
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonds of halonium ions
Lotta
Turunen
 and
Máté
Erdélyi
*
and
Máté
Erdélyi
*
Department of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. E-mail: mate.erdelyi@kemi.uu.se
First published on 25th March 2020
Abstract
Due to their electron deficiency, halonium ions act as particularly strong halogen bond donors. By accepting electrons in both lobes of their empty p-orbital, they are capable of simultaneously interacting with two Lewis bases. The interaction presumes the formation of three molecular orbitals and is accordingly typically entitled as a three-center halogen bond. In analogy to the [D–H–D]+ hydrogen bonds, which are at times entitled as short and strong bonds, the [D–X–D]+ halogen bonds of halonium ions show Bondi normalized interatomic distances of 0.6–0.7 and possess both charge transfer and electrostatic characteristics. The three-center halogen bond of halonium ions shows distinct differences in its properties from coordinative bonds of transition metals and is therefore applicable as a complementary synthon in supramolecular chemistry. The three-center halogen bond modulates the reactivity of halonium ions and is hence a useful tool for synthetic organic chemistry. Following the discussion of the nature and properties of halonium ions’ halogen bonds, this tutorial review provides an overview of their current applications to stimulate future developments.
 Lotta Turunen | Lotta Turunen received her PhD in 2017 from the University of Jyväskylä, Finland, under the supervision of Prof. Kari Rissanen. In 2018, she joined the research group of Prof. Mate Erdelyi at Uppsala University, Sweden. Her current research interests are focused on three-center halogen bonds and their applications in synthetic organic chemistry. |
 Máté Erdélyi | Mate Erdelyi graduated at Semmelweis University, Hungary, and obtained his PhD in organic chemistry under the guidance of Prof. A. Gogoll (Uppsala University, Sweden, 2004). After postdoctoral research in physical organic chemistry with Prof. C. L. Perrin (University of California, USA), he moved to the Max Planck Institute for Biophysical Chemistry to learn NMR spectroscopy from Prof. C. Griesinger. Following an additional postdoctoral project in structural biology under the mentorship of Prof. T. Carlomagno, in 2009 he initiated his independent career at the University of Gothenburg, Sweden, where he was appointed professor of organic chemistry in 2017. He moved to Uppsala University in 2017, and carries out research on halogen bonding, NMR method development, natural product chemistry and medicinal chemistry. |
Introduction
Halogen bonding (XB) is the directional, non-covalent interaction of an electron-poor region of a halogen and a Lewis base.1 It resembles hydrogen bonding and has accordingly been proposed to be applicable as a complementary tool for the rational modulation of molecular recognition events in chemistry and biology.2 Being a weak interaction, halogen bonding is typically difficult to detect, especially in disordered phases.3 The past two decades of halogen bonding research has consequently focused on the investigation of strong and thus comparably easy to detect complexes, in which a large electrophilic area on the halogen, typically called a σ-hole, has been promoted by perfluorination of the nearby carbon atoms (Fig. 1a).3 An alternative to this classical approach that involves a positively charged halogen, X+, formally a halonium ion or halogen(I), as a halogen bond donor has also been introduced (Fig. 1b) recently, yet so far has scarcely been discussed.4 In a conventional halogen bond,2 D1–X⋯D, a neutral halogen, X, typically covalently attached to a carbon (D1), interacts with an electron donor, D, Lewis base. In contrast, the halonium ions, X+, tend to form three-center halogen bonds, in which they simultaneously interact with two Lewis bases, each contributing a lone pair of electrons to the interaction.4–7 This charge transfer to the “p-holes”, the empty p-orbital of X+, results in the formation of a linear [D1⋯X⋯D]+ arrangement.4 Here, the two Lewis bases, D and D1, may or may not be identical. The halogen and the two electron donors are held together by four electrons and thus form a three-center, four-electron bond that is remarkably short (RXB = 0.65–0.69, where RXB = dXB/(XvdW + BvdW))6,8 and strong (up to 180 kJ mol−1).3,9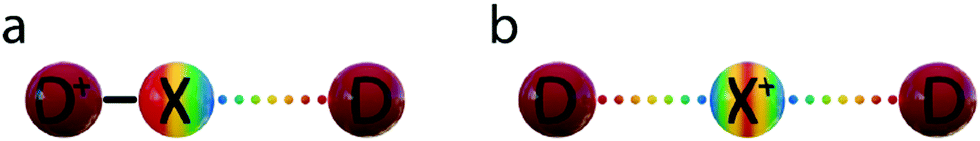 | ||
| Fig. 1 Schematic description of (a) conventional and (b) three-center halogen bonds. The former halogen, X, is neutral and is covalently bound, typically to a carbon, and has a σ-hole that may receive electron density from a Lewis base, D. The latter, X+, in contrast, has an empty p-orbital and consequently a positive charge and two partially positively charged regions, p-holes. It may receive electron density from two Lewis bases simultaneously and thereby form a three-center halogen bond in which the X⋯D interactions possess both covalent and secondary characteristics. | ||
Similar to most halogen bond complexes,2 these were initially often characterized as charge-transfer complexes.10
The main aim of this tutorial review is to summarize and interpret the past decade's studies of halonium ions’ halogen bonding, review its utility in supramolecular chemistry and discuss its emerging applicability, for instance in synthetic chemistry. Despite its short history, the investigations of halonium ions’ halogen bonds have provided unique insights into the fundamentals of chemical bonding. This bond has been proven to be among the strongest halogen bonds, and has consequently rapidly found applications.
The nature of halonium ions’ halogen bonds
Out of the four halogens, iodine(I), bromine(I) and chlorine(I) are capable of forming three-center, four-electron bonds, [D⋯X⋯D]+, whereas fluorine(I) has been demonstrated to prefer conventional halogen bonds, D+–F⋯D, with a distinct short and strong covalent D–F bond and a second, weaker and longer, D⋯F halogen bond (Fig. 2).8 Fluorine centered halogen bonds are rare and only form when fluorine is directly attached to a highly electron withdrawing functionality, such as positively charged nitrogen, which enhances its σ-hole. The former complexes may be formed with variety of Lewis bases including nitrogenous, sulphurous, selenious and tellurious electron donors, which may be of sp3, sp2 or sp character.5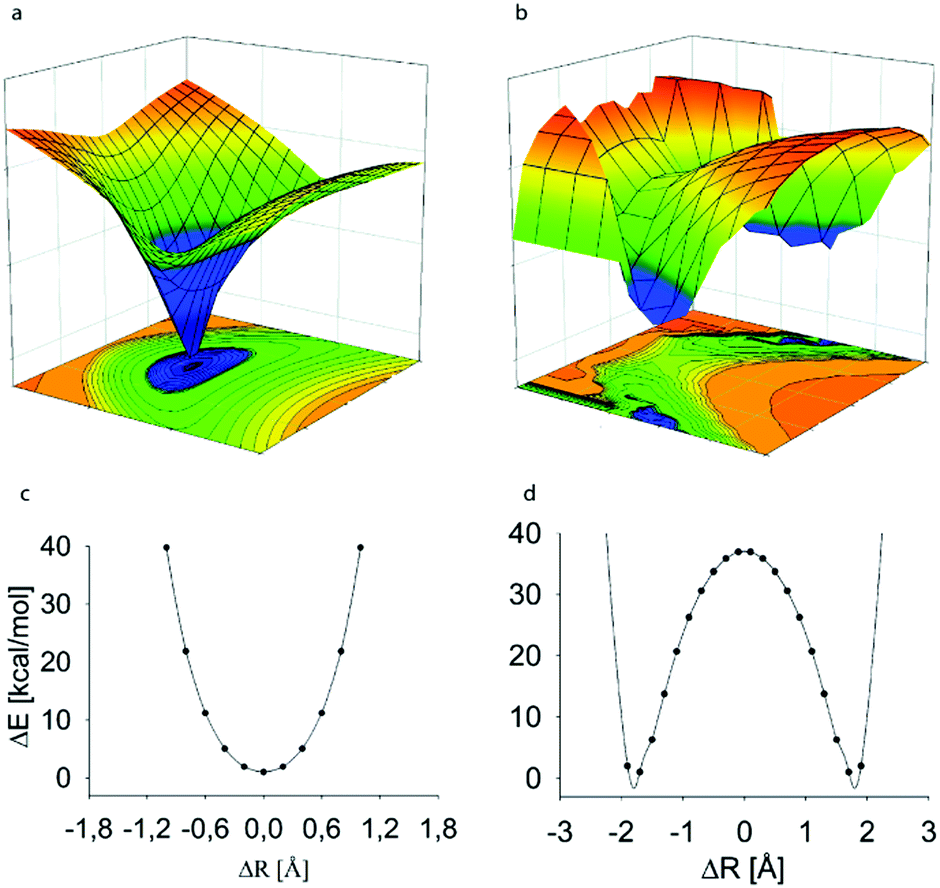 | ||
| Fig. 2 The computed potential energy surface of the halogen-bonded complexes of halonium ions indicates the preference for (a) symmetric [D⋯X⋯D]+ geometries for the complexes of iodonium, bromonium and chloronium ions, whereas (b) asymmetric D+–X⋯D arrangement for the fluoronium ion complexes. The potential energy curves (c and d), that is the 2D-slices corresponding to the D–D distance with the global energy minimum of the complexes, possess a single energy minimum for the larger halogens, whereas that of the fluoronium complex shows two equal minima with a high energy barrier. Here, the potential energy is shown as a function of the donor–donor, rDD, and the halogen–donor, D–X, distance, where ΔR is the difference between the D1–X and D2–X distances at a given rNN. The data of [bis(pyridine)iodine(I)]+ and [bis(pyridine)fluorine(I)]+ complexes are shown as representative examples.8 This figure has been adapted from ref. 8 with permission from the Royal Society of Chemistry, copyright 2014. | ||
A classical, two-center halogen bond is commonly seen to involve two atoms, namely the halogen, X, and the electron donor, D. Its halogen is in oxidation state 0 or formally −1, and possesses two vastly different bonds: a conventional two-center covalent, D–X, and a classical two-center halogen bond, D⋯X (Fig. 1a). In contrast, the halogen of three-center, four-electron halogen bonds involves a halogen(I) in oxidation state +1, which forms two D⋯X bonds that simultaneously possess covalent and secondary bond characteristics (Fig. 1b). The D⋯X bond distances remain in between the sum of the van der Waals radii and the sum of the covalent radii of the participating atoms. They are similar in length, within <10% difference between the D⋯X distances. Breaking one of them typically destabilizes the second D⋯X bond as well, yielding a reactive halonium ion and two equivalents of Lewis bases.
Viewing from an orbital perspective, the three-center halogen bond is formed by the empty p-orbital of an X+ simultaneously accepting electron density from two Lewis bases, with the valence shell of the central X+ having an s2p4 electronic configuration.11 Induced by the electrostatic field of the two Lewis bases, its p-orbitals are occupied in the spin paired px2py2pz0 arrangement. The two lobes of its empty pz0 orbital are available for interaction with the electron pairs of two Lewis bases, D, in a linear D⋯X⋯D fashion. These three atomic orbitals combine into three molecular orbitals that hold four electrons, and thus act as a three-center, four-electron system (Fig. 3). The three-center bond of halonium ions has been computationally thoroughly assessed,8,12–16 and concluded to be dominantly of covalent and electrostatic characteristics, with a smaller contribution from dispersion forces. The origin of the covalency is charge transfer, whereas the electrostatic characteristic is the result of Coulomb attraction and polarization. The partial covalent character of the three-center halogen bond increases as the size of the halogen decreases,8,13 and simultaneously, the strength of the bond decreases as the strength mainly originates from the electrostatic contribution (Fig. 4). This trend has been demonstrated both experimentally and computationally for [N⋯X⋯N]+ systems, for which the chloronium-centered bond was observed to be the weakest despite its largest covalent character, whereas the least covalent iodonium-centered system had the strongest halogen bond.8 The correlation of covalency and the three-center character of halogen bonding has been investigated at the CCSD(T) level of theory.16
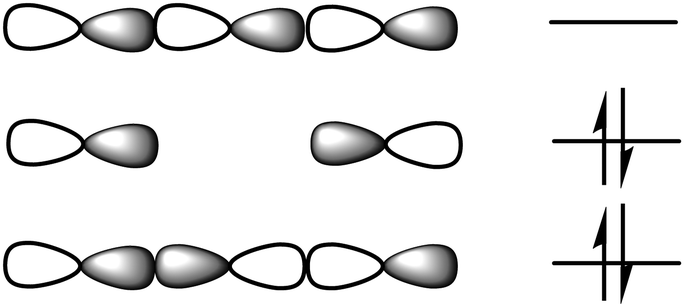 | ||
| Fig. 3 The molecular orbital system of three-center, four-electron bonds, [D⋯X⋯D]+. Two of the electrons are on the bonding orbital, whereas two are on the non-bonding level, and are thus predominantly located on the terminal electron donor atoms, which act as halogen bond acceptors. The central atom, here a halogen, is electron poor. | ||
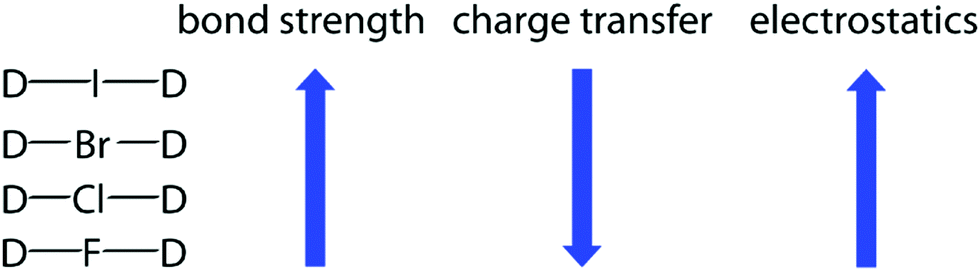 | ||
| Fig. 4 The halogen dependence of the nature and characteristics of the three-center halogen bond: the larger the halogen, the stronger and the more electrostatic it is. With a smaller halogen, the bond is shorter and more covalent. | ||
Three-center halogen bond complexes have been proposed to be termed “coordination complexes”5,12,17 to emphasize the dative nature of their ionic bond.5 It has also been suggested that only the bond of I+ should be termed coordinative, whereas that of Br+ and Cl+ should rather be seen as bonds with covalent character, and the analogous complexes of F+ considered as ion–molecule complexes.12 A combined NMR, X-ray diffraction and computational study has, however, indicated that I+ complexes behave differently to typical transition metal complexes, such as those of Ag(I),18 regarding solvation and counterion coordination. Therefore, the term “coordination complex” for such halogen bond complexes is misleading and should not be used.19
[D⋯X⋯D]+ compounds may also be interpreted as hypercoordinate, hypervalent, or 10-X-2 complexes of halogen(I).20 Historically hypervalent bonding has been considered to involve the expansion of the octet valence electrons, whether through σ-hole interactions, electrostatic interactions or charge transfer, but this expansion is now viewed as being largely formal. The hypervalent bonding model has the advantage of allowing a smooth comparison of the structure and reactivity of electronically related complexes of different research fields.
Bond symmetry
The three-center halogen bond is isoelectronic to the [D–H–D]+ hydrogen bond,4 as both have 4 electrons shared over 3 atomic centers. The three-center hydrogen bond has been postulated to be symmetric, to possess unusually short D–H bond length and to have exceptional bond strength. Accordingly, it is often entitled as a short, strong, or low barrier bond. The strength and symmetry of such hydrogen bonds has, however, been controversial. Using solution NMR, Perrin has shown that such hydrogen bonds are typically asymmetric in solution and do not possess unusual bond strength.21 The interpretation of Perrin's NMR data has been questioned, for example, by Singleton.22 Whereas the nature of the [D–H–D]+ hydrogen bonds has been debated for decades, studies of the analogous [D–X–D]+ halogen bond have been initiated within the last 10 years.9,23A three-center halogen bond may prefer (a) a static and centrosymmetric, [D⋯X⋯D]+ geometry with the central halogen forming two equally strong and long D⋯X bonds (Fig. 5). The motion of the halogen in such a system is best described by a single-well energy potential. Alternatively, the halogen bond of halonium ions may be asymmetric, [D–X⋯D]+, with the halogen forming one shorter and stronger D–X bond, and a weaker and longer D⋯X bond to the other donor. Compared to the centrosymmetric geometry, the covalent character of the D–X and D⋯X bonds of the asymmetric complex is increased and decreased, respectively. The motion of the halogen in such a system may be described by (b) a symmetric double-well potential, (c) an asymmetric single-well potential, or (d) an asymmetric double-well potential. Analogous to the term prototropy, which has been coined to describe tautomeric exchange processes involving H+ transfer, the motion of X+ in such halogen-bonded systems is called halotropy.24 A symmetric double-well potential (b) reflects a pair of asymmetric isomers, [D–X⋯D]+ and [D⋯X–D]+. When the Lewis basicity of the electron donors is comparable and the asymmetric geometries are separated by a shallow energy barrier, these may interconvert and exist as a dynamic mixture, [D–X⋯D]+ ⇄ [D⋯X–D]+. A system described by a symmetric double-well potential with a high energy barrier results in a static asymmetric geometry, D+–X⋯D, which corresponds to a conventional weak σ-hole halogen bond complex. If the electron donors have slightly different Lewis basicities, the complex may become static and asymmetric (c), with the halogen showing a higher preference to one of the electron donors. Its motion between the electron donors is described by an asymmetric single-well. Alternatively, it forms asymmetric geometries that are of different energy (d), and the halogen motion is then described by an asymmetric double-well potential, where one of the minima is of lower energy and of higher stability. The symmetry of the halogen bond of halonium ions in the solid state, in solution and in silico has been reviewed in 2015, using systems possessing nitrogen Lewis bases as a representative example.4 There has been significant progress since then, with several experimental and computational studies addressing the factors possibly influencing halogen bond symmetry.8,15,19,24,26
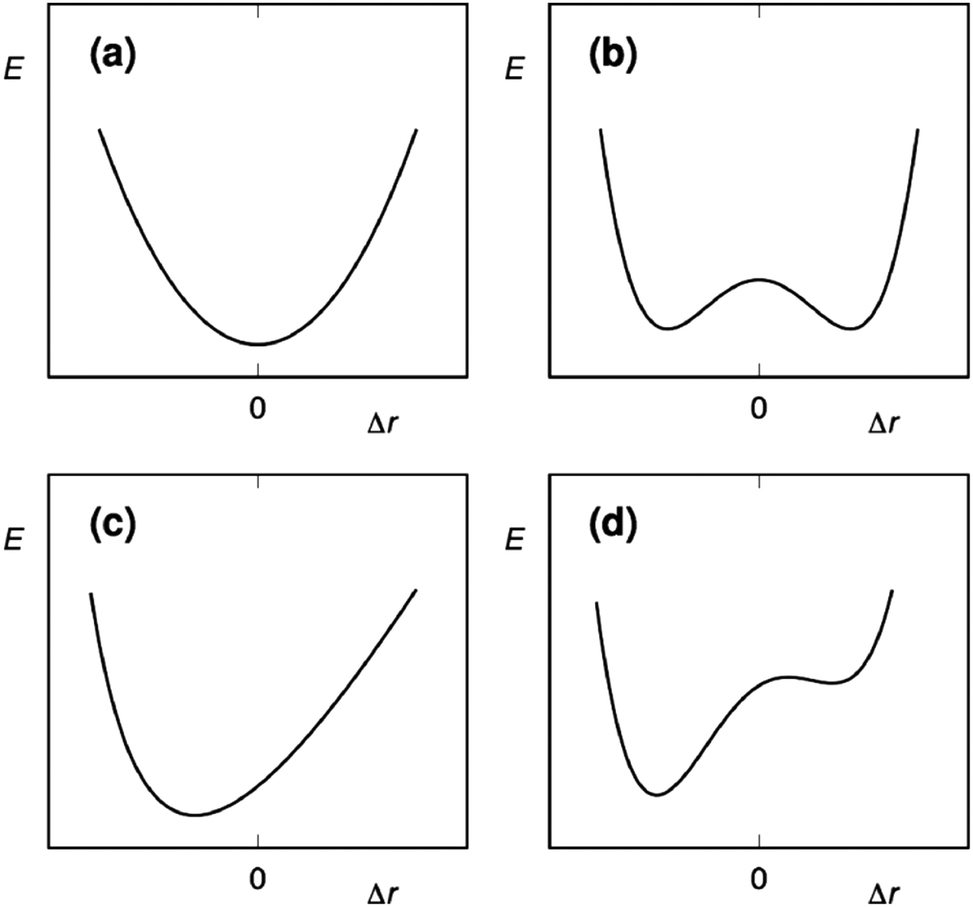 | ||
| Fig. 5 The possible energy potentials of halogen motion in a three-center [D⋯X⋯D]+ halogen bond: (a) a symmetric single-well energy potential describes the halogen motion as a static and symmetric [D⋯X⋯D]+ system, in which the D⋯X bonds are equal. A (b) symmetric double-well potential may either reflect a pair of asymmetric isomers, [D⋯X–D]+ ⇄ [D–X⋯D]+, in dynamic equilibrium, when the energy barrier between the minima is shallow. Each form of this geometry has a shorter and stronger D–X and a longer and weaker D⋯X bond. If the energy barrier between the two minima is high, the system becomes static and asymmetric. For systems possessing Lewis bases, D, with different electron densities, the halogen motion may either follow (c) an asymmetric single-well potential, or (d) an asymmetric double-well potential. The latter has a strong preference for a shorter and stronger bond towards one of the electron donors. The potential energy variation is shown here as a function of Δr, the displacement of X+ from the symmetrical position.24 This figure has been adapted from ref. 24 with permission from the American Chemical Society, copyright 2018.25 | ||
Halogen bond symmetry has been studied on [bis(pyridine)halogen(I)]+ complexes and their geometrically restrained [(1,2-bis(pyridin-2-ylethynyl)benzene)halogen(I)]+ analogues (Fig. 6). The bromine(I) and iodine(I) centered model systems prefer static, symmetric geometries in solution, as demonstrated by the lower temperature dependence of their secondary isotope effects, as compared to the analogous dynamic [N–H⋯N]+ ⇄ [N⋯H–N]+ hydrogen bond complexes, using the isotopic perturbation of equilibrium method of Siehl.27 These results were corroborated by 1H, 13C and 15N NMR spectroscopy, and by computations at the DFT level.9,13 [Bis(pyridine)iodine(I)]+ and the analogous bromine(I) complexes are symmetric also in the solid state,4,7,19 as shown by X-ray diffraction, although these data have originally mistakenly been interpreted as displaying asymmetric structures9 due to the minor, <5%, difference in N–X bond lengths. This difference is explained by crystal packing forces and is insignificant, and thus the X-ray data show symmetric complexes in the solid state. The analogous [bis(pyridine)chlorine(I)]+ complex was shown to also prefer a symmetric geometry in dichloromethane solution by DFT/MP2 studies (Fig. 7), supported by 1H and 13C NMR studies.8 This complex is only stable below −70 °C, and undergoes rapid relaxation (T1 0.08 s of the proton closest to the [N⋯Cl⋯N]+ bond), preventing isotopic perturbation of equilibrium and 15N NMR studies. Complex formation is corroborated by the quick relaxation of the nearby pyridine protons of the chlorine(I) complex (T1 0.08 s) as compared to that of the corresponding Ag+ (T1 1.49 s) complex of pyridine (T1 1.61 s). This quick relaxation of nearby protons to the chlorine may be explained by scalar relaxation of the second kind (SC2). The corresponding fluorine-centered bis(pyridine) complex was demonstrated to prefer a static asymmetric N–F⋯N geometry, with a strongly covalent N–F bond and a weak, yet detectable, fluorine-centered N⋯F halogen bond, as demonstrated by diffusion NMR and by 2–3 ppm 15N and 19F NMR coordination shifts, i.e. chemical shift changes upon complexation (Fig. 8).8
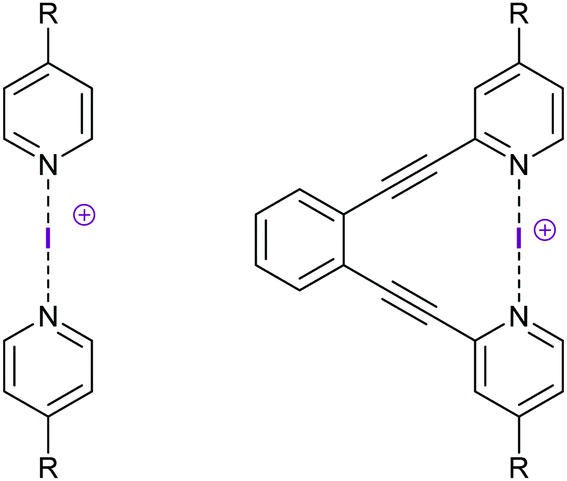 | ||
| Fig. 6 [Bis(4-R-pyridine)iodine(I)]+ BF4−, and geometrically restrained [1,2-bis((4-R-pyridine-2-ylethynyl)benzene)iodine(I)]+ BF4− complexes were used as model systems for assessment of the influence of electron density alterations on the [N–I–N]+ halogen bond. The bis(pyridine)-type complex (left) permits free rotation and adjustment of N–I distances for the most favorable interaction, whereas the 1,2-diethynylbenzene backbone of the complex (right) inhibits rotation around the N–I–N axis and imposes some strain in the N–I bonds to reach a geometrically optimal [N–I–N]+ interaction. A systematic alteration of the electron density of the [N–I–N]+ halogen bond was achieved by variation of the R-substituents, resulting in complexes with an increasing electron density in the order NO2 < CF3 < H < F < Me < OMe < NMe2. Symmetric substitution led to centrosymmetric complexes with varying halogen bond strengths,26 whereas asymmetric substitution led to static asymmetric complexes.24 | ||
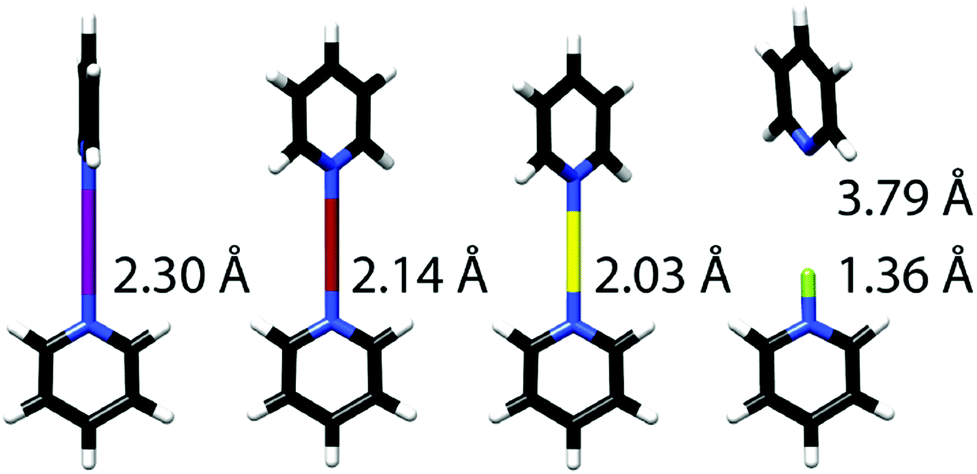 | ||
| Fig. 7 The optimized (B3LYP/LANL08d) geometries of the I, Br, Cl and F-centred [bis(pyridine)halogen(I)]+ complexes, shown from left to right.8 Formation of the three-centred bond results in Bondi normalized interatomic distances 0.65 (N⋯I), 0.63 (N–Br) and 0.62 (N–Cl), with thus >30% shortening of the sum of van der Waals radii of the involved atoms indicating the exceptional strengths of these halogen bonds. This figure has been adapted from ref. 8 with permission from the Royal Society of Chemistry, copyright 2014. | ||
 | ||
| Fig. 8 The overlaid 1H, 15N HMBC of N-fluoropyridinium tetrafluoroborate (black), of pyridine (red) and of their halogen bond complex (green). Observation of −3 ppm 15N coordination shift of the pyridine nitrogen, along with 0 ppm 15N but 1.1 ppm 19F coordination shift of the N-fluoropyridinium ion, indicates formation of a weak F-centred conventional halogen bond.8 The different chemical shifts of the two nitrogens of the complex are compatible with a static asymmetric N+–F⋯N bond, which was also corroborated by DFT calculations. This figure has been adapted from ref. 8 with permission from the Royal Society of Chemistry, copyright 2014. | ||
The preference for a symmetric three-center bond was demonstrated to be independent of solvent polarity.11 Hence, the iodine(I) and bromine(I)-centered complexes showed comparable 15N NMR chemical shifts (2–6% alteration) in dichloromethane and acetonitrile solution,11 whereas the corresponding [bis(pyridine)silver]+ complex, for example, showed 48% 15N NMR shift alteration upon this solvent change.18 DFT (B3LYP/LANL08d) calculations indicated neither solvent dependence in the extent of charge transfer (28% positive charge on Br+ and 41% on I+, respectively) or in bond lengths (2.14 and 2.30 Å), corroborating the experimental evidence.
Besides solvent polarity, the interaction with an asymmetrically positioned counterion (Fig. 9) has been proposed as a plausible inductor of halogen bond asymmetry.19 A solution NMR investigation, including isotopic perturbation of equilibrium, diffusion and 15N NMR studies of iodine-centered complexes possessing spherical and weakly coordinating BF4−, ClO4−, PF6−, SbF6−, moderately coordinating OTf− and OTs−, and small and strongly coordinating NO3− and CF3COO− counterions, suggested that the preferred halogen bond symmetry is retained independent of the size, charge distribution or coordination strength of the counterion.19 These complexes showed a centrosymmetric structure in the solid state as well, with their geometry having been confirmed by DFT calculations.
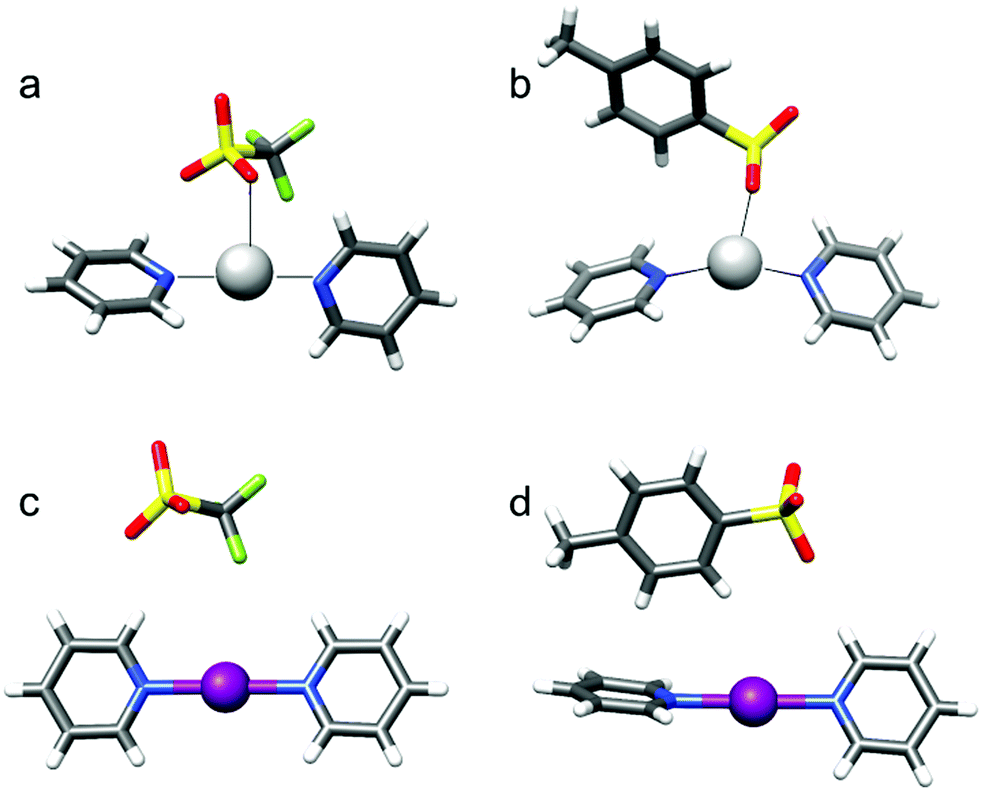 | ||
| Fig. 9 The solid state structures of the [bis(pyridine)silver]+ (a and b) and [bis(pyridine)iodine(I)]+ (c and d) complexes possessing triflate (a and c) and tosylate (b and d), obtained by X-ray crystallography, reveal direct coordination of the counterion to silver(I) but not to iodine(I). The latter forms linear, bis-coordinated complexes that remain symmetric along the N–I–N three-center bond, even when the counterion is asymmetrically positioned. This figure has been adapted from ref. 26 with permission from the Royal Society of Chemistry, copyright 2016. | ||
It has further been studied whether the alteration of the electron density of the three-center halogen bond, other than upon changing the halogen,8 but by instead altering the electron density of the Lewis bases, may influence its geometry and strength.24,26 In case the Lewis bases involved have a low electron density, the central halogen(I) may be unable to efficiently accept electrons from both donors simultaneously and choose to compensate for its electron depletion by forming a strong covalent bond to one of the donors, and a weak halogen bond to the second, rather than forming a three-center halogen bond involving both. This would result in an asymmetric geometry. Alternatively, upon an increased electron density of the Lewis bases involved, the halogen(I) may prefer to form a strong bond to one electron donor, as it may better compensate for its electron depletion by connecting to one Lewis base covalently, than to two through a secondary bond, resulting in an asymmetric geometry. Changing the electron density by symmetric alteration of the pyridine para-hydrogen to NO2, CF3, F, Me, OMe, NMe2 functionalities (Fig. 6), the centrosymmetric three-center halogen bond geometry is retained both in solution and in the solid state, as demonstrated by NMR, X-ray and DFT computations.26 Whereas the bond symmetry, the N–I bond lengths and the 15N NMR coordination shifts remained virtually unaltered, the [N⋯I⋯N]+ bond strength varied by ∼100 kJ mol−1 upon altering the substituents, for both the [bis(pyridine)iodine(I)]+ and the [1,2-bis((pyridine-2-ylethynyl)benzene)iodine(I)]+ model systems (Table 1). This suggests that any general conclusions on the correlation of bond strength with bond lengths, and bond strength with NMR coordination shifts should be made with utmost caution.
| 4-R | r(NI) (Å) | r(NN) (Å) | ∠(NIN) (°) | δ 15Ncoord | ΔEstab (kJ mol−1) |
|---|---|---|---|---|---|
| H | 2.3034 | 4.5934 | 175.7 | −101.0 | 0 |
| Me | 2.3011 | 4.5875 | 175.4 | −101.0 | 11.6 |
| OMe | 2.2982 | 4.5832 | 175.7 | −100.0 | 39.8 |
| NMe2 | 2.2930 | 4.5715 | 175.4 | −100.7 | 69.2 |
| F | 2.3027 | 4.5921 | 175.7 | −101.4 | 16.9 |
| CF3 | 2.3041 | 4.5939 | 175.5 | −106.0 | −11.4 |
| NO2 | 2.3043 | 4.5952 | 175.7 | N.A. | −19.1 |
Upon asymmetric modulation of the substituents of [(1,2-bis(pyridin-2-ylethynyl)benzene)iodine(I)]+, by introduction of a CH3 and a CF3 group in the para-position to the pyridine nitrogens (Fig. 6 and 10a), a static asymmetric [N–I⋯N]+ bond geometry (Fig. 5c) was obtained, with the halogen being closer to the nitrogen of higher electron density (ΔdN–I = 4%).24 This was demonstrated by the experimentally observed 20 ppm difference of the 15N NMR coordination shifts of the two nitrogens, and by the low temperature dependence of the secondary deuterium isotope effects of the pyridine carbons, as compared to those of the reference model system involved in a dynamic equilibrium. This [N–I⋯N]+ complex is slightly destabilized (9.7 kJ mol−1) as compared to its symmetric and unsubstituted analogue. The asymmetric three-center bond remains strong and the complex is stable in solution at room temperature for days. The above conclusions on the substituent effect were confirmed by an independent theoretical investigation.14,15
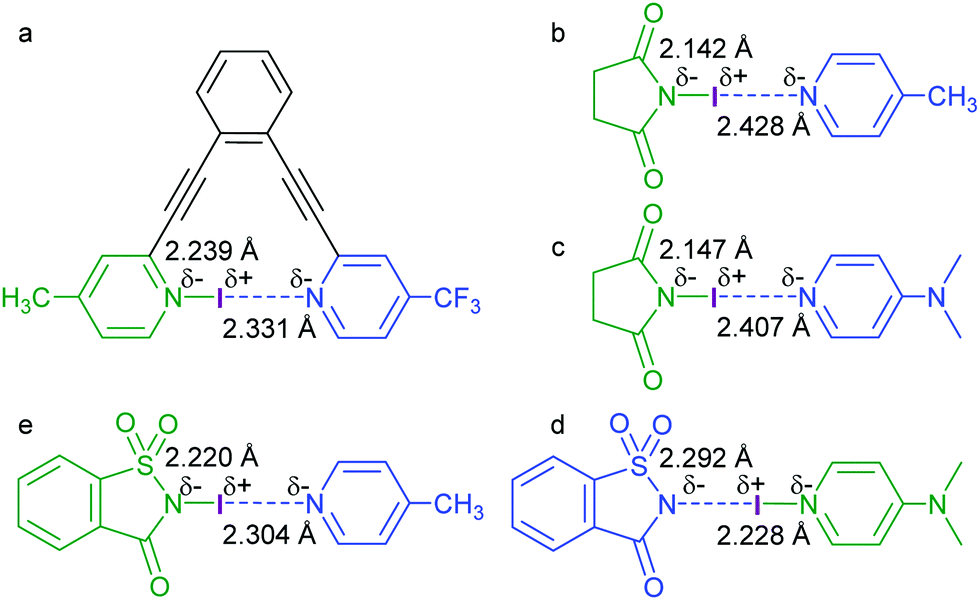 | ||
| Fig. 10 The N–I distances (Å) of a variety of asymmetric N–I–N complexes, determined by DFT calculations and X-ray diffraction studies.24,28 | ||
In an analogous study,29 the halogen position within a [D–X–D]+ bond was assessed using pyridine complexes of N-halosuccinimides and N-halosaccharins (Fig. 10b–e). Due to the comparable Lewis basicity of imides and electron rich pyridines, the position of X+ (Fig. 6) is modulated by a slight adjustment of the electron density of the participating nitrogens. Co-crystallization of N-iodosuccinimide with 4-picoline and 4-dimethylaminopyridine (Fig. 10b and c) yielded complexes, in which the I+ is closer to the nitrogen of succinimide whereas it is further away from the pyridine nitrogen (ΔdN–I = 12–13%). As a consequence of the lower electron density of the saccharin anion as compared to the succinimide anion, the analogous N-iodosaccharin–4-dimethylaminopyridine complex showed a shorter N–I bond to the pyridine nitrogen and a longer bond to the imine nitrogen. Decreasing the electron density of the pyridine by NMe2 to CH3 substitution (Fig. 9d and e) inverted the geometry, with I+ moving closer to the saccharin nitrogen (ΔdN–I = 4%). It should be noted that the difference of N–I bond lengths of the latter two complexes, ΔdN–I = 3–4% (Fig. 10d and e), is comparable to that of the asymmetric [(4-methyl-2-((2-((4-(trifluoromethyl)-pyridin-2-yl)ethynyl)phenyl)ethynyl)pyridine)iodine(I)]+ complex (4%)24 (Fig. 10a). Both N–I bonds of these complexes are undoubtedly part of a three-center complex, and thus possess both covalent and secondary characteristics.8,23 Formally, the shorter bonds can be seen to possess a “more covalent” whereas the longer a “more secondary” bond character. The central halogen of such complexes carries a partial positive charge; however, much of the formal charge of the halogen(I) is distributed into the conjugated electronic system of the coordinating Lewis bases.8,23 Such complexes may be assigned “more ionic” or “more neutral” character,29 although the alteration of the charge distribution is minor. In addition to the Lewis basicity of the halogen bond acceptors, crystal packing forces were suggested to influence the position of the halogen for the N-iodosaccharin–pyridine complex.28,29 Interestingly, whereas the I+ of the N-iodosaccharin–4-dimethylaminopyridine is slightly (ΔdN–I = 4%) closer to the imide nitrogen, the Br+ of the analogous N-bromosaccharin complex shows the opposite preference (dNimide–Br = 2.231 Å, dNpyridine–Br = 2.301 Å, ΔdN–I = 4%).28 This was explained to be a consequence of the crystalline molecular environment that was computationally predicted to have a 26% larger influence on the Br+-centered complex. In agreement with previous literature (Fig. 4),8 the [N–Br⋯N]+ complex was shown (DFT) to have a larger covalent character than the corresponding [N–I⋯N]+ complex.28
Halotropy, that is the low barrier [D–X⋯D]+ ⇄ [D⋯X–D]+ interconversion of asymmetric halogen bond geometries, has a fundamental importance not just for the understanding of the bonding phenomenon itself, but also for that of reactions involving halogen transfer. Electrophilic halogenations (Fig. 11), for instance, involve an analogous X+ transfer by the initial weakening of a C–X, N–X or X–X covalent bond of the halogen transfer agent while forming a conventional halogen bond to an alkene. This is followed by formation of a three-center complex consisting of a nucleophile, a halogen(I) center and the leaving group. The three-center intermediate collapses to the product by forming a covalent halogen–nucleophile bond and eliminating the leaving group. The [D–X⋯D]+, [D⋯X⋯D]+, and [D⋯X–D]+ species can be seen to represent the stages of any halogen transfer process from one halogen bond acceptor to another. These have varying D–X bond lengths and possess a varying degree of covalency. Understanding whether a halogen transfer process between two halogen bond acceptors follows a single-well or double-well potential with a shallow barrier, whether it takes place at all, and whether the dynamic mixture of asymmetric geometries, the symmetric transition geometry or any of the asymmetric forms has the highest stability, is of fundamental interest.30–33
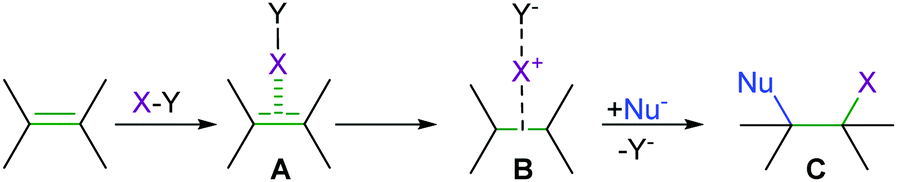 | ||
| Fig. 11 The mechanism of electrophilic halogenation. The reaction is initiated by formation of a weak halogen bond with the alkene acting as a halogen bond acceptor (A). This is followed by formation of an intermediate (B) in which the central halogen possesses two halogen bonds, to Y- and to the π electrons of the double bond, and thus forms an analogous complex to a classical three-centre bond. The reaction reaches completion (C) by disconnection of the leaving group Y, which can be any group that binds a halogen X. | ||
To model the halogen transition between two halogen bond acceptors, and thus establish a dynamic mixture [D–X⋯D]+ ⇄ [D⋯X–D]+, the energy barrier between the two complexes must be shallow. This may be obtained with molecular systems that have a longer-than-optimal donor–donor distance, as predicted by DFT computations.24 For [N–I–N]+-type systems, the global energy minimum is centrosymmetric and is at 4.51 Å nitrogen–nitrogen distance (Fig. 12). Upon increasing this distance above 4.88 Å, the asymmetric [N–I⋯N]+ and [N⋯I–N]+ arrangements become energetically more favorable than the alternative symmetric [N⋯I⋯N]+ one. With an increasing nitrogen–nitrogen distance, the energy barrier of the [N–I⋯N]+ ⇄ [N⋯I–N]+ interconversion increases just as the energy of the most stable asymmetric geometries does, as compared to that of the geometry at electron donor–electron donor distance to a value above the global energy minimum (rNN = 4.51 Å). A promising approach to obtain a model system in which an iodine can move between two halogen acceptors is thus to adjust the bifurcation point (4.88 Å), yet below the distance that would provide too high an energy barrier for interconversion of the asymmetric geometries. This has been confirmed by further computations of Scheiner, who used H3N–X+–NH3 and H3N–X+–OH2 complexes to estimate transfer barriers for various donor–donor distances in [D–X–D]+ systems.35 2,2′-(9,10-Dimethoxyphenanthrene-3,6-diyl)dipyridine24 was proposed as a bidentate ligand for a dynamic system; however, as a result of its suboptimally large N–N distance, 5.24 Å, this system was shown to be unstable as a monomeric complex and showed preference for the formation of a dimer instead, which encompasses two static and symmetric [N⋯I⋯N]+ bonds.
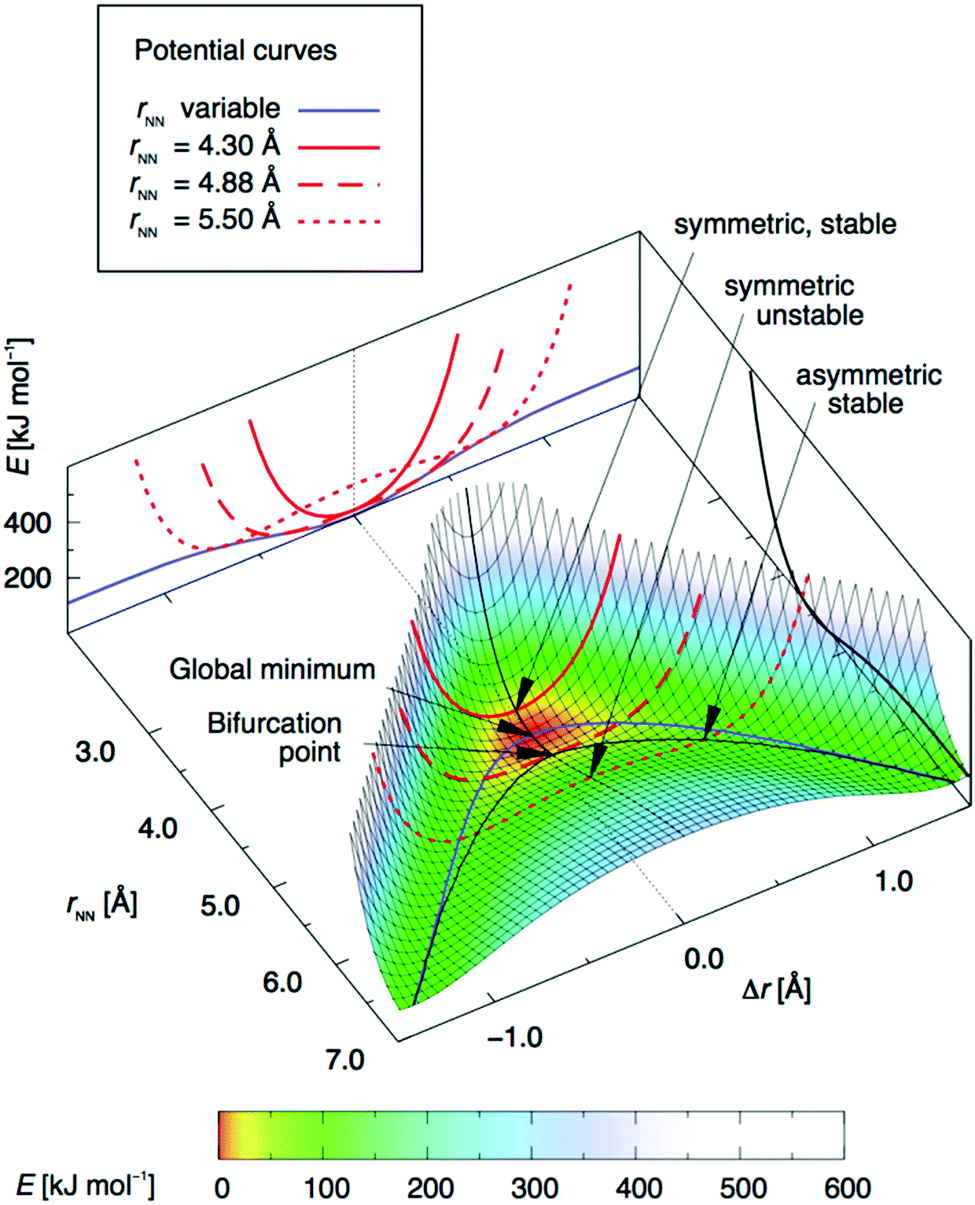 | ||
| Fig. 12 The variation of electronic energy as a function of the position of the iodine in an [N⋯I⋯N]+ halogen bond, upon varying the N–N distance, rNN (Å), and the position of the iodine as described by Δr, the elongation of the iodine from the geometrical midpoint of the nitrogen–nitrogen distance. Thus, at Δr = 0 the iodine is centered between the nitrogens, whereas at Δr = 0.5 Å it is 1 Å closer to one of them. The blue line shows the potential curve, corresponding to Fig. 1a, for variable rNN, whilst the red lines show the potential curves for rNN kept fixed at 4.30 Å (solid), 4.88 Å (long-dashed) and 5.50 Å (short-dashed), respectively, shown both in the 3D plot and projected on the rear border plane. The black line on the right border plane shows the dissociation curve of the [N⋯I⋯N]+ bond model system. DFT calculations were performed using the M06 exchange and correlation functional and a mixed-level (double zeta/triple-zeta/augmented triple-zeta) basis set. This figure has been adapted from ref. 24 with permission from the American Chemical Society, copyright 2018.25 | ||
Analogous elongation of the covalent bond of the interhalogens ICl (7%) and IBr (4%) was reported upon their halogen bonding to the sulfur of N-methylbenzothiazole-2-thione and 2(3)H-benzothiazole-thione (Fig. 13).34 These complexes are slightly asymmetric (ΔdS–I/I–X = 3–7%), with their S⋯I halogen bonds being remarkably strong (−50 to −82 kJ mol−1) as indicated by their S⋯I distances being shortened to 66–72% of the sum of the van der Waals radii of the participating atoms. The interacting iodine of these complexes carries 0.323 (ICl) and 0.187 (IBr) charge, which is somewhat lower than the 0.41 charge that was reported for the analogous [bis(pyridine)iodine(I)]+ complexes.8 Natural bond critical point analyses of these complexes showed |VBCP|/GBCP values from 1.31 to 1.60 (here, V is the potential energy density and G the kinetic energy density) for the S⋯I contacts, indicating them to have a partially covalent character (|VBCP|/GBCP ratio typically has values <1 for non-covalent interactions and >2 for covalent bonds).
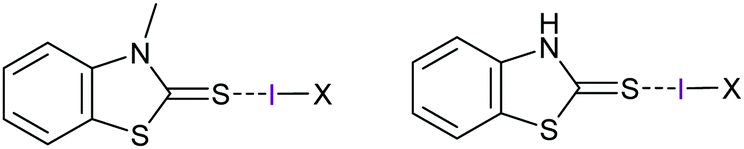 | ||
| Fig. 13 The structure of the halogen bond complexes of N-methylbenzothiazole-2-thione (left) and 2(3)H-benzothiazole-thione (right), where X is Cl or Br.34 For the N-methylbenzothiazole-2-thione complexes, the S–I bond halogen bond lengths were reported to be 2.5485 Å and 2.706 Å, respectively, whereas the I–Cl and I–Br bonds were 2.6179 and 2.6308 Å. | ||
A three-center [S–I–S]+ halogen bond was reported based on the crystallographic and computational investigation of [bis(imidazolidinethione)iodine(I)]+ with an RXB ratio of 0.7, dS–I = 2.6297 Å, |VBCP|/GBCP ratio of 1.58 and interaction energy of −60 kJ mol−1.36 The partial covalent character and the three-center nature of the [S–I–S]+ halogen bond was further confirmed by the Ω(I,S) 0.79 delocalization index, suggesting a considerable amount of electron sharing between the iodine and the two sulfur atoms (the delocalization index describes the average number of shared electrons between (bonding) atoms at the bond critical point). Importantly, in these complexes iodine(I) is not only involved in halogen bonding, but also in hydrogen bonding, donating electrons to the nearby NH protons (Fig. 14). Hence, the empty p-orbital of this I+ receives electrons from two sulfur donors, whereas one of its filled p-orbitals donates electrons to two protons, and therewith, thanks to its anisotropic electron density distribution, it simultaneously acts as a halogen bond donor (60 kJ mol−1) and a hydrogen bond acceptor (9 kJ mol−1). In such ‘hydrogen-bonded halogen bonds’, the two interactions mutually strengthen each other.37
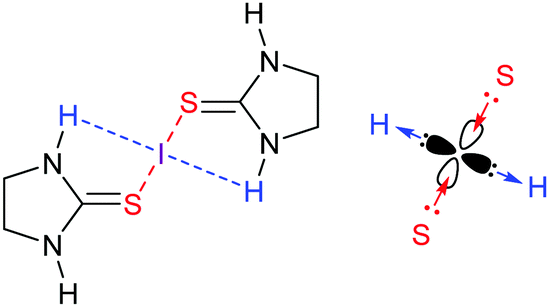 | ||
| Fig. 14 2-Imidazolidinethione forms a strong (60 kJ mol−1) three-center halogen bond complex with iodine(I), which in turn participates in weak hydrogen bonds to the same ligand. Such hydrogen-bonded halogen bonds form due to the anisotropic electron distribution of a halogen, and the two interactions mutually strengthen each other. | ||
Thiones and thioamides, possessing sp2-hybridized sulfur, are the most commonly used electron donors in the formation of [S⋯X⋯S]+ halogen bonds. These ligands efficiently delocalize the positive charge of the halogen, and are often reported as charge-transfer complexes. 2-Imidazolidinethione, for example, was reported to form [(2-imidazolidinethione)2 iodine(I)]+·1/2I−·1/2[I3]−, with the counterions I− and [I3]− being generated throughout the heterolytic cleavage of I2.36 The resulting [S⋯X⋯S]+ bond was reported to be −60 kJ mol−1 strong and partially covalent, and thus the complex similar to the analogous [N–I–N]+ complexes was found to be stable in the solid state, in solution as well as in the gas phase. Moreover, linear three-center iodine(I) complexes were obtained upon crystallization of N-methylbenzothiazole-2(3H)-thione or N-methylbenzothiazole-2(3H)-selone in the presence of I2.38
Although so far less studied, selenium39,40 and tellurium41 may also act as Lewis bases in linear three-center [D⋯X⋯D]+ halogen bonds. These complexes, typically studied in the solid state, are formed by the reaction of the electron donors with dihalogens, often yielding several different counterions.
Phosphine selenides form a mixture of molecular But3PSe–I–I and ionic (But3PSe)2I+ I3− upon interaction with I2.39 A slight excess of I2 transforms the molecular adduct into the linear [Se⋯X⋯Se]+ three-center complex. Similar [Te⋯X⋯Te]+ complex species were also reported.41
The [N–I–O]− bond of N-iodosaccharin complexes with N-oxides shows directionality (∼178°), length (RXB ∼ 0.66) and strength (15–25 kJ mol−1) characteristic of three-center halogen bonds.42 These bonds are slightly asymmetric (Fig. 15) with ∼8% difference in the N–I and O–I bond lengths. The −N–I+⋯−O–N+ motif is not fully linear and the N–O bond, apart from it providing high electron density to its oxygen, does not contribute to the high stabilization of the system.
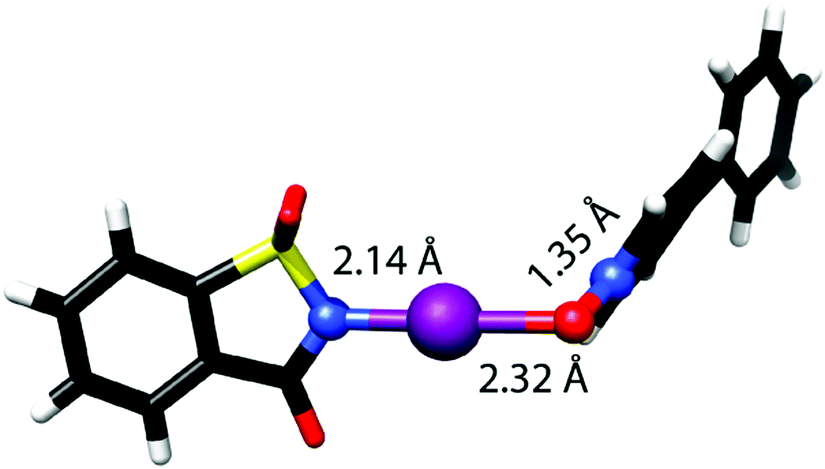 | ||
| Fig. 15 The complex of N-iodosaccharin and 4-phenylpyridine N-oxide possesses an −N–I+⋯−O–N+ motif encompassing a linear [N–I–O]− bond that shows the characteristics (bond strength and length) of a three-center halogen bond.42 | ||
Trihalide ions represent a subgroup of three-center halogen bonds. Early discussions of the nature of their bonding43 have vastly contributed to the understanding of three-center bonds in general.20 Depending on their environment, trihalide ions may exist as X2⋯X− adducts, with a conventional σ-hole mediated halogen bond, or as [X⋯X⋯X]− complexes with a three-center bond, involving the p-hole of their central X+.44
Applications in supramolecular chemistry
Conventional, two-centered, halogen bonding is nowadays commonly used as a driving force in self-assembly processes in supramolecular chemistry. Even though the three-center halogen bond is a well-suited building block with its predictable bond characteristics, e.g. its high linearity and robustness, and with its potential for use with different types of Lewis bases to create well-defined complex structures, halonium ions haven’t yet been widely utilized in supramolecular chemistry. Only of late has the three-center [D⋯X⋯D]+ halogen bond been introduced as a supramolecular synthon in self-assemblies.17,45–47Multiple [N⋯I⋯N]+ halogen bonds were used in the formation of dimeric capsules from ethylene-bridged tetrakis(3-pyridyl)cavitands (Fig. 16).45 Halogen-bonded capsules were achieved by adding molecular iodine to the solution of analogous dimeric Ag+-coordinated capsules. Precursor Ag+-coordinated capsules were shown to exist as a mixture of at least two different dimeric capsules due to the coordinative nature of Ag+, whereas only one symmetric [N⋯I⋯N]+ halogen-bonded dimeric capsule was observed. The capsules were characterized in solution by DOSY NMR, which confirmed the formation of the larger assemblies. Compared to the free cavitands (7.5 × 10−10 m2 s−1 and 6.9 × 10−10 m2 s−1) significantly lower diffusion coefficients (4.8 × 10−10 m2 s−1 and 4.6 × 10−10 m2 s−1) were observed for the complexes. The capsules were also characterized by ESI-MS that further confirmed the formation of the four symmetrical [N⋯I⋯N]+ halogen bond stabilized supramolecular capsules.
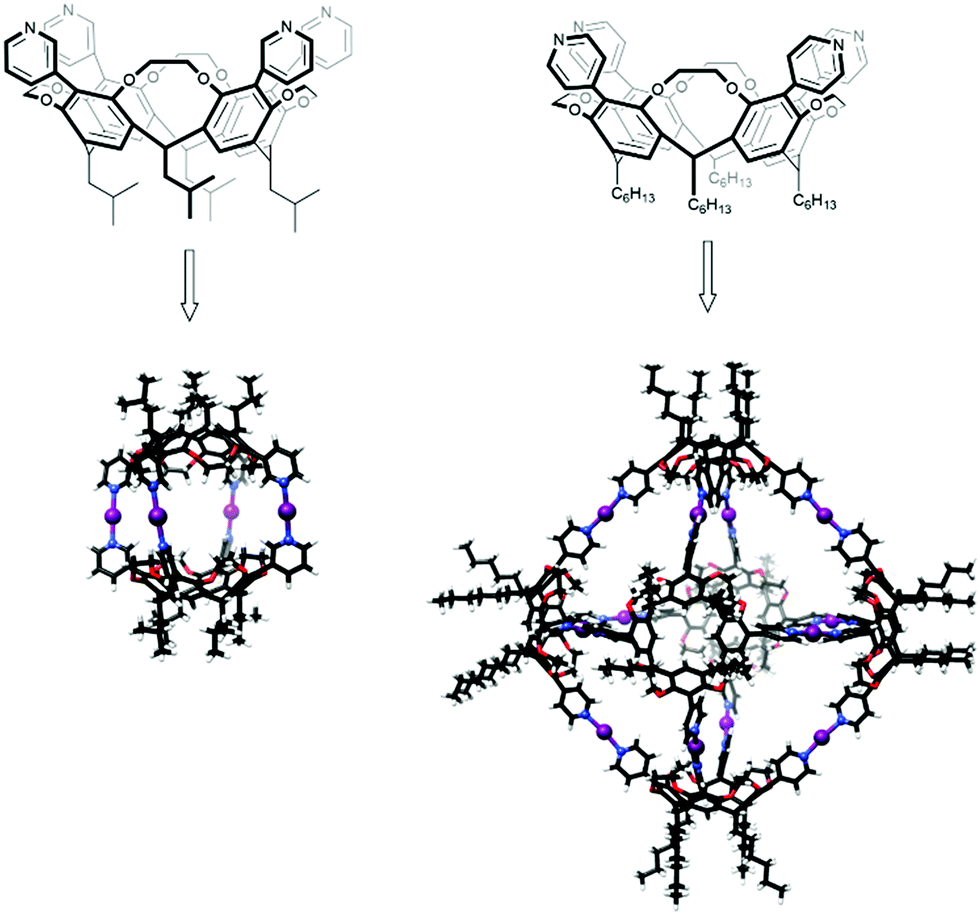 | ||
| Fig. 16 Three-center halogen-bonded dimeric and hexameric capsules.46,48 | ||
In the formation of capsular assemblies through three-center [D⋯X⋯D]+ halogen bonds, the geometry and the spatial arrangement of the Lewis bases play a critical role. Due to the linearity of the three-center [D⋯X⋯D]+ systems, a small structural difference in the Lewis base can enable a construction of assemblies with completely different geometries. Ethylene-bridged tetrakis(pyridyl)cavitands were found to provide a suitable core structure to adjust the position of the electron donor, D, in the way that the construction of capsular assemblies with different geometries and sizes could be achieved.46 By changing the position of the pyridyl N-atoms of the ethylene-bridged cavitands from meta to para (Fig. 16), [N⋯I⋯N]+ halogen-bonded octahedral hexameric capsules could be constructed. DOSY measurements confirmed the formation of a single larger species with a diffusion coefficient of 2.2 × 10−10 m2 s−1. ESI-MS measurements supported the selective formation of the halogen-bonded capsules with twelve [N⋯I⋯N]+ bonds.46 The structures of the [N⋯I⋯N]+ halogen-bonded dimeric and hexameric capsules were further confirmed and studied by a combination of ESI-MS, DT-IMS and theoretical calculations.17 Rearrangement of the hexameric into pentameric capsules upon changing the solvent from chloroform to dichloromethane was observed.17 This stimuli-responsive rearrangement was rationalized by a combination of microsolvation, space-filling, and entropic contributions.
Tripodal N-donor ligands were used to form supramolecular M3L2 and M6L4 [N⋯I⋯N]+ halogen-bonded cages.47 A dimeric M3L2 halogen-bonded cage was synthesized from the flexible 1,3,5-tris(imidazole-1-ylmethyl)-2,4,6-trimethylbenzene ligand. Even though this ligand is capable of adapting different geometries due to the flexibility of the imidazole arms, selective formation of the M3L2 [N⋯I⋯N]+ halogen-bonded cage was observed in solution by NMR spectroscopy as well as in the solid state by single crystal X-ray diffraction (Fig. 17).
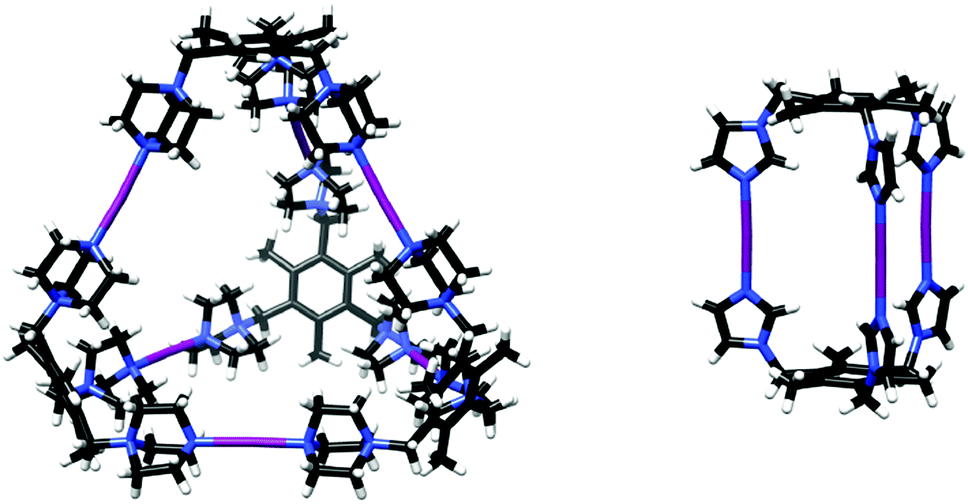 | ||
| Fig. 17 The solid state structures of M6L4 (left) and M3L2 (right) [N–I–N] cages.49 | ||
A tetrameric M6L4 cage was achieved using a rigidified cationic tris(1-methyl-1-azonia-4-azabicyclo[2.2.2]octane)-mesitylene ligand. Interestingly, even though the formation of the analogous Ag+-coordination cage was not confirmed in either solution or in the gas phase, the subsequent [N⋯I⋯N]+ halogen-bonded cage was successfully generated from the precursor solution of the Ag+-complex. The structure of the tetrameric [N⋯I⋯N]+ halogen-bonded cage was confirmed by single crystal X-ray diffraction (Fig. 17).
Halonium-ion-based single and double turn helical complexes were synthesized using oligo-aryl/pyridylene-ethynylene backbones (Fig. 18),50 and characterized by X-ray crystallography and solution NMR spectroscopy, corroborated by DFT calculations. The helical structures were stabilized by three-center halogen bonds, and by π–π interactions. These were shown to preserve their helicity in solution by NOESY measurements, as indicated by the proximity of distinct pyridyl groups. The helices exhibited homochiral self-sorting upon crystallization, and were shown to transfer iodonium ions to alkenes, demonstrated by iodocyclization of 4-penten-1-ol as a model reaction. As the larger helix has a remarkably short distance between its two iodonium ions (0.1 Å less than the sum of the van der Waals radii), it may provide inspiration for the development of highly compact halonium transfer agents with an unusually high halonium ion density.
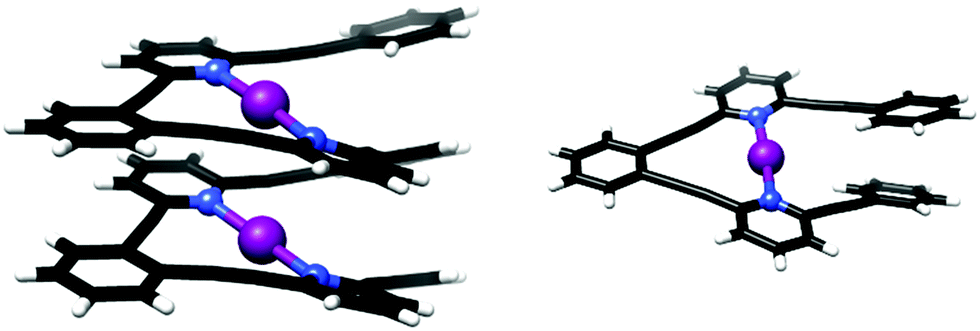 | ||
| Fig. 18 The solid state structures of the three-center halogen-bonded helices.50 | ||
Applications in organic synthesis
[Bis(pyridine)iodine(I)]+ tetrafluoroborate has been introduced by Barluenga as a mild iodine transfer reagent,51 and has been widely applied for alkenes’ 1,2-iodofunctionalization52 and iodocyclizations53–55 as well as for alcohol oxidations,56 among other applications. Due to the inertness of the tetrafluoroborate counteranion, this reagent allows halofunctionalizations with a variety of nucleophiles (Fig. 19). In the absence of added nucleophiles, 1,2-iodofluorination takes place.57 A number of halocyclisation reactions have been reported, in which [bis(pyridine)iodine(I)]+ acts as a mild I+ transfer agent (Fig. 20). Halogen bond-stabilized halonium transfer reactions have recently been reviewed by Sekar et al.58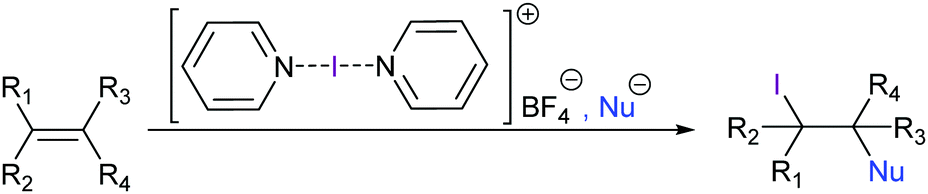 | ||
| Fig. 19 Barluenga's reagent, [bis(pyridine)iodine(I)] BF4, is a mild iodine transfer agent that allows the introduction of an iodine and an optional nucleophile into an alkene. | ||
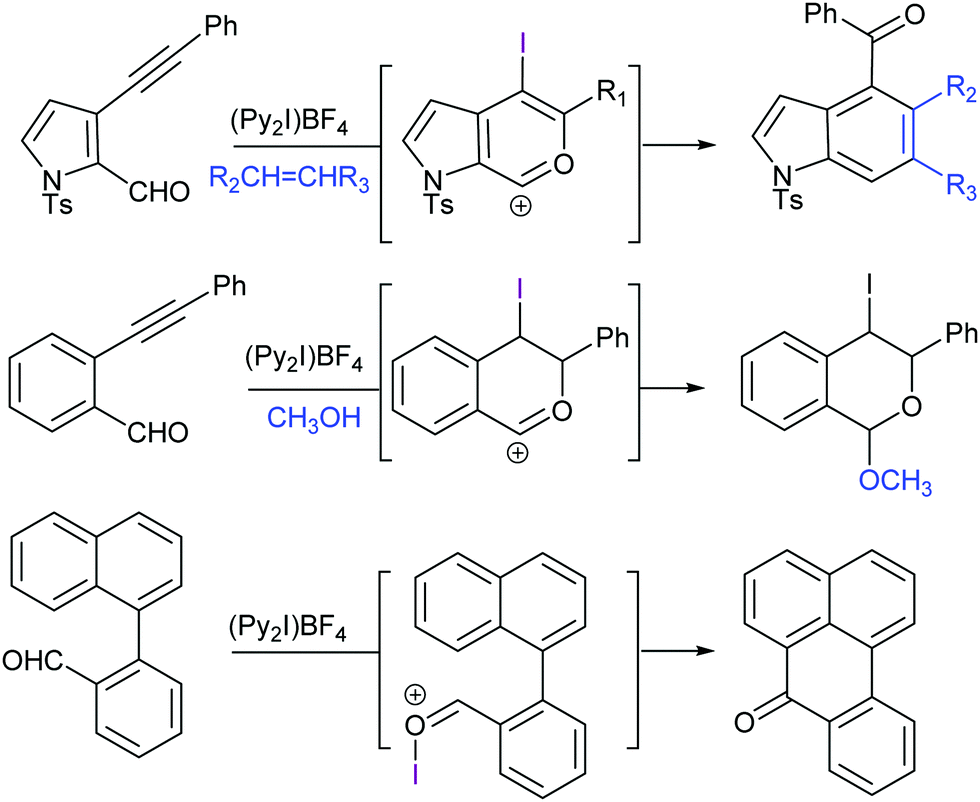 | ||
| Fig. 20 Halocyclization reactions using Barluenga's reagent are useful in the generation of complex molecules, such as indoles.58 | ||
Kinetics studies of halonium transfer from [bis(pyridine)-halogen]+ complexes to alkenes59 (Fig. 21) suggest that the reactions proceed by reversible dissociation of the three-center complex into free pyridine and an N-halopyridinium ion, which is subsequently captured by an alkene, leading to formation of a new halogen-bonded complex, shown as intermediate A in Fig. 11 (where Y is pyridine). The subsequent steps follow the general scheme depicted in Fig. 11 to provide various halofunctionalized products. Comparative kinetic investigation of substituted [bis(pyridine)iodine(I)]+ complexes revealed that the halogen transfer from electron rich complexes, such as that of 4-dimethylamino-pyridine, is slow and most likely proceeds through a different mechanism.
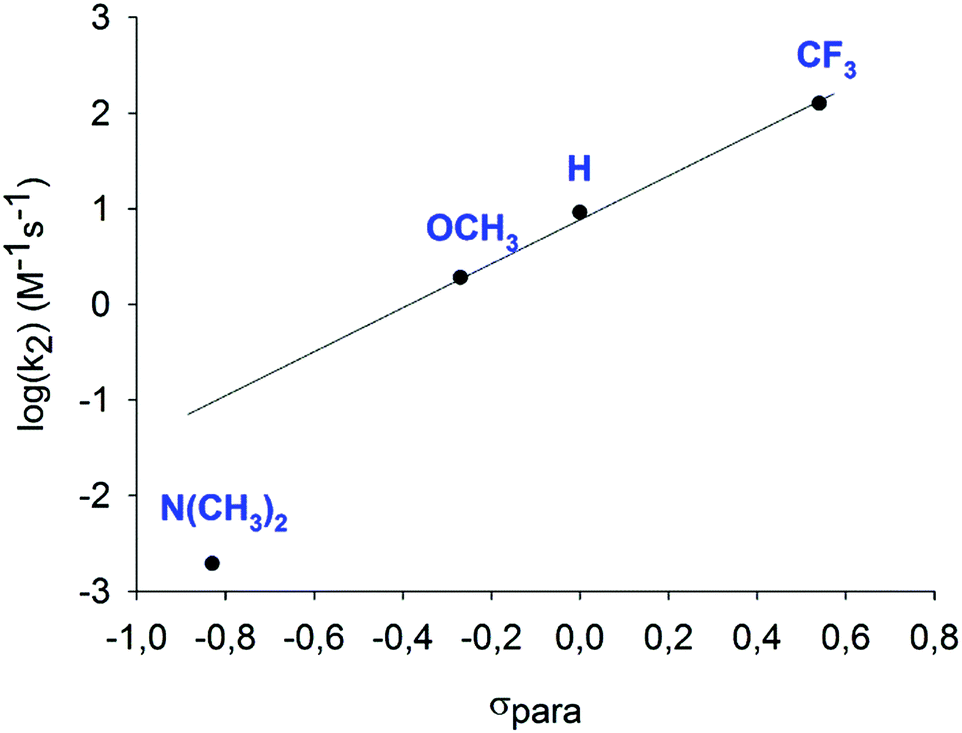 | ||
| Fig. 21 The second-order rate constants (M−1 s−1) of iodocyclization reactions of [bis(4-R-pyridine)iodine(I)]BF4 (R = H, OMe, NMe2 or CF3) reagents with 4-penten-1-ol, normalized to 1 mM, obtained in dry dichloroethane, shown as a function of the Hammett σpara constants for the 4-substituents. A linear correlation is seen for all, but the most electron rich complex suggests that the latter provides I+ transfer through a different mechanism.26 | ||
In addition to the complexes shown in Fig. 6, 2,4,6-collidine has been introduced as a Lewis base to form mild iodine(I) and bromine(I) transfer agents for halocyclisations (Fig. 22).60 [Bis(pyridine)iodine(I)]+ and [bis(collidine)iodine(I)]+ were reported to be superior for some transformations as compared to N-iodosuccinimide.60 Analogous dioxoiodane reagents [R-COO–I–OOC-R]+ possessing a three-center halogen bond with oxygen as a halogen bond acceptor, [O–I–O]−, have also been reported to be applicable in halocyclization reactions.61 Furthermore, the [N–F–N]+-type halogen bond of pyridine was reported as a plausible intermediate in fluorination initiated by Selectfluor.62
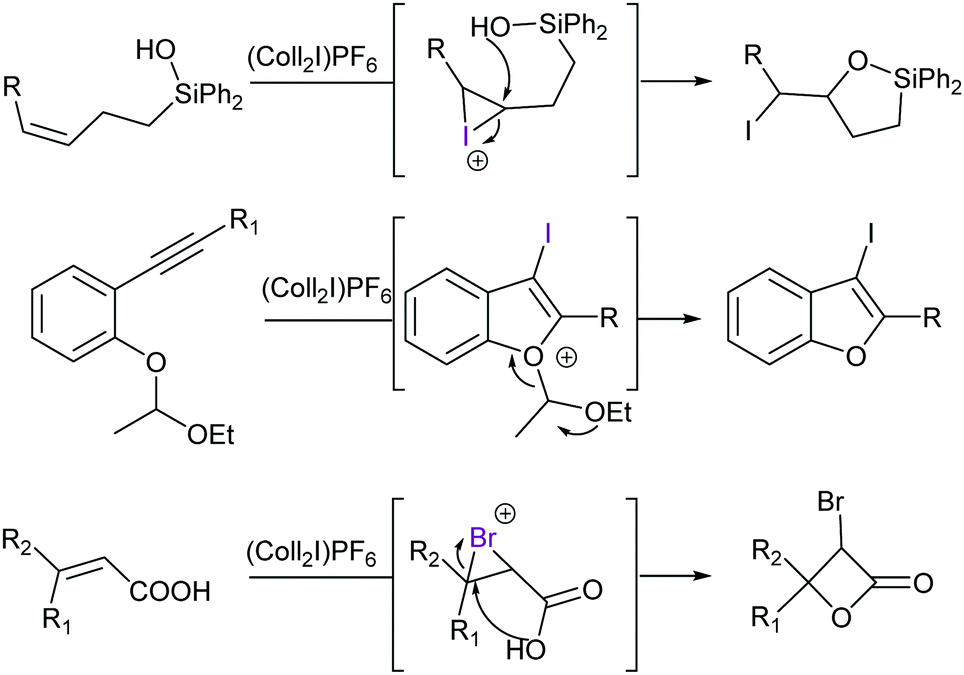 | ||
| Fig. 22 Examples of halogen(I) transfer reactions of synthetic utility, using [bis(2,4,6-collidine)iodine(I)] PF6 as a reagent (Coll = Collidine).58 | ||
Asymmetric halocyclisations using 3-center halogen bond complexes have also been attempted, yet have so far mostly given low enantioselectivities (<15%, Fig. 23),63,64 most likely due to the large distance of the reaction centre, that is I+ (A and B in Fig. 11), from the chiral moieties which in turn also had pronounced flexibility. Using chiral phosphoric acid as an anion phase-transfer catalyst, in combination with an alkyl-DABCO-type [N–I–N]+ or [N–Br–N]+ complex, up to 99% ee has been achieved. Here, however, the chiral phosphate anion was proposed to be the active catalyst, likely involving halogen bonding with an oxygen donor.66 Asymmetric halocyclisation (93% ee) has also been achieved with an [S–X–N]+-type complex as the active halonium transfer (Fig. 24).65
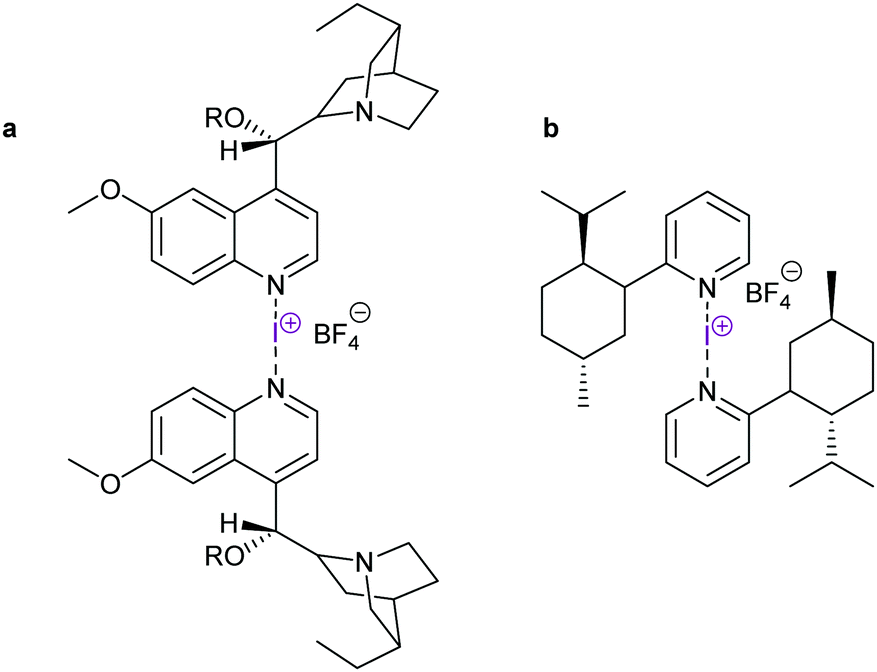 | ||
| Fig. 23 Chiral [bis(pyridine)iodine(I)] reagents have so far given low enantioselectivities in halocyclizations.63,64 | ||
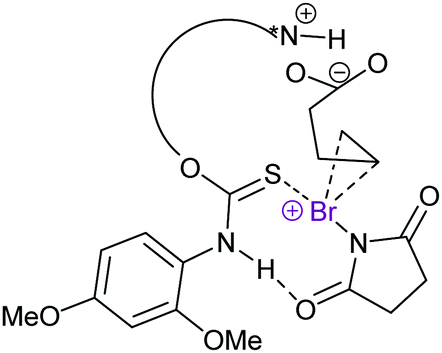 | ||
| Fig. 24 The proposed mechanism of an enantioselective halocyclisation involving an [S–X–N]+ halonium transfer agent.65 | ||
Chiral electrophilic olefin halocyclizations have also been achieved by using N-haloamides, for example, in the presence of a chiral ligand in catalytic amounts.67,68 In these examples the halonium transfer catalyst was generated upon formation of a three-center halogen bond complex (Fig. 25), with a halonium ion subsequently being transferred to a chiral phosphate catalyst.
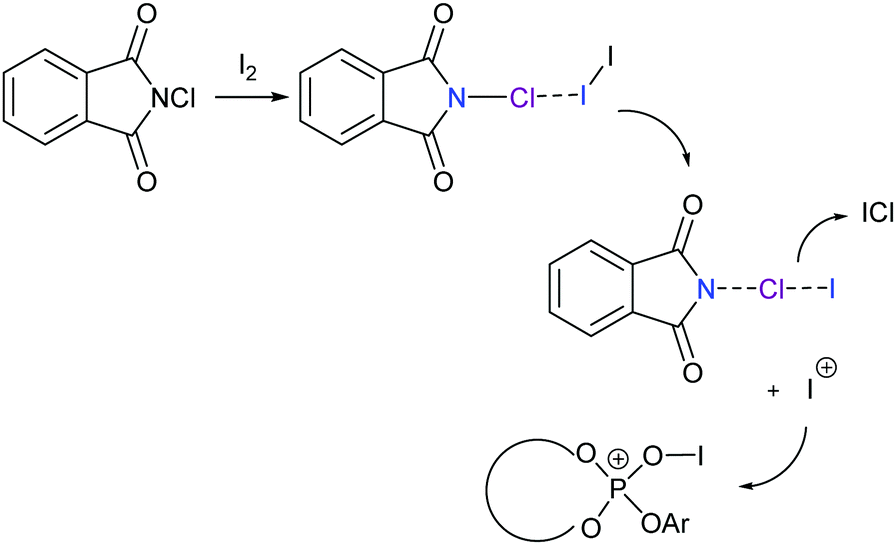 | ||
| Fig. 25 The proposed mechanism of an enantioselective halocyclisation involving an I+ generated from N-chlorosuccinimide and iodine through formation of a three-center halogen bond.67,68 | ||
Conclusions and future perspectives
Although the fundamentals of the three-center halogen bonding of halonium ions have been described already 70 years ago,43 its experimental investigations have lately intensified and its applications have just begun to take off. The nature of their three-center bonding has over time been interpreted with a variety of theoretical models,20 contributing to our current understanding of chemical bonding. The investigation of the symmetry of halonium ion's halogen bonds, that is the factors determining the position of a halonium ion in between two Lewis bases, has started to shed light on the factors governing halonium transfer processes. Subtle differences between the behavior of I+, Br+, Cl+ and F+ in three-center bonds have been reported, providing useful knowledge about their structure, stability and hence reactivity enabling novel future applications and opening up for studies aiming for a deeper understanding.Over the past decade the factors possibly influencing the geometry and the stability of this bond have been systematically studied with spectroscopic techniques, providing the ground for future developments. Halonium ions’ halogen bonds are remarkably strong and along with their directionality have been demonstrated to provide a robust tool for the design of complex supramolecular structures, such as capsules, cages and helices. Halogen bonds stabilize halonium ions, whose influence can be utilized by rational alteration of the electron density of the three-center system, to thereby modulate halonium ions’ reactivity. Synthetic applications have initially been explored by Barluenga and have begun to flourish.58 Accordingly, novel synthetic applications utilizing the gained knowledge for the development of selective halonium transfer agents and protocols are to be expected. Establishment of a conceptually new, general strategy for asymmetric halofunctionalization, with a detailed understanding of its mechanism will likely be one of the useful contributions of this research field to a major breakthrough in synthetic organic chemistry. Future applications of halonium ions’ halogen bonds in supramolecular systems are expected to provide halonium transfer agents with an unusually high halonium ion loading, and to vastly complex supramolecular architectures with applications in a wide range of scientific fields.
Similar to the recent developments within the wide field of secondary bonding,69,70 a quick transfer of knowledge from three-center halogen bonds of halonium ions to related systems is to be expected. Accordingly, three-center tetrel bonds have recently been discussed.44,71 While analogies to “short, strong” hydrogen bonds have been valuable for the discussion of the three-center halogen bonds of halonium ions, the more useful will be understanding their differences, and the differences from other types of hypervalent secondary bonds. This will provide a deeper understanding of the fundamentals of bonding, and allow identification of the most beneficial applications of each as well as utilizing their complementarity.
Overall, applications of the three-center bond of halonium ions’ are at a very initial stage. Significant knowledge of their nature and properties that has lately been collected is expected to provide the solid ground for a quickly growing number of well-thought future applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Research Council (grant 2016-03602) and FORMAS (grant 2017-01173) are gratefully acknowledged for funding.Notes and references
- G. R. H. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS
.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC
- S. B. Hakkert and M. Erdélyi, J. Phys. Org. Chem., 2015, 28, 226–233 CrossRef CAS
-
K. Rissanen and M. Haukka, in Halogen Bonding II: Impact on Materials Chemistry and Life Sciences, ed. P. Metrangolo and G. Resnati, Springer International Publishing, Cham, 2015, pp. 77–90 Search PubMed
- R. W. Troff, T. Makela, F. Topic, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS
- A.-C. C. Carlsson, A. X. Veiga and M. Erdelyi, Top. Curr. Chem., 2015, 359, 49–76 CrossRef CAS PubMed
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Gräfenstein and M. Erdelyi, Chem. Sci., 2014, 5, 3226–3233 RSC
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdélyi, Chem. Commun., 2012, 48, 1458–1460 RSC
- N. W. Alcock and G. B. Robertson, J. Chem. Soc., Dalton Trans., 1975, 2483–2486 RSC
- A.-C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Gräfenstein and M. Erdélyi, CrystEngComm, 2013, 15, 3087–3092 RSC
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, Aust. J. Chem., 2013, 66, 1179–1188 CrossRef CAS
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed
- H. Razmazma and A. Ebrahimi, J. Mol. Graphics Modell., 2018, 84, 134–144 CrossRef CAS PubMed
- A. Ebrahimi, H. Razmazma and H. S. Delarami, Phys. Chem. Res., 2016, 4, 1–15 Search PubMed
- V. Oliveira, E. Kraka and D. Cremer, Phys. Chem. Chem. Phys., 2016, 18, 33031–33046 RSC
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343–8351 RSC
- R. Kleinmaier, S. Arenz, A. Karim, A.-C. C. Carlsson and M. Erdelyi, Magn. Reson. Chem., 2013, 51, 46–53 CrossRef CAS PubMed
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topic, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdelyi, Chem. Sci., 2015, 6, 3746–3756 RSC
- R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720–1729 RSC
- C. L. Perrin, P. Karri, C. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2012, 134, 7766–7772 CrossRef CAS PubMed
- X. S. Bogle and D. A. Singleton, J. Am. Chem. Soc., 2011, 133, 17172–17175 CrossRef CAS PubMed
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed
- A. a. https://pubs.acs.org/doi/10.1021/jacs.8b09467, Further permissions related to the material excerpted should be directed to the ACS.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdelyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed
- H. U. Siehl, Adv. Phys. Org. Chem., 1987, 23, 63–163 CrossRef CAS
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigué, Faraday Discuss., 2017, 203, 389–406 RSC
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigue, Faraday Discuss., 2017, 203, 389–406 RSC
- S. B. Hakkert and M. Erdelyi, J. Phys. Org. Chem., 2015, 28, 226–233 CrossRef CAS
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigue, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS
- K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC
- L. Koskinen, P. Hirva, A. Hasu, S. Jääskeläinen, J. Koivistoinen, M. Pettersson and M. Haukka, CrystEngComm, 2015, 17, 2718–2727 RSC
- J. Lu and S. Scheiner, Chem. Phys. Lett., 2019, 731 Search PubMed
- L. Koskinen, P. Hirva, E. Kalenius, S. Jääskeläinen, K. Rissanen and M. Haukka, CrystEngComm, 2015, 17, 1231–1236 RSC
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. Carlsson, A. K. Rappe, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed
- F. Demartin, P. Deplano, F. A. Devillanova, F. Isaia, V. Lippolis and G. Verani, Inorg. Chem., 1993, 32, 3694–3699 CrossRef CAS
- E. Seppälä, F. Ruthe, J. Jeske, W.-W. du Mont and P. G. Jones, Chem. Commun., 1999, 1471–1472 RSC
- W.-W. du Mont, M. Bätcher, C. Daniliuc, F. A. Devillanova, C. Druckenbrodt, J. Jeske, P. G. Jones, V. Lippolis, F. Ruthe and E. Seppälä, Eur. J. Inorg. Chem., 2008, 4562–4577 CrossRef CAS
- G. M. de Oliveira, E. Faoro and E. S. Lang, Inorg. Chem., 2009, 48, 4607–4609 CrossRef PubMed
- R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC
- G. C. Pimentel, J. Chem. Phys., 1951, 19, 446–448 CrossRef CAS
- P. Deplano, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, Coord. Chem. Rev., 1999, 188, 71–95 CrossRef CAS
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed
- J. Barluenga, J. M. González, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1985, 24, 319–320 CrossRef
- J. Barluenga, Pure Appl. Chem., 1999, 71, 431–436 CAS
- D. S. T. Scott, A. Snyder and A. P. Brucks, Aldrichimica Acta., 2011, 2, 27–40 Search PubMed
- J. Barluenga, H. Vázquez-Villa, A. Ballesteros and J. M. González, J. Am. Chem. Soc., 2003, 125, 9028–9029 CrossRef CAS PubMed
- T. Okitsu, S. Yumitate, K. Sato, Y. In and A. Wada, Chem. – Eur. J., 2013, 19, 4992–4996 CrossRef CAS PubMed
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Chem. – Eur. J., 2004, 10, 4206–4213 CrossRef CAS PubMed
- J. Barluenga, J. M. Gonzalez, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1985, 24, 319–320 CrossRef
- S. Guha, I. Kazi, A. Nandy and G. Sekar, Eur. J. Org. Chem., 2017, 5497–5518 CrossRef CAS
- A. A. Neverov and R. S. Brown, J. Org. Chem., 1998, 63, 5977–5982 CrossRef CAS PubMed
- T. Okitsu, D. Nakazawa, R. Taniguchi and A. Wada, Org. Lett., 2008, 10, 4967–4970 CrossRef CAS PubMed
- K. Muniz, B. Garcia, C. Martinez and A. Piccinelli, Chem. – Eur. J., 2017, 23, 1539–1545 CrossRef CAS PubMed
- A. M. Hua, S. L. Bidwell, S. I. Baker, H. P. Hratchian and R. D. Baxter, ACS Catal., 2019, 9, 3322–3326 CrossRef CAS
- X.-L. Cui and R. S. Brown, J. Org. Chem., 2000, 65, 5653–5658 CrossRef CAS PubMed
- R. B. Grossman and R. J. Trupp, Can. J. Chem., 1998, 76, 1233–1237 CrossRef CAS
- L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y. Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474–15476 CrossRef CAS PubMed
- Y. M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928–12931 CrossRef CAS PubMed
- H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974–6977 CrossRef CAS PubMed
- Y. Lu, H. Nakatsuji, Y. Okumura, L. Yao and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 6039–6043 CrossRef CAS PubMed
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC
- A. C. Legon, Phys. Chem. Chem. Phys., 2017, 19, 14884–14896 RSC
- A. Karim, N. Schulz, H. Andersson, B. Nekoueishahraki, A.-C. C. Carlsson, D. Sarabi, A. Valkonen, K. Rissanen, J. Gräfenstein, S. Keller and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 17571–17579 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |