
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular radical carboamination of alkenes
Heng
Jiang
a and
Armido
Studer
 *ab
*ab
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
bKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou, Fujian 350002, P. R. China
First published on 14th February 2020
Abstract
Vicinal alkene carboamination is a highly efficient and practical synthetic strategy for the straightforward preparation of diverse and valuable amine derivatives starting from simple compounds. During the last decade that approach has found continuous research interests and various practical methods have been developed using transition-metal catalysis. Driven by the renaissance of synthetic radical chemistry, intermolecular radical alkene carboamination comprising a C–C bond and a C–N bond forming step has been intensively investigated recently culminating in novel strategies and improved protocols which complement existing methodologies. Radical alkene carboamination can be achieved via three different reaction modes. Such cascades can proceed through N-radical addition to an alkene with subsequent C–C bond formation leading to 2,1-carboamination products. Alternatively, the C–C bond can be installed prior to the C–N bond via initial C-radical addition to the alkene with subsequent β-amination resulting in 1,2-carboamination. The third mode comprises initial single electron oxidation of the alkene to the corresponding alkene radical cation that gets trapped by an N-nucleophile and the cascade is terminated by radical C–C bond formation. In this review, the three different conceptual approaches will be discussed and examples from the recent literature will be presented. Further, the reader will get insights into the mechanism of the different transformations.
 Heng Jiang | Heng Jiang received his diploma from Jiangnan University in 2010. He then pursued his PhD degree in Nanjing University from Sept. 2010 under supervision of Prof. Yan Zhang and Prof. Shouyun Yu. During Oct. 2014 to Oct. 2015, he learnt Pd catalysed C–H bond activation as a joint PhD student at the Scripps Research Institute (La jolla, USA) under the direction of Prof. Jin-Quan Yu. After receiving his PhD degree in Mar. 2016, he stayed at Nanjing University as a research assistant for half a year in collaboration with Prof. Shouyun Yu. From Jan. 2017, he continued his research at the Westfälische Wilhelms-University (Münster, Germany) as an Alexander von Humboldt scholar in collaboration with Prof. Armido Studer. His current research interests focus on radical chemistry and transition metal catalysed C–H activation. |
 Armido Studer | Armido Studer received his Diploma in 1991 and his PhD in 1995 from ETH Zürich under the direction of Prof. Dieter Seebach. He continued his scientific education as a Swiss National Science Foundation Postdoctoral Fellow at the University of Pittsburgh with Prof. Dennis P. Curran. In 1996 he started his independent career at the ETH Zürich. In 2000, he was appointed as Associate Professor of Organic Chemistry at the Philipps-University in Marburg, Germany and in 2004 as Full Professor of Organic Chemistry at the Westfälische Wilhelms-University in Münster, Germany. His current research interests focus on the development of new synthetic methods. In addition, he also works on living radical polymerizations, on the preparation of functional polymers and on the development of methods for the chemical modification of surfaces. |
1. Introduction
Alkenes are important and versatile synthons that engage in diverse organic transformations. Difunctionalization of alkenes by constructing two different vicinal chemical bonds is a valuable synthetic strategy that has gained great interests during the last few decades.1–12 Among the various vicinal difunctionalizations known, alkene carboamination comprising a C–N bond and a C–C bond formation to provide structurally diverse amine derivatives in a straightforward manner is of particular interest. Initial studies on vicinal alkene carboamination focused on aza-Diels–Alder reactions of electron-rich alkenes with conjugated imines.13,14 The resulting cycloadducts can be easily transformed into multi-substituted pyridines, an approach that was successfully applied to the synthesis of natural products such as (−)-mappicine15 and (+)-camptothecin.16 In addition, Pd or Cu catalyzed alkene carboamination with tethered nitrogen nucleophiles has been achieved through aza-Wacker cyclization followed by transition metal mediated or radical C–C coupling to provide substituted pyrrolidines with high stereoselectivity.17–20 It is worth noting that transition metal catalyzed three component alkene carboamination has also been reported.21–25 For instance, Rovis and co-workers disclosed a Rh-catalyzed intermolecular carboamination of alkenes by using a bifunctional reagent21 and Engle et al. used a directing group to conduct Pd-catalyzed three component alkene carboamination.22Along with these pericyclic and metal-catalyzed carboaminations, radical alkene carboamination has also been achieved. For example, carboamination comprising an N-radical cyclization followed by an intermolecular C–C bond formation to provide pyrrolidine or pyrrolidin-2-one scaffolds has been intensively explored using redox catalysis.26–33 As compared to these cyclizing carboaminations, intermolecular vicinal radical alkene carboamination is more challenging and accordingly less well explored. Based on the nature of the initially attacking species, intermolecular alkene radical carboamination can be categorized into three different classes: (1) initial formation of the C–N bond through intermolecular addition of an N-centered radical to an alkene followed by C-radical trapping; (2) initial formation of the C–C bond through addition of a C-radical to an alkene followed by C-amination; (3) single-electron-transfer (SET) oxidation of the alkene to generate a radical cation that gets trapped with an N-nucleophile resulting in a C-radical that eventually engages in a C–C bond forming step (Scheme 1).
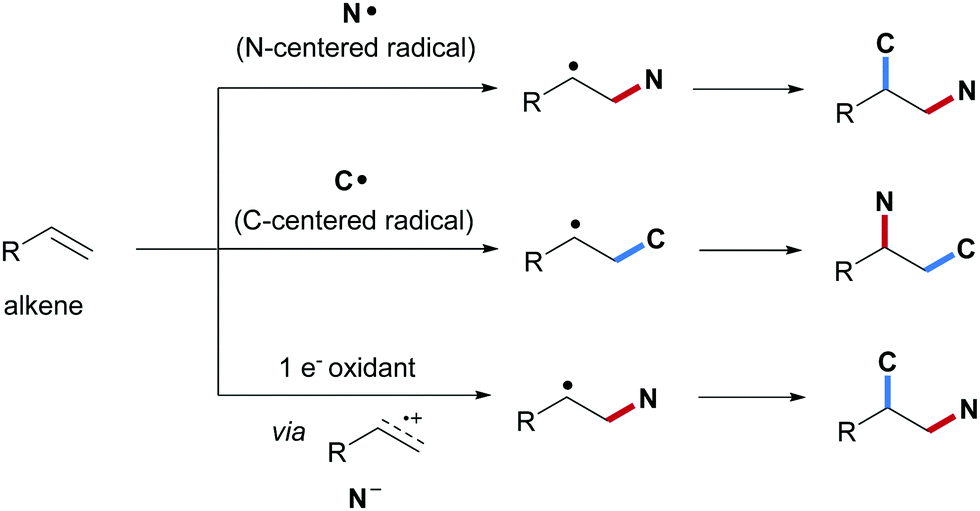 | ||
| Scheme 1 Three different reaction modes to conduct intermolecular radical alkene carboamination. | ||
Considering the first class and an alkene as a C-radical acceptor, the reactivity of two different alkenes has to be controlled and the alkene carboamination selectivity is governed by polar effects.34 Since N-radicals are electrophilic species,35–41 initial C–N bond formation occurs highly chemoselective with an electron-rich alkene such as an aliphatic alkene, a vinyl ether or an enamide to give the corresponding nucleophilic C-radical adduct that gets trapped either via reaction with a π-acceptor or via oxidation and subsequent nucleophilic trapping. Transition metal mediated C–C bond formation is also possible. Looking at the second class, mostly aryl alkenes are used as acceptors for the C-radical and the benzylic adduct radicals generated are readily oxidized to benzylic cations.42 C–N bond formation is finally achieved via nucleophilic trapping of the cation. Alternatively, the C-radical adducts can directly be trapped by a radical amination reagent or C–N bond formation is mediated with a transition metal catalyst. In the third class, an alkene substrate undergoes single-electron oxidation by a photo-excited redox catalyst, producing an alkene radical cation intermediate that can be captured intermolecularly by a nucleophilic amide to give the corresponding C-radical.43 C–C bond formation then proceeds via a radical cyclization reaction to a π-acceptor.
It is obvious that these three different reaction classes nicely complement each other providing an overall broad scope concerning the targeted carboamination products. Nevertheless, pros and cons of the three approaches can clearly be identified. Considering the first reaction class, amines or amides cannot be directly used as the N-radical precursors. In general, N-heteroatom compounds that in most cases are not commercial and have to be prepared ahead are used in such transformations as N-radical precursors. This leads to an increase of the overall costs of the process. Moreover, in many cases, sulfonamidyl radicals have been applied to the carboamidation reaction. The sulfonyl group offers high reactivity to the N-radical but as a disadvantage, it is well known that the sulfonyl group is not an ideal N-protecting group in organic synthesis.
Considering the second reaction class, C–N bond formation is generally achieved with cheap and commercially available “N-donors” such as sulfonyl azides, Me3SiN3, NaN3, nitriles, amines and amides among others. Moreover, also the C-radical precursors are readily available and in many cases at low cost. However, since the two first reaction classes lead to different product regioisomers, the replacement of one strategy by the other to reduce costs is not an option.
The third approach proceeding via alkene radical cation formation is the least general strategy in terms of substrate scope. Only alkenes that are readily oxidized to the corresponding radical cations can be applied. Accordingly, only a few examples can be provided along these lines. With respect to the regiochemistry, the third class provides the same regiosiomer as the first reaction class (see Scheme 1).
2. Carboamination via N-radical addition
Both amidyl and azidyl radicals are electrophilic species which undergo efficient radical addition to electron rich alkenes to generate the corresponding C-radical adducts via C–N bond formation. Subsequent C-radical trapping leads to the targeted 2,1-carboamination products. Generation of the N-radicals has to proceed under mild conditions that are compatible with the subsequent C–C bond formation. Along these lines, various N–X reagents and oxime derivatives have been exploited for amidyl radical generation35–39 and the azidyl radical can be mildly accessed through SET oxidation of commercial TMSN3.44,452.1 Radical 2,1-carboamidation of alkenes
In 2001, Oshima and co-workers reported an elegant radical [3+2] annulation of alkenes with N-allyl-N-chlorotosylamide (1) as N-radical precursor for the preparation of pyrrolidine derivatives 2 (Scheme 2).46 This cascade proceeds through a radical chain process and features a broad scope with respect to the alkene component. Hence, aliphatic alkenes, styrenes, vinyl acetates and even benzofuran are eligible acceptors yielding the corresponding multi-substituted pyrrolidines in high yields albeit with low diastereoselectivity. Chain initiation is achieved with Et3B/O2 to give the amidyl radical 3 which undergoes intermolecular radical addition to the alkene acceptor to provide the C-radical 4. Subsequent 5-exo radical cyclization leads to the primary alkyl radical 5 which undergoes chlorine atom abstraction from 1 to yield the desired pyrrolidine 2 along with the chain carrying amidyl radical 3.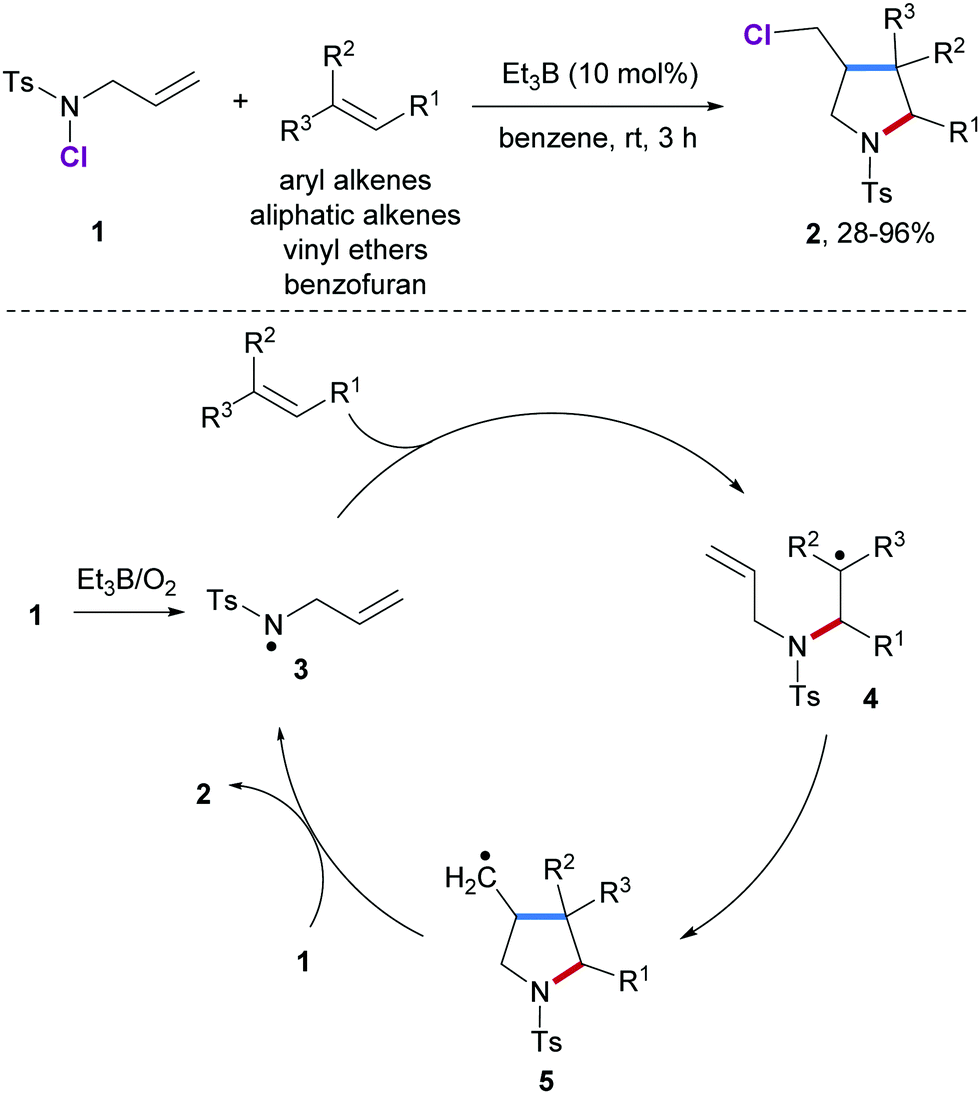 | ||
| Scheme 2 Radical annulation of N-allyl-N-chlorotosylamide with alkenes. | ||
In 2013, Kanai and co-workers described a Cu-catalyzed intermolecular carboamination of aliphatic alkenes by using N-fluorobenzenesulfonimide47 (NFSI) as the amidyl radical precursor (Scheme 3).48 A variety of terminal and internal alkenes were shown react with NFSI smoothly in the presence of a Cu(I)-catalyst in combination with an amide ligand to provide six-membered ring sultams 6 in moderate to satisfactory yields. In the proposed catalytic cycle, the Cu(III)-species 7 existing in equilibrium with Cu(II)-stabilized N-centered radical species 8 is firstly generated through oxidative addition of NFSI to Cu(I). Subsequent N-radical addition to an alkene produces the radical adduct 9 along with a Cu(II)-species. Cyclization onto the arene leads to the cyclohexadienyl radical 10 that gets oxidized by the Cu(II)-species to the stabilized cation 11, thereby regenerating the Cu(I)-catalyst. Deprotonation eventually leads to the sultam 6.
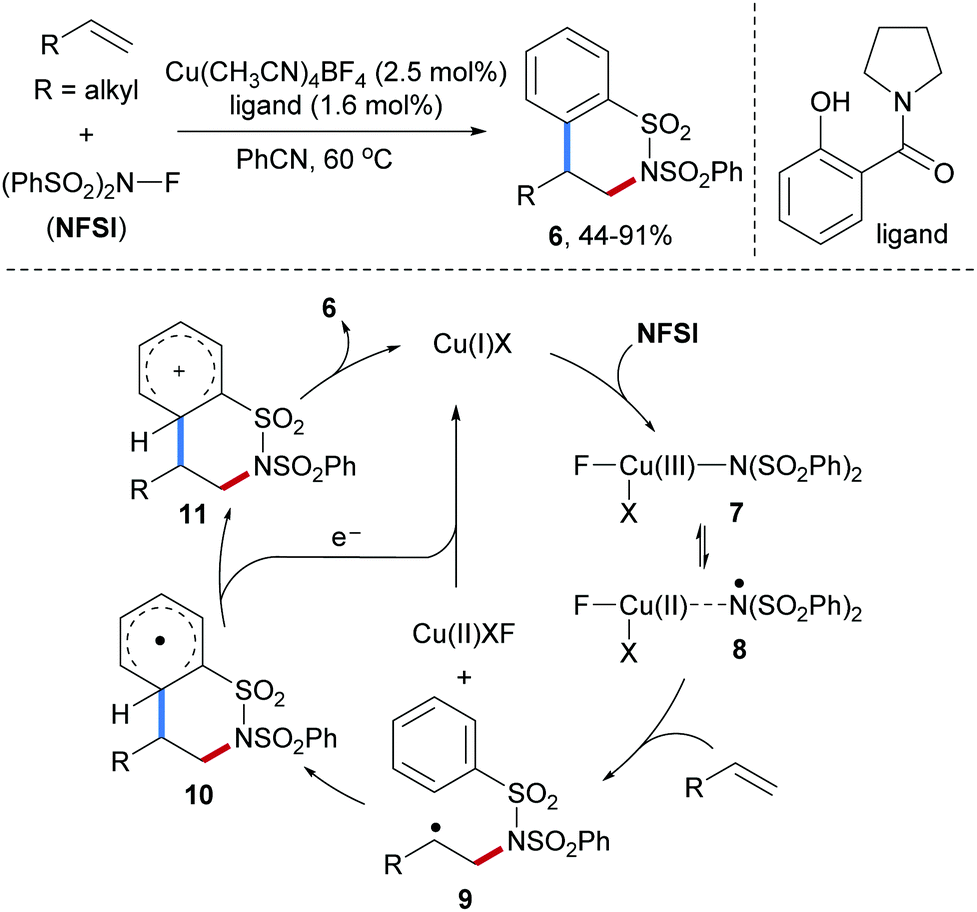 | ||
| Scheme 3 Cu-Catalyzed intermolecular carboamination of alkenes with NFSI. | ||
The Zhang group reported Cu-catalyzed three-component radical aminocyanation of styrenes with NFSI and trimethylsilyl cyanide (TMSCN).49 Styrenes as the amidyl radical acceptors provide the desired β-amino cyanides 12 in high yields, however, other alkenes such as vinyl ethers and aliphatic alkenes afforded the product cyanides in low yields only (Scheme 4). Mechanistically, the disulfonamidyl radical 13 generated by reduction of NFSI with the Cu(I)-catalyst is captured by an alkene to give the benzylic radical 14. Trapping of 14 by the intermediately generated Cu(II)-species provides the Cu(III)-intermediate 15, which undergoes transmetalation with TMSCN to form the Cu(III)-CN complex 16. Reductive elimination finally provides the desired aminocyanation product 12 along with the Cu(I)-catalyst. Radical clock experiments support the radical nature of this process.
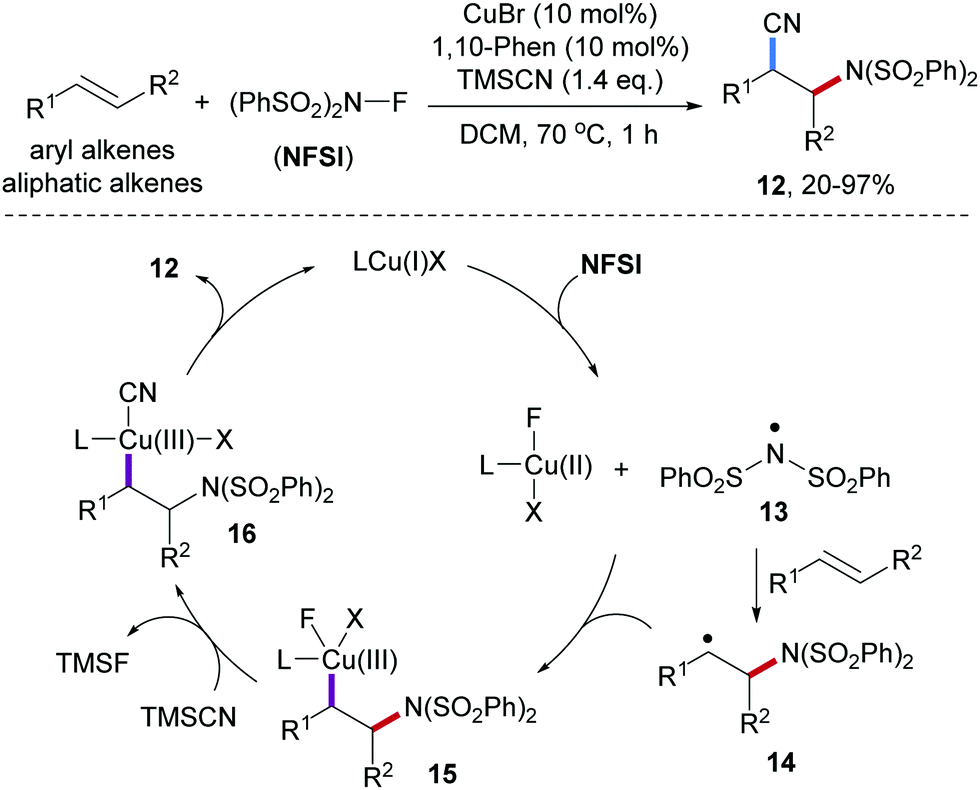 | ||
| Scheme 4 Copper-catalyzed intermolecular aminocyanation of alkenes. | ||
A Cu-catalyzed enantioselective radical aminocyanation of styrenes was achieved by Liu and co-workers in 2017 (Scheme 5).50 The benzylic radical formed through radical addition of the amidyl radical derived from NFSI to a styrene derivative is captured by a chiral Box/Cu(II) cyanide complex, providing the corresponding chiral Cu(III)-complex which undergoes reductive elimination to afford the nitrile 17 in moderate to high yields and good to excellent enantioselectivity.
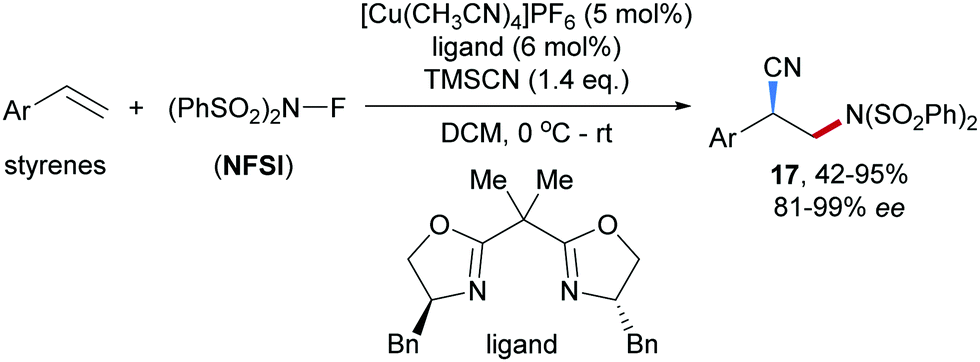 | ||
| Scheme 5 Enantioselective Cu-catalyzed intermolecular aminocyanation of styrenes. | ||
Fluoroamide reagents were also tested as amidyl radical precursors in a Cu-catalyzed aminoarylation of styrenes.51 Systematic variation of the substituents in the N–F reagent revealed that N-fluoro-N-methylbenzenesulfonamide 18, an easily prepared compound, to be the optimal amidyl radical precursor for this transformation (Scheme 6). Alkene aminoarylation was achieved by using a Box/Cu(I)-catalyst to provide the desired sulfonamides 19 in moderate to good yields and satisfactory enantioselectivity. Electronic effects at the arene moiety of the boronic acid and the styrene component are weak and both electron-rich and electron-deficient systems worked well. Mechanistically, single electronic reduction of 18 by Cu(I) generates the amidyl radical 20 along with the formation of Cu(II)–X species which in the presence of an arylboronic acid further reacts to a Cu(II)–Ar-complex. Radical addition of the amidyl radical 20 to a styrene provides the benzylic radical 21, which is trapped by the Cu(II)–Ar species to yield the desired optically enriched 2,2-diarylethylamine 19 thereby regenerating the starting Box/Cu(I)-catalyst. Arylation likely proceeds via the Cu(III)Ar intermediate upon stereoselective reductive elimination. As an application to document the potential of the method, the β-diaryl amine derivative 19a was transformed to the tetraisoquinoline 22.
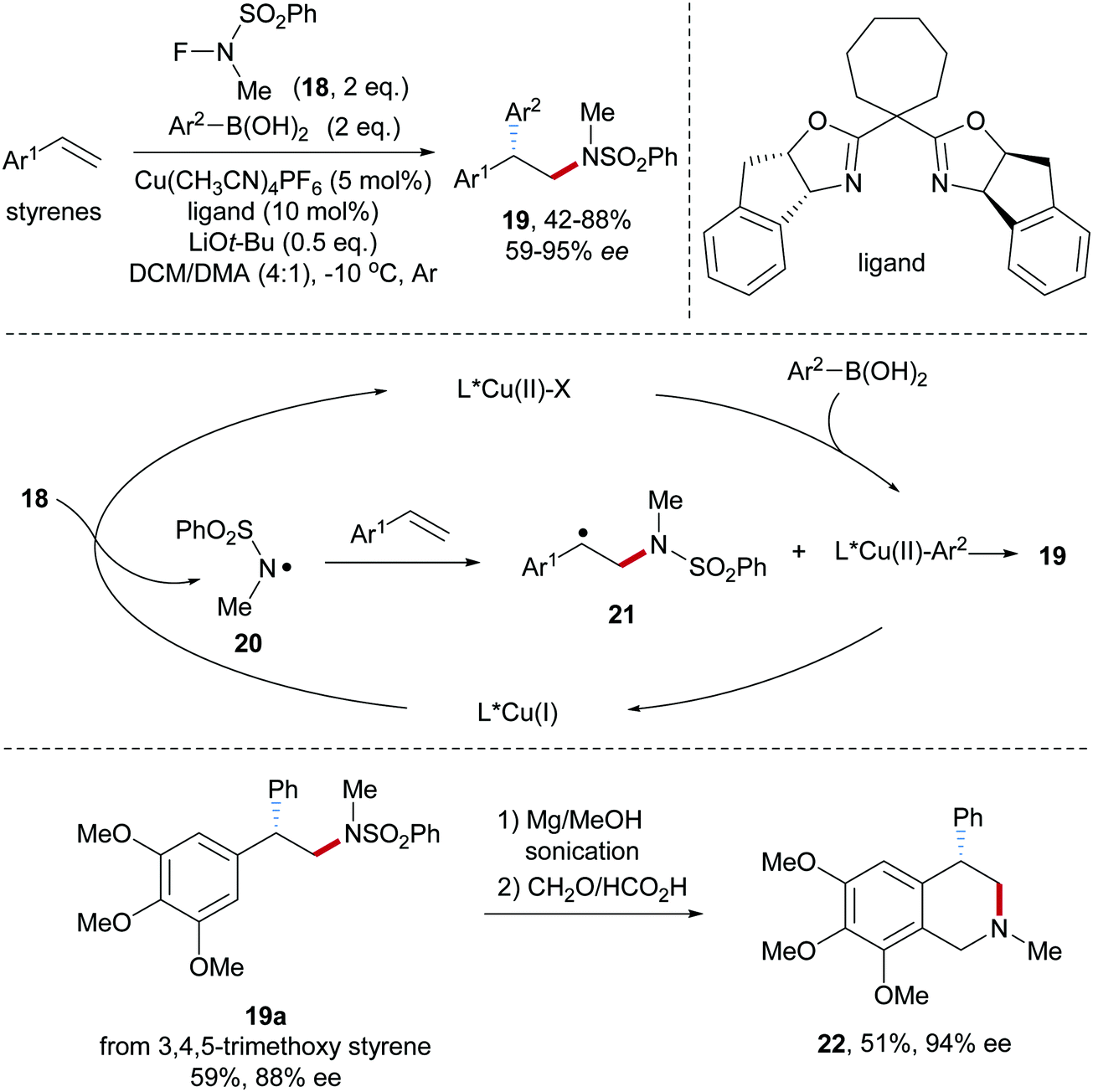 | ||
| Scheme 6 Asymmetric Cu-catalyzed intermolecular aminoarylation of styrenes. | ||
An unprecedented Cu-catalyzed radical 1,2-aminotrifluoromethylation of alkenes by using N–F reagents of type 23 as amidyl radical precursors and (bpy)-Zn(CF3)2 (bpy = 2,2′-bipyridineis) as the CF3 donor was recently achieved by Li and co-workers (Scheme 7).52 Interestingly, the reaction efficiency is significantly improved upon simultaneous addition of Zn(OTf)2 and Zn(OAc)2 to the reaction system. Electrophilic amidyl radicals, generated through single-electron reduction of the easily prepared N–F sulfonamides by Cu(I), were found to react with diverse unsaturated systems including styrenes (24a–d), aliphatic alkenes (24e) and vinyl acetates (24f). The proposed catalytic cycle starts by transmetalation of the CF3-anion from zinc to copper, by which the Cu(I)–CF3 intermediate is generated. This Cu(I)-species reduces the N–F reagent 23 to generate the sulfamidyl radical 25 and a Cu(II)–CF3 intermediate. The C-radical 26 generated through initial radical addition of the electrophilic radical 25 to an alkene is trapped by the Cu(II)–CF3 complex either via direct CF3-group transfer or formation of a Cu(III)-intermediate followed by reductive elimination to eventually provide the 1,2-aminotrifluoromethylation product and a Cu(I)-catalyst.
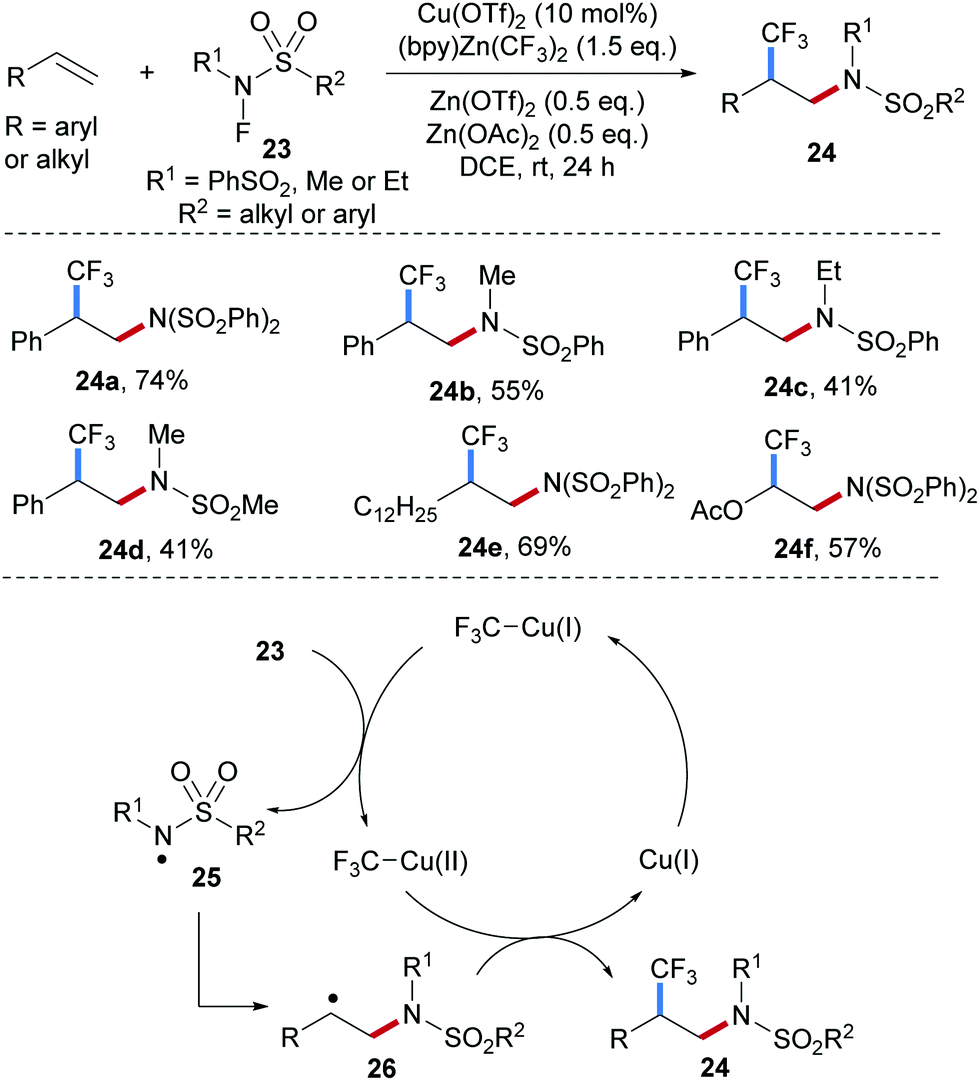 | ||
| Scheme 7 Cu-Catalyzed radical aminotrifluoromethylation of alkenes. | ||
An inevitable drawback of using RSO2N–F reagents as the amidyl radical precursors (especially NFSI) is the harsh conditions that have to be applied for deprotection of the product sulfonamides to access the corresponding free amines, which are generally the final targets. Therefore, the development of novel reagents that allow generating amidyl radicals bearing common N-protecting groups such as Boc, Cbz and Phth is of great importance. Along these lines, several novel and practical amidyl radical precursors have been developed in recent years. In 2018, Feng and co-workers reported the group transfer radical addition of O-vinylhydroxylamine derivatives 27 onto unactivated alkenes to give the carboamination products 28 (Scheme 8).53 This valuable radical alkene carboamination reaction is applicable to electron-rich alkenes as well as to unactivated aliphatic alkenes and works with low initiator loading (0.2 mol%), providing the desired phthaloyl-protected amines 28 in moderate to good yields.
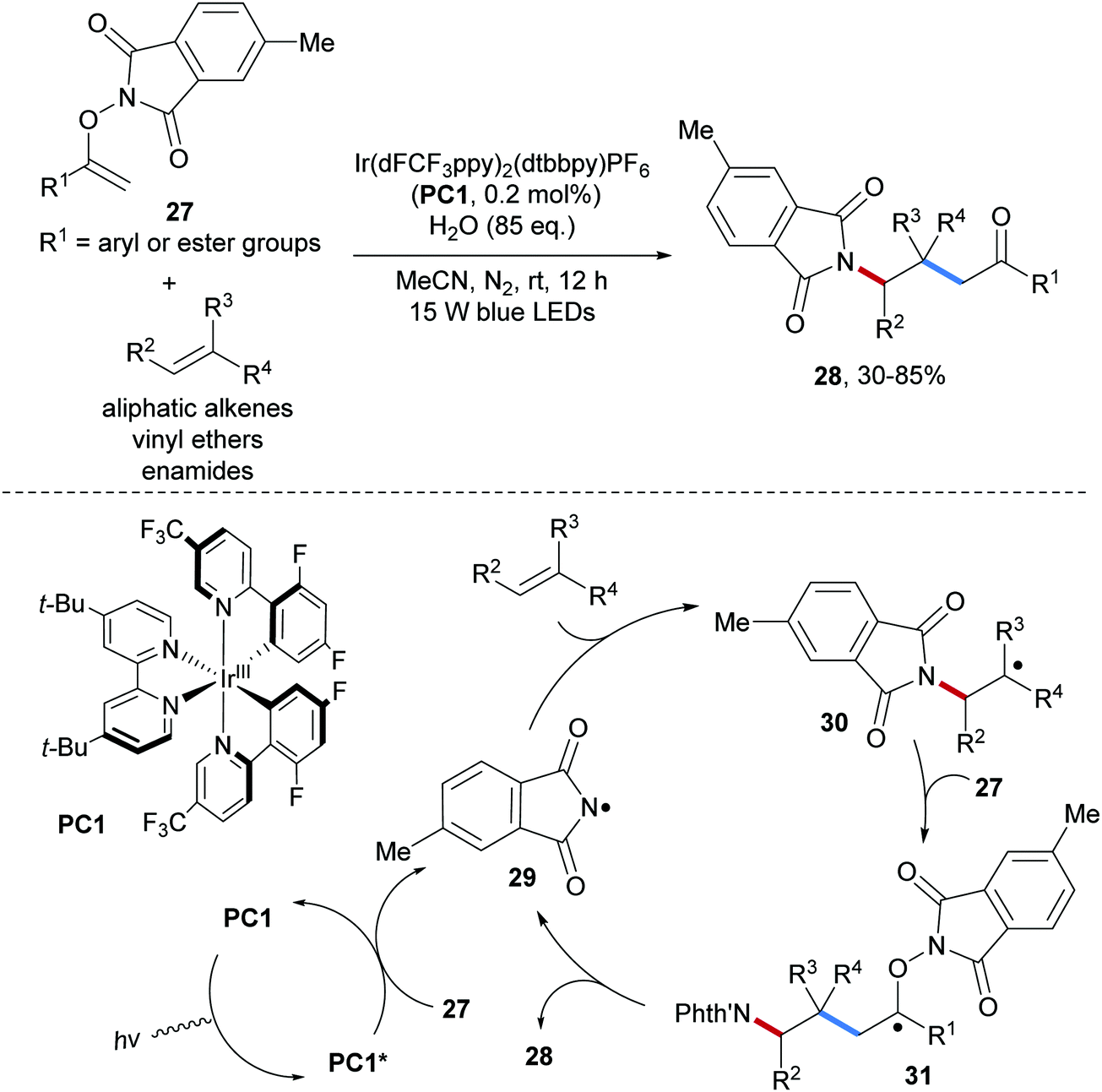 | ||
| Scheme 8 Intermolecular carboamination of unactivated alkenes. | ||
As initiation step, energy transfer from the photo-excited Ir(III)-complex to the O-vinylhydroxylamine derivative 27 leads to homolytic cleavage of the weak N–O bond to give the amidyl radical 29.54 The electrophilic amidyl radical 29 then adds to the alkene generating the alkyl radical 30 which is chemoselectively captured by 27 to generate the tertiary C-radical 31. Fragmentation of 31 through homolytic cleavage of the N–O bond provides the desired alkene carboamination product 28 along with the chain carrying amidyl radical 29. This radical cascade convinces with excellent atom economy and features high chemoselectivity, thus offering a practical approach for the preparation of diverse primary amines.
Radical carboamination of vinyl ethers by photoredox catalysis55–57 was achieved by Yu and co-workers in 2018 (Scheme 9).58 Diverse β-amino alcohol derivatives 33 were obtained in high yields from the O-acyl hydroxylamine derivative 32, benzyl vinyl ether and phenylacetylenes under mild conditions. Upon visible light irradiation, the easily prepared O-acyl hydroxylamine 32 is reduced by a photoexcited Ir(III)-complex to provide the electrophilic amidyl radical 34, which undergoes chemoselective radical addition to the electron-rich benzyl vinyl ether to give the α-oxy-alkyl radical 35. This C-radical in turn reacts with an arylacetylene to the vinyl radical intermediate 36 that further reacts via 1,5-hydrogen atom transfer to give the stabilized C-radical 37, which can be oxidized by Ir(IV) to close the catalytic cycle generating the carbenium ion 38. Trapping with water and half acetal cleavage finally provide 33.
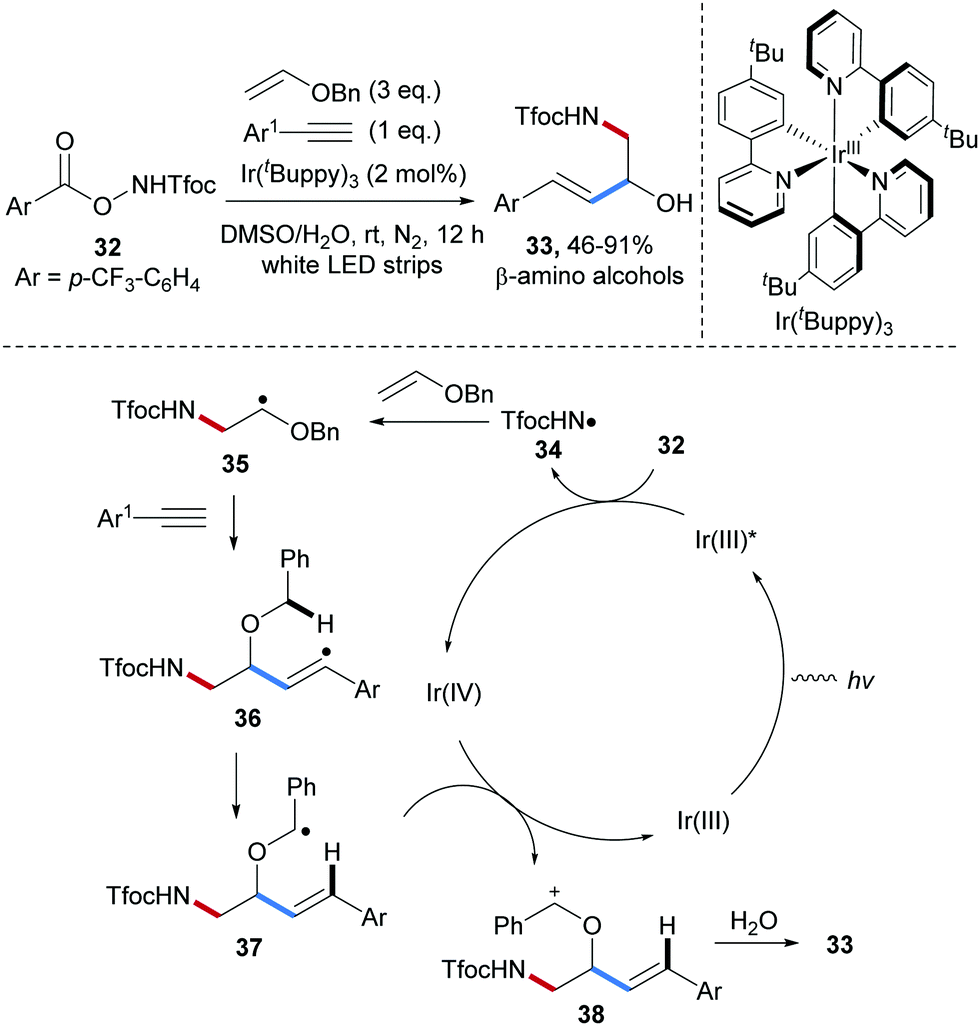 | ||
| Scheme 9 Three-component radical carboamination of enols. | ||
Three-component radical carboamination of electron-rich alkenes with O-acyl hydroxylamines and styrenes was accomplished by the same group (Scheme 10).59 Electrophilic amidyl radicals, reductively generated through single electron reduction of O-acyl hydroxylamines by a photo-excited Ir(III)-complex, chemoselectively react with electron-rich enolethers to give the adduct C-radicals that are then trapped by styrenes with high chemoselectivity. The benzylic radicals thus formed are eventually oxidized to the corresponding cations and trapping with DMSO in a Kornblum oxidation finally provides the targeted alkene carboamination products with exclusive chemoselectivity. Diverse β-amino alcohol derivatives 39 were obtained in moderate to high yields by using vinyl ethers as amidyl radical acceptors. Moreover, by using enamides bearing commonly used N-protecting groups in place of the enol ethers, the protected 1,2-diamines 40 were formed in moderate to good yields.
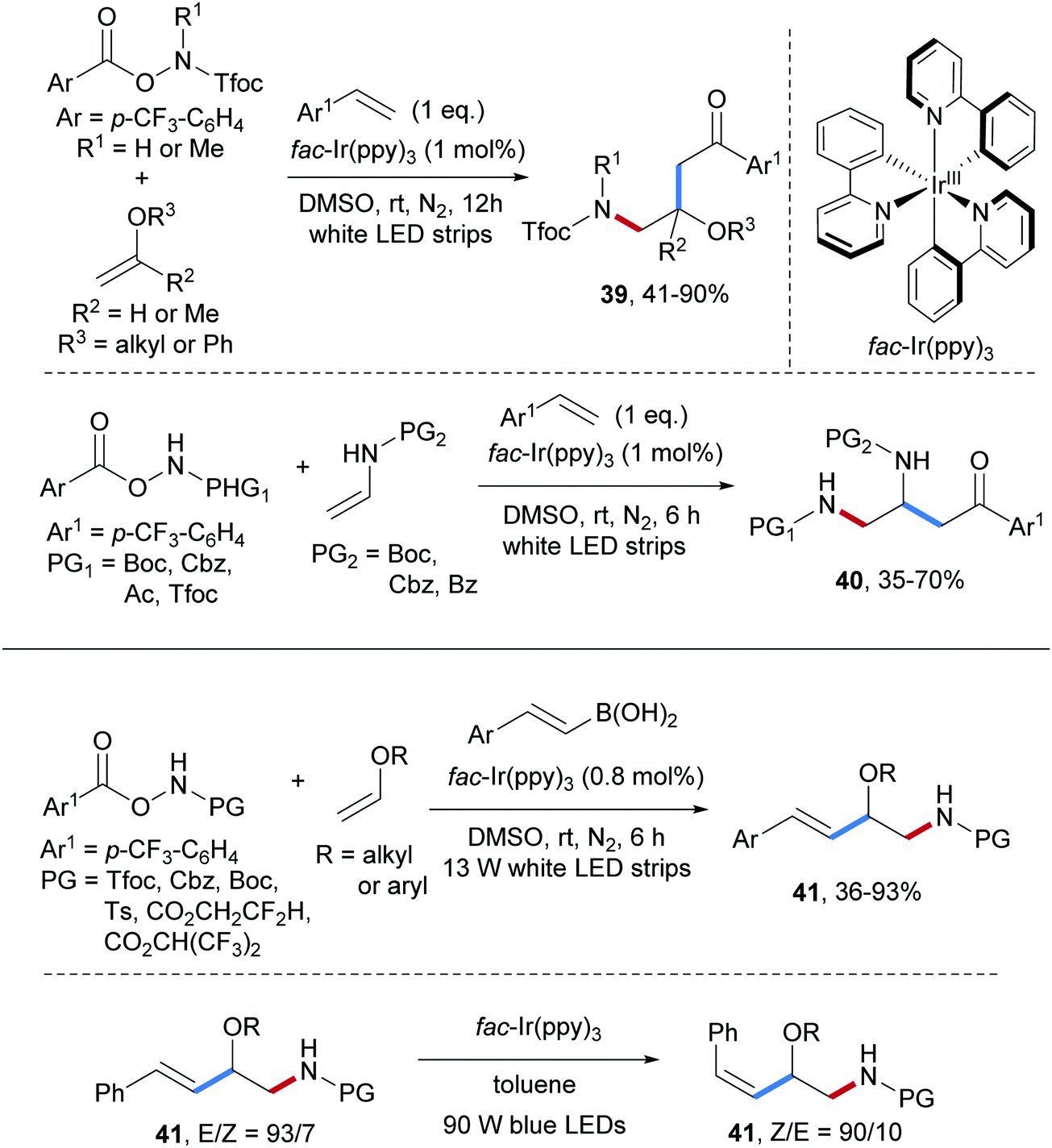 | ||
| Scheme 10 Three-component radical carboamination of electron-rich alkenes with styrenes or alkenyl boronic acids. | ||
Recently, the photoredox catalyzed three-component radical carboamination of enols with O-acyl hydroxylamines and styryl boronic acids was reported by the Yu group (see Scheme 10).60 Various N-protecting groups in the O-acyl hydroxylamine component including Tfoc, Cbz, Boc, Ts are tolerated and the amidyl radicals, reductively generated by a photo-excited Ir(III), undergo efficient radical addition to vinyl ethers to give the corresponding adduct C-radicals which further react with alkenyl boronic acids to provide fully protected β-amino alcohol derivatives 41 in 36–93% yields with high E-selectivity. Notably, isomerization to the thermodynamically less stable Z-isomer can be achieved upon blue light irradiation in the presence of fac-Ir(ppy)3 as an energy transfer catalyst.
Intermolecular radical carboamination of unactivated alkenes is of great importance but still not well established. In 2018, our group developed a transition-metal free process for aminoalkynylation of various unactivated alkenes (Scheme 11).61 In this transformation, the α-amido-oxy acids 42 were used as amidyl radical precursors that can be oxidized by an excited photocatalyst.62,63 By using ethynylbenziodoxolone (EBX) reagents as the alkyl radical acceptors, various β-amino alkynes could be obtained from the corresponding alkenes. The scope with respect to the amidyl radical acceptor was amazingly broad and alkenes bearing different functional groups and substitution patterns including mono-, 1,1-di-, and 1,2,2-tri-substituted alkenes, cyclic alkenes, vinyl ethers and enamides engaged in this valuable transformation (43a–g). Moreover, upon varying the I(III)-reagent, 1,2-amidocyanation (44) and 1,2-amido-alkenylation (45) were accomplished by using the corresponding EBX reagents, albeit with lower yields.
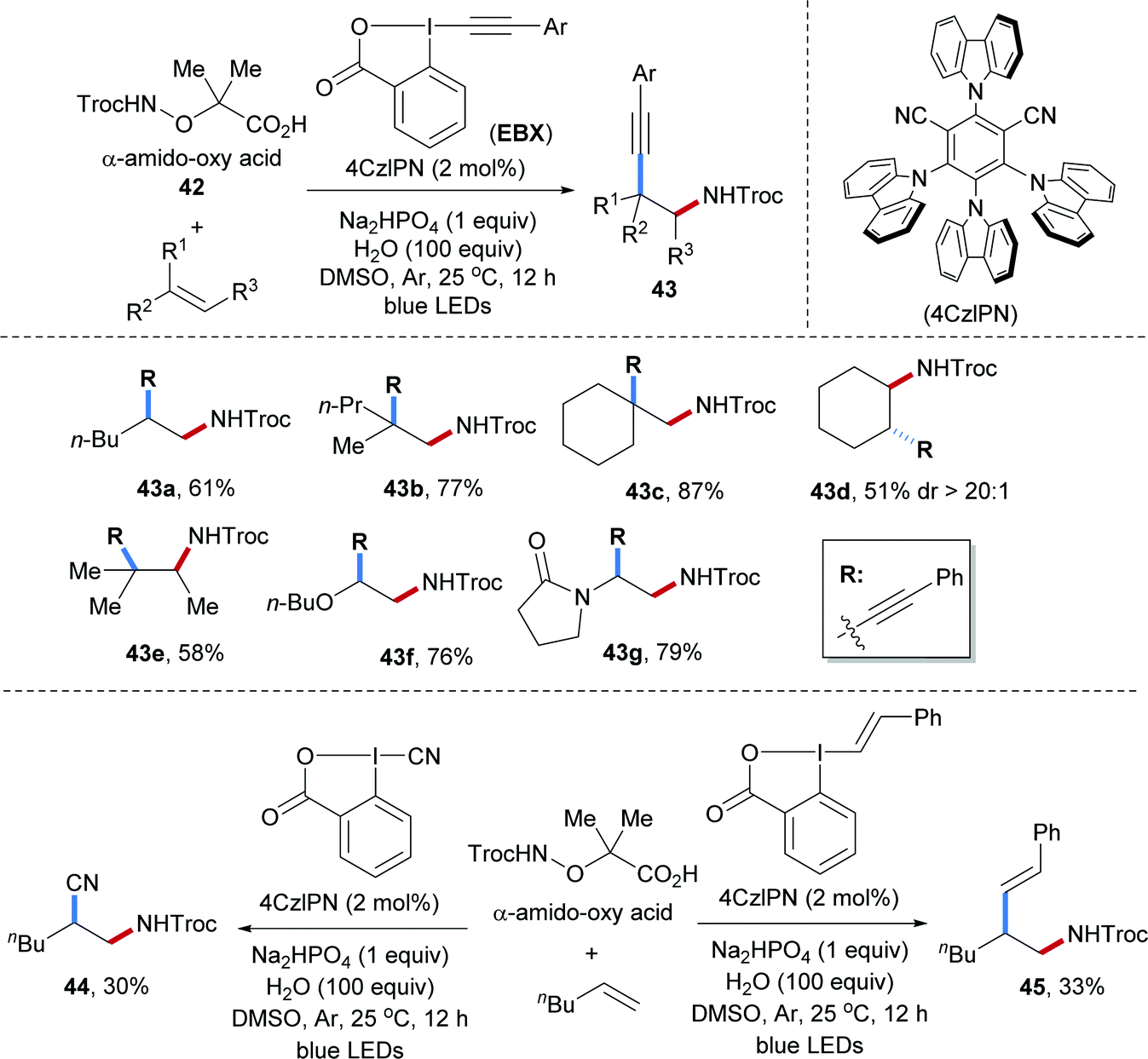 | ||
| Scheme 11 Three-component radical aminoalkynylation of unactivated alkenes. | ||
The proposed catalytic cycle of the radical alkene 1,2-aminoalkynylation commences with photo-excitation of 4CzlPN upon visible-light irradiation to generate the excited 4CzlPN*, which oxidizes carboxylate 46, formed by deprotonation of α-amido-oxy acid, to generate the carboxyl radical 47 along with the radical anion of the photoredox catalyst (4CzlP−˙) (Scheme 12). Sequential fragmentation of 47 with the extrusion of CO2 and acetone generates the amidyl radical 48, which then adds to the carbon–carbon double bond of the alkene to provide the adduct radical 49. EBX reagent traps the C-radical 49 to give alkene amidoalkynylation product 43 with concomitant formation of iodanyl radical 50. Transient radical 50 is then reduced by 4CzlP−˙ to give ortho-iodobenzoate and 4CzlPN, thereby closing the catalytic cycle.
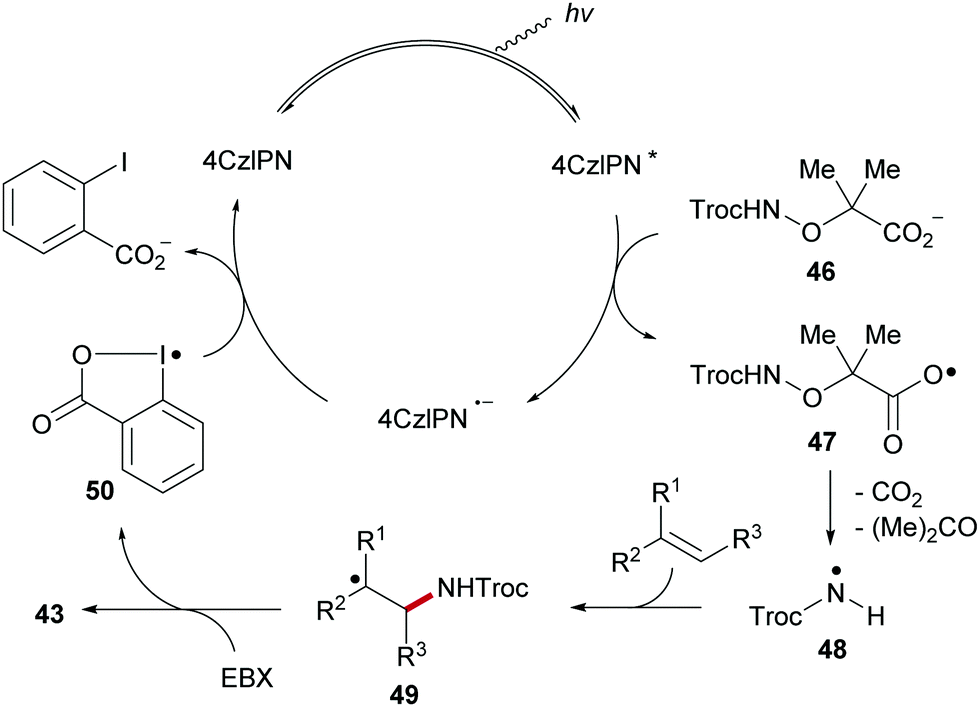 | ||
| Scheme 12 Proposed mechanism for alkene aminoalkynylation. | ||
Three-component radical carboamination including amidyl radicals, unactivated alkenes and Michael acceptors was established very recently by us (Scheme 13).64 Commonly used N-protecting groups installed at the starting α-amido-oxy acids were tolerated and the corresponding carboamination products were obtained in satisfactory yields (51a–h). N-Methyl α-amido-oxy acids bearing electron deficient protecting groups (Troc or Tfoc) also worked well as N-radical precursors in this transformation (51i, 51j). This radical cascade features broad scope with respect to the alkene and Michael acceptors. For example, mono-, di-, tri- and tetra-substituted aliphatic alkenes and electron rich alkenes including vinyl ethers, vinylsilanes and enamides engaged in the cascade to provide the corresponding carboamination products in good to satisfactory yields (51k–r). The valuable amino-C-glycoside 51s could be obtained with high diastereoselectivity (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) by using a glycal as the amidyl radical acceptor. Diverse Michael acceptors including acrylates, vinyl ketones, acrylamides, vinyl phosphates, methacrolein, acrylonitrile and even electron deficient styrenes were found to be eligible coupling partners for this three-component radical cascade (51t–51ad).
:1) by using a glycal as the amidyl radical acceptor. Diverse Michael acceptors including acrylates, vinyl ketones, acrylamides, vinyl phosphates, methacrolein, acrylonitrile and even electron deficient styrenes were found to be eligible coupling partners for this three-component radical cascade (51t–51ad).
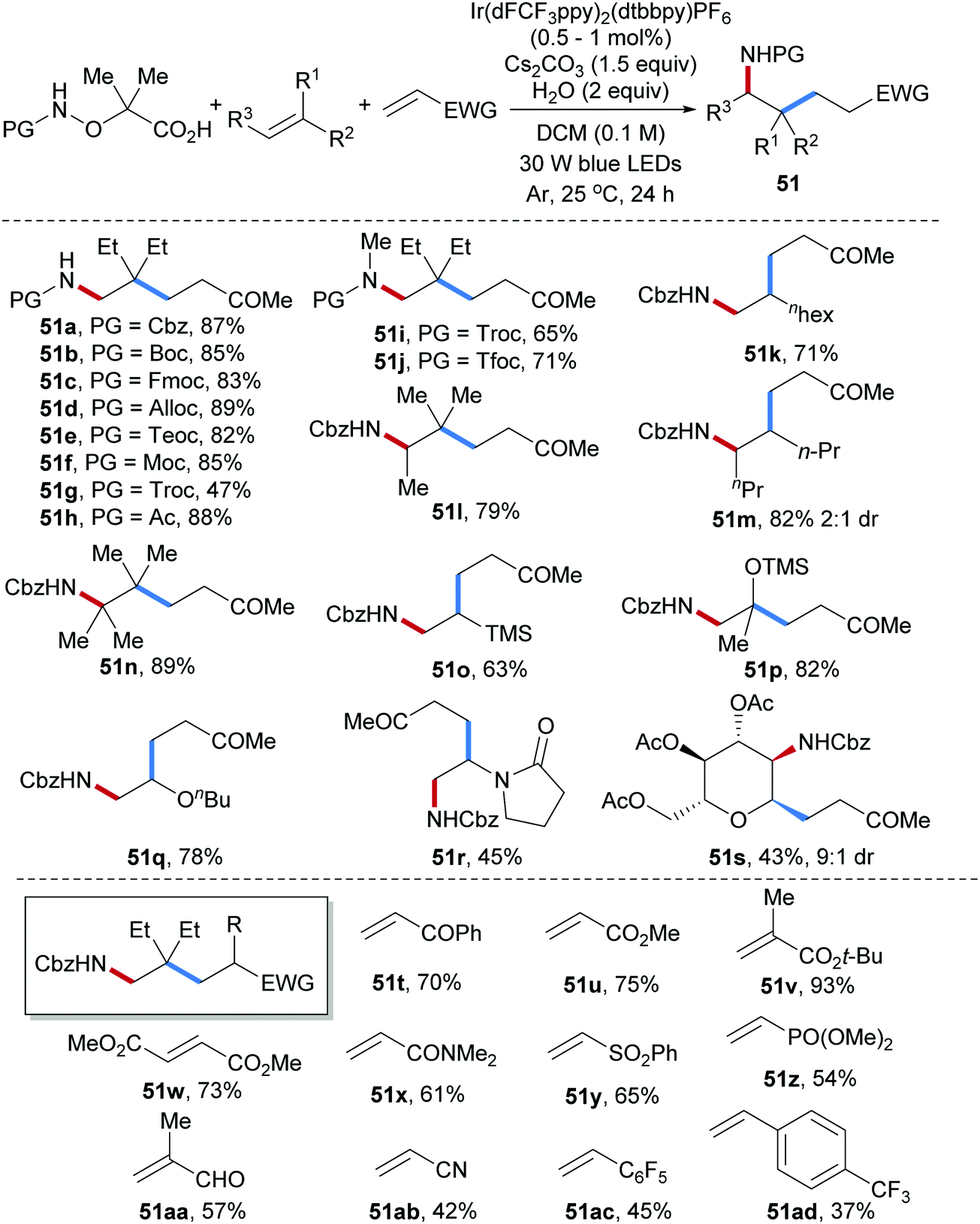 | ||
| Scheme 13 Three-component radical Giese reaction for alkene carboamination. | ||
The catalytic cycle starts by photo-excitation of Ir(III) to generate the excited Ir(III)*-complex which oxidizes carboxylate 52 to the carboxyl radical 53 (Scheme 14). Sequential fragmentation with the extrusion of CO2 and acetone generates the electrophilic amidyl radical 54. Governed by polar effects, the amidyl radical chemoselectively adds to the CC double bond of the electron-richer alkene to provide the adduct radical 55. Michael acceptor then traps the thus generated nucleophilic C-radical 55 to give 56, which is reduced by the Ir(II)-complex to afford the enolate 57, thereby closing the catalytic cycle. Protonation eventually gives the carboamination product 51.
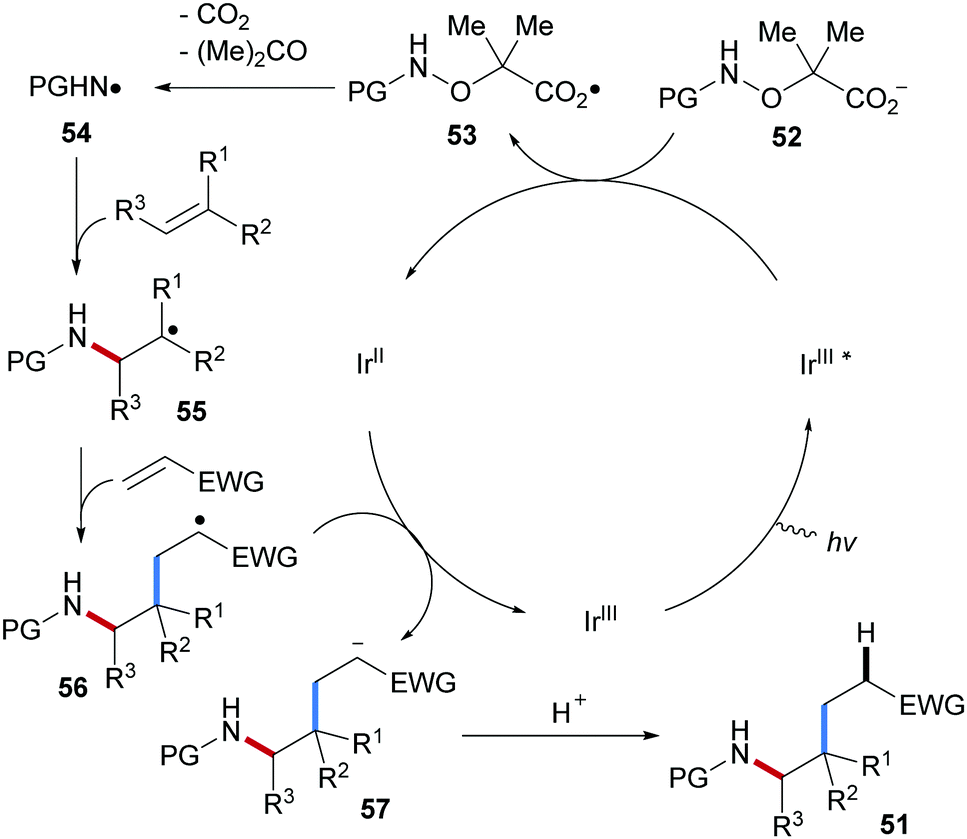 | ||
| Scheme 14 Proposed mechanism for three-component Giese reaction. | ||
N-Aminopyridinium salts 58, that were previously used as amidyl radical precursors for radical arene C–H amidation, were successfully applied as bifunctional reagents to the 1,2-aminopyridylation of electron-rich alkenes (Scheme 15).65 This radical alkene carboamination protocol features high atom economy and proceeds under mild conditions with the use of an organic dye as a photo-sensitizer to initiate the process. Various electron-rich alkenes including vinyl ethers and enamides were found to be eligible acceptors for this carboamination and diverse functional groups at the pyridine ring are tolerated. The resulting sulfonyl protected secondary amines 59 were obtained in good to satisfactory yields (59a–h).
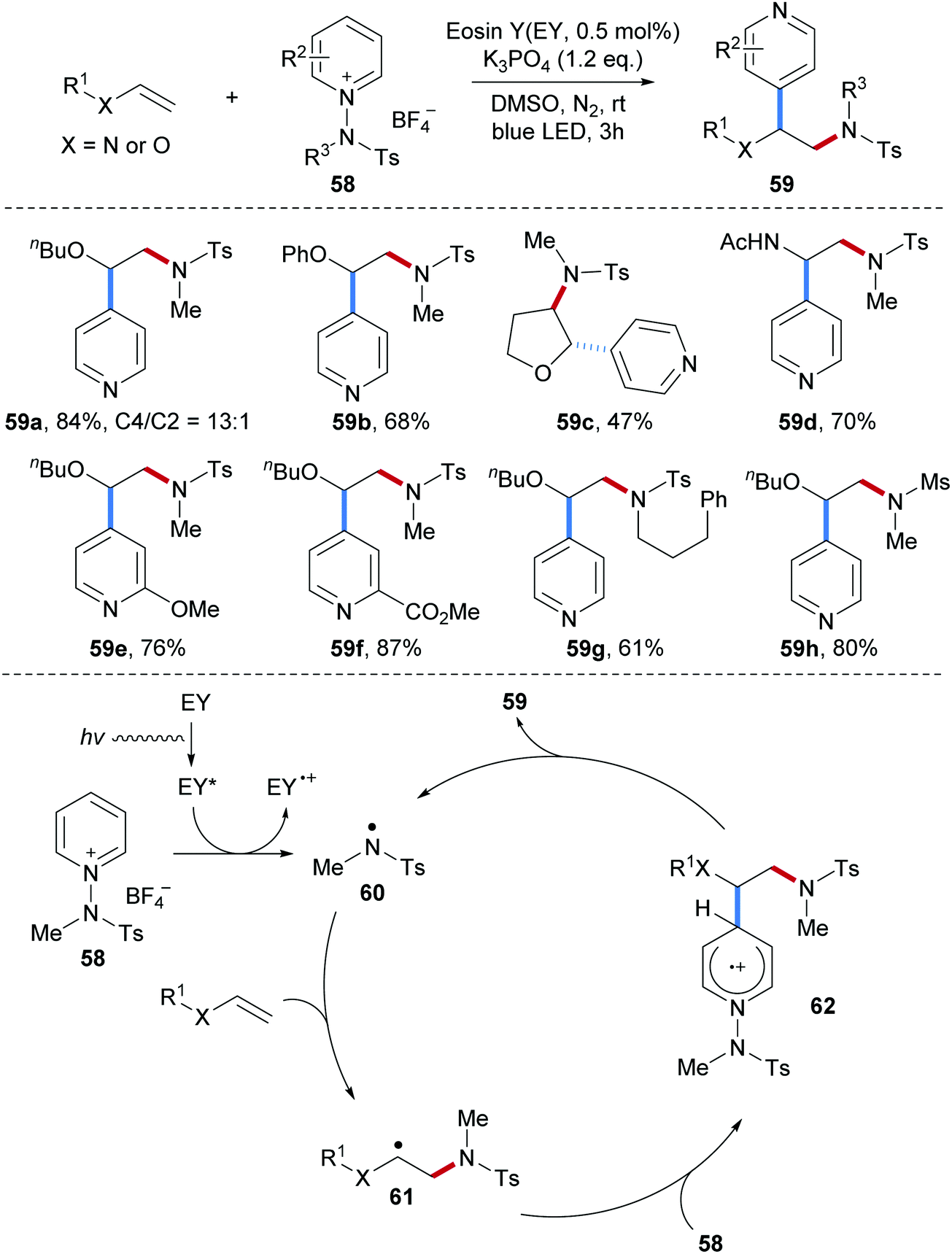 | ||
| Scheme 15 Radical alkene aminopyridylation using N-aminopyridium salts as bifunctional reagents. | ||
The authors proposed a radical chain reaction. Initiation proceeds via single-electron reduction of the N-aminopyridinium salt 58 by a photo-excited organic dye to provide an electrophilic sulfonamidyl radical 60, which is captured by an electron-rich alkene to generate the alkyl radical intermediate 61. Addition of the C-radical 61 to the N-aminopyridinium salt 58 occurs with complete para-selectivity to deliver the arene radical cation 62. Deprotonation and N–N-bond cleavage finally afford the alkene aminopyridylation product 59 along with the chain carrying sulfonamidyl radical 60.
Recently, our group achieved an asymmetric radical aminoarylation of vinyl acetamides by merging chiral Brønsted acid catalysis and photoredox catalysis (Scheme 16).66 Four protected 1,2-diamine derivatives 64 could be obtained in high yields and enantioselectivities through a three-component Minisci reaction using the O-acyl hydroxylamine 63, vinyl acetamide and quinolines as components. In the proposed mechanism, photo excitation of Ir(III) provides Ir(III)* which can be oxidatively quenched to Ir(IV) by the O-acyl hydroxylamine 63 to generate the corresponding amidyl radical by reductive N–O bond cleavage. Amidyl radical addition to vinyl acetamide leads to the adduct radical 65 which undergoes an acid promoted Minisci type reaction to give the aminoarylation product 64. It is assumed that hydrogen bonding as suggested in structure 66 helps orienting the reaction partners to give the addition intermediate 67 in a reversible reaction. The stereodetermining step is likely deprotonation of 67 to give 68 and oxidation with Ir(IV) finally provides the isolated quinoline 64.
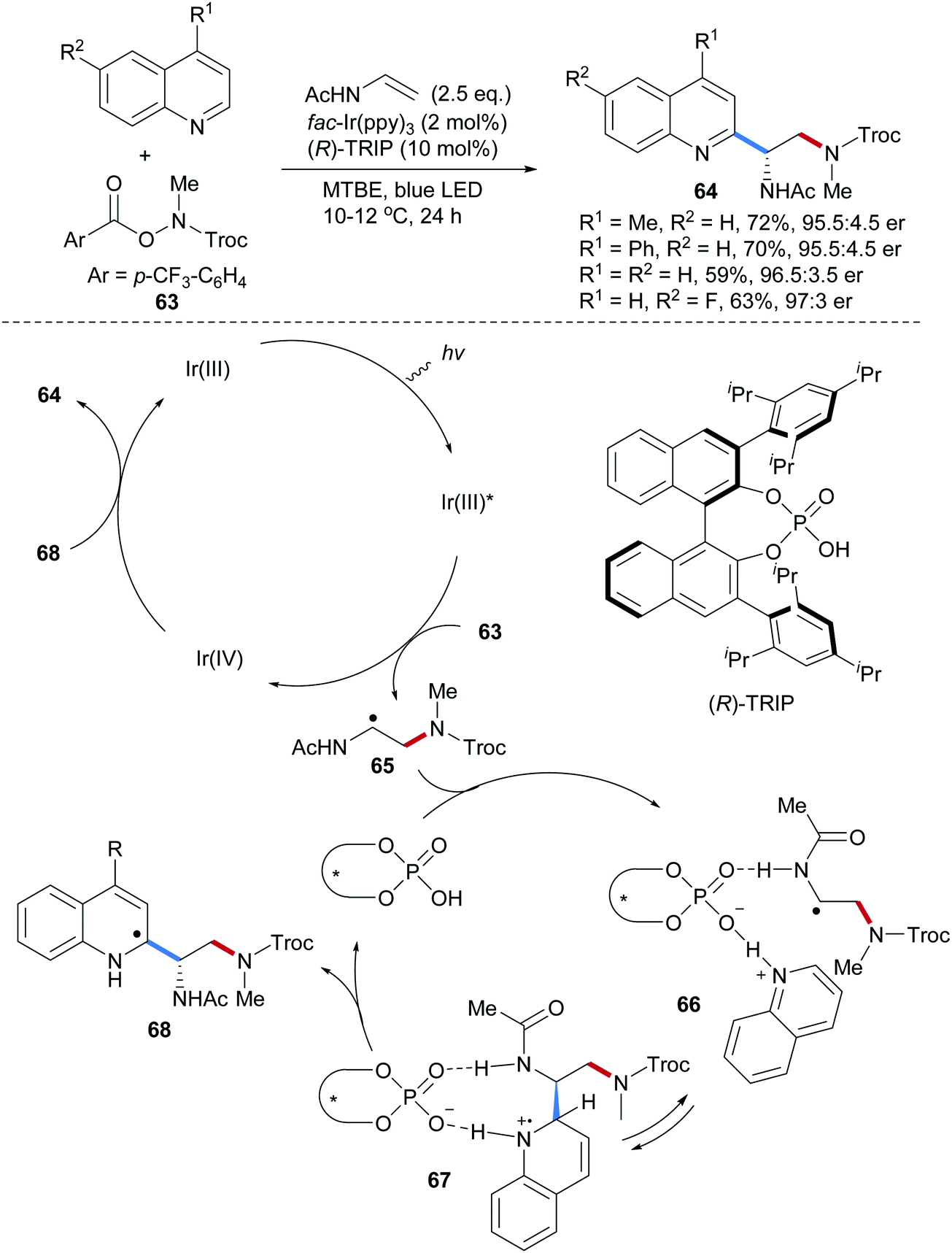 | ||
| Scheme 16 Asymmetric radical aminoarylation of enamides. | ||
2.2 2,1-Carboazidation of alkenes
Wang and co-workers developed a Cu-catalyzed azidocyanation of aryl alkenes with TMSCN and TMSN3 in the presence of an I(III)-reagent as an oxidant (Scheme 17).67 Firstly, the reaction of TMSN3 with PhI(OAc)2 produces PhI(N3)2 which through single-electron reduction by Cu(I) delivers an azidyl radical. Addition of the electrophilic azidyl radical to the aryl alkene provides the C-radical 70, which is oxidized by Cu(II) to give the benzylic cation 71, thereby regenerating the starting Cu(I)-complex. Nucleophilic trapping of the cation 71 with TMSCN finally affords the azidocyanation product 69 in moderate to good yield.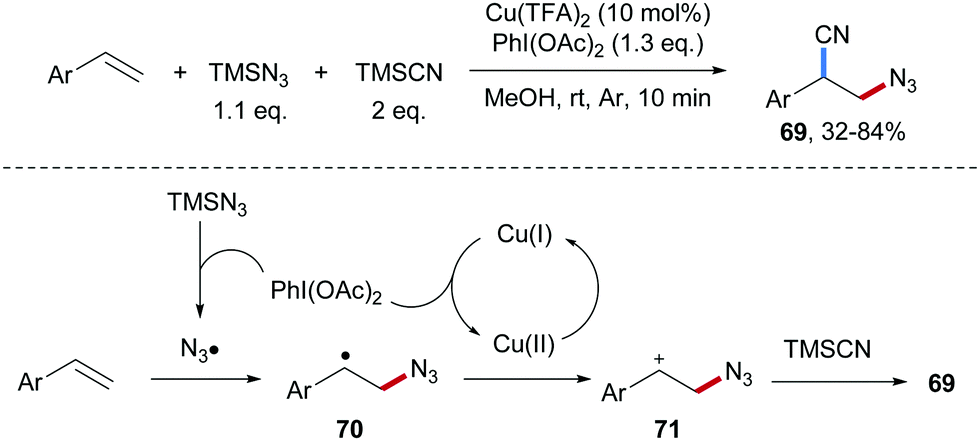 | ||
| Scheme 17 Cu-Catalyzed three-component radical azidocyanation of alkenes. | ||
The asymmetric azidocyanation of aryl alkenes through a Cu-catalyzed radical cascade was achieved by the Liu group in 2017 (Scheme 18).50 A chiral box-type ligand was used in combination with CuCl2 and various enantiomerically enriched β-azido alkylnitriles 72 were obtained with good to excellent enantioselectivity. These products are highly valuable as documented by follow-up chemistry on azide 72a to the thiazole derivative 73, bocylated β-amino alkylnitrile 74 and triazole 75.
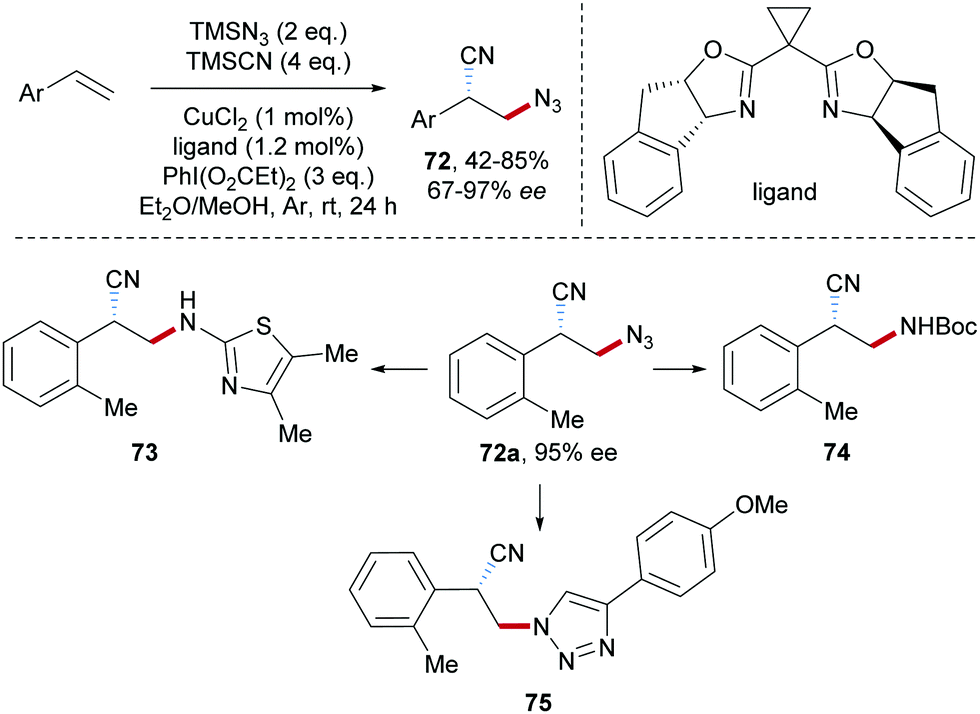 | ||
| Scheme 18 Enantioselective azidocyanation of alkenes. | ||
Amidoheteroarylation of aliphatic alkenes through a radical cascade process using TMSCN, aliphatic alkenes and heteroarenes as reaction components was reported by Liu and co-workers (Scheme 19).68 PhI(N3)277 is generated in situ from PhI(OAc)2 and TMSN3. Upon heating, homolytic cleavage of the I–N3 bond in 77 occurs generating an azidyl radical which adds to the alkene to give the C-radical 78. Minisci type C–H alkylation proceeds through radical addition of the alkyl radical 78 to a TFA activated heteroarene to give the radical cation intermediate 79, which undergoes single-electron oxidation by PhI(OAc)2 to eventually afford the desired azidoarylation product 76. Various heteroarenes including quinolines (76a and 76b), isoquinolines (76c), pyridines (76d), phenanthridines (76e), pyrazines (76f), quinoxalines (76g), quinazolines (76h), pyrimidines (76i), pyrido[3,4-b]pyrazines (76j), pyrido[2,3-b]pyrazines (76k) and phthalazines (76l) engaged in this transformation to afford the corresponding alkene azidoheteroarylation products in good yields.
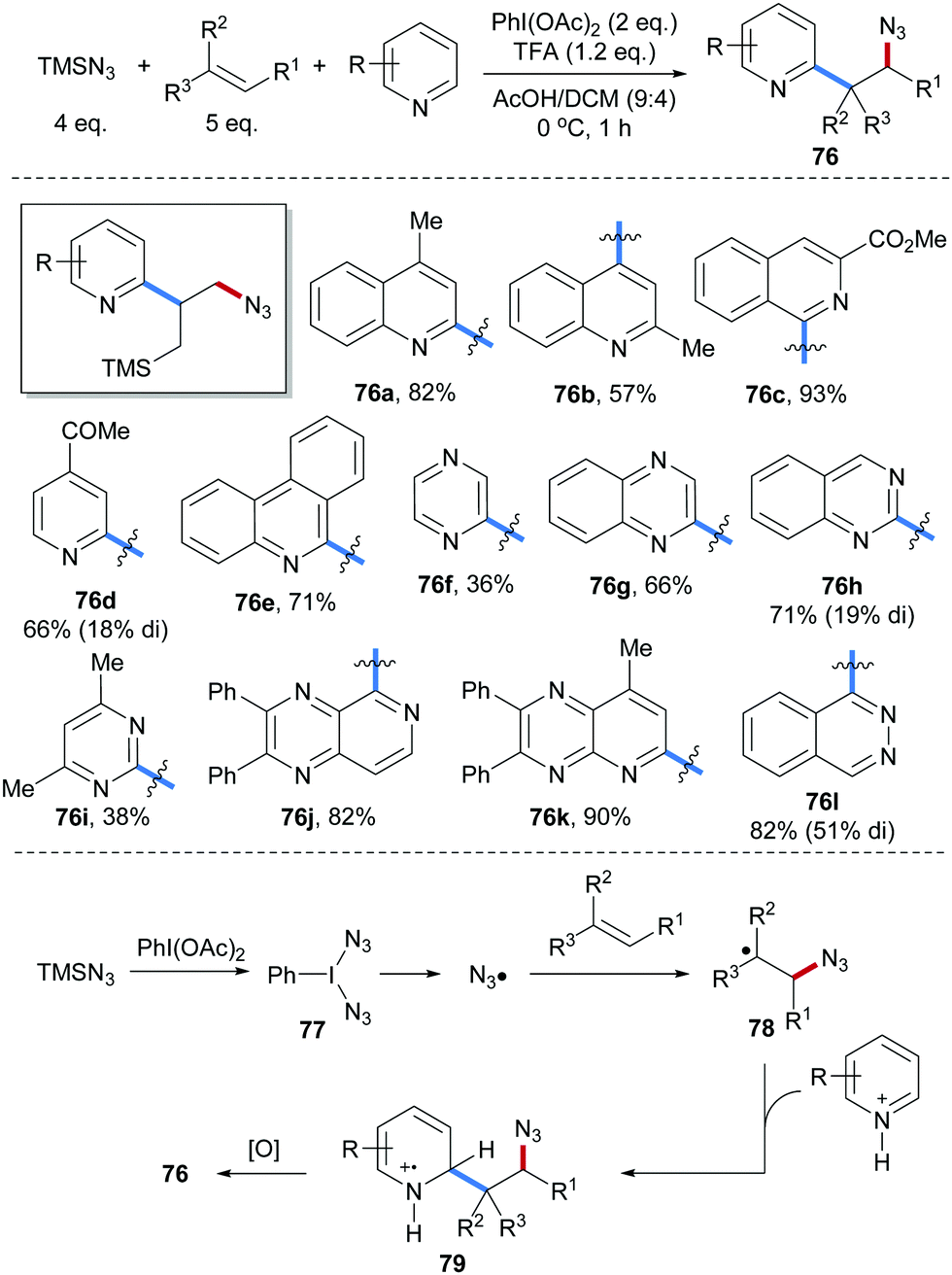 | ||
| Scheme 19 Three-component radical azidoheteroarylation of alkenes. | ||
Lu and co-workers reported a photoredox catalyzed three-component carboazidation of alkenes with TMSN3 and acrylonitrile (Scheme 20).69 Diverse δ-azido nitriles 80 could be efficiently accessed from styrenes and aliphatic alkenes. It was suggested that single-electron oxidation of TMSN3 by the photo-excited Ir(III) complex leads to the electrophilic azidyl radical which adds to the alkene to give the corresponding adduct C-radical 81. Steered by polar effects, 81 reacts chemoselectively with acrylonitrile to 82. Single electron reduction of 82 by Ir(II) closes the catalytic cycle thereby generating the anion 83 that gets finally protonated to afford the isolated carboazidation product 80.
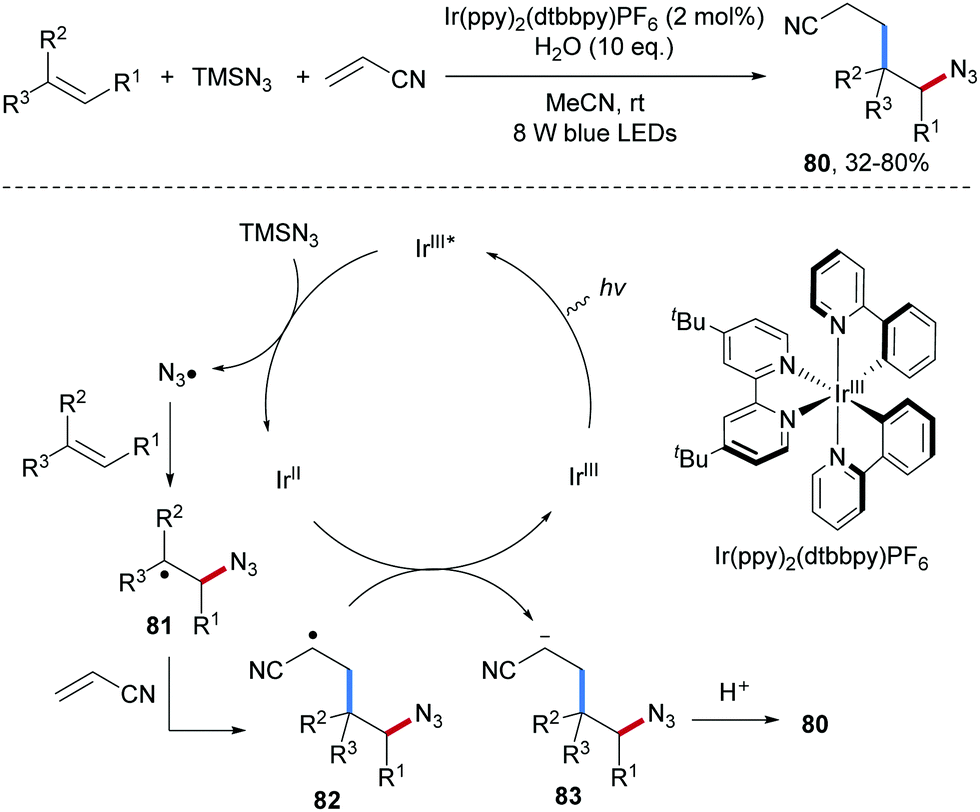 | ||
| Scheme 20 Three-component carboazidation of unactivated alkenes with TMSN3 and acrylonitrile. | ||
3. Carboamination via C-radical addition
An early study on an intermolecular formal carboamination proceeding via alkyl radical addition to an alkene followed by C–N bond formation was reported by the Renaud group in 2002.70 α-Bromo acetic acid ethyl ester was used as a C-radical precursor, di-tert-butylhypodinitrite as an initiator, (Bu3Sn)2 as a chain carrier and phenylsulfonyl azide as a radical azidation reagent. Governed by the polar effect, the electrophilic α-ester radical 85 adds to an unactivated alkene to form a nucleophilic alkyl radical 86. Azidyl group transfer from phenylsulfonyl azide to the alkyl radical 86 provides the carboazidation product 84 along with the phenylsulfonyl radical (Scheme 21). Reaction of the latter radical with (Bu3Sn)2 leads to the tributyl tin radical which abstracts the bromine atom from the α-bromo-ester to give the corresponding α-ester radical 85, sustaining the chain. Along with the bromo ester, the corresponding iodide (see 87), xanthogenate and phenylselenide were successfully used as C-radical precursors in this sequence. Notably, chain initiation can also be achieved with Et3B/O2 in place of di-tert-butylhypodinitrite.71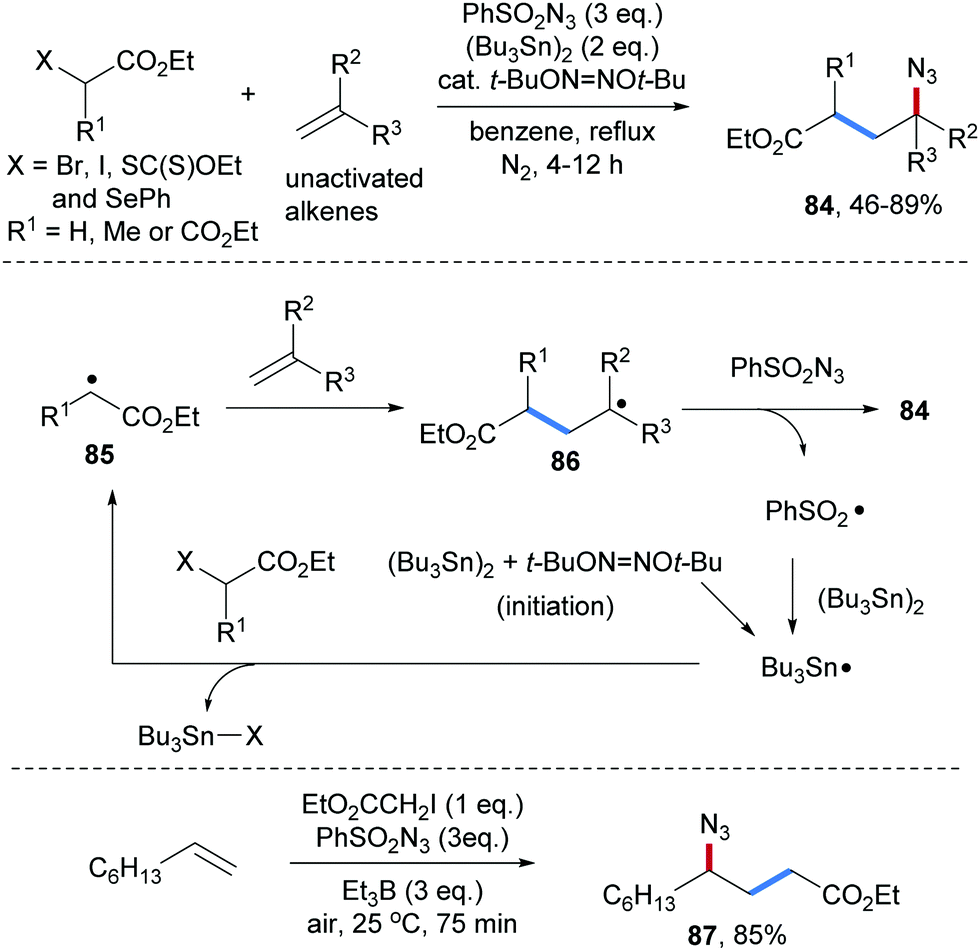 | ||
| Scheme 21 Three-component radical carboazidation of alkenes through a chain process. | ||
Several alkaloids including (±)-cylindricine C,72 hyacinthacine A1,73 (−)-indolizidine 167B74 and (±)-lepadiformine75 were synthesized by using this three-component radical carboazidation as the key step (Scheme 22).
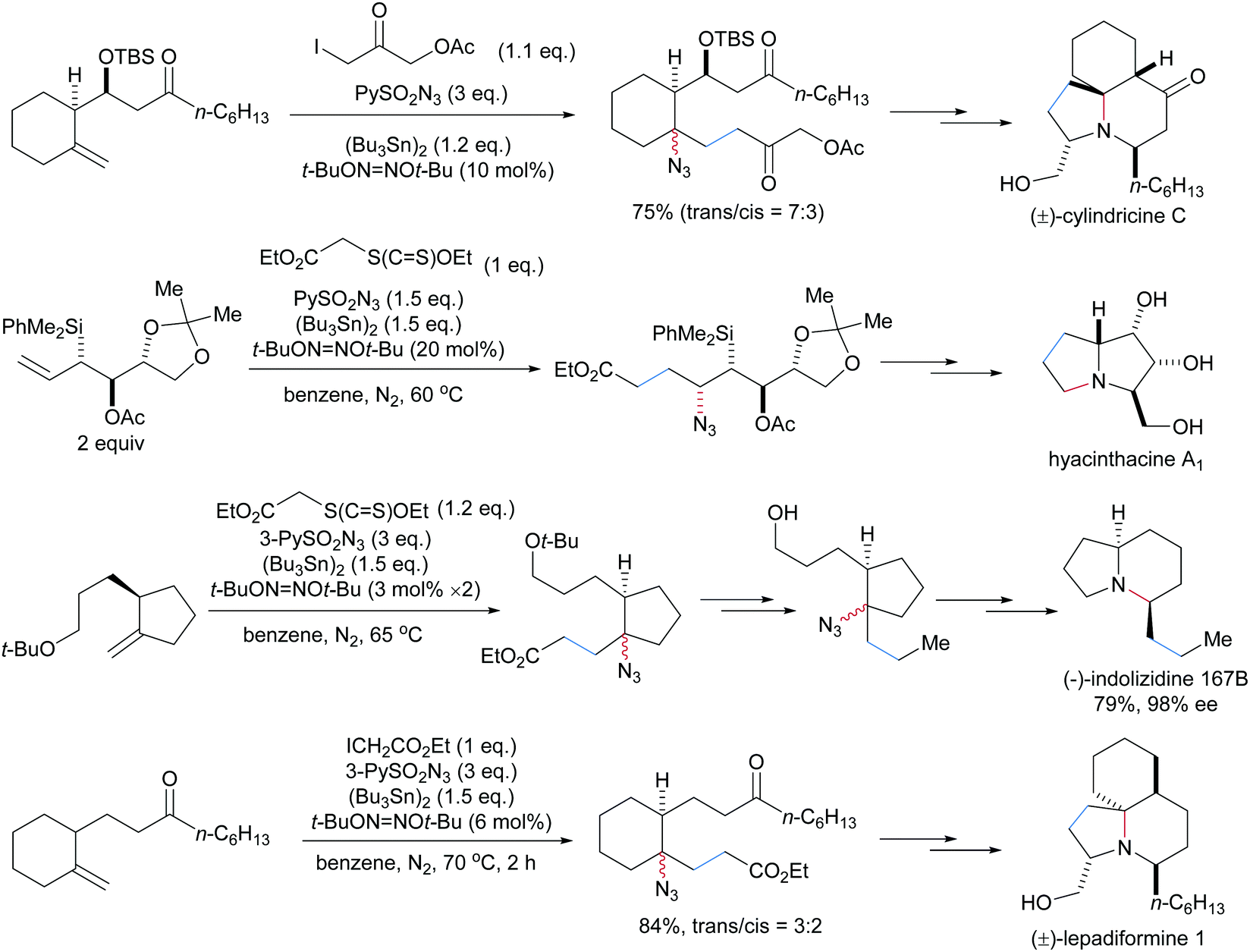 | ||
| Scheme 22 Natural product synthesis through three-component radical alkene carboazidation. | ||
An atom economic alkene carboazidation reaction was reported by the same group in 2010 (Scheme 23).76 The sulfonyl azide 88 was used as a bifunctional reagent allowing to transfer both an alkyl and an azidyl group to an unactivated alkene. Initiation was achieved with di-tert-butyldiazene upon light irradiation leading to the generation of the alkylsulfonyl radical 90. Fragmentation of SO2 generates the electrophilic C-radical 91, which can be trapped by an aliphatic alkene to form the adduct radical 92. Azidyl group transfer from the sulfonyl azide 88 to the alkyl radical 92 furnishes the desired carboazidation product 89 along with the sulfonyl radical 90 that propagates the chain. By using the Weinreb amide 93 or trichloromethylsulfonyl azide 95 as bifunctional reagents, the corresponding carboazidaton products 94 and 96 were obtained in good yields applying the same conditions.
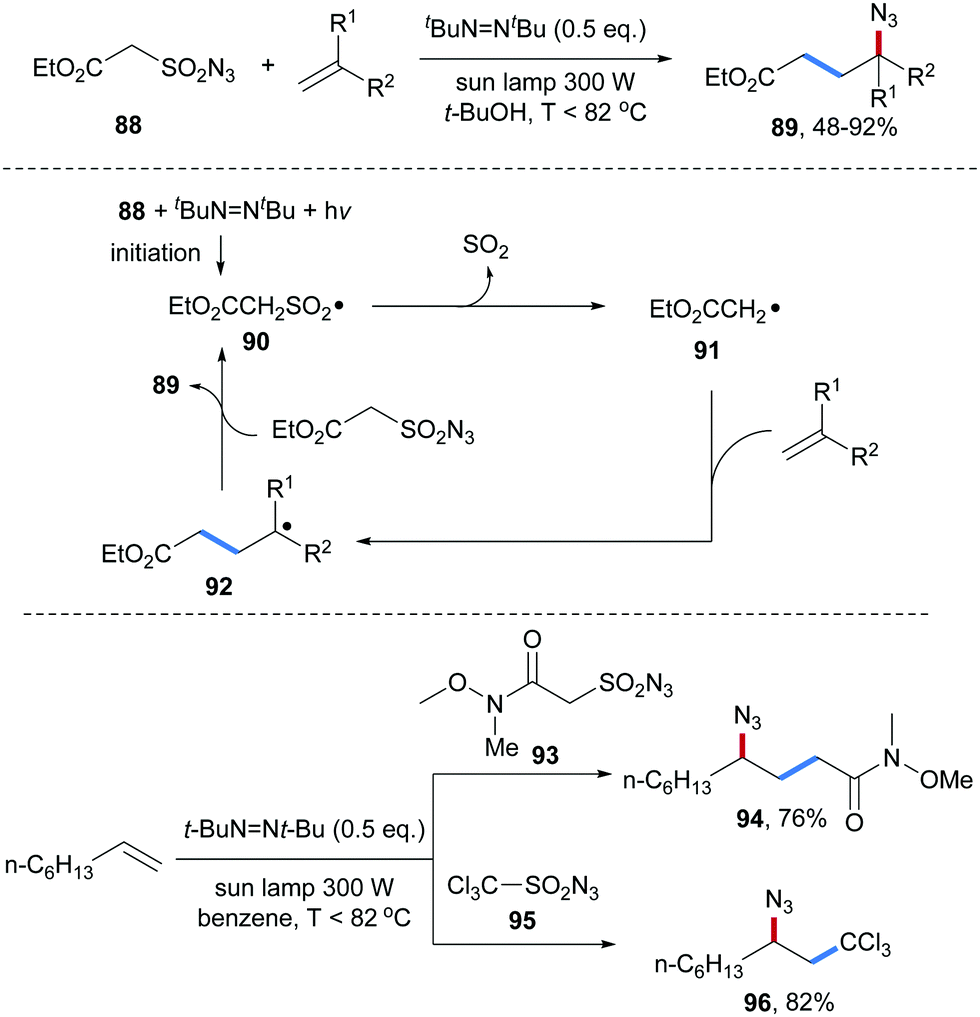 | ||
| Scheme 23 Carboazidation of alkenes using a radical desulfonylative azide transfer process. | ||
The incorporation of CF3 or F-containing functional groups into bioactive compounds heavily impacts their physical and chemical properties.77 Due to the ubiquity of amino groups in bio-active compounds and drug candidates, the development of alkene carboamination reactions with concomitant introduction of fluorinated alkyl groups is of great importance. Photoredox catalysis has been successfully used to conduct radical fluoroalkylamination reactions. An early protocol along these lines was reported by Akita and co-workers in 2013 (Scheme 24).78 A CF3-radical generated through SET reduction of the Umemoto reagent by a photo-excited Ru-complex is trapped by a styrene derivative to form the benzylic radical 98. The Ru(III)-complex generated through oxidative quenching oxidizes 98 to the benzylic cation 99 thereby regenerating the starting Ru(II)-species. Ritter type amination of 99 with CH3CN finally leads to the desired aminotrifluoromethylation product 97 that could be isolated in good to excellent yields.
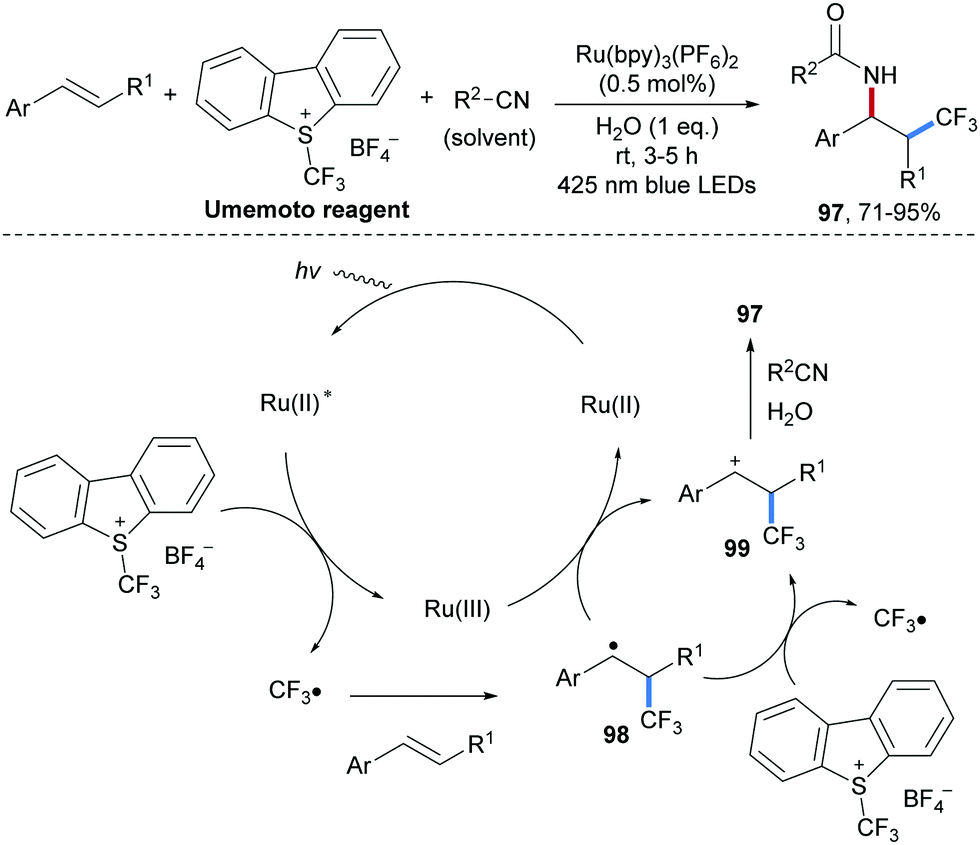 | ||
| Scheme 24 Three-component radical aminotrifluoromethylation of alkenes using photoredox catalysis. | ||
Shortly after, a photoredox catalyzed alkene aminotrifluoromethylation was also disclosed by the Masson group, in which again the Umemoto reagent was used as the CF3-radical precursor in combination with anilines or TMSN3 as nucleophiles to trap the intermediate benzylic cation, providing aminotrifluoromethylation product 100 and azidotrifluoromethylation product 101 respectively (Scheme 25).79
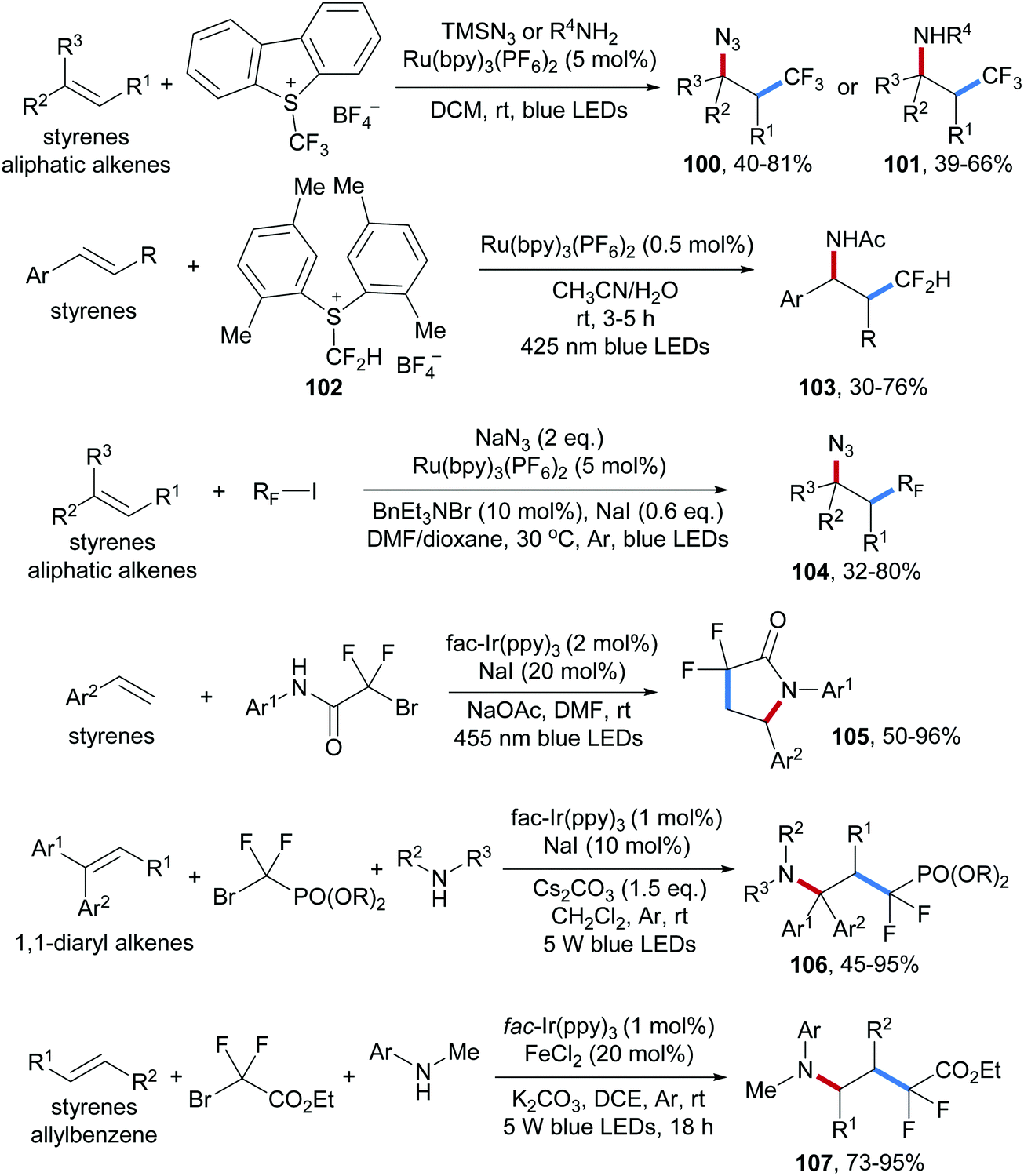 | ||
| Scheme 25 Radical aminoperfluoroalkylation of alkenes. | ||
Several approaches on three-component radical azidofluoroalkylation reactions by using photoredox catalysis have been reported recently (Scheme 25). As above, all these reactions proceed via radical/ionic cross over and various electrophilic fluoroalkylation reagents were used to generate the corresponding fluoroalkyl radicals. Radical addition is followed by oxidation to the corresponding cations and C–N bond formation occurs through nucleophilic trapping by amines or azides. For instance, The S-(difluoromethyl)sulfonium reagent 102, a difluoromethyl radical precursor that can be readily prepared in three steps from 2,5-dimethyl thiophenol, was used for aminodifluoromethylation of aryl alkenes (see 103).80 The Jiao group reported three-component radical azidoperfluoroalkylation of alkenes (104), in which the perfluoroalkyl radicals are generated from the corresponding iodides which can be reduced by a Ru(I)-complex.81 Several examples on three-component aminodifluoroalkylation of alkenes were presented by using α-bromo-α,α′-difluoro-amides,82 -esters83 and -phosphonates84 as difluoroalkyl radical precursors, yielding the corresponding alkene aminodifluoroalkylation products 105–107 under mild conditions in good to excellent yields.
Li and co-workers reported a silver mediated radical carboamination of alkenes with alkyl nitriles and amines to prepare diverse γ-amino alkyl nitriles 108 (Scheme 26).85 The use of Ag2CO3 as an SET oxidant was shown to be necessary. The yield of this cascade could significantly be increased upon using a catalytic amount of Fe(OTf)3 as a Lewis acid. The scope of this transformation with respect to the alkene is restricted to aryl alkenes and the alkyl nitriles that serve as alkyl radical precursors are used as solvents. Various nucleophiles including primary and secondary amines and sulfonamides are competent for the formation of the C–N bond through nucleophilic trapping. In the proposed mechanism, the cyano group in CH3CN is firstly coordinated with Ag2CO3 to form the complex 109, in which the acidity of the adjacent C–H bond is increased significantly. Deprotonation of 109 provides AgCH2CN 110, which undergoes SET oxidation by Ag(I) to give the radical 111 along with Ag(0). Subsequently, the α-cyano carbon radical 111 adds to the styrene derivative to generate the benzylic radical 112 which is then oxidized by Ag(I) to the corresponding benzylic cation 113. Nucleophilic trapping with an amine finally provides the γ-amino alkyl nitrile 106 in moderate to high yield.
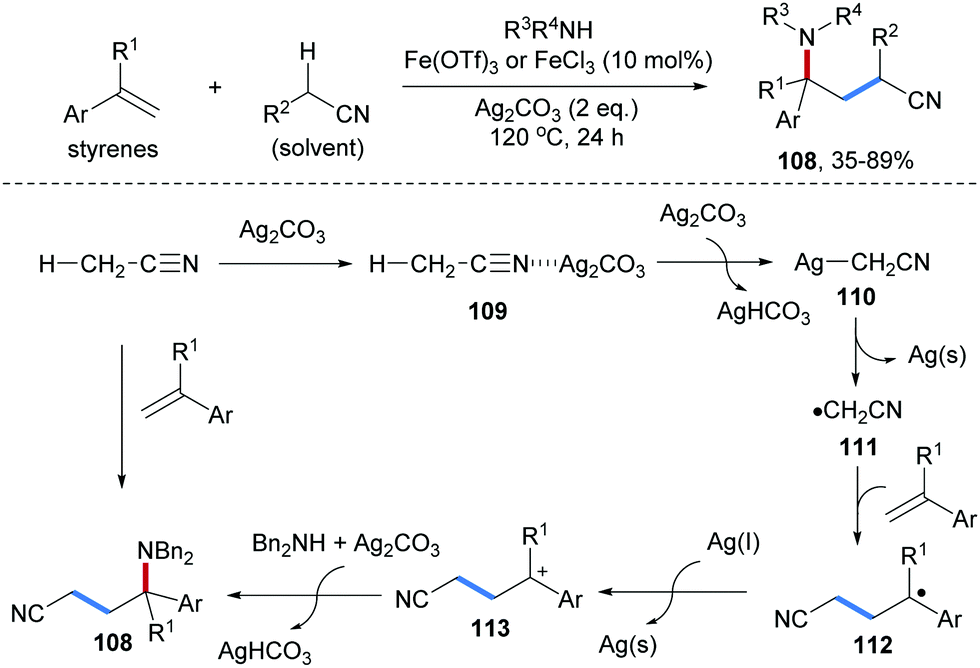 | ||
| Scheme 26 Oxidative radical carboamination of alkenes with alkyl nitriles and amines. | ||
Cu-Catalyzed three-component radical carboazidation of aryl alkenes was reported by the Zhu group (Scheme 27).86 Mono- and 1,1-disubstituted aryl alkenes serve as radical acceptors and the reaction features good functional group tolerance, providing diverse γ-azido alkyl nitriles 114 in moderate to good yields. Mechanistically, coordination of CH3CN to Cu(II) mediates deprotonation of CH3CN to give an alkyl-Cu(II)-complex 115, which undergoes Cu-alkyl bond homolysis to give an electrophilic radical 116. Addition of 116 to an alkene provides the adduct radical 117. N3-group transfer proceeds via capture of 117 by CuLN3 to give a Cu(III)-complex which upon reductive elimination eventually provides the targeted carboazidation product 114 with the concurrent formation of the starting Cu(I)-complex. Direct azidyl group transfer from CuLN3 to 117 is also feasible. Finally, SET-oxidation of Cu(I) by a Mn(III)/DTBP-couple regenerates the Cu(II)-species. An alternative pathway proceeding via single electron oxidation of the C-radical 117 to give cation 118 that gets trapped by NaN3 to 114 is less likely since competing trapping with MeOH, which is used as a co-solvent, should occur.
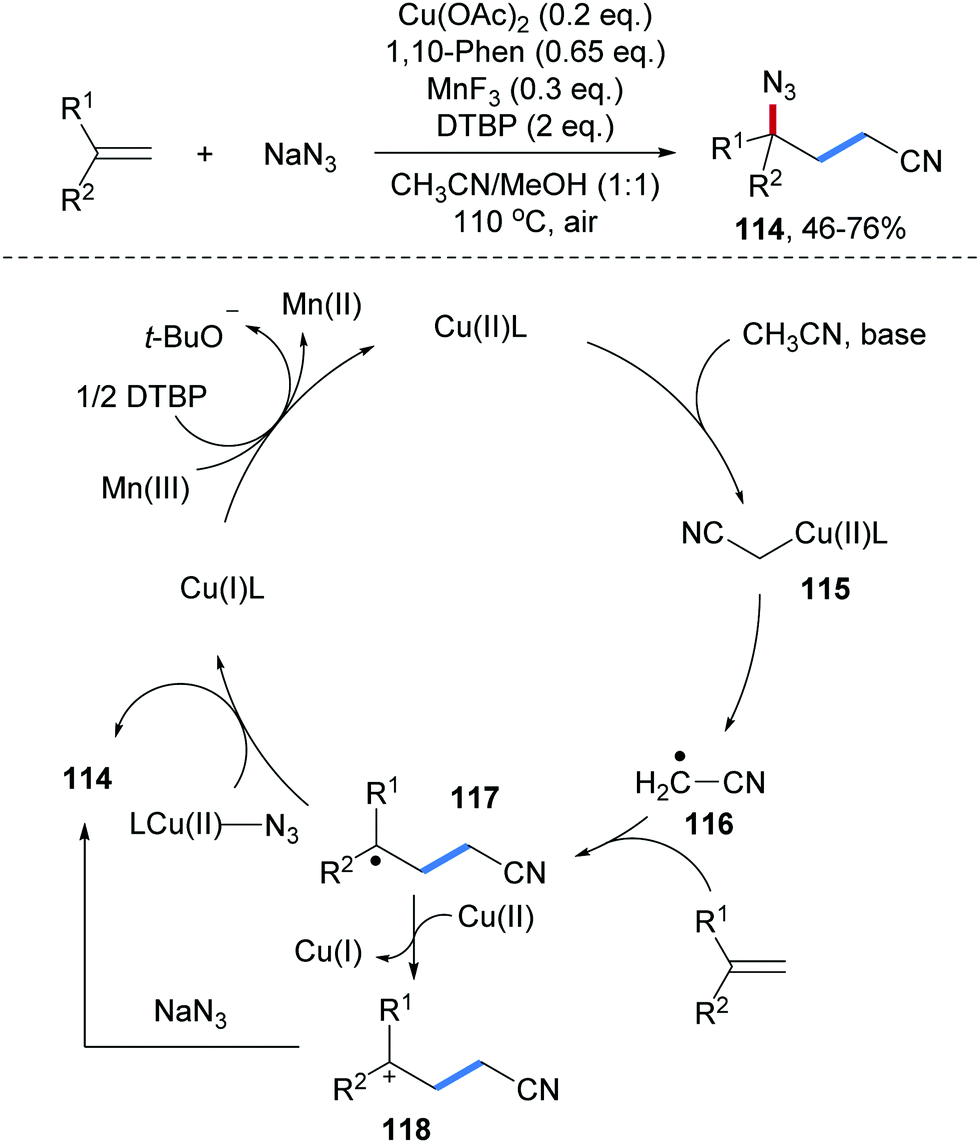 | ||
| Scheme 27 Cu-catalyzed three-component carboazidation of alkenes with acetonitrile and sodium azide. | ||
Bao and co-workers also used acetonitrile as an alkyl radical precursor and amination reagent for radical alkene carboamination reactions where a Cu(I)-complex acted as a redox catalyst (Scheme 28).87 Their efficient alkene carboamination protocol features broad substrate scope, and diverse alkenes including aryl alkenes, enynes and nonconjugated alkenes provided the desired carboamination products in good yields with high diastereoselectivity (119a–e). Other commercial alkyl nitriles were found to be competent reaction partners in this radical cascade. For example, isobutyronitrile used as a solvent delivered in the reaction with styrene the γ-amino alkyl nitrile 119f in 47% yield. The proposed catalytic cycle starts by single electron oxidation of Cu(I) by BPO to give a Cu(II)-species along with the phenyl carboxyl radical 120. Decarboxylation of 120 generates the phenyl radical which subsequently abstracts an H-atom from CH3CN to give the C-radical 121. Radical addition of 121 to styrene provides the benzylic radical 122. Single-electron oxidation of 122 by Cu(II) to 123 and subsequent Ritter reaction provide the γ-amino alkyl nitrile 119 thereby regenerating the Cu(I)-catalyst.
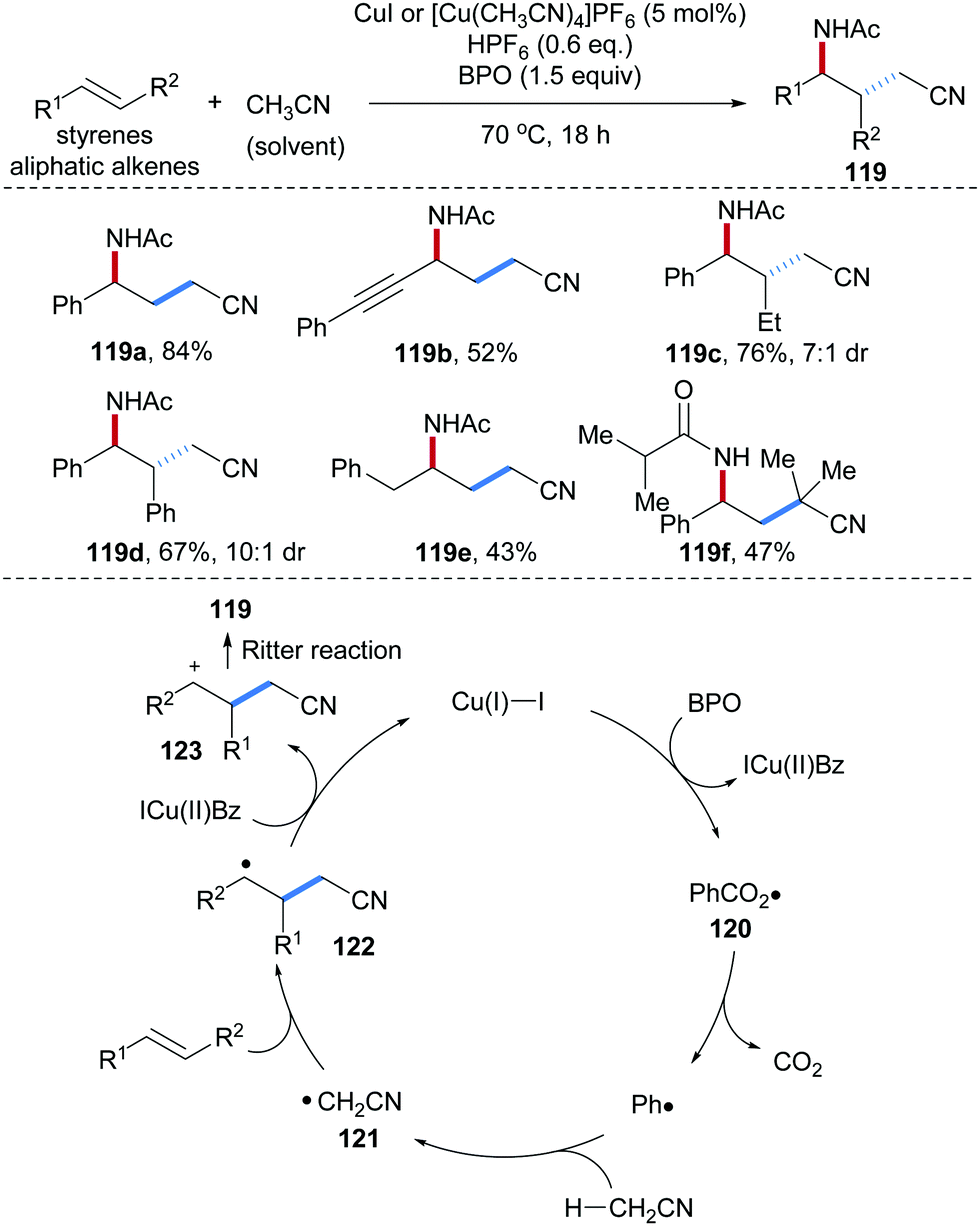 | ||
| Scheme 28 Cu-Catalyzed carboamination of alkenes with acetonitrile. | ||
Fe-Catalyzed three-component radical carboamination of alkenes with peroxides and alkyl nitriles was reported by Bao and co-workers (Scheme 29).88 Readily prepared aliphatic peracids were used as precursors to generate alkyl radicals 129 through single-electron reduction by an Fe(II)-complex. Radical addition of 129 to an alkene generates the C-radical 130, which further undergoes single electron oxidation by Fe(III) to generate the carbon cation 131. Ritter amidation of 131 with acetonitrile yields the targeted carboamidation product 126. Considering the aminomethylation and aminoethylation, the peresters 125 were used as methyl respectively ethyl radical precursors. By using methyl cinnamate derivatives as alkyl radical acceptors, a set of β-amino acid derivatives 128 could be obtained in moderate to good yields. Notably, the Ritter reaction occurred with high diastereoselectivity and the stereochemical outcome of the cascade could be understood based on computational studies.
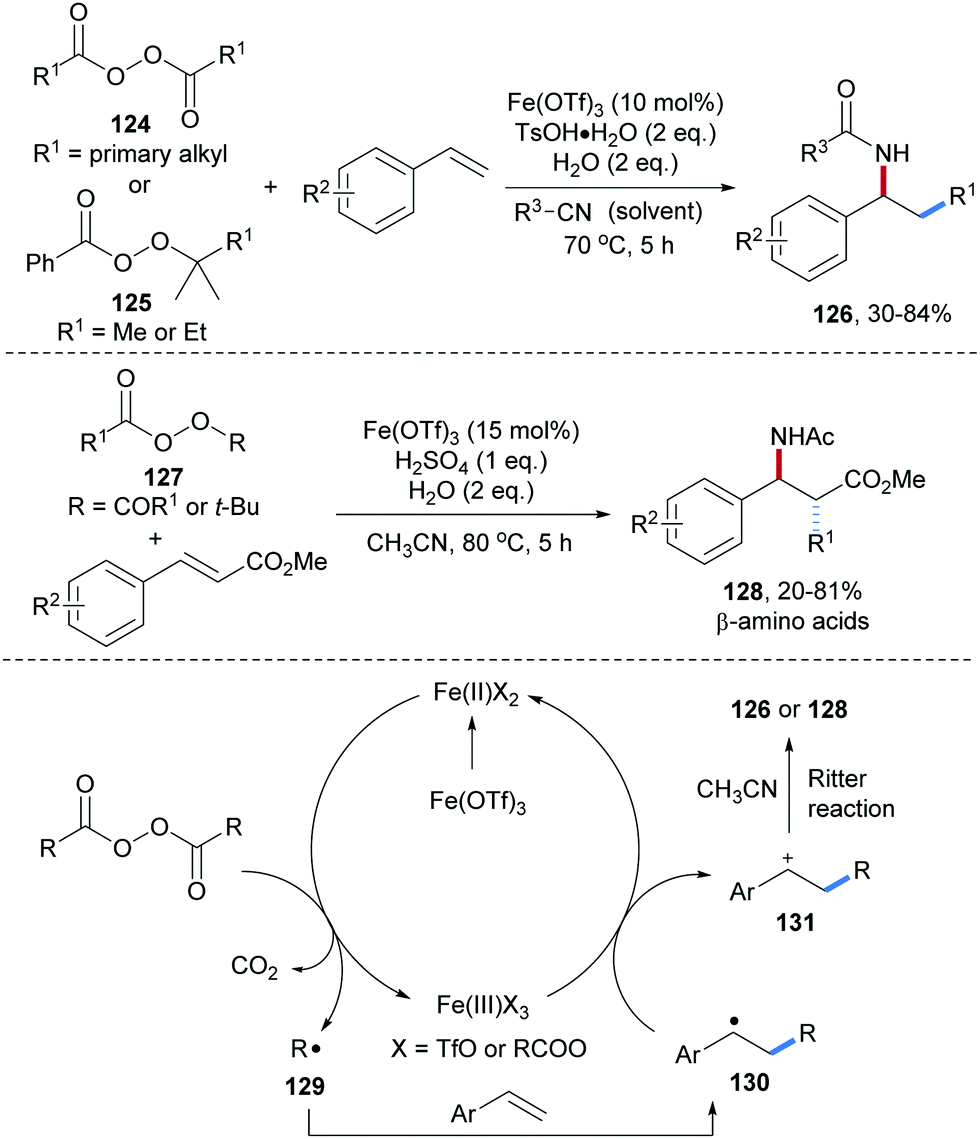 | ||
| Scheme 29 Fe-Catalyzed carboamination of alkenes. | ||
Later, Cu catalyzed three-component radical azidomethylation of aryl alkenes was reported by the Zhu group (Scheme 30).89 Di-tert-butyl peroxide first gets reduced by Cu(I) via SET to give a tert-butyloxyl radical 133, which undergoes fragmentation to generate a methyl radical 134 that adds to an alkene to give the C-radical 135. Cu(II)-mediated azide transfer provides the desired azidomethylation product 132 along with the starting Cu(I)-complex. The targeted alkyl azides 132 were obtained with high efficiency and very broad functional group tolerance in good to very good yields.
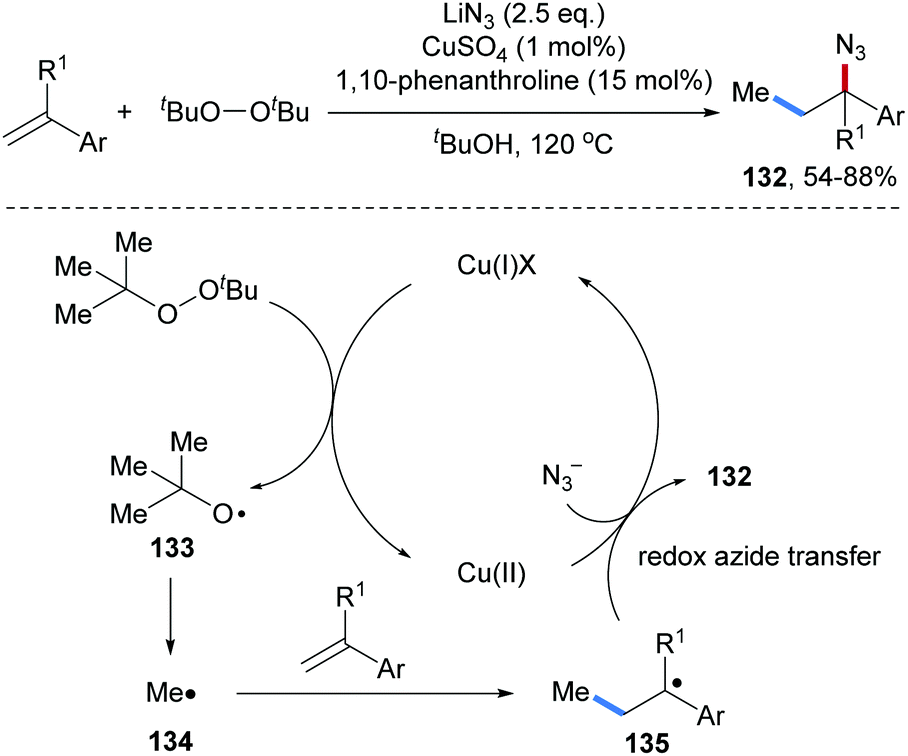 | ||
| Scheme 30 Cu-Catalyzed radical azidomethylation of styrenes. | ||
Copper-catalyzed intermolecular carboamination of alkenes with α-haloacid derivatives and amines were reported by Hull and co-workers (Scheme 31).90 Considering the styrene derivatives as acceptors, both electron donating and withdrawing groups at the aryl group are tolerated and diverse halides including α-bromoesters (137a,b,d and 136a), α-bromoamides (137c) and bromomalonates (137e) were used as C-radical precursors, providing the corresponding carboamination products in good yields. Aliphatic alkenes were found to be competent acceptors for this cascade, as documented by the preparation of the lactam 136b. Notably, both Z and E-β-methylstyrene provided the cis-product as major isomer with high selectivity. This result indicates that the thermodynamically more stable trans-oxocarbenium intermediate 138 is generated. Stereoselective nucleophilic ring-opening of 138 with butylamine followed by lactamization yields the corresponding cis-product.
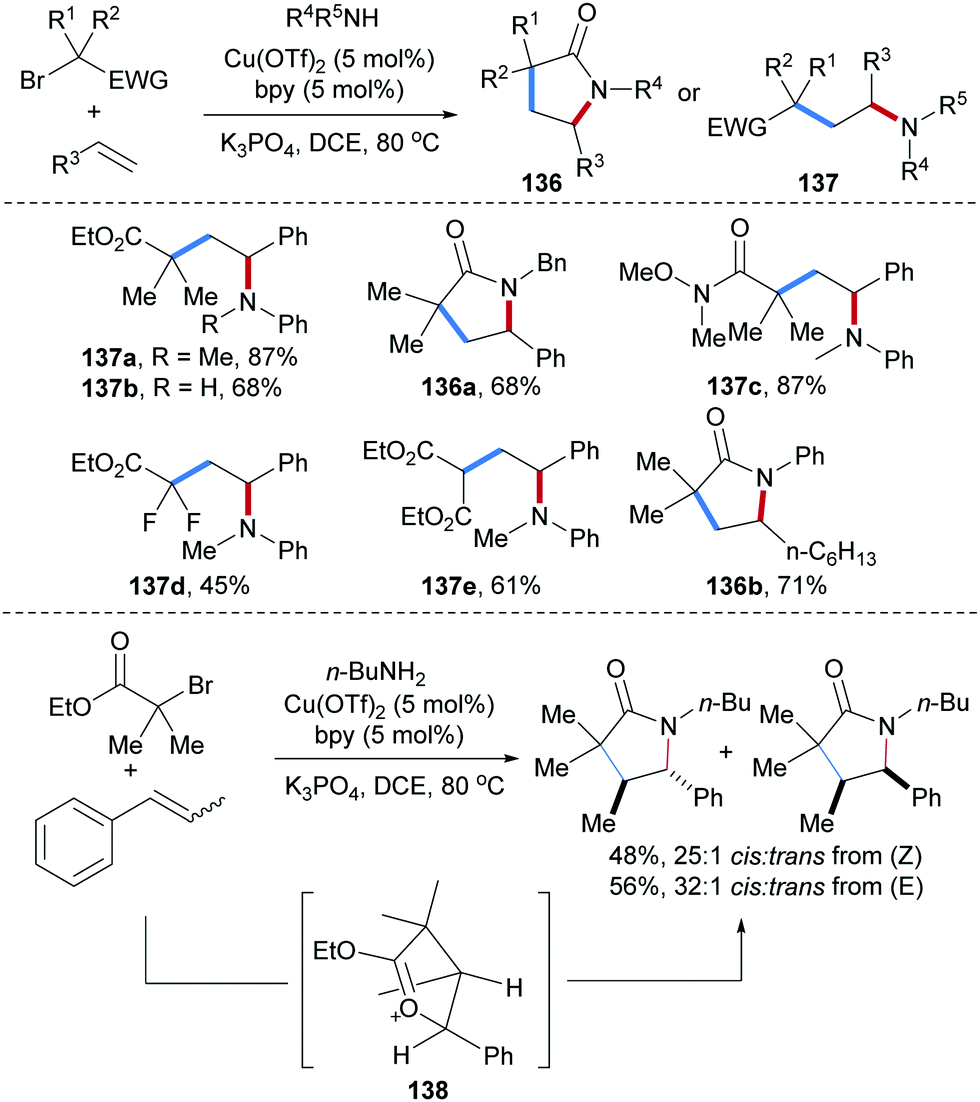 | ||
| Scheme 31 Cu-Catalyzed three-component radical carboamination of alkenes for the synthesis of lactams. | ||
Zhang and co-workers reported Cu-catalyzed three-component radical carboamination of alkenes with secondary amines and alkyl halides upon blue light irradiation (Scheme 32).91 As N-donors, carbazoles, indoles and indazoles were found to participate in this radical cascade in combination with aryl alkenes and alkyl halides, providing the carboamination products 139 in moderate to satisfactory yields. This cascade features broad scope with respect to the halide component and alkyl iodides, α-bromo esters, secondary alkyl halides and even dichloromethane were found to be eligible C-radical precursors. It is noteworthy that upon using anilines and aliphatic amines as nucleophiles, rac-BINOL was required as an additive (140). The authors proposed that the photo-excited Cu(I)-complex 141 or 146 is capable of reducing the substrate halides to the corresponding C-radicals 142. Subsequent radical addition of 142 to an alkene followed by Cu(II) (143 or 147) mediated radical C–N bond formation affords the desired carboamination product thereby regenerating the Cu(I)-species. The radical nature of this cascade was supported by radical clock experiments.
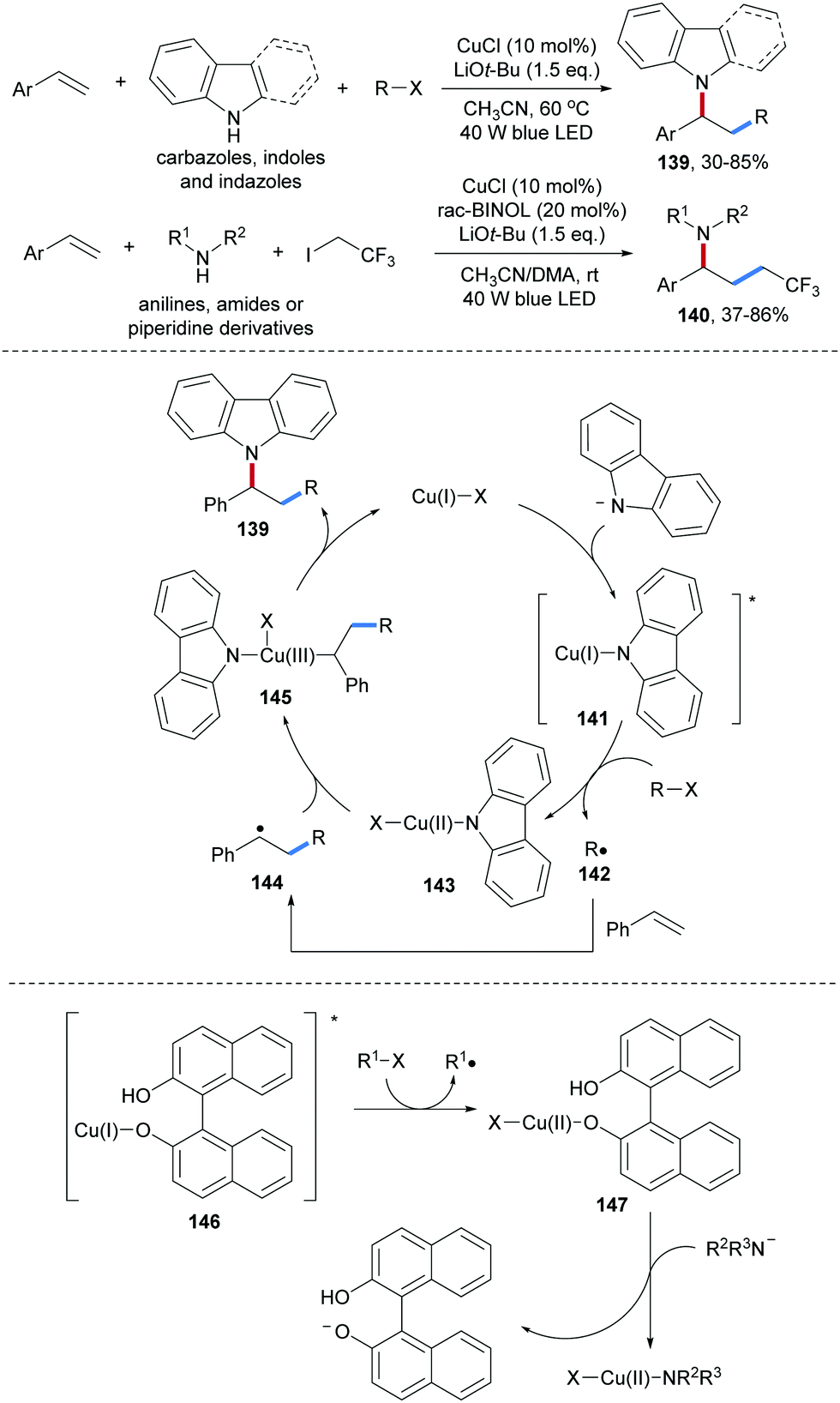 | ||
| Scheme 32 Cu-Catalyzed intermolecular carboamination of alkenes induced by visible light. | ||
An efficient method for radical alkene malonylamination with amines and iodonium ylides 148 was recently disclosed by Wang and co-workers (Scheme 33).92 A catalytic amount of succinimide was used as a “proton shuttle” to conduct the radical cascade and the interesting transformation proceeds in the absence of any transition-metal catalyst. Diverse unactivated alkenes and enamides qualify as radical acceptors and the malonyl radical is generated from 148 through electron-transfer. Primary and secondary anilines were used as aminating reagents to afford the γ-amino acid derivatives 149 in low to high yields. Halogen bonding between iodonium ylide 148 and the amine component leads to a complex that undergoes intramolecular SET to generate an ammonium radical cation and the iodanyl radical anion 150. Protonation of 150 by succinimide gives the neutral iodanyl radical 151, which undergoes fragmentation to the malonyl radical and iodobenzene. Radical addition of the malonyl radical to an alkene generates an alkyl radical which is trapped by the ammonium radical cation to give the ammonium salt 152. Deprotonation of 152 by the succinimide anion eventually provides the desired carboamination product 149 and succinimide is regenerated.
 | ||
| Scheme 33 Succinimide-catalyzed radical malonylamination of alkenes. | ||
Photoredox catalyzed carboamination of aryl alkenes with alkyl N-hydroxyphthalimide esters 153 and anilines was reported by Li and co-workers (Scheme 34).93 In the presence of B(C6H5)3 as a Lewis acid, single electron reduction of 153 by a photo-excited Ru(II)-complex provides the corresponding C-radical 155, which gets trapped by an alkene to give the adduct radical 156. The Ru(III)-complex generated in the initial oxidative quenching of Ru(II)* then oxidizes 156 to the benzylic cation 157, which is trapped by an aniline to afford the final aminoalkylation product 154 in moderate to good yield.
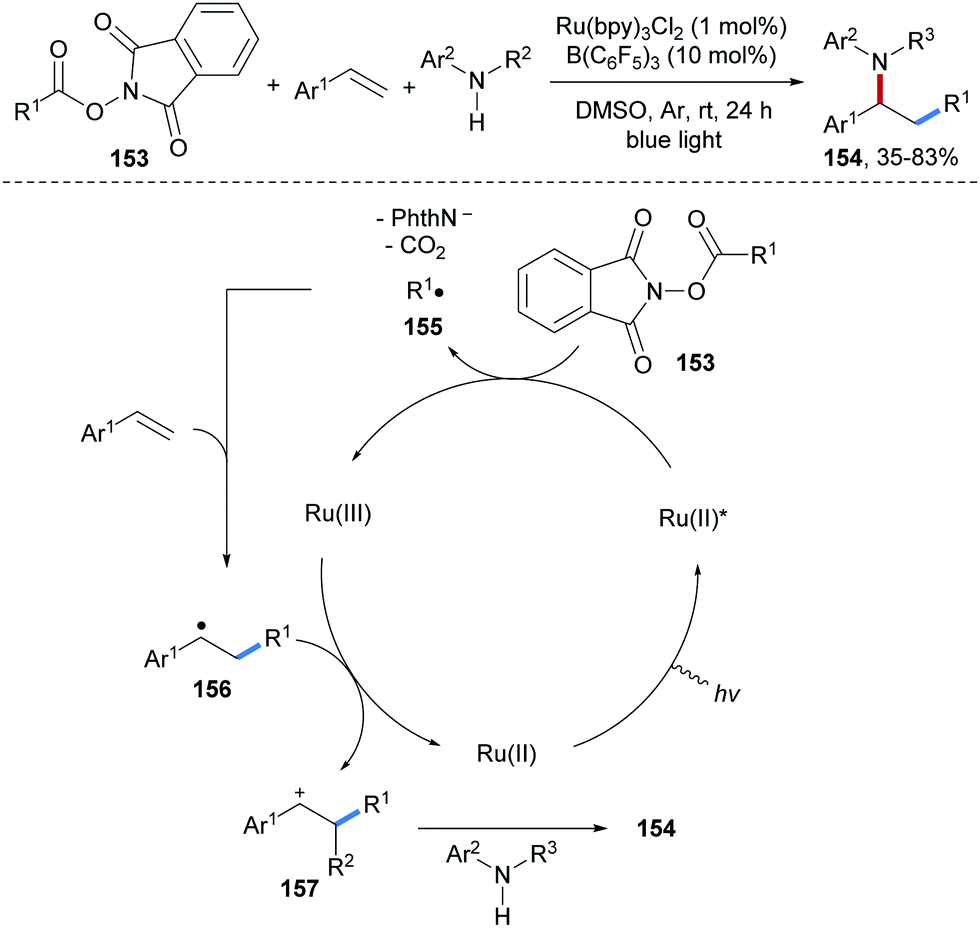 | ||
| Scheme 34 Three-component alkylamination of styrenes with alkyl N-hydroxyphthalimide esters and amines. | ||
Three-component radical aminotrifluoromethylation of unactivated alkenes with aryldiazonium salts and sodium triflinate was disclosed by the Xiao group (Scheme 35).94 A Ru(II)-complex is irradiated to give excited Ru(II)*, which is oxidatively quenched by an aryldiazonium salt to give the corresponding Ru(III)-complex. The Ru(III)-species in turn oxidizes sodium triflinate to give after SO2-fragmentation a trifluoromethyl radical, which adds to an alkene to provide the adduct radical 159 that gets trapped by an aryldiazonium salt to provide the azo radical cation 160. The isolated diazene product 158 obtained in moderate to very good yield results through single-electron reduction of 160 by the photo-excited Ru(II)-complex closing the redox catalysis cycle. Noteworthy, the product diazenes are readily transferred to indoles 161 and amides 162 illustrating the synthetic potential of the method. It should be pointed out that an Ag/H2O2-mediated radical cascade comprising alkenes, aryldiazonium salts and sodium triflinate as reaction partners to afford indoles was reported by the Antonchick group, in which diazo compounds of type 160 were also suggested as intermediates.95
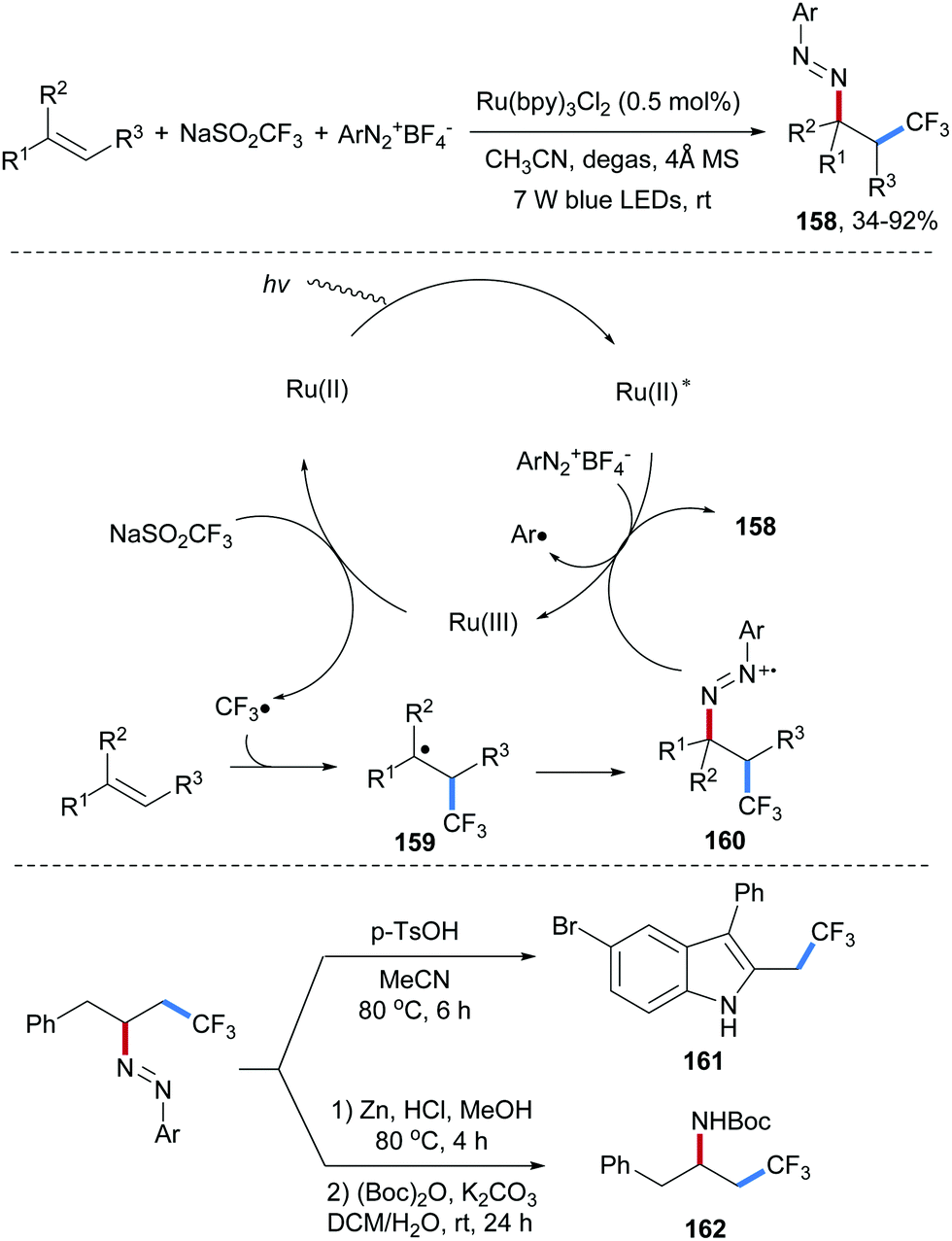 | ||
| Scheme 35 Azotrifluoromethylation of alkenes with aryldiazonium salts and sodium triflinate. | ||
The MacMillan group reported an enantioselective 3+2 coupling of the N-protected β-amino aldehyde 163 and various activated alkenes to access multi-substituted pyrrolidines 164 with moderate to high diastereoselectivity and high enantioselectivity (Scheme 36).96 Styrenes (164a–d), conjugated linear (164d) and cyclic (164e) dienes engage in the cascade to provide the desired pyrrolidines 164 with high efficiency. Mechanistically, the enamine 165 generated through condensation of 163 and the MacMillan catalyst undergoes single electron oxidation by Fe(III) to generate a radical cation intermediate 166. Stereoselective addition of 166 to an activated alkene leads to the distonic radical cation 167. Single electron oxidation of 167 by Fe(III) generates the corresponding benzylic or allylic cation 168, which undergoes cyclization to provide after iminium ion hydrolysis the functionalized pyrrolidine 164.
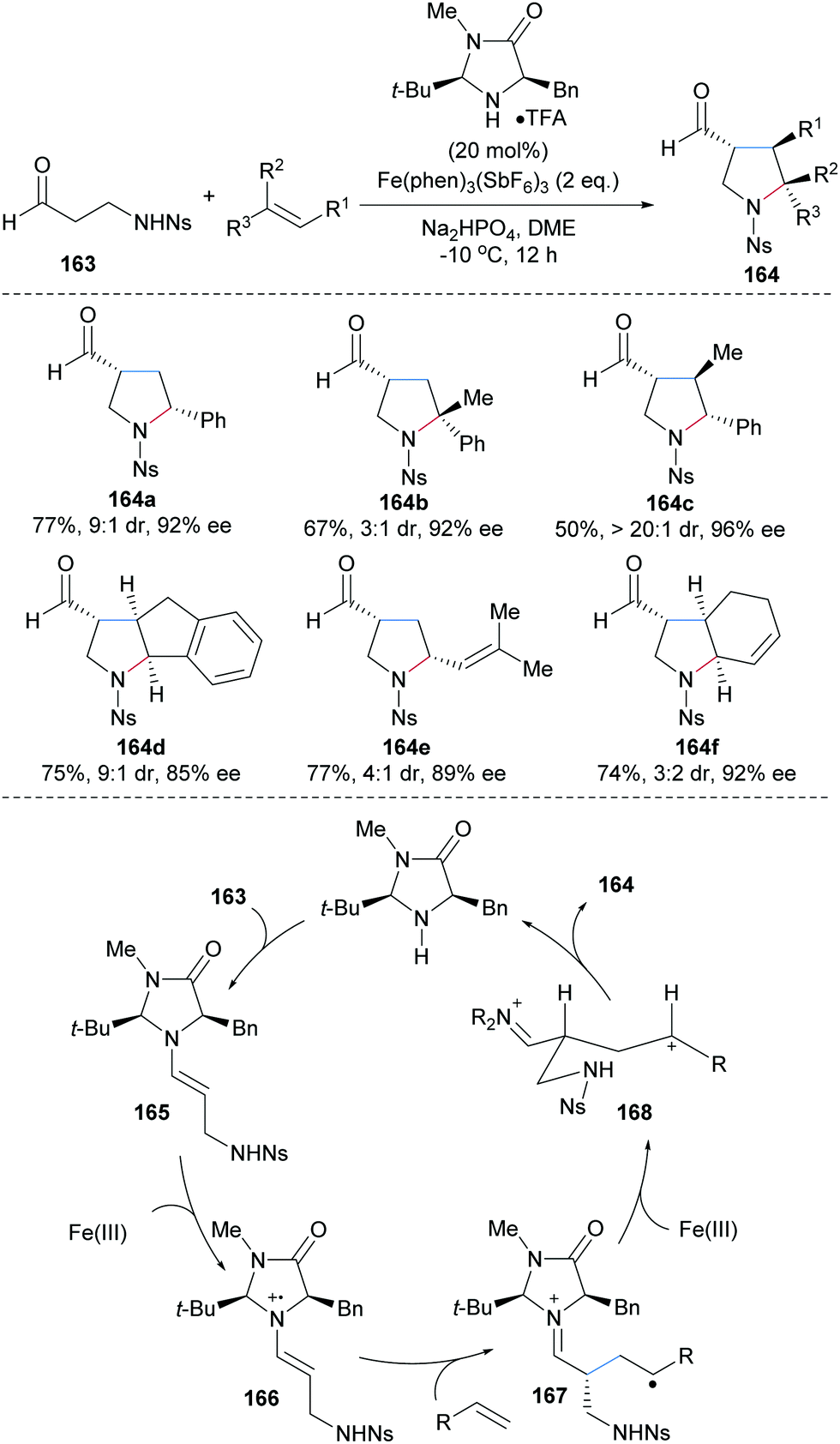 | ||
| Scheme 36 Enantioselective radical alkene carboamination for the preparation of pyrrolidines. | ||
Chemler and co-workers developed an efficient method for the preparation of pyrrolidines 170 through Cu-catalyzed radical coupling of alkenes with potassium N-carbamoyl-β-aminoethyltrifluoroborates 169 (Scheme 37).97 Single electron oxidation of the trifluoroborate 169 by Cu(II) produces the corresponding C-radical 171, which reacts with an alkene to provide the adduct radical 172. Trapping of 172 by the Cu(II)-complex and ligand exchange afford the Cu(III) species 173 which undergoes reductive elimination to give the desired pyrrolidine 170. Alternatively, single-electron oxidation of 172 by Cu(II) leads to the cation 174 which directly cyclizes to afford 170.
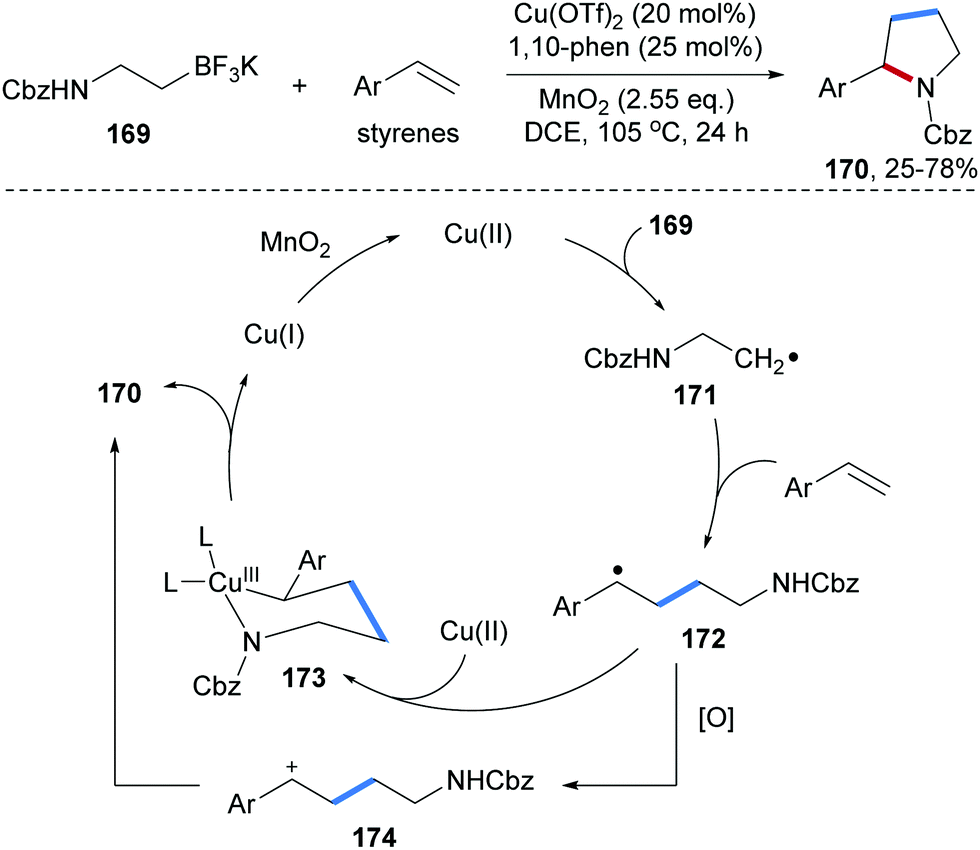 | ||
| Scheme 37 Cu-Catalyzed radical carboamination of alkenes with potassium β-aminoethyl trifluoroborates for the preparation of pyrrolidines. | ||
A photoredox-catalyzed Meerwein addition with styrenes as acceptors and subsequent Ritter type amidation was reported by the König group (Scheme 38).98 An aryl diazonium salt is first reduced by the photo-excited Ru(II)-complex to generate an aryl radical 176. Intermolecular radical addition of 176 to an alkene provides the corresponding adduct C-radical 177 which gets oxidized by the Ru(III)-complex to generate a carbon cation 178 along with the Ru(II)-catalyst. By using alkyl nitriles as the solvent, Ritter amidation via179 is achieved to eventually give the amidoarylation product 175 in moderate to high yield. Later, a transition metal free protocol for Meerwein-type aminoarylation was disclosed by Heinrich and co-workers, in which the diazonium salt is used as aryl radical precursor and also as a radical amination reagent to afford a β-aryl diazene 180 as the final product.99
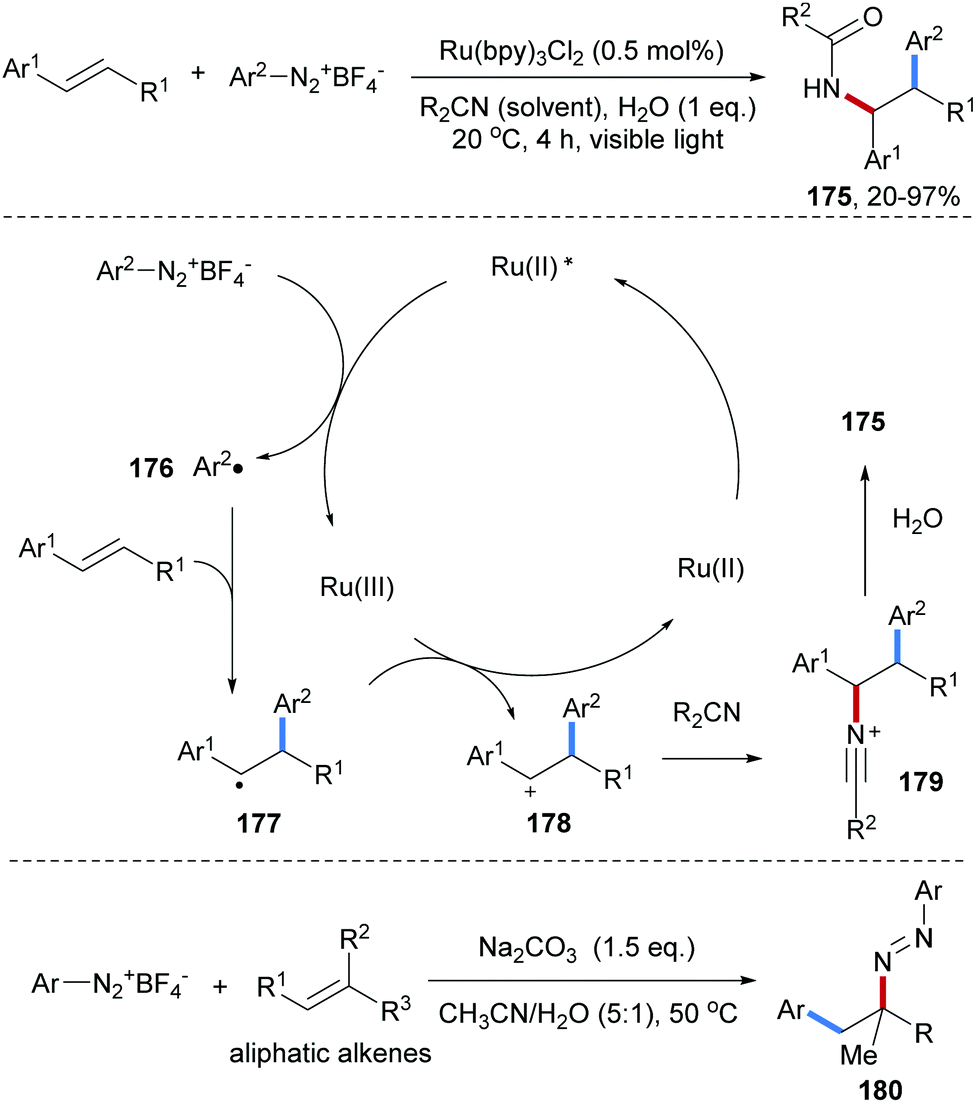 | ||
| Scheme 38 Photoredox-catalyzed Meerwein addition and subsequent Ritter reaction and transition metal free alkene aminoarylation. | ||
4. Carboamination by oxidation of alkenes
The Nicewicz group reported a photoredox and thiol co-catalyzed carboamination of readily oxidizable electron-rich alkenes with α,β-unsaturated amides to provide γ-lactams 181 with good steroselectivity in moderate to good yields (Scheme 39).100N-Bocylated cinnamyl amine also engages in this cascade to afford multi-substituted pyrrolidines 182 with good diastereoselectivity. The proposed mechanism starts by single-electron oxidation of the electron-rich alkene by the photo-excited acridinium salt to give the alkene radical cation 183 and the reduced acridine radical. Nucleophilic addition of an unsaturated amide to 183 leads after deprotonation to the C-radical 184, which undergoes 5-exo-cyclization to the C-radical 185. Reduction of 185 by thiophenol affords the desired γ-lactam 181 and a thiyl radical. The redox catalysis cycle gets closed by SET reduction of the thiyl radical with the acridine radical.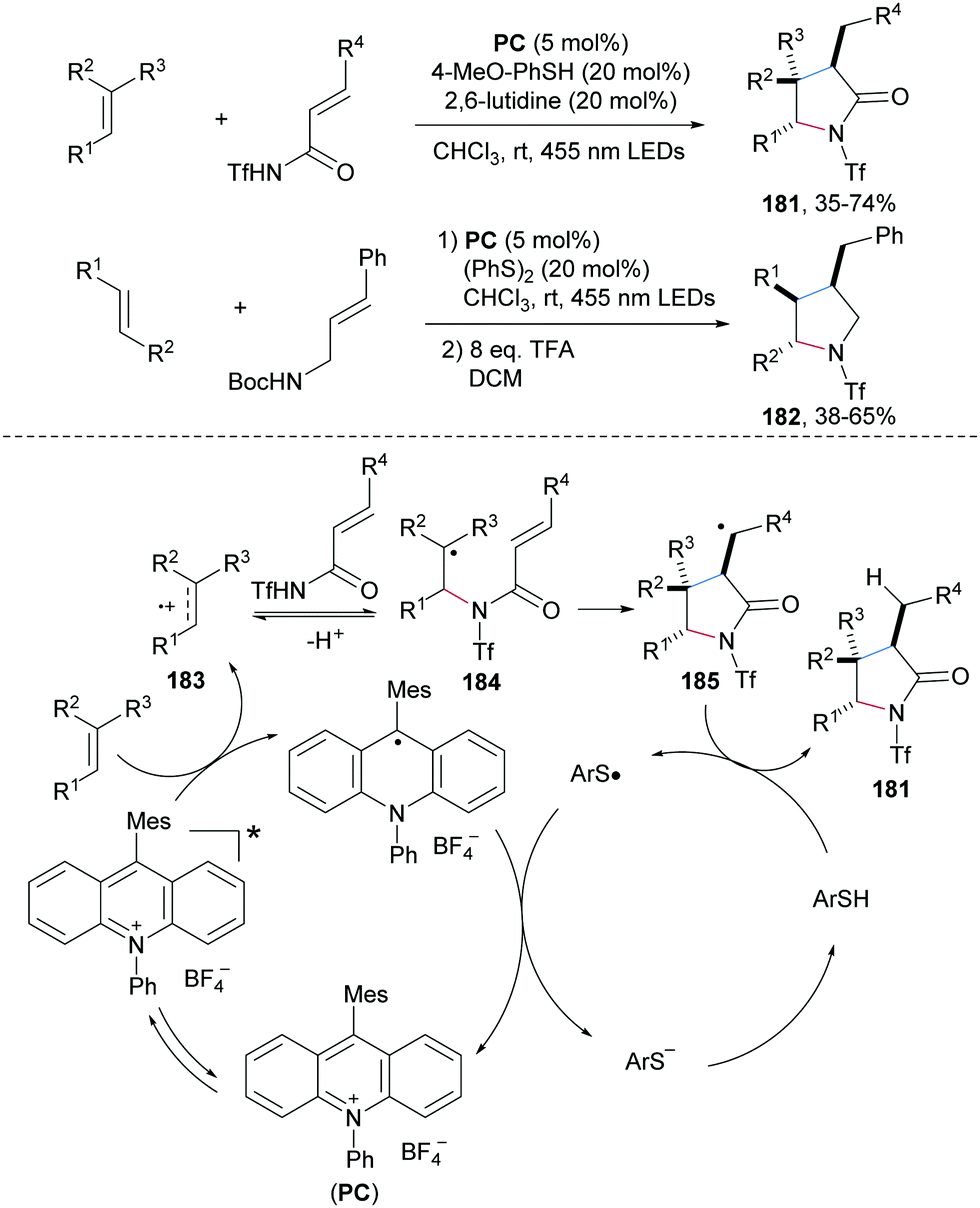 | ||
| Scheme 39 Alkene carboamination through polar radical crossover cycloaddition. | ||
An elegant photoredox catalyzed aminoarylation of electron-rich styrenes with N-arylsulfonyl-acetamides 186 was developed by the Stephenson group (Scheme 40).101 In the presence of a photoredox catalyst, the arylsulfonylacetamides serve as bifunctional reagents to add across an alkene to form structurally useful β-diaryl amides of type 187. Noteworthy, internal alkenes provide cis addition and targeted amides were formed with excellent diasteroselectivity (187d–h). Considering the mechanism, single-electron oxidation of the electron-rich alkene by a photo-excited Ir(III)-complex provides radical cation 188, which undergoes nucleophilic trapping by the arylsulfonylacetamide 186 to give the benzylic radical 189. 1,4-Radical aryl migration from sulfur to the benzylic radical via cyclohexadienyl radical 190 leads to the translocated S-radical 191. Reduction of 191 by the Ir(II)-complex and SO2-fragmenation eventually afford the isolated carboamidation product 187.
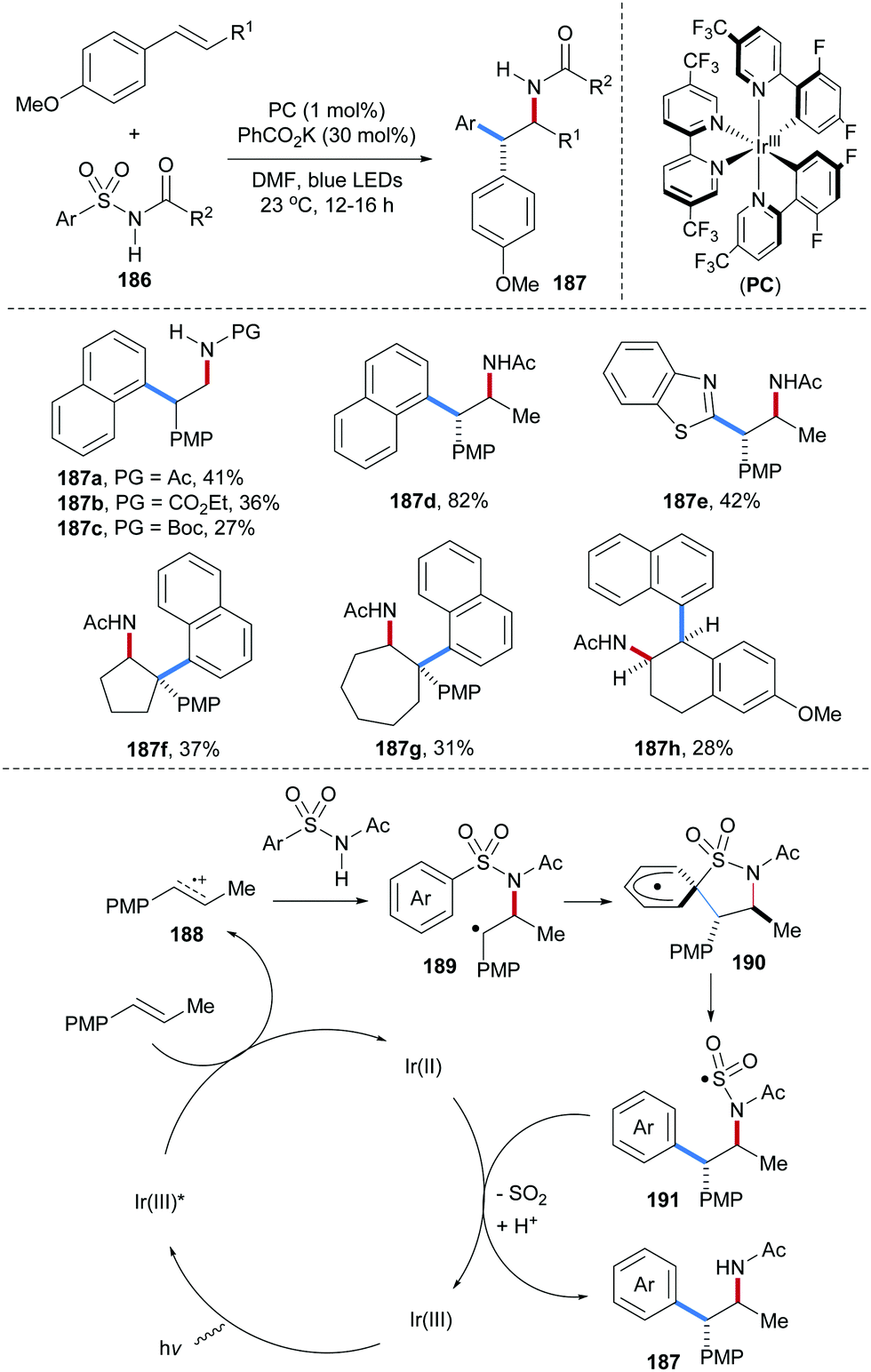 | ||
| Scheme 40 Alkene aminoarylation by using N-arylsulfonylacetamides as bifunctional reagents. | ||
5. Summary and outlook
In this review different approaches to conduct intermolecular radical alkene carboamination have been discussed. Considering non-radical alkene carboaminations, the radical processes nicely complement these methods and accordingly have been successfully used for the preparation of various valuable amine derivatives. Such radical cascades can be conducted following three different reaction modes: (a) by an N-radical adding to an alkene, (b) by a C-radical adding to an alkene or (c) by oxidation of an alkene with subsequent nucleophilic amination. Considering the first approach, readily accessed oxime derivatives, N-halo compounds, N-pyridinium salts and TMSN3 have been used as N-radical precursors, providing the corresponding N-radicals or the azidyl radical through a single-electron transfer pathway or via homolytic cleavage of a weak covalent bond. Focusing on the second reaction mode, various C-radical precursors have been implemented in alkene carboaminations in combination with various amination reagents. In many of these processes, single electron reduction and oxidation steps are involved and consequently radical carboamination is often conducted with the help of a redox catalyst. In the ideal case, the overall sequence is a redox neutral process improving economy of the cascade.In both strategies (a and b), formation of the second σ-bond can proceed via a free radical addition reaction. Alternatively, the C-radical resulting from N- or C-radical addition to the alkene can be oxidized to the corresponding carbenium ion that can eventually be trapped by C- or N-type nucleophiles. Both, the radical trapping and also the ionic pathway are difficult to run enantioselectively. Considering this great challenge, C-radical trapping can also be catalyzed by a transition metal in a so-called radical/transition metal cross-over process. This has been well documented for C–C bond formation and also for C–N bond formation mainly using Cu-catalysis. As advantage of this latter cross-over strategy, enantioselective trapping is feasible and indeed has been realized in few cases. In particular, in this area we see great potential and future activities of this emerging field.
Carboamination via alkene radical cations according to reaction mode (c) is underdeveloped. This is mainly due to the fact that alkene single electron oxidation is currently restricted to electron-rich activated alkenes. This problem has to be solved and then this strategy will gain further importance.
By looking at radical alkene carboamination more generally, the full potential of radical chemistry has to be harnessed in future in particular considering the mild reaction conditions mostly used to run such radical cascades. This ensures high functional group tolerance and radical carboamination should be well applicable to very complex alkenes at a late stage of the synthesis. Therefore, we expect the novel methodologies to be applied more often in complex natural product synthesis.
Conflicts of interest
There are no conflicts to declare.References
- X. Wu, S. Wu and C. Zhu, Tetrahedron Lett., 2018, 59, 1328 CrossRef CAS.
- R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314 CrossRef CAS PubMed.
- E. Godineau and Y. Landais, Chem. – Eur. J., 2009, 15, 3044 CrossRef CAS PubMed.
- G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS.
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036 CrossRef CAS PubMed.
- M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef.
- T. Koike1 and M. Akita, Chem, 2018, 4, 409 Search PubMed.
- X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821 CrossRef CAS.
- Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727 CrossRef CAS.
- G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed.
- Z. Liu, Y. Gao, T. Zeng and K. M. Engle, Isr. J. Chem., 2019, 59 DOI:10.1002/ijch.201900087.
- M.-H. Cao, N. J. Green and S.-Z. Xu, Org. Biomol. Chem., 2017, 15, 3105 RSC.
- G. Masson, C. Lalli, M. Benohoud and G. Dagousset, Chem. Soc. Rev., 2013, 42, 902 RSC.
- D. L. Boger and J. Y. Hong, J. Am. Chem. Soc., 1998, 120, 1218 CrossRef CAS.
- B. S. J. Blagg and D. L. Boger, Tetrahedron, 2002, 58, 6343 CrossRef CAS.
- S. R. Chemler and P. H. Fuller, Chem. Soc. Rev., 2007, 36, 1153 RSC.
- S. R. Chemler, S. D. Karyakarte and Z. M. Khoder, J. Org. Chem., 2017, 82, 11311 CrossRef CAS PubMed.
- A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC.
- M. Schultz and J. P. Wolfe, Synthesis, 2012, 351 Search PubMed.
- T. Piou and T. Rovis, Nature, 2015, 527, 86 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261 CrossRef CAS PubMed.
- A. Lerchen, T. Knecht, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 15166 CrossRef CAS PubMed.
- J. Cheng, X. Qi, M. Li, P. Chen and G. Liu, J. Am. Chem. Soc., 2015, 137, 2480 CrossRef CAS PubMed.
- Z. Hu, X. Tong and G. Liu, Org. Lett., 2016, 18, 1702 CrossRef CAS PubMed.
- K. M. Nakafuku, S. C. Fosu and D. A. Nagib, J. Am. Chem. Soc., 2018, 140, 11202 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273 CrossRef CAS PubMed.
- G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226 CrossRef CAS PubMed.
- S.-H. Cai, J.-H. Xie, S. Song, L. Ye, C. Feng and T.-P. Loh, ACS Catal., 2016, 6, 5571 CrossRef CAS.
- J. Jia, Y. A. Ho, R. F. Bülow and M. Rueping, Chem. – Eur. J., 2018, 24, 14054 CrossRef CAS PubMed.
- S. Zheng, Á. Gutiérrez-Bonet and G. A. Molander, Chem, 2019, 5, 1 Search PubMed.
- L. Angelini, J. Davies, M. Simonetti, L. Malet-Sanz, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2019, 58, 5003 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- H. Jiang and A. Studer, CCS Chem., 2019, 1, 38 CAS.
- X.-D. An and S. Yu, Tetrahedron Lett., 2018, 59, 1605 CrossRef CAS.
- M. D. Kärkäs, ACS Catal., 2017, 7, 4999 CrossRef.
- T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069 RSC.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC.
- S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC.
- L. Stella, Angew. Chem., Int. Ed. Engl., 1983, 22, 337 CrossRef.
- X. Bao, J. Li, W. Jiang and C. Huo, Synthesis, 2019, 51, 4507 CrossRef CAS.
- K. A. Margrey and D. Nicewicz, Acc. Chem. Res., 2016, 49, 1997 CrossRef CAS PubMed.
- M. Minozzi, D. Nanni and P. Spagnolo, Chem. – Eur. J., 2009, 15, 7830 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751 CrossRef CAS.
- T. Tsuritani, H. Shinokubo and K. Oshima, Org. Lett., 2001, 3, 2709 CrossRef CAS PubMed.
- Y. Li and Q. Zhang, Synthesis, 2015, 159 Search PubMed.
- K. Kaneko, T. Yoshino, S. Matsunaga and M. Kanai, Org. Lett., 2013, 15, 2502 CrossRef CAS PubMed.
- H. Zhang, W. Pu, T. Xiong, Y. Li, X. Zhou, K. Sun, Q. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2013, 52, 2529 CrossRef CAS PubMed.
- D. Wang, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 2054 CrossRef CAS PubMed.
- D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed.
- H. Xiao, H. Shen, L. Zhu and C. Li, J. Am. Chem. Soc., 2019, 141, 11440 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, L. Tang, H.-J. Tang, L. Wang, C. Zhu and C. Feng, J. Am. Chem. Soc., 2018, 140, 10695 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- X.-D. An, Y.-Y. Jiao, H. Zhang, Y. Gao and S. Yu, Org. Lett., 2018, 20, 401 CrossRef CAS PubMed.
- X.-D. An and S. Yu, Synthesis, 2018, 3387 CAS.
- X.-D. An, H. Zhang, Q. Xu, L. Yu and S. Yu, Chin. J. Chem., 2018, 36, 1147 CrossRef CAS.
- H. Jiang and A. Studer, Chem. – Eur. J., 2019, 25, 516 CAS.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 10707 CrossRef CAS PubMed.
- S. P. Morcillo, E. D. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 CrossRef CAS PubMed.
- H. Jiang, G. Seidler and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 16528 CrossRef CAS PubMed.
- Y. Moon, B. Park, I. Kim, G. Kang, S. Shin, D. Kang, M.-H. Baik and S. Hong, Nat. Commun., 2019, 10, 4117 CrossRef PubMed.
- D. Zheng and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 15803 CrossRef CAS PubMed.
- L. Xu, X.-Q. Mou, Z.-M. Chen and S.-H. Wang, Chem. Commun., 2014, 50, 10676 RSC.
- Z. Liu and Z.-Q. Liu, Org. Lett., 2017, 19, 5649 CrossRef CAS PubMed.
- B. Yang, X. Ren, X. Shen, T. Li and Z. Lu, Chin. J. Chem., 2018, 36, 1017 CrossRef CAS.
- P. Renaud, C. Ollivier and P. Panchaud, Angew. Chem., Int. Ed., 2002, 41, 3460 CrossRef CAS PubMed.
- P. Panchaud and P. Renaud, J. Org. Chem., 2004, 69, 3205 CrossRef CAS PubMed.
- G. Lapointe, K. Schenk and P. Renaud, Org. Lett., 2011, 13, 4774 CrossRef CAS PubMed.
- L. Chabaud, Y. Landais and P. Renaud, Org. Lett., 2015, 7, 2587 CrossRef PubMed.
- A. Kapat, E. Nyfeler, G. T. Giuffredi and P. Renaud, J. Am. Chem. Soc., 2009, 131, 17746 CrossRef CAS PubMed.
- P. Schär and P. Renaud, Org. Lett., 2006, 8, 1569 CrossRef PubMed.
- K. Weidner, A. Giroult, P. Panchaud and P. Renaud, J. Am. Chem. Soc., 2010, 132, 17511 CrossRef CAS PubMed.
- K. Mgller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed.
- G. Dagousset, A. Carboni, E. Magnier and G. Masson, Org. Lett., 2014, 16, 4340 CrossRef CAS PubMed.
- N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375 RSC.
- X. Geng, F. Lin, X. Wang and N. Jiao, Org. Lett., 2017, 19, 4738 CrossRef CAS PubMed.
- M. Zhang, W. Li, Y. Duan, P. Xu, S. Zhang and C. Zhu, Org. Lett., 2016, 18, 3266 CrossRef CAS PubMed.
- Q. Yang, C. Li, Z.-C. Qi, X.-Y. Qiang and S.-D. Yang, Chem. – Eur. J., 2018, 24, 14363 CrossRef CAS PubMed.
- R. Xu and C. Cai, Org. Biomol. Chem., 2019, 17, 8541 RSC.
- Y.-Y. Liu, X.-H. Yang, R.-J. Song, S. Luo and J.-H. Li, Nat. Commun., 2017, 8, 14720 CrossRef PubMed.
- A. Bunescu, T. M. Ha, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 10555 CrossRef CAS PubMed.
- N. Zhu, T. Wang, L. Ge, Y. Li, X. Zhang and H. Bao, Org. Lett., 2017, 19, 4718 CrossRef CAS PubMed.
- B. Qian, S. Chen, T. Wang, X. Zhang and H. Bao, J. Am. Chem. Soc., 2017, 139, 13076 CrossRef CAS PubMed.
- X. Bao, T. Yokoe, T. M. Ha, Q. Wang and J. Zhu, Nat. Commun., 2018, 9, 3725 CrossRef PubMed.
- S. N. Gockel, T. L. Buchanan and K. L. Hull, J. Am. Chem. Soc., 2018, 140, 58 CrossRef CAS PubMed.
- Y. Xiong, X. Ma and G. Zhang, Org. Lett., 2019, 21, 1699 CrossRef CAS PubMed.
- L. Zhang, X. Kong, S. Liu, Z. Zhao, Q. Yu, W. Wang and Y. Wang, Org. Lett., 2019, 21, 2923 CrossRef CAS PubMed.
- X.-H. Ouyang, Y. Li, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 6659 CrossRef CAS PubMed.
- X.-L. Yu, J.-R. Chen, D.-Z. Chen and W.-J. Xiao, Chem. Commun., 2016, 52, 8275 RSC.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 11960 CrossRef CAS PubMed.
- N. T. Jui, J. A. O. Garber, F. G. Finelli and D. W. C. MacMillan, J. Am. Chem. Soc., 2012, 134, 11400 CrossRef CAS PubMed.
- C. Um and S. R. Chemler, Org. Lett., 2016, 18, 2515 CrossRef CAS PubMed.
- D. P. Hari, T. Hering and B. König, Angew. Chem., Int. Ed., 2014, 53, 725 CrossRef.
- S. Kindt, K. Wicht and M. R. Heinrich, Org. Lett., 2015, 17, 6122 CrossRef CAS PubMed.
- M. A. Zeller, M. Riener and D. A. Nicewicz, Org. Lett., 2014, 16, 4810 CrossRef CAS PubMed.
- T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Science, 2018, 361, 1369 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |