
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of yttrium in medical imaging and therapy: historical background and future perspectives
Ben J.
Tickner
 a,
Graeme J.
Stasiuk
b,
Simon B.
Duckett
a and
Goran
Angelovski
*c
a,
Graeme J.
Stasiuk
b,
Simon B.
Duckett
a and
Goran
Angelovski
*c
aCentre for Hyperpolarisation in Magnetic Resonance, Department of Chemistry, University of York, Heslington, UK
bDepartment of Imaging Chemistry and Biology, School of Biomedical Engineering and Imaging, King's College London, London, UK
cMR Neuroimaging Agents, Max Planck Institute for Biological Cybernetics, Tuebingen, Germany. E-mail: goran.angelovski@tuebingen.mpg.de
First published on 23rd July 2020
Abstract
Yttrium is a chemically versatile rare earth element that finds use in a range of applications including lasers and superconductors. In medicine, yttrium-based materials are used in medical lasers and biomedical implants. This is extended through the array of available yttrium isotopes to enable roles for 90Y complexes as radiopharmaceuticals and 86Y tracers for positron emission tomography (PET) imaging. The naturally abundant isotope 89Y is proving to be suitable for nuclear magnetic resonance investigations, where initial reports in the emerging field of hyperpolarised magnetic resonance imaging (MRI) are promising. In this review we explore the coordination and radiochemical properties of yttrium, and its role in drugs for radiotherapy, PET imaging agents and perspectives for applications in hyperpolarised MRI.
 Ben J. Tickner | Ben. J. Tickner received his MChem degree from the University of York, UK in 2017. He spent time working in the group of Priv.-Doz. Dr Goran Angelovksi at the Max Planck Institute for Biological Cybernetics, Germany before starting his PhD studies with Prof. Simon Duckett at the Center for Hyperpolarisation in Magnetic Resonance at the University of York in 2017. His current research is focused on using para-hydrogen to non hydrogenatively hyperpolarize target molecules for applications in biomedical imaging, chemosensing and reaction monitoring. |
 Graeme J. Stasiuk | Graeme J. Stasiuk received his MChem (2006) and PhD (2010) degrees from the University of Leicester, UK. Following a year at the French Alternative Energies and Atomic Energy Commission (CEA) in Grenoble, he joined Prof. Nick Long's group at Imperial College, London in 2011 as a PDRA. In 2014 he was appointed as a Lecturer in Molecular Imaging at the University of Hull. In 2020 he moved his research group to the Department of Imaging Chemistry and Biology at King's College London. His research focuses on the design, synthesis and validation of molecular imaging agents for all modalities. |
 Simon B. Duckett | Simon B. Duckett received his PhD from the University of York, UK in 1990 and afterwards undertook postdoctoral work with Prof. W. D. Jones and Prof. R. Eisenberg at the University of Rochester, USA. He returned to the University of York in 1993 and was promoted to Professor of Chemistry in 2005. Since 2010 he has been the Director of the Centre for Hyperpolarization in Magnetic Resonance at the University of York. His group studies and develops hyperpolarization techniques using para-hydrogen with a view to translation into the clinic and he has authored over 100 publications on such topics. |
 Goran Angelovski | Goran Angelovski holds a diploma degree in chemistry from the University of Belgrade, Serbia (2000) and PhD degree in chemistry from the University of Dortmund, Germany (2004). Since 2005, he has been a researcher at the MPI for Biological Cybernetics. In 2012, he obtained venia legendi (Habilitation) from the Faculty of Natural Sciences, University of Tübingen. Dr Angelovski is currently in the process of moving to the newly established Center for Excellence in Brain Science and Intelligence Technology (CEBSIT) of the Chinese Academy of Sciences (CAS) in Shanghai, to lead the Laboratory of Molecular and Cellular Neuroimaging and continue his research on the development of bioresponsive MRI contrast agents. |
Key learning points1. Versatility of yttrium coordination chemistry results in a vast number of complexes with variable physicochemical features.2. The properties of a range of yttrium isotopes enable their use in radiochemistry. 3. Yttrium radioisotopes exploited for medical imaging and radiotherapy applications. 4. Suitability of yttrium for NMR investigations and new perspectives for yttrium-based hyperpolarized MRI. |
Introduction
Since its discovery in the late 18th century, yttrium has been used as a material in lasers, superconductors, electrodes, and LEDs. The wide range of applications of yttrium-based materials has now extended into a variety of medical applications. As early as the 1960s, there were reports of using yttrium-doped lasers to remove lesions and 90Y-labelled microspheres for radioembolisation. Since then, the use of 90Y-based materials for therapy has increased and they are now used clinically to treat a range of diseases including Cushing's disease, acromegaly, haemophilia and a wide range of cancers. Rapid technological advances have given additional momentum to its use in various medicinal diagnostic and therapeutic methods. Imaging techniques especially have benefited, giving rise to the development of methodologies for the early stage detection of disease or early treatment response. Developments in the chemistry of yttrium have contributed significantly to this progress, providing valuable solutions through the preparation of contrast agents for different imaging modalities, or radiochemicals and other materials as therapeutics. Indeed, there are not many elements, especially not metal ions, which feature in the production of imaging agents or therapeutics as extensively as yttrium. Owing to the existence of various isotopes, yttrium and its complexes have found use in a wide range of diagnostics and therapy applications in different medical methodologies. The versatility of yttrium isotopes, which include 86Y and 90Y, has allowed for PET and radiotherapy, respectively, underpinned by significant research efforts and consequent reports over the past three decades. Very recently, 89Y and its complexes have received noteworthy attention in an emerging field of HP-MRI, a methodology which has now been proven able to track real-time metabolism in vivo. In turn, yttrium has shown to be an excellent platform for the preparation of diverse agents that can be used for multiple medical purposes. In this review, we aim to provide a brief historical overview of the role of yttrium in therapy and medical imaging applications, anticipating possible advances in the years to come.Coordination properties of yttrium
The element yttrium has atomic number 39 and is grouped as a transition metal in the periodic table, with electronic configuration Kr4d15s2. It exists naturally as the 89Y isotope in 100% abundance, and has a nuclear spin quantum number I of 1/2 which means it can produce very narrow peaks in corresponding 89Y NMR spectra.Yttrium has often been regarded as a rare earth element because it bears little chemical similarity to its counterparts in the d-block. Namely, while it can form metal carbon bonds (various organoyttrium species have been reported),1 its chemistry is most similar to that of the lanthanide elements. The main reason for this behaviour is the phenomenon called the lanthanide contraction, a result of poor shielding of nuclear charge by 4f electrons throughout elements in the lanthanide series (their general electronic configuration is Xe4fn5dm6s2, where n = 1–14 and m = 0–1).2 Consequently, both yttrium and the lanthanides do not exhibit the common properties of the transition metals; in fact, by strongly resembling the chemical properties of the lanthanides, yttrium itself is often regarded as a member of this series.
Some of the properties valid for yttrium and other members of this series are:
• Their overall level of reactivity is greater than that of transition metals, being instead more similar to the group 2 elements.
• They exhibit a wide range of coordination numbers.
• They form labile ionic complexes, which are prone to the facile exchange of ligands.
• Their hydroxides are insoluble at neutral pH.
• Their coordination properties are determined by the steric factors of ligands rather than crystal-field effects.
• They prefer oxidation state +3.
• They have a preference for anionic ligands with donor atoms of rather high electronegativity.
The oxidation state +3 is the most common for yttrium, although oxidation states of 0, +1 and +2 have been reported.3,4 Due to the above mentioned chemical properties, yttrium can exist in a number of complexes in which the central Y3+ ion can assume an array of different CN of up to 9 within the inner sphere. In this review, we will not cover all of the different types of complexes that can be made with yttrium, as this topic has been well explored in previous reviews and book chapters.2,5,6 Here, we will briefly mention its complexes with the most commonly used classes of multidentate ligands as the resulting products exhibit high stability, which makes them suitable for medical applications.
Common ligands used for coordination to yttrium
Yttrium and its analogous lanthanide ions exhibit toxicity in vivo.7 For this reason, it is important to ensure that complexes exhibit high thermodynamic stability, as well as being kinetically inert. This behaviour can be achieved by using multidentate ligands (Fig. 1), usually polyaminocarboxylic acids such as EDTA, DTPA, DO3A and DOTA (all abbreviations used are defined in the notes and references section at the end of the manuscript). It is important to consider ligand denticity and the chelate formation/dissociation kinetics when anticipating applications for these chelated systems. For instance, all of these ligands rapidly form complexes, which is particularly important for the production of radiopharmaceuticals. However, hexadentate EDTA complexes have neither sufficient thermodynamic stability nor the inertness required for medical utilisation. On the other hand, both octadentate DTPA and DOTA complexes exhibit high thermodynamic stability and their derivatives are widely used for the production of radiopharmaceuticals (see below).8,9 Still, one should note that macrocyclic complexes based on the DOTA ligand have considerably higher thermodynamic stability than those based on the acyclic DTPA (Table 1). Namely, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values for Y-EDTA and Y-DTPA are found to increase from 18.5 to 22.5 due to a change in denticity of the chelating ligand from 6 to 8. logK increases further to 24.3 when DOTA, an octadentate macrocyclic ligand, is employed. However, the stability is lower if the complex with the heptadentate macrocyclic ligand DO3A is formed (logK = 21.1).
K values for Y-EDTA and Y-DTPA are found to increase from 18.5 to 22.5 due to a change in denticity of the chelating ligand from 6 to 8. logK increases further to 24.3 when DOTA, an octadentate macrocyclic ligand, is employed. However, the stability is lower if the complex with the heptadentate macrocyclic ligand DO3A is formed (logK = 21.1).
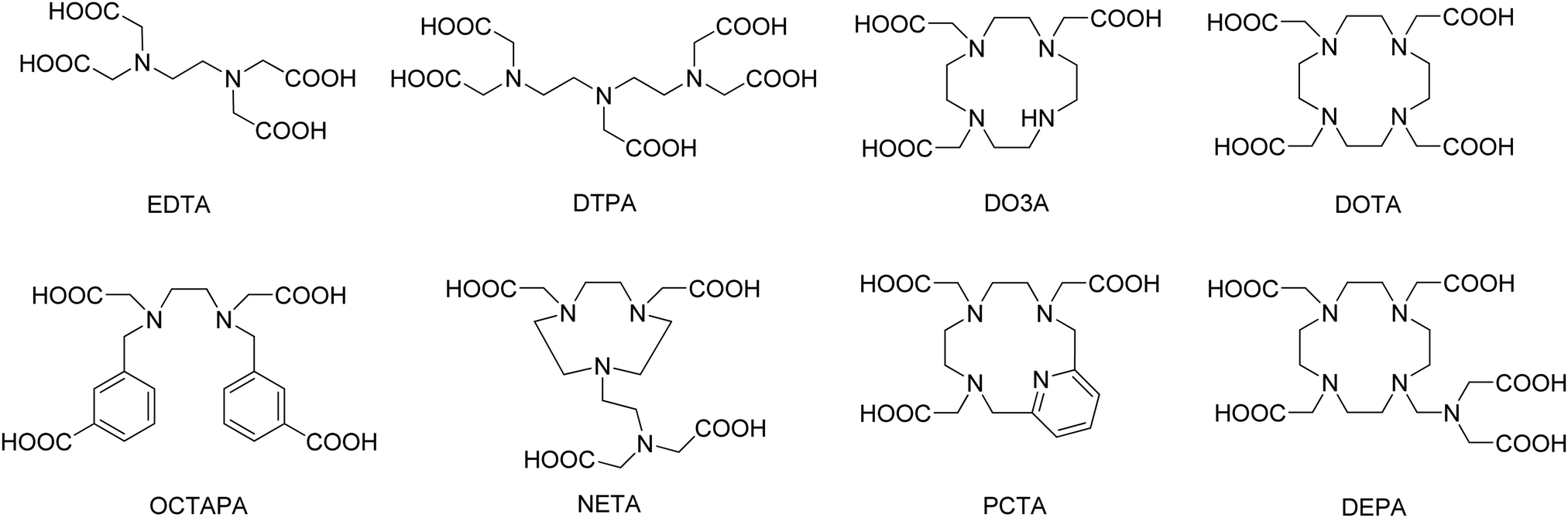 | ||
| Fig. 1 Common ligands reported for chelation to yttrium that are discussed in this work. | ||
Similarly, the kinetic stability of the complexes formed with macrocyclic ligands is significantly higher than those of their acyclic counterparts. In a recent study, the acid-assisted dissociation of Y-DOTA and Gd-DOTA complexes was followed in 2 M and 4 M HCl by HILIC and ICP-MS.10 The obtained half-life was 639 min and 312 min in 2 M HCl for the Y-DOTA or Gd-DOTA complexes respectively, while these values dropped to 230 min and 88 min respectively in 4 M HCl. These values are commensurate with high stability in harsh conditions. In parallel, the studied acyclic complexes show a lack of stability at less acidic conditions compared to the macrocyclic complexes. Specifically, at pH 6 and in the presence of an excess of competing cation (Zn2+), these complexes undergo dissociation, resulting in half-lives of 47 min and 96 min in the presence of 5 mM Zn2+ for Y-DTPA and Gd-DTPA respectively, which reduced to 20 min and 52 min in the presence of 10 mM Zn2+.
The stability of these metal ion complexes can be increased by synthesising analogues with greater backbone rigidity.11 For example, CHX-A′′-DTPA is a common derivative of DTPA that contains a rigid cyclohexane group in the DTPA backbone resulting in an increase in logK from 22.5 to 24.7 for Y-DTPA and Y-CHX-A′′-DTPA respectively.12
The geometry of the Y3+ complex is also affected by CN and ligand denticity. Complexes based on octadentate DOTA or DTPA typically adopt CN of 9 with an inner sphere water molecule providing an additional donor site. These coordination geometries can span distorted dodecahedron, TTP and SAP/TSAP for Y-EDTA, Y-DTPA and Y-DOTA complexes, respectively.2,9 TTP geometries contain ligand donor atoms arranged in triangles in three different planes, whereas SAP and TSAP isomers contain donor sites oriented in rectangles in two different planes with an additional ligand (usually water) occupying a ‘capped’ position (Fig. 2a). SAP and TSAP geometries are distinguished based on the angle between the two planes of donor ligands, which is typically smaller in TSAP structures. Information on these geometries comes most readily from X-ray crystal structures (Fig. 2b).9,13,14 Moreover, the presence of nitrogen donor atoms, and the different binding conformations exhibited by these polyaminocarboxylate ligands results in formation of multiple product isomers which can often interconvert at room temperature.8 For DTPA complexes, eight such isomers exist with the TTP geometry, which can interconvert through nitrogen inversion.15 On the other hand, macrocycle formation reduces the degrees of freedom exhibited in DOTA complexes, thus resulting in four possible isomers that assume the above mentioned SAP and TSAP structures. The latter present more restricted coordination environments, which often result in longer distances between the metal ion and any coordinated water molecules.9 Consequently, isomers existing in a TSAP geometry exhibit faster water exchange rates.8 In other words, the size and steric congestion of the central metal ion and groups attached to the DOTA ligand influence the isomers formed, which affects both the NMR and MRI properties of the desired complexes. These multiple species often display quite complex spectra in corresponding 1H NMR measurements provided isomer exchange is slow on the NMR timescale. However, low temperature NMR studies have been used to slow dynamic behaviour and investigate such isomers in solution.15
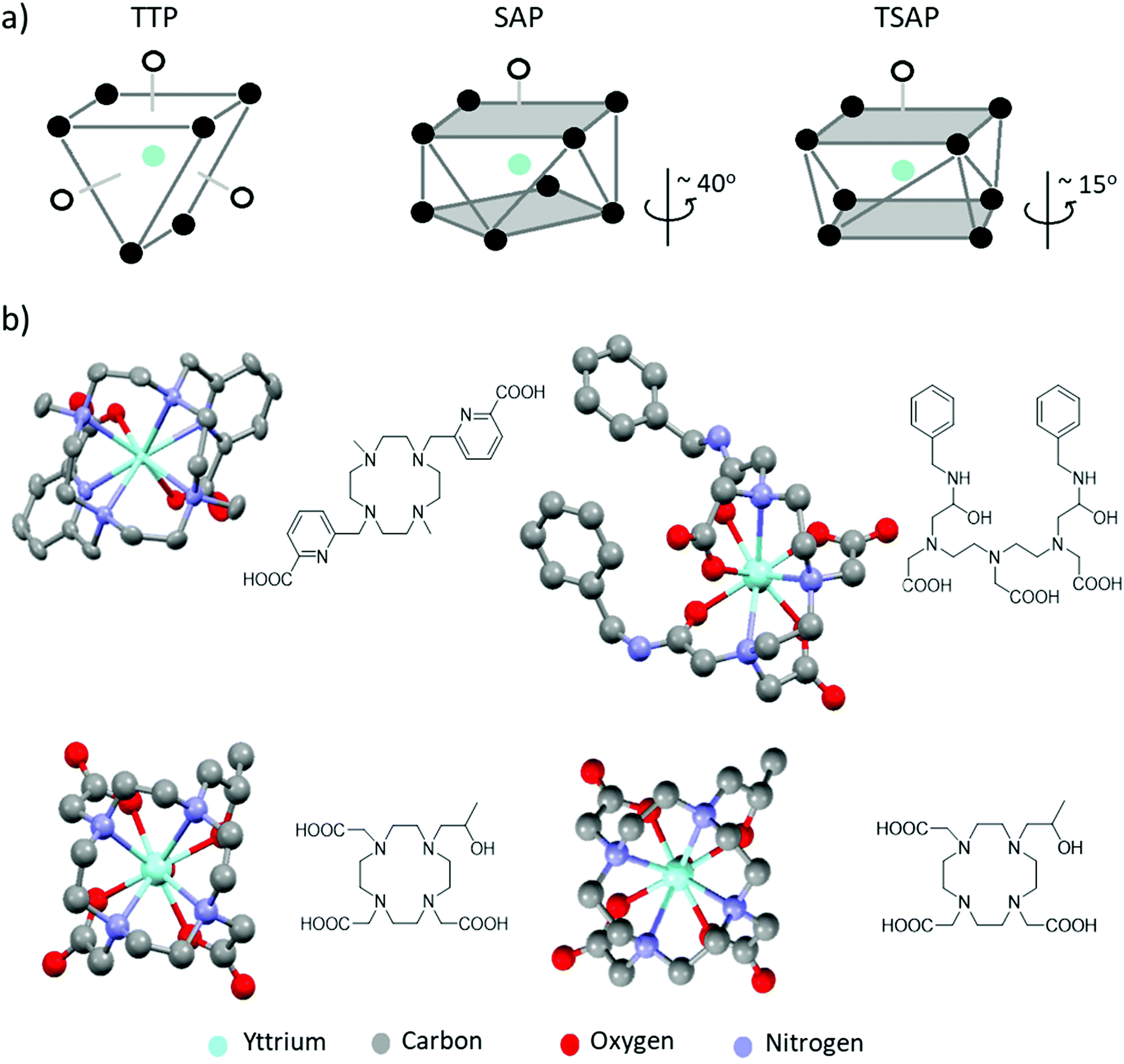 | ||
| Fig. 2 (a) Representative TTP, SAP and TSAP geometries. In SAP geometries the angle of rotation between the two planes is larger than that in TSAP isomers (b) X-ray crystal structures of a range of yttrium complexes: examples of a yttrium cyclen derived complex with CN = 8 (upper left),13 Y-DTPA-derived complex with CN = 9 adopting a TTP structure (upper right)14 and a Y-DOTA derivative existing in both TSAP (lower left) and SAP (lower right) geometries in the same unit cell.9 The coordinated water molecule in the latter three examples is partially obscured by the central yttrium atom. Note that all solvent of crystallisation, counterions and non-hydrogen atoms have been removed for visual clarity. Thermal ellipsoids are shown with 50% probability for those examples with atomic displacement parameters available from the crystallographic database. Otherwise, structures are shown as a ball and stick model. | ||
The ability of DOTA and CHX-A′′-DTPA to form stable complexes with yttrium has led to their widespread use in radiopharmaceutical applications. In fact, of all the reported examples of yttrium containing radio drugs or imaging agents (see below), the significant majority are based on DOTA or CHX-A′′-DTPA. Despite this, many researchers are developing new ligands that form stable yttrium complexes with fast radiolabelling conditions. These are typically based on PCTA, DEPA, OCTAPA or NETA ligands (Fig. 1).12 A number of derivatives, variations, and combinations of these common ligand scaffolds are possible, although a full account of all ligands reported for coordination to yttrium is beyond the scope of this manuscript and has been addressed previously.2,5,6,16 The stability or inertness of these complexes is typically determined by reference to a range of measures including thermodynamic (logK)9 and kinetic (k)17 complex formation constants, dissociation half-lives (ι)10 or pM values,16 which have been described for these and related ligands elsewhere. Beside optimising their stability to match the desired metal ion, these molecules are further developed to serve as so-called bifunctional chelators. Such systems concurrently chelate the metal, while also possessing functional groups that allow further functionalization to e.g. targeting vectors, resulting in a target specific tracer or therapeutic drug (see below).16
Considering the similarities in coordination chemistry between lanthanides and yttrium, the general properties of these ions listed previously should be noted for different potential medical imaging and therapy applications of yttrium. Principally, selecting the ligands with sufficient thermodynamic stability, fast complex formation kinetics and good inertness is sufficient if anticipating the use of yttrium complexes for preparation of radiopharmaceuticals. Nevertheless, the coordination chemistry of the Y3+ complex must also be considered if the purpose is MRI application. Namely, it is well known that highly paramagnetic Gd3+ is routinely used as a MRI contrast agent in its chelated forms with DTPA-, DO3A- or DOTA-type ligands.8 Due to similar ionic radii (90.0 pm and 93.8 pm for Y3+ and Gd3+ respectively), many studies have replaced gadolinium by yttrium and investigated the coordination properties of the resulting complexes by means of 1H NMR.17 However, the most recent applications of yttrium complexes in 89Y NMR combined with hyperpolarisation (see below) mean a more detailed consideration of its coordination properties is warranted.
Radiochemical properties and production of yttrium isotopes
Yttrium is found in many rare earth minerals rather than as the free element and is much more common in the earth's crust than many of the transition metals such as gold and silver. It was first isolated in its elemental form in 1828 and is now commonly obtained from its ores by dissolution in sulphuric acid and fractionation of the different metal ions by ion exchange chromatography. Subsequent addition of oxalic acid allows for the isolation of yttrium oxalate precipitates. From this, oxidation can yield yttrium oxide while further reaction with hydrogen halides yield yttrium halide salts.2 In 2014, around 7000 tonnes of yttrium oxide were produced each year, most of this by China. Yttrium is found in one naturally occurring isotope: 89Y, although other isotopes including 90Y and 91Y are found as waste products from uranium fission. While 89Y is found in many rare-earth minerals and can be found in trace amounts in living systems, it has no known role in biological processes. Studies on the toxicology of 89YCl3 in rats showed an increase in blood calcium concentration upon yttrium addition suggesting the replacement of calcium in bone by yttrium.7 Calcium deposits in the liver and spleen suggest these organs are the primary targets of intravenously injected yttrium.Currently, no fewer than 34 radionuclides of yttrium, from 76Y to 109Y, have been synthesized and observed.18 While 86Y, 87Y, 88Y, 90Y and 91Y have half-lives of 14.7 hours, 79.8 hours, 106.6 days, 64.1 hours, and 58.5 days respectively, all other yttrium isotopes (excluding the naturally abundant 89Y) are much shorter lived with half-lives of less than a few hours. The main isotopes of yttrium can be grouped into two types depending on their main decay processes (β− or β+) and their half-lives (Table 2).
| Isotope | Abundance | Half live | Main decay process | Main decay product | Applications | Ref. |
|---|---|---|---|---|---|---|
| 85Y | Synthetic | 2.7 hours | β+ | 85Sr | N/A | 18 |
| 86Y | Synthetic | 14.7 hours | β+ | 86Sr | PET | 18 and 20 |
| 87Y | Synthetic | 3.4 days | β+ | 87Sr | PET | 18 and 20 |
| 88Y | Synthetic | 106.6 days | β+ | 88Sr | PET | 18 and 20 |
| 89Y | Natural | Stable | N/A | N/A | Lasers, HP-MRI | 18 |
| 90Y | Synthetic | 2.7 days | β− | 90Zr | Radiotherapeutics | 18 and 19 |
| 91Y | Synthetic | 58.5 days | β− | 91Zr | N/A | 18 |
| 92Y | Synthetic | 3.5 hours | β− | 92Zr | N/A | 18 |
| 93Y | Synthetic | 10.2 hours | β− | 93Zr | N/A | 18 |
The neutron rich isotopes (90Y and above) decay predominantly via β− decay (electron emission) in which a neutron is converted into a proton releasing an electron and an anti-neutrino to form zirconium species. The kinetic energy released from rapid deceleration of these high energy electrons through interaction with surrounding matter produces radiation, which can be converted into photons. Alternatively, decay of those proton rich isotopes (88Y and below) can occur via β+ decay (positron emission), in which protons convert into neutrons releasing a positron and a neutrino to form strontium species. Those radioisotopes that exhibit β+ decay can be used for PET imaging, which typically includes isotopes such as 11C, 13N, 15O, 18F, 68Ga or 89Zr.
90Y is a good candidate for radiotherapy as it releases high energy β− particles that have strong penetration of the surrounding tissues (11 mm).12 These particles can affect cell viability by causing direct damage to the structure of DNA, or they can cause indirect cell death by increasing the concentration of toxic free radicals present in cells.6 The conjugation of such radioisotopes to antibodies or other cancer directing groups help restrict secondary damage and focus the benefits to areas of cancer; this forms the basis of radiopharmaceuticals which are discussed later.
Many radioactive yttrium isotopes have important medical uses with 86Y being used for PET imaging and 90Y for radiotherapeutics. The synthesis, separation, and isolation of these isotopes is clearly very important. A summary of the first report of each synthetic yttrium isotope, and its initial synthesis, is given by Nystrom et al.18 Briefly, 90Y isotopes were first made in 1937 from high energy neutron bombardment of 89Y, although separation of 90Y from 89Y is extremely challenging. For in vivo applications, 90Y is now more commonly made from 90Sr, one of the waste products from the fission of 235U (Scheme 1).6,19
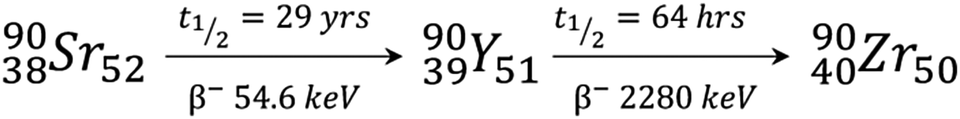 | ||
| Scheme 1 Radiochemical reaction showing 90Y formation from its parent 90Sr isotope and its decay into stable 90Zr. | ||
For medical applications, it is important that 90Y can be synthesised free from its parent 90Sr isotope. 90Sr displays similar behaviour to calcium and is also known to be deposited in bone causing a range of health concerns including leukaemia and bone cancers. Separation of this toxic precursor is therefore essential for any use of 90Y as an in vivo probe. Methods were developed to separate these two ions as early as the 1950s, using ion exchange principles to exploit the different charges and preferred oxidation states of strontium and yttrium salts.19
Lighter radioactive isotopes of yttrium including 86Y and 88Y are often produced by neutron bombardment of 89Y.18,20 There are three main precursors used for the synthesis of isotopes such as 86Y, which include enriched 86Sr, 88Sr, and natural Rb (Scheme 2). Bombardment of Rb with alpha particles to form 86Y and the (p,3n) reaction in which 88Sr nuclei absorb a proton and release 3 neutrons are not commonly used due to the high 30–55 keV energies of the alpha particle (α) or protons necessary. These energies are higher than those that can be produced in small synchrotrons.20 Commonly, the (p,n) reaction from an enriched 86Sr precursor is used due to the high purity of the resulting 86Y.20 Bombardment of a typical 86SrCO3 target in the presence of HCl, NH4OH, H2O, and La3+ yields mixtures containing 86Y(OH)3, which are typically separated using centrifugation and ion exchange chromatography. Subsequent addition of α-hydroxybutyrate and heating yields carrier-free 86Y.20 Alternatively, separation of Sr(OH)3 and Y(OH)3 can occur via electrochemical methods.20
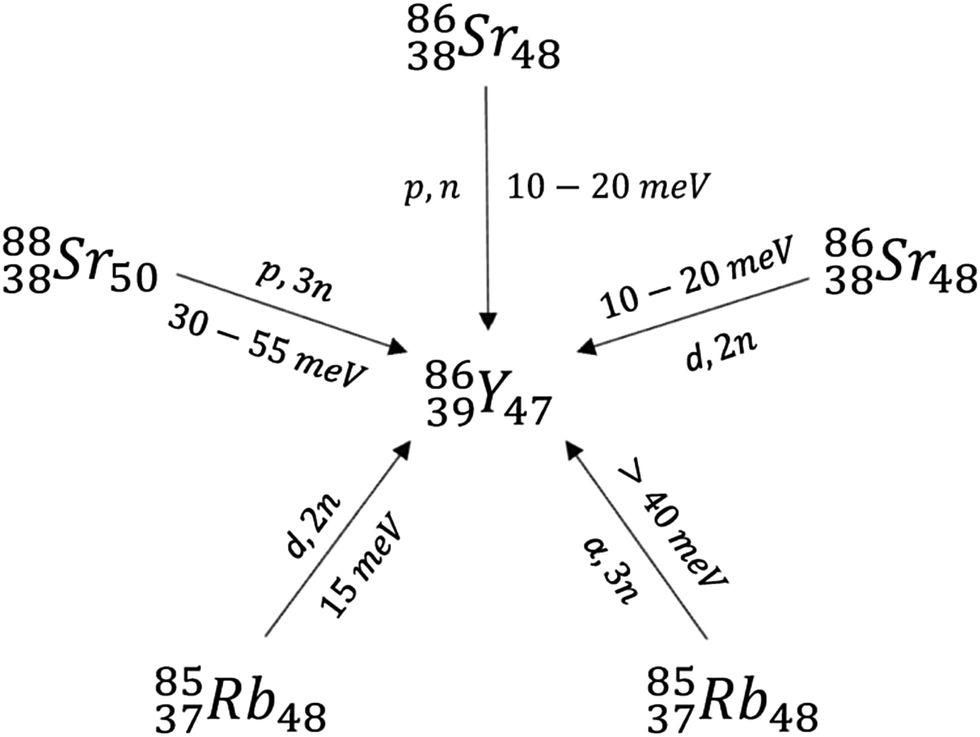 | ||
| Scheme 2 Routes that lead to the formation of 86Y isotopes. | ||
Similar isotopes including 88Y can also be produced by irradiation of a SrCl2 target and subsequent separation using a cation exchange resin. These separation techniques are highly effective and can give batch yields of 3.5 GBq and 35 MBq for 86Y and 88Y respectively with low impurities (<3% 87Y and <0.01 ppm Sr).20 The production of many different yttrium isotopes using cyclotron-based approaches, and their separation is an active area of research.
Yttrium-containing materials for medical purposes
Yttrium has a wide range of medical uses including as radiopharmaceuticals, lasers, and implant coatings. A range of yttrium containing materials including yttrium fluoride nanoparticles have antibacterial properties and can be used to reduce bacterial colonisation of implanted surfaces.21 The use of yttrium stabilised zirconia as materials for dental implants has also been reported (Fig. 3a).22 Here, yttrium finds use in stabilising cubic polymorphs of zirconia which are usually unstable because of the large changes in volume that result upon moving between different polymorphs. This is prevented by substitution of Y3+ into the crystal lattice of the smaller Zr4+ ion to produce doped zirconia that is stable over a wider temperature range.22 | ||
| Fig. 3 The use of yttrium in materials for medical purposes. (a) Radiographic control of a yttrium stabilised zirconia dental implant 24 months after surgery. Reproduced with permission from ref. 22. Copyright © 2012, Medicina Oral S. L. (b) and (c) Examples of typical scanning electron microscope (SEM) images of yttrium-containing microspheres. Reproduced with permission from ref. 25. Copyright © 2014, Springer Nature. | ||
The addition of yttrium to enhance material properties is also observed in the operation of neodymium doped yttrium aluminium garnets (Nd:YAG), which were first demonstrated in 1964 and have since been used to treat a wide range of medical ailments.23 Nd:YAG lasers emitting light at 1064 nm are most commonly used for thermotherapy, in which lesions can be removed from a surface through laser ablation.23 Lasers of this type can cut tissue with high precision, without causing thermal damage to the surrounding areas due to the absorption of laser light by water in nearby tissues. In the 1980s, bladder, penile, and oesophageal tumours were treated with endoscopic laser therapy and the range of cancers treated using this approach has increased since then.23 Such lasers are also used in minor cosmetic procedures ranging from laser hair removal and wart removal to liposuction.
Other yttrium based therapies have been developed that rely not on the addition of naturally occurring 89Y to enhance the properties of materials such as lasers or ceramic coatings, but rather to exploit the radiochemical properties of radioactive 90Y in materials or complexes. Some of the earliest uses of 90Y agents include its incorporation into microspheres for radioembolisation, which has been used clinically since the 1960s. Radioembolisation is a non-invasive method used to treat cancers or internal bleeding, in which a radionuclide in a microsphere is used both to block blood delivery to a site of cancer, and to deliver the β− radiation necessary to promote cancer cell death. This is commonly used to treat unresectable cancers (which cannot be completely removed by surgery), in organs such as the liver and involves injection of the microspheres into the hepatic artery.24 Currently, microspheres are made from either glass24 or resin (Fig. 3b and c).22,25 The diameter and therefore the properties and radioactive doses of these microspheres can be altered to achieve the desired properties. Commonly, these microspheres are synthesised via sol–gel chemistry or spraying techniques to achieve beads containing yttrium oxides of a desired diameter.25 Subsequent neutron bombardment yields the radionuclide 90Y.
90Y can be incorporated into a range of other materials including needles and rods. 90Y rods have been implanted into the pituitary gland in order to treat Cushing's disease.26 This disease results from heightened levels of the hormone cortisol caused usually by steroid overuse or adrenal or pituitary tumours. Implantation of 90Y can have therapeutic effects on such pituitary tumours26 and reduce the effects of acromegaly, a hormonal condition caused by excess production of growth hormone by the pituitary gland.27 Other 90Y containing materials such as 90Y needles have been used for synvectomy. This technique removes synovial tissues around joints which often become inflamed in conditions like rheumatoid arthritis. This inflamed tissue can be removed surgically or by using drugs or radiotherapeutics such as 90Y to reduce the mass of inflamed tissue.28 Injection of 90Y into joints of haemophilia patients has been found to induce fibrosis and reduce internal bleeding in joints.29 Such treatments have previously used radioactive isotopes such as 198Au, although isotopes including 90Y, 186Re and 169Er are preferred. Agents such as 90Y silicate have a particle size of 0.1 μm which is around 50 times larger than those of the previously used colloidal 198Au system which prevents unwanted spread of the agent to other sites via the lymphatic system.29
Yttrium-containing complexes for therapy
Clinically approved radioagents based on 90Y have been used for the treatment of various types of cancers. These are based on the conjugation of 90Y chelators such as DTPA or DOTA to cell receptor targeting motifs (peptides/antibodies) to direct the therapy to the cancer cells. For example, the conjugation of DTPA or DOTA to different peptides has been used to target the chelate ions to different sites in the body. Similarly, Gd3+ complexes based on DOTA and its analogues have been conjugated to peptides for use as targeted MRI contrast agents. 86Y complexes have been used to image the biodistribution and uptake of the analogous Gd3+ or 90Y complex (see below).10 Several excellent reviews on some of these agents, and the clinical studies that have been performed, exist.5,6 Hence, here we provide only a general overview of the different types of 90Y-radiopharmeceuticals and how they work.There are several key factors in the selection of a chelator with targeting peptide/antibody for effective therapy:
• Radiolabelling conditions for the chelator.
• Kinetic/thermodynamic stability of the complex.
• Radiation half-life compared to biological half-life.
Radiolabelling conditions for 90Y complexes vary with the choice of chelator. DOTA-based chelators all use conditions between pH 4.5 to 6, with heating as high as 100 °C for 15 minutes to form stable complexes (Tables 3 and 4), with a high molecular activity (45–75 GBq μmol−1). These harsh conditions are tolerated by small peptide targeting vectors, indicated by the use of DOTA in somatostatin radiotherapy (discussed below). DTPA has been shown to be labelled with 90Y at room temperature at pH 5.5 between 5–30 minutes, giving molecular activities of around 781 MBq mg−1. The resulting complex shows sufficient stability to be utilised in vivo. The key here is that the complex is formed at room temperature, so it can be used for antibody labelling, which is more sensitive to elevated temperatures. The half-life of 90Y is 2.7 days (Table 2), which is consistent with the biological half-life of antibodies; therefore, DTPA is more suited for conjugation to antibodies and radiolabelling with 90Y.
| Name | Chelator | Bioconjugate | Target | Labelling conditions | Dose | Disease | Studies | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Any abbreviations used are defined in the notes and references section at the end of the manuscript. | ||||||||
| Microspheres | N/A | N/A | N/A | Neutron bombardment of 89Y microspheres for 4 days.22 | — | Hepatocellular carcinoma | Human | 24 |
| 100–150000 rad |
Cushing's disease | 26 | ||||||
| — | Acromegaly | 27 | ||||||
| 185 MBq | knee arthritis | 28 | ||||||
| 185–2597 mBq (knee) | Haemophilia | 29 | ||||||
| 74–111 mBq (elbow) | ||||||||
| 90Y-DOTATOC | DOTA | DPhe1-Tyr3-octreotide | Somatostatin | 0.4 M NH4OAc or 0.2 M NaOAc pH 5 90 °C 25 min30 | 6–32 GBq m−2 | Neuroendocrine tumours | Human | 30 |
| 90Y-DOTATATE | DOTA | Tyr3-Thr8-octreotate | ||||||
| 90Y-DOTALAN | DOTA | lanreotide | 30 and 32 | |||||
| 90Y-DOTANOC | DOTA | 1-NaI3-octreotide | 33 | |||||
| 90Y-DOTAOC | DOTA | Octreotide | 33 | |||||
| 90Y-SMT487 | DOTA | DPhe1-Tyr3-octreotide | — | — | Primates | 35 | ||
| 90Y-Citrate | Citrate | N/A | N/A | — | 1 MBq | Prostate cancer (bone metastases) | Human | 35 |
| 90Y-EDTMP | EDTMP | N/A | N/A | — | 1 MBq | Prostate cancer (bone metastases) | Human | 35 and 48 |
| 90Y-Cetuximab | DOTA | Cetuximab | EGFR | — | 1 MBq | Head and neck cancers | Human | 37 |
| 90Y-DOTA-panitumumab | DOTA | Panitumumab | — | 3.7 MBq | Head and neck cancers | Human | 38 | |
| 90Y-CHX-A′′-DTPA-trastuzumab | DTPA | Trastuzumab | HER2 | 0.2 M NH4OAc pH 5.5 37 °C 1 hour | ∼3.7 MBq | HER2 tumours/ovarian cancer | Mice | 39 |
| 90Y-Octapa-trastuzumab | Octapa | Trastuzumab | ||||||
| 90Y-3p-C-NETA-trastuzumab | NETA | Trastuzumab | 0.25 M NH4OAc pH 5.5 RT 5 min | — | Breast cancer | Mice | 12 | |
| Zevalin | Tiuxetan | Ibritumomab | CD20 | 50 mM NaOAc RT 5 min | <1.2 GBq | Non-Hodgkin's lymphoma | Human | 36 |
| 90Y-Rituximab | DOTA | Rituximab | — | 14.8 MBq kg−1 | Nodular lymphocyte predominant Hodgkin lymphoma | 40 | ||
| Name | Chelator | Bioconjugate | Target | Labeling conditions | Dose | Disease | Studies | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Any abbreviations used are defined in the notes and references section at the end of the manuscript. | ||||||||
| 86Y-Citrate | Citrate | N/A | N/A | — | 100 MBq | Prostate cancer (bone metastases) | Human | 10 and 35 |
| 86Y-DTPA | DTPA | N/A | N/A | Citrate buffer pH 6 80 °C 5 min | 11.8–14.7 MBq | — | Rats | 10 |
| 86Y-DOTA | DOTA | N/A | N/A | Citrate buffer pH 6 80 °C, 30 min | 11.8–14.7 MBq | — | Rats | 10 |
| 86Y-EDTMP | EDTMP | N/A | N/A | — | 130–295 MBq | Prostate cancer (bone metastases) | Human | 35 and 48 |
| 86Y-SMT487 | DOTA | DPhe1-Tyr3-octreotide | Somatostatin | — | 10–40 MBq | Neuroendocrine tumours | Primates | 35 |
| 86Y-DOTATOC | DOTA | DPhe1-Tyr3 -octreotide | 0.15 M NH4OAC pH 4.5 100 °C 15 min | 77–186 MBq | Neuroendocrine tumours | Human | 35 and 44 | |
| 86Y-CHX-A′′-DTPA-octreotide | CHX-A′′-DTPA | CHX-A′′ | — | — | — | Neuroendocrine tumours | Mice | 44 |
| 86Y-DOTA-ReCCMSH(Arg11) | DOTA | ReCCMSH(Arg11) | Alpha-melanocyte stimulating hormone | — | — | Melanoma | Mice | 44 |
| 86Y-MP2346 | DOTA | MP2346 | Gastrin releasing peptide receptor | — | — | PC-3 tumours | Mice | 44 |
| 86Y-CHX-A′′-DTPA-cetuximab | CHX-A′′-DTPA | Cetuximab | HER1 receptors | 0.1 M NH4OAC pH 5–6 RT 30–60 min | 2 GBq mg−1 | Mesothelioma Tumors | Mice | 12 |
| 86Y-CHX-A′′-DTPA-panitumumab | CHX-A′′-DTPA | Panitumumab | HER1 receptors | — | 2 MBq m−2 | HER1 tumours | Mice | 44 |
| 86Y-1B4M-DTPA-trastuzumab | DTPA | Trastuzumab | Ovarian cancer cells | — | — | Ovarian and colorectal tumours | Mice | 44 |
| 86Y-CHX-A′′-DTPA-antimindin/RG-1 | CHX-A′′-DTPA | Antimindin/RG-1 | Mindin/RG-1 | 0.1 M NH4OAC pH 5–6 RT 30–60 min | 29.6–39.6 MBq mg−1 | LNCaP tumours | Mice | 12 and 46 |
| 86Y-CHX-A′′-DTPA-bevacizumab | CHX-A′′-DTPA | Bevacizumab | VEGF-A | 0.15 M NH4OAC pH 7 RT 30 min | 1.8–2.0 MBq | Colorectal carcinoma | Mice | 12 and 47 |
| 87Y-EDTMP | EDTMP | N/A | N/A | — | 2 MBq | — | Mice | 49 |
90Y based small molecule agents utilising the coordination chemistry properties of yttrium, rather than its incorporation into bulk materials, have been developed to deliver the β− radiotherapy to a specific cell type. Cancerous cells are known to overexpress receptors on their cell surfaces for a wide range of different proteins. This has formed the basis of drug and peptide receptor radionuclide therapy (PRRT). One such overexpressed receptor is that for somatostatin, a growth hormone-inhibiting hormone, which inhibits the release of a range of hormones including insulin and glucagon via its interaction with G-coupled protein receptors.30 Conjugation of radionuclides to somatostatin analogues allow for incorporation of the agent into the cancerous cell, localising β− emission in the area of cancer. Somatostatin was an important synthetic target for many years and many groups used solid phase approaches to synthesise this natural product. A more potent somatostatin analogue called octreotide was first synthesised in 1979 and derivatives have since been conjugated to 90Y coordinating motifs such as DTPA and DOTA derivatives. Currently, 90Y-DOTATOC is the most commonly reported 90Y radiotherapy that targets somatostatin receptors (Fig. 4).30 It has commonly been used to treat tumours of the endocrine or nervous systems although other cancers such as glioma and meningioma have also been targeted.30 Similar systems such as 90Y-DOTATATE,3190Y-DOTALAN,32 and 90Y-DOTANOC33 have also been reported, which involve the conjugation of 90Y to other octreotide-like peptides that target somatostatin receptors. The selection of either the chelator or the final charge of the complex can alter the biodistribution of the therapeutic agent (or imaging agent, see later), resulting in different levels of accumulation in target sites.
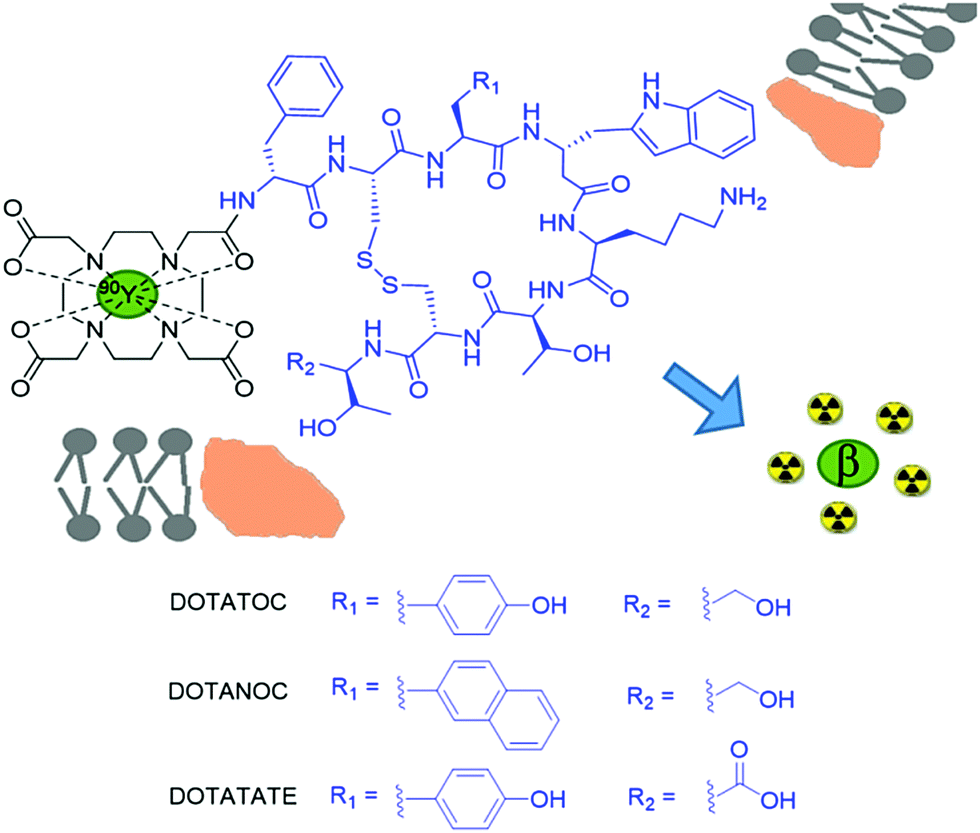 | ||
| Fig. 4 90Y complexes for targeted therapy. Conjugates of 90Y and its macrocyclic chelator DOTA (black) with an octreotide-like peptide (blue) can bind to over expressed somatostatin receptors on cancer cell surface membranes. Upon transport into the cancerous cell, the emitted β radiation has therapeutic effects. | ||
It must be stated that there has been a switch in the clinic towards 177Lu in PRRT, which is due to the β− range decreasing from 11 mm to 2 mm, thus reducing non-specific cross fire side effects.12 This move has also brought the use of combination therapy, using both 90Y-DOTATATE and 177Lu-DOTATATE, which aid these dosimetry calculations, although this matched pair has advantages compared to using just 90Y-DOTATATE.31 The same is valid for the 90Y-DOTATOC analogue.34
An accurate prediction of the radiation dose provided by exposure to these 90Y agents is challenging. In the 1990s analogous 111In agents were used although this isotope is now rarely deployed due to differences in the biochemical behaviours of 90Y and 111In that have since been observed.12 Attempts to reduce the dose administered (3.7 GBq m−2) of 90Y PRRT, have resulted in the match pair isotope 86Y being more commonly used to map where the therapy will go in vivo via PET (see below). This allows clinicians to monitor in each patient where the somatostatin positive tumours and the non-specific localisation of the tracers are, such that an appropriate dose of 90Y can be adminstered.35
In recent years, many other over expressed surface receptors on cancer cells have been targeted for radiotherapy with 90Y complexes. Many lymphomas express specific antigens (CD20) that are not expressed on normal cell surfaces. This provides a route for targeted radiotherapy in which radionuclides are attached to antibodies specific to cancer cells. Since the development of 90Y-DOTATOC, antibody conjugates such as 90Y-ibritumomab tiuxetan (Zevalin) have been developed for this purpose, consisting of a 90Y coordinating group (tiuxetan) and the anti-CD20 antibody ibritumomab (Fig. 5a).36 In addition to 90Y loaded microspheres and 90Y-DOTATOC, Zevalin has also received FDA approval for human use (in 2002) and has been shown to improve survival rates in patients (over a 7 year period).36
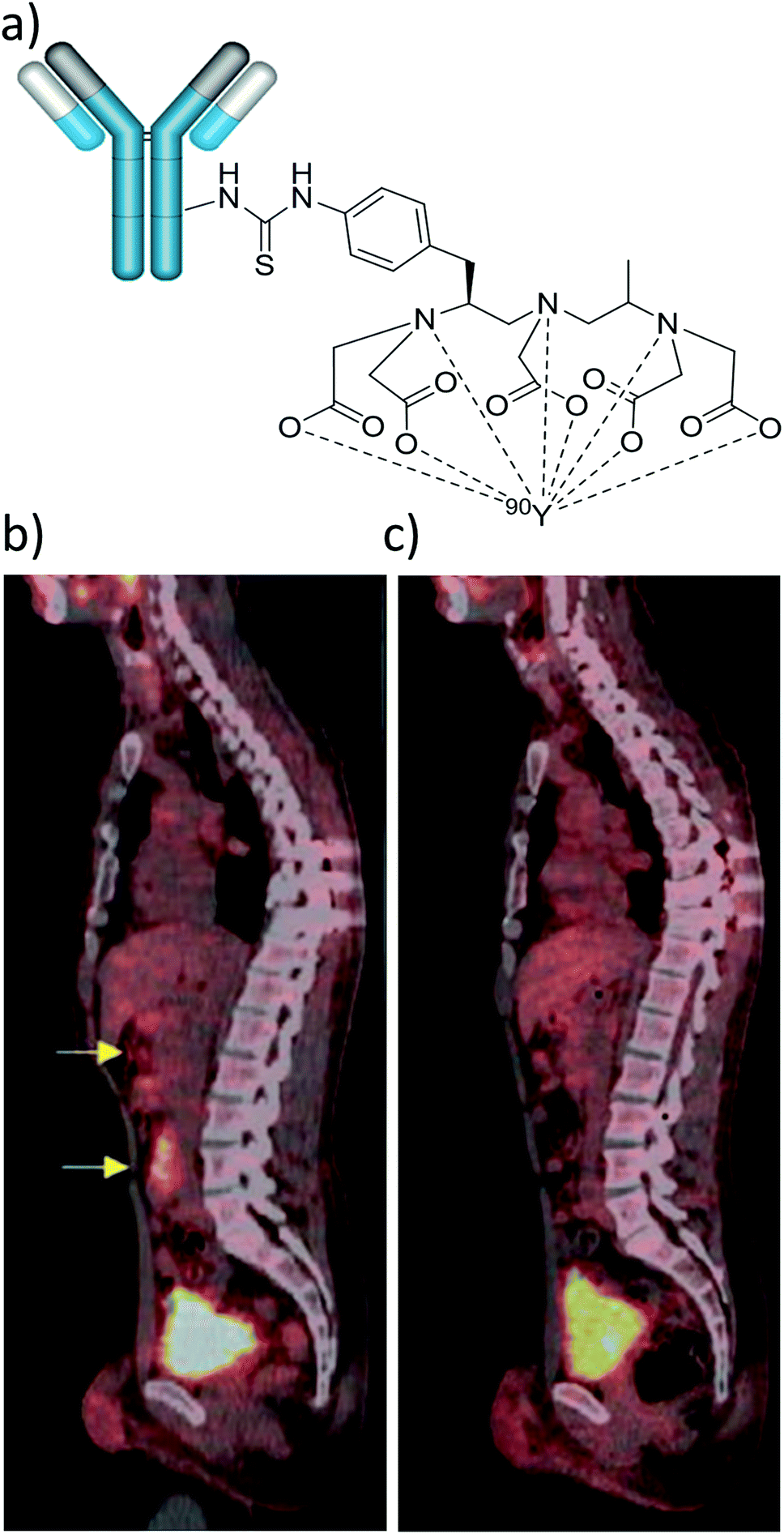 | ||
| Fig. 5 Applications of 90Y labelled antibody-based therapy. (a) Depiction of Zevalin, a conjugate of anti-CD20 antibody ibritumomab and the DTPA-derived 90Y chelator tiuxetan. (b) and (c) Example of 18FDG PET/CT sagittal slices showing retroperitoneal lymph nodes (yellow arrows) of a 38 year old male patient with nodular lymphocyte-predominant Hodgkin lymphoma (b) before and (c) 6 months after treatment with 90Y-rituximab radioimmunotherapy. Adapted with permission from ref. 40. Copyright © 2018 John Wiley & Sons A/S. | ||
Over recent years many other antibody based 90Y therapies have been developed involving cetuximab,37 panitumumab,38 trastuzumab39 and rituximab.40 While rituximab also targets CD20, other therapies have targeted receptors such as EGFR (cetuximab and panitumumab) or HER2 (trastuzumab) (Table 3). This is largely due to the matching half-life of 90Y (2.7 days) and the biological half-life of antibodies.39 Such agents are typically used to treat non-Hodgkin lymphoma, a group of blood cancers that develop from white blood cells. The conjugation of panitumumab to a 90Y-DOTA or 90Y-DTPA based group has been used to treat head and neck cancers in a mouse model.38 The related cetuximab is able to cross the blood brain barrier. Upon the administration of 90Y labelled cetuximab, accumulation in brain tumours was observed and studies have investigated its effect on human cancer cell lines.3790Y radiolabelling was found not to affect the action of cetuximab. Radiotherapy using 90Y labelled rituximab has yielded complete metabolic remission for patients with Nodular lymphocyte-predominant Hodgkin lymphoma40 (Fig. 5b and c) and the use of new 90Y labelled antibody conjugates of this type, and others, are expected to increase in the future.
PET imaging using yttrium-based tracers
Imaging techniques such as PET can take advantage of those radionuclides that exhibit β+ decay. The production of high energy positrons during this decay can be detected indirectly through the production of two gamma photons formed from annihilation of positrons with surrounding electrons in tissues. This annihilation of antiparticles will occur when the high energy positrons have travelled from the source of the radiotracer and have lost sufficient kinetic energy to interact with surrounding electrons. Encounters between positron and electron will produce two gamma photons moving in opposite directions. These are distinguished from background photons by detecting photon pairs arriving at a camera placed around the patient within short time intervals. The most common radiolabelled probe used clinically is 18F-fluorodeoxyglucose (18FDG), which allows glucose uptake to be imaged using PET.41Imaging the uptake and biodistribution of 90Y agents is very important in determining the dosage and side effects of such therapies. Dual therapies in which injected agents have the therapeutic effects of 90Y based agents whilst retaining the potential to be simultaneously imaged inside the body are clearly essential. Imaging 90Y is a challenge and there have been many attempts to address this including imaging the Bremsstrahlung radiation emitted from the β+ decay of 90Y.5 While 90Y decays predominantly by β+ processes, it has been known since the 1950s that minor decay via positron emission (34 ppm, 0.003%) is possible. In the past, the most common approach has been to administer the same 90Y based agent coordinated to a different radionuclide that undergoes positron emission and can be imaged by PET. Co-administration of other radionuclides such as 44Sc, 68Ga, 111In or 177Lu is commonly used as a route to image the progress, uptake, dosage, or biodistribution of the analogous 90Y radiotherapeutic agent at the target site or in off-site organs via PET or SPECT (isotope dependent).31,34
It has recently been shown that 90Y distribution can be quantified using direct positron emission from 90Y.42 This imaging route faces challenges from low positron emission and high background signals from Bremsstrahlung radiation, although despite this it can give some route to imaging 90Y distribution.42 Direct imaging of 90Y via either Bremsstrahlung radiation or β+ decay is feasible. For instance, combined Bremsstrahlung SPECT and 90Y PET/CT imaging of 90Y labelled microspheres was achieved by injecting them into an area of metastatic cholangiocarcinoma in the left hepatic lobe (Fig. 6).43 A PET image using 18FDG is also included for comparative purposes. Here, Bremsstrahlung SPECT imaging gives a diffuse response in the area of the tumour while 90Y PET/CT gives a more localised response.43 In this case there is poor uptake of 90Y agent into the tumour and signal is much more localised in the cancerous target upon imaging with 18FDG.
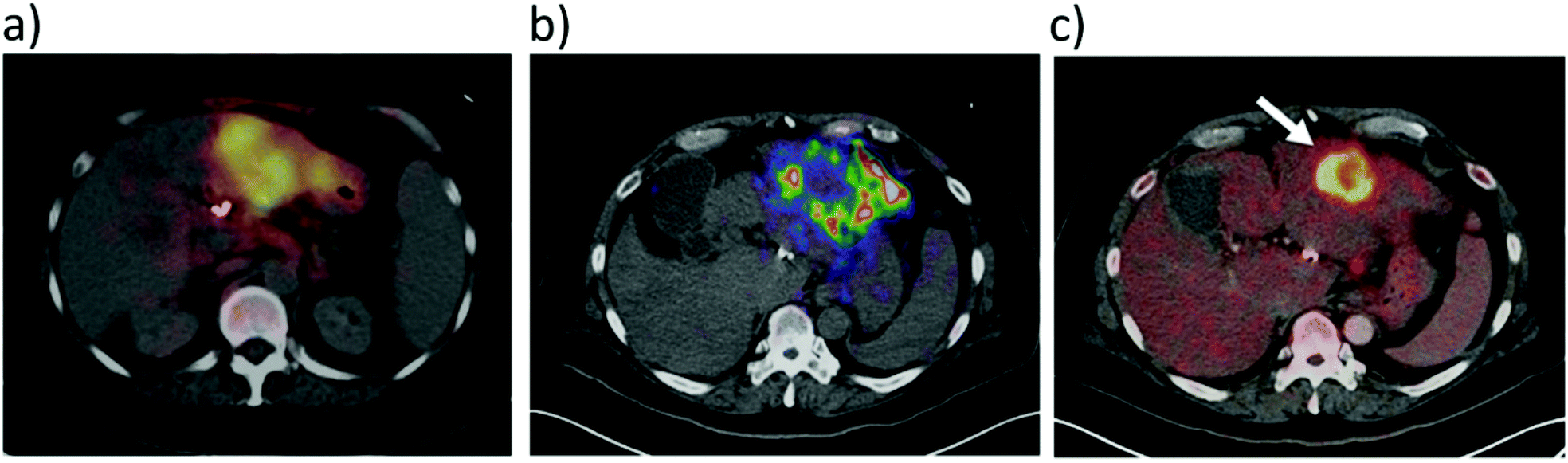 | ||
| Fig. 6 Use of 90Y for SPECT and PET imaging. 90Y labelled microspheres were injected into an area of metastatic cholangiocarcinoma in the left hepatic lobe and the imaging was performed either using: (a) Bremsstrahlung SPECT, (b) 90Y PET/CT, or (c) 18F PET using 18FDG prior to radioembolisation. Reproduced with permission from ref. 43. Copyright © 2014, Pasciak, Bourgeois, McKinney, Chang, Osborne, Acuff and Bradley. | ||
Increasingly, those yttrium isotopes such as 86Y that do decay predominantly via β+ emission (half-life 14.7 hours) are being investigated as potential PET imaging agents.4486Y is a non-ideal isotope for PET imaging because 67% of its β+ decay processes are accompanied by release of gamma rays. Simultaneous γ ray emission can fall into the accepted energy window of PET scanners (350–650 keV), which results in background signals. Such background noise can lead to increased false positive detection by the PET scanner and therefore erroneous quantification of 86Y concentration.
Methods are being developed to apply background corrections to 86Y PET images to help alleviate these interference issues.45 Consequently, noise in PET images of 86Y-DOTATOC can be reduced upon background subtraction (Fig. 7).45 In comparison, decay from the more commonly used 18F isotope consists of only β decay.
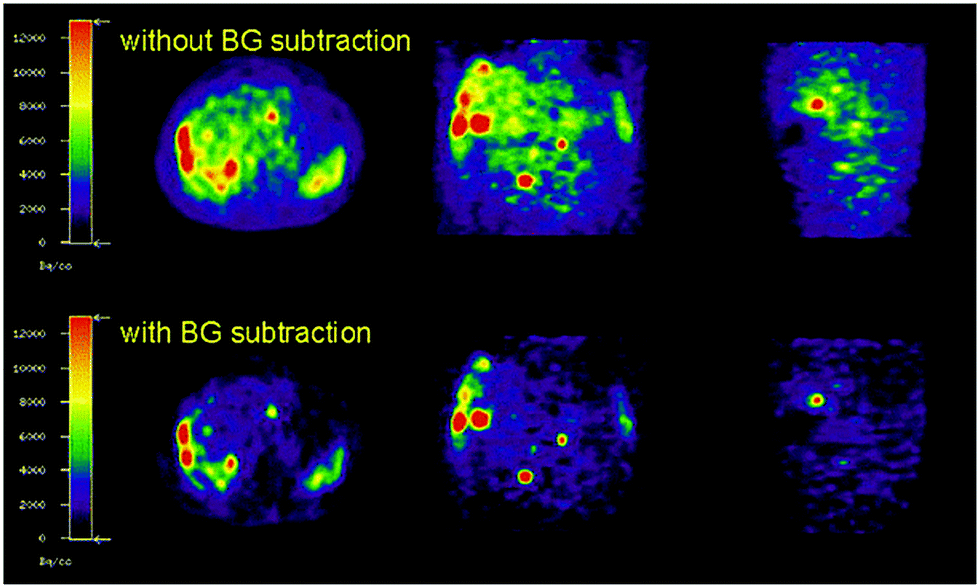 | ||
| Fig. 7 Effect of background subtraction on 86Y PET imaging. PET imaging of 86Y-DOTATOC with and without background subtraction. Reproduced with permission from ref. 45. Copyright © 2003, Springer Nature. | ||
Despite these challenges, the most promising use for 86Y radiotracers is in combination with 90Y radiotherapy analogues as matched pair isotopes to predict the distribution and dosimetry of subsequently injected radiotherapeutics in the same way as other PET agents (i.e.68Ga).46,47 In the case of 86Y/90Y mixed pairs, the biological distribution/pharmacokinetics of the two isotopes will be identical as the chemical properties of the PET tracer and the radiotherapeutic are the same.48 This will give clinicians the ability to predict where the treatment will go on a patient by patient basis. This means that if there is significant off site organ uptake, the optimum radioactivity dose can be given, which is expressed as maximum dose at target tissues versus tolerable dose at healthy organs. The appropriate doses of the radiotherapeutic can be given to lower the cross fire or side effects of the radiotherapy and make it more effective as a therapeutic. The standard doses of PET imaging agents administered to patients lies in the 1–300 MBq range, while for therapeutic isotopes it is in the 6–32 GBq m−2 range (Table 4). Typically, this results in a picomolar concentration range of PET tracers required to give these radiation doses. A similar approach has used PET imaging of 86Y labelled analogues to predict the biodistribution and clearance pathways of analogous gadolinium based MRI contrast agents. It has been known that the use of Gd-based contrast agents can be connected with the nephrogenic system fibrosis disease, which is related to deposition of Gd3+ in the kidneys of patients with poor renal clearance, or can lead to deposits in the brain. In fact, it is extremely challenging to measure the biodistribution of Gd-based agents, and T1-weighted MRI is unable to measure residual concentrations of this ion. To this end, PET imaging of 86Y-DTPA has been used to track the clearance pathways of the analogous Gd-DTPA in rats (Fig. 8).10 These images show that the injected agents are predominantly excreted through the kidneys in less than 4 hours, although a small amount of agent is retained by the kidneys and excreted over a longer time scale (images recorded up to 48 hours post injection).10 PET images are even able to show the presence of secondary excretion pathways. For example, PET images acquired 4 hours post injection showed that the agent was present in the large intestines suggesting that hepatic clearance is also occurring (Fig. 8d).10 The use of 87Y and 88Y as potential SPECT and PET tracers respectively has also been investigated.49 In an attempt to show the broadness of different 90Y- and 86Y-based radiotracers for medical imaging and therapy, we have summarized their use along with the chelators, type of bioconjugates, desired targets, labelling conditions, dosages and specific applications (Tables 3 and 4).
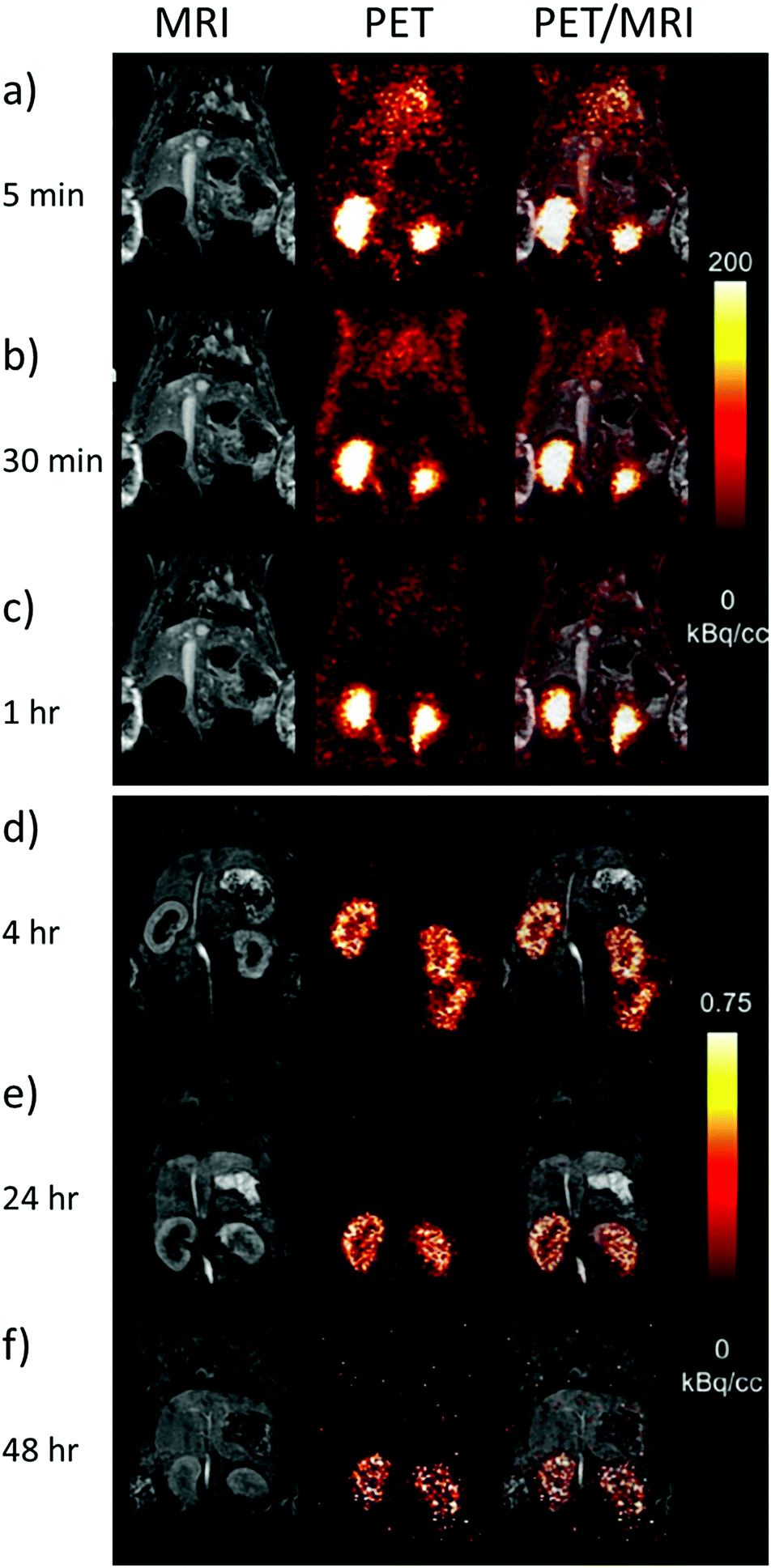 | ||
| Fig. 8 PET imaging of 86Y complexes as surrogates for Gd MRI contrast agents. Example MRI, PET and overlaid PET/MRI images of rats taken (a) 5 min (b) 10 min (c) 1 h (d) 4 h (e) 24 h and (f) 48 h after the injection of 86Y-DTPA (11.8 MBq) and Gd-DTPA (0.6 mmol kg−1). Note that the intensity scale for (a)–(c) and (d)–(f) are not the same. Reproduced with permission from ref. 10. Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Hyperpolarised 89Y complexes as MRI probes
In contrast to PET, MRI uses magnetic field gradients and radiofrequency pulses to produce spatially and temporally resolved images of living systems. This technique involves the use of non-ionising radiation and does not require specialist facilities like those necessary for PET. Instead, it routinely detects NMR-active nuclei, which in the majority of cases are water protons, where different environments are distinguished by their longitudinal (T1) or transverse (T2) relaxation time constants.8 It is the low inherent sensitivity of MRI that necessitates reliance on the bulk 1H2O signal to convey this information. MR sensitivity is derived from small perturbations in the population of states occupying closely spaced nuclear spin energy levels in a magnetic field. While relaxation inducing agents often provide a route to improving contrast, many researchers are turning their attention to hyperpolarisation as a non-toxic alternative to address insensitivity.50Hyperpolarisation techniques such as dynamic nuclear polarisation (DNP) create non-Boltzmann population distributions across nuclear spin energy levels and can lead to MR signal gains of up to five orders of magnitude.50 DNP can achieve this effect in heteronuclei such as 89Y through transfer of polarisation from unpaired electrons. This involves microwave irradiation of a frozen glass matrix containing the 89Y agent and a free radical in a magnetic field (∼3 T) at low temperature (1–2 K). This technique of transferring the inherently greater electron polarisation to that of a proton was first suggested by Albert Overhauser in 1953 and experimentally validated by Carver and Slichter in the same year.50 It took until 2003 for dissolution DNP (d-DNP), which involves the rapid melting of hyperpolarised solids with superheated solvent, to offer a proven route to dramatically enhance the MR signals of small molecules in solution.50 This breakthrough has created the excitement needed to drive further development. Consequently, d-DNP has now been used in a number of clinical studies associated with the production of hyperpolarised 13C tracers, of which pyruvate is the most widely reported.50 Indeed, the in vivo injection and metabolic imaging of hyperpolarised 13C pyruvate into humans has been used as a method to diagnose cancer.50
Despite the current successes of hyperpolarised 13C probes, hyperpolarisation techniques can target many other heteronuclei, including 89Y. 89Y is an interesting nucleus for study by MR because it is non quadrupolar (I = 1/2) with 100% natural abundance. As living cells have no known use for 89Y, there is no inherent background response and consequently its use as a medical probe provides an exciting opportunity for clinical applications. However, one should note that 89Y has a gyromagnetic ratio that is around 20 times smaller than that of 1H and its corresponding MR receptivity is 10000 times smaller than 1H, albeit similar to that of 13C. On the other hand, early studies in the 1970s demonstrated that 89Y salts exhibit long T1 times that can exceed 100 seconds.13 The incorporation of 89Y into metal complexes was found to elongate relaxation times even further.13 Collectively, this makes Boltzmann polarised 89Y MR studies challenging as low receptivity must be overcome by signal averaging which necessitates long measurement times to overcome the long relaxation lifetime. These measurement times for thermally polarised samples can be reduced by the addition of relaxation inducing dopants, which overcome the latter effect. In contrast, long relaxation times provide the potential to detect hyperpolarised 89Y nuclei in vivo before the hyperpolarised state decays back to its Boltzmann populated state and may provide many advantages. For example, DNP has been used to successfully hyperpolarise 89Y with enhanced 89Y NMR signals detected in just a single scan (Table 5).13 Achieving this result is a substantial breakthrough as measurement of Boltzmann derived signals is challenging for the reasons given above and the DNP process is itself costly, time consuming and single shot in nature. In addition, whilst most DNP machines contain an in-built NMR spectrometer that is able to monitor 13C signal intensity as a function of microwave frequency and subsequent signal growth over increasing irradiation time, such systems are unable to monitor changes in 89Y signals in a similar way. Therefore, finding the optimum microwave frequency and irradiation time required for optimum polarisation of 89Y nuclei is challenging and involves the rapid melting and ejection of many separate samples for the buildup and NMR detection of 89Y signal intensity. These technical challenges are reflected in the range of different 89Y polarisation conditions reported in the literature to date (Table 5).13,51–53
| Y material | Polarizing agent | Glassing matrix | Microwave frequency | Irradiation time | Microwave power | Temperature | Magnetic field | Dissolution medium | ε/fold | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| YCl3 (54 mM) | TEMPO (30 mM) | D2O/glycerol-d8 (10 μL, 60/40, v/v) | 93.89 GHz | 5 hours | 30 mW | 1.15 K | 3.35 T | D2O (5 mL) | N/A | 17 |
| Y-DOTP (176 mM) and Y-DO3A-NTs (143 mM) | OX063 (15 mM) | H2O/glycerol(160 μL, 75/25, v/v) | 94.125 GHz | N/A | 100 mW | 1.4 K | 3.35 T | H2O (4 mL) | 3000 | 54 |
| Y-DOTA (400 mM) | TEMPO (50 mM) | 50/50 D2O/glycerol-d8 | N/A | 3 hours | N/A | 1.05 K | 5 T | N/A | 3250 | 55 |
| Various Y complexes (∼200 mM) | OX063 (15 mM) | H2O/glycerol (160 μL, 75/25, v/v) | N/A | 3 hours | N/A | 1.4 K | N/A | N/A | N/A | 13 |
| Various Y complexes (700–1500 mM) | Trityl radical (16.6 mM) | H2O/glycerol (20–40 μL, 50/50, v/v) | 94.118 GHz | 75 min–2.5 hours | 100 mW | 1.4 K | N/A | EDTA in H2O (4 mL, 850 μM) | 246–1527 | 53 |
| Y-DOTA (140–280 mM) | OX063 (15 mM) | H2O/glycerol (40 μL, 50/50, v/v) | ∼94 GHz | Up to 10 hours | 100 mW | 1.4 K | 3.35 T | H2O (4 mL) | 65,000 | 51 |
| Y-13CDOTA (325 mM) | OX063 (15 mM) | H2O/glycerol (40 μL, 50/50, v/v) | 94.16 GHz | 5–7 hours | 100 mW | 1.4 K | 3.35 T | H2O (4 mL) | 387–8040 | 52 |
Optimisation of these polarisation conditions can include modification of several factors including the organic radical used for polarisation transfer, the glassing matrix, concentrations, and irradiation time and frequency. Lumata et al. have performed a detailed study of the effect of these factors on 89Y signal gains.51 It was reported that using organic radicals with narrow electron paramagnetic resonance linewidths (such as trityl rather than nitroxyl radicals) yields more efficient polarisation transfer from the unpaired electron to the 89Y target of interest.51 Moreover, addition of an electron relaxing agent such as a Gd3+ salt can yield higher 89Y polarisation by up to a factor of five (Fig. 9a).51 This effect has been reported for other heteronuclei such as 13C. Performing these optimisation steps is time consuming and challenging as 89Y signal growth cannot be monitored in situ, as previously discussed. For example, optimisation of polarising frequency and irradiation time involved d-DNP of 12 and 7 separate 89Y-DOTA samples respectively.51 Despite these challenges, by combination of various optimisation steps, including using optimum microwave frequencies, irradiation times, radical, Gd3+ additive and viscous glass forming solvents, 89Y signals enhanced by a factor of 65000 were achieved.51
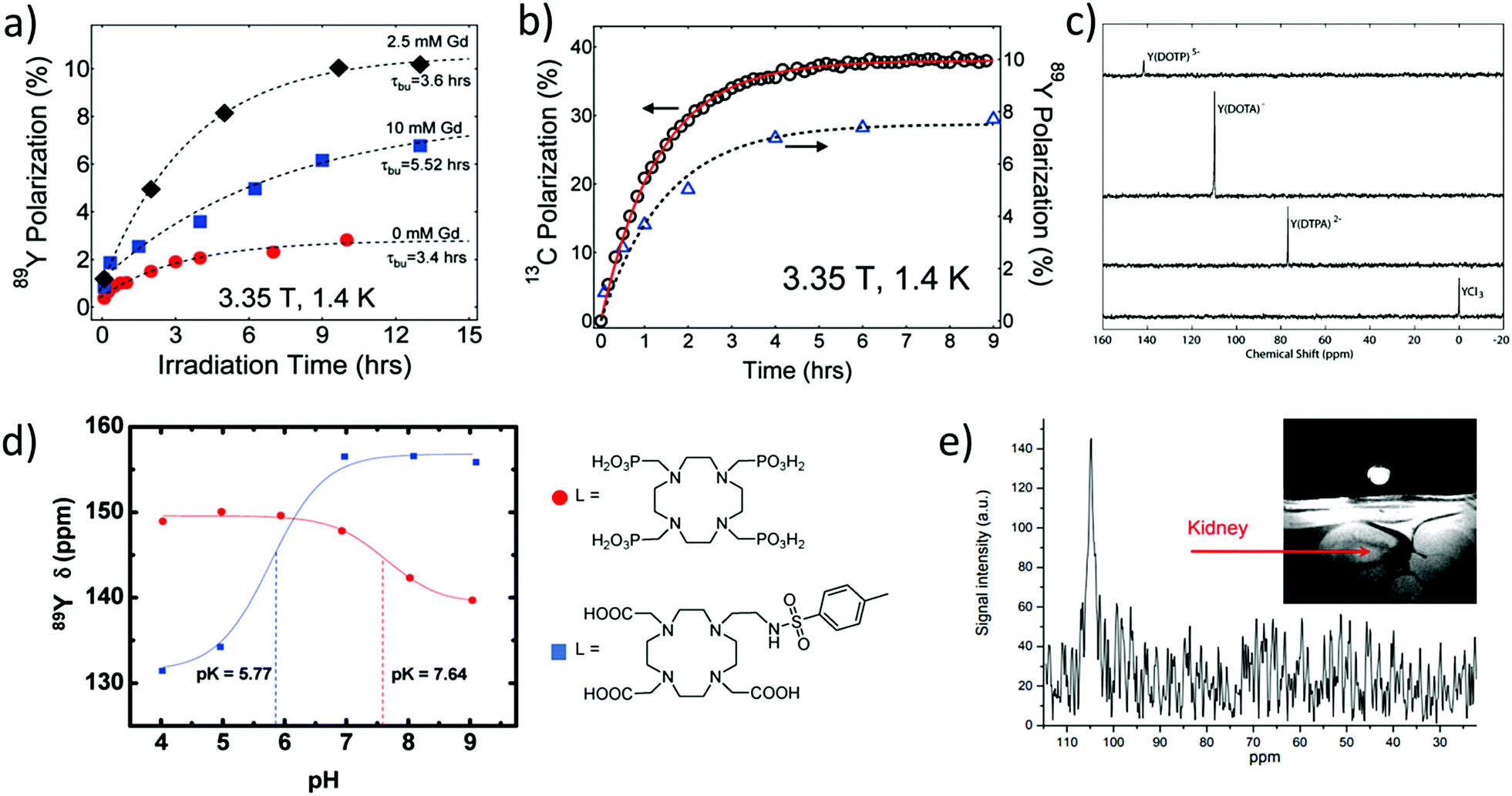 | ||
| Fig. 9 Applications of hyperpolarised 89Y complexes in MR. (a) Example polarisation buildup curves of 0.28 M Y-DOTA samples in a 1:1 glycerol/water glassing matrix doped with 15 mM trityl radical and Gd3+ (0, 2.5, and 10 mM). (b) Polarisation buildup of samples with 1-13C pyruvate (left axis) and 89Y-DOTA (right axis) doped with 15 mM trityl OX063 and 2.5 mM Gd3+ at 1.4 K and 3.35 T. Note that each data point in the 89Y buildup curves in (a) and (b) represents a separate DNP experiment. (c) 89Y NMR spectra collected at 29.4 MHz using a 14.1 T magnet of hyperpolarised yttrium complexes recorded with a 10° excitation pulse ∼30 s after transfer from the DNP polariser to a 8 mm NMR tube. (d) 89Y chemical shift dispersion of hyperpolarised Y-DOTP and YDO3A-NTs as a function of pH (9.4 T and 25 °C) and the corresponding structures of their ligands. (e) Y-DOTA hyperpolarised using DNP has been injected and imaged in a rat kidney. Reproduced with permission from (a) and (b) ref. 51. Copyright © 2011 American Chemical Society (c) ref. 53. Copyright © 2007 American Chemical Society (d) ref. 54. Copyright © 2010 American Chemical Society and (e) ref. 55. Copyright © 2010 International Society for Magnetic Resonance in Medicine. | ||
Interestingly, the co-polarisation of sodium pyruvate-1-13C or polarisation of 13C-enriched 89Y-DOTA has been used to provide information on the mechanism of signal enhancement. In these systems the growth of 13C polarisation can be monitored in situ and this is correlated to the 89Y signal gain (Fig. 9b).51,52 The co-polarisation of 89Y and 13C nuclei may provide a way to indirectly monitor the growth of 89Y polarisation and reliably predict the 89Y signal gain upon rapid melting and ejection of the sample.
The hyperpolarisation of a wide range of yttrium containing complexes has been reported using d-DNP (Fig. 9c and Table 5).11,53 These examples show that 89Y chemical shift is highly dependent on its coordination environment with large (∼80 ppm) shift differences reported for hyperpolarised 89Y nuclei coordinated to various different macrocyclic ligands. Such high sensitivity of 89Y chemical shift to molecular environment has been exploited to design hyperpolarised 89Y pH sensitive probes. Examples contain phosphonate and carboxylate groups, whose protonation state changes with pH, thereby affecting the molecular environment around the coordinated 89Y ion and its subsequent chemical shift (Fig. 9d).54 Other studies have utilised the differences in hyperpolarised 89Y chemical shift that exist between free and complexed ions to measure complexation rates to the macrocyclic DOTAM ligand and the associated binding kinetics.17
It was not until 2016 that a quantitative link between 89Y chemical shift and environment was outlined through studies on 23 complexes containing polyaminocarboxylate ligands. In this seminal work, an empirical equation was derived to accurately relate 89Y chemical shift to the number and type of coordinating atoms.13 Good agreement was found between 89Y shifts predicted from this empirical equation and those observed for both thermal and hyperpolarised samples. Hyperpolarised 89Y studies can therefore provide important information on complex kinetics and protonation states. It should also be noted that typical concentrations of 89Y complexes detected in vitro in single scan hyperpolarised NMR spectra lie in the range 100 μM–7 mM, which are significantly higher than those which feature as PET tracers (see above). We are not aware of any detection limit for 89Y HP-MRI, but note that this can be improved by future development of experimental set up, RF coil detection, and increased 89Y signal gains.
Research into the hyperpolarisation of 89Y is currently at the level of preliminary in vivo imaging studies. In an initial report, Y-DOTA was administered in a rat kidney to provide the first 89Y MR images (Fig. 9e);55 further examples in this direction are still pending. However, 89Y chemical shift has proven to be highly sensitive to the molecular environment and it is expected that future uses of 89Y as a probe for such changes in molecular environment can be envisaged. It has already been shown that 89Y chemical shift of Y-EDTA can change by as much as ∼10 ppm in the presence of sodium lactate at pH 6.4 and 25 °C at 9.4 T.13 Therefore, 89Y complexes may serve a unique role as an in vivo probe for such biomolecules. Currently, DNP is a time consuming and expensive technique available only to a few research institutions. While no reports of 89Y hyperpolarised with techniques other than DNP have been reported, advances in other hyperpolarisation techniques including the cheaper para-hydrogen based methods50 may reflect exciting opportunities to target the hyperpolarisation of 89Y.
Conclusions
The role of yttrium in medical applications, especially radiotherapy and medical imaging, has been frequently neglected. Yttrium can form a large number of complexes owing to its specific coordination and radiochemical properties which show a wide variety of features. To this end, yttrium complexes can provide a route to multimodal imaging in which the same ligands can be coordinated to 86Y and used for PET, 90Y and used for radiotherapy, and 89Y and used for HP-MRI, with 89Y chemical shifts extremely sensitive to molecular environment. The former two methods have already found a number of applications in modern medical diagnostic and therapeutic procedures, which are briefly overviewed in this work. Nonetheless, owing to its versatile properties, yttrium's mostly abundant isotope 89Y possesses properties suitable for NMR and MRI applications. Combined with hyperpolarisation, an emerging technique in modern NMR, yttrium offers a whole new avenue for molecular imaging studies based on metal chelates. All these methods together highlight the substantial contribution that yttrium has had, and will have, to biomedical research and clinical routines towards improving human health. With the most recent advances, the palette of its applications is expanding and further exciting progress is expected in the years to come.Abbreviations
| 1B4M-DTPA | 2-(p-SCN-benzoyl)-6-methyl-DTPA |
| CHX-A′′-DTPA | trans-(S,S)-Cyclohexane-1,2-diamine-N,N-N′,N′,N′′N′′-pentaaceticacid |
| CN | Coordination number |
| CT | Computed tomography |
| DEPA | 7-2-(Bis-carboxymethylamino)-ethyl-4,10-biscarboxymethyl-1,4,7,10-tetraazacyclododec-1-yl-acetic acid |
| DNP | Dynamic nuclear polarization |
| DTPA | Diethylenetriaminepentaacetic acid |
| DO3A | 1,4,7,10-Tetraazacyclododecane-1,4,7-tricarboxylic acid |
| DO3A-NT | 1-2′-(4-methylphenylsulfonylamino)ethyl-4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecane |
| DO3A-NTs | A DO3A derived ligand whose structure is shown in Fig. 9d (lower) |
| DOTA | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetracetic acid |
| DOTALAN | DOTA-lanreotide |
| DOTAM | 1,4,7,19-Tetrakis(caramoylmethyl)-1,4,7,10-tetraazacyclododecane |
| DOTANOC | DOTA-1-NaI3-octreotide |
| DOTATATE | DOTA-Tyr3-octreotide |
| DOTATOC | DOTA-Phe1-Tyr3-octreotide |
| DOTP | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetra(methylene phosphonic acid) |
| EDTA | Ethylenediaminetetraacetic acid |
| EDTMP | Ethylenediamine tetra(methylene phosphonic acid) |
| EGFR | Epidermal growth factor receptors |
| 18FDG | Fluorodeoxyglucose (18F) |
| HER2 | A human epidermal growth factor receptors |
| HILIC-ICP-MS | Hydrophilic interaction chromatography and inductively coupled plasma mass spectrometry |
| HP-MRI | Hyperpolarised magnetic resonance imaging |
| LNCaP | Androgen sensitive human prostate adenocarcinoma cell line |
| MP2346 | A Gastrin-releasing peptide receptors targeted peptide |
| MR | Magnetic resonance |
| NETA | 2-{4,7-Biscarboxymethyl(1,4,7)triazacyclonona-1-yl-ethyl}carbonylmethylaminoacetic acid |
| NMR | nuclear magnetic resonance |
| OCTAPA | N,N′-bis(6-carboxy-2-pyridylmethyl)-ethylenediamine-N,N′-diacetic acid |
| OX063 | Tris8-carboxyl-2,2,6,6-benzo(1,2-d:4,5-d)-bis(1,3)dithiole-4-ylmethyl sodium salt |
| PCTA | 3,6,9,15-Tetraazabicyclo9.3.1pentadeca-1-(15),11,13-triene-3,6,9-triacetic acid |
| PET | Positron emission tomography |
| PRRT | Peptide receptor radionuclide therapy |
| ReCCMSH | A cyclic analogue of α-melanocyte stimulating hormone |
| RF | Radiofrequency |
| SAP | Monocapped square antiprism |
| SEM | Scanning electron microscope |
| SPECT | Single-photon emission CT |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl or (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl |
| TSAP | Monocapped twisted square antiprism |
| TTP | Tricapped trigonal prism |
| VEGF-A | Vascular endothelial growth factor A |
| YAG | Yttrium aluminium garnet |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Simon Cotton for help in obtaining the literature on lanthanide and actinide chemistry and Prof. Gyula Tirsco for helpful discussions on kinetic inertness. B. J. T. would like to thank the Wild Overseas Scholar's Fund (University of York) for financial support. G. J. S. would like to thank the MRC for funding (MR/T002573/1). Open Access funding provided by the Max Planck Society.Notes and references
- H. Schumann, A. Heim, J. Demtschuk and S. H. Mühle, Organometallics, 2003, 22, 118–128 CrossRef CAS.
- S. Cotton, Lanthanide and actinide chemistry, John Wiley & Sons Ltd., Chichester, 2006 Search PubMed.
- N. B. Mikheev, L. Auerman, I. A. Rumer, A. N. Kamenskaya and M. Z. Kazakevich, Russ. Chem. Rev., 1992, 61, 990 CrossRef.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–24 RSC.
- C. L. Wright, J. Zhang, M. F. Tweedle, M. V. Knopp and N. C. Hall, BioMed Res. Int., 2015, 2015, 1–11 Search PubMed.
- T. H. Bokhari, M. B. Butt, S. Hina, M. Iqbal, M. Daud and M. Imran, J. Radioanal. Nucl. Chem., 2017, 314, 1487–1496 CrossRef CAS.
- S. Hirano, N. Kodama, K. Shibata and K. T. Suzuki, Toxicol. Appl. Pharmacol., 1993, 121, 224–232 CrossRef CAS PubMed.
- A. S. Merbach, L. Helm and E. Toth, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley & Sons Ltd., 2013 Search PubMed.
- K. Kumar, C. A. Chang, L. Francesconi, D. Dischino, M. Malley, J. Gougoutas and M. F. Tweedle, Inorg. Chem., 1994, 33, 3567–3575 CrossRef CAS.
- M. Le Fur, N. J. Rotile, C. Correcher, V. Clavijo Jordan, A. W. Ross, C. Catana and P. Caravan, Angew. Chem., Int. Ed., 2019, 59, 1474–1478 CrossRef PubMed.
- Z. Baranyai, G. Tircsó and F. Rösch, Eur. J. Inorg. Chem., 2020, 36–56 CrossRef CAS.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- Y. Xing, A. K. Jindal, M. Regueiro-Figueroa, M. Le Fur, N. Kervarec, P. Zhao, Z. Kovacs, L. Valencia, P. Pérez-Lourido, R. Tripier, D. Esteban-Gómez, C. Platas-Iglesias and A. D. Sherry, Chem. – Eur. J., 2016, 22, 16657–16667 CrossRef CAS PubMed.
- D. Parker, K. Pulukkody, F. C. Smith, A. Batsanov and J. A. K. Howard, Dalton Trans., 1994, 689–693 RSC.
- C. F. G. C. Geraldes, A. M. Urbano, C. Carmen Alpoim, M. A. Hoefnagel and J. A. Peters, J. Chem. Soc., Chem. Commun., 1991, 656–658 RSC.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- P. Miéville, S. Jannin, L. Helm and G. Bodenhausen, J. Am. Chem. Soc., 2010, 132, 5006–5007 CrossRef PubMed.
- A. Nystrom and M. Thoennessen, At. Data Nucl. Data Tables, 2012, 98, 95–119 CrossRef CAS.
- J. Wike, C. Guyer, D. Ramey and B. Phillips, Int. J. Radiat. Appl. Instrum., Part A, 1990, 41, 861–865 CrossRef CAS.
- K. Kettern, K.-H. Linse, S. Spellerberg, H. H. Coenen and S. M. Qaim, Radiochim. Acta, 2002, 90, 845–849 CAS.
- J. Lellouche, A. Friedman, A. Gedanken and E. Banin, Int. J. Nanomed., 2012, 7, 5611–5624 CAS.
- A.-E. Borgonovo, A. Fabbri, V. Vavassori, R. Censi and C. Maiorana, Med. Oral Patol. Oral Cir. Bucal, 2012, 17, e981–e987 CrossRef PubMed.
- E. Schena, P. Saccomandi and Y. Fong, J. Funct. Biomater., 2017, 8, 19 CrossRef PubMed.
- P. Hilgard, M. Hamami, A. E. Fouly, A. Scherag, S. Müller, J. Ertle, T. Heusner, V. R. Cicinnati, A. Paul and A. Bockisch, Hepatology, 2010, 52, 1741–1749 CrossRef CAS PubMed.
- Z. Nosrati, A. R. Khanchi and S. Sheybani, J. Radioanal. Nucl. Chem., 2014, 301, 373–382 CrossRef CAS.
- A. S. Mahmoud-Ahmed and J. H. Suh, Pituitary, 2002, 5, 175–180 CrossRef CAS PubMed.
- J. Rosenstock, R. Jung, F. Doyle, K. Mashiter and G. Joplin, J. R. Soc. Med., 1982, 75, 209–210 CAS.
- W. Taylor, M. Corkill and C. Rajapaske, Br. J. Rheumatol., 1997, 36, 1100–1105 CrossRef CAS PubMed.
- M. Heim, E. Goshen, Y. Amit and U. Martinowitz, Haemophilia, 2001, 7, 36–39 CrossRef PubMed.
- L. Nisa, G. Savelli and R. Giubbini, Ann. Nucl. Med., 2011, 25, 75–85 CrossRef CAS PubMed.
- J. Kunikowska, L. Królicki, A. Hubalewska-Dydejczyk, R. Mikołajczak, A. Sowa-Staszczak and D. Pawlak, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 1788–1797 CrossRef CAS PubMed.
- P. M. Smith-Jones, C. Bischof, M. Leimer, D. Gludovacz, P. Angelberger, T. Pangerl, M. Peck-Radosavljevic, G. Hamilton, K. Kaserer and A. Kofler, Endocrinology, 1999, 140, 5136–5148 CrossRef CAS PubMed.
- D. Wild, J. S. Schmitt, M. Ginj, H. R. Mäcke, B. F. Bernard, E. Krenning, M. De Jong, S. Wenger and J.-C. Reubi, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 1338–1347 CrossRef CAS PubMed.
- L. Villard, A. Romer, N. Marincek, P. Brunner, M. T. Koller, C. Schindler, Q. K. Ng, H. R. Mäcke, J. Müller-Brand and C. Rochlitz, J. Clin. Oncol., 2012, 30, 1100–1106 CrossRef CAS PubMed.
- F. Rösch, H. Herzog and S. M. Qaim, Pharmaceuticals, 2017, 10, 56 CrossRef PubMed.
- F. Morschhauser, J. Radford, A. Van Hoof, B. Botto, A. Z. S. Rohatiner, G. Salles, P. Soubeyran, H. Tilly, A. Bischof-Delaloye and W. L. J. van Putten, J. Clin. Oncol., 2013, 31, 1977–1983 CrossRef CAS PubMed.
- M. Saki, M. Toulany, W. Sihver, M. Zenker, J. M. Heldt, B. Mosch, H. J. Pietzsch, M. Baumann, J. Steinbach and H. P. Rodemann, Strahlenther. Onkol., 2012, 188, 823–832 CrossRef CAS PubMed.
- Z. Liu, Y. Liu, B. Jia, H. Zhao, X. Jin, F. Li, X. Chen and F. Wang, Mol. Cancer Ther., 2010, 9, 1535–7163 Search PubMed.
- E. W. Price, K. J. Edwards, K. E. Carnazza, S. D. Carlin, B. M. Zeglis, M. J. Adam, C. Orvig and J. S. Lewis, Nucl. Med. Biol., 2016, 43, 566–576 CrossRef CAS PubMed.
- S. C. Golstein, K. Muylle, M. Vercruyssen, C. Spilleboudt, A. de Wind and D. Bron, Eur. J. Haematol., 2018, 101, 415–417 CrossRef CAS PubMed.
- S. S. Gambhir, J. Czernin, J. Schwimmer, D. H. Silverman, R. E. Coleman and M. E. Phelps, J. Nucl. Med., 2001, 42, 1S–93S CAS.
- A. A. Attarwala, F. Molina-Duran, K.-A. Büsing, S. O. Schönberg, D. L. Bailey, K. Willowson and G. Glatting, PLoS One, 2014, 9, e110401 CrossRef PubMed.
- A. S. Pasciak, A. C. Bourgeois, J. M. McKinney, T. T. Chang, D. R. Osborne, S. N. Acuff and Y. C. Bradley, Front. Oncol., 2014, 4, 38 Search PubMed.
- T. K. Nayak and M. W. Brechbiel, Med. Chem., 2011, 7, 380–388 CrossRef CAS PubMed.
- H. G. Buchholz, H. Herzog, G. J. Förster, H. Reber, O. Nickel, F. Rösch and P. Bartenstein, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 716–720 CrossRef CAS PubMed.
- D. W. Schneider, T. Heitner, B. Alicke, D. R. Light, K. McLean, N. Satozawa, G. Parry, J. Yoo, J. S. Lewis and R. Parry, J. Nucl. Med., 2009, 50, 435–443 CrossRef CAS PubMed.
- T. K. Nayak, K. Garmestani, K. E. Baidoo, D. E. Milenic and M. W. Brechbiel, Int. J. Cancer, 2011, 128, 920–926 CrossRef CAS PubMed.
- F. Rösch, H. Herzog, C. Plag, B. Neumaier, U. Braun, H.-W. Müller-Gärtnere and G. Stöcklin, Eur. J. Nucl. Med., 1996, 23, 958–966 CrossRef PubMed.
- G. Sgouros, Med. Phys., 1998, 25, 1487–1490 CrossRef CAS PubMed.
- P. Nikolaou, B. M. Goodson and E. Y. Chekmenev, Chem. – Eur. J., 2015, 21, 3156–3166 CrossRef CAS PubMed.
- L. Lumata, A. K. Jindal, M. E. Merritt, C. R. Malloy, A. D. Sherry and Z. Kovacs, J. Am. Chem. Soc., 2011, 133, 8673–8680 CrossRef CAS PubMed.
- L. Lumata, M. Merritt, C. Malloy, A. D. Sherry and Z. Kovacs, Appl. Magn. Reson., 2012, 43, 69–79 CrossRef CAS.
- M. E. Merritt, C. Harrison, Z. Kovacs, P. Kshirsagar, C. R. Malloy and A. D. Sherry, J. Am. Chem. Soc., 2007, 129, 12942–12943 CrossRef CAS PubMed.
- A. K. Jindal, M. E. Merritt, E. H. Suh, C. R. Malloy, A. D. Sherry and Z. Kovács, J. Am. Chem. Soc., 2010, 132, 1784–1785 CrossRef CAS PubMed.
- M. E. Merritt, M. Mishkovsky, T. Cheng, A. Jindal, Z. Kovacs, C. Malloy, R. Gruetter, A. D. Sherry and A. Comment, Proc. Intl. Soc. Mag. Reson. Med., 2010, 18, 1030 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |