
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of O2 and NO in heme-copper oxidases – mechanistic insights from computational modelling
Margareta R. A.
Blomberg

Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91, Stockholm, Sweden. E-mail: margareta.blomberg@su.se; Tel: +46-816-2616
First published on 2nd October 2020
Abstract
Heme-copper oxidases are transmembrane enzymes involved in aerobic and anaerobic respiration. The largest subgroup contains the cytochrome c oxidases (CcO), which reduce molecular oxygen to water. A significant part of the free energy released in this exergonic process is conserved as an electrochemical gradient across the membrane, via two processes, electrogenic chemistry and proton pumping. A deviant subgroup is the cytochrome c dependent NO reductases (cNOR), which reduce nitric oxide to nitrous oxide and water. This is also an exergonic reaction, but in this case none of the released free energy is conserved. Computational studies applying hybrid density functional theory to cluster models of the bimetallic active sites in the heme-copper oxidases are reviewed. To obtain a reliable description of the reaction mechanisms, energy profiles of the entire catalytic cycles, including the reduction steps have to be constructed. This requires a careful combination of computational results with certain experimental data. Computational studies have elucidated mechanistic details of the chemical parts of the reactions, involving cleavage and formation of covalent bonds, which have not been obtainable from pure experimental investigations. Important insights regarding the mechanisms of energy conservation have also been gained. The computational studies show that the reduction potentials of the active site cofactors in the CcOs are large enough to afford electrogenic chemistry and proton pumping, i.e. efficient energy conservation. These results solve a conflict between different types of experimental data. A mechanism for the proton pumping, involving a specific and crucial role for the active site tyrosine, conserved in all CcOs, is suggested. For the cNORs, the calculations show that the low reduction potentials of the active site cofactors are optimized for fast elimination of the toxic NO molecules. At the same time, the low reduction potentials lead to endergonic reduction steps with high barriers. To prevent even higher barriers, which would lead to a too slow reaction, when the electrochemical gradient across the membrane is present, the chemistry must occur in a non-electrogenic manner. This explains why there is no energy conservation in cNOR.
 Margareta R. A. Blomberg | Margareta R. A. Blomberg received her PhD in the field of quantum chemistry at the Department of Physics, Stockholm University in 1983. After a postdoctoral period at the IBM San Jose research laboratory with Bowen Liu, she returned to Stockholm University. Most of her scientific work has been devoted to the elucidation of the reaction mechanisms of transition-metal systems. In recent years her research has focused on biochemical systems, in particular redox-active metalloenzymes. |
1. Introduction
An important group of enzymes involved in cellular energy conservation is the superfamily of heme-copper oxidases. The superfamily is defined by amino acid sequence homology in a core subunit, and it contains both cytochrome c oxidases (CcO) and the divergent nitric oxide reductases (NOR).1 The CcOs are found in mitochondria and bacteria, and use electrons from soluble cytochrome c to reduce molecular oxygen to water as the last step in the respiratory chain in aerobic organisms according to eqn (1):| O2 + 4H+ + 4ecytc− → 2H2O | (1) |
The NORs are found in denitrifying bacteria and reduce nitric oxide to nitrous oxide as one of the steps in the nitrogen cycle. The best characterized NORs use electrons from soluble cytochrome c (cNOR), with a reduction reaction described in eqn (2):
| 2NO + 2H+ + 2ecytc− → N2O + H2O | (2) |
One of the most intriguing issues in the field of bioenergetics concerns when and how the free energy released in exergonic reactions is conserved, to be used at a later stage by the organisms. The overall exergonicities of the reduction processes described above are determined by the difference in reduction potential (midpoint potential) between the electron donor, soluble cytochrome c, and the electron acceptor, molecular oxygen or nitric oxide. The reduction potential of soluble cytochrome c is 0.25 V, the potential for the reduction of molecular oxygen to water is 0.8 V (per electron) and the reduction of nitric oxide to nitrous oxide and water has a potential of 1.177 V (per electron). This means that both reactions are quite exergonic, eqn (1) by 50.7 kcal mol−1 (2.2 V), and eqn (2) by 42.8 kcal mol−1 (1.854 V). A significant part of the free energy in the oxygen reduction process is conserved as an electrochemical gradient across the mitochondrial or bacterial membrane, in which the CcO enzymes are located.2 The gradient is used by the enzyme ATP-synthase to make ATP, the energy currency of the cell. In contrast, it has been found that none of the free energy is conserved in the reduction of nitric oxide taking place in the bacterial membrane.3–5 An interesting observation is that in some of the heme-copper oxidases there is a cross-reactivity, which means that they can reduce both substrates, O2 and NO.6–11
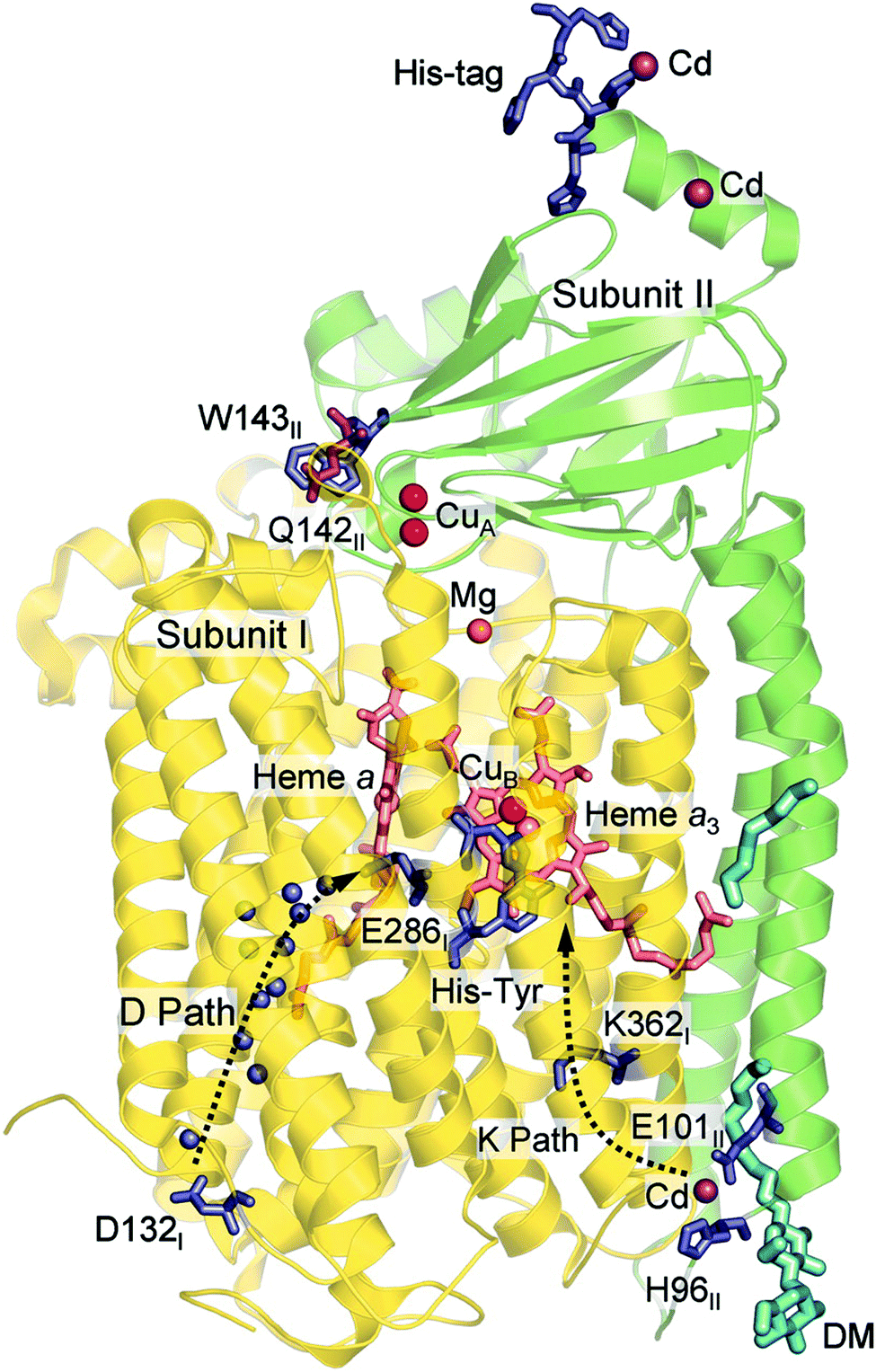
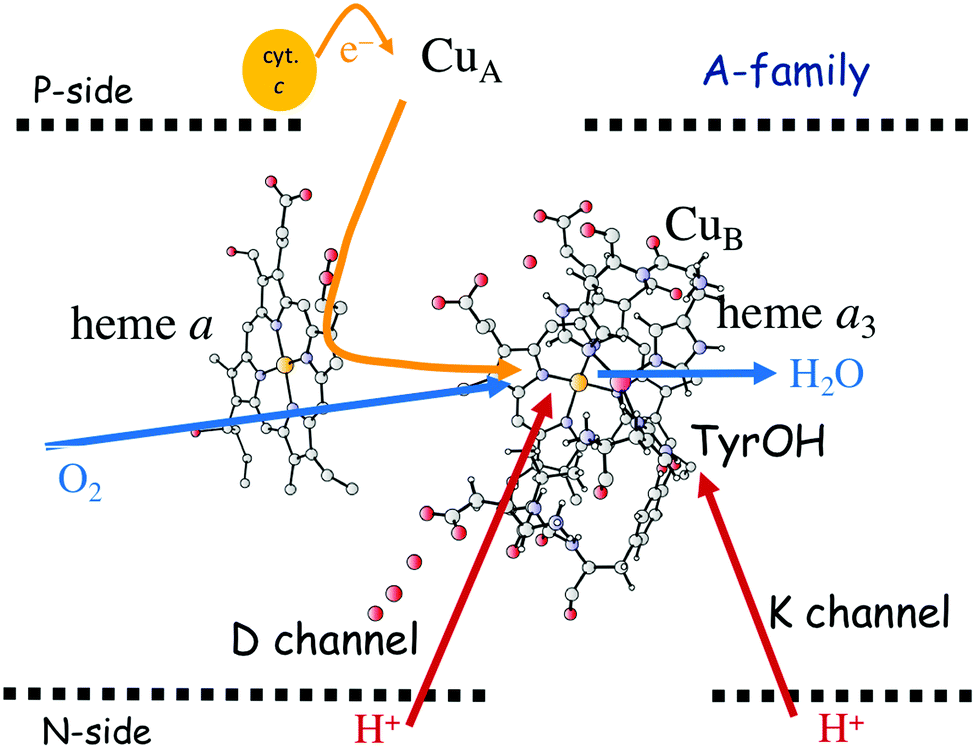
In Fig. 1 an overview is given of the two types of heme-copper oxidases to be discussed in the present review, CcO and cNOR. The active site, where the reduction chemistry takes place, is similar in all heme-copper oxidases, and it is referred to as the binuclear center (BNC). The BNC is composed of two redox-active metal ions: a high-spin heme iron and a non-heme metal. The CcOs have a copper ion as the non-heme metal, CuB, and in the cNORs the copper ion is replaced by a non-heme iron, FeB, see Fig. 1 and 2. The reduced soluble cytochrome c, located on the positive side of the membrane, delivers the electrons to the BNC via a number of cofactors, which are Cu-complexes and/or low-spin heme groups. Regarding the protons needed for the chemistry there is an important difference between the types of heme-copper oxidases. In the CcOs the chemical protons are taken from the opposite side of the membrane, compared to the electrons, the negative side, which means that the chemical process corresponds to a charge separation across the membrane, referred to as an electrogenic reaction. In the cNORs, on the other hand, the protons are taken from the same side of the membrane as the electrons, the positive side, the chemistry is non-electrogenic. In the CcOs there is also a second process that contribute to the charge separation, the chemistry is coupled to a translocation of protons across the entire membrane, referred to as proton pumping.2 Thus, in the CcOs there are two processes contributing to the energy conservation in terms of building up an electrochemical gradient across the membrane, electrogenic chemistry and proton pumping. In the cNORs there is neither electrogenic chemistry nor proton pumping, i.e. no energy conservation, compare Fig. 1. The most essential questions concerning the heme-copper oxidases include how the proton pumping is achieved, i.e. how one electron can trigger the uptake of two protons, but also why there is no energy conservation in the cNOR enzymes.
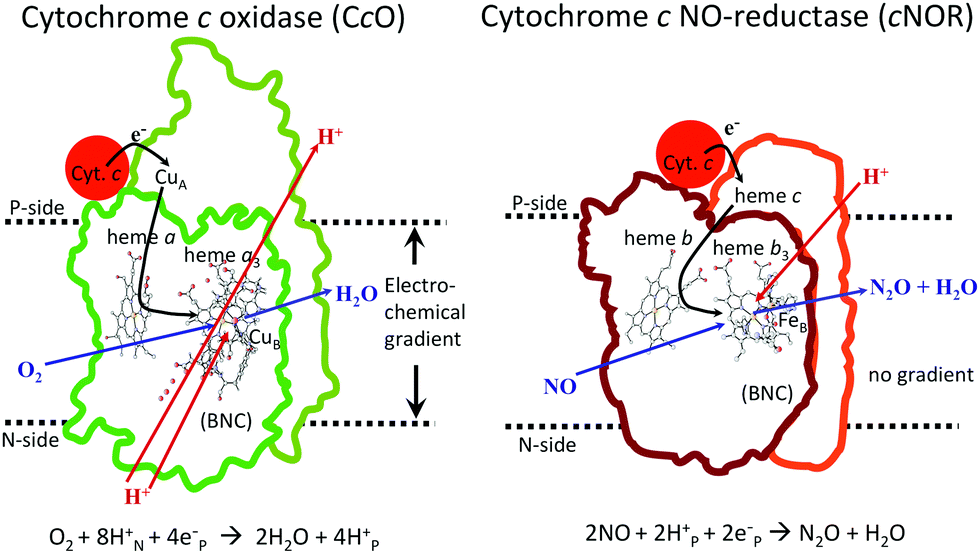 | ||
| Fig. 1 Overview of the overall main reactions in CcO (left, showing the situation in the A family) and cNOR (right), with the binuclear active site (BNC) and the other redox-active cofactors indicated. The electron and proton uptake to the BNC is indicated with arrows, as well as the proton pumping across the membrane in CcO. The figure is reprinted from ref. 12. Copyright (2018), with permission from Elsevier. | ||
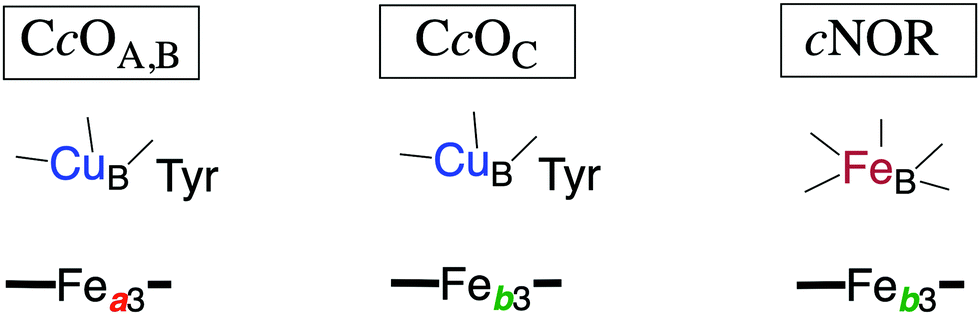 | ||
| Fig. 2 Sketch of the redox-active cofactors in the BNC active sites of the heme-copper oxidase subfamilies, indicating the type of high-spin heme group (a3 or b3) and the type of non-heme metal (CuB or FeB) in each subfamily. The tyrosine in the CcOs is cross-linked to one of the histidine ligands on CuB. The A, B and C families are denoted by the CcO subindices. | ||
A large amount of knowledge about the structure and function of the heme-copper oxidases has been obtained from experimental investigations, both for the CcOs1,13–16 and the cNORs.17–19 However, many questions regarding the details of the reaction mechanisms and the energetics of particular reaction steps are better answered by computational studies, or rather by a combination of experimental and computational data. Quantum mechanical calculations (using hybrid density functional theory) on cluster models of the BNC active site in the heme-copper oxidases are well suited for investigation of the mechanisms for cleavage or formation of covalent bonds, the structure of different intermediates and the energetics of individual steps in the catalytic cycles, but also certain aspects of the mechanisms for the coupling between electron and proton transfer. In this review, results from such studies will be discussed. The purpose is to illustrate how quantum chemical studies can contribute to a better understanding of this family of enzymes.
The discussion about the heme-copper oxidases is divided in five main sections. A short description of the quantum chemical methodology is given in the first section. In the second section different aspects of the oxygen reduction reaction in the CcOs are presented, and in the third section the reduction of nitric oxide in the cNORs is discussed. In the fourth section results for the cross reactions are presented, and finally in the last section the main conclusions are summarized.
2. Methods and models
In the present review mechanistic insights regarding the heme-copper oxidase family of enzymes gained from density functional theory (DFT) cluster calculations are presented. In the cluster approach to enzyme modeling a limited number of atoms are selected from the enzyme to represent the active site, using available crystal structures. All atoms in the selected cluster are treated quantum mechanically at the highest possible level. Cluster models of the heme-copper oxidases contain the two metal complexes with coordinated amino acids constituting the BNC active site, plus occasionally a few nearby amino acids, which means 150–200 atoms. For this size of the cluster model, density functional theory is the only possible choice. In recent years the accuracy of density functional theory methods have developed to a stage where the accuracy in relative energies in most cases is good enough, also for transition metal systems. In particular the introduction of fractions of exact exchange was a major improvement, resulting in so-called hybrid density functional methods.20 As shown in a recent review the hybrid DFT cluster approach is the most common methodology used to study reaction mechanisms of redox-active metalloenzymes.21The cluster approach implies that the protein surrounding the chosen model is not described at an atomistic level. With the large cluster models that are possible to use today, the major effects from the omitted protein are normally of steric and electrostatic character. Those effects are still described in the cluster approach, but in a simplified way. The steric effects are taken into account by fixing the coordinates of some atoms near the truncations to their positions in the crystal structure, a procedure that has been tested carefully for a large number of cases.22 Polarization effects from the surrounding protein on the calculated relative energies, also referred to as solvent effects, are obtained using the self-consistent reaction field method. The dielectric constant is set to 4.0.23 For relative energies between intermediates with the same charge it has been found that the solvent effects usually are small, and therefore the choice of dielectric constant is not very crucial.
Furthermore, zero-point corrections are obtained from the calculated Hessians, using the harmonic approximation. The fixation of certain atomic coordinates during the geometry optimization prevents calculating accurate entropy effects from the Hessians. However, by projecting out the frequencies corresponding to the fixed coordinates it is possible to obtain approximate estimates of the entropy difference between different intermediate structures. In most cases the entropy effects on relative energies are found to be small, and can therefore often be neglected. The exception is for small molecules (like O2 and NO) entering or leaving the active site of the enzyme, which leads to large changes in entropy, and which can be approximated by the translational entropy of the free molecule in gas phase (on the order of 10 kcal mol−1 at room temperature). It is also a general experience that computational studies using the cluster approach yield total free energies that agree better with experiments, as compared to the partitioning into enthalpy and entropy contributions. The reason for this is possibly that the source for this partitioning is not always located in the active site.21,24,25
The DFT functional most used in studies of metalloenzymes is the B3LYP functional by Becke.20 It is a hybrid functional, which in addition to the normal exchange–correlation part of DFT functionals, contains a fraction (20%) of exact exchange. It has been found that a decrease of the fraction of exact exchange to 15% often improves the redox energies,26 generating the B3LYP* notation. A recent study varying the amount of exact exchange indicates that the B3LYP* functional describes the heme-copper oxidases most accurately.27 An important improvement of the DFT methodology made it possible to include also dispersion effects in a simple way for the energies, giving rise to functionals like B3LYP-D3.28
To obtain an energy diagram for the full catalytic cycle of the reduction processes in the heme-copper oxidases, the energetics of the reduction steps has to be calculated, which means that the energy cost of the electron and proton uptake from the donors has to be estimated. Since calculated absolute reduction potentials would not be accurate enough, a procedure has been developed where the overall exergonicity of one catalytic cycle as obtained from the experimental reduction potentials is reproduced.29–32 The energy cost of each reduction step (transfer of one electron from cytochrome c and one proton from the bulk) is set to a value which, in combination with the free energy for the chemistry occurring in the reduction process, reproduces the experimental overall exergonicity. With this procedure the relative energies for each of the reduction steps in the catalytic cycle can be obtained, in combination with the calculated energies of the different intermediates. If the individual energy costs for the electron and the proton are needed, a parameter can be introduced, chosen to fit some other experimental information about the reduction process, apart from the overall energy.
As mentioned above, the accuracy of results from the B3LYP-type of hybrid functionals is high enough for the description of most transition metal containing enzyme active sites. However, there are a few well known cases for which an independent theoretical description of the energetics is not possible, because the accuracy is not high enough. A carefully combination of the calculated results with experimental information is therefore needed. One such case concerns certain properties of heme-groups, which are known not to be accurately reproduced by DFT, e.g. state-splittings.33–35 In line with this experience, it has been found that the reduction potential of the ferric heme comes out significantly too small in the DFT calculations on both CcO and cNOR.36,37 Therefore a correction (about 9 kcal mol−1) has been introduced for the Fe(III)OH to Fe(II)OH2 reduction potential in the more recent studies on both CcO and cNOR reactions, to obtain an overall description that agrees with experimental information. Notably, the calculations reproduce the difference between CcO and cNOR for this reduction potential, which means that the same correction works for both systems. Obviously, when the ferrous heme iron is oxidized, i.e. when the hyponitrite is formed in cNOR and the superoxide in CcO, a corresponding (but opposite) correction has to be applied.
Examples of models of the BNC active site in the heme-copper oxidases used in hybrid DFT cluster calculations are shown in Fig. 3 and 4. Fig. 3 shows a model of the BNC in the A family CcOs based on the X-ray coordinates for the Rhodobacter sphaeroides aa3 CcO,38 and a model of the C family CcOs based on the X-ray coordinates for the Pseudomonas strutzeri cbb3 CcO.39Fig. 4 shows a model of the BNC in cNOR based on the crystal structure from Pseudomonas (Ps) aeruginosa.40 All models include the high-spin heme a3/b3 group, the non-heme metal, CuB or FeB, plus four histidines, one coordinated to the high-spin heme and three coordinated to the non-heme metal. The CcO models include the redox-active tyrosine residue that is cross-linked to one of the histidine ligands on CuB. The cNOR model includes a glutamate coordinated to FeB plus another glutamic acid hydrogen bonding to the glutamate. For the C family CcO model, the negatively charged glutamate, which is hydrogen bonding to the proximal histidine is included. To avoid artificial effects from a negative group on the border of the model, a tryptophan that is also hydrogen bonding to the glutamate, is included as well. The number of substituents kept on the high-spin heme varies between models used in different applications. The models shown in Fig. 3 and 4 include all substituents, except the propionate groups plus the long tail of the farnesyl group on heme a3. The models described so far correspond to basic requirements for modeling the BNC active site in the heme-copper oxidases.
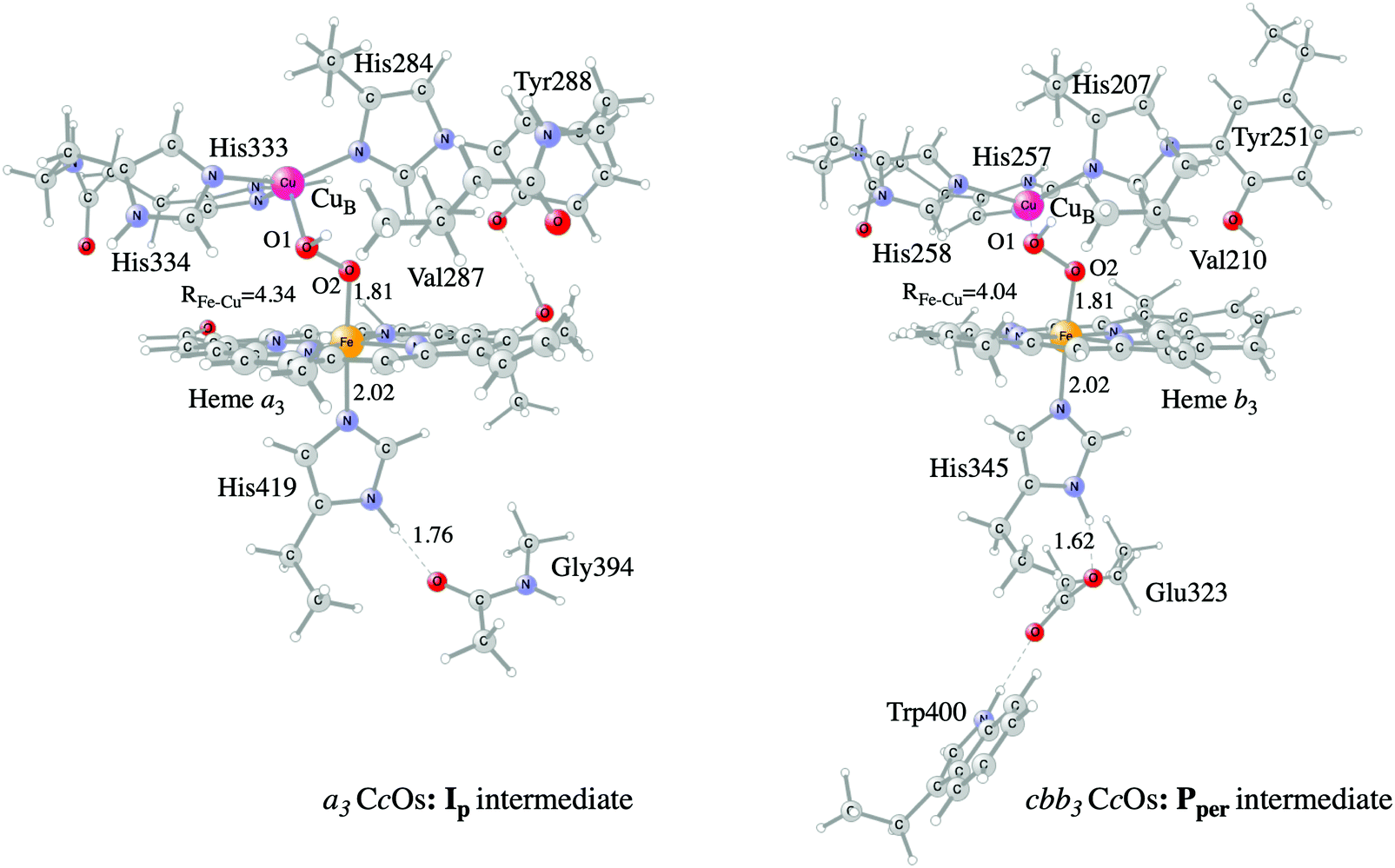 | ||
| Fig. 3 Models of the BNC active sites in a3 (A and B family) and cbb3 (C family) oxidases, respectively. For the a3 model the optimized structure of the Ip state is shown, and for the cbb3 model the optimized structure of the Pper state is shown.50 | ||
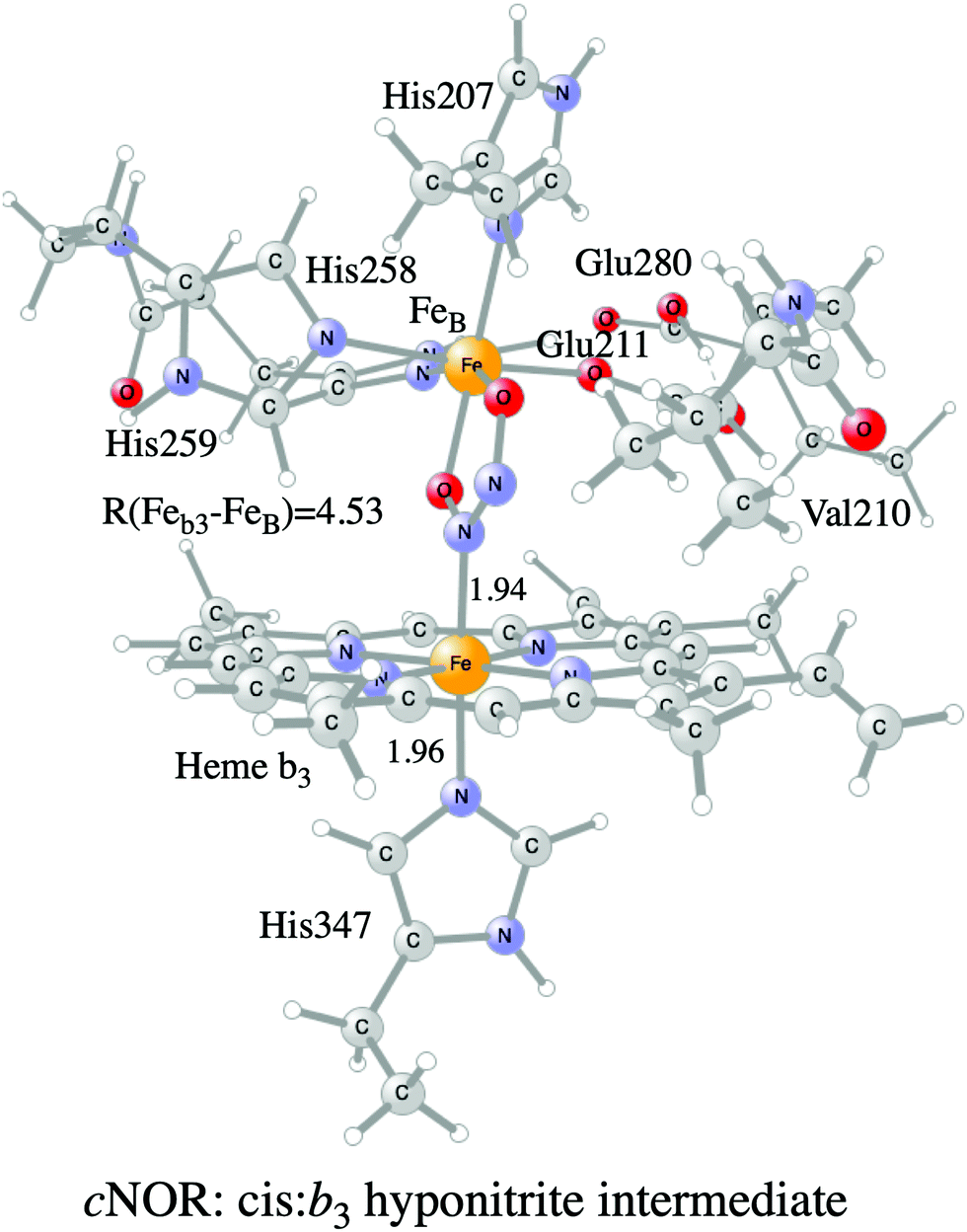 | ||
| Fig. 4 Model of the BNC active site in cNOR showing the cis:b3 hyponitrite intermediate. | ||
In certain applications it has been found that a conserved valine near the active site in the CcO plays a role for the reaction energetics, therefore a valine is included in all models shown in the figures. The coordinates of a few atoms near the truncations are fixed from the crystal structures during optimization of the geometries of the different intermediates. The fixed atoms are typically the alpha carbons on all amino acids, together with the hydrogen atoms replacing the peptide bonds. The proximal histidine in the C family CcO model is an exception. Since the position of this histidine is fixed by its hydrogen bonding to the glutamate, no coordinates are fixed. To make the CcO model for the A family more equivalent to the C family model, a glycine hydrogen bonding to the proximal histidine was included in some cases, as shown in Fig. 3, such that the proximal histidine coordinates could be left unfixed also in the A family model. Finally, if proton transfer within the active site is studied, one or more water molecules must be added. The basic models shown in the figures contain no water molecules (apart from those formed during the reactions), since the number and positions of water molecules within the active site are uncertain and difficult to determine with the methods described here.
The purpose of the present review is to demonstrate that the computational approach described here can produce interesting and reliable information about complicated enzymatic reaction mechanisms. As pointed out in several places, above and below, there are still uncertainties in the obtained results. Clearly, limitations in both the models and the methods used may create inaccuracies in the results. The limitations in the models mainly affect which types of questions that can be attacked. As long as the chemistry occurring in the active site of an enzyme is studied, the cluster approach, omitting large parts of the protein, is expected to give reliable results. With the presently available computational tools, large enough cluster models can be handled, ensuring that all amino acids affecting the energetics of the chemistry can be included in the model. The limitations in the accuracy of the methods used also affect which problems can be approached, mainly meaning that very small energy differences can normally not be expected to be reproduced by the calculations. As discussed above, recent improvements of the DFT methods, mainly inclusion of fractions of exact exchange and dispersion, have increased the accuracy in calculated relative energies, such that differences in calculated barrier heights on the order of 5–10 kcal mol−1 can be considered as quite safe for judging which mechanism is the most likely one. In this context it is important to note that different DFT functionals may give very different relative energies. However, a consistent application of the same functional in a large number of studies, together with a systematic variation of single parameters within the same type of functional, provide a possibility to assess the accuracy of the calculated relative energies. A careful combination with experimental data can then be used to correct for inevitable errors in the calculated results.
3. Oxygen reduction in cytochrome c oxidases
The oxygen reducing heme-copper oxidases can be classified in three main subfamilies, A, B and C. The subfamilies differ in structural details, such as the number and type of proton channels, but also in the number and type of electron transfer cofactors.41–43 The A family includes mitochondrial and bacterial cytochrome c oxidases (CcOs) and the quinol reducing oxidases (such as bo3). The A family CcOs is the largest of the CcO subfamilies, and also the most studied, both experimentally and computationally. The most investigated member of the B-family is the ba3 CcO from Thermus thermophilus. The C-family is represented by the cbb3 oxidases, which are the most distant and least understood CcOs.The BNC active site of all CcOs comprise a high-spin heme group, a copper complex, which is referred to as CuB, and there is also a redox-active tyrosine, which is cross-linked to one of the CuB histidine ligands, see Fig. 2. The reduced soluble cytochrome c is located on the positive side of the membrane, and it delivers the electrons one by one to the BNC via a set of cofactors, which are low-spin heme groups and/or Cu-complexes. For an overview see Fig. 1. The protons needed for the chemistry are transferred via one or two proton channels from bulk water on the negative side of the membrane to the BNC. The A and B family CcOs have a high-spin heme a3 in the BNC (Fig. 2), and the structure of the active site is very similar in these two families, as shown by the different crystal structures. The C family (cbb3 oxidases) has a high-spin heme b3 in the BNC (Fig. 2), and another difference, compared to the A and B families, is that the proximal histidine on the high-spin heme is hydrogen bonding to a negatively charged glutamate. In the A family there are two proton channels, labeled the D and K channel, leading from the negative side of the membrane to the BNC. The D channel ends near the center of the BNC, and the K channel ends at the redox-active tyrosine. The B and the C families have only one proton channel, ending at the redox-active tyrosine, and referred to as the K analogue. The protons to be pumped are all transferred from the negative side to a pump-loading site near the BNC, and there are no well defined pathways from the pump-loading site to the positive side of the membrane. In the A family all protons to be pumped are transferred via the D channel, while one or two of the chemical protons are transferred to the BNC via the K channel.
A number of intermediates appearing during the catalytic cycle of oxygen reduction have been observed, and the general view of the entire reduction process is depicted in Fig. 5. Molecular oxygen binds to the reduced BNC, the O–O bond is cleaved in one of the steps, borrowing electrons from the BNC cofactors, and the rest of the catalytic cycle consists of four proton coupled reduction steps, reducing the BNC cofactors. The exact structures of the different intermediates are not known. Using spectroscopic methods it is easier to determine the positions of the electrons than those of the protons.1 Energy conservation occurs in each reduction step via the electrogenic chemistry, and also via proton pumping. It is generally agreed that the A family CcOs have an efficient energy conservation, which means that four protons are pumped per oxygen molecule, one per electron.14 A remaining issue concerns the efficiency of the energy conservation in the B and the C families of CcOs, for which there are different opinions on the efficiency, corresponding to either two or four protons pumped per oxygen molecule.15,44
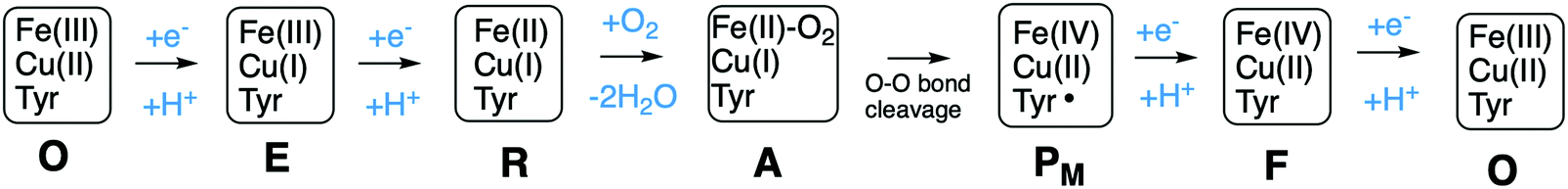 | ||
| Fig. 5 General view of the catalytic cycle in the CcO enzymes, indicating the one-letter notation used for the spectroscopically observed intermediates, together with the proposed oxidation states of the BNC cofactors. | ||
Computational studies, using hybrid density functional cluster calculations, have been performed to investigate a number of different issues regarding the mechanisms for oxygen reduction in different families of CcOs. There are many crystal structures available for different types of CcOs, the first ones for the A family appeared already in 1995, both bovine45 and bacterial,38,46 and later also for the B47 and the C39 families. As an example, the structure of the Rhodobacter sphaeroides aa3 CcO,38 which is used to construct the model used in several computational studies reported here, is shown in Fig. 6. Based on crystal structures, models of the BNC active site have been built, including the high-spin heme, CuB, the cross-linked tyrosine and a varying number of other amino acids in the vicinity. As mentioned above, the BNC active sites are essentially identical for the A and B families, which means that the same model is expected to describe both these families, while the C family needs a different model. The results obtained from such computational studies will be discussed in three different subsections below. In the first one the details for the O–O bond cleavage step are reported. In the next subsection the reduction potentials for the active site cofactors and the corresponding energy profiles for the entire catalytic cycle (Fig. 5) are discussed. In the third subsection some aspects of the proton pumping mechanism are discussed.
 | ||
| Fig. 6 Structure of aa3 CcO from Rhodobacter sphaeroides. The figure is reproduced from ref. 38. Copyright (2006) National Academy of Sciences. | ||
3.1 Mechanism for O–O bond cleavage in CcOs
Molecular oxygen binds to the R state, which is the reduced form of the BNC with a ferrous high-spin heme and a cuprous CuB, forming a new complex labeled A, see Fig. 5. The first intermediate observed after compound A was labled P because it was considered to be a peroxide, until it was finally shown experimentally that the O–O bond is actually cleaved in the P intermediate.48,49 A number of computational studies have investigated the details of the mechanism for the O–O bond cleavage steps, and the results from some of the more recent ones will be described below. The early experiments were performed on the A family of CcOs, but it has generally been assumed that the first step in the oxygen reduction process is the same in all oxidase families.15 The computational results give a different picture, indicating that the O–O bond cleavage in the C family proceeds in a different way.50 Therefore the A and the B families are discussed in the first subsection below and the C family is discussed in a separate subsection.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and one by CuB, going from cuprous to cupric, and it was suggested that the fourth electron is delivered by an amino acid, presumably the tyrosine cross-linked to one of the histidine ligands on CuB.48,51,52 Thus, the O–O bond cleavage step can be described by the following equation:
O and one by CuB, going from cuprous to cupric, and it was suggested that the fourth electron is delivered by an amino acid, presumably the tyrosine cross-linked to one of the histidine ligands on CuB.48,51,52 Thus, the O–O bond cleavage step can be described by the following equation:| Fea3(II)–CuB(I)–TyrOH + O2 → Fea3(IV)O–CuB(II)OH-TyrO˙ |
Based on the experimental data it was originally suggested that the O–O bond cleavage occurs via a hydrogen atom transfer from the cross-linked tyrosine, yielding oxoferryl, CuB(II)-hydroxyl and a tyrosyl radical.48 Early computational studies using hybrid density functional calculations and rather small models indicated that such a hydrogen atom transfer from the tyrosine to the heme coordinated O2 molecule, although thermodynamically feasible, would not be kinetically possible, due to a too high energy barrier.53 Instead it was suggested that a Fea3(III)OOH–CuB(II) type of intermediate had to be formed before the actual O–O bond cleavage could take place.53,54 Those early studies involved an initial activation of an active site water molecule,53 which was not in accordance with experimental data. Somewhat later calculations suggested a more realistic mechanisms, in which the O–O bond cleavage is initiated by proton transfer from the cross-linked tyrosine, via one or two water molecules, to form the type of peroxide intermediate labeled IP in Fig. 7, followed by the actual O–O bond cleavage in a second step.29,55 This type of two-step mechanism with small variations is the one that is still proposed, and it will therefore be discussed more in detail below.
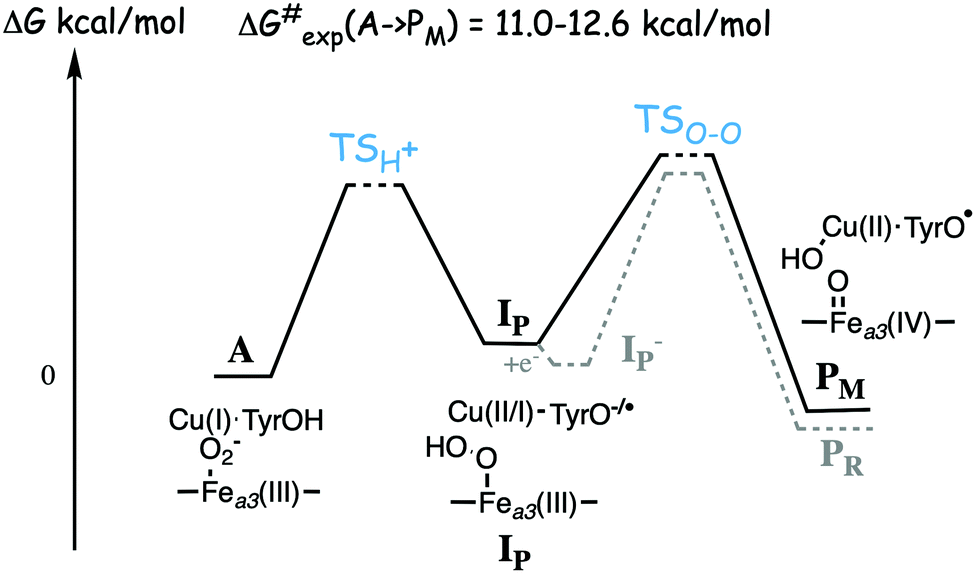 | ||
| Fig. 7 Sketch of the energy profile for the suggested two step mechanism for the O–O bond cleavage in A and B family CcOs. Starting from the observed compound A with molecular oxygen bound to the high-spin heme a3 a proton is transferred from the active site tyrosine, together with an electron, to form the hydroperoxo intermediate labeled IP. The electron is taken either from the tyrosine or from CuB. In the mixed-valence enzyme the O–O bond is cleaved forming the observed PM intermediate. In the fully reduced enzyme an electron is transferred from the low-spin heme forming IP− with a tyrosinate, from which the O–O bond cleavage yields the observed PR intermediate, with a tyrosinate. | ||
Experimentally the binding of molecular oxygen to the reduced active site (R to A step) has been found to be fast, and at least for the A family it is faster than the following O–O bond cleavage step (A to P).56–58 For the A family the R to A step is reversible, indicating a low binding energy of the oxygen molecule, while for the B family the O2 molecule is stronger bound.56,59 The experimental life-time of compound A in the mixed-valence form of the enzyme is found to be 200–300 μs for the A family57 and 18–140 μs for the B family.25,60 These life-times correspond to rate-limiting barriers of 11.0–12.6 kcal mol−1 for the A to PM reaction step, using transition state theory. Although the differences in life-time appear large from an experimental point of view, the corresponding differences in barrier heights of less than two kcal mol−1 is small from a computational point of view, and it would not be meaningful to try to explain those differences on the basis of density functional cluster calculations.
A number of quantum chemical calculations have been performed on the A to PM reaction step, and they all give a qualitative picture that agrees with the sketch in Fig. 7.25,29,50,55,61–63 The A intermediate is often referred to as oxy-ferrous, but according to the calculated electronic structure it should rather be described as a ferric-superoxide complex. Its ground state is singlet, with antiferromagnetic coupling between Fea3(III) in a low-spin state, and the superoxide.21,25,63 In the first step the proton on the cross-linked tyrosine moves, via one or more water molecules to the superoxide, forming a hydroperoxide, Fea3(III)OOH, labeled IP in Fig. 7. The proton transfer is coupled to an electron transfer from the CuB-tyrosine complex, and as indicated in the figure the electronic structure of IP can be either CuB(II) in combination with a tyrosinate, or CuB(I) in combination with a tyrosyl radical, or a mixture of the two. The exact electronic structure depends on the model and the computational level, which indicates that the reduction potentials are rather similar for CuB and the cross-linked tyrosine.25,63 In the second step the O–O bond is cleaved and the PM intermediate is formed, with Fea3(IV)O plus CuB(II)OH–TyrO˙. The oxo-ferryl has a triplet ground state, but the coupling between/to the other two unpaired electrons (one on CuB(II)OH and one on the tyrosyl radical) is weak, why the total spin of the PM intermediate is not well determined by calculations, i.e. the energy difference between the singlet, triplet and quintet states is so small that it is within the uncertainty of the calculations.
As mentioned above, most experiments have been performed on the so-called fully reduced state of CcOs, which means that when O2 binds to the reduced active site, there are two more electrons available, in the low-spin heme and in CuA. In this case the PM intermediate is never observed, instead electron transfer from the low-spin heme into the BNC takes place and an intermediate labeled PR is formed, with a tyrosinate instead of the tyrosyl radical in PM. The rate of PR formation from compound A is slightly higher than that of PM formation, with barriers about 10.2 kcal mol−1 for the B family,25,60 and 11.8 kcal mol−1 for the A family.57 A recent combined experimental and computational study of the fully reduced state of the ba3 oxidase, confirms the picture of the O–O bond cleavage reaction shown in Fig. 7.25 The calculated electron affinities of the A and IP states indicate that the electron transfer from the low-spin heme to the BNC cannot occur until the IP state is formed, and the experiments show that at low temperature the oxidation of the low-spin heme (i.e. electron transfer to the BNC) occurs faster than the formation of the PR intermediate.25 These results indicate a two-step procedure, where an IP− intermediate is formed before the O–O bond cleavage. An exergonic electron transfer from the low-spin heme to IP, forming a lower lying IP− intermediate would also explain the experimental observation that the O–O bond cleavage is faster in the fully reduced enzyme than in the mixed valence enzyme.57,60 The calculations show that the intrinsic O–O bond cleavage barrier is about the same from the IP and IP− intermediates,25 see Fig. 7. These results also indicate that the second step is rate limiting for the entire A to PM reaction step, at least at low temperature. However, this latter conclusion is in contrast to recent computational results indicating that the proton transfer barrier is higher than the actual O–O bond cleavage barrier.62
A recently calculated value of the overall barrier for the bond cleavage step, i.e. the energy of TSO–O relative to A (see Fig. 7), is 16.8 kcal mol−1, using the model shown in Fig. 3.50 This value is thus 4.2–5.8 kcal mol−1 higher than the experimental values of 11.0–12.6 kcal mol−1. In another recent computational study by Schaefer et al., a significantly lower value of 11.6 kcal mol−1 is reported for the same barrier.62 The model used by Schaefer et al. is similar to the one shown Fig. 3, the most important difference is that there is an extra water molecule added to the BNC. Some details of the energetic results obtained in the two studies are given in Table 1.50,62 From the table it can be seen that the water molecule is reported to lower the barrier for the O–O cleavage step from 16.2 to 11.6 kcal mol−1, and that the effect of the water molecule is to lower the relative energy of the IP state (called H in that study) by 4.0 kcal mol−1. The intrinsic barrier from IP is essentially unchanged by the water molecule, which means that the intrinsic barrier from IP to TSO–O is similar in the two studies included in Table 1, about 8 kcal mol−1.50,62 In both the initial O2 bound complex and in the TSO–O the extra water molecule binds only to the cross-linked tyrosine (according to the supporting information in ref. 62), and it was not shown that this is the optimal position of a single water molecule for each structure. It is quite possible that water molecules in the BNC active site are important for the overall O–O bond cleavage barrier, but it should be noted that it is difficult to estimate the effects of water molecules on the energetics in this type of calculations, and artificial effects can easily be obtained. Clearly there are water molecules present, but different crystal structures have a varying number and positions of water molecules in the active site.64
| Model | I P (kcal mol−1) | Δ (kcal mol−1) | TS O–O (kcal mol−1) | PM (kcal mol−1) |
|---|---|---|---|---|
| a Model shown in Fig. 3.50 b Model with an extra water molecule.62 c Model without extra water.62 d Model shown in Fig. 3 plus an extra water molecule.50 e It should be noted that in ref. 50 an empirical correction of −4.4 kcal mol−1 is added to the reported energies for IP and TSO–O to make the a3 results agree with experiments. f In this case the energies are relative to a peroxide intermediate formed from the A state. | ||||
| A and B family | ||||
| a 3 | 8.3 | +8.5 | 16.8 | −6.8 |
| {1F}bf | 3.8 | +7.8 | 11.6 | −11.4 |
| {0}cf | 7.8 | +8.4 | 16.2 | −9.6 |
| C family | ||||
| cbb 3 | 12.4 | +12.8 | 25.2 | −3.7 |
| cbb 3-wde | 11.7 | +13.0 | 24.7 | −3.7 |
For the proton transfer barrier TSH+ (see Fig. 7) a mechanism was suggested by Schaefer et al., where the proton on the cross-linked tyrosine is first moved, via a water molecule, to the oxygen atom closest to Fea3 in the initial complex with O2 bound to the heme, and thereafter moved further to the other oxygen atom, which is closest to CuB, to form the IP intermediate (labeled H in that study).62 The total barrier for the proton transfer was calculated to be 13.7 kcal mol−1, as compared to 11.5 kcal mol−1 for the actual O–O bond cleavage, making the proton transfer rate limiting for the entire A to PM step in Fig. 7.62 This result is also in accordance with the experimental observations of a slowing down of the rate of PM formation in D2O.57 A similar result was obtained in another quantum chemical study using non-hybrid DFT functionals, in which very low barriers were found for both steps in the A to PM reaction, with a rate limiting proton transfer barrier of 7–9 kcal mol−1.63
Finally, the exergonicity of the A to PM step was calculated to be 6.8 kcal mol−1 using the model in Fig. 3,50 and 11.4 kcal mol−1 in the study by Schaefer et al. using a similar model but with an extra water molecule (as reported in supporting information in ref. 62), see Table 1. It is noted that in the Schaefer et al. study the starting structure for the O–O bond cleavage is described as a peroxide,62 and the energy difference between compound A and the peroxide is not discussed. In a computational study by Sharma et al. a value of 26 kcal mol−1 was obtained for the exergonicity of the A to PM step.65 Such a large exergonicity does not seem plausible, considering the efficiency in energy conservation in the A family of oxidases.
An alternative scenario for the C family of oxidases could be that a proton coupled reduction step occurs before the O–O bond cleavage takes place. In this case a hydroperoxide intermediate is formed without using the electron and proton from the tyrosine, given the notation Pper. Such a mechanism was investigated using the model in Fig. 3 (showing the Pper intermediate), and it was estimated that the overall barrier for the O–O bond cleavage may be about 6 kcal mol−1 lower than if the O–O bond cleavage takes place from the A state without preceding reduction.50 Including the same empirical correction, the estimated barrier would be 14.8 kcal mol−1, in reasonable agreement with the experimental observations for the turnover rate, and the rate of oxidation of the low-spin hemes. The initial product of the O–O bond cleavage occurring after a reduction step would be Fea3(IV)O–CuB(II)OH–TyrOH, which would gain some 10 kcal mol−1 by rearranging to Fea3(IV)O–CuB(I)OH2–TyrO˙, the normal form of the F intermediate,50 see further below.
The A to PM energy profile for the cbb3 oxidase has also been investigated in a computational study by Sharma et al.65 In contrast to the results described above, Sharma et al. found the IP intermediate to be about 6 kcal mol−1 lower in cbb3 than in the a3 enzyme.65 In the same study the PM intermediate was found to be 26 kcal mol−1 below compound A for both cbb3 and a3, and no O–O bond cleavage barriers were reported.65
3.2 Reduction potentials and catalytic cycles of CcOs
To understand the mechanisms for energy conservation in the CcO families, including the mechanisms for proton pumping, it is necessary to know the details of the entire catalytic cycle of oxygen reduction, including the reduction steps. A description of the catalytic cycle includes a characterization of each intermediate in the reaction scheme in Fig. 5 in terms of geometric and electronic structures, but also the energetics of the individual reaction steps. The difference in reduction potential between the active site cofactor that is reduced in a specific step and the ultimate electron donor, soluble cytochrome c with a potential of 0.25 V, determines the driving force of each of the four reduction steps. It was found for the A family that the experimental reduction potentials for the BNC cofactors are quite different, two are in the range 0.8 to 1.0 V, and the other two between 0.3 and 0.4 V.66 If these experimental potentials are used to determine the exergonicity of each of the four reduction steps, the exergonicity of the entire catalytic cycle, including also a calculated value for the exergonicity for the O–O bond cleavage step, will be significantly smaller than the expected 50.7 kcal mol−1 (2.2 V), with as much as about 0.6 V missing.13 Furthermore, the electrochemical gradient across the membrane, created by the electrogenic chemistry and proton pumping, has the effect that charge motion against the gradient becomes less exergonic, or even endergonic, and the proton transfer barriers become higher. Thus, each step of electrogenic chemistry and proton pumping during the reduction process contribute to make the reaction less exergonic when the gradient is present. The low experimental reduction potentials for two of the BNC cofactors, 0.3–0.4 V for CuB(II) and Fea3(III),66 would make the corresponding reduction steps significantly endergonic when the electrochemical gradient is present during catalytic turnover. Together with the barrier present for each proton coupled reduction step this would make the overall reaction too slow, in particular if there is proton pumping in all four reduction steps, which is found to be the case in the A family, also at a high gradient.67,68 Thus, there seem to be severe unsolved conflicts between different experimental data.The question is if quantum chemical calculations may shed some light on these puzzles. The proton coupled reduction potentials of the active site cofactors are directly related to the chemistry that take place in the BNC. Starting from the PM state, Fea3(IV)O–CuB(II)OH–TyrO˙, each reduction step, uptake of one electron and one proton, corresponds to the formation of an O–H bond in the active site, finally resulting in a neutral tyrosine and two water molecules. Furthermore, in connection with the formation of each of the O–H bonds, one of the active site cofactors is reduced. This means that an excellent way to estimate the reduction potentials of the different cofactors in the CcO active site is to use the quantum chemical cluster approach to calculate the strength of each of the different O–H bonds. Since the total charge does not change when an O–H bond is formed, satisfactory results can be obtained using cluster models of a feasible size (150–200 atoms). An advantage with the computational approach is that the oxygen reduction chemistry is followed step by step during the catalytic cycle. This means that the structure of the different intermediates used to estimate the reduction potentials is most likely the ones occurring during catalytic turnover, with respect to both the ligands on the metals and the protonation state. In contrast, experimental measurements of the different reduction potentials are made under conditions that differ from the working enzymes during oxygen reduction. Therefore, may the experimental midpoint potentials obtained not correspond to the cofactors in the form they have in the relevant reduction steps during catalytic turnover.
Using the models shown in Fig. 3 the reduction potentials involved in the oxygen reduction process have been calculated for both the a3 and cbb3 type of CcOs. The schematic structures of the optimized intermediates are shown in Fig. 8, and the corresponding calculated proton coupled reduction potentials are summarized in Table 2.69 It is first noted that for three of the cofactor potentials, Fe(IV), Tyr˙ and Fea3(III) (A and B families) the calculations agree very well with the experimental values of 0.8–1.0 V and 0.3–0.4 V, respectively.13,66 The most interesting result in Table 2 is for CuB, with a calculated potential of 1.0 V, as compared to the experimental values of 0.3–0.4 V, which thus can be used to explain the missing 0.6 V in the sum of reaction energies of the catalytic cycle. The calculated CuB potential is furthermore supported by more reliable CCSD(T) calculations on a smaller model of the copper-complex.64 The calculations thus indicate that three of the BNC reduction potentials are in the range 0.8–1.0 V, which seems to solve the problem with the missing energy in the overall energetics and also, at least partly, the problem with the too low exergonicity for proton pumping. The latter issue will be discussed further below.
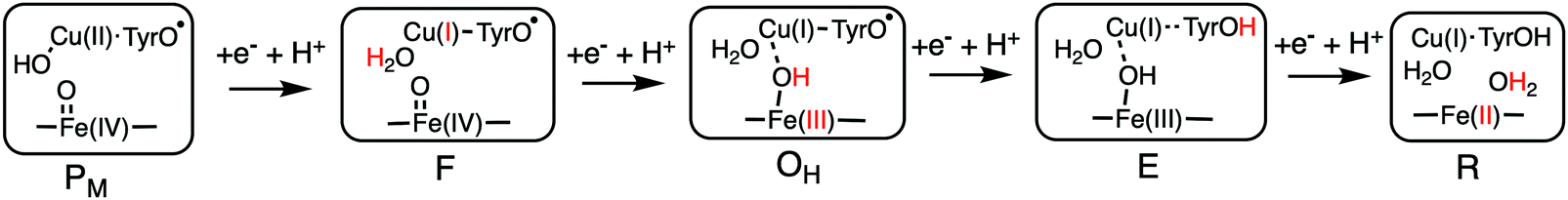 | ||
| Fig. 8 Optimal structures of each intermediate in the catalytic cycle of CcOs used to calculate the proton coupled reduction potentials. The proton and the oxidation state in red indicate the changes that occurred in the preceding reduction step.69 | ||
| Transition in the BNC | Reduction process | Em,7 (V) a3 | Em,7 (V) cbb3 |
|---|---|---|---|
| P M → F | Cu(II) → Cu(I) | 1.0 | 1.0 |
| F → OH | Fe(IV) → Fe(III) | 0.8 | 0.9 |
| O H → E | Tyr˙ → Tyr | 0.8 | 0.8 |
| E → R | Fe(III) → Fe(II) | 0.3 | 0.3 |
The large calculated CuB potential indicates that the real problem is not that the CuB reduction potential is too low during catalytic turnover, but rather how to explain the low potential obtained in experimental measurements. In an attempt to solve the puzzle with the low experimental reduction potentials for the oxidized O state, it was postulated that there exists a “high-energy” metastable state called OH, and that some of the energy from the previous two reduction steps was conserved in this state.70 It was further suggested that the OH state was formed immediately after the F state was reduced, and that after a certain time it would relax to the O state, for which the low CuB(II) and Fe(III) potentials have been observed.71 It was finally suggested that OH state differs in geometrical structure from the relaxed O state, resulting in an elevated midpoint potential of CuB(II). Computational studies were also interpreted to support the suggestion of a structurally metastable “high-energy” OH state, suggesting a water molecule in a non-optimal position.72,73 In contrast, the results for the reduction potentials discussed above, and also a more recent computational investigation of possible structures of the oxidized intermediate show that the geometrically relaxedO state actually has a high reduction potential.69,74 However, relative to the observed oxidized O state with a low experimental CuB reduction potential, the O state involved during catalytic turnover, with a high reduction potential, could still be considered as activated. It should therefore be appropriate to use the OH notation for this state, as in Fig. 8 and Table 2. However, the “deactivation” of the OH state that occurs during relaxation to the O state must be of a more chemical nature, rather than a pure structural relaxation. One suggestion that has been put forward is that there is a slow proton transfer, possibly internal, into the BNC occurring when the turnover electron flow has ceased.74,75 Calculations show that with an extra proton in the active site the proton coupled reduction potential of CuB decreases significantly. The calculations also show that the O–O bond cleavage creates several sites with a high proton affinity, which may be filled via proton transfer from the surrounding protein, slowly enough not to interfere during the catalytic turnover.74,75 This suggestion remains to be confirmed experimentally, which apparently is not an easy task. Several experimental investigations on different A-type of CcOs, designed to prepare the OH state, could not find an increase in the CuB(II) midpoint potential as compared to the resting O state.76–78
The discussion so far has mainly been concerned with the reduction potentials in the A family. Another puzzling experimental result is the very low Feb3(III) reduction potentials observed for the cbb3 type of CcOs.79 For the cbb3 C family there is much less experimental data available for the BNC cofactors, but for the Feb3(III) midpoint potential there are a number of experimental values, varying between −0.12 and 0.27 V.79 It has been suggested that a lower Feb3(III) potential in the C family compared to the Fea3(III) potential in the A and the B families, may be a result of changing the high-spin heme in the BNC from a3 type to b3 type, and comparisons have been made to the cNORs, which also has a heme b3 in the active site (Fig. 2), with a rather low midpoint potential of 0.06 V.79–81 As shown in Table 2, the calculations indicate that there is no big difference between the CcO families, the Fea3(III) and Feb3(III) potentials are about the same.69 Furthermore, as will be discussed below, the same type of calculations reproduces the lower potential in cNOR. It was therefore concluded that the type of non-heme metal in the BNC is more important for high-spin heme Fe(III) potential than the type of heme.69 Again an explanation for a low experimental reduction potential is needed. A possible explanation could again be a proton motion into the BNC, this time into the one electron reduced E state. Calculations show that for this intermediate in the C family, a proton is moving from the high-spin heme b3 proximal histidine to the negatively charged glutamate (see Fig. 3), and that the negative charge thus created on the histidine increases the proton affinity in the BNC.69 Also this extra proton in the BNC would decrease the reduction potential, since the next reduction would not correspond to the full formation of a O–H bond.
With the structures of the intermediates in the catalytic cycle at hand, together with the energetics of all reaction steps it is possible to construct energy profiles for the entire catalytic cycles.36,50 The suggested energy profiles will be discussed separately for each family below.
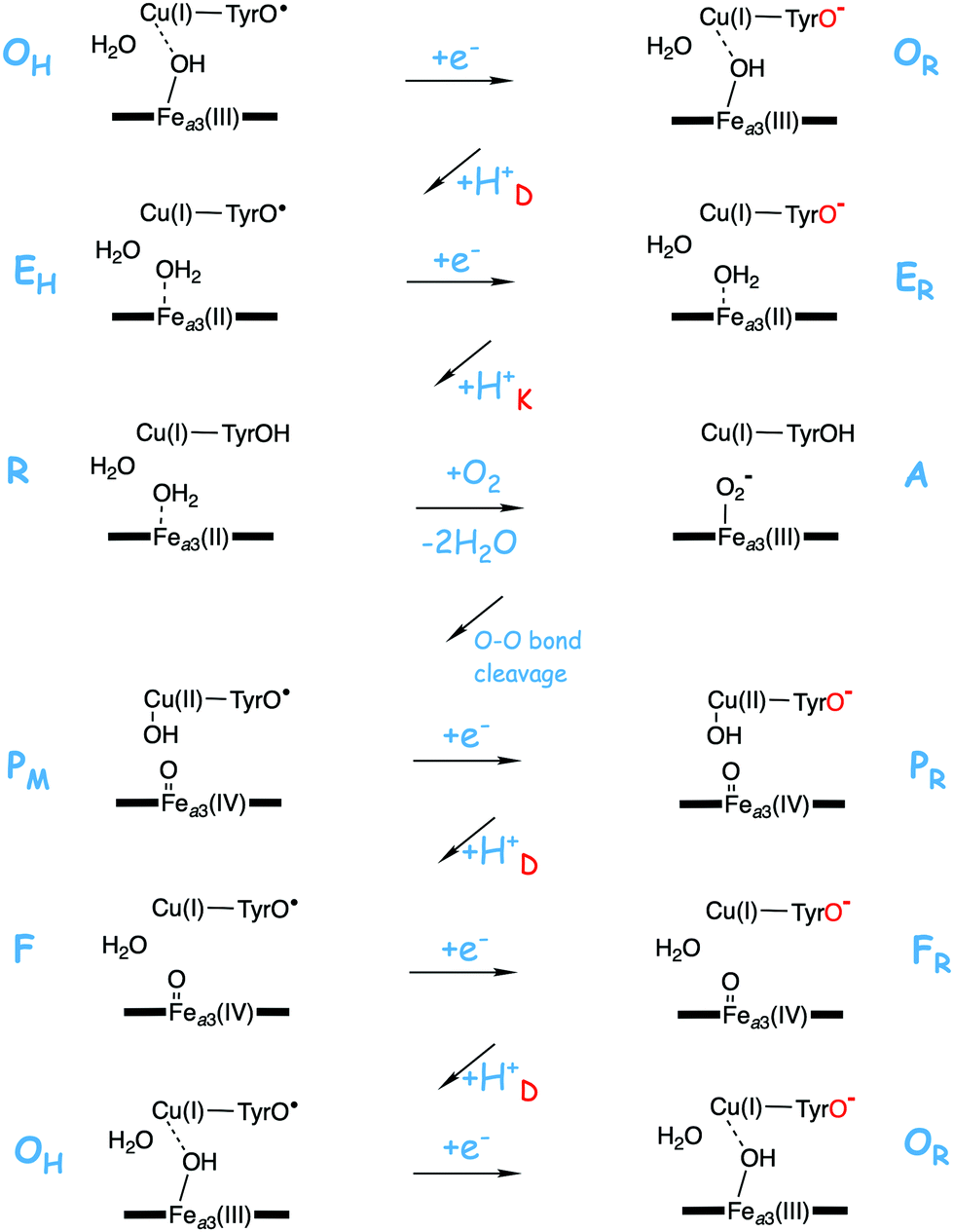 | ||
| Fig. 9 Suggested reaction mechanism during enzyme turnover for the aa3 A family oxidases, showing that the initial electron transfer to the BNC reduces the tyrosyl radical into tyrosinate in every reduction step. The channel suggested to be used for the following proton uptake is indicated with a D or K subindex on the protons.36 The electronic structure of the activated EH state has a fraction of tyrosinate plus Fe(III) mixed in. | ||
 | ||
| Fig. 10 Active site of the aa3 A family oxidases showing the two proton channels from the N-side of the membrane to the active site. The D channel ends near the center of the BNC and the K channel ends at the redox-active tyrosine. The B and the C families have only one proton channel, the K analogue, located in a similar position as the K channel. | ||
Fig. 11 shows an energy profile for the A family, including both the O–O bond cleavage and the reduction steps, following the mechanism described above.64 The energy diagram in Fig. 11 is taken from an earlier study yielding similar but slightly different energetics compared to the reduction potentials discussed above.64 The calculated black curve in Fig. 11 corresponds to the situation without any gradient across the membrane, and each reduction step is shown as a single step, including both electron and proton transfer. Since the low-spin heme a has a reduction potential close to that of the ultimate cytochrome c donor,78 the electron transfer step from cytochrome c to low-spin heme a is not shown separately, but included implicitly in each reduction step, with electron transfer from cytochrome c to the BNC. All barriers associated with the reduction steps are mainly due to proton motion through the protein, and in each reduction step the transfer of both chemical and pumped protons are shown as one single barrier. Experimental rate data is used to estimate the height of these rate limiting barriers. The black curve in Fig. 11 shows that the reaction mechanism in Fig. 9 makes all reduction steps significantly exergonic when there is no gradient present. An estimate of the effects of the gradient on the energy profile can be obtained in a simple way by just adding the endergonicities of moving charges against the gradient to the calculated energy profile. This yields the orange energy profile in Fig. 11, showing that even with a significant gradient present there are no prohibitively high barriers.64 Thus, the mechanism for oxygen reduction depicted in Fig. 9 and 11 allows for efficient energy conservation with proton pumping in all four reduction steps, also at a high gradient, in line with experimental observations.67,68 An important characteristic of this mechanism is that the redox-active tyrosine is unprotonated (possibly a tyrosyl radical) in all intermediates except R and A, which will be further discussed below in connection with the proton pumping mechanism.
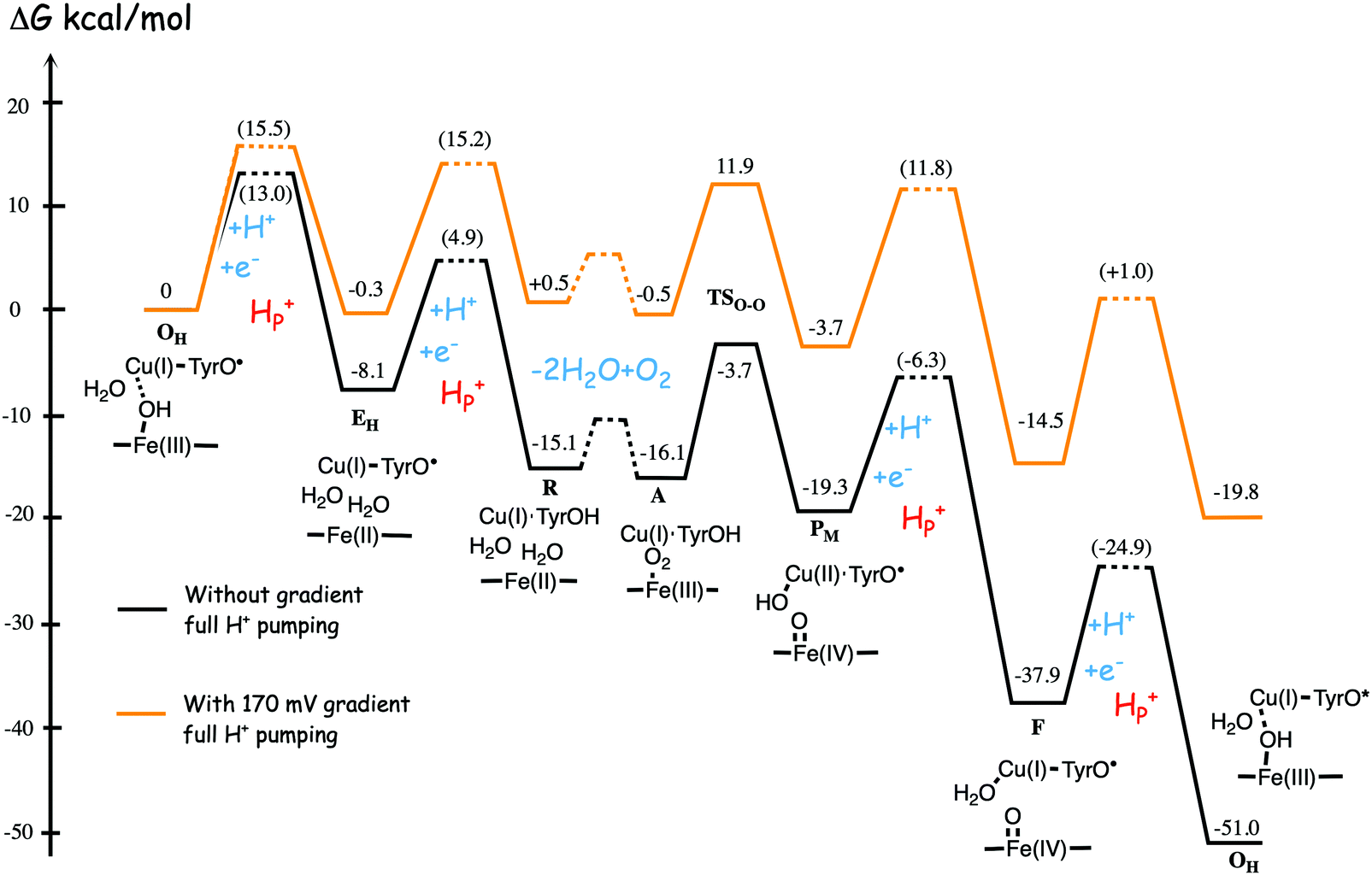 | ||
| Fig. 11 Calculated energy profile for one catalytic cycle in aa3 A family oxidases.64 The black curve corresponds to the situation without gradient. The barriers for proton and electron transfer into the BNC are estimated from experimental data, and the TSO–O barrier is adjusted to experimental rate information. The notation HP+ corresponds to pumped protons. The orange curve include estimated effects on the energetics from a gradient of 170 mV (85% of maximum84) present across the membrane. The figure is reprinted from ref. 64. Copyright (2015), with permission from Elsevier. | ||
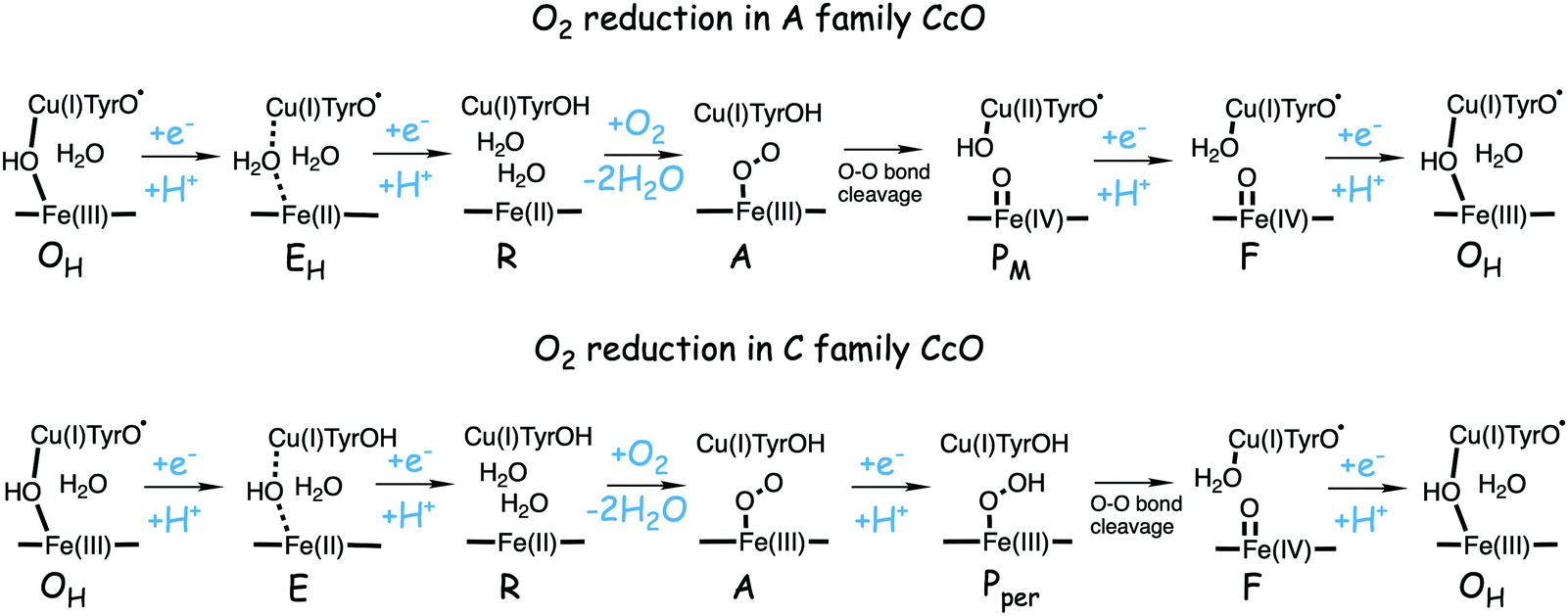 | ||
| Fig. 12 Suggested mechanisms for oxygen reduction in the C family CcOs, compared to the suggested mechanism for the A family.50 | ||
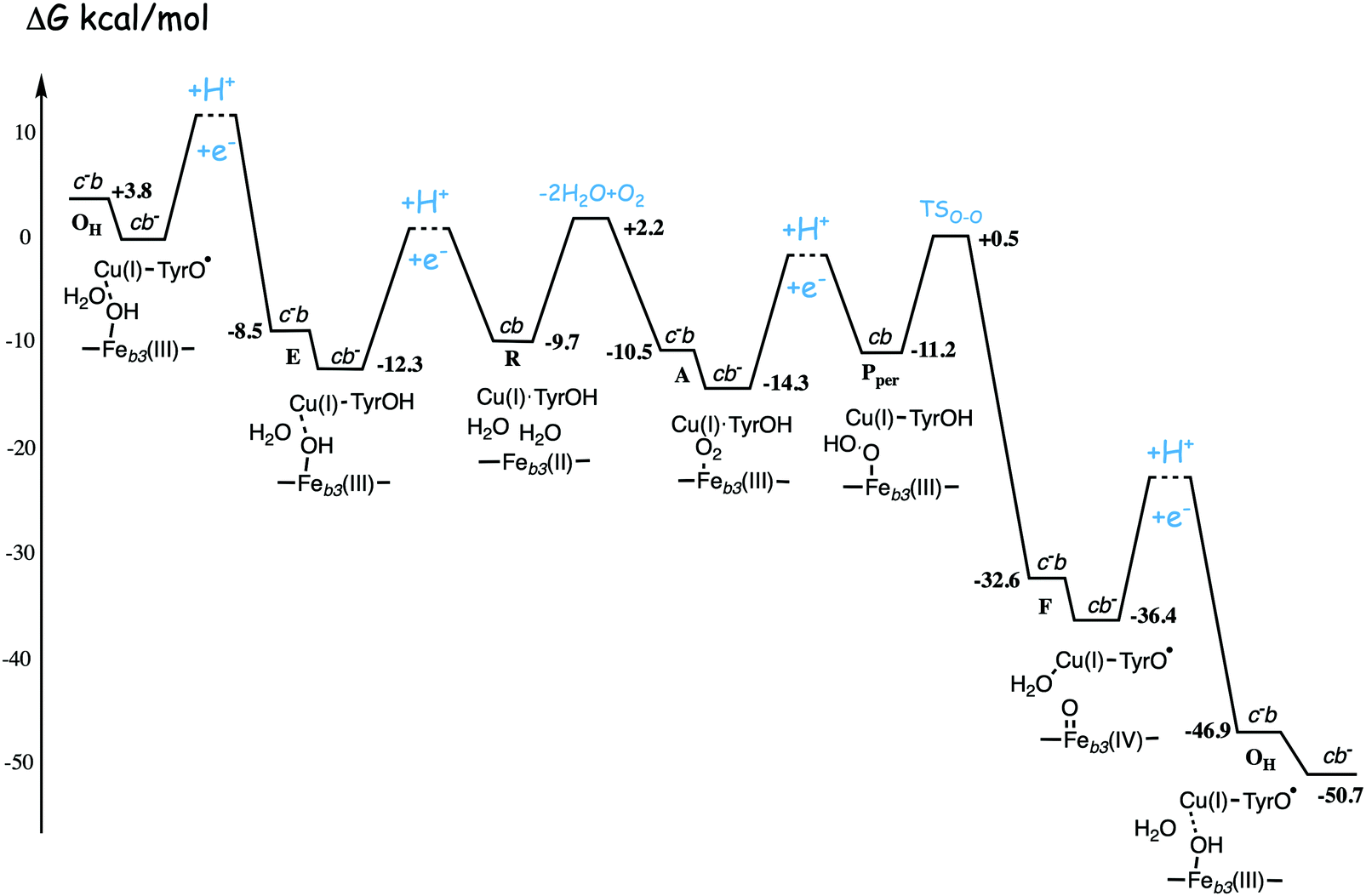 | ||
| Fig. 13 Calculated energy profile for one catalytic cycle in cbb3 C family oxidases.50 The notation c−b means that the ultimate donor, the soluble cytochrome c is reduced. The cb− notation means that the electron has moved to the low-spin heme b, the immediate donor to the BNC. The oxygen binding barrier is obtained from experiment.85 The barriers for the proton and electron transfer steps are only sketched, in accord with general experimental data. The figure is reprinted from ref. 50. Copyright (2020), with permission from Elsevier. | ||
A rather different quantum chemical study of the catalytic cycle in the ba3 B family oxidase has been performed by Noodleman et al.86 In that study a catalytic reaction wheel diagram is constructed using large models of the BNC active site, including parts of the surrounding suggested to be involved in the proton pumping. A few DFT functionals were used to calculate the structures and free energies of at least 14 different intermediates in the catalytic cycle, yielding the energy profiles shown in Fig. 14B. The pump-protons are explicitly included in the calculations, which means that each intermediate included in the energy profiles for the A and C families discussed above, are in Fig. 14 described by several points in the energy profile. The profiles in Fig. 14 show the energetics from the R state (2H in the figure) to the O state (13, 14 and 1 in the figure). The effects of the gradient are included in the energetics of the intermediates, but the kinetics, in terms of transition states, have not been addressed in detail.86 The study is built on an earlier similar study, see Fig. 14A,87 and it is concluded that a new improved reaction path is found by modifying the sequence of proton uptake and proton transfer events, leading to a smoother energy pathway for the catalytic reaction cycle in Fig. 14B.86
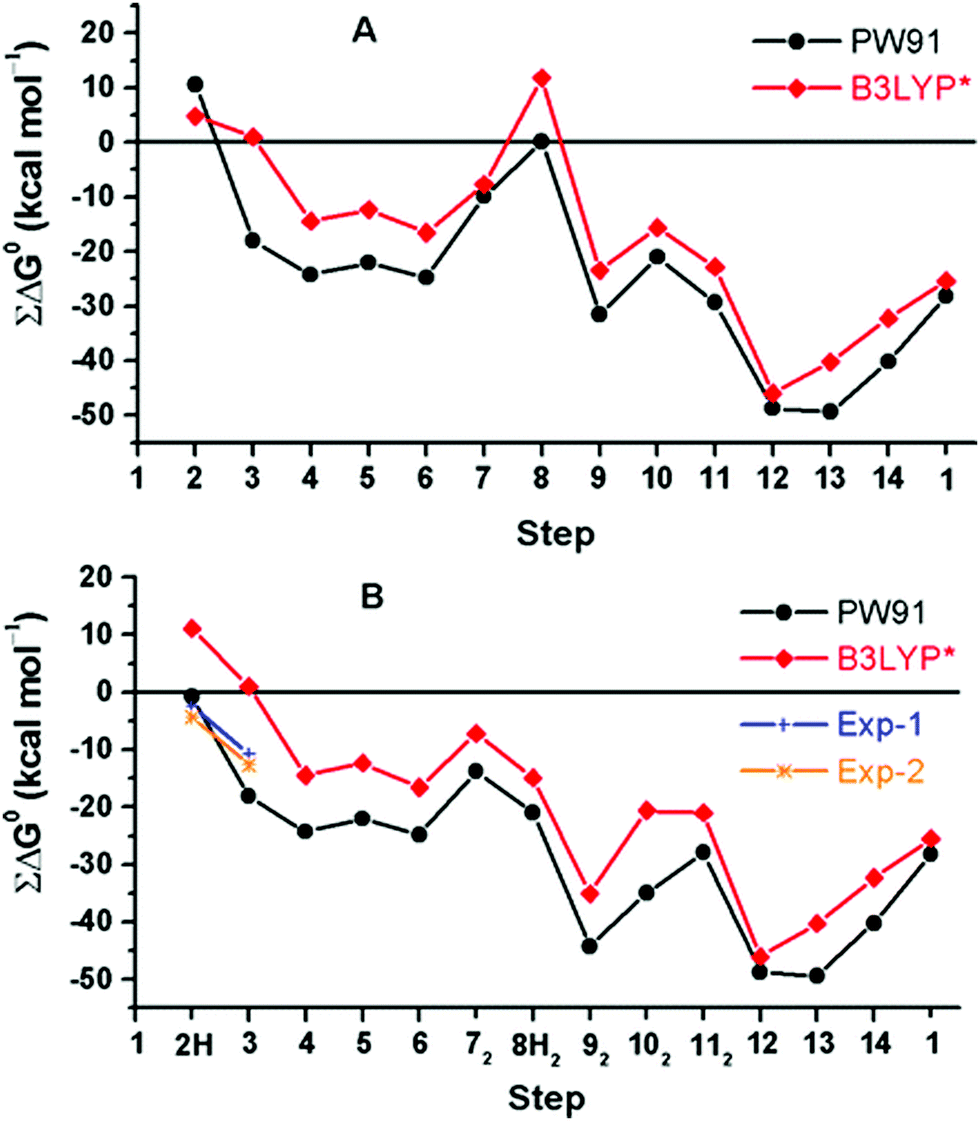 | ||
| Fig. 14 Calculated free energy plot for a detailed reaction mechanism from the R state (2H) to the O state (1) in the ba3 B family using two different DFT functionals.86 The upper curves, A, are from an earlier study,87 and the lower curves, B, are from a more recent study.86 The figure is reprinted from ref. 86 with permission from ACS. | ||
3.3 Energy conservation and proton pumping mechanism in CcOs
In the previous section, suggested mechanisms for the entire catalytic cycle of oxygen reduction in different CcO families were presented. An important prerequisite for efficient energy conservation was discussed, namely the exergonicity of the individual reaction steps. The energy conservation occurs via, electrogenic chemistry and proton pumping. Although considerable knowledge has been achieved about these processes, the detailed mechanisms are still not fully understood.1,14–16,64,88 Both the electrogenic chemistry and the proton pumping imply that charges (mainly protons) have to move against the electrochemical gradient across the membrane in the working enzyme. One important mechanistic aspect pertaining to both the chemical and the pumped protons concerns how to prevent the protons from moving in the wrong direction. Such a gating of the protons must be achieved by flexible barriers outside the BNC active site, which means that it cannot easily be studied by quantum chemical methods, too large and unmanageable models would be needed. A few computational studies have still been performed to evaluate certain aspects of different suggested gating mechanisms, and most of them were reviewed already in 2012.89 The other important mechanistic aspect concerns the driving force for the pumped protons: How can the transfer of one single electron to the BNC active site be coupled in a secure way to the uptake of both the chemical proton and the proton to be pumped? It is generally believed that there exist a so-called pump loading site (PLS) located inside the protein, where the proton to be pumped is temporarily stored. In the most accepted scheme there is an initial coupling between the transfer of an electron into the BNC and the transfer of a proton to the PLS,90,91 see the left panel in Fig. 15. The electron in the BNC, triggers the uptake of the chemical proton, and electrostatic repulsion between the two protons results in the ejection of the proton in the PLS,92,93 see the right panel in Fig. 15. This mechanistic suggestion is in accord with the mechanism for oxygen reduction suggested in Fig. 9 above, with electron transfer to the BNC occurring before proton transfer to the BNC. Each reduction step is initiated by electron transfer into the immediate electron donor to the BNC, the low-spin heme, from soluble cytochrome c. To achieve the first part of the pumping mechanism, it is suggested that the negative charge in the low-spin heme increases the proton affinity of a site located close to both the low-spin heme and the BNC, the PLS, which induce the uptake of a proton to the PLS, from the N-side of the membrane. With a positive charge in the PLS the electron affinity of the BNC increases and the electron moves from the low-spin heme into the BNC (see the left panel in Fig. 15). The exact position of the PLS is not known but it is generally considered to be close to the propionates of the high-spin heme.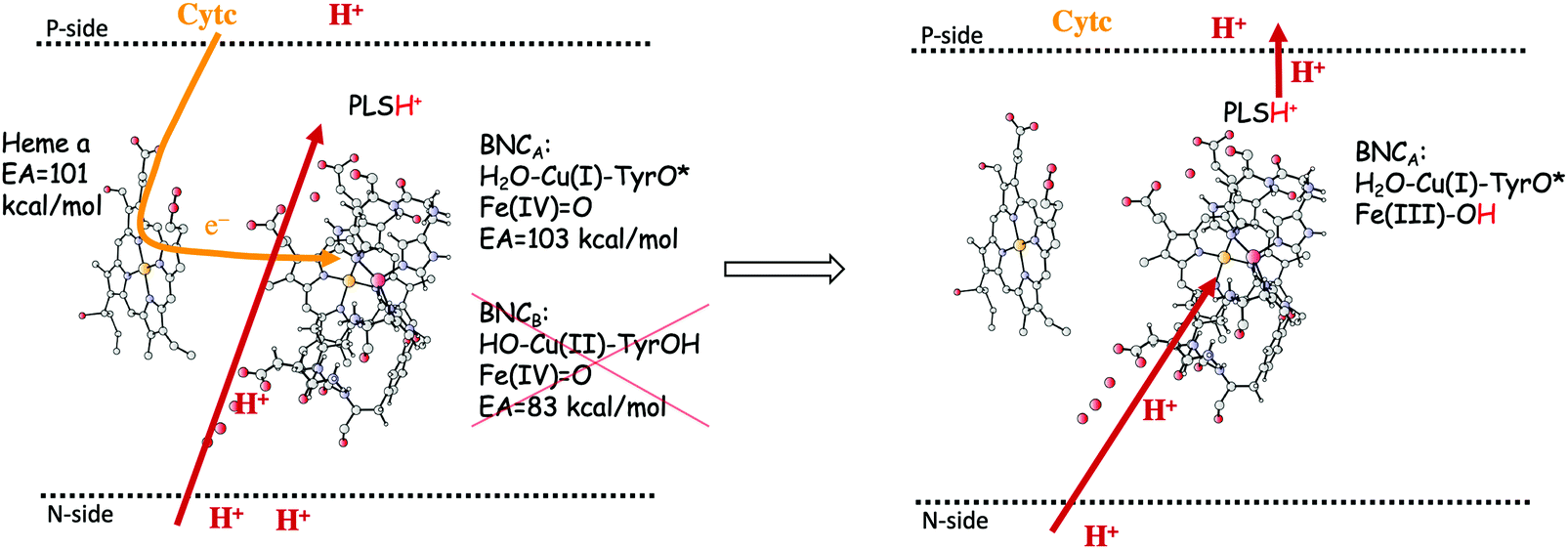 | ||
| Fig. 15 Proton pumping mechanism suggested for aa3 A family CcOs.36 Electron transfer from the soluble cytochrome c, via the low-spin heme a to the BNC is coupled to proton uptake from the N-side to a pump loading site (PLS) close to the active site. This occurs only for the structure of the BNC with a tyrosyl radical, i.e. the one that has the higher electron affinity (left side of the figure). The electron in the BNC triggers the uptake of the chemical proton from the N-side and the proton in the PLS is expelled to the P-side (right side of the figure). | ||
The timing of the two reaction steps shown in the left panel in Fig. 15, the electron transfer into the BNC and the proton transfer into the PLS, is not well determined, but a few crucial points can be made regarding the requirements on the energetics. First, it should not be possible for the electron to move into the BNC before the proton moves into the PLS, because then there would be no need for the pump-proton. This means that the electron affinity of the BNC must not be too high before the PLS is loaded. Second, the structure of the active site in the CcOs implies that the PLS and the BNC electron acceptor cannot be located very close to each other. From this follows that the suggested electrostatic effects between the BNC and the PLS most likely are rather small, on the order of a few kcal mol−1. This means that the electron affinity of the BNC must not be too low before the pump-proton arrives. Third, the combined energetics of the proton transfer into the PLS and the electron transfer into the BNC must be exergonic. Otherwise both the electron and the proton may return back before the chemical proton arrives in the BNC, which would prevent a stable proton pumping mechanism. It can be concluded from these points that the electron affinity of the BNC must not be much lower than that of the low-spin heme, since in such case the chemical proton must be taken up to the BNC (endergonically) before or concerted with the electron transfer into the BNC, and there would be no proton pumping. Such a mechanism has been suggested for the cNOR reduction reaction, where no proton pumping occurs, and also for NO reduction in CcO.12,37,94
The considerations above imply that electron affinities of the BNC at different stages are important for the proton pumping mechanism, and this is where quantum chemical calculations can play a role. As discussed in the previous section, two types of structures with, the same number of electrons and protons, can be constructed for most of the intermediates in the catalytic cycle. One of the transferred chemical protons can be placed either on the redox-active tyrosine, or in the center of the BNC, as exemplified in the left panel in Fig. 15 for the F intermediate. In a study of the A family aa3 oxidase it was found that the two types of structures have quite different affinities for the next electron to be transferred into the BNC,36 as summarized in Table 3. The BNC electron affinity was in all cases found to be significantly higher for the structures with an unprotonated tyrosine as compared to those where the tyrosine is protonated, with a difference in the range 10 to 20 kcal mol−1.36 The high electron affinity is due either to the presence of a tyrosyl radical, or to the presence of an extra proton in the center of the BNC for the cases where the electronic structure is rather to be described as tyrosinate in combination with CuB(II). It was suggested, that only for the intermediates with the higher electron affinity will the electrostatic effect from the proton in the PLS be enough to trigger the electron to move from the low-spin heme into the BNC. In the reaction mechanism suggested on an energy basis for the A family, and shown in Fig. 11, most intermediates have a tyrosyl radical (or unprotonated tyrosine), which means that the succeeding reduction step can be coupled to proton pumping.36 This means that for the aa3 CcOs it is possible to pump one proton in each reduction step, in agreement with experimental information on the aa3 oxidases.70,71 Furthermore, the suggested role for the redox-active tyrosine is in line with its conservation in all types of CcOs.95
| State | aa 3 A family | cbb 3 C family | ||
|---|---|---|---|---|
| Relative energy (kcal mol−1) | EA (kcal mol−1) | Relative energy (kcal mol−1) | EA (kcal mol−1) | |
| A (O2-TyrOH) | 0 | — | 0 | 64.4 |
| P M (TyrO˙) | −5.2 | 102.0 | −3.7 | 92.9 |
| F′ (TyrOH) | +7.6 | 82.6 | +5.9 | 71.9 |
| F (TyrO˙) | 0 | 102.7 | 0 | 92.9 |
| O H′ (TyrOH) | +5.2 | 89.7 | +2.4 | 71.6 |
| O H (TyrO˙) | 0 | 102.6 | 0 | 89.6 |
| E H (TyrO˙) | +4.9 | 99.7 | +7.2 | 90.6 |
| E (TyrOH) | 0 | 86.2 | 0 | 75.7 |
As mentioned above, there is no consensus among researchers about the stoichiometry of the proton pumping in cbb3 C family of oxidases. Experiments on cbb3 from a number of different species led to the conclusion that only two protons are pumped per oxygen molecule in the cbb3 oxidases.15,96 In contrast, it was later claimed that four protons per oxygen molecule are pumped also in the cbb3 oxidases, at least when the enzyme is fully reduced, which was based on a set of experiments on cbb3 oxidase from Rhodobacter sphaeroides.44,97 It is therefore interesting to apply the same type of criteria for proton pumping described above for the A family also to the cbb3 C family, which was done in a recent computational study.50 The calculated electron affinities for the two types of intermediates in the cbb3 C family are also summarized in Table 3. The Table shows that the same difference in electron affinity between the two types of structure is obtained for the cbb3, the lower absolute values in this case are mainly due to the difference in total charge of the two BNC models used in the calculations.50 The type of structures with a tyrosyl radical (unprotonated tyrosine) have a calculated electron affinity that is 15–20 kcal mol−1 larger than those where the tyrosine is protonated. However, according to the mechanism for oxygen reduction in the C family suggested above, see Fig. 12 and 13, only two of the intermediates with a tyrosyl radical are involved during the catalytic cycle. The PM state is not involved due to a too high barrier for O–O bond cleavage from the A intermediate. Instead the first reduction step occurs starting from the A intermediate, which according to Table 3 has a very low electron affinity. Furthermore, since the only proton channel leading from the N-side to the BNC in the C family is the K-analogue, ending at the tyrosine, the EH state can not be involved, instead the reduction leading to the R state has to start from the E state, with a low electron affinity. It was therefore concluded that the computational results indicate that the cbb3 enzymes most likely has a proton pumping stoichiometry that is lower than four per oxygen molecule.50 The reason for this conclusion is that the calculations indicate that less than four of the intermediates in the catalytic cycle has an unprotonated tyrosine, i.e. an intermediate that has an electron affinity high enough to enable a stable proton pumping.
4. NO reduction in cytochrome c dependent NO reductases
Bacterial NO reductases (NOR) catalyse the reduction of nitric oxide (NO) to nitrous oxide (N2O). This is the N–N bond forming step in the denitrification pathway, in which nitrate (NO3−) is transformed to dinitrogen (N2). This is also denoted anaerobic respiration. The product of the NO reduction, the nitrous oxide, which can be released from the cells due to incomplete denitrification is a potent greenhouse gas, and the radical nature of the NO molecule itself makes it a potent cytotoxic that can harm all cell types. It is thus of great interest to thoroughly understand the NO reduction process in the NORs. At least three different subgroups of NORs belonging to the heme-copper oxidases have been identified: the cytochrome c dependent NORs (cNOR), the quinol dependent NORs (qNOR) and a group reported to be dependent on electrons from menaquinol or cytochrome c551 (qCuANOR).98 The cNORs have been extensively studied experimentally, and it seems to be the only type of NOR for which there are computational (quantum chemical) investigations published. Therefore, only the cNORs will be discussed in this review. There are also other types of NO reductases, such as flavodiiron NORs (FNOR) and P450NORs, which do not belong to the heme-copper oxidases.As shown in Fig. 2 the BNC active site in the cNORs has a high-spin heme b3, like the C family CcOs, and a non-heme iron, FeB, instead of CuB in the CcOs. The chemistry occurring in the cNORs is slightly more complicated than the O2 reduction in the CcOs, since it includes both bond formation (N–N) and bond cleavage (N–O). Three types of reaction mechanisms have been suggested for cNORs, which mainly differ in the way that the N–N bond is formed,17,18 see Fig. 16. The first mechanism is labeled cis:b3, in which the first NO molecule binds to the high-spin heme b3 iron. The second NO molecule attacks the bound NO molecule, directly forming the N–N bond. The second mechanism is labeled trans, and the two NO molecules bind more or less simultaneously, one to each of the two iron-ions in the BNC. The N–N bond is formed by coupling the two coordinated NO molecules. The third mechanism is labeled cis:FeB, and in this case both NO molecules are initially bound to FeB. Historically the cis:FeB-mechanism seems to be the first one suggested,99 the trans-mechanism was suggested a few years later,100 and the cis:b3-mechanism is the most recently suggested type of mechanism.4 None of the early experimental studies giving rise to the suggested mechanisms contained any clear information about the structure of the initial intermediates. The first explicit structural information was reported in 2004 based on a time-resolved EPR spectrum. The spectrum was considered to show that an intermediate was initially formed, in which one NO molecule is bound to each of the two metal ions in the BNC.101 This result has been considered to support the suggested trans-mechanism.19,101,102
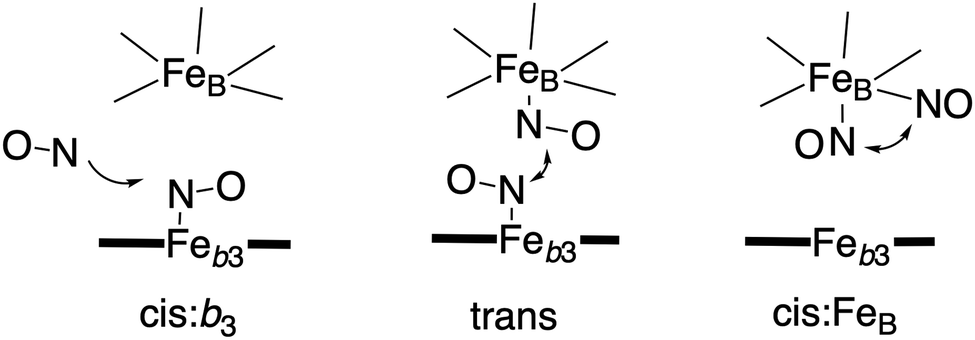 | ||
| Fig. 16 Suggested mechanisms for N–N bond formation in cNOR. | ||
A description of the reaction mechanism for NO reduction in cNOR, summarized in eqn (2), must include both a specification of how the bond formation (N–N) and bond cleavage (N–O) steps occur, and a description of the reduction steps, i.e. the electron and proton uptake to the BNC. As mentioned in the introduction the electrons for the reduction are delivered by reduced cytochrome c located on the N-side of the bacterial membrane, via a low-spin heme b as the immediate donor to the BNC, and the protons are taken up from the same side as the electrons, from bulk water on the N-side, see Fig. 1. The result of this non-electrogenic organization of the electron and proton transfer to the BNC is that no energy is conserved. The lack of energy conservation is surprising, since the reaction is quite exergonic, in fact more exergonic per electron than the oxygen reduction in CcO, for which a significant part of the free energy is conserved, via both electrogenic chemistry and proton pumping.3–5
It has not been possible to determine neither the mechanism for the chemical part of the reaction nor why none of the released free energy is conserved in cNOR on the basis of pure experimental results. On the other hand, computational studies using density functional cluster calculations on models of the BNC, in combination with certain basic experimental information, have suggested a detailed mechanism for NO reduction in cNOR that agrees well with a major part of the experimental information available, and also an explanation for the non-electrogenicity of the reduction part. The report of the cNOR crystal structure in 2010,40 see Fig. 17, was important for the computational studies, since it made it possible to construct reliable active site models for the calculations. The results from the computational studies on the cNOR reaction will be discussed in two subsections below, the first one presents results for the chemical part with bond formation and bond cleavage, and the second one presents results for the entire catalytic cycle and a discussion on the lack of energy conservation.
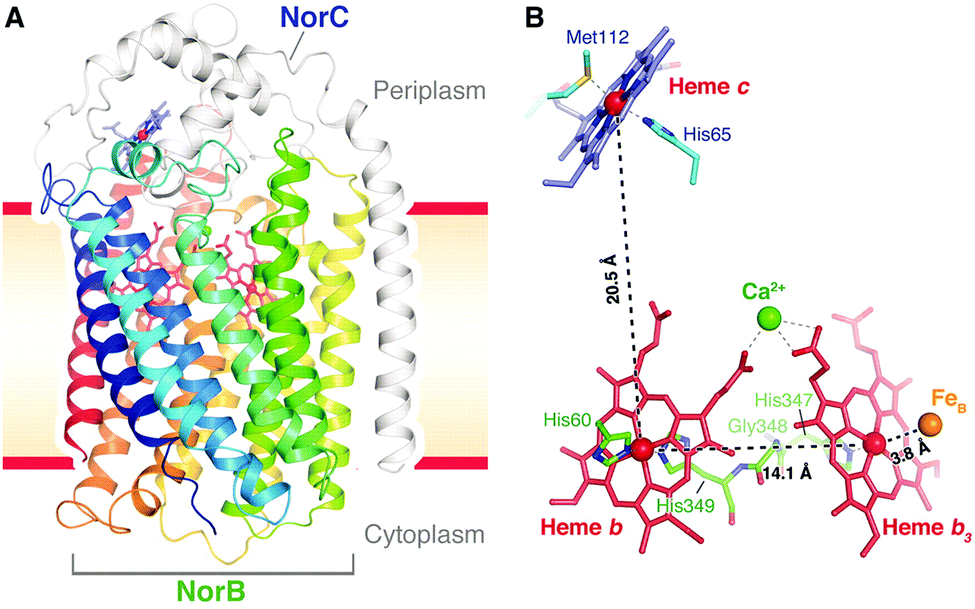 | ||
| Fig. 17 Structure of cNOR from Pseudomonas (Ps) aeruginosa. From ref. 40. Reprinted with permission from AAAS. | ||
4.1 Mechanism for N–N bond formation and N–O bond cleavage in cNORs
A number of computational studies using density functional theory have been performed to investigate the mechanism for the bond formation and bond cleavage in cNOR.32,37,103–107 In fact all these computational studies favor various forms of the cis:b3-mechanism. Before any cNOR crystal structure was available, BNC models for quantum chemical calculations had to rely on information from enzyme models based on the sequence homology between the cNOR and CcO enzymes to construct BNC models, and already a study using such models indicated that a cis:b3-mechanism was most favorable energetically.105 When the crystal structure appeared in 2010, better BNC models could be constructed, and the type of cis:b3-mechanism shown in Fig. 18 was identified.32 The first NO molecule binds to the high-spin heme b3 in the reduced BNC, and this mono nitrosyl complex is taken as the starting point in the energy profile in Fig. 18. The second NO molecule approaches the nitrogen atom of the bound NO molecule forming an N–N bond and dianionic hyponitrite, via electron transfer from both metal ions in the BNC. The barrier for this step is mainly due to the entropy loss of the originally free second NO molecule. The most stable structure for the hyponitrite intermediate is a five-membered ring with the two negatively charged oxygens coordinating to FeB, as shown in Fig. 18. To cleave one N–O bond directly from this intermediate is not possible since it would lead to a too high barrier. Instead a rotation of the hyponitrite has to occur, making one of the oxygen atoms bridging between the two metals, as shown in the figure. The barrier for rotation of the hyponitrite is not very high, on the order of 11–12 kcal mol−1. From the structure with a rotated hyponitrite, one N–O bond can be cleaved with a low barrier, yielding free nitrous oxide, together with the oxidized form of the BNC with a bridging oxo group and two ferric ions.32,37,103 Interestingly, in a later computational study a similar type of hyponitrite rotation has been found to be essential in the reaction mechanism for a model system mimicking a flavindiiron NO reductase.108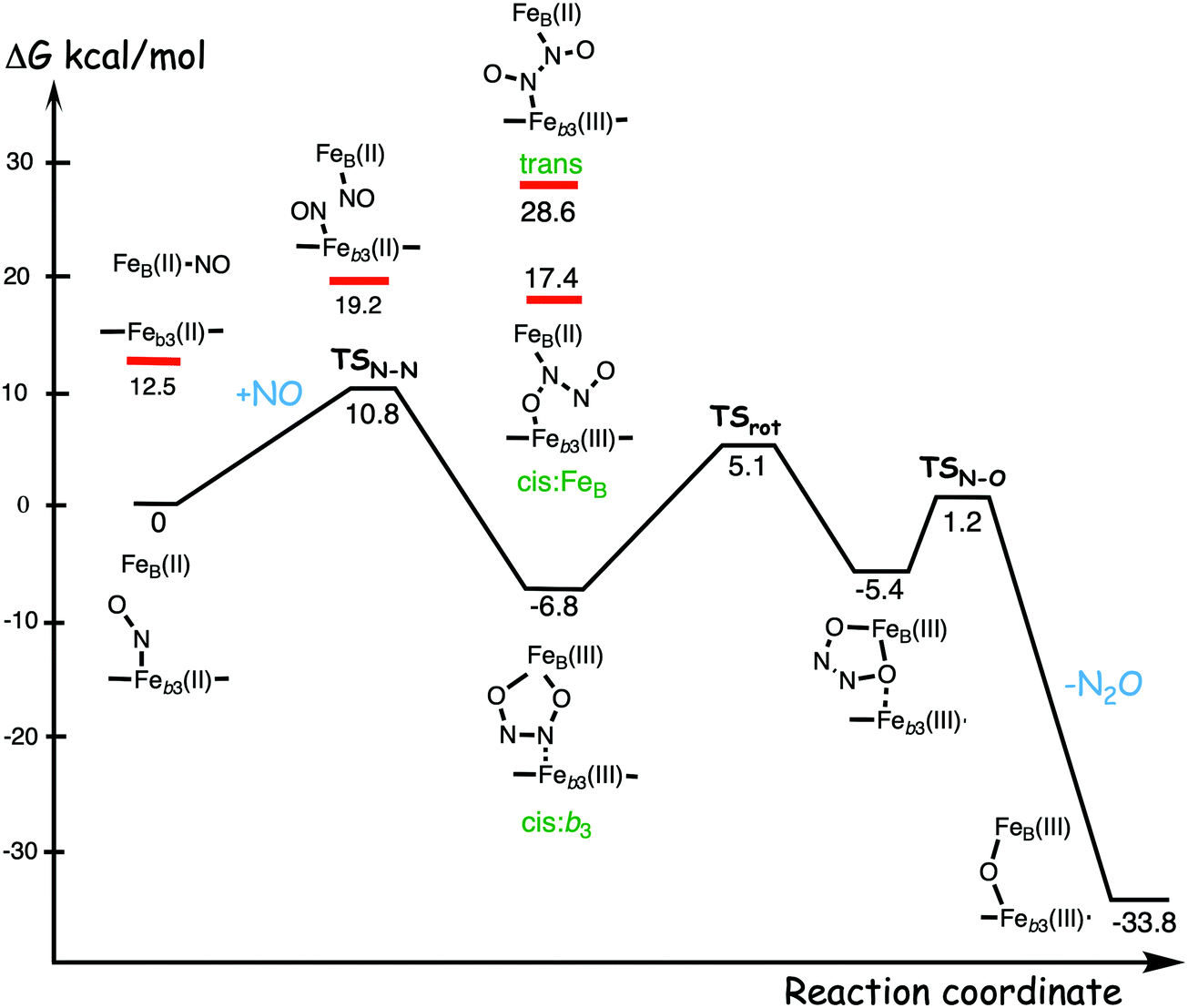 | ||
| Fig. 18 Calculated energy profile for the suggested cis:b3-mechanism for N–N bond formation in cNOR with the mono nitrosyl complex as the starting point. The energetic positions of a few structures supposed to be involved in other suggested mechanisms for N–N bond formation are also shown. The energy values in the graph are obtained from a combination of results from a few different studies.12,103 | ||
Results from a recent experimental study on FeB depleted cNOR can be taken to support the mechanism for N2O formation described in Fig. 18.109 The NO binding to the fully reduced wild-type cNOR was observed to occur in two phases, and the rate of the second phase corresponds to the barrier for binding of the second NO in the energy profile in Fig. 18, i.e. the overall barrier for N2O formation.109 Furthermore, in the FeB depleted variant, there was only one phase of NO binding, illustrating the involvement of FeB in the N–N bond formation.
Two computational studies of QM/MM type (using DFT for the QM part) on cNOR also support a cis:b3-type of mechanism, but argue that the five-membered ring structure of the hyponitrite intermediate must be avoided because it is too stable, and they propose a more open structure with only one oxygen atom coordinating to FeB.37,107 However, it was not demonstrated how the unstable structure should be prevented from falling down to the five-membered ring structure, which is some 20 kcal mol−1 lower in energy than the open structure.103 Results from a combined experimental and computational UV-resonance Raman study on cNOR were interpreted to show the presence of a hyponitrite intermediate.110 The calculations were focused on the vibrational spectroscopy and not on the energetics, why no conclusions could be drawn about the actual structure of the detected hyponitrite intermediate.110 However, since the reported N–N frequency, including isotope effects, agrees very well with the calculated harmonic frequencies of the five-membered ring hyponitrite intermediate, the results may be taken as support for the cis:b3-mechanism.37
In the study where the cis:b3-mechanism shown in Fig. 18 was first suggested, it was also shown that a hyponitrite intermediate corresponding to the trans-mechanism was on the order of 30 kcal mol−1 higher in energy, i.e. too high to be involved in the catalytic reaction, and also that the first NO binds much weaker to FeB than to heme b3.32 Still, the cNOR reaction mechanism continued to be highly debated, and the trans-mechanism was strongly advocated for cNOR, in particular on the basis of experimental results obtained for engineered NOR-models, constructed by insertion of a non-heme iron into myoglobin.111–113 Furthermore, some publications reporting electrochemical measurements on cNOR favoured the cis:FeB-mechanism.114,115 This situation urged a new more comprehensive computational investigation, in which a large number of possible intermediate structures with one or two NO molecules in the cNOR BNC.103 A few of the results from that study are indicated in Fig. 18. First, the suggested precursor for the N–N bond formation in the trans-mechanism, the structure with one NO molecule bound to each iron was shown to be 19.2 kcal mol−1 higher in energy than the mono nitrosyl structure, and also significantly higher than the transition state for N–N bond formation in the cis:b3-mechanism, see Fig. 18. Such an iron-nitrosyl dimer is therefore highly unlikely to be an intermediated in the catalytic reaction in cNOR, and it was suggested that the EPR signals interpreted to show such a structure, and which has been taken as support for the trans-mechanism,101 most likely should be reinterpreted.103 Interestingly, a very recent experimental study utilizing time-resolved spectroscopy rejects the iron-nitrosyl dimer as an intermediate in the NO reduction in cNOR.116 The trans-hyponitrite intermediate was found to be as much as 35.4 kcal mol−1 above the cis:b3-hyponitrite with a five-membered ring structure. In fact, this result is not surprising from a chemical point of view considering the simple hyponitrite molecule itself. The negative charge on the hyponitrite dianion is to a large extent located on the oxygen atoms, and both in an ionic and a covalent picture, bonds to the oxygens give much more stable structures than bonds to the nitrogens.103 Also intermediates related to the cis:FeB-mechanism were found to be too high in energy to be involved in the catalytic reaction mechanism. Binding the single NO molecule to FeB instead of Feb3 was found to be 12.5 kcal mol−1 higher in energy, and the best possible hyponitrite intermediate that could represent the cis:FeB-mechanism was found to be 24.2 kcal mol−1 above the cis:b3-hyponitrite, see Fig. 18.103
In summary, all computational data on the cNOR reaction strongly supports the cis:b3-mechanism and speaks against the two other mechanisms. Interestingly, in a recent combined experimental and computational study on a mono-nuclear heme system it was concluded that the results provide support for the cis:b3-mechanism in cNOR.117 Furthermore, results from a recent experimental study on NOR-models obtained by engineering a zinc ion as the non-heme metal into myoglobin was interpreted to be more in line with the cis:b3-hyponitrite than with the trans-mechanism.118
4.2 The catalytic cycle and the lack of energy conservation in cNORs
To describe reaction mechanisms in redox-active enzymes, such as the heme-copper oxidases, it is not enough to characterize only part of the reaction, as was done in the previous section, but a complete and realistic picture of the entire catalytic cycle has to be obtained. As noted above in the section on methods and models, it is a challenge to obtain accurate results for these transition metal containing systems, and the only possibility is a careful combination of computational and experimental data. In particular this is true for heme-containing systems, since it is well-known that density functional theory, the only choice for the large models needed, has limitations with regard to accuracy in certain cases. A number of energy profiles for the entire catalytic cycle of NO reduction in cNOR have been published, and the main reason for the changes introduced into the newer ones is a better understanding of how to combine computational data with experimental data to give the most reliable picture.12,32,37,103–105 There were also improvements in the pure computational methodology, but with smaller effects on the results.37 A scheme for the suggested mechanism for NO reduction in cNOR is shown in Fig. 19, and a corresponding calculated energy profile for the entire catalytic cycle is shown in Fig. 20. A model of the BNC active site in cNOR used in some of the calculations is shown in Fig. 4.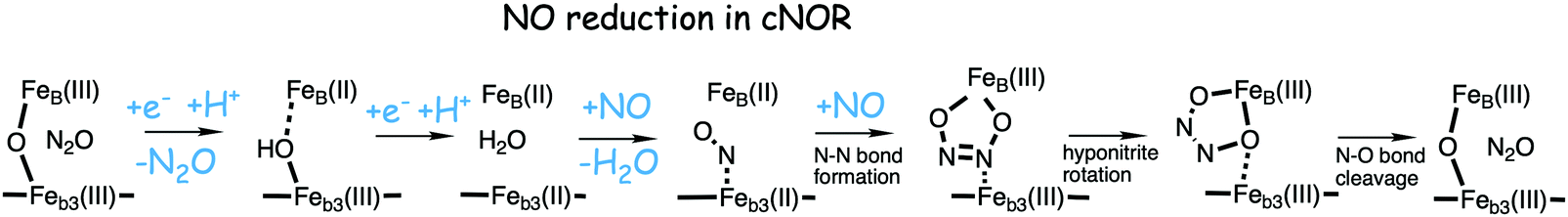 | ||
| Fig. 19 Suggested catalytic cycle for NO reduction in cNOR following the cis:b3-mechanism.32,37,103 | ||
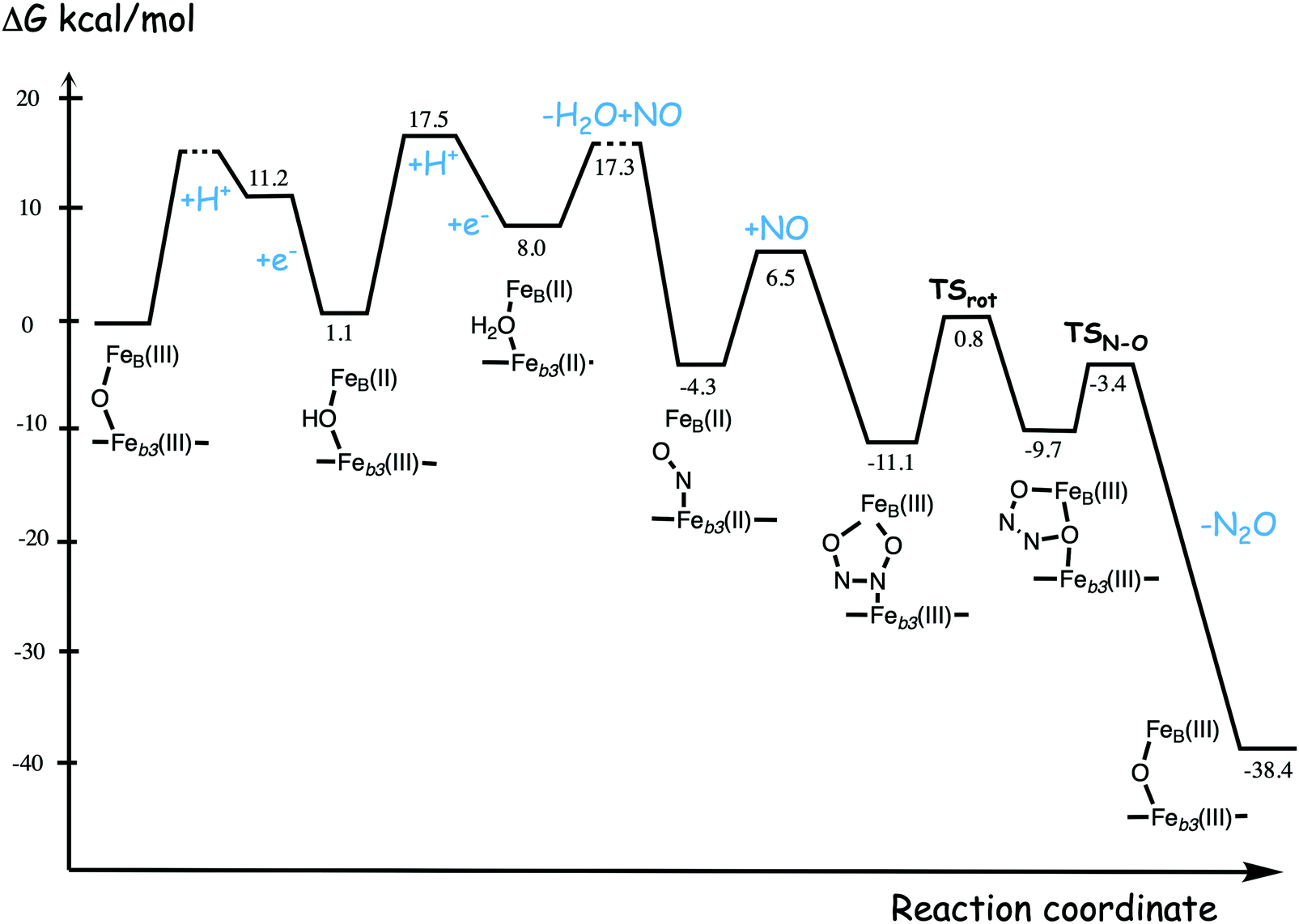 | ||
| Fig. 20 Calculated energy profile for one catalytic cycle of NO reduction in cNOR, with energetics relative to the immediate electron donor low-spin heme b with a reduction potential of 0.345 V. The profile is constructed using results from a few different studies.12,37 | ||
The mechanism shown in Fig. 19 suggests that the NO reduction reaction in cNOR can be divided into two parts, with two proton coupled reduction steps in one part, and the chemistry with bond formation and bond cleavage in the second part. This is in contrast to previous suggestions that proton uptake to the BNC is essential for the N–O bond cleavage,19,101,102 but the calculations indicate that the mechanism shown in Fig. 19 and 20 is the most likely one.32,37,103 Results from flow-flash experiments performed on the four-electron reduced cNOR supply important information for comparisons to the calculated results.119 In those experiments the reaction with NO is initiated from the reduced intermediate FeB(II)–H2O–Feb3(II), which also has two electrons in the low-spin hemes. When the oxidized state FeB(III)–O–Feb3(III) is reached after reaction with two NO molecules, there are two more electrons available to start a new cycle. The rates of oxidation of the low-spin hemes, corresponding to the two reduction steps in the next catalytic cycle are observed.119 The starting point of the mechanism described in Fig. 19, and for the calculated energy profile in Fig. 20, is the resting oxidized state, in which two high-spin ferric irons are antiferromagnetically coupled into a singlet state, via an oxo-bridge, FeB(III)–O–Feb3(III), in accordance with experimental observations for the resting oxidized state in cNOR.120,121 The reaction of the oxidized state starts with two reduction steps, and the calculations show that both steps are initiated by endergonic rate limiting proton transfer followed by exergonic electron transfer.32,37 This result is in agreement with the flow-flash experiments on the four-electron reduced cNOR showing that the oxidations of both low-spin heme cofactors are pH dependent.119 The barriers for the reduction steps can also be determined from the observed rates for the oxidation steps.119 The second reduction step, which is the slowest one, has a barrier estimated to 17.5 kcal mol−1 from the calculations, in good agreement with the experimental rate corresponding to a barrier of 16–17 kcal mol−1.37,119 The organization of the reduction steps in cNOR, with proton uptake to the BNC before (or possibly concerted with) the electron transfer to the BNC, is in contrast to the CcOs, where, in general, the electron transfer to the BNC precedes the proton transfer. The arrangement in the CcOs was found to be important for the proton pumping, and it was found to depend on the involvement of the redox-active tyrosine in the BNC.36
The second part of the reaction mechanism shown in Fig. 19 and 20 starts with the reduced BNC with a water molecule bound, FeB(II)–H2O–Feb3(II). The first step is to replace the water molecule with an NO molecule, yielding the mono nitrosyl complex used as starting point for the discussion in the previous section, and for the energy profile in Fig. 18. In the flow-flash experiments starting from the reduced cNOR it was observed that there is no proton uptake during the first part of the reaction,119 which supports the type of reaction mechanism shown in Fig. 19 and 20, where the reduction steps occur separately from the chemistry. The experiments also show that there is substrate inhibition in cNOR,119 and the calculations suggest that NO may react with the oxidized state to form nitrite (NO2−), which could explain the inhibition.32
The energy profile in Fig. 20 shows that the reduction steps are rate determining for the entire catalytic cycle, with a barrier of about 17 kcal mol−1 for formation of the reduced state, starting from the oxidized state, as compared to about 12 kcal mol−1 for the formation of N2O, starting from the reduced state. It is also noted that the reduction steps are endergonic, which is a result of the low reduction potentials of the BNC cofactors. The experimental midpoint potentials are 0.320 V for FeB(III) and 0.06 V for Feb3(III), which should be compared to 0.345 V for the low-spin heme b, since the energy profile in Fig. 20 is constructed relative to the immediate electron donor.122 Relative to the low-spin heme b, the two reduction steps should be endergonic by 0.6 kcal mol−1 and 6.6 kcal mol−1, respectively, which is close to the computational results of 1.1 kcal mol−1 and 6.9 kcal mol−1, respectively.37 As discussed in the methods section, a correction is introduced for the Feb3(III) potential to obtain agreement with experiment for the overall energetics, and the same correction, but in opposite direction, is applied when Feb3(II) is oxidized.37
The energy profile in Fig. 20, constructed on the basis of the computational results, corresponds to the situation without any gradient across the bacterial membrane in which the cNOR enzymes are located. Since other processes creates an electrochemical gradient across the membrane, also the cNOR enzymes have to work with a gradient present during catalytic turnover. This means that if the reaction occurred in an electrogenic way, the two reduction steps, endergonic already without gradient, would become even more endergonic. Furthermore, the proton transfer barriers connected with the reduction steps are high already without the gradient, and if the protons would have to move against the gradient these barriers would become even higher. Thus, both the increased endergonicity and the raised intrinsic barriers would contribute to increase the overall rate limiting barriers for the entire reduction process, if the reaction was electrogenic. The conclusion is therefore that the low reduction potentials of the BNC makes it necessary for the reduction process to occur in a non-electrogenic way, since with an electrogenic reaction the rereduction, and thereby the entire cycle, would become too slow.104 On the other hand, the exergonicity and most likely also the rate of the chemical part of the reaction, increase by the low reduction potentials, which secures a fast quenching of the toxic NO molecules. These results indicate that the reduction potentials of the cNOR BNC cofactors are quite optimal, making the entire catalytic cycle as fast as possible. If the reduction potentials were higher, the reduction steps would be faster, but the driving force for the chemical steps would become lower, which most likely would make these steps slower. If the reduction potentials of the BNC were even lower, this would probably increase the rate of the chemical steps, but it would make the reduction steps too slow.
In contrast, the situation is quite different for the oxygen reduction reaction in the CcOs, in particular for the A family, where not only the chemistry is electrogenic, there is also proton pumping in all four reduction steps. As shown in Fig. 11, the much larger reduction potentials of the BNC cofactors in CcO make all four reduction steps significantly exergonic without gradient, which means that the energy cost of charge motion against the gradient due to both electrogenic chemistry and proton pumping can be afforded.
5. Cross-reactivity in heme-copper oxidases
Some of the heme-copper oxidases has a so-called cross-reactivity, which means that both O2 and NO can be used as substrate in the reduction reaction, which offers a possibility for further mechanistic insights. Comparative computational studies where different members of the heme-copper oxidase family react with both substrates are informative, and they have the potential to elucidate which enzyme properties are crucial for efficient cellular energy conservation. The experimental information about these cross-reactivities is essential for the evaluation of the computational results. In the case of the cNORs, they have been found to be capable of oxygen reduction,7–9 and in the case of the CcOs, it has been found that some CcOs are capable of NO-reduction, while others are not. For the largest CcO subfamily, the A family, enzymes from several different species have been found not to reduce NO with a measurable rate,123 while at least one member has been shown to reduce NO at a slow rate.10 At least one member of each of the B and the C families have also been shown to slowly reduce NO.6,10,11 Computational results for the cross-reactivity are discussed in two subsections below, the first one dealing with oxygen reduction in cNOR, and the second one with NO reduction in the different CcO families.5.1 O2 reduction in cNOR
One computational study using the DFT cluster approach has been performed on the reduction of molecular oxygen in cNOR.94 The purpose of the study was to explain the observations obtained in a flow-flash experiment on the fully reduced cNOR9 and to elucidate the mechanism for oxygen reduction in cNOR. Furthermore, comparing the O2 reduction mechanisms in the two enzymes, CcO and cNOR, allows for a better understanding of the mechanisms for cellular energy conservation.The energy profile for one catalytic cycle of O2 reduction in cNOR was constructed from a combination of computational and experimental results, see Fig. 21.94 To simplify the comparison to the same reaction in CcO, this energy profile is calculated relative to the ultimate donor cytochrome c, which is in contrast to the energy profile for NO reduction in cNOR discussed above. The energy profile in Fig. 21 agrees well with the experimental observations. For example, the experimental rate of oxygen reduction in cNOR (2–10 electrons per second9) corresponds to a barrier of about 17.5 kcal mol−1, which compares well with the overall rate-limiting barrier, the first proton uptake after oxygen binding, of 18.5 (16.9 + 1.6) kcal mol−1 in the energy profile. Furthermore, a recent experimental study showed that in the reaction of O2 with a fully reduced FeB depleted variant of cNOR, oxidation of the low-spin hemes occurred with a small amplitude.109 This observation supports the mechanism in Fig. 21, in which molecular oxygen binds to the high-spin heme and one reduction step occurs without involvement of FeB.
 | ||
| Fig. 21 Energy profile for one catalytic cycle of reduction of molecular oxygen in cNOR, obtained from a combination of experimental and computational data. Under each intermediate the accumulated number of electrons transferred into the BNC is indicated. The letters ct and b denote cytochrome c and low-spin heme b, respectively. The figure is reprinted from ref. 94. Copyright (2017), with permission from Elsevier. | ||
A scheme for the reaction mechanism for oxygen reduction in cNOR as elucidated from the energy profile is given in Fig. 22, which includes also the oxygen reduction mechanism in the A family CcOs for comparison.94 The most apparent difference between the two schemes concerns the involvement of the redox-active tyrosine in the CcO mechanism. The active site tyrosine is lacking in cNOR, which causes several differences in the reaction mechanisms. First, after the binding of molecular oxygen to the BNC in cNOR, one of the reduction steps has to occur, forming a hydrogen peroxide intermediate, before the O–O bond can be cleaved with a low enough barrier. In CcO the tyrosine can deliver the electron and the proton needed. However, as discussed in Section 3.1 above, it is suggested that the O–O bond cleavage in CcO occurs in a two-step manner, with a hydrogen peroxide intermediate labeled IP (Fig. 7), which resembles the PN intermediate in the scheme in Fig. 22, before the O–O bond cleavage occurs. It can also be noted that the local barrier for the O–O bond cleavage relative to the hydrogen peroxide is similar in the two cases, 9.4 kcal mol−1 in cNOR (Fig. 21) and 8.5 kcal mol−1 in a3 CcO (Table 1).
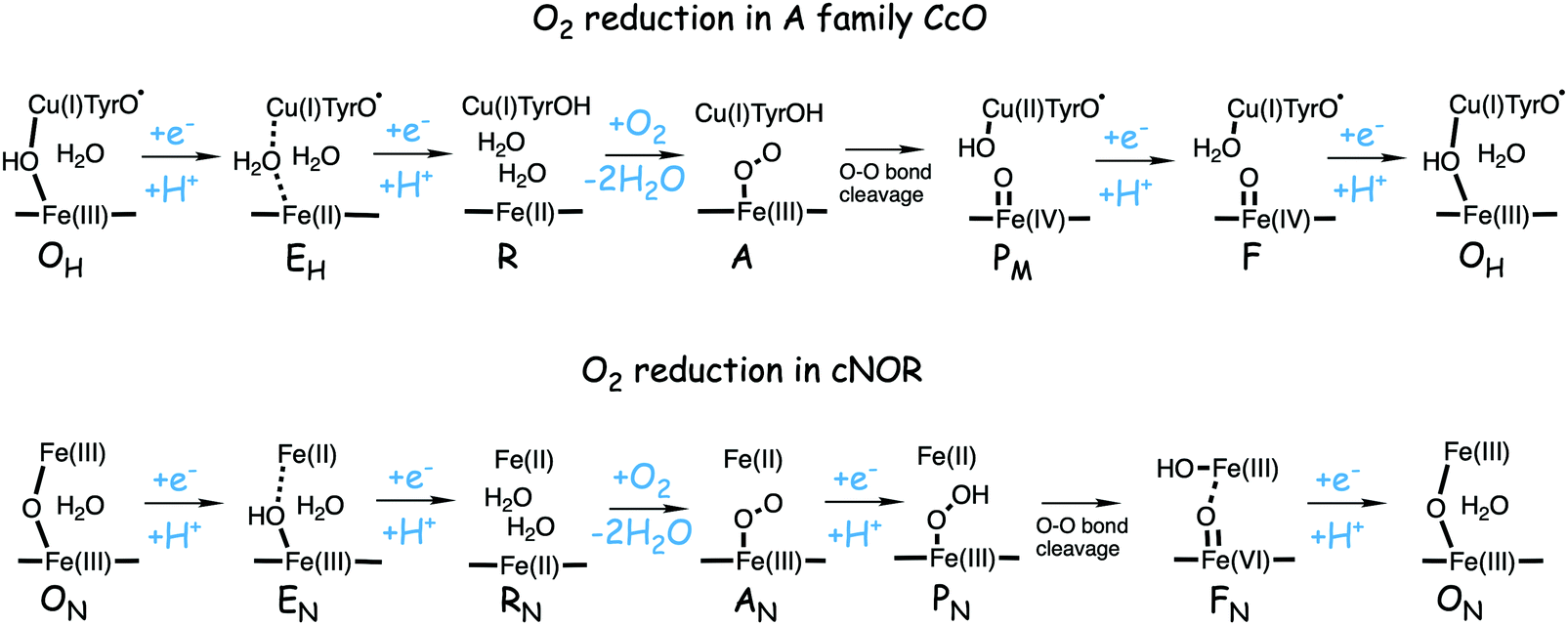 | ||
| Fig. 22 Suggested mechanisms for oxygen reduction in the A family CcOs and in cNORs. The notations for the intermediates in the cNOR reactions was introduced to simplify the comparisons to the CcO reaction.94 | ||
As shown in Fig. 22 the lack of a tyrosine in the cNOR active site results in an important difference between all intermediates in the catalytic cycles for the two enzymes. In the CcO reduction process the tyrosine plays a large role, since it is suggested to stay unprotonated until the last reduction step before the next O2 molecule enters. This form of the intermediates makes it possible for each reduction step to start by electron transfer to the BNC, followed by proton uptake to the BNC, which was suggested to be crucial for the proton pumping, i.e. for efficient energy conservation.36 In contrast, for oxygen reduction in cNOR, it is suggested that the reduction steps are initiated by proton transfer to the BNC, followed by electron transfer, which is similar to the situation for NO reduction in cNOR discussed above. In fact, the two reduction steps from ON to RN in Fig. 22 are more or less identical to the corresponding steps for the NO reduction in cNOR, see Fig. 19, which means that, due to the low reduction potentials of the BNC cofactors they are endergonic, and also that they have high barriers. This is also at variance with oxygen reduction in CcO, where the corresponding reduction steps are significantly exergonic, compare Fig. 11.
In summary, from the computational study on oxygen reduction in the cNOR enzyme, which is known not to contribute to cellular energy conservation, some mechanistic insights can be derived. First, the low reduction potentials of the cofactors in the BNC of cNOR prevents energy conservation via electrogenic chemistry, as occurs in CcO, since some of the reduction steps are quite slow already without an electrochemical gradient present. Second, due to the lack of a tyrosine in the active site of cNOR, the mechanism for the proton coupled reduction steps is different compared to in CcO. This shows how important the redox-active tyrosine in the CcO active site is for energy conservation via proton pumping.36
5.2 NO reduction in CcOs
In a computational study from 2013 it was concluded that the difference in the reduction potentials of the BNC cofactors between CcO and cNOR was decisive for the difference in their capability for energy storage.104 The low potentials of the cNOR cofactors secure a fast reaction with the toxic NO molecule, to the price of endergonic reduction steps, while the larger potentials of the CcO cofactors secure exergonic reduction steps and efficient energy storage in the reaction with O2.37,104 Those conclusions suggest that NO should react slower or not at all with CcO, which is in line with experimental observations. The mitochondrial (bovine) enzyme and several other CcOs from bacteria belonging to the A family have been shown not to reduce NO with a measurable rate,123 while it has been shown that one member of the A family, the Thermus (T.) thermophilus caa3, reduce NO at about 30 mol NO/(mol caa3 × min).10 Among the B-type oxidases, at least the T. thermophilus ba3, has been shown to slowly reduce NO, about 3 mol NO/(mol ba3 × min).10 The fastest NO reduction in a CcO has been found to occur in the C-type cbb3 enzymes, with a rate of about 100 mol NO/(mol cbb3 × min).6,11 The NO reduction in cNORs is much faster 300–4500 mol NO/(mol enzyme × min).10 On the other hand, computational studies have shown that the proton coupled reduction potentials for all different CcOs families are quite similar,69 which makes it more difficult to explain the variation in reactivity with NO among CcOs from different species.Two computational studies have been performed on NO reduction in different types of CcOs with the purpose of comparing possible mechanisms both between cNOR and CcOs, and between the different CcOs.12,125 One of the computational studies used a BNC model taken from a CcO from the A family, but since the BNC active site is essentially identical in the A and the B families, this model was assumed to represent both families, and referred to as the a3 model.12 The other study used a model of the BNC active site from a cbb3 oxidase.125 Two different mechanisms were investigated, labeled mechanism I and II, shown in Fig. 23. Mechanism I is identical to the cis:b3 mechanism, which was previously found to be the most likely mechanism for NO reduction in cNOR (see Fig. 19, which starts from the oxidized state),32,37,103 and in mechanism II a proton coupled reduction step occurs after formation of the hyponitrite intermediate, and before the N–O bond cleavage. The description of the mechanisms and the energy profiles in this section use the reduced BNC active site as starting point, in congruence with the experimental investigations on these reactions.
 | ||
| Fig. 23 Possible mechanisms for NO reduction in CcO. The upper scheme describes mechanism I, which is identical to the cis:b3-mechanism in cNOR as suggested from computational studies (see Fig. 19). Mechanism II, bottom scheme, is an alternative mechanism for NO reduction in CcO. The subindex “x3” on the heme-iron denotes “a3” for the A and B families and “b3” for the C family. | ||
In Fig. 24 the calculated energy profile for reduction of NO in the cbb3-type of CcO following mechanism I, i.e. the cis:b3-mechanism, is compared to the corresponding energy profile for NO reduction in cNOR.125 The energy profile for cbb3 shown in the figure, furthermore, was found to be very similar to the corresponding energy profile obtained for the a3 model describing the A and B families in the other study.12 Therefore, Fig. 24 shows that the energy profiles for NO reduction in all CcOs are quite different from the corresponding profile for the cNOR reaction. In cNOR the formation of the hyponitrite intermediate is exergonic with a low barrier for N2O formation, and the rate-limitation for turnover is the succeeding reduction steps, while in the CcOs the formation of hyponitrite is endergonic with a high barrier for N2O formation. The low reduction potentials of both FeB (0.32 V) and the high-spin heme Fe (0.06 V) contribute to the exergonicity of the hyponitrite formation in cNOR, while the high reduction potentials of both CuB (1.0 V) and high-spin heme Fe (0.3 V) results in endergonic hyponitrite formation in the CcOs. In addition, a valine near the active site in the CcOs was found to have an effect, increasing the barriers for N2O formation a few kcal mol−1, while no such effect was found from a valine in a similar position in cNOR.12 These results, showing a large difference in NO reactivity between CcO and cNOR, are thus in accordance with the expectations, the high barriers for N2O formation in the CcOs result in slow or non-observable reduction of NO.
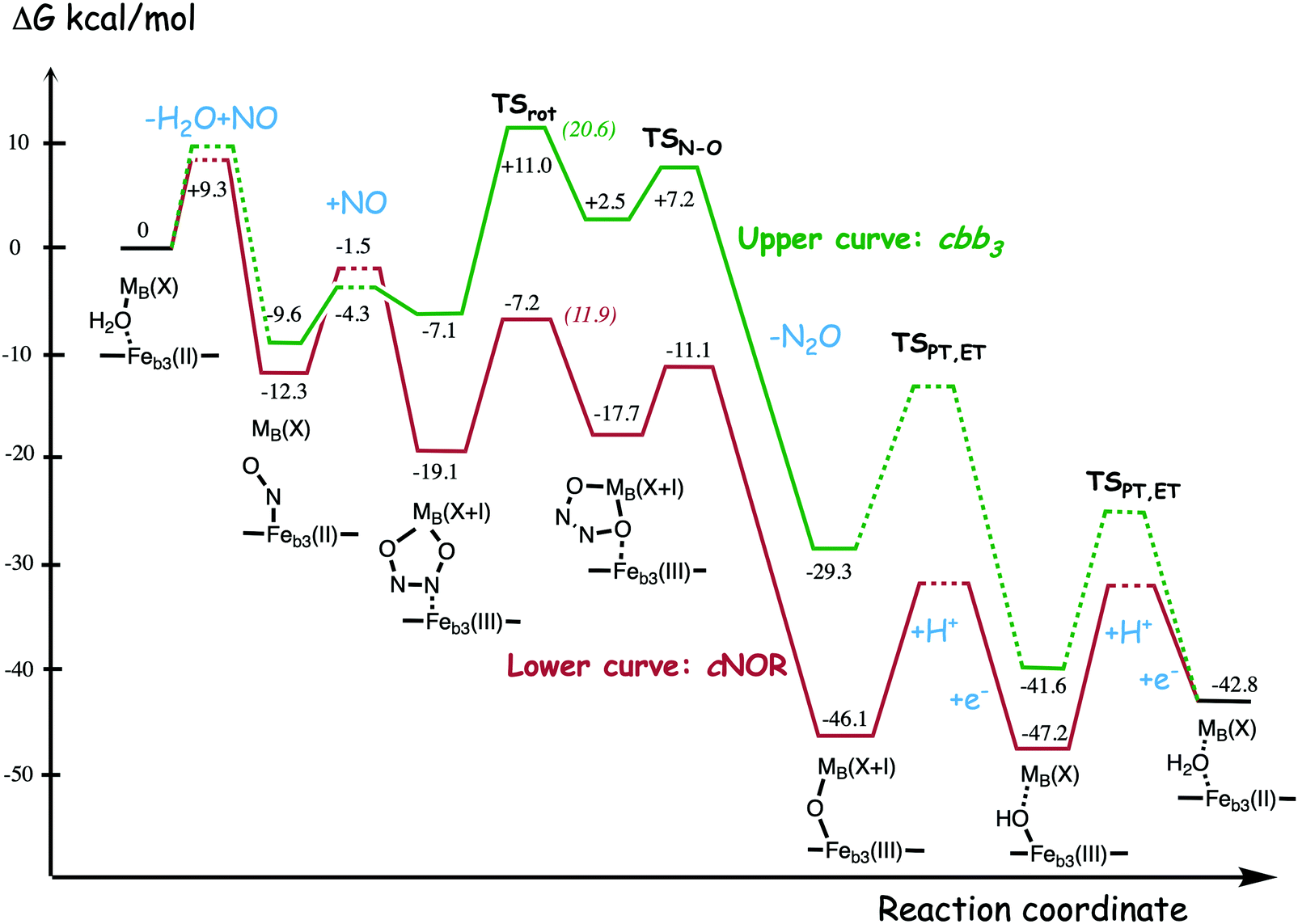 | ||
| Fig. 24 Calculated free energy profile for one full catalytic cycle of NO reduction in cNOR12 and in cbb3 CcO using mechanism I. The notation M(X)B is used for FeB(II) in the cNOR case and for CuB(I) in the cbb3 case, while the notation M(X+1)B is used for FeB(III) and CuB(II), respectively. The dashed parts of the curves were not studied. The cNOR curve is here calculated relative to the ultimate electron donor cytochrome c, which is different from Fig. 20. The figure is reprinted with permission from ref. 125. Copyright (2020) American Chemical Society. | ||
To understand the experimental result that the various CcOs react differently with NO, a closer comparison of the computational results for the different CcOs is needed. Since the CcO with the fastest NO reduction, the cbb3 oxidases, is the CcO that is most similar to the cNORs, it has been speculated that the higher rate of NO reduction in the cbb3 oxidases may be due to structural similarities of the BNC to the cNORs, and that the properties of the high-spin heme determines the NO reactivity in the CcOs (compare Fig. 2).11,124 In Fig. 25 (left part) the energy profile for mechanism I in cbb3 oxidases is compared to the corresponding profile for the a3 oxidases, starting from the nitrosyl complex.125 As was shown in Fig. 24 there is no particular similarity in the energy profiles for the cbb3 oxidases and the cNORs, and in contrast, Fig. 25 emphasize the similarity in the energy profiles for all CcOs. There is, however, a small difference in the heights of the rate determining barriers, with the cbb3 barrier 2.6 kcal mol−1 lower than the a3 barrier, which may explain the faster reaction, although the difference is within the uncertainty of the calculations.
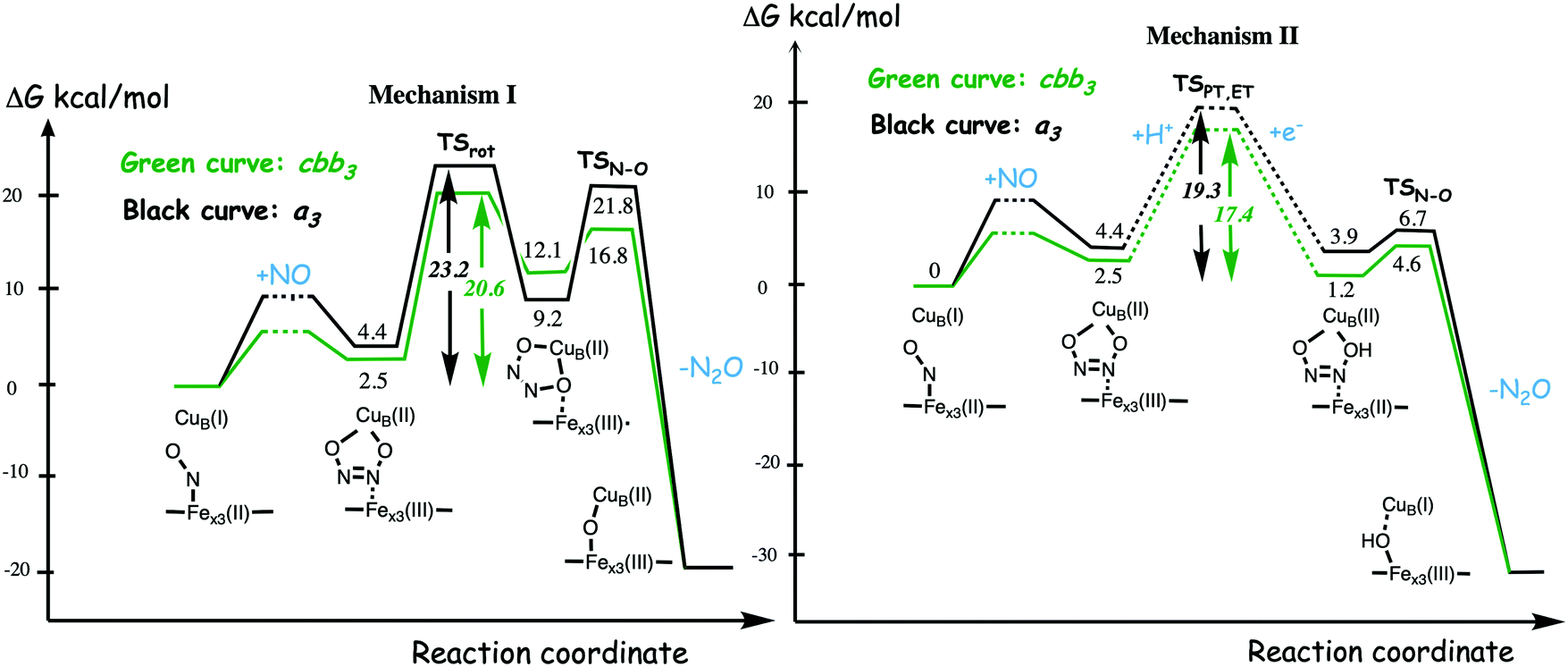 | ||
| Fig. 25 Rate determining steps for N2O formation in the catalytic cycle of NO reduction following mechanism I (left part) or mechanism II (right part) in cbb3 CcO (green curve), compared to a3 CcO (black curve). The calculated energies are given relative to the lowest previous point, the nitrosyl complex. The subindex “x3” on the heme-iron denotes “a3” for the A and B families and “b3” for the C family. | ||
If the energy profile to the left in Fig. 25 may explain the slightly faster rate of NO reduction in cbb3 compared to other CcOs, it cannot explain the differences among the various a3 oxidases. Therefore mechanism II was introduced, which coincides with mechanism I until formation of the hyponitrite intermediate, but involves a proton coupled reduction before the N–O bond cleavage, see Fig. 23. The energy profiles for mechanism II are compared in Fig. 25 (right side), and again it can be seen that the CcOs have very similar energy profiles. In mechanism II for NO reduction, the rate limiting step for N2O formation is the proton transfer into the BNC as part of the reduction step, which means that properties outside the BNC determine the reduction rate.12,125 The barriers for proton transfer cannot be calculated, and therefore they are only sketched in Fig. 25. This suggests that for some CcOs the proton transfer barrier is too high to give any observable NO reduction, while in other CcOs the proton transfer barriers are lower, and of slightly varying heights, resulting in slow NO reduction with slightly different rates. For the non-reacting CcOs the barrier is about 21 kcal mol−1 or higher,12 and for the ones actually reducing NO, the barriers vary between 17 and 19 kcal mol−1, with the lowest value for cbb3. Considering the differences in the protein structures surrounding the BNC active site in the various oxidases, including differences in the number and structure of the proton pathways, this should be a reasonable explanation to the variation in NO reactivity among the CcOs.12,125
The calculations indicate that the cbb3 oxidases may follow the same cis:b3 mechanism as cNOR, although with a very different shape of the energy profile. However, the experimental observation that, contrary to the cNOR reaction,119 the NO reduction in cbb3 oxidases is not substrate-inhibited,11 indicates that the fully oxidized state with an oxo-bridge is not involved when NO is reduced in cbb3 oxidases, which supports mechanism II going directly to the one-electron reduced hydroxyl-bridged intermediate after the N–O bond cleavage, see Fig. 23.125 A computational study on NO reduction in cNOR suggested that the substrate-inhibition is caused by a reaction of the NO molecules with the oxidized BNC with an oxo-bridge, while the same study showed that no reaction occurs with the hydroxyl-bridged one electron reduced intermediate.32 It is furthermore known that NO is a potent inhibitor of the oxygen reaction in both the bovine CcO, which do not reduce NO,126 and cbb3 CcO which do reduce NO.127 This observation can be explained by NO reacting with the oxidized BNC active site, which is an intermediate in all CcOs during the reaction with O2.
The computational results indicate that the main features of the mechanism and the energetics of NO reduction in heme-copper oxidases are determined by the type of non-heme metal, CuB in CcOs or FeB in cNOR, resulting in a large difference between CcO and cNOR. The difference in the type of high-spin heme (a3 or b3) among the CcOs may lead to small differences in energetics, but the main differences in the reaction rates for NO reduction in the various CcOs, including those that are too slow to be observed, are most likely explained by differences in the protein surrounding the BNC active site, including the number and composition of proton pathways.
6. Conclusions
Computational studies of O2 and NO reduction in heme-copper oxidases are reviewed. The main approach in the studies is to apply hybrid DFT calculations to cluster models of the BNC active sites, and to describe entire catalytic cycles, including the reduction steps with electron and proton transfer from donors outside the protein. To achieve a reliable overall picture of the reactions, the computational results are combined with certain experimental information. Mechanistic understanding has been obtained of the bond cleavage and bond formation parts of the reactions, as well as of the energy conservation.For O2 reduction in CcO, the O–O bond cleavage occurs in a two-step reaction. In the first step after O2 binding to the reduced BNC, a hydrogen peroxide bound to the high-spin heme is formed. Normally this step is endergonic and the peroxide intermediate is not observable. The O–O bond is cleaved in the next step forming an oxoferryl at the high-spin heme, a cupric hydroxide and a tyrosyl radical in the A and B family CcOs, where the conserved active site tyrosine supplies the electron and the proton for the peroxide formation. The calculations indicate that for the C family CcOs the same reaction mechanism leads to a too high overall O–O cleavage barrier, and an alternative process is suggested, where one proton coupled reduction step occurs to form the hydrogen peroxide intermediate, followed by the O–O bond cleavage. A similar mechanism as in the C family is used in cNOR, which is known to reduce also molecular oxygen, and where there is no tyrosine available in the active site. Thus, in all cases of oxygen reduction in the heme-copper oxidases a hydrogen peroxide intermediate is formed before the O–O bond cleavage.
For the NO reduction to N2O the chemistry is slightly more complicated, with both bond formation (N–N) and bond cleavage (N–O) before the N2O product is released. The calculations show that the so-called cis:b3 mechanism is the only one of the suggested mechanisms for NO reduction in cNOR that is energetically feasible. The so-called trans mechanism, which has been strongly favored by experimentalists, involves unfavorable energetics. In the cis:b3 mechanism the first NO molecule binds to the high-spin heme b3 in the reduced BNC, and the second NO molecule forms an N–N bond with the first NO. The two BNC metals are oxidized and a hyponitrite dianion is formed in an exergonic step. Rotation of the hyponitrite and cleavage of one of the N–O bonds result in release of N2O. This part of the reaction is found to have a rather low barrier (calculated to be about 12 kcal mol−1) and this step is not rate-limiting for the entire catalytic cycle. In contrast, NO reduction in CcO following the same mechanism would lead to a much higher barrier, which makes this mechanism less likely as an explanation of the actually observed NO reduction in some CcO species. The high barrier is caused by the significantly larger reduction potentials of the BNC cofactors in the CcOs, compared to cNOR, in particular for the non-heme metal. An alternative mechanism is suggested, in which a proton coupled reduction step preceding the N–O bond cleavage is rate-limiting, implying that differences in NO reactivity among the CcOs may be explained by properties outside the BNC.
Regarding energy conservation in the heme-copper oxidases there are several issues, on which the computational studies have shed some light. The calculations on the CcOs show that the proton coupled reduction potential of the non-heme metal in the BNC, CuB, is significantly larger during catalytic turnover than equilibrium measurements indicate. This result solves a problem with a discrepancy between the overall exergonicity of the catalytic cycle, and the sum of exergonicites based on the individual experimental reduction potentials. The result also explains how the A family can afford to pump four protons per oxygen molecule. The calculations further suggest a reaction mechanism for O2 reduction in CcO, in which the tyrosyl radical formed in the O–O bond cleavage step remains in several intermediates during the catalytic cycle. The tyrosyl radical is suggested to be crucial for the proton pumping, since it ensures the coupling between the transfer of one single electron into the active site and the uptake of two protons, one to the BNC for the chemistry and one to be pumped. The function of the tyrosyl radical is to adjust the electron affinity of the BNC in each intermediate, such that a proton must be transferred to a pump-loading site before the chemical proton arrives in the BNC. The calculations further indicate that the oxygen reduction mechanism in the C family of CcOs involves fewer intermediates with a tyrosyl radical, implying a lower stoichiometry of proton pumping in line with some experimental observations.
Finally, the calculations show that the proton coupled reduction potentials of the BNC cofactors in cNOR, which are significantly lower than the corresponding ones in CcO, are optimized for a fast elimination of the toxic NO radical, which leads to endergonic reduction steps. Together with high barriers, the endergonic proton coupled reduction steps would make electrogenic chemistry, i.e. taking electrons and protons from opposite sides of the membrane, too slow when there is a gradient present across the membrane. Therefore, no energy conservation can be accomplished in cNOR.
Conflicts of interest
There are no conflicts of interest.References
- P. Brzezinski and R. B. Gennis, Cytochrome c oxidase: Exciting progress and remaining mysteries, J. Bioenerg. Biomembr., 2008, 40, 521–531 CrossRef CAS.
- M. K. F. Wikström, Proton pump coupled to cytochrome c oxidase in mitochondria, Nature, 1977, 266, 271–273 CrossRef.
- J. P. Shapleigh and W. J. Payne, Nitric oxide-dependent proton translocation in various denitrifiers, J. Bacteriol., 1985, 163, 837–840 CrossRef CAS.
- J. H. Hendriks, A. Jasaitis, M. Saraste and M. I. Verkhovski, Proton and electron pathways in the bacterial nitric oxide reductase, Biochemistry, 2002, 41, 2331–2340 CrossRef CAS.
- J. Reimann, U. Flock, H. Lepp, A. Honigmann and P. Ädelroth, A pathway for protons in nitric oxide reductase from Paracoccus denitrificans, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 362–373 CrossRef CAS.
- Y. Huang, J. Reimann, H. Lepp, N. Drici and P. Ädelroth, Vectorial proton transfer coupled to reduction of O2 and NO by a heme-copper oxidase, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20257–20262 CrossRef CAS.
- T. Fujiwara and Y. Fukumori, Cytochrome cb-type nitric oxide reductase with cytochrome c oxidase activity from Paracoccus denitrificans Atcc 35512, J. Bacteriol., 1996, 178, 1866–1871 CrossRef CAS.
- G. Butland, S. Spiro, N. J. Watmough and D. J. Richardson, Two conserved glutamates in the bacterial nitric oxide reductase are essential for activity but not assembly of the enzyme, J. Bacteriol., 2001, 183, 189–199 CrossRef CAS.
- U. Flock, N. J. Watmough and P. Ädelroth, Electron/Proton Coupling in Bacterial Nitric Oxide Reductase during Reduction of Oxygen, Biochemistry, 2005, 44, 10711–10719 CrossRef CAS.
- A. Giuffrè, G. Stubauer, P. Sarti, M. Brunori, W. G. Zumft, G. Buse and T. Soulimane, The heme-copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: Evolutionary implications, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14718–14723 CrossRef.
- E. Forte, A. Urbani, M. Saraste, P. Sarti, M. Brunori and A. Giuffrè, The cytochrome cbb3 from Pseudomonas stutzeri displays nitric oxide reductase activity, Eur. J. Biochem., 2001, 268, 6486–6491 CrossRef CAS.
- M. R. A. Blomberg and P. Ädelroth, Mechanisms for enzymatic reduction of nitric oxide to nitrous oxide - a comparison between nitric oxide reductase and cytochrome c oxidase, Biochim. Biophys. Acta, 2018, 1859, 1223–1234 CrossRef CAS.
- V. R. I. Kaila, M. I. Verkhovsky and M. Wikström, Proton-Coupled Electron Transfer in Cytochrome Oxidase, Chem. Rev., 2010, 110, 7062–7081 CrossRef CAS.
- M. Wikström, K. Krab and V. Sharma, Oxygen Activation and Energy Conservation by Cytochrome c Oxidase, Chem. Rev., 2018, 118, 2469–2490 CrossRef.
- H. Han, J. Hemp, L. A. Pace, H. Ouyang, K. Ganesan, J. Hyeob Roh, F. Daldal, S. R. Blanke and R. B. Gennis, Adaption of aerobic respiration to low O2 environments, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14109–14114 CrossRef CAS.
- P. R. Rich, Mitochondrial cytochrome c oxidase: catalysis, coupling and controversies, Biochem. Soc. Trans., 2017, 45, 813–829 CrossRef CAS.
- W. G. Zumft, Nitric oxide reductase of prokaryotes with emphasis on the respiratory heme-copper oxidase type, J. Inorg. Biochem., 2005, 99, 194–215 CrossRef CAS.
- P. Moënne-Loccoz, Spectroscopic characterization of heme iron-nitrosyl species and their role in NO reductase mechanisms in diiron proteins, Nat. Prod. Rep., 2007, 24, 610–620 RSC.
- T. Hino, S. Nagano, H. Sugimoto, T. Tosha and Y. Shiro, Molecular structure and function of bacterial nitric oxide reductase, Biochim. Biophys. Acta, 2012, 1817, 680–687 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. R. A. Blomberg, T. Borowski, F. Himo, R.-Z. Liao and P. E. M. Siegbahn, Quantum Chemical Studies of Mechanisms for Metalloenzymes, Chem. Rev., 2014, 114, 3601–3658 CrossRef CAS.
- P. E. M. Siegbahn and T. Borowski, Modeling Enzymatic Reactions Involving Transition Metals, Acc. Chem. Res., 2006, 39, 729–738 CrossRef CAS.
- M. R. A. Blomberg, P. E. M. Siegbahn and G. T. Babcock, Modeling electron transfer in biochemistry: A quantum chemical study of charge separation in Rhodobacter sphaeroides and photosystem II, J. Am. Chem. Soc., 1998, 120, 8812–8824 CrossRef CAS.
- G. V. Isaksen, J. Åqvist and B. O. Brandsdal, Protein surface softness is the origin of enzyme cold-adaptation of trypsin, PLoS Comput. Biol., 2014, 10, e1003813 CrossRef.
- F. Poiana, C. von Ballmoos, N. Gonska, M. R. A. Blomberg, P. Ädelroth and P. Brzezinski, Splitting of the O–O Bond at the Heme-copper Catalytic Site of Respiratory Oxidases, Sci. Adv., 2017, 3, e1700279 CrossRef.
- M. Reiher, O. Salomon and B. A. Hess, Reparameterization of hybrid functionals based on energy differences of states of different multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- P. E. M. Siegbahn and M. R. A. Blomberg, A Systematic DFT Approach for Studying Mechanisms of Redox Active Enzymes, Front. Chem., 2018, 6, 644 Search PubMed.
- S. Grimme, J. Anthony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- M. R. A. Blomberg and P. E. M. Siegbahn, Quantum Chemistry Applied to the Mechanisms of Transition Metal Containing Enzymes - cytochrome c Oxidase a Particularly Challenging Case, J. Comput. Chem., 2006, 27, 1373–1384 CrossRef CAS.
- P. E. M. Siegbahn and M. R. A. Blomberg, Quantum chemical studies of proton-coupled electron transfer in metalloenzymes, Chem. Rev., 2010, 110, 7040–7061 CrossRef CAS.
- P. E. M. Siegbahn, Water oxidation mechanism in photosystem II, including oxidations, proton release pathways, O–O bond formation and O2 release, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Mechanism for N2O Generation in Bacterial Nitric Oxide Reductase: A Quantum Chemical Study, Biochemistry, 2012, 51, 5173–5186 CrossRef CAS.
- M. Radon and K. Pierloot, Binding of CO, NO, and O2 to Heme by Density Functional and Mulitireference ab Initio Calculations, J. Phys. Chem. A, 2008, 112, 11824–11832 CrossRef CAS.
- S. Vancoillie, H. Zhao, M. Radon and K. Pierloot, Performance of CASPT2 and DFT for Relative Spin-State Energetics of Heme Models, J. Chem. Theory Comput., 2010, 6, 576–582 CrossRef CAS.
- J. Olah and J. N. Harvey, NO bonding to Heme Groups: DFT and Correlated av Initio Calculations, J. Phys. Chem. A, 2009, 113, 7338–7345 CrossRef CAS.
- M. R. A. Blomberg, The mechanism of oxygen reduction in cytochrome c oxidase and the role of the active site tyrosine, Biochemistry, 2016, 55, 489–500 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Improved free energy profile for reduction of NO in cytochrome c dependent nitric oxide reductase (cNOR), J. Comput. Chem., 2016, 37, 1810–1818 CrossRef CAS.
- L. Qin, C. Hiser, A. Mulichak, R. M. Gavarito and S. Ferguson-Miller, Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16117–16122 CrossRef CAS.
- S. Buschmann, E. Warkentin, H. Xie, J. D. Langer, U. Ermler and H. Michel, The structure of cbb3 cytochrome oxidase provides insights into proton pumping, Science, 2010, 329, 327–330 CrossRef CAS.
- T. Hino, Y. Matsumoto, S. Nagano, H. Sugimoto, Y. Fukumori, T. Murata, S. Iwata and Y. Shiro, Structural Basis of Biological N2O Generation by Bacterial Nitric Oxide Reductase, Science, 2010, 330, 1666–1670 CrossRef CAS.
- M. M. Pereira, M. Santana and M. Teixeira, A novel scenario for the evolution of haem-copper oxygen reductases, Biochim. Biophys. Acta, 2001, 1505, 185–208 CrossRef CAS.
- J. Hemp and R. B. Gennis, Diversity of the heme-copper superfamily in archaea: Insights from genomics and structural modeling, Results Probl. Cell Differ., 2008, 45, 1–31 CAS.
- H. J. Lee, J. Reimann, Y. Huang and P. Ädelroth, Functional proton transfer pathways in the heme-copper oxidase superfamily, Biochim. Biophys. Acta, 2012, 1817, 537–544 CrossRef CAS.
- V. Rauhamäki, D. A. Bloch and M. Wikström, Mechanistic stoichiometry of proton translocation by cytochrome cbb3, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7286–7291 CrossRef.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Structures of Metal Sites of Oxidized Bovine Heart cytochrome c Oxidase at 2.8 Å, Science, 1995, 269, 1069–1074 CrossRef CAS.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans, Nature, 1995, 376, 660–669 CrossRef CAS.
- T. Soulimane, G. Buse, G. P. Bourenkov, H. D. Bartunik, R. Huber and M. E. Than, Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus, EMBO J., 2000, 19, 1766–1777 CrossRef CAS.
- D. A. Proshlyakov, M. A. Pressler and G. T. Babcock, Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8020–8025 CrossRef CAS.
- M. Fabian, W. W. Wong, R. B. Gennis and G. Palmer, Mass spectrometric determination of dioxygen bond splitting in the “peroxy” intermediate of cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13114–13117 CrossRef CAS.
- M. R. A. Blomberg, The mechanism for oxygen reduction in the C family cbb3 cytochrome c oxidases - Implications for the proton pumping stoichiometry, J. Inorg. Biochem., 2020, 203, 110866 CrossRef CAS.
- R. B. Gennis, Multiple proton-conducting pathways in cytochrome oxidase and a proposed role for the active-site tyrosine, Biochim. Biophys. Acta, 1998, 1365, 241–248 CrossRef CAS.
- E. A. Gorbikova, I. Belevich, M. Wikström and M. I. Verkhovsky, The proton donor for O–O bond scission by cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10733–10737 CrossRef CAS.
- M. R. A. Blomberg, P. E. M. Siegbahn, G. T. Babcock and M. Wikström, Modeling cytochrome oxidase - a quantum chemical study of the O–O bond cleavage mechanism, J. Am. Chem. Soc., 2000, 122, 12848–12858 CrossRef CAS.
- Y. Yoshioka, H. Kawai and K. Yamaguchi, Theoretical study of role of H2O molecule on initial stage of reduction of O2 molecule in active site of cytochrome c oxidase, Chem. Phys. Lett., 2003, 373, 45–52 CrossRef.
- M. R. A. Blomberg, P. E. M. Siegbahn and M. Wikström, A metal-bridging mechanism for O–O bond cleavage in cytochrome c oxidase, Inorg. Chem., 2003, 42, 5231–5243 CrossRef CAS.
- M. I. Verkhovsky, J. E. Morgan and M. Wikström, Oxygen binding and activation in the reaction of oxygen with cytochrome c oxidase, Biochemistry, 1994, 33, 3079–3086 CrossRef CAS.
- M. Karpefors, P. Ädelroth, A. Namslauer, Y. Zhen and P. Brzezinski, Formation of the “Peroxy” Intermediate in cytochrome c Oxidase is Associated with Internal Proton/Hydrogen Transfer, Biochemistry, 2000, 39, 14664–14669 CrossRef CAS.
- I. Szundi, C. Funatogawa, J. A. Fee, T. Soulimane and O. Einarsdottir, CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21010–21015 CrossRef CAS.
- V. Rauhamäki and M. Wikström, The causes of reduced proton-pumping efficiency in type B and C respiratory heme-copper oxidases, and some mutated variants of type A, Biochim. Biophys. Acta, 2014, 1837, 999–1003 CrossRef.
- I. Szundi, C. Funatogawa, T. Soulimane and O. Einarsdottir, The reactions of O2 and NO with mixed-valence ba3 cytochrome c oxidase from Thermus thermophilus, Biophys. J., 2020, 118, 386–395 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Quantum chemistry as a tool in bioenergetics, Biochim. Biophys. Acta, 2010, 1797, 129–142 CrossRef CAS.
- A. W. Schaefer, A. C. Roveda Jr., A. Jose and E. I. Solomon, Geometric and Electronic Structure Contributions to O–O Cleavage and the Resultant Intermediate Generated in Heme-Copper Oxidases, J. Am. Chem. Soc., 2019, 141, 10068–10081 CrossRef CAS.
- W.-G. Han Du, A. W. Götz, L. Yang, R. C. Walker and L. Noodleman, A broken-symmetry density functional study of structures, energies, and protonation states along the catalytic O–O bond cleavage pathway in ba3 cytochrome c oxidase from Thermus thermophilus, Phys. Chem. Chem. Phys., 2016, 18, 21162–21171 RSC.
- M. R. A. Blomberg and P. E. M. Siegbahn, How cytochrome c oxidase can pump four protons per oxygen molecule at high electrochemical gradient, Biochim. Biophys. Acta, 2015, 1847, 364–376 CrossRef CAS.
- V. Sharma, M. Wikström and V. R. I. Kaila, Stabilization of the peroxy intermediate in the oxygen splitting reaction in cytochrome cbb3, Biochim. Biophys. Acta, 2011, 1807, 813–818 CrossRef CAS.
- M. Wikström and J. E. Morgan, The dioxygen cycle. Spectral, kinetic, and thermodynamic characteristics of ferryl and peroxy intermediates observed by reversal of the cytochrome oxidase reaction, J. Biol. Chem., 1992, 267, 10266–10273 Search PubMed.
- S. J. Ferguson, ATP synthase: From sequence to ring size to the P/O ratio, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16755–16756 CrossRef CAS.
- M. Wikström and G. Hummer, Stoichiometry of proton translocation by respiratory complex I and its mechanistic implications, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4431–4436 CrossRef.
- M. R. A. Blomberg, Active site midpoint potentials in different cytochrome c oxidase families - a computational comparison, Biochemistry, 2019, 58, 2028–2038 CrossRef CAS.
- M. I. Verkhovsky, A. Jasaitis, M. L. Verkhovskaya, J. E. Morgan and M. Wikström, Proton translocation by cytochrome c oxidase, Nature, 1999, 400, 480–483 CrossRef CAS.
- D. Bloch, I. Belevich, A. Jasaitis, C. Ribacka, A. Puustinen, M. I. Verkhovsky and M. Wikström, The catalytic cycle of cytochrome c oxidase is not the sum of its two halves, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 529–533 CrossRef CAS.
- V. Sharma, K. D. Karlin and M. Wikström, Computational study of the activated OH state in the catalytic mechanism of cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16844–16849 CrossRef CAS.
- M. Wikström and V. Sharma, Cytochrome c Oxidase – Remaining Questions About the Catalytic Mechanism, in Oxygen Production and Reduction in Artificial and Natural Systems, ed. J. Barber, A. V. Ruban and P. J. Nixon, World Scientific Publishing, Singapore, 2019, pp. 135–145 Search PubMed.
- M. R. A. Blomberg, The structure of the oxidized state of cytochrome c oxidase - experiments and theory compared, J. Inorg. Biochem., 2020, 206, 111020 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Protonation of the binuclear active site in cytochrome c oxidase decreases the reduction potential of CuB, Biochim. Biophys. Acta, 2015, 1847, 1173–1180 CrossRef CAS.
- D. Jancura, V. Berka, M. Antalik, J. Bagelova, R. B. Gennis, G. Palmer and M. Fabian, Spectral and kinetic equivalence of oxidized cytochrome c oxidase as isolated and “activated” by reoxidation, J. Biol. Chem., 2006, 281, 30319–30325 CrossRef CAS.
- S. E. Brand, S. Rajagukguk, K. Ganesan, L. Geren, M. Fabian, D. Han, R. B. Gennis, B. Durham and F. Millett, A new ruthenium complex to study single-electron reduction of the pulsed OH state of detergent-solubilized cytochrome oxidase, Biochemistry, 2007, 46, 14610–14618 CrossRef CAS.
- J. Vilhjámsdóttir, R. B. Gennis and P. Brzezinski, The electron distribution in the “activated” state of cytochrome c oxidase, Sci. Rep., 2018, 8, 7502 CrossRef.
- F. Melin, H. Xie, T. Meyer, Y. O. Ahn, R. B. Gennis, H. Michel and P. Hellwig, The unusual redox properties of C-type oxidases, Biochim. Biophys. Acta, 2016, 1857, 1892–1899 CrossRef CAS.
- V. Rauhamäki, D. A. Bloch, M. I. Verkhovsky and M. Wikström, Active site of cytochrome cbb3, J. Biol. Chem., 2009, 284, 11301–11308 CrossRef.
- V. Sharma, M. Wikström and V. R. I. Kaila, Redox-coupled proton transfer in the active site of cytochrome cbb3, Biochim. Biophys. Acta, 2010, 1797, 1512–1520 CrossRef CAS.
- J. Vilhjálmsdóttir, I. Albertsson, M. R. A. Blomberg, P. Ädelroth and P. Brzezinski, Proton transfer in uncoupled variants of cytochrome c oxidase, FEBS Lett., 2020, 594, 813–822 CrossRef.
- M. L. Björck and P. Brzezinski, Control of transmembrane charge transfer in cytochrome c oxidase by the membrane potential, Nat. Commun., 2018, 9, 3187 CrossRef.
- P. Brzezinski, Redox-driven membrane-bound proton pumps, Trends Biochem. Sci., 2004, 29, 380–387 CrossRef CAS.
- H. J. Lee, R. B. Gennis and P. Ädelroth, Entrance of the proton pathway in cbb3-type heme-copper oxidases, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17661–17666 CrossRef CAS.
- L. Noodleman, W.-G. Han Du, J. A. Fee, A. W. Götz and R. C. Walker, Linking Chemical Electron-Proton Transfer to Proton Pumping in Cytochrome c Oxidase: Broken-Symmetry DFT Exploration of Intermediates along the Catalytic Reaction Pathway of the Iron–Copper Dinuclear Complex, Inorg. Chem., 2014, 53, 6458–6472 CrossRef CAS.
- J. A. Fee, D. A. Case and L. Noodleman, Toward a Chemical Mechanism of Proton Pumping by the B-Type Cytochrome c Oxidase: Application of Density Functional Theory to Cytochrome ba3 of Thermus thermophilus, J. Am. Chem. Soc., 2008, 130, 15002–15021 CrossRef CAS.
- D. M. Popovic, I. V. Leontyev, D. G. Beech and A. A. Stuchebrukhov, Similarity of cytochrome c oxidases in different organisms, Proteins, 2010, 78, 2691–2698 CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, The Mechanism for Proton Pumping in Cytochrome c Oxidase from an Electrostatic and Quantum Chemical Perspective, Biochim. Biophys. Acta, 2012, 1817, 495–505 CrossRef CAS.
- J. E. Morgan, M. I. Verkhovsky and M. Wikström, The histidine cycle: A new model for proton translocation in the respiratory heme-copper oxidases, J. Bioenerg. Biomembr., 1994, 26, 599–608 CrossRef CAS.
- P. R. Rich, B. Meunier, R. Mitchell and A. J. Moody, Coupling of charge and proton movement in cytochrome c oxidase, Biochim. Biophys. Acta, 1996, 1275, 91–95 CrossRef.
- H. Michel, The mechanism of proton pumping by cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12819–12824 CrossRef CAS.
- H. Michel, Cytochrome c Oxidase: Catalytic Cycle and Mechanisms of Proton Pumping-A Discussion, Biochemistry, 1999, 38, 15129–15140 CrossRef CAS.
- M. R. A. Blomberg and P. Ädelroth, The mechanism for oxygen reduction in cytochrome c dependent nitric oxide reductase (cNOR) as obtained from a combination of theoretical and experimental results, Biochim. Biophys. Acta, 2017, 1858, 884–894 CrossRef CAS.
- J. Hemp, D. E. Robinson, K. B. Ganesan, T. J. Martinez, N. L. Kelleher and R. B. Gennis, Evolutionary Migration of a Post-Translationally Modified Active-Site Residue in the Proton-Pumping Heme-Copper Oxygen Reductases, Biochemistry, 2006, 45, 15405–15410 CrossRef CAS.
- R. Murali, G. G. Yildiz, F. Daldal and R. B. Gennis, Stoichiometry of proton pumping by the cbb3 oxygen reductase in whole cells of Rhodobacter capsulatus at pH 7 is about 0.5 H+ per electron, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2144 CrossRef CAS.
- V. Rauhamäki, D. A. Bloch and M. Wikström, Reply to Murali et al.: Proton translocation stoichiometry of cbb3-type cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2145 CrossRef.
- S. Al-Attar and S. de Vries, An electrogenic nitric oxide reductase, FEBS Lett., 2015, 589, 2050–2057 CrossRef CAS.
- R. W. Ye, B. A. Averill and J. A. Tiedje, Denitrification: Production and Consumption of Nitric Osixe, Appl. Environ. Microbiol., 1994, 60, 1053–1058 CrossRef CAS.
- P. Girsch and S. de Vries, Purification and initial kinetic and spectroscopic characterization of NO reductase from Paracoccus denitrificans, Biochim. Biophys. Acta, 1997, 1318, 202–216 CrossRef CAS.
- H. Kumita, K. Matsuura, T. Hino, S. Takahashi, H. Hori, Y. Fukumori, I. Morisima and Y. Shiro, NO Reduction by Nitric-oxide Reductase from Denitrifying Bacterium Pseudomonas aeruginosa, J. Biol. Chem., 2004, 279, 55247–55254 CrossRef CAS.
- Y. Shiro, H. Sugimoto, T. Tosha, S. Nagano and T. Hino, Structural basis for nitrous oxide generation by bacterial nitric oxide reductases, Philos. Trans. R. Soc., B, 2012, 367, 1195–1203 CrossRef CAS.
- M. R. A. Blomberg, Can reduction of NO to N2O in cytochrome c dependent nitric oxide reductase (cNOR) proceed through a trans-mechanism?, Biochemistry, 2017, 56, 120–131 CrossRef CAS.
- M. R. A. Blomberg and P. E. M. Siegbahn, Why is the reduction of NO in cytochrome c dependent nitric oxide reductase (cNOR) not electrogenic?, Biochim. Biophys. Acta, 2013, 1827, 826–833 CrossRef CAS.
- L. M. Blomberg, M. R. A. Blomberg and P. E. M. Siegbahn, Reduction of Nitric Oxide in Bacterial Nitric Oxide Reductase – A Theoretical Model Study, Biochim. Biophys. Acta, 2006, 1757, 240–252 CrossRef CAS.
- M. Shoji, K. Hanaoka, D. Kondo, A. Sato, H. Umeda, K. Kamiya and K. Shiraishi, A QM/MM study of nitric oxide reductase-catalysed N2O formation, Mol. Phys., 2014, 112, 393–397 CrossRef CAS.
- A. A. A. Attia and R. Silaghi-Dumitrescu, Bacterial nitric oxide reductase: a mechanism revisited by an ONIOM (DFT:MM) study, J. Mol. Model., 2015, 21, 1–12 CrossRef CAS.
- C. Van Stappen and N. Lehnert, Mechanism of N–N Bond Formation by Transition Metal–Nitrosyl Complexes: Modeling Flavodiiron Nitric Oxide Reductases, Inorg. Chem., 2018, 57, 4252–4269 CrossRef CAS.
- M. Kahle, M. R. A. Blomberg, S. Jareck and P. Ädelroth, Insights into the mechanism of nitric oxide reductase from a FeB-depleted variant, FEBS Lett., 2019, 593, 1351–1359 CrossRef CAS.
- V. Daskalakis, T. Ohta, T. Kitagawa and C. Varotsis, Structure and properties of the catalytic site of nitric oxide reductase at ambient temperature, Biochim. Biophys. Acta, 2015, 1847, 1240–1244 CrossRef CAS.
- H. Matsumura, T. Hayashi, S. Chakraborty, Y. Lu and P. Moënne-Loczz, The Production of Nitrous Oxide by the Heme/Nonheme Diiron Center of Engineered Myoglobins (FeBMbs) Proceeds through a trans-Iron-Nitrosyl Dimer, J. Am. Chem. Soc., 2014, 136, 2420–2431 CrossRef CAS.
- S. Chakraborty, J. Reed, M. Ross, M. J. Nilges, I. D. Petrik, S. Ghosh, S. Hammes-Schiffer, J. T. Sage, Y. Zhang, C. E. Schulz and Y. Lu, Spectrosopic and Computational Study of a Nonheme Iron Nitrosyl Center in a Biosynthetic Model of Nitric Oxide Reductase, Angew. Chem., Int. Ed., 2014, 53, 2417–2421 CrossRef CAS.
- S. Chakraborty, J. Reed, J. T. Sage, N. C. Brangan, I. D. Petrik, K. D. Minor, M. Y. Hu, J. Zhao, E. E. Alp and Y. Lu, Recent Advances in Biosynthetic Modeling of Nitric Oxide Reductase and Insights Gained from Nuclear Resonance Vibrational and other Spectrosopic Studies, Inorg. Chem., 2015, 54, 9317–9329 CrossRef CAS.
- C. M. Cordas, A. G. Duarte, J. J. G. Moura and I. Moura, Electrochemical behaviour of bacterial nitric oxide reductase – Evidence of low redox potential non-heme FeB gives new perspectives on the catalytic mechanism, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 233–238 CrossRef CAS.
- A. G. Duarte, C. M. Cordas, J. J. G. Moura and I. Moura, Steady-state kinetics with nitric oxide reductase (NOR): New considerations on substrate inhibition profile and catalytic mechanism, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 375–384 CrossRef CAS.
- H. Takeda, T. Kimura, T. Nomura, M. Horitani, A. Yokota, A. Matsubayashi, S. Ishii, Y. Shiro, M. Kubo and T. Tosha, Timing of NO Binding and Protonation in the Catalytic Reaction of Bacterial Nitric Oxide Reductase as Established by Time-Resolved Spectroscopy, Bull. Chem. Soc. Jpn., 2020, 93, 825–833 CrossRef CAS.
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, Lewis Acid Activation of the Ferrous Heme-NO Fragment toward the N–N Coupling Reaction with NO To Generate N2O, J. Am. Chem. Soc., 2018, 140, 4204–4207 CrossRef CAS.
- S. Sabuncu, J. H. Reed, Y. Lu and P. Moënne-Loccoz, Nitric Oxide Reductase Activity in Heme–Nonheme Binuclear Engineered Myoglobins through a One-Electron Reduction Cycle, J. Am. Chem. Soc., 2018, 140, 17389–17393 CrossRef CAS.
- P. Lachmann, Y. Huang, J. Reimann, U. Flock and P. Ädelroth, Substrate control of internal electron transfer in bacterial nitric oxide reductase, J. Biol. Chem., 2010, 285, 25531–25537 CrossRef CAS.
- P. Moënne-Loccoz and S. de Vries, Structural Characterization of the Catalytic High-Spin Heme b of Nitric Oxide Reductase: A Resonance Raman Study, J. Am. Chem. Soc., 1998, 120, 5147–5152 CrossRef.
- P. Moënne-Loccoz, O.-M. H. Richter, H. Huang, I. M. Wasser, R. A. Ghiladi, K. D. Karlin and S. de Vries, Nitric Oxide Reductase from Paracoccus denitrificans Contains an Oxo-Bridged Heme/Non-Heme Diiron Center, J. Am. Chem. Soc., 2000, 122, 9344–9345 CrossRef.
- K. L. C. Grönberg, M. D. Roldan, L. Prior, G. Butland, M. R. Cheessman, D. J. Richardson, S. Spiro, A. J. Thomson and N. J. Watmough, A low redox potential heme in the dinuclear center of bacterial nitric oxide reductase: implications for the evolution of energy-conserving heme-copper oxidases, Biochemistry, 1999, 38, 13780–13786 CrossRef.
- G. Stubauer, A. Giuffrè, M. Brunori and P. Sarti, Cytochrome c Oxidase Does Not Catalyze the Anaerobic Reduction of NO, Biochem. Biophys. Res. Commun., 1998, 245, 459–465 CrossRef CAS.
- A. Bhagi-Damodarana, J. H. Reedb, Q. Zhuc, Y. Shid, P. Hosseinzadeh, B. A. Sandovala, K. A. Harndena, S. Wanga, M. R. Sponholtzb, E. N. Mirtse, S. Dwaraknatha, Y. Zhang, P. Moënne-Loccoz and Y. Lu, Heme redox potentials hold the key to reactivity differences between nitric oxide reductase and heme-copper oxidase, Proc. Natl. Acad. Sci. U. S. A., 2018, 110, 6195–6200 CrossRef.
- M. R. A. Blomberg, Role of the Two Metals in the Active Sites of Heme-Copper Oxidases – A Study of NO Reduction in cbb3 Cytochrome c Oxidase, Inorg. Chem., 2020, 59, 11542–11553 CrossRef CAS.
- P. Sarti, E. Forte, D. Mastronicola, A. Guffre and M. Arese, Cytochrome c oxidase and nitric oxide in action: Molecular mechanisms and pathophysiological implications, Biochim. Biophys. Acta, 2012, 1817, 610–619 CrossRef CAS.
- D. Arjona, M. Wikström and P. Ädelroth, Nitric oxide is a potent inhibitor of the cbb3-type heme-copper oxidases, FEBS Lett., 2015, 589, 1214–1218 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |