
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive targeting of gold nanoparticles as cancer therapeutics
Zoë Rachael
Goddard
 a,
María J.
Marín
b,
David A.
Russell
b and
Mark
Searcey
*a
a,
María J.
Marín
b,
David A.
Russell
b and
Mark
Searcey
*a
aSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: M.Searcey@uea.ac.uk
bSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
First published on 22nd October 2020
Abstract
Gold nanoparticles (AuNPs) are of increasing interest for their unique properties and their biocompatability, minimal toxicity, multivalency and size tunability make them exciting drug carriers. The functionalisaton of AuNPs with targeting moieties allows for their selective delivery to cancers, with antibodies, proteins, peptides, aptamers, carbohydrates and small molecules all exploited. Here, we review the recent advances in targeted-AuNPs for the treatment of cancer, with a particular focus on these classes of targeting ligands. We highlight the benefits and potential drawbacks of each ligand class and propose directions in which the field could grow.
 Zoë Rachael Goddard | Zoë Goddard completed her PhD in medicinal chemistry in 2020 at the University of East Anglia (UK), investigating the synthesis of actively targeted gold nanoparticles, including the study of multiple classes of targeting moieties. She is currently a postdoctoral research associate in the School of Pharmacy at the University of East Anglia where she continues to study targeted cancer therapeutics. |
 María J. Marín | María J. Marín is a Lecturer in Analytical Chemistry at the University of East Anglia, Norwich, UK. Her research focuses on the synthesis and application of nanomaterials to develop diagnostic tools and intracellular sensors, and as drug delivery systems for the targeted treatment of cancer. In 2014, she was awarded the Roscoe Gold Medal for Chemistry and the Westminster Medal at the ‘SET for Britain’ held at the UK Houses of Parliament. She is member of the Royal Society of Chemistry East Anglia Analytical Division Committee, the ASCOS Advisory Committee and the Executive Group of the Norwich Cancer Research Network. |
 David A. Russell | David Russell is an Emeritus Professor of Chemistry at the School of Chemistry, University of East Anglia. David's research is focused on the design and application of novel functionalised nanoparticles. Such nanoparticles have been used to develop innovative diagnostic devices – leading to the formation of two spin-out companies, Intelligent Fingerprinting and Iceni Diagnostics – and for the delivery of photosensitiser agents for targeted photodynamic therapy of cancer. David was awarded the SAC silver medal by the Royal Society of Chemistry. In 2018 the University of East Anglia made David an Innovation Fellow in recognition of the impact of his research. |
 Mark Searcey | Mark Searcey is Pro Vice Chancellor for Science and Chair of Medicinal Chemistry at the University of East Anglia. His research interests lie in the general area of targeted therapeutics for cancer, particularly focusing on DNA binding agents and protein–protein interactions. His laboratory combines solution and solid phase small molecule and peptide chemistry to generate new agents and to investigate their biological properties. |
1. Introduction
Over 100 years ago, Paul Ehrlich described the concept of a ‘magic bullet’ for chemotherapy, in which drugs can be delivered directly to their desired target, removing devastating off-target effects.1,2 While there are many ways to go about the development of a ‘magic bullet’, one such method is the combination of nanotechnology and nanomedicine, an emerging field of cancer therapeutics involving sub-one hundred nanometre delivery vehicles (1–100 nm) to transport the desired therapeutic to its target. Nanomedicine involves the use of nanoparticles, with their nanoscale size providing significantly altered physical, chemical and biological properties from that of the bulk material, with these altered properties favourable for their use as delivery vehicles.Nanoparticles have been shown to display passive targeting towards tumours through the enhanced permeability and retention (EPR) effect (Fig. 1).3 The EPR effect occurs due to the fact that tumours have a high demand for blood flow to provide the necessary nutrients and oxygen for their uncontrolled cell growth. As they rapidly expand in size, tumours form new blood vessels to provide for this excess need, and these blood vessels tend to be poorly formed and ‘leaky’. As nanoparticles are relatively large in size compared to natural small molecules and growth factors, they rarely pass through the walls of properly formed blood vessels in normal tissue. The leaky vasculature in tumours, however, allows for the passage of nanoparticles through their walls and leads to an accumulation of nanoparticles within the tumour. Tumours also display poor lymphatic drainage meaning that the nanoparticles that pass through into the tumour via the leaky blood vessels are not carried away as efficiently from cancerous tissue as from normal tissue, increasing this accumulation in tumours. This passive accumulation of nanoparticles in cancerous tissues highlights their ability to act as ‘magic bullets’.
 | ||
| Fig. 1 The passive targeting of nanoparticles towards tumours through the EPR effect. | ||
A second benefit of nanoparticles is their large surface area to volume ratio, meaning that one nanoparticle can carry a large quantity of a payload to their target, providing an attractive method for drug delivery. This large surface area also allows for the attachment of multiple different payloads to one nanoparticle,4 allowing for their co-delivery to a target, which has many therapeutic benefits. Diagnostic tools can also be attached to nanoparticles alongside payloads to elicit a theranostic effect, where the nanoparticle system can be used to diagnose and treat cancers simultaneously.5,6 Nanoparticles have been seen to improve stability, solubility and circulatory half-lives of drugs, along with improved drug efficacy.7–9 They have also been designed to release their payload upon internalisation into cancerous cells due to an internal stimulus such as pH or a reducing environment, restricting drug release to within cancer cells and improving the pharmacokinetics of a drug.10,11 The benefits of nanoparticle delivery systems highlight their applicability for the delivery of payloads to cancerous tissue.
Many types of nanoparticle have been developed for drug delivery, including liposomes,12 polymeric nanoparticles,13 carbon dots,14 upconverting nanoparticles15 and inorganic nanoparticles.16 Inorganic structures include gold, silica and iron oxide nanoparticles and quantum dots.16 While these all have their benefits, gold nanoparticles (AuNPs) highlight themselves as ideal drug carriers; they are chemically inert and minimally toxic, meaning they can pass through the body without eliciting any adverse reactions.17,18 For intravenous use as drug carriers, AuNPs are often coated in a layer of polyethylene glycol (PEG) ligands. PEG is clinically approved for intravenous use and its amphiphilic nature stabilises nanoparticles within biological media, making AuNPs dispersible within aqueous environments.19 PEG also increases the circulatory half-life of AuNPs by blocking the adsorption of opsonins and serum proteins, which enable the uptake and clearance of nanoparticles through the reticuloendothelial system.19–21 AuNPs are non-porous nanoparticles, and as such they are commonly loaded with payloads through chemical bonding or adsorption to the surface ligands of the AuNPs.
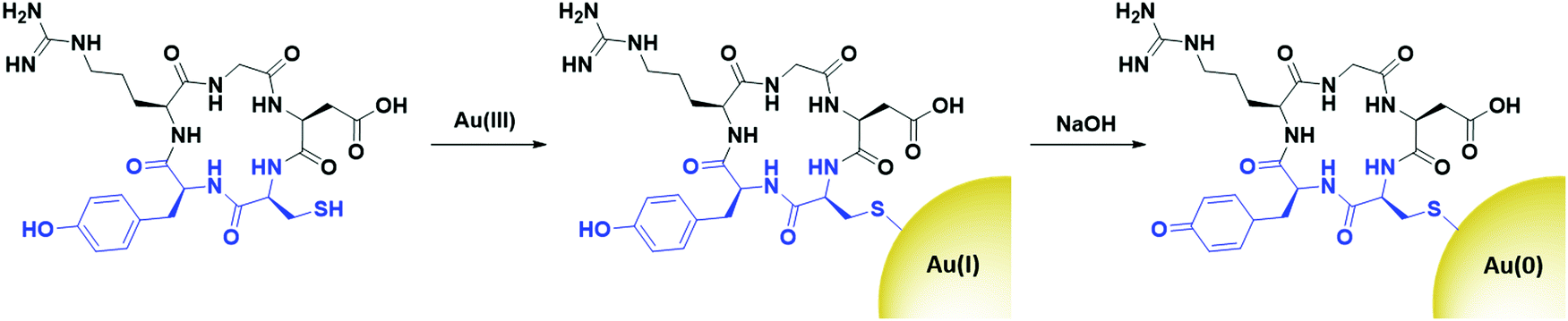
The surface of AuNPs is easily functionalised through the formation of strong gold–sulphur (Au–S) bonds that will spontaneously form through thiol surface adsorption.22 Au–S bonds are non-labile and result in AuNPs that are stable to physiologically relevant pH and salt concentrations.23 The synthesis of AuNPs involves the reduction of Au(III) to Au(0), which initiates the nucleation of AuNPs. Tight control over the reaction conditions means that the size and shape of AuNPs can be selected and varied to fit the desired purpose.24 Different nanostructures are beneficial for different purposes, and their uptake and potential therapeutic properties vary from shape to shape.25 The ability to form these different structures is a benefit of AuNPs and this is not as achievable with other nanosystems. The synthesis of AuNPs of differing shapes, such as nanosquares, nanostars (AuNSs) and nanorods (AuNRs) is usually completed through a seeded growth method.26 Here, small AuNPs are synthesised, generally 4–5 nm in size, and the addition of these seeds to Au(III) solutions containing different reducing and capping agents can influence the shape of the nucleated nanosystems.27
The strength of the reducing agent has a strong influence on the size of the AuNPs produced.28 For example, the use of a strong reducing agent such as sodium borohydride (NaBH4) results in sub-10 nm AuNPs, whereas for larger AuNPs milder reducing agents such as trisodium citrate or ascorbic acid are commonly used.28,29 The synthesis of AuNPs also relies on the presence of a stabilising agent, and the choice of stabilising agent can influence the size and shape of the resulting AuNPs through steric hindrance, and reduce the polydispersity of the synthesised AuNPs.30–32 For example, the addition of thiolated ligands upon reduction of Au(III) leads to the formation of strong Au–S bonds that cap the nanoparticles at a small and relatively monodisperse size, but control over the gold![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio determines the exact size of the AuNPs synthesised.33 While it is known that the size and shape of AuNPs can affect their uptake in cells,34,35 this review will focus on the ligands used for active targeting and not the effect of the core itself. Due to the lack of degradation of AuNPs in vivo, particular care must be taken when choosing the size of the core and the surface ligands to avoid organ toxicity. AuNPs can accumulate in the liver, spleen and lymph nodes if not correctly functionalised to allow filtration by the reticuloendothelial system. The pharmacokinetics of AuNPs have been reviewed elsewhere36 and are beyond the scope of this review.
:ligand ratio determines the exact size of the AuNPs synthesised.33 While it is known that the size and shape of AuNPs can affect their uptake in cells,34,35 this review will focus on the ligands used for active targeting and not the effect of the core itself. Due to the lack of degradation of AuNPs in vivo, particular care must be taken when choosing the size of the core and the surface ligands to avoid organ toxicity. AuNPs can accumulate in the liver, spleen and lymph nodes if not correctly functionalised to allow filtration by the reticuloendothelial system. The pharmacokinetics of AuNPs have been reviewed elsewhere36 and are beyond the scope of this review.
AuNPs display a strong surface plasmon absorption due to the oscillation of electrons upon exposure to light. AuNPs absorb visible light with extinction coefficients orders of magnitude higher than those of many strongly absorbing organic dyes, and this surface plasmon absorption band is dependent on the size and shape of the AuNPs.37 The presence of this surface plasmon absorption band provides a unique property to AuNPs as the strong absorption of light means that the nanoparticle itself can be used as a therapeutic agent. When AuNPs are irradiated with light matching the wavelength of their surface plasmon absorption band they rapidly heat and can destroy cells through photothermal ablation, known as photothermal therapy (PTT).38
The biocompatibility, minimal cytotoxicity, stability, ease of synthesis and functionalisation, passive targeting and the unique surface plasmon properties of AuNPs highlight their exciting potential as delivery systems for cancer therapeutics.
While passive targeting of AuNPs relying on the EPR effect has been explored,39–42 as the interest in personalised medicine has grown in recent years, the focus has turned towards the active targeting of gold nanoparticles to a particular site. Active targeting of AuNPs involves the attachment of a targeting moiety, specific towards a desired surface receptor, onto the nanoparticle surface, alongside the payload. In cancer, the targeting moiety used often recognises a receptor that is overexpressed by tumour cells, but examples also exist of AuNPs targeted towards receptors that are cryptic or not expressed on healthy cells. A plethora of targeting moieties have been explored and these can be split into the general categories of antibodies, proteins, peptides, aptamers, carbohydrates and small molecules.
While there are many reviews highlighting the benefits of targeted AuNPs, this review describes the recent progress towards the utilisation of each class of targeting moiety with respect to cancer therapeutics with the aim of collating these targeting moieties to determine the benefits of each class of ligands for targeted-AuNP therapies.
2. Antibody directed gold nanoparticles
2.1 IgG antibodies
When the idea of actively targeting nanoparticles towards a known oncogene was first developed, antibodies presented themselves as an ideal targeting moiety. Antibodies are highly specific towards a receptor, to which they display a high affinity. While there are many types of antibodies, most antibodies used for therapeutics are IgG antibodies. IgG antibodies have a Y-shaped structure consisting of a constant (Fc) region that is unchanged in all IgG antibodies and a variable (Fab) region that is unique to each antibody (Fig. 2).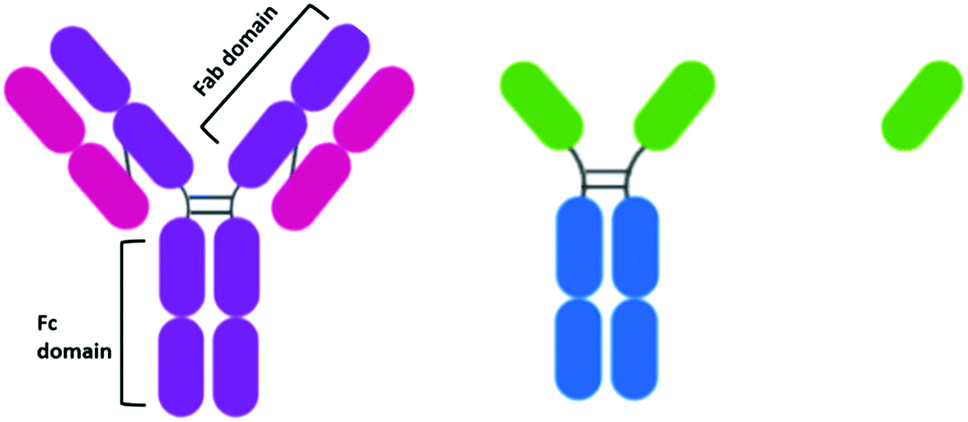 | ||
| Fig. 2 (L-R) the structures of an IgG antibody consisting of two heavy chains (purple) and two light chains (pink) highlighting the Fab and Fc regions, a camelid IgG heavy-chain antibody with variable recognition domains (green) and a single-domain nanobody derived from the variable domain of a camelid IgG. | ||
The development of antibodies as clinically approved therapeutics in their own right may have encouraged the development of antibody-AuNPs. The antibodies trastuzumab and cetuximab have been clinically approved to target HER2 overexpressing breast cancers43 and EGFR overexpressing colorectal cancer respectively.44 As these antibodies are clinically approved, their affinity to their target has already been validated and therefore they have both been extensively investigated for the targeted delivery of AuNPs. For example, cetuximab and trastuzumab have been used to direct AuNPs towards cancers for enhanced radiotherapy.45–48 AuNPs can act as radiosensitisers, increasing the effect of radiation therapy on tumours as they release photoelectrons and Auger electrons upon irradiation with X-ray and near-IR radiation.49 Antibody-functionalised AuNPs have also been extensively studied for targeted PTT of a multitude of cancers.50–52 Consistently, increased cytotoxicity and selective uptake of these targeted AuNPs is observed.
Antibody-functionalised AuNPs were first used for the targeted delivery of anticancer agents in 2008, when gemcitabine (Gem) was delivered to pancreatic adenocarcinoma by cetuximab-functionalised AuNPs with increased cytotoxicity observed over non-targeted Gem-AuNPs.53 Since then, AuNPs have been used to deliver chemotherapeutic drugs such as doxorubicin (Dox),54,55 oxaliplatin56 and docetaxel.4 These chemotherapeutic drugs are either bound to the AuNP core via reversible Au–N bonds, adsorbed to the core through hydrophobic interactions or are conjugated onto PEG ligands (Fig. 3) to form a mixed monolayer on the nanoparticle surface alongside the antibody.54–56
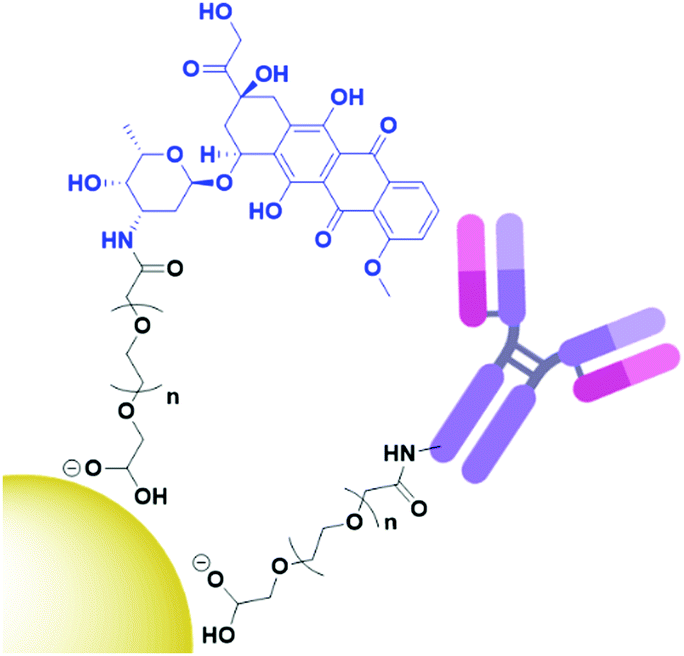 | ||
| Fig. 3 Structure of antibody directed doxorubicin (blue) AuNPs, with both the payload and antibody conjugated through a PEG (black) linker (adapted from ref. 54). | ||
Few examples exist of conjugating the delivered drug onto the antibody itself,57 potentially due to the relative fragility of antibodies and the relative ease of conjugating payloads to the nanoparticle over the antibody. One example of this is the addition of the radionuclide 131I to cetuximab post-conjugation of this antibody onto AuNPs for radioimmunotherapy.58 The decision to radiolabel the antibody itself likely stems from the fact that radiolabelled cetuximab has been widely investigated as an agent for radioimmunotherapy.59–61 These AuNPs were seen to display a targeted decrease in cell viability of A549 cells, with this cytotoxicity higher than that of 131I at the same dosage,58 highlighting the ability of AuNPs to increase the potency of a payload.
The uptake of antibodies into cells often relies on receptor mediated endocytosis. This process provides a target-specific internalisation mechanism for antibody-AuNPs; however, the conjugation of antibodies is notorious for altering the pharmacokinetics of the antibody. It has been observed that the endocytosis of cetuximab-AuNPs is in fact accelerated from that of free cetuximab, and the mechanism by which this endocytosis occurs is altered upon conjugation. This altered internalisation mechanism leads to differing subcellular localisation between cetuximab and its resulting gold nanoconjugate.62 It is also observed that not all antibodies are internalised upon receptor binding. It is still under debate whether trastuzumab is internalised upon binding to HER2,63,64 however it has been shown that the internalisation of trastuzumab is increased upon cross-linking, possibly due to multivalent binding. HER2 overexpressing cells show increased internalisation of trastuzumab-AuNPs compared to non-conjugated trastuzumab at the same concentration.65 These reports highlight a key consideration for the synthesis of antibody-AuNPs; the kinetics and uptake of the antibody is likely to be altered upon conjugation. While in the reported examples these alterations appear to be beneficial for increased uptake, this may not always be the case and the pharmacokinetics of conjugated antibodies may warrant further investigation to help select the best antibodies possible for delivering AuNPs to the corresponding target.
While many different antibody conjugated AuNP systems have been reported, the vast majority of them rely on common chemistry for the addition of the antibody to the nanoparticle system. Firstly, most of these nanosystems are stabilised with a thiolated linker such as a PEG chain, which is highly water soluble and approved for medical use. These PEG chains are often terminated with a carboxylic acid or activated succinimidyl ester for the addition of an antibody through the formation of random amide bonds with free amine residues on the surface of the antibody.45–47,54–56,58,65–72 While many variants of these linkers exist, the chemistry of the antibody conjugation is identical. This ubiquity of functionalisation chemistry, while concentrations of both coupling agents and antibody may need to be varied depending on the desired system, perhaps highlights the versatility of antibodies as targeting agents for AuNPs. It can be imagined that, if the same process is used for antibody conjugation time and time again, antibodies can be switched in and out to target the same system to any oncogene required. While most conjugates in the literature use some variation of this amide bond formation involving lysines on the antibody, a PEG linker terminated in a hydrazide can be used, which allows for the cross-linking of AuNPs with carboxylic acids on the surface of antibodies.73 This chemistry, while utilising different residues on the antibody is still completely random and may lead to even more variations as there are on average more carboxylic acids present on antibodies than amines.74
While antibodies display high affinity towards their target, this affinity relies on the antigen binding sites remaining non-functionalised and unhindered. The commonly used random amide bond formation for the addition of antibodies may result in some of the amide bonds being formed in or near the active site of the antibody, or orientating this active site towards the gold core, and thus reducing the binding ability of these conjugates. It has been shown that protein G, an Fc region binding protein, can be used to control the orientation of an antibody on the AuNP surface and therefore to maintain optimal activity.75 While no comparison is made to AuNPs conjugated with antibody without the presence of protein G, there is increased uptake of these EGFR targeted AuNPs compared to a non-targeted control and selective PTT is observed.53,75 This perhaps provides a sensible alternative to commonly used antibody functionalisation techniques if the desired activity is not observed, however protein G is immunogenic and therefore it may provide other complexities.
2.2 Antibody fragments and nanobodies
While antibodies present such high affinity towards receptors, their size may inhibit their penetration into tumours,76 and it has been suggested that this effect is also observed for antibody-conjugated AuNPs.77 The size-dependent penetration issues have led to the investigation of antibody fragments as targeting moieties. The reduction of the disulphide bonds between the heavy chains of an antibody yields two functional antibody fragments. Notable for AuNPs, these antibody fragments possess free thiols which can bind to the gold core. This conjugation strategy also ensures the active site of the antibody is pointing away from the gold core and thus, is accessible. The conjugation of EGFR antibody fragments to AuNPs for PTT displayed cytotoxicity upon irradiation that varied with EGFR expression.50 While the use of antibody fragments reduces the size of the targeting moiety attached to the AuNPs, these half-antibodies still display an immunogenic effect upon the body due to the presence of the Fc region. Antibody fragments known as Fab fragments have been developed to remove the Fc region, simultaneously reducing the size of the antibody and forming a non-immunogenic targeting moiety. Interestingly, there are no examples of the use of Fab fragments to target therapeutic AuNPs for cancer drug delivery, and research into this area may provide exciting results.While antibody fragments have been investigated to account for the sheer size of antibodies, nanobodies are even smaller than antibody fragments and have recently gained extensive attention as targeting moieties.78 Camelids possess IgG antibodies that consist of only heavy chains and are two thirds of the size of human IgGs (Fig. 2).79 These camelid heavy-chain antibodies possess a variable domain that can be cloned and expressed in bacteria to give a monomeric, single-domain antigen-binding antibody fragment, named a nanobody for its small size (Fig. 2).79 Nanobodies are ca. 15 kDa, one tenth of the size of an antibody, and therefore display higher tumour penetration. Alongside this, they are non-immunogenic and display higher stability than antibodies.80
An anti-HER2 nanobody has been shown to selectively target AuNSs towards HER2 positive ovarian cancer. The nanobody was shown to selectively internalise into HER2 overexpressing SKOV3 cells, with targeted PTT observed.81 This nanobody was modified in production to express a cysteine residue on the C-terminus to allow for site-specific attachment onto AuNSs through a maleimide. Both antibody fragments and nanobodies may provide solutions to the issues of antibody penetration into solid tumours. Further work is needed, however, to confirm the benefit of these targeting moieties, with studies to compare the uptake, selectivity and tumour penetration of these fragments to that of whole antibodies to determine whether this effect is also observed when conjugated to an AuNP core.
3. Protein directed gold nanoparticles
The use of native proteins as targeting agents is relatively unexplored compared to the wealth of research into antibody targeted therapies. Proteins selected as targeting moieties are either natural ligands for a receptor or lectins – carbohydrate binding proteins often isolated from fruit or vegetables. It is perhaps understandable that human native protein ligands towards receptors have not been extensively explored as targeting moieties as, by nature of their abundance in the human body, there will be a large number of competing ligands that are not carrying AuNPs. These proteins will display the same affinity towards the desired receptor and possibly lead to a reduced targeting efficiency. That said, the use of human proteins and growth factors removes any immunogenic response towards these AuNPs and may be worth exploring. One growth factor that has been exploited for targeted AuNP therapy is the epidermal growth factor (EGF) which has been used to target AuNPs towards epidermal growth factor receptor (EGFR) overexpressing breast cancer. A disulphide bond in EGF was reduced to allow for its attachment onto AuNPs, then the EGF itself was radiolabelled with 111In (Fig. 4). These nanoconjugates showed high uptake into EGFR overexpressing breast cancer cells and minimal uptake into cells with low EGFR expression. A competition assay with non-conjugated EGF showed that this uptake was due to EGF recognition by the cells and EGFR selective cytotoxicity was observed.82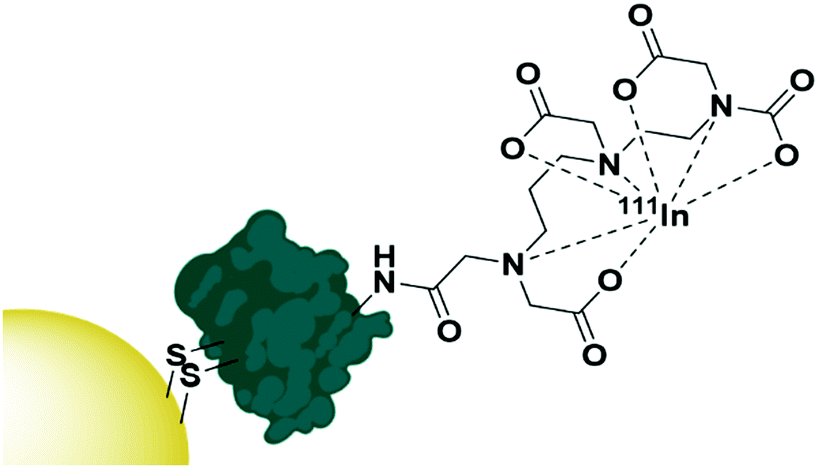 | ||
| Fig. 4 The functionalisation of AuNPs with 111In radiolabelled EGF through disulphide reduction.82 | ||
The protein transferrin has been shown to target AuNRs towards transferrin receptor expressing lung cancers. While transferrin directed AuNRs carrying doxorubicin displayed lower cytotoxicity than doxorubicin alone, the cytotoxicity of these transferrin targeted AuNRs was selective towards transferrin receptor expressing cells, and toxicity was shown to vary with the concentration of transferrin receptor expressed by cell lines.83
While these examples use the receptors’ native proteins, the use of recombinant proteins has been explored. Employing mutations and variations to wild type proteins for a ligand can advantageously alter their characteristics and lead to enhanced properties for these recombinant proteins over the body's native proteins. For example, a recombinant fibroblast growth factor 1 (FGF1) was engineered with four point mutations to protect FGF1 against proteolysis and therefore increase its circulatory half-life over that of native FGF1.84 FGF1 targets all four variations of the fibroblast growth factor receptor that are overexpressed in many cancers including lung cancer.85 FGF1 was also altered to attach a short peptide chain to the N-terminus containing a cysteine for conjugation onto AuNPs, and the native cysteine in the protein was removed to allow for site specific conjugation onto the AuNPs. FGFR negative cells were transfected with FGFR and the uptake of these nanocarriers confirmed to be due to endocytosis by FGFR. These AuNPs were used for PTT, with photothermal cytotoxicity only observed in FGFR expressing cell lines.86 The benefits of this recombinant protein over native FGF1, however, have not been explored in this research as no comparison has been made to the native ligand to determine the advantage of this increased circulatory half-life.
Lectins are more commonly used for biosensors than for targeted cancer therapies, however the lectin Jacalin has been employed to target a zinc phthalocyanine towards Thomsen–Friedenreich antigen (T-antigen) expressing cells for photodynamic therapy (PDT). The T-antigen is expressed in ca. 90% of cancers and is usually cryptic on healthy cells. Jacalin was conjugated onto a PEG shell through random amide-bond formation and cytotoxicity was observed in T-antigen expressing cells, whereas non-conjugated AuNPs displayed negligible cytotoxicity. Pre-incubation with Jacalin binding glycoproteins reduced the phototoxicity, confirming the observed activity was due to the selectivity of Jacalin.67,87 Jacalin is non-immunogenic, and its affinity towards the ubiquitously oncologically expressed T-antigen makes it an exciting targeting moiety. Jacalin has, however, been shown to bind to carbohydrate moieties on IgA antibodies,88,89 which may limit its use as a targeting ligand.
4. Peptide directed gold nanoparticles
Peptides are relatively short polymers of amino acids that can be fully characterised and chemically synthesised to a designed specification. While they generally display lower affinity towards receptors than antibodies and other proteins, peptides are gaining attention as targeting moieties due to their simplicity and rapid uptake kinetics. Many examples of using peptides to target AuNPs towards various cancers for imaging have been reported in the literature,90–95 while surprisingly few examples exist of peptide-directed AuNP-based cancer therapeutics.Peptides have been used to direct AuNPs carrying cytotoxic payloads to their targets. Pancreatic ductal adenocarcinoma (PDAC) has been targeted using a plectin-1 targeting peptide (KTLLPTP). Plectin-1 is expressed on the surface of PDAC but only within the cytoplasm of healthy cells.96 The modification of this peptide with a tyrosine and a cysteine residue (KTLLPTPYC) allows for the use of this peptide to simultaneously reduce gold(III) chloride under basic conditions to initiate the nucleation of AuNPs (Tyr), and to cap the resulting nanoparticles with the thiol side chain of cysteine. This addition of a dipeptide that simultaneously nucleates and caps AuNPs is a unique modality for peptides over all other targeting agents. These AuNPs were further functionalised with the chemotherapeutic drug gemcitabine (Gem), which adsorbs to the AuNP core through hydrophobic interactions, as well as forming a reversible Au–N bond.97 These AuNPs show higher cytotoxicity towards PDAC cell lines than Gem alone and display excellent selectivity for PDAC cells over healthy tissue in mouse models.97 This use of the YC dipeptide to simultaneously nucleate and cap AuNPs has also been used to form AuNPs for use as radiosensitisers. These AuNPs are targeted towards αvβ3 integrin expressing cells through the cyclic peptide c(RGD) (Fig. 5).98
 | ||
| Fig. 5 The synthesis of cRGD directed AuNPs using a YC dipeptide (blue) to nucleate and cap the AuNPs (adapted from ref. 98). | ||
Arg-Gly-Asp (RGD), and its cyclic derivative c(RGD), can be used to direct AuNPs to a wide variety of cancers as αvβ3 integrin is expressed by proliferating endothelial cells involved in angiogenesis.99 The enhanced rate of angiogenesis in cancerous tissues means there is a high expression of αvβ3 integrin in most endothelial cancers. RGD is possibly the most well-known targeting peptide and this tripeptide highlights the ability to produce very small targeting peptides towards receptors while maintaining selectivity. Both RGD and cRGD have been shown to selectively direct AuNPs towards breast cancers and glioblastomas, where these AuNPs can act as radiosensitisers.100,101 c(RGD) has been used to deliver siRNA to cervical cancer models to silence E6, an oncoprotein that inactivates p53.102 The cRGD peptide is conjugated to a PEG-poly-lysine block copolymer, which is utilised to bind the siRNA through steric repulsion and ionic parings. Here, the ligand itself acts as the drug carrier and increased siRNA is observed intracellularly for cells treated with cRGD functionalised siRNA-AuNPs over cRAD functionalised siRNA-AuNPs; a substituted peptide sequence used as a control. The targeting ability of cRGD was further confirmed by pre-incubating cells with cRGD before the addition of cRGD-siRNA-AuNPs, with a much lower siRNA uptake observed.103
The peptide CRGDK is specific towards neuropilin-1 (Nrp-1), a transmembrane glycoprotein that acts as a co-receptor for many ligands and regulates the internalisation of membrane receptors. CRGDK has been used to direct AuNPs carrying the therapeutic peptide p12 (TSFAEYWNLLSP) towards Nrp-1 expressing breast cancers,104 and CRGDK-AuNPs carrying a platinum(IV) agent have been directed towards prostate cancers.105 p12 inhibits the binding of MDM2/MDMX to p53, a tumour suppressor, but it cannot penetrate the cell membrane, while Pt(IV) acts as a chemotherapeutic. Increased cytotoxicity was observed with increased Nrp-1 expression, and pre-treatment of cells with an Nrp-1 antibody resulted in reduced uptake of these nanoparticles, confirming the CRGDK peptide is targeting these AuNPs towards Nrp-1 overexpressing cells,104,105 highlighting the ability of short peptide sequences to selectively target an oncogenic receptor.
The EGFR targeting peptide GE11 (YHWYGYTPQNVI) has been used to deliver AuNPs carrying the photosensitiser Pc4 to EGFR overexpressing glioblastoma cells.106,107 PEG-AuNPs were synthesised and conjugated with GE11 before Pc4 was adsorbed onto the surface of the AuNP. These nanoconjugates displayed minimal dark toxicity and significant phototoxicity in glioblastoma cells. Interestingly, it was found that few nanoparticles were accumulating within the cells, however the concentration of Pc4 internalised by these cells was seen to be dependent on the binding of GE11. Pre-incubation of the cells with GE11 reduced uptake of Pc4 into these glioblastoma cells, and therefore it was hypothesised that while the AuNPs themselves were not being internalised, it is thought that the increased interaction of these directed AuNPs with the surface of glioblastoma cells allows for the Pc4 to desorb from the nanoparticle and accumulate within glioblastoma cells.106,107 As these nanoparticles are designed to pass through the blood brain barrier (BBB), it is vital that their size is maintained as small as possible. The relatively small size of peptides over antibodies and other proteins is beneficial here, and GE11 shows an affinity for the EGFR receptor only 10-fold lower than that of its natural ligand EGF, highlighting the applicability of peptides as targeting agents. The small size and fully characterised structure of peptides provides a benefit over antibodies and other proteins. The relative stability of peptides over antibodies and other proteins means they can either be conjugated to linkers after their attachment to AuNPs or before their use for ligand exchange. Again, as with antibodies and other proteins, the use of EDC/NHS and their derivatives is popular for peptide conjugation.104–107 As peptides are synthetically produced, their structures can be easily modified for attachment of reactive moieties. This allows for the attachment of thiols through cysteine residues that have been used to conjugate peptides directly to the gold core or to conjugate a peptide onto a linker.100,101,108 Most notably, the use of peptides allows site specific conjugation of the targeting moiety to AuNPs. This ensures that the peptides are attached to the AuNPs in a way that maintains their binding capability towards their target.
5. Aptamer directed gold nanoparticles
Aptamers are short, single stranded DNA or RNA sequences selected from a random pool of oligonucleotides. They form secondary structures through complementary base pairings that allow for selective binding towards specific receptors, proteins and small molecules (Fig. 6), with an affinity for their target similar to that of an antibody.109–111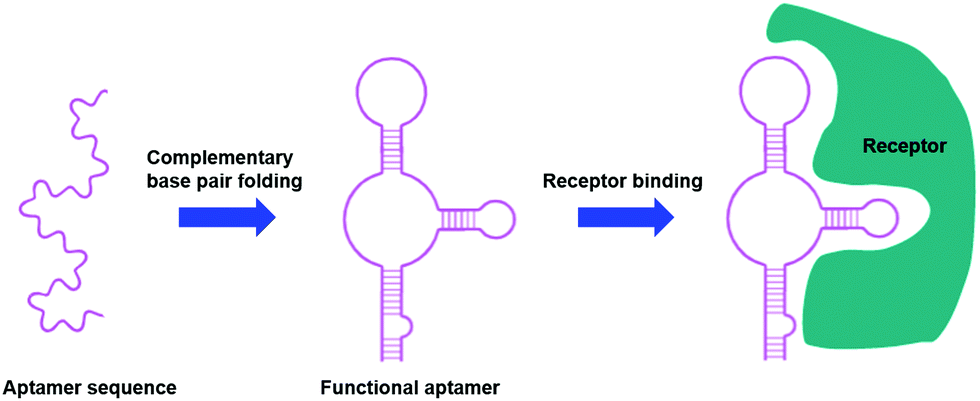 | ||
| Fig. 6 Aptamers as targeting moieties: the folding of aptamers through complementary base pairings results in secondary structures that are highly specific towards target receptors (adapted from ref. 111). | ||
Aptamers can be chemically synthesised and easily modified to improve their pharmacokinetics and stability, and therefore, since their development in the 1990s, have been viewed as an exciting alternative to antibodies.112 The synthetic production of aptamers means that most reports of aptamer-functionalised AuNPs use thiolated aptamers for direct attachment to the gold core,113–118 however one example does exist of amide bond formation to a PEG shell.119
Numerous aptamers have been designed for various targets, however only a select few of these have been applied to the delivery of AuNPs. The most widely explored aptamer for AuNP targeting is AS1411 – a 26-base guanine rich aptamer that targets nucleolin, a phosphoprotein overexpressed by cancerous cells.120 Nucleolin is mainly expressed on the nucleus of healthy cells, but malignant mutation often leads to increased localisation of this receptor onto the surface of cancerous cells.120 The guanine rich sequence of AS1411 leads to the formation of a G-quadruplex structure that is selective towards nucleolin. AS1411 induces an anti-proliferative effect within cells itself, and this in combination with its targeting ability increases the efficacy of a therapy. AS1411 has been used for the delivery of AuNPs for PTT,119 radiotherapy121 and to deliver AuNPs carrying Dox with nucleolin selective cell death observed.113 As AS1411 forms a G-quadruplex in the presence of potassium, the aptamer can be used to bind the photosensitiser N-methylmesoporphyrin IX (NMM), a G-quadruplex DNA binding ligand. The thiolation of this aptamer allows for its direct addition to the surface of AuNPs and these nanoconjugates were seen to be selectively internalised by cancer cells overexpressing nucleolin, with no uptake observed in nucleolin negative normal cell lines.114 This example is particularly interesting as it utilises the aptamer sequence not only as a targeting moiety but also as a drug carrier. A second example of this involves the conjugation of the photosensitiser chlorin e6 (Ce6) to the aptamer sgc8c, a protein tyrosine kinase 7 (PTK7) selective aptamer, to selectively target it towards leukaemia cell lines. Ce6 is conjugated to the 3′-end of the sgc8c aptamer, which is conjugated to a poly-T chain at the 5′-end, which, in turn, is conjugated to a sgc8c complementary DNA (cDNA) sequence. The sgc8c cDNA is further conjugated to AuNRs (Fig. 7). This means that in the absence of PTK7, the sgc8c is hybridised with the cDNA sequence, holding Ce6 close to the AuNR and quenching its fluorescence. Upon binding to PTK7, the sgc8c forms a hairpin, losing affinity for its complementary sequence and moving the Ce6 away from the AuNR, ‘switching on’ its fluorescence and therefore its photodynamic activity, as shown in Fig. 7. Significant targeted photodynamic activity was observed in PTK7 expressing cells, with increased cytotoxicity observed in combination with PTT.115
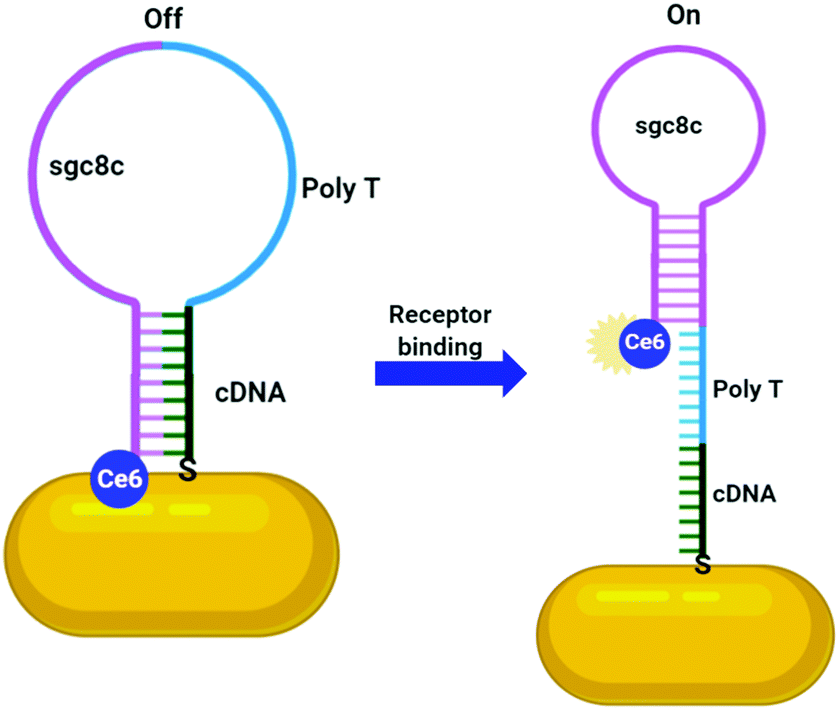 | ||
| Fig. 7 Activatable Ce6 AuNR targeted towards leukaemia cells through sgc8c aptamer (adapted from ref. 115). | ||
As aptamers are oligonucleotides, chemotherapeutic drugs that act by intercalating DNA can also intercalate these aptamers. Dox is a reversible DNA intercalator and therefore can be delivered to its target by binding to aptamers, notably by sgc8c and A9, a prostate specific membrane antigen (PSMA) specific aptamer.116,117 Here, AuNPs act as delivery systems to increase the concentration of payload delivered – multiple drug-aptamer complexes can be delivered at once, increasing the potency of one ligand finding its target. This effect has also been observed with the HER2 aptamer HApt; a trimeric aptamer that displays cytotoxicity towards HER2 overexpressing breast cancers by cross-linking HER2 receptors and sorting them for degradation. The attachment of this aptamer onto AuNSs has been shown to increase its uptake and therefore the therapeutic value of HApt.118
Aptamers, while successful targeting moieties when used individually, have also been used in combination to target multiple receptors on a specific cancer and therefore increase the selectivity towards these cells. AS1411 has been utilised for targeting AuNPs in conjunction with sgc8c, highlighting one of the advantages of aptamers as targeting moieties. As aptamers can be synthetically designed and built to a desired specification, a polyvalent aptamer system can be designed containing both AS1411 and sgc8c.122 The formation of a polyvalent aptamer for dual targeting displays remarkable benefits over the attachment of two individual targeting moieties as the ratio of these targeting ligands can be completely controlled with no concerns that one aptamer may have a higher affinity for AuNPs than another. It can be envisaged that ligands bearing multiple aptamers, or an uneven ratio of two aptamers could be synthesised to further increase the number of receptors targeted or alter the ratio of the targeting ligands in a controlled manner. This polyvalent aptamer was attached to AuNPs through electrostatic interactions, followed by the addition of daunorubicin (Dau), a DNA intercalating chemotherapeutic. A small amount of Dau binds to the aptamer due to its DNA intercalating ability, but the majority is bound to the AuNP surface through electrostatic interactions. These AuNPs displayed selective cytotoxicity towards cancer cell lines expressing both nucleolin and PKT7. A synergistic effect was observed for the use of this polyvalent aptamer over AuNPs targeted with just the sgc8c aptamer,122 highlighting a benefit of these chemically synthesised targeting moieties.
6. Carbohydrate directed gold nanoparticles
Cancer cells have been found to differentially express lectins on their surface compared to healthy cells,123 and the affinity of carbohydrates towards these lectins can be exploited to target these cells.124 The use of carbohydrates as targeting moieties for AuNP-based therapeutics is a relatively new concept, with literature exploring this possibility only disclosed within the last six years. As this is such a new field, few carbohydrate ligands have been explored.Hyaluronic acid (HA, Fig. 8a) is a naturally occurring polysaccharide consisting of a repeating unit of D-glucuronic acid and N-acetyl-D-glucosamine. HA selectively targets the CD44 receptor which is overexpressed by various cancers with a kD ≈ 0.3 nM.125 The polymeric structure of HA means that there are multiple free carboxylic acids that can be used for drug functionalisation and this has been exploited for the delivery of chemotherapeutic drugs and photosensitisers to their targets.126 A porphyrin photosensitiser has been conjugated onto HA alongside the fluorescent imaging agent cresyl violet and cystamine, which adds a thiol functional group for conjugation of this linker onto AuNPs (Fig. 8b). Here, HA acts as both the linker and targeting moiety. When attached to AuNPs, the fluorescence of both the porphyrin and cresyl violet are quenched. Upon uptake by CD44 overexpressing cell lines, hyaluronidase enzymes can degrade the HA, releasing the attached fluorophores and allowing for both the imaging of CD44 overexpressing cancers and their targeted PDT. The uptake and cytotoxicity of these nanoconjugates was observed to be selective towards CD44 overexpressing cancer cell lines.126 The highly repeated sequence of the HA polysaccharide allows HA to act as both a linker and targeting moiety, as well as allowing for functionalisation with a variety of drugs without the loss of targeting ability, a unique property of these polysaccharide targeting moieties. The photosensitiser pheophorbide-A and the chemotherapeutic drug metformin have both been conjugated onto HA with selective uptake observed in CD44 overexpressing lung and liver cancers, respectively.127,128
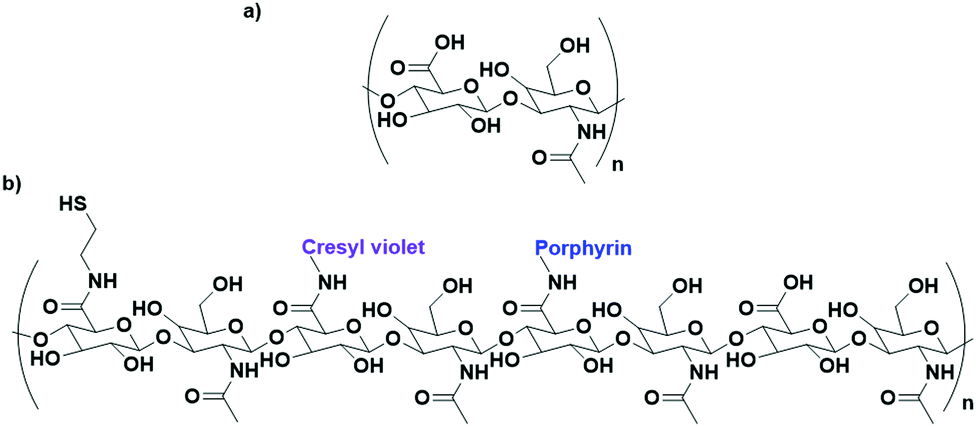 | ||
| Fig. 8 (a) the repeating D-glucuronic acid and N-acetyl-D-glucosamine unit of HA and (b) substituted HA ligand for AuNPs for the dual imaging and PDT of CD44 overexpressing cancer cells.126 | ||
While HA is sparsely described in the literature as a targeting agent for therapeutic AuNPs, it is by far the most investigated carbohydrate ligand. One of the few examples of alternative carbohydrate ligands is that of glucose, a monosaccharide that has been shown to bind to glucose transporter-1 (GLUT1) which is overexpressed by several cancer cells. Glucose functionalised AuNPs have been used to target siRNA carrying AuNPs towards GLUT1 overexpressing breast cancers.129 As glucose is a monosaccharide, the technique of conjugating drugs directly to the carbohydrate as used for HA cannot be utilised. Instead, this work builds a glucose-capped ligand containing a PEG chain, followed by a 40-unit poly-lysine (PLL) chain, and terminated in lipoic acid (Fig. 9). The PEG chain acts as a spacer and provides water dispersibility, while the lipoic acid provides thiols for binding to the AuNP surface. The PLL chain acts to bind siRNA through electrostatic interactions as the PLK1 siRNA used in this study contains 40 negative charges. PLK1 plays a vital role in the cell cycle of cancer cells and its knockdown leads to antitumour activity. This system was observed to specifically target GLUT1 overexpressing breast cancer cells, with its specificity confirmed through a competition assay with the GLUT1 inhibitor phloretin.129
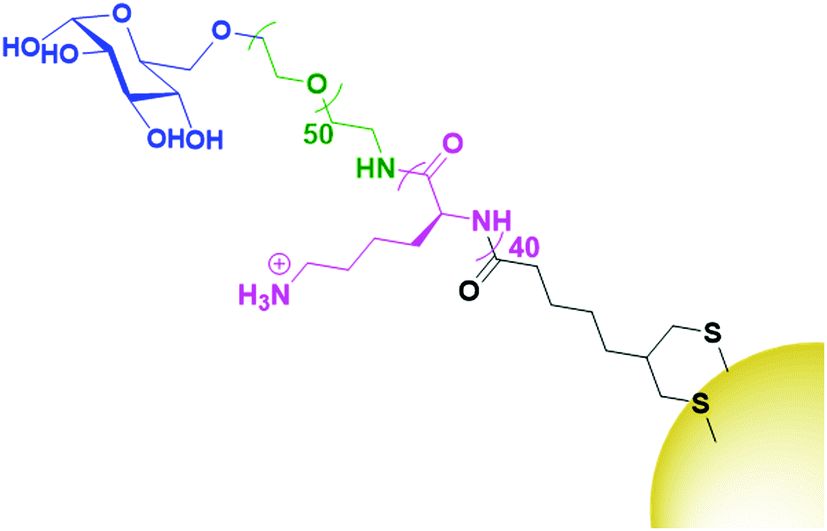 | ||
| Fig. 9 The attachment of glucose (blue) onto AuNPs through a PEG (green), PLL (pink) and lipoic acid (black) chain.129 | ||
Lactose, a disaccharide that is recognised by the lectin galectin-1, has also been investigated as a targeting moiety for AuNP-based cancer therapeutics.130 It has been used to target AuNPs carrying a phthalocyanine photosensitiser towards breast cancer cells for PDT. Lactose was conjugated onto a short, thiolated PEG linker and functionalised onto the gold core in a mixed monolayer with the photosensitiser. While uptake and phototoxicity was specific towards malignant breast cells, the uptake was not determined by the presence of galectin-1.130 This work, while demonstrating the possibility of the use of carbohydrate based ligands as targeting moieties, requires additional studies to determine how this ligand is selectively targeting malignant cells.
7. Small molecule directed gold nanoparticles
The final class of targeting moieties explored for AuNPs is small molecules; low molecular weight organic compounds that display affinity towards cell surface receptors. Benefits of small molecules include the fact that they can be relatively cheap to synthesise and generally display higher stability than the other targeting moieties discussed above. Due to their size, small molecules easily penetrate through tumours to deliver payloads to their target. Folic acid, also known as folate, is the most commonly utilised small molecule for targeted AuNPs. It is a natural ligand towards the folate receptors, which are only accessible to the blood stream in the kidneys on cancerous cells and therefore are useful oncogenic targets.131 The structure of folic acid contains two carboxylic acids which can be used for conjugation onto AuNPs. Due to the small size of folic acid, it is not possible to use this ligand to carry the payload and to conjugate it onto the AuNP itself. Folate is most commonly conjugated onto a linker such as PEG or polyethylenimine (PEI, Fig. 10) through amide bonds for attachment onto AuNPs, but examples of using electrostatic interactions to bind folate to AuNPs do exist.132 Covalently bound folic acid has been used to deliver AuNPs carrying siRNA, photosensitisers and chemotherapeutic drugs to various cancers.133–135 The use of folic acid-AuNPs to deliver chemotherapeutics often relies on electrostatic interactions between the drug, such as Dox, and the surrounding ligands, such as pectin.136 The release of Dox from these AuNPs has been shown to increase at lower pH due to the protonation of the negatively charged pectin shell and therefore a loss of the electrostatic interaction. The pH of tumour cells is pH 5.4, compared to 7.4 of the bloodstream, so this is a favourable characteristic for drug release in the tumour site. While in this case the AuNPs act solely as a delivery system, an increased cytotoxicity is observed for AuNP bound Dox compared to that of free Dox.136 As well as increasing the potency of chemotherapeutic effects, AuNPs allow for combination therapies between chemotherapeutics and PTT, with the synergistic effect resulting in increased cytotoxicity, and the folate ligand providing selectivity towards folate receptor positive tumours.135,137 Interestingly, for the development of folate-targeted AuNP therapies, there are examples that display increased cytotoxicity upon the addition of folic acid,134,136 however no control folate receptor negative cell line or competition assay is run to prove that this increase in cytotoxicity is indeed receptor mediated and selective towards folate receptor overexpressing cancers. To assess the true benefits of folate-targeting AuNPs it is vital that the selectivity of these nanoconjugates is fully assessed, as this increase in cytotoxicity may be due to increased passive penetration and therefore these approaches may not provide any gain over non-targeted AuNPs.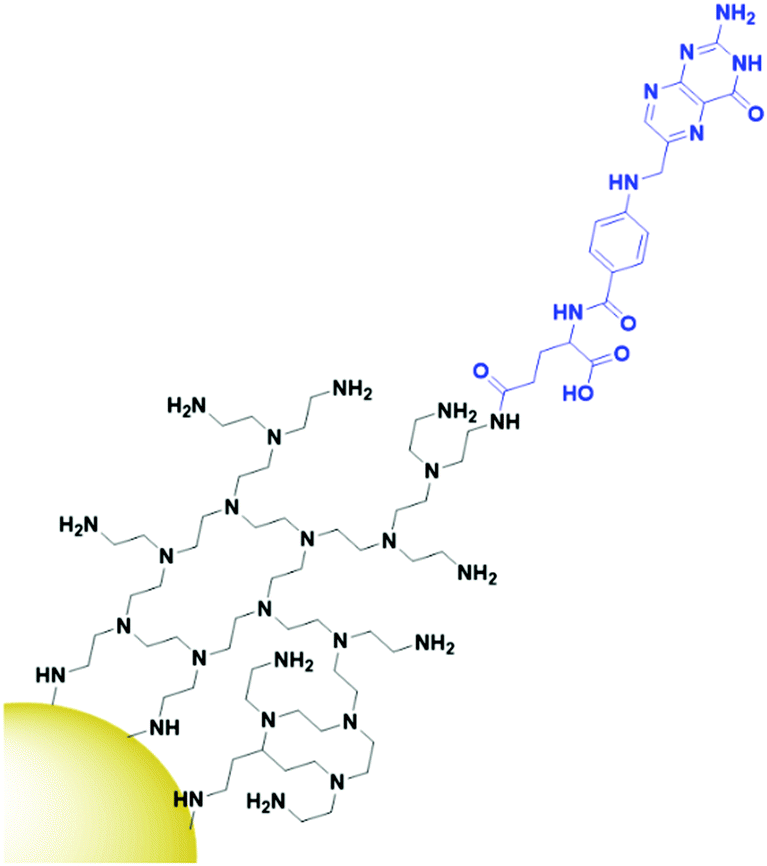 | ||
| Fig. 10 The conjugation of folic acid (blue) onto PEI (black) functionalised AuNPs. | ||
While most examples of small molecule targeted AuNP therapies use folate as their targeting moiety, examples exist using other small molecules. Anisamide is known to bind to the Sigma receptor which is overexpressed in prostate cancer and has been used to deliver AuNPs carrying siRNA to their target. Anisamide is a synthetically produced small molecule derived from anisole, a naturally occurring molecule found in aniseed oil.138 The use of anisamide highlights another advantage of small molecules – they can be easily modified through structure–activity relationship studies to optimise their binding towards a target and remove unnecessary complexities from molecules that are not involved in the binding to a receptor. As with folic acid, anisamide lacks conjugation sites, meaning the siRNA is attached through electrostatic interactions with a PEI coating on the gold core (Fig. 11). The anisamide itself is synthesised by conjugating anisic acid to PEI ligands on these nanoparticles. For siRNA delivery, non-covalent attachment appears to be the most efficient delivery system as the siRNA can diffuse away upon internalisation by cancerous cells where the pH is decreased, and the PEI becomes deprotonated. Anisimide-AuNPs-siRNA are shown to trigger apoptosis due to mRNA knockdown, with this cytotoxicity observed to be receptor mediated.139,140
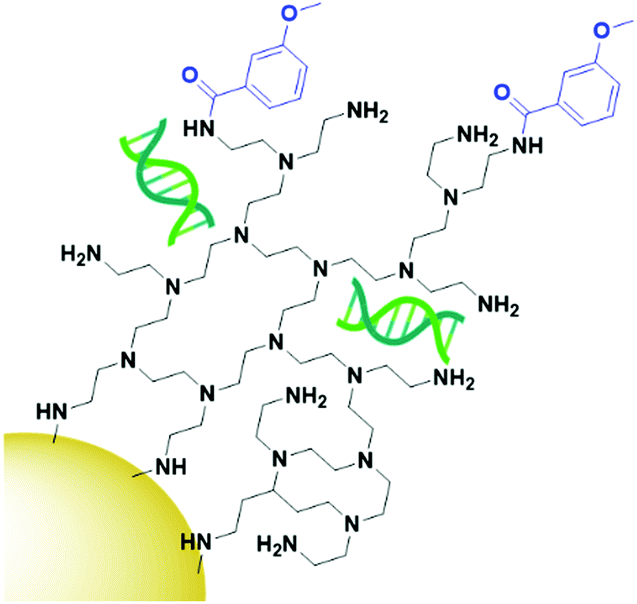 | ||
| Fig. 11 Anisamide (blue) conjugated onto AuNPs through PEI (black), with the siRNA payload (green) electrostatically coordinated to the PEI.139,140 | ||
α- and β-bicalutamide are antiandrogens, small molecules known to bind the androgen-sensing G protein coupled receptor GPRC6A and the membrane androgen receptor (MAR), both expressed by prostate cancers. These antiandrogens are currently clinically used for chemotherapy of prostate cancer, but their attachment onto PEGylated AuNPs (Fig. 12) has shown selective cytotoxicity in MAR/GPRC6A positive chemo-resistant prostate cells.141 In the work reported, the use of a selective chemotherapeutic drug as the targeting moiety for AuNP delivery displays the benefits of AuNPs as delivery vehicles. This ability of AuNP conjugation to increase the uptake of already selective chemotherapeutic drugs through multivalent binding is an exciting prospect.
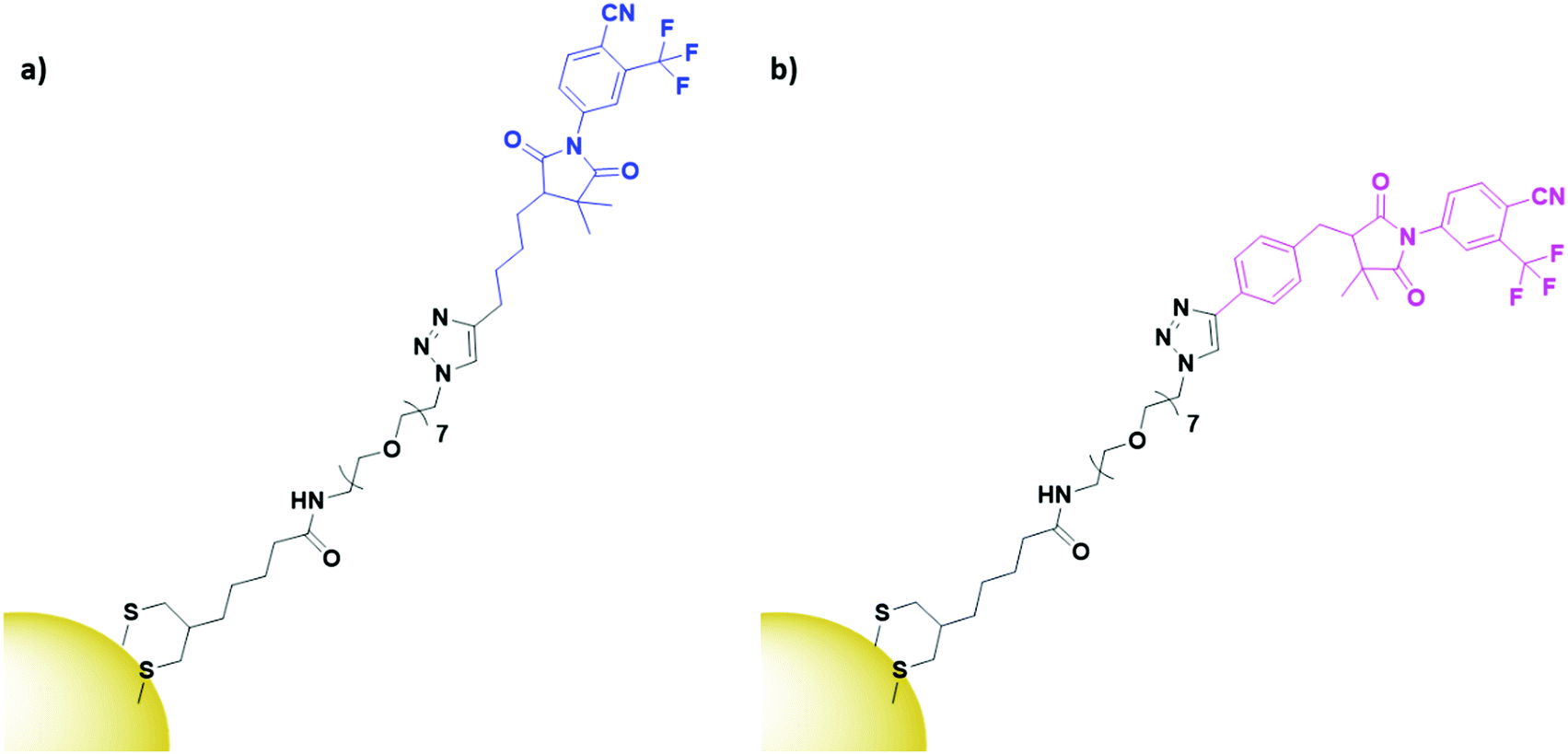 | ||
| Fig. 12 Structures of (a) α-bicalutamide (blue) and (b) β-bicalutamide (pink) AuNPs, conjugated through a PEG linker (black).141 | ||
8. Gold nanoparticles with multiple targeting modalities
The multivalency of AuNPs provides the opportunity to functionalise the surface with multiple directing moieties to target multiple receptors, and these targeting moieties do not have to be of the same class. Gold nanostars have been functionalised with a mixed monolayer of the aptamer A10 and the peptide DUP-1.142 A10 is PSMA specific and DUP-1 binds to an unknown site on prostate carcinomas. It was found that the combination of these targeting modalities allowed for selective targeted PTT of prostate cancers, regardless of their PSMA expression.142AuNPs carrying the ribosome inactivating protein curcin have been targeted towards glioblastoma using a combination of folic acid and an anti-transferrin antibody.143 Glioblastomas overexpress both the FRα and the transferrin receptors. Increased uptake was observed for the dual targeted AuNPs compared to AuNPs functionalised with just folic acid, highlighting the benefit of dually targeting these nanocarriers. Selective cytotoxicity was observed in glioblastoma cells, and the use of curcin was seen to display a synergistic cytotoxicity when delivered by AuNPs that were subsequently used for PTT.143 These examples highlight the benefits of targeting multiple receptors to either widen the therapeutic value of an AuNP system to a higher percentage of a certain cancer type, or to use the synergistic targeting abilities of two moieties to increase specificity towards a target. Most importantly here, these results show that these targeting modalities can be mixed and matched to enhance the targeting ability of AuNPs without interfering with each other, allowing for a large variety in targeting combinations to establish the best possible treatment for specific cancers.
9. Conclusions and future prospects
AuNPs present themselves as ideal drug carriers due to their biocompatibility, facile functionalisation with various ligands, their easily tuned size and shape and their enhanced absorption characteristics due to their surface plasmon absorption band. While AuNPs display passive targeting towards tumours through the EPR effect, with the current trend in developing personalised medicines, the active targeting of AuNPs towards tumours is growing in popularity. Oncogenes expressed on the surface of cancer cells can be directly targeted by antibodies, proteins, peptides, aptamers, carbohydrates and small molecules, and their attachment onto AuNPs leads to selective uptake through receptor mediated endocytosis. The receptors and types of targeting moieties exploited for actively targeting AuNPs to cancers are summarised in Table 1.| Receptor | Targeting moiety | Cancer type |
|---|---|---|
| EGFR | Antibody | Head and neck,47,60 breast,48,70 bladder,50 pancreatic,53,58 skin59 |
| Protein | Breast82 | |
| Peptide | Glioblastoma106,107 | |
| HER2 | Antibody | Breast,45,46,48,57,67,68,72,75 gastric73 |
| Nanobody | Ovarian81 | |
| Aptamer | Breast118 | |
| CD30 | Antibody | Hodgkin lymphoma51 |
| MUC-1 | Antibody | Breast52 |
| hERG1 | Antibody | Pancreatic54 |
| DR5 | Antibody | Colorectal56 |
| TROP-2 | Antibody | Cervical66 |
| CD44 | Antibody | Gastric71 |
| Carbohydrate | Glioblastoma,126 lung,127 liver128 | |
| Transferrin receptor | Protein | Lung83 |
| Antibody | Glioblastoma143 | |
| FGFR | Protein | Lung84 |
| T antigen | Protein | Colon67,87 |
| Plectin-1 | Peptide | Pancreatic97 |
| αvβ3 integrin | Peptide | Cervical,98,103 breast,100 glioblastoma101 |
| Neuropilin-1 | Peptide | Breast,104 prostate105 |
| Nucleolin | Aptamer | Melanoma,113 breast,119,121 cervical114 |
| PSMA | Aptamer | Prostate117,142 |
| PTK7 | Aptamer | Leukaemia115,116,122 |
| Glucose transporter-1 | Carbohydrate | Breast129 |
| Galectin-1 | Carbohydrate | Breast130 |
| Folate receptor | Small molecule | Bile duct,132 cervical,134 breast,135,137 colon,136 glioblastoma143 |
| Sigma receptor | Small molecule | Prostate139,140 |
| GPRC6A/MAR | Small molecule | Prostate141 |
Antibodies are most commonly conjugated onto AuNPs through water soluble ligands, with the payload conjugated to the core instead of to the antibody itself. Antibodies are perhaps the most widely investigated targeting moiety to functionalise AuNPs due to their extremely high selectivity and affinity towards their target receptors. The use of antibodies is, however, more expensive than the majority of other targeting moieties and antibodies display low tumour penetration due to their large size. Antibody fragments and nanobodies are now widely investigated to combat this low penetration, however this decrease in size reduces the circulatory lifetime.
Aptamers display receptor affinity close to that of an antibody and therefore potentially provide a good alternative to antibodies. Aptamers display higher stability than antibodies towards organic solvents and pH which is ideal for the synthesis of nanocarriers. Aptamers are also chemically synthesised which allows for easier modification and characterisation than antibodies, and payloads can be directly attached to these targeting moieties. These synthetic targeting moieties allow for the synthesis of ligands bearing multiple targeting moieties, which could allow for the targeting of multiple receptors with one nanocarrier. Aptamers can themselves elicit anti-tumour effects, and therefore can act as both a targeting moiety and a drug. Payloads can also be directly attached to aptamers without effecting their affinity to a receptor, providing an alternative method for payload delivery on AuNPs as they do not have to be directly bound to the gold core. While these benefits highlight aptamers as a beneficial targeting moiety, they display low stability in circulation due to nucleases in blood plasma.144 Due to the inherent negative charge of aptamers, they cannot be developed to target receptors that display negatively charged surfaces, providing severe limitations to these targeting moieties.
Protein ligands are also investigated as targeting moieties, displaying a high affinity towards their target. The synthesis of therapeutic protein-AuNPs usually involves the protein and payload separately conjugated to the core through linkers. Their affinity towards receptors is generally lower than that of antibodies and aptamers and their relative abundance in the body perhaps reduces their efficacy as there is a lot of natural competition for a binding site.
The low penetration of antibodies and other proteins into tumours has led to the investigation of peptides as targeting moieties. While peptides generally show a lower affinity towards the target receptor, the small size of peptides means they can be synthesised, which allows for ease of modification and control over the binding of these peptides onto nanocarriers. This allows for site-specific conjugation of peptides onto AuNPs and ensures that the amino acids vital for receptor binding remain exposed, a benefit over antibodies and proteins where the conjugation onto AuNPs can be completely random and these binding sites could be blocked. Peptides are also more stable towards pH and organic solvents than proteins, which increases the ease of synthesis of peptide targeted nanocarriers. While their uptake and versatility is high, peptides display a low circulatory half-life due to peptidases in blood plasma,145 and their half-life is similar to that of aptamers.
The expression of lectins on the surface of cancerous tissue has recently led to the investigation of carbohydrates as targeting moieties for AuNPs. Carbohydrate ligands are either attached directly to the gold core or conjugated through linkers. Carbohydrates display high affinities towards their targets and, while their synthesis may be more complicated, they can be modified to fit the desired purpose prior to attachment onto a nanocarrier system. Highly polymeric carbohydrates such as hyaluronic acid allow for the attachment of payloads onto the targeting ligand itself and, as with aptamers, this provides an interesting alternative to directly binding a payload to the gold core, which may display significant advantages for some payloads. The relative lack of investigation into these targeting moieties means that it is hard to conclude on the impact these ligands may have, however some carbohydrates display affinity towards multiple receptors,146 and this may decrease the efficacy of carbohydrate directed therapeutics and lead to off-target effects.
Small molecules are low molecular weight organic compounds that display increased circulatory stability over other targeting moieties as they are not degraded by peptidases or nucleases found in the blood stream. Due to the size of small molecules, they have limited conjugation sites, so both the small molecules and payloads tend to be conjugated onto AuNPs through linkers. The size of small molecules means that they generally display high tumour penetration, and small molecules can themselves elicit a therapeutic effect upon internalisation. Small molecules, however, generally display a lower affinity and selectivity towards their target than antibodies, proteins and aptamers.
The active targeting of AuNPs has allowed for the development of previously unknown cancer therapies with exciting potential. While each of the targeting modalities presented in this review display their own unique characteristics and advantages, there is little work comparing the ability of these modalities to deliver AuNPs. This lack of comparison makes it difficult to conclude whether the benefits of one targeting moiety outweighs the disadvantages of another, and to continue to push this field forward, comparisons between different targeting modalities for the same receptor are needed to determine the most effective approach. These comparisons could allow for the development of highly potent ‘magic bullets’ for the treatment of cancers, with the specificity observed for actively targeted nanoconjugates showing that AuNPs have a bright future in cancer therapeutics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of East Anglia, PDT Norfolk and the Anthony Long Trust for the financial support of this work.Notes and references
- P. Ehrlich, Folia Serol., 1911, 7, 697–714 CAS.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Tan, N. Hou, Y. Liu, B. Liu, W. Cao, D. Zheng, W. Li, Y. Liu, B. Xu, Z. Wang and D. Cui, Nanomedicine, 2020, 27, 102192 CrossRef CAS.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
- R. Guo, L. Zhang, H. Qian, R. Li, X. Jiang and B. Liu, Langmuir, 2010, 26, 5428–5434 CrossRef CAS.
- S. Karve, M. E. Werner, R. Sukumar, N. D. Cummings, J. A. Copp, E. C. Wang, C. Li, M. Sethi, R. C. Chen, M. E. Pacold and A. Z. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8230–8235 CrossRef CAS.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS.
- K. Y. J. Lee, Y. Wang and S. Nie, RSC Adv., 2015, 5, 65651–65659 RSC.
- X. Wang, X. Cai, J. Hu, N. Shao, F. Wang, Q. Zhang, J. Xiao and Y. Cheng, J. Am. Chem. Soc., 2013, 135, 9805–9810 CrossRef CAS.
- T. Olusanya, R. Haj Ahmad, D. Ibegbu, J. Smith and A. Elkordy, Molecules, 2018, 23, 907 CrossRef.
- B. Begines, T. Ortiz, M. Pérez-Aranda, G. Martínez, M. Merinero, F. Argüelles-Arias and A. Alcudia, Nanomaterials, 2020, 10, 1403 CrossRef CAS.
- Q. Zeng, D. Shao, X. He, Z. Ren, W. Ji, C. Shan, S. Qu, J. Li, L. Chen and Q. Li, J. Mater. Chem. B, 2016, 4, 5119–5126 RSC.
- G. Jalani, V. Tam, F. Vetrone and M. Cerruti, J. Am. Chem. Soc., 2018, 140, 10923–10931 CrossRef CAS.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS.
- R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS.
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef.
- C. M. Cobley, J. Chen, E. Chul Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC.
- J. Gao, X. Huang, H. Liu, F. Zan and J. Ren, Langmuir, 2012, 28, 4464–4471 CrossRef CAS.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, Prog. Mater. Sci., 2016, 83, 152–190 CrossRef CAS.
- J. Penders, M. Stolzoff, D. J. Hickey, M. Andersson and T. J. Webster, Int. J. Nanomed., 2017, 12, 2457–2468 CrossRef CAS.
- S. Tokonami, Y. Yamamoto, H. Shiigi and T. Nagaoka, Anal. Chim. Acta, 2012, 716, 76–91 CrossRef CAS.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- C. Daruich De Souza, B. Ribeiro Nogueira and M. E. C. M. Rostelato, J. Alloys Compd., 2019, 798, 714–740 CrossRef CAS.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- L. M. Liz-Marzán, Chem. Commun., 2013, 49, 16–18 RSC.
- R. Herizchi, E. Abbasi, M. Milani and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 596–602 CrossRef CAS.
- L. Freitas De Freitas, G. Henrique Costa Varca, J. Gabriel, S. Batista and A. B. Lugão, Nanomaterials, 2018, 8, 939 CrossRef.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- X. Ma, Y. Wu, S. Jin, Y. Tian, X. Zhang, Y. Zhao, L. Yu and X.-J. Liang, ACS Nano, 2011, 5, 8629–8639 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- Z. Lin, N. A. Monteiro-Riviere and J. E. Riviere, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 189–217 CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217–228 CrossRef.
- Y. J. Gu, J. Cheng, C. W. Y. Man, W. T. Wong and S. H. Cheng, Nanomedicine, 2012, 8, 204–211 CrossRef CAS.
- S. D. Brown, P. Nativo, J. A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS.
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Biomaterials, 2015, 60, 100–110 CrossRef CAS.
- M. Camerin, M. Moreno, M. J. Marín, C. L. Schofield, I. Chambrier, M. J. Cook, O. Coppellotti, G. Jori and D. A. Russell, Photochem. Photobiol. Sci., 2016, 15, 618–625 RSC.
- K. Mckeage and C. M. Perry, Drugs, 2001, 1, 209–243 Search PubMed.
- D. J. Jonker, C. J. O’Callaghan, C. S. Karapetis, J. R. Zalcberg, D. Tu, H.-J. Au, S. R. Berry, M. Krahn, T. Price, R. J. Simes, N. C. Tebbutt, G. van Hazel, R. Wierzbicki, C. Langer and M. J. Moore, N. Engl. J. Med., 2007, 357, 2040–2048 CrossRef CAS.
- N. Chattopadhyay, Z. Cai, Y. L. Kwon, E. Lechtman, J.-P. Pignol and R. M. Reilly, Breast Cancer Res. Treat., 2013, 137, 81–91 CrossRef CAS.
- N. Chattopadhyay, Z. Cai, J. P. Pignol, B. Keller, E. Lechtman, R. Bendayan and R. M. Reilly, Mol. Pharmaceutics, 2010, 7, 2194–2206 CrossRef CAS.
- A. Popovtzer, A. Mizrachi, M. Motiei, D. Bragilovski, L. Lubimov, M. Levi, O. Hilly, I. Ben-Aharon and R. Popovtzer, Nanoscale, 2016, 8, 2678–2685 RSC.
- S. Yook, Z. Cai, J. J. Jeong, Y. Lu, M. A. Winnik, J. P. Pignol and R. M. Reilly, Mol. Pharmaceutics, 2020, 17, 1226–1236 CrossRef CAS.
- A. Mesbahi, Reports Pract. Oncol. Radiother., 2010, 15, 176–180 CrossRef.
- C. H. Chen, Y.-J. Wu and J.-J. Chen, Front. Biosci., Landmark Ed., 2016, 21, 1211–1221 CrossRef CAS.
- X. Qu, C. Yao, J. Wang, Z. Li and Z. Zhang, Int. J. Nanomed., 2012, 7, 6095–6103 CrossRef CAS.
- D. C. Zelasko-Leon, C. M. Fuentes and P. B. Messersmith, PLoS One, 2015, 10, e0128756 CrossRef.
- C. R. Patra, R. Bhattacharya, E. Wang, A. Katarya, J. S. Lau, S. Dutta, M. Muders, S. Wang, S. A. Buhrow, S. L. Safgren, M. J. Yaszemski, J. M. Reid, M. M. Ames, P. Mukherjee and D. Mukhopadhyay, Cancer Res., 2008, 68, 1970–1978 CrossRef CAS.
- J. Spadavecchia, D. Movia, C. Moore, C. M. Maguire, H. Moustaoui, S. Casale, Y. Volkov and A. Prina-Mello, Int. J. Nanomed., 2016, 11, 791–822 CrossRef CAS.
- A. Das, E. Soehnlen, S. Woods, R. Hegde, A. Henry, A. Gericke and S. Basu, J. Nanopart. Res., 2011, 13, 6283–6290 CrossRef CAS.
- S. Tummala, M. N. S. Kumar and S. K. Pindiprolu, Drug Delivery, 2016, 23, 3505–3519 CrossRef CAS.
- E. Cruz and V. Kayser, Cancers, 2019, 11, 870 Search PubMed.
- H. W. Kao, Y. Y. Lin, C. C. Chen, K. H. Chi, D. C. Tien, C. C. Hsia, M. H. Lin and H. E. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 3180–3185 CrossRef CAS.
- D. Rades, C. Wolff, R. Nadrowitz, C. Breunig, S. E. Schild, M. Baehre and B. Meller, Int. J. Radiat. Oncol., Biol., Phys., 2009, 75, 1226–1231 CrossRef CAS.
- G. Niu, X. Sun, Q. Cao, D. Courter, A. Koong, Q.-T. Le, S. Sam Gambhir and X. Chen, Clin. Cancer Res., 2010, 16, 2095–2105 CrossRef CAS.
- S. Shirvani-Arani, A. Bahrami-Samani, A. R. Jalilian, A. Shirvani-Arani and M. Ghannadi-Maragheh, J. Label. Compd. Radiopharm., 2012, 55, 103–107 CrossRef CAS.
- S. Bhattacharyya, R. Bhattacharya, S. Curley, M. A. McNiven and P. Mukherjee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14541–14546 CrossRef CAS.
- C. D. Austin, A. M. De Mazière, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman and R. H. Scheller, Mol. Biol. Cell, 2004, 15, 5268–5282 CrossRef CAS.
- A. M. Hommelgaard, M. Lerdrup and B. van Deurs, Mol. Biol. Cell, 2004, 15, 1557–1567 CrossRef CAS.
- Z. Cai, S. Yook, Y. Lu, D. Bergstrom, M. A. Winnik, J. P. Pignol and R. M. Reilly, Pharm. Res., 2017, 34, 579–590 CrossRef CAS.
- T. Liu, J. Tian, Z. Chen, Y. Liang, J. Liu, S. Liu, H. Li, J. Zhan and X. Yang, Nanotechnology, 2014, 25, 345103 CrossRef.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Photochem. Photobiol. Sci., 2015, 14, 737–747 RSC.
- T. Stuchinskaya, M. Moreno, M. J. Cook, D. R. Edwards and D. A. Russell, Photochem. Photobiol. Sci., 2011, 10, 822–831 RSC.
- G. J. Kim, S. R. Park, G. C. Kim and J. K. Lee, Plasma Med., 2011, 1, 45–54 CrossRef.
- M. Zhang, H. S. Kim, T. Jin and W. K. Moon, J. Photochem. Photobiol., B, 2017, 170, 58–64 CrossRef CAS.
- S. Liang, C. Li, C. Zhang, Y. Chen, L. Xu, C. Bao, X. Wang, G. Liu, F. Zhang and D. Cui, Theranostics, 2015, 5, 970–984 CrossRef CAS.
- O. Penon, M. J. Marín, D. A. Russell and L. Pérez-García, J. Colloid Interface Sci., 2017, 496, 100–110 CrossRef CAS.
- T. Kubota, S. Kuroda, N. Kanaya, T. Morihiro, K. Aoyama, Y. Kakiuchi, S. Kikuchi, M. Nishizaki, S. Kagawa, H. Tazawa and T. Fujiwara, Nanomedicine, 2018, 14, 1919–1929 CrossRef CAS.
- E. W. Bassett, S. W. Tanenbaum, K. Pryzwansky, S. M. Beiser and E. A. Kabat, J. Exp. Med., 1965, 122, 251–261 CrossRef CAS.
- X. Sun, G. Zhang, R. S. Keynton, M. G. O’Toole, D. Patel and A. M. Gobin, Nanomedicine, 2013, 9, 1214–1222 CrossRef CAS.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS.
- M. Melancon, W. Lu and C. Li, Mater. Res. Bull., 2009, 34, 415–421 CrossRef CAS.
- S. Oliveira, R. Heukers, J. Sornkom, R. J. Kok and P. M. P. Van Bergen En Henegouwen, J. Controlled Release, 2013, 172, 607–617 CrossRef CAS.
- S. Muyldermans, T. N. Baral, V. C. Retamozzo, P. De Baetselier, E. De Genst, J. Kinne, H. Leonhardt, S. Magez, V. K. Nguyen, H. Revets, U. Rothbauer, B. Stijlemans, S. Tillib, U. Wernery, L. Wyns, G. Hassanzadeh-Ghassabeh and D. Saerens, Vet. Immunol. Immunopathol., 2009, 128, 178–183 CrossRef CAS.
- V. Cortez-Retamozo, M. Lauwereys, G. Hassanzadeh Gh., M. Gobert, K. Conrath, S. Muyldermans, P. De Baetselier and H. Revets, Int. J. Cancer, 2002, 98, 456–462 CrossRef CAS.
- B. Van de Broek, N. Devoogdt, A. D’Hollander, H.-L. Gijs, K. Jans, L. Lagae, S. Muyldermans, G. Maes and G. Borghs, ACS Nano, 2011, 5, 4319–4328 CrossRef CAS.
- L. Song, N. Falzone and K. A. Vallis, Int. J. Radiat. Biol., 2016, 92, 716–723 CrossRef CAS.
- N. Amreddy, R. Muralidharan, A. Babu, M. Mehta, E. V. Johnson, Y. D. Zhao, A. Munshi and R. Ramesh, Int. J. Nanomed., 2015, 10, 6773–6788 CAS.
- W. S. M. E. Theelen, L. Mittempergher, S. M. Willems, A. J. Bosma, D. D. G. C. Peters, V. van der Noort, E. J. Japenga, T. Peeters, K. Koole, T. Šuštić, J. L. Blaauwgeers, C. J. van Noesel, R. Bernards and M. M. van den Heuvel, J. Pathol.: Clin. Res., 2016, 2, 223–233 CAS.
- N. Cihoric, S. Savic, S. Schneider, I. Ackermann, M. Bichsel-Naef, R. A. Schmid, D. Lardinois, M. Gugger, L. Bubendorf, I. Zlobec and C. Tapia, Br. J. Cancer, 2014, 110, 2914–2922 CrossRef CAS.
- A. Szlachcic, K. Pala, M. Zakrzewska, P. Jakimowicz, A. Wiedlocha and J. Otlewski, Int. J. Nanomed., 2012, 7, 5915–5927 CAS.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 6158–6162 CrossRef CAS.
- K. Hozumi, K. Kunihiko and H. Keiji, Mol. Immunol., 1987, 24, 1219–1222 CrossRef.
- G. Fassina, M. Ruvo, G. Palombo, A. Verdoliva and M. Marino, J. Biochem. Biophys. Methods, 2001, 49, 481–490 CrossRef CAS.
- F. Biscaglia, S. Rajendran, P. Conflitti, C. Benna, R. Sommaggio, L. Litti, S. Mocellin, G. Bocchinfuso, A. Rosato, A. Palleschi, D. Nitti, M. Gobbo and M. Meneghetti, Adv. Healthcare Mater., 2017, 6, 1700596 CrossRef.
- E. J. Simpson, P. Gobbo, F. C. Bononi, E. Murrell, M. S. Workentin and L. G. Luyt, J. Interdiscip. Nanomed., 2017, 2, 174–187 CrossRef CAS.
- X. Xu, L. Zhao, X. Li, P. Wang, J. Zhao, X. Shi and M. Shen, Biomater. Sci., 2017, 5, 2393–2397 RSC.
- S. Jha, F. Ramadori, S. Quarta, A. Biasiolo, E. Fabris, P. Baldan, G. Guarino, M. Ruvoletto, G. Villano, C. Turato, A. Gatta, F. Mancin, P. Pontisso and P. Scrimin, Bioconjugate Chem., 2017, 28, 222–229 CrossRef CAS.
- H.-Q. Yin, D.-S. Mai, F. Gan and X.-J. Chen, RSC Adv., 2014, 4, 9078–9085 RSC.
- S. Avvakumova, E. Galbiati, L. Pandolfi, S. Mazzucchelli, M. Cassani, A. Gori, R. Longhi and D. Prosperi, Bioconjugate Chem., 2014, 25, 1381–1386 CrossRef CAS.
- D. Bausch, S. Thomas, M. Mino-Kenudson, C. Fernández-del Castillo, T. W. Bauer, M. Williams, A. L. Warshaw, S. P. Thayer and K. A. Kelly, Clin. Cancer Res., 2011, 17, 302–309 CrossRef CAS.
- K. Pal, F. Al-Suraih, R. Gonzalez-Rodriguez, S. K. Dutta, E. Wang, H. S. Kwak, T. R. Caulfield, J. L. Coffer and S. Bhattacharya, Nanoscale, 2017, 9, 15622–15634 RSC.
- G. Liang, X. Jin, S. Zhang and D. Xing, Biomaterials, 2017, 144, 95–104 CrossRef CAS.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS.
- P.-H. Wu, T. Hashimoto, H. Shirato, J.-M. Nam, Y. Onodera, Y. Ichikawa, Y. Watanabe, Y. Ichikawa, W. Qian, E. B. Rankin, A. J. Giaccia, H. Shirato, J.-M. Nam, H. Shirato and J.-M. Nam, Int. J. Nanomed., 2017, 12, 5069–5085 CrossRef CAS.
- S. Wang, K. J. Chen, T. H. Wu, H. Wang, W. Y. Lin, M. Ohashi, P. Y. Chiou and H. R. Tseng, Angew. Chem., Int. Ed., 2010, 49, 3777–3781 CrossRef CAS.
- B. A. Werness, A. J. Levine and P. M. Howley, Science, 1990, 248, 76–79 CrossRef CAS.
- Y. Yi, H. J. Kim, P. Mi, M. Zheng, H. Takemoto, K. Toh, B. S. Kim, K. Hayashi, M. Naito, Y. Matsumoto, K. Miyata and K. Kataoka, J. Controlled Release, 2016, 244, 247–256 CrossRef CAS.
- A. Kumar, H. Ma, X. Zhang, K. Huang, S. Jin, J. Liu, T. Wei, W. Cao, G. Zou and X. J. Liang, Biomaterials, 2012, 33, 1180–1189 CrossRef CAS.
- A. Kumar, S. Huo, X. Zhang, J. Liu, A. Tan, S. Li, S. Jin, X. Xue, Y. Zhao, T. Ji, L. Han, H. Liu, X. Zhang, J. Zhang, G. Zou, T. Wang, S. Tang and X. J. Liang, ACS Nano, 2014, 8, 4205–4220 CrossRef CAS.
- Y. Cheng, J. D. Meyers, R. S. Agnes, T. L. Doane, M. E. Kenney, A.-M. M. Broome, C. Burda and J. P. Basilion, Small, 2011, 7, 2301–2306 CrossRef CAS.
- J. D. Meyers, Y. Cheng, A. M. M. Broome, R. S. Agnes, M. D. Schluchter, S. Margevicius, X. Wang, M. E. Kenney, C. Burda and J. P. Basilion, Part. Part. Syst. Charact., 2015, 32, 448–457 CrossRef CAS.
- M. M. Saber, S. Bahrainian, R. Dinarvand and F. Atyabi, Int. J. Pharm., 2017, 517, 269–278 CrossRef CAS.
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CrossRef CAS.
- P. R. Bouchard, R. M. Hutabarat and K. M. Thompson, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 237–257 CrossRef CAS.
- H. Sun, X. Zhu, P. Y. Lu, R. R. Rosato, W. Tan and Y. Zu, Mol. Ther.-Nucleic Acids, 2014, 3, 1–14 Search PubMed.
- K. Han, Z. Liang and N. Zhou, Sensors, 2010, 10, 4541–4557 CrossRef CAS.
- A. Latorre, C. Posch, Y. Garcimartín, A. Celli, M. Sanlorenzo, I. Vujic, J. Ma, M. Zekhtser, K. Rappersberger, S. Ortiz-Urda and Á. Somoza, Nanoscale, 2014, 6, 7436–7442 RSC.
- J. Ai, Y. Xu, B. Lou, D. Li and E. Wang, Talanta, 2014, 118, 54–60 CrossRef CAS.
- J. Wang, G. Zhu, M. You, E. Song, M. I. Shukoor, K. Zhang, M. B. Altman, Y. Chen, Z. Zhu, C. Z. Huang and W. Tan, ACS Nano, 2012, 6, 5070–5077 CrossRef CAS.
- Y.-L. Luo, Y.-S. Shiao and Y.-F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS.
- D. Kim, Y. Y. Jeong and S. Jon, ACS Nano, 2010, 4, 3689–3696 CrossRef CAS.
- H. Lee, D. H. M. Dam, J. W. Ha, J. Yue and T. W. Odom, ACS Nano, 2015, 9, 9859–9867 CrossRef CAS.
- E. J. Hong, Y.-S. Kim, D. G. Choi and M. S. Shim, J. Ind. Eng. Chem., 2018, 67, 429–436 CrossRef CAS.
- C. M. Berger, X. Gaume and P. Bouvet, Biochimie, 2015, 113, 78–85 CrossRef CAS.
- F. Ghahremani, D. Shahbazi-Gahrouei, A. Kefayat, H. Motaghi, M. A. Mehrgardi and S. H. Javanmard, RSC Adv., 2018, 4249–4258 RSC.
- S. M. Taghdisi, N. M. Danesh, P. Lavaee, A. S. Emrani, K. Y. Hassanabad, M. Ramezani and K. Abnous, Mater. Sci. Eng., C, 2016, 61, 753–761 CrossRef CAS.
- R. Lotan and A. Raz, Ann. N. Y. Acad. Sci., 1988, 551, 385–398 CrossRef CAS.
- N. Yamazaki, S. Kojima, N. V. Bovin, S. André, S. Gabius and H. J. Gabius, Adv. Drug Delivery Rev., 2000, 43, 225–244 CrossRef CAS.
- L. Y. Bourguignon, V. B. Lokeshwar, X. Chen and W. G. Kerrick, J. Immunol., 1993, 151, 6634–6644 CAS.
- Y. Song, Z. Wang, L. Li, W. Shi, X. Li and H. Ma, Chem. Commun., 2014, 50, 15696–15698 RSC.
- S. H. Kang, M. Nafiujjaman, M. Nurunnabi, L. Li, H. A. Khan, K. J. Cho, K. M. Huh and Y. K. Lee, Macromol. Res., 2015, 23, 474–484 CrossRef CAS.
- C. S. Kumar, M. D. Raja, D. S. Sundar, M. Gover Antoniraj and K. Ruckmani, Carbohydr. Polym., 2015, 128, 63–74 CrossRef CAS.
- Y. Yi, H. J. Kim, M. Zheng, P. Mi, M. Naito, B. S. Kim, H. S. Min, K. Hayashi, F. Perche, K. Toh, X. Liu, Y. Mochida, H. Kinoh, H. Cabral, K. Miyata and K. Kataoka, J. Controlled Release, 2019, 295, 268–277 CrossRef CAS.
- P. García Calavia, I. Chambrier, M. J. Cook, A. H. Haines, R. A. Field and D. A. Russell, J. Colloid Interface Sci., 2018, 512, 249–259 CrossRef.
- E. I. Sega and P. S. Low, Cancer Metastasis Rev., 2008, 27, 655–664 CrossRef CAS.
- N. Ngernyuang, W. Seubwai, S. Daduang, P. Boonsiri, T. Limpaiboon and J. Daduang, Mater. Sci. Eng., C, 2016, 60, 411–415 CrossRef CAS.
- J. Guo, C. M. O’Driscoll, J. D. Holmes and K. Rahme, Int. J. Pharm., 2016, 509, 16–27 CrossRef CAS.
- L. Zhao, T. H. Kim, H. W. Kim, J. C. Ahn and S. Y. Kim, Mater. Sci. Eng., C, 2016, 67, 611–622 CrossRef CAS.
- B. Feng, Z. Xu, F. Zhou, H. Yu, Q. Sun, D. Wang, Z. Tang, H. Yu, Q. Yin, Z. Zhang and Y. Li, Nanoscale, 2015, 7, 14854–14864 RSC.
- R. M. Devendiran, S. K. Chinnaiyan, N. K. Yadav, G. K. Moorthy, G. Ramanathan, S. Singaravelu, U. T. Sivagnanam and P. T. Perumal, RSC Adv., 2016, 6, 29757–29768 RSC.
- H. Banu, D. K. Sethi, A. Edgar, A. Sheriff, N. Rayees, N. Renuka, S. M. Faheem, K. Premkumar and G. Vasanthakumar, J. Photochem. Photobiol., B, 2015, 149, 116–128 CrossRef CAS.
- A. Dasargyri, C. D. Kümin and J.-C. Leroux, Adv. Mater., 2017, 29, 1603451 CrossRef.
- K. A. Fitzgerald, K. Rahme, J. Guo, J. D. Holmes and C. M. O’Driscoll, J. Mater. Chem. B, 2016, 4, 2242–2252 RSC.
- X. Luan, K. Rahme, Z. Cong, L. Wang, Y. Zou, Y. He, H. Yang, J. D. Holmes, C. M. O’Driscoll and J. Guo, Eur. J. Pharm. Biopharm., 2019, 137, 56–67 CrossRef CAS.
- E. C. Dreaden, B. E. Gryder, L. A. Austin, B. A. Tene Defo, S. C. Hayden, M. Pi, L. D. Quarles, A. K. Oyelere and M. A. El-Sayed, Bioconjugate Chem., 2012, 23, 1507–1512 CrossRef CAS.
- H. Jo, H. Youn, S. Lee and C. Ban, J. Mater. Chem. B, 2014, 2, 4862–4867 RSC.
- M. S. Mohamed, S. Veeranarayanan, A. C. Poulose, Y. Nagaoka, H. Minegishi, Y. Yoshida, T. Maekawa and D. S. Kumar, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1657–1669 CrossRef CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- R. Dings, M. Miller, R. Griffin and K. Mayo, Int. J. Mol. Sci., 2018, 19, 905 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |