
Charge separation and molecule activation promoted by Pd/MIL-125-NH2 hybrid structures for selective oxidation reactions†
Ting
Wang
a,
Xueqin
Tao
a,
Yi
Xiao
a,
Ganhua
Qiu
a,
Yun
Yang
 b and
Benxia
Li
*a
b and
Benxia
Li
*a
aDepartment of Chemistry, School of Science, Zhejiang Sci-Tech University, Second Avenue 928, Xiasha Higher Education Zone, Hangzhou 310018, P.R. China. E-mail: libx@mail.ustc.edu.cn
bNanomaterials and Chemistry Key Laboratory, Wenzhou University, Xueyuan road 276, Wenzhou, Zhejiang 325027, P.R. China
First published on 18th November 2019
Abstract
While heterogeneous photocatalysis is regarded as a sustainable strategy to achieve organic reactions for the production of high-value-added organics, photocatalytic efficiency is generally limited by poor charge separation, dull surface catalytic activity and the consequently caused insufficient generation of active species. Herein, the hybrid structures of metal–organic frameworks (MOFs) incorporated with Pd ultrasmall nanocrystals are synthesized, and their photocatalytic performance is evaluated by the oxidative coupling of benzylamine as a model reaction. The photocatalytic activity of the Pd/MIL-125-NH2 hybrid material with an optimal Pd loading is greatly superior to that of the pristine MIL-125-NH2, achieving a high benzylamine conversion rate of 3136 μmol h−1 gcat−1 at 94.08% conversion. The enhanced charge transfer and separation in Pd/MIL-125-NH2 are verified by photoluminescence spectroscopy and photoelectrochemical experiments. Photogenerated reactive oxygen species are probed by 3,3′,5,5′-tetramethylbenzidine (TMB) as an indicator and the reactive intermediates in the photocatalytic systems are identified by in situ ESR detection. The results confirm that the optimized Pd/MIL-125-NH2 hybrid catalyst possesses higher ability for molecule activation and that both superoxide radical (˙O2−) and singlet oxygen (1O2) are the dominant oxidative species in the present photocatalytic system. Therefore, the dual advantages of Pd/MIL-125-NH2 hybrid structures for promoting the photogenerated charge transfer/separation and providing highly catalytic sites for molecule activation contribute to higher photocatalytic activity.
Introduction
In the past decades, in order to improve the conversion rate and product selectivity of organic transformations, high temperature and/or high pressure were usually required in thermally-driven catalytic reactions.1,2 A great deal of non-renewable energy sources are consumed and some toxic or corrosive chemicals are involved in these processes. Considering the increasing interest in green and sustainable chemistry, it is highly desirable that organic reactions with a high conversion rate and product selectivity can be achieved through readily available and environmentally benign reactants (such as O2 in air) as well as at the low cost of traditional energy consumption.3–7 The development of photocatalysts has offered great possibilities for the realization of this goal.8–11 In recent years, more and more efforts are devoted to improving the photocatalytic transformations of organics, such as the selective oxidation of amines into imines and their derivatives, which are high-value-added organic pharmaceutical intermediates.12–14 However, the photocatalytic selective oxidation of organics still suffers from low conversion rate or/and poor selectivity, because of the sluggish photocatalytic activity or the presence of the reactive oxygen species (ROSs) having strong oxidizing power (such as ˙OH).15,16 Among various photogenerated ROSs, superoxide radicals (˙O2−) and singlet oxygen (1O2) are regarded as the most appropriate candidates for the selective oxidation of organics due to their moderate oxidation ability.17–20 Therefore, developing photocatalysts being adequate in moderate ROS production and organic molecule activation is the key point to boost the selective oxidation of organics with molecular oxygen under ambient conditions.Recently, the rapid development of metal–organic frameworks (MOFs) has captured a great deal of research interest in exploiting MOF-based materials as types of promising photocatalysts.21–23 The high porosity and large specific surface area of MOFs endow them with efficient adsorption and gathering of reactant molecules in pores as well as the relatively rapid charge migration to the pore surface for reactions.24 Nonetheless, the limited active sites in pristine MOFs actually hinder their performance in photocatalytic reactions. Various modification methods were adopted to improve the photocatalytic efficiency, such as incorporating metal particles,25,26 coupling with inorganic semiconductors,27,28 grafting photoactive metal complexes,29 and constructing MOF–MOF (bi-MOF) heterostructures.30
Pd nanocrystals have been demonstrated to be effective for the activation of molecular oxygen (O2) to form 1O2 through electron transfer from Pd to the antibonding π* orbital of O2.31 Moreover, the Pd–semiconductor hybrid configuration enabled the improvement of O2 activation under appropriate illumination due to the increased electron density on the Pd surface in the hybrid structures, which is significant for enhancing the efficiency and selectivity of organic oxidation reactions.32 In a similar way, the integration of Pd nanocrystals into semiconductor-like MOFs is expected to cause enhanced photocatalytic performance due to the transfer and separation of the photoinduced carriers in MOFs as well as the adsorption and activation of O2 and organic molecules. Moreover, the Pd nanocrystals with ultrasmall sizes (<2 nm) often present prominent catalytic activity because their dramatically high surface-to-volume ratios offer abundant available active/reactive sites.33,34 The rich ordered pores in MOFs make them suitable solids for confining the ultrasmall Pd nanocrystals, ensuring the uniform dispersions of the Pd nanocrystals during preparation and catalytic reactions.35 The small-sized Pd nanoclusters/nanocrystals encapsulated inside MOFs have showed superior catalytic activity for thermally-driven oxidation reactions,36 hydrogenation,37 hydrogen absorption,38 Suzuki coupling and Heck arylation reactions,39,40 whereas the synergistic effect between Pd nanocrystals and MOFs on their photocatalysis for the selective oxidation of organics needs to be further studied. In this work, Pd nanocrystals with ultrasmall sizes (<2 nm) are embedded in porous MIL-125-NH2 (Ti8O8(OH)4(BDC-NH2)6, BDC-NH2 = 2-aminobenzene-1,4-dicarboxylate) to form hybrid nanostructures. The Pd(1.0)/MIL-125-NH2 hybrid catalyst with an optimal Pd loading content exhibits much higher photocatalytic activity toward the selective oxidation of organic molecules than the pristine MIL-125-NH2. The enhanced photocatalysis of Pd(1.0)/MIL-125-NH2 results from the superior charge separation efficiency and the enhanced active species generation due to the charge transfer from MIL-125-NH2 to Pd ultrasmall nanocrystals upon photoexcitation.
Experimental section
Materials
2-Aminobenzene-1,4-dicarboxylate (BDC-NH2), titanium(IV) isopropoxide, polyvinylpyrrolidone (PVP, K-88, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000), ammonium tetrachloropalladate ((NH4)2PdCl4), ascorbic acid (AA), 4-fluorobenzylamine, 4-methoxybenzylamine, 4-chlorobenzylamine, 4-methylbenzylamine, benzylamine, β-carotene, catalase (≥200000 unit per g), mannitol, superoxide dismutase (SOD) (≥1400 units per mg dry weight), 3,3′,5,5′-tetramethylbenzidine (TMB), acetate (HAc), sodium acetate anhydrous (NaAc), 5,5-dimethyl-pyrroline-N-oxide (DMPO), 2,2,6,6-tetramethylpiperidine (TEMP), potassium hydrogen phthalate (C8H5KO4), potassium iodide (KI), methanol and acetonitrile. All of the chemicals were of analytical grade and purchased from Aladdin Industrial Corporation.
300000), ammonium tetrachloropalladate ((NH4)2PdCl4), ascorbic acid (AA), 4-fluorobenzylamine, 4-methoxybenzylamine, 4-chlorobenzylamine, 4-methylbenzylamine, benzylamine, β-carotene, catalase (≥200000 unit per g), mannitol, superoxide dismutase (SOD) (≥1400 units per mg dry weight), 3,3′,5,5′-tetramethylbenzidine (TMB), acetate (HAc), sodium acetate anhydrous (NaAc), 5,5-dimethyl-pyrroline-N-oxide (DMPO), 2,2,6,6-tetramethylpiperidine (TEMP), potassium hydrogen phthalate (C8H5KO4), potassium iodide (KI), methanol and acetonitrile. All of the chemicals were of analytical grade and purchased from Aladdin Industrial Corporation.
Synthesis of MIL-125-NH2
MIL-125-NH2 was synthesized by a previously reported procedure with minor modification.41 Typically, 0.628 g BDC-NH2 and 0.531 mL titanium(IV) isopropoxide were dissolved in the mixed solvent consisting of 15 mL DMF and 15 mL MeOH by magnetic stirring for 30 min. Then, the solution was transferred into a 50 mL Teflon-lined autoclave, which was sealed and heated at 150 °C for 15 h. Finally, the yellow powder of MIL-125-NH2 was collected, washed with DMF and MeOH, and dried at 60 °C overnight under vacuum.Synthesis of Pd/MIL-125-NH2 hybrid structures
The ultrasmall Pd nanocrystals were embedded in MIL-125-NH2 through mild reduction with ascorbic acid as a reducing agent. 100 mg of the as-prepared MIL-125-NH2 powder was ultrasonically dispersed in 20 mL of PVP aqueous solution (0.50 g L−1). After 1 h stirring, a certain volume of (NH4)2PdCl4 solution (0.10 M) was injected into the above suspension, followed by another 8 h stirring. The solid particles were collected by centrifugation, and then re-dispersed in 20 mL deionized water. 0.24 g of ascorbic acid was added in this suspension, and the mixture was then stirred at 60 °C for 2 h. The precipitate was collected by centrifugation and washed with deionized water several times. The final product was dried at 60 °C overnight in a vacuum. The Pd(x)/MIL-125-NH2 hybrid structures with different Pd loading contents (x wt%) were obtained by varying the injected volume of (NH4)2PdCl4 solution, and denoted as Pd(0.2)/MIL-125-NH2, Pd(0.5)/MIL-125-NH2, Pd(1.0)/MIL-125-NH2 and Pd(1.5)/MIL-125-NH2, respectively.It should be noted that the Pd loading amounts (x wt%) herein are the theoretical values calculated based on the assumption that all the injected Pd2+ ions were transformed into Pd nanocrystals in the Pd(x)/MIL-125-NH2 hybrid structures. The actual Pd loading amounts should be much lower than the corresponding theoretical values.
Characterization
Powder X-ray diffraction (XRD) was conducted on an X-ray diffractometer (Japan Shimadzu XRD-6000) with Cu Kα radiation. The morphology and microstructures were observed by field emission scanning electronic microscopy (FESEM, Zeiss, Ultra 55) and high resolution transmission electron microscopy (HRTEM, Hitachi S4800). The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method on the basis of N2 adsorption/desorption measurement on a Micromeritics ASAP 2020 analyzer, and the pore size distribution was obtained by the Barrett–Joyner–Halenda (BJH) method. X-ray photoelectron spectroscopy (XPS) analysis was conducted using a Thermo Scientific ESCALAB 250Xi X-ray photoelectron spectrometer using Al Kα radiation as the excitation source. UV-vis diffuse reflection spectra (DRS) were recorded on a spectrophotometer (UV-2700, Shimadzu). Photoluminescence (PL) spectroscopy was analyzed a HORIBA HuoroMax-4P spectrometer. The photoelectrochemical experiments were carried out on an electrochemical workstation (CHI660E, Shanghai Chenhua, China) with a standard three electrode system and under irradiation (360–780 nm) using a 300 W xenon lamp (CEL-HXF300). The electron spin resonance (ESR) signals were obtained on an ESR JES-FA200 spectrometer, according to the photocatalytic conditions, with DMPO for the detection of ˙O2− and TEMP for the detection of 1O2, respectively.Evaluation of photocatalytic properties
The photocatalytic properties of the as-prepared catalysts were evaluated by the oxidative coupling of benzylamine under a 300 W Xe lamp equipped with a reflector that could afford irradiation mainly within the wavelength range of 360–780 nm, as shown in Fig. S1.† The optical power density that irradiated on the reaction solution is 100 mW cm−2. In a typical reaction, 5 mg catalyst was suspended in 2 mL CH3CN containing 0.05 mmol benzylamine in a quartz tube with a magnetic stirrer, using molecular O2 from atmospheric air as the oxidizing agent. After being stirred in the dark for 30 min, the reaction suspension was exposed to irradiation for a certain time. Finally, the reaction solution was filtered through a syringe filter with a 0.22 μm polymer membrane to remove the catalyst particles, and the concentrations of reactants and products were analyzed by gas chromatography (GC, Shimadzu 2014C).Molecular oxygen activation tests
O2 activation was tested by 3,3′,5,5′-tetramethylbenzidine (TMB) oxidation according to the evolution in the spectral absorption of TMB solution along the irradiation time.31 In a typical test, 15 mg TMB was suspended in 8 mL deionized water, and 24 mL HAc/NaAc buffer solution (pH = 4.7) was added in the suspension. Subsequently, 100 μL of the photocatalyst aqueous suspension (5 g L−1) was injected into the TMB suspension. After the adsorption equilibrium in the dark, the mixture was exposed to the Xe lamp irradiation of 360–780 nm. The reaction was probed by recording the absorbance around 370 nm on a UV-vis spectrophotometer.To verify the generation of different reactive oxygen species, the contrast experiments with adding the related scavenger were conducted similarly according to the above experimental procedure. The added amounts of scavengers are listed as follows: catalase (3500 unit per mL, 200 μL), superoxide dismutase (2000 unit per mL, 2 mL), mannite (10 mg), and carotene (5 mg).
Results and discussion
Characterization of MIL-125-NH2 and Pd(x)/MIL-125-NH2 materials
The Pd(x)/MIL-125-NH2 hybrid structures are synthesized through a two-step process (Fig. 1a), where MIL-125-NH2 is synthesized through a solvothermal method and Pd nanocrystals are then embedded in MIL-125-NH2 by a mild reaction with ascorbic acid as the reducing agent. The XRD pattern of the as-prepared MIL-125-NH2 sample (Fig. 1b) is in good agreement with the simulated pattern of MIL-125-NH2 which has a quasi-cubic tetragonal structure.42 After loading Pd, the crystal structure of MIL-125-NH2 remains unchanged, but the diffraction peaks of Pd nanocrystals are unobservable in the XRD patterns of Pd(x)/MIL-125-NH2 hybrid structures (Fig. 1b and S2†), suggesting the ultrasmall size and high dispersion of Pd nanocrystals in MIL-125-NH2. The MIL-125-NH2 sample consists of well-dispersed hexahedron-shaped nanocrystals with a length/width of around 600 nm and thickness of about 150 nm (Fig. S3a and b†). Fig. 1c–f show the SEM and TEM images of the Pd(1.0)/MIL-125-NH2 sample which was optimized to display the best photocatalytic performance. The Pd(1.0)/MIL-125-NH2 hybrid structures maintain well the morphology of MIL-125-NH2. The HRTEM image (Fig. 1e) reveals that the ultrasmall Pd nanocrystals with an average size of ∼1.6 nm are highly dispersed in MIL-125-NH2. The distribution of Pd element is further demonstrated by HAADF-STEM imaging and EDX element mapping (Fig. 1f and g). During the synthesis process, Pd2+ ions were readily adsorbed onto the pores of MIL-125-NH2 after the long-time (8 h) stirring at room temperature due to the capillary force and the interaction between Pd2+ ions and amino groups in MIL-125-NH2. The subsequent reduction of Pd2+ ions to Pd nanocrystals occurred in the MOF pores, which prevented the migration and aggregation of Pd nanocrystals. As a result, the Pd nanocrystals with ultrasmall sizes were produced and stabilized in MIL-125-NH2. The Pd nanocrystals of the Pd(0.5)/MIL-125-NH2 sample also presents a similar ultrasmall particle size of around 1.5 nm (Fig. S3c†). However, superfluous Pd2+ ions in the solution would lead to the formation of larger Pd nanocrystals attached on the surface of MIL-125-NH2. The Pd particle sizes of Pd(1.5)/MIL-125-NH2 are estimated to be around 7.0 nm (Fig. S3d†). In addition, a contrast sample of Pd/MIL-125-NH2 (Fig. S4†) prepared by the direct reduction-deposition of Pd nanocrystals onto MIL-125-NH2, without the long-time adsorption before ascorbic-acid reduction, presents the larger particle sizes (∼3.9 nm) and inferior distribution uniformity of Pd nanocrystals, which indirectly proves that the ultrasmall Pd nanocrystals of the Pd(x)/MIL-125-NH2 samples synthesized by our absorption–reduction method are highly dispersed and embedded inside the pores of MIL-125-NH2.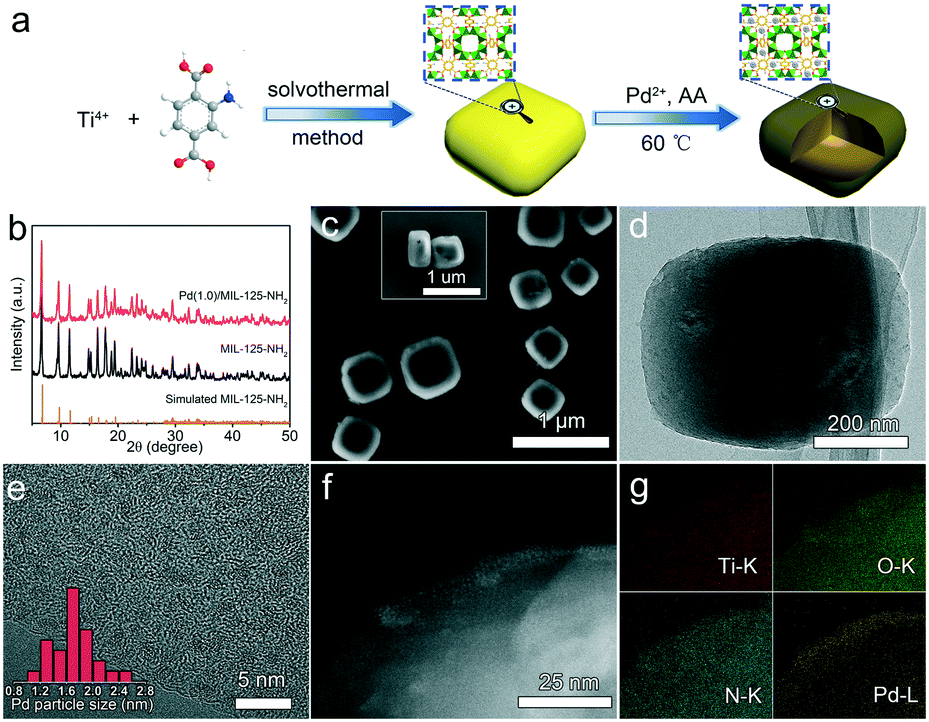 | ||
| Fig. 1 (a) Schematic illustration for the synthesis of Pd/MIL-125-NH2 hybrid structures. (b) Powder XRD patterns of the simulated MIL-125-NH2, the as-synthesized MIL-125-NH2 and Pd(1.0)/MIL-125-NH2. (c) SEM image (inset: lateral view), (d) TEM image, (e) HRTEM image (inset: size distribution of Pd nanocrystals), (f) HAADF-STEM image and (g) the corresponding elemental mapping images of Pd(1.0)/MIL-125-NH2. | ||
The specific surface areas and porous structures of MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 are determined using the N2 adsorption–desorption isotherms (Fig. 2). Both samples exhibit type IV isotherms with obvious hysteresis loops, suggesting their mesoporous structures. The specific surface areas of MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 are calculated by the BET method to be 857.1 and 789.7 m2 g−1, as listed in Table S1,† and their average pore sizes are 2.3 and 2.8 nm, respectively. The slight decrease in the specific surface area of Pd(1.0)/MIL-125-NH2 possibly results from the presence of Pd nanocrystals.
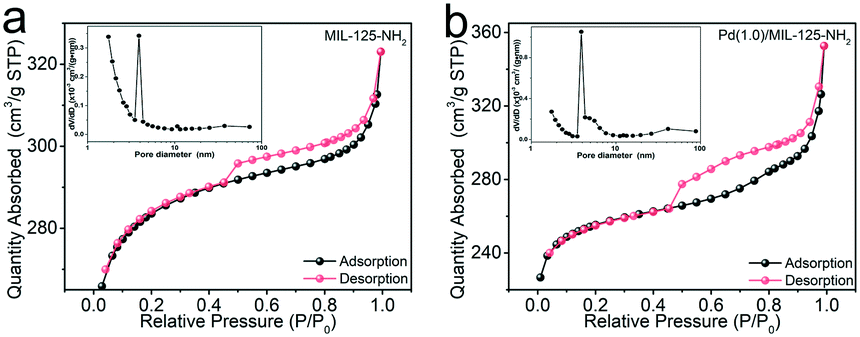 | ||
| Fig. 2 N2 adsorption–desorption isotherms and pore size distributions (inset) of (a) MIL-125-NH2 and (b) Pd(1.0)/MIL-125-NH2. | ||
XPS characterization techniques are applied to analyse the chemical states of surface elements as well as their interactions in MIL-125-NH2 before and after loading Pd nanocrystals. The survey spectra (Fig. S5†) indicate that the additional Pd signal emerges in Pd(1.0)/MIL-125-NH2 by comparison with that of the pristine MIL-125-NH2 sample. The XPS spectra (Fig. 3a–c) of Ti 2p, O 1s, and N 1s of Pd(1.0)/MIL-125-NH2 present negative shifts with respect to those of pristine MIL-125-NH2. The Ti 2p spectra (Fig. 3a) of Pd(1.0)/MIL-125-NH2 involve two pairs of Ti 2p3/2 and Ti 2p1/2 peaks, corresponding to Ti(IV) 2p (458.78 and 464.61 eV) and Ti(III) 2p (457.67 and 462.88 eV), respectively, which indicates that the introduction of Pd ultrasmall nanocrystals engenders Ti(III) in MIL-125-NH2 due to the oxygen vacancies and linker defects.43 The O 1s spectra (Fig. 3b) of both MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 samples are divided into three peaks centered at 529.95 (529.55), 531.55 (531.17), and 533.40 (533.05) eV, respectively, corresponding to O–Ti bonds in the titanium-oxo cluster, O–C and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups in the framework.44 In the N 1s spectra of both samples (Fig. 3c),the fitted two peaks at 399.36 (399.20) and 402.28 (400.84) eV can be ascribed to the –N+ and –NH−+ groups, respectively.45,46 Moreover, the N 1s binding energy for –NH−+ of Pd(1.0)/MIL-125-NH2 presents a 1.44 eV shift towards lower values, which can be attributed to the interaction between Pd and the amidogen of MIL-125-NH2. The Pd 3d spectrum (Fig. 3d) of Pd(1.0)/MIL-125-NH2 shows a pair of spin–orbit doublet of Pd 3d5/2 and Pd 3d3/2 peaks at 338.07 and 343.25 eV, respectively, corresponding to Pd(II).47 The positive shift in the binding energies of Pd 3d confirms the strong interaction between Pd and MIL-125-NH2 in the hybrid structures, which is consistent with the negative shifts in the Ti 2p, O 1s, and N 1s peaks of Pd(1.0)/MIL-125-NH2 in comparison to the pristine MIL-125-NH2.36 The electron transfer from Pd ultrasmall nanocrystals to MIL-125-NH2 reduces more Ti(III) sites and oxygen vacancies, which are beneficial to catalysis by promoting charge transfer and providing more active sites.39
C groups in the framework.44 In the N 1s spectra of both samples (Fig. 3c),the fitted two peaks at 399.36 (399.20) and 402.28 (400.84) eV can be ascribed to the –N+ and –NH−+ groups, respectively.45,46 Moreover, the N 1s binding energy for –NH−+ of Pd(1.0)/MIL-125-NH2 presents a 1.44 eV shift towards lower values, which can be attributed to the interaction between Pd and the amidogen of MIL-125-NH2. The Pd 3d spectrum (Fig. 3d) of Pd(1.0)/MIL-125-NH2 shows a pair of spin–orbit doublet of Pd 3d5/2 and Pd 3d3/2 peaks at 338.07 and 343.25 eV, respectively, corresponding to Pd(II).47 The positive shift in the binding energies of Pd 3d confirms the strong interaction between Pd and MIL-125-NH2 in the hybrid structures, which is consistent with the negative shifts in the Ti 2p, O 1s, and N 1s peaks of Pd(1.0)/MIL-125-NH2 in comparison to the pristine MIL-125-NH2.36 The electron transfer from Pd ultrasmall nanocrystals to MIL-125-NH2 reduces more Ti(III) sites and oxygen vacancies, which are beneficial to catalysis by promoting charge transfer and providing more active sites.39
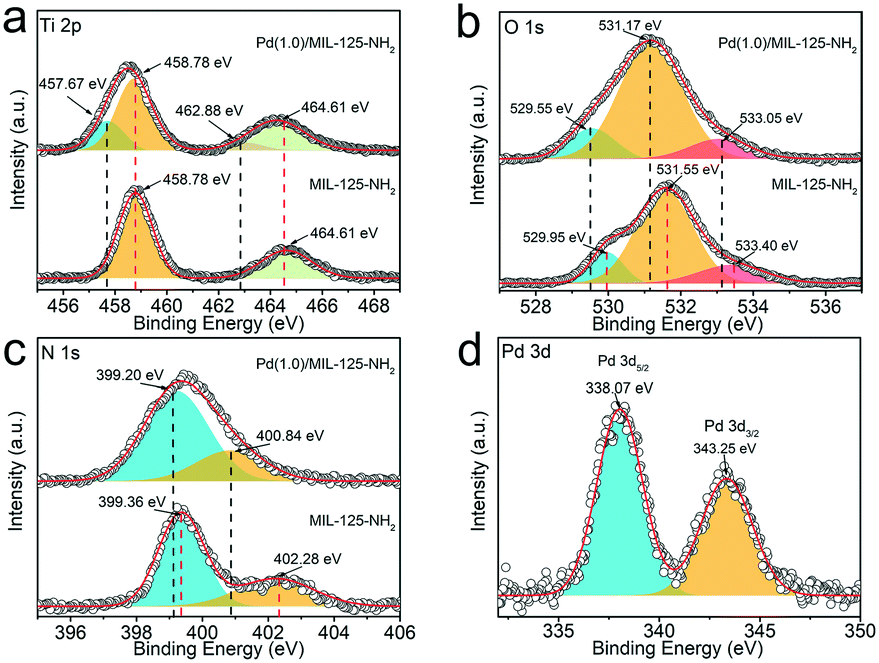 | ||
| Fig. 3 High-resolution XPS spectra of (a) Ti 2p, (b) O 1s, (c) N 1s and (d) Pd 3d for MIL-125-NH2 and Pd(1.0)/MIL-125-NH2, respectively. | ||
Photoabsorption and photogenerated carrier separation
The photoabsorption properties of the MIL-125-NH2 sample and different Pd(x)/MIL-125-NH2 hybrid materials are determined by DRS analysis (Fig. 4a). The pristine MIL-125-NH2 presents strong absorption in the range of wavelengths less than ca. 475 nm. The absorption bands of Pd(x)/MIL-125-NH2 hybrid structures show a little shift towards longer wavelengths due to interband transition and light scattering of Pd nanocrystals.48,49 The HOMO/LUMO gap of the MIL-125-NH2 sample is calculated to be 2.71 eV by a Tauc plot (Fig. S6†). To elucidate the semiconductor-like character of MIL-125-NH2, its Mott–Schottky plot was measured (Fig. 4b), and the positive slope of the plot demonstrates the n-type semiconductor feature of MIL-125-NH2. The flat band potential (Vfb) of MIL-125-NH2 was determined from the intersection value to be −1.09 V vs. Ag/AgCl. Accordingly, the LUMO potential of MIL-125-NH2 was determined to be −0.89 V vs. NHE, and the HOMO potential was calculated to be 1.81 V vs. NHE, on the basis of the HOMO/LUMO gap of 2.71 eV.50,51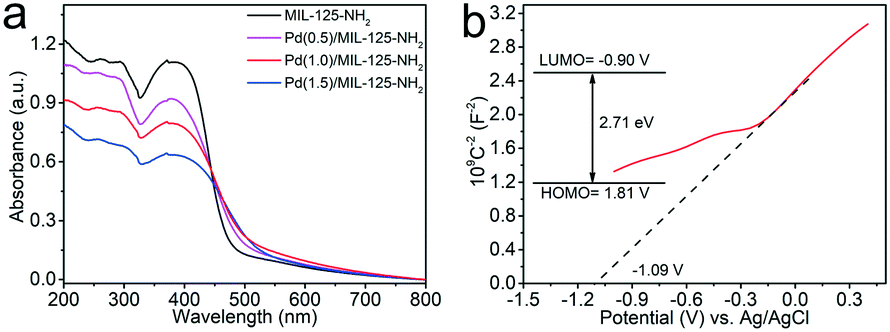 | ||
| Fig. 4 (a) UV-vis DSR of MIL-125-NH2 and different Pd(x)/MIL-125-NH2 samples, (b) Mott–Schottky plots and HOMO/LUMO levels (inset) of the pristine MIL-125-NH2. | ||
The PL spectra of MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 are measured to reflect the photogenerated carrier separation of Pd/MIL-125-NH2 hybrid structures. Under the excitation wavelength of 300 nm, the Pd(1.0)/MIL-125-NH2 hybrid structures show a quenched fluorescence emission intensity compared with pure MIL-125-NH2 (Fig. 5a), suggesting the suppressed recombination of photogenerated electron–hole pairs due to the efficient electron transfer from MIL-125-NH2 to Pd NPs. The time-resolved photoluminescence (TRPL) decay profiles of the two samples are shown in Fig. 5b, to probe their photogenerated carrier dynamics. The TRPL decay profiles can be fitted by the biexponential decay function:
| I(t) = A1exp(−t/τ1) + A2exp(−t/τ2) | (1) |
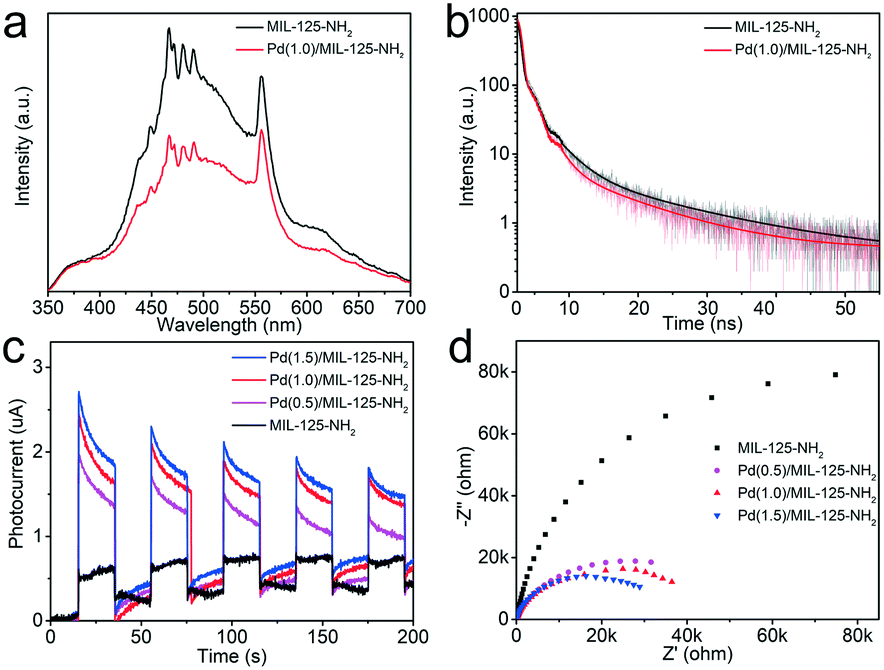 | ||
| Fig. 5 (a) Steady PL spectra under the excitation wavelength of 300 nm and (b) time-resolved photoluminescence (TRPL) decay profiles of MIL-125-NH2 and Pd(1.0)/MIL-125-NH2. (c) Photocurrents and (d) EIS Nyquist plots of MIL-125-NH2 and Pd(x)/MIL-125-NH2 with different Pd loading amounts. | ||
Photocatalytic selective oxidation of benzylamines
The above analyses predicted the great potential of Pd(x)/MIL-125-NH2 hybrid materials as photocatalysts. The photocatalytic performance of Pd(x)/MIL-125-NH2 with varied Pd loading amounts is assessed by the selectively oxidative coupling of benzylamine to imine (Fig. 6a). Photocatalytic reactions were carried out under ambient conditions and with an irradiation of 360–780 nm for 3 h. As illustrated in Fig. 6b and Table S3,† the pristine MIL-125-NH2 shows a moderate activity to achieve a benzylamine conversion of 68.82% and a dissatisfactory selectivity of 86.01% for producing the corresponding imine. With introducing ultrasmall Pd nanocrystals, Pd(0.2)/MIL-125-NH2, Pd(0.5)/MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 hybrid photocatalysts exhibit both the incremental activity and the excellent selectivity (>99%) for the oxidative coupling of benzylamine. The highest benzylamine conversion of 94.08% was achieved in the presence of the Pd(1.0)/MIL-125-NH2 catalyst after 3 h reaction, corresponding to a conversion rate of 3136 μmol h−1 gcat−1, which is about 1.37 times that of pristine MIL-125-NH2. However, with further increasing Pd-loading amount, the Pd(1.5)/MIL-125-NH2 sample exhibited a much decreased benzylamine conversion (62.46%) in spite of maintaining the excellent selectivity. The variations in the photocatalysis of Pd(x)/MIL-125-NH2 hybrid structures along with their varied Pd loading are possibly due to that the properly increased Pd content provides more active sites in the Pd(1.0)/MIL-125-NH2 catalyst, but excess Pd loading leads to the formation of larger Pd nanocrystals which blocks the structural pores of MIL-125-NH2 and reduces the efficient active sites. As a result, the Pd(1.0)/MIL-125-NH2 sample exhibits the best photocatalytic activity for selective oxidation. Then the photocatalytic performance of Pd(1.0)/MIL-125-NH2 is determined under different conditions (Fig. 6c and Table S4†). The oxidation of benzylamine cannot proceed in the absence of either catalyst or light irradiation. Under visible light irradiation (λ > 420 nm), the Pd(1.0)/MIL-125-NH2 hybrid catalyst also shows higher benzylamine conversion (77.41% after 3 h) and product selectivity (99.70%) than pristine MIL-125-NH2 (52.61% for conversion and 82.61% for selectivity), confirming the significant role of Pd ultrasmall nanocrystals in improving the photocatalytic activity and selectivity. In addition, the result for photocatalytic selective oxidation of benzylamine in air is almost identical to that obtained under an O2 atmosphere, indicating the high oxygen activation ability of the Pd(1.0)/MIL-125-NH2 hybrid catalyst. Therefore, selective oxidation reactions are carried out in air in view of the sustainable chemistry. When the photocatalytic reaction is conducted in a N2 atmosphere, the benzylamine conversion decreases to 42.74% after the same time, indicating that the molecular O2 is crucial for the oxidative coupling of benzylamine to imine. Furthermore, the photocatalytic benzylamine conversion over Pd(1.0)/MIL-125-NH2 is investigated under different monochromatic lights from the Xe lamp with optical filters. As shown in Fig. 6d and Table S5,† the variation tendency of benzylamine conversion under various monochromatic wavelengths from 365 to 700 nm coincides with the photoabsorption intensity of the Pd(1.0)/MIL-125-NH2 catalyst at the corresponding wavelengths. The photocatalytic efficiency of Pd(1.0)/MIL-125-NH2 is dominated in the wavelength range of 365–450 nm corresponding to its intensive photo-absorption in this region. Moreover, the selective oxidation of benzylamine can proceed slowly in the visible light region from 500 to 700 nm, in agreement with the visible light harvesting capability of this photocatalyst. The photoabsorption of Pd(1.0)/MIL-125-NH2 hybrid structures covers almost the whole UV-visible region of the solar spectrum, which contributes to achieving high utilization of solar energy for photocatalytic reactions.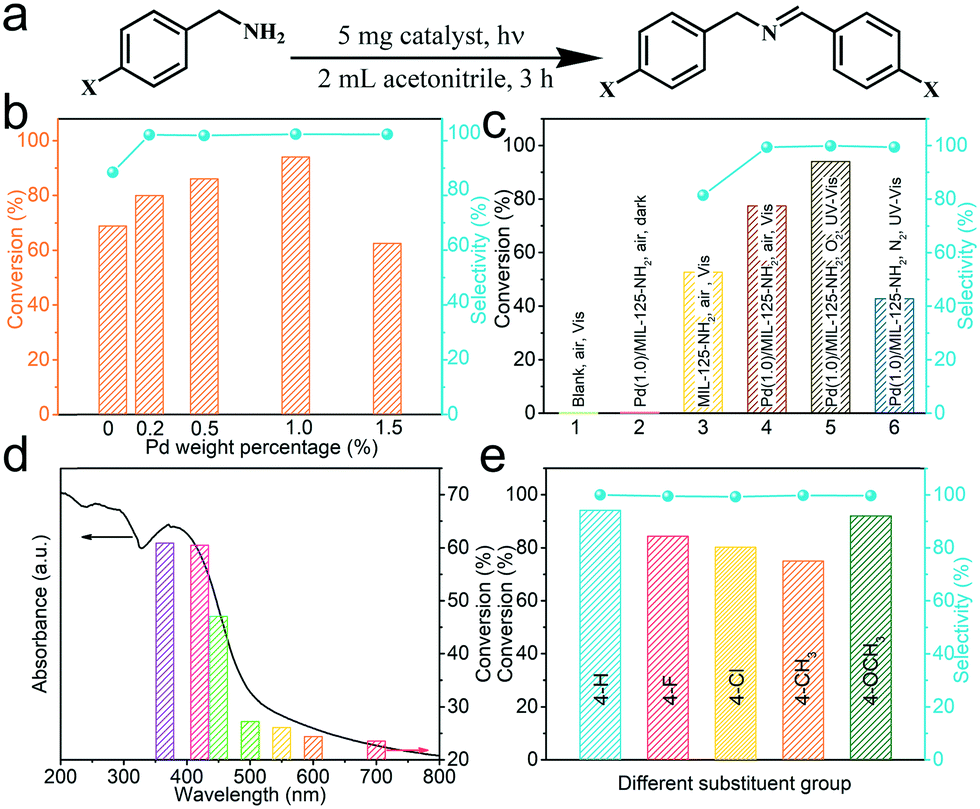 | ||
| Fig. 6 (a) Photocatalytic oxidative coupling of benzylamines to corresponding imines, (b) conversion and selectivity of benzamine oxidation after 3 h over the Pd(x)/MIL-125-NH2 photocatalyst with different Pd loading amounts, (c) the results of selective benzylamine oxidation under different conditions, (d) the relationship between conversion of benzylamine and light harvesting of Pd(1.0)/MIL-125-NH2, and (e) photocatalytic selective oxidation of benzylamines with different substituent groups catalyzed by Pd(1.0)/MIL-125-NH2. | ||
On the basis of the optimal photocatalytic performance of the Pd(1.0)/MIL-125-NH2 hybrid catalyst, the selective oxidation of benzylamines with various substituents is carried out under the same conditions for 3 h (Fig. 6e and Table S6†). Among various substrates, unsubstituted benzylamine holds the highest conversion rate (94.08%), compared with benzylamines with 4-methoxy (91.76%), 4-methyl (77.22%), 4-F (84.82%) and 4-Cl (80.22%) for producing the corresponding imines with high selectivity (>99%), indicating that the universality of the Pd(1.0)/MIL-125-NH2 photocatalyst for the oxidative coupling of amines. The differences in the conversion rates of various substrates are determined by both steric and electronic effects of the reactants. Finally, the recyclability of the Pd(1.0)/MIL-125-NH2 photocatalyst is tested by the selective oxidation of benzylamine under the same irradiation (Fig. S7a and Table S7†). The cyclic experiment demonstrates that the activity and selectivity of the Pd(1.0)/MIL-125-NH2 catalyst are well maintained after 3 cycles except a slight decrease in the activity due to the loss of the photocatalyst during the separation using a centrifuge. Moreover, the XRD patterns (Fig. S7b†) of Pd(1.0)/MIL-125-NH2 before and after the three cycles remained unchanged, suggesting that the crystal structure of the catalyst are well preserved and it has good stability to undergo a long-term reaction. In addition, the Pd(x)/MIL-125-NH2 catalysts present the same variation tendency in their photocatalysis toward selective oxidation of benzyl alcohol (Table S8†) along with the varied Pd loading contents, and the selectivity for producing benzaldehyde is 100%. Meanwhile, benzyl alcohol oxidation needs a much longer reaction time due to the moderate oxidizing power of the present photocatalytic system, which can avoid the overoxidation of organic molecules.
Insight into the photocatalytic mechanism
The oxygen activation process plays an important role in the selective oxidation reactions with molecular oxygen as an oxidant. To account for the enhanced activity and selectivity of Pd(1.0)/MIL-125-NH2 hybrid structures toward the selective oxidation of benzylamine, molecular oxygen activation was probed by TMB oxidation through monitoring the evolution in the spectral absorption of TMB solution along the irradiation time. As shown in Fig. S8,† upon one-electron oxidation by the reactive oxygen species (ROS) generated from the Pd(1.0)/MIL-125-NH2 photocatalyst, the colorless TMB (λmax = 285 nm) gradually turns into a blue charge-transfer complex (λmax = 370, 650 nm) of the parent diamine and the diimine oxidation product.17,54 The charge-transfer complex can both exist in rapid equilibrium with the radical cation. The time-dependent absorption spectra of TMB solution (Fig. 7a) reflect the higher O2 activation efficiency of Pd(1.0)/MIL-125-NH2 than that of pristine MIL-125-NH2. Subsequently, in order to identify the type of ROS generated in the photocatalytic system, different scavengers including superoxide dismutase (SOD), carotene, mannitol, and catalase were introduced into the TMB solution to consume superoxide (˙O2−), singlet oxygen (1O2), hydroxyl radicals (˙OH), and hydrogen peroxide (H2O2), respectively (Fig. 7b, S9 and S10†). For MIL-125-NH2, TMB oxidation is significantly suppressed by the addition of SOD but little influenced by other scavengers, suggesting that ˙O2− is the primary ROS generated during photocatalytic O2 activation over MIL-125-NH2. In the case of the Pd(1.0)/MIL-125-NH2 hybrid catalyst, TMB oxidation can be remarkably restrained by both SOD and carotene, indicating that ˙O2− and 1O2 are dominated together in this reaction system. In consequence, the incorporation of Pd ultrasmall nanocrystals into MIL-125-NH2 can enhance the O2 activation efficiency as well as facilitate the generation of 1O2 species, thereby greatly improving the photocatalytic activity and selectivity toward selective oxidation of benzylamines.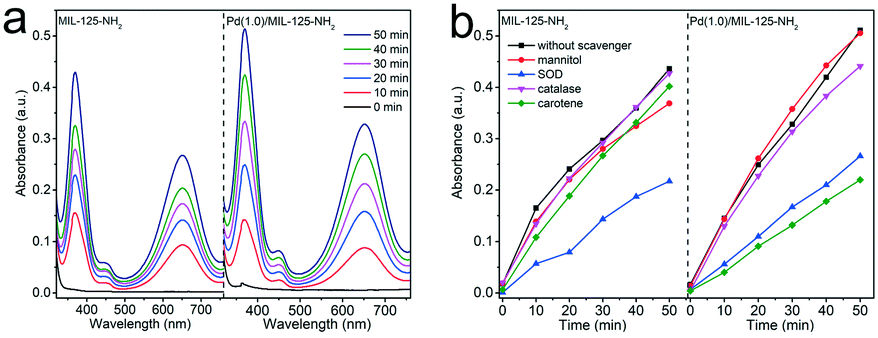 | ||
| Fig. 7 (a) Time-dependent absorption spectra of TMB oxidation with MIL-125-NH2 and Pd 1.0/MIL 125-NH2 in air, and (b) the absorbance evolutions monitored for TMB oxidation at 370 nm over MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 in the presence of different scavengers. | ||
Furthermore, ESR detections provide more specific information about the photogenerated ROS and other radical intermediates during the photocatalytic oxidative coupling of benzylamine. With TEMP as a trapping agent, the 1:1:1 triplet signals are the characteristic of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), corresponding to the generation of 1O2.20 With MIL-125-NH2 as a photocatalyst, the weak triple peaks remained the same whether in the dark or under irradiation (Fig. 8a), which indicates that the pristine MIL-125-NH2 cannot effectively produce 1O2 by photoexcitation. The emergence of weak triple signals in the dark is possibly due to the inevitable oxidation of TEMP upon exposure to air.55 However, in the case of the Pd(1.0)/MIL-125-NH2 hybrid photocatalyst, the triple-signal intensity is significantly intensified upon illumination (Fig. 8b), confirming the photogeneration of 1O2 on Pd(1.0)/MIL-125-NH2. After introducing benzylamine into the reaction system, the triple signal is weakened because of the consumption of 1O2 by the selective oxidation of benzylamine. In addition, both MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 can create the sextet signal (Fig. 8c) under illumination, which proves the formation of DMPO–OOH, a spin adduct derived from DMPO–˙O2−.56 Moreover, Pd(1.0)/MIL-125-NH2 exhibits a stronger sextet signal than that generated by MIL-125-NH2 under the same conditions, demonstrating the higher ability of Pd(1.0)/MIL-125-NH2 for producing ˙O2− radical. The addition of benzylamine into the system also remarkably suppresses the DMPO–˙O2− signal (●) from Pd(1.0)/MIL-125-NH2 (Fig. 8d), and a set of strong ESR signals (♦) appear simultaneously, which can be assigned to the benzyl-type nitrogen-centered or carbon-centered radicals.57 In contrast, pristine MIL-125-NH2 shows a very low activity to generate the benzyl-type nitrogen-centered or carbon-centered radicals. Therefore, the Pd(1.0)/MIL-125-NH2 hybrid photocatalyst possesses higher abilities for activating both O2 and benzylamine molecules, which jointly boosted the photocatalytic efficiency for benzylamine oxidation.
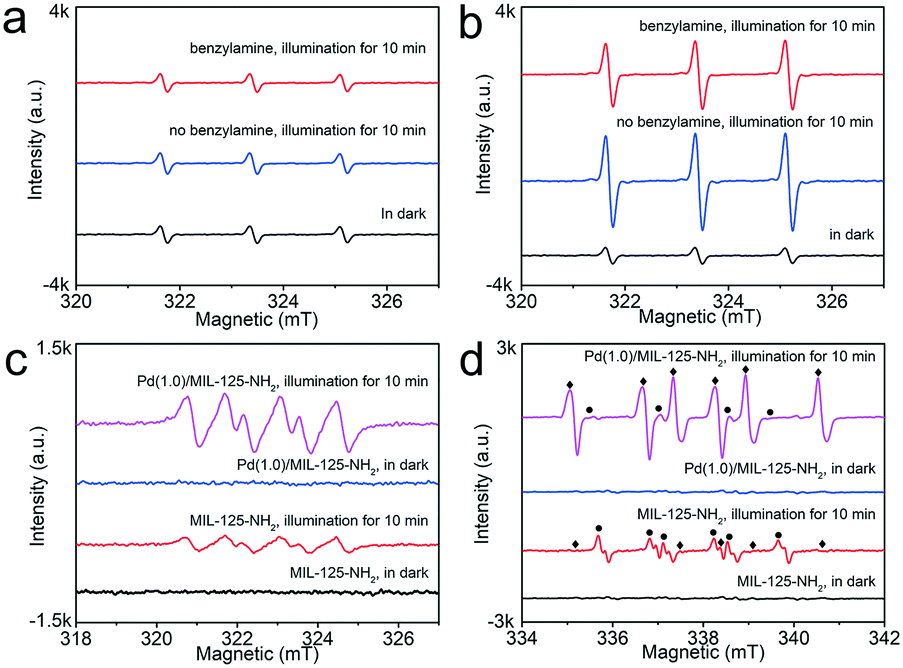 | ||
| Fig. 8 ESR detection of 1O2 generated by (a) MIL-125-NH2 and (b) Pd(1.0)/MIL-125-NH2 with TEMP as the 1O2 trapping agent. (c) ESR detection of ˙O2− generated by MIL-125-NH2 and Pd(1.0)/MIL-125-NH2 with DMPO as the ˙O2− trapping agent. (d) ESR signals of DMPO–˙O2− (●) and DMPO–nitrogen (or carbon)-centered radicals (♦). | ||
Based on the above results, the photocatalytic mechanism for selective oxidation of benzylamine to imine is illustrated in Fig. 9. Upon excitation under light irradiation, the electron–hole pairs are generated in MIL-125-NH2 by ligand-to-metal charge transfer (LMCT) where the electron transfer occurs from an organic ligand to Ti4+, similar to the generation of electrons and holes in semiconductors.41 Ti4+ is reduced to Ti3+ after accepting electrons, leaving holes in the organic ligand. Ti3+ is an active site for the absorption and reduction of O2 to form ˙O2− radicals.58 Simultaneously, the holes left in ligands oxidize benzylamine to PhCH2NH2˙+. Proton transfer takes place from PhCH2NH2˙+ to ˙O2−, resulting in the appearance of PhCH2NH˙ or PhCH˙NH2 and HO2˙, and the subsequent transformation into PhCHNH and H2O2. The final product of imine is produced by the condensation reaction of a PhCHNH intermediate with a benzylamine molecule. In the case of anchoring Pd ultrasmall nanocrystals into MIL-125-NH2, the electrons of Ti3+ sites in MIL-125-NH2 readily flow to the Fermi level of Pd, increasing the electron intensity on the surface of Pd nanocrystals, which promotes the activation of O2 to 1O2 on Pd nanocrystals.32 The generated 1O2 having high electrophilicity can oxidize amine to a PhCHNH intermediate, along with the release of H2O2.55,59 As a consequence, both ˙O2− and 1O2 are the dominating reactive oxygen species contributing the selective oxidation of benzylamine in the presence of Pd(1.0)/MIL-125-NH2 as a photocatalyst. H2O2 production has been detected in the photocatalytic reaction solution of benzylamine oxidation (Fig. S11†). The evolution trends of both benzylamine conversion and H2O2 concentration are coincident, suggesting that the benzylamine oxidation process is accompanied by H2O2 production. Moreover, the H2O2 amount produced by the Pd(1.0)/MIL-125-NH2 hybrid catalyst is much higher than that by pristine MIL-125-NH2, confirming the high efficiency of O2 activation on Pd(1.0)/MIL-125-NH2 catalyst. Therefore, the Pd/MIL-125-NH2 hybrid nanostructures have been verified to possess remarkable superiority in the spatial separation of photoinduced electron–hole pairs as well as the activation of O2 and benzylamine molecules. These abilities endow the Pd/MIL-125-NH2 hybrid material with excellent photocatalytic activity toward the selective oxidation of organics.
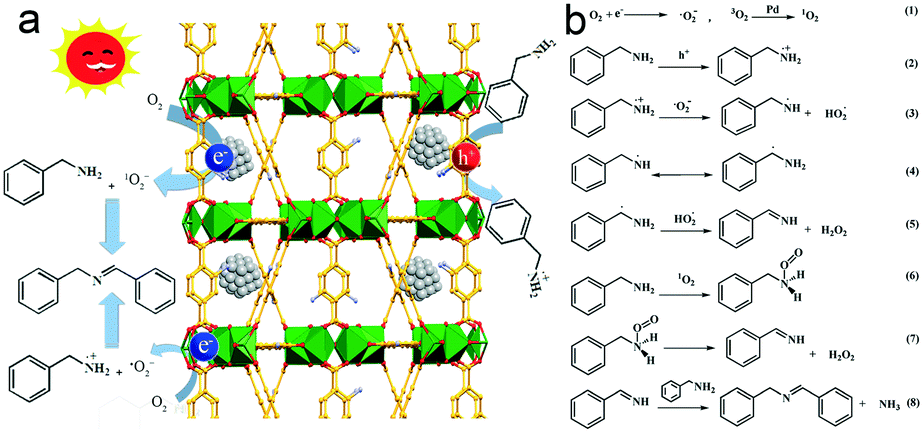 | ||
| Fig. 9 (a) Schematic illustration for the photocatalytic mechanism of Pd/MIL-125-NH2 hybrid structures, and (b) the possible pathway of oxidative coupling of benzylamine. | ||
Conclusions
In summary, Pd/MIL-125-NH2 hybrid structures are synthesized by controllably anchoring Pd ultrasmall nanocrystals (<2 nm) in the structural pores of MIL-125-NH2. The optimal photocatalyst of Pd(1.0)/MIL-125-NH2 exhibits excellent activity, selectivity and recyclability in the oxidative coupling of aromatic amines under ambient conditions, far surpassing the pristine MIL-125-NH2. The introduction of ultrasmall Pd nanocrystals plays a significant role in promoting the generation and separation of photo-induced electron–hole pairs as well as the catalytic activation of substrate molecules. The enhancement in charge-carrier kinetics and ROS generation in the system of the Pd/MIL-125-NH2 hybrid catalyst are demonstrated by the joint observations from PL spectroscopy, photoelectrochemical tests, and TMB oxidation experiments. The simultaneous generation of ˙O2− and 1O2 is dominated in this system. The reactive intermediates generated during the photocatalytic oxidative coupling of benzylamine are further verified by the in situ ESR technique and H2O2 detection, providing an in-depth understanding on the photocatalytic processes. Benefiting from the dual advantages of promoted charge separation and efficient O2 activation, the Pd/MIL-125-NH2 hybrid material with an appropriate Pd content is identified as a promising photocatalyst in solar-driven selective oxidation of organics. This work not only demonstrates the potential of Pd/MOF hybrid structures as photocatalysts for selective organic transformations but also provides an in-depth understanding on charge separation and molecule activation in such hybrid photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21471004), the Natural Science Foundation of Zhejiang Province of China (No. LY19B010005), and the Science Foundation of Zhejiang Sci-Tech University (No. 17062002-Y).Notes and references
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhofer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Bruckner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS.
- I. Sorribes, L. Liu and A. Corma, ACS Catal., 2017, 7, 2698–2708 CrossRef CAS.
- J. Long, L. Wang, X. Gao, C. Bai, H. Jiang and Y. Li, Chem. Commun., 2012, 48, 12109–12111 RSC.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- B. Zhao, Y. Huang, D. Liu, Y. Yu and B. Zhang, Sci. China: Chem., 2019 DOI:10.1007/s11426-019-9620-1.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2019 DOI:10.1093/nsr/nwz146.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS.
- J. Kou, C. Lu, J. Wang, Y. Chen, Z. Xu and R. S. Varma, Chem. Rev., 2017, 117, 1445–1514 CrossRef CAS.
- Y. Xin, Y. Huang, K. Lin, Y. Yu and B. Zhang, Sci. Bull., 2018, 63, 601–608 CrossRef CAS.
- B. Yuan, R. Chong, B. Zhang, J. Li, Y. Liu and C. Li, Chem. Commun., 2014, 50, 15593–15596 RSC.
- H. Chen, C. Liu, M. Wang, C. Zhang, N. Luo, Y. Wang, H. Abroshan, G. Li and F. Wang, ACS Catal., 2017, 7, 3632–3638 CrossRef CAS.
- H. Wei, Z. Guo, X. Liang, P. Chen, H. Liu and H. Xing, ACS Appl. Mater. Interfaces, 2019, 11, 3016–3023 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- H. Hao, L. Zhang, W. Wang and S. Zeng, Catal. Sci. Technol., 2018, 8, 1229–1250 RSC.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS.
- H. Wang, S. Chen, D. Yong, X. Zhang, S. Li, W. Shao, X. Sun, B. Pan and Y. Xie, J. Am. Chem. Soc., 2017, 139, 4737–4742 CrossRef CAS.
- X. Sun, X. Luo, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 3797–3801 CrossRef CAS.
- J. D. Xiao and H. L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS.
- G. Li, S. Zhao, Y. Zhang and Z. Tang, Adv. Mater., 2018, 30, 1800702 CrossRef.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Adv. Mater., 2017, 1703663 Search PubMed.
- Q. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- J. D. Xiao, L. Han, J. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS.
- H.-Q. Xu, S. Yang, X. Ma, J. Huang and H.-L. Jiang, ACS Catal., 2018, 8, 11615–11621 CrossRef CAS.
- J. W. Yoon, D. H. Kim, J.-H. Kim, H. W. Jang and J.-H. Lee, Appl. Catal., A, 2019, 244, 511–518 CrossRef CAS.
- Y. Isaka, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Sci. Technol., 2019, 9, 1511–1517 RSC.
- L. Liu, L. Zhang, F. Wang, K. Qi, H. Zhang, X. Cui and W. Zheng, Nanoscale, 2019, 11, 7554–7559 RSC.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS.
- R. Long, K. Mao, M. Gong, S. Zhou, J. Hu, M. Zhi, Y. You, S. Bai, J. Jiang, Q. Zhang, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 3205–3209 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS.
- X. Pei, Y. Deng, Y. Li, Y. Huang, K. Yuan, J.-F. Lee, T.-S. Chan, J. Zhou, A. Lei and L. Zhang, Nanoscale, 2018, 10, 14719–14725 RSC.
- L. Chen, W. Huang, X. Wang, Z. Chen, X. Yang, R. Luque and Y. Li, Chem. Commun., 2017, 53, 1184–1187 RSC.
- P. Zhang, C. Chen, X. Kang, L. Zhang, C. Wu, J. Zhang and B. Han, Chem. Sci., 2018, 9, 1339–1343 RSC.
- X. Qiu, J. Chen, X. Zou, R. Fang, L. Chen, Z. Chen, K. Shen and Y. Li, Chem. Sci., 2018, 9, 8962–8968 RSC.
- A. Malouche, G. Blanita, D. Lupu, J. Bourgon, J. Nelayah and C. Zlotea, J. Mater. Chem. A, 2017, 5, 23043–23052 RSC.
- D. Sun and Z. Li, J. Phys. Chem. C, 2016, 120, 19744–19750 CrossRef CAS.
- K. Titov, D. B. Eremin, A. S. Kashin, R. Boada, B. E. Souza, C. S. Kelley, M. D. Frogley, G. Cinque, D. Gianolio, G. Cibin, S. Rudić, V. P. Ananikov and J.-C. Tan, ACS Sustainable Chem. Eng., 2019, 7, 5875–5885 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- A. P. Smalley, D. G. Reid, J. C. Tan and G. O. Lloyd, CrystEngComm, 2013, 15, 9368–9371 RSC.
- B. Zhang, J. Zhang, X. Tan, D. Shao, J. Shi, L. Zheng, J. Zhang, G. Yang and B. Han, ACS Appl. Mater. Interfaces, 2018, 10, 16418–16423 CrossRef CAS PubMed.
- S. Venkateswarlu, A. Panda, E. Kim and M. Yoon, ACS Appl. Nano Mater., 2018, 1, 4198–4210 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng, Z. Wu, L. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS.
- R. Bibi, H. Huang, M. Kalulu, Q. Shen, L. Wei, O. Oderinde, N. Li and J. Zhou, ACS Sustainable Chem. Eng., 2018, 7, 4868–4877 CrossRef.
- F. R. Fortea-Pérez, M. Mon, J. Ferrando-Soria, M. Boronat, A. Leyva-Pérez, A. Corma, J. M. Herrera, D. Osadchii, J. Gascon, D. Armentano and E. Pardo, Nat. Mater., 2017, 16, 760–766 CrossRef.
- S. Sarina, H.-Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., Int. Ed., 2014, 53, 2935–2940 CrossRef CAS.
- R. Wang, B. Li, Y. Xiao, X. Tao, X. Su and X. Dong, J. Catal., 2018, 364, 154–165 CrossRef CAS.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- S. Gao, W. Cen, Q. Li, J. Li, Y. Lu, H. Wang and Z. Wu, Appl. Catal., A, 2018, 227, 190–197 CrossRef CAS.
- X. Wang, C. Liow, A. Bisht, X. Liu, T. C. Sum, X. Chen and S. Li, Adv. Mater., 2015, 27, 2207–2214 CrossRef CAS.
- M. V. Dozzi, A. Candeo, G. Marra, C. D'Andrea, G. Valentini and E. Selli, J. Phys. Chem. C, 2018, 122, 14326–14335 CrossRef CAS.
- P. Josephy, T. Eling and R. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CAS.
- C. Xu, H. Liu, D. Li, J. H. Su and H. L. Jiang, Chem. Sci., 2018, 9, 3152–3158 RSC.
- H. Wang, D. Yong, S. Chen, S. Jiang, X. Zhang, W. Shao, Q. Zhang, W. Yan, B. Pan and Y. Xie, J. Am. Chem. Soc., 2018, 140, 1760–1766 CrossRef CAS.
- R. Wang, G. Qiu, Y. Xiao, X. Tao, W. Peng and B. Li, J. Catal., 2019, 374, 378–390 CrossRef CAS.
- D. Sun, L. Ye and Z. Li, Appl. Catal., A, 2015, 164, 428–432 CrossRef CAS.
- E. Baciocchi, T. Del Giacco, O. Lanzalunga and A. Lapi, J. Org. Chem., 2007, 72, 9582–9589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy01690b |
| This journal is © The Royal Society of Chemistry 2020 |