
Understanding the molecular properties of the E1 subunit (SucA) of α-ketoglutarate dehydrogenase complex from Vibrio vulnificus for the enantioselective ligation of acetaldehydes into (R)-acetoin†
Pil-Won
Seo‡
a,
Hye-Jin
Jo‡
b,
In Yeub
Hwang‡
c,
Ha-Yeon
Jeong
b,
Jun-Hong
Kim
a,
Ji-Won
Kim
a,
Eun Yeol
Lee
*c,
Jin-Byung
Park
 *b and
Jeong-Sun
Kim
*a
*b and
Jeong-Sun
Kim
*a
aDepartment of Chemistry, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: jsunkim@chonnam.ac.kr
bDepartment of Food Science and Engineering, Ewha Womans University, Seoul 03760, Republic of Korea. E-mail: jbpark06@ewha.ac.kr
cDepartment of Chemical Engineering, Kyung Hee University, Gyeonggi-do, 17104, Republic of Korea. E-mail: eunylee@khu.ac.kr
First published on 28th October 2019
Abstract
Escherichia coli SucA, a decarboxylating E1 component of the α-ketoglutarate dehydrogenase complex, has been reported to possess another catalytic activity in carboligating two acetaldehyde molecules to form acetoin in a thiamine diphosphate (ThDP)-dependent manner. Here, we examined acetoin formation activity from acetaldehyde using SucAs from various organisms, as well as the Zymomonas pyruvate decarboxylase (ZmPDC) and the formaldehyde-ligating formolase. Among the studied SucA enzymes, Vibrio vulnificus SucA (VvSucA) exhibited the highest ligating activity against acetaldehyde with an excellent enantioselectivity of the product in contrast to ZmPDC and formolase of poor enantioselectivity. The revealed VvSucA structure shows a dimeric assembly with the bound ThDP within the active sites at the two-subunit interface. Non-covalently bound glycolaldehyde molecules suggest the two substrate-binding sites within the carboligating active site for acetaldehyde. Structure-guided mutants reveal residues critical for regiospecificity of VvSucA. The obtained biochemical and structural data help to understand the high enantioselective ligation of two acetaldehyde molecules by VvSucA.
Introduction
The (R)-acetoin [(R)-3-hydroxy-2-butanone] is widely used as a flavor and fragrance in the food industry.1 It is also extensively used as an intermediate for chemical synthesis and as a multifunctional compound.2–4 Because of its broad applications, acetoin was classified as a promising bio-based platform chemical.5Acetoin is commercially produced by chemical means from fossil feedstocks such as 2,3-butanediol, butanone, and diacetyl.1,6 However, the chemical processes require harsh reaction conditions and thus cause environmental problems. An alternative method may include microbial fermentation.2 Acetoin is produced from pyruvate through two enzyme-reaction steps involving acetolactate synthase and α-acetolactate decarboxylase. The acetolactate synthase catalyzes carboligation of two pyruvate molecules into α-acetolactate, which is then converted into (R)-acetoin by α-acetolactate decarboxylase.7–9 However, the by-products (e.g., 2,3- butanediol, acetic acid, and lactic acid) can be formed during the fermentation, making the downstream processes complicated.
Acetoin can be also produced by enzyme catalysis.2,9–18 One example is to produce acetoin from acetaldehyde by using a formaldehyde-ligating formolase or pyruvate decarboxylase.10,14,15 However, these enzymes usually generate racemic products, which are different from natural acetoin [(R)-3-hydroxy-2-butanone]. Alternatively, the E1 component of the α-ketoglutarate dehydrogenase complex (i.e., SucA) of Escherichia coli (EcSucA) was reported to produce (R)-acetoin as a major product (Scheme S1†) when acetaldehyde was provided as a sole substrate.17,18 However, the molecular features to identify the enantioselectivity remained undiscovered.
Most of the carboligating enzymes including acetolactate synthase, formolase, pyruvate decarboxylase, and SucA belong to the thiamine diphosphate (ThDP)-dependent enzymes.19 These enzymes possess the common enzymatic mechanism, containing generation of carbanion or ylide of the bound ThDP cofactor, aided by enzymes. The initial carbanion is formed by dissociation of the proton from the C2–H of the thiazolium ring of ThDP, which generates the anion at the C2 atom (ylide) that consequently attacks the donor substrate to form a covalent bond.20–22 The second carbanion, for example, formation of a covalent bond between two carbon atoms of two substrates, is formed at the carbon added to the thiazolium ring of ThDP from the first substrate, which induces the nucleophilic attack of the carbonyl moiety of the second substrate, resulting in the covalent bond formation of the two carbon atoms of the two substrates. However, enantioselectivities of the enzymes for the ligation of two molecules of acetaldehyde into acetoin are significantly different from each other.9–18,23
Therefore, we have investigated the carboligating activities of the bacterial SucAs including E. coli (EcSucA), Mycobacterium bovis (MbSucA), Vibrio vulnificus (VvSucA), Methylomicrobium alcaliphilum 20Z (MaSucA), and Methylomonas sp. DH-1 (MtSucA, MtputSucA). Furthermore, their reaction characteristics were compared with those of the formolase23 and pyruvate decarboxylase14 in order to understand the molecular features that determine the enantioselectivity for the synthesis of (R)-acetoin from acetaldehyde. This approach may contribute to the production of (R)-acetoin from low-value acetaldehyde that can be used as a flavor and/or pharmaceutical intermediates.
Results and discussion
Vibrio SucA is an efficient enantioselective carboligase of acetaldehyde
With an aim to understand the reaction properties of the bacterial SucAs for ligation of two molecules of acetaldehyde into (R)-acetoin (Scheme S1†), we prepared the SucAs from E. coli (EcSucA), M. bovis (MbSucA), V. vulnificus (VvSucA), M. alcaliphilum 20Z (MaSucA), and Methylomonas sp. DH-1 (MtSucA, MtputSucA) via expression in E. coli BL21(DE3) (Tables S1 and S2†) and purification by affinity chromatography on a Ni-NTA gel matrix. Most enzymes were expressed in a soluble form in E. coli and purified as a single band on SDS-PAGE (Fig. S1†). Among the tested SucA enzymes, the VvSucA produced the largest amount of acetoin from acetaldehyde in 10 h after biotransformation (Fig. 1), indicating that VvSucA is the most active for the carboligation of acetaldehyde. The low product formation by the SucAs from M. alcaliphilum 20Z and Methylomonas sp. DH-1 remained to be investigated.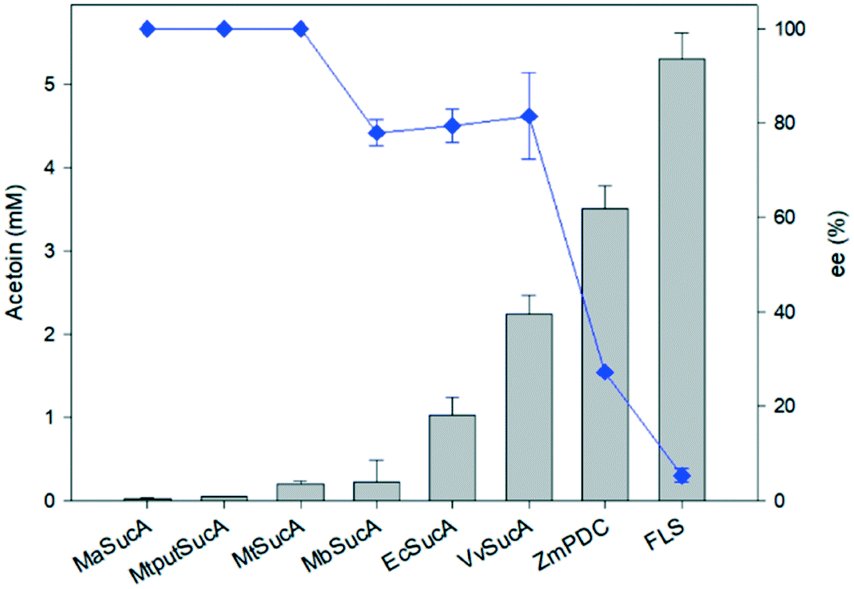 | ||
| Fig. 1 Acetoin formation by carboligating enzymes. The acetoin concentrations and the enantiomeric excesses were measured 10 h after biotransformation of acetaldehyde (25 mM) with the enzymes (1 mg mL−1). The cofactors such as magnesium(II) ion and thiamine pyrophosphate were added to a concentration of 0.1 mM and 2 mM, respectively, into the reaction medium. The error bars indicated the standard deviation. | ||
We also carried out biotransformation of acetaldehyde by the formolase, the engineered variant of benzaldehyde lyase from Pseudomonas fluorescens biovar I (FLS),23 and the pyruvate decarboxylase from Zymomonas mobilis (ZmPDC),14 which were reported for their carboligating activities of acetaldehyde, under the conditions identical to the reaction by the SucAs. FLS and ZmPDC allowed higher acetoin production with 140% and 57% more acetoin formed than that of VvSucA, respectively (Fig. 1).
Analysis of the stereoisomers of the produced acetoin has shown that the SucAs displayed rather a high enantioselectivity toward R-conformation (Fig. 1); we observed the highest R-form acetoin formation in VvSucA (92 ee%), compared to 86 and 80 ee (%) from EcSucA and MbSucA, respectively (Fig. S2 to S4†). However, FLS generated a racemic mixture as a major product, whereas ZmPDC displayed a slightly greater preference for (S)-conformation (Fig. S5 and S6†).
The enantioselectivities of the SucA carboligases appeared to be inversely dependent upon the biotransformation activities (Fig. 1). Therefore, we investigated the biotransformation rates of the enzymes in more detail. When the carboligation rates of the enzymes were measured, FLS displayed significantly greater specific reaction rates than did the others (Fig. 2). However, the substrate conversion remained below 30%, which might be due to large KM value of FLS (i.e., 58 mM9). On the other hand, VvSucA showed the highest reaction rates among the SucAs examined. This result indicated that the enantioselectivities of the enzymes were correlated somehow inversely with the carboligation rates.
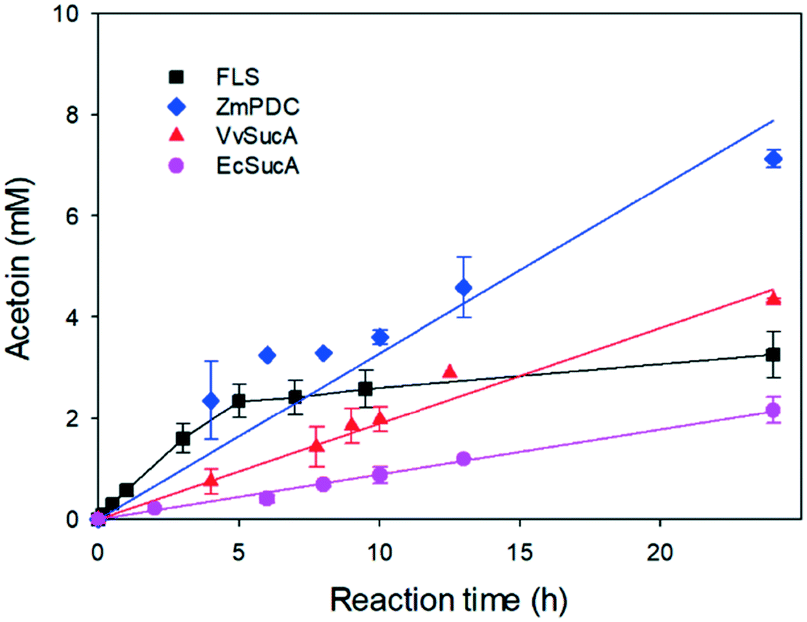 | ||
| Fig. 2 Time course of the biotransformations of acetaldehyde into acetoin by FLS, ZmPDC, VvSucA, and EcSucA. FLS was added to 0.1 mg mL−1 while the other enzymes were added to 1 mg mL−1 in the reaction medium containing 25 mM acetaldehyde and the cofactors (i.e., 0.1 mM thiamine pyrophosphate and 2 mM magnesium chloride). The error bars indicated the standard deviation. | ||
Taken together, EcSucA and VvSucA have shown meaningful catalytic activities, 20% and more than 40% activity, respectively, compared to FLS. Especially, (R)-stereoisomeric acetoin production was highest in VvSucA (92 ee%) among the SucAs with reasonable acetaldehyde-ligating activities, suggesting its possible usage for selective (R)-acetoin formation from acetaldehyde without using any extra energy.
Structural features of VvSucAΔ84
In order to get a structural background for the carboligase activity of VvSucA from acetaldehyde, we elucidated the crystal structures of VvSucAΔ84 (Asp85-Asp941) with and without a substrate analogue (Fig. 3). Most of the cloned residues of VvSucAΔ84 have been traced, except for five residues at the extreme C-terminus (Glu937-Asp941) and the region between Gly288 and Gly294. Besides the two protein molecules, two ThDP cofactors and two Mg2+ ions are also observed in the refined structure (Table S3†).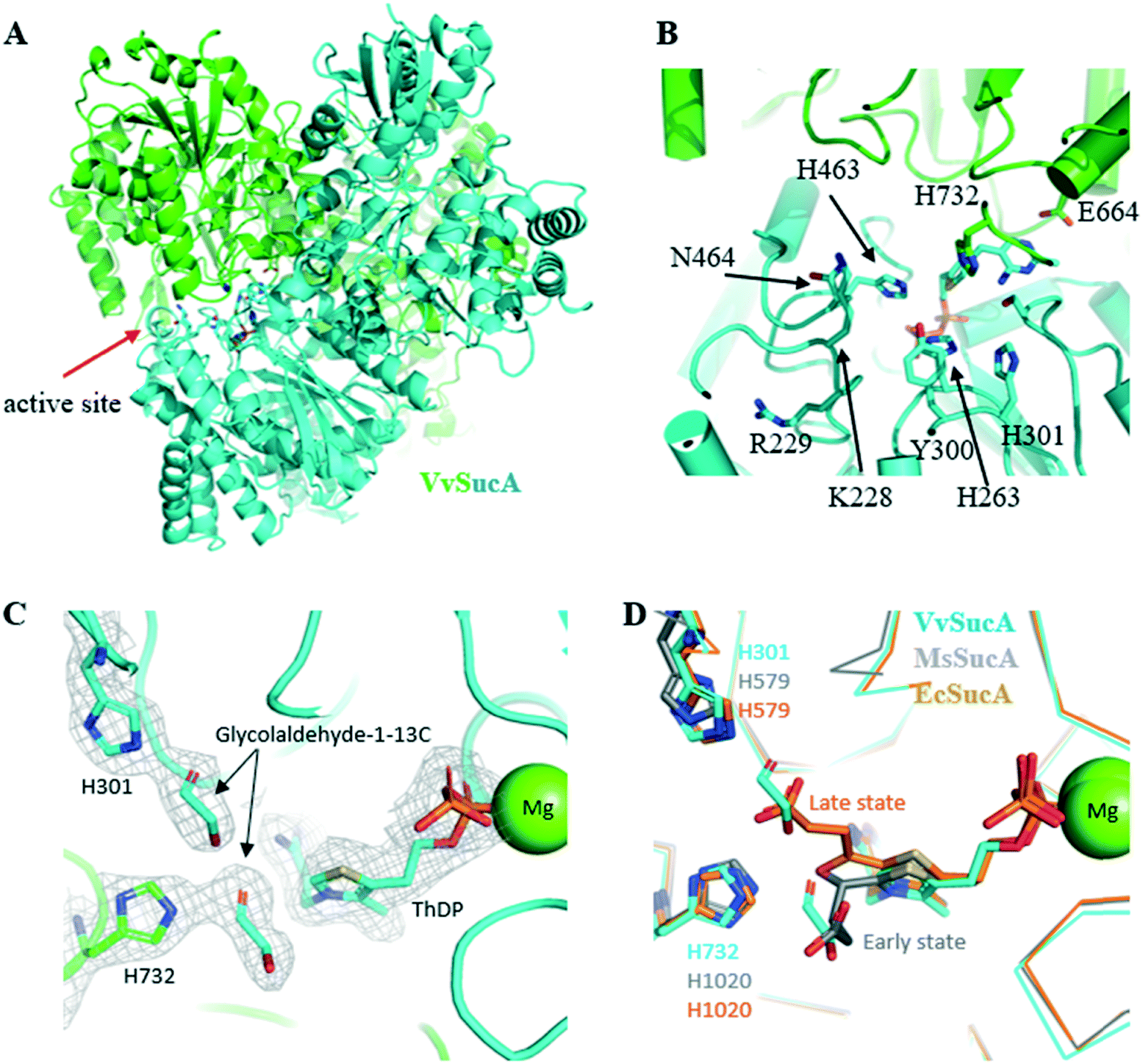 | ||
| Fig. 3 Crystal structure of VvSucA. (A) Overall structure of VvSucA. Two molecules were differentiated by green and cyan. Some residues at the active site formed at the intermolecular interface are displayed with stick models. (B) The active site of VvSucA. The α-helices and β-strands were depicted as cylinders and arrows, respectively. The bound ThDP cofactor and some conserved residues are displayed with stick models. (C) Two glycolaldehyde molecules bound to the active site of VvSucA. The bound glycolaldehyde-1-13C molecules are depicted with a stick model in the 2Fo-Fc map contoured at 1.0σ level, along with some key residues of stick models and a magnesium ion of sphere. (D) Superimposition of glycolaldehyde-1-13C-bound VvSucA (cyan) and the decarboxylated adduct of α-ketoglutarate to ThDP in Mycobacterium smegmatis SucA (MsSucA, early state, gray, PDB ID 3ZHS; late state, orange, PDB ID 2YID). | ||
The monomeric VvSucAΔ84 is composed of three similarly-folded globular domains, each of which has a central β-sheet with a number of α-helices at both sides. The α-helices of one domain interact with those of another to form an elongated monomeric structure. A dimeric structure is formed by associating three sequential domains with each other and the bound ThDP cofactor is found at the interfaces formed by the first two domains of both molecules (Fig. 3A). The diphosphate moiety of the bound ThDP interacts with the side-chain atoms of His263, Arg264, and Asn323 and the peptidyl amide nitrogen atoms of Ser360 and Ala361, whereas the pyrimidine ring moiety makes a hydrophilic interaction with the peptidyl carbonyl oxygen of Asn323 and the side chain atom of Glu662 from the neighboring monomer. The magnesium ions interact with the diphosphate moiety of ThDP and with the side-chain atom of Asp359.
Structural similarity search using the DALI server (http://ekhidna2.biocenter.helsinki.fi/dali/)24 suggests that VvSucA has a close structural relationship with other functionally related proteins, for example, α-KG dehydrogenase E1 component including E. coli and M. genitalium, pyruvate dehydrogenase E1 component, 2-oxoisovalerate dehydrogenase α-subunit, transketolase, xylulose-5-phosphate/fructose-6-phosphate phosphoketolase, oxalate oxidoreductase α-subunit, benzoylformate decarboxylase, FLS, and acetolactate synthase, with root-mean-square-deviation (RMSD) values of less than 3 Å.
In the next step to get a clue to how VvSucA interacts with a substrate, we determined the crystal structure of VvSucAΔ84 in complex with glycolaldehyde-1-13C, an acetaldehyde mimicker, at a resolution of 2.4 Å (Table S3†). Notably, the acetaldehyde-resembling glycolaldehyde was less effectively converted into a C4 product (erythrulose) than acetaldehyde by VvSucA (Fig. S7†), a fourth of the produced acetoin amount from acetaldehyde in 10 h. Two glycolaldehyde-1-13C molecules in the complex structure are found near the ThDP cofactor at the active site of VvSucAΔ84. One glycolaldehyde interacts with the side-chain atoms of His301 and Ser324 (Fig. 3C), whose spatial position coincides with the decarboxylated α-KG intermediate covalently attached to ThDP in the early state of MsSucA (Fig. 3D).25 The other glycolaldehyde hydrogen-bonds with the side-chain atoms of His732 and Gln688 (Fig. 3C) and its position also spatially matches with the carboxylate moiety of the decarboxylated α-KG that is attached to ThDP in the late state of MsSucA (Fig. 3D).25 The absence of a non-covalent adduct to the ThDP cofactor in the complex structure may be related with an insufficient incubation time of glycolaldehyde-1-13C into the packed VvSucA crystal. However, soaking a crystal for more than 30 min disordered the crystal packing, which made it impossible to collect the diffraction data (data not shown). Nonetheless, the observed interaction between the glycolaldehyde-1-13C and the protein residues is not likely to afford favourable access to the ThDP cofactor and explains VvSucA as a poor catalyst for glycolaldehyde, which is why a fairly small amount of the erythrulose product from the glycolaldehyde-1-13C substrate was obtained in solution by VvSucA (Fig. S7†).
Collectively, the observed structural features, including the ThDP cofactor and the ThDP-activating Glu residue at the active site, suggest that VvSucA shares the common enzymatic mechanism for its catalysis of carboligating two acetaldehyde molecules with other functionally related formolase and PDC proteins.
The entrance part to the active site is related to the carboligating activity of VvSucA
All the tested SucA enzymes have exclusively produced an (R)-acetoin from acetaldehyde, which contradicts the results from FLS producing a racemic mixture of acetoin and the ZmPDC preferentially producing an (S)-form acetoin (Fig. S2 to S6†). Especially, VvSucAΔ84 functions as the most efficient carboligase against acetaldehydes with the best enantioselectivity of (R)-acetoin among the tested enzymes (Fig. 1).The revealed crystal structure of VvSucAΔ84 is a dimeric structure with a bound ThDP cofactor at the intermolecular interface. As indicated from the high sequence identity between the three compared SucA enzymes (Fig. S8†), the overall structure of VvSucAΔ84 is almost the same as those of the reported MsSucA and EcSucA (RMSD values of less than 1 Å) (Fig. S9†) and several residues critical for decarboxylation are spatially conserved at the active site (Fig. 3D). Upon analysis of the aligned sequences, MsSucA has inserted residues at three regions that are far apart from the active site in the structures. Besides, the deviated regions of Lys223-Ser231 and His460-Glu467 of VvSucA form separate loops that interact with each other near the active site in VvSucA, but are disordered in EcSucA.
Based on the elucidated VvSucAΔ84 structures, together with comparison with the known MsSucA fragment structures in complex with the reaction intermediates,25 a gallery of mutations has been generated in order to get a clue to explain the high stereospecificity of VvSucAΔ84. Introducing a single-point mutation near the bound ThDP cofactor at the active site (N464Q, S324T, and Y300H) did not significantly change the (R)-specific enantioselectivity of the enzyme, despite their changed initial reaction rates (Fig. 4). Replacement of four successive residues between Val298 and His301 with three consecutive Gly residues (V298-H301-to-GGG), which forms a loop at the entrance to the active site, reduced its enzymatic activity. Interestingly, this derivative has more than 30% of the wild-type enzyme, its enantioselectivity for (R)-acetoin production is almost close up to 100% (Fig. 4).
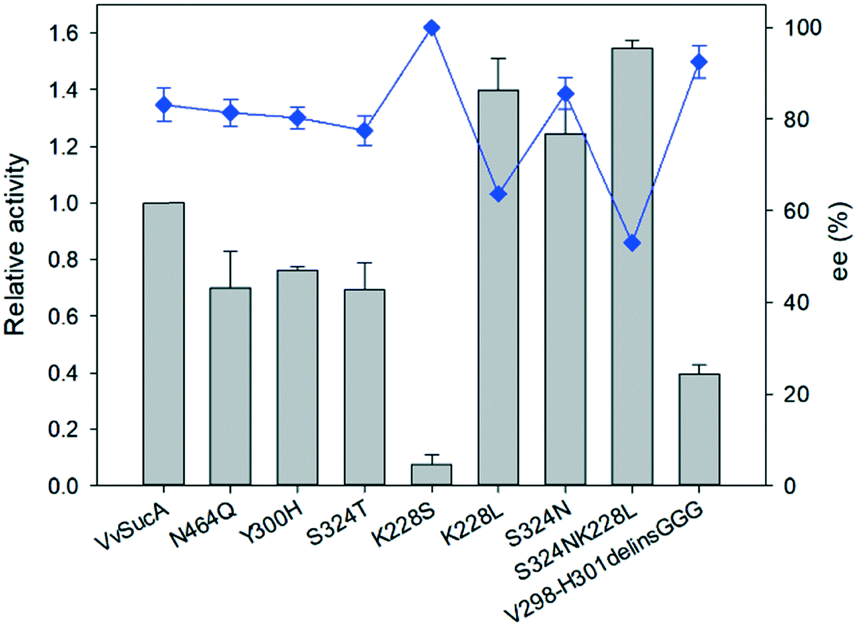 | ||
| Fig. 4 Relationship between carboligating activity and enantioselectivity of the VvSucA variants. The acetoin concentrations and the enantiomeric excesses were measured 10 h after biotransformation of acetaldehyde (25 mM) with the enzymes (1 mg mL−1). The cofactors such as magnesium(II) ion and thiamine pyrophosphate were added to a concentration of 0.1 mM and 2 mM, respectively, in the reaction medium. The error bars indicated the standard deviation. | ||
Another mutation at the entrance part to the active site (Lys228) has also shown contradictory results. Introduction of a small hydrophilic Ser residue almost eliminated its catalytic activity, whereas its replacement with the hydrophobic Leu residue increased its catalytic activity with the reduction of enantioselectivity (Fig. 4). Also, the Asn residue introduced at the Ser324 position of the active site increased the product amount with the decreased (R)-isomer, but its replacement with a Thr residue decreased the catalytic efficiency of the enzyme (Fig. 4). When a double mutation (K228L, S324N) was introduced at these two sites, the amount of the observed product was increased with the reduced amount of (R)-acetoin product (Fig. 4). These mutation studies indicate that these two residues are closely related to the enzyme catalysis with their partial relationship with the enantioselectivity of VvSucA and confirm the relationship to the enantioselectivities of the enzymes with the carboligation rates.
Two residues of Lys228 and Ser324 are well conserved among the compared SucAs (Fig. S8†). Ser324 residues occupy spatially equivalent positions in all three compared structures of EcSucA, MsSucA, and VvSucA, but the spatial positions of the conserved Lys residues are diverse: Lys225 of EcSucA was not traced and the MsSucA Lys504 belongs to the region with various spatial positions (a stretched loop or a single turn α-helix) upon complex with the reaction intermediates.25 This MsSucA lysine residue rotates around 90 degrees, when the decarboxylated intermediate is attached to the ThDP, resulting in the interaction between the Arg505 residue of MsSucA and the carboxylate moiety of the reaction intermediate. It is worthwhile to mention that the Arg505 residue-containing region of MsSucA is sequentially aligned to the Lys228 residue-including region of VvSucA (Fig. S8†) and they are located at the same space in the superposed structures (Fig. S9†), suggesting a different role of this region in decarboxylation and carboligation reactions of SucA enzymes.
Taken together, these obtained results from the structure and mutational studies strongly suggest that the incoming acetaldehyde molecule interacts with the Lys228 residue of VvSucA and some loops at the entrance part to the active site and this interaction probably affects the substrate orientation, together with Ser324 at the active site, to predominantly produce (R)-form acetoin.
Structural basis for the enantioselective carboligating activity of ZmPDC, FLS, and VvSucA
Our present work has shown that VvSucA, ZmPDC, and FLS successfully introduced a covalent bond between two carbon atoms of acetaldehyde molecules without consuming extra energy. These three enzymes are intrinsically different, in that VvSucA, composed of more than 800 residues, is a dimeric structure, and ZmPDC and FLS, with less than 600 residues, form a tetrameric structure.26,27 Even though these three enzymes have shown a different stereoisomeric product as well as a catalytic efficiency, they have common structural features that an indispensable ThDP cofactor and a magnesium ion within the active site are found at the interface of two subunits, indicating that there is a common catalytic mechanism for carboligation of acetaldehyde.The different catalytic efficiencies of the three enzymes against the acetaldehyde substrate appear to be related to their surface property: a relatively hydrophobic surface is formed at the entrance to the active site in FLS, while a positively charged surface is distributed in VvSucA (Fig. 5, upper panel). On the other hand, the optimal surface potential of ZmPDC cannot exactly be calculated, because of its complete closed structure. Nonetheless, it has a slightly positively charged surface near the active site. This surface potential matches well, for example, with the original substrate of FLS, the hydrophobic benzaldehyde, and of SucA, the negatively charged α-KG. Therefore, the initial reaction rate for carboligating acetaldehydes by these three enzymes is likely to depend on the affinity between acetaldehyde and the surfaces of the relevant enzymes. Of course, more favorable interaction between the acetaldehyde and the hydrophobic surface of FLS is expected than between the substrate and the positively charged surfaces of SucAs and ZmPDC. Notably, ZmPDC and formolase have shown a much faster initial reaction rate than that of VvSucA (Fig. 2). It would also be worthwhile to mention that introduction of a hydrophobic residue (K228L) at the entrance of the active site of VvSucA increased the activity, but replacement with a Ser residue almost depleted the catalytic activity (Fig. 4), indicating a relationship of the surface property of the enzyme with the initial reaction rate in VvSucA.
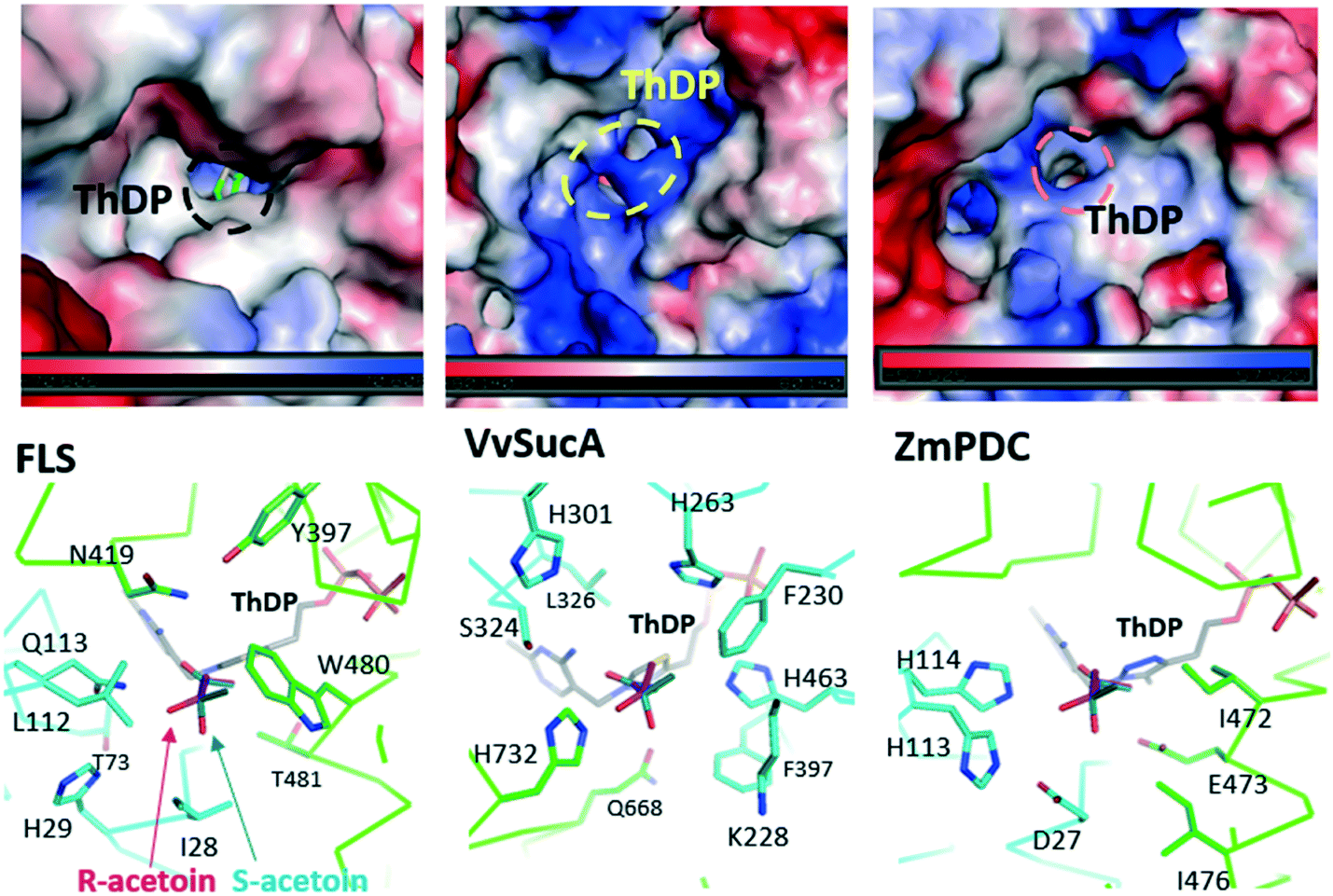 | ||
| Fig. 5 Structural comparison of FLS, ZmPDC, and VvSucA. The three structures are similarly oriented with regard to the bound ThDP cofactor. (upper panel) Surface potential mapping of the compared proteins. The entrance part to the active site of each enzyme is magnified with the bound ThDP of stick models and its entrance to the active site is indicated with dotted circles. The positively and negatively charged surfaces are colored in blue and red, respectively (lower panel). Close-up view of the active site of each protein. Protein chains are differentiated by colors. Residues at the active sites are displayed with stick models and the modelled (R)-acetoin (magenta) and (S)-acetoin (green) are located near the ThDP cofactors. | ||
The different enantiomeric product preferences of the three enzymes can be partially explained by the respective active site environment and the modelled product (Fig. 5, lower panel). In FLS, the Gln113 appears to equally interact with both conformations. Similarly, His114 of ZmPDC and the nearby local hydrophobic area by Ile471 and Ile476 can play an important role in a rather dominant formation of (S)-acetoin. On the other hand, His732 of VvSucA, together with several aromatic residues, is located in a position to predominantly interact with (R)-acetoin. The S326N mutation in VvSucA that increased (S)-acetoin is likely to interrupt this favourable interaction between the (R)-acetoin and His732. However, we should agree that the exact substrate binding mode within the active site of VvSucA and the regiospecific enzymatic mechanism of VvSucA as a carboligase of acetaldehyde will be clarified by the subsequent VvSucA structures in complex with the substrate.
Conclusions
In this study, we have shown that SucA enzymes successfully converted acetaldehydes into acetoin. Unlike FLS and ZmPDC, an acetoin product by SucAs is enriched as an (R)-isomer (Fig. 1) that can be more economically used than can the racemic mixtures. Among the tested SucAs, VvSucA has shown the highest acetaldehyde–carboligating activity with the best (R)-enantioselectivity (Fig. 1). Furthermore, its regioselectivity could be improved, as exemplified from a derivative of VvSucA (V298-H301-to-GGG) that has produced almost up to 100% (R)-acetoin from acetaldehyde (Fig. 4). Of course, we should agree that a carboligating activity can be lowered when better enantioselectivity was observed in either a native enzyme (Fig. 1) or a variant (Fig. 4). Nonetheless, this approach, using a regiospecific carboligation activity of VvSucA and its engineered derivative with a reasonable capacity and an improved regioselectivity, may contribute to the production of (R)-acetoin from low-value acetaldehyde.Accession number
The coordinates of the VvSucA structures together with the corresponding structure factors have been deposited in the Protein Data Bank and released immediately upon publication (PDB code: 6KM9 and 6KMA).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray diffraction experiments were performed with Beamline 11C at the Pohang Accelerator in Korea. This work was supported by C1 Gas Refinery Research Center (NRF Grant Number: NRF-2018M3D3A1A01055735) of the National Research Foundation (NRF) of Korea funded by Ministry of Science and ICT.References
- Z. Xiao and J. R. Lu, Biotechnol. Adv., 2014, 32, 492–503 CrossRef CAS PubMed.
- L. Zhang, Q. Liu, Y. Ge, L. Li, C. Gao, P. Xu and C. Ma, Green Chem., 2016, 18, 1560–1570 RSC.
- C. Zhu, T. Shen, D. Liu, J. Wu, Y. Chen, L. Wang, K. Guo, H. Ying and P. Ouyang, Green Chem., 2016, 18, 2165–2174 RSC.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, United States, 2004, DOI:10.2172/15008859.
- X. J. Ji, H. Huang and P. K. Ouyang, Biotechnol. Adv., 2011, 29, 351–364 CrossRef CAS PubMed.
- Z. Xiao and P. Xu, Crit. Rev. Microbiol., 2017, 33, 127–140 CrossRef.
- Y. Liu, Y. Li and X. Wang, Appl. Microbiol. Biotechnol., 2016, 100, 8633–8649 CrossRef CAS PubMed.
- X. Jia, L. Ying and H. Yejun, Sci. Rep., 2017, 7, 4333 CrossRef.
- L. Zhang, R. Singh, Z. Guo, J. Li, F. Chen, Y. He, X. Guan, Y. C. Kang and J. K. Lee, Green Chem., 2018, 20, 230–242 RSC.
- Z. Cui, Y. Zhao, Y. Mao, T. Shi, L. Lu, H. Ma, Z. Wang and T. Chen, J. Chem. Technol. Biotechnol., 2019, 94, 2547–2554 CrossRef CAS.
- Y. He, F. Chen, M. Sun, H. Gao, Z. Guo, H. Lin, J. Chen, W. Ji, Y. Yang, L. Zhang and J. Yuan, Molecules, 2018, 23, 691 CrossRef.
- C. Gao, L. Zhang, Y. Xie, C. Hu, Y. Zhang, L. Li, Y. Wang, C. Ma and P. Xu, Bioresour. Technol., 2013, 137, 111–115 CrossRef CAS PubMed.
- P. Siegert, M. J. McLeish, M. Baumann, H. Iding, M. M. Kneen, G. L. Kenyon and M. Pohl, Protein Eng., Des. Sel., 2005, 18, 345–357 CrossRef CAS PubMed.
- H. G. David, J. Chem. Soc., Perkin Trans. 1, 1993, 3, 309–311 Search PubMed.
- T. Gerhards, U. Mackfeld, M. Bocola, E. Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- M. Müller, D. Gocke and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef.
- M. Beigi, S. Loschonsky, P. Lehwald, V. Brecht, S. L. A. Andrade, F. J. Leeper, W. Hummeld and M. Müller, Org. Biomol. Chem., 2013, 11, 252–256 RSC.
- P. P. Giovannini, O. Bortolini and A. Massi, Eur. J. Org. Chem., 2016, 26, 4441–4459 CrossRef.
- F. J. Alvarez, J. Ermer, G. Huebner, A. Schellenberger and R. L. Schowen, J. Am. Chem. Soc., 1991, 113, 8402–8409 CrossRef CAS.
- D. Kern, G. Kern, H. Neef, K. Tittmann, M. Killenberg-Jabs, C. Wikner, G. Schneider and G. Hübner, Science, 1997, 275, 67–70 CrossRef CAS.
- R. Kluger and K. Tittmann, Chem. Rev., 2008, 108, 1797–1833 CrossRef CAS.
- H. C. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS.
- J. B. Siegel, A. L. Smith, S. Poust, A. J. Wargacki, A. Bar-Even, C. Louw, B. W. Shen, C. B. Eiben, H. M. Tran, E. Noor, J. L. Gallaher, J. Bale, Y. Yoshikuni, M. H. Gelb, J. D. Keasling, B. L. Stoddard, M. E. Lidstrom and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3704–3709 CrossRef CAS.
- L. Holm and M. L. Laura, Nucleic Acids Res., 2016, 44, W351–W355 CrossRef CAS PubMed.
- T. Wagner, N. Barilone, P. M. Alzari and M. Bellinzoni, Biochem. J., 2014, 457, 425–434 CrossRef CAS.
- D. Dobritzsch, S. König, G. Schneider and G. Lu, J. Biol. Chem., 1998, 273, 20196–20204 CrossRef CAS.
- T. G. Mosbacher, M. Mueller and G. E. Schulz, FEBS J., 2005, 272, 6067–6076 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for construction of expression plasmids; purification of the recombinant proteins; crystallization of VvSucA; structure determination and refinement of VvSucA; enzymatic assays; product analysis using gas chromatography and mass spectroscopy; structural comparison. See DOI: 10.1039/c9cy01566c |
| ‡ These three authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2020 |