
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solving the COF trilemma: towards crystalline, stable and functional covalent organic frameworks
Frederik
Haase
*a and
Bettina V.
Lotsch
 *bcd
*bcd
aInstitute of Functional Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Bldg. 330, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: frederik.haase@kit.edu
bNanochemistry Department, Max Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany. E-mail: b.lotsch@fkf.mpg.de
cDepartment of Chemistry, University of Munich (LMU), Butenandtstrasse 5-13, 81377 Munich, Germany
dExzellenzcluster E-conversion, Lichtenbergstrasse 4a, 85748 Garching, Germany
First published on 6th November 2020
Abstract
Covalent organic frameworks (COFs) have entered the stage as a new generation of porous polymers which stand out by virtue of their crystallinity, diverse framework topologies and accessible pore systems. An important – but still underdeveloped – feature of COFs is their potentially superior stability in comparison to other porous materials. Achieving COFs which are simultaneously crystalline, stable, and functional is still challenging as reversible bond formation is one of the prime prerequisites for the crystallization of COFs. However, as the COF field matures new strategies have surfaced that bypass this crystallinity – stability dichotomy. Three major approaches for obtaining both stable and crystalline COFs have taken form in recent years: Tweaking the reaction conditions for reversible linkages, separating the order inducing step and the stability inducing step, and controlling the structural degrees of freedom during assembly and in the final COF. This review discusses rational approaches to stability and crystallinity engineering in COFs, which are apt at overcoming current challenges in COF design and open up new avenues to new real-world applications of COFs.
 Frederik Haase | Frederik Haase received his PhD from the LMU Munich while working at the Max-Planck Institute for Solid State Research in 2018 in the group of Prof. Bettina Lotsch. After a postdoctoral period at the laboratory of Prof. Shuhei Furukawa in Kyoto, he returned to Germany where he is currently working as a YIG Prep Pro fellow at the Karlsruhe Institute of Technology. His current research focusses on linkage modifications in metal organic and covalent organic frameworks. |
 Bettina V. Lotsch | Bettina V. Lotsch is the Director of the Nanochemistry Department at the Max Planck Institute for Solid State Research in Stuttgart, Germany, and honorary professor at the Universities of Munich (LMU) and Stuttgart. She studied Chemistry at the Universities of Munich and Oxford and received her PhD from LMU Munich in 2006. After a postdoctoral stay at the University of Toronto she became professor at LMU Munich in 2009 and was appointed Director at MPI-FKF in 2017. Bettina's research explores the rational synthesis of new materials for energy conversion and storage by combining the tools of molecular, solid-state and nanochemistry. Bettina was awarded an ERC Starting Grant (2014) and has been elected a Fellow of the Royal Society of Chemistry in 2014. Her work has been recognized by the EU-40 Materials Prize 2017 of the European Materials Research Society. |
1. Introduction
As COFs are coming of age, conceptual advances in their design and the diversity of synthetic protocols have established COFs at the forefront of modern porous materials research. While in principle COFs have proven competitive to metal organic frameworks and zeolites in terms of their functional group tolerance, structural tunability and stability, achieving these features within a covalently connected, crystalline backbone is still a formidable challenge.1 The advantage of covalent connectivity is that it makes COFs robust and creates a versatile platform for tuneable materials design, as the building blocks assemble in a predictable manner. However, the combination of covalent connectivity with crystallinity in COFs requires special functional groups and reaction conditions, which typically are designed for reversible covalent bonds. When invoking dynamic covalent chemistry (DCC) for COF design based on reversible bond forming reactions, a trade-off is usually observed between crystallinity and stability,2 which has been a bottleneck to the development of new COFs for applications: We call this ubiquitous feature of COF chemistry the “crystallinity – stability dichotomy”. But even engineering crystallinity and stability individually is challenging in itself. COFs are typically obtained as powders with crystallite sizes in the range of several hundred nanometres.3 Within one crystallite, crystallinity is defined by the amount of disorder and defects, and hence the porosity of COFs. While the possibility to precisely position functional groups within the crystal lattice and the pore is a hallmark of COFs, this feature is lost if disorder prevents structural control.Crystallinity is also central to characterizing COFs, which is usually done by powder X-ray diffraction (PXRD) in combination with structure modelling and Pawley or Rietveld refinement, or very rarely by single crystal diffraction.4 Even for characterization techniques that do not rely on crystallinity such as solid-state NMR, a reduced line width in highly crystalline materials can improve the information that can be extracted from the measurements,5 thereby allowing for a better understanding and prediction of properties.
1.1. The crystallinity, stability and functionality trilemma in COFs
In COFs the three features of crystallinity, stability and functionality are competing with each other, if the COF is synthesized by reversible reactions (Fig. 1). Crystallinity and stability are inversely related to each other, as an increased reversibility leads to higher crystallinity and lower stability, and vice versa. By improving the reversibility, the building blocks and the COF linkages can rearrange to adopt the thermodynamic minimum structure, consistent with improved crystallinity. At the same time, the reversibility negatively influences the stability, as the cleavage of the COF linkage is simply the reverse of its formation, such as hydrolysis of a linkage that is formed in a condensation reaction. Both crystallinity and stability are also competing with functionality in a less obvious way. Stability is a feature that has to be assessed in the context of the operating temperature and conditions of a desired application.6,7 If a material can be operated at significantly milder conditions than those applied during its synthesis, then reversibility during the synthesis has little impact on the stability. This is the case for materials such as diamond, which is an extremely stable material at room temperature (although metastable) while it is synthesized at a temperature above 2000 K.8 Synthesizing stable COFs with the help of reactions that are only reversible at significantly higher temperatures or under harsher conditions9,10 limit the types of functional groups and functionalities that can be installed in a COF due to their chemical and thermal decomposition during the synthesis.11 Thus, a trilemma exists for the formation of COFs in terms of achieving crystallinity, stability and complexity (implying functionality) at the same time, if one limits oneself to only reversible reactions. A way to escape this trilemma (Fig. 1) involves strategies that lead to ordered structures, but are not completely dependent on reversible reactions (see Section 3.2).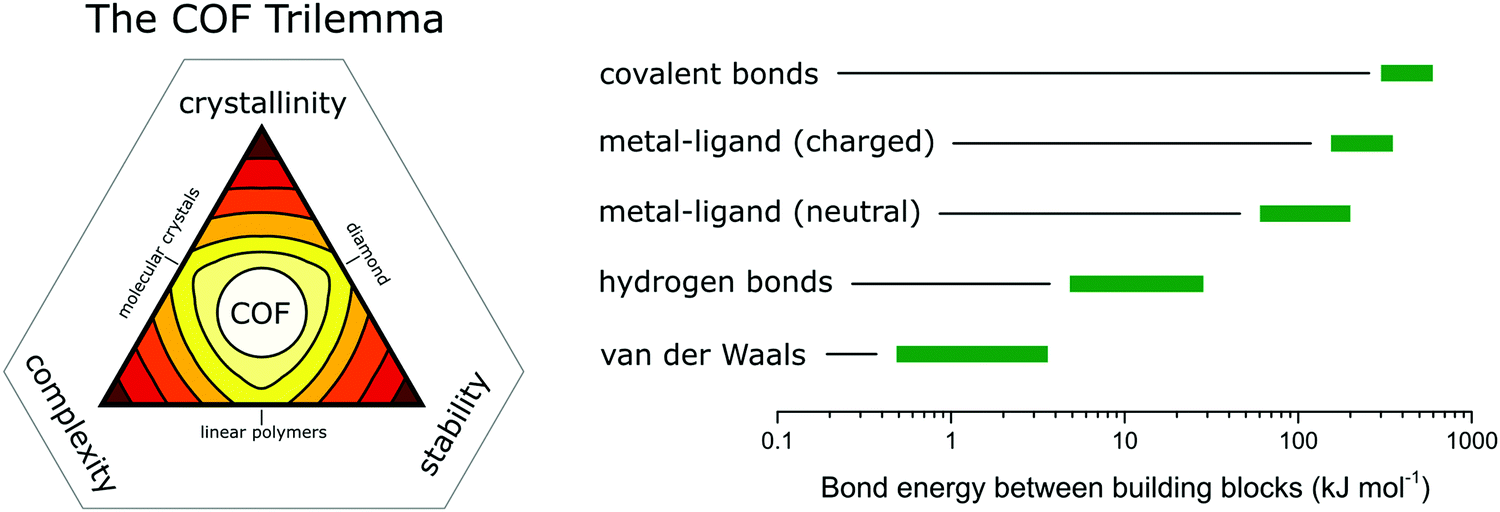 | ||
| Fig. 1 The trilemma of materials design for COF synthesis based on reversible reactions (left). Strengths of bonds that are used for the synthesis of molecularly defined materials (right).12–14 | ||
Since balancing crystallinity, stability and complexity in COFs has played an important role since the inception of this material class, several strategies have been developed from early on in the field to improve the stability while maintaining crystallinity. Stability and the lack thereof can be linked to three pathways by which important COF features such as order, porosity and connectivity are lost (Fig. 2): bond breakage, stacking breakdown, or flexible pore collapse. How to avoid these failure modes will be addressed throughout this review. The bond breakage of COFs can be addressed by creating strong covalent bonds or stabilizing them, as discussed in Section 3.1 and throughout this review, stacking breakdown15 can be mitigated by designing the stacking interactions as described in Section 6.3.4, and the flexible pore collapse16 can be avoided by reducing the number of conformers as described in Section 6.3. Central to escaping this apparently inescapable challenge is to understand the formation principles that govern the synthesis of complex ordered and stable materials as will be introduced in the following and elaborated on further below.17,18
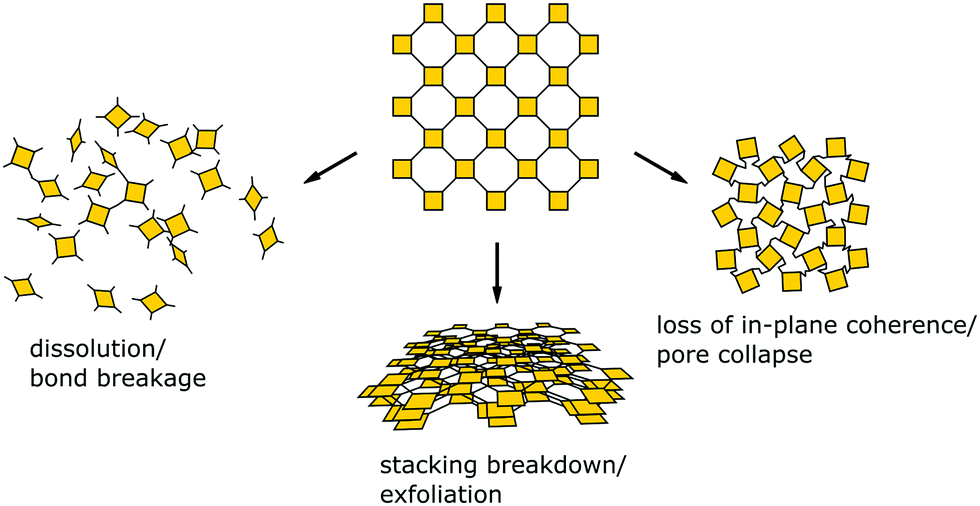 | ||
| Fig. 2 Common modes for loss of crystallinity and porosity in COFs: bond breakage, loss of stacking order, and pore collapse. | ||
1.2. Formation principles of COFs
The general design of COFs has been discussed extensively in a number of excellent reviews.1,19–21 A typical approach as proposed by Diercks et al.1 involves the retrosynthetic disassembly of the target topology into individual building blocks and matching these with molecular equivalents to construct the target COF. Alternately, possible nets and structures are constructed starting from a given molecular building block carrying a desired functionality. Central to both strategies are the number of functional groups used in a building block to propagate the growth of a COF (points of extension), its geometry and the specific combination of different building blocks. The target net with its connectivity has to be able to accommodate the target building blocks without significant distortion to enable the facile synthesis of the material.22,23 In addition, the building blocks need to be connected by a covalent COF linkage that is compatible with the synthesis of a crystalline material, which is usually achieved by a reversible reaction.In recent years, strategies for the formation of ordered materials by other means than relying on reversible reactions have gained attention. New synthetic approaches to ordered covalently connected materials promise improved crystallinity and stability in COFs, and at the same time tolerate chemical functionalities that would be incompatible with reversible reactions. The approaches to obtain crystalline COFs can be classified into three strategies (Fig. 3): reversible reaction, pre-orientation of building blocks and the single reaction pathway. In each class, examples exist that only use a single strategy to obtain a crystalline material; however, the strategies are often used in conjunction with each other, or design principles derived from one strategy can be generalized for other COF syntheses.
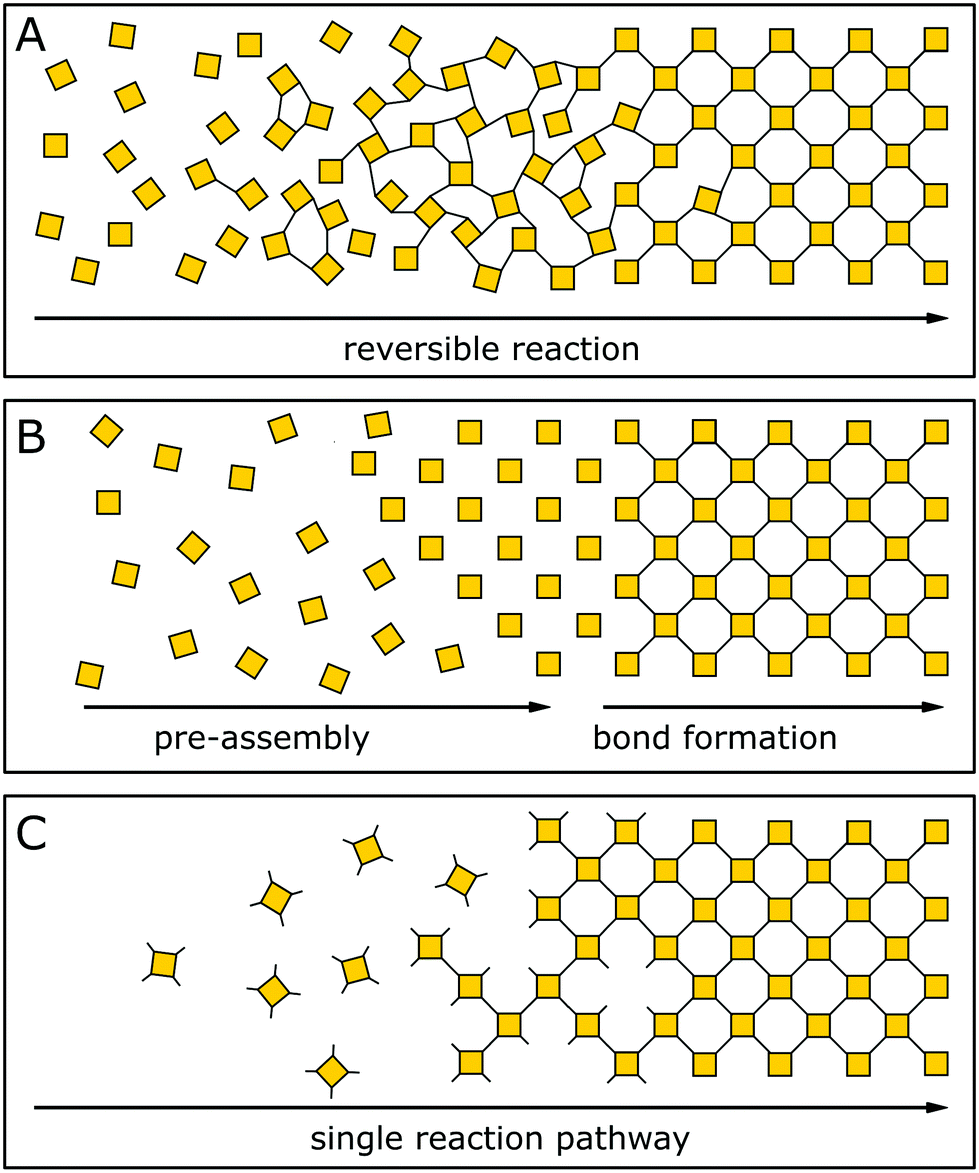 | ||
| Fig. 3 Three approaches to forming a crystalline covalently bound polymer: (A) reversible reactions; (B) pre-orientation; (C) single reaction pathway. | ||
In the following, we will briefly summarize the key features of each strategy which will then be discussed in detail in the following sections, along with pertinent examples:
Crystallization of COFs by reversible covalent bond formation (see Section 4) is the most prominent strategy for constructing highly crystalline COFs. A reversible reaction enables continuous bond formation and breakage and, hence, defect healing during the assembly of the building blocks, such that the system can finally arrive at the thermodynamic equilibrium structure. Using reversible covalent bonds for the crystallization of COFs is the most prevalent mechanism in producing ordered materials and is the generally accepted rationale for crystallinity in COFs.3,24–26
Pre-orientation of building blocks (see Section 5) separates the assembly step from the formation of strong covalent bonds. Building blocks are first aligned by using weak interactions that make the ordering step highly reversible and allow for facile crystallization. In a second step, the building blocks are connected with each other by stronger bonds. This strategy ranges from the formation of molecular crystals which are then crosslinked photochemically, to crystallizing COFs by reversible reactions and subsequently transforming the labile linkage into a more stable linkage by an irreversible reaction.
The formation of COFs by a single reaction pathway (see Section 6) describes the conformational design of COFs and their constituent building blocks in such a way that attachment of building blocks can only occur in only one – the correct – position and orientation, making reversibility of the connecting linkage optional. Building blocks are typically aligned by using a strong connection to the other building blocks and order is achieved by a low number of conformational degrees of freedom. By reducing the number of possible conformers, this strategy has also been successfully applied in improving the crystallinity in COFs based on reversible reactions. Notably, this strategy allows for the formation of COFs based on completely irreversible reactions – a strategy that complements and extends the chemistry of COF formation significantly.
2. Reversible covalent bond formation
The typical approach to designing crystalline COFs takes advantage of dynamic covalent chemistry (Fig. 4 and 5), i.e. it relies on reactions that allow for the reversible covalent bond formation during the synthesis. The reversible bond enables the building blocks to arrange in the thermodynamic minimum configuration (Fig. 4). If the structure is not crystalline or contains defects, dangling bonds and off-equilibrium structures are produced, which are energetically less favourable than the corresponding ordered structure. This energy gain drives the formation and crystallization of the structure.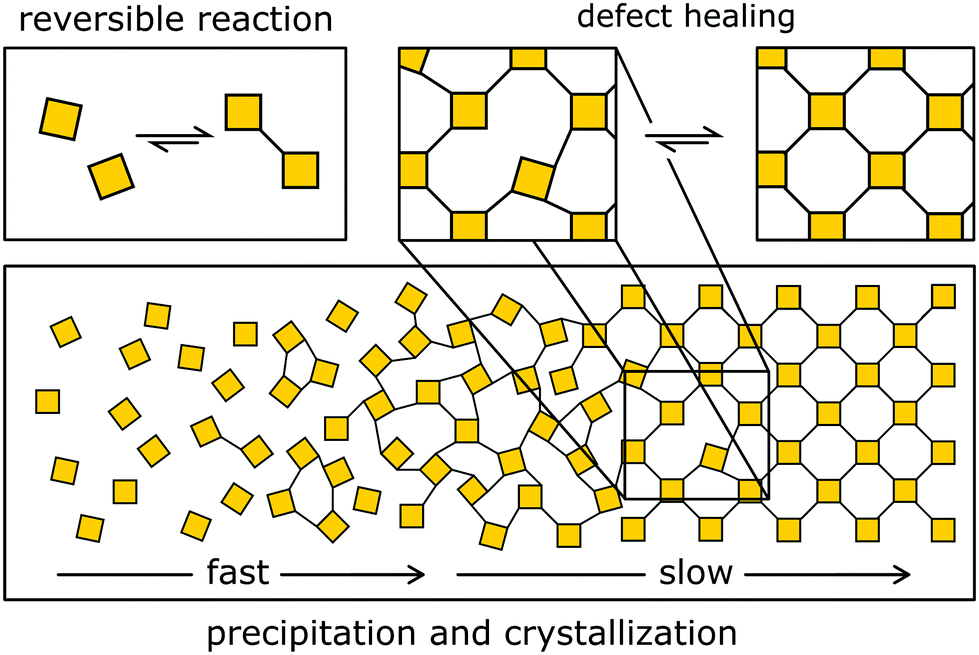 | ||
| Fig. 4 Reversible COF formation by slow crystallization. First, an amorphous gel is formed that then slowly crystallizes. This crystallization mechanism corresponds to the formation of imine COFs. | ||
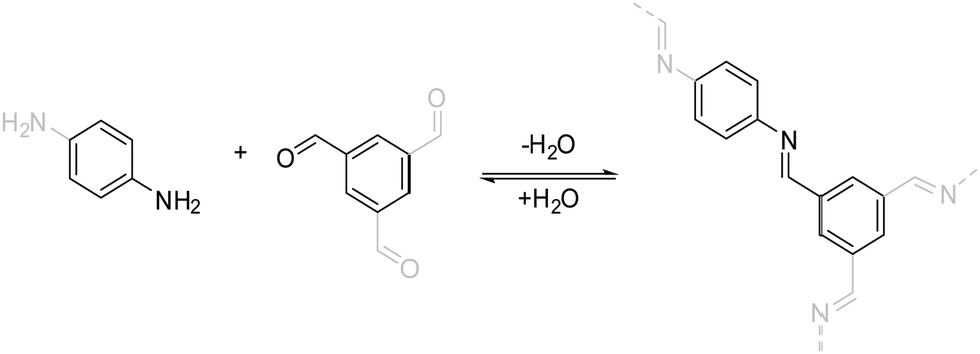 | ||
| Fig. 5 The imine condensation reaction, which is a prototypical reversible reaction that is used for the formation of COFs. | ||
Dichtel and co-workers have investigated the formation mechanisms of COFs that help understand and design the reaction process to improve the crystallinity in COFs. COFs are built from strong covalent bonds, which can lead to a large driving force for bond formation and quick kinetic trapping of the building blocks in a disordered state.18 In boronic ester COFs, sheet-like oligomers and crystallites form first under reversible conditions in solution, and then precipitate by stacking and aggregation, after which further crystallization is kinetically limited.18 In the case of imine COFs, however, the mechanism follows a different route: The quick formation of an amorphous polymer is followed by the slow transformation into a crystalline material.2,17
Based on the paradigm of reversible covalent bond formation governing the synthesis of crystalline covalent organic materials, many new reactions have been developed since the first COFs were demonstrated, which will be discussed in the following.
2.1. Reversible reactions for the formation of COFs
The linkage responsible for connecting the individual building blocks to form the COF is the so called “COF linkage” (Fig. 5). Many types of reversible covalent bond forming reactions are known from DCC, but in the original field of molecular DCC, the requirements on rigidity are not as stringent. COFs, however, rely on a rigid and ordered backbone, which is why many of the reactions found in DCC cannot directly be applied to the COF synthesis. Rigidity and directionality play an important role in the crystallization of the COF and will be discussed separately in Section 6.A range of reactions (Fig. 6) has been explored and reported for the synthesis of COFs since the seminal report by O. Yaghi and co-workers in 20053 who used boronic ester and boronic acid anhydride formation to design the first COF. The COF field was initially focused on borate COFs, but quickly expanded to include imine linkages since their development in 2009, which currently dominate the COF field. Linkages based on the formation of carbon–carbon double bonds are experiencing a surge of interest since their development in 2016. Beyond these archetypical COF linkages, many more have been developed over the years, while many have only been demonstrated in one or very few COF systems.
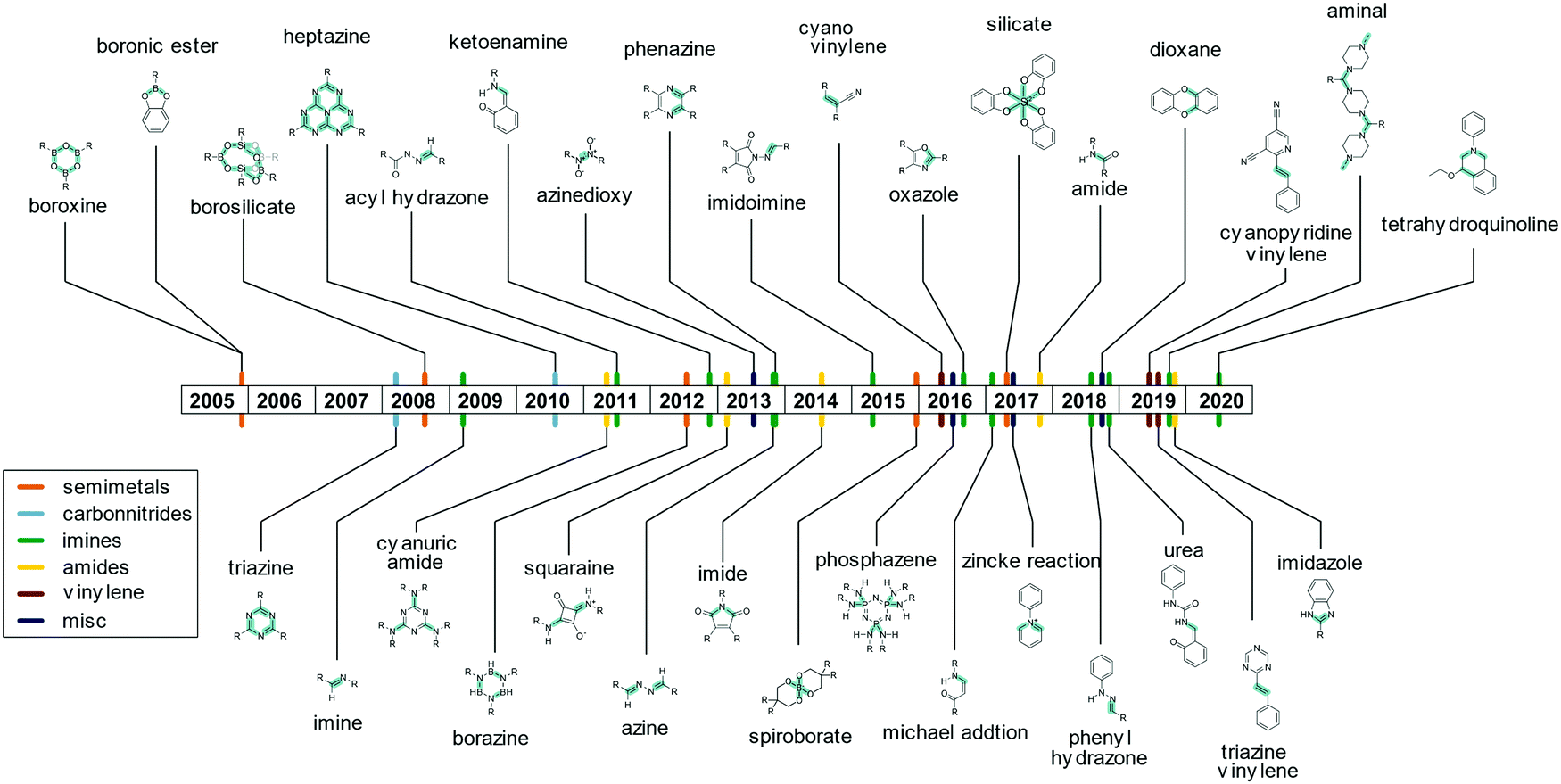 | ||
| Fig. 6 Different COF producing linkages based on reversible covalent bond formation and their time of emergence in the literature. The bond that is formed by the COF linkage formation are indicated by the cyan overlay. | ||
The boronic ester and boronic acid COFs were attractive as a first target for the formation of COFs due to their very high reversibility which led to much more crystalline materials than their imine counterparts that followed them.3 However, it is precisely this improved reversibility that also made them prone to hydrolysis under ambient conditions, thus putting their suitability for certain applications at stake.27 Based on the excellent reversibility of bonds formed with boronates or anionic silicon species such heteroatom containing building blocks have remained of interest to the COF community ever since. The most prominent examples include the formation of borosilicate25 and borazine,24 as well as ionic spiroborate28 and silicate linkages.26 These linkages present a borderline case towards metal organic frameworks, and as such share improved crystallinity and hydrolytic lability with them.29
The currently most common COF linkage is based on the formation of a nitrogen–carbon double bond from a variety of carbonyl compounds and amines. The archetype of this reaction is the imine formation from an aromatic amine, like aniline, and an aromatic aldehyde. Many variations of this motif have been developed that differ in their crystallinity and stability. Adjacent functional groups and the type of the used nitrogen functionality alter the properties of the imine linkage significantly, such as bulky hydrophobic side chains that stabilize imine COFs by blocking the attack by water.30,31 Deviations from the prototype imine linkage have been explored that often improve the stability of the bonds towards hydrolytic cleavage, but at the expense of the crystallinity.
Several variations of the imine linkage relying on a H2N–N motif have been reported, which lead to an increase in hydrolytic stability. Examples include hydrazine giving rise to an azine linkage,32 aminoamide forming the imidoimine linkage,33 and the hydrazone linkage formed from carboxylic acid hydrazides or from phenyl hydrazines.34,35 Phenazine formation from ortho diones with ortho diamines has been used for the formation of COFs.36,37 This leads to the formation of two imine bonds in one COF linkage and the formation of an aromatic ring that improves the stability of these systems significantly in comparison to imines, while they often display poor crystallinity. Imine linkages have also been stabilized by the introduction of an ortho hydroxy group to the aldehyde that leads to keto–enol tautomerism and intramolecular hydrogen bonding that stabilizes the bond.38–40 The most popular linker with this functional group, triformyl phloroglucinol, was developed by the Banerjee group.38–40 This tridentate linker was used for a wide range of COFs due to the good stability of the formed COFs.40 The enhanced robustness is associated with intramolecular hydrogen bonding in the favoured keto form, which is obtained by a facile switching from the enol to the keto form on reaction with the amine. Instead of an aromatic ortho hydroxy aldehyde, a Michael system can also be used to stabilize imine linkages through tautomerization and intramolecular hydrogen bonding.41 A special case of the imine condensation is the formation of an aminal-linked COF by the use of a secondary rather than a primary amine and an aldehyde.42 As the piperazine molecules connect two adjacent aldehyde moieties, this type of COF linkage can also be considered to be the first example of a one dimensional secondary building unit, a feature frequently encountered in metal organic frameworks but not in COFs.43 Instead of a typical amine, an urea functional group can be used in combination with an ortho hydroxy aldehyde, leading to COFs with good stability towards acids.44 Imine linkages can be further stabilized by reacting them in situ to the oxazole linkage, which leads to higher stability in comparison to the amine, but also lower crystallinity, as will be discussed in Section 5.2. Similar to the formation of the oxazole linkage, the tetrahydroquinoline linkage is formed by the in situ cascade Povarov reaction.45 Here, an amine and an aldehyde form an imine, which further reacts with ethyl vinyl ether to form the tetrahydroquinoline COF linkage. The linkage confers significant stability towards even strong acids and bases. Since the linkage is formed in situ and not in a two-step process, the crystallinity of the obtained COFs is limited.
Amide linkages have been used to form COFs even though they typically suffer from low reversibility of the amide bond,46 due to their higher resistance towards acid and bases compared to imines. The reduced reversibility can be compensated by the use of high reaction temperatures of >200 °C.9 Other examples of amide type linkages are imides,47 squaraines48 and cyanuric amide,49 where the latter reaction can also be considered a nucleophilic aromatic substitution of a triazine. A special case of the amide linkages are the imidazole linkages, which can be formed from an ortho diamine and a carboxylic acid synthesized in pyrophosphoric acid, where the highly acidic conditions ensure the reversibility of the reaction.50 This linkage offers highly stable materials, but the harsh reaction conditions limit the substrate scope for this linkage.
Linkages based on aromatic systems composed of alternating carbon and nitrogen atoms can be used for the synthesis of COFs. These “carbon nitride” type linkages are based on the triazine and heptazine building blocks that are characteristic of the family of materials known as carbon nitrides.51,52 The most common example is the triazine linkage, giving rise to covalent triazine frameworks (CTFs), which are typically formed either by cyclotrimerization of nitriles by zinc chloride or from carboxylic acid amides with phosphorous pentoxide at high temperatures of up to 500 °C.51,53 These extremely harsh conditions lead to partial carbonization of the materials and low crystallinity.54 Milder conditions are achieved by using superacid catalysis with trifluoromethanesulfonic acid or by the formation of the triazine unit from multifunctional benzyl alcohols that react with amidines, by slow in situ oxidation.54,55 Such conditions lead to very stable materials, but make for poor functional group tolerance. Further, the crystallinity of CTFs is typically limited, and so far only a few out of the many reported CTF materials are crystalline.10,55–58 Heptazine linkages can also be formed by the reaction of nitriles with diamino triazine terminated building blocks.59 The synthetic conditions are similar to carbon nitride synthesis in that ionothermal conditions are used with high temperatures up to 500 °C.10 A reaction that has gained much recent attention is the formation of vinylic carbon–carbon bond by Knoevenagel reaction between an activated CH2 group and an aldehyde, which have been demonstrated with cyanomethyl groups,60 trimethyl triazine,61 and trimethyl dicyanopyridine.62 The vinylene based COF linkages possess more favourable hydrolytic stability in comparison to the corresponding imine COFs.61 The Zincke reaction is a synthesis method that produces quarternized pyridines, which can be employed for the synthesis of COFs. Here, a pyridine is activated by reaction with a dinitro phenyl group and subsequently reacts with aromatic amines replacing an amino dinitro benzene unit, leading to the formation of charged COF backbones.63 As the bond is only reversible under special reaction conditions, the obtained COFs were shown to be stable towards water, acids and bases. Phosphazene linkages have been employed based on the reaction of a hexachloro phosphazene molecules with aromatic amines to form a 2D COF. This COF was shown to be stable towards dilute acids.64 Highly reversible linkages have been reported, which are key to obtain single crystalline COFs, albeit at the expense of stability. An example is the nitrosyl dimerization that can be used to crystallize large single crystalline COFs from multifunctional nitroso compounds. This reaction can be seen as an extreme case of reversibility in COFs as the covalent bond is very weak (20–30 kcal mol−1) and COF assembly from solutions of the precursors is reminiscent of the growth of regular molecular organic crystals.65
2.2. Increasing the reversibility of the COF formation reaction
Drawing on the insights gained from mechanistic studies and the fact that the crystallization of many COFs crucially depends on reversibility, several reaction design principles exist to improve COF crystallinity by improving reversibility. In this context, studies of imine-linked COFs have been at the forefront for improving the crystallinity of COFs due to their wide adoption in the COF community.
Imide COFs can be synthesized under basic conditions catalysed by isoquinoline, which is in contrast to most imine COFs that require Brønsted or Lewis acid catalysis.47 Recently, a new synthetic route to imide COFs has been reported based on ionothermal synthesis using ZnCl2 or eutectic salt mixtures.5
Banerjee and co-workers68 devised a method that favours the back reaction by transforming the amine into a salt of p-toluenesulfonic acid. The p-toluenesulfonic acid hereby acts simultaneously as a catalyst, as a templating agent, as an inhibitor of the COF forming reaction, and as an agent that favours the back reaction. The protonated amine first needs to be deprotonated to be able to participate in the formation of the imine bond in the COF, thereby slowing down the initial reaction. In addition, the presence of this strong acid regulates the equilibrium of imine formation and hydrolysis by favouring hydrolysis through protonation of the free amine. Through this strategy the crystallinity of twelve different COFs was improved, which confirms the wide applicability of this method. This so-called organic “terra cotta” process allowed for improved surface areas by two to three times in comparison to the conventional solvothermal process.
The use of modulators for the synthesis of COFs as discussed above has laid the grounds for the work of Wang, Sun, Yaghi and co-workers who were able to grow COF single crystals large enough to be analysed by single crystal X-ray diffraction (Fig. 8).4 The reaction equilibrium of the imine forming reaction was tuned by addition of a monodentate aniline to such an extent that the X-ray single crystal structure solution of 3D imine COFs was successful for the very first time – a milestone in the structural chemistry of COFs. This study revealed the atomic structure of two 3D imine frameworks and their degree of interpenetration. The monofunctional amines used in the reaction compete with the linker in the formation of the imine product, thus slowing down the reaction and increasing the back reaction. In order to obtain crystallinity enhancements by the addition of the modulator, several anilines were tested and only those that matched the reactivity of the linkers led to crystallinity enhancements. Otherwise, reduced crystallinity was observed or even amorphous materials were obtained.
3. Pre-orientation of building blocks
The synthesis of amorphous materials based on strong covalent bonds under non-reversible conditions is straightforward, as well as the crystallization of materials by using only weak interactions, due to the high reversibility. The pre-orientation strategy separates the ordering and the (irreversible) covalent bond formation into separate steps that are performed in sequence. By this approach, the best of both worlds can be achieved, obtaining highly crystalline and highly stabile COFs.In the order inducing step, the building blocks are oriented and ordered by a highly reversible reaction (Fig. 9, left). This pre-oriented intermediate may show different degrees of fragility, depending on the type of reaction used to form the ordered state, ranging from non-covalently bonded molecular crystals (Fig. 9A) all the way to weakly covalently bonded solids (Fig. 9B). In the stability inducing step, the building blocks of the previously formed ordered assembly are linked with strong bonds often by an irreversible reaction (Fig. 9, right). By the choice of the reaction and the reaction conditions, the previously obtained order is maintained and locked in place.
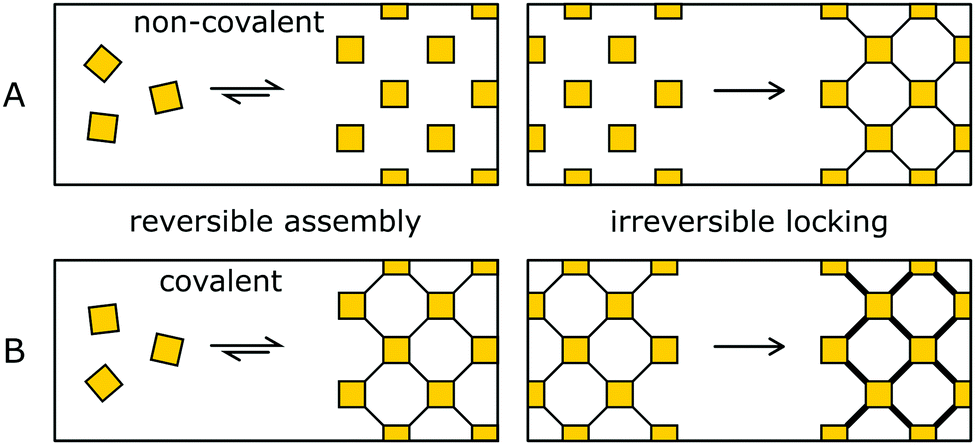 | ||
| Fig. 9 Pre-orientation in COFs: two step formation of stable COFs by the reversible pre-orientation of building blocks, followed by the irreversible locking of the ordered state achieved in the first step. (A) Pre-orientation of the building blocks in a non-covalent fashion and subsequent bond formation. (B) Formation of a reversible covalent bond in the first step, followed by irreversible locking of the labile bond. | ||
Pre-orientation has been used in a variety of material classes, but has recently gained momentum in the COF field due to works showing the feasibility of linkage conversion in COFs. The underlying principle of pre-orientation is closely related to classical templating which has been explored for the synthesis of mesoporous silica73,74 and two-dimensional polymers.75,76 These examples however demonstrate only long range order while the materials are commonly disordered on molecular length scales. By influencing the interactions of reactive building blocks on a molecular level, it is also possible to induce short range order and produce crystalline materials that would be difficult to synthesize by other means. In zeolites this approach has been used to synthesize ‘unfeasible’ zeolites77 by pre-orienting exfoliated crystalline layers by the addition of ionic stacking modulators that influence the stacking sequence. This pre-oriented assembly is then locked by irreversibly reacting the sheets with each other through reactive silanes. This approach allows for the synthesis of crystalline zeolites that would be unfeasible to synthesize by direct solvothermal approaches.77,78 Similarly, in the synthesis of linear polyamides, significant improvements in crystallinity have been obtained by first pre-orienting the precursors as molecular salts based on ionic ammonium and carboxylate groups. These pre-oriented materials are subsequently heated to generate the polyamide, thus transferring the pre-oriented crystalline state into the stable polyamide.79 These examples demonstrate how pre-orientation can be used from the meso down to the molecular scale to obtain crystalline solids.
In self-assembled cages this approach has been used to lock the linkages within the cage that where formed under reversible conditions. It has been demonstrated that the linking groups can be transformed into different functional groups with varying stability and rigidity. A first example is the reduction of imine linkages to amines which improves the stability of the cage, but reduces their rigidity.80 Even multistep reactions can be performed on these cage linkages, for example with an ortho hydroxy group participating in a multistep procedure of irreversible reactions to form a stabilized carbamate linkage (Fig. 10A).80 The reduced amine can also be transformed into a pure hydrocarbon linkage by the extrusion of nitrogen with the help of isoamyl nitrite (Fig. 10B).81
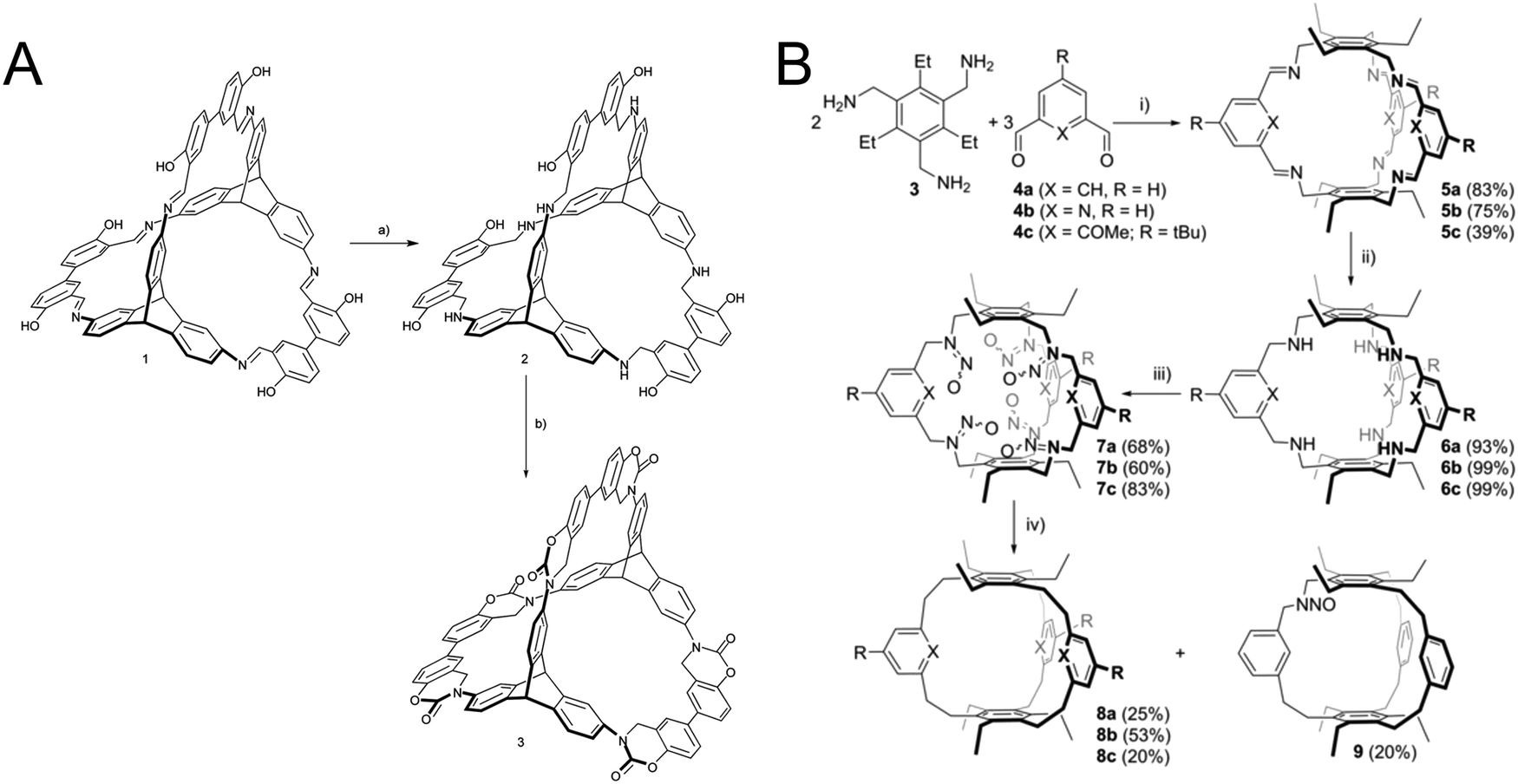 | ||
| Fig. 10 Locking chemistry that has been explored in organic cages before the advent of locking in COFs: (A) locking imine linkages as a carbamate. (B) Transforming an imine linkage into an ethane linkage. (A) Reprinted with permission from ref. 80 Copyright (2017) Royal Society of Chemistry. (B) Reprinted with permission from ref. 81. Copyright (2019) The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA. | ||
This widespread pre-orientation strategy is dependent on four prerequisites (Fig. 11): The reversible assembly does not disassemble under the conditions of the stability inducing step; the building blocks are oriented correctly to allow the reaction to occur; the order in the material needs to withstand forces generated during or because of the transformation; the reagents and catalysts need to reach the COF linkage and need to be able to perform the reaction irrespective of the sterically challenging environment.
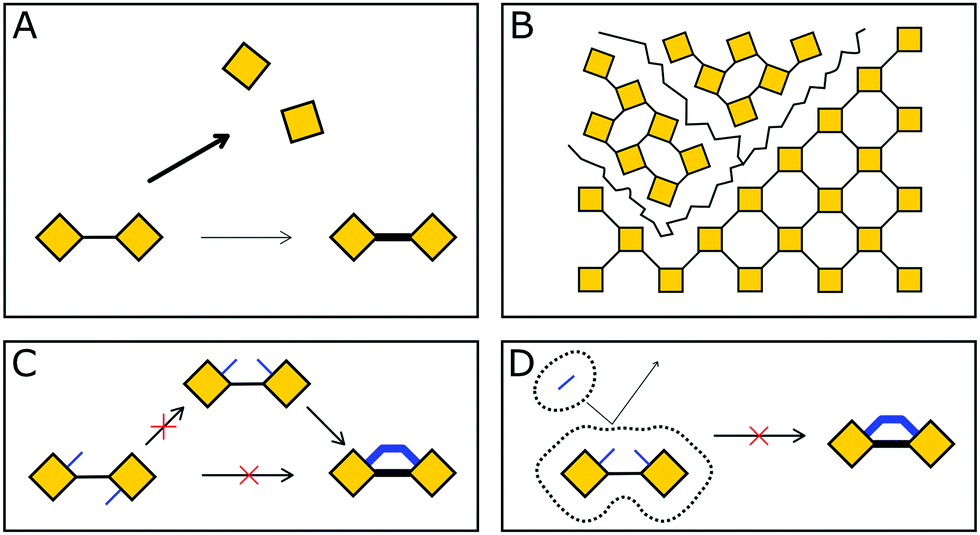 | ||
| Fig. 11 Four challenges in locking a COF linkage: (A) bond breakage instead of locking. (B) Transformation induced strain and fracturing. (C) Wrong orientation of building blocks for the desired reaction. (D) Steric hindrance preventing access of the reactants. | ||
In order to stabilize a system ordered by comparably weaker interactions, disassembly during the stability inducing reaction needs to be circumvented. In ordered crystals of molecules, which are only connected by weak interactions, this is often achieved by using a photochemical reaction in the solid state.82–84 This way the order of the crystal is not affected by solvents or reagents. When transforming a COF based on reversible linkages into a material connected by irreversible linkages, it is often beneficial that most COF linkages are formed by condensation reactions, and thus by excluding water during the locking reaction it is possible to avoid disassembly.
In examples based on weak intermolecular forces, obtaining correct orientation of the building blocks in the solid state to enable the stability inducing step is not trivial and often not amenable to design. Here, sometimes extensive screening of crystallization conditions, such as solvents are required.82 In COFs the strong covalent bonds between the building blocks lead to very well-defined orientations of the building blocks that allow the tailored design of reactive sites and reactions in the solid state, when locking the COF linkages with an irreversible reaction. Furthermore, it was shown that even in the solid state the orientation of linkers or the imine group are to some extent dynamic and hence can change, making the reaction possible even with initially misaligned building blocks.16 Nevertheless, if multiple orientations of a building block are possible, it may limit the conversion of the COF linkage.
The transformation of the pre-oriented assembly can lead to strain on the material due to changes in the bond length and angles by the transformation itself.82 For example, the formation of an oxazole or a thiazole from an imine linkage by oxidative cyclization leads to a significant change of the angle in the backbone of the COF (Fig. 16), which in turn results in a contraction of the unit cell.2 In molecular crystals, which are mechanically fragile, even relatively small changes in the unit cell dimensions can lead to the fracturing of the crystal and thereby the loss of order.82 This is especially true if the stability inducing step follows a nucleation and growth mechanism, as even a small change in bond angles or unit cell size would build up considerable strain that could lead to the fragmentation of the crystallites. However, if the transformation occurs randomly within the crystallite or if the crystallite can withstand the strain build-up during the linkage transformation, the order remains. A further challenge can be the loss of rigidity during the linkage transformation, as seen with the reduction of the COF linkage from an imine to an amine. This can lead to a loss of crystallinity as observed in a 2D COF while the topology of the connection was maintained.16 This was proven by the re-rigidification of the COF linkage leading to the recovery of the crystallinity. In contrast, the crystallinity was maintained after the reduction to the amine in other 2D and 3D COFs, which clearly shows that the effect of losing crystallinity depends on the framework.85,86
3.1. Pre-orientation in COFs based on weak interactions
Pre-orientation has been used in the synthesis of crystalline two-dimensional polymers by creating molecular crystals of precursors that can in a second step be crosslinked in a topotactic single-crystal-to-single-crystal transformation (Fig. 12). This approach enables the synthesis of covalently crosslinked crystalline polymers by means of an irreversible reaction. This approach has been demonstrated with the photochemical dimerization of anthracenes or by the cycloaddition between anthracenes and alkynes.82–84,87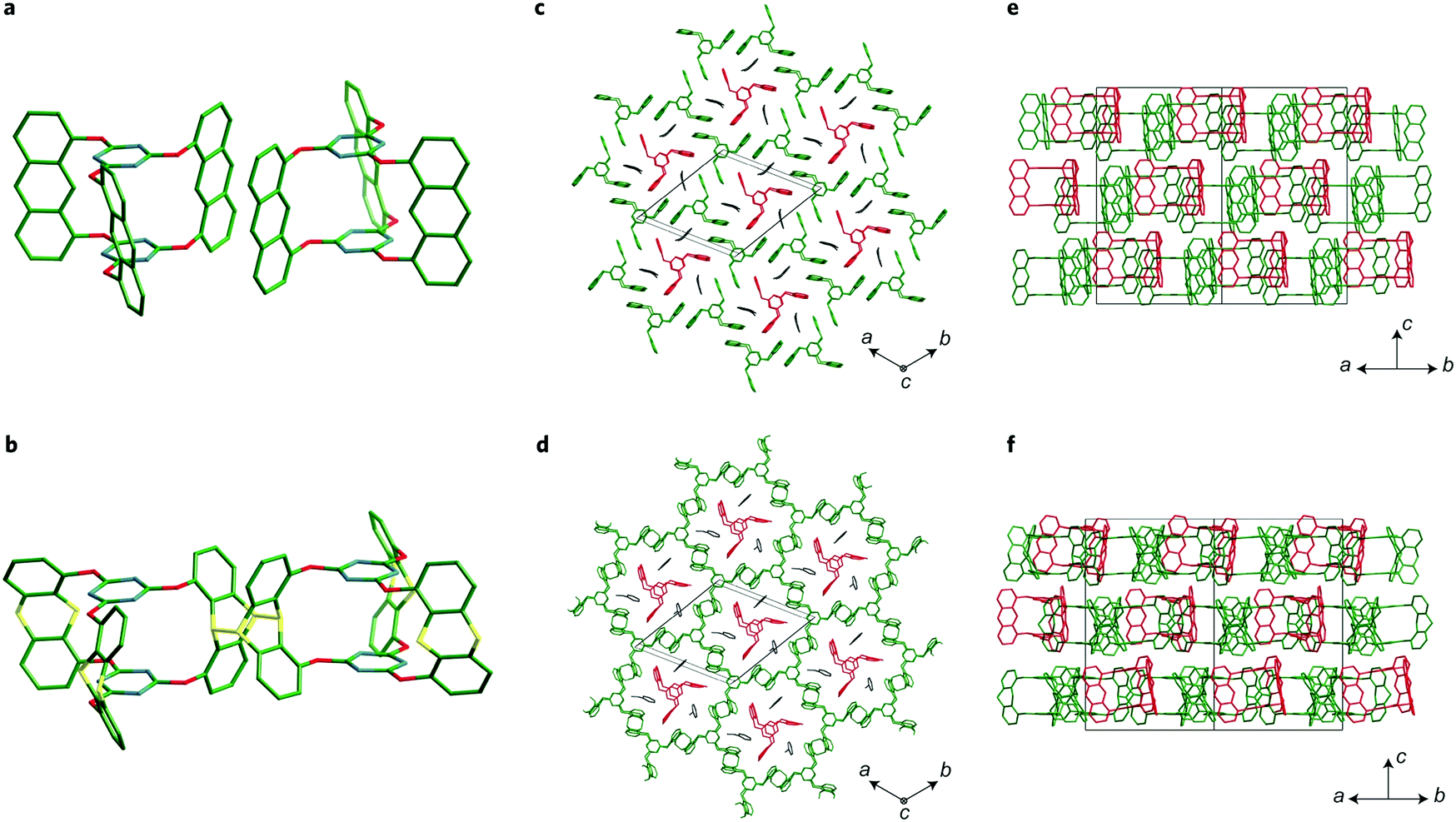 | ||
| Fig. 12 Transforming a molecular crystal of a propeller-shaped trianthracene into a two-dimensional covalently connected polymer.82 The figure is reprinted from ref. 82. Copyright (2014) Springer Nature. | ||
This elegant approach allows for the synthesis of highly crystalline, extended covalently linked polymers with millimetre sized single crystals.82 These two-dimensional polymers additionally possess nanopores.87 However, the feasibility of this method is highly dependent on the molecular orientation of the molecular units and reactive functional groups in the solid state, which currently cannot be predicted in a facile manner, thus making these materials difficult to design from scratch.
It is precisely this predictability inherent to COFs that has been one of the driving forces in making them so popular among organic and solid-state chemists alike. To benefit from these strategies in the synthesis of COFs, a number of approaches have been developed to implement strategies of pre-orientation into the synthesis of COF.
A directional and specific interaction between molecules has been designed for the two-step formation of a 3D COF, where linkers with multiple vinyl-pyridinium groups are assembled into a non-covalent 3D lattice by the help of the host–guest chemistry of cucurbituril (Fig. 13).88 This assembly is then covalently linked by photodimerization of two vinyl groups, leading to a framework that is covalently connected in three dimensions. This approach shows that weak interactions can be designed to place reactive functional groups in close spatial proximity, allowing their reaction with each other.
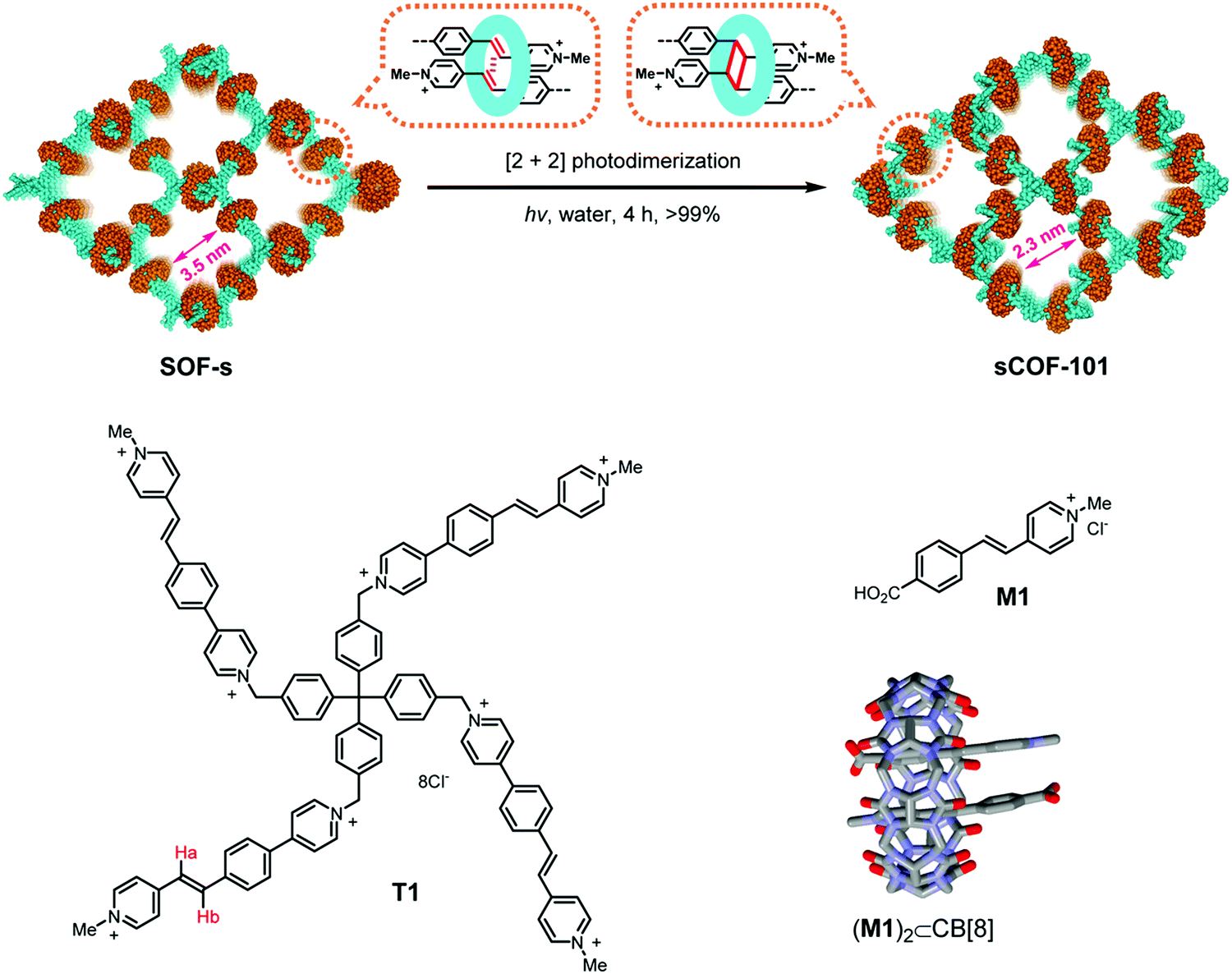 | ||
| Fig. 13 Pre-orientation of a cationic tetraphenyl methane derivative with vinyl functional groups that forms an inclusion compound with cucurbituril with each of its four appendages. This 3D supramolecular network is then transformed into a covalent framework by [2+2] photodimerization of the vinyl functional groups that have been pre-oriented within the cucurbituril.88 Adapted with permission from ref. 88. Copyright (2020) American Chemical Society. | ||
The assembly of precursor molecules in the solid state can be used as a template for the oriented growth of 2D imide COFs (Fig. 14).89,90 The assembly of porphyrin building blocks at the air/water interface with the help of surfactants can successfully pre-orient the building blocks for the formation of a crystalline COF. The orientation of the COF follows the orientation of the precursors and is dependent on the used surfactants. The use of sulfonate surfactants promoted the assembly parallel to the interface, while carboxylic acid surfactants led to growth of the precursors and COFs perpendicular to the surface. This approach led to micron sized COF crystallites that are only 2 nm thick. The pre-orientation of the precursors at the interface allowed the COF forming reaction to proceed under exceptionally mild conditions, especially for imide COFs, with the reaction occurring at room temperature over a course of 7 days.
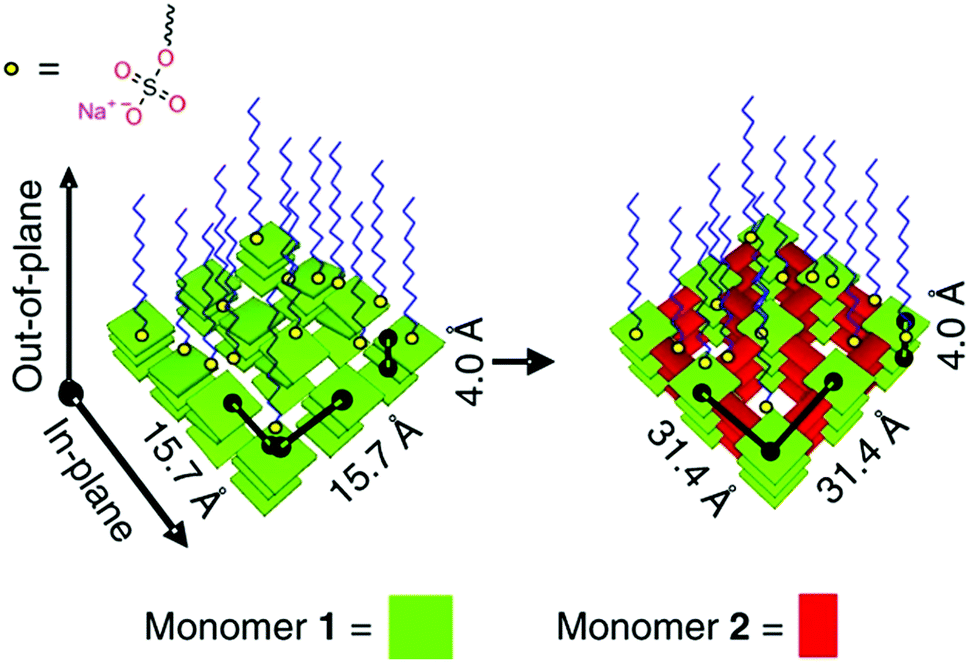 | ||
| Fig. 14 Pre-orientation of molecules in the interfacial synthesis of an imide based COF.89 The figure is reprinted from ref. 89. Copyright (2019) Springer Nature. | ||
The crystallization of amine precursors as p-toluenesulfonic acid salts serves as another means to pre-orient the amine molecules for the reaction to an imine based COF.68 As the distance between individual amines in the salt crystal structure matches well with the distance of the aldehyde precursor, the lattice of the tosylate salt is transformed into the COF lattice by replacing the tosylate molecules by aldehyde building blocks (Fig. 15). According to the proposed mechanism, the amine building block gets deprotonated and the weak hydrogen bond is replaced with a stronger covalent bond resulting in an imine-linked framework. The crystallinity only becomes evident after washing the COF, indicating residual acid and precursors in the pores, which further underscores the mechanism.
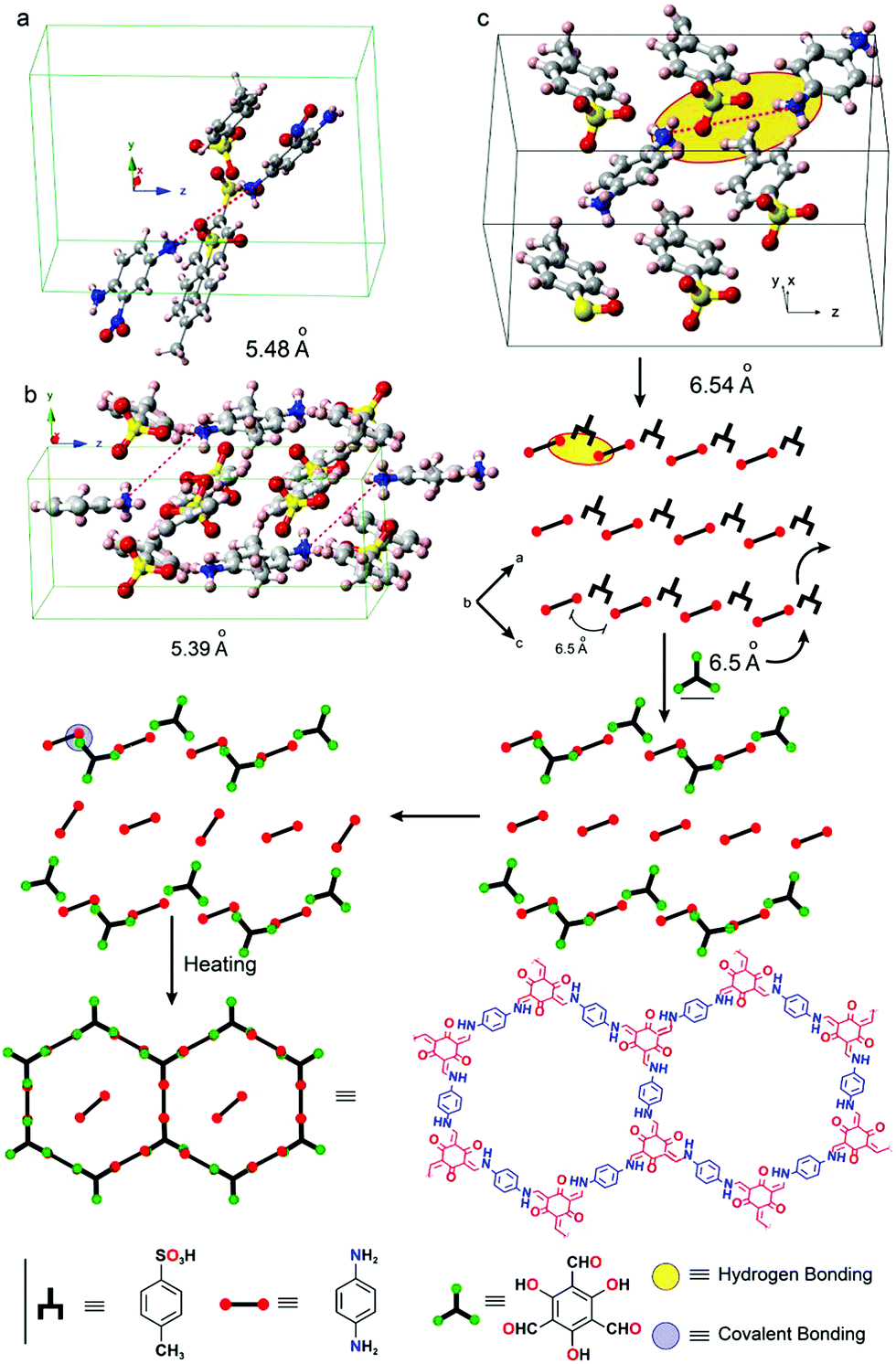 | ||
| Fig. 15 Pre-orientation of building blocks by formation of a p-toluenesulfonic acid salt of the amine precursors used in the COF formation reaction. The transformation of the tosyl salt into the COFs proceeds in a stepwise fashion. First the amine linker is reacted to a salt crystal with p-toluenesulfonic acid. In a second step this crystal is transformed into the COF by reaction with an aldehyde linker. Reprinted with permission from ref. 68. Copyright (2017) American Chemical Society. | ||
“Woven” COFs are composed of linear covalently connected chains that are oriented relative to each other by metal coordination bonds.91,92 The synthesis of these COFs utilizes the coordination bonds to pre-orient sections of the linear chain, relative to each other, drawing on the chemistry developed for catenates.93 These pre-oriented chain sections are then linked by the COF linkages into an ordered covalently connected lattice. The connectivity within the lattice ensures mechanical interlock between the woven strands. The metal required for the pre-orientation can be removed after the polymerization and the topology, but the crystalline order of the strands is not preserved. It was shown that the chain sections need to be pre-oriented in a separate step to arrive at an ordered lattice, since if the orientation of the building blocks is performed during the crystallization, no ordered structure is obtained.92
Pre-orientation can also be used after the formation of a two-dimensional COF to form a 3D COF by photodimerization of anthracene building blocks placed in adjacent layers.94 The face-to-face arrangement that is achieved through the stacking of the individual COF layers enables the dimerization without significant rearrangement. This pre-orientation of functional groups allows the transformation of a van der Waals-stacked structure into one that is connected covalently in 3D. In a similar manner, vinyl groups in a 2D COF can react in a 2+2 photoaddition to connect the layers of the COFs with each other.61
3.2. Pre-orientation based on covalent bonds for the locking of COF linkages
The pre-orientation in COFs shown above were primarily used to improve the crystallinity and not focused on improving the stability. In contrast, chemical transformation of a reversible into an inert COF linkage is a powerful tool for drastically enhancing the stability of COFs. The COF linkages are ideal targets for a pre-orientation strategy, since the assembly of COFs by reversible reactions is now well established, while the chemistry of the COF linkages is well known from their molecular counterparts.The strategies for locking of COF linkages can be classified by either the type of locking procedure or by the generality of the approach. The type of transformation can be differentiated into two categories: The first is altering the chemical nature of the COF linkage itself, without the introduction of additional bonds between the linkers, such as with the reduction of an imine linkage to an amine linkage (Fig. 16, red).16,85 The second strategy creates additional bonds between the linkers that are parallel to the existing COF linkage, as with the cyclization of an imine to a thiazole, where the sulphur atom formally creates an additional connection between the amine and the aldehyde building blocks (Fig. 16, blue).2 The generality of the locking reaction depends on whether a reactive functional group needs to be predesigned into the building block, or whether a typical unfunctionalized COF can be used in a locking procedure (Fig. 16, grey). The general methods are those that alter the nature of the COF linkage, such as the coordination of a pyridine Lewis base to a boronic ester,95 the reduction of an imine to an amine,16,85 or the oxidation of an imine to an amide.46 Among the general strategies are also bridging reactions that do not require special functional groups such as the formation of a thiazole with sulfur2 or of a pyridine with an acetylene96 from the corresponding imine. Building block-specific reactions are the formation of an oxazole97,98 or a thiazole97 from the ortho hydroxy and ortho thiol amines, respectively. Both functional groups can also be introduced into the COF via post-synthetic linker exchange, however the challenging synthesis of these functional groups makes these locking procedures less accessible. The post-synthetic locking can also be performed as a multistep reaction if the above-mentioned criteria are maintained for each step.16 An ortho methoxy imine COF was transformed into a carbamate or thiocarbamate by first removing the methyl group, exposing a free hydroxyl group, and subsequently cyclizing the COF linkage to the final (thio)carbamate linkage. Another reaction based on a building block predesigned for locking is the oxidative cyclization of an imine COF with pendant thiophene groups that allows the locking of the imine group into an extended heterocyclic aromatic system.99
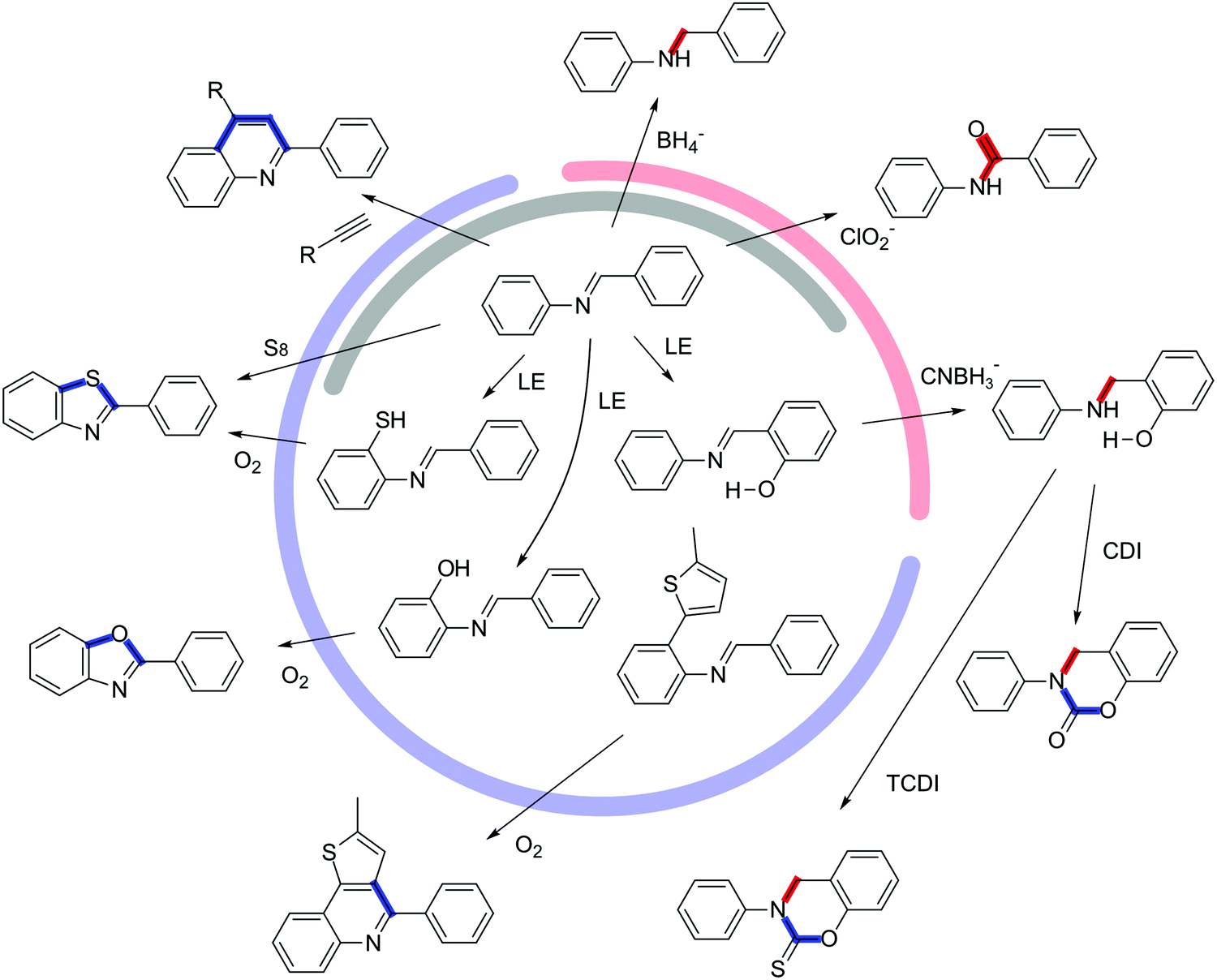 | ||
| Fig. 16 Overview of the reactions used to lock the imine linkage. The reactions can be separated into different categories: Reactions changing the nature of the linkage (red) and reactions bridging the COF linkage (blue), and reactions that can be performed on any unfunctionalized aromatic imine (grey). 1,1′-Carbonyldiimidazole (CDI), 1,1′-thiocarbonyldiimidazole (TCDI); linker exchange (LE). | ||
The transformation of the COF linkage, especially in the case of the imine transformations, was generally accompanied by the retention of crystallinity and porosity. While in some cases the porosity was reduced by the transformation,46 in other cases it increased, possibly due to the transformation reaction cleaning the pores.2 In all reported cases, the stability of the COFs was drastically improved with regards to their parent compounds, but also with regard to other reported COFs. The locked COFs were shown to be stable against strong aqueous acids, bases, nucleophiles, electrophiles, reducing and oxidizing conditions. These results nicely demonstrate that locking the COF linkage can solve the stability issues in COFs.
The stabilization inducing step can also be made by a linker exchange reaction, within the preformed COF network such as by the formation of an imine COF where the aldehyde linker is subsequently linker exchanged with an orthohydroxy aldehyde building block, leading to stabilization of the imine linkage by intramolecular hydrogen bonding and the keto-enamine tautomerization.100 While it is possible to directly synthesize crystalline imine COFs based on orthohydroxy aldehyde building blocks, the separation into two steps is able to produce materials with a much higher crystallinity.
4. Single reaction pathway
Besides reversible reactions and pre-orientation, a third strategy can be distinguished based on the reaction of highly rigid molecules with only few conformational degrees of freedom.101,102 The assembly of these building blocks allows for only very few conformational degrees of freedom when forming a framework, leading to an ordered lattice even in the absence of reversibility (Fig. 17). In other words, the ordered arrangement of the building blocks is the most probable or the only possible outcome of the reaction.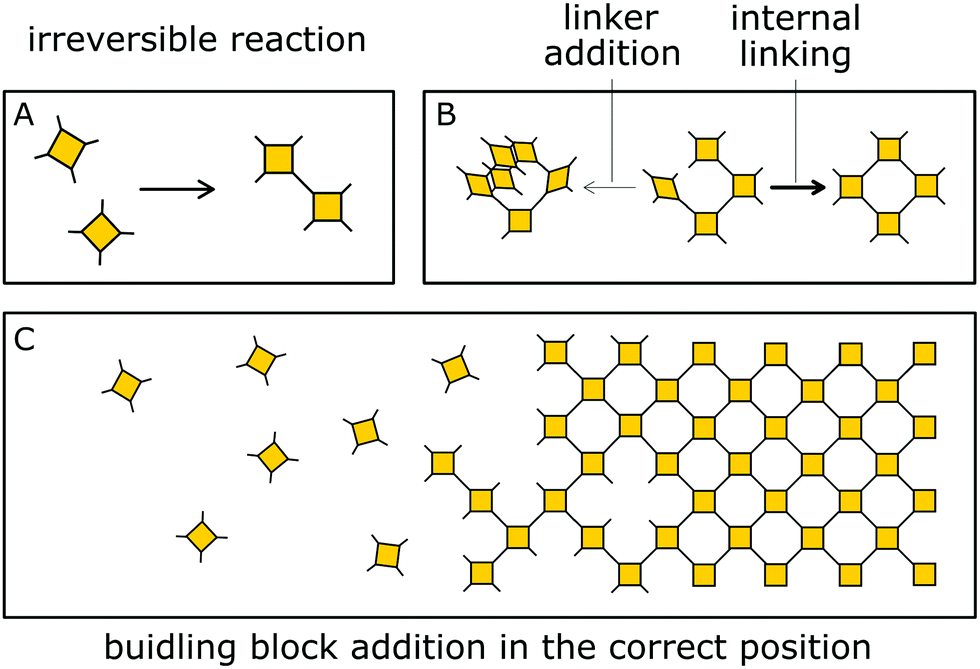 | ||
| Fig. 17 Formation of COFs by the single reaction pathway is characterized by irreversible bond formation between linker molecules and by building block addition in the correct position, favouring the internal linking over linker addition. | ||
Normally, the reversibility of the reaction allows for defect healing and ordering under thermodynamic control, during the formation of a crystalline material. But such error correction is not always available with irreversible reactions or even in some COFs based on reversible reactions. The predesigned COF linkage ensures that attachment of a new building block occurs at the correct position, but as soon as two building blocks are attached at adjacent positions on the crystallite two possibilities arise from the available unsaturated functional groups (Fig. 17B): adjacent building blocks can react with each other (internal linking) or a new building block can be added to one of the free functional groups. If the addition occurs at the same site that could be used for internal linking, it creates a defect site. Multiple additions with less internal linking could be likened to a dendrimer type growth of the structure that leads to a reduction in order, because the building block addition can trap the incompatible conformer.103 The possibility of dendrimer type growth depends on the flexibility and possible conformers of the surface attached building blocks, which are not compatible with the desired structure. The flexibility of the building block therefore plays a major role in determining the crystallinity of a COF based on irreversible and to a lesser extent in COFs based on reversible reactions. Strategies for reducing the number of conformers and degrees of freedom in building blocks and in COFs will be discussed in detail in Section 6.3. Another consequence of the competition of building block addition with internal linking is the dependence of the formation of crystalline materials by irreversible reactions on the concentration of the building blocks during the synthesis. The rate of internal linking is concentration independent, but the rate of addition increases with the building block concentration. The slow supply of building block can reduce trapping of misaligned building blocks, and additionally favours growth at the existing particles instead of reacting in solution leading to larger crystallites (Fig. 18).104 As COFs based on irreversible reactions were often obtained by controlling the rate at which the precursor molecules reach the growing crystallite, this suggests that the slow addition is an essential strategy in obtaining crystalline materials from irreversible reactions (see Section 6.1). Most of the examples of materials synthesized by irreversible reactions using a single reaction pathway are 2D structures, which add degrees of freedom for the stacking arrangement, and therefore, disorder is possible (see Section 6.3.4).
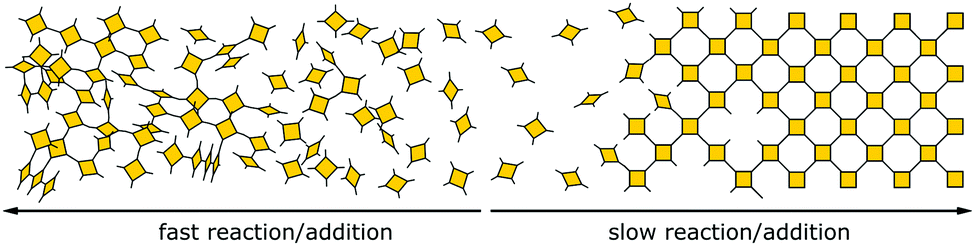 | ||
| Fig. 18 Schematic showing how the speed of addition influences the order in a two- or three-dimensional polymer. | ||
4.1. Direct synthesis of COFs based on irreversible reactions
By following the above outlined strategies for designing COFs and their synthesis several COFs based on irreversible reactions have been realized.105–108 This shows that a reversible reaction is not a necessary prerequisite for obtaining crystalline COFs. However, obtaining crystalline COFs from irreversible reactions is not straightforward, as is attested by the vast literature on amorphous polymers based on the same rigid building blocks that are used to make COFs. The benefit of using irreversible reactions is the improved stability of these COF materials that result from the lack of reversibility.Using highly rigid building blocks to obtaining crystalline COFs by an irreversible reaction was nicely demonstrated by Loh and co-workers,106,107 who were able to couple several flat and rigid aromatic bromo precursors in an uncatalyzed Ullman coupling at high temperatures to form COFs (Fig. 19).
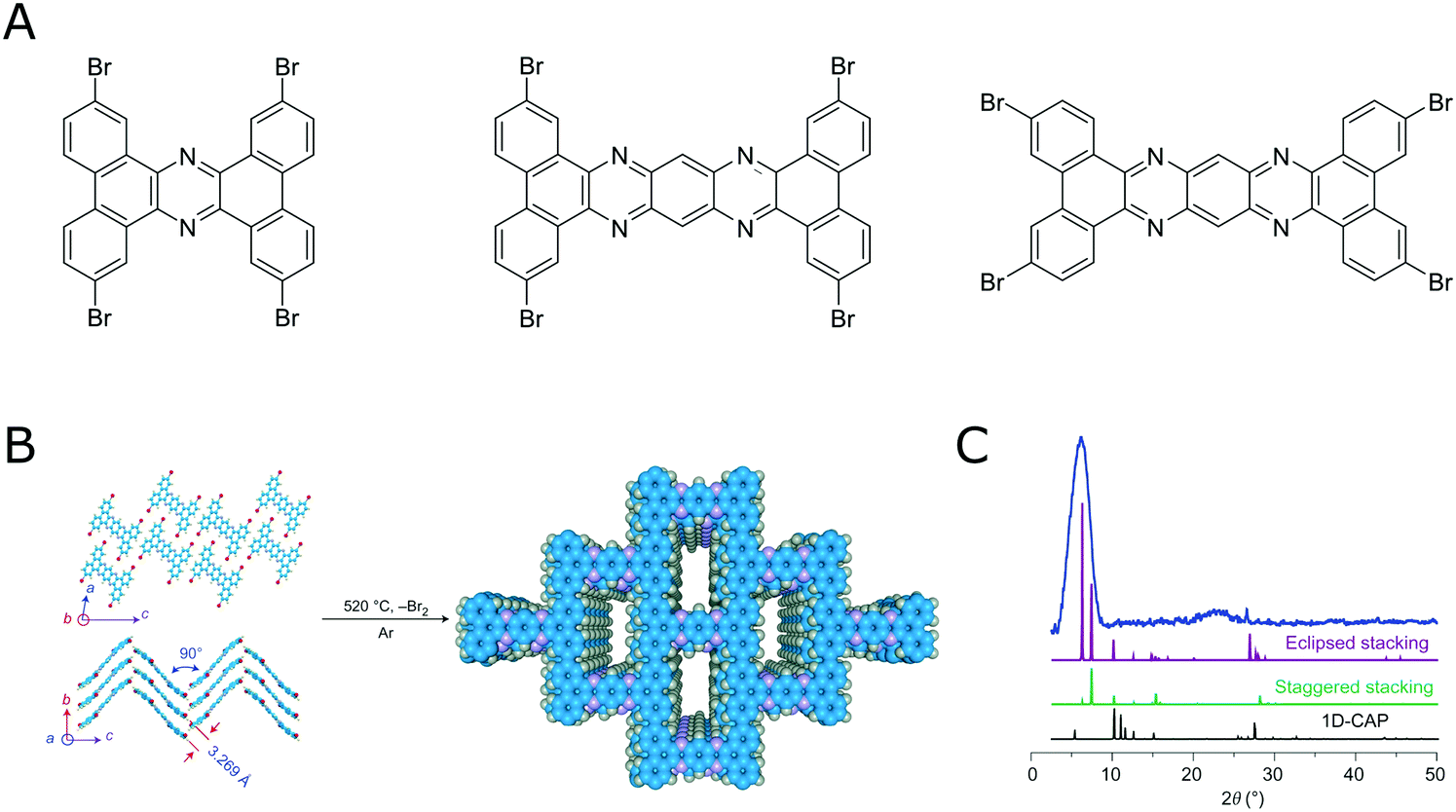 | ||
| Fig. 19 Direct coupling bromo aromatic compounds at high temperatures for the synthesis of COFs. (A) Molecules that have been demonstrated for this transformation.106,107 (B) Transformation of a tetrabromo compound into a crystalline COF by an irreversible reaction.106 (C) PXRD pattern of the material shown in B. Reprinted with permission from ref. 106. Copyright (2017) Springer Nature. | ||
In this example the rigidity of the building blocks and linkages compensates for a lack of error correction and healing that would be available by reversible reaction. By designing highly rigid building blocks, the degrees of freedom during the formation of the framework are severely restricted. A growing number of examples use this design principle to form crystalline COFs based on irreversible reactions directly in solution,102,106,107,109,110 however, many examples use special experimental setups to enable the formation of crystalline COFs.
A COF with very low degrees of freedom that is based on irreversible reactions can be grown at an interface. Carbon–carbon cross-coupling reactions, which are known to be kinetically controlled and essentially irreversible, such as the Suzuki and Glaser couplings, were shown to lead to crystalline sheets by a synthesis at the liquid–liquid interface.111 Similarly, a COF carbon allotrope graphdiyne was synthesized from benzene hexa-alkyne at the liquid-gas and the liquid–liquid interface, with micron sized crystallites in the intralayer direction while being only a few layers thick.112 The method used an interface to construct the nanosheets but also the slow diffusion of the catalyst to the reaction site to slow down the reaction and to allow the formation of a crystalline material.
Electrochemical deposition of a porphyrin framework was obtained by electrochemical coupling of aminophenyl porphyrin on an electrode. The aminophenyl groups couple by electrochemical oxidation to form a phenazine group resulting in crystalline COF films. The oxidative coupling led to the growth of dendritic COF structures, which nevertheless revealed facets.113 This method avoids the reaction of the precursors in solution, which would normally compete with the growth of the already formed crystallites, thus leading to overall larger crystallites by a preferred reaction of the molecules at the surface of the COF attached to the electrode.
The irreversible Glaser coupling can be used to obtain ordered materials if the feed rate and hence the reaction rate is slowed down.103 This was shown by using two different reaction protocols that differ significantly in their reaction rates: a Glaser coupling based on copper(I) chloride and a Sonogashira-like coupling catalysed by palladium. While the faster reaction catalysed by palladium yielded only an amorphous polymer, it was possible to obtain a somewhat ordered material with the slower Glaser coupling.
4.2. Slow supply of building block in COFs based on reversible COF linkages
In COFs based on reversible reactions, the importance of slowly providing the reactants was already observed in the first COF paper by Yaghi and co-workers.3 Diffusion of the precursors was controlled by adjusting the solubility of the linkers by choice of the solvents. The choice of solvents in the synthesis of COFs is of paramount importance when screening for conditions that produce crystalline COFs, showing that the control of the feed rate might be important for the formation of all COFs in addition to its role in controlling the stacking equilibrium.114 These results indicate that the reaction speed can be modulated by the choice of solvent also as a means of controlling the driving force of π-stacked COFs.Investigation of the mechanism of formation of boronate COFs showed that once precipitation of oligomers and COF sheets occurs, only little improvement of crystallinity can be observed, indicating that further crystallization is hindered.18 This mechanism indicates that a slowed down initial reaction rate helps avoiding kinetic trapping of a COF of low crystallinity. Based on these results, approaches have been developed for slowing down the initial reaction speed. Dichtel and co-workers27 demonstrated that the protection of amine precursors such as benzophenone imines is able to slow down the initial reaction speed as the amine first needs to be deprotected before it can participate in the formation of the COF, thereby leading to increased crystallinity.
Slowly supplying solutions of boronic acid and diol precursor monomers by a syringe pump has been shown by Dichtel and co-workers for the formation of boronic acid COFs and imine COFs.115,116 By this approach the nucleation and growth of the particles could be separated leading to only homoepitaxial growth of the COF crystallites.104 This method was then successfully applied to grow COFs in suspension featuring larger single crystalline dimensions than what is normally possible. These results have similarly been shown with the growth of imine COF thin films, where only by slow addition of the precursors crystallinity could be achieved.117
CTFs based on the reaction between aldehydes and amidines were synthesized in a crystalline form by supplying a benzyl alcohol instead of an aldehyde. The slow oxidation by air to the aldehyde during the reaction served as a means to supply the precursor at a controlled rate, enabling the formation of a crystalline framework.54 Alternately, crystalline CTFs were obtained from amidine and aldehyde building blocks by slow addition to the reaction mixture by controlling the speed of a peristaltic pump.118 The crystallinity increased with slower feed rates and in the TEM large ordered crystallites are visible in addition to the PXRD, while the direct solution synthesis produced a material with a much lower crystallinity (Fig. 20).
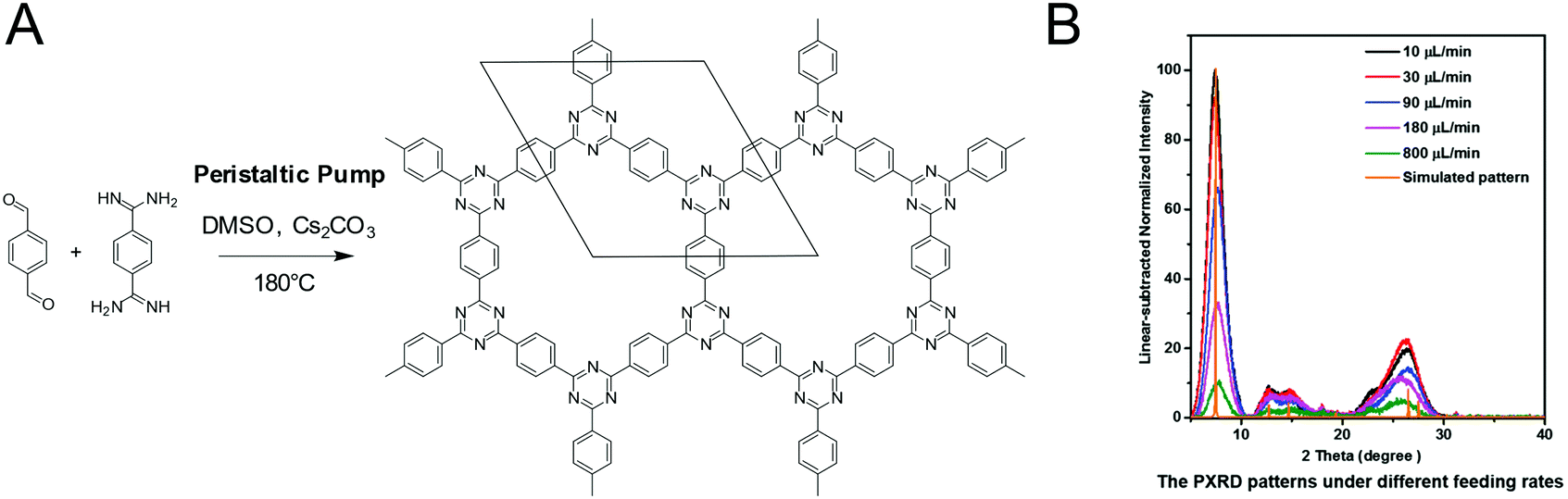 | ||
| Fig. 20 Influencing the crystallinity of a CTF by controlling the monomer feed rate. (A) Reaction scheme of the CTF. (B) PXRD patterns at different feed rates118 Reprinted with permission from ref. 118. Copyright (2019) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
4.3. Designing COFs with a low number of structural degrees of freedom
It is known from molecules that compounds with many competing conformational polymorphs within a small energy window do not crystallize well and often form disordered solids.119 At the same time molecules with low conformational freedom often show reduced solubility, forcing a molecule to crystallize.82 Transferring these design strategies to COFs can be used to improve their crystallinity. The structural degrees of freedom and the effect they have on the crystallization of COFs can be easily understood by two example COF systems (Fig. 21A and C). The first COF is based on the pyrazine formation reaction with straight and highly rigid linkers37 while the second is based on the boronic ester formation with an angled and more flexible linker.120 Both structures are completely flat in their equilibrium shape, but possess different propensity for out-of-plane bending due to the rigidity of their building blocks and the COF linkage (Fig. 21B and D). During the formation of the COF, the linkers are reacting with each other to form oligomers (Fig. 21A and C). The oligomers of the boronic ester COF have more unique conformers for a given oligomer size in comparison to the phenazine COF. All oligomers of the phenazine COF are compatible with the “ideal” periodic structure, meaning that no conformational rearrangement or reversible reattachment is necessary to ensure the correct positioning. The boronic ester oligomers on the other hand have multiple conformers located in close proximity on the potential energy surface. Only some of these conformers are compatible with the final COF structure, requiring defect healing to arrive at an ordered structure. Additionally, the out-of-plane rotation around the COF linkage is possible in the case of the boronic ester with a relatively small energy barrier.114 In the phenazine system such an out-of-plane bending is accompanied by a larger energy penalty and the full rotation is only possible with bond breakage as the system is annulated. The rotation around the COF linkage or the bending of the linkers themselves can result in out-of-plane defects such as spirals in the stacking direction or 5- or 7-membered rings instead of 6 membered rings.114 Although a comparison of these two COFs with regard to their crystallinity is difficult due to their different chemistries, systems based on the same chemistry clearly show improvement in crystallinity upon reduction of conformational degrees of freedom (see below).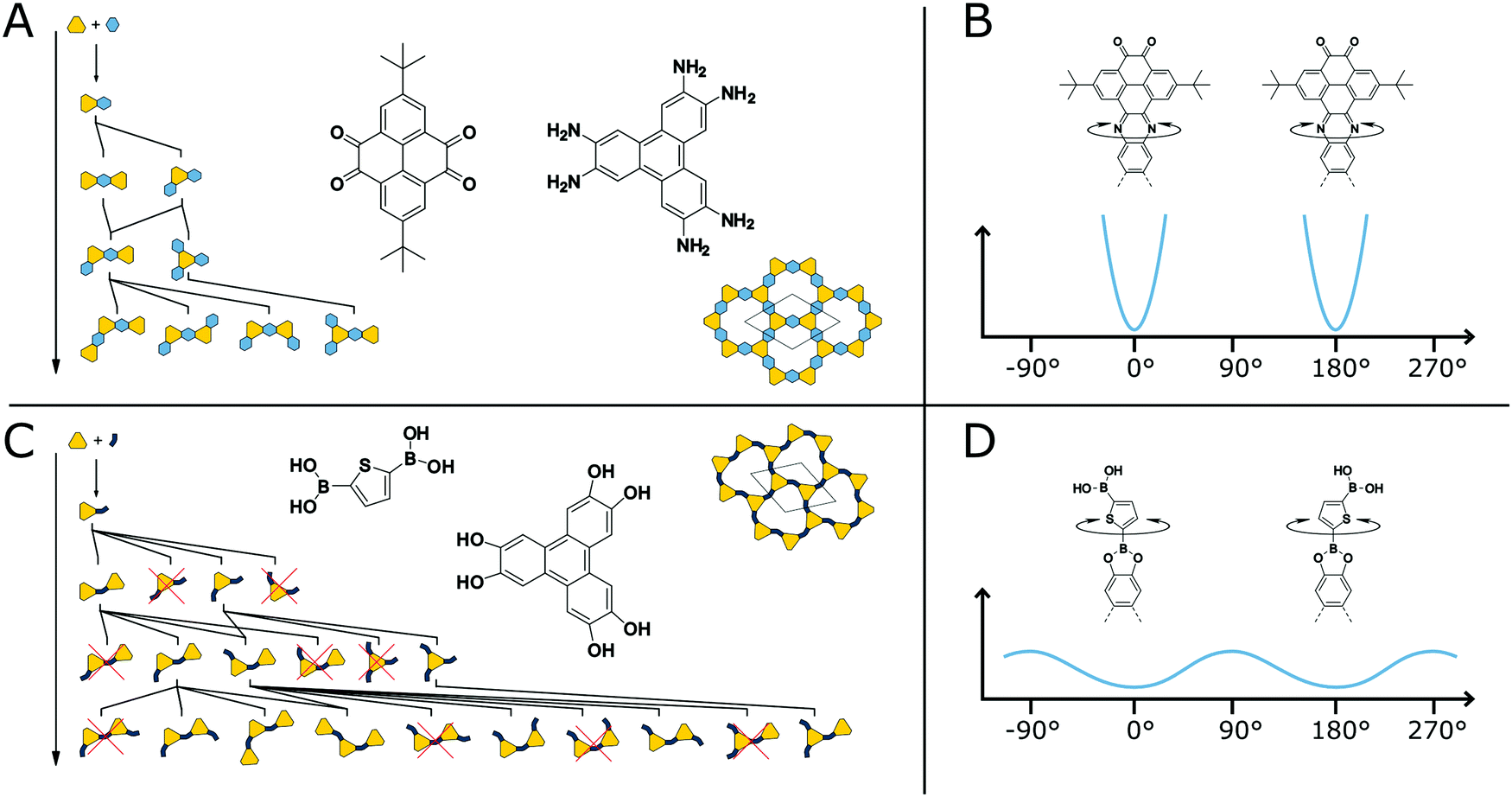 | ||
| Fig. 21 Comparison of conformational degrees of freedom in two COFs (A and C). A highly rigid COF based on the phenazine linkage with annulated and straight linkers (A and B).37 A COF based on a bent linker produces many possible orientational conformers during the polymerization of the building blocks (C and D).120 | ||
The number of possible conformers affects the degrees of freedom on the local building block level and as such, the growth of the oligomers and the formation of the final COF, primarily in the form of disorder and flexibility. Thus, reducing the degrees of freedom on the building block level can have large implications for the growth of a COF and its crystallinity. The first reported COFs, based on the boronic ester and boronic acid anhydride linkage, are structures with very low conformational degrees of freedom, rendering crystallization more facile.3 In COF reactions with only limited reversibility, a low number of conformers might even be essential in leading to crystallinity as exemplified by the examples of COFs based on irreversible reactions (see Section 6.1). The cyclotrimerization of nitriles to form CTFs has a low reversibility,121 consequently only a few CTFs have been reported to be crystalline, and all are based on low conformer building blocks,10,56–58 while others based on building blocks with more conformers are amorphous.122–125
The structural degrees of freedoms that appear in COFs because of the flexibility of building blocks are largely tuneable due to the wide range of possible structures, while flexibility of the COF linkages, which are limited in number, make additional conformational degrees of freedom sometimes unavoidable. If multiple different structures are possible based on the same composition or the same building blocks, this presents an additional “topological” degree of freedom and the competition between these phases can also lead to low crystallinity. Finally, in 2D COFs the stacking is a strong source for structural degrees of freedom, as the stacking occurs by weak interactions of the layers. In this section notable examples and strategies for reducing structural degrees of freedoms in COFs are discussed, which open up more subtle, yet efficient means of crystallinity control in COFs.
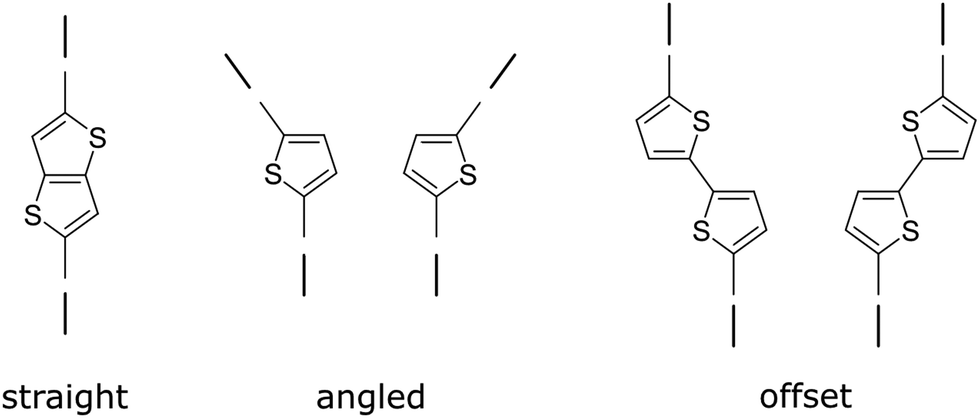 | ||
| Fig. 22 Geometry of linkers with two points of extension. The symmetry of the linker affects the degrees of freedom in the COF, where lower symmetry can lead to more degrees of freedom. | ||
Some of the building block's inherent flexibility can be avoided by using highly rigid molecules. Highly annulated building blocks based on a triphenylene or pyrene core are extremely rigid as they can only bend out of shape with a high energy penalty but possess no rotational degrees of freedom, as opposed to building blocks that possess a connecting C–C single bond as in biphenyl (Fig. 23). The conformational polymorphism depends also on the equilibrium dihedral angle between two adjacent rings as these flexible molecules have multiple conformational possibilities within a small energy window. In the biphenyl case an optimum dihedral angle of ∼44127 is observed, which leads to four energy minima for just one biphenyl linkage that can complicate the formation of the COF structure.
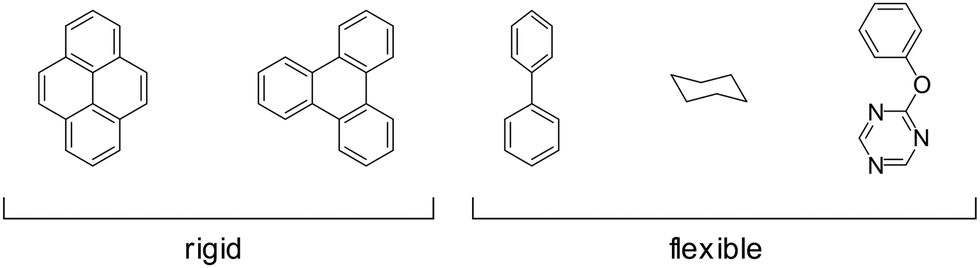 | ||
| Fig. 23 Examples of rigid and flexible structural fragments that have been used in COFs. | ||
The dihedral angle between two adjacent aromatic rings can be controlled by managing the repulsion between hydrogen atoms of adjacent aryl rings. For 2D COFs low dihedral angles are often desired, as they can improve stacking order and, hence, crystallinity.128 Low dihedral angles can be achieved by combining a phenyl ring with heteroatomic rings, that do not possess hydrogen atoms in ortho position to the connecting bond between both rings, such as structures based on boroxine, triazine, bipyridine, pyridine, pyrimidine or tetrazine.3,129,130 The use of a five membered ring can also enable flat structures such as those based on thiophene.131 The repulsion of hydrogen atoms on adjacent phenyl rings can also be reduced by introducing a separating alkyne group.132
Common design principles of COF building blocks include the use of highly rigid building blocks that are composed of only sp2- or sp-carbons in their backbone to reduce the number of conformers and to increase porosity.133–135 These building blocks are based on polyaromatic units together with alkyne or vinyl groups.20,21,136 Typically, the employed building blocks have high symmetry as these tend to produce higher crystallinity in COFs,1 while asymmetric building blocks tend to increase disorder in the COF.129 Nevertheless, there is a rich literature in asymmetric building blocks that have been used for COF design as well.129,137–139 Linkers with sp3-carbons are known as well and are especially prominent for tetrahedral building blocks based on tetraphenylmethane or adamantane.4,9,65,140–145 These building blocks are especially important for the construction of three-dimensional COFs, as the number of rigid three-dimensional nodes other than those mentioned above are generally limited. The examples based on sp3-carbons still represent highly rigid building blocks as the numbers of possible conformers are restricted.28,146 In addition, some COFs with highly flexible linkers are known such as those based on cyclohexane9 and piperazine.49 In general, however, the overall low number of reported COF structures with flexible building blocks suggests that the formation of crystalline and porous COFs with such linkers is more challenging.147
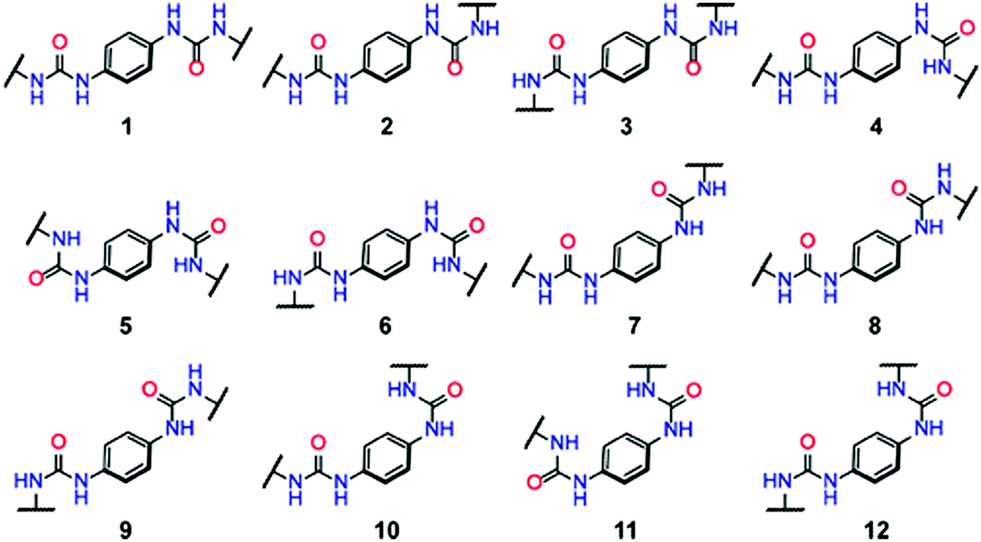 | ||
| Fig. 24 Possible conformers of a urea-linked COF.44 Reprinted with permission from ref. 44. Copyright (2018) American Chemical Society. | ||
The imine linkage entails two orientational isomers forming flat structures (Fig. 26A) that could lead to reduced order in the COF. The flexibility of the imine linkage has been directly implicated in the flexibility observed in several COFs, thus showing their ability, in principle, to form several structural conformers.4,148,149 This has been demonstrated with the control over the 3D structure of a COF where slightly altered synthetic conditions lead either to a dense, non-porous structure or a porous COF, while both structures were crystalline. The structures could be switched based on the presence of water, which leads to a significant contraction of the unit cell due to changes in hydrogen bonding.4,150 Similar switchable structures are observed with the breathing effect in a 3D COF with THF (Fig. 25),149 leading to conformational changes in the structure which influence the distance between the interpenetrated nets as a result of the imine rotation that can be seen as a cause of this flexibility. Similarly, flexibility of the imine linkage has been detected in early examples of 3D COFs where temperature dependent symmetry variations led to changes in the pore shape from rectangular to square.4,148
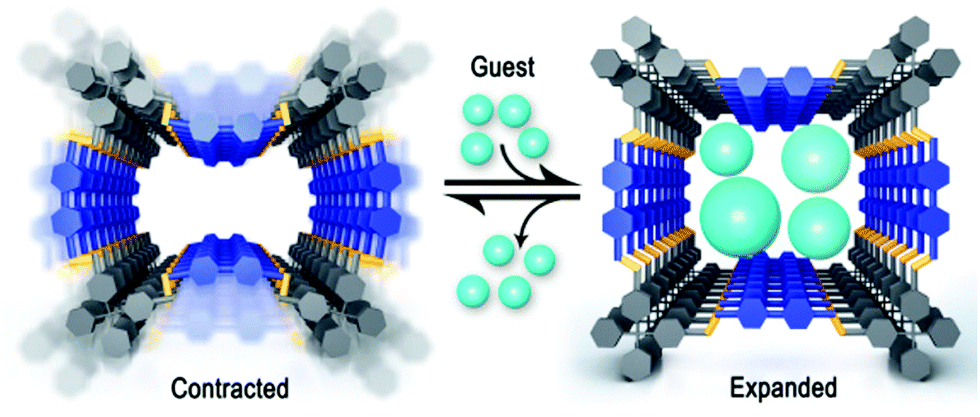 | ||
| Fig. 25 Structural transformation in a COF due to the presence of guest molecules, which emphasizes the flexibility of the framework.149 Reprinted with permission from ref. 149. Copyright (2017) American Chemical Society. | ||
There are however strategies to conformationally lock imine linkages in place so that the number of conformers is reduced from two to one, by using an ortho hydroxy aldehyde linker in combination with the amine, which leads to intramolecular hydrogen bonding that directs the imine bond (Fig. 26B).38 The opposite conformational locking can be induced by using an ortho methoxy substituted aldehyde, which leads to imine groups facing away from the methoxy group (Fig. 26C).16
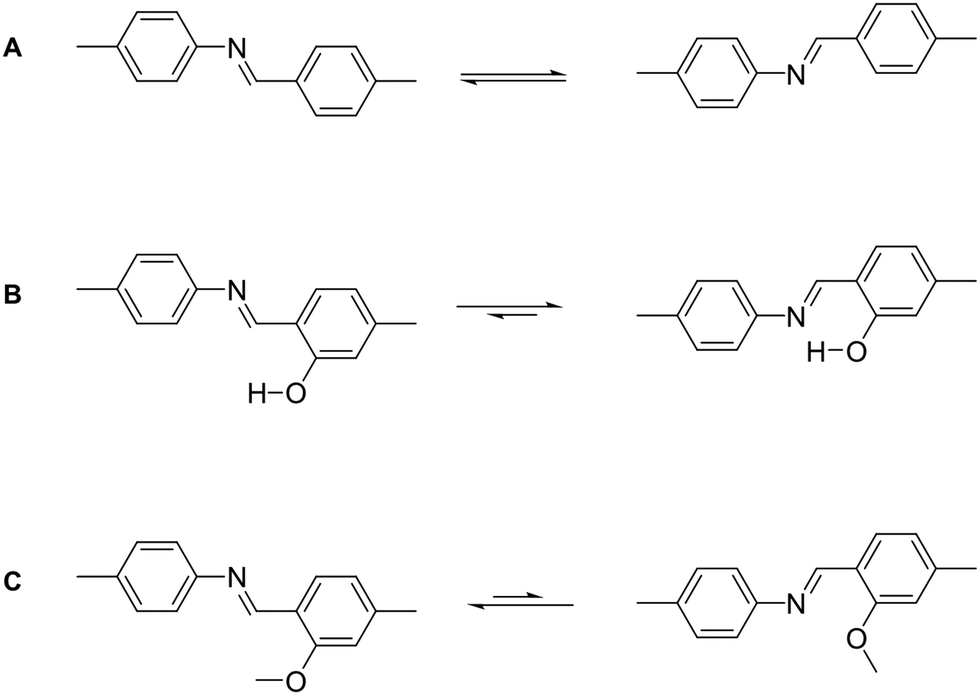 | ||
| Fig. 26 (A) Conformational isomers of the imine linkage. (B) Conformational locking of the imine linkage by favouring intramolecular hydrogen bonding or (C) by steric repulsion between the methoxy group and the imine nitrogen. | ||
However, in many cases the lock-in by intramolecular hydrogen bonding does not reduce the number of conformers, as the COF fragments containing the imine linkage can rotate such that two conformers are still available (Fig. 27A). This issue can be circumvented by building block design, when the hydrogen bonding locks the direction of the imine linkage so that a straight COF fragment is formed (Fig. 27B).151 By designing a building block where the imine is locked either on opposing (Fig. 26A) or the same side (Fig. 26B), the conformational offset found in the trans case is eliminated in the cis case. The linker then still has two different conformers (Fig. 27B), which might influence the stacking and thereby the crystallinity. As the orientation of the linker can alternate between layers, favourable dipole–dipole interactions between layers could occur.152
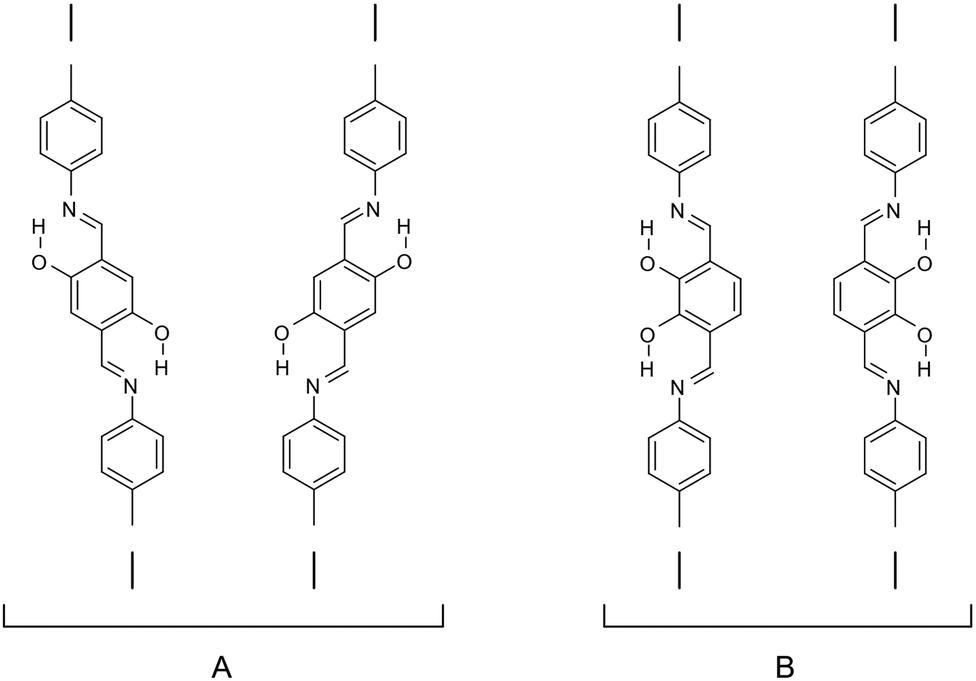 | ||
| Fig. 27 Locking the conformers by hydrogen bonding in an imine-linked fragment of a COF. (A) Offset in the linking axis due to two possible orientations of the structural fragment with hydroxy groups on opposite sites; (B) straight linkage axis of the COF fragment due to the placement of the hydroxy groups on the same side.151 | ||
Esteves and co-workers153 were able to show that by introducing benzene trialdehydes with different amounts of ortho hydroxy groups the crystallinity and porosity of the COFs could be improved, implying that the locking of a conformation can improve the crystallinity by reducing the number of conformers (Fig. 28).
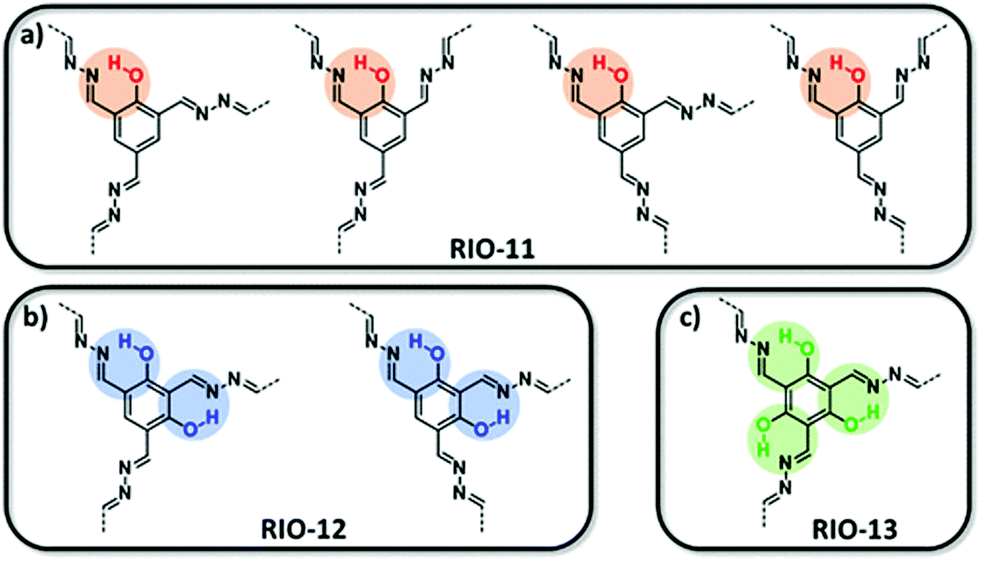 | ||
| Fig. 28 Conformational locking in an azine COF. With a larger number of hydroxy groups in the linker, the number of conformers is reduced.153 Reprinted with permission from ref. 153. Copyright (2018) American Chemical Society. | ||
When an imine is transformed into an amine by reduction, additional flexibility is created as a new sp3 bond is formed. The resulting flexibility can be so substantial that the structural integrity is lost and the structure loses its order as detectable by PXRD and porosity measurements.16 Since the topology of the structure is maintained during this transition, it can be recovered by re-rigidifying the structure, by further reaction to a carbamate. This shows how the flexibility of the linkage groups plays an essential role for the crystallinity of COFs.
Annulated linkages such as the phenazine or the dioxane type linkage are prime examples for linkage groups that lead to only a single conformer. They possess a very rigid structure that improves the crystallinity of the structure even though the reaction has a comparatively low reversibility (Fig. 29).154
 | ||
| Fig. 29 Formation of dioxin type linkages that have very low reversibility, but are very rigid.154 Reprinted with permission from ref. 154. Copyright (2018) American Chemical Society. | ||
An even higher rigidity of the COF linkage is observed in phthalocyanine COFs synthesized from ortho dinitriles. This highly rigid and essentially irreversible reaction still give rise to crystalline materials, as observed by PXRD and TEM, including ordered stacking reflections. The exceptionally rigid structure of a phthalocyanine linkage makes this a prime example for a single reaction pathway.155
 | ||
| Fig. 30 Interpenetrating structures with hexagonal nets: (A) interpenetration in simulated nuclei of COF-5;114 (B) interpenetration as found in a 2D HOF.163 (A) Reprinted with permission from ref. 114. Copyright (2018) American Chemical Society. (B) Reprinted with permission from ref. 163. Copyright (2015) Royal Society of Chemistry. | ||
The design of nets is normally straightforward, but there are exceptions where competing interactions hinder the formation of the desired net.164,165 This was initially demonstrated with the unexpected formation of a bex net instead of the desired tbo net, driven by π–π interactions.23 In this case, a 2D framework was obtained instead of a 3D framework from nominally a tetraamine and a trialdehyde building block. The bex topology was only formed because the pyrene building block simultaneously acted in a dual role – as a four connected and as a two connected node in the structure. In similar systems based on pyrene or tetraphenylethylene building blocks combined with trigonal building blocks multiple sub-stoichiometric structure were obtained.22,166–168 This shows that even in carefully designed systems, structural polymorphism or multiple competing phases are a possibility.
Conformers of a structure cannot only lead to decreased crystallinity and increased disorder, but also to polymorphism based on the same or different nets, when multiple configurations of conformers can form different periodic structures. In surface COFs (sCOFs) the different conformers can be resolved by STM at the local scale.169 By this method it was possible to show the existence and co-existence of three distinct polymorphs of a sCOF, based on a four-connected pyrene and a linear linker linked by imine groups. The polymorphs differ in the net of the corresponding sCOF, where two polymorphs were found featuring a square lattice and one with a kagome lattice. This results from the fact that the 60° and 120° bite angles of the pyrene linker enable the formation of both a square and a kagome lattice without deformation of bonds (Fig. 31). The second differentiating criterium was the orientation of the imines. Two polymorphs in the square lattice could be differentiated based on whether the imines were oriented in a “homodromous” or “heterodromous” fashion, meaning that the imines were either rotated in the same direction or in different directions. These two arrangements lead to slightly different lattice parameters.
 | ||
| Fig. 31 sCOFs with three different polymorphs synthesized from the same imine and aldehyde precursors.169 Reprinted with permission from ref. 169. Copyright (2017) American Chemical Society. | ||
Similar polymorphs can be imagined for bulk COFs based on the same or similar linkers. While the differences between the polymorphs based on the rotation of the imines might not be resolvable by PXRD or be disordered at room temperature, the kagome versus square lattice would be clearly differentiable by X-ray powder diffraction. Two linkers are prominently used for square lattice and kagome COFs that both possess the 120° + 60° binding motif: pyrene and tetraphenylethylene. For both linkers both nets would be possible. Interestingly, COFs using the tetraphenylethylene building block crystallize in the kagome net,158,170–172 while the pyrene based COFs crystallize with a square net.32,173–175 The fact that not both polymorphs are observed for each material could be explained with the different stacking behaviour of these materials, which showed that linkers like tetraphenylethylene tend to stack with zero offset,158 while the pyrene linkers lead to constant offset stacking.176 Whereas the zero offset stacking might be possible for a square lattice of tetraphenylethylene, a constant offset stacking would be difficult to reconcile with a kagome lattice. The pyrene linker tends to stack with an offset along its long axis. As the constant offset stacking has to agree between all the linkers, only one pyrene in a triangle stacks in the favoured directions, while the two others would be forced in directions that are not optimal. These examples suggest that polymorphism in COFs might be influenced by the stacking as well.
Both kagome and square lattice could be shown in a tetraphenyl ethylene based COF, by using the side chains on a second linear linker to influence the net formation (Fig. 32).177 By introducing bulky side chains onto the linker, the formation of the kagome lattice was suppressed and the square lattice was formed, since the triangular pores of the kagome lattice where too small for the side chains. Only when no alkyl side chain was used, the kagome structure formed. Additional control over the formation of a square lattice or a kagome lattice can be exerted by the introduction of intramolecular hydrogen bonding178 or by the choice of the solvent.179 While these examples show well-resolved cases of polymorphism, in other cases were the different polymorphs cannot be separated or crystallized independently, polymorphism might lead to a mixture or a decrease in order if multiple crystal structures compete with each other and co-exist.
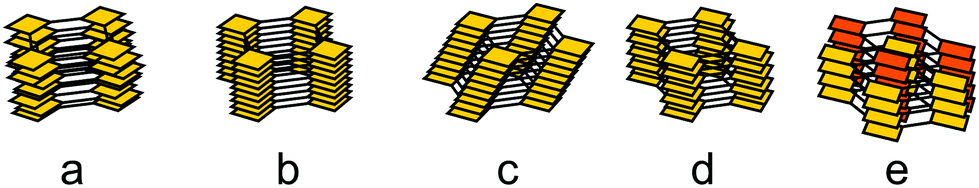 | ||
| Fig. 33 Stacking modes of 2D COFs: (a) random stacking with a small offset; (b) eclipsed stacking; (c) unidirectional slip-stacking; (d) alternating stacking with a small offset; (e) staggered stacking. | ||
Investigation of the stacking in COFs is often limited by the information that can be extracted from the available powder X-ray diffraction data. As such, the most prominent stacking types that are identified from PXRD are the so-called eclipsed stacking, which is likely to be an approximate to the real stacking type only,182 and the completely offset, staggered stacking, all others being rarely discerned.3,128,183–185 In addition to these simple descriptions of stacking, works on varied forms of stacking and their influence on structure and properties of COFs exist,186 but here we will focus on the challenges of stacking for crystallinity in COFs. In order to reduce the disorder generated by stacking in COFs, the non-directional forces involved during stacking can be used and rationally altered to improve the stacking order and thereby the overall crystallinity of the COF. When considering the stacking landscape of only two isolated layers, often multiple minima within a small energy window are observed (Fig. 34).128,183
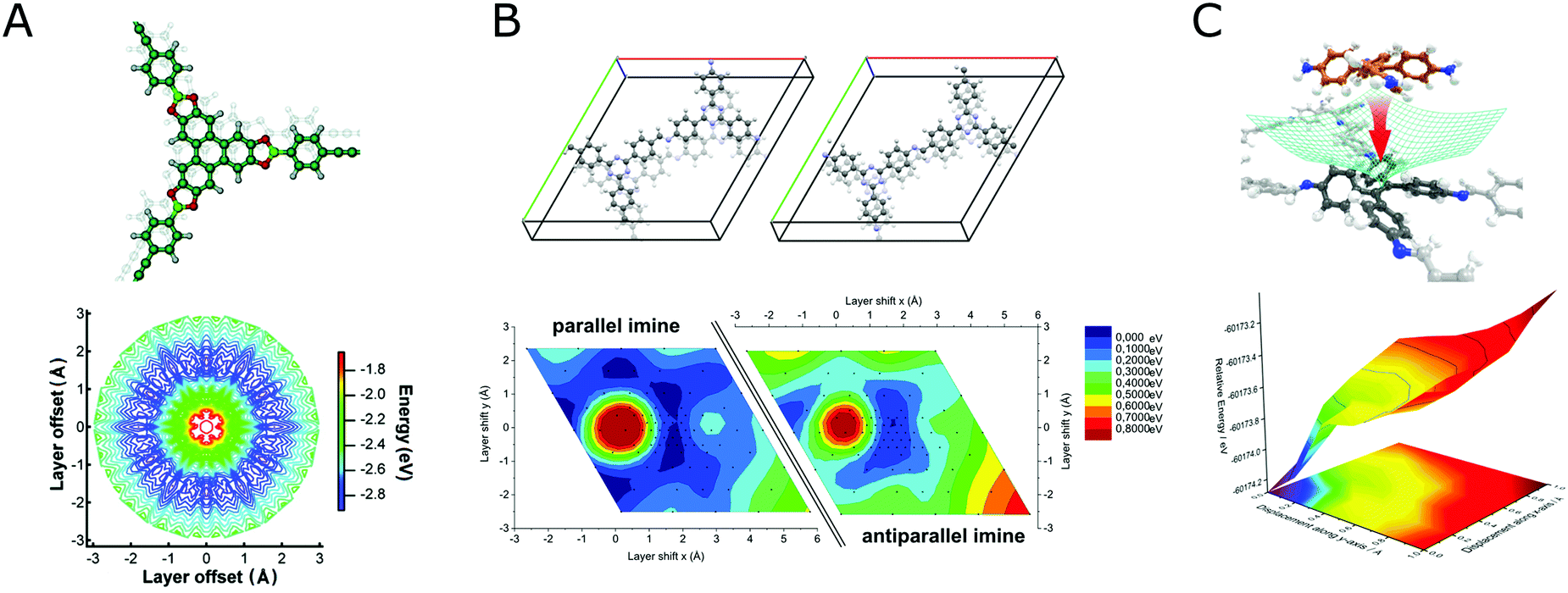 | ||
| Fig. 34 (A) Stacking energy landscape for an eclipsed boronic ester COF showing a distributed minimum in the potential energy landscape (Reprinted with permission from ref. 183. Copyright (2011) American Chemical Society); (B) potential energy landscape of a 2D imine COF, with and without the dipolar interaction of the layers (Reprinted with permission from ref. 128 Copyright (2017) Royal Society of Chemistry); (C) potential energy landscape of a COF with molecular docking sites, showing a single minimum at the zero offset mark (Reprinted with permission from ref. 158 Copyright (2016) Springer Nature). | ||
Several design strategies have been developed to improve the stacking and thereby the crystallinity of 2D COFs.186 One approach to reduce the number of minima in the stacking landscape is to introduce molecular docking sites, for example by using propeller shaped building blocks that result in only a single minimum for the staking energy landscape, thereby leading to eclipsed stacking.183 Several minima within in a small energy window in the stacking landscape can lead to disorder. So-called lock-in configurations can introduce interactions beyond next neighbours can lift this degeneracy and lead to improved stacking. The lock-in can be achieved by out-of-plane features such as twisted of phenyl rings, methyl groups on a truxene, or bulky isopropyl sidechains that lead to slip-stacking or in some cases staggered stacking.30,128,176,187 These are strategies based on the shape and geometry of the building block that lead to self-complementary van-der-Waals interactions. Since the lock-in of the layers leads to steeper energetic minima in the stacking landscape, the structure of the COFs is more well-defined and thereby these COFs are more crystalline.128 Alternatively, stronger interlayer interactions are introduced to improve the layer stacking.
The tuning of donor–acceptor interactions between layers can be used to improve the crystallinity by the introduction of electron deficient and electron rich building blocks, such as fluorinated and nonfluorinated compounds.128,188–191 Since many functionalized COFs possess dipole moments perpendicular to the stacking direction, these dipoles need to be carefully managed as parallel aligned dipoles lead to repulsion that can make the stacking more difficult, resulting in lower crystallinity.192 The inherent dipole moment of the COF linkage can be the source of such an interaction. The dipole of the imine linkage can be reduced by the introduction of a methoxy group adjacent to the imine bond192 or by designing the structure such that the imines can stack in an antiparallel fashion in adjacent layers.128 Antiparallel dipoles can also be introduced by dipole bearing building blocks that stack in an alternating fashion, such as with a pyrene dione building block.152 Even stronger layer interactions can be introduced based on hydrogen bonding, which is possible with amide functional groups that twist out of plane and lead to continuous hydrogen bonded chains since each amide is able to provide one hydrogen bond donor group and one hydrogen bond acceptor group, leading to high crystallinity in the resulting COF.9,193 Less directional, but strong interlayer forces have been used in a viologen based COF that contains cationic pyridinium groups.63 These ionic groups provide strong interlayer interactions, as the chloride counterions are sandwiched between the staggered layers, providing the structure with a strong interaction across the layers. In an achiral imine COF, the stacking was influenced by the use of a chiral monoamine during the synthesis of the COF (Fig. 35). The chiral molecule was able to induce chirality in the stacked COF by the arrangement of the layers and building blocks within the layer, even though the chiral molecule was completely removed after the synthesis. The approach was shown to produce a range of chiral frameworks which could be proven by circular dichroism.
 | ||
| Fig. 35 Influencing the stacking in an imine COF by the use of a chiral auxiliary during the synthesis of the COF.194 (A) Different chiralities of the structural motif forming the COF. (B) Examples of chiral COFs that were synthesized from achiral building blocks by chiral catalytic induction. Reprinted with permission from ref. 194. Copyright (2018) Springer Nature. | ||
All these strategies that are used to improve the stacking can be used to improve the crystallinity of 2D COFs, however, these interactions are difficult to introduce in a purposeful manner and therefore are difficult to design.
5. Conclusion and outlook
The multitude of examples shown in this review that have explored paths of COFs formation underline the importance and the desire of the COF community to improve the crystallinity and stability in COFs (Fig. 36). Three paths were differentiated that can lead to crystalline covalently connected materials: reversible bond formation, pre-orientation and reduction of conformers. Following the proposed design strategies enables even the synthesis of crystalline COFs based on completely irreversible reactions. | ||
| Fig. 36 Overview of the strategies used to enable crystallization of COFs. | ||
We expect these three major trends to continue to improve the crystallinity and stability in COFs further, opening the door to entirely new COF designs where crystallinity and stability are not at odds with each other.
Identifying new reversible reaction pathways continues to produce chemical reactions that are capable of forming crystalline and covalently linked materials. Desired new reactions are reversible under mild conditions to allow for a wide range of functional groups, but ideally these reactions need very specific conditions or catalysts for reversibility, so that the resulting COFs are stable under the conditions relevant for applications. We expect that reactions will be identified where stable and highly crystalline COFs with high functional group tolerance can be synthesized directly. Possible examples include reactions that require special catalysts such as alkene metathesis, where removal of the catalyst completely arrests the reaction.
The separation of crystallization and stabilization will benefit from developing reactions that are highly reversible, but are followed by reactions that lock the labile linkage in place in a facile manner. Ideally, it will be possible to obtain single crystals of a material that can then be locked into a highly ordered and at the same time extremely stable material. This field will benefit especially from the investigation of the mechanism of COF formation and the development of new COF linkage locking strategies. The latter may lead to a resurgence in popularity of linkage groups that have attracted less attention recently due to their lower stability, such as boronic ester COFs, if their bonds can be suitably stabilized.
The formation of COFs from irreversible reactions through the design of low-conformer frameworks and tailored reaction conditions is still only in its infancy. We expect this field to act as a nucleation site for a rich new COF chemistry, as it promises highly robust and functional materials that can be synthesized in a single step. As the mechanism of formation of these materials is only partially related to the traditional reversible way in which COFs are formed, new conceptual groundwork and mechanistic insights will be needed to establish a more generalized and unified framework for the growth of crystalline and stable COFs based on reversible and irreversible reactions alike.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open Access funding provided by the Max Planck Society. F. H. thanks the Young Investigator Group Preparation Program (YIG PrepPro) for the awarded fellowship. Financial support by an ERC Starting Grant (project COF Leaf, Grant No. 639233), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation – project number 358283783 – SFB 1333), the Max Planck Society, the Cluster of Excellence e-conversion (Grant No. EXC2089), and the Center for Nanoscience (CeNS) is gratefully acknowledged.References
- C. S. Diercks and O. M. Yaghi, The Atom, the Molecule, and the Covalent Organic Framework, Science, 2017, 355(6328), eaal1585, DOI:10.1126/science.aal1585.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Topochemical Conversion of an Imine- into a Thiazole-Linked Covalent Organic Framework Enabling Real Structure Analysis, Nat. Commun., 2018, 9, 2600, DOI:10.1038/s41467-018-04979-y.
- A. P. Cote, Porous, Crystalline, Covalent Organic Frameworks, Science, 2005, 310(5751), 1166–1170, DOI:10.1126/science.1120411.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Single-Crystal x-Ray Diffraction Structures of Covalent Organic Frameworks, Science, 2018, 361(6397), 48–52, DOI:10.1126/science.aat7679.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Ionothermal Synthesis of Imide-Linked Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2020, 59(36), 15750–15758, DOI:10.1002/anie.202007372.
- T. Banerjee, T. Bennett, K. Butler, T. L. Easun, M. Eddaoudi, R. Forgan, L. Gagliardi, C. Hendon, M. Jorge, C. Lamberti, J.-S. M. Lee, K. Leus, J. Li, W. Lin, M. Ranocchiari, N. Rosi, J. G. Santaclara, S. Shevlin, K. Svane, V. Ting, M. Veen, P. V. D. van der; Voort, A. Walsh, D. Woods, O. M. Yaghi and G. Zhu, Electronic, Magnetic and Photophysical Properties of MOFs and COFs: General Discussion, Faraday Discuss., 2017, 201, 87–99, 10.1039/C7FD90043K.
- F. Carraro, K. Chapman, Z. Chen, M. Dincă, T. Easun, M. Eddaoudi, O. Farha, R. Forgan, L. Gagliardi, F. Haase, D. Harris, S. Kitagawa, J. Knichal, C. Lamberti, J.-S. M. Lee, K. Leus, J. Li, W. Lin, G. Lloyd, J. R. Long, C. Lu, S. Ma, L. McHugh, J. P. H. Perez, M. Ranocchiari, N. Rosi, M. Rosseinsky, M. R. Ryder, V. Ting, M. Veen, P. V. D. van der; Voort, D. Volkmer, A. Walsh, D. Woods and O. M. Yaghi, Catalysis in MOFs: General Discussion, Faraday Discuss., 2017, 201, 369–394, 10.1039/C7FD90046E.
- F. P. Bundy, W. A. Bassett, M. S. Weathers, R. J. Hemley, H. U. Mao and A. F. Goncharov, The Pressure-Temperature Phase and Transformation Diagram for Carbon; Updated through 1994, Carbon, 1996, 34(2), 141–153, DOI:10.1016/0008-6223(96)00170-4.
- D. Stewart, D. Antypov, M. S. Dyer, M. J. Pitcher, A. P. Katsoulidis, P. A. Chater, F. Blanc and M. J. Rosseinsky, Stable and Ordered Amide Frameworks Synthesised under Reversible Conditions Which Facilitate Error Checking, Nat. Commun., 2017, 8, 1102, DOI:10.1038/s41467-017-01423-5.
- P. Kuhn, M. Antonietti and A. Thomas, Porous, Covalent Triazine-Based Frameworks Prepared by Ionothermal Synthesis, Angew. Chem., Int. Ed., 2008, 47(18), 3450–3453, DOI:10.1002/anie.200705710.
- A. M. Evans, M. R. Ryder, W. Ji, M. J. Strauss, A. Corcos, E. Vitaku, N. C. Flanders, R. P. Bisbey and W. Dichtel, Trends in the Thermal Stability of Two-Dimensional Covalent Organic Frameworks, Faraday Discuss., 2020 10.1039/D0FD00054J.
- J. Jiang, Y. Zhao and O. M. Yaghi, Covalent Chemistry beyond Molecules, J. Am. Chem. Soc., 2016, 138(10), 3255–3265, DOI:10.1021/jacs.5b10666.
- M. T. Rodgers and P. B. Armentrout, Noncovalent Metal–Ligand Bond Energies as Studied by Threshold Collision-Induced Dissociation, Mass Spectrom. Rev., 2000, 19(4), 215–247, DOI:10.1002/1098-2787(200007)19:4<215::AID-MAS2>3.0.CO;2-X.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular Synthesis and the Design of New Materials, Nature, 2003, 423(6941), 705–714, DOI:10.1038/nature01650.
- T. Sick, J. M. Rotter, S. Reuter, S. Kandambeth, N. N. Bach, M. Döblinger, J. Merz, T. Clark, T. B. Marder, T. Bein and D. D. Medina, Switching on and off Interlayer Correlations and Porosity in 2D Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(32), 12570–12581, DOI:10.1021/jacs.9b02800.
- S. J. Lyle, T. M. Osborn Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, Multistep Solid-State Organic Synthesis of Carbamate-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(28), 11253–11258, DOI:10.1021/jacs.9b04731.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Insight into the Crystallization of Amorphous Imine-Linked Polymer Networks to 2D Covalent Organic Frameworks, Chem. Commun., 2016, 52(18), 3690–3693, 10.1039/C5CC10221A.
- B. J. Smith and W. R. Dichtel, Mechanistic Studies of Two-Dimensional Covalent Organic Frameworks Rapidly Polymerized from Initially Homogenous Conditions, J. Am. Chem. Soc., 2014, 136(24), 8783–8789, DOI:10.1021/ja5037868.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent Organic Frameworks: Design, Synthesis, and Functions, Chem. Rev., 2020, 120(16), 8814–8933, DOI:10.1021/acs.chemrev.9b00550.
- S.-Y. Ding and W. Wang, Covalent Organic Frameworks (COFs): From Design to Applications, Chem. Soc. Rev., 2013, 42(2), 548–568, 10.1039/C2CS35072F.
- X. Feng, X. Ding and D. Jiang, Covalent Organic Frameworks, Chem. Soc. Rev., 2012, 41(18), 6010, 10.1039/c2cs35157a.
- B. Zhang, H. Mao, R. Matheu, J. A. Reimer, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, Reticular Synthesis of Multinary Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(29), 11420–11424, DOI:10.1021/jacs.9b05626.
- T. Banerjee, F. Haase, S. Trenker, B. P. Biswal, G. Savasci, V. Duppel, I. Moudrakovski, C. Ochsenfeld and B. V. Lotsch, Sub-Stoichiometric 2D Covalent Organic Frameworks from Tri- and Tetratopic Linkers, Nat. Commun., 2019, 10, 2689, DOI:10.1038/s41467-019-10574-6.
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Targeted Synthesis of a Porous Borazine-Linked Covalent Organic Framework, Chem. Commun., 2012, 48(70), 8823, 10.1039/c2cc33583b.
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Côté and O. M. Yaghi, Reticular Synthesis of Covalent Organic Borosilicate Frameworks, J. Am. Chem. Soc., 2008, 130(36), 11872–11873, DOI:10.1021/ja805064f.
- J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Anionic Silicate Organic Frameworks Constructed from Hexacoordinate Silicon Centres, Nat. Chem., 2017, 9(10), 977–982, DOI:10.1038/nchem.2771.
- E. Vitaku and W. R. Dichtel, Synthesis of 2D Imine-Linked Covalent Organic Frameworks through Formal Transimination Reactions, J. Am. Chem. Soc., 2017, 139(37), 12911–12914, DOI:10.1021/jacs.7b06913.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S.-H. Lee and W. Zhang, Ionic Covalent Organic Frameworks with Spiroborate Linkage, Angew. Chem., Int. Ed., 2016, 55(5), 1737–1741, DOI:10.1002/anie.201509014.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, A Titanium–Organic Framework as an Exemplar of Combining the Chemistry of Metal– and Covalent–Organic Frameworks, J. Am. Chem. Soc., 2016, 138(13), 4330–4333, DOI:10.1021/jacs.6b01233.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, Control Interlayer Stacking and Chemical Stability of Two-Dimensional Covalent Organic Frameworks via Steric Tuning, J. Am. Chem. Soc., 2018, 140(47), 16124–16133, DOI:10.1021/jacs.8b08452.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, Enhanced Hydrolytic Stability of Self-Assembling Alkylated Two-Dimensional Covalent Organic Frameworks, J. Am. Chem. Soc., 2011, 133(35), 13975–13983, DOI:10.1021/ja203807h.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, An Azine-Linked Covalent Organic Framework, J. Am. Chem. Soc., 2013, 135(46), 17310–17313, DOI:10.1021/ja4103293.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chemical Sensing in Two Dimensional Porous Covalent Organic Nanosheets, Chem. Sci., 2015, 6(7), 3931–3939, 10.1039/C5SC00512D.
- C. Liang, H. Lin, Q. Wang, E. Shi, S. Zhou, F. Zhang, F. Qu and G. Zhu, A Redox-Active Covalent Organic Framework for the Efficient Detection and Removal of Hydrazine, J. Hazard. Mater., 2020, 381, 120983, DOI:10.1016/j.jhazmat.2019.120983.
- S. He, B. Yin, H. Niu and Y. Cai, Targeted Synthesis of Visible-Light-Driven Covalent Organic Framework Photocatalyst via Molecular Design and Precise Construction, Appl. Catal., B, 2018, 239, 147–153, DOI:10.1016/j.apcatb.2018.08.005.
- Y. Ma, X. Liu, X. Guan, H. Li, Y. Yusran, M. Xue, Q. Fang, Y. Yan, S. Qiu and V. Valtchev, One-Pot Cascade Syntheses of Microporous and Mesoporous Pyrazine-Linked Covalent Organic Frameworks as Lewis-Acid Catalysts, Dalton Trans., 2019, 48(21), 7352–7357, 10.1039/C8DT05056B.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Conjugated Organic Framework with Three-Dimensionally Ordered Stable Structure and Delocalized π Clouds, Nat. Commun., 2013, 4, 2736, DOI:10.1038/ncomms3736.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, Construction of Crystalline 2D Covalent Organic Frameworks with Remarkable Chemical (Acid/Base) Stability via a Combined Reversible and Irreversible Route, J. Am. Chem. Soc., 2012, 134(48), 19524–19527, DOI:10.1021/ja308278w.
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Enhancement of Chemical Stability and Crystallinity in Porphyrin-Containing Covalent Organic Frameworks by Intramolecular Hydrogen Bonds, Angew. Chem., Int. Ed., 2013, 52(49), 13052–13056, DOI:10.1002/anie.201306775.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, Chemically Stable Multilayered Covalent Organic Nanosheets from Covalent Organic Frameworks via Mechanical Delamination, J. Am. Chem. Soc., 2013, 135(47), 17853–17861, DOI:10.1021/ja408121p.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, Conjugated Covalent Organic Frameworks via Michael Addition–Elimination, J. Am. Chem. Soc., 2017, 139(6), 2421–2427, DOI:10.1021/jacs.6b12005.
- S.-Y. Jiang, S.-X. Gan, X. Zhang, H. Li, Q.-Y. Qi, F.-Z. Cui, J. Lu and X. Zhao, Aminal-Linked Covalent Organic Frameworks through Condensation of Secondary Amine with Aldehyde, J. Am. Chem. Soc., 2019, 141(38), 14981–14986, DOI:10.1021/jacs.9b08017.
- A. Schoedel, M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Structures of Metal–Organic Frameworks with Rod Secondary Building Units, Chem. Rev., 2016, 116(19), 12466–12535, DOI:10.1021/acs.chemrev.6b00346.
- C. Zhao, C. S. Diercks, C. Zhu, N. Hanikel, X. Pei and O. M. Yaghi, Urea-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(48), 16438–16441, DOI:10.1021/jacs.8b10612.
- C. Li, Y. Ma, H. Liu, L. Tao, Y. Ren, X. Chen, H. Li and Q. Yang, Asymmetric Photocatalysis over Robust Covalent Organic Frameworks with Tetrahydroquinoline Linkage, Chin. J. Catal., 2020, 41(8), 1288–1297, DOI:10.1016/S1872-2067(20)63572-0.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, Chemical Conversion of Linkages in Covalent Organic Frameworks, J. Am. Chem. Soc., 2016, 138(48), 15519–15522, DOI:10.1021/jacs.6b08377.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Designed Synthesis of Large-Pore Crystalline Polyimide Covalent Organic Frameworks, Nat. Commun., 2014, 5, 4503, DOI:10.1038/ncomms5503.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, A Squaraine-Linked Mesoporous Covalent Organic Framework, Angew. Chem., Int. Ed., 2013, 52(13), 3770–3774, DOI:10.1002/anie.201300256.
- H. Zhao, Z. Jin, H. Su, X. Jing, F. Sun and G. Zhu, Targeted Synthesis of a 2D Ordered Porous Organic Framework for Drug Release, Chem. Commun., 2011, 47(22), 6389, 10.1039/c1cc00084e.
- K. C. Ranjeesh, R. Illathvalappil, S. D. Veer, J. Peter, V. C. Wakchaure, Goudappagouda, K. V. Raj, S. Kurungot and S. S. Babu, Imidazole-Linked Crystalline Two-Dimensional Polymer with Ultrahigh Proton-Conductivity, J. Am. Chem. Soc., 2019, 141(38), 14950–14954, DOI:10.1021/jacs.9b06080.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Phenyl-Triazine Oligomers for Light-Driven Hydrogen Evolution, Energy Environ. Sci., 2015, 8(11), 3345–3353, 10.1039/C5EE02574E.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, Graphitic Carbon Nitride Materials: Variation of Structure and Morphology and Their Use as Metal-Free Catalysts, J. Mater. Chem., 2008, 18(41), 4893–4908, 10.1039/B800274F.
- S.-Y. Yu, J. Mahmood, H.-J. Noh, J.-M. Seo, S.-M. Jung, S.-H. Shin, Y.-K. Im, I.-Y. Jeon and J.-B. Baek, Direct Synthesis of a Covalent Triazine-Based Framework from Aromatic Amides, Angew. Chem., Int. Ed., 2018, 57(28), 8438–8442, DOI:10.1002/anie.201801128.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Crystalline Covalent Triazine Frameworks by In Situ Oxidation of Alcohols to Aldehyde Monomers, Angew. Chem., 2018, 130(37), 12144–12148, DOI:10.1002/ange.201806664.
- M. Liu, X. Wang, J. Liu, K. Wang, S. Jin and B. Tan, Palladium as a Superior Cocatalyst to Platinum for Hydrogen Evolution Using Covalent Triazine Frameworks as a Support, ACS Appl. Mater. Interfaces, 2020, 12(11), 12774–12782, DOI:10.1021/acsami.9b21903.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Covalent Triazine Frameworks Prepared from 1,3,5-Tricyanobenzene, Chem. Mater., 2013, 25(9), 1542–1548, DOI:10.1021/cm303751n.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Rational Extension of the Family of Layered, Covalent, Triazine-Based Frameworks with Regular Porosity, Adv. Mater., 2010, 22(19), 2202–2205, DOI:10.1002/adma.200903436.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Porous, Fluorescent, Covalent Triazine-Based Frameworks Via Room-Temperature and Microwave-Assisted Synthesis, Adv. Mater., 2012, 24(17), 2357–2361, DOI:10.1002/adma.201200751.
- M. J. Bojdys, S. A. Wohlgemuth, A. Thomas and M. Antonietti, Ionothermal Route to Layered Two-Dimensional Polymer-Frameworks Based on Heptazine Linkers, Macromolecules, 2010, 43(16), 6639–6645, DOI:10.1021/ma101008c.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, A Two-Dimensional Conjugated Polymer Framework with Fully Sp2 -Bonded Carbon Skeleton, Polym. Chem., 2016, 7(25), 4176–4181, 10.1039/C6PY00561F.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Vinylene-Linked Covalent Organic Frameworks by Base-Catalyzed Aldol Condensation, Angew. Chem., Int. Ed., 2019, 58(42), 14865–14870, DOI:10.1002/anie.201905886.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Two-Dimensional Semiconducting Covalent Organic Frameworks via Condensation at Arylmethyl Carbon Atoms, Nat. Commun., 2019, 10, 2467, DOI:10.1038/s41467-019-10504-6.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J.-C. Olsen and A. Trabolsi, Viologen-Based Conjugated Covalent Organic Networks via Zincke Reaction, J. Am. Chem. Soc., 2017, 139(28), 9558–9565, DOI:10.1021/jacs.7b02836.
- S. Zhang, X. Zhao, B. Li, C. Bai, Y. Li, L. Wang, R. Wen, M. Zhang, L. Ma and S. Li, “Stereoscopic” 2D Super-Microporous Phosphazene-Based Covalent Organic Framework: Design, Synthesis and Selective Sorption towards Uranium at High Acidic Condition, J. Hazard. Mater., 2016, 314, 95–104, DOI:10.1016/j.jhazmat.2016.04.031.
- D. Beaudoin, T. Maris and J. D. Wuest, Constructing Monocrystalline Covalent Organic Networks by Polymerization, Nat. Chem., 2013, 5(10), 830–834, DOI:10.1038/nchem.1730.
- H. Li, A. D. Chavez, H. Li, H. Li, W. R. Dichtel and J.-L. Bredas, Nucleation and Growth of Covalent Organic Frameworks from Solution: The Example of COF-5, J. Am. Chem. Soc., 2017, 139(45), 16310–16318, DOI:10.1021/jacs.7b09169.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, Rapid, Low Temperature Formation of Imine-Linked Covalent Organic Frameworks Catalyzed by Metal Triflates, J. Am. Chem. Soc., 2017, 139(14), 4999–5002, DOI:10.1021/jacs.7b01240.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, Constructing Ultraporous Covalent Organic Frameworks in Seconds via an Organic Terracotta Process, J. Am. Chem. Soc., 2017, 139(5), 1856–1862, DOI:10.1021/jacs.6b08815.
- S. Karak, S. Kumar, P. Pachfule and R. Banerjee, Porosity Prediction through Hydrogen Bonding in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(15), 5138–5145, DOI:10.1021/jacs.7b13558.
- E. L. Spitler, M. R. Giovino, S. L. White and W. R. Dichtel, A Mechanistic Study of Lewis Acid-Catalyzed Covalent Organic Framework Formation, Chem. Sci., 2011, 2(8), 1588–1593, 10.1039/C1SC00260K.
- J. Tan, S. Namuangruk, W. Kong, N. Kungwan, J. Guo and C. Wang, Manipulation of Amorphous-to-Crystalline Transformation: Towards the Construction of Covalent Organic Framework Hybrid Microspheres with NIR Photothermal Conversion Ability, Angew. Chem., Int. Ed., 2016, 55(45), 13979–13984, DOI:10.1002/anie.201606155.
- M. Calik, T. Sick, M. Dogru, M. Döblinger, S. Datz, H. Budde, A. Hartschuh, F. Auras and T. Bein, From Highly Crystalline to Outer Surface-Functionalized Covalent Organic Frameworks—A Modulation Approach, J. Am. Chem. Soc., 2016, 138(4), 1234–1239, DOI:10.1021/jacs.5b10708.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism, Nature, 1992, 359(6397), 710–712, DOI:10.1038/359710a0.
- D. Zhao, Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores, Science, 1998, 279(5350), 548–552, DOI:10.1126/science.279.5350.548.
- S. I. Stupp, S. Son, H. C. Lin and L. S. Li, Synthesis of Two-Dimensional Polymers, Science, 1993, 259(5091), 59–63, DOI:10.1126/science.259.5091.59.
- T. Takami, H. Ozaki, M. Kasuga, T. Tsuchiya, A. Ogawa, Y. Mazaki, D. Fukushi, M. Uda and M. Aono, Periodic Structure of a Single Sheet of a Clothlike Macromolecule(Atomic Cloth) Studied by Scanning Tunneling Microscopy, Angew. Chem., Int. Ed. Engl., 1997, 36(24), 2755–2757, DOI:10.1002/anie.199727551.
- M. Mazur, P. S. Wheatley, M. Navarro, W. J. Roth, M. Položij, A. Mayoral, P. Eliášová, P. Nachtigall, J. Čejka and R. E. Morris, Synthesis of ‘Unfeasible’ Zeolites, Nat. Chem., 2016, 8(1), 58–62, DOI:10.1038/nchem.2374.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Čejka, A Family of Zeolites with Controlled Pore Size Prepared Using a Top-down Method, Nat. Chem., 2013, 5(7), 628–633, DOI:10.1038/nchem.1662.
- B. Baumgartner, M. J. Bojdys and M. M. Unterlass, Geomimetics for Green Polymer Synthesis: Highly Ordered Polyimides via Hydrothermal Techniques, Polym. Chem., 2014, 5(12), 3771–3776, 10.1039/C4PY00263F.
- X.-Y. Hu, W.-S. Zhang, F. Rominger, I. Wacker, R. R. Schröder and M. Mastalerz, Transforming a Chemically Labile [2 + 3] Imine Cage into a Robust Carbamate Cage, Chem. Commun., 2017, 53(61), 8616–8619, 10.1039/C7CC03677A.
- T. H. G. Schick, J. C. Lauer, F. Rominger and M. Mastalerz, Transformation of Imine Cages into Hydrocarbon Cages, Angew. Chem., Int. Ed., 2019, 58(6), 1768–1773, DOI:10.1002/anie.201814243.
- M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Gram-Scale Synthesis of Two-Dimensional Polymer Crystals and Their Structure Analysis by X-Ray Diffraction, Nat. Chem., 2014, 6(9), 779–784, DOI:10.1038/nchem.2007.
- R. Z. Lange, G. Hofer, T. Weber and A. D. Schlüter, A Two-Dimensional Polymer Synthesized through Topochemical [2+2]-Cycloaddition on the Multigram Scale, J. Am. Chem. Soc., 2017, 139(5), 2053–2059, DOI:10.1021/jacs.6b11857.
- P. Kissel, R. Erni, W. B. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Götzinger, A. D. Schlüter and J. Sakamoto, A Two-Dimensional Polymer Prepared by Organic Synthesis, Nat. Chem., 2012, 4(4), 287–291, DOI:10.1038/nchem.1265.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Covalent Organic Frameworks Linked by Amine Bonding for Concerted Electrochemical Reduction of CO2, Chem, 2018, 4(7), 1696–1709, DOI:10.1016/j.chempr.2018.05.003.
- L. Grunenberg, G. Savasci, M. Terban, V. Duppel, I. Moudrakovski, M. Etter, R. E. Dinnebier, C. Ochsenfeld and B. Lotsch, Amine-Linked Covalent Organic Frameworks as a Powerful Platform for Post-Synthetic Modification: Structure Interconversion and Combined Linkage- and Pore-Wall-Modification, 2020, DOI:10.26434/chemrxiv.12985907.v1.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, A Nanoporous Two-Dimensional Polymer by Single-Crystal-to-Single-Crystal Photopolymerization, Nat. Chem., 2014, 6(9), 774–778, DOI:10.1038/nchem.2008.
- Z.-Z. Gao, Z.-K. Wang, L. Wei, G. Yin, J. Tian, C.-Z. Liu, H. Wang, D.-W. Zhang, Y.-B. Zhang, X. Li, Y. Liu and Z.-T. Li, Water-Soluble 3D Covalent Organic Framework That Displays an Enhanced Enrichment Effect of Photosensitizers and Catalysts for the Reduction of Protons to H2, ACS Appl. Mater. Interfaces, 2020, 12(1), 1404–1411, DOI:10.1021/acsami.9b19870.
- K. Liu, H. Qi, R. Dong, R. Shivhare, M. Addicoat, T. Zhang, H. Sahabudeen, T. Heine, S. Mannsfeld, U. Kaiser, Z. Zheng and X. Feng, On-Water Surface Synthesis of Crystalline, Few-Layer Two-Dimensional Polymers Assisted by Surfactant Monolayers, Nat. Chem., 2019, 11(11), 994–1000, DOI:10.1038/s41557-019-0327-5.
- H. Qi, H. Sahabudeen, B. Liang, M. Položij, M. A. Addicoat, T. E. Gorelik, M. Hambsch, M. Mundszinger, S. Park, B. V. Lotsch, S. C. B. Mannsfeld, Z. Zheng, R. Dong, T. Heine, X. Feng and U. Kaiser, Near–Atomic-Scale Observation of Grain Boundaries in a Layer-Stacked Two-Dimensional Polymer, Sci. Adv., 2020, 6(33), eabb5976, DOI:10.1126/sciadv.abb5976.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gandara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Weaving of Organic Threads into a Crystalline Covalent Organic Framework, Science, 2016, 351(6271), 365–369, DOI:10.1126/science.aad4011.
- Y. Zhao, L. Guo, F. Gándara, Y. Ma, Z. Liu, C. Zhu, H. Lyu, C. A. Trickett, E. A. Kapustin, O. Terasaki and O. M. Yaghi, A Synthetic Route for Crystals of Woven Structures, Uniform Nanocrystals, and Thin Films of Imine Covalent Organic Frameworks, J. Am. Chem. Soc., 2017, 139(37), 13166–13172, DOI:10.1021/jacs.7b07457.
- M. Cesario, C. O. Dietrich-Buchecker, J. Guilhem, C. Pascard and J. P. Sauvage, Molecular Structure of a Catenand and Its Copper(I) Catenate: Complete Rearrangement of the Interlocked Macrocyclic Ligands by Complexation, J. Chem. Soc., Chem. Commun., 1985,(5), 244, 10.1039/c39850000244.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, A Photoresponsive Smart Covalent Organic Framework, Angew. Chem., Int. Ed., 2015, 54(30), 8704–8707, DOI:10.1002/anie.201503902.
- Y. Du, K. Mao, P. Kamakoti, P. Ravikovitch, C. Paur, S. Cundy, Q. Li and D. Calabro, Experimental and Computational Studies of Pyridine-Assisted Post-Synthesis Modified Air Stable Covalent–Organic Frameworks, Chem. Commun., 2012, 48(38), 4606, 10.1039/c2cc30781b.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Facile Transformation of Imine Covalent Organic Frameworks into Ultrastable Crystalline Porous Aromatic Frameworks, Nat. Commun., 2018, 9, 2998, DOI:10.1038/s41467-018-05462-4.
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, Conversion of Imine to Oxazole and Thiazole Linkages in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(29), 9099–9103, DOI:10.1021/jacs.8b05830.
- J.-M. Seo, H.-J. Noh, H. Y. Jeong and J.-B. Baek, Converting Unstable Imine-Linked Network into Stable Aromatic Benzoxazole-Linked One via Post-Oxidative Cyclization, J. Am. Chem. Soc., 2019, 141(30), 11786–11790, DOI:10.1021/jacs.9b05244.
- Y. Wang, H. Liu, Q. Pan, C. Wu, W. Hao, J. Xu, R. Chen, J. Liu, Z. Li and Y. Zhao, Construction of Fully Conjugated Covalent Organic Frameworks via Facile Linkage Conversion for Efficient Photoenzymatic Catalysis, J. Am. Chem. Soc., 2020, 142(13), 5958–5963, DOI:10.1021/jacs.0c00923.
- M. C. Daugherty, E. Vitaku, R. L. Li, A. M. Evans, A. D. Chavez and W. R. Dichtel, Improved Synthesis of β-Ketoenamine-Linked Covalent Organic Frameworks via Monomer Exchange Reactions, Chem. Commun., 2019, 55(18), 2680–2683, 10.1039/C8CC08957D.
- K. Baek, G. Yun, Y. Kim, D. Kim, R. Hota, I. Hwang, D. Xu, Y. H. Ko, G. H. Gu, J. H. Suh, C. G. Park, B. J. Sung and K. Kim, Free-Standing, Single-Monomer-Thick Two-Dimensional Polymers through Covalent Self-Assembly in Solution, J. Am. Chem. Soc., 2013, 135(17), 6523–6528, DOI:10.1021/ja4002019.
- K. Baek, I. Hwang, I. Roy, D. Shetty and K. Kim, Self-Assembly of Nanostructured Materials through Irreversible Covalent Bond Formation, Acc. Chem. Res., 2015, 48(8), 2221–2229, DOI:10.1021/acs.accounts.5b00067.
- D. Schwarz, A. Acharjya, A. Ichangi, Y. S. Kochergin, P. Lyu, M. V. Opanasenko, J. Tarábek, J. Vacek Chocholoušová, J. Vacek, J. Schmidt, J. Čejka, P. Nachtigall, A. Thomas and M. J. Bojdys, Tuning the Porosity and Photocatalytic Performance of Triazine-Based Graphdiyne Polymers through Polymorphism, ChemSusChem, 2019, 12(1), 194–199, DOI:10.1002/cssc.201802034.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Seeded Growth of Single-Crystal Two-Dimensional Covalent Organic Frameworks, Science, 2018, 361(6397), 52–57, DOI:10.1126/science.aar7883.
- S. Thomas, H. Li, R. R. Dasari, A. M. Evans, I. Castano, T. G. Allen, O. G. Reid, G. Rumbles, W. R. Dichtel, N. C. Gianneschi, S. R. Marder, V. Coropceanu and J.-L. Brédas, Design and Synthesis of Two-Dimensional Covalent Organic Frameworks with Four-Arm Cores: Prediction of Remarkable Ambipolar Charge-Transport Properties, Mater. Horiz., 2019, 6, 1868–1876, 10.1039/C9MH00035F.
- W. Liu, X. Luo, Y. Bao, Y. P. Liu, G.-H. Ning, I. Abdelwahab, L. Li, C. T. Nai, Z. G. Hu, D. Zhao, B. Liu, S. Y. Quek and K. P. Loh, A Two-Dimensional Conjugated Aromatic Polymer via C–C Coupling Reaction, Nat. Chem., 2017, 9(6), 563–570, DOI:10.1038/nchem.2696.
- W. Liu, M. Ulaganathan, I. Abdelwahab, X. Luo, Z. Chen, S. J. Rong Tan, X. Wang, Y. Liu, D. Geng, Y. Bao, J. Chen and K. P. Loh, Two-Dimensional Polymer Synthesized via Solid-State Polymerization for High-Performance Supercapacitors, ACS Nano, 2018, 12(1), 852–860, DOI:10.1021/acsnano.7b08354.
- D. L. Pastoetter, S. Xu, M. Borrelli, M. Addicoat, B. P. Biswal, S. Paasch, A. Dianat, H. Thomas, R. Berger, S. Reineke, E. Brunner, G. Cuniberti, M. Richter and X. Feng, Synthesis of Vinylene-Linked Two-Dimensional Conjugated Polymers via the Horner–Wadsworth–Emmons Reaction, Angew. Chem., Int. Ed. DOI:10.1002/anie.202010398.
- S. Jhulki, J. Kim, I.-C. Hwang, G. Haider, J. Park, J. Y. Park, Y. Lee, W. Hwang, A. A. Dar, B. Dhara, S. H. Lee, J. Kim, J. Y. Koo, M. H. Jo, C.-C. Hwang, Y. H. Jung, Y. Park, M. Kataria, Y.-F. Chen, S.-H. Jhi, M.-H. Baik, K. Baek and K. Kim, Solution-Processable, Crystalline π-Conjugated Two-Dimensional Polymers with High Charge Carrier Mobility, Chem, 2020, 6(8), 2035–2045, DOI:10.1016/j.chempr.2020.05.026.
- S. Thomas, H. Li, R. R. Dasari, A. M. Evans, I. Castano, T. G. Allen, O. G. Reid, G. Rumbles, W. R. Dichtel, N. C. Gianneschi, S. R. Marder, V. Coropceanu and J.-L. Brédas, Design and Synthesis of Two-Dimensional Covalent Organic Frameworks with Four-Arm Cores: Prediction of Remarkable Ambipolar Charge-Transport Properties, Mater. Horiz., 2019, 6(9), 1868–1876, 10.1039/C9MH00035F.
- D. Zhou, X. Tan, H. Wu, L. Tian and M. Li, Synthesis of C–C Bonded Two-Dimensional Conjugated Covalent Organic Framework Films by Suzuki Polymerization on a Liquid–Liquid Interface, Angew. Chem., Int. Ed., 2019, 58(5), 1376–1381, DOI:10.1002/anie.201811399.
- R. Matsuoka, R. Sakamoto, K. Hoshiko, S. Sasaki, H. Masunaga, K. Nagashio and H. Nishihara, Crystalline Graphdiyne Nanosheets Produced at a Gas/Liquid or Liquid/Liquid Interface, J. Am. Chem. Soc., 2017, 139(8), 3145–3152, DOI:10.1021/jacs.6b12776.
- E. Tavakoli, A. Kakekhani, S. Kaviani, P. Tan, M. M. Ghaleni, M. A. Zaeem, A. M. Rappe and S. Nejati, In Situ Bottom-up Synthesis of Porphyrin-Based Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(50), 19560–19564, DOI:10.1021/jacs.9b10787.
- V. Nguyen and M. Grünwald, Microscopic Origins of Poor Crystallinity in the Synthesis of Covalent Organic Framework COF-5, J. Am. Chem. Soc., 2018, 140(9), 3306–3311, DOI:10.1021/jacs.7b12529.
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Controlled Growth of Imine-Linked Two-Dimensional Covalent Organic Framework Nanoparticles, Chem. Sci., 2019, 10(13), 3796–3801, 10.1039/C9SC00289H.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, Colloidal Covalent Organic Frameworks, ACS Cent. Sci., 2017, 3(1), 58–65, DOI:10.1021/acscentsci.6b00331.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, Rapid and Efficient Redox Processes within 2D Covalent Organic Framework Thin Films, ACS Nano, 2015, 9(3), 3178–3183, DOI:10.1021/acsnano.5b00184.
- M. Liu, K. Jiang, X. Ding, S. Wang, C. Zhang, J. Liu, Z. Zhan, G. Cheng, B. Li, H. Chen, S. Jin and B. Tan, Controlling Monomer Feeding Rate to Achieve Highly Crystalline Covalent Triazine Frameworks, Adv. Mater., 2019, 31(19), 1807865, DOI:10.1002/adma.201807865.
- J. E. Carpenter and M. Grünwald, Heterogeneous Interactions Promote Crystallization and Spontaneous Resolution of Chiral Molecules, J. Am. Chem. Soc., 2020, 142(24), 10755–10768, DOI:10.1021/jacs.0c02097.
- G. H. V. Bertrand, V. K. Michaelis, T.-C. Ong, R. G. Griffin and M. Dinca, Thiophene-Based Covalent Organic Frameworks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(13), 4923–4928, DOI:10.1073/pnas.1221824110.
- N. Tahir, C. Krishnaraj, K. Leus and P. Van Der Voort, Development of Covalent Triazine Frameworks as Heterogeneous Catalytic Supports, Polymers, 2019, 11(8), 1326, DOI:10.3390/polym11081326.
- X. Liu, H. Li, Y. Zhang, B. Xu, H. S. Xia and Y. Mu, Enhanced Carbon Dioxide Uptake by Metalloporphyrin-Based Microporous Covalent Triazine Framework, Polym. Chem., 2013, 4(8), 2445, 10.1039/c3py00083d.
- J. Artz, S. Mallmann and R. Palkovits, Selective Aerobic Oxidation of HMF to 2,5-Diformylfuran on Covalent Triazine Frameworks-Supported Ru Catalysts, ChemSusChem, 2015, 8(4), 672–679, DOI:10.1002/cssc.201403078.
- A. Bhunia, V. Vasylyeva and C. Janiak, From a Supramolecular Tetranitrile to a Porous Covalent Triazine-Based Framework with High Gas Uptake Capacities, Chem. Commun., 2013, 49(38), 3961, 10.1039/c3cc41382a.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, Highly Stable Nanoporous Covalent Triazine-Based Frameworks with an Adamantane Core for Carbon Dioxide Sorption and Separation, J. Mater. Chem. A, 2013, 1(47), 14990, 10.1039/c3ta13407e.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Sulfone-Containing Covalent Organic Frameworks for Photocatalytic Hydrogen Evolution from Water, Nat. Chem., 2018, 10(12), 1180–1189, DOI:10.1038/s41557-018-0141-5.
- J. C. Sancho-García and J. Cornil, Anchoring the Torsional Potential of Biphenyl at the Ab Initio Level:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The Role of Basis Set versus Correlation Effects, J. Chem. Theory Comput., 2005, 1(4), 581–589, DOI:10.1021/ct0500242.
The Role of Basis Set versus Correlation Effects, J. Chem. Theory Comput., 2005, 1(4), 581–589, DOI:10.1021/ct0500242. - F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Tuning the Stacking Behaviour of a 2D Covalent Organic Framework through Non-Covalent Interactions, Mater. Chem. Front., 2017, 1(7), 1354–1361, 10.1039/C6QM00378H.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, A Tunable Azine Covalent Organic Framework Platform for Visible Light-Induced Hydrogen Generation, Nat. Commun., 2015, 6, 8508, DOI:10.1038/ncomms9508.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, Identification of Prime Factors to Maximize the Photocatalytic Hydrogen Evolution of Covalent Organic Frameworks, J. Am. Chem. Soc., 2020, 142(21), 9752–9762, DOI:10.1021/jacs.0c02633.
- S. Li, Y. Liu, L. Li, C. Liu, J. Li, S. Ashraf, P. Li and B. Wang, Enhanced Proton Conductivity of Imidazole-Doped Thiophene-Based Covalent Organic Frameworks via Subtle Hydrogen Bonding Modulation, ACS Appl. Mater. Interfaces, 2020, 12(20), 22910–22916, DOI:10.1021/acsami.0c04002.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, Diacetylene Functionalized Covalent Organic Framework (COF) for Photocatalytic Hydrogen Generation, J. Am. Chem. Soc., 2018, 140(4), 1423–1427, DOI:10.1021/jacs.7b11255.
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, Isoreticular Two-Dimensional Covalent Organic Frameworks Synthesized by On-Surface Condensation of Diboronic Acids, ACS Nano, 2012, 6(8), 7234–7242, DOI:10.1021/nn302363d.
- J. Dong, Y. Wang, G. Liu, Y. Cheng and D. Zhao, Isoreticular Covalent Organic Frameworks for Hydrocarbon Uptake and Separation: The Important Role of Monomer Planarity, CrystEngComm, 2017, 19(33), 4899–4904, 10.1039/C7CE00344G.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, Mechanochemical Synthesis of Chemically Stable Isoreticular Covalent Organic Frameworks, J. Am. Chem. Soc., 2013, 135(14), 5328–5331, DOI:10.1021/ja4017842.
- A. A. Olajire, Recent Advances in the Synthesis of Covalent Organic Frameworks for CO 2 Capture, J. CO2 Util., 2017, 17, 137–161, DOI:10.1016/j.jcou.2016.12.003.
- S.-L. Cai, K. Zhang, J.-B. Tan, S. Wang, S.-R. Zheng, J. Fan, Y. Yu, W.-G. Zhang and Y. Liu, Rationally Designed 2D Covalent Organic Framework with a Brick-Wall Topology, ACS Macro Lett., 2016, 5(12), 1348–1352, DOI:10.1021/acsmacrolett.6b00805.
- Y. Zhu, S. Wan, Y. Jin and W. Zhang, Desymmetrized Vertex Design for the Synthesis of Covalent Organic Frameworks with Periodically Heterogeneous Pore Structures, J. Am. Chem. Soc., 2015, 137(43), 13772–13775, DOI:10.1021/jacs.5b09487.
- Y. Zhao, W. Dai, Y. Peng, Z. Niu, Q. Sun, C. Shan, H. Yang, G. Verma, L. Wojtas, D. Yuan, Z. Zhang, H. Dong, X. Zhang, B. Zhang, Y. Feng and S. Ma, A Corrole-Based Covalent Organic Framework Featuring Desymmetrized Topology, Angew. Chem., 2020, 132(11), 4384–4389, DOI:10.1002/ange.201915569.
- C. Wang, Y. Wang, R. Ge, X. Song, X. Xing, Q. Jiang, H. Lu, C. Hao, X. Guo, Y. Gao and D. Jiang, A 3D Covalent Organic Framework with Exceptionally High Iodine Capture Capability, Chem. – Eur. J., 2018, 24(3), 585–589, DOI:10.1002/chem.201705405.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, 3D Porous Crystalline Polyimide Covalent Organic Frameworks for Drug Delivery, J. Am. Chem. Soc., 2015, 137(26), 8352–8355, DOI:10.1021/jacs.5b04147.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, Covalent Organic Frameworks as Exceptional Hydrogen Storage Materials, J. Am. Chem. Soc., 2008, 130(35), 11580–11581, DOI:10.1021/ja803247y.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, A Pyrene-Based, Fluorescent Three-Dimensional Covalent Organic Framework, J. Am. Chem. Soc., 2016, 138(10), 3302–3305, DOI:10.1021/jacs.6b00652.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, 3D Porphyrin-Based Covalent Organic Frameworks, J. Am. Chem. Soc., 2017, 139(25), 8705–8709, DOI:10.1021/jacs.7b04141.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, 3D Microporous Base-Functionalized Covalent Organic Frameworks for Size-Selective Catalysis, Angew. Chem., Int. Ed., 2014, 53(11), 2878–2882, DOI:10.1002/anie.201310500.
- J.-T. Yu, Z. Chen, J. Sun, Z.-T. Huang and Q.-Y. Zheng, Cyclotricatechylene Based Porous Crystalline Material: Synthesis and Applications in Gas Storage, J. Mater. Chem., 2012, 22(12), 5369, 10.1039/c2jm15159f.
- L. Xu, S.-Y. Ding, J. Liu, J. Sun, W. Wang and Q.-Y. Zheng, Highly Crystalline Covalent Organic Frameworks from Flexible Building Blocks, Chem. Commun., 2016, 52(25), 4706–4709, 10.1039/C6CC01171C.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, A Crystalline Imine-Linked 3-D Porous Covalent Organic Framework, J. Am. Chem. Soc., 2009, 131(13), 4570–4571, DOI:10.1021/ja8096256.
- Y.-X. Ma, Z.-J. Li, L. Wei, S.-Y. Ding, Y.-B. Zhang and W. Wang, A Dynamic Three-Dimensional Covalent Organic Framework, J. Am. Chem. Soc., 2017, 139(14), 4995–4998, DOI:10.1021/jacs.7b01097.
- D. M. Fischbach, G. Rhoades, C. Espy, F. Goldberg and B. J. Smith, Controlling the Crystalline Structure of Imine-Linked 3D Covalent Organic Frameworks, Chem. Commun., 2019, 55(25), 3594–3597, 10.1039/C8CC09571J.
- X. Chen, N. Huang, J. Gao, H. Xu, F. Xu and D. Jiang, Towards Covalent Organic Frameworks with Predesignable and Aligned Open Docking Sites, Chem. Commun., 2014, 50(46), 6161–6163, 10.1039/C4CC01825G.
- L. M. Salonen, D. D. Medina, E. Carbó-Argibay, M. G. Goesten, L. Mafra, N. Guldris, J. M. Rotter, D. G. Stroppa and C. Rodríguez-Abreu, A Supramolecular Strategy Based on Molecular Dipole Moments for High-Quality Covalent Organic Frameworks, Chem. Commun., 2016, 52(51), 7986–7989, 10.1039/C6CC02170K.
- R. A. Maia, F. L. Oliveira, M. Nazarkovsky and P. M. Esteves, Crystal Engineering of Covalent Organic Frameworks Based on Hydrazine and Hydroxy-1,3,5-Triformylbenzenes, Cryst. Growth Des., 2018, 18(9), 5682–5689, DOI:10.1021/acs.cgd.8b01110.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, Crystalline Dioxin-Linked Covalent Organic Frameworks from Irreversible Reactions, J. Am. Chem. Soc., 2018, 140(40), 12715–12719, DOI:10.1021/jacs.8b08374.
- P. Peng, L. Shi, F. Huo, S. Zhang, C. Mi, Y. Cheng and Z. Xiang, In Situ Charge Exfoliated Soluble Covalent Organic Framework Directly Used for Zn–Air Flow Battery, ACS Nano, 2019, 13(1), 878–884, DOI:10.1021/acsnano.8b08667.
- S. Bureekaew and R. Schmid, Hypothetical 3D-Periodic Covalent Organic Frameworks: Exploring the Possibilities by a First Principles Derived Force Field, CrystEngComm, 2013, 15(8), 1551–1562, 10.1039/C2CE26473K.
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Synthesis of Metallophthalocyanine Covalent Organic Frameworks That Exhibit High Carrier Mobility and Photoconductivity, Angew. Chem., Int. Ed., 2011, 50(6), 1289–1293, DOI:10.1002/anie.201005919.
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Molecular Docking Sites Designed for the Generation of Highly Crystalline Covalent Organic Frameworks, Nat. Chem., 2016, 8(4), 310–316, DOI:10.1038/nchem.2444.
- S. B. Alahakoon, C. M. Thompson, A. X. Nguyen, G. Occhialini, G. T. McCandless and R. A. Smaldone, An Azine-Linked Hexaphenylbenzene Based Covalent Organic Framework, Chem. Commun., 2016, 52(13), 2843–2845, 10.1039/C5CC10408D.
- W.-D. Fessner, B. A. R. C. Murty, J. Wörth, D. Hunkler, H. Fritz, H. Prinzbach, W. D. Roth, P. Schleyer, R. von, A. B. McEwen and W. F. Maier, Dodecahedranes from[1.1.1.1]Pagodanes, Angew. Chem., Int. Ed. Engl., 1987, 26(5), 452–454, DOI:10.1002/anie.198704521.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Cubane: 50 Years Later, Chem. Rev., 2015, 115(14), 6719–6745, DOI:10.1021/cr500523x.
- T. Ma, J. Li, J. Niu, L. Zhang, A. S. Etman, C. Lin, D. Shi, P. Chen, L.-H. Li, X. Du, J. Sun and W. Wang, Observation of Interpenetration Isomerism in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(22), 6763–6766, DOI:10.1021/jacs.8b03169.
- C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. FitzGerald and J. L. C. Rowsell, High Surface Area and Z′ in a Thermally Stable 8-Fold Polycatenated Hydrogen-Bonded Framework, Chem. Commun., 2015, 51(58), 11642–11645, 10.1039/C5CC04219D.
- J. Hu, S. K. Gupta, J. Ozdemir and M. H. Beyzavi, Applications of Dynamic Covalent Chemistry Concept toward Tailored Covalent Organic Framework Nanomaterials: A Review, ACS Appl. Nano Mater., 2020, 3(7), 6239–6269, DOI:10.1021/acsanm.0c01327.
- F. Haase, G. A. Craig, M. Bonneau, K. Sugimoto and S. Furukawa, Pseudo-5-Fold-Symmetrical Ligand Drives Geometric Frustration in Porous Metal–Organic and Hydrogen-Bonded Frameworks, J. Am. Chem. Soc., 2020, 142(32), 13839–13845, DOI:10.1021/jacs.0c04450.
- H. L. Nguyen, C. Gropp and O. M. Yaghi, Reticulating 1D Ribbons into 2D Covalent Organic Frameworks by Imine and Imide Linkages, J. Am. Chem. Soc., 2020, 142(6), 2771–2776, DOI:10.1021/jacs.9b13971.
- Q. Liao, W. Xu, X. Huang, C. Ke, Q. Zhang, K. Xi and J. Xie, Donor-Acceptor Type [4 + 3] Covalent Organic Frameworks: Sub-Stoichiometric Synthesis and Photocatalytic Application, Sci. China Chem., 2020, 63(5), 707–714, DOI:10.1007/s11426-019-9696-3.
- Y. Xiong, Q. Liao, Z. Huang, X. Huang, C. Ke, H. Zhu, C. Dong, H. Wang, K. Xi, P. Zhan, F. Xu and Y. Lu, Ultrahigh Responsivity Photodetectors of 2D Covalent Organic Frameworks Integrated on Graphene, Adv. Mater., 2020, 32(9), 1907242, DOI:10.1002/adma.201907242.
- Y.-P. Mo, X.-H. Liu and D. Wang, Concentration-Directed Polymorphic Surface Covalent Organic Frameworks: Rhombus, Parallelogram, and Kagome, ACS Nano, 2017, 11(11), 11694–11700, DOI:10.1021/acsnano.7b06871.
- Z.-F. Pang, S.-Q. Xu, T.-Y. Zhou, R.-R. Liang, T.-G. Zhan and X. Zhao, Construction of Covalent Organic Frameworks Bearing Three Different Kinds of Pores through the Heterostructural Mixed Linker Strategy, J. Am. Chem. Soc., 2016, 138(14), 4710–4713, DOI:10.1021/jacs.6b01244.
- T.-Y. Zhou, S.-Q. Xu, Q. Wen, Z.-F. Pang and X. Zhao, One-Step Construction of Two Different Kinds of Pores in a 2D Covalent Organic Framework, J. Am. Chem. Soc., 2014, 136(45), 15885–15888, DOI:10.1021/ja5092936.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, Highly Emissive Covalent Organic Frameworks, J. Am. Chem. Soc., 2016, 138(18), 5797–5800, DOI:10.1021/jacs.6b02700.
- M. G. Rabbani, A. K. Sekizkardes, Z. Kahveci, T. E. Reich, R. Ding and H. M. El-Kaderi, A 2D Mesoporous Imine-Linked Covalent Organic Framework for High Pressure Gas Storage Applications, Chem. – Eur. J., 2013, 19(10), 3324–3328, DOI:10.1002/chem.201203753.
- W. Leng, Y. Peng, J. Zhang, H. Lu, X. Feng, R. Ge, B. Dong, B. Wang, X. Hu and Y. Gao, Sophisticated Design of Covalent Organic Frameworks with Controllable Bimetallic Docking for a Cascade Reaction, Chem. – Eur. J., 2016, 22(27), 9087–9091, DOI:10.1002/chem.201601334.
- Y. Wu, H. Xu, X. Chen, J. Gao and D. Jiang, A π-Electronic Covalent Organic Framework Catalyst: π-Walls as Catalytic Beds for Diels–Alder Reactions under Ambient Conditions, Chem. Commun., 2015, 51(50), 10096–10098, 10.1039/C5CC03457D.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, Synchronized Offset Stacking: A Concept for Growing Large-Domain and Highly Crystalline 2D Covalent Organic Frameworks, J. Am. Chem. Soc., 2016, 138(51), 16703–16710, DOI:10.1021/jacs.6b09787.
- Z.-F. Pang, T.-Y. Zhou, R.-R. Liang, Q.-Y. Qi and X. Zhao, Regulating the Topology of 2D Covalent Organic Frameworks by the Rational Introduction of Substituents, Chem. Sci., 2017, 8(5), 3866–3870, 10.1039/C6SC05673C.
- H. R. Abuzeid, A. F. M. EL-Mahdy and S.-W. Kuo, Hydrogen Bonding Induces Dual Porous Types with Microporous and Mesoporous Covalent Organic Frameworks Based on Bicarbazole Units, Microporous Mesoporous Mater., 2020, 300, 110151, DOI:10.1016/j.micromeso.2020.110151.
- R.-R. Liang, F.-Z. Cui, A. Ru-Han, Q.-Y. Qi and X. Zhao, A Study on Constitutional Isomerism in Covalent Organic Frameworks: Controllable Synthesis, Transformation, and Distinct Difference in Properties, CCS Chem., 2020, 2(2), 139–145, DOI:10.31635/ccschem.020.201900094.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z.-H. Chen, H.-S. Xu, W. Tang, K. Leng, G.-H. Ning, J. Wu, Q.-H. Xu, S. Y. Quek, Y. Lu and K. P. Loh, Tuneable near White-Emissive Two-Dimensional Covalent Organic Frameworks, Nat. Commun., 2018, 9, 2335, DOI:10.1038/s41467-018-04769-6.
- Y. Du, D. Calabro, B. Wooler, Q. Li, S. Cundy, P. Kamakoti, D. Colmyer, K. Mao and P. Ravikovitch, Kinetic and Mechanistic Study of COF-1 Phase Change from a Staggered to Eclipsed Model upon Partial Removal of Mesitylene, J. Phys. Chem. C, 2014, 118(1), 399–407, DOI:10.1021/jp4097293.
- A. M. Pütz, M. W. Terban, S. Bette, F. Haase, R. E. Dinnebier and B. V. Lotsch, Total Scattering Reveals the Hidden Stacking Disorder in a 2D Covalent Organic Framework, Chem. Sci., 2020 10.1039/D0SC03048A.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, A 2D Covalent Organic Framework with 4.7-Nm Pores and Insight into Its Interlayer Stacking, J. Am. Chem. Soc., 2011, 133(48), 19416–19421, DOI:10.1021/ja206242v.
- B. T. Koo, W. R. Dichtel and P. Clancy, A Classification Scheme for the Stacking of Two-Dimensional Boronate Ester-Linked Covalent Organic Frameworks, J. Mater. Chem., 2012, 22(34), 17460, 10.1039/c2jm32009f.
- B. Lukose, A. Kuc and T. Heine, The Structure of Layered Covalent-Organic Frameworks, Chem. – Eur. J., 2011, 17(8), 2388–2392, DOI:10.1002/chem.201001290.
- S. B. Alahakoon, S. D. Diwakara, C. M. Thompson and R. A. Smaldone, Supramolecular Design in 2D Covalent Organic Frameworks, Chem. Soc. Rev., 2020, 49(5), 1344–1356, 10.1039/C9CS00884E.
- Y. Fan, Q. Wen, T.-G. Zhan, Q.-Y. Qi, J.-Q. Xu and X. Zhao, A Case Study on the Influence of Substitutes on Interlayer Stacking of 2D Covalent Organic Frameworks, Chem. – Eur. J., 2017, 23(24), 5668–5672, DOI:10.1002/chem.201700915.
- W. A. Braunecker, K. E. Hurst, K. G. Ray, Z. R. Owczarczyk, M. B. Martinez, N. Leick, A. Keuhlen, A. Sellinger and J. C. Johnson, Phenyl/Perfluorophenyl Stacking Interactions Enhance Structural Order in Two-Dimensional Covalent Organic Frameworks, Cryst. Growth Des., 2018, 18(7), 4160–4166, DOI:10.1021/acs.cgd.8b00630.
- S. B. Alahakoon, G. T. McCandless, A. A. K. Karunathilake, C. M. Thompson and R. A. Smaldone, Enhanced Structural Organization in Covalent Organic Frameworks Through Fluorination, Chem. – Eur. J., 2017, 23(18), 4255–4259, DOI:10.1002/chem.201700412.
- S. B. Alahakoon, G. Occhialini, G. T. McCandless, A. A. K. Karunathilake, S. O. Nielsen and R. A. Smaldone, Experimental and Theoretical Insight into the Effect of Fluorine Substituents on the Properties of Azine Linked Covalent Organic Frameworks, CrystEngComm, 2017, 19(33), 4882–4885, 10.1039/C7CE00598A.
- X. Chen, M. Addicoat, S. Irle, A. Nagai and D. Jiang, Control of Crystallinity and Porosity of Covalent Organic Frameworks by Managing Interlayer Interactions Based on Self-Complementary π-Electronic Force, J. Am. Chem. Soc., 2013, 135(2), 546–549, DOI:10.1021/ja3100319.
- H. Xu, J. Gao and D. Jiang, Stable, Crystalline, Porous, Covalent Organic Frameworks as a Platform for Chiral Organocatalysts, Nat. Chem., 2015, 7(11), 905–912, DOI:10.1038/nchem.2352.
- S. B. Alahakoon, K. Tan, H. Pandey, S. D. Diwakara, G. T. McCandless, D. I. Grinffiel, A. Durand-Silva, T. Thonhauser and R. A. Smaldone, 2D-Covalent Organic Frameworks with Interlayer Hydrogen Bonding Oriented through Designed Nonplanarity, J. Am. Chem. Soc., 2020, 142(30), 12987–12994, DOI:10.1021/jacs.0c03409.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Chiral Induction in Covalent Organic Frameworks, Nat. Commun., 2018, 9, 1294, DOI:10.1038/s41467-018-03689-9.
| This journal is © The Royal Society of Chemistry 2020 |