
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule recognition of disease-relevant RNA structures†
Samantha M.
Meyer
,
Christopher C.
Williams
,
Yoshihiro
Akahori
,
Toru
Tanaka
 ,
Haruo
Aikawa
,
Yuquan
Tong
,
Jessica L.
Childs-Disney
and
Matthew D.
Disney
*
,
Haruo
Aikawa
,
Yuquan
Tong
,
Jessica L.
Childs-Disney
and
Matthew D.
Disney
*
Department of Chemistry, The Scripps Research Institute, 130 Scripps Way, Jupiter, FL 33458, USA. E-mail: disney@scripps.edu
First published on 25th September 2020
Abstract
Targeting RNAs with small molecules represents a new frontier in drug discovery and development. The rich structural diversity of folded RNAs offers a nearly unlimited reservoir of targets for small molecules to bind, similar to small molecule occupancy of protein binding pockets, thus creating the potential to modulate human biology. Although the bacterial ribosome has historically been the most well exploited RNA target, advances in RNA sequencing technologies and a growing understanding of RNA structure have led to an explosion of interest in the direct targeting of human pathological RNAs. This review highlights recent advances in this area, with a focus on the design of small molecule probes that selectively engage structures within disease-causing RNAs, with micromolar to nanomolar affinity. Additionally, we explore emerging RNA-target strategies, such as bleomycin A5 conjugates and ribonuclease targeting chimeras (RIBOTACs), that allow for the targeted degradation of RNAs with impressive potency and selectivity. The compounds discussed in this review have proven efficacious in human cell lines, patient-derived cells, and pre-clinical animal models, with one compound currently undergoing a Phase II clinical trial and another that recently garnerd FDA-approval, indicating a bright future for targeted small molecule therapeutics that affect RNA function.
 Samantha M. Meyer | Samantha M. Meyer received her BS in Biochemistry and Molecular Biology from the University of Wisconsin – Eau Claire in 2019. The following fall she began doctoral studies under the guidance of Prof. Matthew D. Disney at The Scripps Research Institute in Jupiter, Florida. Her current research focuses on targeting disease-causing RNAs with small molecules, with an emphasis on expanding the versatility of RIBOTACs. |
 Christopher C. Williams | Christopher C. Williams received his BA in Chemistry from Hamilton College (2017). He conducts his graduate studies under the guidance of Prof. Matthew D. Disney at The Scripps Research Institute in Florida, where he explores RNA-small molecule binding interactions in the context of infectious disease. |
 Yoshihiro Akahori | Yoshihiro Akahori received his PhD in Synthetic Organic Chemistry (2014) from the Nagoya City University, supervised by Prof. Seiichi Nakamura. While there he worked on synthesizing oxygenated natural terpenoids. He then joined Daiichi Sankyo Co., Ltd where he engaged in drug discovery research. Since 2020, he has worked at The Scripps Research Institute with Prof. Matthew D. Disney. His current research focuses on developing small molecules that target RNA. |
 Toru Tanaka | Toru Tanaka received his PhD in Organic Chemistry (2016) from Kyoto Pharmaceutical University under the guidance of Prof. Masayuki Yamashita, where he worked on using sulfoxonium methylide skeletal transformation reactions to open cyclopropane and cyclobutene rings. Since 2020, he has worked as a postdoctoral fellow with Prof. Matthew D. Disney. His current research focuses on the design and synthesis of small molecules that bind RNA. |
 Haruo Aikawa | Haruo Aikawa was awarded his PhD in Chemistry from Tohoku University, Japan, in 2009, under Drs. Yoshinori Yamamoto and Naoki Asao. Dr. Aikawa also worked on peptide chemistry in the lab of Dr. Hirokazu Tamamura at Tokyo Medical and Dental University. His research on nucleic acid-binding molecules began in 2013 in the lab of Dr. Kazuhiko Nakatani at Osaka University. In 2018, he joined Dr. Matthew D. Disney's lab at The Scripps Research Institute in Florida as a postdoctoral fellow. His current research themes are regulation of RNA biology by small molecules targeting repeat expansion disorders and miRNAs. |
 Yuquan Tong | Yuquan Tong received his BS in Chemical and Biomolecular Engineering from the University of Illinois at Urbana – Champaign in 2019. Under the supervision of Prof. Matthew D. Disney, he started his doctoral studies at The Scripps Research Institute in Florida, working on small molecules targeting RNA. His current research focuses on targeting mRNAs of “undruggable” disease-causing proteins, including alpha synuclein and tau. |
1. Introduction
RNA is a critical component of the Central Dogma, best known for its roles in transcription and translation. However, non-coding (nc) RNAs play important functions critical for the regulation of cell homeostasis and normal biology.1 These ncRNAs, such as microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), small nuclear RNAs (snRNAs), transfer RNAs (tRNAs), long non-coding RNAs (lncRNAs), etc. (Fig. 1) are highly structured1 and offered the first clues that RNA structures may play vital roles in human biology beyond the encoding and synthesis of protein. Indeed, this hypothesis has been proven true as RNA structures have been linked to both normal biology and disease pathology.2,3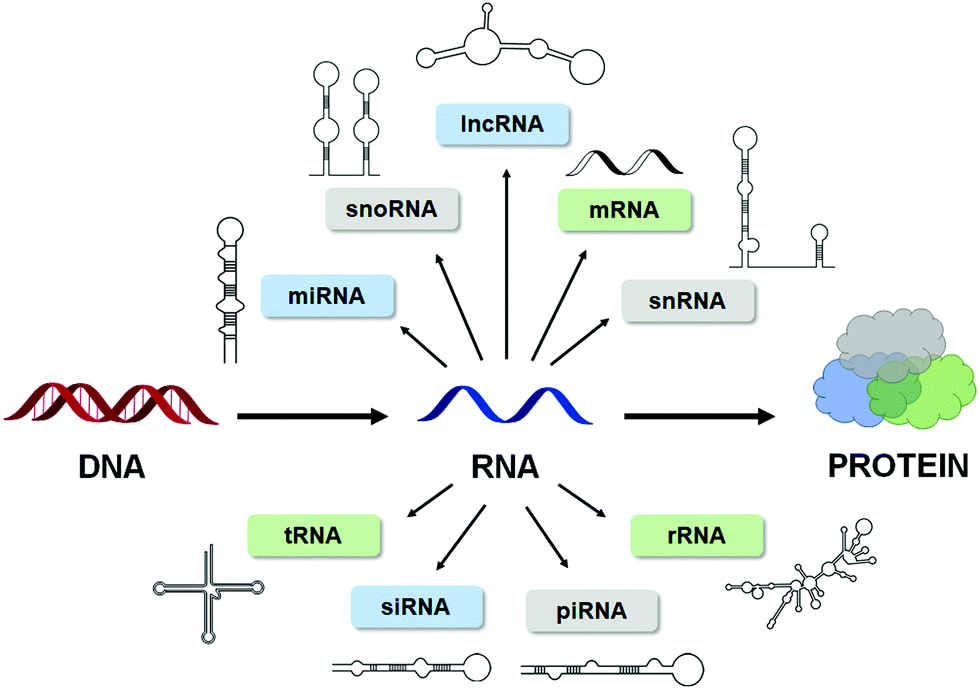 | ||
| Fig. 1 RNA is highly structured. The Central Dogma of biology, showcasing the numerous types of structured RNAs that have been identified to date. | ||
The variability and complexity of RNA structures has been widely explored, leading to the appreciation that RNAs range from being largely disordered (dynamic) to adopting simple structures such as loops and bulges (secondary structure) to creating highly intricate pseudoknots, G-quadruplexes, and coaxial stacking (tertiary structure). The influence of these structures has been explored in the context of bacterial gene expression and riboswitches4 and in viral replication and infection.5 In the context of human biology, structured RNAs influence translational regulation,6–8 alternative splicing,9,10 and even enzymatic catalysis,11–14 further demonstrating their intimate involvement in maintaining healthy biology. As these topics will not be reviewed in depth here, we direct the reader to the references cited above for additional detail.
Predictably, disruption of RNA structure via mutation, formation of unnatural RNA structures, e.g., by insertions or expansions, or aberrant expression, leads to dysregulation of cellular processes, resulting in disease. For example, dysregulation of miRNAs, short regulatory RNAs that modulate gene expression via the RNA-induced silencing complex (RISC),7 have been associated with, among others, cardiovascular disease, inflammatory disorders, and cancer.7,15,16 Additionally, structured RNAs have been implicated in several neurological disorders, as reviewed in Bernat et al.,17 a well-known example being RNA repeat expansion/microsatellite disorders. This class of disorders is responsible for over 30 human diseases including Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), fragile X-associated tremor and ataxia syndrome (FXTAS), and myotonic dystrophies type 1 and 2 (DM1 and DM2).17 The biological consequences of these repeat expansions will be reviewed in detail below.
To date, two main therapeutic strategies have been employed to target disease-causing RNAs: antisense oligonucleotides (ASOs) and small molecules.18 ASOs are single-stranded nucleotide sequences designed to complementarily base pair a target RNA's primary sequence. ASOs, which often contain modified phosphate backbones and sugar motifs to protect against cellular degradation, either repress translation by sterically blocking ribosomal loading onto the RNA or induce degradation of the target RNA via Ribonuclease H (RNase H).19 RNase H recognizes the RNA–DNA heteroduplex and hydrolyzes the phosphodiester bonds of the RNA strand, cleaving it.19 Conversely, small molecules are designed to target RNA structure instead of sequence, much like how small molecules are designed to target proteins via structure-based recognition. Small molecule binding of an RNA target can modulate disease biology, thus creating avenues to further explore RNA-disease biology and potential therapeutics against RNA-associated disorders.18
This review provides a general overview of recently developed RNA-targeting small molecules, highlighting advances in the field that continue to push towards the development of potent and selective small molecule lead therapeutics. A focus is placed on small molecules targeting miRNA biogenesis, lncRNAs, mRNAs encoding intrinsically disordered proteins (IDPs), and repeat expansion disorders. This review details both the pathomechansims caused by the RNA's structure and how small molecules can alleviate this pathology. Additionally, emergent modalities such as RNA-targeted cleaver and degrader compounds, including ribonuclease targeting chimeras (RIBOTACs), are reviewed in detail, highlighting the selectivity and potency of these compounds. There is still much to be learned about small molecules targeting RNA before these probes can be converted into lead medicines, but a solid foundation has been laid to enable clinical advancement across multiple indications. (See Table 1 for a complete list of diseases mentioned in this review and the abbreviations used to define them.) A tutorial on targeting RNA structures derived from sequence with small molecules can be found in ref. 20.
| Disease | Abbreviation |
|---|---|
| Triple negative breast cancer | TNBC |
| Fragile X-associated tremor and ataxia syndrome | FXTAS |
| Spinal muscular atrophy | SMA |
| Frontotemporal dementia | FTD |
| Parkinsonism linked to chromosome 17 | FTDP-17 |
| Myotonic dystrophy type 1 | DM1 |
| Myotonic dystrophy type 2 | DM2 |
| C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis | c9FTD/ALS |
| Parkinson's disease | PD |
2. Small molecule targeting of miRNAs
MiRNAs are short 20–25 nucleotide (nt) sequences of RNA that negatively regulate gene expression through translational repression of their mRNA targets, dictated by sequence complementarity to the 3′ untranslated region (UTR) of the mRNA target. These RNAs are actively involved in regulation of cellular processes including proliferation, development, differentiation, and apoptosis. Like other types of RNAs, miRNAs are transcribed as primary transcripts (pri-miRs) that are processed into precursor miRNAs (pre-miRs), both of which fold into hairpin structures. These structures are cleaved sequentially by the nucleases Drosha and Dicer to produce the final, single-stranded mature miRNA (Fig. 2A).7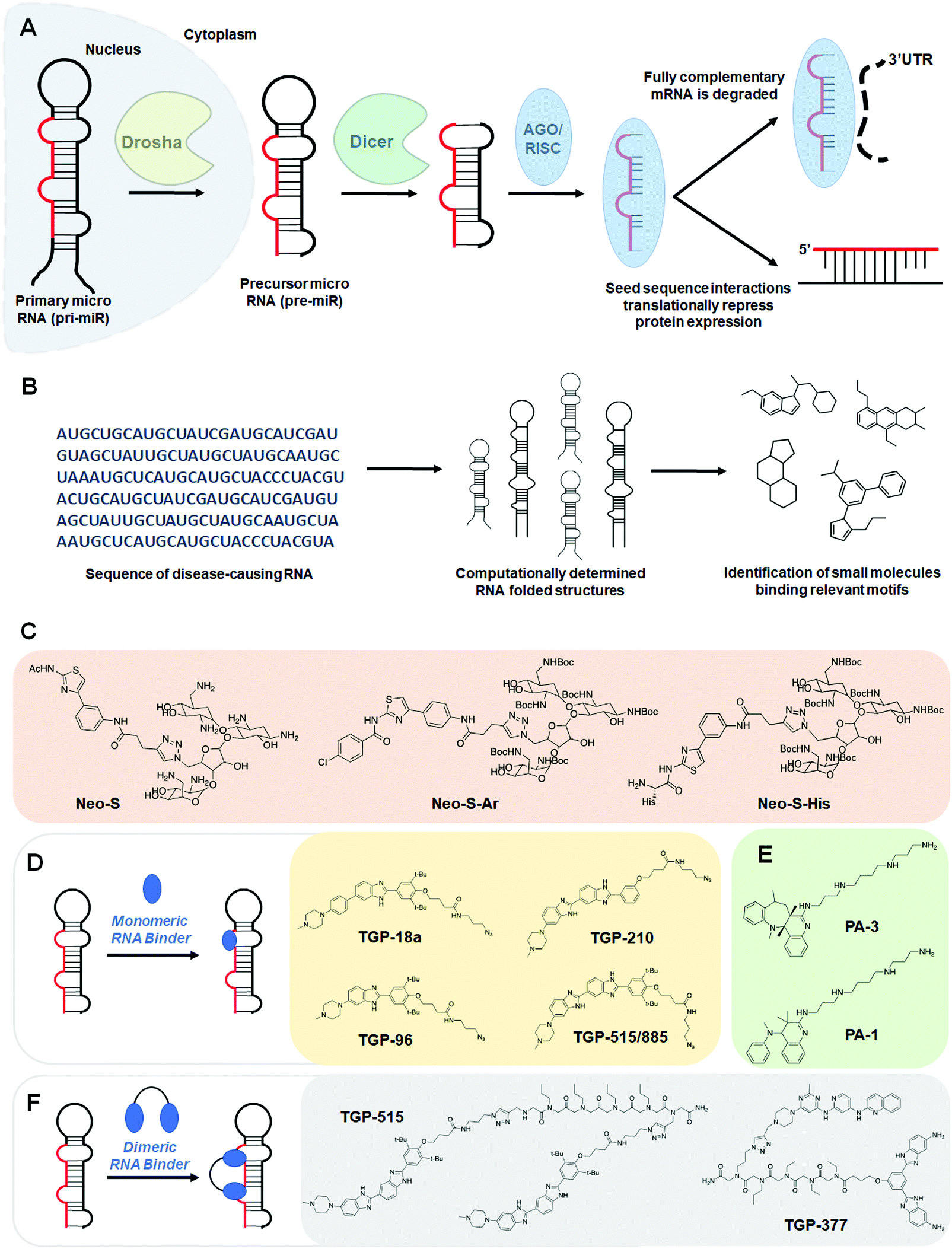 | ||
| Fig. 2 Small molecule targeting of miRNAs. (A) Schematic of the biogenesis of miRNAs. Primary miRNAs (pri-miR) are processed by the nuclear RNase III Drosha and exported to the cytoplasm, affording precursor miRNAs (pre-miR), which are then processed by the RNase III endonuclease Dicer. The miRNA duplex is loaded into the AGO/RISC complex, where the duplex is dissociated and acts through either translational repression or mRNA degradation to downregulate target proteins. (B) Workflow schematic of the Inforna hit identification process. (C) Structures of neomycin conjugates that inhibit miRNA biogenesis. (D) Schematic of monomeric RNA binder mode of action, blocking functional processing sites on miRNA. Representative chemical structures of these monomers are also shown. (E) Structures of polyamines that inhibit miRNA biogenesis. (F) Dimeric RNA binders have improved potency and selectivity by binding to a functional site and nearby druggable motif simultaneously. Structures of representative miRNA-targeting dimers are shown. | ||
Biogenesis begins with the Drosha:DiGeorge syndrome critical region 8 (DGCR8) microprocessor complex that excises a portion of the pri-miR at the open stranded end of the hairpin, yielding a pre-miR of ∼70 nucleotides in length. The pre-miR is then exported from the nucleus via exportin 5.21 In the cytoplasm, pre-miR is further processed at the hairpin loop by the enzyme Dicer, which acts as a molecular ruler, yielding a double stranded miRNA.22 The miRNA is then loaded into the argonaute (AGO)/RISC complex where the guide strand stays successfully loaded and the complementary strand is degraded.23 After biogenesis, the RISC complex regulates gene expression either through translational inhibition via steric hinderance of ribosomal loading or via stimulation of complete mRNA decay.24–26
Due to the complexity of miRNA interaction networks (i.e., multiple miRNAs often act upon one mRNA, and one miRNA can regulate multiple mRNAs),27,28 dysregulation of miRNA expression has been associated with a variety of human diseases, especially cancer.29–31 Examples of how RNA-binding small molecules have been designed and optimized to bring about potent and selective regulators of miRNA function are discussed in detail below.
2.1 Neomycin–nucleobase conjugates targeting oncogenic miRNAs
Neomycin–nucleobase conjugates are small molecules that target disease-causing miR-372 and -373 (Fig. 2C and Table S1, ESI†).32 These bifunctional conjugates consist of (i) an artificial nucleobase designed to specifically recognize an RNA base pair of the double-stranded region of pre-miRNA and (ii) an aminoglycoside shown to have strong binding affinity to stem-loop RNA motifs. Artificial nucleobases engage in the formation of Hoogsteen-type triplex DNA helices,33 and when conjugated to basic amino acids, form compounds with high affinity and selectivity for the stem loop structure of human immunodeficiency virus type 1 (HIV-1) transactivation response element (TAR) RNA.34 Aminoglycoside antibiotics, which alone constitute a class of widely prescribed medicines targeting the decoding A-site in prokaryotic rRNA to inhibit protein translation, bind stem-loop structured RNAs along the major groove of the RNA duplex.On the basis of these findings, the Duca lab rationalized conjugation of the aminoglycoside neomycin with an artificial nucleobase would yield chemical matter capable of binding the stem-loop sequences of miR-372 and -373.32 These first-generation conjugate compounds fortuitously bound the Dicer processing sites of pre-miR-373 and pre-miR-372, inhibiting their biogenesis in vitro, as determined by a cell-free Förster resonance energy transfer (FRET) based assay, and reduced oncogenic burden in cells. The Neo-S conjugate inhibited Dicer cleavage in vitro and rescued expression of the miRNA-regulated protein Large Tumor Suppressor homologue 2 (LATS2). However, Neo-S was not entirely selective and affected expression of miR-17-5p, -21, and -200b in a dose-dependent manner, albeit to a lesser extent than miR-372 and -373 (Table S1, ESI†).
Through medicinal chemistry efforts, Vo et al.35 synthesized and evaluated the properties of second generation compounds with the aim of improving potency and selectivity. Using a cell-free FRET based assay, they learned that select modifications of the artificial nucleobase motif yielded little to no improvement in inhibitory activity. Extended linker length proved deleterious and between a selection of other aminoglycosides, neomycin still remained the best at inhibiting Dicer processing. Preliminary evaluation of compounds offered two new structures for examination in further studies, the first of which quickly fell out of favor due to unspecific binding to both the stem and loop regions of pre-miR-372 and evidence of binding to double-stranded DNA and tRNA. The second structure, Neo-S-Ar, with an improved IC50 relative to Neo-S (1.0 μM versus 2.4 μM for Neo-S), decreases miR-372 and -373 levels in cells in a dose-dependent manner, but much like Neo-S, inhibits Dicer processing of pre-miR-17 and -21 and affects levels of miR-200b in AGS (human Caucasian gastric adenocarcinoma) cells (Table S1, ESI†). The authors noted that Neo-S and Neo-S-Ar only elicit a phenotypic response in AGS cells, which overexpress miR-372 and -373, and not in MKN74 (human gastric tubular adenocarcinoma) cells, which do not overexpress these oncogenic miRNAs. Despite imperfect selectivity, these efforts provided a lead for further drug optimization.
With the aim of improving potency and selectivity for the miR-372 and -373 targets, Vo et al.36 reasoned that because amino acids are natural ligands of RNA and easily interact with negatively charged RNA structures/sequences, appending one such amino acid could improve potency and selectivity.37 The lab had also shown that basic amino acids, including arginine, lysine, and histidine are particularly effective in the design of selective RNA ligands,34,38 inspiring Neo-S-His, which had improved selectivity for pre-miR-372 over previous generations (Neo-S and Neo-S-Ar) (Table S1, ESI†).36 Conjugation of different amino acids appended at various positions on the neomycin–nucleobase scaffold were synthesized, but Neo-S-His was the only compound selective for pre-miR-372 over DNA. Treatment of AGS cells with Neo-S-His showed the compound inhibited cell growth by ∼40% and even though the compound also affected expression levels of oncogenic miR-21, aberrant expression of miR-21 has been shown to have no effect on the proliferation of AGS cells, indicating this off-target did not contribute to the observed anti-proliferative effects. Furthermore, expression levels of other miRNAs, including miR-371, -373, -17, and -200b, were not affected by Neo-S-His, unlike previous generations of the compound (Table S1, ESI†).
2.2 Polyamines targeting oncogenic miRNAs
In addition to neomycin–nucleobase conjugates, Staedel et al.39 screened a 640-member library for inhibition of Dicer-mediated pre-miR-372 processing to identify novel scaffolds with enhanced potency and selectivity. The top hits were all polyamines, the most potent of which, PA-1, inhibited growth of AGS cells, but not MKN74 cells (Fig. 2E and Table S1, ESI†). Treatment of AGS cells with PA-1 also resulted in a dose-dependent accumulation of the downstream protein LATS2, much like the first-generation neomycin–nucleobase conjugate series. PA-1, however, binds and affects expression levels of miRs other than miR-372 in AGS cells (Table S1, ESI†). Binding studies of PA-1 revealed that RNA binding was enhanced most significantly by interactions between the polyamine chain and the RNA phosphate backbone, and less significantly by π–π interactions between the dihydroquinoline motif and specific nucleotides.40 Therefore, a strained analog of PA-1, PA-3, featured a fused benzazepine-dihydroquinoline motif appended to the polyamine chain. Using the previously employed cell-free FRET based assay, it was shown that PA-3 inhibited Dicer processing of pre-miR-372 twice as well as PA-1. Compared to PA-1 and other newly synthesized analogs, PA-3 showed (i) the most selective inhibition of Dicer-mediated processing of miR-372, (ii) the most selective binding of pre-miR-372 in the presence of a large excess of tRNA and DNA, and (iii) the greatest selectivity for pre-miR-372 and pre-miR-373 over other pre-miRNAs in terms of activity and affinity (Table S1, ESI†). Furthermore, thermodynamic binding profiles of the polyamine/pre-miR-372 complex revealed that PA-3 bears the highest enthalpic contribution.2.3 Design of monomeric small molecules targeting disease-causing miRNAs
Additional small molecules targeting miRNAs have been identified by the lead identification strategy dubbed Inforna.41 (For a more in-depth, tutorial review of Inforna and its utilization please see ref. 20.) Inforna comprises a database of experimentally selected RNA motif-small molecule interactions and mines the structural motifs in a chosen disease-related RNA target, deduced from its sequence, for overlap with the database (Fig. 2B). Inforna allows for transcriptome-wide probing of bioactive small molecules that target RNA without target bias (a target agnostic approach). This “bottom-up” strategy has enabled the design of modularly assembled small molecules that bind RNAs linked to human disease, proving particularly successful in the targeting of disease-causing miRNAs. One such example includes the design of Targapremir-18a (TGP-18a), named for its![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif)
![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif) eting of
eting of ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif)
![[- with combining low line]](https://www.rsc.org/images/entities/char_002d002d_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif)
![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) (Fig. 2D and Table S1, ESI†).42In vitro studies showed that TGP-18a inhibits Dicer processing of multiple members of the miR-17-92 cluster, namely pre-miR-17, pre-miR-18a, and pre-miR-20a, which share a common bulge at the Dicer site and adjacent structural similarity. Using RT-qPCR, these in vitro results were corroborated in DU145 prostate cancer cells, in which miR-18a is overexpressed. Importantly, inhibition de-represses serine/threonine protein kinase 4 (STK4) and rescues phenotype, i.e., triggers apoptosis. Interestingly, these studies used Inforna to identify potential miRNA off-targets, that is other miRNAs with binding sites for TGP-18a, albeit with less avidity. The potential off-targets are expressed at much lower levels than the miRNAs in the miR-17-92 cluster, on average about 10-fold less (Table S1, ESI†). Not only were these miRNAs unaffected by TGP-18a, target engagement studies show that they were not bound by the small molecule, demonstrating that differences in target expression level can be exploited to enhance the observed selectivity.
(Fig. 2D and Table S1, ESI†).42In vitro studies showed that TGP-18a inhibits Dicer processing of multiple members of the miR-17-92 cluster, namely pre-miR-17, pre-miR-18a, and pre-miR-20a, which share a common bulge at the Dicer site and adjacent structural similarity. Using RT-qPCR, these in vitro results were corroborated in DU145 prostate cancer cells, in which miR-18a is overexpressed. Importantly, inhibition de-represses serine/threonine protein kinase 4 (STK4) and rescues phenotype, i.e., triggers apoptosis. Interestingly, these studies used Inforna to identify potential miRNA off-targets, that is other miRNAs with binding sites for TGP-18a, albeit with less avidity. The potential off-targets are expressed at much lower levels than the miRNAs in the miR-17-92 cluster, on average about 10-fold less (Table S1, ESI†). Not only were these miRNAs unaffected by TGP-18a, target engagement studies show that they were not bound by the small molecule, demonstrating that differences in target expression level can be exploited to enhance the observed selectivity.
Another example of a miRNA target proven druggable through the use of Inforna is miR-96 (Table S1, ESI†).43 Velagapudi et al.43 showed the compound Targaprimir-96 (TGP-96) reduces miR-96 levels (via inhibition of Drosha processing) at least as selectively as a locked nucleic acid (LNA). In one example, when dosed at concentrations high enough to silence approximately 90% of miR-96 expression, the miR-96 LNA also silenced ∼50% of miR-183 expression, owing to the overlapping seed sequences of the two miRNAs (only the first nucleotide differs). In contrast, TGP-96 only silenced miR-182 expression by ∼15% when dosed at concentrations that silenced miR-96 expression by ∼90% (Table S1, ESI†). Inhibition of miR-96 biogenesis by TGP-96 de-repressed downstream protein expression of Forkhead box O1 (FOXO1), a putative tumor suppressor regulated by miR-96,44 and stimulated apoptosis in MCF-7 breast cancer cells. Importantly, these studies confirmed that TGP-96 acts along the miR-96-FOXO1 circuit by knocking down FOXO1 expression with an siRNA. Indeed, knockdown of FOXO1 reduces TGP-96 activity.
In complementary studies, Costales et al.45 designed TGP-210, a miR-210 binding small molecule that inhibits Dicer processing (Table S1, ESI†). MiR-210 controls hypoxia inducible factors (HIFs) through the negative regulation of glycerol-3-phosphate dehydrogenase 1-like (GPDL1).46In vitro and in triple negative breast cancer (TNBC) MDA-MB-231 cells, cultured under hypoxic conditions, TGP-210 dose-dependently inhibited Dicer processing of pre-miR-210. In cells, this inhibition resulted in rescue of GPDL1 expression, reduction of levels of HIF1-α, and triggering of apoptosis. TGP-210 was selective across a panel of hypoxia-associated miRNAs, as determined by RT-qPCR of treated MDA-MB-231 cells (Table S1, ESI†).
A technique termed Chemical-Cross Linking and Isolation by Pull Down (Chem-CLIP)47 was then used to confirm direct target engagement of pre-miR-210 by TGP-210. In this technique, the small molecule of interest (in this case TGP-210) was appended with cross-linking (ex. chlorambucil) and purification (ex. biotin) modules. Upon compound binding to the target RNA, proximity-induced cross-linking occurs, which results in a complex that can be purified via pull-down with streptavidin beads. The RNAs enriched in the pull-down fraction, relative to the starting lysate, can be determined either through RT-qPCR or RNA-seq to confirm the compound's cellular target. Although expression levels of other mature miRNAs had been shown to be unaffected by TGP-210, Chem-CLIP experiments demonstrated binding does occur to other miRNAs. These studies showed that binding to a functional site is required for bioactivity and confirmed the observation by Velagapudi et al.42 that expression level influences the degree of target occupancy. In vivo studies in NOD/SCID mice showed that treatment with TGP-210 effectively reduces tumor growth via inhibition of miR-210 levels, de-repression of GPDL1, and reduction of HIF1-α levels.
2.4 Design of dimeric small molecules targeting disease-causing miRNAs
The observed selectivity for TGP-210 was fortuitous, as off-targets were bound significantly less avidly and/or at non-functional sites and their expression levels were significantly lower than the desired target. However, such factors are unlikely to align for most targets. Therefore, facile methods to enhance small molecule potency and selectivity would be beneficial. As a test case, Costales et al.48 explored TGP-515/885, a monomeric compound designed using Inforna that binds with dual selectively to the Drosha processing sites of both miR-515 and -885 (Fig. 2D and Table S1, ESI†). While both hairpin miRNA structures bear similar sequences at the Drosha processing sites, miR-515 folds with an additional internal loop adjacent to the Drosha processing site that also binds TGP-515/885. Dimerization of TGP-515/885 yields TGP-515, which binds both the Drosha processing site and the adjacent internal loop to confer selectivity for pri-miR-515 over pri-miR-885 (Fig. 2F). Indeed, TGP-515 bound miR-515 avidly while discriminating against pri-miR-885 in vitro and in cells (Table S1, ESI†). Its >200-fold enhancement in affinity compared to TGP-515/885 translated into a >10-fold boost in potency in cells. Experiments in MCF-7 cells revealed that across all miRNAs, the entire transcriptome, and the proteome, TGP-515 was selective for its RNA target.Interestingly, cellular inhibition of miR-515 biogenesis de-repressed sphingosine kinase 1 (SK1), responsible for the synthesis of the second messenger sphingosine 1-phosphate (S1P), both of which were upregulated by TGP-515 treatment. Activation of this circuit triggers migratory and proliferative characteristics of MCF-7 cells. However, it also enhances levels of human epidermal growth factor receptor 2 (HER2) levels, sensitizing HER2 negative cells (MCF-7 cells) to HER2-targeting precision medicines. Taken all together, this study shows that Inforna can inform how to design a specific compound from a dual-selective monomeric fragment.
In addition to the design of homodimeric molecules, Inforna can be used to design heterodimeric compounds which bind avidly to miRNAs.49 Vascular endothelial growth factor A (VEGFA) stimulates angiogenesis in human endothelial cells and is a sought after target in the treatment of heart failure.50–52 MiR-377 regulates VEGFA expression, and repression of miR-377 by an antisense oligonucleotide has been shown to rescue VEGFA expression and stimulate angiogenesis.53,54 Inforna-based design afforded TGP-377, which binds pre-miR-377 at the Dicer site and another bulge directly adjacent (Fig. 2F and Table S1, ESI†).49 Expression levels of miR-377 from human umbilical vein endothelial cells (HUVEC) treated with TGP-377 were knocked down with an IC50 of ∼500 nM, 10-fold more potently than the lead small molecule monomer (Table S1, ESI†). Accumulation of pre-miR-377 was also observed, demonstrating TGP-377 acts through inhibition of Dicer processing and correspondingly rescues VEGFA expression. A miRNA profiling experiment showed that TGP-377 targets miR-377 selectively, including among miR-377 isoforms (Table S1, ESI†). Global proteomics analysis revealed that TGP-377 affects only 160 of over 4000 unique proteins. A bioinformatic STRING analysis uncovering protein association networks showed, unsurprisingly, cell proliferative pathways including FGFR, Hedgehog, MAP kinase, and ERK were upregulated. Furthermore, TGP-377 induced a pro-angiogenic phenotype in HUVEC cells as evidenced by increased tubule branching density by ∼50% relative to control. As gene therapy is the only known treatment strategy to increase VEGFA expression, TGP-377 represents the first small molecule to do so.50–52,55
3. Small molecule recognition of lncRNAs
LncRNAs are eukaryotic transcripts >200 nt in length that do not encode a protein.56 These RNAs play key regulatory roles in cellular processes such as proliferation, differentiation, and development, the aberrant expression of which can lead to cancer,57 neurodegenerative58 and neuromuscular59 disorders, and immune disorders.60,61 LncRNAs are promising therapeutic targets because of their differential expression between cancerous and normal tissues and their important roles in carcinogenesis.62 Not surprisingly, small molecule screening against lncRNAs has been attracting attention.63–65 In this section, we describe examples of small molecule regulation of lncRNAs.3.1 Small molecule recognition of the lncRNA HOTAIR
The lncRNA HOX transcript antisense RNA (HOTAIR) is involved in several cellular processes associated with carcinogenesis, such as those affecting cell mobility, proliferation, apoptosis, invasion, aggression, and metastasis.66 Additionally, HOTAIR recruits chromatin-modifying complexes, such as polycomb repressive complex 2 (PRC2) and lysine-specific histone demethylase 1 (LSD1) to modulate the cancer epigenome and suppress tumor suppressor genes.67Ren et al.68 used in silico high-throughput screening to identify ADQ as a potent small molecule binder of HOTAIR (Fig. 3A and Table S1, ESI†). In multiple cancer cell lines, ADQ increased expression of nemo like kinase (NLK), a transcriptional target of HOTAIR, in a luciferase assay. Electrophoretic mobility shift assay (EMSA) confirmed ADQ directly binds HOTAIR. To further confirm the functional domains of ADQ, full-length HOTAIR, the 5′ domain, or a mutant 5′ domain construct were stably transfected into U87 and MDA-MB-231 cells. The ADQ-mediated dissociation of HOTAIR and enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) was confirmed using full-length HOTAIR but was not observed with the mutant 5′ domain in which the ADQ binding site was ablated (Fig. 3B).
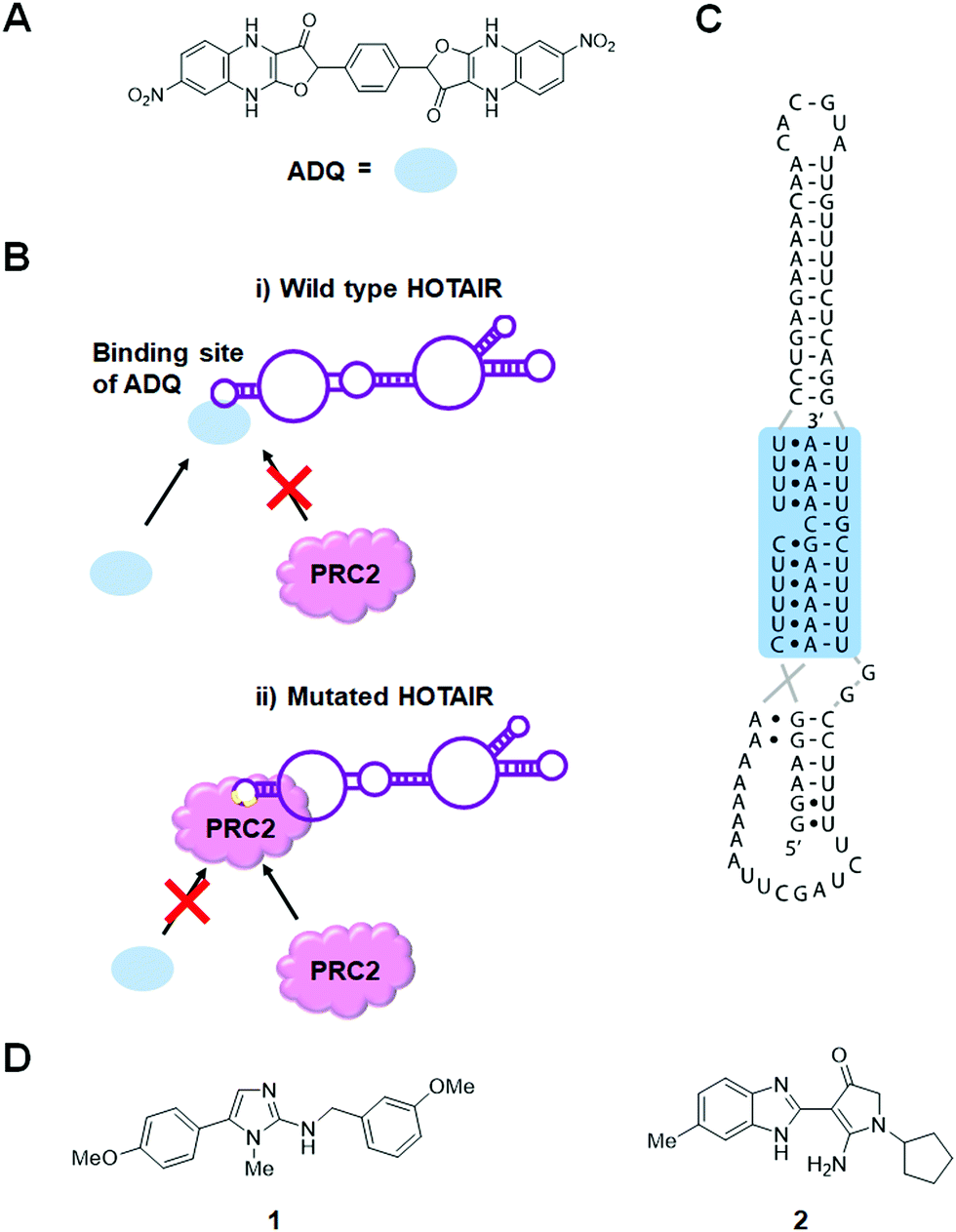 | ||
| Fig. 3 Small molecule inhibition of lncRNAs. (A) Chemical structure of ADQ. (B) ADQ binds to the 5′ domain of HOTAIR and suppresses trimethylation of histone H3 lysine 27 (H3K27) in the promoter region of nemo like kinase (NLK) by weakening HOTAIR's ability to recruit and bind enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), the enzymatic component of the PRC2 complex, thus restoring expression of NLK. (C) MALAT1 triple helix structure. (D) Chemical structures of MALAT1 small molecule binders. | ||
3.2 Small molecule recognition of the lncRNA MALAT1
The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has recently been identified to be upregulated and coupled to tumorigenesis in several cancers.69 MALAT1 has been linked to several physiological processes, including alternative splicing, nuclear organization, and epigenetic modulation of gene expression.70 A study in colorectal cancer cells showed that an ∼1500 nt segment at the evolutionarily conserved 3′ end of MALAT1 was sufficient to increase invasion and proliferation, implying that this region enables its oncogenic function.71 The recent structural characterization of a 74 nt region at the 3′ end of MALAT1 by X-ray diffraction confirmed a unique, bipartite triple helix where the U-rich stem-loop sequesters the A-rich tail, a phenomenon proposed to prevent exonucleolytic degradation (Fig. 3B).72,73 Notably, the deletion of this segment decreased accumulation of the MALAT1 transcript. A comparable decrease in accumulation was also observed upon mutation of a Hoogsteen-positioned uridine, thought to disrupt the triple-helix structure, indicating that subtle alterations in the stability of this structure can lead to significant changes in transcript level.Donlic et al.74 have identified small molecule binders of MALAT1 through in vitro assays. They used furamidine, the tunable diphenylfuran (DPF)-based scaffold, as a starting point because furamidine is known to bind to triple helix structures of various DNAs.75,76 They synthesized a DPF scaffold-based small molecule library, diversified in subunit composition and positioning, to explore the recognition of MALAT1.
Using a small molecule microarray (SMM) strategy, Abulwerdi et al.64 reported the discovery of two structurally unrelated derivatives (1 and 2) that target the triplex region of MALAT1 (Fig. 3C and Table S1, ESI†). Compound 1 was selective for MALAT1 and nuclear paraspeckle assembly transcript 1 (NEAT1), which has a similar structure to MALAT1 (Table S1, ESI†). FRET, isothermal titration calorimetry (ITC) and nuclear magnetic resonance (NMR) spectroscopic experiments confirmed 1 binds to MALAT1 in vitro. However, understanding of the inhibitory mechanism of 1 is limited by the lack of knowledge surrounding the actual mechanism of triplex-mediated protection. Additional research in this area will prove advantageous for the design of therapeutics targeting oncogenic lncRNAs and provide further support for target engagement.
4. Small molecule rescue of repeat-associated transcriptional repression in fragile X syndrome
Currently without a cure, fragile X syndrome (FXS) is the most common hereditary disorder that causes mental retardation, resulting from >200 CGG triplet repeats [full mutation allele; r(CGG)exp] in the 5′ UTR of the fragile X mental retardation 1 (FMR1) gene on the X chromosome.77 The FMR1 promoter is epigenetically silenced through elevated levels of DNA CpG methylation and repressive histone marks H3K9me2, H3K9me3, H3K27me3, and H4K20me3, as well as lower levels of active histone marks H3K9ac, H3K4me2, and H4K16ac. Silencing progresses during embryonic development with the consequent loss of fragile X mental retardation protein (FMRP) encoded by the FMR1 gene.77 Although the mechanism of disease progression of FXS is not fully understood at present, a small molecule targeting the FXS RNA has been discovered that rescued repeat-associated epigenetic silencing.4.1 Small molecule prevents the formation of RNA:DNA hybrids
Colak et al.78 reported that treatment of human embryonic stem cells (hESCs) from FXS patients with 1a, which was discovered using Inforna,79 can prevent epigenetic silencing during neuronal differentiation (Table S1, ESI†). Knockdown of FMR1 mRNA in hESCs decreased silencing histone marks, suggesting the FMR1 transcript is involved in the gene silencing process of its own gene. In the presence of 1a, repressive histone marks induced by differentiation also decreased. Since the compound thermodynamically stabilizes the r(CGG)exp hairpin by binding to its 1 × 1 GG internal loops, it was speculated that the unfolded FMR1 mRNA is responsible for epigenetic silencing. To support this hypothesis, they performed chromatin isolation by RNA purification, a technique used to identify DNA sequences which bind to a specific RNA sequence. These studies showed the FMR1 DNA adjacent to the genomic CGG repeat is highly enriched only in the absence of 1a. Furthermore, treatment with RNase H, which selectively digests RNA:DNA duplexes, significantly reduced the enrichment of the FMR1 promoter. Based on these results, a mechanism was proposed by which FMR1 mRNA containing extended CGG repeats binds to complementary DNA to form the RNA:DNA duplex that induces epigenetic silencing of the FMR1 promoter (Fig. 4A). It was also suggested that 1a promotes CGG stem-loop formation of the FMR1 transcript and thus prevents the formation of the RNA:DNA duplex. In addition, because silencing decreases FMR1 mRNA expression, RNA:DNA duplex formation would be engaged only at the initiation of silencing. In fact, 1a has no effect on silenced cells, as subsequent gene silencing is maintained by other factors.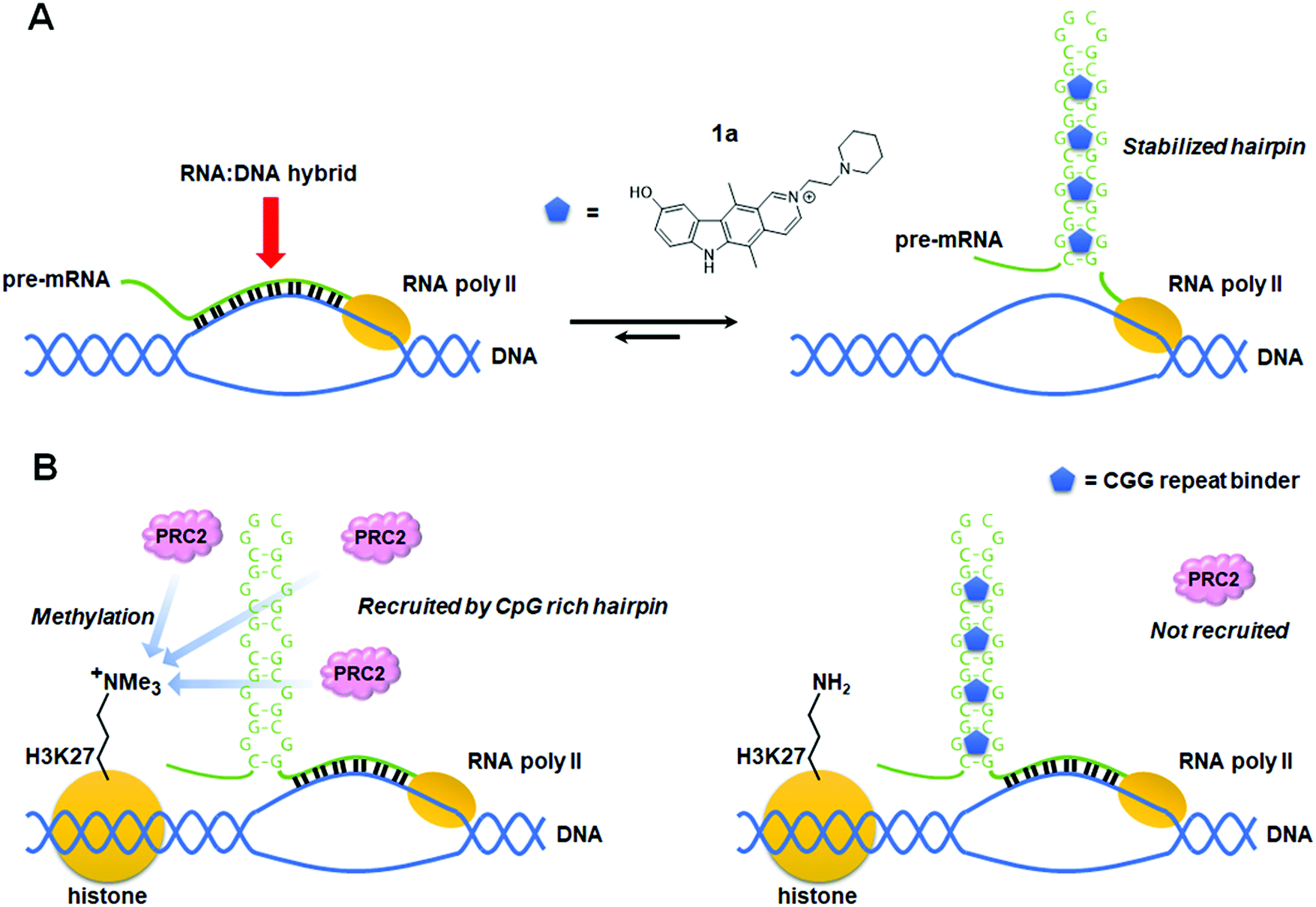 | ||
| Fig. 4 Proposed mode of action of an r(CGG)exp repeat binder that prevents epigenetic silencing of the FMR1 promoter in fragile X syndrome (FXS). (A) Schematic mechanism showing stabilized r(CGG)exp hairpins restrict formation of the RNA:DNA hybrids responsible for epigenetic silencing of FMR1. (B) Schematic mechanism showing binding of a small molecule to the r(CGG)exp hairpin preventing recruitment of polycomb repressive complex 2 (PRC2), a H3K27 methylation enzyme complex. | ||
4.2 Small molecule targeting of r(CGG)exp in combination with 5-azadeoxycytidine
In 2016, Kumari et al.80 proposed that 1a also has an inhibitory effect on histone methyltransferase polycomb repressive complexes 2 (PRC2) recruitment. Treatment of FXS patient cells with 5-azadeoxycytidine (AZA), an inhibitor of DNA methyltransferase 1, has been reported to demethylate the FMR1 promoter and reactivate the FMR1 gene.81,82 Although AZA withdrawal causes re-silencing of the FMR1 gene, this can be greatly delayed in the presence of 1a, but not by inhibiting the RNA:DNA hybrid. Rather, 1a inhibits association of r(CGG)exp with PRC2, interfering with its recruitment to unmethylated CpG motifs and thus slowing FMR1 resilencing in FXS patient cells (Table S1, ESI†).79,83 It is assumed that H3K27 in the FMR1 promoter is methylated by the aberrantly recruited histone methyltransferase. Indeed, it was observed that inhibitors of EZH2, the enzymatic component of PRC2, affect the maintenance of the reactivated state similar to how 1a does. Knockdown of FMR1 mRNA also reduced EZH2 levels associated with the FMR1 gene. Taken together, these data support that 1a is a dual functioning compound, preventing DNA:RNA hybrid formation and the recruitment of PRC2 by binding to the r(CGG)exp stem-loop (Fig. 4B).5. Small molecules modulate alternative splicing
Alternative splicing is a complex, elegant cellular process that allows for variation in protein isoforms to modulate protein function.84 During the splicing process, exons can be included or excluded, giving rise to a variety of splicing isoforms afforded from a single pre-mRNA.84 Mutations that change splicing patterns unsurprisingly cause human diseases including muscular atrophy,85,86 tauopathies,87 β-thalassemia,88 progeria,89 and Pompe disease.905.1 Small molecules modulate SMN2 splicing
Spinal muscular atrophy (SMA) is caused by mutations in the SMN1 gene that decrease levels of survival motor neuron (SMN) protein produced in the spinal cord.86 In humans, SMN1 and SMN2 are the two genes that encode for SMN, but the majority of SMN protein is translated from the full-length mRNA produced from the SMN1 pre-mRNA.91 Due to a C-to-U transition at position 6 on exon 7, exon 7 skipping is dominant in the splicing of SMN2 pre-mRNA,92 producing a truncated SMN protein with a reduced half-life.93 Currently, there are three treatment options for SMA: nusinersen, an ASO that regulates SMN2 splicing to produce the full-length SMN protein;94 onasemnogene abeparvovec, an adeno-associated virus (AAV) carrying the normal SMN1 gene;95 and risdiplam (PTC/Roche), an orally avaliable small molecule that was recently FDA-approved.96,97 Another small molecule therapeutic candidate, branaplam (Novartis), is also currently undergoing clinical trials (Fig. 5A and Table S1, ESI†).96Risdiplam and branaplam generate the SMN protein via regulation of SMN2 splicing. Since both compounds were discovered from phenotypic screening, a series of studies on their modes of action (MOAs) were reported and are discussed below.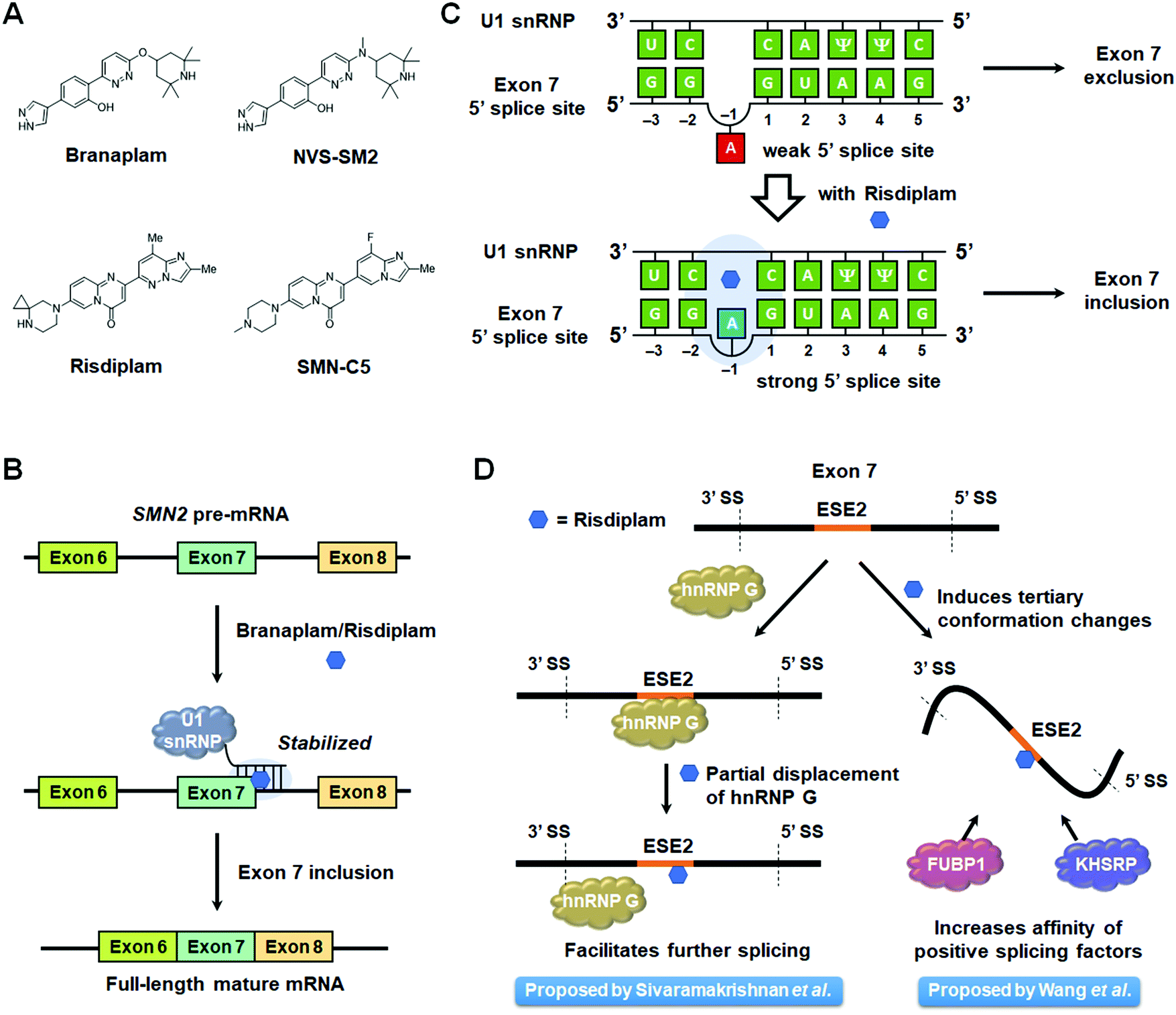 | ||
| Fig. 5 Mode of action of small molecule splicing modulators targeting SMN2 pre-mRNA. (A) Structures of small molecule splicing modulators targeting SMN2 pre-mRNA and the derivatives used to study their mechanisms of action. (B) Schematic mechanism of small molecules facilitating SMN2 exon 7 inclusion by stabilizing the complex between SMN2 exon 7, the 5′ splice site (SS), and the U1 snRNP. (C) Schematic representation of 5′ splice site bulge repair mediated by risdiplam. (D) Competing modes of action proposed for risdiplam. | ||
Risdiplam's MOA was defined using a derivative dubbed SMN-C5 and a duplex model of the 5′ splice site/U1 snRNP complex (Table S1, ESI†).103 The model consisted of 11 nt of the 5′ splice site hybridized to 11 nt of the U1 snRNA. The three-dimensional structure of this model with and without SMN-C5 was defined by NMR spectroscopy, constrained by nuclear Overhauser effects (NOEs). In the binding model of the apo form, the unpaired adenine in the 5′ splice site is located in the minor groove. Upon SMN-C5 binding to the RNA's major groove, the bulged adenine is pushed back into the duplex, stabilized by the hydrogen bond formed between the carbonyl group of SMN-C5 and the amino group of the adenine. Previous structural studies have shown that the U1 snRNP zinc finger stabilizes the minor groove at exon–intron junction of RNA duplexes.101,102 Modeling the apo duplex in the zinc finger produces an obvious steric clash between the bulged adenine and the zinc finger. In contrast, the SMN-C5-bound duplex alleviates this clash, improving the accessibility of the minor groove. Collectively, these studies suggest that SMN-C5 improves splice site recognition by U1 snRNP, facilitating exon 7 inclusion and expression of functional, full length SMN protein (Table S1, ESI†).
The authors then sought to identify potential protein components that may be contributing to SMN2 exon 7 skipping using a pull-down experiment. Ten proteins were enriched only in the presence of SMN-C5, among them heterogenous nuclear ribonucleoprotien (hnRNP) G, a known positive splicing factor that interacts with ESE2.106 Unexpectedly, SMN-C5 partially competes with hnRNP G for ESE2 binding, and small molecule binding alters the RNA structure of the region to which hnRNP G normally binds. Thus, one hypothesis is that partial displacement of hnRNP G by SMN-C5 facilitates the progression of the splicing process (Fig. 5D).
Wang et al.107 also reported that SMN-C2 and SMN-C3, derivatives of risdiplam, act on ESE2. SMN2 exon 7 is known to form two stem-loops, terminal stem-loop 1 (TSL1) and terminal stem-loop 2 (TSL2), that have an inhibitory effect on splicing.108 Both cell-free and cell-based selective 2′ hydroxyl acylation analyzed by primer extension (SHAPE) analysis showed that the addition of SMN-C2 altered the reactivity of some bases in TSL1, suggesting this compound induces conformational changes of this inhibitory loop. Further, proteomics analysis using a photo-cross-linking probe revealed enrichment of far upstream element binding protein 1 (FUBP1)109 and far upstream element binding protein 2 (KHSRP).110 Fluorescence polarization assays with SMN-C2 and recombinant FUBP1 induced higher polarization in the presence of ESE2. These results indicated that SMN-C2, FUBP1, and exon 7 form a ternary complex. Furthermore, EMSA showed the formation of FUBP1–exon 7 complexes are enhanced in a dose-dependent manner by SMN-C3. Based on these results, it was concluded that derivatives of risdiplam interact with ESE2 to induce conformational changes in exon 7 and improve the binding affinity of positive splicing factors (Fig. 5D).
In summary, risdiplam has been proposed to have two modes of action: (i) stabilizing the RNA duplex of exon 7 5′ splice site and U1 snRNP and (ii) inducing conformational changes of exon 7 ESE2 to facilitate the formation of a complex with positive splicing factors. These two modes of action may contribute to risdiplam's high selectivity.
5.2 Small molecule modulation of MAPT pre-mRNA splicing
The small molecules described above direct SMN2 splicing towards exon 7 inclusion. However, many diseases are caused by aberrant exon inclusion and therapeutic benefit is achieved by exclusion of exons. One such example is tauopathies, caused by aggregation of the protein tau, a regulator of microtubule stability that is highly expressed in neurons.111 The microtubule-associated protein tau (MAPT) gene encoding tau is composed of 16 exons and is known to produce six isoforms by alternative splicing.87 Exclusion of exon 10 produces the 3R isoform, with three microtubule binding domains (MBDs), while inclusion produces the aggregation-prone 4R isoform, with four MBDs.112 The ratio of 3R tau to 4R tau is nearly equal in healthy adults (Fig. 6A).113 However, in frontotemporal dementia (FTD) and Parkinsonism linked to chromosome 17 (FTDP-17), genetic mutations of the MAPT gene increase the rate of exon 10 inclusion, and hence the ratio of 4R/3R tau.114 This causes aggregation of tau proteins and ultimately neuronal death.114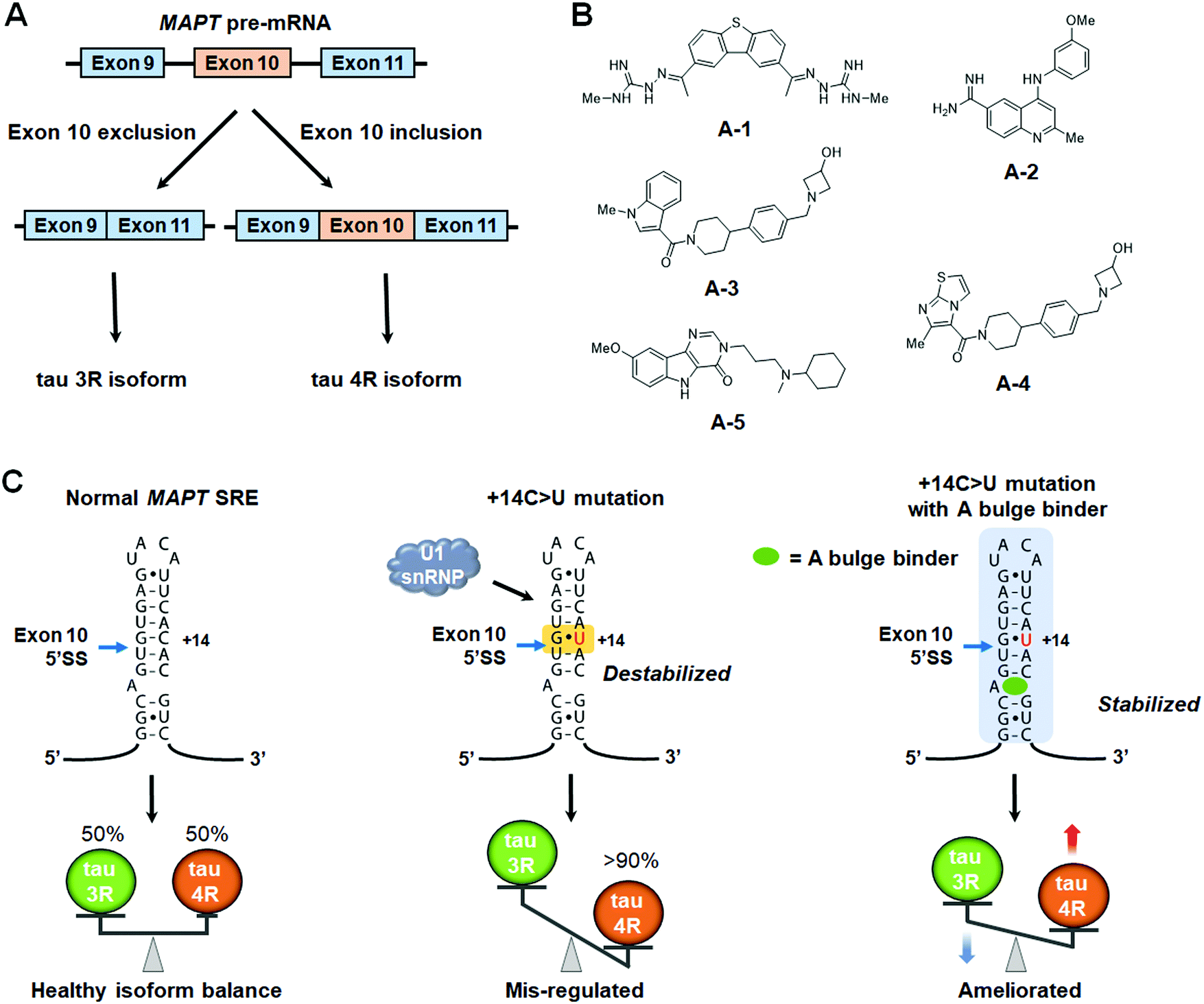 | ||
| Fig. 6 Small molecule modulation of MAPT pre-mRNA splicing. (A) Alternative splicing of MAPT exon 10 yields tau 3R and 4R isoforms. (B) Structures of small molecule splicing modulators that bind to the A-bulge of the MAPT splicing regulatory element (SRE). (C) Schematic representations showing the effect of U1 snRNP accessibility to the MAPT SRE on tau 3R/4R isoform balance. | ||
The 5′ splice site of MAPT exon 10 forms a stem-loop, known as a splicing regulatory element (SRE).115,116 Genetic mutations in the SRE destabilize its structure, increasing the rate of exon 10 inclusion in the mature transcript.115–117 For example, DDPAC is an intronic mutation in which the 14th C downstream from the 5′ splice site is mutated to U, therefore mutating a GC base pair to a GU base pair, thermodynamically destabilizing the SRE by 1.2 kcal mol−1.117 This results in an ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 4R:3R tau isoforms. Thus, thermodynamic stabilization of the tau SRE via small-molecule targeting could be a viable therapeutic option.
:1 ratio of 4R:3R tau isoforms. Thus, thermodynamic stabilization of the tau SRE via small-molecule targeting could be a viable therapeutic option.
One of the first studies to demonstrate the ligandability of the SRE in tau exon 10 showed the anticancer drug, mitoxantrone (MTX) binds and stabilizes the SRE, resulting in decreased production of the tau 4R isoform (Table S1, ESI†).118 Zheng et al.118 reported the NMR structure of the tau pre-mRNA-MTX complex, showing MTX interacts with the bulged region of the SRE stem-loop. The elucidation of this structure highlighted the importance of structure-based recognition between RNA and small molecule ligands as it showed the three-dimensional shape of the RNA was necessary for binding to MTX.118 Additional structure–activity relationships (SAR) were used to optimize MTX's ability to decrease exon 10 inclusion, leading to compounds with enhanced affinity for tau pre-mRNA and increased potency for reducing the levels of 4R tau (Table S1, ESI†).119
More recently, Chen et al.120 reported that stabilizing the SRE by small molecule binding to the A bulge present in the SRE structure could inhibit recognition by U1 snRNP and promote exon 10 exclusion in wild-type (WT) and DDPAC tau. Tanimoto score-based similarity searching using a previously reported Inforna hit121 as a query identified A-1 as a modulator of the 4R/3R tau ratio (Fig. 6B and Table S1, ESI†). To improve physical properties of A-1, in silico-based hit expansions were conducted. As a result, A-2, A-3, and A-4 were obtained from the pharmacophore modeling of A-1, and A-5 was obtained from structure-based design using the three-dimensional structure of the SRE (Table S1, ESI†).
All five compounds not only decrease the 4R/3R ratio by 50% at 10–25 μM, but also had improved physicochemical properties, including potential for blood–brain barrier (BBB) penetrance, compared to A-1 (Table S1, ESI†). In particular, the average central nervous system multiparameter optimization (CNS-MPO) score for A-3, A-4, and A-5 was 4.8; CNS-MPO scores >4 indicate high potential for brain pentrance.122
Target engagement studies using Chem-CLIP confirmed A-5 binds directly to the MAPT pre-mRNA SRE. Furthermore, melting curve analysis showed that hit compounds specifically increased the melting temperature (Tm) of tau SRE, providing experimental evidence that small molecule binding to the A bulge indeed thermodynamically stabilizes the tau SRE and prevents recognition by U1 snRNP (Fig. 6C). To further elucidate the binding mode, three-dimensional structures of the apo-SRE and the compound bound SRE (A-1, A-2, and A-5) were characterized by NMR spectroscopy. In both cases, the RNA duplex was consistent with an A-form helical structure, and all compounds bound to a cavity around the bulged adenine, despite having different binding modes. Altogether, these studies demonstrated that compounds identified using Inforna can be converted to more potent and drug-like compounds possessing the designed RNA-centric mechanism of action.
As is presented here, small molecules can modulate the alternative splicing of pre-mRNAs selectively, either by binding to RNA structural motifs or stabilizing complexes between pre-mRNA and splicing factors. Since aberrant alternative splicing has been associated with various diseases, including rare hereditary diseases,123 central nervous system disorders,124,125 and cancers,126,127 further studies in this field could lead to the development of potent and selective small molecules that can direct splicing outcomes.
6. Small molecules targeting RNA repeat expansion disorders
RNA repeat expansion, or microsatellite, disorders are characterized by long abnormal stretches of repeating RNA nucleotides that can be harbored in intronic, coding, or untranslated regions of pre-mRNAs. These expanded repeats often fold into hairpin structures that interfere with normal RNA processing, leading to disease. Indeed, RNA repeat expansions are responsible for over 30 human diseases, with a large majority being neurodegenerative and neuromuscular in nature.17 Repeats contribute to disease via various mechanisms, including: (i) RNA gain-of-function in which RNA-binding proteins (RBPs) are sequestered and inactivated; (ii) formation of nuclear foci; and (iii) production of toxic proteins, either as a result of canonical translation of an open reading frame (ORF) or as a result of repeat-associated non-ATG (RAN) translation (discussed in Section 8, “Small Molecules Targeting RNA Repeat Expansions Inhibit RAN Translation”).A common RNA gain-of-function pathomechanism in microsatellite disorders is the formation of RNA–protein complexes between the hairpin structures of repeating RNA and RBPs. However, there are various ways by which these complexes lead to disease (Fig. 7). For example, the sequestration of endogenous splicing factors by RNA repeats leads to deregulation of alternative pre-mRNA splicing that affects overall cellular protein levels and homeostasis. Additionally, RNA–protein complexes aggregate in the nucleus in toxic RNA foci, affecting nucleocytoplasmic transport. Thus, the driving idea behind small molecule therapeutics for these disorders is that binding of small molecules competes with RBPs for the disease-causing RNA target, liberating them to fulfill their normal function. In this section, we focus on four neurodegenerative diseases and their associated RNA–protein complexes: (i) the r(CUG)exp–muscleblind-like 1 (MBNL1) complex in myotonic dystrophy type 1 (DM1) where the repeating nucleotides are indicated in parentheses and “exp” denotes “expanded”; (ii) the r(CCUG)exp–MBNL1 complex causative of myotonic dystrophy type 2 (DM2); (iii) the r(CGG)exp–DGCR8 complex that forms a scaffold for splicing regulators Src-associated in mitosis 68 kDa protein (Sam68) and hnRNP in FXTAS; and (iv) the r(G4C2)exp–hnRNP H complex in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis (c9FTD/ALS).
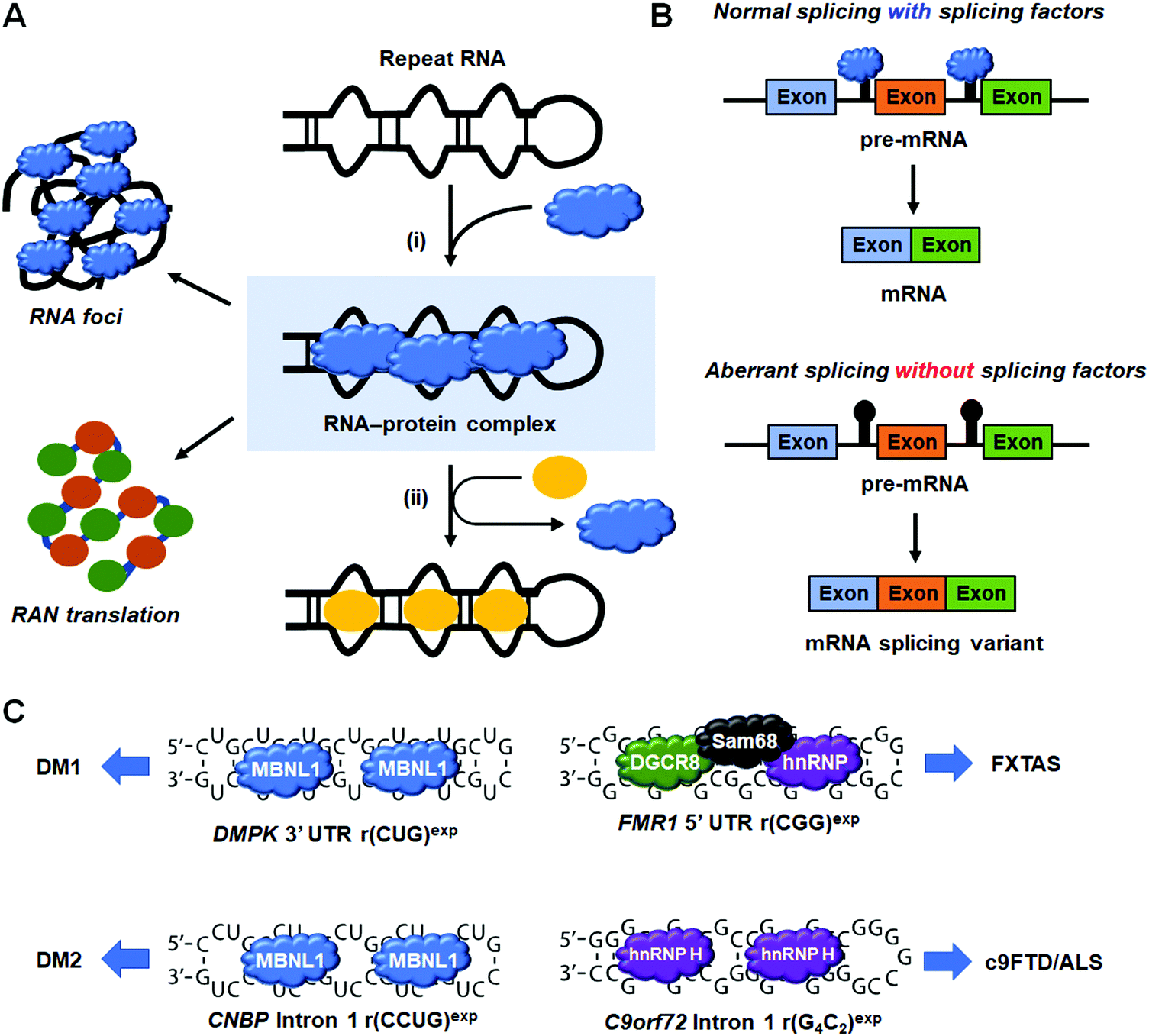 | ||
| Fig. 7 Small molecule binding of RNA repeat expansions releases sequestered RNA-binding proteins (RBPs). (A) Schematic of RBP sequestration by RNA repeat expansions. (i) RBPs, such as splicing factors, are sequestered by RNA repeat expansions, contributing to disease pathology. (ii) Small molecules competitively bind to RNA repeats and release sequestered proteins, resulting in rescue of splicing defects, reduction in RNA foci, and repression of repeat-associated non-ATG (RAN) translation. (B) Schematic of alternative splicing resulting from the presence or absence of endogenous splicing factors. (C) The RNA:protein complexes that contribute to myotonic dystrophy type 1 (DM1), myotonic dystrophy type 2 (DM2), fragile X-associated tremor and ataxia syndrome (FXTAS), and C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis (c9FTD/ALS). | ||
6.1 Small molecule inhibition of the r(CUG)exp–MBNL1 complex in DM1
DM1 is an adult-onset neuromuscular disorder caused by an expanded repeating CUG sequence [r(CUG)exp] in the 3′ UTR of dystrophia myotonica protein kinase (DMPK) mRNA (Fig. 8A). The expanded RNA affects disease biology by folding into a hairpin structure with a periodic array of internal loops that sequester proteins (RNA gain-of-function), such as the splicing factor MBNL1. Sequestration of MBNL1 deregulates the alternative splicing of the protein's natural substrates, which are directly correlated with disease symptoms. For example, MBNL1 regulates the alternative splicing of the muscle-specific chloride ion channel (CLCN1). In DM1-affected cells, CLCN1's aberrant splicing causes loss of the chloride ion channel from the surface of muscle cells, altering conductance and resulting in myotonia. MBNL1 also self-regulates splicing of its own exon 5. In normal cells, exon 5 is included in the mature mRNA sequence ∼45% of the time (Fig. 8B). In DM1-affected cells however, exon 5 is included 85% of the time. In addition, r(CUG)exp–MBNL1 complexes aggregate and form RNA foci in the nucleus that lead to reduction of nucleocytoplasmic transport and result in cytotoxicity.128,129 Being that the r(CUG)exp hairpin plays a key role in DM1 pathology, this structure has become a promising target for small molecule therapeutics.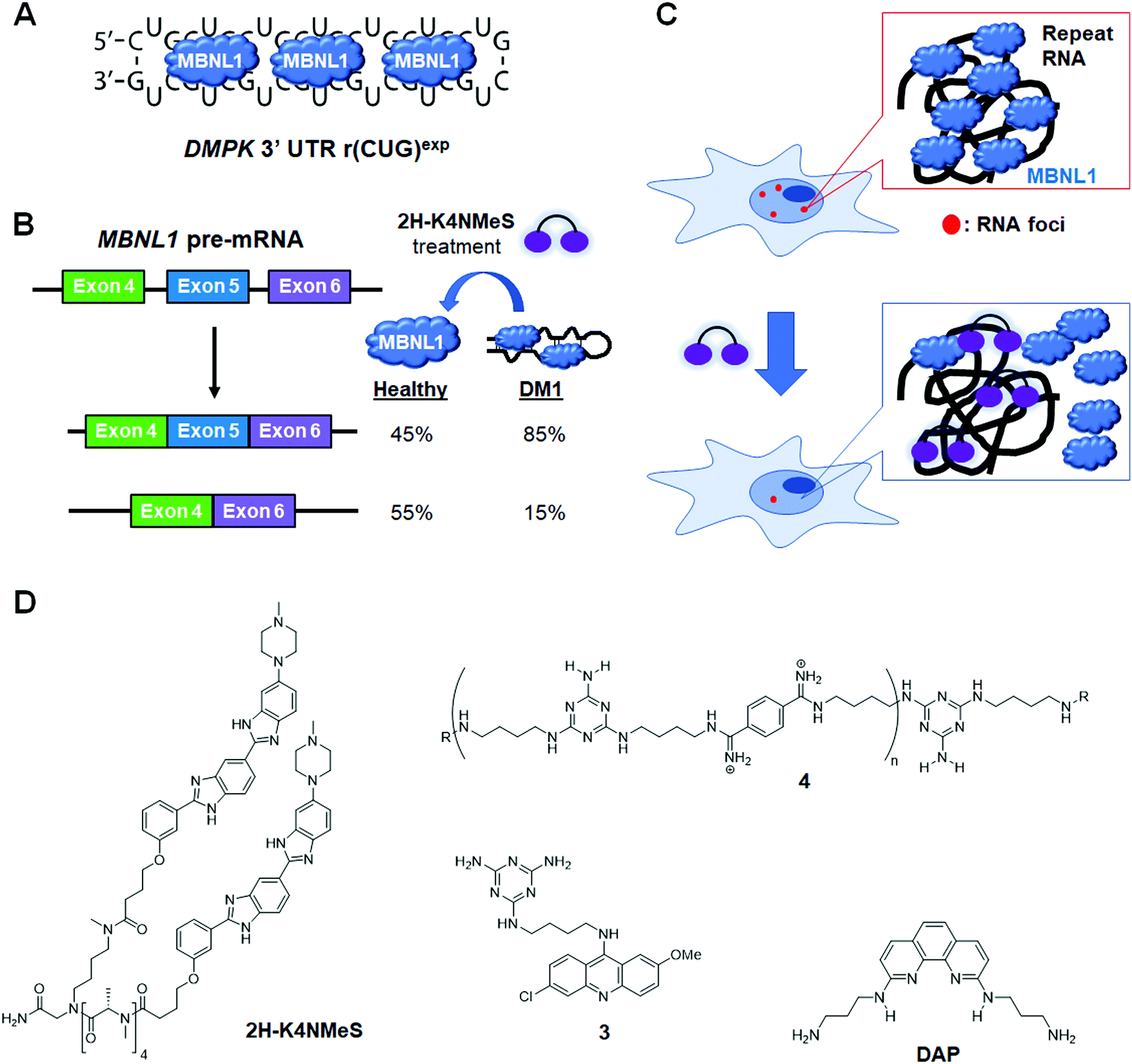 | ||
| Fig. 8 Small molecule targeting of r(CUG)exp, the RNA causative of myotonic dystrophy type 1 (DM1). (A) r(CUG)exp sequesters MBNL1 protein, which regulates alternative pre-mRNA splicing. (B) MBNL1 regulates self-splicing of its own exon 5. Sequestration of MBNL1 by r(CUG)exp results in exon 5 being included in the mature MBNL1 transcript too frequently, contributing to DM1 pathology. (C) Schematic representation of RNA foci formation and disruption by small molecule binding. (D) Structures of compounds that bind r(CUG)exp. | ||
In 2013, Rzuczek et al.130 identified a bis-benzimidazole derivative (H) as a 1 × 1 UU internal loop RNA binder. To target the repeating chain of UU internal loops present in r(CUG)exp, they synthesized a series of H-dimers with various linkers. After assessing rescue of DM1-associated splicing, cellular permeability, cytotoxicity, and proteolytic stability of the compounds, 2H-K4NMe was identified as the most promising ligand (Table S1, ESI†). 2H-K4NMe showed >30-fold binding selectivity to r(CUG)12 over other RNA sequences, with a Kd of 13 nM (Table S1, ESI†). Further, 2H-K4NMe rescued the cardiac troponin T (cTNT) splicing defect at a 5 μM dose.
Based on these findings they developed the dimer 2H-K4NMeS, which displayed enhanced metabolic stability over 2H-K4NMe (Table S1, ESI†).1312H-K4NMeS has Kd's of 280 nM and 12 nM for r(CUG)12 and r(CUG)109, respectively, indicating cooperative binding (Table S1, ESI†). Treatment of DM1-patient-derived cells with as little as 100 nM of 2H-K4NMeS improved the MBNL1 exon 5 pre-mRNA splicing defects (Fig. 8D). 2H-K4NMeS also rescued splicing defects of other MBNL1-regulated splicing events, such as calcium/calmodulin dependent protein kinase II gamma (CAMK2G) exon 14 and nuclear receptor corepressor 2 (NCOR2) exon 45a splicing, and to a similar extent as MBNL1 exon 5. This study clearly showed that RNA-binding small molecules can free MBNL1 from RNA–protein complexes at reasonable concentrations for therapeutic use, thereby normalizing splicing events.
Another mechanism by which RNA–protein complexes contribute to DM1 pathology is by aggregating into RNA foci in the nucleus (Fig. 8C). RNA-binding small molecules are expected to disrupt RNA foci by competitively binding to the RNA, preventing protein binding or releasing bound proteins from the complex. Indeed, 2H-K4NMeS decreased the number of foci present in cells by ∼50% when treated at 1 μM.131 As expected, the activity of 2H-K4NMeS for improving nucleocytoplasmic transport defects was also observed using a firefly luciferase reporter with r(CUG)800 in the 3′ UTR. Disruption of RNA foci was also reported after treatment of cells with compound 3.
To study target engagement, 2H-K4NMeS was converted into a Chem-CLIP probe, 2H-K4NMeS-CA-Biotin.131 This molecule potently rescued splicing defects and reduced the number of nuclear foci when DM1 patient-derived cells were dosed at a 10 nM concentration. In pulled down factions, an ∼13000-fold enrichment of DMPK mRNA was observed, as compared to the starting cell lysate. Using the competitive version of Chem-CLIP, C-Chem-CLIP, in which increasing concentrations of 2H-K4NMeS were co-treated with a constant concentration of 2H-K4NMeS-CA-Biotin, confirmed 2H-K4NMeS and 2H-K4NMeS-CA-Biotin share the same binding site in cells. The specific binding site was further defined by Chem-CLIP-Map,132 confirming binding of the UU internal loops of r(CUG)exp in the DMPK mRNA.
More interestingly, Rzuczek et al.131 demonstrated target-templated oligomerization of an H-dimer in cells (Fig. 9). The designed H-dimer was modified with azide and alkyne moieties at opposite ends of the molecule, allowing oligomerization upon binding r(CUG)exp through click chemistry. This oligomerization only occurred in DM1 cells, as healthy cells lack the repeating RNA necessary to template the reaction. This in situ-produced oligomer rescued splicing defects at concentrations as low as 100 pM in DM1 patient-derived cells.
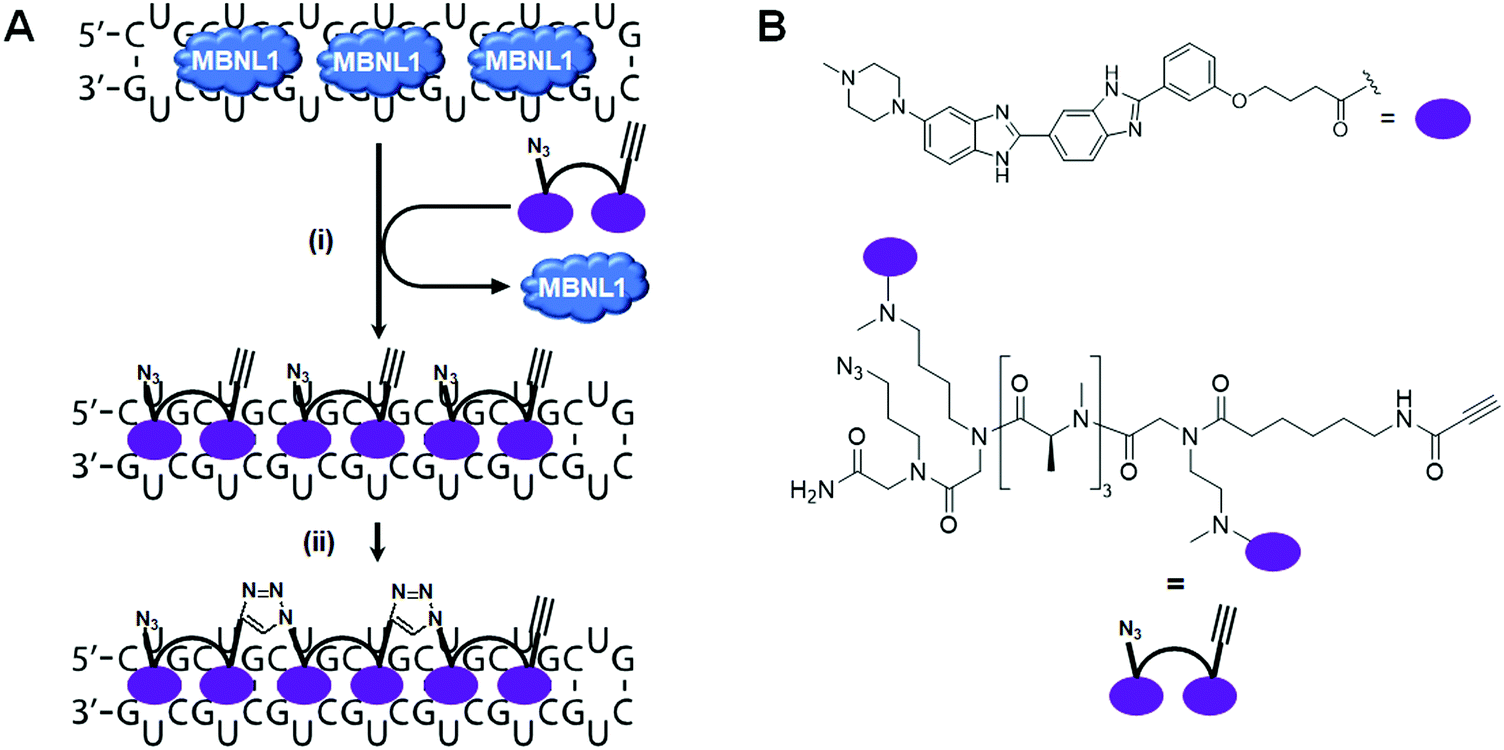 | ||
| Fig. 9 RNA-templated ligand oligomerization catalyzed by r(CUG)exp. (A) In cellulis click chemistry, templated by the RNA repeat expansion, forms an oligomeric compound on-site, that is bound to the r(CUG)exp RNA target. (i) MBNL1 sequestered by r(CUG)exp is released upon binding of the dimeric click compound. (ii) The azide terminus of one dimer reacts with the alkyne terminus of another dimer in close proximity to synthesize an oligomer in cellulis. (B) Structures of the RNA binding motif and dimeric click compound that oligomerizes in cellulis. | ||
Arambula et al.133 developed acridine–triaminotriazine conjugate 3 targeting the r(CUG)exp (Table S1, ESI†). They designed 3 based on the complementary Janus-wedge hydrogen bonding between triaminotriazine and the UU internal loops of r(CUG)exp. This bonding is further stabilized by stacking interactions of the acridine moiety (Fig. 8D). ITC, using a model RNA hairpin, r(CUG)4, revealed 3 has a Kd of 430 nM and 1:1 binding stoichiometry (Table S1, ESI†). However, 3 also binds with similar avidity to d(CTG)2 duplex with a Kd of 390 nM. In vitro, 3 inhibits r(CUG)4–MBNL1 complex formation with an IC50 of 52 μM and a Ki of 6 μM to r(CUG)4, similar to values observed for r(CUG)12. To capitalize on the multiple binding sites (UU internal loops) of the target, bivalent derivatives of 3 were developed.134 A bivalent ligand containing an oligoamino linker was deemed the most superior with improved properties such as aqueous solubility and cell permeability compared to monomeric 3. The dimer inhibited formation of RNA foci in a transfected cellular model of DM1 at 20 μM, and almost complete disruption at 50 μM.
In 2016, Luu et al.135 demonstrated that dimerization of a dimeric compound which has two triaminotriazines linked with bisimidate produced a potent inhibitor of the r(CUG)exp–MBNL1 complex. This intricate “dimer of dimers”, has 1000-fold improved potency in vitro (Ki of 25 nM) compared to the original dimer. This molecule reduced RNA foci by ∼20% when treated at 1 μM in cells, significantly improved splicing defects of insulin receptor (IR) exon 11 (10 μM dose in cells), and alleviated disease phenotypes in a Drosophila model of DM1. However, due to the compounds high molecular weight, it had issues with cellular uptake. To overcome this weakness, Lee et al.136 developed oligomeric ligand 4, composed of triaminotriazine units (targeting the UU internal loops of r(CUG)exp) and bisimidate units (targeting the major groove of RNA) (Fig. 8D and Table S1, ESI†). Although 4 is still too large to permeate the cell membrane, its poly-cationic nature makes it membrane penetrant by endocytosis. Using 200 nM of 4, they showed full rescue of IR mis-splicing and a decrease in foci number in a transfected model of DM1. However, 4 also inhibits transcription of d(CTG)exp, indicating the compound is not specific for the RNA repeat (Table S1, ESI†). They used adult DM1 Drosophila (CTG480) to investigate the in vivo effects of 4 by measuring the improvement of climbing defects observed after treatment with the compound. Approximately 80% of untreated files show significant defects in their ability to climb, but this was rescued by treatment with 4 (80 μM; 37% fail to climb). In addition, in a liver-specific DM1 mouse model containing 960 interrupted CUG repeats, 4 decreased the levels of the transgene, likely due to the compound's inhibitory effect on d(CTG)exp transcription, improved pre-mRNA splicing defects, and reduced RNA foci formation, highlighting the compound's potential in preclinical animal models.
As another example of an r(CUG)exp binding molecule, Li et al.137 designed a 1,10-phenanthroline derivative (DAP) and studied its effect by in vitro translation (Fig. 8D and Table S1, ESI†). Using a transfected template RNA with r(CUG)20 inserted between Renilla luciferase (Rluc) and firefly luciferase (Fluc) showed treatment with DAP suppressed translation of Fluc downstream of the repeat sequence in a concentration-dependent manner. The translation of Rluc was also moderately affected by DAP treatment. The selectivity of DAP was assessed by SPR and melting temperature, revealing DAP shows preferential binding to r(CUG)9 and r(CCG)9 among r(CXG)9 sequences (X = A, U, G, or C) (Table S1, ESI†). Furthermore, electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) analysis showed DAP binds to r(CUG)9 with an RNA:compound ratio of 1:4.
6.2 Small molecule inhibition of the r(CCUG)exp–MBNL1 complex in DM2
DM2 is caused by r(CCUG)exp in intron 1 of CCHC-type zinc finger nucleic acid binding protein (CNBP) pre-mRNA (Fig. 10A). As observed in DM1, r(CCUG)exp sequesters MBNL1, causing MBNL1-dependent splicing defects and RNA foci formation, but also causes aberrant splicing of CNBP intron 1 (intron retention). To target DM2, Lee et al.138 developed a dimeric kanamycin compound (5) that inhibits formation of r(CCUG)12–MBNL1 complexes in vitro with an IC50 of ∼90 nM (∼2500-fold more potent than the monomer; IC50 = ∼220 μM) (Table S1, ESI†). Compound 5 demonstrated good cellular permeability and localized to both the nucleus and cytoplasm. In DM2 fibroblasts, 5 (10 μM) successfully rescued IR splicing defects and significantly reduced the number of RNA foci (Fig. 10B and C).139 These activities were further improved by the incorporation of a cleavage module on the ligand (discussed in Section 10.4, “Targeted Cleavage of r(CCUG)exp by a Small Molecule–Bleomycin A5 Conjugate”).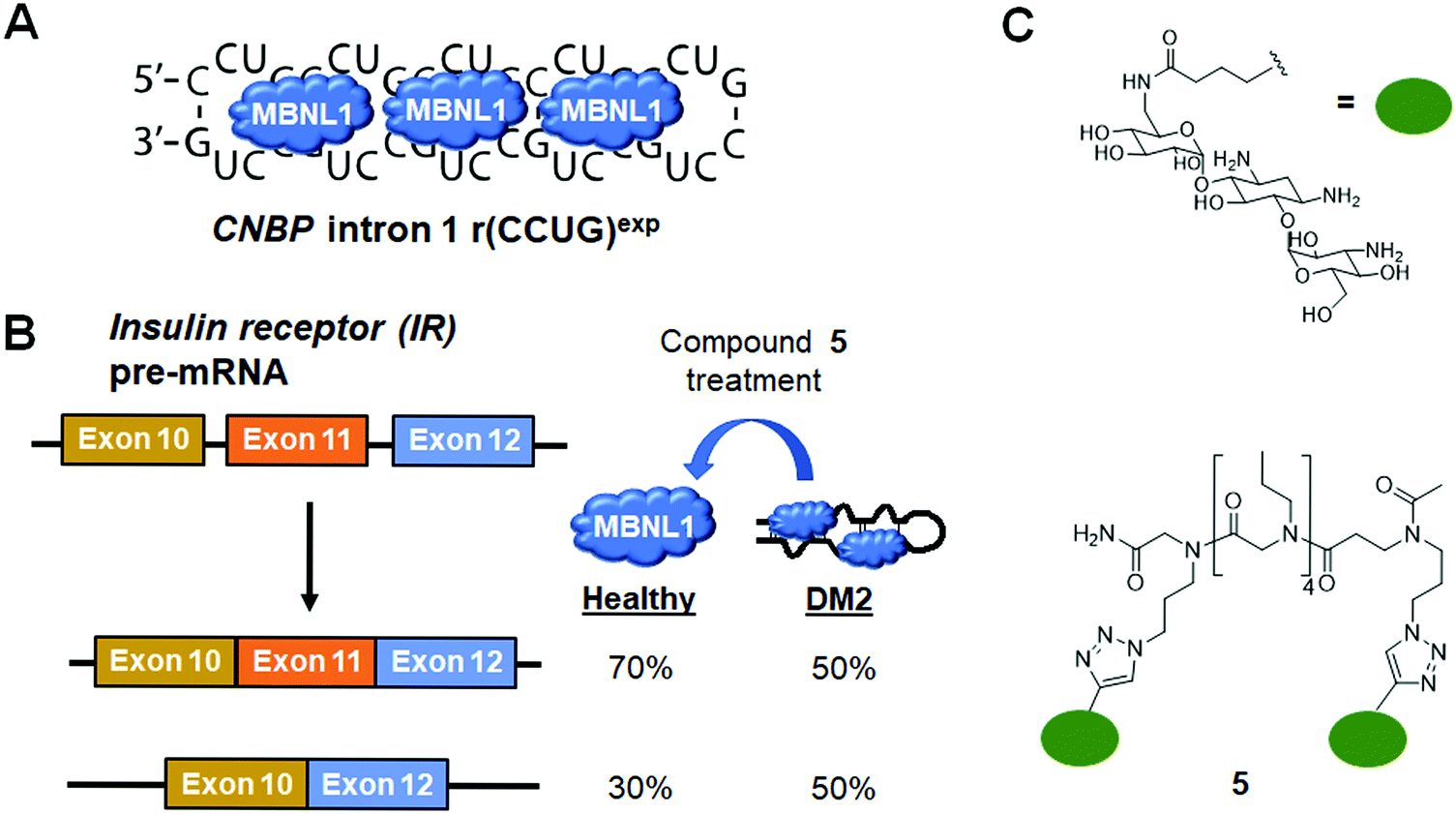 | ||
| Fig. 10 Small molecule targeting of r(CCUG)exp, the causative agent of myotonic dystrophy type 2 (DM2). (A) r(CCUG)exp sequesters MBNL1. (B) MBNL1 regulates splicing of insulin receptor (IR) exon 11. Aberrant splicing results in exon 11 being excluded from the mature IR transcript, contributing to DM2 pathology. (C) Structures of the kanamycin RNA-binding motif and dimeric compound that bind r(CCUG)exp. | ||
Similar to the case shown with DM1 (Fig. 9), incorporation of azide and alkyne moieties into the kanamycin RNA-binding module afforded target-templated oligomerization in DM2 patient-derived cells.140 When DM2 fibroblasts were treated with this clickable molecule (1 μM), the number of foci observed was reduced by ∼45% and IR exon 11 splicing defects were rescued by approximately the same percentage. These results clearly indicated the activity of the compound was far improved by on-site, in situ oligomerization. This oligomeric molecule also affected aberrant splicing of CNBP mRNA by inhibiting binding of MBNL1 to intron 1, thus allowing the intron to be properly spliced out of CNBP pre-mRNA (discussed in Section 7, “Small Molecules Shunt Toxic RNA to Endogenous Decay Pathways”).
6.3 Small molecule inhibition of the r(CGG)exp–protein complexes in FXTAS
In FXTAS, expanded r(CGG) repeats of lengths >55 but <200 (premutation allele) in the 5′ UTR of FMR1 mRNA cause disease (Fig. 11A). The repeat folds into a hairpin structure with repeating 1 × 1 GG internal loops that sequester several proteins, such as DGCR8, Sam68, and hnRNP. Because these proteins have important roles in RNA biogenesis, their sequestration alters pre-mRNA splicing, thus resulting in disease.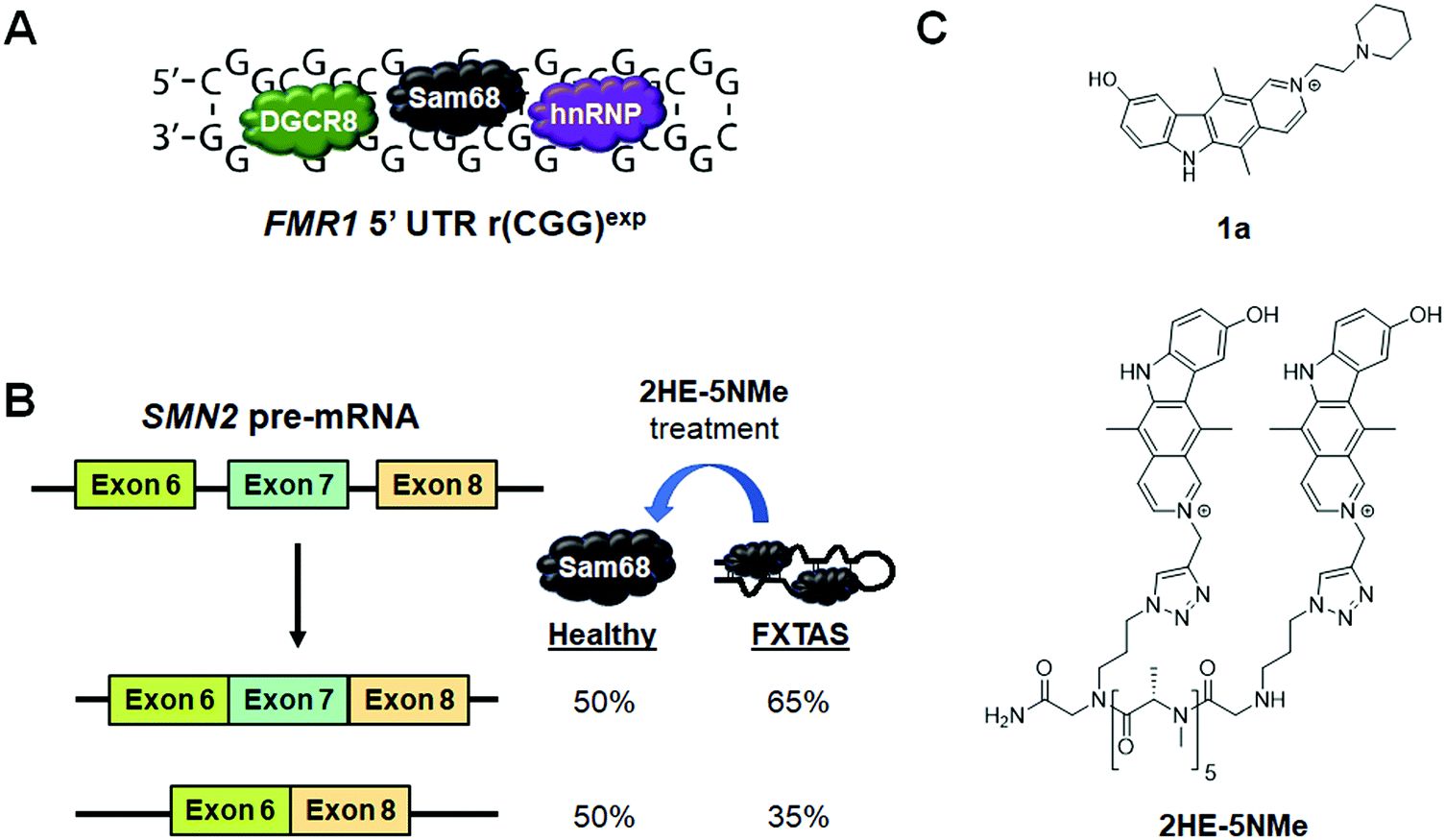 | ||
| Fig. 11 Small molecule targeting of r(CGG)exp, the RNA causative of fragile X-associated tremor ataxia syndrome (FXTAS). (A) r(CGG)exp sequesters proteins such as DGCR8, Sam68, and hnRNP. (B) Sam68 regulates splicing of SMN2 exon 7. Thus, its sequestration results in increased exon 7 inclusion in the mature SMN2 transcript, contributing to FXTAS pathology. (C) Structures of compounds that bind to r(CGG)exp. | ||
Disney et al.79 previously identified compound 1a as a binder to r(CGG)exp by screening compounds using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay that monitors r(CGG)12–DGCR8Δ complex formation and SAR (Fig. 11C and Table S1, ESI†). In particular, 1a disrupted the r(CGG)12–DGCR8Δ complex with an IC50 of 12 μM, in the presence of competitor tRNA. Sequestration of Sam68 by r(CGG)exp dysregulates splicing of SMN2 mRNA, therefore the ability of 1a to improve Sam68-regulated splicing defects was assessed (Fig. 11B). In transfected COS7 cells, r(CGG)exp causes SMN2 exon 7 to be included too frequently (∼70% compared to 30% in healthy cells). Upon treatment of these cells with 1a (20 μM), improvement of the SMN2 splicing defect was observed while improvement of another Sam68-regulated splicing event, apoptosis regulator Bcl-X (Bcl-x) exon 2, was observed upon treatment with 100 μM of 1a (Table S1, ESI†). 1a (10 μM) also reduced the number of RNA foci, as studied by RNA fluorescence in situ hybridization (FISH).79
As discussed previously, dimerization of RNA-binding modules is a powerful and easy method to obtain highly potent and selective compounds. Thus, a dimeric derivative of 1a, 2HE-5NMe, was designed and studied (Fig. 11C and Table S1, ESI†).141 Inhibition of the r(CGG)12–DGCR8Δ complex by 2HE-5NMe was assessed by TR-FRET and revealed the compound inhibits complex formation with 6-fold greater activity than monomeric 1a (IC50 = 3.5 μM in the presence of tRNA). Further, 2HE-5NMe has 16-fold higher affinity for r(CGG)12 (Kd = 50 ± 0.6 nM) than 1a, which translates into a 3-fold higher occupancy in cellulis, as revealed by Chem-CLIP studies (Table S1, ESI†).141
The activity of 2HE-5NMe for rescuing splicing defects was assessed by exon 7 inclusion in SMN2 mRNA. Treatment of 2HE-5NMe at 50 μM rescued exon 7 inclusion levels back to those observed in wild type cells, demonstrating a >10-fold increase in activity over 1a. While 1a can inhibit foci formation but not disrupt existing foci, 2HE-5NMe has the ability to do both (∼70% reduction at 50 μM). It should be noted that most foci in this study were dissolved within 1 h of treatment and fully dissolved after 4 h. Recovery of SMN2 splicing defects were observed in parallel to this time course. The effect of these compounds on RAN translation is discussed in Section 8.2, “Small Molecules Targeting the r(CGG)exp in FMR1 Inhibit RAN Translation”.
6.4 Small molecule inhibition of the r(G4C2)exp–hnRNP H complex in c9FTD/ALS
An expanded repeat of G4C2 [r(G4C2)exp] in intron 1 of C9orf72 mRNA is the most common genetic cause of the neurodegenerative disease c9FTD/ALS (Fig. 12A). The structure of r(G4C2)exp has been well-studied, revealing the repeating RNA can adopt two main structures, a hairpin with an array of internal loops and a G-quadruplex. Because the hairpin form of r(G4C2)exp forms the same 1 × 1 GG internal loops as r(CGG)exp, Su et al.142 hypothesized that 1a might also bind to r(G4C2)exp. Using 1a as a lead, a library of chemically similar compounds was created and screened for binding r(G4C2)8 using a dye displacement assay. The screen revealed 1a and two additional compounds, 6 and 7, bind r(G4C2)exp (Fig. 12B and Table S1, ESI†), with Kds of 9.7, 10, and 16 μM, respectively (Table S1, ESI†). To assess the biological activities of each compound, foci formation was evaluated in r(G4C2)66-expressing COS7 cells. Compounds 1a and 6, but not 7, showed a 3-fold reduction of foci-positive cells. This reduction in foci by 1a can be traced to its direct engagement of r(G4C2)exp, as determined by Chem-CLIP, which revealed an 80-fold enrichment of r(G4C2)66 in the pulled down fractions, as compared to 18S rRNA.142 C-Chem-CLIP studies where r(G4C2)66-expressing COS7 cells were co-treated with 1a and its Chem-CLIP probe verified target engagement by the parent compound.142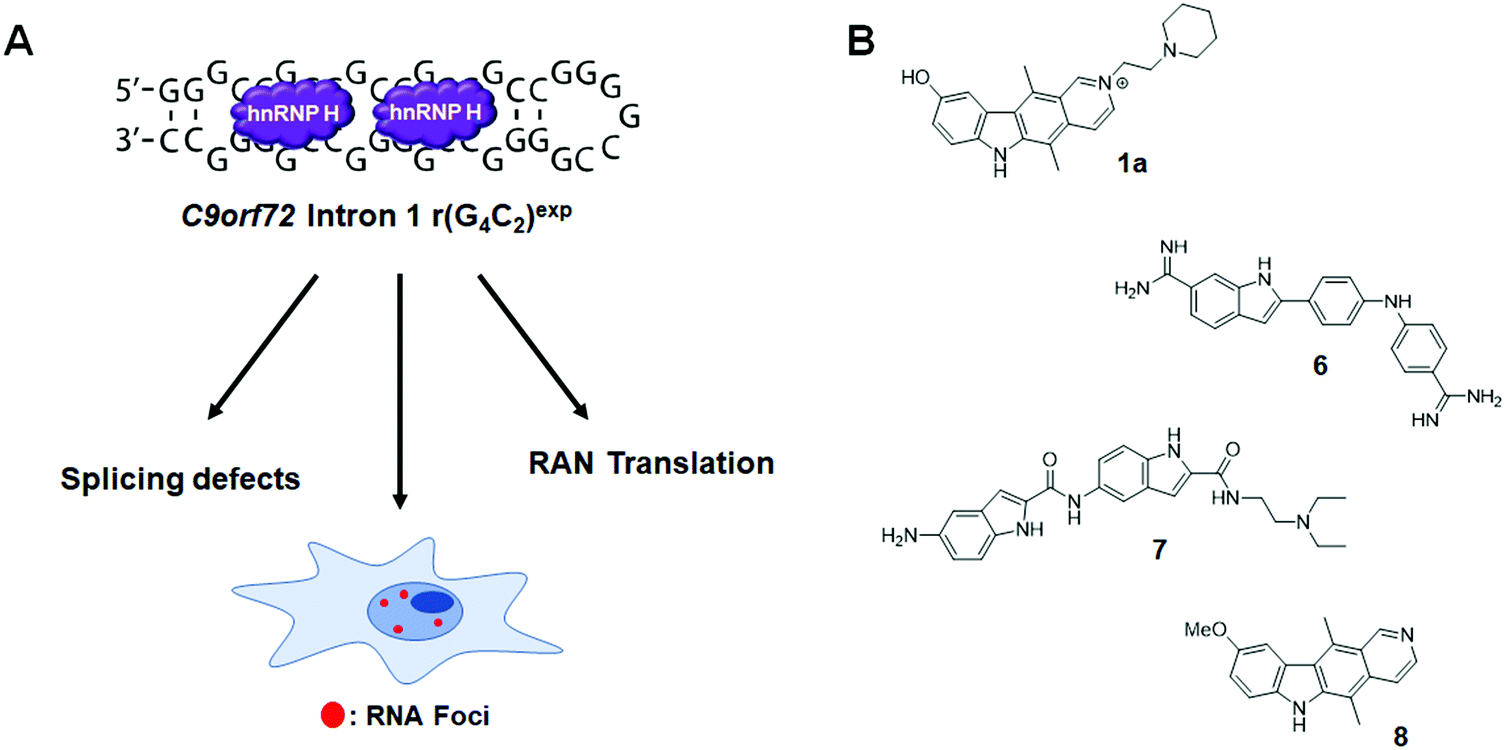 | ||
Fig. 12 Small molecule targeting of r(G4C2)exp, the most common genetic cause of C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis (c9FTD/ALS). (A) r(G4C2)exp sequesters hnRNP H, resulting in splicing defects. The repeat expansion also undergoes RAN translation and forms RNA foci. (B) Structures of compounds that bind to r(G4C2)exp. Compound 1a also binds to r(CGG)exp due to the 5′-C![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) G/3′-GC binding site shared between the two repeats. G/3′-GC binding site shared between the two repeats. | ||
Interestingly, C9orf72 mRNA is bidirectionally transcribed and thus repeats from the sense [r(G4C2)exp] and antisense [r(G2C4)exp] strand are produced. Like the sense strand, r(G2C4)exp also forms nuclear inclusions. However, the antisense foci were not reduced by the treatment of 1a, confirming its selectivity for the sense strand. Furthermore, 1a showed significant reduction of RNA foci-positive cells in three C9ORF72+ induced neuron (iNeuron) lines.
In a subsequent study,1431a was further optimized, affording 8 (Fig. 8 and Table S1, ESI†). Compound 8 binds to r(G4C2)8 with a Kd of 0.26 μM, while showing ∼300-fold weaker binding to antisense r(G2C4)8 and ∼540-fold weaker binding to base-pair control r(G2C2)8 (Table S1, ESI†). With the remarkable binding affinity of 8, the binding mechanism was further investigated by NMR spectroscopy. In brief, 8 stacks between GG internal loops and closing GC base pairs to stabilize the closing base pairs with π–π interactions. In vitro, 8 inhibited the r(G4C2)8-hnRNP H complex with an IC50 of 19 μM and reduced both the number of foci-positive cells and the number of foci present per cell by half in HEK293T cells transfected with a plasmid encoding r(G4C2)66 (5 μM dose). Therefore, 8 is a potent and selective small molecule capable of alleviating disease-associated phenotypes in cellular models of c9ALS/FTD. The inhibition of RAN translation by 8 is discussed in Section 8.3, “Small Molecules Targeting the r(G4C2)exp in C9orf72 Inhibit RAN Translation”. Most importantly, these studies with 8 revealed that the hairpin form of r(G4C2)exp, not the G-quadruplex, is RAN translated.
7. Small molecules shunt toxic RNAs to endogenous decay pathways
As discussed in Section 6.2 (“Small Molecule Inhibition of the r(CCUG)exp-MBNL1 Complex in DM2”), r(CCUG)exp causes CNBP intron 1 retention. Although formation of nuclear foci and splicing defects have been well studied in DM2, intron retention was only recently discovered by the Swanson group (∼40% retained in DM2-affected cells vs. ∼10% in healthy cells) (Fig. 13A).144 Intronic regions of pre-mRNAs are normally subjected to endogenous decay upon liberation, but in DM2 the intron containing the repeat expansion remains present in the mature mRNA transcript.145 Shortly after this discovery, 5, previously reported to target r(CCUG)exp and alleviate DM2-associated defects, was employed as a chemical probe to investigate the mechanism of intron retention (Table S1, ESI†).140 These studies showed that binding of MBNL1 causes intron retention and that small molecules can alleviate this retention by shunting the intron to endogenous decay pathways.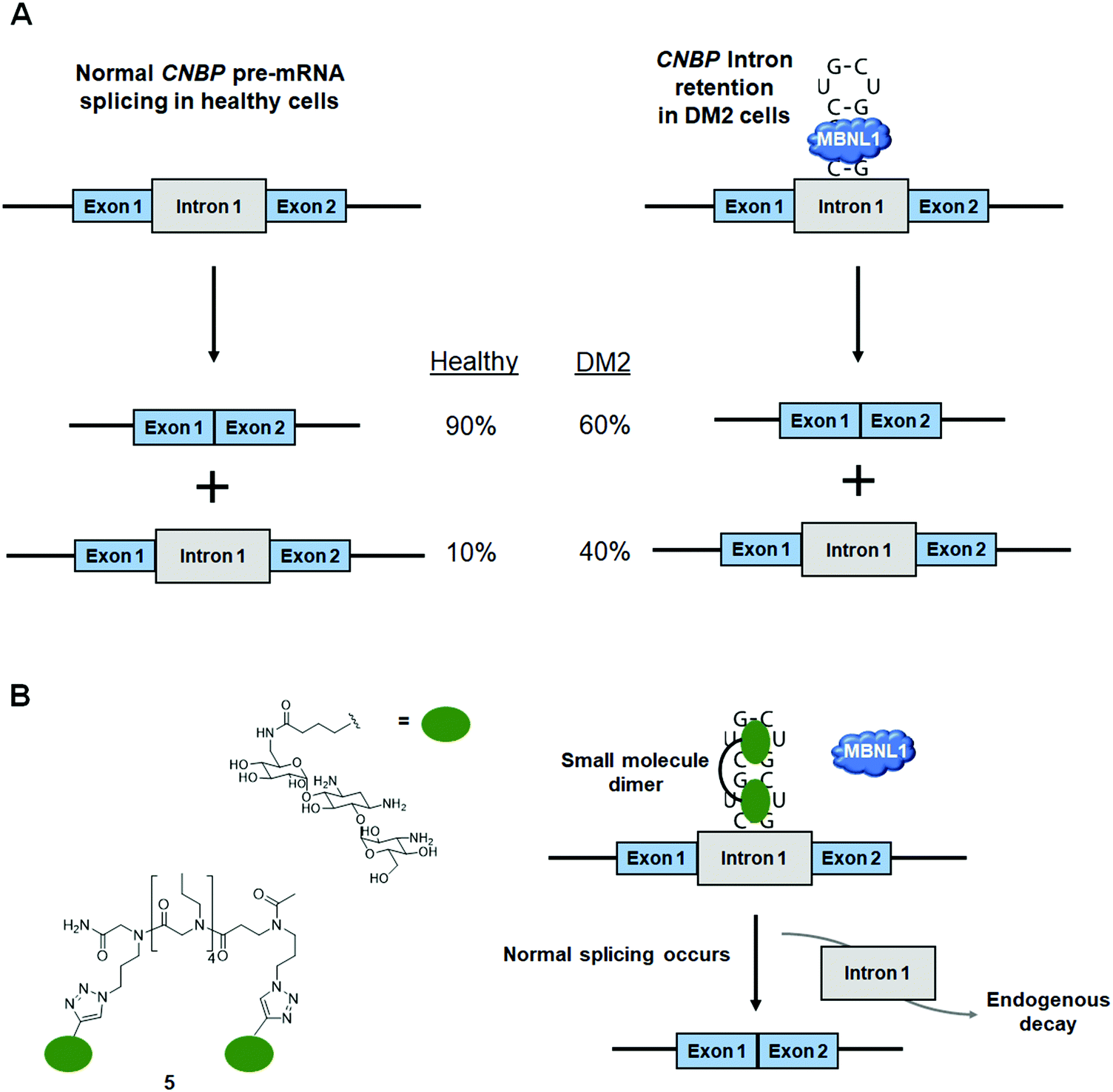 | ||
| Fig. 13 Small molecule binding causes toxic RNAs to be shunted to endogenous decay pathways. (A) MBNL1 sequestration by r(CCUG)exp results in CNBP intron 1 retention. (B) Small molecule binding frees MBNL1 and allows for proper intron splicing to occur. The excised intron is shunted to endogenous decay pathways. | ||
Treatment of DM2 patient-derived cells with 5 (1–10 μM) led to ∼15–20% of the retained intron being eliminated, while no effect was observed on CNBP mature mRNA levels (Fig. 13B),140 suggesting a mechanism by which small molecule binding of the r(CCUG)exp shunts pathogenic RNAs to endogenous quality control pathways. Notably, there are a variety of disease-causing RNA repeats harbored in introns that lead to intron retention, such as c9FTD/ALS caused by r(G4C2)exp and Fuchs endothelial corneal dystrophy (FED) caused by r(CUG)exp. Small molecule intervention in these cases may have similar cooperative effects with endogenous RNA decay mechanisms to be therapeutically advantageous.
8. Small molecules targeting RNA repeat expansions inhibit RAN translation
8.1 RAN translation in microsatellite diseases
An additional pathomechanism found in some neurodegenerative RNA repeat expansion disorders, such at r(CGG)exp and r(G4C2)exp, is RAN translation.146–150 In this phenomenon, repeat expansions serve as non-canonical translation initiation sites, thus giving rise to homopolymeric, as in the case of r(CGG)exp,149,150 or dipeptide repeat (DPR) proteins, as in the case of r(G4C2)exp.146,148 These proteins are intrinsically disordered and form neurotoxic aggregates that contribute to disease pathology.151 Therefore, small molecules that inhibit RAN translation are of high therapeutic importance.8.2 Small molecules targeting the r(CGG)exp in FMR1 inhibit RAN translation
In FXTAS, RAN translation produces the homopolymeric protein poly(G) (Fig. 14A).149,150 As 1a and 2HE-5NMe (Fig. 14B and Table S1, ESI†) were shown to bind r(CGG)exp selectively both in vitro and in cellulis (as determined by Chem-CLIP),79,141 their ability to inhibit RAN translation was also assessed. Interestingly, both 1a and 2HE-5NMe thermally stabilize r(CGG)12 (by 1.4 and 0.9 kcal mol−1 respectively),141 suggesting they may prevent ribosomal readthrough or loading and thereby inhibit RAN translation. In agreement with their similar degree of stabilization of r(CGG)12, 1a and 2HE-5NMe inhibited RAN translation to a similar extent (∼80% inhibition at 50 μM) as well as reduced the number of poly(G) nuclear inclusions.141 Notably, both compounds reduced polysome loading onto r(CGG)88, as hypothesized.79,141 Importantly, neither compound affects mRNA levels or canonical translation of the downstream ORF.79,141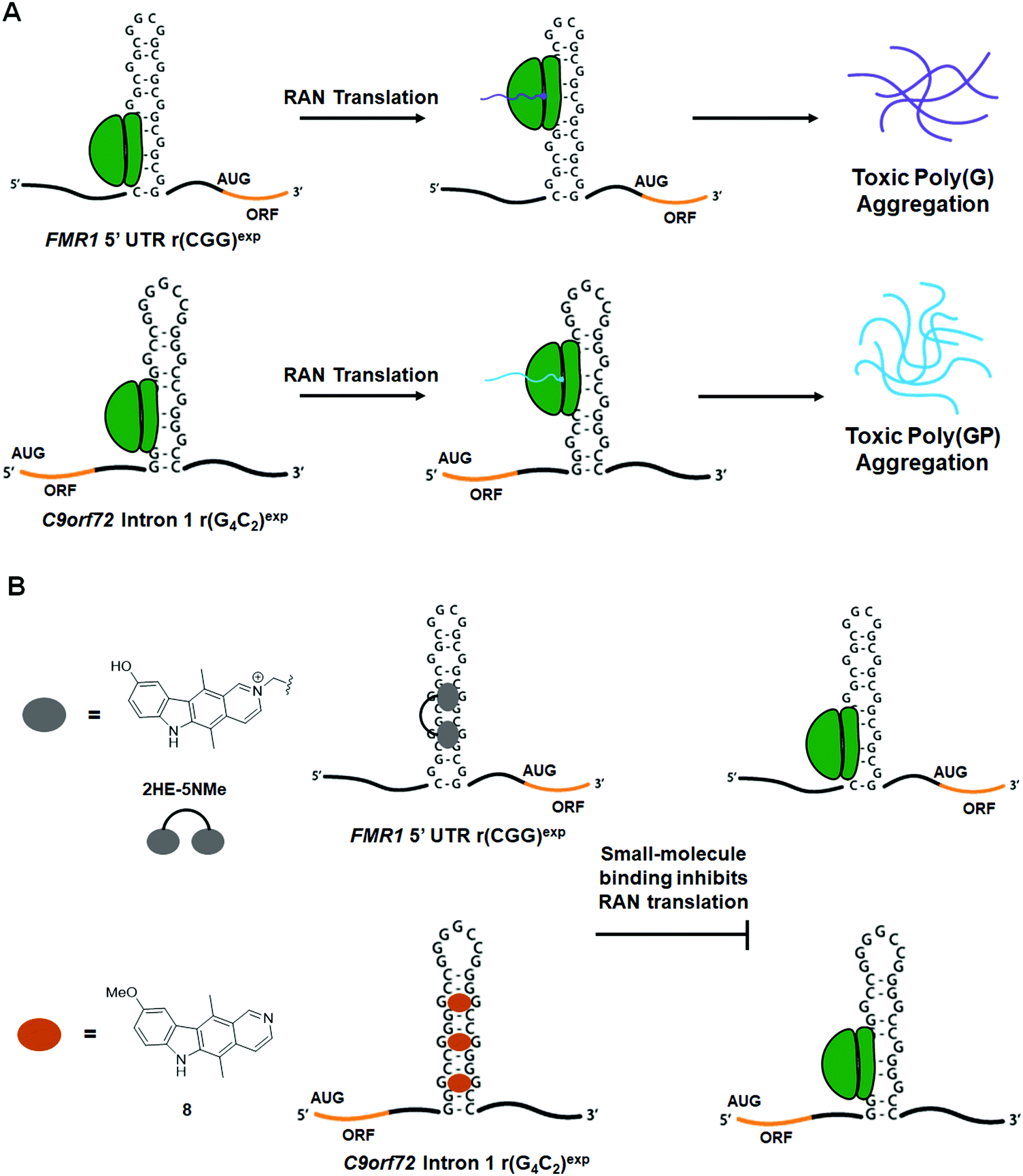 | ||
| Fig. 14 Small molecule targeting of RNA repeat expansions reduces aberrarnt repeat-associated non-ATG (RAN) translation. (A) Schematic of RAN translation of FMR1 due to r(CGG)exp in the 5′ UTR and C9orf72 due to r(G4C2)exp in intron 1. (B) Small molecules targeting r(CGG)exp and r(G4C2)exp inhibit RAN translation. | ||
To date, the most potent inhibitor of r(CGG)exp RAN translation is the covalent cross-linker 2H-5-CA-Biotin (Table S1, ESI†).1522H-5-CA-Biotin selectively engaged the RNA target in cells and inhibited RAN translation at a dose of only 500 nM (∼40% decrease in poly(G) levels) while not affecting canonical translation of the downstream ORF.152 Additionally, polysome profiling indicated that 2H-5-CA-Biotin disrupts polysome loading onto r(CGG)exp-containing transcripts.
8.3 Small molecules targeting the r(G4C2)exp in C9orf72 inhibit RAN translation
In c9FTD/ALS, RAN translation gives rise to five DPRs.151 Poly(GA) and poly(GR) are translated from the sense strand [r(G4C2)exp], while poly(PA) and poly(PR) are translated from the antisense strand [r(G2C4)exp].153 Poly(GP) is RAN translated from both strands and is highly expressed in the central nervous system. Additionally, it is the most soluble of the DPRs, making its detection facile (Fig. 14A).146,151,154 In agreement with its ability to alleviate another c9FTD/ALS-associated defect (nuclear inclusions), 1a dose-dependently reduced levels of poly(GP) by 10%, 18%, and 47% in iNeurons treated at 25, 50, or 100 μM, respectively.142 Likewise, the 1a derivative 8 dose-dependently inhibited RAN translation in HEK293T cells expressing r(G4C2)66 (IC50 = 1.6 ± 0.20 μM), while having no effect on canonical translation (Fig. 14B).143 Polysome profiling upon treatment with 8 showed the amount of r(G4C2)66 transcripts loaded into polysomes was significantly decreased for high and low molecular weight polysomes and monosomes, indicating that 8 works by sterically blocking the assembly of ribosomes onto r(G4C2)exp, thus reducing the levels of toxic DPRs produced.1439. Small molecules inhibit translation of traditionally undruggable proteins
Over the past decades, tremendous efforts have been invested in the development of small molecules targeting disease-causing proteins, and yet only 15% of proteins are considered “druggable” from genome-wide analysis.155,156 One major roadblock in drugging the remaining 85% is their lack of defined structures that can serve as potential small molecule binding pockets.157,158 To overcome this challenge, an alternative strategy, especially useful for proteins with aberrantly high expression levels, is to target their encoding mRNA specifically with small molecules and hence inhibit downstream translation. A recent example of this is the development of a small molecule targeting the α-synuclein mRNA,159 which encodes an intrinsically disordered protein (IDP) key to the pathogenesis of Parkinson's disease (PD).160The α-synuclein protein, encoded by the SNCA gene, can oligomerize to form fibrils across neurons in the brain as well as accumulate in Lewy bodies and Lewy neurites, contributing to the risk of developing PD (Fig. 15A).161,162 Since individuals with multiplication of the SNCA gene locus develop dominantly inherited PD with a gene-dosage effect,163 reducing the level of α-synuclein expression could be a promising disease-alleviating strategy.164,165 As an IDP, α-synuclein is challenging to target. The SNCA mRNA, however, displays a functionally important and structurally defined 5′ UTR with an iron responsive element (IRE) that provides opportunities for small molecule targeting.166,167 Indeed, employment of the sequence-based design and lead identification strategy, Inforna (discussed in Section 2.3, “Design of Monomeric Small Molecules Targeting Disease-Causing miRNAs”),41 yielded a set of small molecules that bind the IRE region of SNCA mRNA. These initial hits were subjected to a western blot screen of α-synuclein inhibition potency in neuroblastoma cells, with the most potent compound, Synucleozid, exhibiting an IC50 ∼500 nM (Fig. 15B and Table S1, ESI†).159 To ensure that inhibition occurs at the translational and not transcriptional level, RT-qPCR was used to confirm the level of SNCA mRNA remained constant upon treatment of Synucleozid.
 | ||
| Fig. 15 RNA-targeted small molecules inhibit translation of traditionally undruggable proteins. (A) Schematic depiction of α-synuclein-mediated disease pathway. (B) Structure of Synucleozid, an Inforna-designed small molecule that binds the A-bulge of the SNCA IRE that regulates translation of the mRNA. (C) Synucleozid targets the IRE structure of SNCA mRNA and represses α-synuclein protein expression by inhibiting ribosomal assembly onto the SNCA transcript. | ||
It should be noted, however, that observation of the expected biological effects can only support, not validate, the putative binding mode of a small molecule. To validate the A bulge of the IRE of SNCA mRNA as the binding site of Synucleozid, competitive binding assays and mutational analyses were performed. Indeed, mutations of the A bulge to either a U/G bulge or a base pair reduced the binding affinity of Synucleozid by 10-fold, while mutations of other non-canonical base pairs had no effect on Synucleozid binding avidity. Furthermore, ASO-Bind-Map18,168 was used to confirm the binding of Synucleozid to the IRE both in vitro and in cellulis. Briefly, ASO-Bind-Map relies on ASO-mediated RNA cleavage, via RNase H, which can be inhibited by small molecule binding of the RNA target. Small molecule binding thermodynamically stabilizes the RNA and impedes ASO binding at the binding site. In vitro, protection from RNase H cleavage can be read out by gel electrophoresis while RT-qPCR or RNA-seq can be used to read out protection by the binding small molecule in cellulis. In this case, treatment of Synucleozid impeded cleavage of the IRE, indicating that Synucleozid indeed binds to the IRE and stabilizes its structure.
In addition to its intrinsic specificity for the binding pocket on RNA, the overall specificity of a small molecule depends on the prevalence of the structured pocket across the entire human transcriptome. In other words, a small molecule that is specific to its target binding pocket would still suffer from off-target effects if this binding pocket is shared by other non-target RNAs. The selectivity of Synucleozid was first assessed by studying its effect on mRNAs expressed in the nervous system that contain known IREs in their UTRs, including amyloid precursor protein (APP), prion protein (PrP), ferritin and the transferritin receptor (TfR). Upon treatment of Synucleozid (1 μM), no effect was observed upon APP, PrP, or TfR levels, but ferritin levels were reduced by ∼50%. This reduction could be the result of an off-target effect of Synucleozid or could be due to compound-mediated rescue of autophagic and lysosomal dysfunction caused by an accumulation of α-synuclein in PD.169 While future studies are necessary to elucidate Synucleozid's effect on ferritin levels, the compound demonstrated overall high selectivity for SNCA mRNA due to its unique structure in the 5′ UTR. Moreover, the targeted 5′-G_G/3′-CAU region was searched across a database of structural elements expressed in the human transcriptome, including miRNA hairpin precursors (7436 motifs) and 2459 other known motifs from rRNA, RNase P RNA, U4/U6 snRNA, and nonredundant tRNAs.159,170 Remarkably, the bulge targeted by Synucleozid only occurs five times among these motifs (0.051%) and fortuitously, the potential miRNA off-targets are not expressed at appreciable levels.170 Not surprisingly, Synucleozid had no effect on their expression. A transcriptome-wide assessment of Synucleozid treatment using RNA-seq revealed very few changes (55/20034 genes changed; 0.3%).159,171 Similarly, a proteome-wide selectivity analysis also showed limited changes (283/3300 proteins changed; 8%). Collectively, these data support the fundamental claim that RNAs can indeed form unique 3D structures suitable for targeting with small molecules, therefore expanding the druggability of proteins broadly. Notably, this assertion is bolstered by studies that direct the splicing outcome of MAPT exon 10 (tau), another IDP.120
10. Targeted cleavage of disease-causing RNAs using bleomycin A5-conjugates
10.1 Bleomycin A5 cleavage of miRNAs
Bleomycin A5 is a well-known, DNA-cleaving natural product172 that also cleaves RNA (Fig. 16A).173,174 Building on the foundational studies of Hecht,173,174 it was recently determined that bleomycin A5 has two preferred RNA cleavage sites, AU rich regions, with longer stretches of AU base pairs correlating with more efficient RNA cleavage and purine-rich sequences.175 Angelbello et al.175 identified a compilation of 13 human miRNAs that contain AU-rich regions, seven of which have been tied to disease. Of these, pri-miR-10b has a 5′AUAUAU/3′UAUAUA sequence, creating a potential recognition site for bleomycin A5 cleavage. Indeed, bleomycin A5 cleaved pri-miR-10b at two locations, the predicted AU-rich region and a 5′GUG/3′CAC site near the Dicer processing site. This finding was not surprising as bleomycin A5 also prefers purine-rich sequences.175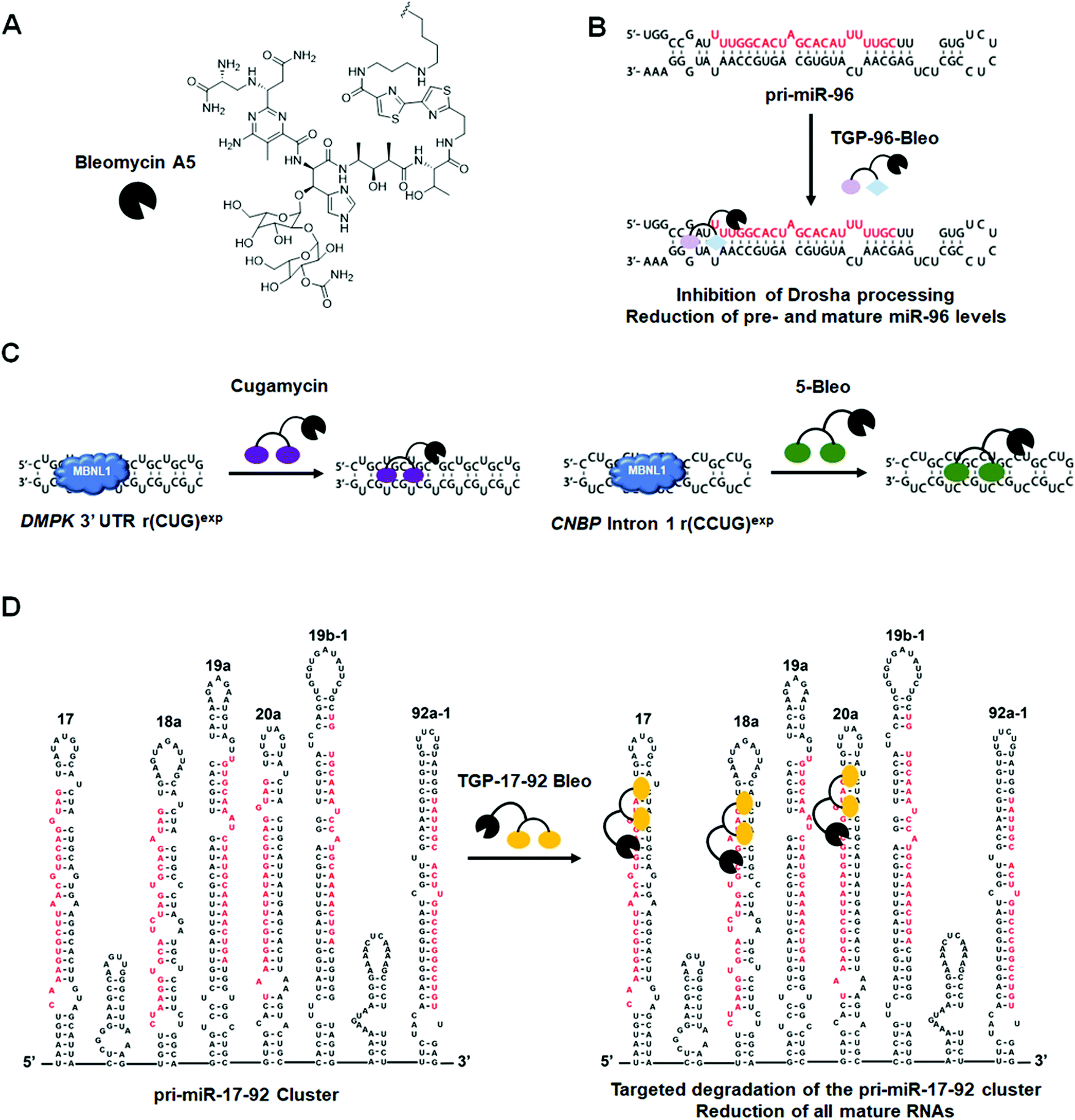 | ||
| Fig. 16 Small molecule-bleomycin A5 conjugates cleave disease-causing RNAs in a targeted manner. (A) Structure of Bleomycin A5. (B) TGP-96-Bleo targets and cleaves oncogenic pri-miR-96. (C) Targeted degradation of r(CUG)exp by Cugamycin and r(CCUG)exp by 5-Bleo. (D) Targeted degradation of the pri-miR-17-92 cluster by TGP-17-92 Bleo. | ||
Bleomycin A5 was then studied for cleavage of pri-miR-10b in two cellular models: (i) HeLa cells transfected with a plasmid encoding pri-miR-10b and (ii) the TNBC cell line MDA-MB-231 in which miR-10b is overexpressed.176 Aberrant levels of miR-10b have been linked to both tumor invasion and metastasis in TNBC.176 At nM concentrations, the compound cleaved pri-miR-10b in both cell types thereby reducing levels of mature miR-10b, as determined by RT-qPCR.
This study highlighted the ability of bleomycin A5 to cleave RNA in cells, opening the door for the development of small molecule-bleomycin conjugates that direct the natural product to cleave a specific RNA target. This work also emphasized that ncRNAs can be targets of known drugs and should therefore be considered in drug discovery screens.
10.2 Targaprimir-96-Bleo (TGP-96-Bleo): Targeted cleavage of pri-miR-96 by a small molecule–bleomycin A5 conjugate
The first example of using small molecule–bleomycin conjugates to cleave miRNAs came from Li et al.,177 in which they used a heterodimeric–bleomycin A5 conjugate to target oncogenic pri-miR-96. Both the bleomycin derivative and its site of conjugation to the small molecule were carefully selected. Bleomycin contains four domains: (i) a metal-binding nucleic acid cleavage domain; (ii) a C-terminal DNA-binding domain; (iii) a linker connecting the cleavage and DNA-binding domains; and (iv) a carbohydrate domain important for cellular uptake of the molecule.172 The derivative bleomycin A5 was chosen for conjugation to RNA-binding small molecules because the DNA-binding domain contains a butyl-1,4-diamine side chain that allows for easy conjugation of small molecules. Additionally, it has been shown that conjugation through bleomycin's C-terminal free amine reduces affinity for DNA via ablation of the amine's positive charge.178,179 These studies suggest that small molecule–bleomycin A5 conjugates have the potential to selectively cleave RNA targets.As discussed previously (in Section 2.3. “Design of Monomeric Small Molecules Targeting Disease-Causing miRNAs”), TGP-9647 was designed using Inforna and is a potent binder of the pri-miR-96 Drosha processing site and adjacent 1 × 1 GG loop. Binding of TGP-96 to pri-miR-96 inhibited the biogenesis of mature miR-96, derepressed FOXO1, and triggered apoptosis in MDA-MB-231 cells.47 To further improve bioactivity, a small molecule cleaver was synthesized by conjugating TGP-96 to bleomycin A5 (TGP-96-Bleo) via the C-terminal amine in the traditional DNA-binding domain of bleomycin A5, thus disrupting key interactions necessary for DNA recognition (Table S1, ESI†).177 As bleomycin A5 has been shown to cleave AU base pairs,175 and pri-miR-96 has AU base pairs in close proximity to TGP-96's binding site, this conjugation strategy had a high potential for success.
Indeed, TGP-96-Bleo bound pri-miR-96 with a Kd of 64 ± 11 nM and cleaved the hairpin at the predicted AU base pairs, while no binding to DNA was observed (Fig. 16B and Table S1, ESI†).177 A control compound lacking the RNA-binding modules cleaved plasmid DNA at levels 5-fold greater than those seen with TGP-96-Bleo,177 indicating conjugation of bleomycin A5 to RNA binding modules reduces its affinity for DNA, lowering the potential for off-targets. This was further supported by visualizing γ-H2AX foci formation in cells, a marker for DNA double stranded breaks. Cells treated with the control compound lacking RNA-binding modules displayed ∼2.3-fold more foci than cells treated with TGP-96-Bleo.177 Notably, the concentrations of TGP-96-Bleo that cleaved DNA and induced double stranded DNA breaks are 20-fold higher than the concentrations that reduced mature miR-96 levels, vide infra.
Based on these promising results, TGP-96-Bleo was compared to TGP-96 in MDA-MB-231 TNBC cells for reducing levels of mature miR-96. RT-qPCR analysis confirmed treatment with TGP-96-Bleo decreased the levels of both pri-miR-96 and mature miR-96.177 As mentioned above, TGP-96 decreased mature miR-96 levels but increased pri-miR-96 levels. These data are consistent with the mechanisms of action for the two compounds; TGP-96-Bleo as an RNA cleaver and TGP-96 as an RNA binder. Target occupancy of TGP-96-Bleo for the predicted pri-miR-96 binding sites was confirmed via a competitive cleavage assay in which cells were co-treated with TGP-96-Bleo and TGP-96, with TGP-96 added in increasing concentrations to compete off a constant concentration of TGP-96-Bleo. Treatment with TGP-96-Bleo also resulted in rescue of FOXO1 expression and subsequent activation of apoptotic pathways in MDA-MB-231 cells, demonstrating rescue of disease phenotypes by TGP-96-Bleo.177TGP-96-Bleo did not have an effect on any other miRNAs predicted to target FOXO1.180
To further profile the selectivity of TGP-96-Bleo, small molecule nucleic acid profiling by cleavage applied to RNA (RiboSNAP) was utilized against the 349 miRNAs expressed in MDA-MB-231 cells.177 The results of this profiling showed miR-96 levels were the most drastically and significantly affected by TGP-96-Bleo treatment, highlighting the selectivity of this small molecule RNA cleaver (Table S1, ESI†). Additionally, this experiment showed DNA off-targets of bleomycin A5 can be ablated by conjugation to an RNA-binder and that small molecule cleaver compounds can be successfully used for cellular profiling. A variation of RiboSNAP, RiboSNAP-Map, in which cleavage fragments are analyzed to determine the exact binding site of a small molecule, was also debuted in this paper.177 RiboSNAP-Map uses a gene specific forward primer and universal reverse primer to amplify cleavage products, which are then sequenced to determine the site of cleavage.177 This method confirmed the TGP-96-Bleo cleavage site is in close proximity to the known binding sites of TGP-96, positioning bleomycin A5 to cleave the proximal AU base pairs of pri-miR-96.
The data presented in this paper demonstrate the utility of conjugating bleomycin A5 to selective RNA-binding small molecules for the targeted degradation of disease-relevant RNAs and introduces novel methods for cellular profiling and target engagement validation using these compounds.
10.3 Cugamycin: Targeted cleavage of r(CUG)exp by a small molecule–bleomycin A5 conjugate
Small molecule–bleomycin A5 conjugates can be used to target structured RNAs other than miRNAs. For example, another class of important RNA targets is the hairpin structures characteristic to microsatellite/repeat expansion disorders. The dimeric compound 2H-K4NMeS, described above, that reverses DM1-associated defects was appended with bleomycin A5 to yield the small molecule cleaver, Cugamycin (Fig. 16C and Table S1, ESI†).181In vitro binding studies demonstrated Cugamycin's selectivity for r(CUG)exp (EC50 = 365 nM) over DNA, and cleavage studies confirmed bleomycin A5′s reduction in affinity for DNA when conjugated to an RNA-binding small molecule, as DNA cleavage was reduced by 4-fold as compared to bleomycin A5 (Table S1, ESI†).181In DM1 patient-derived myotubes, Cugamycin localized to the nucleus and cleaved ∼70% of r(CUG)exp-containing DMPK transcripts (dosed at 1 μM), while having no effect on wild-type DMPK transcripts that contain only a few r(CUG) repeats (non-pathogenic).181 Further, Cugamycin demonstrated allele selectivity; that is, it only cleaved the mutant DMPK allele (1300 repeats). In conjunction with this cleavage, Cugamycin rescued MBNL1-dependent alternative splicing by ∼40%, leading to a ∼30% reduction in MBNL1-r(CUG)exp nuclear foci when treated at 1 μM. Cugamycin did not have an effect on NOVA-mediated splicing, indicating selectivity for the r(CUG)exp target.181 Additionally, selectivity was profiled by measuring cleavage of five additional mRNAs that contain short r(CUG) repeats (<20 repeats). All five transcripts were unaffected by Cugamycin treatment.181 Of note, an antisense LNA gap-mer complementary to r(CUG)exp reduced the levels of all five r(CUG) repeat-containing mRNAs, as well as wild-type DMPK levels.181 Modeling of RNA structures present in these mRNAs, as well as r(CUG)exp, indicated the hairpin structure adopted by r(CUG)exp is not recapitulated by shorter repeat lengths, bolstering the notion that structure-binding small molecules can indeed be selective in patient-derived cells and that selectivity translates in vivo (discussed below).
Off-target DNA cleavage in DM1 myotubes was assessed by visualizing γ-H2AX foci after treatment with Cugamycin, a control compound lacking the RNA-binding modules, or bleomycin A5, all tested at concentrations at which Cugamycin cleaved r(CUG)exp and improved DM1-associated defects. Both the control compound and bleomycin A5 caused formation of γ-H2AX foci, while Cugamycin had no effect.181 This again demonstrates that conjugation of bleomycin A5 through its C-terminal amine ablated affinity for DNA.
Cugamycin was also tested in vivo using the HSALR mouse model of DM1.181 This model contains 250 CTG repeats driven by the human skeletal actin (HSA) promoter and recapitulates DM1 disease phenotypes such as dysregulation of MBNL1-dependent splicing, loss of the muscle-specific chloride ion channel (CLCN1), and myotonia.182,183Cugamycin was i.p. injected every other day, at a dose of 10 mg kg−1, for a total of 8 days. After treatment, an ∼40% reduction in the levels of the HSA transgene [r(CUG)250] was observed in tibialis anterior (TA) and gastrocnemius muscles, indicating that Cugamycin engaged its RNA target in vivo.181 Lung fibrosis, a common side effect of bleomycin,184 was not observed with Cugamycin treatment.
Rescue of aberrant alternative splicing in the TA and gastrocnemius muscles were also studied upon treatment with Cugamycin, showing that MBNL1-dependent splicing events Mbnl1 exon 7 and Clcn1 exon 7A were rescued by ∼50%, while alternative splicing of integrin β-1 precursor (Itgb1) exon 17 and capping actin protein of muscle z-line subunit β (Capzb) exon 8, non MBNL1-dependent events, were unaffected.181 Loss of the CLCN1 protein, due to aberrant alternative splicing and exon 7A inclusion contributes directly to myotonia.183 Therefore, recuse of MBNL1-dependent splicing should increase CLCN1 protein expression on the cell surface, leading to a rescue of disease phenotype. Indeed, upon Cugamycin treatment, the levels of CLCN1 in TA muscle plasma membranes increased and an ∼40% reduction in myotonia was observed.181 These results were consistent across TA, gastrocnemius, and quadriceps muscles, indicating Cugamycin reaches DM1-affected tissues and rescues disease-associated phenotypes broadly.
Interestingly, Cugamycin's parent compound, 2H-K4NMeS, when delivered at the same dose and route of administration was unable to rescue MBNL1-dependent splicing or myotonia, indicating the cleavage capacity of Cugamycin is essential for in vivo efficacy.181 These data also suggest that a cleavage-driven mechanism of action can provide a more prolonged and potent effect in vivo than a simple binding mode of action.
The ability of Cugamycin to rescue MBNL1-associated alternative splicing defects broadly and selectively was assessed by transcriptome-wide analysis of splicing events. By comparison to wild-type mice, the extent of the dysregulation of each splicing event in HSALR was measured. Angelbello et al.181 identified 138 exons that are deregulated, reported as percent spliced in (Ψ), using mixture of isoforms (MISO)185 analysis. Of these 138 exons, 134 of them showed Ψ values shifted back towards wild-type upon treatment with Cugamycin.181 These data indicate that through Cugamycin's selective recognition of r(CUG)exp, the compound was able to globally rescue aberrant MBNL1-dependent alternative splicing in a mouse model of DM1. In addition to changes in alternative splicing, transcriptomic changes are also observed in DM1 mice. In particular, 326 genes are significantly dysregulated in HSALR mice.181 Treatment with Cugamycin resulted in rescue of expression of 177 of these genes, highlighting the ability of the compound to normalize the transcriptome.181Cugamycin had no effect on the transcriptome of wild-type mice, as measured by RNA-seq, again highlighting the selectivity of this small molecule cleaver compound.
This study validated the strategy of using a small molecule-bleomycin A5 conjugate, Cugamycin, to target and cleave RNA repeats selectively in microsatellite/repeat expansion disorders, including in preclinical mouse models. Cugamycin showed remarkable potency in vitro and rescued DM1-associated phenotypes both in cells and in vivo. Additionally, the compound showed high selectivity for the DMPK mutant allele harboring r(CUG)exp and demonstrated the ability to rescue MBNL1-dependent alternative splicing transcriptome-wide with no significant off-targets. The data presented in this study indicate the cleavage-mediated mechanism of action of Cugamycin is highly effective at rescuing DM1-associated disease phenotypes in a mouse model, suggesting Cugamycin is a strong lead candidate for further optimization into a preclinical compound.
10.4 Targeted cleavage of r(CCUG)exp by a small molecule-bleomycin A5 conjugate
After the success observed with Cugamycin, a small molecule-bleomycin A5 conjugate was created to target the r(CCUG)exp in intron 1 of CNBP, causative of DM2.139 Building off a previously designer dimer (5)139 (discussed in Sections 6.2 and 7), Benhamou et al.139 conjugated bleomycin A5 through the natural product's C-terminal amine to afford 5-Bleo (Table S1, ESI†). In vitro studies showed 5 and 5-Bleo bind r(CCUG)10 with similar affinities (∼100 nM) and that 5-Bleo cleaved RNA between 5′-GC steps in base paired regions adjacent to the compound's binding site (Fig. 16C and Table S1, ESI†).139 This compound also demonstrated selectivity for the RNA target over DNA, consistent with the results from previous bleomycin A5 conjugates.139,181While the dimer and cleaver compounds display similar binding affinities for r(CCUG)exp (∼100 nM) in vitro, in DM2 fibroblasts the cleaver reduced levels of intron 1-containing r(CCUG)exp transcripts by an ∼2.5-fold greater extent than the dimer, the binding of which shunts the intron down endogenous decay pathways (i.e., has a different mode of action).139 Mature CNBP mRNA levels were also reduced upon treatment of 5-Bleo. Thus, the bleomycin A5 conjugate was able to more effectively reduce levels of mutant CNBP mRNA, compared to the binder, and cleaves the r(CCUG)exp target. Off-target DNA cleavage was again assessed by visualizing the formation of γ-H2AX foci in DM2 fibroblasts. Treatment with 5-Bleo did not result in a significant increase in foci at the active concentration, demonstrating the compound's selectivity in cellulis.139
Target engagement of 5-Bleo was confirmed in cells using a competitive cleavage assay in which increasing concentrations of the dimer were co-treated with a constant concentration of 5-Bleo and the levels of mature CNBP mRNA were measured. Increasing the concentration of the simple binding dimer led to an increase in CNBP mRNA levels (a reduction in cleavage), indicating 5-Bleo and the dimer share the same RNA target and that the mechanism of 5-Bleo is through direct cleavage of r(CCUG)exp.139
Further cellular studies demonstrated 5-Bleo's enhanced ability to rescue MBNL1-dependent IR pre-mRNA splicing defects compared to the dimer. 5-Bleo rescued splicing by ∼50% at 5 μM, while the dimer only rescued splicing defects by ∼20% at 10 μM, a 2-fold higher concentration.139 Additionally, an ∼50% reduction in r(CCUG)exp-containing foci was observed upon treatment of 5-Bleo. Evaluation of mature CNBP mRNA levels and IR splicing in healthy fibroblasts after treatment with 5-Bleo showed no changes, confirming 5-Bleo's allele selectivity for the disease-causing r(CCUG)exp as the shorter r(CCUG) repeats present in healthy fibroblasts were unaffected.139
11. Targeted degradation of disease-causing RNAs using RIBOTACs
The advent of proteolysis targeting chimeras (PROTACs)186 demonstrated the ability to trigger protein degradation with small molecules. This concept has been broadened to other biomolecules, such as RNAs, as is the case of RIBOTACs.187 RIBOTACs mediate RNA decay by recruiting endogenous RNases to degrade specific transcripts. In particular, RIBOTACs have been developed that recruit RNase L, a component of the antiviral immune response. RNase L is present in minute quantities in all cells as an inactive monomer. Upon viral infection, it is upregulated, dimerized and activated by 2′–5′ oligoadenylate [2′–5′ poly(A)].188 RNase L is thus an intriguing enzyme for small molecule recruitment and targeted RNA destruction. That is, an RNA-binding small molecule coupled to an RNase L-recruiting module could locally recruit and activate RNase L to cleave the target selectively, without activation of the immune system.11.1 Targaprimir-96 RIBOTAC (TGP-96 RIBOTAC): Targeting pri-miR-96 for degradation
As previously described in Section 2.3 (“Design of Monomeric Small Molecules Targeting Disease-Causing miRNAs”), TGP-96 is a dimeric small molecule that binds pri-miR-96 and inhibits its biogenesis, thereby derepressing the transcription factor FOXO1 and triggering apoptosis in TNBC cells.47TGP-96 was converted into a RIBOTAC (TGP-96 RIBOTAC) by appending a short 2′–5′ A4 oligonucleotide as the RNase L recruiting module onto the compound (Fig. 17 and Table S1, ESI†).187In vitro binding assays confirmed the recruiting module does not affect the avidity of the compound for pri-miR-96's Drosha processing site (Kd = 20 nM) (Table S1, ESI†).187In vitro cleavage assays demonstrated the ability of TGP-96 RIBOTAC to recruit and dimerize RNase L, leading to the selective cleavage of pri-miR-96.187 This cleavage was inhibited when TGP-96 was added as a competitor, validating the binding sites of TGP-96 and TGP-96 RIBOTAC are the same.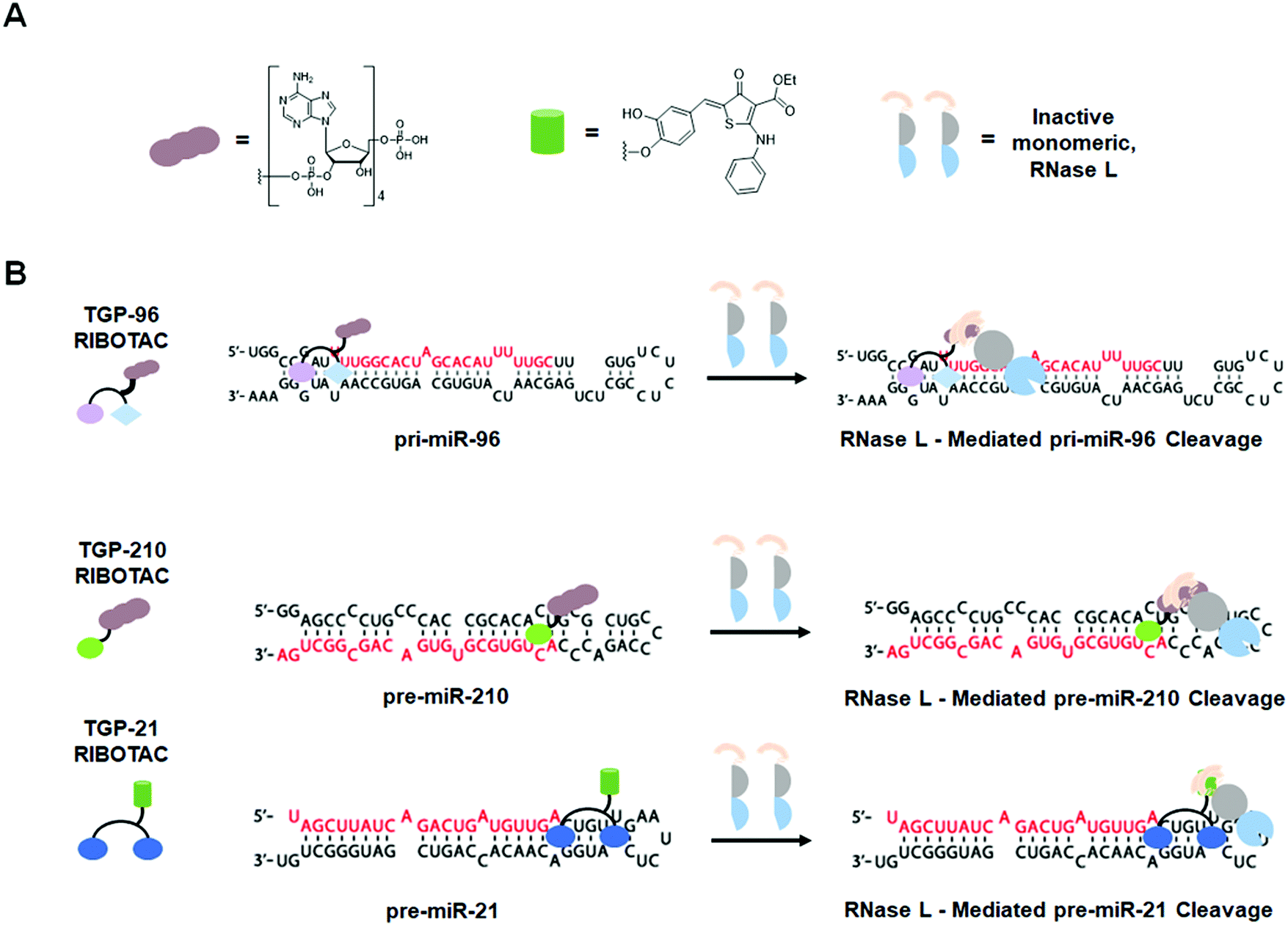 | ||
| Fig. 17 Ribonuclease targeting chimeras (RIBOTACs) degrade disease-causing RNAs in a targeted manner. (A) Structures of first- and second-generation RNase L recruiting modules and schematized monomeric RNase L. (B) RIBOTAC-mediated degradation of oncogenic pri-miR-96, pre-miR-210, and pre-miR-21. | ||
In MDA-MB-231 cells, despite having ∼2-fold reduced cellular permeability compared to TGP-96, TGP-96 RIBOTAC reduced the levels of pri-miR-96 and mature miR-96, confirming compound mode of action.187 RNase L-dependent cleavage was verified in multiple experiments: (i) immunoprecipitation with an RNase L antibody confirmed ternary complex formation between pri-miR-96, RNase L, and TGP-96 RIBOTAC; (ii) competitive cleavage between TGP-96 RIBOTAC and a derivative lacking the RNase L-recruiting module showed a dose-dependent decrease in cleavage of pri-miR-96, validating the ability of TGP-96 RIBOTAC to locally dimerize RNase L; and (iii) siRNA knockdown of RNase L ablated TGP-96 RIBOTAC's ability to degrade pri-miR-96.187
Treatment of TGP-96 RIBOTAC in MDA-MB-231 cells resulted in modulation of the invasive phenotype associated with miR-96 in cancer. An ∼2-fold increase in FOXO1 expression was observed upon treatment, consistent with inhibition of miR-96 biogenesis, and thus resulted in apoptosis.187 This effect can be reversed upon over expression of pri-miR-96. TGP-96 RIBOTAC has no effect on apoptosis in MCF-10a cells (healthy breast epithelial cells).187 Further, TGP-96 RIBOTAC stimulated apoptosis to the same extent as its parent binding compound at a 2.5-fold lower dose. Combined with the decreased uptake of the compound, RNase L recruitment by TGP-96 RIBOTAC enhances the compound's activity by ∼5-fold compared to TGP-96.187 Most importantly, TGP-96 RIBOTAC acts catalytically and in a substoichiometric fashion to recruit RNase L for targeted RNA degradation, cleaving 3.1 pri-miR-96 molecules per molecule of RIBOTAC.187
11.2 Targapremir-210 RIBOTAC (TGP-210 RIBOTAC): Targeting pre-miR-210 for degradation
As discussed in Section 2.3 (“Design of Monomeric Small Molecules Targeting Disease-Causing miRNAs”), TGP-210, designed by Inforna, binds the Dicer processing site of pre-miR-210, inhibits its biogenesis and normalizes proteins in this circuit, ultimately inducing apoptosis in hypoxic cancer cells.45 Costales et al.189 optimized TGP-210 by appending a 2′–5′ A4 RNase L recruiting module to yield TGP-210 RIBOTAC (Fig. 17 and Table S1, ESI†). In vitro cleavage assays showed TGP-210 RIBOTAC cleaved pre-miR-210 and binding assays demonstrated TGP-210 RIBOTAC is more selective than TGP-210; an ∼10-fold difference in affinity is observed between the Dicer processing site of pre-miR-210 and DNA while only an ∼5-fold difference was observed for TGP-210 (Table S1, ESI†).189 Thus RNA degraders can show enhanced selectivity over their simple binding counterparts.In hypoxic MDA-MB-231 cells, TGP-210 RIBOTAC decreased the levels of both pre-miR-210 and mature miR-210, consistent with its mode of action.189 Upon treatment with TGP-210 RIBOTAC, GPD1L mRNA levels were significantly increased and HIF1α mRNA levels were decreased.189 As a result of deactivation of the oncogenic circuit, TGP-210 RIBOTAC triggered apoptosis in hypoxic cancer cells, to a similar extent as TGP-210. However, because TGP-210 RIBOTAC is half as cell permeable as TGP-210, these results demonstrate the RIBOTAC has ∼2-fold enhanced activity over TGP-210.189
Specific RNase L recruitment was confirmed via: (i) competitive cleavage assays, in which co-treatment of increasing amounts of TGP-210 with TGP-210 RIBOTAC resulted in a dose-dependent decrease in pre-miR-210 cleavage; (ii) overexpression of RNase L, resulting in increased cleavage; (iii) overexpression of pre-miR-210, resulting in decreased cleavage; and (iv) siRNA ablation of RNase L, resulting in no TGP-210 RIBOTAC-mediated cleavage of pre-miR-210.189 Additionally, immunoprecipitation of RNase L showed enrichment for pre-miR-210 only in cells treated with TGP-210 RIBOTAC.
RNA-seq and RT-qPCR profiling experiments revealed TGP-210 RIBOTAC has no significant off-targets trancriptome-wide (Table S1, ESI†).189 Combined with its catalytic and substoichiometric mode of action, TGP-210 RIBOTAC demonstrates that enhanced activity and selectivity can be achieved through the targeted recruitment of nucleases via RIBOTAC compounds.
11.3 Targapremir-21 RIBOTAC (TGP-21 RIBOTAC): Targeting pre-miR-21 for degradation
A RIBOTAC targeting pre-miR-21, dubbed TGP-21 RIBOTAC, was recently reported that is based on the dimeric binding compound TGP-21 (Table S1, ESI†).190TGP-21 was first validated in MDA-MB-231 TNBC cells, reducing levels of mature miR-21 and increasing levels of pre-miR-21, in accordance with its mechanism of inhibiting Dicer processing (Table S1, ESI†).190 The binding dimer also increased expression of programmed cell death protein 4 (PDCD4) and phosphatase and tension homolog (PTEN), two proteins that are translationally repressed by miR-21.190 Additionally, invasion assays confirmed TGP-21's ability to inhibit the invasive phenotype of MDA-MB-231 cells.190TGP-21 was optimized by appending a heterocyclic small molecule recruiter of RNase L to create TGP-21 RIBOTAC (Fig. 17 and Table S1, ESI†). Previous studies by Thakur et al.191 demonstrated the ability of a heterocyclic small molecule to recruit and activate RNase L in place of the traditional 2′–5′ poly(A) substrate. Extensive optimization of this structure by Costales et al.190 yielded the small molecule RNase L-recruiting heterocycle that was incorporated in TGP-21 RIBOTAC.
In MDA-MB-231 cells, TGP-21 RIBOTAC showed a 20-fold enhancement in activity for reducing mature miR-21 levels compared to TGP-21 and reduced pre-miR-21 levels in a substoichiometric manner (Table S1, ESI†).190 RNase L recruitment was confirmed using the same assays as described for TGP-210 RIBOTAC, further supporting the mechanism of small molecule-targeted degradation.190 A time course experiment in which reduction of mature miR-21 levels were monitored up to 96 h post-treatment revealed TGP-21 RIBOTAC has a more potent and prolonged effect than TGP-21.190
Importantly, TGP-21 RIBOTAC did not trigger an innate immune response, as monitored by mRNA and protein levels of innate immunity-associated biomarkers.190 In contrast, transfection of 2′–5′ A4 into MDA-MB-231 cells resulted in upregulation of several innate immunity biomarkers, such as interferon gamma (IFN-γ), 2′–5′-oligoadenylate synthase 1 (OAS1), retinoic acid-inducible gene 1 (RIG-1), and melanoma differentiation-associated protein 5 (MDA5).190 These results support the hypothesis that TGP-21 RIBOTAC locally recruits and activates RNase L, instead of triggering a global antiviral innate immune response.190
Selectivity was also assessed miRNome-wide and quantified by calculating Gini coefficients. Gini coefficients were first introduced as a metric of biological selectivity by Graczyk192 as a measure of kinase inhibitor selectivity, however the metric can be broadly applied to any biomolecule and small molecule modulator. A Gini coefficient of 0 represents a nonselective compound while a Gini coefficient of 1 represents an exquisitely selective compound. Gini coefficients for the monomer that comprises TGP-21, TGP-21, and TGP-21 RIBOTAC, by measuring their effects on the miRNome, are 0.52, 0.68, and 0.84 respectively, highlighting the increase in selectivity that is achieved by dimerization and by RNase L-mediated targeted degradation of RNA.190 Proteome-wide analysis of the effects of TGP-21 RIBOTAC in MDA-MB-231 cells confirmed the selectivity of the compound with only 47 of 4181 proteins being significantly affected.190
TGP-21 RIBOTAC was also evaluated in vivo in a mouse model of metastatic breast cancer (NOD/SCID mice i.v. injected with MDA-MB-231-Luc cells). Treatment with TGP-21 RIBOTAC (10 mg kg−1, every other day for 6 weeks) inhibited metastasis to lung, as evident by a significant decrease in the number of lung nodules present in TGP-21 RIBOTAC-treated mice.190 Additionally, tissues from mice in the RIBOTAC treatment group displayed decreased hematoxylin and eosin (H&E) staining, decreased pre-miR-21 and mature miR-21 levels (as assayed by FISH and RT-qPCR), and increased levels of PDCD4 (as assayed by immunohistochemistry).190 Thus, TGP-21 RIBOTAC selectivity and potently modulates the miR-21 pathway in a preclinical mouse model, resulting in inhibition of breast cancer metastasis.
12. Case study: direct comparison of bleomycin A5-mediated cleavage versus RNase L-mediated degradation of the pri-miR-17-92 cluster
The pri-miR-17-92 cluster is a direct target of the transcription factor c-MYC193 and is upregulated in human diseases ranging from cancers194–196 to fibrosis.197 For each disease, the mature miRNA deregulated from the cluster and its downstream effects can be unique or overlap,198 and the mature miRNAs can act either individually or synergistically to affect disease biology.193,199 Through the use of Inforna and subsequent optimization steps, Liu et al.200 developed a dimeric small molecule that binds to three miRNAs in the pri-miR-17-92 cluster (miR-17, miR-18a, and miR-20a) that share structural commonalities at and adjacent to their Dicer processing sites. They then appended the small molecule with either bleomycin A5, yielding Targaprimir-17-92 Bleo (TGP-17-92 Bleo) or the heterocyclic RNase L-recruiting module, yielding TGP-17-92 RIBOTAC (Fig. 16D and Table S1, ESI†).200 The ability of these two compounds to reduce levels of pri-miR-17-92 was evaluated in MDA-MB-231 TNBC cells and DU-145 prostate cancer cells.200TGP-17-92 Bleo reduced pri-miR-17-92 levels and hence functionally inhibited all six miRNAs it encodes (Table S1, ESI†). Further, pre-miR17, pre-miR-18a, and pre-miR-20a levels were reduced. Cleavage of the cluster and the three pre-miRNAs that TGP-17-92 Bleo binds is consistent with the compound's cellular localization. In contrast, TGP-17-92 RIBOTAC only reduced levels of pre-miR17, pre-miR-18a, and pre-miR-20a and their mature miRNAs while not affecting the primary transcript (Table S1, ESI†).200 That is, the RIBOTAC only cleaved the pre-miRNAs that bind TGP-17-92 because of the co-localization of the RIBOTAC, RNase L, and the pre-miRNA, the three components required for cleavage, in the cytoplasm.200 Thus, these studies devised a facile design strategy to remove an entire pri-miRNA cluster, of importance since ∼25% of all miRNAs are transcribed in clusters, or individual members of the cluster simply by careful selection of the cleavage module and by exploiting differences in cellular localization.13. Conclusion
As this review showcases, various types of RNAs have been successfully targeted with small molecules including miRNAs, lncRNAs, splicing modifiers, repeat expansion disorders, and structured elements found within disease-causing RNAs. Since only 1–2% of the genome is translated into protein but ∼80% is transcribed into RNA, it is not surprising that RNA is rapidly emerging as a promising target of small molecule therapeutics.3,18,201 As illustrated by the examples discussed herein, RNA-targeting small molecules can display impressive potency (nM) and selectivity for their RNA targets, often rivaling those seen with traditional protein-targeting small molecules.Current chemotherapeutic options come with a myriad of side-effects due to off-target effects of the drug. However, compounds such as TGP-96 RIBOTAC, TGP-210 RIBOTAC, TGP-21 RIBOTAC and TGP-17-92 Bleo demonstrate proteome- and transcriptome-wide selectivity while decreasing levels of oncogenic miRs. Thus, these compounds could offer a starting place for the development of novel anti-cancer treatments with fewer side effects.
In addition, this review highlights several RNA-targeting small molecules that affect neurodegenerative disease biology, further highlighting the potential of these small molecules to act as novel therapeutics. More importantly, RNA-targeting small molecules have unlocked therapeutic avenues against proteins of neurological relevance that were traditionally viewed as “undruggable,” such as the case of Synucleozid for α-synuclein and A-5 for tau. Examples such as these highlight the importance of targeting RNA over traditional protein targets as a means of investigating the vast space of disease biology that has remained elusive to drug development. Additionally, RNA cleavers and degraders, such as bleomycin A5-conjugates and RIBOTACs, offer novel therapeutic modalities to reduce levels of disease-causing RNAs, whether oncogenic or neurodegenerative, further expanding the types of diseases that can be targeted with small molecules.
In comparison to antisense oligonucleotide (ASO) technology, the current leading therapeutic option for targeting RNA, small molecules are an attractive alternative due to their synthetic accessibility, on- and off-target optimization via structure–activity relationships, mode of action, and ease of administration. ASOs recognize RNA sequence, while small molecules recognize RNA structure; this essential difference gives small molecules many advantages over ASOs. For example, ASOs targeting repeat expansion disorders are often designed to target the coding region of the gene harboring the expansion, not the expansion itself due to off-target effects on transcripts that contain short repeats.43,139,181 Thus, ASOs have to be specifically designed for each disease, even if the diseases are caused by the same RNA repeat expansion. In contrast, a single small molecule recognizing a disease-causing structure could be a potential treatment for all diseases that harbor the same repeat expansion. For example, DM1, Fuchs endothelial corneal dystrophy (FECD),202 and Huntington's disease-like 2 (HDL-2)203 are all caused by r(CUG)exp. Therefore, small molecules targeting r(CUG)exp could be applied broadly across these diseases as shorter, non-pathogenic repeats found in other transcripts typically lack structure. Another advantage of small molecules over ASOs is the route of therapeutic administration. ASOs are injected intrathecally, a painful and sometimes dangerous procedure for patients to endure. However, RNA-targeting small molecules have shown efficacy in mouse models with intraperitoneal injection181 and success with oral bioavailability in clinical trials.96
Looking at the various classification of RNA-targeting small molecules that have emerged it is interesting to note the differences in affinity and bioactivity that result from appending bleomycin A5 or an RNase L-recruiting module to simple RNA-binding small molecules. While the conjugates tend to decrease the compounds’ binding affinities for the target RNA, the overall biological effect observed upon treatment with the cleaving/degrading compounds far exceeds that of the simple RNA binder (Table S1, ESI†). This is demonstrated in the case of TGP-96 (Kd = 39 nM) and TGP-96 Bleo (Kd = 64 nM), where TGP-96 Bleo demonstrates a greater biological effect than TGP-96. This trend was also observed for TGP-210 (Kd = 160 nM) versusTGP-210 RIBOTAC (Kd = 340 nM) and 2H-K4NMeS (Kd = 12 nM) versusCugamycin, the bleomycin conjugate of 2H-K4NMeS (EC50 = 365 nM). In all of these cases, the small decrease in affinity that results from appending a cleaving or degrading module to the compound is made up for by the large increase in biological activity (up to 20-fold) due to target RNA ablation, rather than simple binding.
Selectivity is also improved when an RNA-binding small molecule is converted to either a RIBOTAC or bleomycin A5 conjugate. Again, looking at the case of TGP-210 and TGP-210 RIBOTAC, converting the small molecule to a RIBOTAC resulted in an increase in the selectivity window over DNA from 5-fold with the RNA binder to 10-fold with the degrader, despite TGP-210 RIBOTAC having decreased affinity for pre-miR-210.189 Additionally, appending bleomycin A5 to RNA-binding small molecules decreases both the affinity of bleomycin A5 and the RNA binder itself for DNA, further enhancing the compound's selectivity. Thus, RNA-targeting small molecules demonstrate a novel avenue for achieving highly selective RNA-centric therapeutics.
The field of RNA-targeting small molecules is still in its infancy, as such the only FDA-approved small molecules on the market are anti-bacterials (targeting bacterial RNA), noting that risdiplam, which targets an RNA–protein interface, recently received FDA approval for the treatment of SMA (approved August 7th, 2020).97 Additionally, branaplam (Novartis)204 is also undergoing a Phase II clinical trial for the treatment of SMA. Both risdiplam and branaplam are administered orally, further supporting the advantages of using small molecules to target RNA. Despite the lack of branded “RNA-targeting” FDA-approved small molecules it is important to note that many protein-targeting FDA-approved drugs, such as kinase and topoisomerase inhibitors, have been found to also bind RNA as off-targets. Thus, the future is exciting for the field of RNA-targeted small molecule therapeutics and will undoubtedly contribute to the advancement of modern, precision medicines.
Conflicts of interest
Matthew D. Disney is a founder of Expansion Therapeutics.References
- X.-D. Fu, Natl. Sci. Rev., 2014, 1, 190–204 CrossRef CAS.
- C. M. Connelly, M. H. Moon and J. S. Schneekloth, Cell Chem. Biol., 2016, 23, 1077–1090 CrossRef CAS.
- T. A. Cooper, L. Wan and G. Dreyfuss, Cell, 2009, 136, 777–793 CrossRef CAS.
- R. R. Breaker, Cold Spring Harbor Perspect. Biol., 2018, 10, a032797 CrossRef CAS.
- Z. A. Jaafar and J. S. Kieft, Nat. Rev. Microbiol., 2019, 17, 110–123 CrossRef CAS.
- A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727–4741 CrossRef CAS.
- L. A. Macfarlane and P. R. Murphy, Curr. Genomics, 2010, 11, 537–561 CrossRef CAS.
- K. Leppek, R. Das and M. Barna, Nat. Rev. Mol. Cell Biol., 2018, 19, 158–174 CrossRef CAS.
- M. B. Warf and J. A. Berglund, Trends Biochem. Sci., 2010, 35, 169–178 CrossRef CAS.
- E. Buratti and F. E. Baralle, Mol. Cell. Biol., 2004, 24, 10505–10514 CrossRef CAS.
- A. J. Zaug and T. R. Cech, Science, 1986, 231, 470–475 CrossRef CAS.
- B. C. Stark, R. Kole, E. J. Bowman and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3717–3721 CrossRef CAS.
- E. A. Doherty and J. A. Doudna, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 457–475 CrossRef CAS.
- S. Müller, B. Appel, D. Balke, R. Hieronymus and C. Nübel, F1000Research, 2016, 5, 1511 Search PubMed.
- Z. Huang, J. Shi, Y. Gao, C. Cui, S. Zhang, J. Li, Y. Zhou and Q. Cui, Nucleic Acids Res., 2019, 47, 1013–1017 CrossRef.
- A. M. Ardekani and M. M. Naeini, Avicenna J. Med. Biotechnol., 2010, 2, 161–179 CAS.
- V. Bernat and M. D. Disney, Neuron, 2015, 87, 28–46 CrossRef CAS.
- M. D. Disney, J. Am. Chem. Soc., 2019, 141, 6776–6790 CrossRef CAS.
- X.-H. Liang, H. Sun, J. G. Nichols and S. T. Crooke, Mol. Ther., 2017, 25, 2075–2092 CrossRef CAS.
- A. Ursu, J. L. Childs-Disney, R. J. Andrews, C. A. O’Leary, S. M. Meyer, A. J. Angelbello, W. N. Moss and M. D. Disney, Chem. Soc. Rev., 2020 10.1039/D0CS00455C.
- M. M. Chong, G. Zhang, S. Cheloufi, T. A. Neubert, G. J. Hannon and D. R. Littman, Genes Dev., 2010, 24, 1951–1960 CrossRef CAS.
- L. F. R. Gebert and I. J. MacRae, Nat. Rev. Mol. Cell Biol., 2019, 20, 21–37 CrossRef CAS.
- T. A. Rand, S. Petersen, F. Du and X. Wang, Cell, 2005, 123, 621–629 CrossRef CAS.
- H. A. Meijer, Y. W. Kong, W. T. Lu, A. Wilczynska, R. V. Spriggs, S. W. Robinson, J. D. Godfrey, A. E. Willis and M. Bushell, Science, 2013, 340, 82–85 CrossRef CAS.
- T. Fukaya, H. O. Iwakawa and Y. Tomari, Mol. Cell, 2014, 56, 67–78 CrossRef CAS.
- A. Fukao, Y. Mishima, N. Takizawa, S. Oka, H. Imataka, J. Pelletier, N. Sonenberg, C. Thoma and T. Fujiwara, Mol. Cell, 2014, 56, 79–89 CrossRef CAS.
- M. Selbach, B. Schwanhausser, N. Thierfelder, Z. Fang, R. Khanin and N. Rajewsky, Nature, 2008, 455, 58–63 CrossRef CAS.
- S. Uhlmann, H. Mannsperger, J. D. Zhang, E. A. Horvat, C. Schmidt, M. Kublbeck, F. Henjes, A. Ward, U. Tschulena, K. Zweig, U. Korf, S. Wiemann and O. Sahin, Mol. Syst. Biol., 2012, 8, 570 CrossRef.
- M. Esteller, Nat. Rev. Genet., 2011, 12, 861–874 CrossRef CAS.
- S. Lin and R. I. Gregory, Nat. Rev. Cancer, 2015, 15, 321–333 CrossRef CAS.
- C. P. Bracken, H. S. Scott and G. J. Goodall, Nat. Rev. Genet., 2016, 17, 719–732 CrossRef CAS . 31.
- D. D. Vo, C. Staedel, L. Zehnacker, R. Benhida, F. Darfeuille and M. Duca, ACS Chem. Biol., 2014, 9, 711–721 CrossRef CAS.
- V. Malnuit, M. Duca and R. Benhida, Org. Biomol. Chem., 2011, 9, 326–336 RSC.
- M. Duca, V. Malnuit, F. Barbault and R. Benhida, Chem. Commun., 2010, 46, 6162–6164 RSC.
- D. D. Vo, T. P. Tran, C. Staedel, R. Benhida, F. Darfeuille, A. Di Giorgio and M. Duca, Chem. – Eur. J., 2016, 22, 5350–5362 CrossRef CAS.
- D. D. Vo, C. Becquart, T. P. A. Tran, A. Di Giorgio, F. Darfeuille, C. Staedel and M. Duca, Org. Biomol. Chem., 2018, 16, 6262–6274 RSC.
- S. Kumar and S. Maiti, Biochimie, 2013, 95, 1422–1431 CrossRef CAS.
- J. P. Joly, G. Mata, P. Eldin, L. Briant, F. Fontaine-Vive, M. Duca and R. Benhida, Chem. – Eur. J., 2014, 20, 2071–2079 CrossRef CAS.
- C. Staedel, T. P. A. Tran, J. Giraud, F. Darfeuille, A. Di Giorgio, N. J. Tourasse, F. Salin, P. Uriac and M. Duca, Sci. Rep., 2018, 8, 1667 CrossRef.
- C. Becquart, M. Le Roch, S. Azoulay, P. Uriac, A. Di Giorgio and M. Duca, ACS Omega, 2018, 3, 16500–16508 CrossRef CAS.
- M. D. Disney, A. M. Winkelsas, S. P. Velagapudi, M. Southern, M. Fallahi and J. L. Childs-Disney, ACS Chem. Biol., 2016, 11, 1720–1728 CrossRef CAS.
- S. P. Velagapudi, Y. Luo, T. Tran, H. S. Haniff, Y. Nakai, M. Fallahi, G. J. Martinez, J. L. Childs-Disney and M. D. Disney, ACS Cent. Sci., 2017, 3, 205–216 CrossRef CAS.
- S. P. Velagapudi, S. M. Gallo and M. D. Disney, Nat. Chem. Biol., 2014, 10, 291–297 CrossRef CAS.
- I. K. Guttilla and B. A. White, J. Biol. Chem., 2009, 284, 23204–23216 CrossRef CAS.
- M. G. Costales, C. L. Haga, S. P. Velagapudi, J. L. Childs-Disney, D. G. Phinney and M. D. Disney, J. Am. Chem. Soc., 2017, 139, 3446–3455 CrossRef CAS.
- T. J. Kelly, A. L. Souza, C. B. Clish and P. Puigserver, Mol. Cell. Biol., 2011, 31, 2696–2706 CrossRef CAS.
- S. P. Velagapudi, M. D. Cameron, C. L. Haga, L. H. Rosenberg, M. Lafitte, D. R. Duckett, D. G. Phinney and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5898–5903 CrossRef CAS.
- M. G. Costales, D. G. Hoch, D. Abegg, J. L. Childs-Disney, S. P. Velagapudi, A. Adibekian and M. D. Disney, J. Am. Chem. Soc., 2019, 141, 2960–2974 CrossRef CAS.
- H. Haniff, L. Knerr, X. Liu, G. Crynen, J. Boström, D. Abegg, A. Adibekian, M. Lemurell and M. Disney, Nat. Chem., 2020 DOI:10.1038/s41557-020-0514-4.
- N. Ferrara, H. P. Gerber and J. LeCouter, Nat. Med., 2003, 9, 669–676 CrossRef CAS.
- N. Sun, B. Ning, K. M. Hansson, A. C. Bruce, S. A. Seaman, C. Zhang, M. Rikard, C. A. DeRosa, C. L. Fraser, M. Wagberg, R. Fritsche-Danielson, J. Wikstrom, K. R. Chien, A. Lundahl, M. Holtta, L. G. Carlsson, S. M. Peirce and S. Hu, Sci. Rep., 2018, 8, 17509 CrossRef.
- S. Yla-Herttuala, T. T. Rissanen, I. Vajanto and J. Hartikainen, J. Am. Coll. Cardiol., 2007, 49, 1015–1026 CrossRef.
- Z. Wen, W. Huang, Y. Feng, W. Cai, Y. Wang, X. Wang, J. Liang, M. Wani, J. Chen, P. Zhu, J. M. Chen, R. W. Millard, G. C. Fan and Y. Wang, PLoS One, 2014, 9, 104666 CrossRef.
- D. Joladarashi, V. N. S. Garikipati, R. A. Thandavarayan, S. K. Verma, A. R. Mackie, M. Khan, A. M. Gumpert, A. Bhimaraj, K. A. Youker, C. Uribe, S. Suresh Babu, P. Jeyabal, R. Kishore and P. Krishnamurthy, J. Am. Coll. Cardiol., 2015, 66, 2214–2226 CrossRef CAS.
- Z. Taimeh, J. Loughran, E. J. Birks and R. Bolli, Nat. Rev. Cardiol., 2013, 10, 519–530 CrossRef CAS.
- X. Guo, L. Gao, Y. Wang, D. K. Y. Chiu, T. Wang and Y. Deng, Briefings Funct. Genomics, 2015, 15, 38–46 CrossRef.
- F. Zhang, L. Zhang and C. Zhang, Tumor Biol., 2016, 37, 163–175 CrossRef CAS.
- C.-W. Wei, T. Luo, S.-S. Zou and A.-S. Wu, Front. Behav. Neurosci., 2018, 12, 348–358 Search PubMed.
- M. V. Neguembor, M. Jothi and D. Gabellini, Skeletal Muscle, 2014, 4, 8 CrossRef.
- S. Dahariya, I. Paddibhatla, S. Kumar, S. Raghuwanshi, A. Pallepati and R. K. Gutti, Mol. Immunol., 2019, 112, 82–92 CrossRef CAS.
- J. Chen, L. Ao and J. Yang, Ann. Transl. Med., 2019, 7, 494 CrossRef CAS.
- M. J. Delás and G. J. Hannon, Open Biol., 2017, 7, 170121 CrossRef.
- R. Pedram Fatemi, S. Salah-Uddin, F. Modarresi, N. Khoury, C. Wahlestedt and M. A. Faghihi, J. Biomol. Screening, 2015, 20, 1132–1141 CrossRef.
- F. A. Abulwerdi, W. Xu, A. A. Ageeli, M. J. Yonkunas, G. Arun, H. Nam, J. S. Schneekloth, T. K. Dayie, D. Spector, N. Baird and S. F. J. Le Grice, ACS Chem. Biol., 2019, 14, 223–235 CrossRef CAS.
- N. F. Rizvi and E. B. Nickbarg, Methods, 2019, 167, 28–38 CrossRef CAS.
- M. Hajjari and A. Salavaty, Cancer Biol. Med., 2015, 12, 1–9 CAS.
- K. Zhang, X. Sun, X. Zhou, L. Han, L. Chen, Z. Shi, A. Zhang, M. Ye, Q. Wang, C. Liu, J. Wei, Y. Ren, J. Yang, J. Zhang, P. Pu, M. Li and C. Kang, OncoTargets Ther., 2015, 6, 537–546 CrossRef.
- Y. Ren, Y.-F. Wang, J. Zhang, Q.-X. Wang, L. Han, M. Mei and C.-S. Kang, Clin. Epigenet, 2019, 11, 29 CrossRef.
- T. Gutschner, M. Hämmerle and S. Diederichs, J. Mol. Med., 2013, 91, 791–801 CrossRef CAS.
- R. Yoshimoto, A. Mayeda, M. Yoshida and S. Nakagawa, Biochim. Biophys. Acta, Gene Regul. Mech., 2016, 1859, 192–199 CrossRef CAS.
- C. Xu, M. Yang, J. Tian, X. Wang and Z. Li, Int. J. Oncol., 2011, 39, 169–175 CAS.
- J. A. Brown, D. Bulkley, J. Wang, M. L. Valenstein, T. A. Yario, T. A. Steitz and J. A. Steitz, Nat. Struct. Mol. Biol., 2014, 21, 633–640 CrossRef CAS.
- J. E. Wilusz, C. K. JnBaptiste, L. Y. Lu, C.-D. Kuhn, L. Joshua-Tor and P. A. Sharp, Genes Dev., 2012, 26, 2392–2407 CrossRef CAS.
- A. Donlic, B. S. Morgan, J. L. Xu, A. Liu, C. Roble Jr. and A. E. Hargrove, Angew. Chem., Int. Ed., 2018, 57, 13242–13247 CrossRef CAS.
- W.-Y. Yang, R. Gao, M. Southern, P. S. Sarkar and M. D. Disney, Nat. Commun., 2016, 7, 11647 CrossRef CAS.
- J. B. Chaires, J. Ren, D. Hamelberg, A. Kumar, V. Pandya, D. W. Boykin and W. D. Wilson, J. Med. Chem., 2004, 47, 5729–5742 CrossRef CAS.
- D. Kumari, I. Gazy and K. Usdin, Brain Sci., 2019, 9, 39–57 CrossRef CAS.
- D. Colak, N. Zaninovic, M. S. Cohen, Z. Rosenwaks, W. Y. Yang, J. Gerhardt, M. D. Disney and S. R. Jaffrey, Science, 2014, 343, 1002–1005 CrossRef CAS.
- M. D. Disney, B. Liu, W. Y. Yang, C. Sellier, T. Tran, N. Charlet-Berguerand and J. L. Childs-Disney, ACS Chem. Biol., 2012, 7, 1711–1718 CrossRef CAS.
- D. Kumari and K. Usdin, Hum. Mol. Genet., 2016, 25, 3689–3698 CrossRef CAS.
- P. Chiurazzi, M. G. Pomponi, R. Willemsen, B. A. Oostra and G. Neri, Hum. Mol. Genet., 1998, 7, 109–113 CrossRef CAS.
- B. Coffee, F. Zhang, S. T. Warren and D. Reines, Nat. Genet., 1999, 22, 98–101 CrossRef CAS.
- D. Kumari and K. Usdin, Hum. Mol. Genet., 2014, 23, 6575–6583 CrossRef CAS.
- Y. Lee and D. C. Rio, Annu. Rev. Biochem., 2015, 84, 291–323 CrossRef CAS.
- M. R. Lunn and C. H. Wang, Lancet, 2008, 371, 2120–2133 CrossRef.
- S. Lefebvre, L. Bürglen, S. Reboullet, O. Clermont, P. Burlet, L. Viollet, B. Benichou, C. Cruaud, P. Millasseau, M. Zeviani, D. Le Paslier, J. Frézal, D. Cohen, J. Weissenbach, A. Munnich and J. Melki, Cell, 1995, 80, 155–165 CrossRef CAS.
- K. R. Brunden, J. Q. Trojanowski and V. M. Lee, Nat. Rev. Drug Discovery, 2009, 8, 783–793 CrossRef CAS.
- M. Busslinger, N. Moschonas and R. A. Flavell, Cell, 1981, 27, 289–298 CrossRef CAS.
- A. Shilo, F. A. Tosto, J. W. Rausch, S. F. J. Le Grice and T. Misteli, Nucleic Acids Res., 2019, 47, 5922–5935 CrossRef CAS.
- A. J. Bergsma, S. L. In't Groen, F. W. Verheijen, A. T. van der Ploeg and W. W. M. P. Pijnappel, Mol. Ther. – Nucleic Acids, 2016, 5, 361 CrossRef.
- C. F. Rochette, N. Gilbert and L. R. Simard, Hum. Genet., 2001, 108, 255–266 CrossRef CAS.
- C. L. Lorson, E. Hahnen, E. J. Androphy and B. Wirth, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6307–6311 CrossRef CAS.
- B. G. Burnett, E. Muñoz, A. Tandon, D. Y. Kwon, C. J. Sumner and K. H. Fischbeck, Mol. Cell. Biol., 2009, 29, 1107–1115 CrossRef CAS.
- T. Gidaro and L. Servais, Dev. Med. Child Neurol., 2019, 61, 19–24 CrossRef.
- S. M. Hoy, Drugs, 2019, 79, 1255–1262 CrossRef CAS.
- A. N. Calder, E. J. Androphy and K. J. Hodgetts, J. Med. Chem., 2016, 59, 10067–10083 CrossRef CAS.
- U.S. Food & Drug Administration, https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy, (accessed August 2020).
- J. Palacino, S. E. Swalley, C. Song, A. K. Cheung, L. Shu, X. Zhang, M. Van Hoosear, Y. Shin, D. N. Chin, C. G. Keller, M. Beibel, N. A. Renaud, T. M. Smith, M. Salcius, X. Shi, M. Hild, R. Servais, M. Jain, L. Deng, C. Bullock, M. McLellan, S. Schuierer, L. Murphy, M. J. Blommers, C. Blaustein, F. Berenshteyn, A. Lacoste, J. R. Thomas, G. Roma, G. A. Michaud, B. S. Tseng, J. A. Porter, V. E. Myer, J. A. Tallarico, L. G. Hamann, D. Curtis, M. C. Fishman, W. F. Dietrich, N. A. Dales and R. Sivasankaran, Nat. Chem. Biol., 2015, 11, 511–517 CrossRef CAS.
- S. J. Kolb and J. T. Kissel, Arch. Neurol., 2011, 68, 979–984 CrossRef.
- E. Goina, N. Skoko and F. Pagani, Mol. Cell. Biol., 2008, 28, 3850–3860 CrossRef CAS.
- D. A. Pomeranz Krummel, C. Oubridge, A. K. Leung, J. Li and K. Nagai, Nature, 2009, 458, 475–480 CrossRef CAS.
- Y. Kondo, C. Oubridge, A. M. van Roon and K. Nagai, eLife, 2015, 4, e04986 CrossRef.
- S. Campagne, S. Boigner, S. Rudisser, A. Moursy, L. Gillioz, A. Knorlein, J. Hall, H. Ratni, A. Clery and F. H. Allain, Nat. Chem. Biol., 2019, 15, 1191–1198 CrossRef CAS.
- M. Sivaramakrishnan, K. D. McCarthy, S. Campagne, S. Huber, S. Meier, A. Augustin, T. Heckel, H. Meistermann, M. N. Hug, P. Birrer, A. Moursy, S. Khawaja, R. Schmucki, N. Berntenis, N. Giroud, S. Golling, M. Tzouros, B. Banfai, G. Duran-Pacheco, J. Lamerz, Y. Hsiu Liu, T. Luebbers, H. Ratni, M. Ebeling, A. Cléry, S. Paushkin, A. R. Krainer, F. H. Allain and F. Metzger, Nat. Commun., 2017, 8, 1476 CrossRef.
- B. J. Blencowe, Trends Biochem. Sci., 2000, 25, 106–110 CrossRef CAS.
- A. Moursy, F. H. Allain and A. Cléry, Nucleic Acids Res., 2014, 42, 6659–6672 CrossRef CAS.
- J. Wang, P. G. Schultz and K. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, e4604–e4612 CrossRef CAS.
- N. N. Singh, R. N. Singh and E. J. Androphy, Nucleic Acids Res., 2007, 35, 371–389 CrossRef CAS.
- L. Debaize and M. B. Troadec, Cell. Mol. Life Sci., 2019, 76, 259–281 CrossRef CAS.
- P. Briata, D. Bordo, M. Puppo, F. Gorlero, M. Rossi, N. Perrone-Bizzozero and R. Gherzi, Wiley Interdiscip. Rev.: RNA, 2016, 7, 227–240 CrossRef CAS.
- D. W. Cleveland, S. Y. Hwo and M. W. Kirschner, J. Mol. Biol., 1977, 116, 207–225 CrossRef CAS.
- M. Goedert, M. G. Spillantini, M. C. Potier, J. Ulrich and R. A. Crowther, EMBO J., 1989, 8, 393–399 CrossRef CAS.
- Q. Gao, J. Memmott, R. Lafyatis, S. Stamm, G. Screaton and A. Andreadis, J. Neurochem., 2000, 74, 490–500 CrossRef CAS.
- M. Hong, V. Zhukareva, V. Vogelsberg-Ragaglia, Z. Wszolek, L. Reed, B. I. Miller, D. H. Geschwind, T. D. Bird, D. McKeel, A. Goate, J. C. Morris, K. C. Wilhelmsen, G. D. Schellenberg, J. Q. Trojanowski and V. M. Lee, Science, 1998, 282, 1914–1917 CrossRef CAS.
- A. Grover, H. Houlden, M. Baker, J. Adamson, J. Lewis, G. Prihar, S. Pickering-Brown, K. Duff and M. Hutton, J. Biol. Chem., 1999, 274, 15134–15143 CrossRef CAS.
- L. Varani, M. Hasegawa, M. G. Spillantini, M. J. Smith, J. R. Murrell, B. Ghetti, A. Klug, M. Goedert and G. Varani, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8229–8234 CrossRef CAS.
- C. P. Donahue, C. Muratore, J. Y. Wu, K. S. Kosik and M. S. Wolfe, J. Biol. Chem., 2006, 281, 23302–23306 CrossRef CAS.
- S. Zheng, Y. Chen, C. P. Donahue, M. S. Wolfe and G. Varani, Chem. Biol., 2009, 16, 557–566 CrossRef CAS.
- Y. Liu, E. Peacey, J. Dickson, C. P. Donahue, S. Zheng, G. Varani and M. S. Wolfe, J. Med. Chem., 2009, 52, 6523–6526 CrossRef CAS.
- J. L. Chen, P. Zhang, M. Abe, H. Aikawa, L. Zhang, A. J. Frank, T. Zembryski, C. Hubbs, H. Park, J. Withka, C. Steppan, L. Rogers, S. Cabral, M. Pettersson, T. T. Wager, M. A. Fountain, G. Rumbaugh, J. L. Childs-Disney and M. D. Disney, J. Am. Chem. Soc., 2020, 142, 8706–8727 CrossRef.
- Y. Luo and M. D. Disney, ChemBioChem, 2014, 15, 2041–2044 CrossRef CAS.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 CrossRef CAS.
- M. M. Scotti and M. S. Swanson, Nat. Rev. Genet., 2016, 17, 19–32 CrossRef CAS.
- G. Biamonti, A. Amato, E. Belloni, A. Di Matteo, L. Infantino, D. Pradella and C. Ghigna, Aging: Clin. Exp. Res., 2019 DOI:10.1007/s40520-019-01360-x.
- M. Hecker, A. Rüge, E. Putscher, N. Boxberger, P. S. Rommer, B. Fitzner and U. K. Zettl, Autoimmun. Rev., 2019, 18, 721–732 CrossRef CAS.
- S. C. Lee and O. Abdel-Wahab, Nat. Med., 2016, 22, 976–986 CrossRef CAS.
- L. M. Urbanski, N. Leclair and O. Anczuków, Wiley Interdiscip. Rev.: RNA, 2018, 9, e1476 CrossRef.
- N. P. Mastroyiannopoulos, M. L. Feldman, J. B. Uney, M. S. Mahadevan and L. A. Phylactou, EMBO Rep., 2005, 6, 458–463 CrossRef CAS.
- J. L. Childs-Disney, J. Hoskins, S. G. Rzuczek, C. A. Thornton and M. D. Disney, ACS Chem. Biol., 2012, 7, 856–862 CrossRef CAS.
- S. G. Rzuczek, Y. Gao, Z. Z. Tang, C. A. Thornton, T. Kodadek and M. D. Disney, ACS Chem. Biol., 2013, 8, 2312–2321 CrossRef CAS.
- S. G. Rzuczek, L. A. Colgan, Y. Nakai, M. D. Cameron, D. Furling, R. Yasuda and M. D. Disney, Nat. Chem. Biol., 2017, 13, 188–193 CrossRef CAS.
- W. Y. Yang, H. D. Wilson, S. P. Velagapudi and M. D. Disney, J. Am. Chem. Soc., 2015, 137, 5336–5345 CrossRef CAS.
- J. F. Arambula, S. R. Ramisetty, A. M. Baranger and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16068–16073 CrossRef CAS.
- A. H. Jahromi, Y. Fu, K. A. Miller, L. Nguyen, L. M. Luu, A. M. Baranger and S. C. Zimmerman, J. Med. Chem., 2013, 56, 9471–9481 CrossRef CAS.
- L. M. Luu, L. Nguyen, S. Peng, J. Lee, H. Y. Lee, C. H. Wong, P. J. Hergenrother, H. Y. Chan and S. C. Zimmerman, ChemMedChem, 2016, 11, 1428–1435 CrossRef CAS.
- J. Lee, Y. Bai, U. V. Chembazhi, S. Peng, K. Yum, L. M. Luu, L. D. Hagler, J. F. Serrano, H. Y. E. Chan, A. Kalsotra and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8709–8714 CrossRef CAS.
- J. Li, J. Matsumoto, L.-P. Bai, A. Murata, C. Dohno and K. Nakatani, Chem. – Eur. J., 2016, 22, 14881–14889 CrossRef CAS.
- M. M. Lee, A. Pushechnikov and M. D. Disney, ACS Chem. Biol., 2009, 4, 345–355 CrossRef CAS.
- R. I. Benhamou, A. J. Angelbello, R. J. Andrews, E. T. Wang, W. N. Moss and M. D. Disney, ACS Chem. Biol., 2020, 15, 485–493 CrossRef CAS.
- R. I. Benhamou, A. J. Angelbello, E. T. Wang and M. D. Disney, Cell Chem. Biol., 2020, 27, 223–231 CrossRef CAS.
- W. Y. Yang, F. He, R. L. Strack, S. Y. Oh, M. Frazer, S. R. Jaffrey, P. K. Todd and M. D. Disney, ACS Chem. Biol., 2016, 11, 2456–2465 CrossRef CAS.
- Z. Su, Y. J. Zhang, T. F. Gendron, P. O. Bauer, J. Chew, W. Y. Yang, E. Fostvedt, K. Jansen-West, V. V. Belzil, P. Desaro, A. Johnston, K. Overstreet, S. Y. Oh, P. K. Todd, J. D. Berry, M. E. Cudkowicz, B. F. Boeve, D. Dickson, M. K. Floeter, B. J. Traynor, C. Morelli, A. Ratti, V. Silani, R. Rademakers, R. H. Brown, J. D. Rothstein, K. B. Boylan, L. Petrucelli and M. D. Disney, Neuron, 2014, 83, 1043–1050 CrossRef CAS.
- Z. F. Wang, A. Ursu, J. L. Childs-Disney, R. Guertler, W. Y. Yang, V. Bernat, S. G. Rzuczek, R. Fuerst, Y. J. Zhang, T. F. Gendron, I. Yildirim, B. G. Dwyer, J. E. Rice, L. Petrucelli and M. D. Disney, Cell Chem. Biol., 2019, 26, 179–190 CrossRef CAS.
- Ł. J. Sznajder, J. D. Thomas, E. M. Carrell, T. Reid, K. N. McFarland, J. D. Cleary, R. Oliveira, C. A. Nutter, K. Bhatt, K. Sobczak, T. Ashizawa, C. A. Thornton, L. P. W. Ranum and M. S. Swanson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4234–4239 CrossRef.
- J. R. Hesselberth, Wiley Interdiscip. Rev.: RNA, 2013, 4, 677–691 CrossRef CAS.
- P. E. Ash, K. F. Bieniek, T. F. Gendron, T. Caulfield, W. L. Lin, M. Dejesus-Hernandez, M. M. van Blitterswijk, K. Jansen-West, J. W. Paul, 3rd, R. Rademakers, K. B. Boylan, D. W. Dickson and L. Petrucelli, Neuron, 2013, 77, 639–646 CrossRef CAS.
- T. F. Gendron, K. F. Bieniek, Y. J. Zhang, K. Jansen-West, P. E. Ash, T. Caulfield, L. Daughrity, J. H. Dunmore, M. Castanedes-Casey, J. Chew, D. M. Cosio, M. van Blitterswijk, W. C. Lee, R. Rademakers, K. B. Boylan, D. W. Dickson and L. Petrucelli, Acta Neuropathol., 2013, 126, 829–844 CrossRef CAS.
- K. Mori, T. Arzberger, F. A. Grasser, I. Gijselinck, S. May, K. Rentzsch, S. M. Weng, M. H. Schludi, J. van der Zee, M. Cruts, C. Van Broeckhoven, E. Kremmer, H. A. Kretzschmar, C. Haass and D. Edbauer, Acta Neuropathol., 2013, 126, 881–893 CrossRef CAS.
- T. Zu, B. Gibbens, N. S. Doty, M. Gomes-Pereira, A. Huguet, M. D. Stone, J. Margolis, M. Peterson, T. W. Markowski, M. A. C. Ingram, Z. Nan, C. Forster, W. C. Low, B. Schoser, N. V. Somia, H. B. Clark, S. Schmechel, P. B. Bitterman, G. Gourdon, M. S. Swanson, M. Moseley and L. P. W. Ranum, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 260–265 CrossRef CAS.
- P. K. Todd, S. Y. Oh, A. Krans, F. He, C. Sellier, M. Frazer, A. J. Renoux, K. C. Chen, K. M. Scaglione, V. Basrur, K. Elenitoba-Johnson, J. P. Vonsattel, E. D. Louis, M. A. Sutton, J. P. Taylor, R. E. Mills, N. Charlet-Berguerand and H. L. Paulson, Neuron, 2013, 78, 440–455 CrossRef CAS.
- T. F. Gendron, V. V. Belzil, Y. J. Zhang and L. Petrucelli, Acta Neuropathol., 2014, 127, 359–376 CrossRef CAS.
- W.-Y. Yang, H. D. Wilson, S. P. Velagapudi and M. D. Disney, J. Am. Chem. Soc., 2015, 137, 5336–5345 CrossRef CAS.
- T. F. Gendron, J. Chew, J. N. Stankowski, L. R. Hayes, Y. J. Zhang, M. Prudencio, Y. Carlomagno, L. M. Daughrity, K. Jansen-West, E. A. Perkerson, A. O'Raw, C. Cook, L. Pregent, V. Belzil, M. van Blitterswijk, L. J. Tabassian, C. W. Lee, M. Yue, J. Tong, Y. Song, M. Castanedes-Casey, L. Rousseau, V. Phillips, D. W. Dickson, R. Rademakers, J. D. Fryer, B. K. Rush, O. Pedraza, A. M. Caputo, P. Desaro, C. Palmucci, A. Robertson, M. G. Heckman, N. N. Diehl, E. Wiggs, M. Tierney, L. Braun, J. Farren, D. Lacomis, S. Ladha, C. N. Fournier, L. F. McCluskey, L. B. Elman, J. B. Toledo, J. D. McBride, C. Tiloca, C. Morelli, B. Poletti, F. Solca, A. Prelle, J. Wuu, J. Jockel-Balsarotti, F. Rigo, C. Ambrose, A. Datta, W. Yang, D. Raitcheva, G. Antognetti, A. McCampbell, J. C. Van Swieten, B. L. Miller, A. L. Boxer, R. H. Brown, R. Bowser, T. M. Miller, J. Q. Trojanowski, M. Grossman, J. D. Berry, W. T. Hu, A. Ratti, B. J. Traynor, M. D. Disney, M. Benatar, V. Silani, J. D. Glass, M. K. Floeter, J. D. Rothstein, K. B. Boylan and L. Petrucelli, Sci. Transl. Med., 2017, 9, eaai7866 CrossRef.
- T. F. Gendron, M. van Blitterswijk, K. F. Bieniek, L. M. Daughrity, J. Jiang, B. K. Rush, O. Pedraza, J. A. Lucas, M. E. Murray, P. Desaro, A. Robertson, K. Overstreet, C. S. Thomas, J. E. Crook, M. Castanedes-Casey, L. Rousseau, K. A. Josephs, J. E. Parisi, D. S. Knopman, R. C. Petersen, B. F. Boeve, N. R. Graff-Radford, R. Rademakers, C. Lagier-Tourenne, D. Edbauer, D. W. Cleveland, D. W. Dickson, L. Petrucelli and K. B. Boylan, Acta Neuropathol., 2015, 130, 559–573 CrossRef CAS.
- M. Clamp, B. Fry, M. Kamal, X. Xie, J. Cuff, M. F. Lin, M. Kellis, K. Lindblad-Toh and E. S. Lander, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19428–19433 CrossRef CAS.
- A. Hopkins and C. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS.
- C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502–508 CrossRef CAS.
- J. Spiegel, P. M. Cromm, G. Zimmermann, T. N. Grossmann and H. Waldmann, Nat. Chem. Biol., 2014, 10, 613–622 CrossRef CAS.
- P. Zhang, H. J. Park, J. Zhang, E. Junn, R. J. Andrews, S. P. Velagapudi, D. Abegg, K. Vishnu, M. G. Costales, J. L. Childs-Disney, A. Adibekian, W. N. Moss, M. M. Mouradian and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1457–1467 CrossRef CAS.
- V. M. Lee and J. Q. Trojanowski, Neuron, 2006, 51, 33–38 CrossRef.
- M. G. Spillantini, M. L. Schmidt, V. M.-Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS.
- K. C. Luk, V. Kehm, J. Carroll, B. Zhang, P. O’Brien, J. Q. Trojanowski and V. M.-Y. Lee, Science, 2012, 338, 949–953 CrossRef CAS.
- A. B. Singleton, M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy and K. Gwinn-Hardy, Science, 2003, 302, 841 CrossRef CAS.
- E. Junn, K. W. Lee, B. S. Jeong, T. W. Chan, J. Y. Im and M. M. Mouradian, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13052–13057 CrossRef CAS.
- D. M. Maraganore, J. Mov. Disord., 2011, 4, 1–7 CrossRef.
- A. L. Friedlich, R. E. Tanzi and J. T. Rogers, Mol. Psychiatry, 2007, 12, 222–223 CrossRef CAS.
- F. Febbraro, M. Giorgi, S. Caldarola, F. Loreni and M. Romero-Ramos, NeuroReport, 2012, 23, 576–580 CrossRef CAS.
- J. L. Childs-Disney, T. Tran, B. R. Vummidi, S. P. Velagapudi, H. S. Haniff, Y. Matsumoto, G. Crynen, M. R. Southern, A. Biswas, Z. F. Wang, T. L. Tellinghuisen and M. D. Disney, Chem, 2018, 4, 2384–2404 CAS.
- J. R. Mazzulli, F. Zunke, O. Isacson, L. Studer and D. Krainc, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1931–1936 CrossRef CAS.
- B. Liu, J. L. Childs-Disney, B. M. Znosko, D. Wang, M. Fallahi, S. M. Gallo and M. D. Disney, BMC Bioinf., 2016, 17, 112 CrossRef.
- H. Pimentel, N. L. Bray, S. Puente, P. Melsted and L. Pachter, Nat. Methods, 2017, 14, 687–690 CrossRef CAS.
- D. L. Boger and H. Cai, Angew. Chem., Int. Ed., 1999, 38, 448–476 CrossRef CAS.
- B. J. Carter, E. de Vroom, E. C. Long, G. A. van der Marel, J. H. van Boom and S. M. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9373–9377 CrossRef CAS.
- A. T. Abraham, J. J. Lin, D. L. Newton, S. Rybak and S. M. Hecht, Chem. Biol., 2003, 10, 45–52 CrossRef CAS.
- A. J. Angelbello and M. D. Disney, ChemBioChem, 2018, 19, 43–47 CrossRef CAS.
- L. Ma, J. Teruya-Feldstein and R. A. Weinberg, Nature, 2007, 449, 682–688 CrossRef CAS.
- Y. Li and M. D. Disney, ACS Chem. Biol., 2018, 13, 3065–3071 CrossRef CAS.
- Q. Ma, Z. Xu, B. R. Schroeder, W. Sun, F. Wei, S. Hashimoto, K. Konishi, C. J. Leitheiser and S. M. Hecht, J. Am. Chem. Soc., 2007, 129, 12439–12452 CrossRef CAS.
- Z.-D. Xu, M. Wang, S.-L. Xiao, C.-L. Liu and M. Yang, Bioorg. Med. Chem. Lett., 2003, 13, 2595–2599 CrossRef CAS.
- V. Agarwal, G. W. Bell, J.-W. Nam and D. P. Bartel, eLife, 2015, 4, e05005 CrossRef.
- A. J. Angelbello, S. G. Rzuczek, K. K. Mckee, J. L. Chen, H. Olafson, M. D. Cameron, W. N. Moss, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7799–7804 CrossRef CAS.
- A. Mankodi, E. Logigian, L. Callahan, C. McClain, R. White, D. Henderson, M. Krym and C. A. Thornton, Science, 2000, 289, 1769–1773 CrossRef CAS.
- A. Mankodi, M. P. Takahashi, H. Jiang, C. L. Beck, W. J. Bowers, R. T. Moxley, S. C. Cannon and C. A. Thornton, Mol. Cell, 2002, 10, 35–44 CrossRef CAS.
- R. H. Blum, S. K. Carter and K. Agre, Cancer, 1973, 31, 903–914 CrossRef CAS.
- Y. Katz, E. T. Wang, E. M. Airoldi and C. B. Burge, Nat. Methods, 2010, 7, 1009–1015 CrossRef CAS.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS.
- M. G. Costales, Y. Matsumoto, S. P. Velagapudi and M. D. Disney, J. Am. Chem. Soc., 2018, 140, 6741–6744 CrossRef CAS.
- R. H. Silverman, J. Virol., 2007, 81, 12720–12729 CrossRef CAS.
- M. G. Costales, B. Suresh, K. Vishnu and M. D. Disney, Cell Chem. Biol., 2019, 26, 1180–1186 CrossRef CAS.
- M. G. Costales, H. Aikawa, Y. Li, J. L. Childs-Disney, D. Abegg, D. G. Hoch, S. Pradeep Velagapudi, Y. Nakai, T. Khan, K. W. Wang, I. Yildirim, A. Adibekian, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2406–2411 CrossRef CAS.
- C. S. Thakur, B. K. Jha, B. Dong, J. Das Gupta, K. M. Silverman, H. Mao, H. Sawai, A. O. Nakamura, A. K. Banerjee, A. Gudkov and R. H. Silverman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9585–9590 CrossRef CAS.
- P. P. Graczyk, J. Med. Chem., 2007, 50, 5773–5779 CrossRef CAS.
- P. Mu, Y. C. Han, D. Betel, E. Yao, M. Squatrito, P. Ogrodowski, E. de Stanchina, A. D'Andrea, C. Sander and A. Ventura, Genes Dev., 2009, 23, 2806–2811 CrossRef CAS.
- K. Conkrite, M. Sundby, S. Mukai, J. M. Thomson, D. Mu, S. M. Hammond and D. MacPherson, Genes Dev., 2011, 25, 1734–1745 CrossRef CAS.
- G. Huang, K. Nishimoto, Z. Zhou, D. Hughes and E. S. Kleinerman, Cancer Res., 2012, 72, 908–916 CrossRef CAS.
- K. Kim, G. Chadalapaka, S. O. Lee, D. Yamada, X. Sastre-Garau, P. A. Defossez, Y. Y. Park, J. S. Lee and S. Safe, Oncogene, 2012, 31, 1034–1044 CrossRef CAS.
- V. K. Topkara and D. L. Mann, Cardiovasc. Drugs Ther., 2011, 25, 171–182 CrossRef CAS.
- E. Mogilyansky and I. Rigoutsos, Cell Death Differ., 2013, 20, 1603–1614 CrossRef CAS.
- S. P. Kabekkodu, V. Shukla, V. K. Varghese, J. D'Souza, S. Chakrabarty and K. Satyamoorthy, Biol. Rev., 2018, 93, 1955–1986 CrossRef.
- X. Liu, H. S. Haniff, J. L. Childs-Disney, A. Shuster, H. Aikawa, A. Adibekian and M. D. Disney, J. Am. Chem. Soc., 2020, 142, 6970–6982 CrossRef CAS.
- M. G. Costales, J. L. Childs-Disney, H. S. Haniff and M. D. Disney, J. Med. Chem., 2020 DOI:10.1021/acs.jmedchem.9b01927.
- E. D. Wieben, R. A. Aleff, X. Tang, M. L. Butz, K. R. Kalari, E. W. Highsmith, J. Jen, G. Vasmatzis, S. V. Patel, L. J. Maguire, K. H. Baratz and M. P. Fautsch, Invest. Ophthalmol. Visual Sci., 2017, 58, 343–352 CrossRef.
- R. L. Margolis and D. D. Rudnicki, Curr. Opin. Neurol., 2016, 29, 743–748 CrossRef CAS.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02268552, accessed July 2020.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00560f |
| This journal is © The Royal Society of Chemistry 2020 |