
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular (directed) electrophilic C–H borylation
S. A.
Iqbal†
,
J.
Pahl†
 ,
K.
Yuan†
and
M. J.
Ingleson
*
,
K.
Yuan†
and
M. J.
Ingleson
*
EastCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: mingleso@ed.ac.uk
First published on 4th June 2020
Abstract
The intramolecular C–H borylation of (hetero)arenes and alkenes using electrophilic boranes is a powerful transition metal free methodology for forming C–B bonds. These C–H borylation reactions are preceded by intermolecular bond (both dative and covalent) formation, with examples proceeding via initial C–B and N–B bond formation dominating this field thus both are discussed in depth herein. Less prevalent intramolecular electrophilic C–H borylation reactions that proceed by intermolecular O–B, S–B and P–B bond formation are also summarised. Mechanistic studies are presented that reveal two mechanisms for C–H borylation, (i) electrophilic aromatic substitution (prevalent with B–X electrophiles); (ii) σ-bond metathesis mediated (prevalent with B–H and B–R electrophiles). To date, intramolecular electrophilic C–H borylation is utilised mainly for accessing boron containing conjugated organic materials, however recent developments, summarized herein alongside early studies, have highlighted the applicability of this methodology for forming synthetically versatile organo-boronate esters and boron containing bioactives. The multitude of synthetic procedures reported for intramolecular electrophilic C–H borylation contain many common features and this enables key requirements for successful C–H borylation and the factors effecting regioselectivity and substrate scope to be identified, discussed and summarized.
 S. A. Iqbal | Saqib A. Iqbal received his undergraduate degree from Cardiff University. He completed his MChem under the supervision of Dr Duncan Browne and Dr Louis Morrill on the study of organocatalysis under mechanochemical conditions. He joined the Ingleson group in 2018 where he is currently working on directed electrophilic C–H borylation using trihaloboranes. |
 J. Pahl | Jürgen Pahl obtained both his BSc and MSc in chemistry at the Friedrich-Alexander Universität Erlangen-Nürnberg. At the same university he focused his studies on group 2 coordination chemistry where he received his PhD degree 2019 under the supervision of Professor Sjoerd Harder. In 2019 he joined the Ingleson Group at the University of Edinburgh where he is investigating new methods for directed electrophilic borylation. |
 K. Yuan | Kang Yuan received his BSc degree in 2013 from Sun Yat-sen University under the guidance of Prof. Ming-Liang Tong. Then, he moved to Canada to explore main group chemistry and obtained his PhD degree under the supervision of Prof. Suning Wang at Queen's University. In 2018, he joined the Ingleson group as a postdoctoral research associate. His current research focuses on developing new methods for constructing novel boron containing molecules. |
 M. J. Ingleson | Mike Ingleson received his PhD. in organometallic chemistry in 2004 working with Prof. Andrew S. Weller. He then pursued postdoctoral studies with Prof. Kenneth G. Caulton at Indiana University and with Professor Matthew J. Rosseinsky at the University of Liverpool. He started his independent career at the University of Manchester in 2008 with a Royal Society University Research Fellowship. At Manchester, he was promoted to Reader in 2012 and Full Professor in 2018. He joined the University of Edinburgh in 2019. His research focuses on main group chemistry, and he has a particular fondness for all things boron. |
1. Introduction
The formation of (sp2)C–B bonds is of significant import across many fields. Historically, this is due to the power of organoboranes in functional group transformations coupled with their low toxicity and ease of handling (relative to other organometallic reagents).1 The past twenty years in particular have seen the utility of organoboranes expand with the incorporation of (sp2)C–B units now established as a useful function imparting tool in sensors2 and organic materials (Fig. 1).3 Concomitantly, there has been a number of boracycles containing (sp2)C–B units identified as bioactive molecules, such as diazaborines and benzoxaboroles including commercialised drugs such as tavaborole (Fig. 1).4 | ||
| Fig. 1 Applications of (sp2)C–B containing molecules focusing on general structures accessed (or potentially accessible) by intramolecular electrophilic C–H borylation. DG = directing group, FG = functional group. | ||
This broad importance has led to the development of many methods to form (sp2)C–B bonds. While the most prevalent synthetic route proceeds via metalation of an organohalide and trapping the organometallic product with B(OR)3, a more efficient approach is the conversion of C–H to C–B. This has been achieved predominantly through (sp2)C–M intermediates via: (i) C–H deprotonation (e.g. with an organolithium) then in situ trapping with a B(OR)3 electrophile and (ii) transition metal catalysed C–H borylation, with iridium catalysis being the most powerful example of the latter.5 C–H acidity and steric factors dominate selectivity via metal mediated C–H borylation routes (i) and (ii). An alternative route that avoids C–M containing intermediates is electrophilic C–H borylation. This affords borylated products often under combined steric and electronic control and via an SEAr mechanism.6 Notably, these three C–H borylation routes generally do not lead to the formation of ortho substituted arylboranes. Instead substrates containing directing groups (DGs) are required, e.g. to enable ortho C–H metalation with subsequent C–M to C–B conversion forming ortho borylated products. For more on metal mediated directed C–H borylation the reader is directed to recent reviews.7
An alternative approach to access ortho-borylated compounds is directed electrophilic C–H borylation. This transformation proceeds via an initial intermolecular step that forms an E–B bond (E = C, N, O, S, P), followed by intramolecular electrophilic C–H borylation. Since its discovery in the 1950s this transformation has been most widely used to make boron containing organic materials containing three and four coordinate boron centres.8 While its use is well established in the organic materials field, intramolecular electrophilic C–H borylation is much more rarely utilised to access organoboranes for use in functional group transformations or for accessing the boracycles found in sensors and bioactive molecules. This is in part due to the misconception that C–H borylation always requires forcing conditions and reactive catalysts (e.g. AlCl3). In fact, the conditions required to effect C–H borylation depend on multiple factors and there are many facile (e.g. proceeding within 1 h at room temperature without catalyst) intramolecular electrophilic C–H borylation reactions reported. Key variables effecting the reaction conditions required for intramolecular electrophilic C–H borylation include: the type of directing group, the nucleophilicity of the (hetero)arene being functionalised, the nature (e.g. ring size) of the boracycle being formed, the electrophilicity of the borane Lewis acid, the presence of an exogenous Brønsted base etc. This review focuses on identifying, discussing and summarizing the key factors required for intramolecular electrophilic (sp2)C–H borylation. It aims to increase the understanding of this topic and thereby facilitate the wider utilisation of directed electrophilic C–H borylation. To maintain focus on these principle objectives this review omits a number of other directed borylation reactions, such as hydroboration9 and (sp3)C–H borylation.10 Throughout the major focus is on discussing the conditions required (and the underlying reasons) for successful directed electrophilic C–H borylation and highlighting key structure–reactivity relationships. When multiple examples are reported proceeding via effectively identical synthetic procedures to access closely related borylated products this review focuses only on the key synthetic observations and does not provide an exhaustive list of products accessed via intramolecular electrophilic C–H borylation. Due to a number of commonalities in mechanism and C–H borylation conditions, this review covers the formation of borylated products that are 3 and 4 coordinate at boron.
2. Intramolecular electrophilic C–H borylation: the early years
The pioneering work on N-directed electrophilic C–H borylation comes from the group of Dewar and was developed concomitantly to intermolecular electrophilic C–H borylation.11 From 1958 onwards Dewar and co-workers published a series of N-directed C–H borylation studies utilising BCl3 and PhBCl2 with AlCl3 as catalyst to form BN analogues of polycyclic aromatic hydrocarbons (PAHs).12 For example, compounds 1–3 (Fig. 2) amongst others, were made by combining aniline derivatives with BCl3 to initially form ArylN(H)BCl2 intermediates post heating (e.g. inset Fig. 2).12,13 The significant B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N multiple bond character in these intermediates considerably reduces the Lewis acidity at boron and thus an additional Lewis acid (e.g. AlCl3) was required as a catalyst to generate a more reactive boron electrophile able to effect C–H borylation. In these reports using AlCl3 as a catalyst intramolecular electrophilic C–H borylation was performed under forcing conditions (140–175 °C). However, this temperature is principally required to access the melt phase and it is important to note that subsequently 1 (Y = Cl) was accessed using BCl3/AlCl3 at a lower temperature by performing the reaction in refluxing benzene.14 The exact origin of the high barrier for C–B bond formation in these examples is unclear, although it should be noted that in concurrent studies Dewar and co-workers showed that 2-vinyl-anilines undergo C–H borylation, e.g. to form 4, under milder conditions (using PhBCl2 or BCl3 without AlCl3 C–H borylation proceeded at 80 °C).15
N multiple bond character in these intermediates considerably reduces the Lewis acidity at boron and thus an additional Lewis acid (e.g. AlCl3) was required as a catalyst to generate a more reactive boron electrophile able to effect C–H borylation. In these reports using AlCl3 as a catalyst intramolecular electrophilic C–H borylation was performed under forcing conditions (140–175 °C). However, this temperature is principally required to access the melt phase and it is important to note that subsequently 1 (Y = Cl) was accessed using BCl3/AlCl3 at a lower temperature by performing the reaction in refluxing benzene.14 The exact origin of the high barrier for C–B bond formation in these examples is unclear, although it should be noted that in concurrent studies Dewar and co-workers showed that 2-vinyl-anilines undergo C–H borylation, e.g. to form 4, under milder conditions (using PhBCl2 or BCl3 without AlCl3 C–H borylation proceeded at 80 °C).15
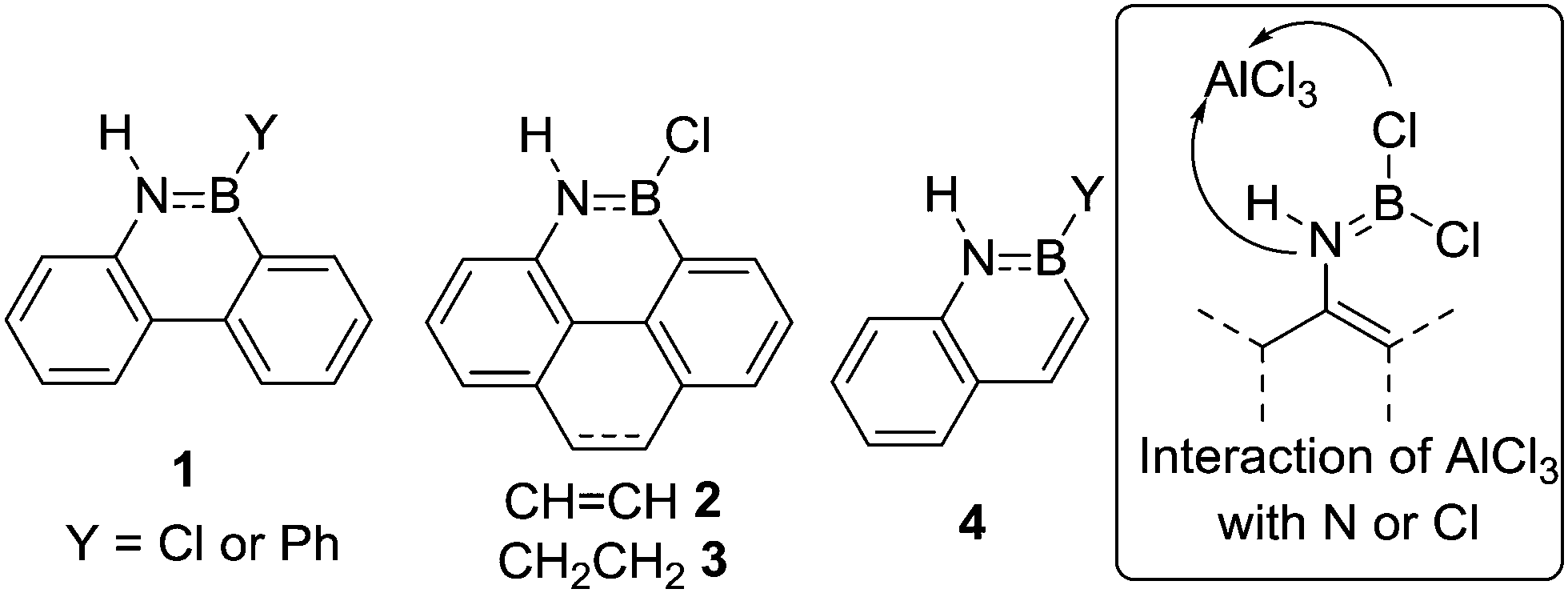 | ||
| Fig. 2 Select products from Dewar's N-directed electrophilic C–H borylation studies. Inset, the reactivity of AlCl3 with R(H)NBCl2 can be at Cl or N. | ||
The active boron electrophile in these arene-borylation examples will be derived from AlCl3 interacting with either N or Cl in the amido-borane (e.g. inset Fig. 2), with species related to the latter proposed as the key electrophiles in intermolecular C–H borylation by Olah and co-workers.16 Compounds containing R2NB(Cl)–(μ-Cl)–AlCl3 and R2(AlCl3)N–BCl2 units can be viewed as containing borenium cation (three coordinate at boron cationic compounds) type sub-units (though formally they are zwitterionic). For discussions on the role of borenium cations (and their functional equivalents) in intermolecular C–H borylation see recent reviews.6c,17
Irrespective of the identity of the active boron electrophile, Dewar's studies highlighted some key considerations controlling the feasibility of R2N-directed electrophilic C–H borylation. For example, while compound 5 (Fig. 3) is accessible from 8-phenyl-tetrahydroquinoline on reaction with BCl3 in the presence of AlCl3 at high temperature, its five membered ring analogue, A, was not formed under identical conditions.18 This can be attributed to the smaller bond angles in the five membered N-heterocyclic analogue creating a less favourable geometry for C–H borylation and thus higher reaction barriers. A related rationale can be applied to the fact that compound B is not accessible from the parent aniline using Dewar's forcing C–H borylation conditions. In this case steric clash in the fjord region will enforce non-planarity and presumably result in high barriers for C–H borylation to form B.
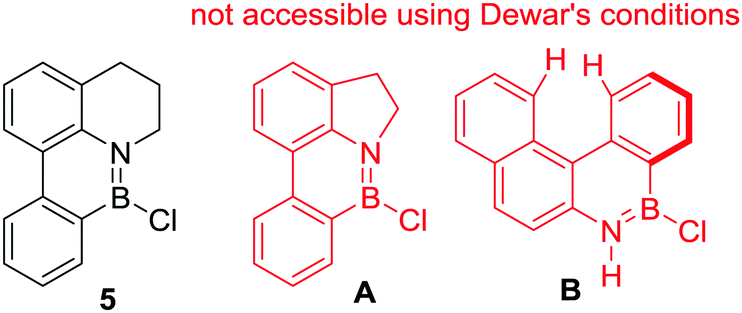 | ||
| Fig. 3 The sensitivity of Dewar's electrophilic C–H borylation conditions to modifications in ring size and increased dihedral angles. | ||
Other notable observations from these early studies include the formation of a single BN containing borylated product when commencing from aniline derivatives containing multiple proximal sites that could feasibly undergo directed electrophilic C–H borylation (Scheme 1, Ha and Hb can both be substituted).13 For example, compound 6 is formed exclusively with no compound C observed. This indicates that steric effects are important in controlling the overall outcome from R2N-directed intramolecular C–H borylation under these conditions, with no products from C–H borylation of the more hindered peri position (Ha) observed despite this position generally being more reactive in SEAr reactions. This suggests that these high temperature reaction conditions are leading to the thermodynamic borylated products. Furthermore, the six membered boracycle containing compound 7 is formed exclusively with no compound D observed.
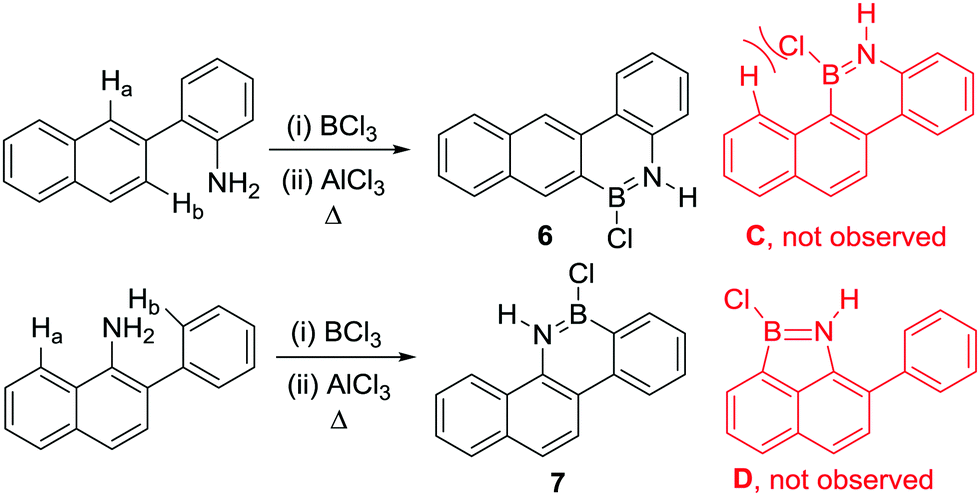 | ||
| Scheme 1 Highly selective directed electrophilic C–H borylation. | ||
Dewar and co-workers also reported the propensity of an isoelectronic biphenyl series containing 2-NH2, 2-OH and 2-SH directing groups to undergo electrophilic C–H borylation.19 In each case combination with BCl3 resulted in loss of HCl (which occurred more readily for the O and S derivatives relative to N) and formation of (biphenylyl)E–BCl2 intermediates (E = NH, O, S). Notably, the subsequent intramolecular C–H borylation step proceeded under drastically different conditions in each case (Scheme 2). While requiring catalysis by AlCl3 in all three reactions, the C–H borylation conditions correlated to the π donor ability of the heteroatom bonded to boron. Hence the sulfur congener underwent C–H borylation most readily presumably as poorer π donation to the boron centre (relative to O and N congeners) generates the most reactive boron electrophile of the series. Related effects were subsequently seen in intermolecular electrophilic C–H borylation and were correlated to relative LUMO energies by calculations.20 The formation of 9 at 20 °C confirms that intramolecular electrophilic C–H borylation can occur at room temperature provided a sufficiently electrophilic borane is generated and the higher temperature conditions required for borylation in RN(H)BCl2/AlCl3 systems (R = aromatic containing hydrocarbyl group) is principally due to the presence of weaker boron electrophiles.
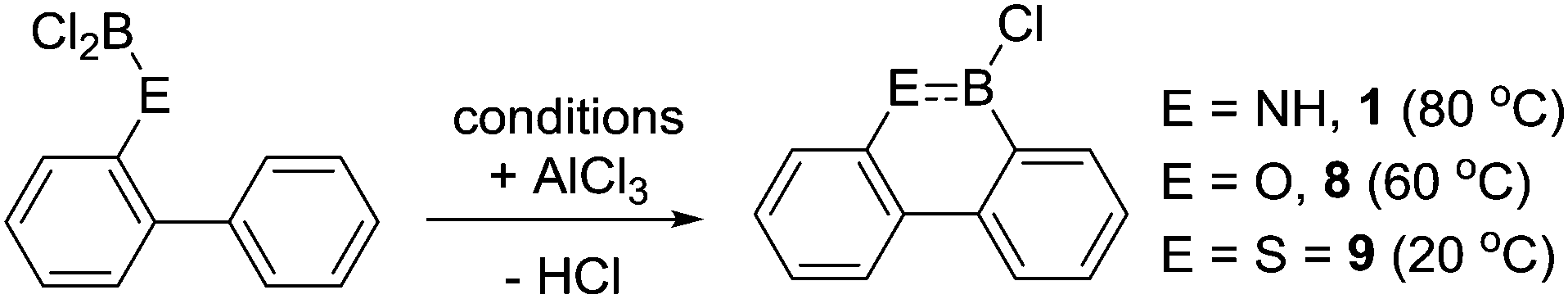 | ||
| Scheme 2 Varying C–H borylation conditions reported for an isoelectronic 2-(EBCl2)-biphenyl series. | ||
Dewar and co-workers also reported the reaction of an imine with BCl3/AlCl3 that ultimately led to intramolecular C–H borylation (Scheme 3).18 This is potentially the earliest example of a dative bonded species (N→BCl3) leading to intramolecular electrophilic C–H borylation. However, the identity of the initial product from C–H borylation is unknown and the species isolated post heating, 10, contained an enamine unit (Scheme 3). Thus, in this case intramolecular C–H borylation may occur pre- or post-transformation of the imine into an enamine. The latter would contain a R2NBCl2 unit and not a datively bonded borane unit.
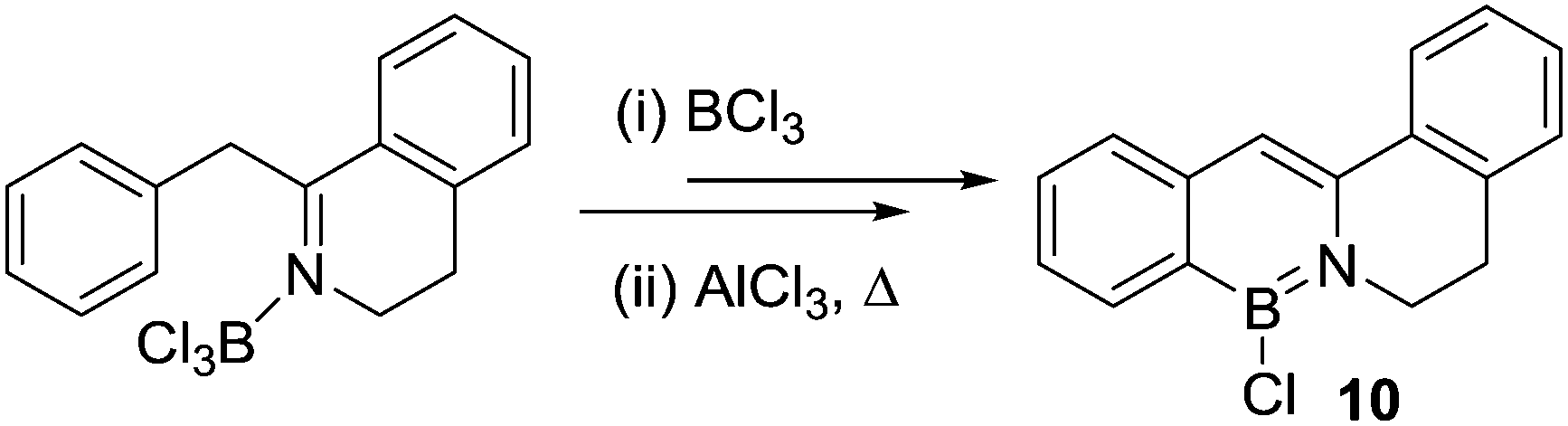 | ||
| Scheme 3 The formation of 10 by N-directed C–H borylation via a Lewis adduct. | ||
In parallel to Dewar's work, from 1959 onwards Köster and co-workers reported another series of intramolecular C–H borylation reactions. These proceeded via B–H electrophiles generated from organoboranes at high temperatures.21 On heating, a range of triorganoboranes underwent retro-hydroboration to produce compounds such as (ArylCH2)B(R)H that then reacted via intramolecular C–H borylation evolving H2. This electrophilic borylation reaction is comparable to Hurd's earlier work on borylating benzene with B2H6 (where H2 is also the only by-product).22 These transformations have also been termed dehydrogenative electrophilic borylation. Köster's work included the formation of 1-boraindanes (11) and 1-boratetralins (12, Scheme 4). A mechanism proceeding through a four membered σ-bond metathesis transition state for C–H borylation was proposed (inset bottom left, Scheme 4). This mechanism was supported by subsequent calculations, which also found that the barrier to aryl-C–H borylation correlated well with Hammett σp values, with electron donating substituents having lower barriers.23 Köster's reports showed that multiple compounds with one aryl containing substituent and one substituent that can undergo retro-hydroboration are amenable to intramolecular C–H borylation. Even an alkoxy group on boron was tolerated, with 13 formed from a R2BOR species via initial retro-hydroboration (albeit at 190 °C).
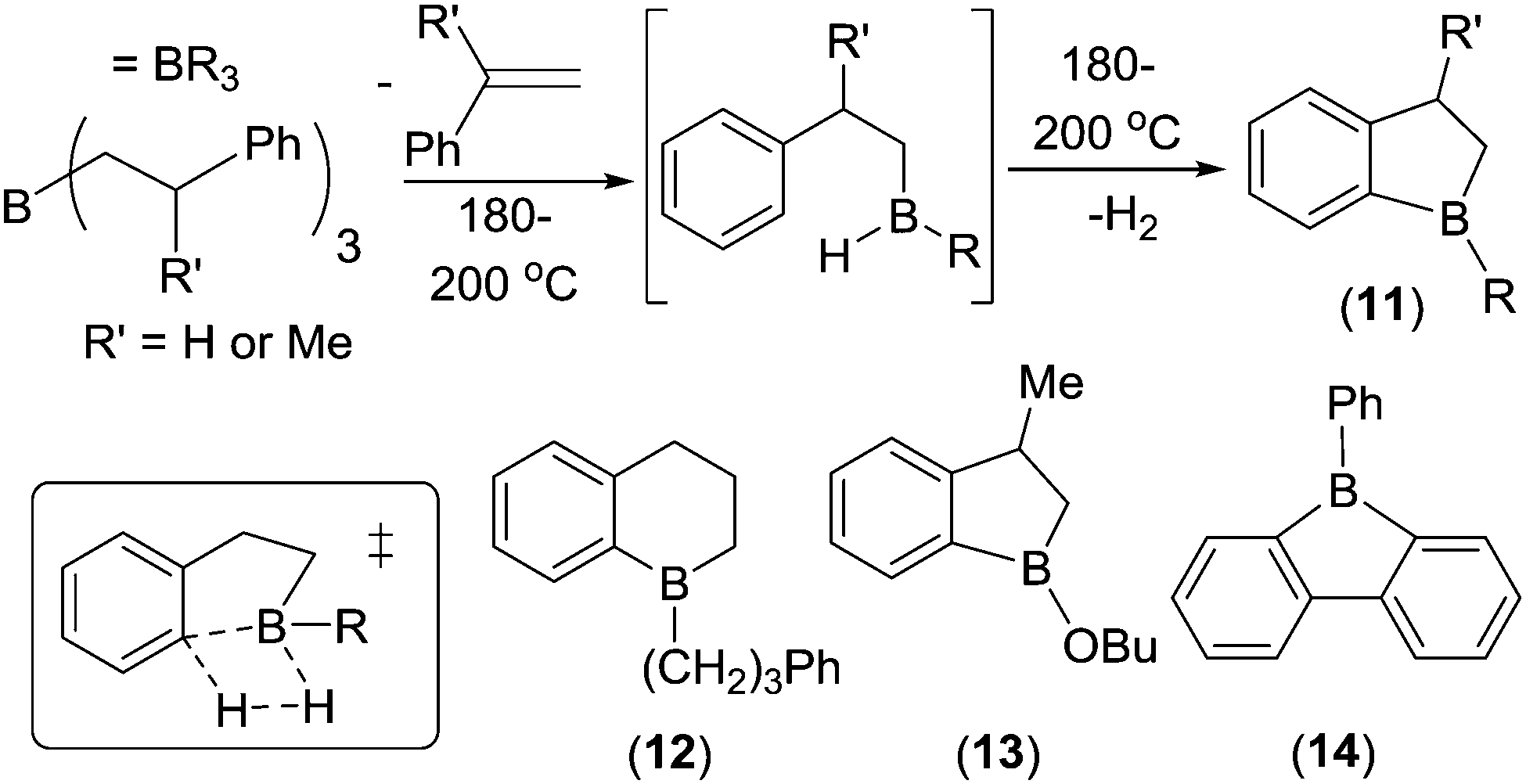 | ||
| Scheme 4 Select examples of Köster and co-workers’ intramolecular C–H borylation studies. Inset bottom left, the proposed transition state for C–H borylation mediated by B–H containing electrophiles. | ||
During these studies it was observed that the loss of RH (R = hydrocarbyl) as the by-product from boracycle formation (via a transition state related to that shown in inset Scheme 4 where B–H is replaced with B–R) had a much higher barrier than the respective borylation involving loss of H2. For example, 2-biphenyldiphenyl borane evolves benzene and forms 9-phenylborafluorene, 14, only at temperatures >280 °C. Whereas Köster and co-workers24 found that the formation of 9-borafluorenes via electrophilic borylation using B–H containing intermediates and evolving H2 proceeded at 120 °C. In most cases the major factor controlling reaction temperature in these studies is accessing the R2BH/RBH2 electrophile that effects C–H borylation. These are generally formed by retro-hydroboration which generally requires high temperature. Lower temperatures can be utilised for C–H borylation by accessing the key HBR2 species via alternative routes; e.g. substituent redistribution between BR3 and H3B–NEt3 or by alkene hydroboration (indeed intramolecular C–H borylation to form 1-boratetralins can occur at only 50 °C, vide infra).25
These early reports from the groups of Dewar and Köster demonstrate that both B–X and B–H electrophiles can effect intramolecular C–H borylation to give single products in good yield. However, after these reports intramolecular electrophilic C–H borylation was only infrequently utilised until the last decade. This review next discusses the developments post Dewar's and Köster's seminal studies and is organised by the type of interaction between the boron electrophile and the directing group (covalent E–BCl2versus dative [E→BCl2]+) and the identity of E in the directing group. It should be noted that there are significant commonalities in the factors controlling the reaction conditions and selectivity across these classes, and these are highlighted where appropriate.
3. Intramolecular C–H borylation preceded via covalent (E–BY2) bond formation
This section discusses reports where intramolecular C–H borylation is preceded by formation of E–BY2 (containing a conventional covalent bond, Y = halide or hydride most commonly). In some cases, the E–BY2 intermediate is observed, in others its presence is assumed based on literature precedence. The two major classes are where E is an amido (R2N) or a hydrocarbyl group, and these are discussed first in separate sections followed by the limited examples involving other heteroatom based directing groups.3.1 Via R2N–BY2 intermediates
To our knowledge, post Dewar's work the next report of N-directed electrophilic C–H borylation is from Letsinger and MacLean.26 The combination of 2-substituted benzimidazoles with BCl3 under forcing conditions (passing a stream of BCl3 through the substrate in the melt phase (>260 °C)) was proposed to form R2NBCl2 species that then led to C–H borylation and, post work up isolation of 15 and 16 (Scheme 5). However, an alternative scenario involving N→BCl3 formation and borylation without R2NBCl2 formation cannot be precluded (vide infra for discussion of the calculations on this system). This is also the case for a related system from Hatakeyama and co-workers where a 2-substituted benzimidazole undergoes double C–H borylation (using each N as a directing group) with BBr3 at 200 °C.27 In Letsinger's and MacLean's work it is also unclear if the high temperatures were essential for C–H borylation in the absence of AlCl3 as no lower temperature reactions were reported.
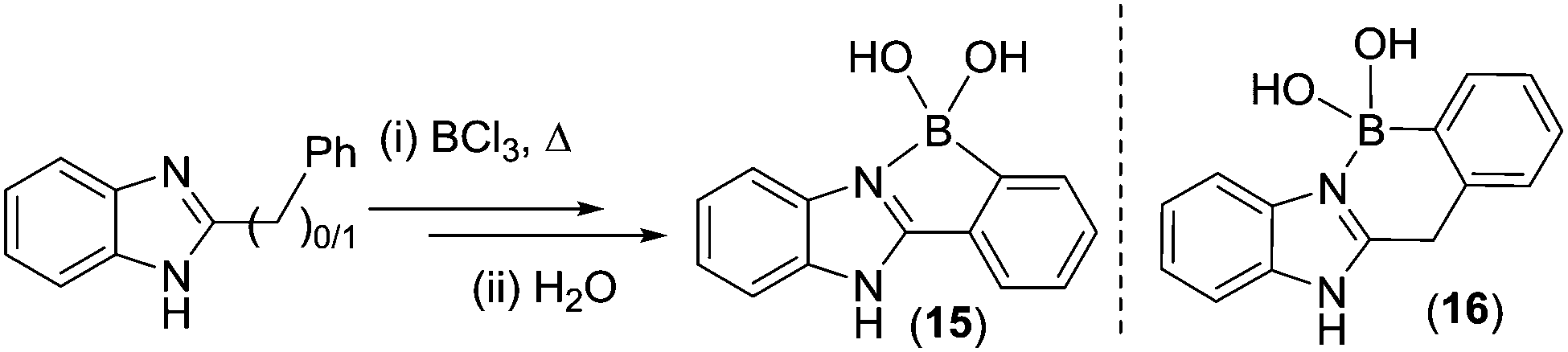 | ||
| Scheme 5 C2-Substituted benzimidazoles undergoing directed C–H borylation. | ||
In 1968, Köster and co-workers reported the first N-directed electrophilic C–H borylation reaction using B–H electrophiles (Scheme 6).28 The reaction of triethylamine-borane with N-methylbenzylamine was followed by H2 loss to produce the amino-borane. Under forcing conditions this formed 17 by dehydrogenative C–H borylation along with substituent redistribution products. Compounds containing six membered boracycles, e.g.18, were also accessible under similar conditions. The forcing conditions (>200 °C) required to access 17 and 18 are consistent with NB multiple bond character reducing the electrophilicity of the key boron species (presumably R2NBH2), as discussed subsequently carbon analogues (RBH2) undergo C–H borylation at temperatures as low as 50 °C.25,28
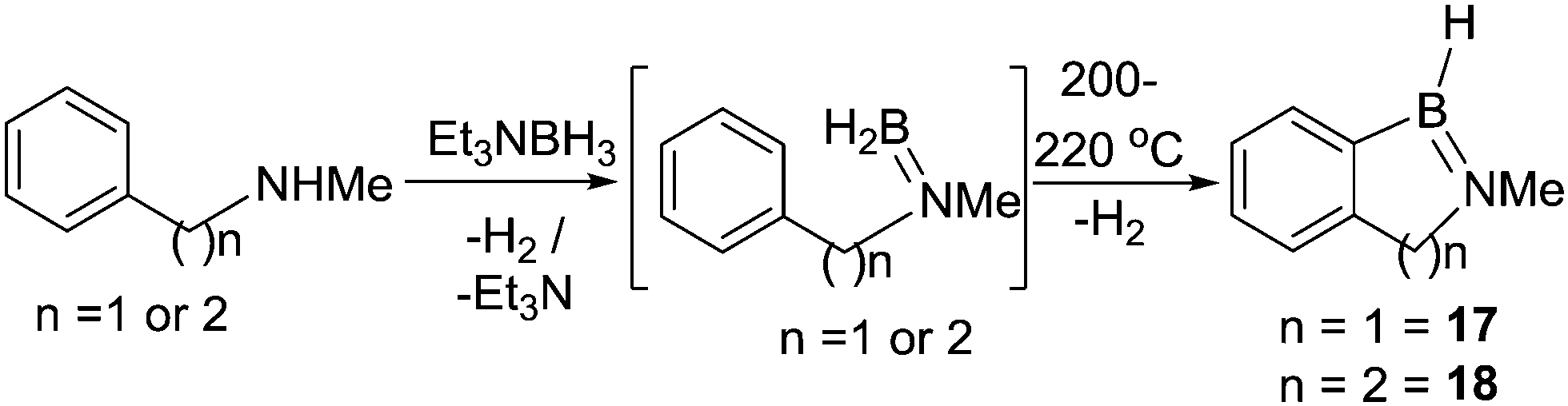 | ||
| Scheme 6 N-Directed electrophilic borylation via a B–H electrophile. | ||
Subsequent to this work, Müller and Grassberger utilised R2N directed electrophilic C–H borylation for the formation of a range of 2,3,1-diazaborines (19x, Scheme 7).29 In these independent reports arylsulfonyl-hydrazones were borylated using excess BX3 (X = Cl or Br), with work up affording 19x. The reaction conditions were relatively mild, proceeding at 60 °C and without requiring AlCl3 as catalyst. Other notable points include BBr3 outperforming BCl3 in terms of the isolated yield of borylated products. This is presumably due to bromo-borane species being more electrophilic than the chloro analogues. Steric effects were found to be important with a single isomer (19d) formed for the naphthalene congener with the more hindered isomer, E (Scheme 7), from borylation at the peri position not observed. The yield of 19b is lower than 19a, c and d under comparable conditions consistent with electron withdrawing groups (positive σpara values) leading to a higher barrier in the SEAr reaction. The milder borylation conditions in this study relative to Dewar's work can be attributed to the electron withdrawing sulfonyl group which presumably enhances the Lewis acidity of the key R2N-substituted boron electrophile that effects C–H borylation.
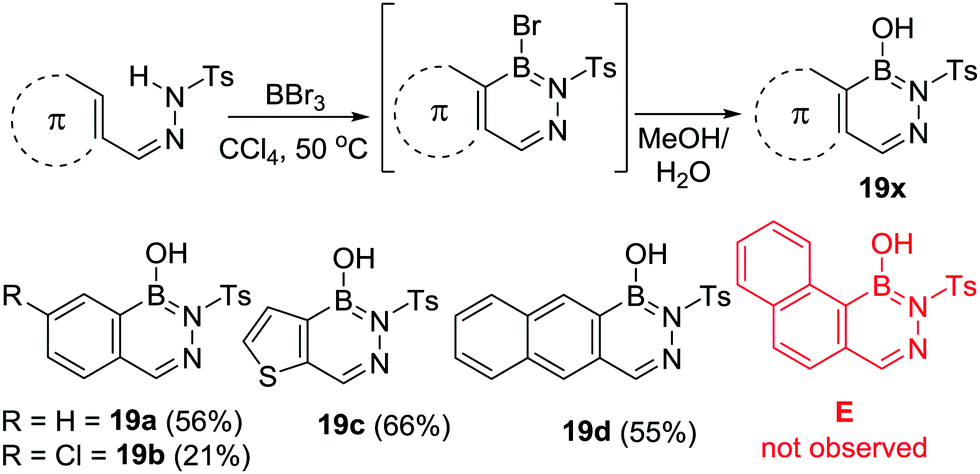 | ||
| Scheme 7 C–H borylation of sulfonyl-hydrazones with BBr3 (isolated yields in parentheses). | ||
The concomitant study by Grassberger and co-workers also noted that a catalyst was not necessary for C–H borylation, although they obtained higher yields when using BX3 in combination with FeCl3 as catalyst. This report contained a comprehensive substrate scope study with Me, F, Cl, Br, NH2, NMe2, NO2 functional groups tolerated. This study revealed, again, that electron withdrawing substituents on the aromatic group being borylated led to much lower yields. Furthermore, attempts to borylate deactivated (towards SEAr) heteroaromatics led to no conversion, this included pyridines, imidazoles and isothiazoles. Intramolecular C–H borylation to form 2,3,1-diazaborines also was shown to be applicable to other sulfonylamides.
Consistent with C–H borylation being dependent on the nucleophilicity of the (hetero)aromatic, Gronowitz and co-workers reported the facile intramolecular C–H borylation of bithiophenes. Amino bithiophenes were borylated using the less electrophilic borane PhBCl2 (compared to BCl3) in refluxing toluene without any Lewis acid activator (Scheme 8).30 The disparity to the biphenyl analogues (e.g.1) studied by Dewar highlights the milder borylation conditions (specifically no requirement for AlCl3 as a catalyst) permitted by moving to more nucleophilic (hetero)aromatics. The preference for thienyl alpha borylation over beta borylation (20 is formed selectively with no F observed, Scheme 8) is consistent with the SEAr of thiophenes.
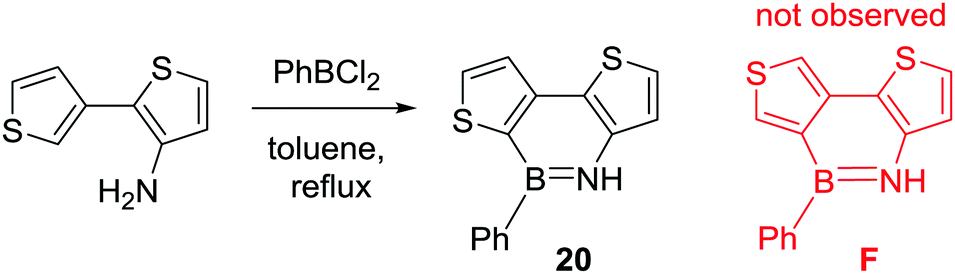 | ||
| Scheme 8 N-Directed C–H borylation of bithiophenes using PhBCl2. | ||
Isoelectronic analogues of 19 were formed by Mikhailov and co-workers, with intramolecular electrophilic C–H borylation providing 4-boraquinazolines 21 and 22.31 Forcing conditions were required to access 21 due to the mechanism proceeding via evolution of R–H during the electrophilic C–H borylation step (analogous to Köster's work). In contrast, the use of chloro-borane electrophiles and a Brønsted base led to milder C–H borylation conditions (enabling formation of 22 at 80 °C Scheme 9). Notably, this is an early example of an exogenous Brønsted base being added in an electrophilic C–H borylation reaction. The base may enable directed C–H borylation of a phenyl ring to proceed at a lower temperature (80 °C) in the absence of AlCl3 by facilitating a deprotonation step, e.g. of an arenium cation intermediate (in weakly basic media the deprotonation of arenium cations can be rate limiting in SEAr, vide infra).6c Subsequent work from Prasad and co-workers reported the closely related compound 23 (post work-up), albeit under more forcing conditions. However, no exploration of lower temperatures or the use of an exogenous base was reported.32
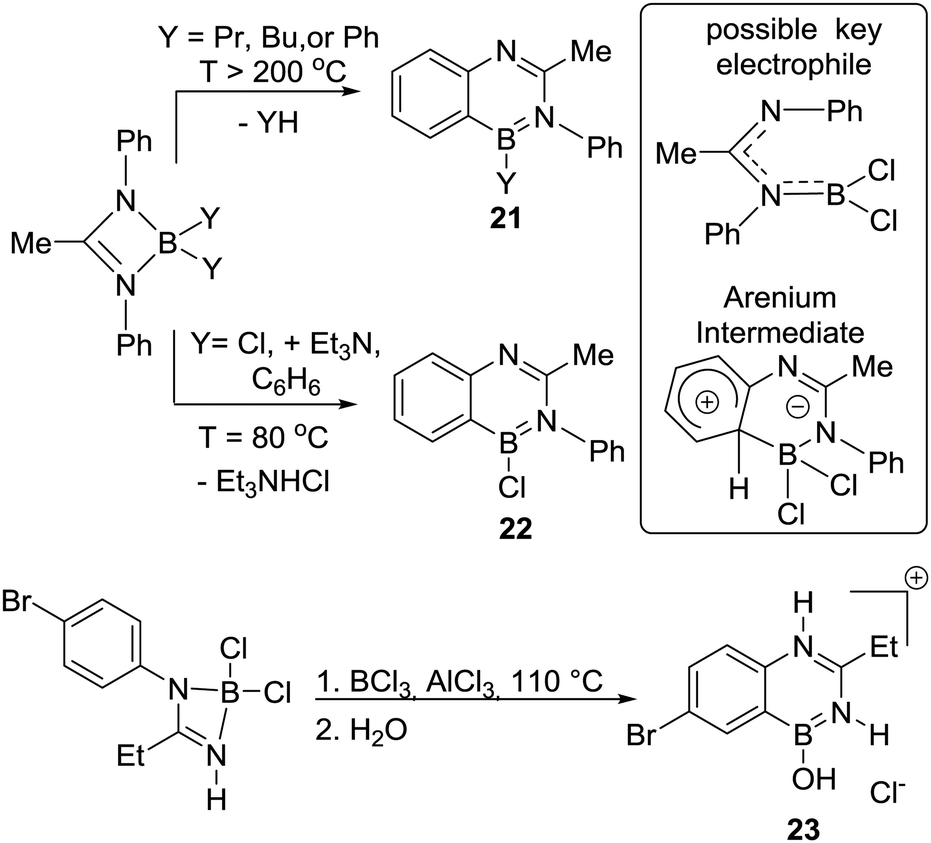 | ||
| Scheme 9 N-Directed C–H borylation of amidines. | ||
Another closely related compound, 24, was produced in good yield during attempts to form a bromo substituted borazine (Scheme 10). Compound 24 was formed on combining 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios of o-toluidine and BBr3 (at 130 °C). In contrast, a borazine product was produced using BCl3 under identical conditions.33 This again indicates that the use of bromo-boranes enables access to more reactive electrophiles and/or generates a more effective base (e.g. possible bases maybe [BBr4]− and [BCl4]−, respectively, with the former a better Brønsted base, vide infra). These factors, individually or combined, enable the intramolecular SEAr reaction starting from BBr3 in contrast to the chloro congener for this system.
:1 ratios of o-toluidine and BBr3 (at 130 °C). In contrast, a borazine product was produced using BCl3 under identical conditions.33 This again indicates that the use of bromo-boranes enables access to more reactive electrophiles and/or generates a more effective base (e.g. possible bases maybe [BBr4]− and [BCl4]−, respectively, with the former a better Brønsted base, vide infra). These factors, individually or combined, enable the intramolecular SEAr reaction starting from BBr3 in contrast to the chloro congener for this system.
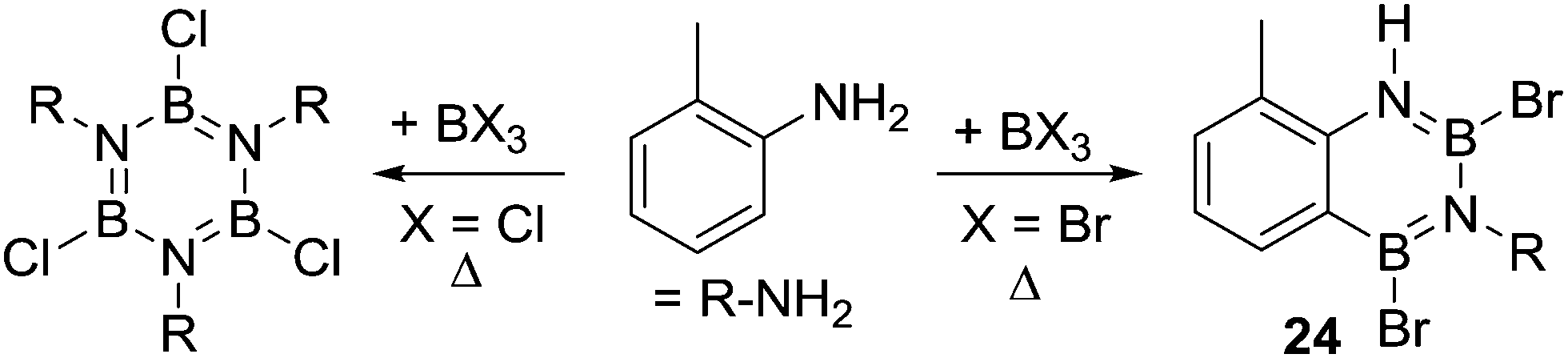 | ||
| Scheme 10 BX3 disparity in the formation of borazine (X = Cl) or 24 (X = Br). | ||
A notable study targeting analogues of 17 using B–Cl electrophiles (instead of B–H) was reported by Nagy and co-workers.34 In this report the addition of AlX3 (X = Cl or Br) to an aminoborane led to C–H borylation occurring at only 0 °C to form 25 (Scheme 11). An in situ NMR spectroscopy study was performed and the authors suggested a borenium cation (G),35 formed by protonation at N, as the key electrophile enabling the low temperature SEAr reaction. The proton source was attributed to adventitious moisture/impure AlX3. The contrast between these borylation conditions and Dewar's conditions, which both use R2NBCl(Ph)/AlCl3 is notable. While the flexibility of the sp3C centre present in G may be a factor facilitating C–H borylation (relative to Dewar's more rigid systems), the formation of a borenium cation and the enhanced electrophilicity (relative to neutral boranes) provided by the unit positive charge may also be vital. The in situ conversion of an aminoborane into an amine-stabilised borenium cation (G) blurs the distinction between directed C–H borylation proceeding via a R2NBX2 unit containing a covalent N–B bond, or [R2(E)N–BX2]+ (E = H for example) species containing a dative N→B bond.
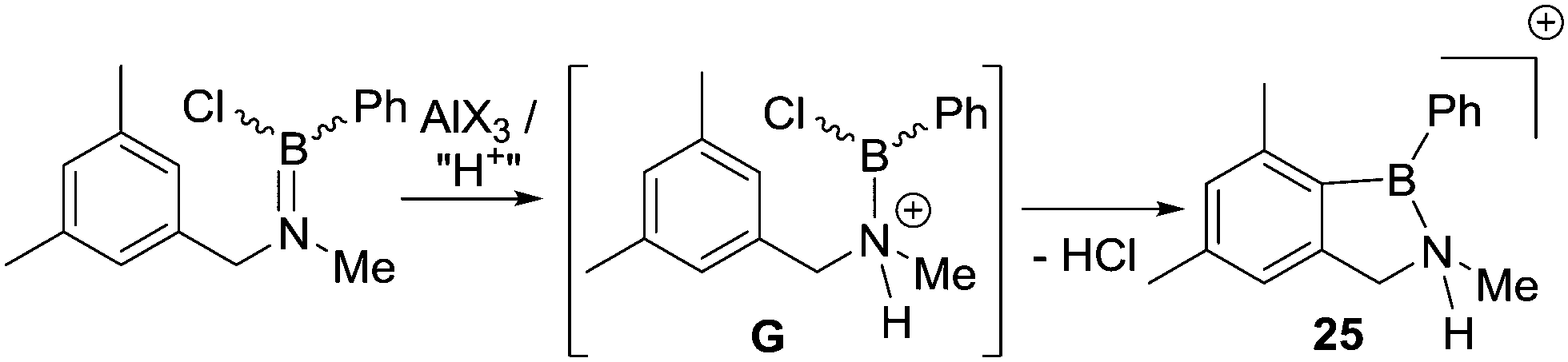 | ||
| Scheme 11 The formation of 25 proposed to proceed via a borenium cation intermediate (G). | ||
Later more detailed studies (see Section 4) confirmed that using borenium cations can lead to low barriers for intramolecular electrophilic C–H borylation. Thus, protonating nitrogen in a R2NBX2 species appears to be an overlooked route to facilitate low barrier intramolecular electrophilic C–H borylation reactions starting from R2NBX2 species. Furthermore, it suggests that AlCl3 is a less effective activator than “H+” with respect to forming strong boron electrophiles derived from R2NBY2 species. This is consistent with Corey and co-workers’ studies developing Lewis acidic borane catalysts by activating oxazaborolidines by binding electrophiles at N (Fig. 4). In these studies, the addition of AlCl3 led to a significantly less active (less Lewis acidic) boron based catalyst than the use of strong Brønsted acids (e.g. HOTf) to protonate at N.36
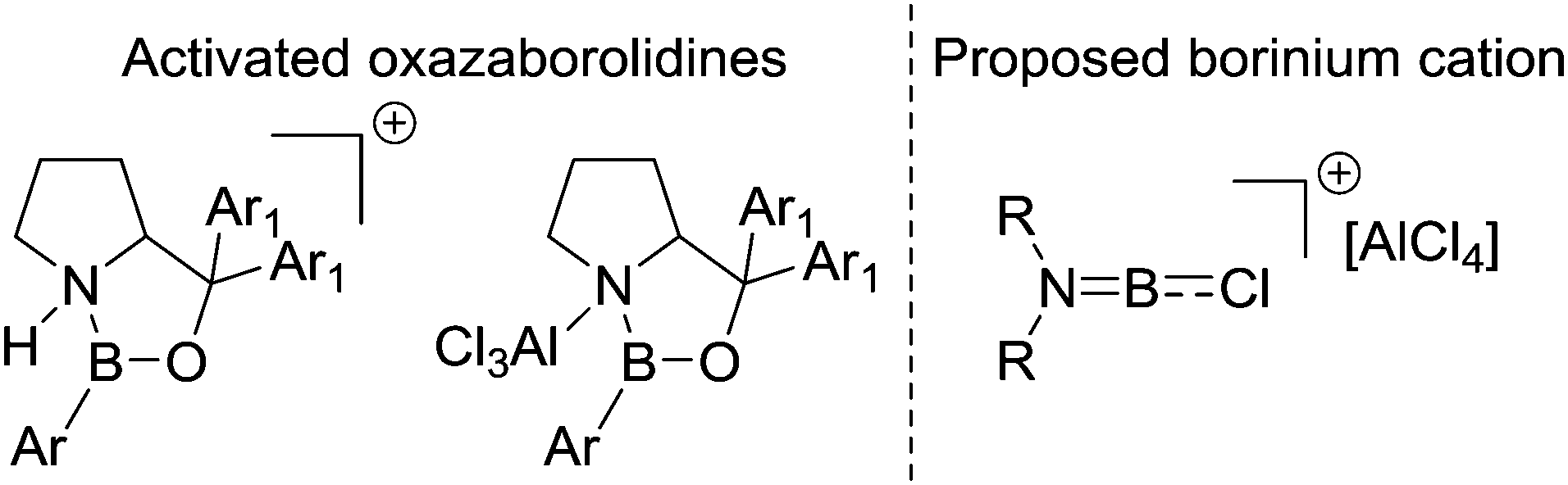 | ||
| Fig. 4 Left, oxazaborolidines activated by a Lewis acid or a Brønsted acid. Right, Dewar's proposed key electrophile for intramolecular electrophilic borylation. | ||
Dewar suggested that the role of AlCl3 in these borylation reactions was to abstract halide from R2NBCl2 to form [R2NBCl][AlCl4] borinium (two coordinate at boron) cations, (Fig. 4, right) which then effect the intramolecular SEAr reaction.37 However, subsequent work has shown that related borinium cations are extremely challenging to access in the condensed phase even using very strong halophilic Lewis acids.38 Indeed, weakly stabilised two and three coordinate (at boron) borocations have very high Lewis acidity towards chloride (often greater than AlCl3) and a propensity to oligomerise.39 While [R2NBCl]+ borinium cations are unlikely to be accessible in these reactions they cannot be precluded. However, it is more likely that these reactions proceed via species such as R2NB(Cl)–(μ-Cl)–AlCl3 or R2(AlCl3)N–BCl2.
Regardless of the active electrophile, there are now many reports on the borylation of 2-aminobiphenyls (and derivatives) using Dewar's conditions (or minor modifications thereof). It is worth noting here that amino-borane mediated C–H borylation reactions often require prolonged heating, however the time can be significantly reduced by using microwave irradiation. For example, using the same reagents and solvents compound 1 could be isolated in moderate yield (42%) after only 15 min under microwave irradiation (115 W).40
The current interest in exploring larger BN-doped polycyclic aromatic hydrocarbons (PAHs) for organic materials applications has led to a growth in the utilisation of amine directed electrophilic C–H borylation. In this context the early work of Dewar in forming 26,41 and that from the group of Philp is noteworthy. The latter performed N-directed diborylation by reacting a t-butyl-(2,6-diamino)biphenyl with PhBCl2 and AlCl3 as catalyst in refluxing xylene to form 27 (Scheme 12, inset).42 The formation of 26 and 27 initially both proceeds by evolution of two equivalents of HCl on heating to form the respective aminoboranes, Ph(Cl)BN(H)aryl, to which AlCl3 is added. Compound 27 is then accessible from this combination at lower temperature than 26, possibly due to the inductive effect of the tBu groups generating a more nucleophilic arene.
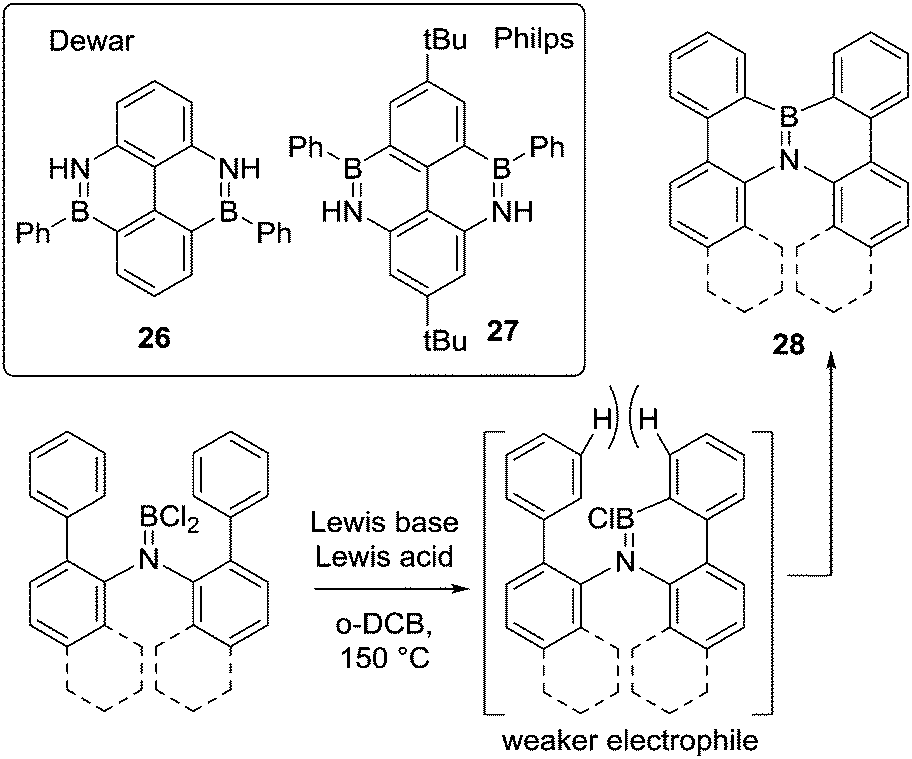 | ||
| Scheme 12 Formation of NB embedded PAHs by directed electrophilic borylation. | ||
Other notable work in this area is the use of a single boron centre in double C–H borylation. In this case of bis(biphenylyl)amines to form 28 (and derivatives, Scheme 12). This was achieved using a Lewis acid activator and a bulky base at 150 °C.43 The requirement for forcing conditions was ascribed to the lower Lewis acidity at boron in the intermediate formed from the first C–H borylation reaction (e.g. bottom right Scheme 12, due to π donation from the aromatic group) and the twisted conformation due to the steric demand of the hydrogens ortho to the borylated carbon atoms. Screening of different Lewis acids and bases as well as different reagent stoichiometry revealed dramatic variations in yield. The optimal reaction conditions utilised 4 eq. of AlCl3 and 1.5 eq. of 2,2,6,6-tetramethylpiperidine (TMP) as a non-nucleophilic Brønsted base. When either the amount of Lewis acid or amine were changed, even by only 0.5 eq., the yield was lowered significantly. It is currently unclear precisely what is leading to these changes in yield, however, the requirement for at least four equivalents of AlCl3 to obtain acceptable yields suggests the higher chloride affinity associated with Al2Cl6 (relative to AlCl3) maybe essential to access the key boron electrophile.44
Subsequently, Pei and co-workers and Zhang and co-workers reported the synthesis of BN–PAHs such as heterocoronene, 29, (Scheme 13).45 The electron rich nature of the aromatic being borylated and the use of a base made it possible to use PhBCl2 without any Lewis acid catalyst (the absence of which was essential to minimise ether cleavage) to achieve C–H borylation. The use of exogenous base even enabled Zhang and co-workers to synthesise a BN-fused perylenediimide (30) by amine directed C–H borylation. This is notable due to the electron deficient nature of perylenediimides (thus they have low energy HOMO) and indicates that directed electrophilic C–H borylation can be extended to deactivated (towards SEAr) aromatics using the appropriate conditions (such as using an exogenous base potentially facilitating deprotonation of an arenium cation).46
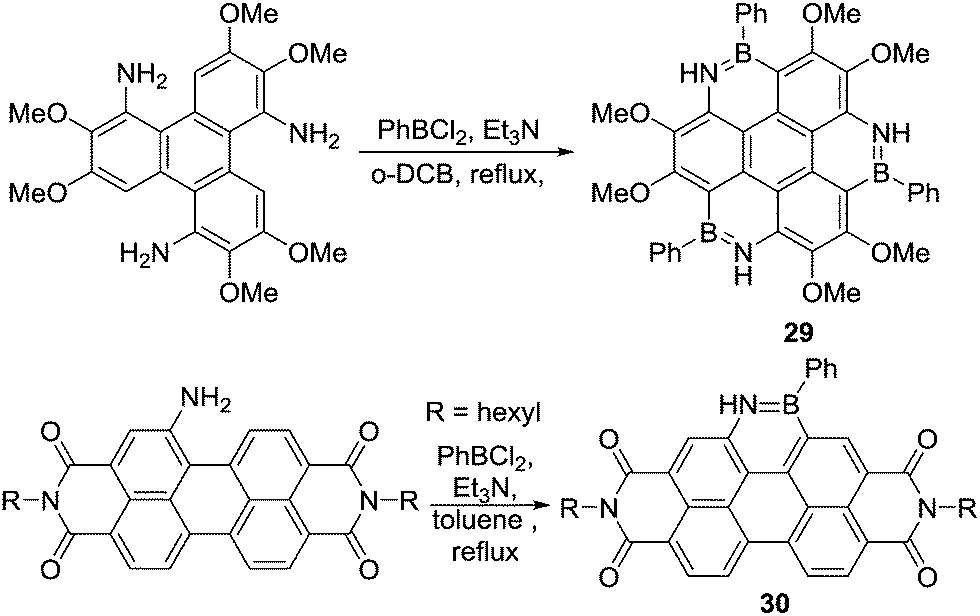 | ||
| Scheme 13 Top, functional group tolerant triple N-directed C–H borylation and bottom, N-directed C–H borylation of a perylene diimide. | ||
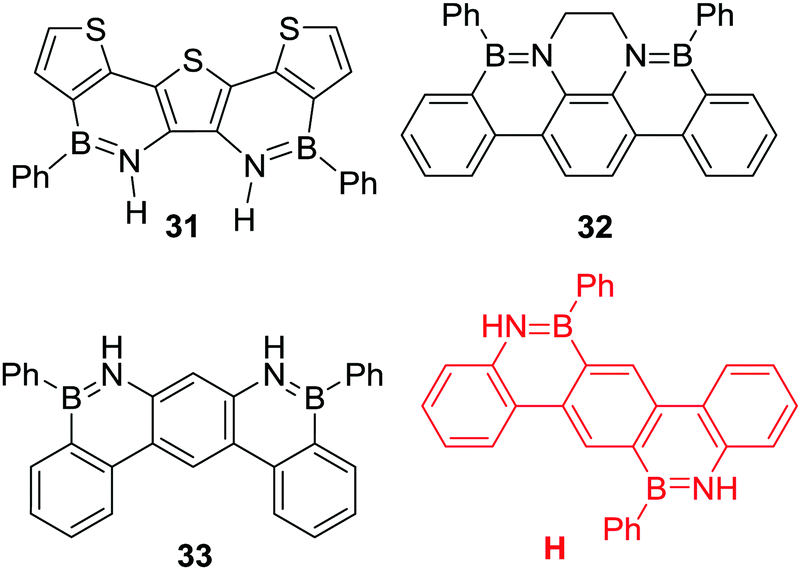
Multiple N-directed C–H borylation reactions have been reported of terphenyl derivatives. While the more nucleophilic thienyl congener 31 was accessible in good yield (89% using PhBCl2/Et3N reflux in PhCl) it is noteworthy that out of the three other BN-terphenyl compounds in Scheme 14, only 32 was accessible from the parent amino-terphenyl under catalyst free conditions (using PhBCl2, 3 eq. NEt3, 130 °C, 24 h).47 More forcing conditions were required to access 33, which could be obtained only after heating the precursor in the presence of PhBCl2 and FeCl3 at 260 °C for 7 h.48 While the origin of this disparity is not clear it may be due to the relative basicity of the respective anilines, with the parent aniline of 32 containing more basic nitrogen centres, which will favour dative bond formation with PhBCl2. This is the first step in directed electrophilic borylation, and without initial dative bond formation no C–H borylation will proceed. It is also notable that compound H was not accessible by heating the parent amino-terphenyl with PhBCl2/Et3N at 180 °C. This was attributed to the challenge of doubly borylating the central 1,4-phenylene ring with the first borylation installing a mesomerically deactivating boron moiety.49 Consistent with previous reports, directed C–H borylation was more readily achieved when the unit being borylated was a more nucleophilic congener of H, e.g. with a thienyl derivative as the central aromatic unit being borylated.
 | ||
| Scheme 14 Select accessible (and inaccessible) products derived from amino-terphenyl directed C–H borylation. | ||
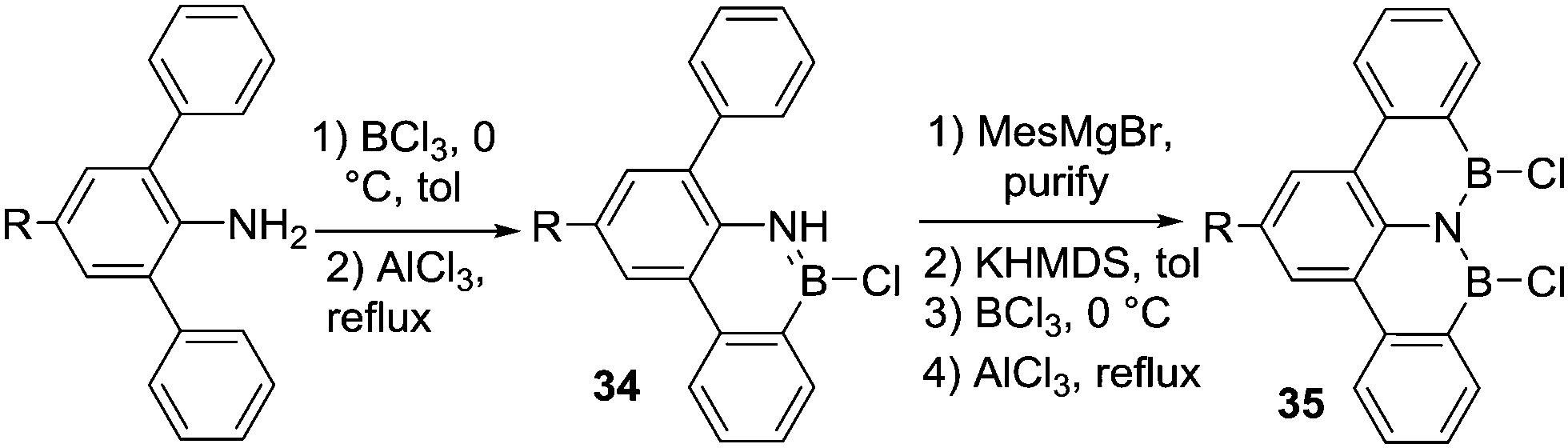
Bettinger and co-workers attempted to access a BNB-benzo-tetracene, 35, via double N-directed C–H borylation of an amino-terphenyl. However, using BCl3 and AlCl3 in refluxing toluene led only to the borylation of one phenyl ring and formation of 34 (Scheme 15).50 This can be attributed to a significant reduction in the basicity of the N centre in 34 by N to B π-donation preventing further BCl3 coordination to this nitrogen (and thus precluding the second C–H borylation). The Lewis basicity of the N-directing group is an important consideration for enabling C–H borylation, as a minimum basicity is required to access R2N(H)–BY3 intermediates. If low Lewis basicity precludes adduct formation (as in 34), then metalation of N–H and a subsequent salt metathesis, e.g. by addition of BCl3, provides a solution and can afford the desired species (e.g. R2NBCl2). Thus, treatment of the B-mesityl derivative of 34 with potassium bis(trimethylsilyl)amide and subsequent reaction with BCl3 followed by AlCl3 addition and heating to 110 °C led to the desired BNB-benzo-tetracene 35 (C–H borylation occurs along with B-Mes to B-Cl conversion).
 | ||
| Scheme 15 Metalation assisted N-directed electrophilic C–H borylation. | ||
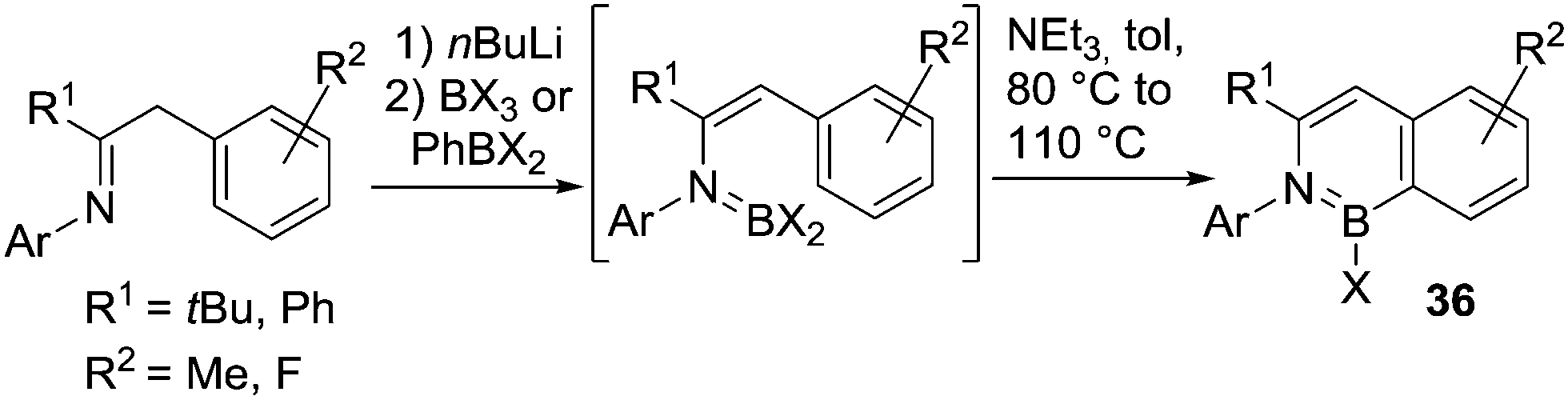
Deprotonation of a N–H substrate with a strong base followed by a trans-metalation reaction (using BX3) is essential in many cases where dative bond formation (e.g. forming R2(H)N–BX3) or where loss of HX post adduct formation to form R2NBX2 is not favoured. In this context, the metalation/BX3 transmetalation approach has been applied by Cui and co-workers for the functionalisation of benzyl imines to form borazanaphthalenes, 36 (Scheme 16).51 Borylation proceeded with no added Lewis acid catalyst and at temperatures <110 °C due to the presence of an exogenous base. Notably, no C–H borylation is observed in the absence of Et3N under these conditions. This suggests that a deprotonation step during SEAr is rate limiting in this example. As expected, the use of more electrophilic borylating agents led to faster borylation reactions that also proceeded at lower reaction temperatures.
 | ||
| Scheme 16 Imine conversion to an enamine for use in subsequent N-directed electrophilic C–H borylation. | ||
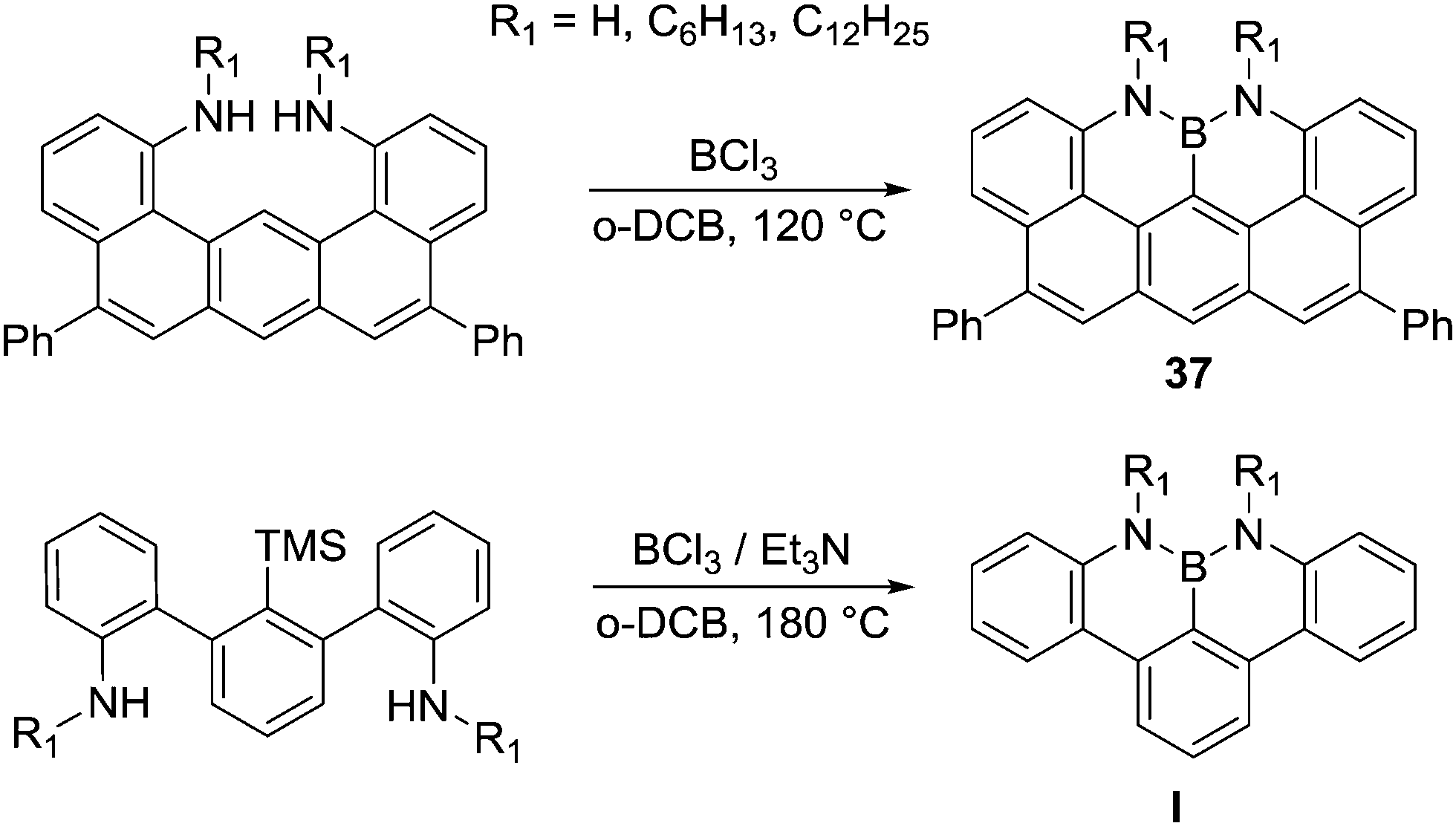
The formation of NBN–PAHs also can be achieved by intramolecular electrophilic C–H borylation. Milder reaction conditions (no AlCl3 required, in contrast to Dewar's conditions) were used to access compound 37, and this was attributed to the extended π-conjugated backbone generating a more nucleophilic aromatic facilitating electrophilic attack (Scheme 17).52 Indeed, a related compound without an extended PAH required installation of trimethylsilyl group to enable subsequent formation of the C–B bond in I, with no electrophilic C–H borylation observed when using a non-silylated precursor.
 | ||
| Scheme 17 Formation of NBN PAHs via N-directed electrophilic borylation. | ||
A method to access NBN containing PAHs was reported by Hatakeyama and co-workers.53 This accompanied directed C–H borylation with demethylation of N,N-dimethylaniline groups in the presence of basic additives (Scheme 18), although it is not currently known if C–H borylation occurs pre or post demethylation. The presence of NaBPh4 as an additive produced higher yields (ca. 70%), and it was proposed that the borate anion acts as a non-coordinating (towards BY3) Brønsted base. Consistent with the importance of base in this reaction 2,2,6,6-tetramethylpiperidine (TMP) also successfully enabled formation of 38 (albeit in lower yield). In the absence of any base conversion was extremely low, again highlighting the importance of exogenous base in enabling certain electrophilic C–H borylation reactions. This work also emphasises the importance of using Brønsted bases that do not strongly coordinate to boron Lewis acids.
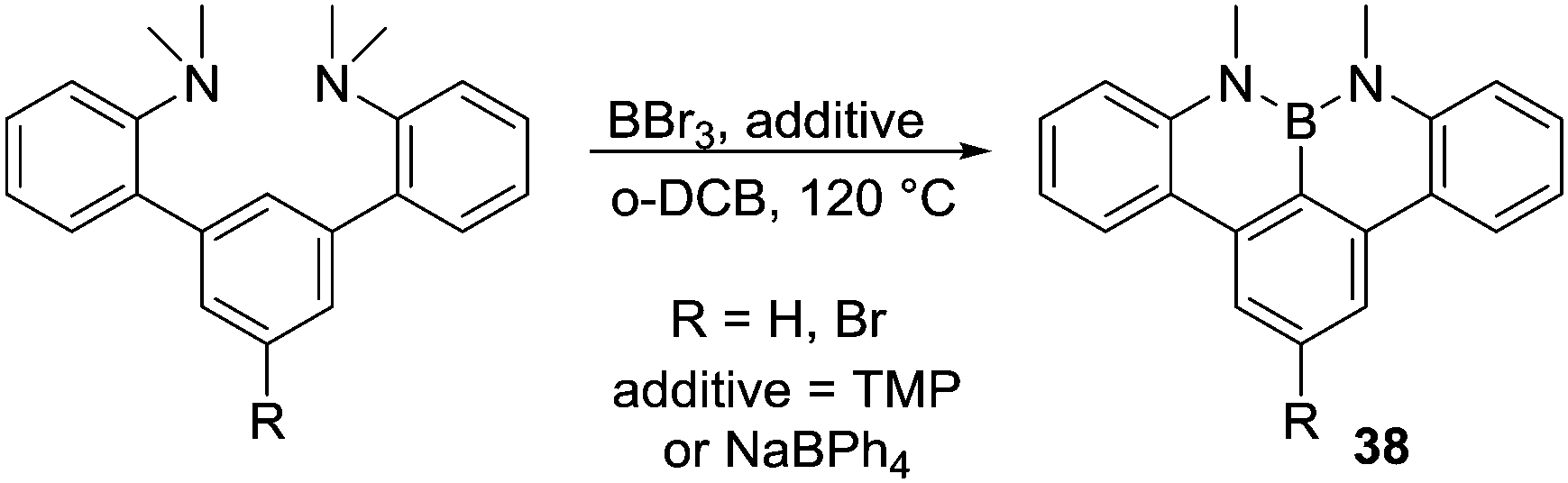 | ||
| Scheme 18 Demethylative N-directed C–H borylation. | ||
Other reactions can be utilised to access the key boron species for N-directed C–H borylation, as exemplified by the synthesis of 39 and 40 (Scheme 19). In these reactions N-directed alkyne trans-haloboration is followed by intramolecular C–H borylation.54 The low yields and the harsh reaction conditions (using AlCl3 along with a sterically encumbered base in refluxing 1,2,4-trichlorobenzene (boiling point 214 °C)) are presumably required in part due to the low electrophilicity of the boron species performing electrophilic borylation (e.g. inset right Scheme 19). As expected, the reaction proceeds under more facile conditions (without AlCl3 and a hindered base) when a more nucleophilic thiophene unit is borylated. The relative ease of thiophenes to undergo electrophilic C–H borylation has been widely exploited in the synthesis of BN-containing PAHs. Similar borylation conditions (PhBCl2 or BX3, NR3, aromatic solvent, reflux) have been found to produce good outcomes (yields >60%) for many N-directed C–H borylation reactions of thiophene containing systems. However, in the interest of brevity we do not discuss these transformations individually herein.55
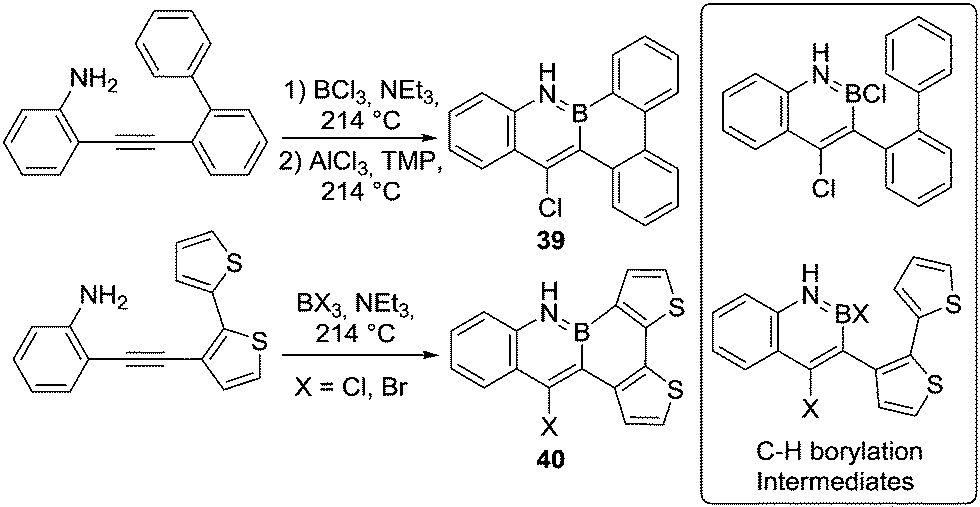 | ||
| Scheme 19 Sequential trans-haloboration and directed C–H borylation reactions. | ||
In addition to activated heteroarenes such as thiophene, non-aromatic π systems (e.g. alkenes) also undergo N-directed C–H borylation reliably under milder (compared to analogous aromatic systems) reaction conditions (e.g. without AlCl3, at temperatures <130 °C) using a variety of boron electrophiles.56 Provided the amine is sufficiently Lewis basic to form the initial N→B dative bond N-directed alkene C–H borylation has been used to synthesise many extended BN containing PAHs.57 Again in cases where amine Lewis basicity is low deprotonation/trans-metalation can be used to access the R2N–BCl2 intermediate. Another consideration is highlighted by the observation that 41 is much less accessible than an N-alkylated analogue 42 (Scheme 20). This emphasises the importance of precluding unwanted side reactions, as formation of a diazaborole (e.g.J) is presumably competing with directed C–H borylation to form 41 but not to form 42.58 This is in contrast to the formation of 32, potentially due to the relatively facile C–H borylation of thienyls and the greater strain in fusing two all sp2 five membered rings through beta positions.
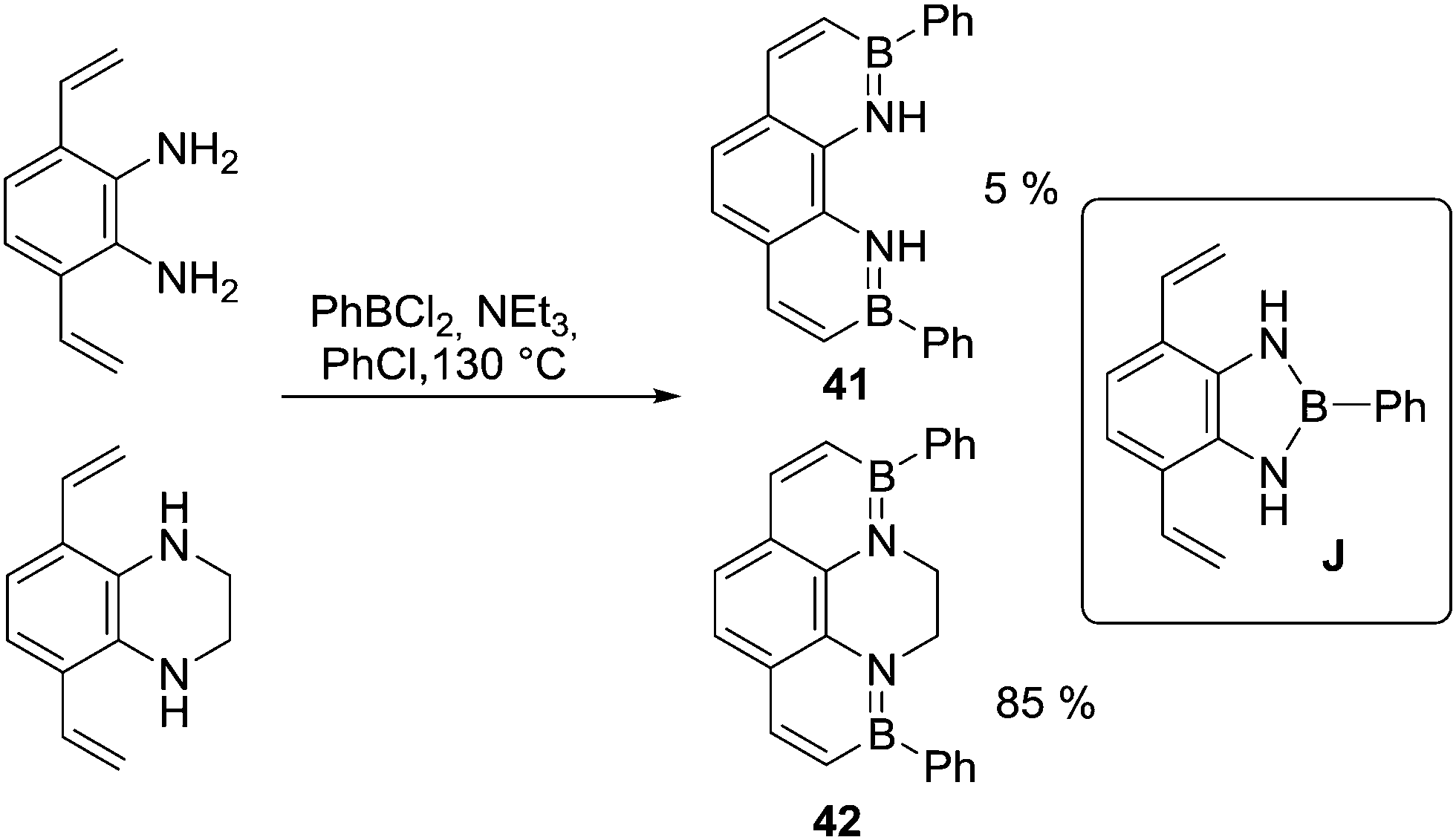 | ||
| Scheme 20 The N-substituent dependent formation of BN-phenanthrenes. | ||
In the area of N-directed alkene C–H borylation the work of Molander and co-workers is notable. They accessed a large variety of 2,1 borazanaphthalenes, 43 (Scheme 21), using combinations of potassium organo-trifluoroborate salts and SiCl4.59 This allowed for the in situ generation of the electrophilic boron reagent (e.g. organoBCl2) by halide exchange between boron and silicon centres. Alkene borylation then proceeded effectively without AlCl3 and at temperatures <60 °C consistent with other reports on intramolecular alkene C–H borylation being facile relative to that of aromatics. An impressive range of functional groups (on B and N substituents) are tolerated under the reaction conditions. However, borylation of deactivated N-heterocycles (e.g. pyridines) was not reported in this work. Rombouts and co-workers adapted Molander's conditions to extend this methodology to borylate 2-vinyl-3-amino-pyridines (Scheme 21, bottom) to access 44.60 The reaction was carried out using microwave assisted heating at 180 °C in the presence of SiCl4 and NEt3. The higher temperature was proposed to be required to enable SEAr of the deactivated π system, while NEt3 may also be essential to facilitate a deprotonation step during C–H borylation. However, Molander and co-workers subsequently suggested that pyridyl (and other Lewis basic N-heterocycles) coordination to main group Lewis acids was preventing N-directed borylation in their previous work.61 Therefore they increased the amount of Lewis acid (SiCl4) to two equivalents (with respect to the N-heterocycle) and included Et3N as a relatively hindered base. Combined this enabled N-directed alkene C–H borylation at only 80 °C including of vinyl-amino-pyridines. This method was used subsequently by Vaquero and co-workers to synthesize an intermediate on the way to a BN-embedded chrysene.62
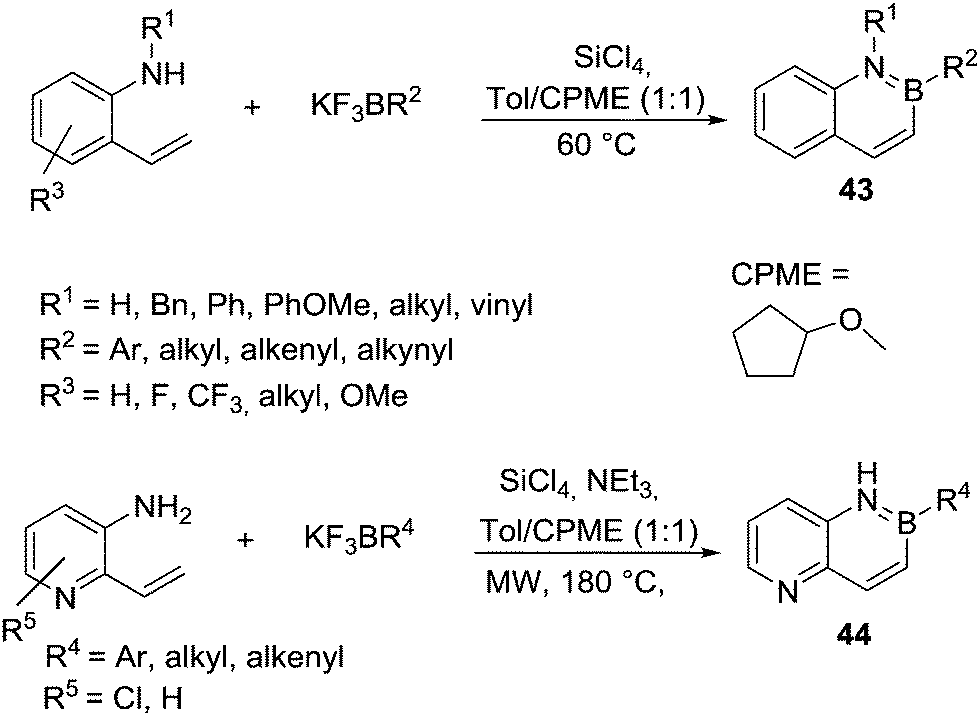 | ||
| Scheme 21 C–H borylation using potassium organo-trifluoroborates/SiCl4. | ||
The group of Yan and co-workers also utilised this synthetic approach to borylate 1-(2-aminophenyl)pyrroles to form 45. In this case C–H borylation is occurring on a heteroarene not an alkene (Scheme 22),63 but borylation conditions are still relatively mild due to the highly nucleophilic nature of pyrroles. Unsurprisingly, the reaction does not proceed in coordinating solvents such as DMF, which will sequester the main group Lewis acids required for electrophilic borylation. BCl3 can be used in place of SiCl4 to access the key electrophile (e.g. ArylBCl2) and more notably BF3–OEt2 also worked as the Lewis acid activator. This suggests that ArylBF2 species are effective for directed C–H borylation under these conditions, at least of highly activated heteroaromatics, via the possible intermediate shown (inset Scheme 22).
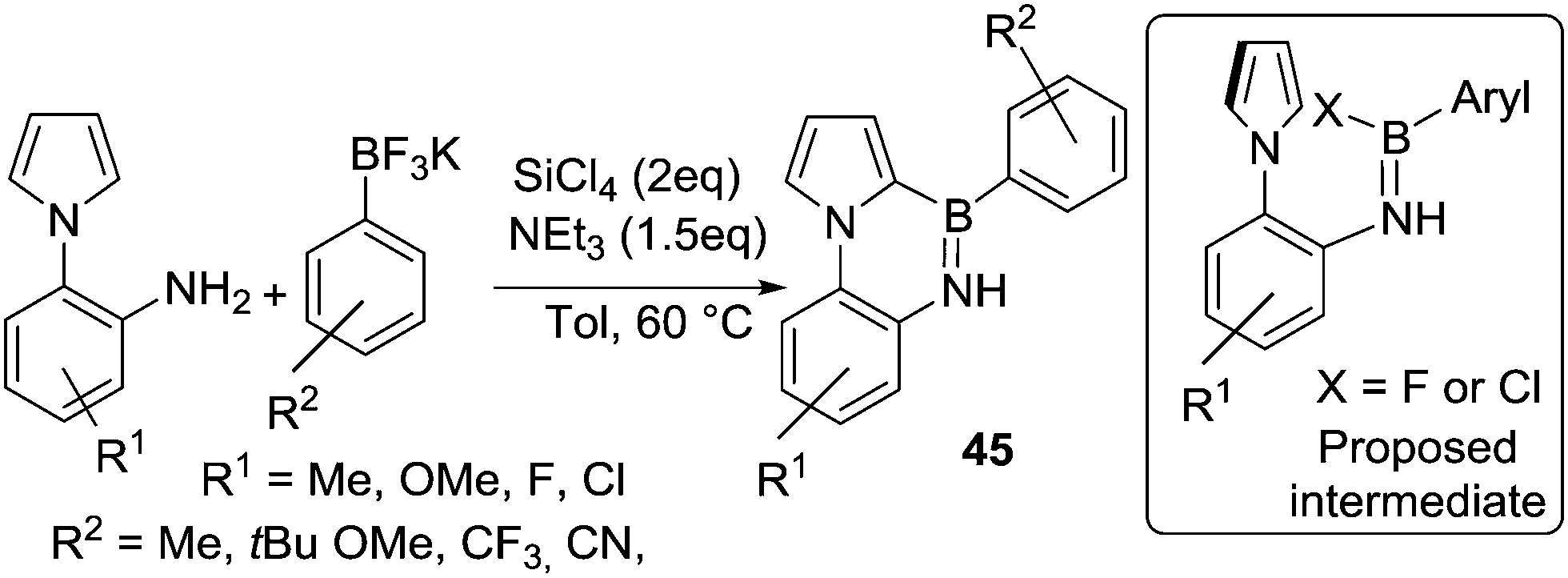 | ||
| Scheme 22 N-Directed C–H borylation of pyrroles using potassium organo-trifluoroborates/SiCl4 to generate boron electrophiles. Inset the proposed intermediate, note that X = F is from replacing SiCl4 with BF3. | ||
As the above demonstrates, N-directed electrophilic C–H borylation is a powerful tool for the functionalisation of arenes, heteroarenes and alkenes. This reaction proceeds selectively to give six membered boracycles in most cases (especially for boracycles containing all sp2 main group atoms) and can proceed under mild conditions with broad functional group tolerance, particularly when borylating more nucleophilic aromatic systems or alkenes. These points indicate it will continue to be a highly useful methodology.
3.2 Borylation via C–BY2 intermediates (Y = H, R or halide)
After N-directed C–H borylation the next most reported is borylation proceeding via initial intermolecular C–B bond formation. While pioneered by Köster and co-workers, their borylation procedures are not practical due to the high temperature conditions required. Shortly after Köster's work Bickelhaupt and co-worker performed pyrolysis on a mixture of tris(2-benzhydylphenyl)boroxine, lithium aluminium hydride and tributoxyborane at 220 °C leading to C–H borylation product 46 (Scheme 23). The reaction may proceed via a primary borane (RBH2, insert Scheme 23) or other derivatives (e.g. RB(H)(OBu)).64a The same product could also be accessed by the pyrolysis of the borane-pyridine adduct at 220 °C.64b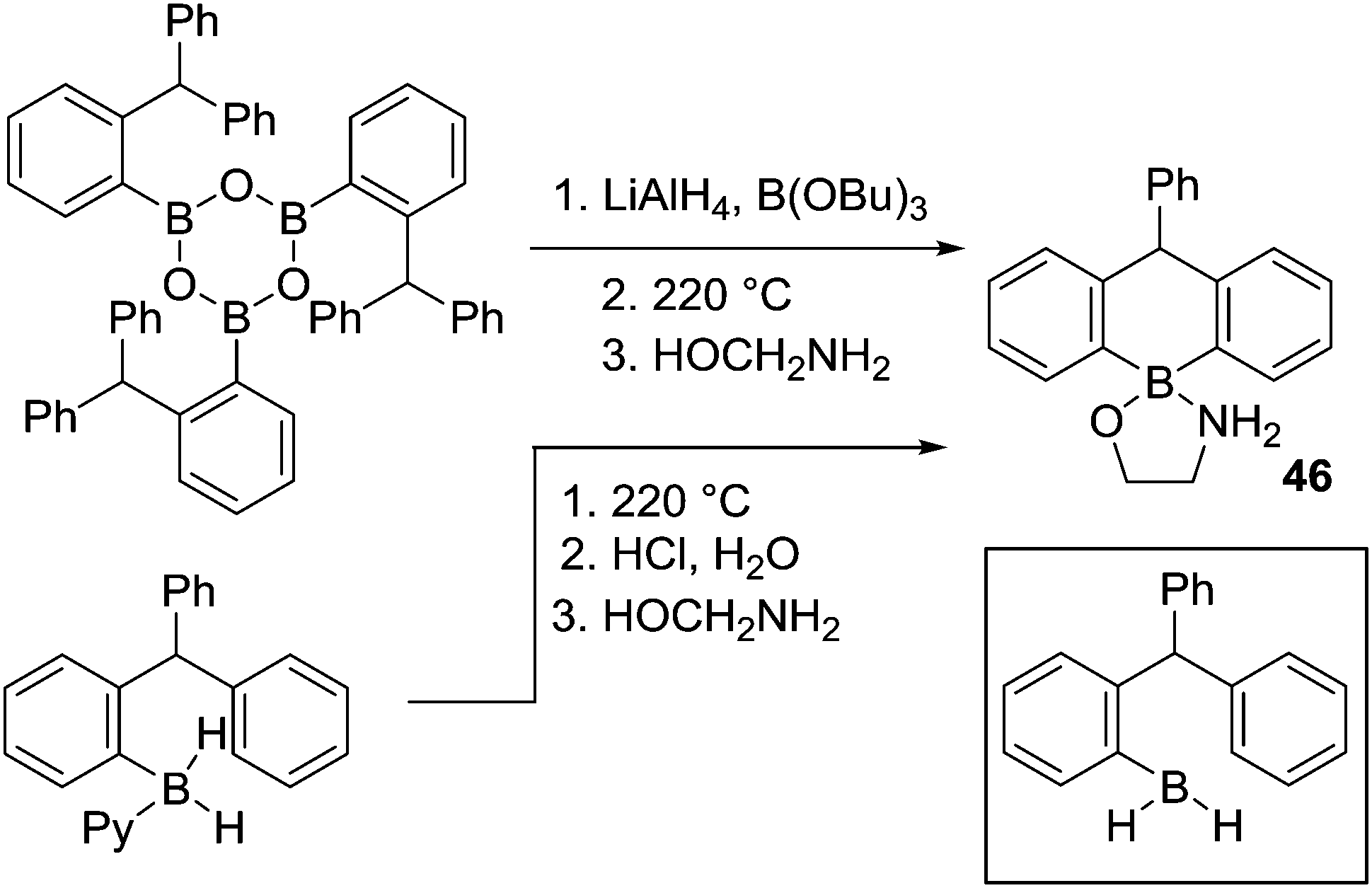 | ||
| Scheme 23 Alternative routes to access B–H species effective for intramolecular C–H borylation. | ||
This early work on C–BY2 mediated C–H borylation required extremely high temperatures, though the high barrier in many cases in Köster's work arises from the retro-hydroboration step required to generate the key boron electrophiles. Thus, lower temperature intramolecular C–H borylation processes using C–B(R)–H based electrophiles are feasible by using more facile routes to access the key electrophile. To our knowledge this was first demonstrated in 1999 when Knochel and co-workers reported the synthesis of cyclic boranes by intramolecular C–H borylation using a B–H electrophile under relatively mild conditions.25 By heating a mixture of a tetra-substituted olefin with a borane at 50 °C both five- and six-membered boracycles, 47 and 48, were obtained after one hour indicating a lower barrier process compared to Köster's systems.25c Notably, only 48 was observed after 24 h at 50 °C (Scheme 24),25a with the formation of specific boracycles confirmed by oxidative workup. While 47 was formed through sequential hydroboration/intramolecular C–H borylation reactions, mechanistic studies indicated that compound 48 was generated from 47via a retro-hydroboration/hydroboration sequence. Calculations showed that 48 was thermodynamically favored over 47 by 5 kcal mol−1, indicating that 47 is the kinetic product but 48 is the thermodynamic product.
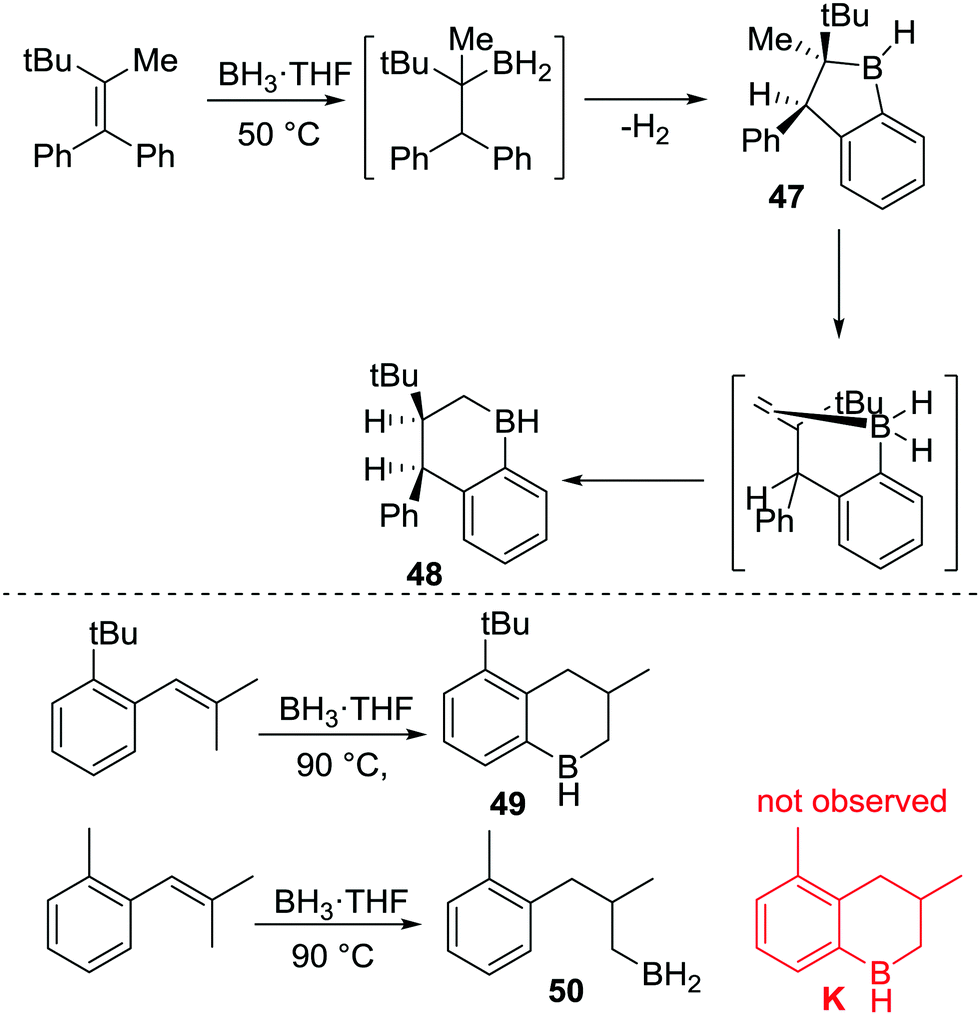 | ||
| Scheme 24 Relatively low temperature intramolecular C–H borylation via B–H intermediates. | ||
This methodology also could be applied to several tri-substituted alkenes, e.g. to form 49, under similar conditions (Scheme 24, bottom). Notably, proximal bulky groups were found to be essential for C–H borylation at these lower temperatures. For example, replacement of a tert-butyl group with a methyl group led to no C–H borylation (K is not formed) and only 50 was produced. The tert-butyl group is proposed to orientate the initial hydroboration product into a productive conformation for C–H borylation, thus enabling low temperature C–H borylation. The importance of bulk proximal to the C–BH2 unit for C–H borylation to proceed was also observed in other related examples, as was the finding that five membered boracycles are the kinetic products derived from the C(sp3)–BH2 intermediates, while six membered boracycles were often the thermodynamic products.25c
In further studies, Knochel and co-workers obtained the five-membered boracycle 51 (Scheme 25) exclusively as an analogous rearrangement to a six-membered boracycle (as per formation of 48) is not possible. In contrast, only hydroboration product 52 was formed, with no six-membered boracycle, L, generated under these lower temperature (50 °C) reaction conditions.25c This is consistent with the slower formation of six membered boracycles in these systems. Hence longer reaction times and higher temperatures are required to form six membered boracycles directly via sp3C–BH2 electrophiles. This is emphasized by only 16% of diol 53 being formed after the C–H borylation step had been heated for 9 days at 90 °C (53 is formed from boracycle 49). This is in contrast to the outcome using an isomeric alkene where initial C–H borylation can proceed via a five membered boracycle (e.g. formation of 49 is high yielding in Scheme 24, bottom). In the formation of five membered boracycles it is unclear if the energetic barrier requiring heating to 50 °C comes from dissociation of THF to form free RBH2 or the C–H borylation step.
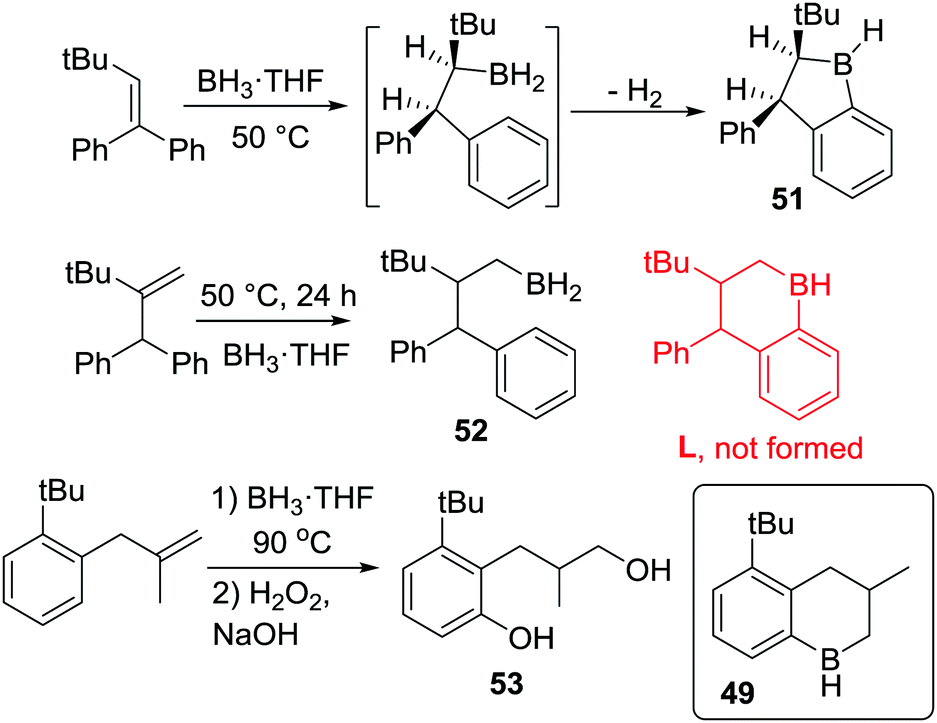 | ||
| Scheme 25 Disparities in C–H borylation reactions proceeding initially to form five or six membered boracycles. | ||
This methodology could also be extended to form boracycles derived from biphenyl substrates (and related molecules), again provided sufficiently bulky tri-substituted alkenes are used.25c The six-membered boracycles 54 are prepared via sequential hydroboration/C–H borylation reactions (Scheme 26). Notably, the borylation reaction was found to be insensitive to the electronic nature of substituents on the meta position (relative to the borylation site) of the phenyl group. Similar yields were obtained for substrates containing H, methoxy or trifluoromethyl at this position under the same reaction conditions. The authors used this to conclude that the C–H borylation step was proceeding via a four membered metathesis type transition state (analogous to Köster's proposal) and not an SEAr mechanism.
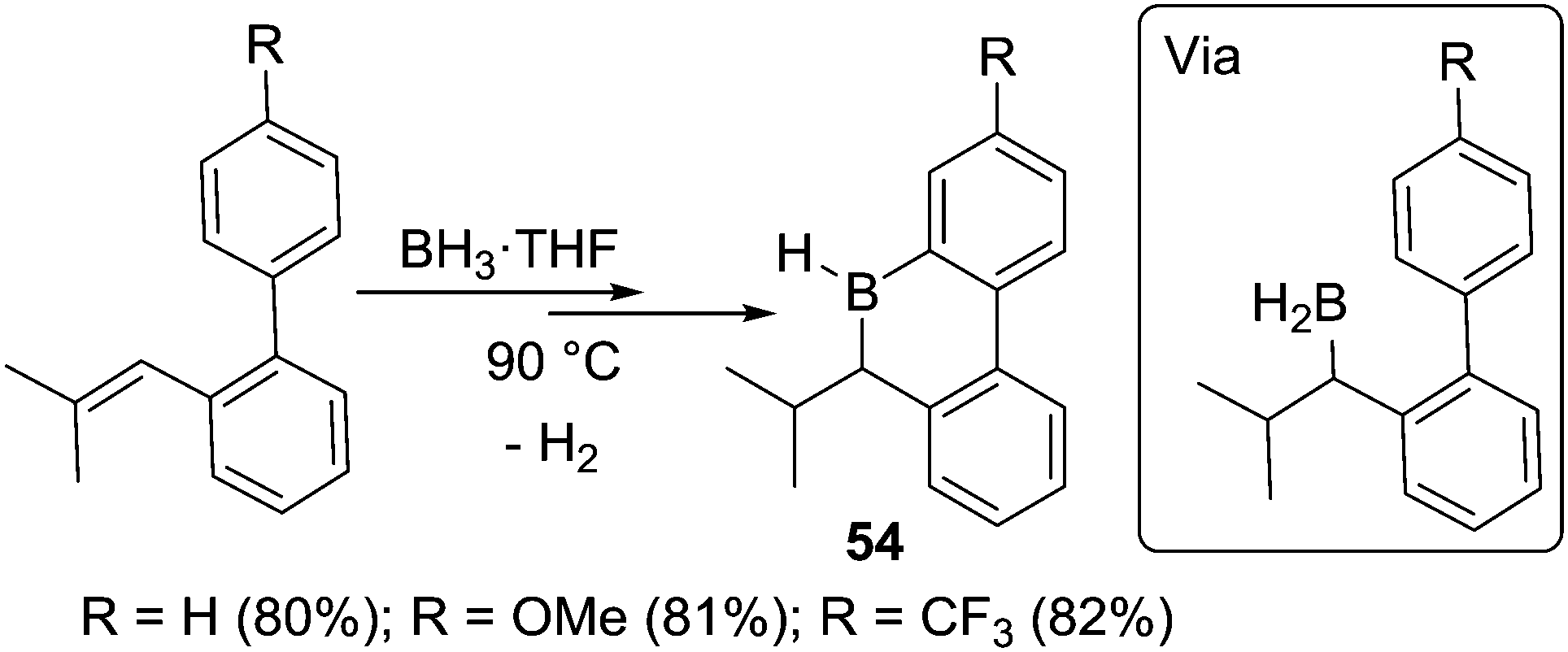 | ||
| Scheme 26 Hydroboration/C–H borylation of biphenyl derivatives. | ||
Subsequent to Knochel's work on directed C–H borylation, Kawachi and co-workers found that o-(hydrosilyl)-(dimesitylboryl)benzene compounds undergo intramolecular H/mesityl exchange in refluxing toluene to afford hydroborane intermediates.65 The rigid skeleton of the intermediate forces the B–H moiety to be in close proximity to the Si-phenyl group and dehydrogenative C–H borylation proceeds under these conditions to form 55. This is an alternative route to alkene hydroboration or alkyl borane retro-hydroboration to form an active (for C–H borylation) B–H intermediate (Scheme 27). Other intramolecular C–H borylation reactions have been reported using B–H containing electrophiles, but as these proceed via NHC–borenium cations they are discussed in Section 4.3 post the detailed discussion on amine-ligated borenium cations in intramolecular C–H borylation.
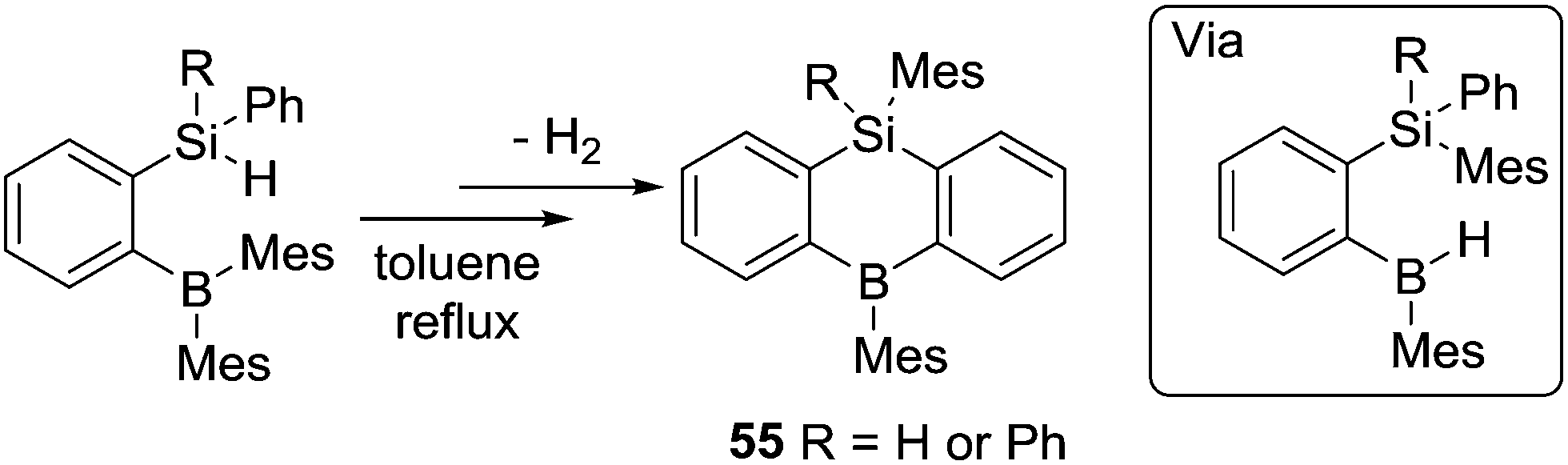 | ||
| Scheme 27 Si/B substituent exchange enabling C–H borylation. | ||
To our knowledge, Kaufmann and co-workers reported the first intramolecular C–H borylation reactions mediated by a C–BX2 species instead of a hydroborane, albeit at very high temperatures.66 At 780 °C, the five membered boracycle 56 was produced via intramolecular electrophilic C–H borylation along with products from substituent redistribution (e.g. inset top right, Scheme 28). Interestingly, a four-membered boracycle, 57, could be generated despite the ring strain. The formation of 57 was confirmed by a trapping reaction with an alkyne to form 58. The high temperatures required was attributed to the lower electrophilicity of chloroboranes (relative to B–Br), and the lack of both an exogenous base and a catalyst (such as AlCl3).
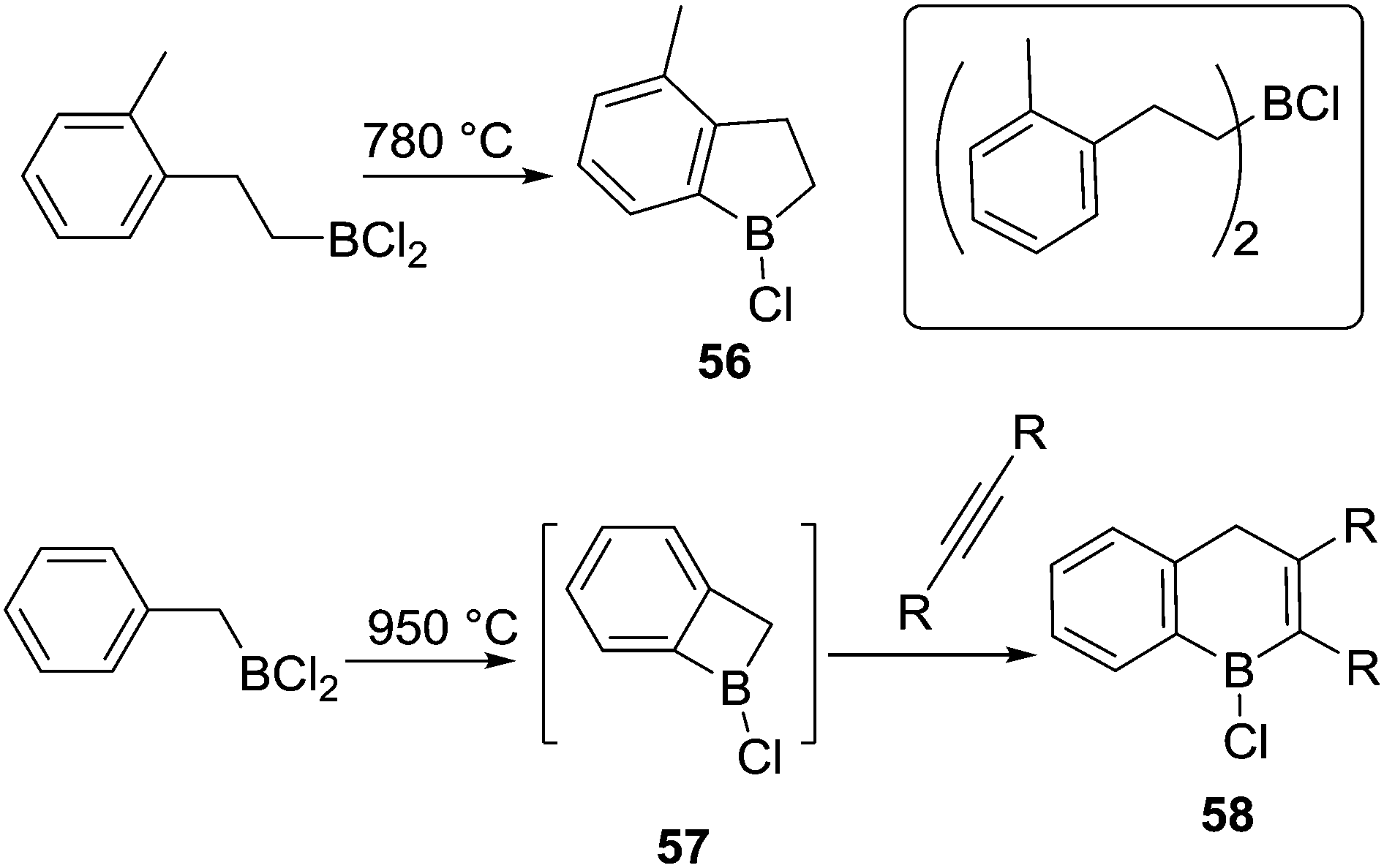 | ||
| Scheme 28 Intramolecular C–H borylation mediated by organoBCl2 species. | ||
In contrast to this earlier work, in 2015 Hatakeyama and co-workers reported a carbon-directed intramolecular C–H borylation reaction that proceeded under much milder conditions (Scheme 29).67 Directed lithiation of 1,3-diphenoxybenzene followed by trans-metalation to boron afforded an arylBBr2 species that in the presence of N,N-diisopropylethylamine led to high yielding double intramolecular C–H borylation at 120 °C to afford compound 59. Notably, the base was crucial for the borylation reaction with only 16% yield obtained without the base. The authors also found that both 1,2,2,6,6-pentamethylpiperidine and N,N-dimethyl-p-toluidine are effective at enabling formation of 59, while 59 was formed in only 13% yield when triethylamine was used. This again emphasizes the need for weakly nucleophilic (towards boranes) Brønsted bases for enabling C–H borylation. It is notable that this reaction proceeds without requiring a Lewis acid activator (e.g. AlX3) and this can be attributed to the presence of an exogenous base, the electronically activating effect of the ortho-OR group, and the fact that ArylBBr2 species are more Lewis acidic than R2NBX2 and RBCl2 species.
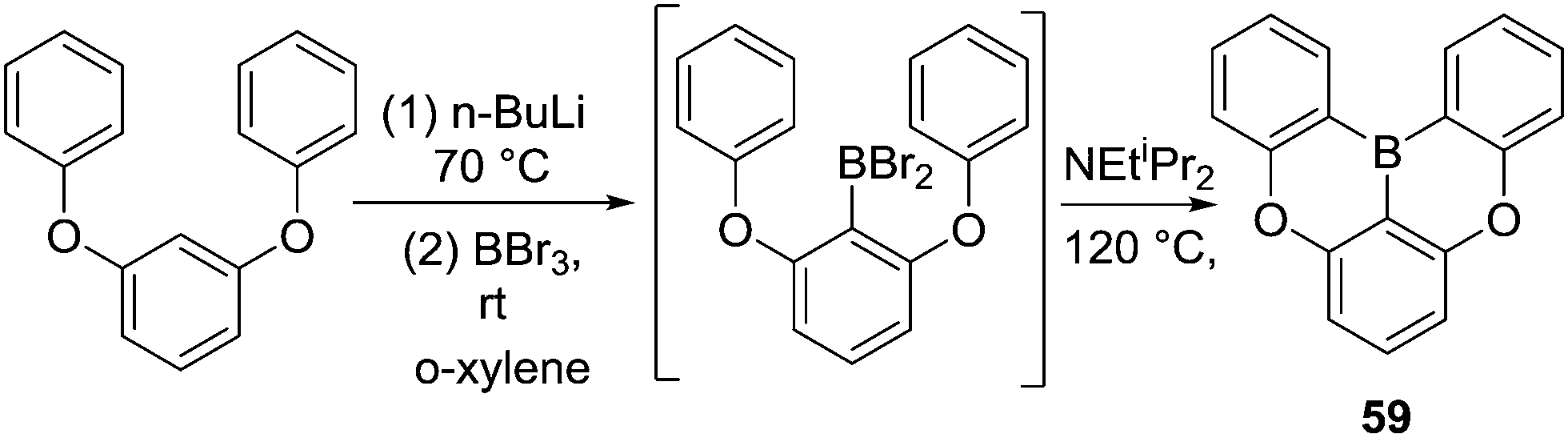 | ||
| Scheme 29 Lithiation/trans-metalation/intramolecular C–H borylation. | ||
Interestingly, for the borylation of 1,3-bis(naphthalen-2-yloxy)benzene, the more hindered product 60 was obtained as the major product while the less congested isomer 61 was only obtained in 2% yield (Scheme 30). This suggests the kinetic product (derived from functionalization of the two alpha-naphthalene positions) is formed under these conditions, possibly due to the presence of a base enabling rapid and irreversible deprotonation (and thus irreversible borylation).
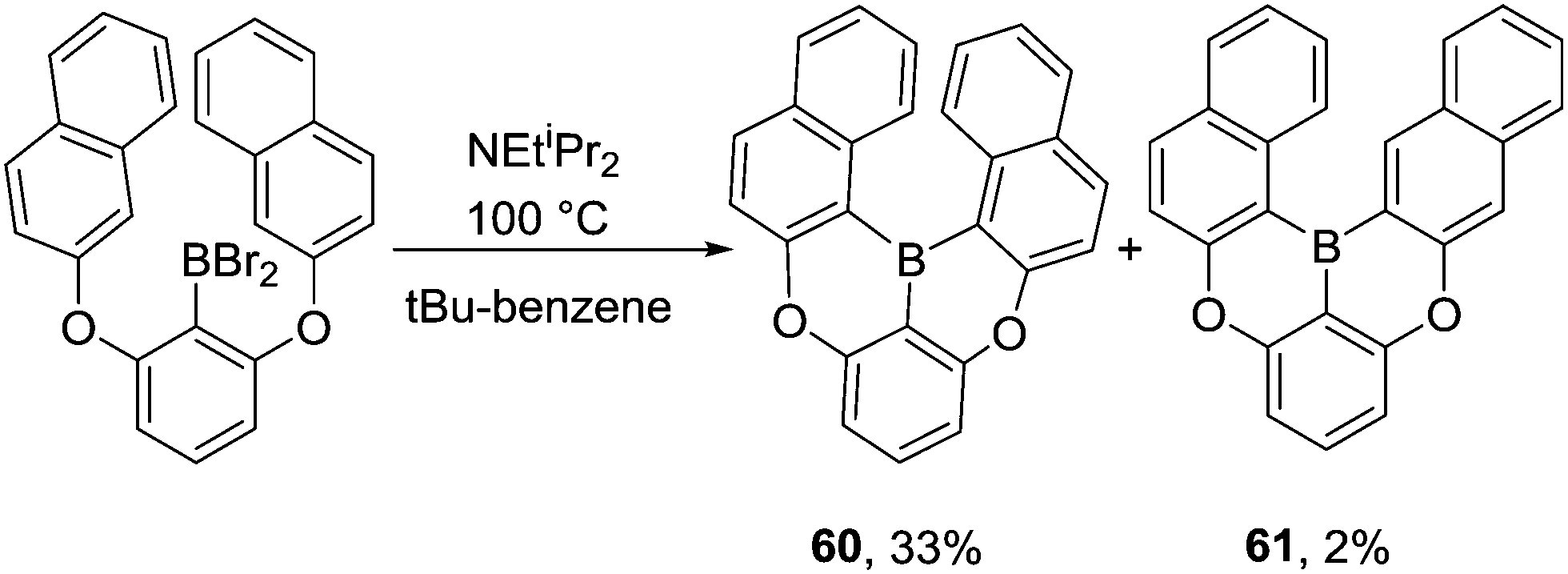 | ||
| Scheme 30 Isomer distribution from C–BBr2 mediated intramolecular electrophilic C–H borylation of a bisnaphthyl derivative. | ||
The lithiation/trans-metalation to BX3/electrophilic C–H borylation strategy has been applied to a variety of substrates using similar reaction conditions. While this has generated many molecules with intriguing properties a comprehensive listing of these is not the focus of this review.68 However, it is noteworthy from a synthetic perspective that this approach also can be used to introduce more than one boron moiety into a PAH. For example, a triple lithiation followed by addition of two equivalents of BBr3 likely affords intermediate 62, which in the presence of 1,2,2,6,6-pentamethylpiperidine, undergoes electrophilic borylation at 160 °C to give 63 (Scheme 31).69 Interestingly, Wang and co-workers prepared closely related compounds via a different method. By heating a mixture of a borinic acid and BBr3 at 120 °C for 1 hour, a substituent exchange reaction occurred (to generate intermediate 65) along with an intramolecular electrophilic C–H borylation. This proceeded in the absence of an exogenous base to afford compound 64 as the major product (Scheme 31, bottom).70 It should be noted that the formation of 64 from the borinic acid also occurred at room temperature (using 5 eq. of BBr3) albeit requiring ca. 20 h for full consumption of the starting material. The addition of Hünig's base did not alter the outcome of this reaction or accelerate the formation of 64, suggesting an alternative Brønsted base is involved in the deprotonation step in this SEAr reaction. The disparity to the conditions required to form 61 is notable and it is feasible that a borate base derived from B-OH/B-Mes/B-Br substituent redistribution reactions is playing a role in the formation of 62 as observed in C–H borylation reactions using [BPh4]− as a Brønsted base (which in some reports is more effective at enabling C–H borylation than hindered amines).53 Regardless of the identity of the base this report confirms that intramolecular electrophilic C–H borylation proceeding via C–BBr2 intermediates can proceed at room temperature.
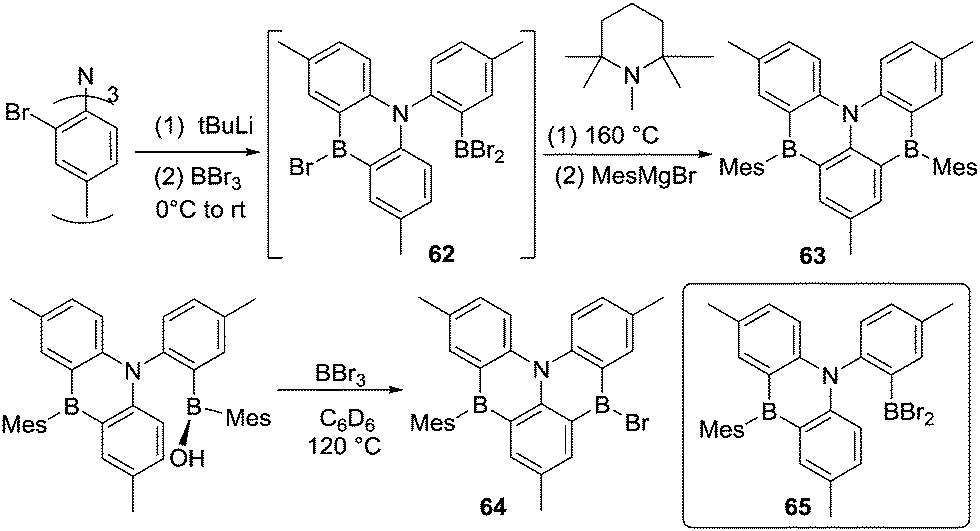 | ||
| Scheme 31 C-Directed borylation of triphenylamine derivatives. | ||
Instead of using organolithium reagents to enable formation of the C–BX2 intermediates, Osuka and co-workers prepared diphenylborane-fused porphyrin 66via boron–silicon exchange using BBr3 and then electrophilic C–H borylation (Scheme 32).71 Presumably, due to the high nucleophilicity of the meso position in these compounds, the C–H borylation reaction occurs at relatively mild conditions without using any exogenous base.
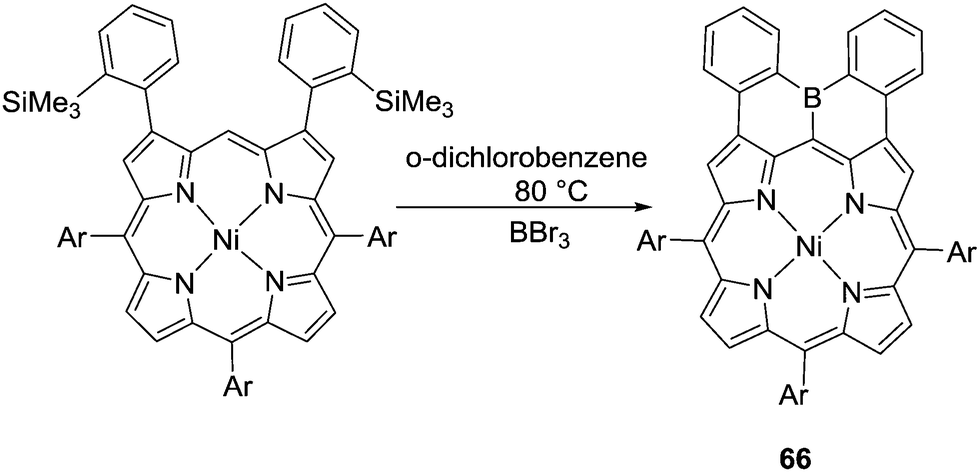 | ||
| Scheme 32 Borodesilylation enabling intramolecular C–H borylation. | ||
Other routes to generate C–BX2 units for subsequent use in intramolecular C–H borylation reactions have been reported. For example, Ingleson and co-workers developed a sequential inter-/intra-molecular electrophilic C–H borylation of an indole substrate using an amine, BCl3 and AlCl3.72 The propensity of the indole C3 position to undergo electrophilic substitution enables facile intermolecular C–H borylation with a borenium cation to give 67. A double intramolecular C–H borylation to form 68 then was achieved by addition of 2,6-dichloropyridine (Cl2-py) and AlCl3 to 67 (proposed to form a reactive borenium cation in situ that is effective for electrophilic borylation at room temperature, inset Scheme 33). There are a number of other reports of intermolecular electrophilic C–H borylation being followed by intramolecular C–H borylation to form boron containing PAHs. These reactions are presumably also proceeding via ArylBX2 species, however, due to the fact that these intermediates are either not observed or that the synthetic conditions used for intramolecular C–H borylation are similar to those already discussed in this section these reports are not discussed individually herein.73
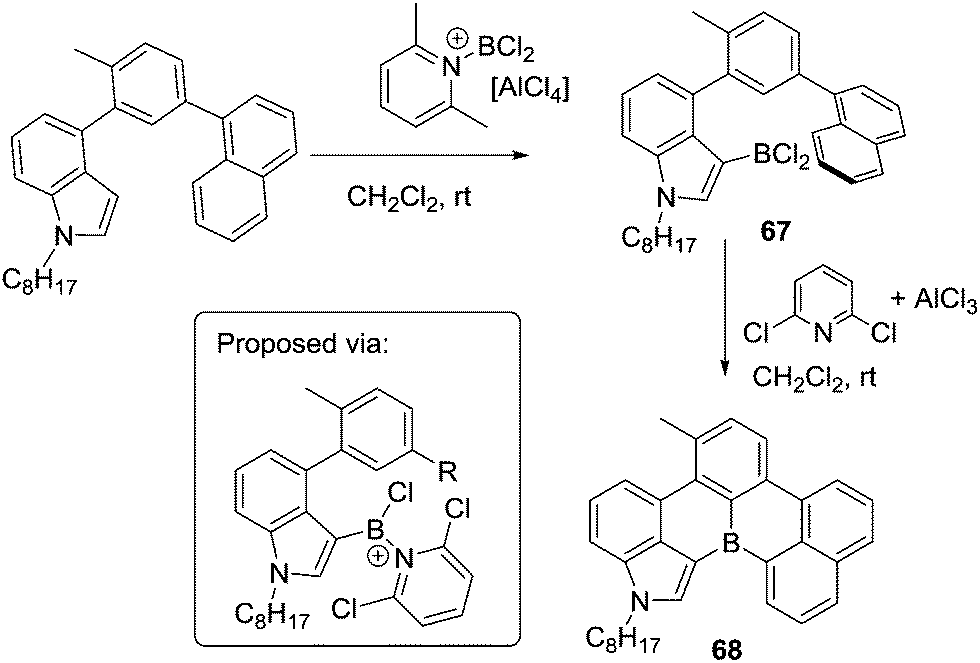 | ||
| Scheme 33 Intermolecular and intramolecular electrophilic C–H borylation. | ||
Another alternative route to install the C–BX2 unit for subsequent intramolecular borylation is borylative cyclisation of 1-(4-phenylbut-1-yn-1-yl)-naphthalene (and derivatives) to give 69 using BCl3/2,4,6-tri-tert-butyl pyridine (TBP).74 Addition of stoichiometric AlCl3 and 2,6-dichloropyridine to 69 enabled intramolecular C–H borylation within 20 minutes at room temperature to afford compound 70 in good yield (Scheme 34). Again, the results clearly demonstrate the key role of AlCl3/amine base in promoting intramolecular electrophilic C–H borylation at room temperature. This is presumably due to borenium cation formation, although these intermediates are not observed in this case.
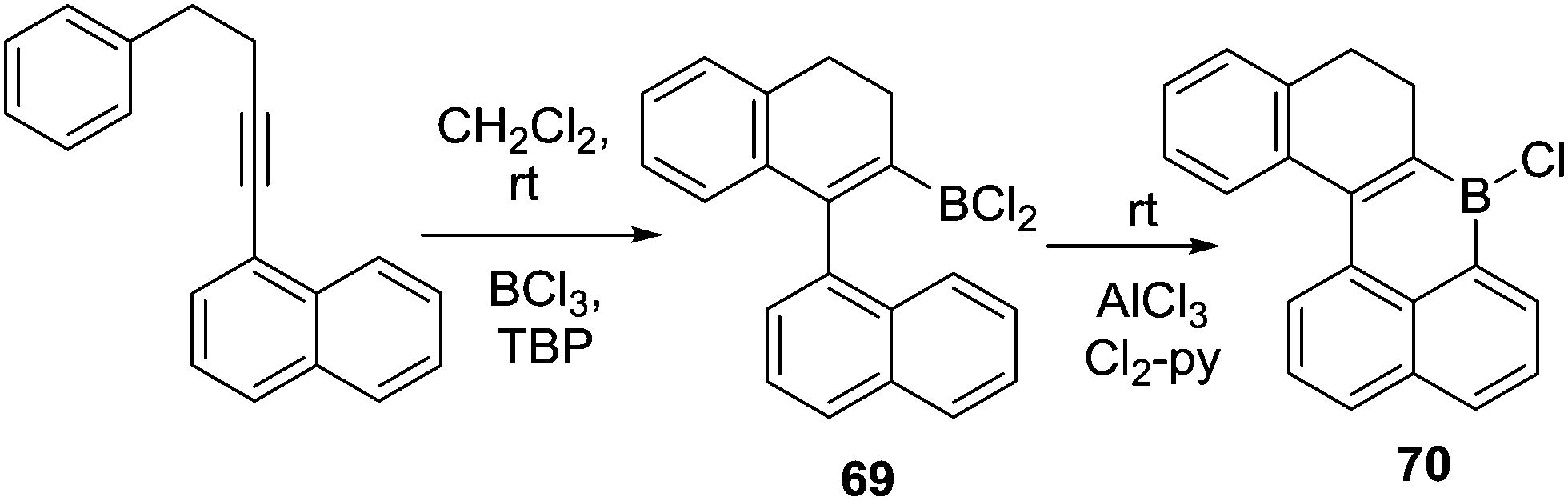 | ||
| Scheme 34 Borylative cyclisation preceding intramolecular C–H borylation. | ||
Other alkyne transformations can be used to install the C–BX2 unit for subsequent intramolecular C–H borylation, including haloboration, both trans-1,2-haloboration and 1,1-haloboration.75 For example, when 1-ethynylnaphthalene was treated with BBr3, a bromoboration/intramolecular C–H borylation reaction sequence proceeded (Scheme 35). The intermediate 71 was formed upon addition of BBr3via alkyne trans-bromoboration, the subsequent intramolecular C–H borylation from the vinylBBr2 species then occurred at room temperature to generate 72 without additional Lewis acid or base. This is in contrast to the reactivity of vinylBCl2 units (e.g.69) which required Lewis acid/base additives (to enable borenium formation). Presumably, this is due to the enhanced Lewis acidity of vinylBBr2 relative to vinylBCl2 combined with the absence of H⋯H steric clash (which is present in the fjord region of 69). However, the identity of the base deprotonating the arenium cation in these reactions is unclear, it is potentially a [BBr4]− species generated in situ. As noted for other C–BX2 containing systems double C–BX2 installation by borylative cyclisation and haloboration can be followed by twofold intramolecular C–H borylation to form B2-containing PAHs.75b
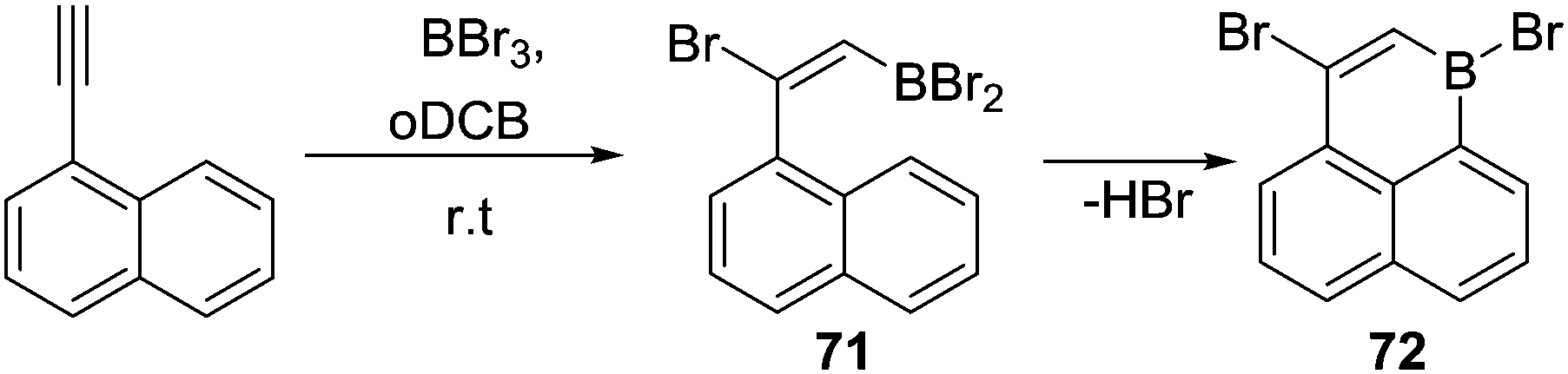 | ||
| Scheme 35 trans-Haloboration followed by intramolecular C–H borylation. | ||
The development of multiple routes to generate C–BX2 intermediates using simple precursors, coupled with the observation of subsequent high yielding, intramolecular C–H borylation clearly indicates the significant utility of the approaches outlined above, particularly in forming PAHs containing C3B units. The preference for forming six membered boracycles in systems containing all sp2 hybridised centres is pronounced. This is presumably due to a combination of more favourable bond angles in six membered all sp2 boracycles and the higher energy of anti-aromatic borole units present in structures when five membered C4B boracycles are formed.
3.3 Other heteroatom–BX2 directed borylations
Dewar's studies into O directed C–H borylation (to form 8 and 9, respectively) were extended by Zhou and co-workers using anisole derivatives in-place of hydroxyl precursors. The reaction of a 2-phenylanisole derivative with BBr3 led to ether cleavage (with evolution of bromomethane) and then room temperature C–H borylation.14 This work also demonstrated a disparity between BBr3versus BCl3. Borylation proceeded rapidly at room temperature with O–BBr2 derivatives 73 to form 74, whereas with the chloro analogues (73-Cl) an additional Lewis acid (AlCl3) was required. It is likely that AlCl3 coordinates at O or Cl in 73-Cl enhancing the electrophilicity at boron. With both systems, there is a dependence on the nucleophilicity of the arene being borylated. For example, using BBr3 only the dimethyl substrate (R1 and R2 = Me, Scheme 36) produced the C–H borylation product 74. While with the BCl3/AlCl3 system the presence of two electron-withdrawing fluorine substituents (R3 and R4 = F) produced no borylated product, but when R3 = Me (located para to the borylation site) C–H borylation occurred even with R4 = F.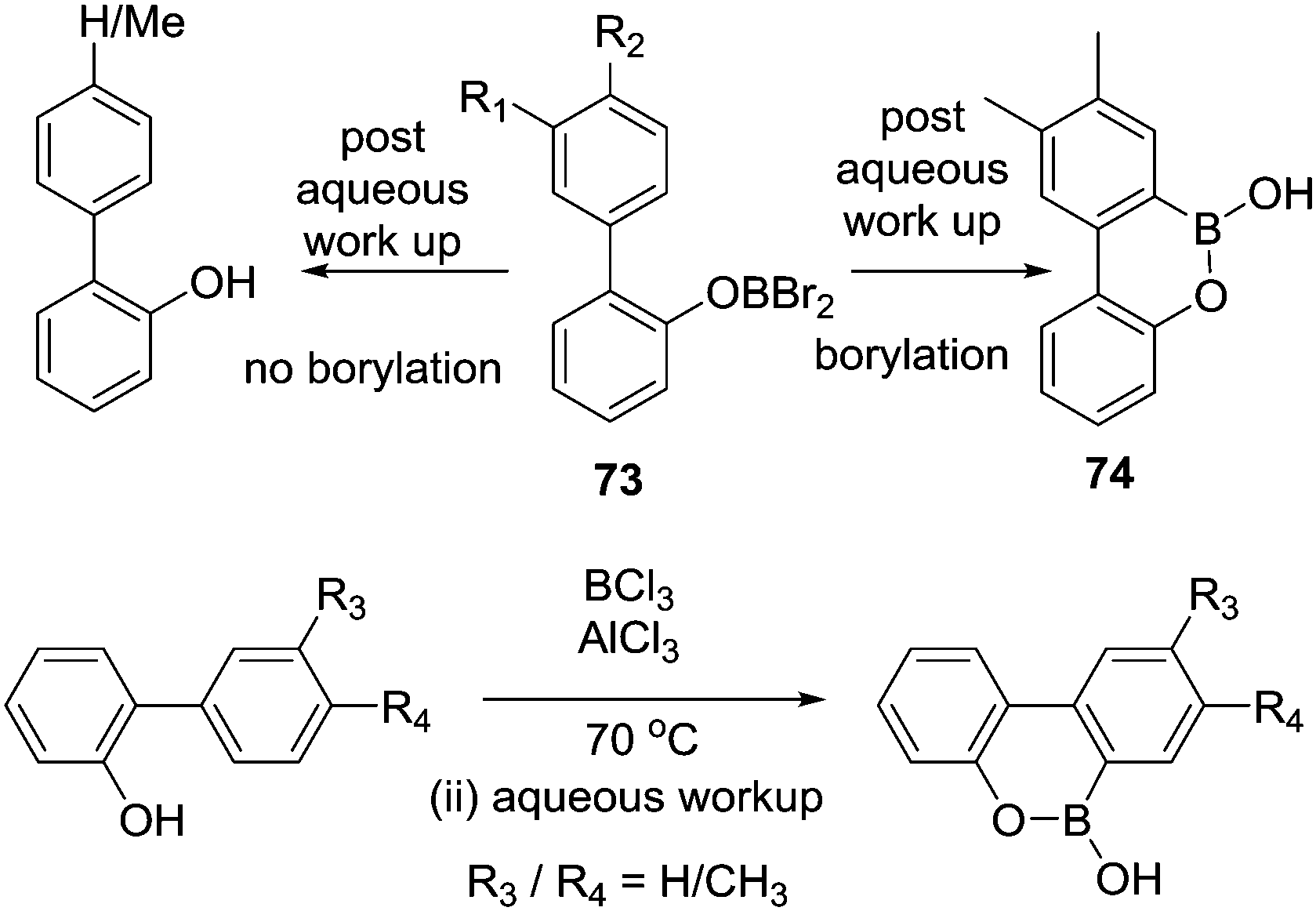 | ||
| Scheme 36 O directed C–H borylation using BBr3 and BCl3/AlCl3. | ||
O and S directed C–H borylation has been employed in the synthesis of larger B-doped PAHs. In this area the cleavage of ArylO-Me with BBr3 to form ArylOBBr2 units that then effect intramolecular C–H borylation is most common, for example, in the synthesis of 75 and 76 (Scheme 37).53 Demethylation proceeded at room temperature to afford the demethylated intermediate (inset Scheme 37) with subsequent heating in the presence of an amine producing 75 in moderate to good yield (without a base 75 is formed in much lower yield). The high temperature required may be in part due to the lower Lewis acidity of the diaryloxy-borane electrophile (e.g. inset) relative to RBBr2 for which borylation proceeds at room temperature.
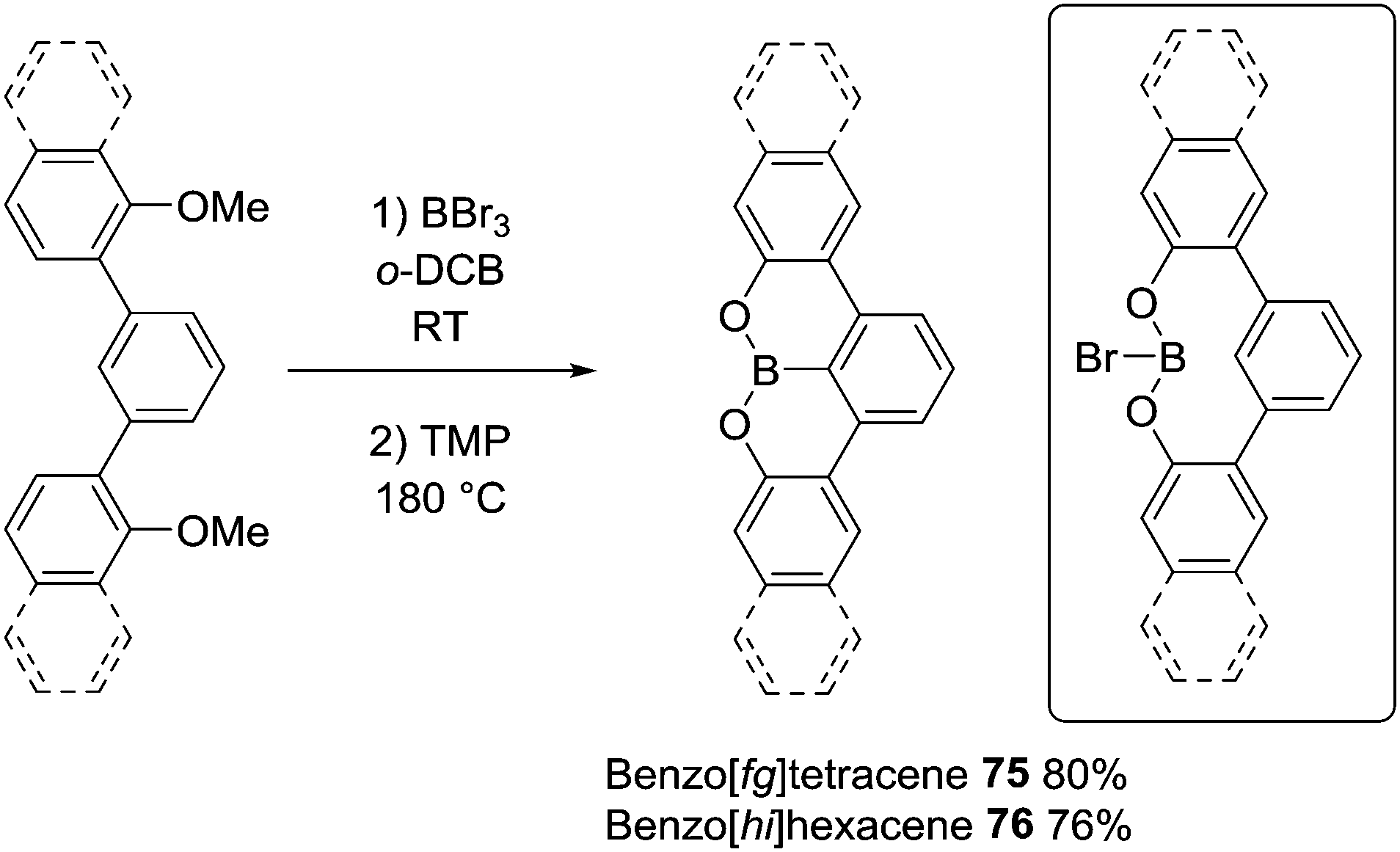 | ||
| Scheme 37 Demethylative borylation using BBr3 to form BO-containing PAHs. | ||
Thioether congeners required more forcing conditions for an analogous demethylative S-directed C–H borylation, in part due to the lower reactivity of thioanisoles towards demethylation with boron electrophiles.76 For example, heating at 200 °C resulted in only a 21% yield of 77 (Scheme 38). Interestingly, the addition of bases to this S directed C–H borylation reaction did not improve the conversion (using either a hindered amine or NaBPh4), suggesting the deprotonation step is not the limiting factor leading to low yields in this case.
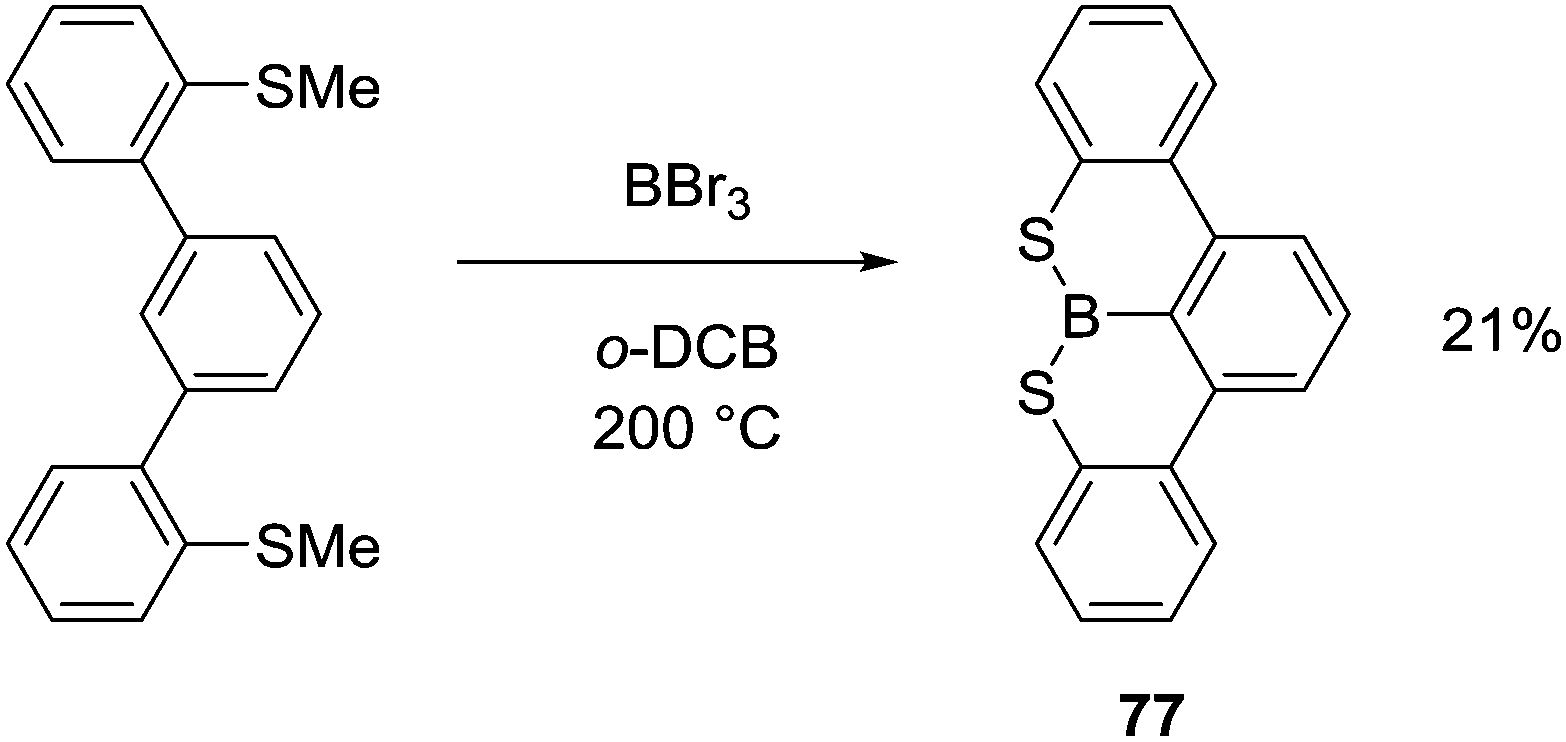 | ||
| Scheme 38 Demethylative thioanisole directed intramolecular C–H borylation. | ||
Demethylative directed C–H borylation of anisole derivatives also has been extended to form B2-PAHs (Scheme 39).77 Addition of BBr3 at room temperature forms the demethylated O–B–O intermediate 78, which then undergoes high yielding C–H borylation at 150 °C to form 79. This methodology also was applied to pyrene derivatives and as double demethylation occurs rapidly (relative to C–H borylation) the selectivity is high in the subsequent C–H borylation step in these systems.78 Müllen and co-workers also prepared the double helicene 80 (using BBr3 at 180 °C) albeit in low yield (Scheme 39) via directed C–H borylation.79 The low yield relative to 79 was attributed to the significant strain during the borylative ring closure.
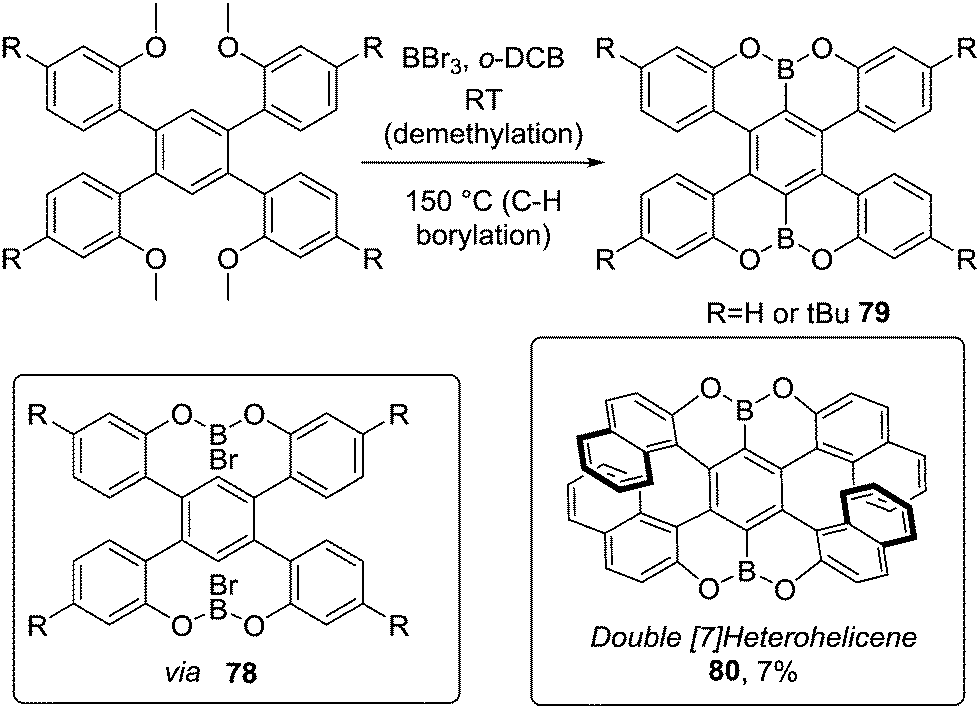 | ||
| Scheme 39 Demethylative double O-directed C–H borylation to produce OBO–PAHs via O–B–O intermediates. | ||
Subsequently, Suga, Mitsudo and co-workers demonstrated a demethylative borylation to yield thiophene-fused 1,2-oxaborine derivatives.80 Notably, the use of PhBCl2 as the borylating electrophile required an iodide salt to facilitate demethylation. Iodide was proposed to be acting as the nucleophile to demethylate the less activated ArO(Me)–borane adduct (less activated due to coordination of Ar(O)Me to a less Lewis acidic borane). Iodide demethylation of 81 initially produced MeI and 82, the latter then underwent C–H borylation at 135 °C in the presence of triethylamine to form 83. This methodology was applicable to a range of other substituted thiophenes and extended to BCl3 as the borane source. As expected, in substrates where multiple six membered boracycles could be formed, the reaction proceeded selectively at the more nucleophilic site. Furthermore, the reaction did not proceed under the same reaction conditions with less nucleophilic aromatics (inset Scheme 40).
 | ||
| Scheme 40 Dithieno-oxaborine synthesis using PhBCl2 and iodide mediated demethylation followed by O-directed C–H borylation. | ||
Finally in this section, in recent patent literature an O-directed electrophilic C–H borylation route to access benzoxaboroles has been reported.81 In this report a benzyl alcohol was reacted with BCl3 at low temperature, proposed to form an RO–BCl2 species (Scheme 41), which on refluxing in toluene led to C–H borylation. Notably, aqueous work up then led to benzoxaboroles, including the commercialised tavaborole, 84, in moderate yields (ca. 40%). The ability of this transformation to proceed in the absence of AlCl3 is, again, consistent with the formation of five membered boracycles having lower reaction barriers for systems containing at least one sp3 centre.
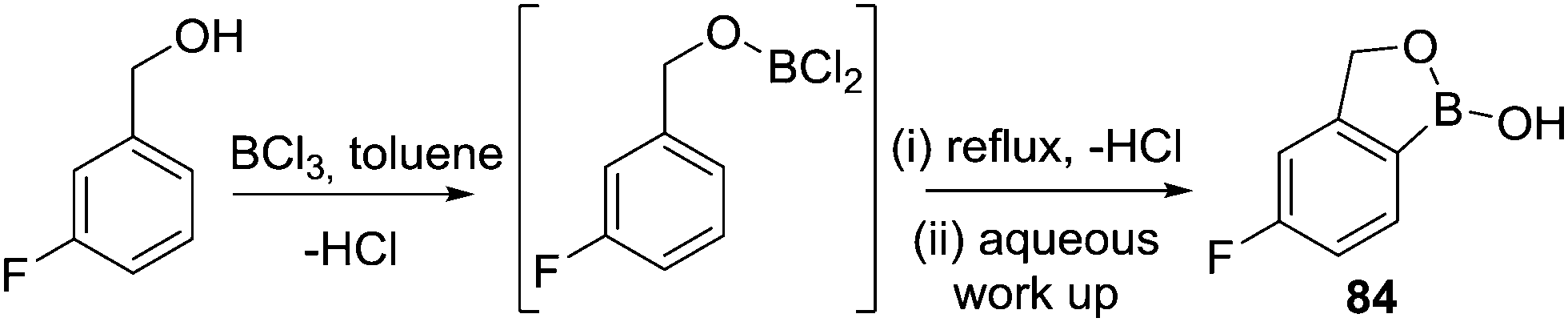 | ||
| Scheme 41 Intramolecular electrophilic C–H borylation to form tavaborole. | ||
While O directed electrophilic C–H borylation has lagged behind N– and C–BX2 mediated intramolecular electrophilic C–H borylation the recent successes in forming BO-containing PAHs and tavaborole (84) confirms the considerable potential that this methodology has. It is also clear that RS–BX2 mediated intramolecular C–H borylation requires further development before it is a reliable, high yielding methodology with broad applicability.
4. Intramolecular C–H borylation via E–BY3 dative bond containing intermediates
This section discusses systems where intramolecular C–H borylation is preceded by formation of a E→BY3 unit (Y = halide or hydride most commonly) and where the dative bond is unlikely to be transformed into a covalent bond (e.g. cannot form a neutral E–BY2 unit by loss of HY). In this section the major class is when E is a nitrogen Lewis base, and this area is discussed first followed by the more limited examples involving other heteroatom centred Lewis bases (e.g. carbonyls).4.1 Via N→BY3 intermediates
An important study into N→B dative bond directed electrophilic C–H borylation was from Vedejs and co-workers in 2009.82 The trityl salt of the robust weakly coordinating anion [B(C6F5)4]− (essential to avoid anion decomposition when combined with borocations) was used to abstract hydride from N,N-dimethylbenzylamine borane. At room temperature this led to the C–H borylation product, 85, with the reaction proposed to proceed via a borenium cation (87, Scheme 42). A limited selection of functionalised benzyl derivatives was utilised successfully in this C–H borylation reaction, e.g. containing halide or Me groups, with halide substituents, again, leading to slower C–H borylation. Borylation could be extended to form 6-membered boracycle analogues in good yield, however, low yields were observed for seven membered boracycle analogues. It is notable that compound 85 could be hydrolysed readily to the boronic acid derivative that contains the core-structure of the glucose sensor shown in Fig. 1.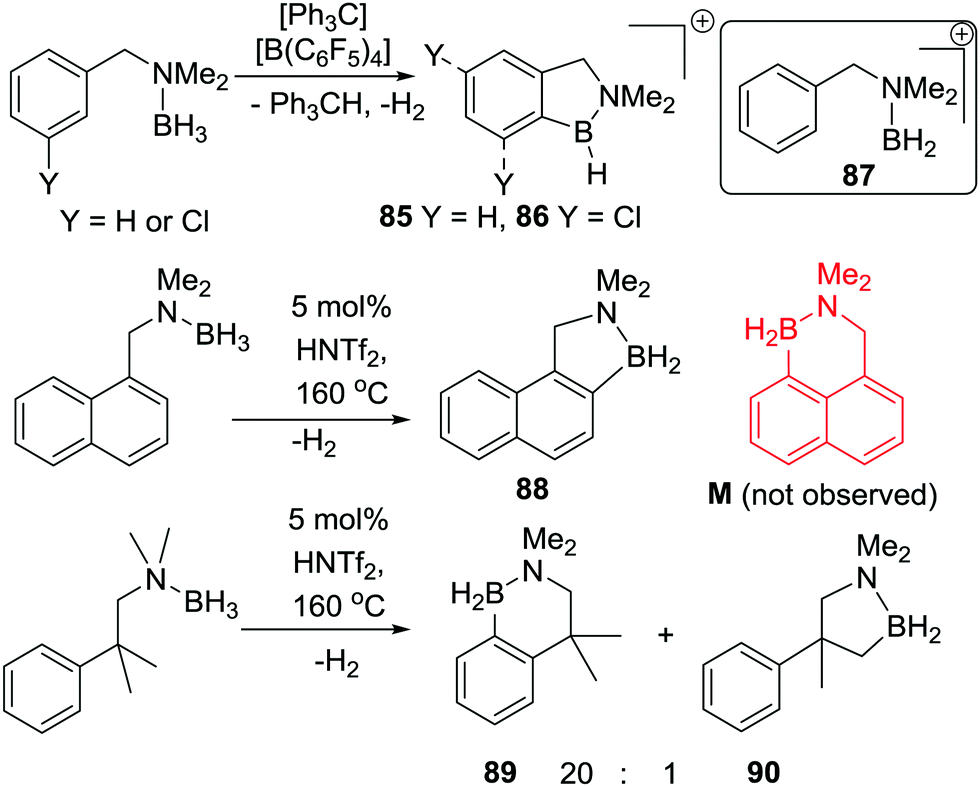 | ||
| Scheme 42 Hydride abstraction leading to: top, room temperature stoichiometric (in activator) borylation; middle, catalytic in activator C–H borylation at raised temperatures; bottom, sp2C–H borylation dominating over sp3C–H borylation. | ||
The use of forcing conditions (160 °C) enabled the borylation to form 85 (and derivatives) to be carried out with only 5 mol% of the hydride abstractor, [Ph3C][B(C6F5)4] or HNTf2. Hydride transfer from BnMe2NBH3 to 85 was proposed as turnover enabling. With catalytic C–H borylation proceeding under kinetic control (i.e. it was shown to be irreversible under the reaction conditions) it is notable that 88 is formed exclusively, with no compound M observed. From these studies in flexible systems (containing multiple sp3 centres) the formation of five membered boracycles is preferred, at least kinetically, over six membered (consistent with observations on covalently bonded C–BH2 directed borylation systems discussed above). Notably, in the formation of 86 the isomer distribution in the catalytic (with HNTf2 at 160 °C) and stoichiometric (with [Ph3C][B(C6F5)4] at ambient temperature) borylation reactions was dramatically different. The purified isomers of 86 did not equilibrate on heating under the borylation conditions thus the isomer ratio is fixed during C–B bond formation/C–H cleavage (i.e. the products are formed under kinetic control). The difference in selectivity was attributed in these reports to different borylating electrophiles generated using NTf2 and [B(C6F5)4], with borylation at 160 °C potentially not proceeding via simple borenium cations such as 87.83,84
Considering the mechanism for the ambient temperature borylation with [B(C6F5)4]− as the counterion, a four membered B–H/C–H metathesis transition state for forming 85 from 87 was calculated. As 85 is isoelectronic to 1-boraindenes (e.g.11) it is notable that the calculated mechanism for these two types of borylation reactions are closely related. In both, post formation of the key electrophile (R–BH2 or isoelectronic [R3N→BH2]+), subsequent C–H borylation can actually be a facile process forming five membered boracycles as the kinetic products. The calculation of a metathesis type transition state leading to 85 also was consistent with the observation of a kinetic isotope effect (KIE) of 2.8 using an ortho-mono-deuterated analogue of BnMe2NBH3. This indicated that the C–H bond at which boron substitution occurs is broken during or before the regioselectivity-determining step. This is in contrast to Friedel–Crafts reactions, in these there is negligible or inverse KIEs.85 This highlights a mechanistic distinction that can occur between C–H borylation with B–X (SEAr mechanism, vide infra) and B–H (σ-bond metathesis type transition state) electrophiles. Related systems to 87 were subsequently shown to lead to intramolecular sp3C–H borylation under forcing conditions (albeit with a different key electrophile proposed).10 These studies also revealed that borylation of sp2C–H sites is preferred over sp3C–H (Scheme 42, bottom 89vs.90).
Shortly after Vedejs’ seminal work Murakami reported a method to borylate 2-aryl-pyridines and derivatives using BBr3 and a bulky tertiary amine (Scheme 43) e.g. to form 91.86 This work was notable as it proceeded in high yield at ambient temperature using simple precursors. The authors proposed borenium cation intermediates and an SEAr mechanism (inset right Scheme 43). During the borylation of a naphthyl substituted pyridine two isomers, 92 and 93, were formed, with the product from borylation at the more nucleophilic peri C–H position dominating. This is in contrast to Dewar's work on borylating naphthyl substituted anilines (Scheme 1) where no peri C–H borylation was observed. This disparity can be attributed to rapid and irreversible borylation (possibly due to rapid deprotonation of the arenium cation either by amine or [BBr4]−) in Murakami's system. This leads to product formation under kinetic control as previously observed in intermolecular C–H borylation reactions proceeding in the presence of an effective Brønsted base.87 In Dewar's system the more forcing conditions and the absence of exogenous base presumably led to slow deprotonation (enabling isomerisation potentially at the arenium cation stage), ultimately forming the thermodynamic product (which is from borylation at the less hindered naphthyl beta position).67
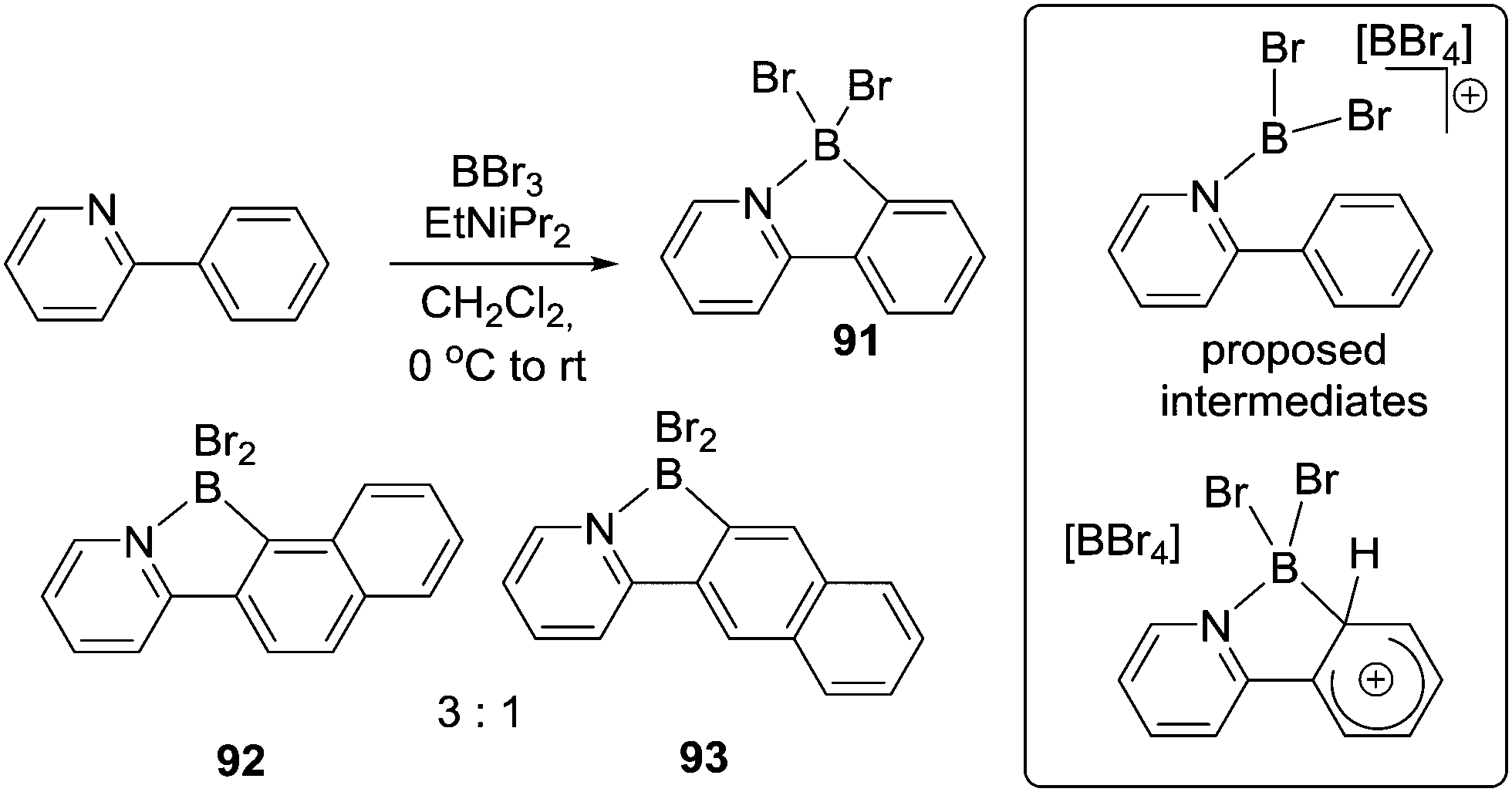 | ||
| Scheme 43 Select examples from Murakami's pyridine directed C–H borylation. | ||
Since this original report Murakami's C–H borylation conditions have been used extensively, and extended to other N-directing groups including: 2-phenoxy-pyridines, imidazolones, pyrazines, pyrimidines, pyrazoles, imidazoles and quinolines.88 It worth noting that with certain ditopic Lewis basic heteroarene directing groups, e.g. pyrazines and pyrimidines, double directed C–H borylation has also been achieved using BBr3 and base, e.g. to form 94 and 95 (Scheme 44, top). Additional points of note from these reports include further evidence of kinetic control; i.e. the selective formation of 96 from borylation at the more nucleophilic naphthyl peri position (Scheme 44, bottom).89,90 In contrast, applying comparable borylation conditions to related systems five membered boracycles are formed exclusively, e.g.97, with no compound O observed (97 forms despite the presence of strain arising from close H⋯H contacts causing non-planarity in the polycyclic aromatic). Indeed, the formation of five membered boracycles using pyridyl directing groups is the most common outcome. Calculations performed on 96 and its isomer N at the M06-2X/6-311G(d,p) PCM (DCM) level and the B3LYP/6-31G(d)/LANL2DZ level (both commonly used in the literature for calculating boracycle structures and their relative energies) found them to be effectively isoenergetic (ΔE < 1.2 kcal mol−1) at both levels. This supports the conclusion that pyridyl directed electrophilic C–H borylation reactions using BBr3/base are proceeding under kinetic control (as only 96 is observed). In summary, these reports indicate that while five membered boracycles are often the kinetically preferred products from pyridyl directed C–H borylation minor changes in the compound being borylated can switch this selectivity from five to six membered boracycle formation.
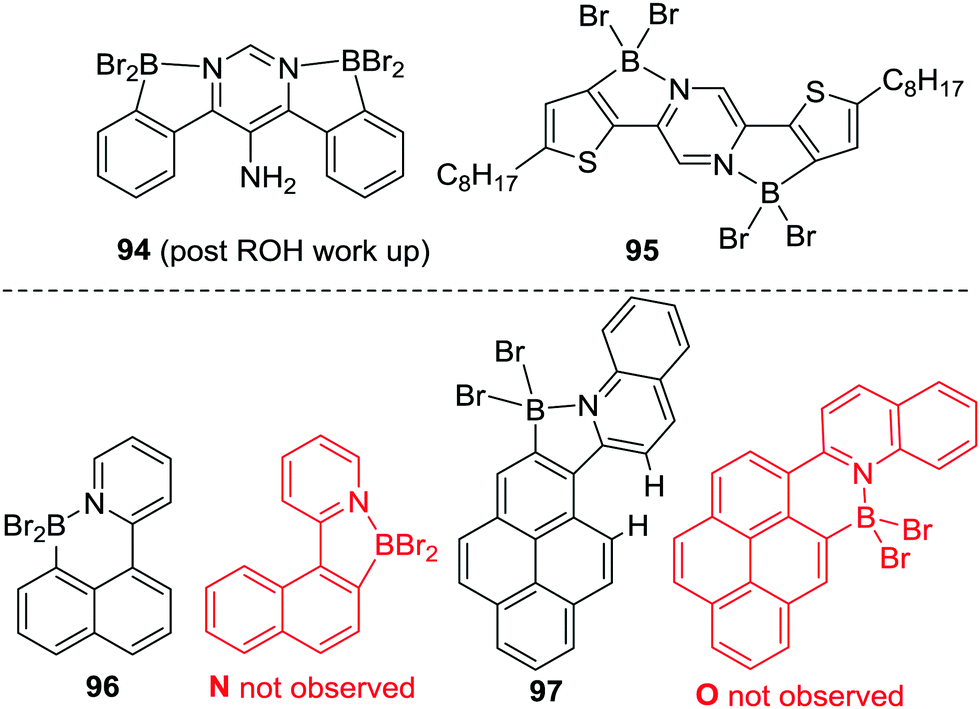 | ||
| Scheme 44 Double directed borylation using Murakami's borylation conditions. Bottom, evidence for kinetic control in borylation using Murakami's conditions. | ||
A pyridyl directed C–H borylation reaction using a related borylation reagent mixture to Murakami's, BI3 and N,N-dimethyl-p-toluidine, led to the formation of a five and a six membered boracycle in a single compound, 98 (Scheme 45) albeit at high temperature (140 °C). This report compared this outcome to the fact that only six membered boracycle formation was observed for the related C–BX2 mediated intramolecular C–H borylation, with compound 99 (Scheme 45, bottom) being the thermodynamic product. This further emphasises the fact that when forming all sp2-containing boracycles six membered rings are the generally observed products. In contrast, due to the presence of a tetrahedral boron centre (preferring bond angles around boron ≪120°) compound P was found to be 4.7 kcal mol−1 higher in energy than 98 and was not observed during borylation.91
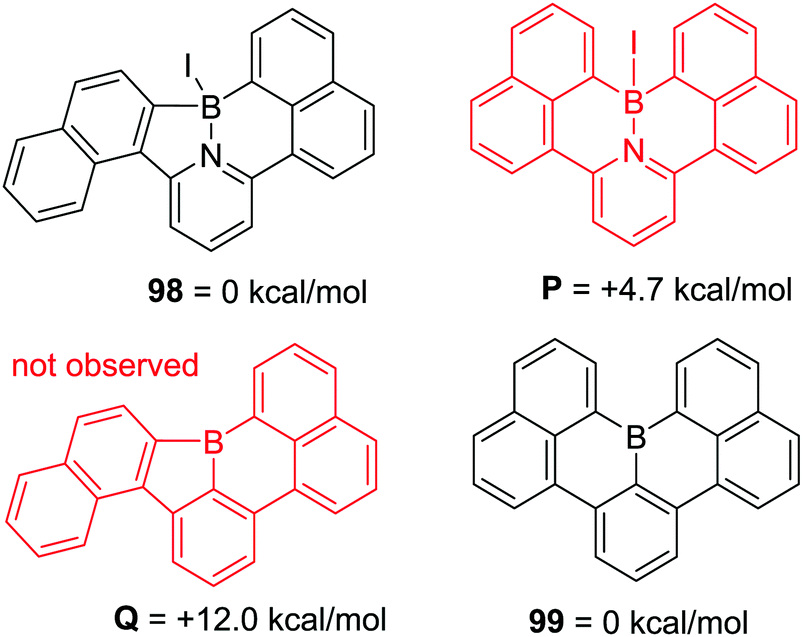 | ||
| Scheme 45 Calculated energies on products from related N and C directed C–H borylation reactions at the B3LYP level of theory with a LANL2DZ basis set for iodine and 6-31G(d) basis set for the other atoms. | ||
The mechanism of N→BX3 mediated intramolecular C–H borylation was explored computationally by Uchiyama and Wang using 2-phenylbenzimidazole.92 Starting from BBr3 and BCl3 borenium cations, 100, were found to be key intermediates on the C–H borylation pathway (boreniums may be more readily accessible in this case due to a BN containing resonance form stabilising the borocation to a greater extent than possible with [pyridylBX2]+,93Scheme 46, inset top right). From the borenium cations a C–H insertion mechanism proceeding via a four membered σ-bond metathesis transition state (related to that calculated for B–H electrophiles) was found to be a high barrier process. Instead Friedel–Crafts type mechanisms had lower energy barriers leading to 101. With the B–Br congener a Wheland intermediate was found, which underwent subsequent deprotonation with [BBr4]−, while for the B–Cl analogue a concerted process was calculated involving formation of the B–C bond concomitantly with deprotonation by [BCl4]− (Scheme 46). The key differences between BCl3/BBr3 thus appears to be that forming the borenium cation 100 is more thermodynamically favoured with the bromo congener and that the [BBr4]− anion enables more facile deprotonation than [BCl4]−. From these observations C–H borylation will be facilitated by: (i) increasing the solution concentration of the borenium cation by using more halophilic Lewis acids (e.g. AlCl3) and (ii) adding exogenous bases to facilitate deprotonation.
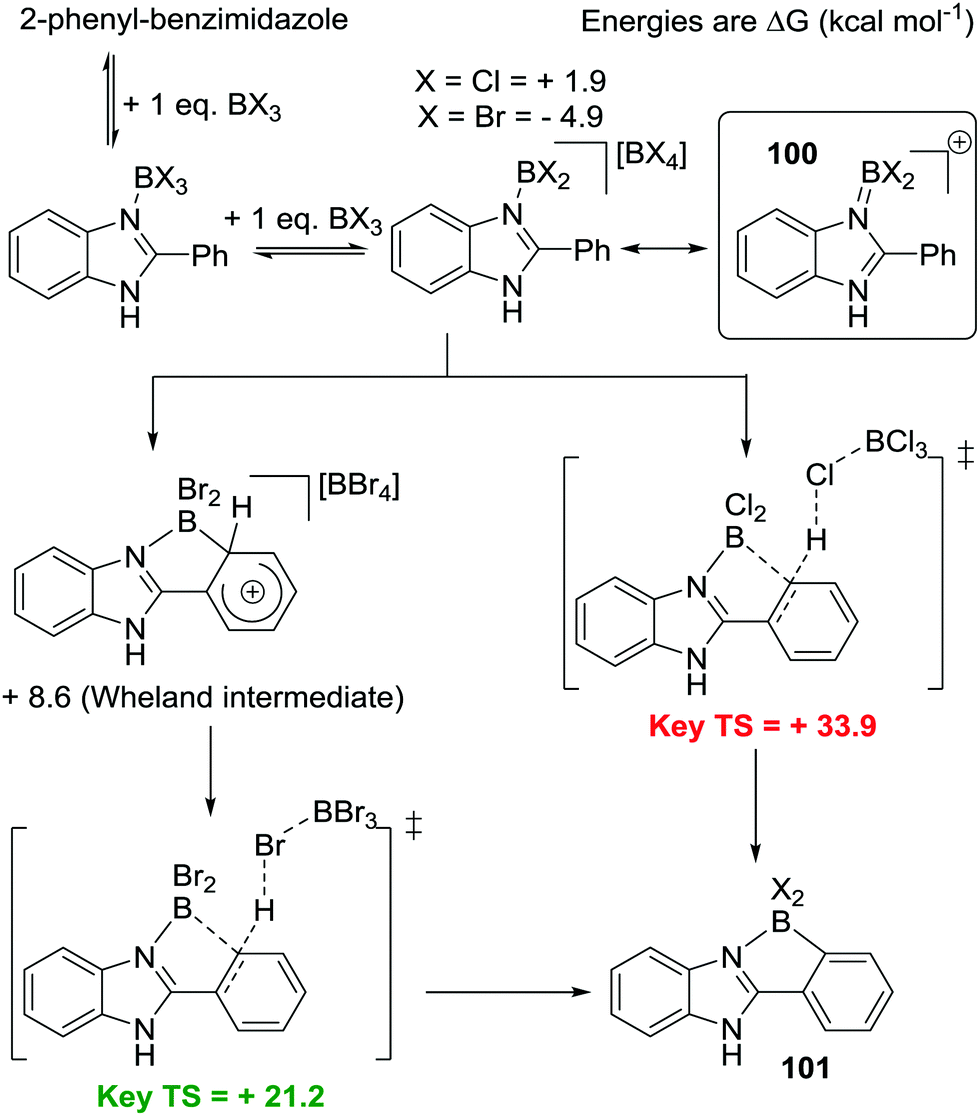 | ||
| Scheme 46 Calculated mechanism for intramolecular C–H borylation of 2-phenyl-benzimidazole highlighting the disparity between the halide congeners. | ||
Notably, 2-aryl pyridines do undergo borylation on addition of excess BCl3 (or PhBCl2)94 even in the absence of an additional base. However, in studies with 2-phenyl pyridine the absence of additional base led to 0.5 eq. of protonated 2-phenyl-pyridine and 0.5 eq. of borylated 2-aryl pyridine. This indicates that some free 2-phenyl-pyridine is present in solution (due to the equilibrium between the pyridyl→BCl3 Lewis adduct and free pyridyl/BCl3) which is then enabling directed C–H borylation by fulfilling the role of Brønsted base (presumably with a lower barrier than if [BCl4]− is the base based on Uchiyama's calculations). On addition of a hindered base (a 2,6-ditertbutyl substituted pyridine to preclude dative bond formation with Lewis acids) 2-phenyl pyridine borylation with BCl3 proceeds slowly at ambient temperature and more rapidly on heating, and forms the C–H borylated product (103) quantitatively. Therefore, pyridyl-directed C–H borylation is feasible with just BCl3 provided there is a suitable Brønsted base present. The combination of a hindered base and equimolar BCl3/AlCl3 also led to borylation to form 103 in this case borylation is rapid at room temperature.93 Borylation is presumably proceeding via the borenium cation (102) expected from adding AlCl3 to py-BCl3 adducts (Scheme 47).95 It should be noted that 103 can transfer chloride to 102, resulting in additional Lewis acid being required to ensure high yielding borylation occurs. The more rapid borylation to form 103 observed in the presence of BCl3/AlCl3/hindered base (vs. with just BCl3/hindered base) is presumably due to a higher solution concentration of the borenium cation than that using BCl3 as halide abstractor (consistent with related borenium cation formation using BCl3 being calculated to be endergonic – Scheme 44).
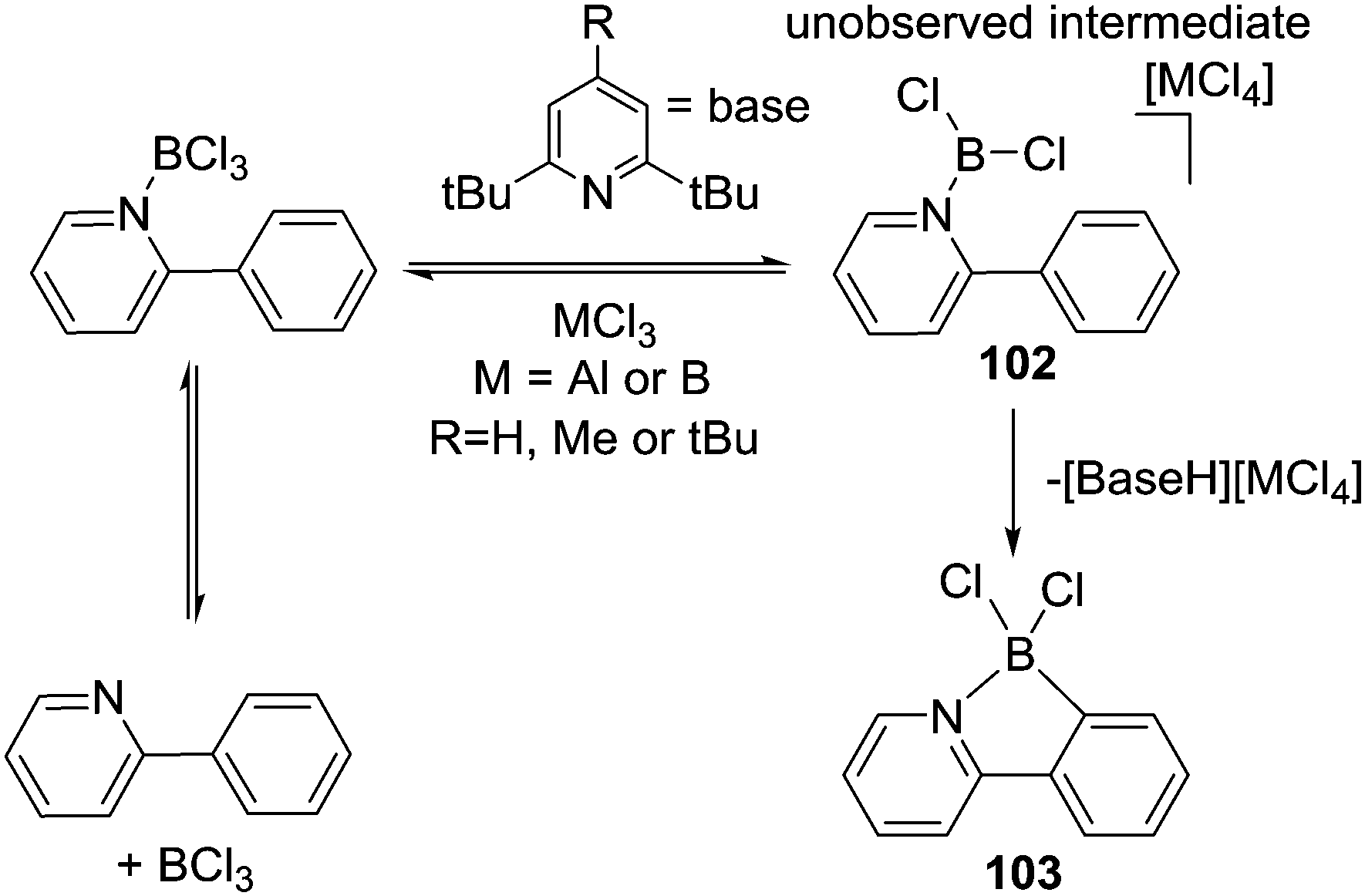 | ||
| Scheme 47 BCl3/hindered base/MCl3 directed borylation. | ||
From these studies a number of key considerations for borenium cation mediated borylation can be defined: (a) ensuring a sufficient concentration of the borenium cation is present in solution; (b) that a competent Brønsted base is present otherwise the deprotonation step can have a high barrier. Thus, the addition of exogenous bases can be key to facilitate borylation via N→B dative bonded species, particularly with BCl3 derived electrophiles due to the less basic nature of [BCl4]− (relative to [BBr4]−). This is also consistent with earlier observations on E–BX2 systems where many borylation reactions were facilitated by exogenous base.
The selectivity in borylation using BCl3/AlCl3/hindered base appears comparable to Murakami's conditions thus is presumably also proceeding under kinetic control, with both five and six membered boracycles accessible depending on the specific aromatic being borylated (e.g.104 and 105 have both been reported, Scheme 48). Again, five membered boracycle formation is generally favoured over six, suggesting that their formation has a lower barrier in the absence of other factors (e.g. steric crowding/ring strain etc.Scheme 48).96 Notably, these borylation conditions can be used to access borylated systems containing considerable strain (e.g.106 in Scheme 48 which has significant distortion in the fused anthracene moiety).
 | ||
| Scheme 48 Five and six membered boracycle formation and right, a significantly strained system generated by electrophilic C–H borylation. | ||
It should also be noted that N→B directed electrophilic C–H borylation also can be performed with electrophiles other than BX3, including with Ph2BCl97 and PhBCl2 in the presence of a base. With PhBCl2 borenium cations, again, are postulated but are not observed even at −70 °C,94 with only the py-BPhCl2 adduct, 107, and the ferrocene borylation product, 108, observable by NMR spectroscopy (Scheme 49). This suggests that the rate limiting step in this example involving borylation with PhBCl2 may well be borenium cation formation, with subsequent C–B bond formation/deprotonation rapid in the presence of the exogenous base. It is notable that this N-directed borylation occurs at very low temperature, which in this case can be attributed to a combination of: (a) the highly nucleophilic nature of ferrocene and (b) the presence of an exogenous base.
 | ||
| Scheme 49 Low temperature pyridyl directed borylation of ferrocene. | ||
Alongside pyridyl, another well studied Lewis basic moiety used in directed electrophilic C–H borylation is 2,1,3-benzothiadiazole (BT). It is notable that in contrast to pyridyl, benzothiadiazole directed C–H borylation reactions are rapid at room temperature with BBr3 and with BCl3 even in the absence of an exogenous base.98 In an open system (where HX is removed with a flow of N2) or on addition of a hindered base, borylation proceeds facilely and rapidly to form 109 (Scheme 50). This occurs when the π system being borylated is thiophene, but also with less nucleophilic aromatics (e.g. fluorene, phenyl). The more rapid borylation (relative to pyridyl analogues) is attributed to benzothiadiazole more effectively facilitating deprotonation (possibly due to the equilibrium for Lewis adduct formation favouring free benzothiadiazole significantly more than free pyridine due to the lower Lewis basicity of BT relative to pyridine). In addition, the borenium cation 110 will be more thermodynamically accessible than borenium 102. This is due to a greater stabilisation of the boron centre by enhanced NBCl2 multiple bond character (see inset) with BT, supported by calculations on borenium cations ligated by both pyridine and BT.93
 | ||
| Scheme 50 Proposed mechanism for C–H borylation directed by benzothiadiazole (BT). | ||
In benzothiadiazole directed electrophilic borylation steric effects again are significant, with steric hinderance disfavouring borylation (see Scheme 51 top). As expected, C–H borylation is more rapid with increasing arene nucleophilicity, which can be used to provide reasonable selectivity during borylation of unsymmetrically substituted derivatives (Scheme 51, top right). Benzothiadiazole directed C–H borylation could be achieved using PhBCl2 and also can be applied to borylate BT containing conjugated polymers using BCl3.99 Directed borylation also could be extended to the Se and N–R analogues of benzothiadiazole (2,1,3-benzoselenodiazole and benzotriazole).100 Notably, while benzothiadiazole contains two nitrogen Lewis basic sites it only directs a single electrophilic C–H borylation. Presumably this is due to the lower Lewis basicity of BT (e.g. relative to pyrimidine where double borylation does proceed e.g. to form 94), which will be further reduced on binding a Lewis acid to one N thereby precluding coordination of a second boron Lewis acid at the remaining nitrogen of BT. Molecules containing two BT groups can undergo double borylation with just BCl3, e.g. to form 111, provided a substituent disfavours BCl3 coordination to the external nitrogen site, which otherwise forms more stable Lewis adducts such as 112 (see Scheme 51, bottom).
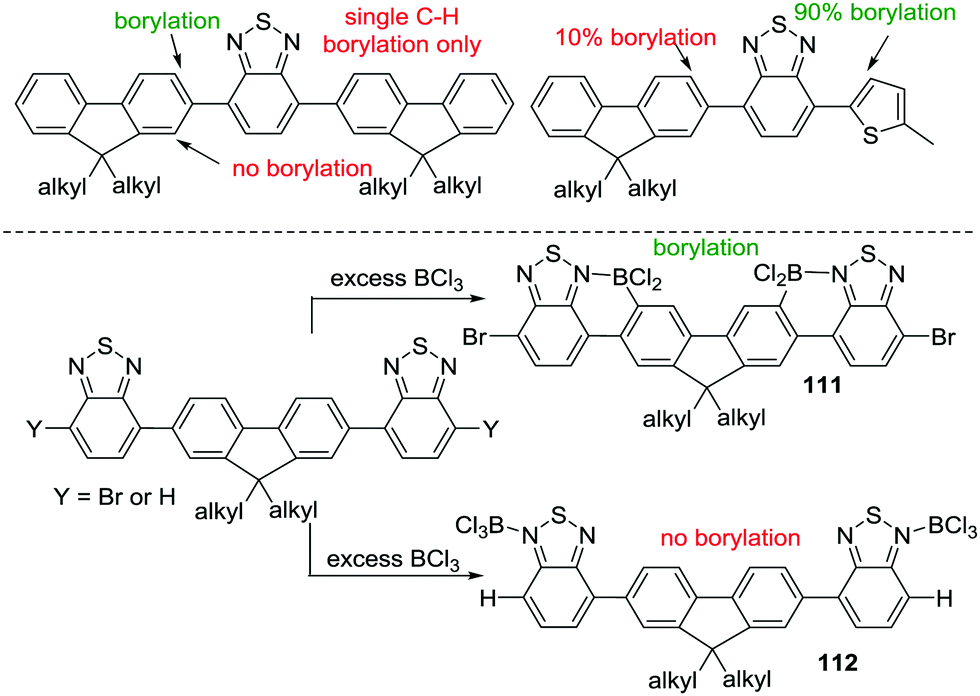 | ||
| Scheme 51 Selectivity/steric effects in BT directed C–H borylation. | ||
Recently, Chang, Park and co-workers have reported the directed C–H borylation of N,N-dimethyl-biphenyl-2-amines using a combination of HB(C6F5)2 and PhSiH3.101 The exact electrophile performing C–H borylation is not known, but it is feasible that borenium salts such as 113 (Scheme 52) generated by hydride abstraction (by a neutral borane) are intermediates consistent with the mechanistic work discussed early in this section. At some point Si–H/C6F5–B exchange occurs, with Si–C6F5 products observed, however, this may occur before or after the C–H borylation step. A functional group tolerance study revealed that while alkyl and halide groups were tolerated the presence of strong boron (and silicon) electrophiles led to decomposition of groups such as OMe and CF3 as expected. Finally, the addition of B(C6F5)3 to 114 enables demethylation, in a process again potentially mediated by a borenium cation, to generate 115 and methane as the by-product.
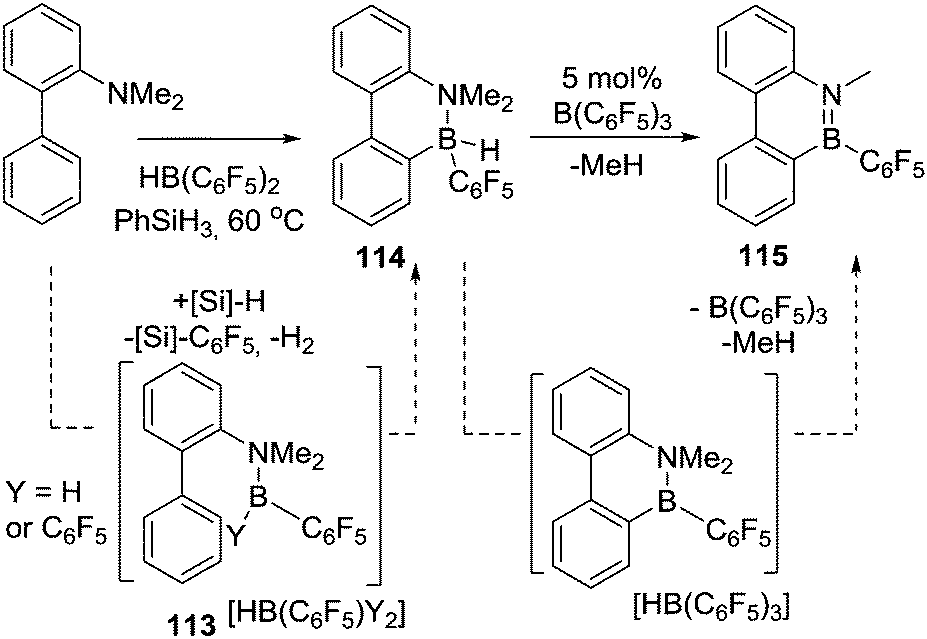 | ||
| Scheme 52 N-Directed borylation using an electrophile derived from HB(C6F5)2. | ||
Directed electrophilic C–H borylation via N→B dative bond formation and borenium cations is the most well studied and understood system in Section 4. As well as providing access to many interesting borylated materials the increased understanding provided by breakthroughs in this area have enabled the expansion of E→BX3 mediated directed C–H borylation to a wider selection of directing groups, including non-nitrogen based systems and these are discussed next.
4.2 Via BY3 coordination to oxygen centred Lewis bases
Nicholson and co-workers reported what is potentially the first example of a carbonyl directed electrophilic C–H borylation reaction.102 Addition of excess BBr3 enabled C–H borylation at room temperature to form 116 (isolated post treatment with H2O, Scheme 53). Notably, C–H borylation was selective for six membered boracycles formation, with no products derived from a five membered boracycle observed (e.g.R). This may indicate boron-enolate formation prior to C–H borylation (as this would lead to an all sp2 system which would now favour six membered boracycles formation), however the sensitivity of electrophilic C–H borylation to substituent effects (such as a deactivating OR group meta to the C–H borylation position, R maybe Me or BBr2 formed through ether cleavage) may also preclude five membered boracycle formation via a carbonyl-BBr3 adduct (and the borenium derived therefrom).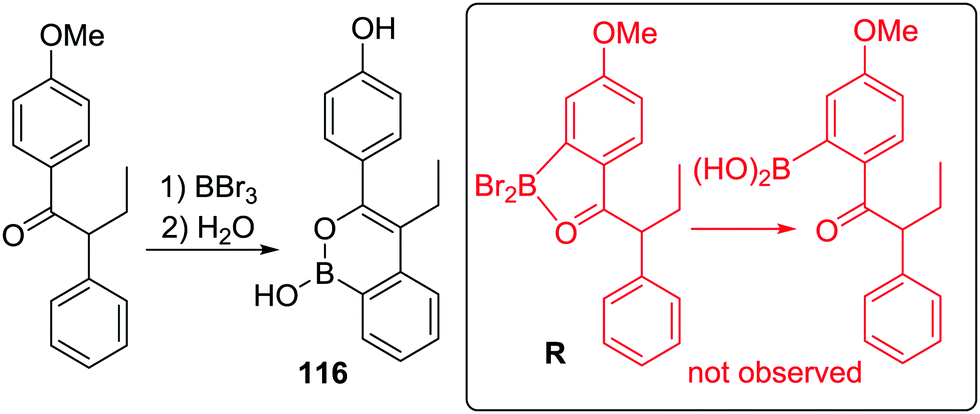 | ||
| Scheme 53 Carbonyl directed borylation for six membered boracycle formation. | ||
An expansion of carbonyl-directed C–H borylation to selective indole C7–H borylation was reported in 2019 independently by Ingleson and co-workers,103 and Shi and co-workers (Scheme 54).104 Steric interactions between the tBu group with the C7–H of indole orients the directing group with the oxygen positioned proximal to the C7 position.105 Mechanistic studies indicated that two eq. of BBr3 are involved in C–H borylation, one coordinating to the acyl group, with the second then abstracting bromide to form borenium salt 117. This is comparable to calculations on N→BX3 mediated directed borylation reactions from Uchiyama and Wang. This intermediate then forms the borylated compounds 118 and 119 by SEAr. These studies also calculated that for these substrates the six membered boracycle is the thermodynamic product. However, the reaction again appears to be proceeding under kinetic control with minor C2-borylation products (e.g.118, containing five membered boracycles) observed with some substrates. Notably, certain substrates that produced a mixture of 118 and 119 reacted further on addition of pinacol/NEt3 to convert 118 to the C7-pinacol boronate-ester product, 120, with the C2 isomer, S, not observed. It is unclear how this reaction proceeds as mixtures of 118 and 119 are stable under the borylation reaction conditions and on heating, thus pinacol appears essential to trigger C2–B cleavage/C7–B bond formation. This highlights the importance of observing primary products during C–H borylation for determining selectivity.
 | ||
| Scheme 54 N-Pivaloyl directed C–H borylation of indoles. | ||
Both teams independently extended this methodology to selectively borylate anilines at the ortho position (Scheme 55 top) to form 121. Calculations by Shi, Houk and co-workers on an aniline substrate indicated a borenium cation mediated directed C–H borylation mechanism. One significant difference between the calculations on the directed electrophilic C–H borylation of aniline and indole is the relative barriers of each step. For indole C7 borylation the barrier to arenium cation formation and subsequent deprotonation (by [BBr4]−) are effectively isoenergetic (δΔG = 0.2 kcal mol−1). However, for aniline C–H borylation the deprotonation step has a significantly larger barrier (δΔG 8.1 kcal mol−1) compared to arenium cation formation, indicating deprotonation is rate limiting in this case (nevertheless the barrier is still relatively low and reactions proceed within 1 h).
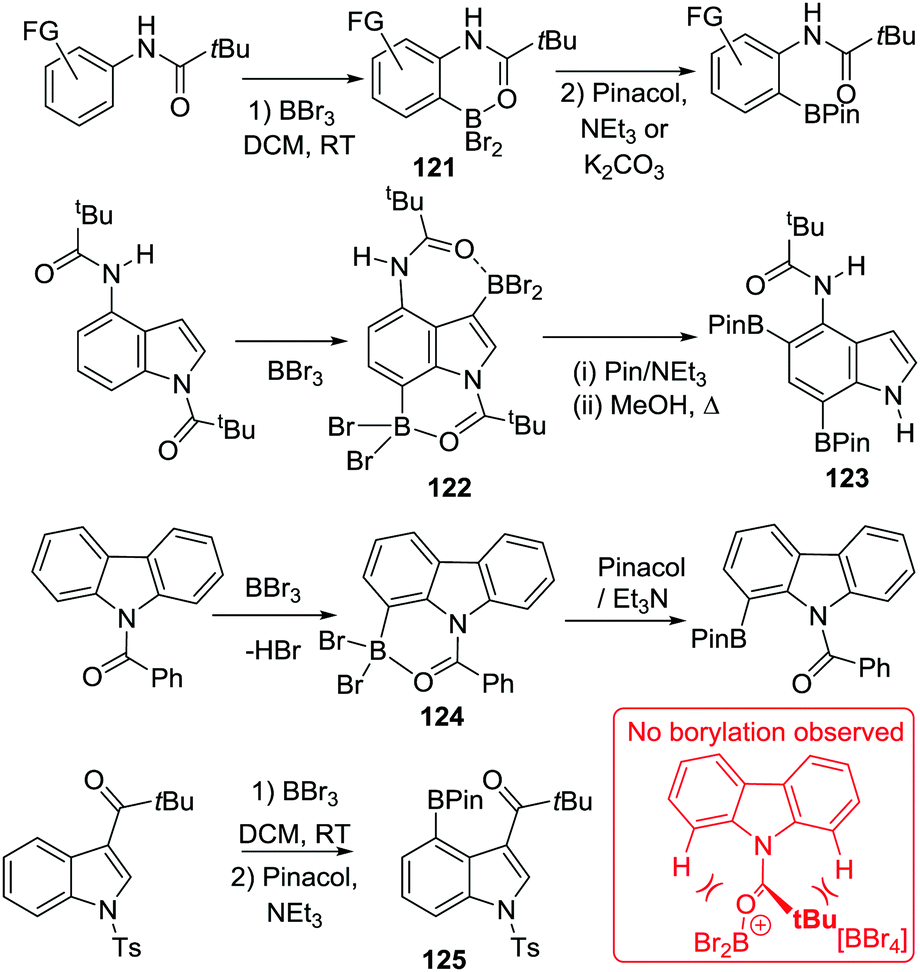 | ||
| Scheme 55 Extending acyl directed electrophilic C–H borylation to anilines, carbazoles and the C4 and C5 borylation of indoles. | ||
Regarding the scope of directed electrophilic borylation of anilines both benzoyl and pivaloyl directing groups were effective and a range of functional groups at the ortho, meta and para positions were tolerated. Notably, directed borylation of anilines could be combined with directed C7-indole borylation to enable a double carbonyl directed C–H borylation of a single substrate using BBr3 to afford 122. Again, the intermediate underwent a C–B cleavage/C–B bond formation process during protection with pinacol to afford 123. Notably, pivaloyl directed borylation of carbazole was not successful, however, a benzoyl directing group enabled carbazole C–H borylation to form 124 (Scheme 55, middle). This can be attributed to the larger steric impact of the pivaloyl group relative to benzoyl that presumably results in the required orientation for C–H borylation having a much higher energy barrier (inset bottom right for a schematic highlighting these steric interactions, Scheme 55). Finally, Shi and co-workers extended this approach to the acyl directed C4 borylation of indoles, again, using just BBr3 (Scheme 55, bottom) to form 125 (post pinacol protection), with a range of functional groups again tolerated. These studies clearly demonstrate that carbonyl directed electrophilic C–H borylation mediated by borenium cations is an underexplored route to access synthetically useful organoboranes.
4.3 Other Lewis base directed borylation
Outside of the use of N and O Lewis bases as directing groups there are a limited number of examples using other Lewis bases to direct electrophilic C–H borylation. One notable exception is the report from Stephan and co-workers on intramolecular electrophilic C–H borylation using an NHC–borenium cation (NHC = N-heterocyclic carbene, Scheme 56). Hydride abstraction from an NHC–borane affords 126 (or a functional equivalent), which subsequently undergoes a dehydrogenative borylation to form 127 at room temperature.106 The mild conditions required for this transformation is consistent with weakly stabilized borenium cations being able to rapidly perform intramolecular C–H borylation reactions. Further dehydrogenative borylation of 127 was achieved, albeit at 130 °C (24 h), affording borenium salt 128. The reduced borylating reactivity of 127 compared with 126 is attributed to the reduced Lewis acidity of the boron center in 127 as a result of enforced planarity (increasing π delocalization) and strain in the transition state due to the fused nature of borenium 127. | ||
| Scheme 56 Intramolecular borylation directed by NHC→B dative bond. | ||
Extending this approach, Würthner and co-workers developed a sequential hydroboration-electrophilic C–H borylation of aryl-alkenes using NHC–borenium equivalents. This enabled access to a wide range of boron containing PAHs.107 The initial borenium cation equivalent (NTf2 is bound to boron in these species) was generated by treating the NHC–borane adduct with HNTf2 at room temperature (evolving H2).108 By heating the mixture of the borenium functional equivalent and an appropriate alkene, e.g. 9,10-distyrylanthracene, at 110 °C a six-membered boracycle containing compound, 129, was obtained via sequential alkene hydroboration – intramolecular electrophilic C–H borylation (Scheme 57). Hydroboration is presumed to precede C–H borylation as in previous work hydroboration using related boron electrophiles proceeded at room temperature.108 The requirement for higher temperatures for C–H borylation to occur than when using [B(C6F5)4]− (e.g. to access 127) may be attributed to the requirement to displace the NTf2 anion in this case to generate the active boron electrophile, [(NHC)B(R)–H]+, which then undergoes C–H borylation. In the formation of 127–129 H2 is the observed by-product, thus, a four membered σ-bond metathesis transition state related to that proposed for C–H borylation using RBH2 and [R3N–BH2]+ electrophiles is presumably key in this C–B bond forming process.
 | ||
| Scheme 57 Sequential hydroboration, intramolecular C–H borylation to form B2-doped PAHs. Inset bottom, selectivity observed during the intramolecular C–H borylation and a plausible mechanism via a five membered boracycles. | ||
Notably, in substrates where 5 or 6 membered boracycles are feasible final products (e.g. naphthyl derivatives) only six membered boracycles are observed (e.g.130 is formed in good yield, with compound T not observed). Several related borylation reactions proceeding via borenium cations were shown to be irreversible and proceed under kinetic control. Thus, the formation of 130 directly from the alkene hydroboration product is unlikely (based on Knochel's work identifying the relatively slow kinetics of six membered boracycle formation in systems with multiple sp3-centres). An alternative mechanism is more probable proceeding via the alternative alkene hydroboration isomer, then formation of a five membered boracycle (U) and finally isomerization via a retrohydroboration/hydroboration sequence in line with Knochel's studies (Scheme 57, bottom).
Phosphorous centred Lewis bases also have been used to direct electrophilic C–H borylation, albeit to our knowledge in only one report.109 Phosphinite boranes were activated to form borenium cations (or functional equivalents) using trityl salts or strong Brønsted acids. When the anion was [B(C6F5)4]− this led to rapid room temperature borylation to form 131 (Scheme 58). Notably, when the anion was Tf2N− instead of [B(C6F5)4]− prolonged heating was required for C–H borylation to proceed, while with −OTf only traces of the borylated product was observed even after prolonged heating. This, again, confirms the importance of accessing a highly electrophilic borenium type intermediate for C–H borylation to proceed, something facilitated by more weakly coordinating anions.
 | ||
| Scheme 58 Phosphorous directed ortho-C–H borylation of phenols. | ||
The primary products (e.g.131) could be reduced to the neutral BH2 analogues by addition of a borohydride salt or converted into the respective ortho-phenol-trifluoro-organoborates, 132. The latter conversion can be viewed as a traceless ortho-C–H borylation of phenols. However, the intermediacy of highly reactive borenium type species limited functional group tolerance while regioselectivity in meta substituted derivatives also was low. As expected, the borylation of a naphthyl derivative led to the selective formation of 133 (post addition of [BH4]−) with no six membered boracycle product, V, observed. This is consistent with the outcome for the isoelectronic amine analogues (compound 88) and further confirms that five membered boracycles are the kinetic products (when containing multiple sp3 centres). While the observation of comparable outcomes for nitrogen and phosphorous derivatives is notable, there are significant differences in the metrics for PB and NB boracycles. Analysis of solid state structures for the related series of compounds 134 and 135 reveal that BP containing systems have smaller bond angles presumably due to the longer B–P and P–C bond distances (Scheme 59).110 Thus, there may be different outcomes in forming PB-boracycles via C–H borylation and further work is required to firmly identify reactivity and selectivity trends with P directing groups.
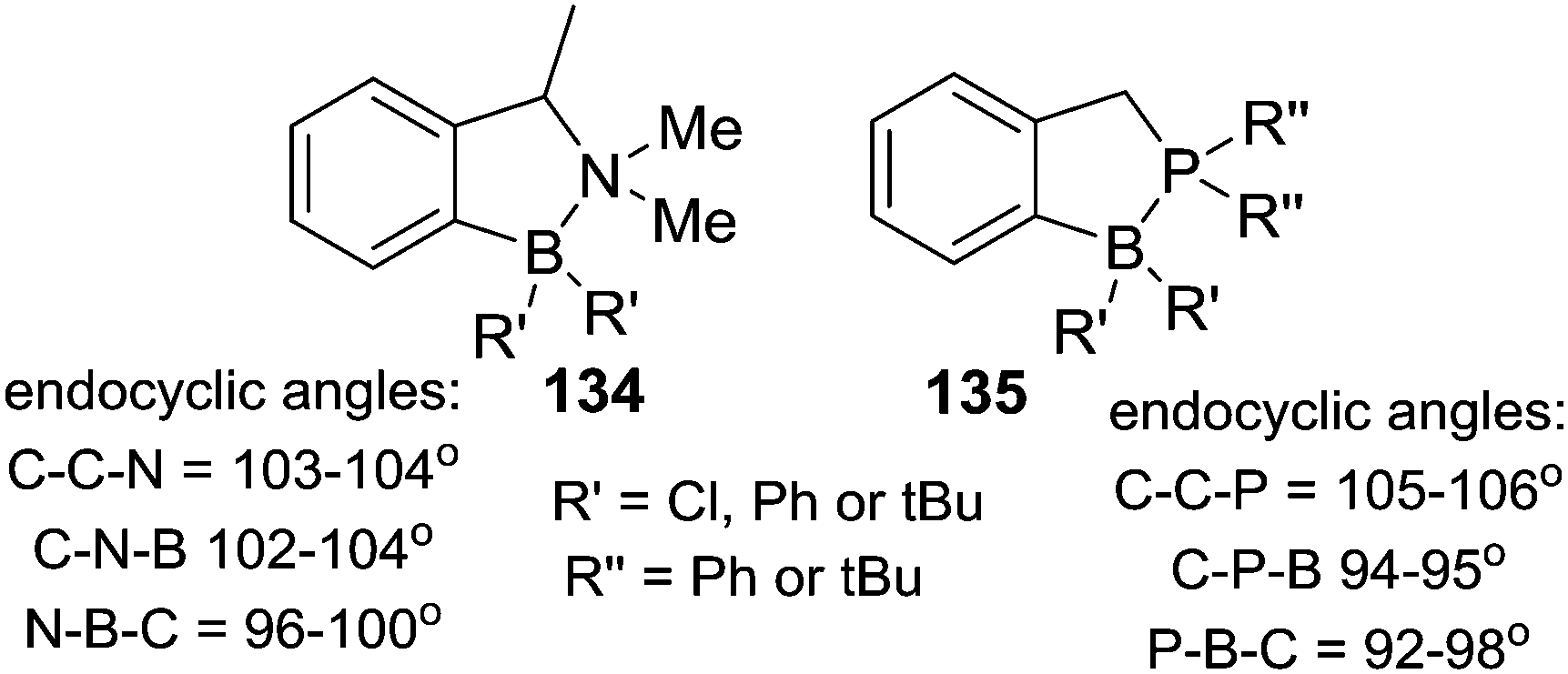 | ||
| Scheme 59 Comparison of boracycle metrics in N and P analogues. | ||
Finally, a B–H bond can also act as a directing group to enable C–H borylation using BI3 to form 136, albeit in low yield (12%, Scheme 60).111 In this unusual example the more Lewis acidic borane BI3 is essential with BBr3 and BCl3 not leading to C–H borylation, possibly as these boranes do not form the initial hydride bridged complex (bottom, Scheme 60). The mechanism of borylation and the precise boron electrophile are unknown in this system, but HI evolution occurs, indicating either iodide or [BI4]− (with H[BI4] then evolving HI) are acting as the base. Post C–H borylation the reaction of HI with a B–H species would then give the observed species (136), but HI was also reported to lead to decomposition of 136 and thus may also result in the low yield of 136.
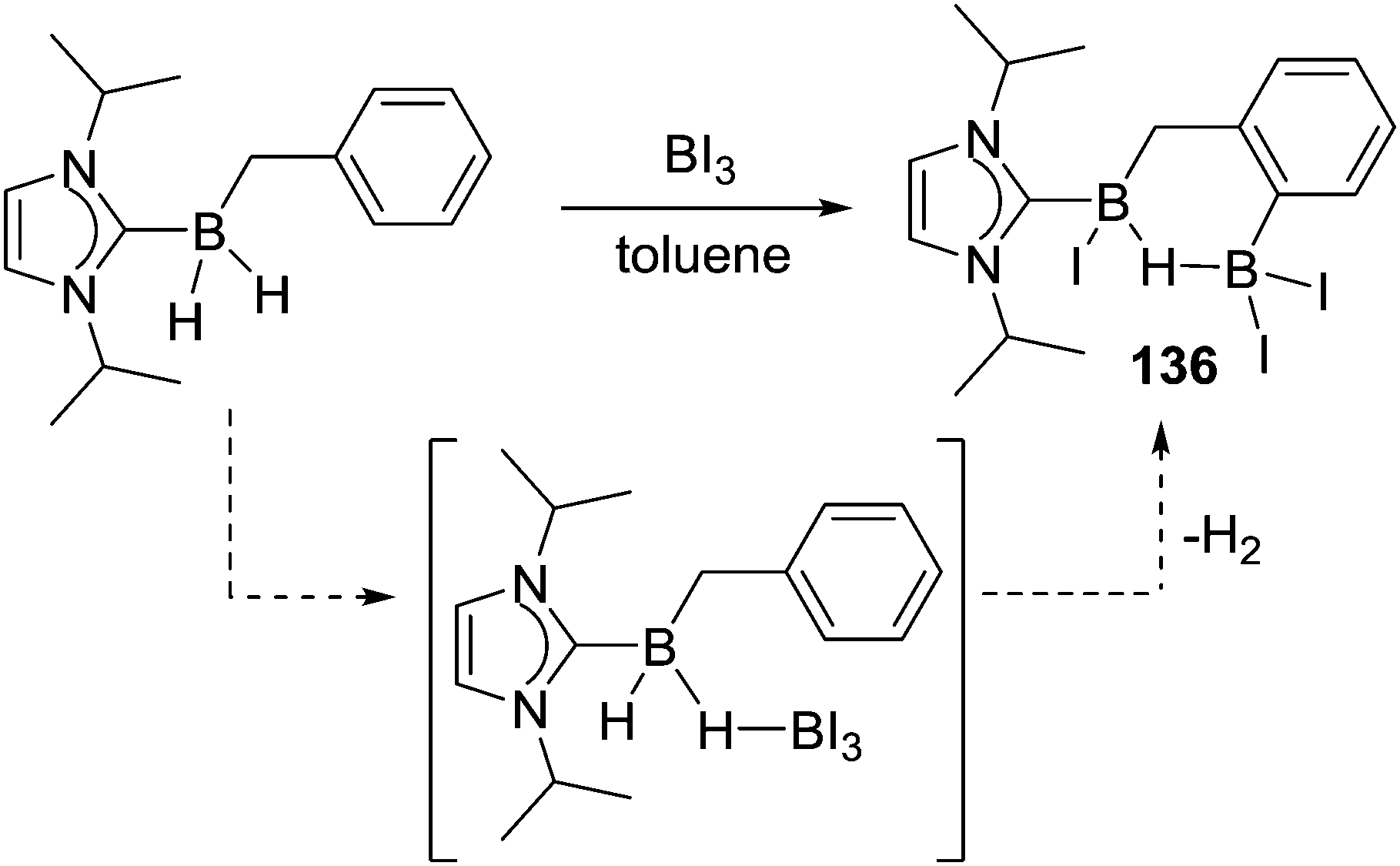 | ||
| Scheme 60 B–H directed intramolecular C–H borylation using BI3. | ||
5. Conclusions
To facilitate summarizing important points this section is split into three covering key considerations regarding: (i) mechanism, (ii) reaction conditions and (iii) scope/selectivity. It should be noted that directed C–H borylation involving third period elements (e.g. S and P based directing groups) is under developed, therefore the discussion below applies only to C, N and O based directing groups.5.1 Mechanistic considerations
There are two mechanisms reported to operate in intramolecular electrophilic C–H borylation, with the lowest energy pathway dependent principally on the identity of the boron electrophile. With B–X (X = halide) containing electrophiles directed C–H borylation generally proceeds via an SEAr mechanism, with borenium cations proposed or calculated as intermediates in multiple cases. However, in other cases the active electrophile is unidentified, and some neutral electrophiles, such as C–BX2 (X = Br or I) species, are also sufficiently reactive to effect intramolecular C–H borylation (even at ambient temperature and below). In some cases, the mechanism proceeds via an arenium cation intermediate, in other cases the C–B bond forming step and deprotonation step are concerted. Furthermore, in a number of cases the rate-determining step has been shown to be the deprotonation of an arenium cation. This is presumably due to the low basicity nature of the borylating medium. Thus, the electrophilicity of the borane and the identity (particularly the basicity) of the Brønsted base are often both important in enabling facile C–H borylation via this mechanism.The second mechanism, sometimes termed dehydrogenative electrophilic borylation, involves intramolecular C–H borylation proceeding via B–H (and to a lesser extent B–R) containing electrophiles. This process proceeds via a four membered σ bond metathesis transition state and evolves H2 (from B–H electrophiles) concomitant with C–B bond formation. It has been found to be the lowest barrier mechanism for C–H borylation mediated by both neutral and cationic B–H containing species and can also proceed rapidly at room temperature.
5.2 Reaction conditions
In many of the cases discussed the key requirement to enable C–H borylation is accessing a sufficiently electrophilic boron centre. Indeed, the greater the electrophilicity at boron the more facile the C–H borylation step generally is. Hence weakly stabilised borenium (e.g. [R3N→BH2]+) cations are particularly effective borylating species. A number of neutral boron electrophiles also are effective for borylation, particularly examples that do not contain good π donors that reduce the Lewis acidity at boron. Thus, intramolecular C–H borylation often proceeds readily with C–BX2 and C–BH2 electrophiles without the addition of Lewis acidic activators. In contrast, in systems containing amide groups, which are good π donors, additional Lewis acids are generally essential to convert the R2NBX2 species into a more electrophilic borane to enable C–H borylation. This can proceed via addition of an electrophile to N, converting the amido borane into a borenium cation or a functional equivalent thereof (e.g. R2(E)NBX2, E = electrophile, and when E = H+ this is a borenium cation). Indeed, intramolecular C–H borylation reactions proceeding through bona-fide borenium cations are amongst the more facile conversions. Therefore, the major consideration in many systems is identifying an appropriate reagent to access the borenium cation.With enhancing of electrophilicity at boron an enabling factor for intramolecular C–H borylation, it is no surprise that bromo-boranes are more effective at C–H borylation than chloro analogues. This is due in part to enhanced electrophilicity e.g. of C–BBr2vs. C–BCl2 species, but also due to borenium cations being more readily accessible through bromide abstraction from Lewis adducts of BBr3 (e.g. by another equivalent of BBr3) compared to Lewis adducts of BCl3 (where chloride abstraction from L→BCl3 with BCl3 is more energetically uphill). It should be noted that accessing borenium cations from L→BH3 species invariably requires stoichiometric quantities of strong hydridophiles that are relatively expensive (e.g. [Ph3C][B(C6F5)4]), thus making these routes less attractive for large scale synthetic applications unless they can be made to turnover (and therefore use sub-stoichiometric amounts of the hydridophile activator).
The Lewis basicity of the directing group, be it involved in a dative bond or a covalent bond to boron, is also crucial. A number of factors need to be considered: (i) the Lewis base has to be sufficiently basic to form a dative bond to the borane reagent, if not this problem can be circumvented e.g. in N–H containing system by using deprotonation and subsequent transmetalation to BX3; (ii) a less basic (be it as a dative bond or π donor to boron) nucleophile leads to a more reactive boron electrophile and thus more facile C–H borylation.
Finally, the role of a basic additive often proves vital in many C–H borylation reactions, particularly those proceeding via an SEAr mechanism (the base is not involved in the rate determining step in the σ-bond metathesis mediated borylation, thus exogenous bases do not facilitate borylation via this mechanism). The repeated observation that bases (both bulky amines and borate anions) dramatically improve C–H borylation outcomes is consistent with calculations on intramolecular C–H borylation reactions that proceed via an SEAr mechanism. In these arenium cation deprotonation is often rate limiting, thus an appropriate base (that does not quench the boron electrophile by adduct formation) that has sufficient Brønsted basicity can result in lower barriers for C–H borylation. In the absence of an exogenous base in situ formed borate anions, such as [BBr4]−, can fulfil this role (and then subsequently release HBr to form BBr3 for further C–H borylation). Therefore, the identity of the by-product from borenium cation formation can also be important in determining if intramolecular C–H borylation reactions proceed.
5.3 Factors controlling substrate scope and selectivity
In both borylation mechanisms the nucleophilicity of the π system has been found to be important. Unactivated and deactivated (towards reaction with an electrophile) (hetero)arenes are challenging to borylate using any directing group. Activated heteroarenes, such as thiophenes, are much more amenable to directed electrophilic C–H borylation, as are alkenes, with both readily borylated even using weaker boron electrophiles (including a fluoroborane electrophile in one case). Functional group tolerance is clearly dependent on the electrophilicity of the borane, thus less Lewis acidic boranes will have greater functional group tolerance but be restricted to borylating only the most nucleophilic π systems. However, with the optimal conditions, directed C–H borylation can occur facilely at room temperature and tolerate a broad range of functional groups. This is particularly the case in the borylation of nucleophilic arenes where borylation is rapid, and thus occur in preference to undesired side reactions. For example, in a very recent report directed electrophilic C–H borylation of aniline derivatives tolerated Me, tBu, F, Cl, Br, I, CF3, esters, CN, OSiR3, SMe, OR, NNPh and heteroaryl functional groups.112
In systems where multiple boracycles are potentially accessible through intramolecular electrophilic C–H borylation the boracycle ring size formed is dependent on the boron electrophile and the unit linking the π system to the directing group. For example, for all sp2 based systems six membered boracycles are the observed products. This is partly due to the more favourable bond angles present in six membered all sp2-boracycles, but also due to the higher energy of five membered all sp2 C4B boracycle units that are formally anti-aromatic. Notably, the introduction of one or more tetrahedral centres into the boracycle forming through the C–H borylation step leads to more complex outcomes. In general, in systems with one or more tetrahedral centres five membered boracycles are the kinetic products from intramolecular C–H borylation, but in some cases these can convert to six membered boracycles (e.g. by retrohydroboration–hydroboration) and these can be the thermodynamic products. The occurrence of this isomerisation depends not only on the relative energy of the five and six membered boracycle but there being a viable isomerisation pathway. In many borylation systems where there is an exogenous base present (for SEAr mechanisms) or if H2 is lost (via dehydrogenative borylation), C–H borylation is irreversible and thus the kinetic product is formed exclusively. Which in most cases is a five membered boracycle (provided at least one sp3 centre is present).
Other factors that can affect the outcome from C–H borylation include ring strain and torsional twisting in PAHs (e.g. to reduce steric effects in fjord regions). Both can lead to C–H borylation being precluded or less common C–H borylation selectivity (e.g. switching from five to six membered boracycles as the kinetic products in pyridyl directed borylation). Finally, most C–H borylation reactions discussed herein show a significant preference to borylate the less sterically hindered C–H position when there are multiple feasible borylation sites present in a molecule. Although in cases where the kinetic and thermodynamic products are different the borylation conditions again will control the outcome. For example, alpha borylation of naphthalenes (the kinetic product) is observed in room temperature irreversible borylation reactions despite this site being more hindered. In contrast, in other cases the thermodynamic product from beta naphthalene C–H borylation is the only product observed e.g. under Dewar's more forcing borylation conditions which appear to be operating under thermodynamic control. However, these are among the exceptions, and in the majority of cases C–H borylation proceeds under combined steric (least hindered) and electronic (most nucleophilic) control enabling there to be high confidence in predicting the borylation reaction outcome a priori.
The future of intramolecular electrophilic C–H borylation will hopefully see many more impressive advances as there remains much scope for applying this methodology in synthetic endeavours, particularly outside of the organic materials field. In addition, the utilisation of directing groups that are not based on nitrogen is still relatively undeveloped. Therefore, further investigations utilising other directing groups, particularly based on the third period (and below) elements, are overdue.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Research Council under the Horizon 2020 Research and Innovation Program (Grant no. 769599). Dr Sven Kirschner is thanked for useful comments.References
- Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed.
- X. Sun, B. M. Chapin, P. Metola, B. Collins, B. Wang, T. D. James and E. V. Anslyn, Nat. Chem., 2019, 11, 768–778 CrossRef CAS PubMed.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (b) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC; (c) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS; (d) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (e) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (f) Z. X. Giustra and S. Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (g) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (h) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (i) X. Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- F. Yang, M. Zhu, J. Zhang and H. Zhou, MedChemComm, 2018, 9, 201–211 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- (a) M. J. Ingleson, Synlett, 2012, 1411–1415 CrossRef CAS; (b) F. G. Fontaine and É. Rochette, Acc. Chem. Res., 2018, 51, 454–464 CrossRef CAS; (c) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- For reviews on B–N containing complexes, including key physical properties see: X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. – Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS.
- A. Prokofjevs and E. Vedejs, J. Am. Chem. Soc., 2011, 133, 20056–20059 CrossRef CAS.
- (a) Z. J. Bujwid, W. Gerrard and M. F. Lappert, Chem. Ind., 1959, 1091 CAS; (b) E. L. Muetterties, J. Am. Chem. Soc., 1959, 81, 2597 CrossRef CAS; (c) E. L. Muetterties, J. Am. Chem. Soc., 1960, 82, 4163–4166 CrossRef CAS; (d) E. L. Muetterties and F. N. Tebbe, Inorg. Chem., 1968, 7, 2663–2664 CrossRef CAS.
- M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073–3076 RSC.
- M. J. S. Dewar and W. H. Poesche, J. Org. Chem., 1964, 29, 1757–1762 CrossRef CAS.
- Q. J. Zhou, K. Worm and R. E. Dolle, J. Org. Chem., 2004, 69, 5147–5149 CrossRef CAS PubMed.
- M. J. S. Dewar and R. Dietz, J. Chem. Soc., 1959, 2728–2730 RSC.
- G. A. Olah, Angew. Chem., Int. Ed. Engl., 1993, 32, 767–788 CrossRef.
- M. J. Ingleson, Top. Organomet. Chem., 2015, 49, 39–71 CrossRef CAS.
- M. J. S. Dewar, C. Kaneko and M. K. Bhattacharjee, J. Am. Chem. Soc., 1962, 84, 4884–4887 CrossRef CAS.
- (a) M. J. S. Dewar and R. Dietz, Tetrahedron Lett., 1959, 1, 21–23 CrossRef; (b) F. A. Davis and M. J. S. Dewar, J. Am. Chem. Soc., 1968, 90, 3511–3515 CrossRef CAS.
- S. A. Solomon, A. Del Grosso, E. R. Clark, V. Bagutski, J. J. W. McDouall and M. J. Ingleson, Organometallics, 2012, 31, 1908–1916 CrossRef CAS.
- This body of work was reviewed in 1964 in English see: (a) R. Köster, Angew. Chem., Int. Ed. Engl., 1964, 3, 174–185 CrossRef ; earlier individual studies were reported in German e.g.: ; (b) R. Köster and K. Reinert, Angew. Chem., Int. Ed. Engl., 1959, 71, 521 Search PubMed.
- D. T. Hurd, J. Am. Chem. Soc., 1948, 70, 2053–2055 CrossRef CAS.
- B. Goldfuss, P. Knochel, L. O. Bromm and K. Knapp, Angew. Chem., Int. Ed., 2000, 39, 4136–4139 CrossRef CAS PubMed.
- R. Köster and G. Benedikt, Angew. Chem., Int. Ed. Engl., 1963, 2, 323–324 CrossRef.
- (a) H. Laaziri, L. O. Bromm, F. Lhermitte, R. M. Gschwind and P. Knochel, J. Am. Chem. Soc., 1999, 121, 6940–6941 CrossRef CAS; (b) J. A. Varela, D. Peña, B. Goldfuss, K. Polborn and P. Knochel, Org. Lett., 2001, 3, 2395–2398 CrossRef CAS PubMed; (c) J. A. Varela, D. Peñà, B. Goldfuss, D. Denisenko, J. Kulhanek, K. Polborn and P. Knochel, Chem. – Eur. J., 2004, 10, 4252–4264 CrossRef CAS PubMed.
- R. L. Letsinger and D. B. MacLean, J. Am. Chem. Soc., 1963, 85, 2230–2236 CrossRef CAS.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562–13565 CrossRef CAS.
- R. Köster, K. Iwasaki, S. Hattori and Y. Morita, Liebigs Ann. Chem., 1968, 720, 23–31 CrossRef.
- (a) B. W. Müller, Helv. Chim. Acta, 1978, 61, 325–327 CrossRef; (b) M. Grassberger, Ger. Offen., 2750878, 1978 Search PubMed; (c) M. A. Grassberger, F. Turnowsky and J. Hildebrandt, J. Med. Chem., 1984, 27, 947–953 CrossRef CAS PubMed.
- (a) S. Gronowitz and I. Ander, Chem. Scr., 1980, 1, 23–26 Search PubMed; (b) S. Gronowitz and I. Ander, Chem. Scr., 1980, 15, 135–144 CAS; (c) S. Gronowitz and I. Ander, Chem. Scr., 1980, 15, 145–151 CAS; (d) S. Gronowitz, I. Ander and P. Zanitaro, Chem. Scr., 1983, 22, 55–59 CAS.
- (a) V. A. Dorokhov, O. F. Boldyreva, M. N. Bochkaxeva and B. M. Mikhailov, Russ. Chem. Bull., 1979, 28, 163–168 CrossRef; (b) O. G. Boldyreva, V. A. Dorokhov and B. M. Mikhailov, Russ. Chem. Bull., 1985, 34, 390–392 CrossRef.
- G. T. Lee, K. Prasad and O. Repič, Tetrahedron Lett., 2002, 43, 3255–3257 CrossRef CAS.
- S. Allaoud and B. Frange, Inorg. Chem., 1985, 24, 2520–2523 CrossRef CAS.
- A. M. Genaev, S. M. Nagy, G. E. Salnikov and V. G. Shubin, Chem. Commun., 2000, 1587–1588 RSC.
- Note that a significant amount of the positive charge in borenium cations such as F will be localised on boron as the least electronegative atom, however as charge is inherently delocalised all charged compounds are represented simply with a unit positive charge.
- (a) D. H. Ryu and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 6388–6390 CrossRef CAS PubMed; (b) D. Liu, E. Canales and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 1498–1499 CrossRef CAS PubMed.
- M. J. S. Dewar, in Progress in Boron Chemistry, ed. H. Steinberg, Pergamon Press, Oxford, 1964, ch. 5, vol. 1, pp. 235–264 Search PubMed.
- (a) P. Koelle and H. Noeth, Chem. Rev., 1985, 85, 399–418 CrossRef CAS; (b) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS.
- (a) E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem. – Eur. J., 2013, 19, 2462–2466 CrossRef CAS; (b) A. Del Grosso, E. R. Clark, N. Montoute and M. J. Ingleson, Chem. Commun., 2012, 48, 7589–7591 RSC.
- M. J. D. Bosdet, C. A. Jaska, W. E. Piers, T. S. Sorensen and M. Parvez, Org. Lett., 2007, 9, 1395–1398 CrossRef CAS PubMed.
- S. S. Chissick, M. J. S. Dewar and P. M. Maitlis, Tetrahedron Lett., 1960, 1, 8–10 CrossRef.
- P. R. Ashton, K. D. M. Harris, B. M. Kariuki, D. Philp, J. M. A. Robinson and N. Spencer, J. Chem. Soc., Perkin Trans. 2, 2001, 2166–2173 RSC.
- (a) T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed; (b) T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS PubMed.
- A. Kraft, J. Beck and I. Krossing, Chem. – Eur. J., 2011, 17, 12975–12980 CrossRef CAS PubMed.
- (a) G. Li, W. W. Xiong, P. Y. Gu, J. Cao, J. Zhu, R. Ganguly, Y. Li, A. C. Grimsdale and Q. Zhang, Org. Lett., 2015, 17, 560–563 CrossRef CAS PubMed; (b) X. Y. Wang, F. D. Zhuang, X. C. Wang, X. Y. Cao, J. Y. Wang and J. Pei, Chem. Commun., 2015, 51, 4368–4371 RSC.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS PubMed.
- (a) M. Lepeltier, O. Lukoyanova, A. Jacobson, S. Jeeva and D. F. Perepichka, Chem. Commun., 2010, 46, 7007–7009 RSC; (b) J. Zhang, F. Liu, Z. Sun, C. Li, Q. Zhang, C. Zhang, Z. Liu and X. Liu, Chem. Commun., 2018, 54, 8178–8181 RSC.
- W. Zhang, F. Zhang, R. Tang, Y. Fu, X. Wang, X. Zhuang, G. He and X. Feng, Org. Lett., 2016, 18, 3618–3621 CrossRef CAS PubMed.
- X. Wang, F. Zhang, J. Gao, Y. Fu, W. Zhao, R. Tang, W. Zhang, X. Zhuang and X. Feng, J. Org. Chem., 2015, 80, 10127–10133 CrossRef CAS PubMed.
- M. Fingerle, C. Maichle-Mössmer, S. Schundelmeier, B. Speiser and H. F. Bettinger, Org. Lett., 2017, 19, 4428–4431 CrossRef CAS PubMed.
- X. Liu, P. Wu, J. Li and C. Cui, J. Org. Chem., 2015, 80, 3737–3744 CrossRef CAS PubMed.
- (a) X. Wang, F. Zhang, K. S. Schellhammer, P. Machata, F. Ortmann, G. Cuniberti, Y. Fu, J. Hunger, R. Tang, A. A. Popov, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2016, 138, 11606–11615 CrossRef CAS PubMed; (b) P. Qiang, Z. Sun, M. Wan, X. Wang, P. Thiruvengadam, C. Bingi, W. Wei, W. Zhu, D. Wu and F. Zhang, Org. Lett., 2019, 21, 4575–4579 CrossRef CAS PubMed.
- (a) M. Numano, N. Nagami, S. Nakatsuka, T. Katayama, K. Nakajima, S. Tatsumi, N. Yasuda and T. Hatakeyama, Chem. – Eur. J., 2016, 22, 11574–11577 CrossRef CAS PubMed; (b) Z. Sun, C. Yi, Q. Liang, C. Bingi, W. Zhu, P. Qiang, D. Wu and F. Zhang, Org. Lett., 2020, 22, 209–213 CrossRef CAS.
- F.-D. Zhuang, J.-M. Han, S. Tang, J.-H. Yang, Q.-R. Chen, J.-Y. Wang and J. Pei, Organometallics, 2017, 36, 2479–2482 CrossRef CAS.
- (a) J. Zhang, F. Liu, Z. Sun, C. Li, Q. Zhang, C. Zhang, Z. Liu and X. Liu, Chem. Commun., 2018, 54, 8178–8181 RSC; (b) X. Wang, F. Zhang, J. Gao, Y. Fu, W. Zhao, R. Tang, W. Zhang, X. Zhuang and X. Feng, J. Org. Chem., 2015, 80, 10127–10133 CrossRef CAS PubMed; (c) X. Wang, F. Zhang, J. Liu, R. Tang, Y. Fu, D. Wu, Q. Xu, X. Zhuang, G. He and X. Feng, Org. Lett., 2013, 15, 5714–5717 CrossRef CAS; (d) X.-Y. Wang, H.-R. Lin, T. Lei, D.-C. Yang, F.-D. Zhuang, J.-Y. Wang, S.-C. Yuan and J. Pei, Angew. Chem., Int. Ed., 2013, 52, 3117–3120 CrossRef CAS PubMed; (e) X.-Y. Wang, F.-D. Zhuang, X. Zhou, D.-C. Yang, J.-Y. Wang and J. Pei, J. Mater. Chem. C, 2014, 2, 8152–8161 RSC; (f) X.-Y. Wang, F.-D. Zhuang, R.-B. Wang and X.-C. Wang, Chem. – Eur. J., 2015, 21, 8867–8873 CrossRef CAS PubMed; (g) C.-J. Sun, N. Wang, T. Peng, X. Yin, S. Wang and P. Chen, Inorg. Chem., 2019, 58, 3591–3595 CrossRef CAS PubMed; (h) Y. Chen, W. Chen, Y. Qiao and G. Zhou, Chem. – Eur. J., 2019, 25, 9326–9338 CrossRef CAS PubMed.
- P. Paetzold, C. Stanescu, J. R. Stubenrauch, M. Bienmüller and U. Englert, Z. Anorg. Allg. Chem., 2004, 630, 2632–2640 CrossRef CAS.
- (a) J. S. A. Ishibashi, J. L. Marshall, A. Mazière, G. J. Lovinger, B. Li, L. N. Zakharov, A. Dargelos, A. Graciaa, A. Chrostowska and S.-Y. Liu, J. Am. Chem. Soc., 2014, 136, 15414–15421 CrossRef CAS PubMed; (b) J. S. A. Ishibashi, A. Dargelos, C. Darrigan, A. Chrostowska and S.-Y. Liu, Organometallics, 2017, 36, 2494–2497 CrossRef CAS.
- L. Zi, J. Zhang, C. Li, Y. Qu, B. Zhen, X. Liu and L. Zhang, Org. Lett., 2020, 22(4), 1499–1503 CrossRef CAS PubMed.
- S. R. Wisniewski, C. L. Guenther, O. A. Argintaru and G. A. Molander, J. Org. Chem., 2014, 79, 365–378 CrossRef CAS PubMed.
- M. R. Sánchez Casado, M. Ciordia Jiménez, M. Ariza Bueno, M. Barriol, J. E. Leenaerts, C. Pagliuca, C. Martínez Lamenca, A. I. De Lucas, A. García, A. A. Trabanco and F. J. R. Rombouts, Eur. J. Org. Chem., 2015, 5221–5229 CrossRef.
- G. H. M. Davies, Z. Z. Zhou, M. Jouffroy and G. A. Molander, J. Org. Chem., 2017, 82, 549–555 CrossRef CAS.
- J. J. Vaquero, D. Sucunza, A. Abengózar, P. García-García, I. Valencia, A. Perez-Redondo, G. Otarola and F. Mendicuti, Chem. Commun., 2020, 56, 3669–3672 RSC.
- (a) Z. An, M. Wu, J. Kang, J. Ni, Z. Qi, B. Yuan and R. Yan, Eur. J. Org. Chem., 2018, 4812–4817 CrossRef CAS; (b) C. Li, Y. Liu, Z. Sun, J. Zhang, M. Liu, C. Zhang, Q. Zhang, H. Wang and X. Liu, Org. Lett., 2018, 20, 2806–2810 CrossRef CAS PubMed.
- (a) R. van Veen and F. Bickelhaupt, J. Organomet. Chem., 1972, 43, 241–248 CrossRef CAS; (b) R. Van Veen and F. Bickelhaupt, J. Organomet. Chem., 1973, 47, 33–38 CrossRef CAS.
- (a) A. Kawachi, H. Morisaki, T. Hirofuji and Y. Yamamoto, Chem. – Eur. J., 2013, 19, 13294–13298 CrossRef CAS PubMed; (b) T. Hirofuji, T. Ikeda, T. Haino, Y. Yamamoto and A. Kawachi, Chem. – Eur. J., 2016, 22, 9734–9739 CrossRef CAS PubMed.
- W. Schacht and D. Kaufmann, Chem. Ber., 1987, 120, 1331–1338 CrossRef CAS.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed.
- (a) F. Miyamoto, S. Nakatsuka, K. Yamada, K. I. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed; (b) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed; (c) S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 CrossRef CAS PubMed; (d) G. Meng, X. Chen, X. Wang, N. Wang, T. Peng and S. Wang, Adv. Opt. Mater., 2019, 7, 1900130 CrossRef; (e) D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311–9314 CrossRef CAS PubMed.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed.
- K. Fujimoto, J. Oh, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 3196–3199 CrossRef CAS PubMed.
- A. Escande, D. L. Crossley, J. Cid, I. A. Cade, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 17160–17167 RSC.
- (a) K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed; (b) K. Mitsudo, K. Shigemori, H. Mandai, A. Wakamiya and S. Suga, Org. Lett., 2018, 20, 7336–7340 CrossRef CAS PubMed; (c) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS; (d) S. Oda, K. Ueura, B. Kawakami and T. Hatakeyama, Org. Lett., 2020, 22, 700–704 CrossRef CAS PubMed; (e) A. John, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 5588–5592 CrossRef CAS PubMed.
- D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969–7977 RSC.
- (a) R. J. Kahan, D. L. Crossley, J. Cid, J. E. Radcliffe and M. J. Ingleson, Angew. Chem., Int. Ed., 2018, 57, 8084–8088 CrossRef CAS PubMed; (b) K. Yuan, R. J. Kahan, C. Si, A. Williams, S. Kirschner, M. Uzelac, E. Zysman-Colman and M. J. Ingleson, Chem. Sci., 2020, 11, 3258–3267 RSC.
- (a) T. M. Kosak, H. A. Conrad, A. L. Korich and R. L. Lord, Eur. J. Org. Chem., 2015, 7460–7467 CrossRef CAS PubMed; (b) A. J. Warner, A. Churn, J. S. McGough and M. J. Ingleson, Angew. Chem., Int. Ed., 2017, 56, 354–358 CrossRef CAS PubMed.
- X. Y. Wang, A. Narita, W. Zhang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 9021–9024 CrossRef CAS PubMed.
- X. Yao, K. Zhang, K. Müllen and X.-Y. Wang, Asian J. Org. Chem., 2018, 7, 2233–2238 CrossRef CAS.
- X. Y. Wang, X. C. Wang, A. Narita, M. Wagner, X. Y. Cao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 12783–12786 CrossRef CAS PubMed.
- K. Shigemori, M. Watanabe, J. Kong, K. Mitsudo, A. Wakamiya, H. Mandai and S. Suga, Org. Lett., 2019, 21, 2171–2175 CrossRef CAS PubMed.
- W. Di and C. Liu, Preparation Method for Tavaborole, Faming Zhuanli Shenqing, CN106467557, 2017 Search PubMed.
- T. S. De Vries, A. Prokofjevs, J. N. Harvey and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14679–14687 CrossRef CAS PubMed.
- A. Prokofjevs, J. Jermaks, A. Borovika, J. W. Kampf and E. Vedejs, Organometallics, 2013, 32, 6701–6711 CrossRef CAS PubMed.
- A. Prokofjevs, Angew. Chem., Int. Ed., 2015, 54, 13401–13405 CrossRef CAS PubMed.
- R. Taylor, Electrophilic Aromatic Substitution, John Wiley and Sons, Chichester, 1990 Search PubMed.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef CAS PubMed.
- For the application of Murakami's C–H borylation conditions see: with pyridyl directing groups (and substituted pyridyls): (a) H. L. Wong, W. T. Wong and V. W. W. Yam, Org. Lett., 2012, 14, 1862–1865 CrossRef CAS PubMed; (b) L. Niu, H. Yang, R. Wang and H. Fu, Org. Lett., 2012, 14, 2618–2621 CrossRef CAS; (c) Z. Zhao, Z. Chang, B. He, B. Chen, C. Deng, P. Lu, H. Qiu and B. Z. Tang, Chem. – Eur. J., 2013, 19, 11512–11517 CrossRef CAS; (d) S. K. Mellerup, K. Yuan, C. Nguyen, Z.-H. Lu and S. Wang, Chem. – Eur. J., 2016, 22, 12464–12472 CrossRef CAS; (e) M. Yusuf, K. Liu, F. Guo, R. A. Lalancette and F. Jäkle, Dalton Trans., 2016, 45, 4580–4587 RSC; (f) B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Chem. – Eur. J., 2016, 22, 15095–15106 CrossRef CAS; (g) K. Matsuo and T. Yasuda, Chem. Commun., 2017, 53, 8723–8726 RSC; (h) Y. J. Shiu, Y. T. Chen, W. K. Lee, C. C. Wu, T. C. Lin, S. H. Liu, P. T. Chou, C. W. Lu, I. C. Cheng, Y. J. Lien and Y. Chi, J. Mater. Chem. C, 2017, 5, 1452–1462 RSC; (i) C. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. G. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Chem. – Eur. J., 2017, 23, 407–418 CrossRef CAS PubMed; (j) M. Stanoppi and A. Lorbach, Dalton Trans., 2018, 47, 10394–10398 RSC; (k) Y. Li, H. Meng, D. Yan, Y. Li, B. Pang, K. Zhang, G. Luo, J. Huang and C. Zhan, Tetrahedron, 2018, 74, 4308–4314 CrossRef CAS; (l) A. F. Alahmadi, R. A. Lalancette and F. Jäkle, Macromol. Rapid Commun., 2018, 39, 1800456 CrossRef PubMed; (m) M. Mamada, G. Tian, H. Nakanotani, J. Su and C. Adachi, Angew. Chem., Int. Ed., 2018, 57, 12380–12384 CrossRef CAS PubMed; (n) H. Jin, H. J. Bae, S. Kim, J. H. Lee, H. Hwang, M. H. Park and K. M. Lee, Dalton Trans., 2019, 48, 1467–1476 RSC; (o) D. Kunchala, S. Sa, P. Nayak, J. S. Ponniah and K. Venkatasubbaiah, Organometallics, 2019, 38, 870–878 CrossRef CAS ; with pyrazine directing groups: ; (p) C. Zhu, Z. H. Guo, A. U. Mu, Y. Liu, S. E. Wheeler and L. Fang, J. Org. Chem., 2016, 81, 4347–4352 CrossRef CAS PubMed; (q) Y. Li, B. Pang, H. Meng, Y. Xiang, Y. Li and J. Huang, Tetrahedron Lett., 2019, 60, 151286 CrossRef CAS; (r) Y. Li, H. Meng, T. Liu, Y. Xiao, Z. Tang, B. Pang, Y. Li, Y. Xiang, G. Zhang, X. Lu, G. Yu, H. Yan, C. Zhan, J. Huang and J. Yao, Adv. Mater., 2019, 31, 1904585 CrossRef CAS PubMed ; with pyrimidine directing groups: ; (s) C. R. Opie, H. Noda, M. Shibasaki and N. Kumagai, Chem. – Eur. J., 2019, 25, 4648–4653 CrossRef CAS PubMed ; with imidazolinone directing groups (and derivatives): ; (t) M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed; (u) M. S. Baranov, K. M. Solntsev, N. S. Baleeva, A. S. Mishin, S. A. Lukyanov, K. A. Lukyanov and I. V. Yampolsky, Chem. – Eur. J., 2014, 20, 13234–13241 CrossRef CAS PubMed; (v) S. Olsen, M. S. Baranov, N. S. Baleeva, M. M. Antonova, K. A. Johnson and K. M. Solntsev, Phys. Chem. Chem. Phys., 2016, 18, 26703–26711 RSC ; with imidazole directing groups: ; (w) K. Dhanunjayarao, S. Sa, B. P. R. Aradhyula and K. Venkatasubbaiah, Tetrahedron, 2018, 74, 5819–5825 CrossRef CAS ; with pyrazole directing groups: ; (x) V. Mukundam, S. Sa, A. Kumari, R. Das and K. Venkatasubbaiah, J. Mater. Chem. C, 2019, 7, 12725–12737 RSC.
- M. Kondrashov, D. Provost and O. F. Wendt, Dalton Trans., 2016, 45, 525–531 RSC.
- A. C. Shaikh, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115–16118 RSC.
- S. Oda, H. Abe, N. Yasuda and T. Hatakeyama, Chem. – Asian J., 2019, 14, 1657–1661 CrossRef CAS PubMed.
- D.-Y. Wang, H. Minami, C. Wang and M. Uchiyama, Chem. Lett., 2015, 44, 1380–1382 CrossRef CAS.
- D. L. Crossley, J. Cid, L. D. Curless, M. L. Turner and M. J. Ingleson, Organometallics, 2015, 34, 5767–5774 CrossRef CAS PubMed.
- J. Chen, R. A. Lalancette and F. Jäkle, Organometallics, 2013, 32, 5843–5851 CrossRef CAS.
- G. E. Ryschkewitsch and J. W. Wiggins, J. Am. Chem. Soc., 1970, 92, 1790–1791 CrossRef CAS.
- For other applications of BCl3/AlCl3/hindered base in directed borylation see: (a) V. Mukundam, S. Sa, A. Kumari, R. Das and K. Venkatasubbaiah, J. Mater. Chem. C, 2019, 7, 12725–12737 RSC; (b) J. S. A. Ishibashi, C. Darrigan, A. Chrostowska, B. Li and S. Y. Liu, Dalton Trans., 2019, 48, 2807–2812 RSC; (c) M. Vanga, R. A. Lalancette and F. Jäkle, Chem. – Eur. J., 2019, 25, 10133–10140 CrossRef CAS PubMed; (d) M. M. Morgan, M. Nazari, T. Pickl, J. M. Rautiainen, H. M. Tuononen, W. E. Piers, G. C. Welch and B. S. Gelfand, Chem. Commun., 2019, 55, 11095–11098 RSC; (e) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2019, 141, 7453–7462 CrossRef CAS PubMed.
- K. Yang, G. Zhang and Q. Song, Chem. Sci., 2018, 9, 7666–7672 RSC.
- (a) D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC; (b) D. L. Crossley, R. Goh, J. Cid, I. Vitorica-Yrezabal, M. L. Turner and M. J. Ingleson, Organometallics, 2017, 36, 2597–2604 CrossRef CAS; (c) D. L. Crossley, P. Kulapichitr, J. E. Radcliffe, J. J. Dunsford, I. Vitorica-Yrezabal, R. J. Kahan, A. W. Woodward, M. L. Turner, J. J. W. McDouall and M. J. Ingleson, Chem. – Eur. J., 2018, 24, 10521–10530 CrossRef CAS PubMed.
- D. L. Crossley, L. Urbano, R. Neumann, S. Bourke, J. Jones, L. A. Dailey, M. Green, M. J. Humphries, S. M. King, M. L. Turner and M. J. Ingleson, ACS Appl. Mater. Interfaces, 2017, 9, 28243–28249 CrossRef CAS PubMed.
- B. P. Dash, I. Hamilton, D. J. Tate, D. L. Crossley, J. S. Kim, M. J. Ingleson and M. L. Turner, J. Mater. Chem. C, 2019, 7, 718–724 RSC.
- J. Zhang, H. Jung, D. Kim, S. Park and S. Chang, Angew. Chem., Int. Ed., 2019, 58, 7361–7365 CrossRef CAS PubMed.
- V. L. Arcus, L. Main and B. K. Nicholson, J. Organomet. Chem., 1993, 460, 139–147 CrossRef CAS.
- S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2019, 58, 15381–15385 CrossRef CAS PubMed.
- J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336–340 CrossRef CAS PubMed.
- T. Fukuda, R. Maeda and M. Iwao, Tetrahedron, 1999, 55, 9151–9162 CrossRef CAS.
- J. M. Farrell and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 5214–5217 CrossRef CAS PubMed.
- (a) J. M. Farrell, D. Schmidt, V. Grande and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 11846–11850 CrossRef CAS PubMed; (b) J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A. M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed.
- A. Prokofjevs, A. Boussonnière, L. Li, H. Bonin, E. Lacôte, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 134, 12281–12288 CrossRef CAS PubMed.
- C. Cazorla, T. S. De Vries and E. Vedejs, Org. Lett., 2013, 15, 984–987 CrossRef CAS.
- Z. M. Heiden, M. Schedler and D. W. Stephan, Inorg. Chem., 2011, 50, 1470–1479 CrossRef CAS PubMed.
- R. Böser, L. Denker and R. Frank, Chem. – Eur. J., 2019, 25, 10575–10579 CrossRef.
- J. Lv, B. Zhao, Y. Yuan, Y. Han and Z. Shi, Nat. Commun., 2020, 1316 CrossRef CAS PubMed.
Footnote |
| † These authors all contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |