2/Al2O3 and CeO2/ZSM-5-15 was also observed for the corresponding pure carriers, indicating that CeO2 in the catalyst formulation is not the cause, but rather the result of coke formation (Fig. 6b), that is caused by the support acidity, as also observed for dehydrochlorination catalysts reported in the literature.20,26 Thirdly, the propensity towards combustion in ethene oxychlorination is likely not only determined by the presence of free or chlorinated vacancies, which are known to reduce combustion to some extent,16 but primarily influenced by the ease of oxygen removal from the surface.16,42 As this property is heavily influenced by the degree of chlorination and thus difficult to quantify under relevant reaction conditions,16 another probe reaction is used to rationalise the selectivity towards combustion products. While ethene oxidation primarily yields CO2 on CeO2, chlorinated hydrocarbons yield CO2 and CO.16 Therefore, the oxidation of VCM (Fig. S3b†) was studied on pre-equilibrated catalysts and correlated with the COx selectivity in ethene oxychlorination, as depicted in Fig. 5c. Therein, the temperature where 20% conversion in VCM oxidation was reached serving as a measure for the ease of combustion, resulting in a linear correlation between bulk CeO2 and CeO2/ZSM-5-15, CeO2/Al2O3, CeO2/ZrO2, and CeO2/SiO2. In contrast to HCl oxidation, CeO2/MgO emerged as the only outlier, surprisingly displaying the least activity in VCM oxidation while exhibiting the highest COx selectivity in ethene oxychlorination, which might be linked to oxidation of ethene or EDC. Nevertheless, the correlation of the other materials exemplifies how strongly dehydrochlorination and oxidation functions are intertwined or influence each other, as catalysts with high VCM selectivity also produce a high fraction of combustion products. Thus, in accordance with studies on bulk ceria,16 a very simplified reaction pathway for nanostructured CeO2 catalysts can be formulated as C2H4 → EDC → VCM → COx, clarifying the need for a catalyst which enables steps one and two, but suppresses step three, while two and three seem to be linked.
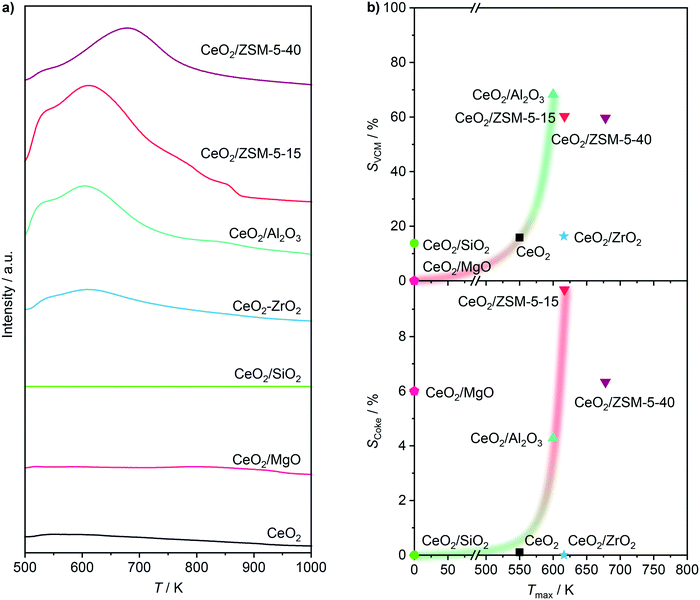 |
| | Fig. 6 a) NH3-TPD profiles of selected catalysts equilibrated in ethene oxychlorination. b) Selectivity to VCM (top) and coke (bottom) in ethene oxychlorination at 673 K as a function of the maximum temperature in NH3-TPD of the used catalysts. Conditions are detailed in Table 2. | |
Inspired by the high activity and high selectivity towards the VCM of CeO2/ZSM5-15, as well as the goal to satisfy the main priority to suppress combustion, i.e. designing an active catalyst that would lie in the lower right corner of Fig. 5c, zeolite supports were further investigated with the aim of understanding the impact of the framework, Si:Al ratio, Ce loading, and calcination temperature on the catalytic performance, as shown in Fig. 3e–h. First, considering different framework types (FER, Y, MOR, and ZSM-5) with as close as possible Si:Al ratios and 10 wt% Ce loading, it becomes evident that their activity at the same reaction temperature is very different, ranging from 4.5 (CeO2/FER-29) to 10 (CeO2/MOR-110) molC2H4 h−1 molCe−1. However, none of the catalysts reached more than 70% VCM selectivity, or fully suppressed combustion or DCE formation. In addition, the observed differences in the conversion–selectivity relationships are marginal considering the hugely different frameworks and the number of changing parameters in the system. Therefore, in the Pareto set of conversion–selectivity curves, the ZSM-5 framework was selected as a carrier material for further studies, due to (i) its good selectivity towards VCM at reasonable conversions and most importantly, (ii) its broad available range of Si:Al ratios. Therein four carriers with Si:Al ratios of 15, 40, 140, and 1040 were selected, of which the corresponding catalysts with 10 wt% Ce loading showed no linear trend in terms of activity. Instead, a maximum could be observed for CeO2/ZSM5-140 (the order with respect to ethene oxychlorination rate: CeO2/ZSM5-140 > CeO2/ZSM5-40 > CeO2/ZSM5-15 ≈ CeO2/ZSM5-1040), which might be attributed to better dispersion of ceria nanoparticles due to optimal acid properties for Ce anchoring.43 In terms of selectivity, a higher Si:Al ratio of the carrier led to lower COx and higher EDC selectivity, while the selectivity of VCM decreased (as evidenced also by the probe reaction in Fig. 5b) and DCE formation, already at very low levels, was not affected. The selectivity to coke decreased with a rising Si:Al ratio as expected, since acidic sites in the zeolite and aromatics formation on its external surface are known to generate coke.44 The fact that strong acidity is necessary for EDC dehydrochlorination, while it seems to promote COx and coke formation, excludes the as-prepared zeolite based catalysts from successful application in ethene oxychlorination to VCM. One remaining strategy to tune the performance of zeolite supported ceria systems might depend on adjusting the ceria particle size and coverage of the acidic carrier through optimisation of Ce loading and calcination temperature (Fig. 3g and h). However, while an increase of the calcination temperature slightly decreased the activity, not affecting the selectivity significantly, lower (5%) or higher (20%) Ce loading negatively affected the performance by strongly decreasing activity, or increasing combustion, respectively.
Overall, considering the observed catalytic performance and the descriptors in Fig. 5, it indicates that all the catalysts promoting VCM formation also promote combustion and coking, which leaves only CeO2/ZrO2 as a candidate that remains highly active and selective towards EDC at low temperatures. In addition to forming no other chlorinated hydrocarbons than EDC, no Cl2 evolved on any catalyst below 673 K, preserving a high chlorine efficiency.
Dual catalytic system for selective VCM production
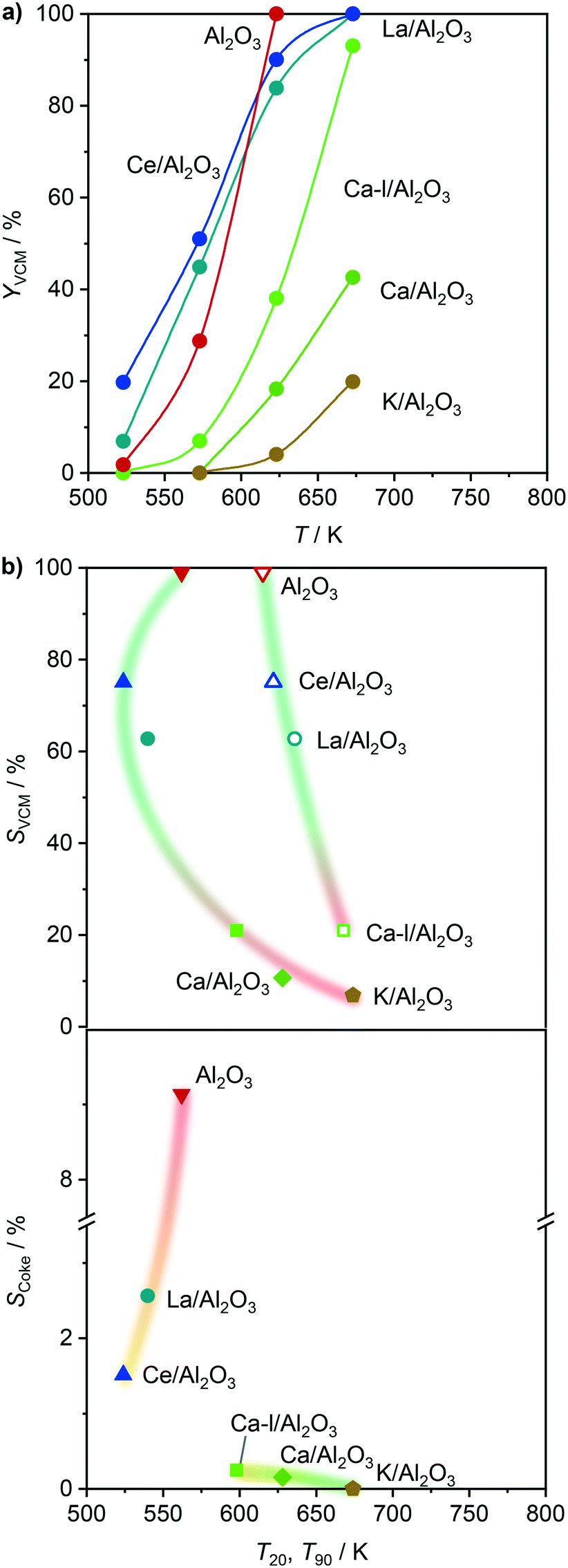
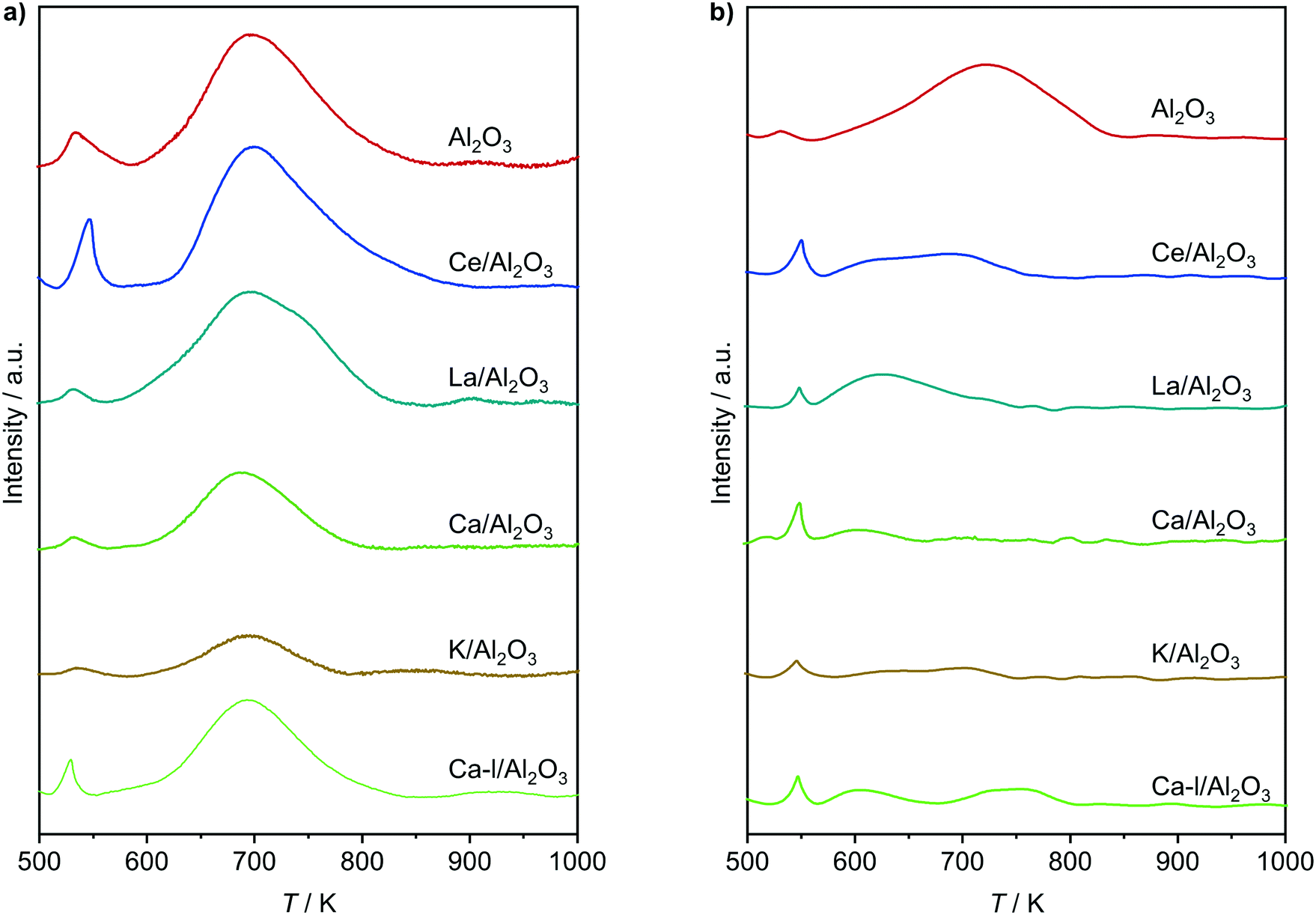
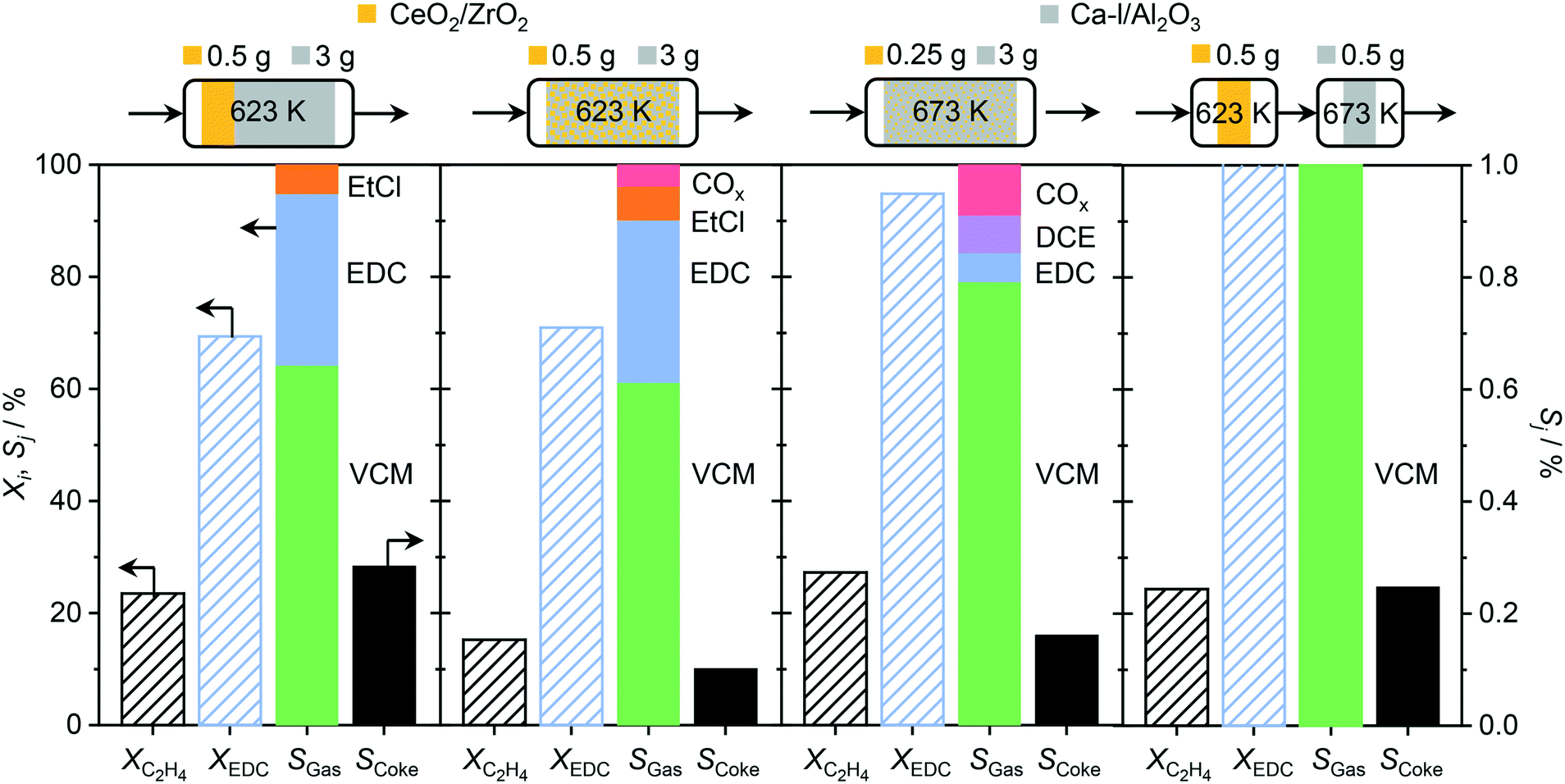
In order to overcome the above described obstacles, while refraining from using thermal but instead catalytic EDC dehydrochlorination, a promising catalyst for the latter was developed. Pure Al2O3 was selected as the starting material (Fig. 7a), since CeO2/Al2O3 was found to coke less in ethene oxychlorination compared to CeO2/ZSM-5-15 (Fig. 3). Moreover, Al2O3 enables the same dehydrochlorination performance as ZSM-5-15, reaching 100% yield at 623 K (Fig. S3c†). In order to reduce this coking, Al2O3 was promoted with additives known to improve selectivity for copper based oxychlorination catalysts,1,2,9,45 in particular Ca, K, La, and Ce. Doping with 2 wt% metal led to reduced coke formation, with an effectiveness order K > Ca > Ce > La, while the VCM yield decreased in the almost opposite order: Ce/Al2O3 > La/Al2O3 > Ca/Al2O3 > K/Al2O3 (Fig. 7a). Using a dual catalyst configuration with 0.5 g CeO2/ZrO2 and a 0.5 g Al2O3-based bed in series in one reactor (similar to illustration in Fig. 9), the VCM selectivity could again be correlated with the temperature of 20% (and 90%) conversion in EDC dehydrochlorination, as shown in Fig. 7b. This suggests that the used dopants mask the acid sites, which are responsible for coking and EDC dehydrochlorination. In addition, the NH3-TPD of fresh dehydrochlorination catalysts (Fig. 8a) evidences a decrease in total acidity upon doping with different cations, which is in accordance with the dehydrochlorination performance (Fig. 7). However, catalysts where coking was reduced to values close to zero did not show high VCM yields in the desired temperature region below 673 K. Therefore, further optimisation was performed by reducing the dopant loading to 0.5 wt% on the intermediate Ca promoted Al2O3, with the aim of masking primarily the strongest sites, while keeping weaker sites available for EDC dehydrochlorination, resulting in Ca-l/Al2O3, which showed a 40% VCM yield at 623 K (Fig. 7a). In addition, reaching 90% conversion in EDC dehydrochlorination was enabled in contrast to Ca/Al2O3. The coke selectivity was slightly increased, yet very limited, following a correlation between acidity availability for EDC dehydrochlorination and coke selectivity in ethene oxychlorination (Fig. 7b). However, as the yield of this catalyst was insufficient for application in VCM selective dual catalytic systems, different strategies to increase the EDC conversion were explored. In the first step, the amount of Ca-l/Al2O3 was increased by a factor of 6, still not yielding full EDC conversion and producing new products, in particular ethyl chloride (EtCl), in parallel with increased coke selectivity (Fig. 9). Tighter integration of the same amounts of the two catalysts in a single, physically mixed bed reduced the overall coke selectivity, likely due to the fact that the VCM is a major contributor to coke formation,20,26 yet also reduced VCM selectivity and increased the formation of combustion products. Increasing the temperature to achieve higher EDC conversion on the dehydrochlorination catalyst, while reducing the amount of the oxychlorination catalyst for keeping similar conversion levels (Fig. 9, panel three), led as expected to higher EDC conversion, but also increased combustion and DCE selectivity. Therefore, the solution to preserve the selective performance of the oxychlorination step, while ensuring selective and full EDC dehydrochlorination to VCM in the second step with minimal coking, was to operate CeO2/ZrO2 at 623 K, coupled to a separate reactor, containing Ca-l/Al2O3 and operated at 673 K. This indeed resulted in full and 100% selective EDC to VCM conversion (at 25% ethene conversion) and a coke selectivity of <0.3% (Fig. 9).
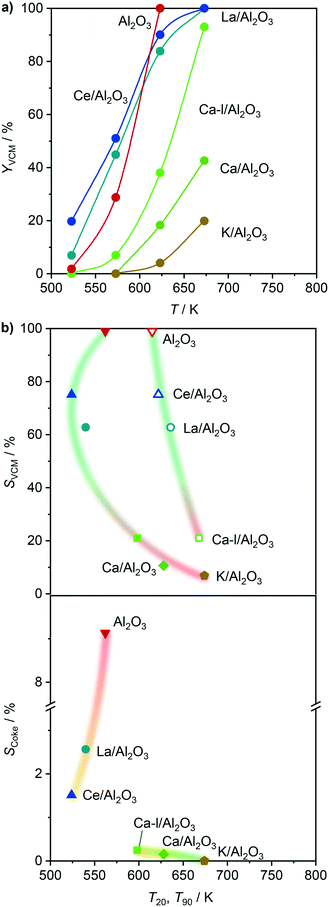 |
| | Fig. 7 a) VCM yield as a function of temperature in EDC dehydrochlorination over the Al2O3-based catalysts. b) Selectivity to VCM (top) and coke (bottom) in a dual-catalyst configuration as a function of the temperature of 20% (solid symbols) and 90% (open symbols) conversion in EDC dehydrochlorination. In the dual-catalyst configuration for ethene oxychlorination, CeO2/ZrO2 was used in the first bed, and the corresponding Al2O3-based catalyst in the second bed, operated at 623 K. Conditions are detailed in Table 2. | |
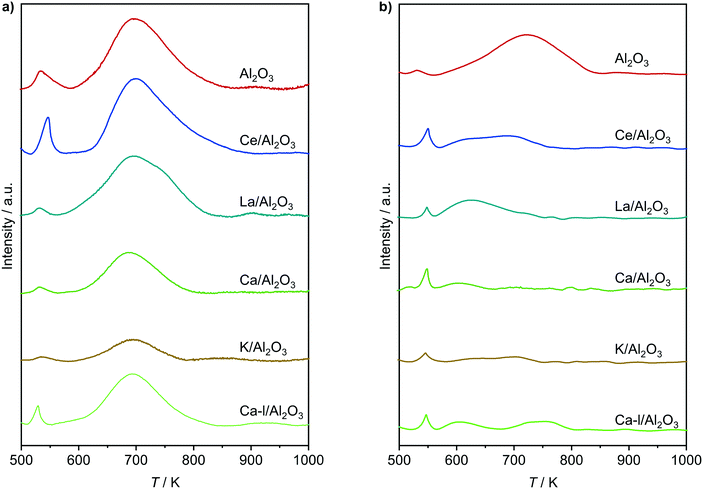 |
| | Fig. 8 NH3-TPD of a) fresh and b) used dehydrochlorination catalysts. Conditions are detailed in Table 2. | |
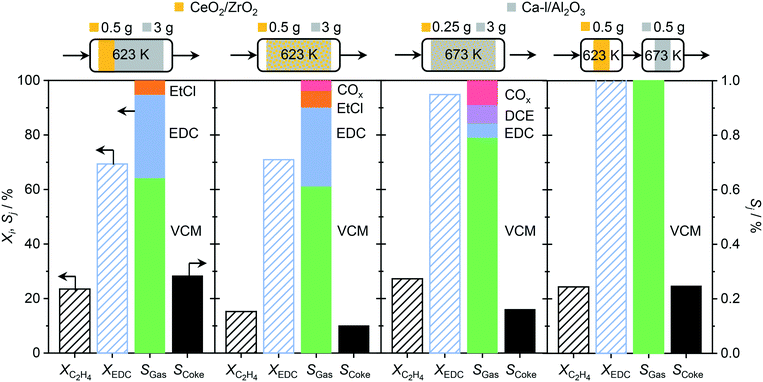 |
| | Fig. 9 Conversion of ethene and in situ produced EDC, and product selectivity for ethene oxychlorination, obtained in different reactor configurations, which are depicted above the plots. CeO2/ZrO2 (yellow) and Ca-l/Al2O3 (grey) are used as oxychlorination and dehydrochlorination catalysts, respectively. The operation temperature and used catalyst amounts are depicted on top of their respective illustrations. Conditions are detailed in Table 2. | |
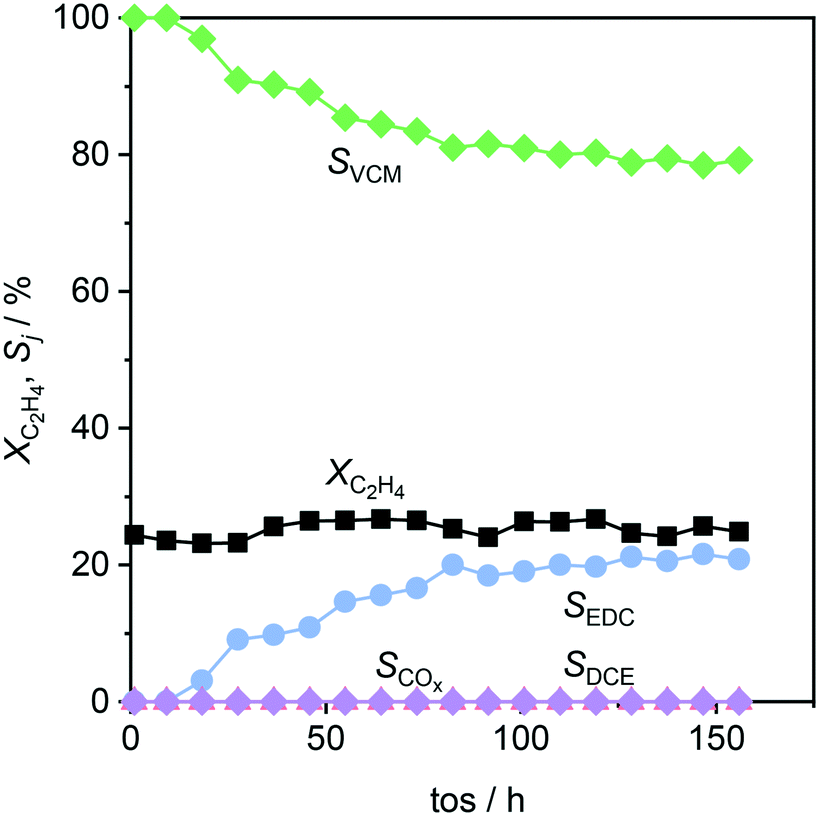
This system was then subjected to a stability test, as depicted in Fig. 10, where the conversion, determined by CeO2/ZrO2, rendered stable for 160 h on stream. However, while no combustion or formation of undesired chlorinated products occurred, the selectivity to VCM, and thus EDC, changed over the course of the test. The observed decrease of VCM selectivity can be linked to a moderate loss of acidity upon use (Fig. 8b), and a decrease in surface area and pore volume (Table 3), consistent with the formation of coke species. Yet, the decrease levelled off, reaching stable values of about 80% VCM and 20% EDC selectivity, which outperforms the current dehydrochlorination yield by 60% at a 100 K lowered temperature.1,2,6
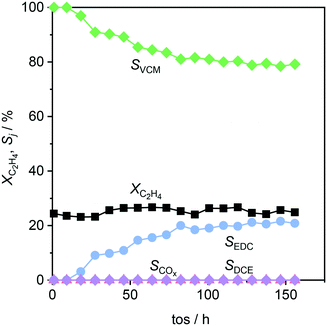 |
| | Fig. 10 Conversion and selectivity as a function of time-on-stream in ethene oxychlorination over a CeO2/ZrO2–Ca-l/Al2O3 system as depicted in the fourth panel of Fig. 9. Conditions are detailed in Table 2. | |
Conclusions
In this work, we developed copper-free dual catalytic systems for direct VCM production from ethene. In order to suppress combustion, chlorinated side-product formation, and coking, while improving the VCM selectivity to make thermal cracking obsolete, nanostructured ceria catalysts were designed in the first step. In-depth rationalisation and exploration of carrier effects led to the result that achieving all the goals is not feasible in a single material. CeO2/ZrO2 proved to be a stable oxychlorination catalyst, exhibiting full selectivity to EDC and extraordinary activity even at low temperature. In order to catalytically dehydrochlorinate EDC to VCM, a dedicated catalyst for this second function was developed, consisting of the promoted solid acid catalyst Ca-l/Al2O3 which led to a minimal coking, and 100% EDC conversion to VCM. Some degree of deactivation was observed during the first 100 h on stream, decreasing the overall selectivity to VCM from 100% to stable 80% at 25% ethene conversion. Nevertheless, the developed dual system outperforms EuOCl as the best ethene oxychlorination catalyst to directly form VCM by a factor of four in terms of space time yield. Thus, this study provides the basis for potential intensification of the current industrial-scale VCM.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Dr. Frank Krumreich and Evgeniya Vorobyeva for help with microscopy analysis by HAADF-STEM, and Lorenz Olbrich for help with synthesis and testing of catalysts depicted in the upper half of Fig. 3 and characterisation by NH3-TPD.
References
-
K. Weissermel and H.-J. Arpe, in Industrial Organic Chemistry, Wiley-VCH, Weinheim, 4th edn, 2008, pp. 217–238 Search PubMed.
-
E.-L. Dreher, K. K. Beutel, J. D. Myers, T. Lübbe, S. Krieger and L. H. Pottenger, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2014, p. 2 Search PubMed.
- K. Mulder and M. Knot, Technol. Soc., 2001, 23, 265–286 CrossRef.
- N. B. Muddada, U. Olsbye, G. Leofanti, D. Gianolio, F. Bonino, S. Bordiga, T. Fuglerud, S. Vidotto, A. Marsella and C. Lamberti, Dalton Trans., 2010, 39, 8437–8449 RSC.
- N. B. Muddada, U. Olsbye, T. Fuglerud, S. Vidotto, A. Marsella, S. Bordiga, D. Gianolio, G. Leofanti and C. Lamberti, J. Catal., 2011, 284, 236–246 CrossRef CAS.
-
J. S. Naworski and E. S. Velez, in Applied Industrial Catalysis, Academic Press, New York, 1983, pp. 239–273 Search PubMed.
-
S. R. L. Süd Chemie Mt, EP 1020222 B1, 2004.
- N. B. Muddada, T. Fuglerud, C. Lamberti and U. Olsbye, Top. Catal., 2014, 57, 741–756 CrossRef CAS.
- N. B. Muddada, U. Olsbye, L. Caccialupi, F. Cavani, G. Leofanti, D. Gianolio, S. Bordiga and C. Lamberti, Phys. Chem. Chem. Phys., 2010, 12, 5605–5618 RSC.
-
L. E. Walko, K. A. Marshall, M. M. Olken, D. A. Hickman, W. D. Clarke, M. E. Jones, D. J. Reed and J. P. Henley, Dow Global Technologies Inc., US 6933417 B1, 2005.
- W. D. Mross, Catal. Rev.: Sci. Eng., 1983, 25, 591–637 CrossRef CAS.
- J. Pérez-Ramírez, C. Mondelli, T. Schmidt, O. F. K. Schlüter, A. Wolf, L. Mleczko and T. Dreier, Energy Environ. Sci., 2011, 4, 4786–4799 RSC.
- A. P. Amrute, C. Mondelli, M. Moser, G. Novell-Leruth, N. López, D. Rosenthal, R. Farra, M. E. Schuster, D. Teschner, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2012, 286, 287–297 CrossRef CAS.
- M. Scharfe, P. A. Lira-Parada, V. Paunovic, M. Moser, A. P. Amrute and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 3068–3072 CrossRef CAS PubMed.
- Flid M. R., Trushechkina M. A and Treger Y. A., J. Thermodyn. Catal., 2017, 08, 1–24 Search PubMed.
- M. Scharfe, M. Capdevila-Cortada, V. A. Kondratenko, E. V. Kondratenko, S. Colussi, A. Trovarelli, N. López and J. Pérez-Ramírez, ACS Catal., 2018, 8, 2651–2663 CrossRef CAS.
- M. Scharfe, P. A. Lira-Parada, A. P. Amrute, S. Mitchell and J. Pérez-Ramírez, J. Catal., 2016, 344, 524–534 CrossRef CAS.
- V. Paunović, G. Zichittella, S. Mitchell, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2018, 8, 291–303 CrossRef.
- Z. A. Qiao, Z. Wu and S. Dai, ChemSusChem, 2013, 6, 1821–1833 CrossRef CAS PubMed.
- S. Bai, Q. Dai, X. Chu and X. Wang, RSC Adv., 2016, 6, 52564–52574 RSC.
- C. Li, G. Zhou, L. Wang, S. Dong, J. Li and T. Cheng, Appl. Catal., A, 2011, 400, 104–110 CrossRef CAS.
- H. A. Miran, M. Altarawneh, Z.-T. Jiang, H. Oskierski, M. Almatarneh and B. Z. Dlugogorski, Catal. Sci. Technol., 2017, 7, 3902–3919 RSC.
- W. Zhao, M. Sun, H. Zhang, Y. Dong, X. Li, W. Li and J. Zhang, RSC Adv., 2015, 5, 104071–104078 RSC.
- A. S. Shalygin, L. V. Malysheva and E. A. Paukshtis, Kinet. Catal., 2011, 52, 305–315 CrossRef CAS.
- D. Carmello, E. Finocchio, A. Marsella, B. Cremaschi, G. Leofanti, M. Padovan and G. Busca, J. Catal., 2000, 191, 354–363 CrossRef CAS.
- D. V. Kazachkin, D. R. Luebke, V. I. Kovalchuk and J. D'Itri, J. Sib. Fed. Univ., Chem., 2008, 4, 303–325 Search PubMed.
- Y. Du, Y. Zhu, S. Xi, P. Yang, H. O. Moser, M. B. H. Breese and A. Borgna, J. Synchrotron Radiat., 2015, 22, 839–843 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. M. Reddy, A. Khan, P. Lakshmanan, M. Aouine, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2005, 109, 3355–3363 CrossRef CAS PubMed.
- B. M. Reddy, P. Bharali, P. Saikia, A. Khan, S. Loridant, M. Muhler and W. Grünert, J. Phys. Chem. C, 2007, 111, 1878–1881 CrossRef CAS.
- J. Strunk, W. C. Vining and A. T. Bell, J. Phys. Chem. C, 2011, 115, 4114–4126 CrossRef CAS.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- A. Trovarelli, F. Zamar, J. Llorca, C. de Leitenburg, G. Dolcetti and J. T. Kiss, J. Catal., 1997, 169, 490–502 CrossRef CAS.
- D. N. Talwar, L. Wan, C. C. Tin and Z. C. Feng, J. Mater. Sci. Eng., 2017, 06, 1000324 Search PubMed.
- A. Wypych, I. Bobowska, M. Tracz, A. Opasinska, S. Kadlubowski, A. Krzywania-Kaliszewska, J. Grobelny and P. Wojciechowski, J. Nanomater., 2014, 124814, DOI:10.1155/2014/124814.
- Y. Inoue, S. Nakashima and A. Mitsuishi, Solid State Commun., 1983, 48, 1071–1075 CrossRef CAS.
- E. Stavrou, Y. Yao, J. M. Zaug, S. Bastea, B. Kalkan, Z. Konôpková and M. Kunz, Sci. Rep., 2016, 6, 1–11 CrossRef.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- R. M. German, JOM, 2016, 68, 878–884 CrossRef.
- M. Moser, G. Vilé, S. Colussi, F. Krumeich, D. Teschner, L. Szentmiklósi, A. Trovarelli and J. Pérez-Ramírez, J. Catal., 2015, 331, 128–137 CrossRef CAS.
- M. Moser, C. Mondelli, T. Schmidt, F. Girgsdies, M. E. Schuster, R. Farra, L. Szentmiklósi, D. Teschner and J. Pérez-Ramírez, Appl. Catal., B, 2013, 132–133, 123–131 CrossRef CAS.
- M. D. Higham, M. Scharfe, M. Capdevila-Cortada, J. Pérez-Ramírez and N. López, J. Catal., 2017, 353, 171–180 CrossRef CAS.
- L. I. van der Wal, K. P. de Jong and J. Zečević, ChemCatChem, 2019, 11, 1–9 CrossRef PubMed.
- B. Liu, D. Slocombe, M. AlKinany, H. AlMegren, J. Wang, J. Arden, A. Vai, S. Gonzalez-Cortes, T. Xiao, V. Kuznetsov and P. P. Edwards, Appl. Petrochem. Res., 2016, 6, 209–215 CrossRef CAS.
-
M. Rossberg, W. Lendle, G. Pfleiderer, A. Tögel, E.-L. Dreher, E. Langer, H. Rassaerts, P. Kleinschmidt, H. Strack, R. Cook, U. Beck, K.-A. Lipper, T. R. Torkelson, E. Löser, K. K. Beutel and T. Mann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01801h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sharon
Mitchell
a,
Sharon
Mitchell
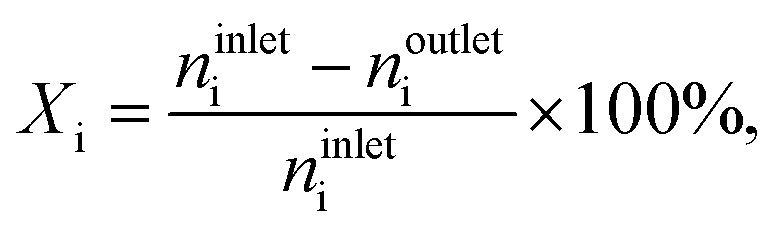
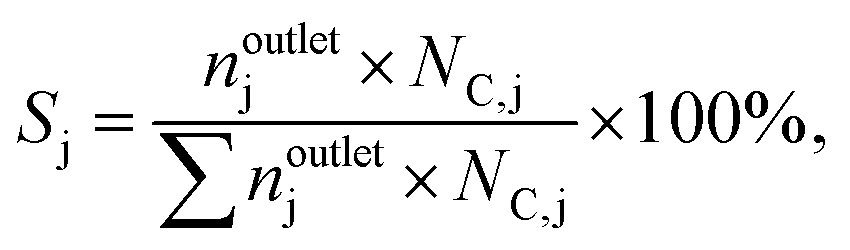
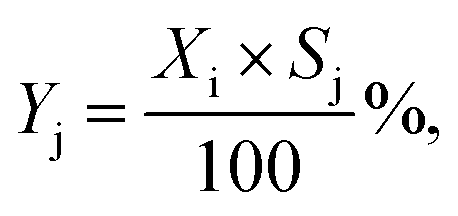
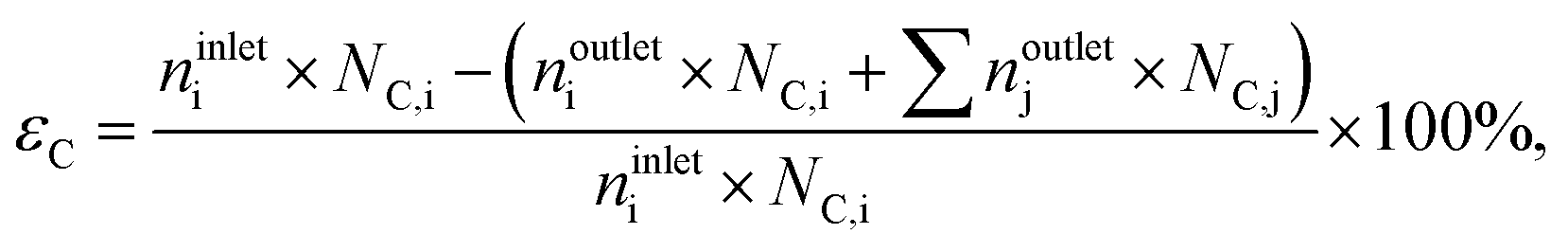
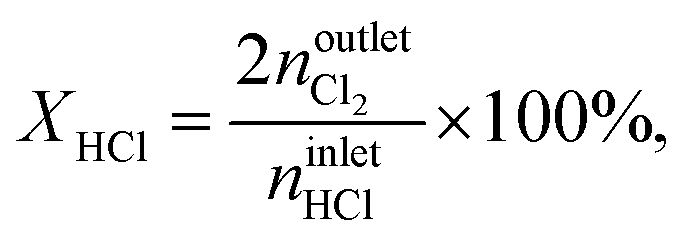
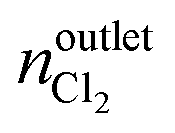 denote the molar flow of HCl and Cl2 at the reactor inlet and outlet, respectively. Calculation of the Weisz–Prater and Mears criteria indicated that all catalytic tests were performed in the absence of heat and mass transport limitations. After the tests, the reactor was quenched to room temperature in a He flow.
denote the molar flow of HCl and Cl2 at the reactor inlet and outlet, respectively. Calculation of the Weisz–Prater and Mears criteria indicated that all catalytic tests were performed in the absence of heat and mass transport limitations. After the tests, the reactor was quenched to room temperature in a He flow.