
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyoxometalates in solution: speciation under spotlight†
Nadiia I.
Gumerova
 and
Annette
Rompel
*
and
Annette
Rompel
*
Universität Wien, Fakultät für Chemie, Institut für Biophysikalische Chemie, Althanstr. 14, 1090 Vienna, Austria. E-mail: annette.rompel@univie.ac.at; Web: http://www.bpc.univie.ac.at
First published on 29th September 2020
Abstract
Polyoxometalates (POMs) are a large group of anionic polynuclear metal–oxo clusters with discrete and chemically modifiable structures. In most aqueous POM solutions, numerous, and often highly negatively charged, species of different nuclearities are formed. It is rather difficult to determine the dominant POM species or their combination, which is responsible for the specific POM activity, during a particular application. Thus, the identification of all individual speciation profiles is essential for the successful implementation of POMs in solution applications. This review article summarizes species that are present in isopoly- and heteropolyvanadates, -niobates, -molybdates and -tungstates aqueous solutions and covers their stability and transformations. The ion-distribution diagrams over a wide pH range are presented in a comprehensive manner. These diagrams are intended for the targeted use of POMs, and in a clear form shows species that are in equilibrium at the given pH value. Thus, the data accumulated in this review can serve as both a starting point and a complete reference material for determining the composition of POM solutions. Some examples are highlighted where the POM speciation studies led to a detailed understanding of their role in applications. In doing so, we aim to motivate the POM community for more speciation studies and to make the subject more comprehensible, both for synthetic POM chemists and for scientists with different backgrounds interested in applying POMs in biological, medical, electrochemical, supramolecular and nanochemistry fields, or as homogeneous catalysts and other water-soluble materials.
 Nadiia I. Gumerova | Nadiia Gumerova is currently a postdoctoral research fellow at the Department of Biophysical Chemistry at the University of Vienna (Austria). She received her PhD degree in Inorganic Chemistry in 2015 from the Vasyl’ Stus Donetsk National University (Ukraine). During her PhD studies, Nadiia Gumerova investigated the solution chemistry of isopoly- and heteropolytungstates, including formation conditions of Anderson type polyoxometalates. In 2017, she received the Lise Meitner Fellowship from Austrian Science Fund and joined the group of Prof. Annette Rompel at the University of Vienna (Austria). She has an enduring interest in the areas of organic–inorganic hybrid polyoxometalate-based materials for their biological application as additives for macromolecular crystallography and as enzyme inhibitors. |
 Annette Rompel | Annette Rompel studied Chemistry at the Westfälische Wilhelms University of Münster where she received her doctoral degree. Besides research at the University of California, Berkeley, and the Lawrence Berkeley National Laboratory, she was a visiting scientist at the RIKEN, Institute of Physical and Chemical Research, Sendai, Japan, and the University of Southern Denmark, Odense. Since 2008, she is the Head of the Department of Biophysical Chemistry at the University of Vienna. Her main research interests are the structure/function elucidation of metalloenzymes and the synthesis and characterization of biologically active polyoxometalates. |
1. Introduction
1.1. Solution and solid-state structural chemistry of polyoxometalates (POMs)
Polyoxometalates (POMs) are a large group of discrete, mostly anionic polynuclear metal–oxo clusters amenable to a variety of chemical transformations.1–3 POMs are generally characterized in the solid state prior to dissolution, and this structural information is used as the framework on which the solution chemistry is developed. The compound isolated in crystalline form may not necessarily be the one with highest abundance in solution. For example, di- and tri-molybdates can be easily crystallized from an acidified molybdate solution at pH ∼ 7,4,5 but they are not represented as discrete solution species, and the dominating anion at this pH is [MoVI7O24]6−.6 In solution, POMs form species that can be protonated and undergo redox processes, which contribute to the utmost importance of the speciation characterization. For the application and/or investigation of POM complexes in aqueous solution, a thorough insight of the solution chemistry by identifying all equilibrium constants and individual speciation profiles is essential in order to understand the reaction mechanism and tune the application conditions accordingly. In rare cases where the solution chemistry is presented in literature, the solution details are often not of the main interest for the authors, and are described only in the supplementary materials. The rapidly growing number of POMs application in solution, especially their catalytic7 and biological ones,8–11 entails the need for a deeper understanding and analysis of the fundamental relationship between POM's structural behavior in the solid state and in solution. This is currently an acute drawback, often leading to an incorrect interpretation and erroneous determination of structure–activity relationships. To greatly facilitate the selection of a suitable POM cluster with classical addenda atoms (VV, NbV, TaV, MoVI and WVI) for any deliberate and purposeful use in solution, this review will serve as a guide for better understanding of the POM behavior in the liquid phase. The POM solution equilibria presented in details can and should be used to interpret the results obtained in POM solution application for fundamental vision and full understanding of the POM nature.1.2. Speciation in chemistry: key factors affecting POMs solution behavior
The most concise definition of chemical speciation is as follows: composition, concentration, and oxidation state of each of the chemical forms of an element present in a sample.12,13 The term “speciation” is also used to describe the distribution of species in a particular sample, where it is synonymous with the “species distribution”.14 This notion has been accepted in such diverse fields as toxicology, clinical chemistry, geochemistry, environmental chemistry, biochemistry and inorganic chemistry. New developments in analytical instrumentation and methodology (electronical, vibrational, X-ray absorption and nuclear magnetic resonance spectroscopy, mass-spectrometry, electrochemistry) allow identifying and quantifying the species present in solution.The key factors affecting POMs speciation and the mechanism of isopoly- and heteropolyanions formation are the added acid and metal concentrations, kind of interactions and the range of chemical conditions (ionic strength, buffer type, presence of potential heteroatoms, type of countercations, etc.) under which the dissolution takes place. It is not the pH alone that determines POMs speciation; rather, it is the ratio of acid (usually strong inorganic acids such as HCl, HNO3, H2SO4) to monomeric oxo-ions (e.g. [MoVIO4]2−, [WVIO4]2− or [VVO4]3−) concentration. This ratio determines the ‘degree of protonation’, Z, which is defined as the average number of protons bound to monomeric oxo-metalate in solution.1 It is widely used in speciation studies, and we will refer to it in the cases where no pH regions are given and Z is used by the authors. Z is defined as the ratio q/p in the general eqn (1):
p[MlOn]k− + qH+ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [(MlOn)pHq](k−q)−, [(MlOn)pHq](k−q)−, | (1) |
The main results of speciation studies are distribution curves that represent the percentages, partial mole fractions (α) or equilibrium concentrations of the different chemical species present in solution under given conditions.15 Concentration distribution curves are generally presented as a function of a single variable, such as pH. For distribution curves of POMs, the ‘degree of protonation’ Z is commonly used as one single variable. The equilibrium concentrations of various species are calculated by solving the mathematical system of mass balance equations constructed for each component, and these mass balance equations are then solved iteratively for the concentrations of the free components.16 It should be noted that the thermodynamic equilibrium constants are based on activities that depend on temperature and pressure. Most reported stability constants for POMs are considered as stoichiometric constants, which are expressed as equilibrium concentration quotients, and thus they are valid only at a given ionic strength (μ, M) and in a given solvent. As an example, the concentration equilibrium constants lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kc for the formation of Ni-centered Anderson-type polyoxomolybdate (POMo) eqn (2):
Kc for the formation of Ni-centered Anderson-type polyoxomolybdate (POMo) eqn (2):
| Ni2+ + 6[MoVIO4]2− + (6 + x)H+ ⇆ Hx[NiII(OH)6MoVI6O18](4−x)−, x = 0–3 | (2) |
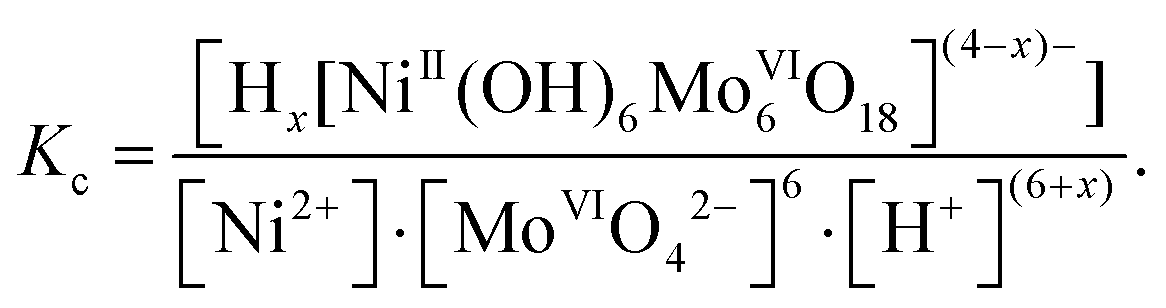 | (3) |
Potentiometric studies (Fig. 1A) allowed to calculate lgKc for each anion in solution and based on these values the ion distribution diagram depending on the acidity [α, mole fraction = f(Z)] was built (Fig. 1B).17
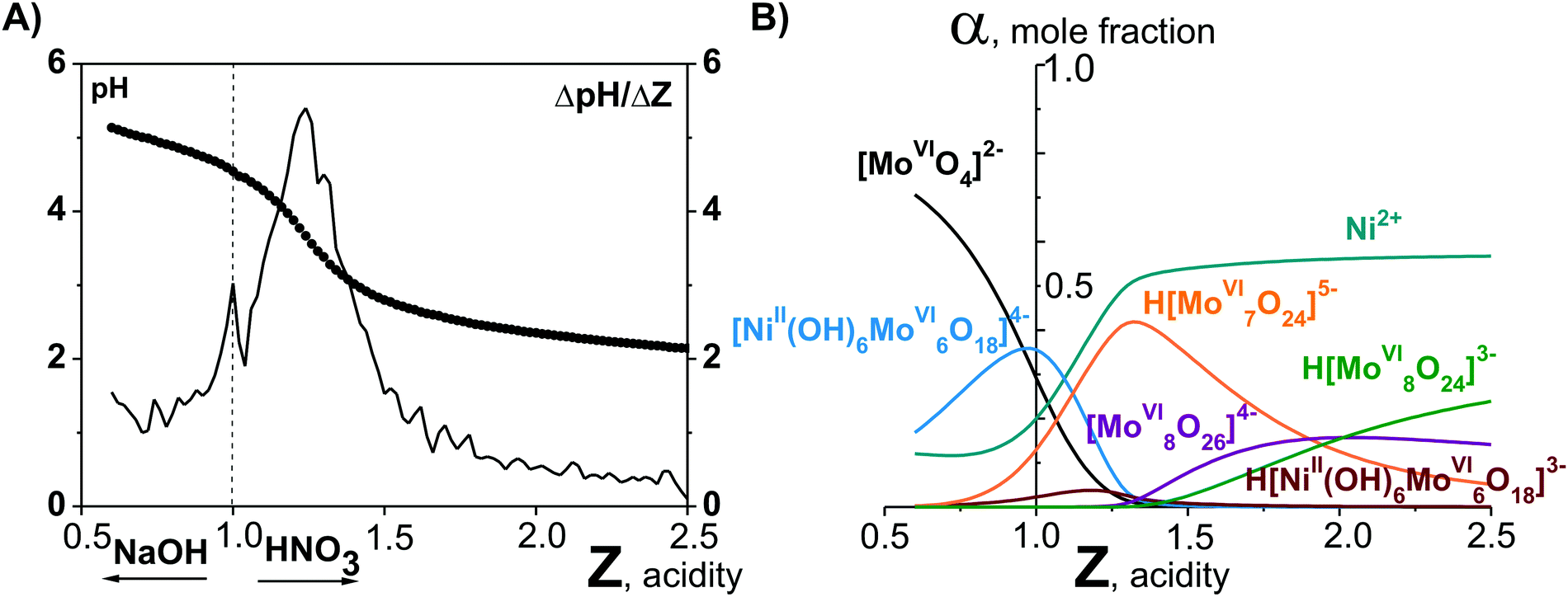 | ||
| Fig. 1 pH-Potentiometry and mathematical modeling in the system Ni2+–[MoVIO4]2−–H+–H2O acidified to mole ratio Z = n(H+)/n([MoVIO4]2−) = 1.00.17 (A) Integral (points, pH = f(Z)) and differential (line, ΔpH/Z = f(Z)) titration curve. (B) Distribution diagrams of ions presented in solution after 60 days aging of the initial Ni2+–[MoVIO4]2−–H+–H2O solution. | ||
To avoid questioning of inconsistent results presented by various authors, throughout this review we deliberately display distribution diagrams without considering concentration or mole fraction and only focus on the pH range in which a particular POM species is stable. As an example, the maximum mole fraction at pH 5.5 for [MoVI7O24]6− is ∼75% reported by Cruywagen et al.,6 and is ∼100% by Maximovskaya et al.18 Only species confirmed by several methods (preferably NMR spectroscopy among them) and/or groups are included to our analysis and as a result to the diagrams. The speciation analysis for POM with VV, NbV, TaV, MoVI and WVI as addenda atoms and for majority of archetypes is provided; however, due to lacking a “data base” for POM anions in solution, some speciation works might have been unintentionally overlooked.
Since the majority of POMs have been formed and studied in water, this review focuses on speciation in aqueous solutions, where many applications take place, e.g. catalysis and biological application. Although the possible supramolecular assembly of POMs with H-binding and counterions is an important topic for POM chemistry and application, this aspect is not covered here. We first discuss the state of the art and methodology in studying polyanions in solution, which often is a complex issue. This section is followed by the detailed description of POM speciation mostly according to the solution pH and organized by their addenda atoms (VV, NbV, TaV, MoVI and WVI). The common trends in POM solution behavior with the aim of predicting stability trends based on the addenda atom type and structure are discussed throughout the text. Some POM applications performed without POM stability proofs are examined.
2. Methods of POM investigation in solution
The ambiguity of POMs equilibria in solution requires appropriate experimental techniques and careful interpretation of the results. Ideally, a number of different complementary and orthogonal experimental techniques are necessary to study and understand the distribution of species. However, this extensive characterization is rarely done. An exceptional example for a detailed species analysis is an investigation of acidified orthotungstate solution using electrospray-ionization mass-spectrometry (ESI-MS), 183W-NMR and Raman spectroscopy.19 The polycondensation product, heptatungstate [WVI7O24]6−, has been proven by NMR and Raman spectroscopies to be the main species in an equilibration mixture at pH < 7, but fails to be detected by ESI-MS due to its ESI-induced dissociation into Lindqvist [WVI6O19]2− anion (for more details see Section 3.4.1). The reason leading to the different results obtained via NMR, Raman and MS is the instability of [WVI7O24]6− upon ionization. This study also shows that ESI-MS is mainly applicable to stable polyanions, and that the method of investigation should be selected based on its strengths and weaknesses and by taking into account the specific characteristics of a particular POM.In order to obtain a spectrum as a fingerprint and assign it to a species, the species must be either homogeneous in solution or solid. The POM solution to be examined requires observation over a varying pH, time range and, if applicable, temperature and concentration range. Currently, solution studies no longer use only potentiometry and vibration spectroscopy as their exclusive methods for characterizing metal oxide systems, but various advanced methods complement these studies, including multinuclear NMR spectroscopy, small-angle X-ray scattering (SAXS), X-ray absorption spectroscopy (XAS, consisting of Extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES)) investigations as well as mass-spectrometry. We briefly summarize the characterization methods in context of their application in POM systems. A detailed description of the basic principles of the method, the recording, evaluation and interpretation of the data can be found in the specialized literature. In the following, the methods for examining POMs in solutions are presented in the order of their chronological appearance.
2.1. Potentiometry
Potentiometric titration was one of the first methods that was used to investigate POM speciation.1 In principle, the method involves measuring the hydrogen ion concentration in polyanion solutions as a function of the acid or base added (Fig. 1A) and the total metal ion concentration.16,20 The values for the formation constants and stoichiometric coefficients are determined by titration, followed by the iterative calculation of the constants of one species in the presence of another and the entire system being described by calculations in an iterative process. The formation constants Kc are the basis for species distribution diagrams (Fig. 1B) showing various types of POM that exist at different pH values. However, great care should be taken to ensure that a true equilibrium has been reached for each measurement, that the activity coefficient quotients are reasonably constant (use of supporting electrolyte) and that the potentials of the liquid transitions can be controlled. At the moment, potentiometry is outdated due to the availability of other advanced methods that provide a more accurate picture of the processes in solution.2.2. Electronic and vibrational spectroscopy
Typically, the addenda ions in POMs have d0 electronic configuration, and as a result, only one absorption band occurs in the UV-vis between 190 and 400 nm due to the oxygen-to-metal charge transfer transition.21,22 The spectra of the reduced “heteropoly blue’’ complexes show intervalence charge-transfer transitions, e.g., MoV → MoVI at ∼700 nm.23 Electronic spectroscopy implies practically no structural information, but it is one of the easiest ways to check POM stability in solution.24,25 For example, at pH 1 the Keggin polyanion [PVWVI12O40]3− ({PW12}) (Fig. 2B) exhibits two intense absorption bands in the UV range, with maxima at about 200 and 263 nm attributed to the pπ–dπ charge-transfer transitions of the Ot → WVI (Ot – terminal oxygen atom), and pπ–dπ charge-transfer transitions of the Ob,c → WVI (Ob,c – bridge oxygen atoms) (Fig. 2A), respectively.25 The absorption band maximum at 263 nm corresponds to the electron transition Ob,c → WVI of the intact Keggin anion and is shifted to 252.5 nm at pH 3.5 corresponding to mono-lacunary form [PVWVI11O39]7− ({PW11}), indicating [PVWVI12O40]3− decomposition (Fig. 2).25 A detailed description of the hydrolytic stability of {PW12} is given in Section 3.4.2.1.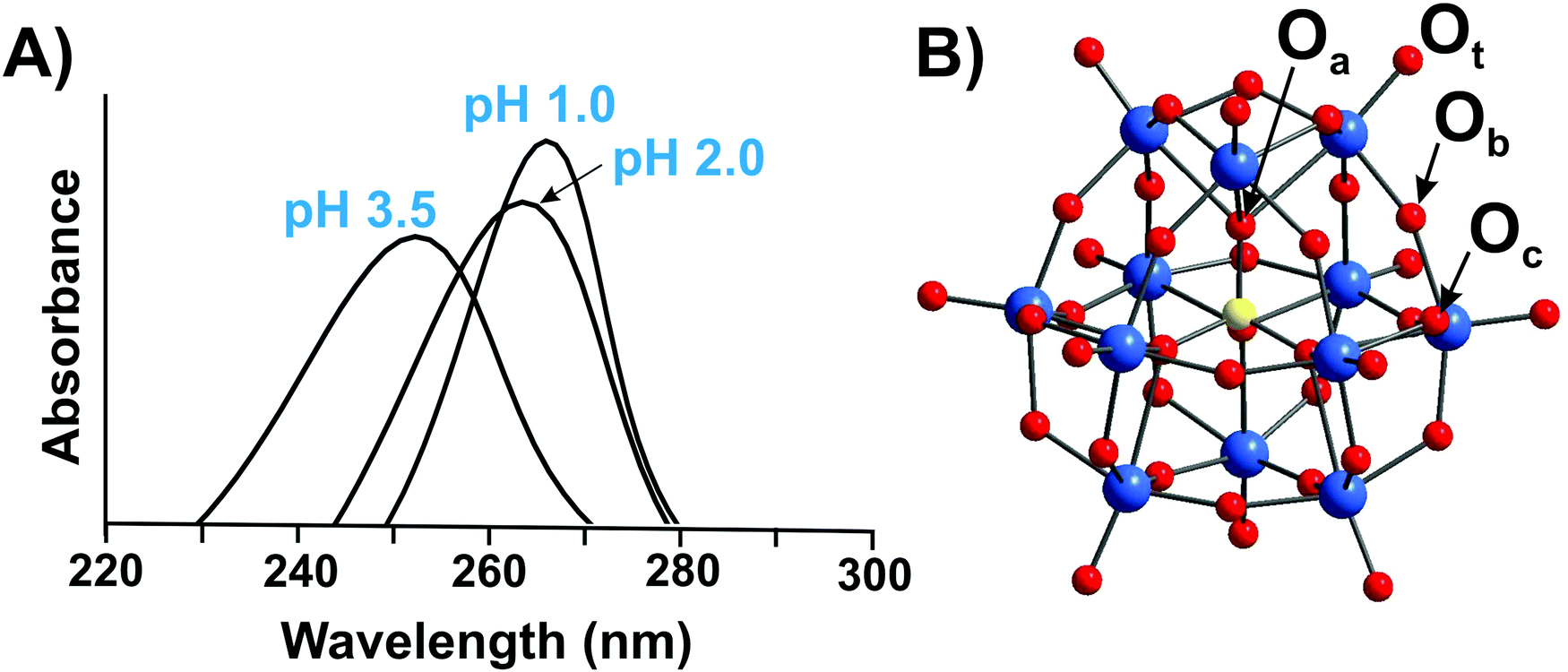 | ||
| Fig. 2 (A) UV-vis spectra of aqueous solutions of H3[PVWVI12O40] recorded at pH 1.0; 2.0 and 3.5.25 The decomposition of Keggin anion can be clearly seen already at pH 3.5 by shifting of the maximum absorption from 263 nm to 252.5 nm. For more details see Section 3.4.2.1. (B) Ball-and-stick representation of an α-Keggin type anion [PVWVI12O40]3− with the indicated types of oxygen atoms: μ3-Oa – oxygen atom connected to heteroatom PV; μ2-Ob and μ2-Oc – two types of bridging atoms in the structure; Ot – terminal oxygen atom. Color code: W, blue; P, yellow; O, red. | ||
Infrared, Raman and resonance Raman spectroscopy are broadly used in POM chemistry as diagnostic fingerprints. Similarities of spectral band positions, shapes and relative intensities for two compounds strongly indicate that both have identical structures. The characteristic spectrum region is between 1000 and 400 cm−1 where absorptions due to metal–oxygen stretching vibrations occur.
A good agreement between the spectra of crystalline and dissolved polyanions shows that the structure of the dissolved anion is the same as that observed in the solid state. Raman spectroscopy is more frequently used for aqueous solution studies of POMs26–29 and IR spectroscopy for studies in both aqueous and nonaqueous solvents.30
2.3. Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance spectroscopy has been carried out on POMs containing NMR-active nuclei, i.e.31P (natural abundance (NA): NA(31P) = 100%; nuclear spin (I): I = 1/2), 51V (NA = 99.75%; I = 7/2), 17O (NA = 0.04%; I = 5/2), 1H (NA = 99.98%; I = 1/2), 29Si (NA = 4.7%; 1/2), and, later, 95Mo (NA = 15.87%; I = 5/2) and 183W (NA = 14.32%, I = 1/2), to investigate their solution structures and dynamics since the 1970s.31,32 For reliable identification of a POM species in solution, it is desirable, whenever possible, to measure NMR spectra of all NMR-active core components.51V NMR. So far, the largest number of measurements for POMs has been carried out at 51V, a core nucleus with relatively high sensitivity, which provides spectra with line widths in the range from ∼10 to ∼800 Hz in diamagnetic polyanions. The chemical shifts (reference VOCl3) in isopoly- and heteropolyvanadates fall in the range between −400 to −600 ppm. Even relatively small structural variations result in separable peaks due to the broad chemical shift range.33,34
95Mo NMR. Despite the existence of two NMR-active isotopes, 95Mo (NA = 15.87%; I = 5/2) and 97Mo (NA = 9.46%, I = 5/2), Mo NMR is less frequently used due to their low natural abundance and their low gyromagnetic ratios: γ(95Mo) = −1.751 × 107 rad s−1 T−1 and γ(97Mo) = −1.788 × 107 rad s−1 T−1. The 95Mo nucleus is generally preferred over 97Mo because of its lower quadrupolar moment. Compared with 183W NMR, the 95Mo NMR signals from a typical asymmetric POMo environment are strongly broadened due to the quadrupole moment, which complicates spectral measurements and their interpretation. The earlier measurements of the 95Mo NMR spectra of isopolymolybdates (IPOMos) [MoVI2O7]2−, [MoVI7O24]6−, [MoVI6O19]2−, and α-/β-[MoVI8O26]4− were mainly carried out in non-aqueous solutions after 95Mo enrichment (96%) and, subsequently, 95Mo NMR was applied to study aqueous MoVI solutions.18
183W NMR. Despite its low sensitivity, the 183W NMR is of unique importance in studying polyoxotungstates (POTs). Narrow NMR lines of 183W with nuclear spin I = 1/2 allow to observe constants of the indirect spin–spin coupling, i.e.2J(W–P), 2J(W–W), which provide structural information. The use of high field spectrometers significantly reduces the sensitivity limitations, although a concentrated sample solution (∼1 mol L−1) and a long acquisition time are still required. A saturated solution of sodium tungstate is recommended as a reference for the chemical shift.32 For POTs, the range of 183W-chemical shifts lays between +260 and −300 ppm and even up to −670 ppm, if the POT-peroxocomplexes are taken into account.32 When looking at reduced POTs or at POTs with incorporated paramagnetic ions even larger chemical shifts from +2500 to −4000 ppm are observed.
93Nb (NA = 100%; I = 9/2) and 181Ta (NA = 99.98%; I = 7/2) NMR spectroscopy is hardly used for POM investigation, due to the excessive line widths as a result of large quadrupole coupling.
1H NMR. The rapid exchange with solvent protons limits the use of high resolution proton NMR for fully inorganic POMs, while integrated resonances of non-labile protons in organic units of hybrid polyoxoanions and countercations are routinely used for analytical purposes.1 However, in some cases separate signals for solvent and polyanions were observed. One of the earlier successful examples was published in 1966, when Pope and Varga demonstrated the presence of two central protons in the metatungstate anion, [H2WVI12O40]6−, using 1H NMR.35
17O NMR is more universal, as oxygen is an indispensable element of all POM clusters. There are two main structural types of oxygen atoms in a polyanion (Fig. 2B): terminal oxygens O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) M with coordination number one and bridging ones M–O–M with coordination numbers from two to six. NMR lines for oxygen atoms of different types are well resolved and the line's form is characteristic for a structural type of oxygen atom.31 Although 17O is difficult to observe on account of its low natural abundance (0.04%) and its negative quadrupole moment Q(17O) = −26 mB, these disadvantages are compensated by its large chemical shift ranging from 1200 to −100.1,31 To overcome the low natural abundance of 17O (0.04%), target POMs can be enriched with H217O. This allows studying the rates of oxygen-isotope exchange between solvent and POM molecules sites and has been proven useful for understanding of POMs equilibria.36,37
M with coordination number one and bridging ones M–O–M with coordination numbers from two to six. NMR lines for oxygen atoms of different types are well resolved and the line's form is characteristic for a structural type of oxygen atom.31 Although 17O is difficult to observe on account of its low natural abundance (0.04%) and its negative quadrupole moment Q(17O) = −26 mB, these disadvantages are compensated by its large chemical shift ranging from 1200 to −100.1,31 To overcome the low natural abundance of 17O (0.04%), target POMs can be enriched with H217O. This allows studying the rates of oxygen-isotope exchange between solvent and POM molecules sites and has been proven useful for understanding of POMs equilibria.36,37
31P NMR. The large number and wide variety of heteropoly compounds with phosphorus as a heteroatom led to a significant development of 31P NMR, which shows a high sensitivity of its chemical shift based on POMs composition. In the 31P NMR spectra of Keggin type H3[PVWVI12O40] (Fig. 2B) and the majority of its derivatives, each form is represented only by one signal in the chemical shift range between −15 and −2.5 ppm (relative to 85% H3PO4), which allows to directly detect several coexisting species and to determine their concentrations.38
Other nuclei. 11B (NA = 80.42%; I = 3/2),3919F (NA = 100%; I = 1/2),4027Al (NA = 100%; I = 5/2)41 and 195Pt (NA = 33.7%; I = 1/2)42 NMR spectroscopy are not as often used, but equally important for POM solution investigation.
If suitable NMR-active cations are present, NMR measurements can also probe cation–POM interactions in solution, which can affect the transformation between POM species.43 Using 7Li,4423Na45 and 133Cs46 NMR spectroscopy, the Nyman group successfully studied the cationic association with various POMs in solution.
2.4. Mass-spectrometry (MS)
Electrospray-ionization mass-spectrometry (ESI-MS) is suitable for the elucidation of solution phase equilibria of stable upon ionization anions, since it enables semi-quantitative detection of both cationic and anionic species in aqueous solvents with excellent detection limits. POMs are ideal candidates for mass-spectrometry studies since they exhibit complex isotopic envelopes resulting from the high number of stable isotopes as for tungsten (182W, 26.5%; 183W, 14.3%; 184W, 30.6%; 186W, 28.4%) or molybdenum (92Mo, 14.8%; 94Mo, 9.3%; 95Mo, 15.9%; 96Mo, 16.7%; 97Mo, 9.6%; 98Mo, 24.1%; 100Mo, 9.6%), and are intrinsically charged.47,48 However, the experiments must be carefully designed in order to obtain reliable data without overinterpretation of gas phase data for the solution chemistry.49 While ESI-MS does not yield any information beyond the mass-to-charge ratio of the analyte, it has high sensitivity and does not impose too many requirements on the system to be analyzed, and time-resolved data can be obtained on very dilute solutions. ESI-MS measurements have been used for comprehensive POM speciation studies with all kind of addenda atoms and significantly contribute to the speciation analysis given in detailed in Section 3.2.5. Small angle X-ray scattering (SAXS)
SAXS is a well-established non-destructive method for probing the size, shape, reactivity, and interactions of dissolved species.50 This method is very powerful, but so far, an underutilized technique to obtain speciation information on POM solutions. SAXS is fundamentally similar to X-ray crystallography, where a sample is irradiated by a collimated monochromatic X-ray beam.51 Like nanoparticles and quantum dots, many POMs exhibit high net charge, contain high electron-density elements (W, Mo and other metals) and therefore scatter X-rays strongly. Since POMs are molecular by definition, solutions in which the clusters are stable must be absolutely monodisperse, and their X-ray scattering data can be simulated very accurately by applying solid-state crystal structure data sets. To date, many POM classes have been thoroughly investigated using SAXS, including POMs of group V (NbV and TaV),50,51 POTs52 and their complexes with actinides,53 large reduced POMs54 as well as POM supramolecular assemblies.55 SAXS can be successfully used to investigate speciation in catalytic systems, as one representative example, the Co-containing POTs speciation as a function of pH, buffer salts, and addition of a chemical oxidant during water oxidation catalysis has been carefully studied.562.6. Other methods
POMs have a rich electrochemistry associated with both reduction of tungsten or molybdenum57 and redox-reaction of heterometals (i.e., incorporated cobalt, ruthenium, iridium, or nickel). These characteristic redox wave peaks can be used to identify the number of terminal oxygen atoms, metastable hydrolysis fragments, new isomers and reduced anions.58,59Extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) are valuable techniques to probe both the local coordination environment and the oxidation state of POM's atoms either in solution or in solid-state materials. Each kind of atom in the POM cluster can be accessed individually and an average spectrum for each element is observed.60 Despite XAS (X-ray absorption spectroscopy) being a powerful technique, there are just a limited number of examples for their usage in POM structure analysis.61,62
Dynamic light scattering (DLS) is aimed to determine whether particles are formed in solutions and, if present, to examine their size.63 DLS has found its broadest usage in monitoring POM stability during catalytic reactions (e.g. water splitting systems64).
3. POM speciation in aqueous solutions
3.1. Polyoxovanadates (POVs)
This section is divided into isopoly- and heteropolyvanadates. First, the general behavior of POVs in solution is described, and second, the characteristics of tri-, tetra-, penta- and decavanadates, as well as polyoxophosphovanadates are detailed in the order of increasing nuclearity.3.1.1.1. Ion-distribution diagram for IPOVS. A great variety of discrete isopolyvanadates (IPOVs) has been characterized in the solid state isolated from aqueous and non-aqueous solutions which include [VV3O9]3− (Fig. 3C),65 [HVV4O12]3− (Fig. 3E)66 and [VV5O14]3− (Fig. 3G)67 with tetrahedrally coordinated vanadium (Table 1) and [VV10O28]6− {V10}68 (Fig. 5), [VV12O32]4−,69 [VV13O34]3−,70 and [VV15O42]9−
71 (Fig. 6) with octahedrally coordinated VV ions. The maximum species concentration and the equilibrium of IPOVs are influenced by pH, ionic strength, temperature, and vanadate concentrations. The most commonly used speciation model (Fig. 4 and Table 1) was first constructed about thirty years ago as a result of rigorous research using 51V NMR investigations in conjunction with potentiometric titration studies.72,73 The proposed speciation model (Fig. 4) postulates, that in aqueous vanadium solutions of pH > 6, polymers with one to five V atoms (Fig. 3) are formed, where many of these IPOVs also undergo several individual protonation steps. In contrast, under acidic conditions, only two major species {V10} and [VVO2]+ (Fig. 4 and Table 1) have been reported.
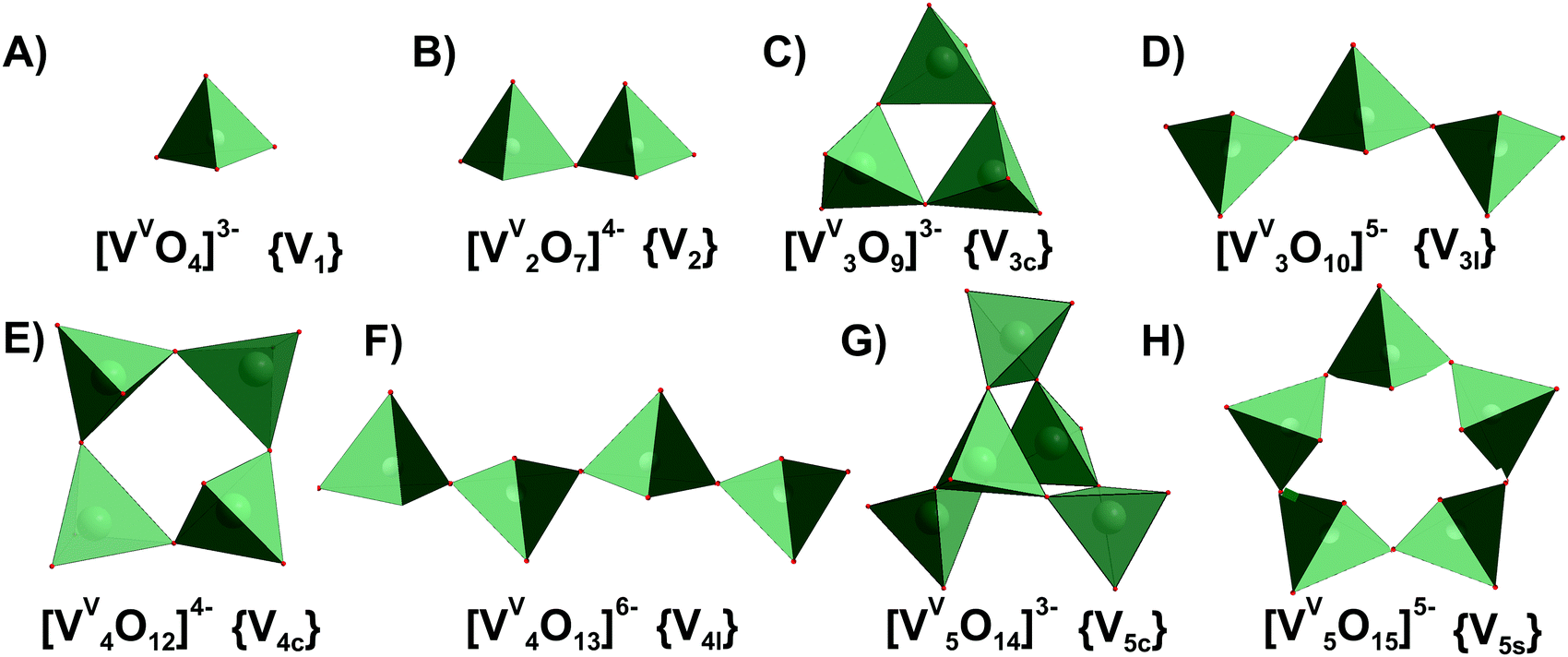 | ||
| Fig. 3 IPOVs with the nuclearity up to 5 addenda atoms and tetrahedral coordination of VV ((A)–(H)), which can be present in aqueous media. Tri- and tetravanadates are presented in two forms – cyclic ({V3c}, (C) and {V4c}, (E)) and linear ({V3l}, (D) and {V4l}, (F)). The linear forms of {V3l} and {V4l} have never been obtained in solid state. Pentavanadate is depicted in two cyclic forms: [VV5O14]3− ({V5c}, (G),67 which was synthesized from non-aqueous solution and [VV5O15]5− ({V5s}, (H),78,79 which has never been crystallized. Color code: {VO4}, green; O, red. In abbreviations the subscript “c” stands for “cyclic”, “l” – for “linear”, “s” – for “star”. | ||
| IPOV species | q, pa | pKa | Isolated in solid state |
δ(51V), ppm80,87b |
Formation constantsa lgKc at 25 °C according to |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sigelc84 |
McCannd85 |
Larsond86 |
Traceyd78 |
Elvingsone79 |
Heathf87 |
|||||
|
a Stoichiometric coefficients and formation constants for the reaction p[H2VVO4]− + qH+ [HzVVpOm]n− + (q/2 + p − z/2)H2O.
b
δ
51V relative to VOCl3.
c Ionic strength μ = 0.6 M (NaCl).
d Without electrolyte addition.
e
μ = 0.15 M (NaCl).
f
μ = 2 M (NaClO4).
|
||||||||||
| [VVO4]3− (Fig. 3A) | −2, 1 | Yes | −541.2 | −21.31 | >−23 | >−22 | ∼−19 | |||
| H[VVO4]2− | −1, 1 | 13.4 | No | −538.8 | −7.91 | −9.02 | −8.75 | −8.8 | −8.17 | −7.1 |
| H2[VVO4]− | 0, 1 | 7.91 | No | −560.4 | ||||||
| [VV2O7]4− (Fig. 3B) | −2, 2 | Yes81 | −561.0 | −15.13 | −19.0 | −18.60 | −16.19 | −12.8 | ||
| H[VV2O7]3− | 1, 2 | 9.74 | No | −563.5 | −5.39 | −7.47 | −7.30 | −5.85 | −3.9 | |
| H2[VV2O7]2− | 0, 2 | 8.29 | No | −572.7 | 2.90 | 2.15 | 2.30 | 2.5 | 2.65 | 3.3 |
| H[VV3O10]4− (Fig. 3D) | −1, 3 | No | ∼−570 | −6.1 | −11.4 | |||||
| [VV4O13]6− (Fig. 3F) | −2, 4 | No | Between −566 and −585 | −8.50 | −16.1 | −9.98 | 28.1 | |||
| H[VV4O13]5− | −1, 4 | 8.9 | No | Between −566 and −585 | 0.4 | |||||
| [VV4O12]4− (Fig. 3E) | 0, 4 | Yes66 | −577.6 | 10.04 | 7.0 | 7.60 | 8.4 | 9.24 | 11.6 | |
| [VV5O15]5− (Fig. 3H) | 0, 5 | No | −586.0 | 12.43 | 7.5 | 11.17 | 14.5 | |||
| [VV10O28]6− (Fig. 5) | 4, 10 | Yes68 | −422, −496, −513 | 51.98 | 50.28 | |||||
| H[VV10O28]5− | 5, 10 | 6.14 | No | −424, −500, −516 | 58.12 | 56.90 | ||||
| H2[VV10O28]4− | 6, 10 | 3.68 | Yes82 | −425, −506, −524 | 61.80 | 61.07 | ||||
| H3[VV10O28]3− | 7, 10 | 1.57 | Yes83 | −427, −515, −534 | 63.37 | 62.93 | ||||
| [VVO2]+ | 2, 1 | Yes | −545.0 | 6.97 | 7.00 | |||||
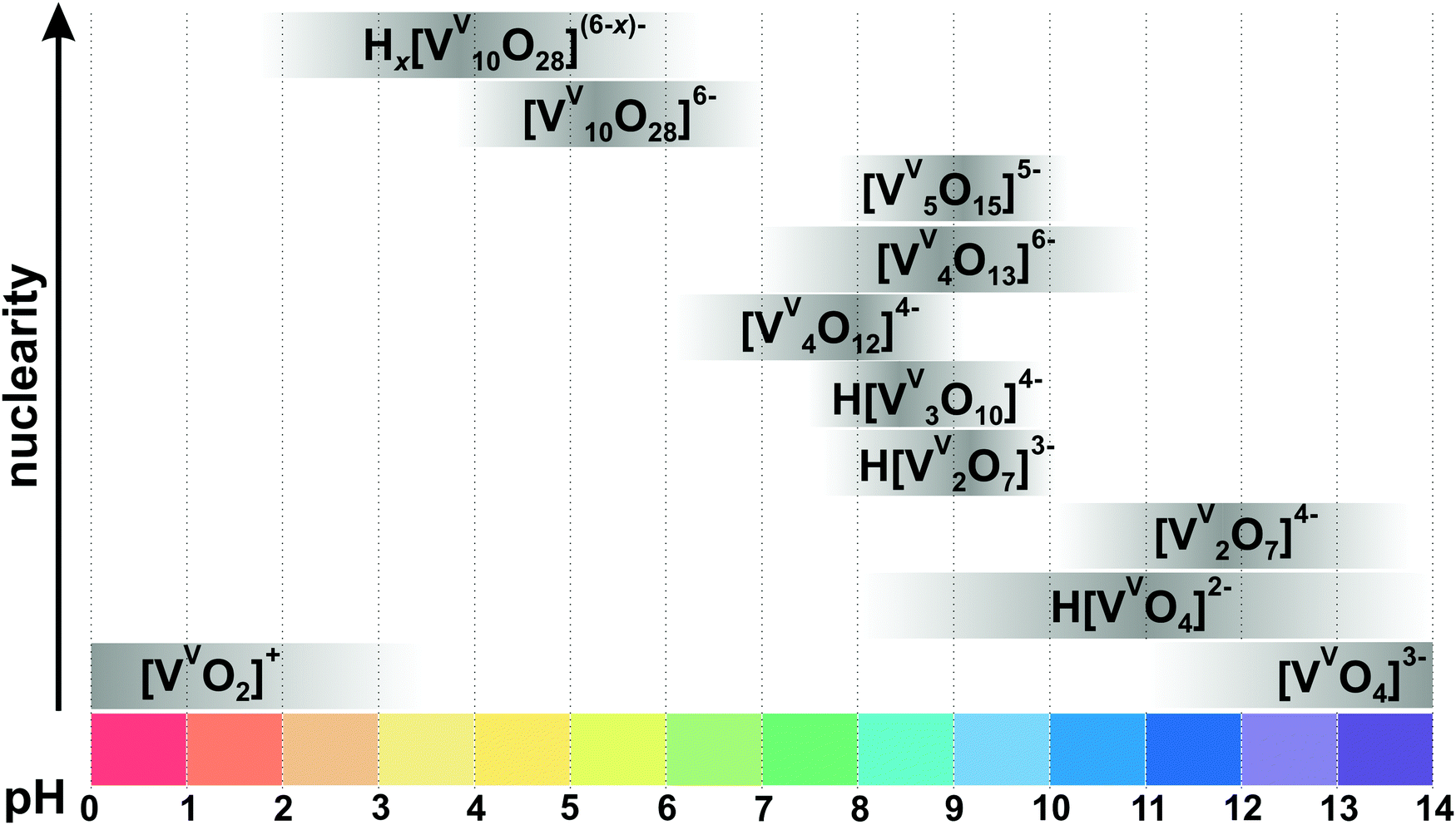 | ||
| Fig. 4 Speciation of IPOVs in an aqueous solution with a total concentration of vanadium more than 0.1 mM based on ref. 34, 72, 78, 79 and 84–88. The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region, e.g. the maximum concentration of [VV2O7]4− is at pH 12. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. x in Hx[VV10O28](6−x)− is 1–3. The molecular structures of the clusters are depicted in Fig. 3 and 5. The 51V chemical shifts, formation constants and pKa values are summarized in Table 1. | ||
Since 51V NMR is a powerful technique to determine POV speciation and easily accessible, the use of other techniques is rarely reported. ESI-MS analysis of ammonium metavanadate (NH4)3[VVO4] aqueous solution at pH 4.574 and 675 revealed a series of known (protonated {V10} and [VVO4]3−) and “previously unknown” polyoxovanadate anions and cations, which might have been formed in gas phase due to ionization.
3.1.1.2. Tri-, tetra- and penta-vanadates. The structures of the trivandate (Fig. 3C and D) and tetravanadate (Fig. 3E and F) in water were debated for a long time and numerous studies lead to contradictory conclusions.76 The various structural suggestions include linear (Fig. 3D and F) or cyclic (Fig. 3C and E) trivanadate or tetravanadate ions exhibiting tetra- or penta-coordinated vanadium. Nowadays, a consensus has been reached that a linear form of tri- and tetranuclear vanadates, [VV3O10]5− and [VV4O13]6− (Fig. 3D and F), does exist in aqueous solution at pH < 10, which was confirmed by 51V and 17O NMR (Fig. 4).77 Up to date only salts with cyclic [VV3O9]3− (Fig. 3C), [HVV4O12]3− (Fig. 3E) and [VV5O14]3− (Fig. 3G) have been obtained from non-aqueous media.65–67 The isolation of the discrete linear tri- and tetravanadate (Fig. 3D and F) from aqueous solution has failed so far because the crystallization always goes hand in hand with the polymerization of the anion to “endless” metavanadate chains ([VVO4]3−)n.
3.1.1.3. Decavanadate [VV10O28]6− stability. {V10} is stable for days at neutral pH;29 while at pH values higher than 7 it, at least in part, turns into structurally and functionally different smaller oxovanadates such as the monomeric [VVO4]3− (Fig. 3A), dimeric [VV2O7]4− (Fig. 3B) or tetrameric [VV4O12]4− (Fig. 3E) with a half-life time depending on vanadium concentration, ionic strength and temperature.89 In the basic pH region, {V10} will ultimately convert to the colorless metavanadates Hx[VVO4](3−x)− (x = 0–2) (Fig. 4), although near neutral pH, this process may take up to three weeks depending on ionic strength and temperature (Fig. 5). At neutral pH, {V10} can be destructed to smaller IPOVs and therewith being removed by boiling the solution.90
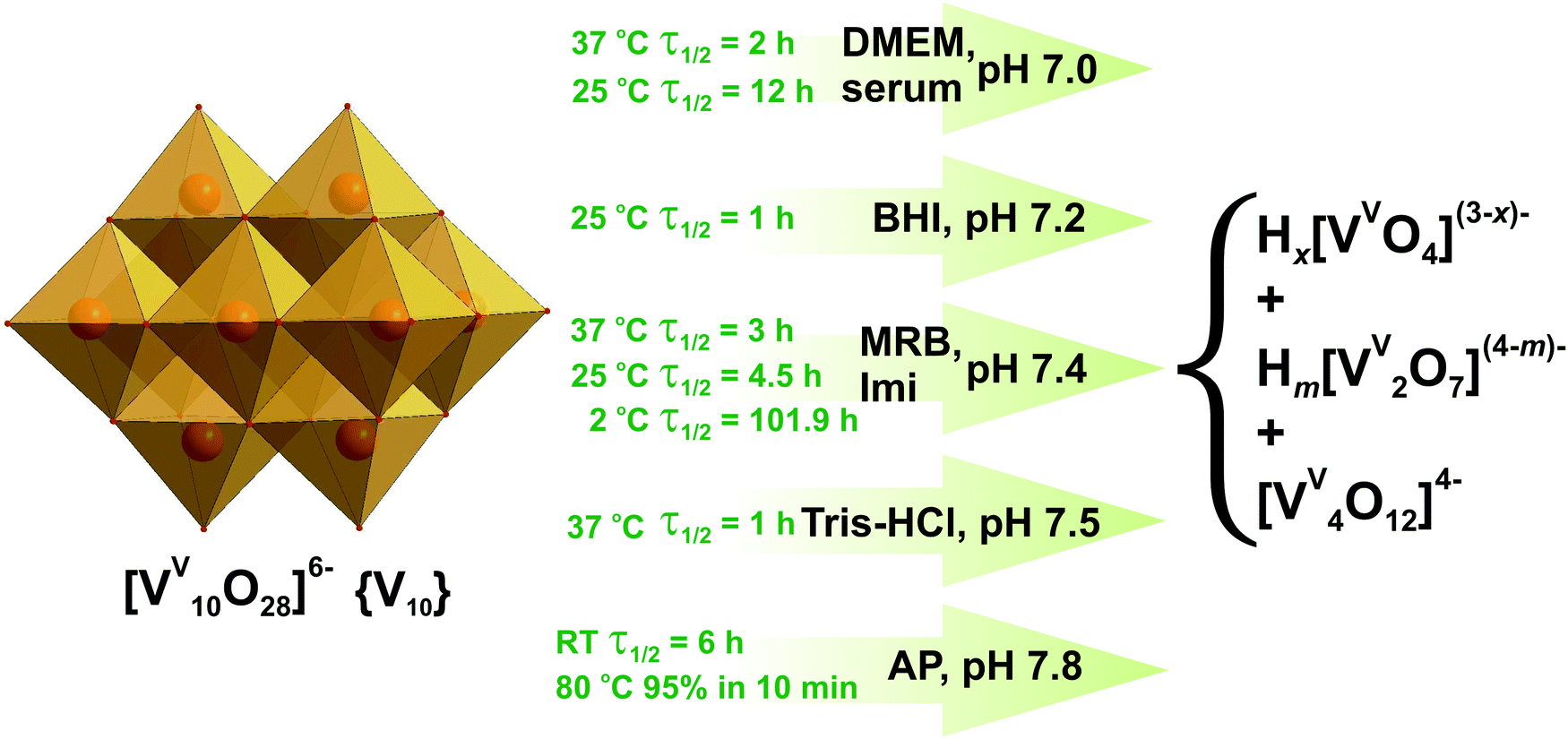 | ||
| Fig. 5 Decavanadate {V10} decomposition in aqueous solutions between pH 7.0 and 7.8 based on 51V NMR, UV-vis and XAS investigations.61,89,91–95 Half-life of {V10} decomposition is given for a total V concentration of 5 mM. DMEM – serum-free Dulbecco's modified Eagle's medium, CaCl2, 0.2 g L−1; KCl, 0.4 g L−1; NaCl, 6.4 g L−1; Na2HPO4, 0.109 g L−1; Na2CO3, 3.7 g L−1; glucose, 1 g L−1, and 20 proteinogenic amino acids); MRB – mitochondrial respiration buffer, sucrose, 0.2 M; KH2PO4, 5 mM; KCl, 10 mM; MgCl2, 5 mM; Tris–HCl, 10 mM; pyruvate 5 mM; malate, 0.5 mM; serum – physiological serum, NaCl, 0.9%; BHI – brain-heart infusion medium (calf brain, 12.5 g L−1; beef heart, 5 g L−1; peptone, 10 g L−1; D-glucose, 2 g L−1; NaCl, 5 g L−1; Na2HPO4, 2.5 g L−1, pH 7.2); Imi – imidazole buffer (KCl, 0.1 M; MgCl2, 5 mM; EGTA, 0.5 mM; imidazole, 10 mM); Tris–HCl – 2 mM Tris–HCl buffer; AP – alkaline phosphatase buffer (NaCl, 5 M; Tris–HCl, 1 M; MgCl2, 1 M). The structures of Hx[VVO4](3−x)− (x = 0–2), Hm[VV2O7](4−m)− (m = 0–2) and [VV4O12]4− are shown in Fig. 3A, B and E. Color code: {VO6}, light orange; O, red. | ||
51V NMR and UV-vis spectroscopic investigations revealed that at pH 7 and 37 °C the half-life of {V10} decomposition is 2 h in serum-free Dulbecco's modified Eagle's medium (DMEM: CaCl2, 0.2 g L−1; KCl, 0.4 g L−1; NaCl, 6.4 g L−1; Na2HPO4, 0.109 g L−1; Na2CO3, 3.7 g L−1; glucose, 1 g L−1, and 20 proteinogenic amino acids).91 At room temperature in physiological serum (pH = 7, NaCl 0.9%) the half-life of 12 h was estimated for {V10}.92 In brain-heart infusion medium (BHI: calf brain, 12.5 g L−1; beef heart, 5 g L−1; peptone, 10 g L−1; D-glucose, 2 g L−1; NaCl, 5 g L−1; Na2HPO4, 2.5 g L−1) at a slightly higher pH of 7.2, {V10} decomposes quickly with measurable decay in as little as one hour and the dominant species after 19 hours is tetrameric [VV4O12]4− (Fig. 3E).93 Almost no {V10} was detected after 42 h while tetravanadate is still the dominating species after 42 h and 66 h (Fig. 5). The half-life of {V10} in aqueous buffered solution (KCl, 0.1 M; MgCl2, 5 mM; ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.5 mM; imidazole, 10 mM) at pH = 7.4 with [{V10}] = 5 mM at 25 °C is 43 h, and under the same conditions with a higher concentration [{V10}] = 50 μM decreases to 19 h (Fig. 5).94 In a view of the significant decomposition of {V10} in diluted solutions at room temperature, it is advisable to perform the experiments involving {V10} in more concentrated solutions and limit them to a few hours when working at room temperature. At increased 37 °C and keeping the same pH of 7.4 however in mitochondrial respiration buffer (MRB: sucrose, 0.2 M; KH2PO4, 5 mM; KCl, 10 mM; MgCl2, 5 mM; Tris–HCl, 10 mM; pyruvate 5 mM; malate, 0.5 mM) 5 mM {V10} solution shows the short half-life of just 3 h.95 A UV-vis kinetics study of the {V10} decay in the presence of alkaline phosphatase buffer (AP: NaCl, 5 M; Tris–HCl, 1 M; MgCl2, 1 M) at pH 7.8, carried out by following the peak disappearance at 430 nm (pπ–dπ charge transfer of the Obridg. → VV), indicated that {V10} degrades by approximately 15% per hour at room temperature (t1/2 = 6 h under first order kinetics) (Fig. 5).93 About 95% of {V10} decomposed in 10 minutes to tetravanadate [VV4O12]4− (Fig. 3E) in the same AP buffer (pH = 7.8) at increased 80 °C.
Under acidic conditions (pH 5.4) in Schneider's insect medium (SIM: Na2HPO4, 0.7 g L−1; MgSO4, 3.7 g L−1; KCl, 1.6 g L−1; KH2PO4, 0.45 g L−1; NaCl, 2.1 g L−1 and 20 proteinogenic amino acids), the decomposition of {V10} is substantially slower and after 2 hours, only 0.06 molar equivalent of {V4} (Fig. 3E) was detectable and the rest remained as {V10}.93
Recently EXAFS and XANES spectroscopic investigations were applied to examine the V oxidation state in {V10} solution.61 Under the physiological temperature of 37 °C {V10} began to decompose after about 1 h at pH 7.5 (2 mM Tris–HCl buffer), while at room temperature {V10} was present for the entire acquisition period (ca. 7 h). According to the XANES analysis the solution of {V10} at pH 7.5 and 37 °C contained 81.3% of [VVO4]3− (Fig. 3A) and 18.7% of the reduced oxidovanadium (VIV) species ([VIVO]2+ and its complexes), evidencing {V10} reduction upon interaction with actin, and meaning that all {V10} species have been completely decomposed in solution visible also by a color change from yellow to colorless. The {V10} stability studies performed in different media and temperatures lead to sometimes contradictory results, and it is recommendable to check the {V10} integrity under the applied conditions.
3.1.1.4. The use of speciation data to understand the biological activities of decavanadate. The applicability of this review will be shown in this section using decavanadate as an example, since among all IPOVs (Fig. 3), {V10} (Fig. 5) demonstrates many important roles in fundamental biological processes.61,90,96–101
Three IPOVs: [VVO4]3− (Fig. 3A), [VV4O12]4− (Fig. 3E) and {V10} (Fig. 5), were tested as antibacterial agents against six strains of Streptococcus pneumoniae (penicillin-intermediate-resistant IID553, IID554 and penicillin-resistant BS225, BS234, BS259, BS269) without the use of any additional antibiotic.102 Prior to the evaluation of the minimum inhibitory concentrations (MIC), all IPOVs were incubated at 37 °C for 20 h in Mueller–Hinton Agar medium (agar, 17 g L−1; beef infusion solids, 2.0 g L−1; casein hydrolysate, 17.5 g L−1; starch, 1.5 g L−1; pH = 7.3), which according to works61,89,93,94 (Fig. 5) should lead to almost complete hydrolysis of {V10}. The ion-distribution diagram for IPOVs (Fig. 4) clearly shows that {V10} is the stable and dominant species between pH 4 and 7, and the additional heating used for incubation only aggravates the {V10} hydrolysis rate. MICs of all three IPOVs, [VVO4]3−, [VV4O12]4− and {V10}, were in the concentration range between 4 and 32 μg mL−1 with slight lower numbers for {V10} solution (4–8 μg mL−1). The authors, without considering the {V10} decomposition into mono-, di- and tetravanadates during the incubation, made a conclusion about the superiority of decavanadate as an antibacterial agent, which in fact definitely requires a more careful study.
An example of the proper use of decavanadate is when applied as an inhibitor for bovine pancreatic ribonuclease A (RNase A).103 Before evaluating the thermodynamic parameters for the {V10} binding to RNase A applying isothermal titration calorimetry (ITC), a stock solution of decavanadate was prepared with a pH between 3 and 5 and checked after 24 hours to verify that equilibrium was established. All experiments have been performed at 25 °C in 30 mM sodium succinate buffer (pH 6.0), which, in accordance with the IPOVs ion-distribution diagram (Fig. 4) and {V10} decomposition scheme (Fig. 5), should not lead to decavanadate decomposition. As a result, it is justified to conclude that the intact cluster interacts with RNase A.
Crans and co-authors recently conducted an even more thorough analysis of decavanadate speciation to study its effect on the growth of Mycobacterium smegmatis (M. smeg) and Mycobacterium tuberculosis (M. tb).10151V NMR spectra were recorded at pH between 5.8 and 6.8, where there should not have been any decavanadate hydrolysis according to Fig. 4. The spectra showed 100% intact decavanadate {V10} in stock solution and growth media with {V10} but without bacterial cells. The addition of growth media containing cells to {V10} stock solution immediately caused some decomposition of {V10}.101 These results lead to the conclusion, that {V10} can interact with some cell components, leading to {V10} decomposition and emphasize the importance to investigate the influence of macromolecules on POM speciation. Ramos et al.97 confirmed the influence of macromolecules on decavanadate stability by investigating the interaction of {V10} solutions with G-actin. Different macromolecules show various effects on decavanadate stability, thus, the {V10} half-life increases from 5 to 27 h in the presence of G-actin and from 5 to 18 h in the presence of sarcoplasmic reticulum vesicles at 22 °C and pH 7.5 (Tris, 2 mM; CaCl2, 0.2 mM; KCl, 100 mM; MgCl2, 2 mM). The addition of ATP to the medium decreases the half-life of {V10} from 27 to 10 h, while in the presence of phosphatidylcholine liposomes or myosin {V10} stability does not change. It is assumed that the decavanadate interaction with G-actin, favored by the G-actin polymerization, stabilizes the presence of intact {V10}.
3.1.2.1. Speciation in phospho-vanadate solutions. Among the variety of heteroatoms present in vanadates only the vanadate-phosphate speciation has been investigated systematically.104–106 In the aqueous H+–H2VVO4−–H2PVO4− solution in the presence of 0.15105 or 0.60104 M NaCl at 25 °C three types of phospho-vanadates – [HkPVVVO7](4−k)− (k = 1–4) ({PV}), [HmPV2VVO10](5−m)− (m = 1, 2) ({P2V}), [HnPVVV14O42](9−n)− (n = 3–5) ({PV14}, Fig. 6A) – have been identified by pH-potentiometry and high-field 31P and 51V NMR spectroscopy to be present between pH 1 and 10 (Fig. 7 and Table 2).
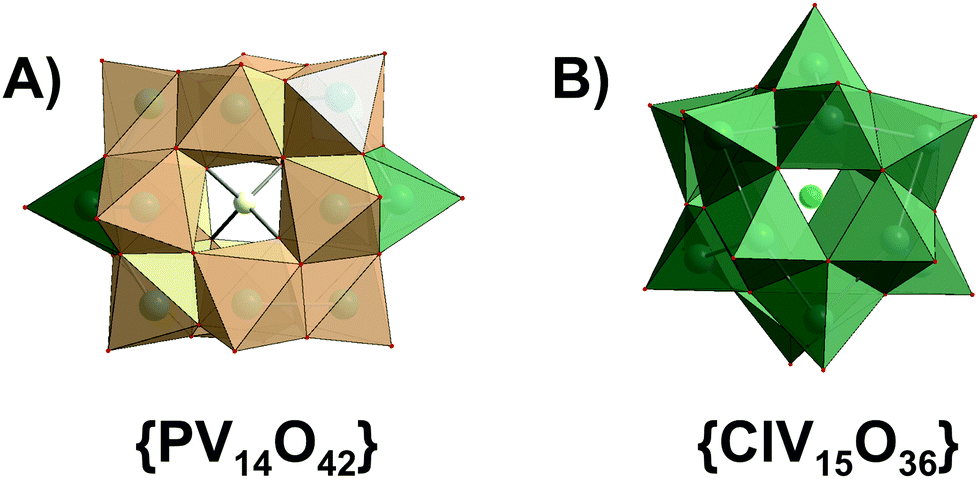 | ||
| Fig. 6 Polyhedral representation of (A) [HnPVVV14O42](9−n)−n = 3–5 and [H6VV12VIV2O38(PVO4)]5− ({PV14O42}) and (B) [VV7VIV8O36(Cl)]6− ({ClV15O36}). Color code: {VO6}, light orange, {VO4} and {VO5}, green; O, red; P, yellow; Cl, green. | ||
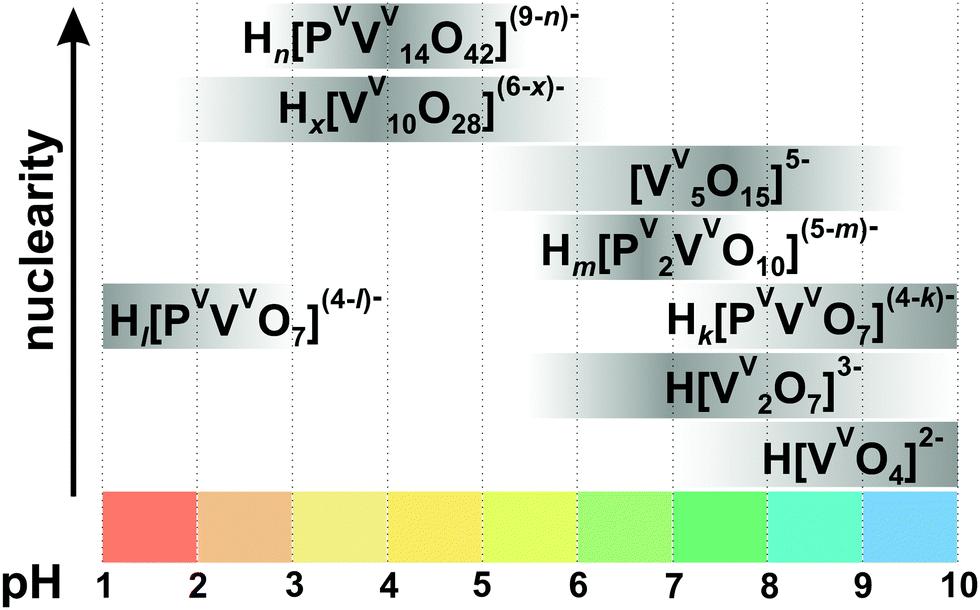 | ||
| Fig. 7 Speciation in the aqueous solution of H+–H2VVO4−–H2PVO4− with 0.15 M NaCl at 25 °C and [V]tot = 5 mM, [P] = 60 mM based on ref. 105. Protonation degree: Hl[PVVVO7](4−l)− (k = 3–4); Hk[PVVVO7](4−k)− (k = 1–2), Hm[PV2VVO10](5−m)− (m = 1, 2), Hn[PVVV14O42](9−n)− (n = 3–5), Hx[VV10O28](6−x)− (x = 1–3). The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. The structures of the species are presented in Fig. 3, 5 and 6. The 51V chemical shifts, formation constants and pKa values are summarized in Table 2. | ||
| Phospho-vanadate species | q, p, ra | pKa105 | Isolated in solid state |
δ(51V), ppmb105 |
lgKc at 25 °Ca,c |
|---|---|---|---|---|---|
|
a Stoichiometric coefficients and formation constants for: p[H2VVO4]− + qH+ + r[H2PO4]− [HzPrVVpOv]n− + (q/2 + r + p − z/2)H2O.
b
δ
51V relative to VOCl3.
c Ionic strength μ(NaCl) = 0.15 M.
|
|||||
| H3[PVVV14O42]6− (Fig. 6A) | 9, 14, 1 | Yes107 | −521, −572, −589 | 89.39 (0.19) | |
| H4[PVVV14O42]5− | 10, 14, 1 | 4.5 | No | −530, −580, −598 | 93.93 (0.04) |
| H5[PVVV14O42]4− | 11, 14, 1 | 2.1 | No | −534, −589, −598 | 96.03 (0.17) |
| H[PVVVO7]3− | −1, 1, 1 | No | −567.1 | −5.68 (0.10) | |
| H2[PVVVO7]2− | 0, 1, 1 | 7.19 | No | −582.5 | 1.51 (0.08) |
| H3[PVVVO7]− | 1, 1, 1 | 3.82 | No | −558.7 | 5.33 (0.07) |
| H4[PVVVO7] | 2, 1, 1 | 3.04 | No | −557.9 | 8.37 (0.07) |
| H[PVVV2O10]4− | −1, 1, 2 | No | −579.4 | −3.9 (0.26) | |
| H2[PV2VVO10]3− | 0, 1, 2 | 6.3 | No | −602.0 | 2.4 (0.12) |
The kinetically labile {PV} and {P2V} have been reported only in solutions and never been crystallized. The predominant equilibrium species, {PV14} (Fig. 6A), which has a trans-bicapped α-Keggin structure, has been known both in solution ([V]/[P] = 0.5–14, pH = 1–6) and solid state.107 Reaching equilibrium in the system H+–H2VVO4−–H2PVO4− is generally fast, except under acidic condition. Here, the formation of {PV14} species (Fig. 6A) is even slower and requires at least three months to establish a complete equilibrium. Naturally, high vanadate total concentrations favor the formation of {PV14} species, but also that of decavanadate {V10}.
3.1.2.2. Speciation of mixed valence HPOVs and the use of speciation data to understand their biological activities. Speciation studies were carried out for mixed valence polyoxovanadates K(NH4)4[H6VV12VIV2O38(PVO4)]·11H2O (Fig. 6A) and [(CH3)4N]6[VV7VIV8O36(Cl)] (Fig. 6B) by 51V NMR and EPR spectroscopic studies in aqueous solution and in Luria-Bertani (LB) medium (pH 7.4; tryptone, 10 g L−1; yeast extract, 5 g L−1; NaCl, 10 g L−1) to understand mixed valence HPOVs chemoprotective activity against the alkylating agent diethylsulphate (DES) in Escherichia coli DH5α cultures.108 [(CH3)4N]6[VV7VIV8O36(Cl)] (Fig. 6B) is more stable in LB than in pure aqueous solution, and is able to react with increasing amounts of DES. According to ion-distribution diagram (Fig. 7), fully oxidized {PV14} (Fig. 6A) should be stable between pH 3.5 and 5.5. The tested reduced analog [H6VV12VIV2O38(PVO4)]5− also decomposes and rearranges rapidly in LB (pH = 7.4), resulting in complete absence of chemoprotective activity against DES. Decomposition products, [VVO4]3− and {V10}, react poorly, or even do not react, with the alkylating agent. The observation of chemoprotective activity for [(CH3)4N]6[VV7VIV8O36(Cl)] (Fig. 6B) or its absence for K(NH4)4[H6VV12VIV2O38(PVO4)]·11H2O (Fig. 6A) against DES is clearly dependent on the chemical nature and stability of the soluble species in the culture medium after addition of the alkylating agent.
3.2. Polyoxoniobates (PONbs) and polyoxotantalates (POTas)
In contrast to POVs, POMos and POTs, polyoxoniobates (PONbs) and polyoxotantalates (POTas) can be stabilized only under basic conditions due to their high negative charge, and their chemistry was initially dominated by the Lindqvist anion [M6O19]8− (M = NbV, TaV) in solution and by its alkali salts in the solid state.1 Owing to the narrow working pH region (>7), the low solubility, and the low reactivity of niobate and tantalate species, the progress of PONb and POTas chemistry is far behind that of POMo or POT chemistry.109 This chapter is devoted only to isopolyniobates (IPONbs) and -tantalates (IPOTas) due to the lack of information about heteropoly PONbs and POTas stability and speciation in solution.3.2.1.1. Ion-distribution diagram. The Lindqvist hexaniobate [NbV6O19]8− ({Nb6}, Fig. 8A)110 and the related decaniobate [NbV10O28]6− ({Nb10}, Fig. 8B)111,112 were for a long time the only known PONbs. Spectroscopic and potentiometric studies have shown that the Lindqvist ion is indeed the dominant species of Nb in solutions at pH higher than 7 and room temperature (Fig. 9).113 The heptaniobate [NbV7O22]9− ({Nb7}, Fig. 8C) has been recently detected only in solution at pH 9 (100 mM H3BO3),114,115 as a part of larger structures. The formation of other Nb or Ta POM archetypes cannot be reproducibly achieved by pH change, because Nb and Ta solutions are difficult to retain except at high pH > 12. Therefore, forming other polyoxoniobate and polyoxotantalate clusters requires finding conditions in which the Nb or Ta precursor is soluble, without {Nb6} being the dominant species. Using hydrothermal synthesis allows the dissolution of NbV2O5 precursor without adding a huge excess of base that favors {Nb6} precipitation. Despite the growing number of new PONbs,109 their solution behavior needs to be investigated in more details. The speciation diagram for IPONbs is rather incomplete and includes four species {Nb6}, {Nb7}, {Nb10} and {Nb24} (Fig. 9 and Table 3). It should be noted that aqueous polyniobate chemistry at pH lower than 5 remains to be explored.
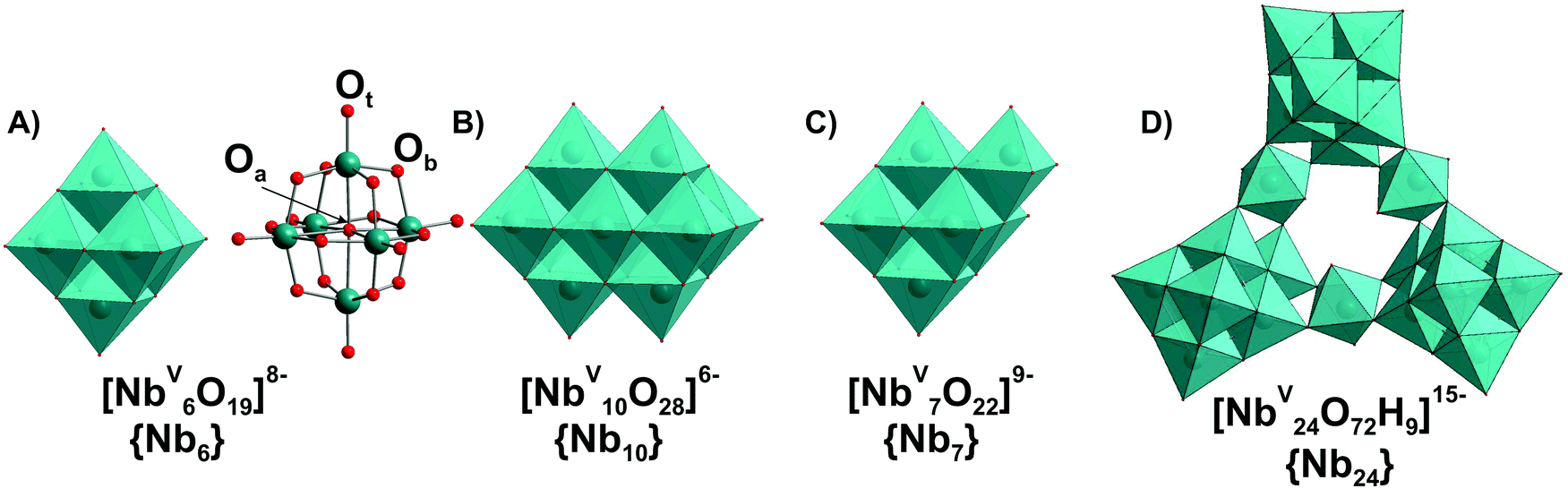 | ||
| Fig. 8 IPONb species ((A)–(D)) with the nuclearity up to 24 addenda atoms, which can be present in aqueous media. The Lindqvist anion {Nb6} is presented in (A) as polyhedral and ball-and-stick form with the indicated types of oxygen atoms: μ6-Oa – hexacoordinated oxygen atom; μ2-Ob – dicoordinated bridging atom; Ot – terminal oxygen atom. Color code: {NbO6}, blue; O, red. | ||
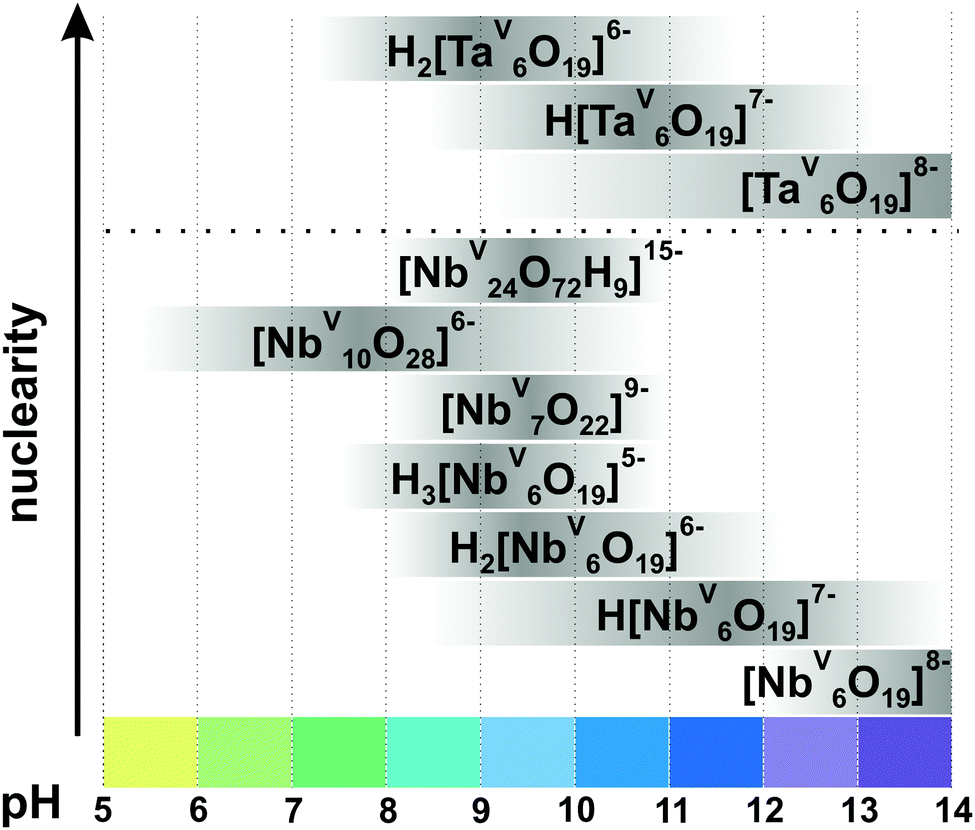 | ||
| Fig. 9 Speciation of IPONbs and IPOTas in an aqueous solution based on works.29,113–115,118,119,126 The range of pH < 5 is not studied well and the precipitation of NbV2O5·nH2O is usually observed. The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. The structures of IPONbs are depicted in Fig. 8 and their formation constants are given in Table 3. | ||
| IPONb species | q, pa | Isolated in solid state | Formation constantsa lgKc at 25 °C according to |
|
|---|---|---|---|---|
| Rozantsevb113 |
Etxebarriac119 |
|||
|
a Stoichiometric coefficient and formation constants for: p[NbV6O19]8− + qH+ [HzNbV6pOv]n− + (q/2 + p − z/2)H2O.
b Ionic strength μ = 0.1 M (KCl).
c
μ = 3 M (KCl).
|
||||
| [NbV6O19]8− (Fig. 8A) | 0, 1 | Yes110 | ||
| H[NbV6O19]7− | 1, 1 | Yes120 | 9.44 | 13.63 |
| H2[NbV6O19]6− | 2, 1 | Yes121 | 15.95 | 23.55 |
| H3[NbV6O19]5− | 3, 1 | Yes122 | 22.12 | 32.90 |
| [NbV7O22]9− (Fig. 8B) | 0.28, 1 | Yes123,124 | ||
| [NbV10O28]6− (Fig. 8C) | 4.4, 1 | Yes111 | ||
| [H9NbV24O72]15− (Fig. 8D) | 4.25, 1 | Yes125 | ||
3.2.1.2. Hexaniobate and -tantalate. The Lindqvist ion {Nb6} (Fig. 8A) was the first PONb studied in both the solid-state and solution. {Nb6} has the highest charge-density of all known POMs and readily associates with alkali metals in solutions. Nyman group studied the alkali metals ion-association of {Nb6} in solution via SAXS and showed that the completely neutralized hexaniobate, A8[NbV6O19] (A = Rb+, Cs+), is the dominant species in AOH solution, while the ‘nude’ anion [NbV6O19]8− dominates in TMAOH (TMA = tetramethylammonium) solutions.116 It is worth mentioning that the hexatantalate [TaV6O19]8− ({Ta6}) exhibits lower solubility in water than the analog hexaniobate salts.109 The dissolution of TBA6[H2TaV6O19] in toluene under prolonged heating leads to the isolation of TBA6[TaV10O28]·6H2O, which has an isostructural anion with decavanadate {V10} (Fig. 5) and decaniobate {Nb10} (Fig. 8B).117
At pH higher than 12 the hexaniobate ion {Nb6} is deprotonated and at lower pH values (Fig. 9), it carries between one and three protons, probably located on the μ2-bridging Ob oxygen atoms (Fig. 8A). According to 17O NMR measurements and density functional theory (DFT) calculations the protonated states of {Ta6} are all observed at lower pH values – mono-protonated species dominate at pH 10.5 and di-protonated at pH 9.118 Hexaniobate {Nb6} (Fig. 8A) in its various protonation states has been characterized in solution and in the solid-state (Table 3). While all hexatantalate protonation states have been observed in solution, only the salt in its di-protonated form was isolated in solid state.109
3.2.1.3. Decaniobate transformation. Aqueous {Nb10} (Fig. 8B) is stable and soluble at neutral pH, converts to {Nb6} with increased pH (pH > 11) and precipitates as NbV2O5 by acidification (pH < 5.5) (Fig. 9).29 The decaniobate ion showed no sign of protonation between pH 6–10, in contrast to the hexaniobate ion which is protonated between pH 8 and 13.115 The conversion of {Nb10} to {Nb6} was studied by 17O NMR, ESI-MS and DFT calculations and proceeds via a “heptaniobate” intermediate, {Nb7} (Fig. 8C).114,115 {Nb7} features three reactive terminal oxo-ligands leading to condensation into larger clusters such as the {Nb24} unit.125 Recently, Nyman and co-authors showed via Raman spectroscopy, ESI-MS, X-ray scattering and computational studies that {Nb10} converts to oligomers of [HxNbV24O72](24−x)− ({Nb24}, Fig. 8D) upon adding only alkali chloride salts, even in buffered (1 M HEPES, pH = 7) neutral solutions.127 The rate of {Nb10} to {Nb24} conversion increases in raw Cs+ > Rb+ > K+ > Na+ > Li+ and cation concentration and indicates that the alkali cations open the compact {Nb10} structure and are primarily responsible for driving the reaction.127
3.3. Polyoxomolybdates (POMos)
Since the 1960s the speciation in acidified solutions of [MoVIO4]2− with different concentration and ionic strength has been a subject of comprehensive investigation by potentiometry, NMR spectroscopy, and ESI-MS and was extensively elaborated by Tytko and Glemser,128 Cruywagen,6 Pettersson,129 Maksimovskaya18 and others. However, in the last decades, this important topic receives less and less attention and for newly synthesized POMos their solution stability and transformation are rarely investigated.3.3.1.1. Ion-distribution diagram for IPOMos. The polymerization processes of [MoVIO4]2− are observed in solution where the MoVI concentration is higher than 10−4 M. In strongly alkaline molybdate(VI) solutions the monomeric [MoVIO4]2− anion is dominating and forms salts with mono- to trivalent cations, whereas in strongly acidic solutions, the predominant species are stable mono-[MoVIO2(OH)(H2O)3]+ or bi-[MoVI2O5(OH)(H2O)5]+ oxocations, which form salts with inorganic anions and complexes with organic ligands.18 It is generally accepted that the first major product that forms upon acidification of aqueous [MoVIO4]2− is heptamolybdate, [MoVI7O24]6− ({Mo7}, Fig. 10A), which shows its maximum concentration at pH ∼ 5 and can be protonated upon further acidification (Fig. 11). The {Mo7} ion, its protonated forms and the octamolybdate ion β-[MoVI8O26]4− (β-{Mo8}, Fig. 10B) are included in most speciation models describing potentiometric results (Table 4). In some cases, H3[MoVI7O24]3− is preferred over β-{Mo8} as the only existing species, but in the majority of models both polyanions are present. There are contradictory opinions about the speciation being present in the pH region between 4.5 and 2.5.
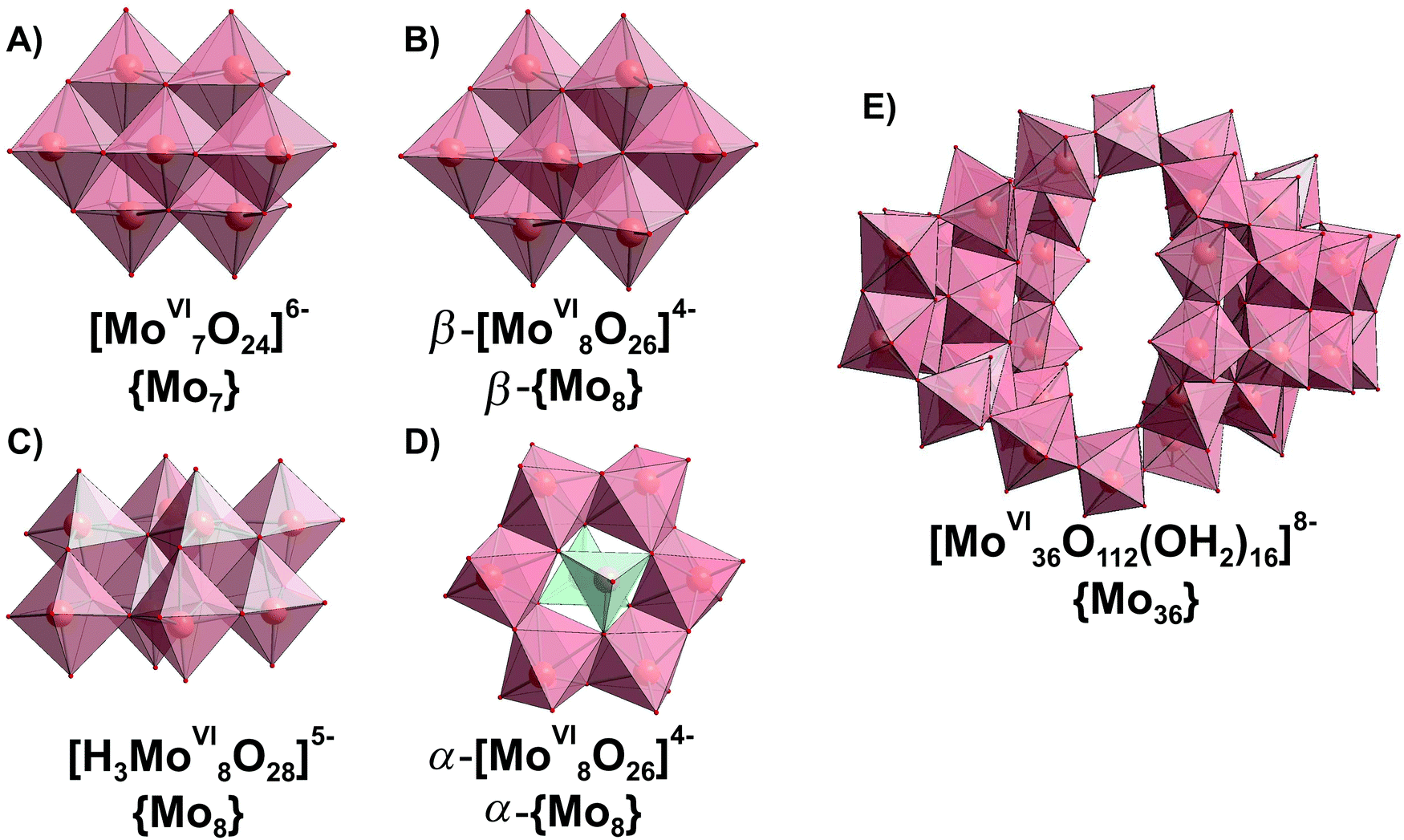 | ||
| Fig. 10 IPOMos with nuclearity of up to 36 addenda atoms, which are confirmed to be present in aqueous media. Color code: {MoO6}, pink; {MoO4}, green; O, red. | ||
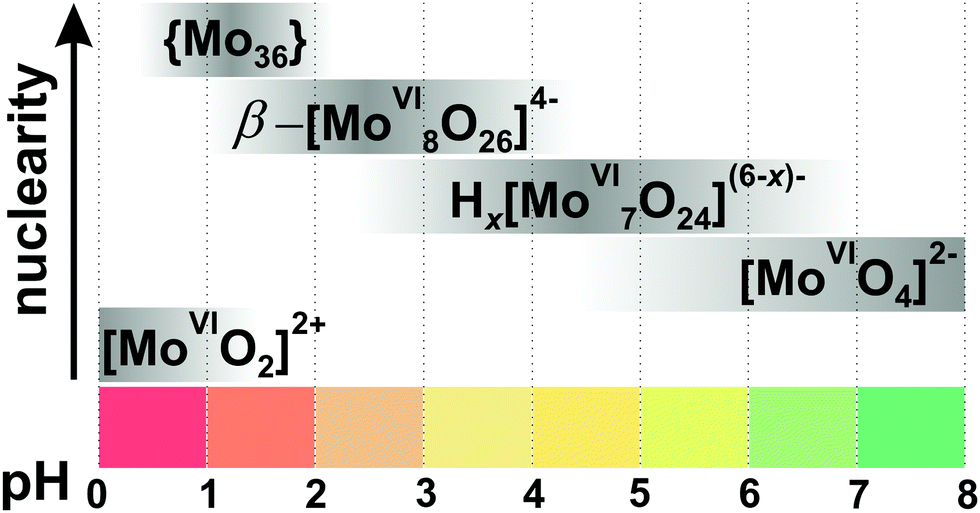 | ||
| Fig. 11 Speciation of IPOMos in an aqueous solution with the concentration of MoVI 0.1–0.4 M based on works.6,18,128,131,132 The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. x in Hx[MoVI7O24](6−x)− is 0–2; {Mo36} is abbreviation for [MoVI36O112(OH2)16]8−. The structures of species are presented in Fig. 10. The 95Mo and 17O chemical shifts, stoichiometry, formation constants and pKa values are summarized in Table 4. | ||
| IPOMo species | p, qa | pKa | Isolated in solid state | δ(95Mo), ppmb | δ(17O), ppmb | Raman bands, cm−1 | Formation constantsa lgKc at 25 °C |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Cruywagenc6 |
Tytkod135 |
Taubee136 |
Cruywagenf137 |
|||||||
|
a Stoichiometric coefficients and formation constants for p[MoVIO4]2− + qH+ [HzMoVIpOm]n− + (q − z/2)H2O δ95Mo relative to Na2MoO4.
b
δ
17O relative to H2O.
c Ionic strength μ = 2 M (LiClO4).
d
μ = 2 M (NaCl).
e
μ = 0.3 M (Na2SO4).
f
μ = 3 M (Na(H)ClO4).
|
||||||||||
| [MoVIO4]2− | 0, 1 | Yes | 896133 | |||||||
| H[MoVIO4]− | 1, 1 | No | 3.48 | 3.40 | ||||||
| [MoVI7O24]6− (Fig. 10A) | 8, 7 | Yes138 | 210, 32, 1518 | 121.7, 338.6, 356.5, 395.2, 745, 754.4, 815129 | 939133 | 54.07 (0.03) | 55.71 | 52.43 | ||
| H[MoVI7O24]5− | 9, 7 | 4.38 | No | 59.01 (0.03) | 60.22 | 57.42 | ||||
| H2[MoVI7O24]4− | 10, 7 | 3.38135 | No | 61.24 | ||||||
| H3[MoVI7O24]3− | 11, 7 | 1.87135 | No | 63.90 | ||||||
| [H3MoVI8O28]5− (Fig. 10C) | 11, 8 | Yes130 | 261, 370, 395, 840, 850, 860129 | 70.30 (0.03) | 71.41 | |||||
| α-[MoVI8O26]4− (Fig. 10D) | 12, 8 | Yes139 | 959, 918133 | |||||||
| β-[MoVI8O26]4− (Fig. 10B) | 12, 8 | Yes140 | 100, 10 18 | 53.5, 290.7, 404.3, 738.2, 861.6, 876, 878, 917.3129 | 971, 943, 915, 904133 | 74.10 (0.03) | 71.52 | |||
| β-H[MoVI8O26]3− | 13, 8 | 1.83135 | No | |||||||
| [MoVI18O56(H2O)8]4− | 32, 18 | half unit of {Mo36}141 | 179.28 | 180.39 | ||||||
| [MoVI36O112(OH2)16]8− (Fig. 10E) | 64, 36 | Yes141 | 983, 957, 899133 | |||||||
| [MoVIO2]2+ | 4, 1 | Yes142 | −62 to −6918 | |||||||
| [MoVIO2(OH)(H2O)3]+ | 3, 1 | 0.9 | No | 8.09 | 8.81 | |||||
| [MoVI2O5(OH)(H2O)5]+ | 5, 2 | ∼0.1–0.2 | No | 18.02 | 18.93 | |||||
| [MoVI2O5(H2O)6]+ | 19.60 | |||||||||
| [MoVI2O4(OH)(H2O)5]+ | 18.98 | |||||||||
In 1990, Howarth and co-authors129 proposed, based on 95Mo and 17O NMR spectroscopic studies, that monoprotonated {Mo7}, instead of being higher protonated, is converted to the intermediate polyanion [H3MoVI8O28]5− (Fig. 10C). The structure of the latter species is believed to be the same as that of the [H2MoVI8O28]6− anion, which was crystallized from aqueous solution with isopropylammonium as countercation.130 Later Maksimovskaya and Maksimov have shown using the same methods, 95Mo and 17O NMR, that only heptamolybdate and octamolybdate co-exist in the pH region from 5.5 to 2.5.18 Moreover, they showed evidence of the di-protonated anion {Mo7} and that chemical exchange occurs between Hx[MoVI7O24](6−x)− (x = 0–2) and β-{Mo8} without any additional intermediate species. Notably, a recent potentiometric study confirms the presence of both octamolybdate and the up to three protonated forms of {Mo7} in solution (CMo = 0.15 M, ionic strength μ = 0.15 M (NaClO4)).131
Salts of the larger anion, [MoVI36O112(OH2)16]8− (Fig. 10E and Table 4) can be crystallized from systems at pH lower than 2.8141 and its identity in solution was confirmed by 95Mo NMR18,132 and Raman133 spectroscopy. The anion is a dimer of [MoVI18O56(H2O)8]4− with inversion center, that is why both {Mo36} and {Mo18} are often included in the ion-distribution diagram6 for MoVI solution in acidic region (Table 4).
Hypothetical anions such as [HMoVI17O55]7−, [HMoVI13O42]5− and [MoVI10O34]8− have been included to theoretical models for better interpretation of potentiometric results obtained at high molybdate concentration (>0.1 M),6,134 but more work is necessary to determine the existence and structure of these species in solution. At lower molybdate concentrations, the presence of the hypothetical species [H2MoVI6O21]4− has been proposed.6 Equilibrium constants obtained by different research groups based on potentiometric and NMR spectroscopic studies under the same conditions agree quite well (Table 4). The ion-distribution diagram presented here (Fig. 11) is based on potentiometric, Raman and NMR spectroscopic investigations (Table 4).
3.3.1.2. IPOMos under physiological conditions. Two stable species, [MoVIO4]2− and {Mo7} (Fig. 10A), have been reported to be simultaneous present at neutral pH, that makes the analysis of their equilibrium important for understanding their function in biological or catalytic application complicated. Ng et al. investigated isopolymolybdate solutions as a function of concentration, time and pH by Raman spectroscopy, light and X-ray scattering,143 showing that significant amounts of both [MoVIO4]2− and {Mo7} are present in equilibrium over a wide pH range (4.5–7) in a 0.1 M solution of MoVI. Quantitative deconvolution of the Raman spectra taken from 0.1 M solution of MoVI demonstrated that even at pH 6.6, only half of all orthomolybdate is in hepta-form and the full conversion to {Mo7} is completed only at pH 4.5. Ng et al.143 did not observe the formation of significant quantities of β-{Mo8} (Fig. 10B), as was suggested by Tytko.135 When studying the sorption of isopolyoxomolybdates into layered double hydroxides by in situ real time infrared spectroscopy, the coexistence of [MoVIO4]2− and {Mo7} was observed only in the pH range from 5.5 to 5.7 in 0.1 M solution of MoVI, whereas at pH above 5.7, the predominant anion is the monomeric one.144
Investigation of the hydrolysis of the phosphodiester bond in the DNA model substrate bis(p-nitrophenyl)phosphate (BNPP, 25 mM) in the presence of Na6[MoVI7O24] (25 mM) at 50 °C in the absence of buffer showed that the maximal cleavage reaction was observed at pH = 5.3.14595Mo NMR confirmed that the predominant species at this pH is {Mo7} (Fig. 10A), which promotes the hydrolysis. Furthermore, 95Mo NMR and Mo K-edge EXAFS investigations of the different hydrolytic reaction stages showed a gradual disappearance of {Mo7} during the hydrolytic reaction and appearance of [PV2MoVI5O23]6− (Fig. 14A), which was the final compound observed at the end of hydrolytic reaction.146
3.3.1.3. Hexamolybdate [MoVI6O19]2− and its stability in aqueous solutions. In addition to the five species shown in the ion-distribution diagram for IPOMos (Fig. 11), the Lindqvist type hexamolybdate anion [MoVI6O19]2− ({Mo6}) exists in solid state and has the same structure as its niobate analog (Fig. 8A). The {Mo6} anion is the only colored IPOMo (yellow, λmax = 325 nm, Ob → MoVI charge transfer) structurally based on six mono-units MoOt (Ot – terminal oxygen atom), rather than three cis MoOt2 units. {Mo6} does not exist in any notable concentration in pure aqueous solution and requires solvents with lower dielectric constants147 or macrocycles132 for stabilization due to its low charge density and rigid symmetry. UV-vis spectroscopy shows that [MoVI6O19]2− is formed in solution containing acetone, acetonitrile, ethanol, dimethyl formamide and 1,4-dioxane with an organic solvent content >30%.147 The addition of a large γ-cyclodextrin macrocycle to the acidified aqueous solution of sodium orthomolybdate leads to the host–guest stabilization of {Mo6} by formation of the complex {Mo6O19@γ-CD}2−, as a new species in the pH range between 1 and 2.5.132 Still the inclusion of the Lindqvist type hexamolybdate in the IPOMos distribution diagram in purely aqueous solutions is reported which should be seen critically.148
3.3.1.4. Mono- and bimolybdyl cationic species in strongly acidic solutions. The successive protonation equilibria of [MoVIO4]2− lead to formation of the cationic species: [MoVIO2(OH)(H2O)3]+, [MoVI2O5(H2O)6]2+, [MoVI2O4(OH)(H2O)6]3+, that are well characterized by potentiometry, and the equilibrium constants have been determined for different ionic media by various authors (Table 4).137,149,150 The total molybdate concentration must be lower than 10−4 M for the mono- and binuclear species to be predominantly present in aqueous solution. Cruywagen has reported the presence of four species, [MoVIO3(H2O)3], [MoVIO2(OH)(H2O)3]+, [MoVI2O5(H2O)6]2+, [MoVI2O4(OH)(H2O)6]3+, under strongly acidic conditions (HClO4 with concentration 0.5–4 M and pH region from −3 to 0.5).137 Later, applying XAS, UV-vis spectroscopy in combination with first-principles calculation methods, all four species were confirmed and the predominant species in 2 M nitric acid are [MoVI2O5(H2O)6]2+ and [MoVI2O4(OH)(H2O)6]3+ (Fig. 12).149
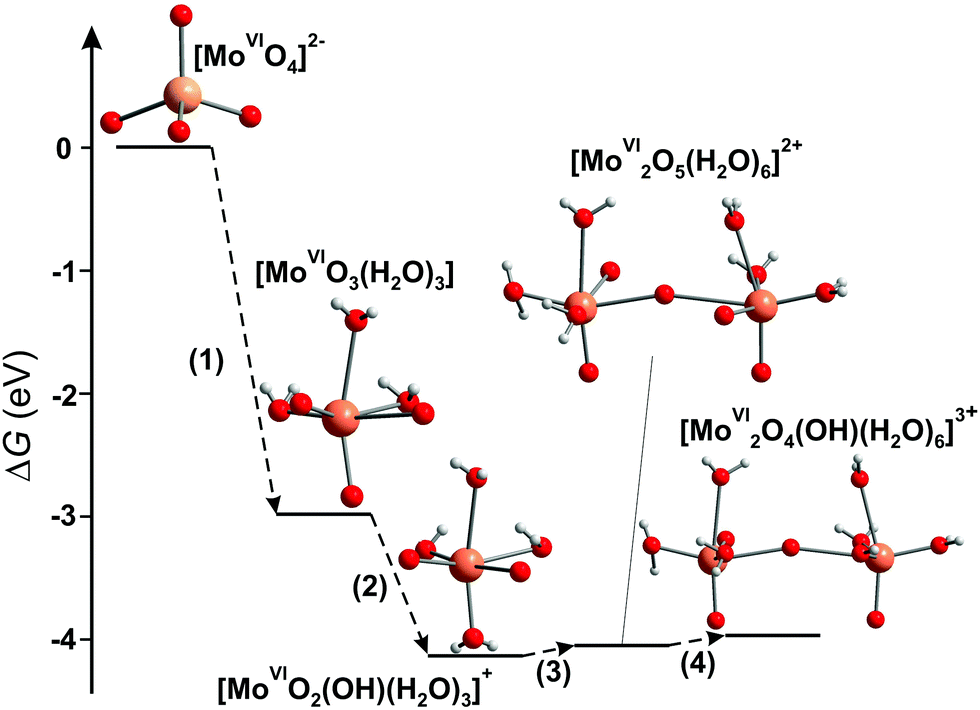 | ||
| Fig. 12 Energy diagram of the free-energy change (ΔG) for the condensation reaction of Mo complexes.149 Color code: Mo, orange; O, red; H, grey. | ||
3.3.1.5. Computational investigation of IPOMos formation. The speciation diagram for IPOMos does not show species with two to six molybdenum ions (Fig. 11). It is possible that these species exist at low concentration as intermediates in the process of forming larger IPOMos. DFT calculations together with ESI-MS experiments provide151,152 insights into the possible formation mechanism of the Lindqvist [MoVI6O19]2− ion (Fig. 8A), which is preferably formed in organic media and is undergoing a transformation in aqueous solution.1 Unstable low-nuclearity species such as [MoVI2O7]2−, [MoVI3O10]2−, [MoVI4O13]2−, [MoVI5O16]2− and [MoVI6O19]2− were detected experimentally by ESI-MS in acetonitrile. Recently, Duarte and co-authors have shown using DFT/PBE calculations that formation of IPOMos with up to four molybdenum atoms is highly favorable in water.153 The formation of the predominant species [MoVI7O24]6− and β-[MoVI8O26]4− involves a reaction between the species with six and seven Mo atoms, respectively, and the monomer [MoVIO4]2−, which are frequently assisted by protons through an aggregation or water condensation mechanism.153
3.3.1.6. Comparison of IPOMos in solution and the solid state. As with many other POMs there are some isopolymolybdates, which occur only in solution, and others only in solid state. However, all prevalent molybdate species ([MoVIO4]2−, [MoVI7O24]6− (Fig. 10A), β-[MoVI8O26]4− (Fig. 10B), [MoVI36O112(OH2)16]8− (Fig. 10E)) exist in solution and in solid state (Table 4). Low-nuclearity IPOMos, which are unstable in solution, tend to form polymeric structures with infinite two-dimensional chains that have been synthesized in solid state reactions154,155 or hydrothermally (Table 5).156,157In situ Raman spectroscopy confirmed that between 170 and 190 °C and pH 7 and 5, chain-like or discrete molecular structures of dimolybdates [MoVI2O7]2− (Fig. 13B) and trimolybdates [MoVI3O10]2− (Fig. 13D) are preferentially formed,158 whereas heptamolybdate {Mo7} dominates under ambient pressure at 25 °C (Fig. 11). Long-term equilibrium in solutions of {Mo7} leads to its transformation and crystallization of different products depending on temperature and time (Fig. 13 and Table 5), which should undoubtedly be taken into account when using heptamolybdate. The addition of structure-directing reagents such as big cage-like organic cations (1,4-diazabicyclo[2.2.2]octane159 or bis[benzidinium(1−)]157) affects the speciation at a given pH value and leads to the isolation of new, sometimes polymerized, structures (Table 5).
| pH or Za | Species isolated from solution | Cation | Ref. |
|---|---|---|---|
| a Z = n(H+)/n(MoO42−). b HT – hydrothermal synthesis. | |||
| >7 | [MoVIO4]2− | Metal ions in ox. state 2+, 3+ | 160 |
| 5–7 | [MoVI7O24]6− (Fig. 10A) | Na+, K+, Cs+, NR4+ (R = H, alkyls, alkenyls, phenyls) | 161 |
| 6–7 (from [MoVI7O24]6− solution at 60 °C after addition of NH4+) | ([MoVI2O7]2−)∞ (Fig. 13B) | NH4+ | 4 |
| 6–7 (aging of [MoVI7O24]6− solution – 12 days) | ([MoVI3O10]2−)∞ (Fig. 13D) | Rb+ | 5 |
| 6–7 (aging of [MoVI7O24]6− solution – weeks or HTb) | ([MoVI8O27]4−)∞ (Fig. 13C) | NH4+, (C4H12N2)+, (NH3(CH2)3NH3)2+, etc. | 162–164 |
| 6–7 (aging of [MoVI7O24]6− solution – under UV irradiation) | [H2MoVI8O28]6− (Fig. 13A) | (C3H10N)+ | 130 |
| 6–7 (aging of [MoVI7O24]6− solution – several days at 80 °C) | [MoVI10O34]8− (constructed from {MoVI8O28} unit connected at corners to two {MoVIO4} tetrahedra) (Fig. 13E) | NH4+ | 165 |
| 5 (HTb from [Mo7O24]6−) | [MoVI5O16]2− (constructed from [(MoVI4O14)n]4n− chains linked through MoO6) | (C12H13N2)+ | 157 |
| 4 (HTb from [MoVI7O24]6−) | ([MoVI3O10]2−)∞ | (C6H13N2)+ | |
| 3 | [MoVI7O24]6− (Fig. 10A) | Zn2+, protonated hexamethylenetetramine | 166 |
| 2–3 (HT from [MoVI7O24]6−) | ([MoVI8O26]4−)∞ or ([MoVI4O13]2−)∞ (constructed from γ-[MoVI8O28] | (C6H13N2)+, NR4+ (R = H, alkyls, alkenyls, phenyls) | 159 |
| Z = 1.8–2.0 | [MoVI36O112(OH2)16]8− (Fig. 10E) | K+ | 141 |
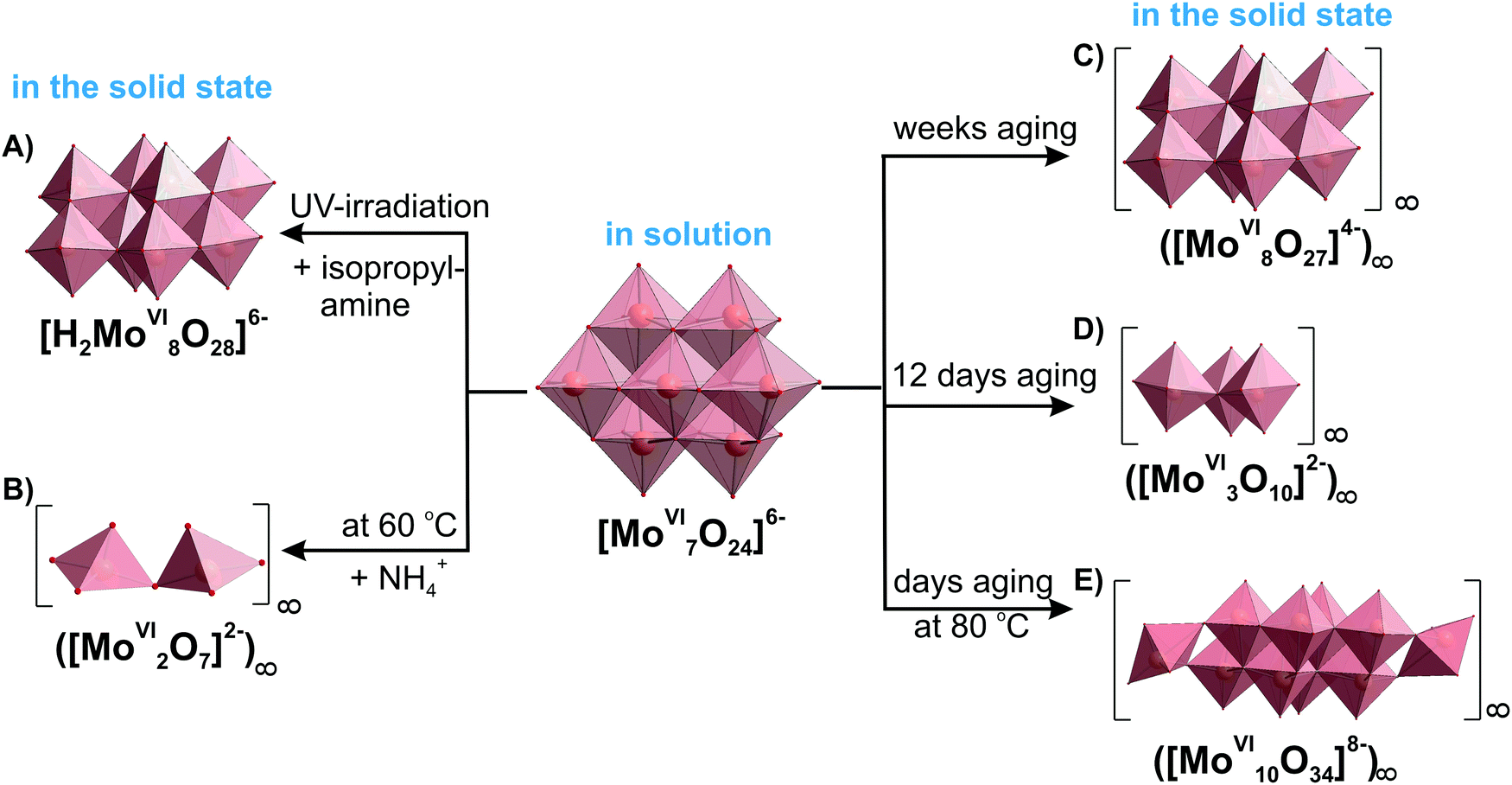 | ||
| Fig. 13 Heptamolybdate {Mo7} transformation at different synthetic conditions showing the conformity of IPOMos in solution and in the solid state based on ref. 4, 5, 130, 141, 162, 165 and 166. The time of aging for the polymeric octa- and decamolybdate has been described as “several” days or weeks in the original works162,165 without exact number. Color code: {MoO6}, pink; {MoO4}, green; O, red. | ||
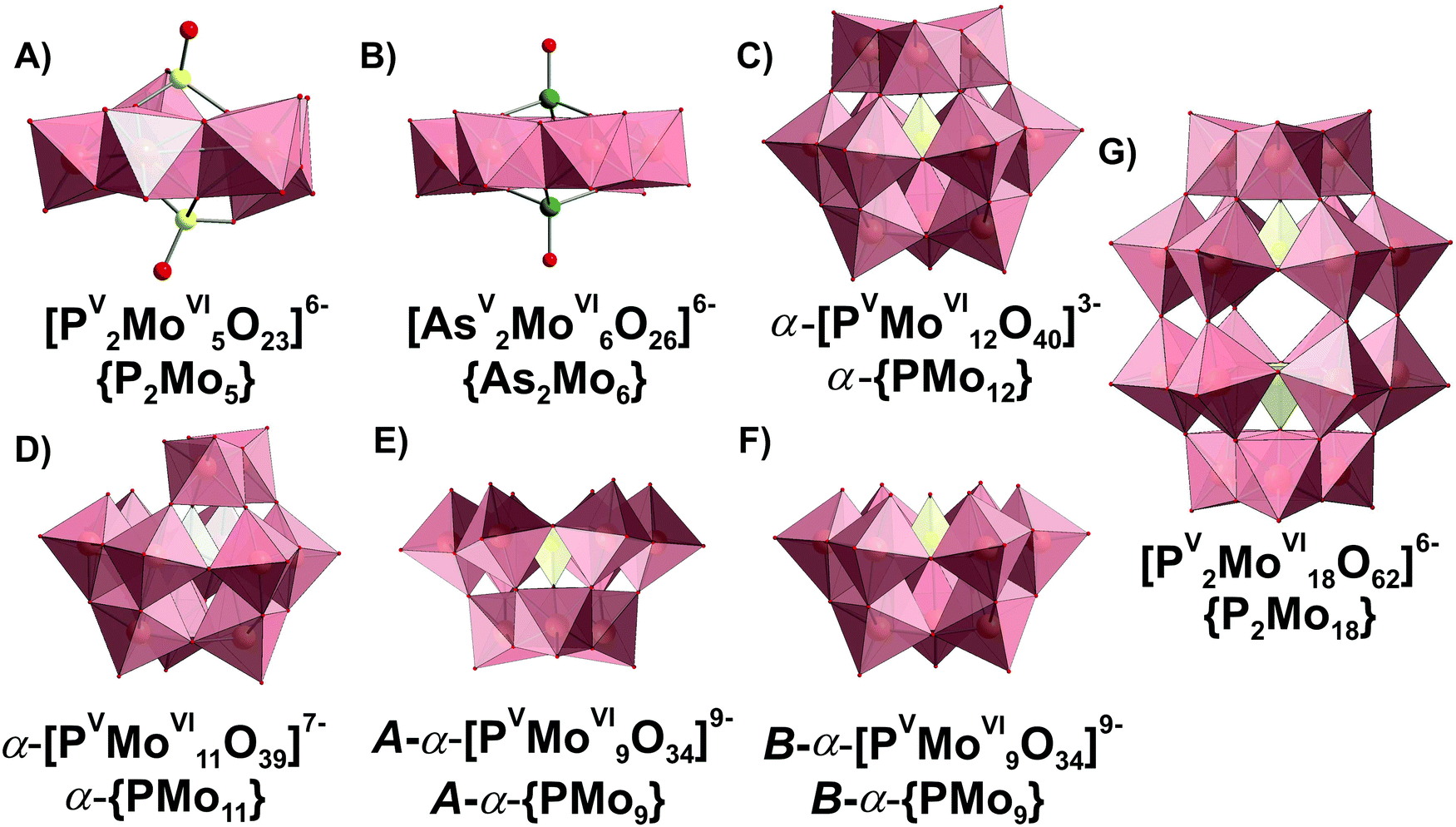 | ||
| Fig. 14 Phospho- and arsenatomolybdates present in aqueous media. The data about structural isomerism of lacunary Keggin anions are given in ref. 1. Color code: {MoO6}, pink; {PO4} and P, yellow; As, green; O, red. | ||
3.3.2.1. Speciation in the phosphate-molybdate system. In the late 1980s Pettersson et al. extensively analyzed speciation in the system [MoVIO4]2−–HPVO42−–H+ using potentiometry and 31P NMR spectroscopy in the pH range from 0.5 to 7 and the concentration range [Mo] = 0.030–0.480 M, [P] = 0.005–0.040 M, where [Mo] and [P] – are the total concentrations of molybdenum(VI) and phosphorus(V).167,168 When the ratio [Mo]/[P] is lower than 2.5 and the pH between 2 and 7, only colorless heteropolyanions Hx[PV2MoVI5O23](6−x)− (x = 0–2, {P2Mo5}, Fig. 14A) are present. The {P2Mo5} unit, co-called Strandberg {X2Mo5} archetype, is built up from five {MoO6} octahedra and two {PO4} tetrahedra, as shown in Fig. 14A, and was isolated in solid state in all protonation states (Table 6).169–171 At [Mo]/[P] > 2.5 and pH below 5.5, other molybdophosphate complexes with 9 ([PVMoVI9O34]9−, {PMo9}, Fig. 14E and F), 11 ([PVMoVI11O39]7−, {PMo11}, Fig. 14D), 12 ([PVMoVI12O40]3−, {PMo12}, Fig. 14C) and 18 ([PV2MoVI18O62]6−, {P2Mo18}, Fig. 14G) MoVI atoms start to form. Besides, the {P2Mo5} (Fig. 14A) and {PMo11} (Fig. 14D) series, protonated trilacunary species {PMo9} (Fig. 14E and F) are also formed in solution. In the acidic region with pH < 1.8 and at high [Mo]/[P] ratios, the Keggin {PMo12} (Fig. 14C) anion is present. During aging in solution [MoVIO4]2−–HPVO42−–H+ at pH lower than 3, the yellow Wells–Dawson {P2Mo18} anion (Fig. 14G) is slowly – up to 1 month – formed. The Wells–Dawson formation is slowed down with increasing [Mo] to [P] ratio. The distribution diagrams for “fresh” (after preparation) and “aged” (up to 1 month) solutions are illustrated in Fig. 15. Fig. 15 clearly demonstrates that the initially high concentration of the Keggin complex [PVMoVI12O40]3− (Fig. 14C) in fresh acidic solutions with [Mo]/[P] = 12 diminishes considerably when the Wells-Dawson anion [PV2MoVI18O62]6− (Fig. 14G) is formed.168
| Molybdophosphate species | Abbreviation | q, p, ra | pKa168 | Isolated in solid state | δ(31P), ppmb | Raman bands, cm−1 | Formation constants lgKc at 25 °Ca,c168 |
|---|---|---|---|---|---|---|---|
|
a Stoichiometric coefficients and formation constants for the equilibrium: p[MoVIO4]2− + qH+ + r[HPVO4]2− (H+)q(MoVIO42−)p(HPVO42−)r.
b
δ
31P relative to H3PO4 (85%).
c Ionic strength μ = 3 M (NaCl).
|
|||||||
| [PV2MoVI5O23]6− | {P2Mo5} (Fig. 14A) | 8, 5, 2 | Yes169 | 2.35167 | 61.97 (2) | ||
| H[PV2MoVI5O23]5− | 9, 5, 2 | 5.10 | Yes170 | 1.94167 | 67.07 (5) | ||
| H2[PV2MoVI5O23]4− | 10, 5, 2 | 3.79 | Yes171 | 1.86167 | 70.86 (8) | ||
| B-α-[PVMoVI9O34]9− | B-{PMo9} (Fig. 14F) | 14, 9, 1 | No | 0.47–0.08168 | 98.21 (7) | ||
| B-α-H[PVMoVI9O34]8− | 15, 9, 1 | 3.83 | No | 102.04 (8) | |||
| B-α-H2[PVMoVI9O34]7− | 16, 9, 1 | 2.85 | No | 104.89 (8) | |||
| B-α-H3[PVMoVI9O34]6− | 17, 9, 1 | 1.53 | No | 106.42 (8) | |||
| A-α-[PVMoVI9O31(OH)(OH2)2]4− | A-{PMo9} (Fig. 14E) | 16, 9, 1 | Yes176 | −1.00 to −1.15168 | 104.71 (8) | ||
| A-α-[PVMoVI9O31(OH2)3]3− | 17, 9, 1 | 2.46 | Yes176 | 968, 854, 716, 641172 | 107.17 (5) | ||
| [PV2MoVI18O62]6− | {P2Mo18} (Fig. 14G) | 34, 18, 2 | Yes177 | −2.53;168 −2.4178 | 971, 962, 707172 | 217.8 (1) | |
| [PVMoVI11O39]7− | {PMo11} (Fig. 14D) | 17, 11, 1 | Yes179 (SXRD only for substituted anions180) | −0.78 to −1.20168 | — | 118.68 (8) | |
| H[PVMoVI11O39]6− | 18, 11, 1 | 4.42 | No | — | 123.10 (5) | ||
| H2[PVMoVI11O39]5− | 19, 11, 1 | 2.95 | No | 974, 969, 957, 875, 763172 | 126.05 (5) | ||
| [PVMoVI12O40]3− | {PMo12} (Fig. 14C) | 23, 12, 1 | Yes181 | −3.20168 | 988, 965, 894, 602172 | 139.7 (1) | |
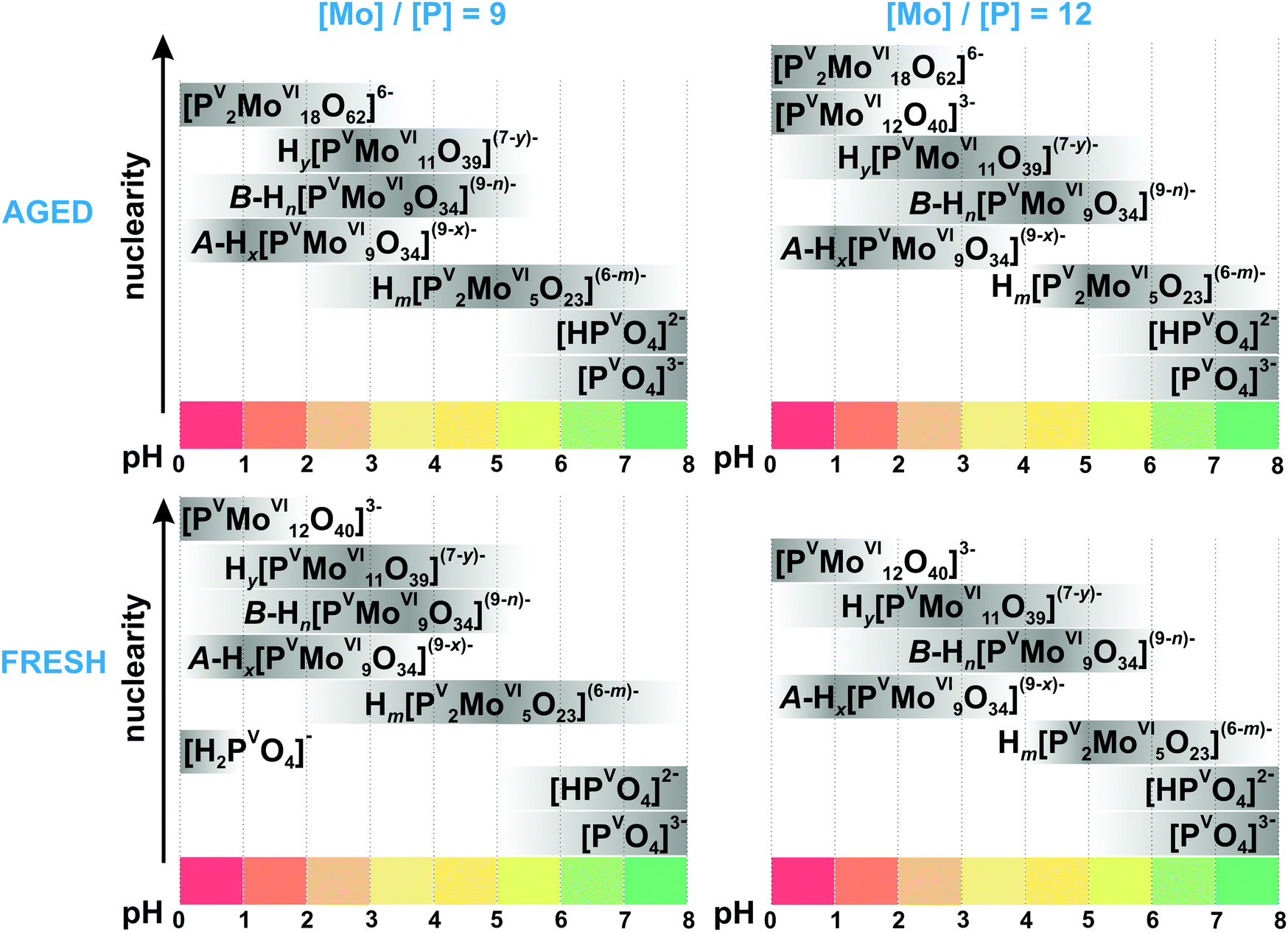 | ||
| Fig. 15 Speciation of phosphomolybdates in freshly prepared and aged (up to 1 month) aqueous solution with the concentration of 0.18 M MoVI ([Mo]/[P] = 9) and 0.24 M ([Mo]/[P] = 12) based on works.167,168 The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. The x value in A-Hx[PVMoVI9O34](9−x)− is 5–6; y in Hy[PVMoVI11O39](7−y)− is 0–2; n in B-Hn[PVMoVI9O34](9−n)− is 0–3; m in Hm[PV2MoVI5O23](6−m)− is 0–2. The structures of species are presented in Fig. 14. The 31P chemical shifts, stoichiometry, formation constants and pKa values are summarized in Table 6. | ||
Mono- and trilacunary Keggin species. Raman spectroscopic studies performed by Ueda et al. confirmed, that the formation of Wells–Dawson type anion (Fig. 14G) takes around 30 h after solution ([P] > 20 mM, [Mo] = 100 mM, pH = 3) preparation and an elevated temperature of about 80 °C.172 However, unlike the results of Pettersson,167,168 they did not detect the {PMo11} complex (Fig. 14D) in the system with [Mo] = 100 mM and [P] > 7 mM, and {PMo12} is directly converted into its trilacunary form {A-PMo9} (Fig. 14E). At the same time, van Veen et al. suggested based on 31P NMR and Raman spectroscopies that {PMo11} is more prominent at higher [Mo]/[P] = 12 than at lower [Mo]/[P] = 9 ratios, while the opposite is true for the {A-PMo9} species.178 The results of ESI-MS studies on the pH-dependence of phosphomolybdate solutions from pH between 1.7 and 10.2 confirm the presence of both mono- and trilacunary species.173 The confusion in the literature about the existence of mono-{PMo11} and trilacunary {A-PMo9} complexes is understandable, since their spectroscopic characteristics (31P NMR shifts and Raman spectra (Table 6)) are indeed very similar.
3.3.2.2. Speciation in the phosphito-molybdate system. The speciation in acidified molybdate solution with trivalent phosphorus anions, such as HPIIIO32−, (C6H5)PIIIO32− or (CH3)PIIIO32−, has been determined from both potentiometric and 31P NMR measurements.174,175 In the system with phosphite, phenyl- and methylphosphonate anions structures similar to the Hx[PV2MoVI5O23](6−x)− (x = 0–2) anion169 (Fig. 14A) dominate.174 It was shown that partial oxidation of phosphite to phosphate occurs, especially in the thin-walled NMR sample tubes when exposed to fluorescent light. In {X2M5}-type HPOMos the phosphite anion HPIIIO32− binds weaker to the MoVI-oxoframework than phosphate or phosphonates.
3.3.2.3. Speciation in arsenato-molybdate system. Aqueous equilibria in the arsenato-molybdate system [AsVO4]3−–[MoVIO4]2− have been studied by potentiometric and spectrophotometric methods (298 K, 3.0 M NaClO4).182 These investigations established the formation of two series of complexes: colorless anions with two As atoms and five or six Mo atoms, and yellow complexes with [As]/[Mo] ratio 1
:9 ({AsMo9}) (the same structure as {PMo9} see Fig. 14E and Table 7). Pettersson suggests for {As2Mo5} the same structure as for {P2Mo5} (Fig. 14A),182 while the {As2Mo6} complex was isolated in all protonated states and characterized by SXRD (Table 7 and Fig. 14B). Applying large-angle X-ray scattering (LAXS) investigations on {AsMo9} complexes showed them to have the same basic trilacunary structure (Fig. 14E) as the [AsVMoVI9O31(OH2)]3− anion present in Na3[AsVMoVI9O31(OH2)3]·13H2O.183 Later Ueda et al. showed,172 that at AsV concentrations lower than 10 mM, in contrast to the phosphate system, the corresponding MoVI–AsV system did not produce the Keggin-type [AsVMoVI12O40]3− anion (the same structure as {PMo12} see Fig. 14C) but a labile complex (tentatively formulated as [AsVMoVI10O37H5]4−) at ambient temperature and higher AsV concentration. The [AsVMoVI10O37H5]4− complex transforms directly into [AsV2MoVI6O26H2]4− (Fig. 14B). The Dawson-type [AsV2MoVI18O62]6− complex (the same structure as {P2Mo18} see Fig. 14G), which was formed upon heating at 80 °C for 5 h, also underwent subsequent transformation into the more stable [AsV2MoVI6O26H2]4− complex (Fig. 14B) at As(V) concentrations higher than 20 mM.
| Molybdoarsenate species | Abbreviation | q, p, ra | pKa | Isolated in solid state | Formation constants lgKc at 25 °Ca,b182 |
|---|---|---|---|---|---|
|
a Stoichiometric coefficients and formation constants for: p[MoVIO4]2− + qH+ + r[HAsVO4]2− (H+)q(MoVIO42−)p(HAsVO42−)r.
b Ionic strength μ = 3 M (NaClO4).
|
|||||
| [AsV2MoVI5O23]6− | {As2Mo5} (Fig. 14A) | 8, 5, 2 | No | 60.92 (0.04) | |
| [AsV2MoVI6O26]6− | {As2Mo6} (Fig. 14B) | 10, 6, 2 | Yes184 | 75.25 (0.04) | |
| H[AsV2MoVI6O26]5− | 11, 6, 2 | Yes185 | 80.73 (0.05) | ||
| H2[AsV2MoVI6O26]4− | 12, 6, 2 | Yes186 | 84.07 (0.07) | ||
| H3[AsVMoVI9O34]6− | {AsMo9} (Fig. 14E and F) | 14, 9, 1 | Yes183 | 100.11 (0.02) | |
| H4[AsVMoVI9O34]5− | 15, 9, 1 | 3.70182 | No | 103.81 (0.05) | |
| H5[AsVMoVI9O34]4− | 16, 9, 1 | 2.33182 | No | 106.14 (0.05) | |
| H6[AsVMoVI9O34]3− | 17, 9, 1 | 1.33182 | No | 107.47 (0.06) | |
| [AsV2MoVI18O62]6− | {As2Mo18} (Fig. 14G) | 34, 18, 2 | Yes187 | — | |
3.3.2.4. Hydrolytic stability of HPOMos with Keggin structure. {PMo12} and {AsMo12}. According to Raman spectroscopic data, the Keggin molybdophosphate (Fig. 14C) firstly appears 1 min and 23 s after the acidified molybdate solution is mixed with the acidified phosphate solution at pH 1, which is the optimal acidity for the {PMo12} formation.188 The UV-vis, IR and 31P NMR spectra of {PMo12} solution recorded at pH values between 0 and 12 manifest that already at pH 1.5 the intact Keggin anion decomposes into lacunary forms, and at pH 7, monomeric molybdate and phosphate dominate in the solution.189–192 {PMo12} co-exists with its monolacunary form {PMo11} between pH 1.5 and 5 (Fig. 15).190
{SiMo12} and {GeMo12}. Already in 1975, it was established that α-H4[SiIVMoVI12O40] ({SiMo12}) acid is formed between pH 3.8 and 4.8 and the β-isomer at lower pH values between 1.0 and 1.8.193,194 The yield of both α- and β-forms (at pH 4.0 and 1.2, respectively) is independent of molybdate concentrations between 0.015 and 0.100 M. Later, using ESI-MS the formation of molybdosilicates was detected between pH 1 and 2.195 The decomposition of {SiMo12} is 50% completed immediately after dissolution at pH 4.1.191 As the pH is increased, the decomposition also proceeds rapidly and at pH 5.2 the acid is completely decomposed. The α-[GeIVMoVI12O40]4− ({GeMo12}) Keggin complex has a stability range similar to {SiMo12} and is reported to form mono-lacunary [GeIVMoVI11O39]8− between pH 4.3 and 4.5. The formation rate of [GeIVMoVI11O39]8− is faster than that of [SiIVMoVI11O39]8−, while the formation rate of {GeMo12} is slower than the one of {SiMo12}.196 Thus, the stability of the lacunary anion relative to the Keggin anion, is higher for molybdogermanates than for molybdosilicates, which leads to a higher number of sandwich derivatives for molybdogermanates.197
At pH above 2, the stability of {XMo12} anions can be increased by the addition of organic solvents.198 Heteropolyanions with protonated amino acids as cations can be stable up to pH 6.2, which can provide valuable insights for correct application of these POMos in medical usage.199
In general, complexes based on lacunary HPOMos are more unstable in aqueous solution than, for example, their tungsten analogs,197 and there is still insufficient data on lacunary HPOMos stability in solution. Heteropolymolybdates follow a generally accepted trend – the substitution of addenda atoms with transition metals in Keggin clusters reduces the net charge and stabilizes therewith the anions (Table 8). Thus, monosubstituted germanomolybdates [GeIVMoVI11M(H2O)O39]6− (M = CoII, NiII and MnII, have the same structure as unsubstituted {GeMo12} see Fig. 14C) and stay intact in aqueous solution between pH 4 and 10.200
| HPOMs species | Stable in the pH region | Analysis method | Fingertip characteristics | Ref. |
|---|---|---|---|---|
| a The data for this table were searched in May 2020 in the scientometric databases Scopus and Web of Science with key words: “sandwich polyoxometalate” and “solution stability”. | ||||
| HPOMos based on lacunary forms of {PMo 12 } | ||||
| [M(PVMoVI11O39)2]10− (M = CeIV, ThIV) | 1–5 | 31P NMR | δ(31P) = 2.55 ppm for CeIV and 2.52 for ThIV | 205 |
| [M(PVMoVI12O40)(PVMoVI11O39)]6− (M = ZrIV, HfIV) | 2–5.3 | 31P NMR | δ(31P) = −3.5 ppm for HfIV and −3. 2 for ZrIV | 205 |
| HPOMos based on lacunary forms of {GeMo 12 } | ||||
| [GeIVMoVI11M(H2O)O39]6− (M = CoII, NiII and MnII) | 4–10 | UV-vis | Absorption at 207, 231 and 568 nm (only for CoII) | 200 |
| [CuII2(β-Y-GeIVMoVI9O33)2]12− | 4.5–10 | UV-vis | Absorption at 208 and 233 nm | 206 |
| [NiII4(H2O)2(α-B-GeIVMoVI9O34)2]12− | 3–10 | UV-vis | Absorption at 206 and 232 nm | 206 |
| HPOTs based on lacunary forms of {PW 12 } | ||||
| [NiII(H2O)PVWVI11O39]5− | pH 4 (at least for 4 h) | ESI-MS and UV-vis | m/z = 683.5 (for anion with net charge −4) | 207 |
| [H2PVWVI9NiII4O34(OH)3(H2O)6]2− | At pH 6 converted to [NiII(H2O)PVWVI11O39]5− within 7 h; at pH < 3 unstable | UV-vis | Shoulder located between 280 and 260 nm | 208 |
| [Na2(UVIO2)2(PVWVI9O34)2]12− | At pH 7; at pH 6 stable for 4 days; at pH 8.1 30% decomposes after 7 days; at pH 9.4 full decomposition in 3 h | 31P and 183W NMR | δ(31P) = −9.7 ppm; δ(183W) = −124.6, −141.1, −148.7, −182.2, −209.6 | 209 |
| [CoII4(H2O)2(α-PVWVI9O34)2]10− | 6–10 | ESI-MS and UV-vis | m/z = 1202.6 for [CoII4(PVWVI9O34)2HNa5]4− | 210 |
| [CoII9(H2O)6(OH)3(HPVO4)2(PVWVI9O34)3]16− | 5.5–11 | SAXS | — | 56 |
| HPOTs based on lacunary forms of {SiW 12 } | ||||
| [NiII(H2O)SiIVWVI11O39]6− | At pH 4 (at least for 4 h) | ESI-MS and UV-vis | m/z = 684.3 for anion with net charge −4 | 207 |
| [CoII(H2O)SiIVWVI11O39]6− | 6–10 | ESI-MS and UV-vis | m/z = 1406.6 for [(CoIISiIVWVI11O39)K2H2]2− | 210 |
| α- and β-[SiIVWVI9O37(M(H2O))3]10− (M = CoII, NiII, MnII, CuII) | 5–10 | UV-vis | No data provided | 211 |
| α- and β-[SiIVWVI9O37(M(H2O))3]7− (M = FeIII, AlIII, GaIII) | 5–10 | UV-vis | No data provided | 211 |
| HPOTs based on lacunary forms of {GeW 12 } | ||||
| [NiII(H2O)GeIVWVI11O39]6− | pH 4 (at least for 4 h) | ESI-MS and UV-vis | m/z = 694.8 (for anion with net charge −4) | 207 |
| HPOTs based on lacunary {As III W 9 } | ||||
| [Na{2-((CH3)2HNCH2)C6H4SbIII}AsIII2WVI19O67(H2O)]10− | 3–7 (at least for 5 h) | UV-vis, 183W NMR | Absorption bands at 200 and 250–254 nm; δ(183W) = −87.2, −95.1, −102.9, −103.3, −105.9, −138.1, −141.1, −157.8, −174.3 ppm | 24 |
3.3.2.5. Hydrolytic stability of HPOMos with Anderson structure. HPOMos with Anderson structure can be synthesized from aqueous solution between pH 4 and 5 and are stable in near neutral aqueous media.1,201 There are only a few papers discussing the hydrolytic stability of the Anderson POMos with AlIII,202 CrIII,203 NiII
17 and GaIII204 as a central heteroatom (Fig. 16A and B). UV-vis studies revealed that [CrIII(OH)6MoVI6O18]3− is stable in a fairly narrow pH range of 4.50 to 5.83 (Fig. 17).203 In solutions with an aluminum concentration of 1 mM and ratio [Mo]/[Al] between 0.1 and 1, [AlIII(OH)6MoVI6O18]3− is the predominate species over IPOMos (octa- and heptamolybdate) between pH 2 and 5 (Fig. 17).202 According to potentiometric and 27Al NMR data, the formation constant for this species is lgKc = 50.95 ± 0.04 in 0.6 M NaCl medium at 25 °C based on reaction (4):| Al3+ + 6[MoVIO4]2− + 6H+ ⇆ [AlIII(OH)6MoVI6O18]3− | (4) |
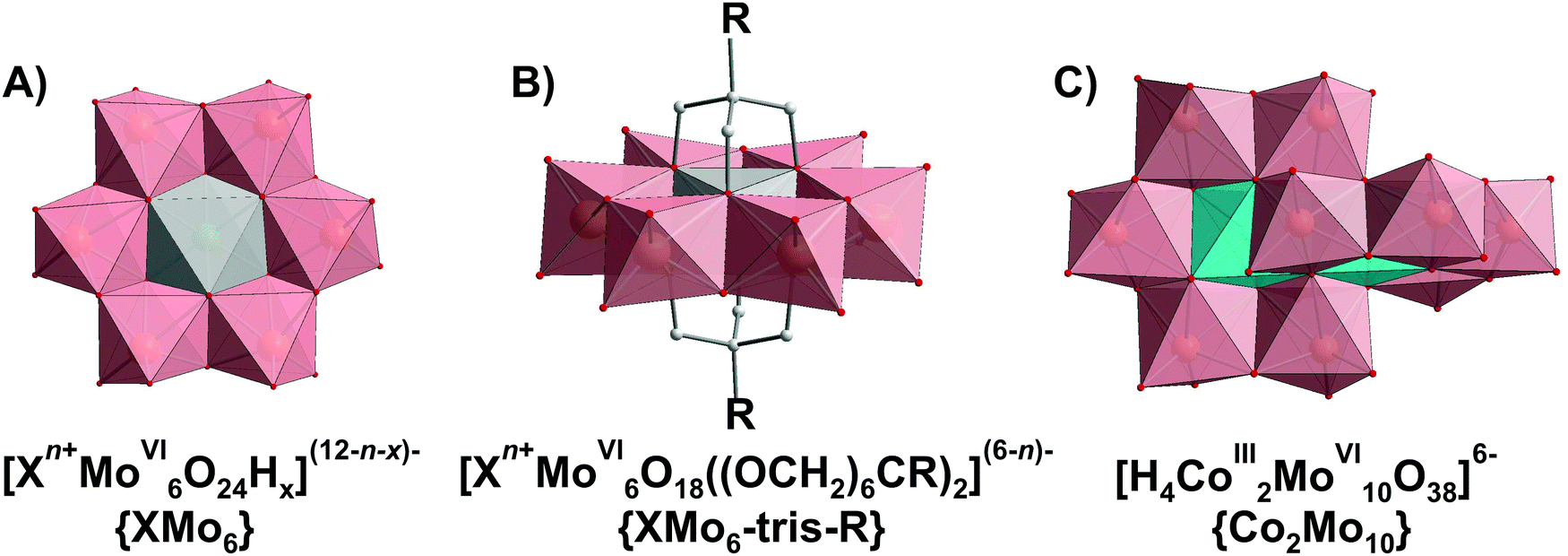 | ||
| Fig. 16 HPOMos with Anderson and Anderson-like strucutures: (A) Anderson type anion [Xn+MoVI6O24Hx](12−n−x)−, where X – heteroion (e.g. NiII,17 AlIII202 or CrIII203), n = 2–6, x = 0–6; (B) functionalized with tris-alkoxo ligands Anderson type anion [Xn+MoVI6O18((OCH2)6CR)2](6−n)−, where X – heteroion (e.g. GaIII204), n = 2–3, R = –NH2, CH2OH, –CH3etc.; (C) Anderson-like decamolybdocobaltate [H4CoIII2MoVI10O38]6−. Color code: {MoO6}, pink; {XO6}, grey; {CoO6}, light blue; C, light grey; O, red. | ||
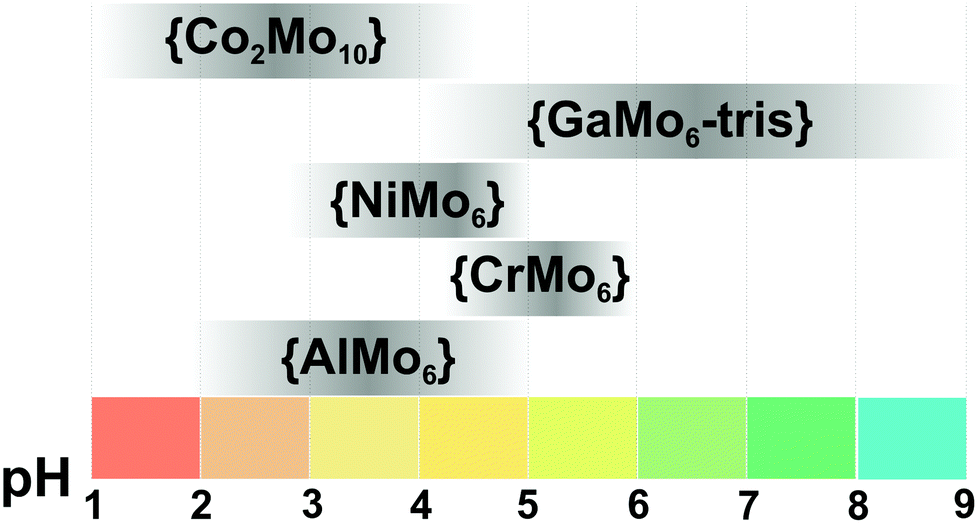 | ||
| Fig. 17 Hydrolytic stability of Anderson based HPOMos based on ref. 17, 202–204 and 212. The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. {AlMo6}–[AlIII(OH)6MoVI6O18]3−, {CrMo6}–[CrIII(OH)6MoVI6O18]3−; {NiMo6}–[NiII(OH)6MoVI6O18]4−; {GaMo6-tris}–[GaIIIMoVI6O18(OH)3{(OCH2)3CCH2OH}]3−; {Co2Mo10}–[H4CoIII2MoVI10O38]6−. | ||
The [NiII(OH)6MoVI6O18]4− {NiMo6} complex forms in 0.1 M NaNO3 medium at 25 °C according to reactions (5) and (6) with formation constants:
| Ni2+ + 6[MoVIO4]2− + 6H+ ⇆ [NiII(OH)6MoVI6O18]4−, lgKc = 44.9 ± 0.06, | (5) |
| Ni2+ + 6[MoVIO4]2− + 7H+ ⇆ H[NiII(OH)6MoVI6O18]3−, lgKc = 48.9 ± 0.10 | (6) |
Potentiometric studies reveal that upon acidification, the [NiII(OH)6MoVI6O18]4− {NiMo6} complex is firstly protonated (Z = n(H+)/n([MoO4]2−) = 1–1.17) (Fig. 1B):
| [NiII(OH)6MoVI6O18]4− + H+ ⇆ H[NiII(OH)6MoVI6O18]3−, lgKc = 4.00, | (7) |
| 7[NiII(OH)6MoVI6O18]4− + 12H+ ⇆ 6H[MoVI7O24]5− + 7Ni2+ + 24H2O, lgKc = 36.1, | (8) |
| [NiII(OH)6MoVI6O18]4− + 6OH− = NiIIMoVIO4↓ + 5[MoVIO4]2− + 6H2O. | (9) |
The described decomposition pathway could be principally applied to Anderson HPOMos with different di- and trivalent ions. Noteworthy, the organic functionalization can significantly increase the stability of Anderson HPOMos. Thus, the alkoxy-functionalized Anderson gallatomolybdate (Fig. 16B) is reported by ESI-MS to be stable up to pH 9 (Fig. 17).204 The stability of the Anderson-like decamolybdocobaltate [H4CoIII2MoVI10O38]6− (Fig. 16C) has been verified by Raman and UV-vis spectroscopies and showed that the Co-containing HPOMos is the predominant species between pH 1 and 4.5 (Fig. 17).212
3.4. Polyoxotungstates (POTs)
Polyoxotungstate species are overall more stable than their molybdate analogues due to the enthalpy factor of the condensation process when the tetrahedrally coordinated WVI or MoVI ions expand their coordination spheres from four to six during POM formation reaction.1–3 Part of this effect can be rationalized in terms of the slightly larger force constant of the W–O bonding as compared to that of the Mo–O bonding.213 Owing to the existence of a number of simultaneous equilibria, some of which are very slow to establish, and the metastability of some anions, speciation in isopolytungstates (IPOTs) solutions has not been studied as extensively as in MoVI or VV systems. However, the solution stability of the phosphotungstate anion [PVWVI12O40]3− ({PW12}, Fig. 2B) was investigated as thoroughly as no other POM species.383.4.1.1. Ion-distribution diagram for IPOTs. Despite the similarity of the solid monomeric precursors for MoVI and WVI, differences do occur in the course of the aggregation reactions, while only one type of isopolyspecies, [MVI7O24]6− (M = MoVI and WVI), is present in both systems and all others vary. The speciation in acidified aqueous solution of [WVIO4]2− has been studied predominantly by potentiometry,128,214–216 Raman spectroscopy,26,28183W, 17O and 1H NMR spectroscopy,217,218 and lately by ESI-MS.19,219 When an aqueous solution of tungstate is acidified, condensation reactions lead to the formation of IPOT anions, of which the following types are certainly present in solution and in the solid phase (Table 9): heptatungstate (paratungstate A) [WVI7O24]6− ({W7}, Fig. 18A); paratungstate B [WVI12O40(OH)2]10− ({W12O42}, Fig. 18B); α-metatungstate [H2WVI12O40]6− ({W12O40}, Fig. 18C) and decatungstate [WVI10O32]4− ({W10}, Fig. 18D). Studies of how the pH affects aqueous solutions of [WVIO4]2− are difficult to perform, partly due to very slow equilibria, and they are often problematic to interpret. Notably, the age of the solution and the total WVI concentration (aggregation does not occur in highly diluted solutions) plays a role.
| IPOT species | p, qa | pKa | Isolated in solid state | δ(183W),b ppm | δ(17O),b ppm | Formation constantsa lgKc at 25 °C |
||
|---|---|---|---|---|---|---|---|---|
| Cruywagen214c |
Rozantsev215d |
Rozantsev216e |
||||||
|
a Stoichiometric coefficients and formation constants for: p[WVIO4]2− + qH+ [HzWVIpOm]n− + (q/2 − z/2) H2O.
b
δ
183W relative to Na2WO4, δ17O relative to H2O.
c Ionic strength μ = 1 M (NaCl).
d
μ = 0.4 M (KNO3).
e
μ = 0.1–0.5 M (NaNO3).
|
||||||||
| [WVIO4]2− | 1, 0 | Yes | ||||||
| [HWVIO4]− | 1, 1 | 4.6214 | No | |||||
| [WVI6O20(OH)]5− | 6, 7 | No | ||||||
| [WVI6O20(OH)2]6− | 6, 6 | No | 49.01 | 51.66 (1.29) | 53.68 (0.28) | |||
| [WVI7O24]6− (Fig. 18A) | 7, 8 | Yes218 | 268, −106, −189217 | 71, 254, 295, 314, 595, 590, 624, 648218 | 65.19 | |||
| H[WVI7O24]5− | 7, 9 | 4.77214 | Yes221 | 69.96 | 72.31 (1.81) | 76.59 (0.7) | ||
| [WVI12O40(OH)2]10− (Fig. 18B) | 12, 14 | Yes222 | −109, −114, −116, −147217 | 115.38 | 119.45 (2.99) | 123.38 (1.27) | ||
| H[WVI12O40(OH)2]9− | 12, 16 | 4.59217 | Yes223 | |||||
| H2[WVI12O40(OH)2]8− | 12, 16 | 129.01 (3.23) | ||||||
| H3[WVI12O40(OH)2]7− | 12, 17 | 133.70 (3.34) | ||||||
| α-[H2WVI12O40]6− (Fig. 18C) | 12, 18 | Yes224 | −123217 | 80, 415, 418, 723217 | 136.98 (3.42) | 149.59 (0.7) | ||
| β-[H2WVI12O40]6− | 12, 18 | No | −112, −127, −136217 | 61, 397, 442, 725217 | ||||
| [WVI10O32]4− (Fig. 18D) | 10, 16 | Yes225 | −105, −118217 | 129.63 (0.98) | ||||
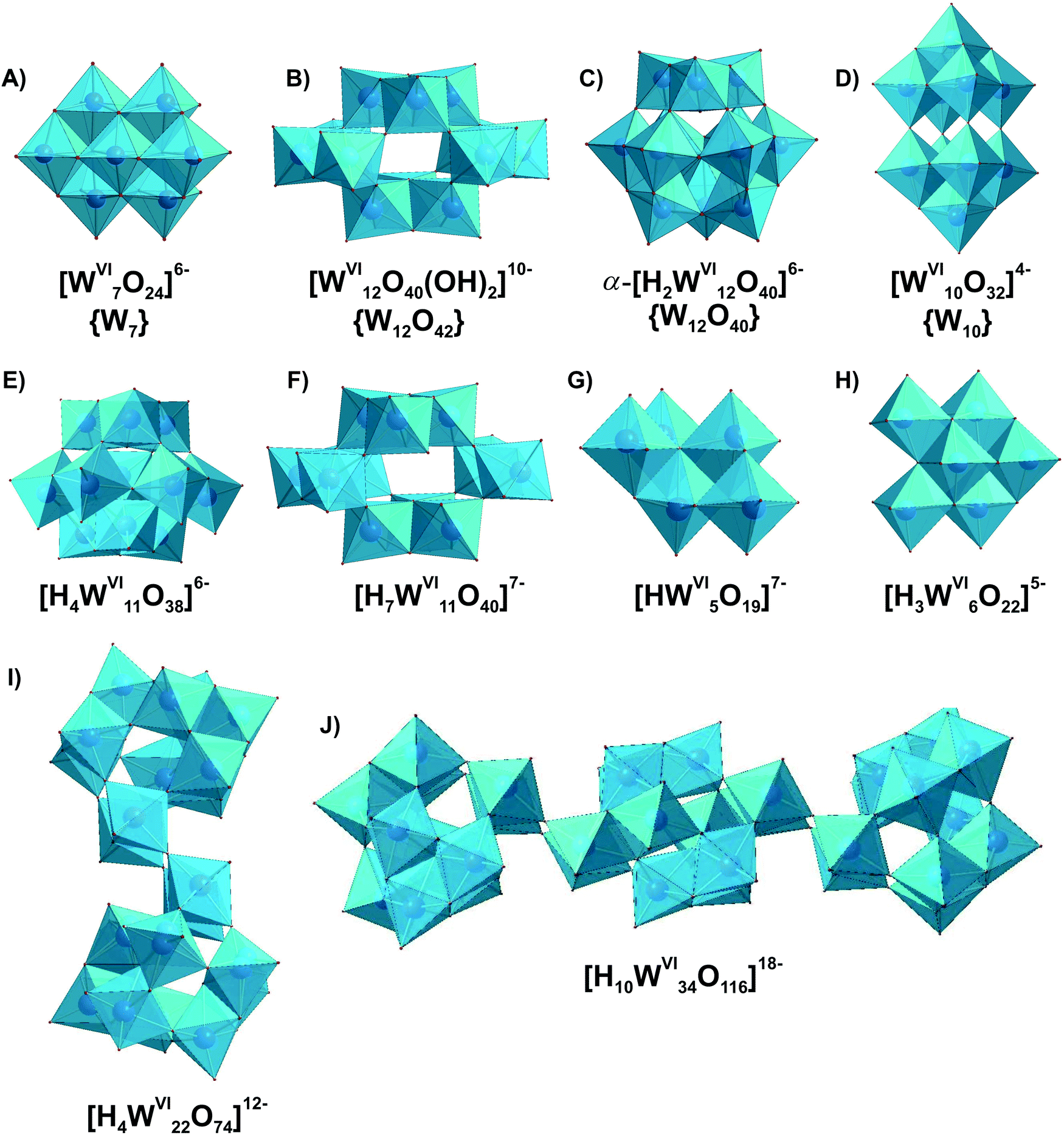 | ||
| Fig. 18 IPOTs with nuclearity up to 34 addenda atoms, which can be present and/or isolated from aqueous media. Color code: {WO6}, pink; O, red. | ||
NMR studies confirm that above pH 5, the major polytungstate species are heptatungstate {W7} (Fig. 18A) and paratungstate B {W12O42} (Fig. 18B), which have been isolated in numerous salts (Fig. 19 and Table 9). At pH lower than 5, equilibria are established very slowly, and most IPOTs anions are metastable, with the exception of the metatungstate anion α-[H2WVI12O40]6− (Fig. 18C). Acidification of paratungstate solutions leads to “pseudometatungstates”, which ultimately form the stable metatungstate anion (Fig. 18C). From solutions suggested to contain “pseudometatungstates”, the salt K6H4[WVI11O38]·11H2O, with an anion of Cs symmetry (Fig. 18E), has been crystallized.220 However, the 183W NMR spectrum of the similar acidified paratungstate solution shows a species with 11 resonances of equal intensity, which has tentatively been assigned to [H7WVI11O40]7−, a lacunary form of paratungstate-B (Fig. 18F), and also two lines attributed to decatungstate {W10} (Fig. 18D), once known as tungstate-Y, a species that is stabilized in nonaqueous solvents.
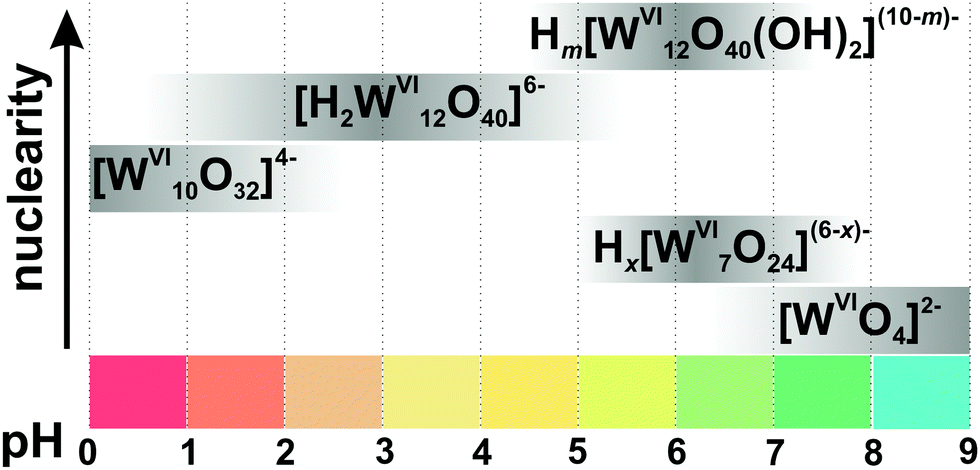 | ||
| Fig. 19 Speciation of isopolyotungstates in an aqueous solution with the concentration of 0.01–0.1 M WVI based on works.132,215,217,218 The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. x in Hx[WVI7O24](6−x)− is 0, 1; m in Hm[WVI12O40(OH)2](10−m)− between 0 and 3. The structures of species are presented in Fig. 18. The 183W and 17O chemical shifts, stoichiometry, formation constants and pKa values are summarized in Table 9. | ||
3.4.1.2. Aging of tungstate solution. Conversions of paratungstates. It is a well-known experimental fact that, from aged fully equilibrated tungstate solutions close to neutral and slightly acidic pH (Fig. 19), the isolation of paratungstate B {W12O42} (Fig. 18B) prevails.218 Potentiometric studies in the [WVIO4]2− – H+ system with different aging times (0–20
000 min from the preparation) show that heptatungstate (Fig. 18A) and possibly unstable hexatungstate transform over time into protonated forms of paratungstate B (Fig. 18B), the existence of which was proven by crystallization,223 according to eqn (10)–(12):215| 2[WVI6O20(OH)2]6− + H2O H[WVI12O40(OH)2]9− + 3OH−, lgKc = −11.87; | (10) |
| 1.71[HWVI7O24]5− + 1.43H2O H2[WVI12O40(OH)2]8− + 0.57OH−, lgKc = −2.93; | (11) |
| 1.71[HWVI7O24]5− + 2.43H2O H3[WVI12O40(OH)2]7− + 1.57OH−, lgKc = −12.24. | (12) |
Taking together the results obtained by ESI-MS, 183W NMR and Raman spectroscopy, it must be concluded that the heptatungstate (Fig. 18A) anion is the main species in freshly prepared solutions under nearly neutral conditions (pH = 5.8 and 6.8).19 During aging of Na6[WVI7O24] solutions, paratungstate B, Na10[WVI12O40(OH)2], crystallizes within an hour after preparation. However, 183W NMR shows a very low concentration of paratungstate B {W12O42} (Fig. 18B) in neutral to slightly acidic solutions, indicating that after conversion paratungstate B tends to crystallize immediately.218 The reverse transition is also possible as upon prolonged heating of a Na10[WVI12O40(OH)2] solution, the paratungstate B (Fig. 18B) anions partly transform to [WVI7O24]6− (Fig. 18A). To conclude, heptatungstate {W7} (Fig. 18A) is more stable in solution, while paratungstate B {W12O42} (Fig. 18B) is more likely to crystallize from solution.
3.4.1.3. Metatungstate [H2WVI12O40]6− in solution. α-Keggin metatungstate has been reported in 4 protonation states [HnWVI12O40](8−n)− (n = 1–4), in which the ‘‘non-acidic” protons occupy the central tetrahedral cavity (Fig. 18C). Launay reported the formation of a monoprotonated species, [H1WVI12O40]7− during the reduction of the diprotonated anion [H2WVI12O40]6−.226 The NMR spectra (183W, 17O, 1H) also reveal a conversion of paratungstate B, [WVI12O40(OH)2]10− (Fig. 18B) to the monoprotonated Keggin anion, [H1WVI12O40]7−, which leads to the formation of di-protonated [H2WVI12O40]6− (Fig. 18C) upon further acidification. The existence of tri-protonated [H3WVI12O40]5− or tetra-protonated [H4WVI12O40]4− has not been widely accepted, because the third or fourth proton is regarded as an external acidic proton. Thus, uncertainty still remains about the external (acidic) and internal (non-acidic) location of the protons. Under non-aqueous or mixed water–solvent conditions, [H3WVI12O40]5− and [H4WVI12O40]4− were reported, with all three or four protons assigned to the internal cryptand tetrahedral cavity.227–229 Although [H3WVI12O40]5− is not formed in aqueous WVI (0.05–0.50 M) solutions, the correct choice of countercation (e.g. tetrabuthylammonium) can lead to its isolation from aqueous WVI (0.05 M) solutions at pH 4.230
3.4.1.4. Comparison of IPOTs in solution and in the solid state. Very slow equilibrium reactions in tungstate solutions result in very long crystallization times and low yields. Along with the IPOTs dominating in acidified aqueous tungstate solution, {W7} (Fig. 18A), {W12O42} (Fig. 18B), {W12O40} (Fig. 18C) and {W10} (Fig. 18D) (Table 9), the following IPOTs could be crystallized. Salts containing fragments of the heptatungstate structure, Na5[H3WVI6O22]231 (Fig. 18H) and K7[HWVI5O19]232 (Fig. 18G), have been isolated from concentrated weakly alkaline aqueous tungstate solutions, but there is no evidence that these anions are stable in solution. These penta- and hexatungstates contain {WO6} octahedra with free octahedra faces showing three terminal oxygen atoms in an unstable fac configuration (Fig. 18H and G). Obviously, [H3WVI6O22]5− and [HWVI5O19]7− react immediately upon dissolution to become a more stable, more condensed heptatungstate ion (Fig. 18A), and therefore might exist in such low concentrations that they cannot be detected experimentally.218
Concentrated tungstate solutions with c([WVIO4]2−) = 1.5 M were used to generate two high nuclearity derivatives of [H4WVI11O38]6− (Fig. 18E), [H4WVI22O74]12− (Fig. 18I) and [H10WVI34O116]18− (Fig. 18J) at pH 3.4 and 2.4, respectively in the presence of Na2SO3.233 [H4WVI22O74]12− consists of two [H4WVI11O38]6− fragments, and in the case of [H10WVI34O116]18− two undecatungstates are connected via one paratungstate B cluster {W12O42} (Fig. 18B). So far, the stability of high nuclearity species was only demonstrated in the gas phase with ESI-MS. The synthetic procedures for synthesizing these IPOTs require the presence of the sulfite anion, and control experiments without sulfite do not allow the isolation of the compounds in high yields. Once again, not only pH and acidity affect the speciation of POMs, but also the substance present in the medium including choice of countercation, aging time and concentration.
3.4.1.5. IPOTs in non-aqueous solutions. The Lindqvist [WVI6O19]2− (isostructural with the corresponding niobate Fig. 8A) and decatungstate anions {W10} (Fig. 18D) are well characterized in non-aqueous solutions. The {W10} complex becomes stable in the presence of many organic solvents such as acetonitrile, 1,4-dioxane, methanol, dimethylsulfoxide and N,N-dimethylformamide.230 All IPOTs that are stable in non-aqueous media, which are α- and β-{W12O40}, {W10} and [WVI6O19]2−, are known to be easily reducible.57
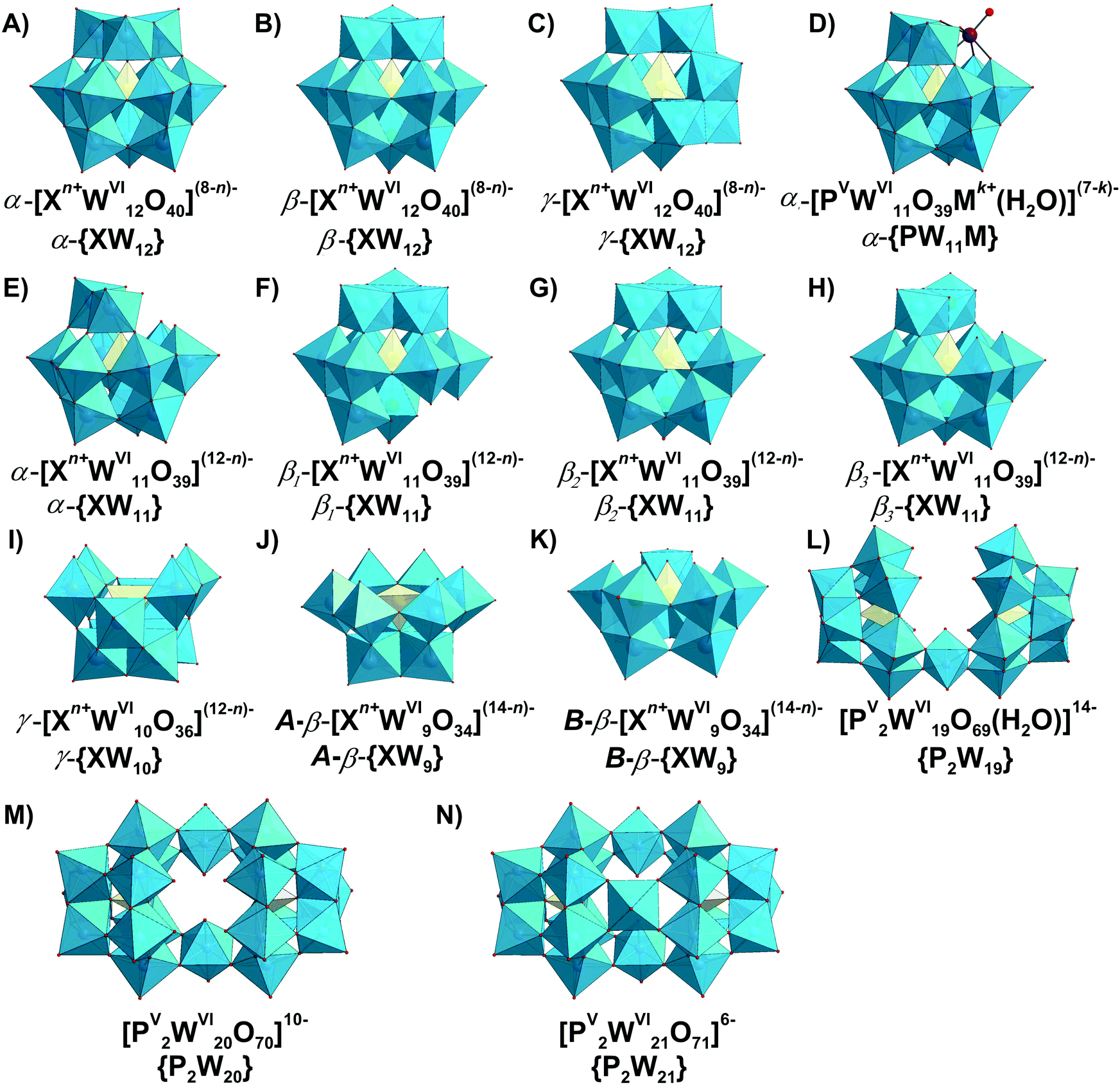 | ||
| Fig. 20 HPOTs with Keggin ([Xn+WVI12O40](8−n)−, where X = PV, SiIV, AlIIIetc.) and lacunary Keggin structures, which can be present and/or isolated from aqueous media ((A)–(N)). In (D) [PVWVI11O39Mk+(H2O)](7−k)− Mk+ is Li+, Na+, K+, or [WVIO2]2+. Color code: {WO6}, blue; {XO4}, yellow; M, purple; O, red. | ||
3.4.2.1. Hydrolytic stability of HPOTs with Keggin structure. The [XWVI12O40]n− anions (Fig. 20A–C) begin to disintegrate with decrease in acidity via interactions with hydroxyls. The stability towards the base hydrolysis increasing with the anion net charge as follows: [PVWVI12O40]3− {PW12} (pH < 1.5); [SiIVWVI12O40]4− {SiW12} (pH < 4.5); [BIIIWVI12O40]5− {BW12} and [AlIIIWVI12O40]5− {AlW12} (pH < 7); [BeIIWVI12O40]6− {BeW12} (pH < 9) (Fig. 22).1,234 Decomposition of the Keggin structure, like its formation, begins with disintegration of the [XWVI12O40]n− anions by the removal of one W
O4+ unit accompanied with the formation of a monolacunary derivative [XWVI11O39](n−4)− (Fig. 20E–H), followed by formation of di- or trilacunary anions (Fig. 20I–K).
{PW12}. The hydrolytic conversions of {PW12} (Fig. 20A) have been studied for more than a century and most recently were thoroughly reviewed by Maksimovskaya and Maksimov.38 The analysis of 95 previously published investigations made it possible to propose the most complete currently existing hydrolysis scheme for the “main” anion of POM chemistry (Fig. 21).38
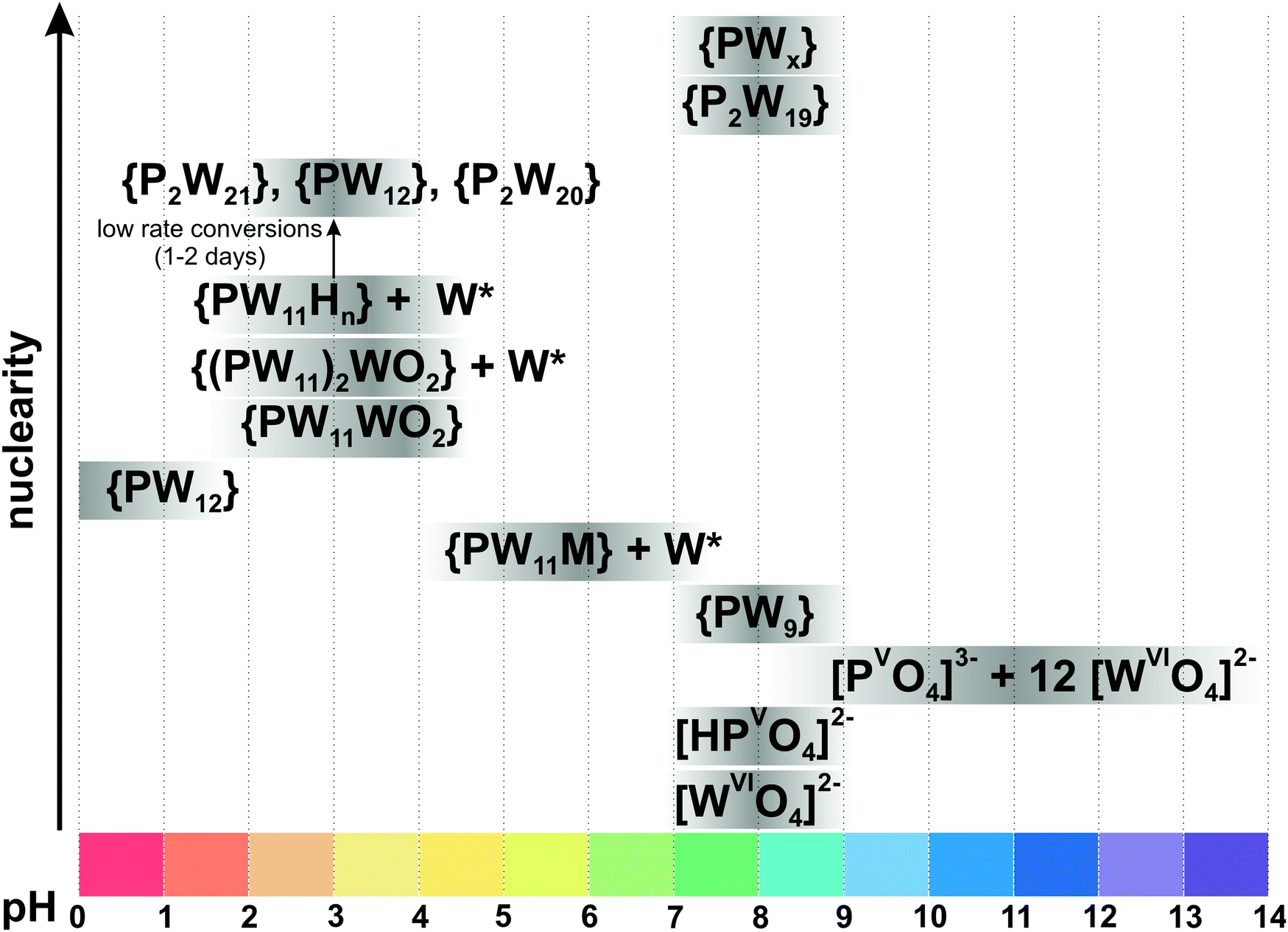 | ||
| Fig. 21 Hydrolytic conversions of H3[PVWVI12O40] (c = 0.01–0.1 M, [W]/[P] = 12) alkalized by MOH (M = Li+, Na+, K+).38 The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. The abbreviation {PW11M} corresponds to the formula [PVWVI11O39M]6−, where M = Li+, K+, Na+; W* – [H2WVI12O40]6− or [WVI12O40(OH)2]10−; {PW11WO2} – [PVWVI11O39(WVIO2)]5−; {(PW11)2WO2} – [(PVWVI11O39)2WVIO2]12−; {HnPW11} – [HnPVWVI11O39](7−n)−, n = 2–3; {P2W21} – [PV2WVI21O71]6−; {P2W20} – [PV2WVI20O70]10−; {PW9} – A-α-, A-β-, B-α-[PVWVI9O34]9−; {P2W19} – [PV2WVI19O69(H2O)]14−; {PWx} – unidentified intermediate species. The structures of species are presented in Fig. 14 and 20. The 31P chemical shifts and stoichiometry are summarized in ref. 38, and no formation constants have been reported for phosphotungstates. | ||
A few explanatory remarks need to be added to this scheme, while a detailed discussion is given in ref. 38. Systematical 31P and 183W NMR studies reveal that {PW12} exists in strongly acidic solutions with pH lower than 1.5 and converts to [PVWVI11O39M]6− (M = Li+, Na+, K+, Fig. 20D) through the intermediates [HnPVWVI11O39](7−n)− (Fig. 20E), n = 2–3 and [PVWVI11O39(WVIO2)]5− (Fig. 20D). In concentrated solutions ([WVI] > 0.01 M) the latter {PW11} (Fig. 20E) anions slowly transform into several more stable species including [PV2WVI21O71]6− (Fig. 20N) and [PV2WVI20O70]10− (Fig. 20M).235 At pH higher than 7 three isomers of the trilacunary anion A-α-, A-β-, B-α-[PVWVI19O34]9− (see isostructural molybdates for α-isomers in Fig. 14E and F and β-forms Fig. 20J and K) and [PV2WVI19O69]14−235 (Fig. 20L), an unstable derivative of A-α-{PW9}, are present.
The gas-phase fragmentation of {PW12} in 50:50 acetonitrile–H2O medium leads to pairs of product ions of general formulas [WVIxO3x+1]2− and [PVWVI12−xO39−3x]− (x = 6–9), with the most prominent pair being [WVI6O19]2− (see analog structure Fig. 8A) and [PVWVI6O21]−.236 The formation and stability of the compact hexatungstate anion is not surprising in mixed water-organic solvent solution and was observed also during ESI-MS investigations of IPOTs solutions.19
{SiW12}. The α-isomer of [SiIVWVI12O40]4− (Fig. 20A) is stable in dilute solutions below pH 4.5, but the β- and γ-isomer (Fig. 20B and C) are metastable, slowly converting to the α-form (Fig. 23).1 In their pioneering work on the hydrolysis of α-[SiIVWVI12O40]4− ({SiW12}), Kepert and Kyle reported that the reaction of silicotungstate with excess base proceeded via three distinct stages, with the intermediates being α-[SiIVWVI11O39]8− (Fig. 20E) and [SiIVWVI9O34]10− (see isostructural molybdates Fig. 14E and F), to finally produce [SiIVO4]4− and [WVIO4]2− (Fig. 23).237183W NMR spectroscopic studies confirmed the formation of monolacunary species and its Na-substituted analog α-[NaSiIVWVI11O39]7− (Fig. 20D) in the pH range of 4–7.5 together with corresponding IPOTs {W7}, {W12O42}, {W12O40} (Fig. 19).238,239 The β-{SiW12} complex upon alkalization forms three short-lived isomers of the β-[SiIVWVI11O39]8− anion (Fig. 20F–H),239β1 (Cs symmetry), β2 (C1), and β3 (Cs), and the isomerization pathway is irreversible in solution and finally yields α-[SiIVWVI11O39]8− (Fig. 20E).
 | ||
| Fig. 22 pH stability regions of intact α-Keggin anions {XW12} with selected heteroatoms. | ||
 | ||
| Fig. 23 Hydrolytic conversions of α-, β- and γ-[SiIVWVI12O40]4− ({SiW12}, [W] = 1.5 × 10−5–0.1 M, [W]/[Si] = 12) alkalized by NaOH based on works.237–240 The maximum intensity of grey color in each box with a single species corresponds to its maximum concentration in the chosen pH region. The grey boxes along the y-axis are positioned according to increasing nuclearity, but do not show the domination over other species at a certain pH range. The abbreviation α-{SiW11} corresponds to the mixture of α-[SiIVWVI11O39Na]7− and α-[SiIVWVI11O39]8−; β-{SiW11} – to the mixture of three isomers β1-, β2-, β3-[SiIVWVI11O39]8−; α-{SiW9} – α-[SiIVWVI9O34]10−; β-{SiW9} – β-[HSiIVWVI9O34]9−; γ-{SiW10} – γ-[HSiIVWVI10O36]7−. The time-dependent conversions of β and γ forms to more stable α isomers are shown by arrows. The structures of species are presented in Fig. 14 and 20. No formation constants have been reported for silicotungstates. | ||
Above pH 8, the trilacunary anions α-[SiIVWVI9O34]10− (see isostructural molybdates Fig. 14E and F) and β-[HSiIVWVI9O34]9− (Fig. 20J and K) originate from their monolacunary analogs. Fresh aqueous solutions of α-[SiIVWVI9O34]10− (see isostructural molybdates Fig. 14E and F) between pH 9 and 10 are metastable, leading to monomeric silicate and tungstate (Fig. 23).238
γ-[SiIVWVI12O40]4− (Fig. 20C) is stable only in non-aqueous or mixed water–ethanol or dioxane media. Upon dissolution of γ-{SiW12} in acidic aqueous media (pH < 4), only its α- and β-forms were detected.240 At pH between 4 and 7, the highly reactive241γ-[HSiIVWVI10O36]7− (Fig. 20I) is present in freshly prepared solutions (Fig. 23). Then the β- and finally the α-isomer of {SiW11} are formed slowly by reaction of γ-{SiW10} with the IPOT species ({W7} or {W12O42}), which was shown by cyclic voltammetry240 and ESI-MS.242 The polarogram of the γ-{SiW10} solution at pH between 4 and 6 shows one wave, which can be tentatively attributed to the intermediate species γ-[HxSiIVWVI11O39](8−x)− in low concentration.240 Even though the γ-11-tungstosilicate polyanion cannot be obtained in a pure state, it has been identified as a subunit of a more complex structure [H2SiIV2WVI23O78]8−.240 Another labile γ lacunary complex has been determined by ESI-MS as an intermediate during the transformation from β-{SiW11} to γ-{SiW10}.243 The unexpected labile precursor γ-{SiW9} is capable of a direct β → γ isomerization via a rotational transformation (Fig. 23).243
{AlW12}. Weinstock et al. comprehensively investigated formation, isomerization, and derivatization of [AlIIIWVI12O40]5− {AlW12} (Fig. 20A) using 27Al and 183W NMR spectroscopy, UV-vis and cyclic voltammetry.41,244 Contrary to {PW12} and {SiW12}, β-forms of {AlW12} (Fig. 20B) and {AlW11} (Fig. 20F–H) are much more stable. Acidification of [WVIO4]2−, followed by addition of AlIII at pH 7, leads to the formation of β-[AlIII(AlIIIOH2)WVI11O39]6− {Al2W11}, with the α derivative as a minor product with very low yield. Further acid addition to pH 0 and refluxing resulted in H5[AlIIIWVI12O40] mostly in its β-form with the α-isomer as a minor product.244 Prolonged (5 days) heating of the β-{Al2W11} solution at pH of 0–0.2 and 100 °C resulted in a mixture containing 92% α-{AlW12} (Fig. 20A) and 8% β-{Al2W11} (Fig. 20B).41 The introductory data on pH stability of some sandwich-like HPOTs archetypes is described in (Table 8).
3.4.2.2. The use of speciation data to understand the biological and catalytic activities of HPOTs with Keggin structure. Apparently, in a physiological pH range as required for most biological investigations, the low charge clusters, [PVWVI12O40]3− and [SiIVWVI12O40]4− (Fig. 20A), should not be considered as intact species which will undergo at least partial hydrolysis. Due to their good synthetic accessibility they are often used for biological applications without performing careful speciation studies under the applied physiological buffer conditions. A rational approach for the use of Keggin type POTs under physiological conditions has been demonstrated by our group.245,246 The combination of 12Be, 27Al, 29Si, 31P and 183W NMR spectroscopies allowed to determine that the enzymatic inhibition behavior of the α-Keggin POTs against AbPPO4247,248 in citric buffer at pH 6.8 is charge-dependent, revealing significant inhibitory effects for POTs with lower net charge ([PVWVI12O40]3− and [SiIVWVI12O40]4− (Fig. 20A)) counterbalanced by their pronounced hydrolysis. A recent theoretical study by Carbo and co-authors also demonstrates that Keggin anions with lower net charge have higher affinity towards proteins.249
The selective peptide bond hydrolysis for human serum albumin, oxidized insulin chain B, myoglobin and cytochrome c by a range of Ce(IV)-substituted and Zr(IV)-substituted Lindqvist, Keggin and Wells–Dawson POMs was investigated by Parac–Vogt's group along with speciation in solution detailed in her recent review.250 For instance, a combination of low-temperature 31P DOSY NMR spectroscopy and theoretical calculations allowed authors to conclude that in an aqueous solution with pD 6.4 the Keggin-type dimer [{α-PVWVI11ZrIV(μ-OH)(H2O)}2]8− used as enzyme mimetic for the hydrolysis of the phosphoester bonds in the DNA model substrate BNPP is present in equilibrium with its monomeric form.251 In the proposed reaction mechanism, BNPP initially coordinates to monomeric {ZrIVPVWVI11} in a monodentate fashion, which results in hydrolysis of the first phosphoester bond in BNPP and formation of nitrophenyl phosphate.
Many reactions applying POM catalysts are conducted in pure water,7 and so the hydrolytic stability must be taken into account. A good example is given by Neumann and co-authors investigating a sandwich Krebs type HPOT Na12[WVIZnIIZnII2(H2O)2(ZnIIWVI9O34)2] (two trilacunary Keggin fragments connected via “belt” of Zn and W ions) as a catalyst for selective alcohol oxidation with hydrogen peroxide in water–alcohol reaction media.252,253 Using 183W NMR under reaction conditions (1 mmol of substrate (various alcohols), 5 mmol of H2O2, 1 mL of water, 85 °C) the presence of intact anion was proved. The performed speciation analysis allowed the authors to determine the active POT species present under the chosen conditions and to suggest a catalytic mechanism.
Supporting the results of solution speciation using theoretical calculations can significantly improve understanding the reaction mechanism. Thus, the results of 31P-NMR-monitoring of mixed-metal Keggin [γ-PVVV2WVI10O40]5− protonation in CH3CN together with DFT calculations of the oxygen transfer process during the aromatic hydroxylation reaction allowed to determine a peroxo complex [γ-PVVV2WVI10O38(O2)]3− as the most active form of the catalyst.254
To illustrate a thorough speciation analysis of POM catalyst during water oxidation, six spectroscopic, scattering, and chemical experiments were used to indicate the stability of [{RuIV4O4(OH)2(H2O)4}(γ-SiIVWVI10O36)2]10− in solution and under catalytic turnover conditions.255 UV-vis, cyclic voltammetry, 183W and 29Si NMR spectroscopy confirmed the stability of anion in aqueous solution at pH 3–4 up to 5 months, while SAXS and DLS in photocatalysis solution (Ru(bipy)32+, 0.5–1.0 mM; Na2S2O8, 2.5–10 mM; [{RuIV4O4(OH)2(H2O)4}(γ-SiIVWVI10O36)2]10−, 1.25–10 μM; sodium phosphate buffer, 20–50 mM; pH 7.2) did not show any particles indicative of RuO2 or other possible decomposition products. Here we emphasize once again that a combination of different methods is the best strategy to get a broad picture of the POM solution behavior in aqueous media.
3.4.2.3. Hydrolytic stability of HPOTs with Wells–Dawson structure. Some aspects of the hydrolytic stability of {X2W18} (Fig. 24) are highlighted in a general review about compounds with Wells–Dawson structure.256 The most common representative of Wells–Dawson HPOTs, [PV2WVI18O62]6− ({P2W18}, Fig. 24A), shows stability at pH lower than 6, and its alkaline degradation leads to the formation of three different lacunary species: [PV2WVI17O61]10− ({P2W17}, Fig. 24B), [PV2WVI15O56]12− ({P2W15}, Fig. 24C) and H2[PV2WVI12O48]12− ({P2W12}, Fig. 24D).257 The {P2W12} species can further assemble to a very stable crown-like POT [H7PV8WVI48O184]33−, which is stable from pH 1 to 8. Since no detailed investigation of speciation was reported for {P2W18}, the hydrolytic conversion is presented as a scheme without exact pH ranges (Scheme 1).
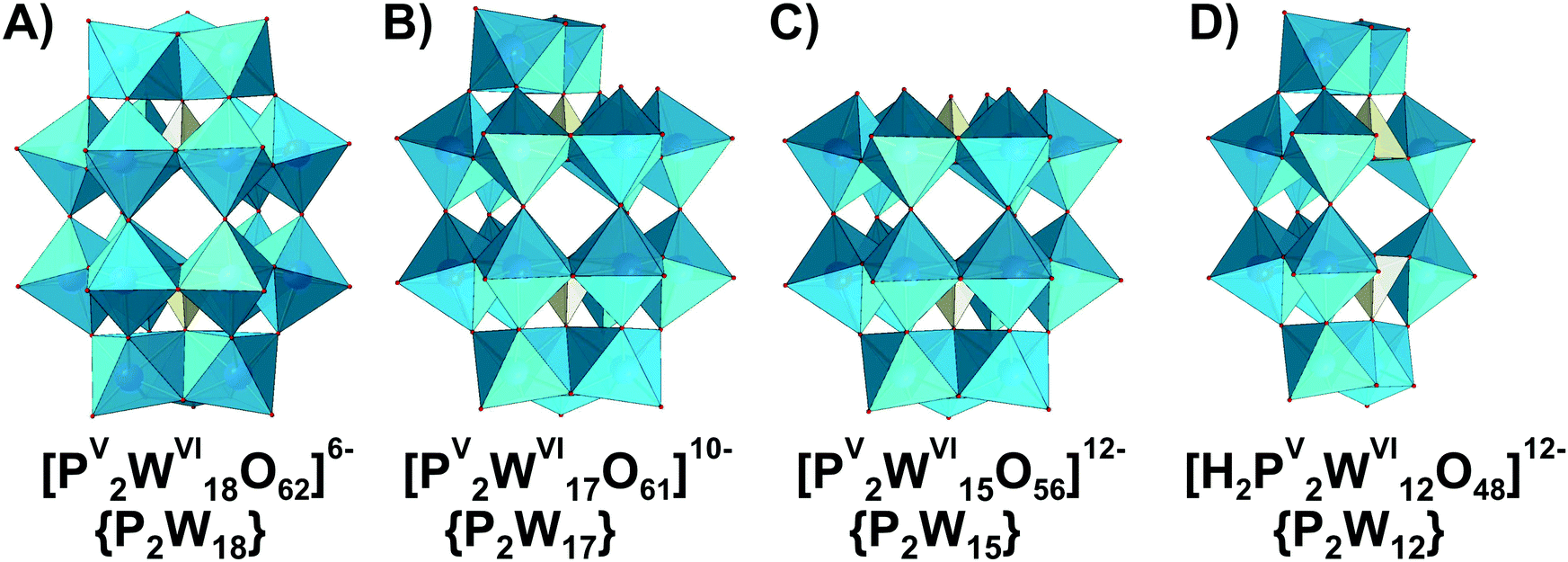 | ||
| Fig. 24 Phosphotungstates with intact (A) and lacunarized (B–D) α-Wells–Dawson structure. Color code: {WO6}, blue; {PO4}, yellow; O, red. | ||
Contant and Thouvenot studied the isomerism and stability of Wells–Dawson tungstates with PV and AsV as heteroions.258 As for the Keggin anions, β- and γ-isomers have structures in which one and two {W3} groups, respectively, have been rotated by 60°. Structures in which the central {W6} belts are staggered rather than eclipsed are denoted with an asterisk, and out of those, only the γ*-isomer has been confirmed.1 Spectroscopic (IR, Raman, UV-vis, 183W and 31P NMR), polarographic and kinetic measurements showed that the α isomers are the only species stable in aqueous solution and all other forms isomerize to it.258 All isomers of {X2W18} (X = PV and AsV) under alkaline hydrolysis firstly form monolacunary compounds. The rate of hydrolysis follows the trend for countercations K+ > Na+ > Li+ and for isomers α < β < γ < γ*.
3.4.2.4. The use of speciation data to understand the biological activities of HPOTs with Dawson structure. The ability of {P2W18} (Fig. 24A), {P2W12} (Fig. 24D) and {P2W15} (Fig. 24C) to affect aquaporin-3 (AQP3) activity and impair melanoma cell migration has been recently tested.259 The careful speciation investigation of respective POT clusters at physiological pH 7.4 (phosphate-buffered saline medium; NaCl, 137 mmol L−1; KCl, 2.7 mmol L−1; Na2HPO4, 10 mmol L−1; KH2PO4, 1.8 mmol L−1) and in water by 31P NMR analysis to address cluster species responsible for interaction in solution was performed. Solutions of the most promising AQP3 inhibitor, {P2W18}, contain intact Wells–Dawson anions, along with the mono-lacunary {P2W17}, while in the spectra of {P2W12} and {P2W15} only mono-lacunary species are present in PBS buffer. Although more investigations are needed to establish structure–activity relationships, it can be assumed that it is the intact phosphotungstate {P2W18} that is responsible for the observed inhibitory effect on AQP3 activity and melanoma cell migration.
3.4.2.5. Hydrolytic stability of HPOTs with Anderson structure. Like their molybdate analogues, {XW6} Anderson archetype (see Fig. 16A for {XMo6} analog) compounds can be synthesized from slightly acidified solutions and are stable in near neutral pH range.1,201 However, the data on Anderson HPOTs stability is very limited and only systems with NiII, CrIII and TeVI have been investigated. The potentiometric studies show that [NiII(OH)6WVI6O18]4− ({NiW6}) forms in 0.1 M NaNO3 medium at 25 °C according to reaction (13) with formation constant:
| Ni2+ + 6[WVIO4]2− + 6H+ ⇆ [NiII(OH)6WVI6O18]4− lgKc = 55.9 ± 0.2, | (13) |
After protonation of {NiW6} in acidified solutions it hydrolyzed to paratungstate B {W12O42} (Fig. 18B) (Z = n(H+)/n([WO4]2− = 1.17) with the release of free NiII ions reaction (14):17
| [NiII(OH)6WVI6O18]4− + H[NiII(OH)6WVI6O18]3− + H+ ⇆ [WVI12O40(OH)2]10− + 2Ni2+ + 6H2O lgKc = 1.9 | (14) |
During the investigation of numerous successful co-crystallizations of [TeVIWVI6O24]6− ({TeW6}) with different proteins, it was proven that [TeVIW6O24]6− is stable in a pH range from 4.5 to 7.5.260–263 In the presence of aurone synthase CgAUS264 {TeW6} was even functionalized by carboxylic fragment of protein glutamic acid on the μ2 oxygen atoms, which proves the reactivity of Anderson POTs and demonstrates a special kind of flexibility with respect to both geometric and functional properties.262 All these examples show the importance of speciation analysis in physiological systems in the presence of biomacromolecules. The double-side tris-functionalized [CrIII((OCH2)3CC2H5)2WVI6O18]3− is a predominant species in neutral pH between 5.7 and 7 almost without decomposition fragments, which was shown by ESI-MS.265
4. Conclusions
In the presented survey, the ion-distribution diagrams in a wide pH range (analog to “phase diagrams”) for POMs of VV, NbV, TaV, MoVI and WVI are given. They sum up our current understanding about POM stability and transformations in aqueous media. A brief timeline of POM speciation in aqueous media highlighting the first and the most important studies is given in Fig. S1 (ESI†). POM species in solution may be different to compounds that can be crystallized from the same solutions. It is advisable to use several orthogonal methods to understand and analyze the speciation. Sometimes, conclusions of the speciation differ according to the methods applied and the research method should be selected by taking into account the characteristics of a particular POM.Throughout, this review factors that affect speciation have been shown. Among the obvious, such as pH (Section 3), ionic strength (Section 3), countercations (Section 3.2.1.3), buffer composition (Sections 3.2.1.3 and 3.4.1.4), aging time (Sections 3.3.1.6 and 3.3.2.1) and physical factors as temperature (Sections 3.2.1.3 and 3.3.2.1), attention should be paid to the presence of interfering substances as macromolecules and substrates of enzymes (Sections 3.1.1.4 and 3.4.2.5). Two intrinsic features of the POMs itself affect the stability in solution: charge density and anion symmetry (Sections 3.4.1.4 and 3.4.2.2). Stability increases with increasing charge density and symmetry of the POM anion. Strategies to stabilize POMs are: partial reduction or the replacement of addenda cation MVI by a cation with lower charge.
We hope as an outcome of this review that special attention will be paid to investigation of POM behavior in solution that will facilitate the deeper understanding of their role in various applications. Reliable and accurate published data are of utmost importance for any type of POM project. For deliberate application of POMs, there is a great need for stability data obtained in non-aqueous solutions, water-organic solvent mixtures, or unusual experimental conditions (pressure, temperature, or at high concentrations), as well as in the presence of amphipathic molecules or other micelle-forming organic substances, and for complex formation processes on the surfaces of solid materials. Virtually nothing is known, about the possible role of enzymes or other physiologically occurring biological macromolecules, as well as their substrates with respect to enhancing the breakdown and/or rearrangements of POM species with some exception investigating decavanadate biomolecule interactions. We are confident that the new era of POM chemistry will be opened once the POM speciation gets the attention it deserves and analyzing the behavior of the solution will become a common tool for understanding each POM application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Austrian Science Fund (FWF): M2203 (N.G.), P27534 (A.R.), P33089 (A.R.) the University of Vienna. We thank Dr Joscha Breibeck and Ian Oesterle, MSc for proofreading of the manuscript.Notes and references
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, New York, 1983 Search PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48, DOI:10.1002/anie.199100341.
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121, 10.1039/B502666K.
- I. Knöpnadel, H. Hartl, W.-D. Hunnius and J. Fuchs, Angew. Chem., Int. Ed. Engl., 1974, 13, 823, DOI:10.1002/anie.197408231.
- H.-U. Kreusler, A. Förster and J. Fuchs, Z. Naturforsch., B: J. Chem. Sci., 1980, 35, 242–244, DOI:10.1515/znb-1980-0223.
- J. J. Cruywagen, A. G. Draaijer, J. B. B. Heyns and E. A. Rohwer, Inorg. Chim. Acta, 2002, 331, 322–329, DOI:10.1016/S0020-1693(02)00700-4.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 11, 4893–4962, DOI:10.1021/cr500390v.
- A. Bijelic and A. Rompel, Acc. Chem. Res., 2017, 50, 1441–1448, DOI:10.1021/acs.accounts.7b00109.
- A. Bijelic and A. Rompel, ChemTexts, 2018, 4, 10, DOI:10.1007/s40828-018-0064-1.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169, 10.1039/c7cc07549a.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed. 2019, 58, 2980–2999 (Angew. Chem., 2019, 131, 3008–3029), DOI:10.1002/anie.201803868, ( DOI:10.1002/ange.201803868).
- T. Kiss and A. Odani, Bull. Chem. Soc. Jpn., 2007, 80, 1691–1702, DOI:10.1246/bcsj.80.1691.
- D. C. Crans, Coord. Chem. Rev., 2017, 352, 398–400, DOI:10.1016/j.ccr.2017.11.008.
- D. M. Templeton, F. Ariese, R. Cornelis, L.-G. Danielsson, H. Muntau, H. P. van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72, 1453–1470, DOI:10.1351/pac200072081453.
- T. Kiss, É. A. Enyedy and T. Jakuscha, Coord. Chem. Rev., 2017, 352, 401–423, DOI:10.1016/j.ccr.2016.12.016.
- I. Ali and H. Y. Aboul-Enein, Instrumental methods in metal ion speciation, Taylor & Francis, Boca Raton, 2006 DOI:10.1201/9781420019407.
- N. I. Gumerova, A. V. Notich, G. M. Rozantsev and S. V. Radio, J. Solution Chem., 2016, 45, 849–860, DOI:10.1007/s10953-016-0471-0.
- R. I. Maksimovskaya and G. M. Maksimov, Inorg. Chem., 2007, 46, 3688–3695, DOI:10.1021/ic061549n.
- L. Fan, J. Cao and C. Hu, RSC Adv., 2015, 5, 83377–83382, 10.1039/c5ra18059g.
- D. C. Crans, K. A. Woll, K. Prusinskas, M. D. Johnson and E. Norkus, Inorg. Chem., 2013, 52, 12262–12275, DOI:10.1021/ic4007873.
- T. Yamase, Chem. Rev., 1998, 98, 307–325, DOI:10.1021/cr9604043.
- H. So and M. T. Pope, Inorg. Chem., 1972, 11, 1441–1443, DOI:10.1021/ic50112a061.
- G. M. Varga Jr, E. Papaconstantinou and M. T. Pope, Inorg. Chem., 1970, 9, 662–667, DOI:10.1021/ic50085a045.
- P. Yang, Z. Lin, G. Alfaro-Espinoza, M. S. Ullrich, C. I. Rat, C. Silvestru and U. Kortz, Inorg. Chem., 2016, 55, 251–258, DOI:10.1021/acs.inorgchem.5b02189.
- I. Holclajtner-Antunović, D. Bajuk-Bogdanović, M. Todorović, U. B. Mioć, J. Zakrzewska and S. Uskoković-Marković, Can. J. Chem., 2008, 86, 996–1004, DOI:10.1139/V08-138.
- P. Häufe, Fresenius' Z. Anal. Chem., 1982, 310, 388–391, DOI:10.1007/BF00483010.
- R. Q. Cabrera, S. Firth, C. S. Blackman, D.-L. Long, L. Cronin and P. F. McMillan, J. Solid State Chem., 2012, 186, 171–176, DOI:10.1016/j.jssc.2011.12.005.
- A. F. Redkin and G. V. Bondarenko, J. Solution Chem., 2010, 39, 1549–1561, DOI:10.1007/s10953-010-9595-9.
- M. Aureliano, C. A. Ohlin, M. O. Vieira, M. P. M. Marques, W. H. Casey and L. A. E. Batista de Carvalho, Dalton Trans., 2016, 45, 7391–7399, 10.1039/C5DT04176G.
- W. G. Klemperer and W. Shum, J. Am. Chem. Soc., 1976, 98, 8291–8293, DOI:10.1021/ja00441a083.
- M. A. Fedotov and R. I. Maksimovskaya, J. Struct. Chem., 2006, 47, 952–978, DOI:10.1007/s10947-006-0413-6.
- Y.-G. Chen, J. Gong and L.-Y. Qu, Coord. Chem. Rev., 2004, 248, 245–260, DOI:10.1016/j.cct.2003.11.003.
- A. S. Tracey, G. R. Willsky and E. S. Takeuchi, Vanadium: Chemistry, Biochemistry, Pharmacology and Practical Applications, CRC Press, 2007, p. 200 Search PubMed.
- D. C. Crans, P. K. Shin and K. B. Armstrong, Adv. Chem., 1996, 246, 303–328, DOI:10.1021/ba-1995-0246.ch011.
- M. T. Pope and G. M. Varga, J. Chem. Soc., Chem. Commun., 1966, 653–654, 10.1039/C19660000653.
- M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Inorg. Chem., 1979, 18, 93–103, DOI:10.1021/ic50191a021.
- C. A. Ohlin and W. H. Casey, Annu. Rep. NMR Spectrosc., 2018, 94, 187–248, DOI:10.1016/bs.arnmr.2018.01.001.
- R. I. Maksimovskaya and G. M. Maksimov, Coord. Chem. Rev., 2019, 385, 81–99, DOI:10.1016/j.ccr.2019.01.014.
- R. I. Maksimovskaya and G. M. Maksimov, Inorg. Chem., 2011, 50, 4725–4731, DOI:10.1021/ic101996f.
- M. Bugnola, R. E. Schreiber, Y. Kaufman, G. Leitus, L. J. W. Shimon and R. Neumann, Eur. J. Inorg. Chem., 2019, 482–485, DOI:10.1002/ejic.201800552.
- J. J. Cowan, A. J. Bailey, R. A. Heintz, B. T. Do, K. I. Hardcastle, C. L. Hill and I. A. Weinstock, Inorg. Chem., 2001, 40, 6666–6675, DOI:10.1021/ic0106120.
- P. Klonowski, J. C. Goloboy, F. J. Uribe-Romo, F. Sun, L. Zhu, F. Gándara, C. Wills, R. J. Errington, O. M. Yaghi and W. G. Klemperer, Inorg. Chem., 2014, 53, 13239–13246, DOI:10.1021/ic502617k.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612, DOI:10.1002/anie.201905600.
- J. Xie, H. A. Neal, J. Szymanowski, P. C. Burns, T. M. Alam, M. Nyman and L. Gagliardi, Inorg. Chem., 2018, 57, 5514–5525, DOI:10.1021/acs.inorgchem.8b00474.
- M. Nyman, F. Bonhomme, T. M. Alam, J. B. Parise and G. M. B. Vaughan, Angew. Chem., Int. Ed., 2004, 43, 2787–2792, DOI:10.1002/anie.200353410.
- D. J. Sures, S. A. Serapian, K. Kozma, P. I. Molina, C. Bo and M. Nyman, Phys. Chem. Chem. Phys., 2017, 19, 8715–8725, 10.1039/C6CP08454K.
- H. N. Miras, E. F. Wilson and L. Cronin, Chem. Commun., 2009, 1297–1311, 10.1039/B819534J.
- C. S. Truebenbach, M. Houalla and D. M. Hercules, J. Mass Spectrom., 2000, 35, 1121–1127, DOI:10.1002/1096-9888(200009)35:9<1121::AID-JMS40>3.0.CO;2-7.
- C. A. Ohlin, Chem. – Asian J., 2012, 7, 262–270, DOI:10.1002/asia.201100763.
- M. Nyman, Coord. Chem. Rev., 2017, 352, 461–472, DOI:10.1016/j.ccr.2016.11.014.
- M. Nyman and L. Fullmer, Small angle X-ray scattering of group V polyoxometalates, Trends in Polyoxometalates Research, Nova Science Publishers Inc., 2015, Book chapter pdf: ISBN 978-163482693-8; 978-163482656-3 Search PubMed.
- A. A. Shmakova, V. V. Volchek, V. Yanshole, N. B. Kompankov, N. P. Martin, M. Nyman, P. A. Abramov and M. N. Sokolov, New J. Chem., 2019, 43, 9943–9952, 10.1039/C9NJ01907C.
- M. Dufaye, S. Duval, G. Stoclet and T. Loiseau, Eur. J. Inorg. Chem., 2019, 4500–4505, DOI:10.1002/ejic.201900732.
- P. Yin, B. Wu, E. Mamontov, L. L. Daemen, Y. Cheng, T. Li, S. Seifert, K. Hong, P. V. Bonnesen, J. K. Keum and A. J. Ramirez-Cuesta, J. Am. Chem. Soc., 2016, 138, 2638–2643, DOI:10.1021/jacs.5b11465.
- P. Yin, T. Li, R. S. Forgan, C. Lydon, X. Zuo, Z. N. Zheng, B. Lee, D. Long, L. Cronin and T. Liu, J. Am. Chem. Soc., 2013, 135, 13425–13432, DOI:10.1021/ja404777g.
- S. Goberna-Ferrón, J. Soriano-López, J. R. Galán-Mascarós and M. Nyman, Eur. J. Inorg. Chem., 2015, 2833–2840, DOI:10.1002/ejic.201500404.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112, DOI:10.1038/s41570-018-0112.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–237, DOI:10.1021/cr960403a.
- T. Ueda, ChemElectroChem, 2018, 5, 823–838, DOI:10.1002/celc.201701170.
- G. Bunker, Introduction to XAFS, Cambridge University Press, Cambridge, 2010 Search PubMed.
- M. P. M. Marques, D. Gianolio, S. Ramos, L. A. E. Batista de Carvalho and M. Aureliano, Inorg. Chem., 2017, 56, 10893–10903, DOI:10.1021/acs.inorgchem.7b01018.
- B. C. Bostick, J. Sun, J. D. Landis and J. L. Clausen, Environ. Sci. Technol., 2018, 52, 1045–1053, DOI:10.1021/acs.est.7b05406.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351, DOI:10.1016/j.jconrel.2016.06.017.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909–933, DOI:10.1021/cs4011716.
- E. E. Hamilton, P. E. Fanwick and J. J. Wilker, J. Am. Chem. Soc., 2002, 124, 78–82, DOI:10.1021/ja010820r.
- J. Fuchs, S. Mahjour and J. Pickardt, Angew. Chem., Int. Ed. Engl., 1976, 15, 374–375, DOI:10.1002/anie.197603741.
- V. W. Day, W. G. Klemperer and O. M. Yaghi, J. Am. Chem. Soc., 1989, 111, 4518–4519, DOI:10.1021/ja00194a068.
- P. Y. Zavalij, T. Chirayil, M. S. Whittingham, V. K. Pecharsky and R. Jacobson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 170–171, DOI:10.1107/S0108270196013480.
- V. W. Day, W. G. Klemperer and O. M. Yaghi, J. Am. Chem. Soc., 1989, 111, 5959–5961, DOI:10.1021/ja00197a077.
- D. Hou, K. S. Hagen and C. L. Hill, J. Am. Chem. Soc., 1992, 114, 5864–5866, DOI:10.1021/ja00040a061.
- D. Hou, K. S. Hagen and C. L. Hill, J. Chem. Soc., Chem. Commun., 1993, 426–428, 10.1039/C39930000426.
- D. C. Crans, Pure Appl. Chem., 2005, 77, 1497–1527, DOI:10.1351/pac200577091497.
- L. Pettersson, Mol. Eng., 1993, 3, 29–42, DOI:10.1007/bf00999622.
- D. K. Walanda, R. C. Burns, G. A. Lawrance and E. I. von Nagy-Felsobuki, Inorg. Chem. Commun., 1999, 2, 487–489, DOI:10.1016/S1387-7003(99)00129-X.
- D. K. Walanda, R. C. Burns, G. A. Lawrance and E. I. von Nagy-Felsobuki, Inorg. Chim. Acta, 2000, 305, 118–126, DOI:10.1016/S0020-1693(00)00122-5.
- D. C. Crans, C. D. Rithner and L. A. Theisen, J. Am. Chem. Soc., 1990, 112, 2901–2908, DOI:10.1021/ja00164a009.
- I. Andersson, L. Pettersson, J. J. Hastings and O. W. Howarth, J. Chem. Soc., Dalton Trans., 1996, 3357–3361, 10.1039/DT9960003357.
- A. S. Tracey, J. S. Jaswal and S. J. Angus-Dunne, Inorg. Chem., 1995, 34, 5680–5685, DOI:10.1021/ic00126a043.
- K. Elvingson, A. González Baró and L. Pettersson, Inorg. Chem., 1996, 35, 3388–3393, DOI:10.1021/ic951195s.
- D. C. Crans, B. Baruah and N. E. Levinger, Biomed. Pharmacother., 2006, 60, 174–181, DOI:10.1016/j.biopha.2006.02.006.
- R. Gopal and C. Calvo, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2491–2493, DOI:10.1107/S0567740874007400.
- S. Nakamura and T. Ozeki, J. Chem. Soc., Dalton Trans., 2001, 472–480, 10.1039/B008128K.
- V. W. Day, W. G. Klemperer and D. J. Maltbie, J. Am. Chem. Soc., 1987, 109, 2991–3002, DOI:10.1021/ja00244a022.
- Vanadium and its role for life; metal ions in biological systems, ed. A. Sigel and H. Sigel, New York, Marcel Dekker, 1995 DOI:10.1016/0307-4412(95)90238-4.
- N. McCann, M. Wagner and H. Hasse, Dalton Trans., 2013, 42, 2622–2628, 10.1039/c2dt31993d.
- J. W. Larson, J. Chem. Eng. Data, 1995, 40, 1276–1280, DOI:10.1021/je00022a030.
- E. Heath and O. W. Howarth, J. Chem. Soc., Dalton Trans., 1981, 1105–1110, 10.1039/DT9810001105.
- D. C. Crans, B. J. Peters, X. Wu and C. C. McLauchlan, Coord. Chem. Rev., 2017, 344, 115–130, DOI:10.1016/j.ccr.2017.03.016.
- O. W. Howarth and M. Jarrold, J. Chem. Soc., Dalton Trans., 1978, 503–506, 10.1039/DT9780000503.
- M. Aureliano and D. C. Crans, J. Inorg. Biochem., 2009, 103, 536–546, DOI:10.1016/j.jinorgbio.2008.11.010.
- S. S. Soares, F. Henao, M. Aureliano and C. Gutiérrez-Merino, Chem. Res. Toxicol., 2008, 21, 607–618, DOI:10.1021/tx700204r.
- R. M. C. Gândara, S. S. Soares, H. Martins, C. Gutiérrez-Merino and M. Aureliano, J. Inorg. Biochem., 2005, 99, 1238–1244, DOI:10.1016/j.jinorgbio.2005.02.023.
- T. L. Turner, V. H. Nguyen, C. C. McLauchlan, Z. Dymon, B. M. Dorsey, J. D. Hooker and M. A. Jones, J. Inorg. Biochem., 2012, 108, 96–104, DOI:10.1016/j.jinorgbio.2011.09.009.
- P. Csermely, A. Martonosi, G. C. Levy and A. J. Ejchart, Biochem. J., 1985, 230, 807–815, DOI:10.1042/bj2300807.
- S. S. Soares, C. Gutiérrez-Merino and M. Aureliano, J. Inorg. Biochem., 2007, 101, 789–796, DOI:10.1016/j.jinorgbio.2007.01.012.
- M. Aureliano, Dalton Trans., 2009, 9093–9100, 10.1039/b907581j.
- S. Ramos, M. Manuel, T. Tiago, R. Duarte, J. Martins, C. Gutiérrez-Merino, J. J. G. Moura and M. Aureliano, J. Inorg. Biochem., 2006, 100, 1734–1743, DOI:10.1016/j.jinorgbio.2006.06.007.
- M. Aureliano, G. Fraqueza and C. A. Ohlin, Dalton Trans., 2013, 42, 11770–11777, 10.1039/c3dt50462j.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902, DOI:10.1021/cr020607t.
- A. Al-Qatati, F. L. Fontes, B. G. Barisas, D. Zhang, D. A. Roess and D. C. Crans, Dalton Trans., 2013, 42, 11912–11920, 10.1039/C3DT50398D.
- N. Samart, Z. Arhouma, S. Kumar, H. A. Murakami, D. C. Crick and D. C. Crans, Front. Chem., 2018, 6, 519, DOI:10.3389/fchem.2018.00519.
- N. Fukuda and T. Yamase, Biol. Pharm. Bull., 1997, 20, 927–930, DOI:10.1248/bpb.20.927.
- J. M. Messmore and R. T. Raines, Arch. Biochem. Biophys., 2000, 381, 25–30, DOI:10.1006/abbi.2000.1951.
- A. Selling, I. Andersson, L. Pettersson, C. M. Schramm, S. L. Downey and J. H. Grate, Inorg. Chem., 1994, 33, 3141–3150, DOI:10.1021/ic00092a021.
- I. Andersson, A. Gorzsás, C. Kerezsi, I. Tóth and L. Pettersson, Dalton Trans., 2005, 3658–3666, 10.1039/B508273K.
- W. Zhang, T. Zhang, G. Lv, X. Cao and H. Zhu, Sep. Purif. Technol., 2019, 218, 164–172, DOI:10.1016/j.seppur.2019.02.025.
- R. Kato, A. Kobayashi and Y. Sasaki, Inorg. Chem., 1982, 21, 240–246, DOI:10.1021/ic00131a045.
- K. Postal, D. F. Maluf, G. Valdameri, A. L. Rüdiger, D. L. Hughes, E. L. de Sá, R. R. Ribeiro, E. M. de Souza, J. F. Soares and G. G. Nunes, RSC Adv., 2016, 6, 114955–114968, 10.1039/c6ra15826a.
- M. Nyman, Dalton Trans., 2011, 40, 8049–8058, 10.1039/c1dt10435g.
- I. Lindqvist, Ark. Kemi, 1953, 5, 247–250 Search PubMed.
- E. J. Graeber and B. Morosin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2137–2143, DOI:10.1107/S0567740877007900.
- C. A. Ohlin, E. M. Villa and W. H. Casey, Inorg. Chim. Acta, 2009, 362, 1391–1392, DOI:10.1016/j.ica.2008.06.006.
- G. M. Rozantsev, O. I. Dotsenko and G. V. Taradina, Russ. J. Coord. Chem., 2000, 26, 247–253 Search PubMed.
- E. M. Villa, C. A. Ohlin, E. Balogh, T. M. Anderson, M. D. Nyman and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 4844–4846, DOI:10.1002/anie.200801125.
- W. G. Klemperer and K. A. Marek, Eur. J. Inorg. Chem., 2013, 1762–1771, DOI:10.1002/ejic.201201231.
- M. R. Antonio, M. Nyman and T. M. Anderson, Angew. Chem., Int. Ed., 2009, 48, 6136–6140, DOI:10.1002/anie.200805323.
- M. Matsumoto, Y. Ozawa, A. Yagasaki and Y. Zhe, Inorg. Chem., 2013, 52, 7825–7827, DOI:10.1021/ic400864e.
- E. Balogh, T. M. Anderson, J. R. Rustad, M. Nyman and W. H. Casey, Inorg. Chem., 2007, 46, 7032–7039, DOI:10.1021/ic700845e.
- N. Etxebarria, L. A. Fernández and J. M. Madariaga, J. Chem. Soc., Dalton Trans., 1994, 3055–3059, 10.1039/DT9940003055.
- A. Goiffon, E. Philippot and M. Maurin, Rev. Chim. Miner., 1980, 17, 466–476 Search PubMed.
- M. Nyman, T. M. Alam, F. Bonhomme, M. A. Rodriguez, C. S. Frazer and M. E. Welk, J. Cluster Sci., 2006, 17, 197–219, DOI:10.1007/s10876-006-0049-x.
- C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 8251–8254, DOI:10.1002/ange.200803688.
- N. P. Martin, E. Petrus, M. Segado, A. Arteaga, L. N. Zakharov, C. Bo and M. Nyman, Chem. – Eur. J., 2019, 25, 10580–10584, DOI:10.1002/chem.201902770.
- P. Müscher-Polzin, C. Näther and W. Bensch, Z. Anorg. Allg. Chem., 2020, 646, 193–198, DOI:10.1002/zaac.201900276.
- R. P. Bontchev and M. Nyman, Angew. Chem., Int. Ed., 2006, 45, 6670–6672, DOI:10.1002/anie.20060220.
- J. R. Black, M. Nyman and W. H. Casey, J. Am. Chem. Soc., 2006, 128, 14712–14720, DOI:10.1021/ja065529w.
- D. Sures, M. Segado, C. Bo and M. Nyman, J. Am. Chem. Soc., 2018, 140, 10803–10813, DOI:10.1021/jacs.8b05015.
- K.-H. Tytko and O. Glemser, Adv. Inorg. Chem. Radiochem., 1976, 19, 239–315, DOI:10.1016/S0065-2792(08)60073-4.
- O. W. Howarth, P. Kelly and L. Patterson, J. Chem. Soc., Dalton Trans., 1990, 81–84, 10.1039/dt9900000081.
- M. Isobe, F. Marumo, T. Yamase and T. Ikawa, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 2728–2731, DOI:10.1107/S0567740878009103.
- J. Torres, L. Gonzatto, G. Peinado, C. Kremer and E. Kremer, J. Solution Chem., 2014, 43, 1687–1700, DOI:10.1007/s10953-014-0231-y.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198–11201, DOI:10.1021/jacs.8b07525.
- S. Himeno, H. Niiya and T. Ueda, Bull. Chem. Soc. Jpn., 1997, 70, 631–637, DOI:10.1246/bcsj.70.631.
- P. L. Brown, M. E. Shyinq and R. N. Sylva, J. Chem. Soc., Dalton Trans., 1987, 2149–2157, 10.1039/DT9870002149.
- K. H. Tytko, G. Baethe and J. J. Cruywagen, Inorg. Chem., 1985, 24, 3132–3136, DOI:10.1021/ic00214a010.
- F. Taube, I. Andersson and L. Pettersson, Molybdate speciation in systems related to the bleaching of kraft pulp, in Polyoxometalate chemistry from topology via self-assembly to applications, ed. M. T. Pope and A. Müller, Springer, Dordrecht, 2001, pp. 161–173 DOI:10.1007/0-306-47625-8.
- J. J. Cruywagen and J. B. B. Heyns, Polyhedron, 2000, 19, 907–911, DOI:10.1016/S0277-5387(00)00331-4.
- I. Lindqvist, Acta Crystallogr., 1950, 3, 159–160, DOI:10.1107/S0365110X50000355.
- J. Fuchs and H. Hartl, Angew. Chem., Int. Ed. Engl., 1976, 15, 375–376, DOI:10.1002/anie.197603751.
- J. Fuchs and I. Knöpnadel, Z. Kristallogr., 1982, 158, 165–179, DOI:10.1524/zkri.1982.158.12.165.
- I. Paulat-Böschen, J. Chem. Soc., Chem. Commun., 1979, 780–782, 10.1039/C39790000780.
- H. Schulz and F. A. Schröder, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 322–333, DOI:10.1107/S0567739473000902.
- K. Y. S. Ng and E. Gulari, Polyhedron, 1984, 3, 1001–1011, DOI:10.1016/S0277-5387(00)84659-8.
- A. Davantès and G. Lefèvre, J. Phys. Chem. A, 2013, 117, 12922–12929, DOI:10.1021/jp408885k.
- E. Cartuyvels, G. Absillis and T. N. Parac-Vogt, Chem. Commun., 2008, 85–87, 10.1039/b714860g.
- G. Absillis, R. Van Deun and T. N. Parac-Vogt, Inorg. Chem., 2011, 50, 11552–11560, DOI:10.1021/ic201498u.
- S. Himeno, N. Ishii, M. Hasegawa, A. Saito and T. Hori, Inorg. Chim. Acta, 1987, 131, 11–13, DOI:10.1016/S0020-1693(00)87899-8.
- K. Murata and S. Ikeda, Spectrochim. Acta, Part A, 1983, 39, 787–794, DOI:10.1016/0584-8539(83)80018-X.
- S. Watanabe, T. Sato, M. Nakaya, T. Yoshida and J. Onoe, Chem. Phys. Lett., 2019, 723, 76–81, DOI:10.1016/j.cplett.2019.02.049.
- K. Yokoi, N. Matsubayashi, T. Miyanaga, I. Watanabe and S. Ikeda, Polyhedron, 1993, 12, 911–914, DOI:10.1016/S0277-5387(00)81545-4.
- L. Vilà-Nadal, E. F. Wilson, H. N. Miras, A. Rodríguez-Fortea, L. Cronin and J. M. Poblet, Inorg. Chem., 2011, 50, 7811–7819, DOI:10.1021/ic200969h.
- M. Marianski, J. Seo, E. Mucha, D. A. Thomas, S. Jung, R. Schlögl, G. Meijer, A. Trunschke and G. von Helden, J. Phys. Chem. C, 2019, 123, 7845–7853, DOI:10.1021/acs.jpcc.8b06985.
- F. Steffler, G. Ferreira de Lima and H. Anderson Duarte, Chem. Phys. Lett., 2017, 669, 104–109, DOI:10.1016/j.cplett.2016.12.010.
- B. M. Gatehouse and P. Leverett, J. Chem. Soc. A, 1971, 2107–2112, 10.1039/j19710002107.
- B. M. Gatehouse and P. Leverett, J. Chem. Soc. A, 1968, 1398–1405, 10.1039/j19680001398.
- P. J. Zapf, R. C. Haushalter and J. Zubieta, Chem. Commun., 1997, 321–322, 10.1039/a607022A.
- S.-F. Zhang, Y.-Q. Suna and G.-Y. Yang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m299–m301, DOI:10.1107/S010827010401090X.
- J. Noack, F. Rosowski, R. Schlögl and A. Trunschke, Z. Anorg. Allg. Chem., 2014, 640, 2730–2736, DOI:10.1002/zaac.201400439.
- V. Coué, R. Dessapt, M. Bujoli-Doeuff, M. Evain and S. Jobic, Inorg. Chem., 2007, 46, 2824–2835, DOI:10.1021/ic0621502.
- H.-J. Lunk and H. Hartl, ChemTexts, 2017, 3, 13, DOI:10.1007/s40828-017-0048-6.
- A. Wutkowski, B. R. Srinivasan, A. R. Naik, C. Schütt, C. Näther and W. Bensch, Eur. J. Inorg. Chem., 2011, 2254–2263, DOI:10.1002/ejic.201001154.
- I. Böschen, B. Buss and B. Krebs, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 48–56, DOI:10.1107/S0567740874002184.
- W. T. A. Harrison, L. L. Dussack and A. J. Jacobson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1075–1077, DOI:10.1107/S0108270195015642.
- S. Chakrabarti and S. Natarajan, Cryst. Growth Des., 2002, 2, 333–335, DOI:10.1021/cg0200210.
- J. Fuchs, H. Hartl, W.-D. Hunnius and S. Mahjour, Angew. Chem., Int. Ed. Engl., 1975, 14, 644, DOI:10.1002/anie.197506441.
- T. Arumuganathan, A. Srinivasarao, T. Vijay Kumar and S. K. Das, J. Chem. Sci., 2008, 120, 95–103, https://www.ias.ac.in/article/fulltext/jcsc/120/01/0095-0103 Search PubMed.
- L. Pettersson, I. Andersson and L.-O. Öhman, Acta Chem. Scand., Ser. A, 1985, A39, 53–58, DOI:10.3891/acta.chem.scand.39a-0053.
- L. Pettersson, I. Andersson and L.-O. Öhman, Inorg. Chem., 1986, 25, 4726–4733, DOI:10.1021/ic00246a028.
- R. Strandberg, Acta Chem. Scand., 1973, 27, 1004–1018, DOI:10.3891/acta.chem.scand.27-1004.
- B. Hedman and R. Strandberg, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 278–284, DOI:10.1107/S0567740879003356.
- B. Hedman, Acta Chem. Scand., 1973, 27, 3335–3364, DOI:10.3891/acta.chem.scand.27-3335.
- S. Himeno, M. Hashimoto and T. Ueda, Inorg. Chim. Acta, 1999, 284, 237–245, DOI:10.1016/S0020-1693(98)00294-1.
- C. E. Easterly, D. M. Hercules and M. Houalla, Appl. Spectrosc., 2001, 55, 1671–1675, DOI:10.1366/0003702011953991.
- M. Hashimoto, I. Andersson and L. Pettersson, Dalton Trans., 2007, 124–132, 10.1039/b613214f.
- M. Hashimoto, I. Andersson and L. Pettersson, Dalton Trans., 2009, 3321–3327, 10.1039/B818530A.
- R. Strandberg, Acta Chem. Scand., Ser. A, 1974, 28, 217–225, DOI:10.3891/acta.chem.scand.28a-0217.
- R. Strandberg, Acta Chem. Scand., Ser. A, 1975, 29A, 350–358, DOI:10.3891/acta.chem.scand.29a-0350.
- J. A. Rob van Veen, O. Sudmeijer, C. A. Emeis and H. de Wit, J. Chem. Soc., Dalton Trans., 1986, 1825–1831, 10.1039/dt9860001825.
- L. A. Combs-Walker and C. L. Hill, Inorg. Chem., 1991, 30, 4016–4026, DOI:10.1021/ic00021a010.
- A. J. Gaunt, I. May, M. J. Sarsfield, D. Collison, M. Helliwell and I. S. Denniss, Dalton Trans., 2003, 2767–2771, 10.1039/b301995k.
- R. Strandberg, Acta Chem. Scand., Ser. A, 1975, 29, 359–364, DOI:10.3891/acta.chem.scand.29a-0359.
- L. Pettersson, Acta Chem. Scand., Ser. A, 1975, 29, 677–689, DOI:10.3891/acta.chem.scand.29a-0677.
- G. Johansson, L. Pettersson and N. Ingri, Acta Chem. Scand., Ser. A, 1978, 32, 681–688, DOI:10.3891/acta.chem.scand.32a-0681.
- X. Zhang, X. Luo, Y. Duan, Y. Huang, N. Zhang, L. Zhao and J. Wu, J. Mol. Struct., 2017, 1141, 245–251, DOI:10.1016/j.molstruc.2017.03.110.
- B. Hedman, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2241–2246, DOI:10.1107/S0567740880008503.
- B.-W. Cong, Z.-H. Su, Z.-F. Zhao, B.-Y. Yu, W.-Q. Zhao, L. Xia, X.-J. Ma and B.-B. Zhou, CrystEngComm, 2017, 19, 2739–2749, 10.1039/c7ce00319f.
- H. Ichida and Y. Sasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 529–533, DOI:10.1107/S0108270183005363.
- K. Murata and S. Ikeda, Polyhedron, 1987, 6, 1681–1685, DOI:10.1016/S0277-5387(00)80771-8.
- D. Bajuk-Bogdanović, I. Holclajtner-Antunović, M. Todorović, U. B. Mioć and J. Zakrzewska, J. Serb. Chem. Soc., 2008, 73, 197–209, DOI:10.2298/JSC0802197B.
- G. B. McGarvey and J. B. Moffat, J. Mol. Catal., 1991, 69, 137–155, DOI:10.1016/0304-5102(91)80110-O.
- A. Jürgensen and J. B. Moffat, Catal. Lett., 1995, 34, 237–244, DOI:10.1007/BF00808338.
- D. Bajuk-Bogdanović, S. Uskoković-Marković, R. Hercigonja, A. Popa and I. Holclajtner-Antunović, Spectrochim. Acta, Part A, 2016, 153, 152–159, DOI:10.1016/j.saa.2015.08.029.
- V. W. Truesdale and C. J. Smith, Analyst, 1975, 100, 203–212, 10.1039/an9750000203.
- V. W. Truesdale and C. J. Smith, Analyst, 1975, 100, 797–805, 10.1039/AN9750000797.
- M. Takahashi, Y. Abe and M. Tanaka, Talanta, 2015, 131, 301–308, DOI:10.1016/j.talanta.2014.07.079.
- H. Katano, T. Osakai, S. Himeno and A. Saito, Electrochim. Acta, 1995, 40, 2935–2942, DOI:10.1016/0013-4686(95)00225-4.
- F. Li and L. Xu, Dalton Trans., 2011, 40, 4024–4034, 10.1039/c0dt00691b.
- R. Massart, R. Contant, J.-M. Fruchart, J.-P. Ciabrini and M. Fournier, Inorg. Chem., 1977, 16, 2916–2921, DOI:10.1021/ic50177a049.
- S. Dianat, S. Tangestaninejad, B. Yadollahi, A.-K. Bordbar, M. Moghadam, V. Mirkhani and I. Mohammadpoor-Baltork, J. Mol. Liq., 2012, 174, 76–79, DOI:10.1016/j.molliq.2012.07.006.
- N. Wang, F. Li, Y. Wang, L. Xu, K. Cui and B. Xu, Eur. J. Inorg. Chem., 2013, 1699–1705, DOI:10.1002/ejic.201201137.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64, DOI:10.1016/j.ccr.2015.07.001.
- L. O. Öhman, Inorg. Chem., 1989, 28, 3629–3632, DOI:10.1021/ic00318a004.
- L. Yang, Z. Zhou, P.-T. Ma, X.-F. Zhang, J.-P. Wang and J.-Y. Niu, J. Coord. Chem., 2013, 66, 1058–1067, DOI:10.1080/00958972.2013.775429.
- A. Blazevic, E. Al-Sayed, A. Roller, G. Giester and A. Rompel, Chem. – Eur. J., 2015, 21, 4762–4771, DOI:10.1002/chem.201405644.
- R. Copping, L. Jonasson, A. J. Gaunt, D. Drennan, D. Collison, M. Helliwell, R. J. Pirttijarvi, C. J. Jones, A. Huguet, D. C. Apperley, N. Kaltsoyannis and I. May, Inorg. Chem., 2008, 47, 5787–5798, DOI:10.1021/ic800101t.
- S. Li, J. Zhao, P. Ma, J. Du, J. Niu and J. Wang, Inorg. Chem., 2009, 48, 9819–9830, DOI:10.1021/ic901330y.
- K. von Allmen, R. Moré, R. Müller, J. Soriano-López, A. Linden and G. R. Patzke, ChemPlusChem, 2015, 80, 1389–1398, DOI:10.1002/cplu.201500074.
- D. Jabbour, B. Keita, I.-M. Mbomekalle, L. Nadjo and U. Kortz, Eur. J. Inorg. Chem., 2004, 2036–2044, DOI:10.1002/ejic.200300830.
- K.-C. Kim and M. T. Pope, J. Am. Chem. Soc., 1999, 121, 8512–8517, DOI:10.1021/ja9909125.
- D. Lieb, A. Zahl, E. F. Wilson, C. Streb, L. C. Nye, K. Meyer and I. Ivanović-Burmazović, Inorg. Chem., 2011, 50, 9053–9058, DOI:10.1021/ic201243n.
- J. Liu, F. Ortéga, P. Sethuraman, D. E. Katsoulis, C. E. Costello and M. T. Pope, J. Chem. Soc., Dalton Trans., 1992, 1901–1906, 10.1039/dt9920001901.
- J. Moreau, O. Delpoux, E. Devers, M. Digne and S. Loridant, J. Phys. Chem. A, 2012, 116, 263–270, DOI:10.1021/jp208534p.
- L. C. W. Baker and D. C. Glick, Chem. Rev., 1998, 98, 3–50, DOI:10.1021/cr960392l.
- J. J. Cruywagen and I. F. J. van der Merwe, J. Chem. Soc., Dalton Trans., 1987, 1701–1705, 10.1039/DT9870001701.
- G. M. Rozantsev, S. V. Radio and N. I. Gumerova, Pol. J. Chem., 2008, 82, 2067–2080 Search PubMed.
- G. M. Rozantsev and O. I. Sazonova, Russ. J. Coord. Chem., 2005, 31, 552–558, DOI:10.1007/s11173-005-0135-x.
- J. J. Hastings and O. W. Howarth, J. Chem. Soc., Dalton Trans., 1992, 209–215, 10.1039/DT9920000209.
- R. I. Maksimovskaya and K. G. Burtseva, Polyhedron, 1985, 4, 1559–1562, DOI:10.1016/S0277-5387(00)87227-7.
- D. K. Walanda, R. C. Burns, G. A. Lawrance and E. I. von Nagy-Felsobuki, J. Cluster Sci., 2000, 11, 5–28, DOI:10.1023/A:1009098627616.
- T. Lehmann and J. Fuchs, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 89–93, DOI:10.1515/znb-1988-0116.
- X.-F. Wang, J. Cao, K.-L. Huang, Y.-Q. Xu, Y.-N. Chi and C.-W. Hu, Eur. J. Inorg. Chem., 2013, 1788–1792, DOI:10.1002/ejic.201200810.
- I. Lindqvist, Acta Crystallogr., 1952, 5, 667–670, DOI:10.1107/S0365110X52001817.
- A. L. Nolan, E. N. Wilkes, T. W. Hambley, C. C. Allen, R. C. Burns and G. A. Lawrance, Aust. J. Chem., 1999, 52, 955–964, DOI:10.1071/CH99070.
- M. Asami, H. Ichida and Y. Sasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 35–37, DOI:10.1107/S0108270184002924.
- J. Fuchs, H. Hartl and W. Schiller, Angew. Chem., Int. Ed. Engl., 1973, 12, 420, DOI:10.1002/anie.197304201.
- J.-P. Launay, M. Boyer and F. Chauveau, J. Inorg. Nucl. Chem., 1976, 38, 243–247, DOI:10.1016/0022-1902(76)80402-2.
- J. Fuchs and E.-P. Flindt, Z. Naturforsch., B: Chem. Sci., 1979, 34, 412–422, DOI:10.1515/znb-1979-0311.
- C. R. Sprangers, J. K. Marmon and D. C. Duncan, Inorg. Chem., 2006, 45, 9628–9630, DOI:10.1021/ic061370c.
- S. Himeno, M. Yoshihara and M. Maekawa, Inorg. Chim. Acta, 2000, 298, 165–171, DOI:10.1016/S0020-1693(99)00436-3.
- S. Himeno, K. Nakajima and K. Eda, Polyhedron, 2010, 29, 2595–2599, DOI:10.1016/j.poly.2010.06.002.
- H. Hartl, R. Palm and J. Fuchs, Angew. Chem., Int. Ed. Engl., 1993, 32, 1492–1494, DOI:10.1002/anie.199314921.
- J. Fuchs, R. Palm and H. Hartl, Angew. Chem., Int. Ed. Engl., 1996, 35, 2651–2653, DOI:10.1002/anie.199626511.
- H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 8420–8423, DOI:10.1002/anie.200802109.
- A. V. Anyushin, A. I. Smolentsev, D. A. Mainichev, C. Vicent, A. L. Gushchin, M. N. Sokolov and V. P. Fedin, Chem. Commun., 2014, 50, 9083–9085, 10.1039/C4CC02456G.
- C. M. Tourné and G. F. Tourné, J. Chem. Soc., Dalton Trans., 1988, 2411–2420, 10.1039/dt9880002411.
- M. T. Ma, T. Waters, K. Beyer, R. Palamarczuk, P. J. S. Richardt, R. A. J. O’Hair and A. G. Wedd, Inorg. Chem., 2009, 48, 598–606, DOI:10.1021/ic8016326.
- D. L. Kepert and J. H. Kyle, J. Chem. Soc., Dalton Trans., 1978, 137–141, 10.1039/dt9780000137.
- B. J. Smith and V. A. Patrick, Aust. J. Chem., 2002, 55, 281–286, DOI:10.1071/ch01092.
- B. J. Smith and V. A. Patrick, Aust. J. Chem., 2003, 56, 283–286, DOI:10.1071/ch01112.
- A. Tézé, J. Canny, L. Gurban, R. Thouvenot and G. Hervé, Inorg. Chem., 1996, 35, 1001–1005, DOI:10.1021/ic951004l.
- B. S. Bassil and U. Kortz, Dalton Trans., 2011, 40, 9649–9661, 10.1039/c1dt10845J.
- Q. Jia, J. Cao, Y. Duan and C. Hu, Dalton Trans., 2015, 44, 553–559, 10.1039/c4dt02723j.
- J. M. Cameron, L. Vilà-Nadal, R. S. Winter, F. Iijima, C. J. Murillo, A. Rodríguez-Fortea, H. Oshio, J. M. Poblet and L. Cronin, J. Am. Chem. Soc., 2016, 138, 8765–8773, DOI:10.1021/jacs.6b02245.
- I. A. Weinstock, J. J. Cowan, E. M. G. Barbuzzi, H. Zeng and C. L. Hill, J. Am. Chem. Soc., 1999, 121, 4608–4617, DOI:10.1021/ja982908j.
- J. Breibeck, N. I. Gumerova, B. B. Boesen, M. Galanski and A. Rompel, Sci. Rep., 2019, 9, 5183, DOI:10.1038/s41598-019-41261-7.
- J. Breibeck, A. Bijelic and A. Rompel, Chem. Commun., 2019, 55, 11519–11522, 10.1039/c9cc05818d.
- M. Pretzler, A. Bijelic and A. Rompel, Sci. Rep., 2017, 7, 1810, DOI:10.1038/s41598-017-01813-1.
- M. Pretzler and A. Rompel, Inorg. Chim. Acta, 2018, 481, 25–31, DOI:10.1016/j.ica.2017.04.041.
- A. Solé-Daura, J. M. Poblet and J. J. Carbó, Chem. – Eur. J., 2020, 26, 5799–5809, DOI:10.1002/chem.201905533.
- L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92–99, DOI:10.1016/j.copbio.2018.11.013.
- T. K. N. Luong, P. Shestakova, T. T. Mihaylov, G. Absillis, K. Pierloot and T. N. Parac-Vogt, Chem. – Eur. J., 2015, 21, 4428–4439, DOI:10.1002/chem.201405810.
- D. Sloboda-Rozner, P. L. Alsters and R. Neumann, J. Am. Chem. Soc., 2003, 125, 5280–5281, DOI:10.1021/ja0344821.
- D. Sloboda-Rozner, P. Witte, P. L. Alsters and R. Neumann, Adv. Synth. Catal., 2004, 346, 339–345, DOI:10.1002/adsc.200303156.
- I. Y. Skobelev, V. Yu. Evtushok, O. A. Kholdeeva, N. V. Maksimchuk, R. I. Maksimovskaya, J. M. Ricart, J. M. Poblet and J. J. Carbó, ACS Catal., 2017, 7, 8514–8523, DOI:10.1021/acscatal.7b02694.
- Y. V. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quiñonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360–17370, DOI:10.1021/ja907277b.
- L. E. Briand, G. T. Baronetti and H. J. Thomas, Appl. Catal., A, 2003, 256, 37–50, DOI:10.1016/S0926-860X(03)00387-9.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614, DOI:10.1021/ic00220a036.
- R. Contant and R. Thouvenot, Inorg. Chim. Acta, 1993, 212, 41–50, DOI:10.1016/S0020-1693(00)92306-5.
- C. Pimpão, I. V. da Silva, A. F. Mósca, J. O. Pinho, M. M. Gaspar, N. I. Gumerova, A. Rompel, M. Aureliano and G. Soveral, Int. J. Mol. Sci., 2020, 21, 2467, DOI:10.3390/ijms21072467.
- S. G. Mauracher, C. Molitor, R. Al-Oweini, U. Kortz and A. Rompel, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70, 263–266, DOI:10.1107/S2053230X14000582.
- S. G. Mauracher, C. Molitor, R. Al-Oweini, U. Kortz and A. Rompel, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 2301–2315, DOI:10.1107/S1399004714013777.
- C. Molitor, A. Bijelic and A. Rompel, Chem. Commun., 2016, 52, 12286–12289, 10.1039/C6CC07004C.
- A. Bijelic, C. Molitor, S. G. Mauracher, R. Al-Oweini, U. Kortz and A. Rompel, ChemBioChem, 2015, 16, 233–241, DOI:10.1002/cbic.201402597.
- C. Molitor, S. G. Mauracher and A. Rompel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1806–E1815, DOI:10.1073/pnas.1523575113.
- N. I. Gumerova, T. Caldera Fraile, A. Roller, G. Giester, M. Pascual-Borràs, A. C. Ohlin and A. Rompel, Inorg. Chem., 2019, 58, 106–113, DOI:10.1021/acs.inorgchem.8b01740.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00392a |
| This journal is © The Royal Society of Chemistry 2020 |