
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic strategies tailoring colours in multichromophoric organic nanostructures
Olesia
Kulyk
 ab,
Lou
Rocard
a,
Laura
Maggini
c and
Davide
Bonifazi
*ac
ab,
Lou
Rocard
a,
Laura
Maggini
c and
Davide
Bonifazi
*ac
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
bSchool of Chemistry, Department of Organic Chemistry, V. N. Karazin Kharkiv National University, Svobody sq. 4, Kharkiv, 61022, Ukraine
cInstitute of Organic Chemistry, Faculty of Chemistry, University of Vienna, Währinger Strasse 38, 1090, Vienna, Austria. E-mail: davide.bonifazi@univie.ac.at
First published on 27th October 2020
Abstract
There has never been a time when colour did not fascinate humanity, inspiring an unceasing manufacturing of a kaleidoscopic variety of dyes and pigments that brought about great revolutions in art, cosmetics, fashion, and our lifestyle as a whole. Over the centuries these tints evolved from raw earths to molecular masterpieces devised by expert chemists whose properties are now being exploited far beyond traditional applications. Mimicking Nature, a timely challenge, regards the preparation of innovative and highly efficient multi-coloured architectures structured at the molecular and nanoscopic scale with specific light-absorbing and light-emitting properties. This tutorial review provides an overview on the chemical strategies developed to engineer and customise these ingenious coloured nanostructures tackling the current performance of organic matter in cutting edge technological sectors, such as solar energy conversion.
 Olesia Kulyk | Olesia Kulyk was born in Kriviy Rih in 1988. After obtaining her Master Degree in Organic Chemistry at V. N. Karazin Kharkiv National University (Ukraine) in 2010, she did her PhD (2014) under the supervision of Dr M. Kolosov working on the functionalization of pyrimidine heterocycles. In 2015 she joined the group of Prof. D. Bonifazi (first at the University of Namur and then at Cardiff University). Her research work focused on the functionalization of BN nanoparticles and synthesis of artificial multichromophoric antenneas. In 2019, she joined V. N. Karazin Kharkiv National University as an assistant professor. |
 Lou Rocard | Lou Rocard was born in Versailles in 1989. She obtained her Master Degree in Biomolecular Chemistry at the University of Montpellier in 2012 working with β-peptides inhibitors under the supervision of Dr Eric at Hoffmann-la-Roche in Basel. In 2012, she joined the group of Prof. Bonifazi as PhD (first at the University of Namur and then at Cardiff University) working on the design of peptidic multichromophoric architectures. In 2018, she enrolled as lecturer at University of Angers and joined the SOMaf team, to work on the synthesis of new chromophores and their optoelectronic characterisation. She recently joined the ENS ParisSaclay as a postdoctoral researcher in the Supramolecular and Macromolecular Photophysics and Photochemistry (PPSM) laboratory to develop multicatalytic reactions controlled by light. |
 Laura Maggini | Laura Maggini obtained her Master Degree in Organic Chemistry at the University of Rome – Sapienza in 2006. She joined the group of Prof. Bonifazi at the University of Namur as a PhD candidate, where she pioneered the supramolecular functionalisation of CNTs. Funded by two consecutive MSCA Fellowships she first joined the group of Prof. De Cola at I.S.I.S., University of Strasbourg, to develop “breakable” silica NPs, and later the Institute for Manufacturing at the University of Cambridge with Dr De Volder to study the hybrid assembly of nanomaterials via manufacturing technologies. Since 2020 she joined the Institute for Organic Chemistry at the University of Vienna as Senior Researcher developing novel manufacturing platforms and scale up production processes for organic supramolecular materials. |
 Davide Bonifazi | Davide Bonifazi obtained the “Laurea” in “Industrial Chemistry” from the University of Parma. He joined the group of Prof. François Diederich as PhD at the ETH Zürich (2000–2004). He was awarded the Silver Medallion of the ETH for his doctoral dissertation (2005). After a one-year postdoctoral fellowship with Prof. Maurizio Prato at University of Trieste, he joined the same University as a research associate first, and then as a part-time Professor (2012–2015). In 2006, he joined the University of Namur (Belgium) as Junior Professor (2006–2011) and as Professor of Organic Chemistry (2012–2015). In 2016 he moved to the School of Chemistry at Cardiff University (UK) as Chair Professor of Organic Supramolecular Chemistry until his current appointment (2020) as Chair Professor of Organic Chemistry at the Institute of Organic Chemistry at the University of Vienna (Austria). His activities are focused on the creation of functional organic molecules in interdisciplinary projects through targeted organic synthesis, self-assembly, and self-organization of architectures in solution and on surfaces, physicalorganic studies, material- and bio-based design. |
Key learning points• Synthetic strategies tailoring colours in multichromophoric systems with tuneable absorptive and emissive properties.• Strength and limitations of covalent and non-covalent bottom-up approaches for the synthesis of light-harvesting systems. • Tailoring the photophysical properties of multichromophoric architectures: structure/property correlations. |
1. Introduction
We live in a vibrant world, in which the kaleidoscopic palette of colours developed by Nature supports life by covertly taking part to vital physiological and biological processes, while also enriching our lives through aesthetic pleasure (Fig. 1). Animals use colours for many different purposes, such as camouflage, and mating. Plants exhibit an incredible variety of colours, which are skilfully exploited not only to interact with their environment (i.e., attract pollinators), but also to harvest energy from sunlight and transform it into chemical energy for later usage (i.e., photosynthesis).1 The scenic green, yellow and orange colours of leaves are due to molecules (chlorophylls, green; xanthophylls, yellow; carotenes, orange) which, arranged in sophisticated architectures templated by proteins, work together to harvest energy from sunlight and transfer it through space to a reaction centre where it is captured in the form of carbohydrates. | ||
| Fig. 1 (a) Colour array; (b) autumnal leaves; (c) peacock feathers; and (d) bioluminescent jellyfish. | ||
Colour is perceived as a consequence of the interaction (e.g., absorption, reflection, refraction, scattering, and diffraction) of matter with the different wavelengths composing white light, or when light is emitted only at specific wavelengths.2 Coloured molecules (all dyes and most pigments) are generally complex organic structures exhibiting a colour-yielding unit known as a chromophore. This moiety absorbs light within a definite range of wavelengths, whilst reflecting and/or refracting the non-absorbed light to manifest colour. For example, chlorophyll is a pigment capable of absorbing the blue and yellow/red regions of the visible light, while reflecting green. This reflection is what confers the leaves their archetypal colour. When chlorophyll degradation occurs during leaf senescence revealing the carotenoids, leaves turn to their emblematic autumnal orange/yellow colour. Structural coloration can occur when colour is produced by interference of microscopically structured surfaces with visible light.2 For example, peacock tail feathers are naturally tinted brown, but their microscopic structure makes them reflect blue, turquoise, and green, as well as appear iridescent. Colour can also be produced by a chemical reaction (i.e., chemiluminescence). Indeed in Nature certain species (e.g., jellyfish, fireflies, etc.) are able to emit coloured light as a result of a reaction between a chromophore and an enzyme (i.e., bioluminescence involving luciferin dye and luciferase enzyme) to divert attacks, as warning, or to illuminate their environment.
Since the beginning of civilisation, humans have been fascinated by colours and promptly developed skills to manufacture manipulable substances to employ as communication media (e.g., indicate a source of food or water, mark territory). Soon enough humans transcended the manipulation of colour for merely practical purposes, and began using colour also to embellish themselves and their surroundings, and as a medium to express their thoughts and feelings.3 Early colours were as simple as coloured earths and charcoal. With time the preparation of colours evolved from the forthright use of natural resources to the transformation of bulk materials into processable dyes and pigments by talented artisans.3 In modern times, the production of colours further evolved to the molecular devising of new chromophores performed by skilful chemists.4 Chromophores have thus become sophisticated functional molecules, with disruptive potential far beyond tinting and painting (Fig. 2), namely in modern organic electronics and solar energy conversion systems (e.g., organic light emitting diodes displays, organic photovoltaics, organic semiconductors, etc.), printing (e.g., ink-jet printing, security printing, etc.), optical data storage, and biotechnology (e.g., as biomarkers, fluorescent sensors, anticancer treatments, etc.). Chemists are thus required to overcome the challenge of tailoring the photophysical and electrochemical properties of chromophores in order to fully exploit the potential of colours in such high-tech applications and meet the increasingly demanding operational specifications.5
 | ||
| Fig. 2 Traditional and frontier applications of colours: (a) tinting; (b) displays; (c) solar cells; (d) security printing; (e) biological staining. | ||
Generally, colours can be identified following two different descriptive models: the subtractive (RYB – red, yellow and blue) and the additive model (RGB – red, green and blue). Both use a small number of primary colours that mix to produce a range of colours, but the way they do is different. The RYB mixing model involves the visual colours obtained by subtracting the absorbed light at certain wavelengths and reflecting the rest of it, whereas in the RGB model the colour derives from the mixing of red, green and blue light emitted directly from a source. Given the fact that current literature in the field is more focused on the properties of excited states, in this review we will mainly refer to the tuning of emissive colours using the RGB model. However, the RYB model is a more suitable descriptive model when the focus is on the absorption properties (e.g., in photovoltaics, black solids are desired to maximise the absorption of the solar irradiation). Finally, for printing technologies, the interested reader should refer to cyan, magenta, yellow and black (CMYK model) as the primary colours (not discussed in this review).
1.1 Chemical strategies to tailor colours
Chemists have developed three main strategies to tune the properties of chromophores at the nanoscale (Fig. 3): (i) modification of the chromophore's molecular structure (e.g., conjugation, doping, etc.); (ii) assembly of chromophores in solid phases (i.e., the pigment strategy); and (iii) engineering of covalent/non-covalent multichromophoric architectures.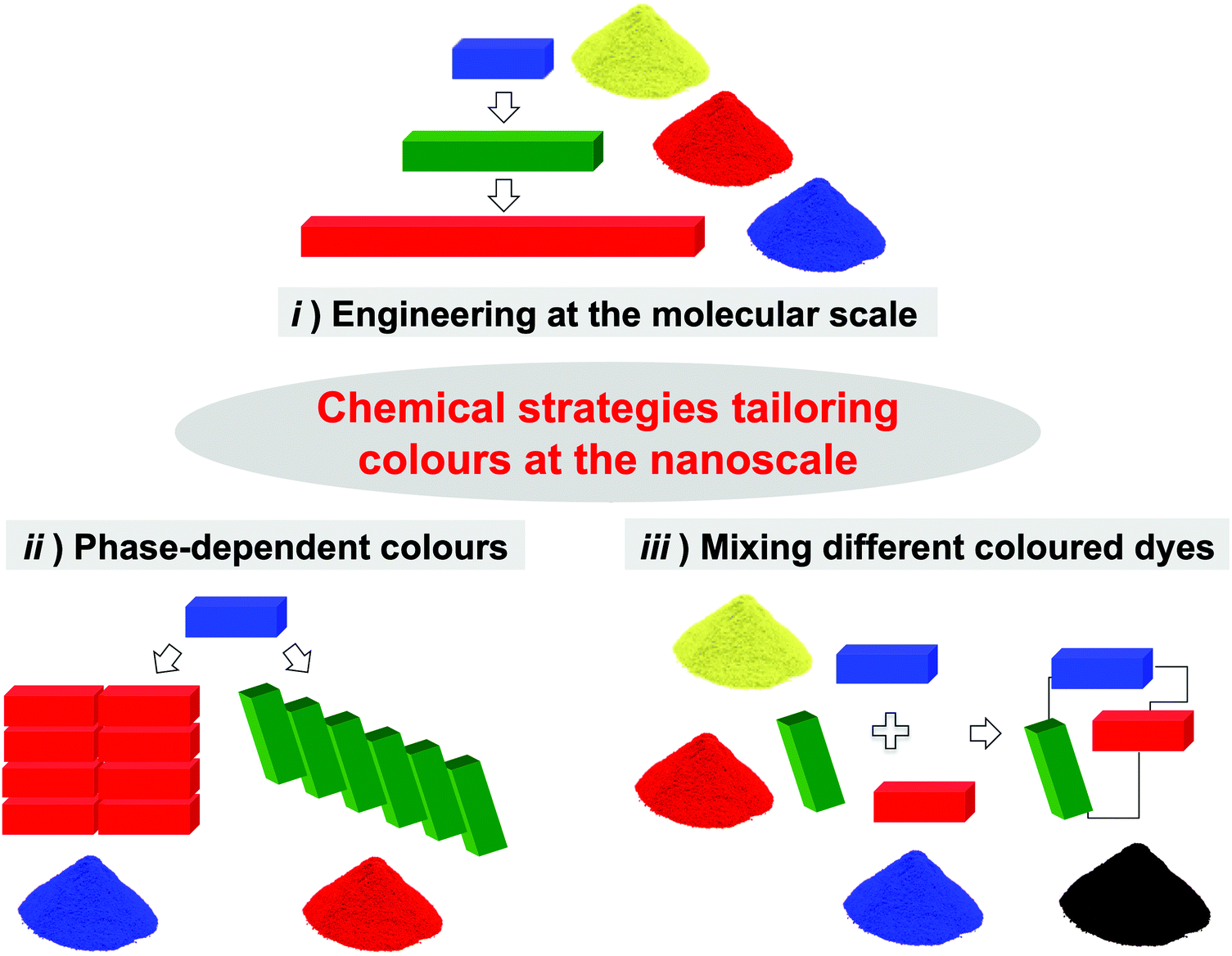 | ||
| Fig. 3 Strategies (i)–(iii) for tailoring colours at the nanoscale. The parallelepipeds in the cartoon represent the molecular entities, whilst the powders represent the final materials. The RGB model is employed for defining the photophysical (absorption/emission) properties of the chromophores while the corresponding material visually appears with its complementary colour (RYB). For the sake of simplicity, we assume the photophysical properties of the chromophores in approach iii) do not change upon assembly. | ||
A chromophore's absorption and emission properties depend on its electronic structure,6 which is in turn determined by molecular features such as size, planarity, π-conjugation, and topology of the given molecule. Chemists have nowadays developed an assorted array of synthetic tools to tailor such features (e.g., addition of electroactive substituents, replacement of C atoms with isostructural/isoelectronic heteroatoms, etc.; approach i, Fig. 3). Yet, the scope of this strategy is often limited by the complexity of the organic synthesis, as not every chromophore's structure allows the whole range of chemical modifications tailoring the required photophysical properties.7 The second strategy entails the assembly of chromophores into bulk solid phases, whose organisation dictates the optical properties of the resulting material (approach ii, Fig. 3). For example, a broad variety of dyes such as cyanines, porphyrins, phthalocyanines and perylene diimides (PDI), can self-assemble forming J-aggregates featuring red-shifted bands in their absorption spectra compared to the monomeric entities in solution.8 Some organic luminophores (e.g., hexaphenylsilole, tetraphenylethene) are non-emissive in dilute solutions, while they become highly luminescent in the solid state due to the aggregation induced emission (AIE) phenomenon.9 Unfortunately, the serendipity of aggregation and the structure-related stability of the assembled phases, limit the capacity of this approach to only certain chromophores, and certain colour shifts.
Taking inspiration from the fundamental chromophoric architectures present in Nature, the third path embroils the bottom-up assembly of different chromophoric modules through the formation of pre-programmed covalent or non-covalent bonds (approach iii, Fig. 3).10 The grouping of the different chromophores into a unique architecture gives rise to materials (represented as the powdery pile in Fig. 3 stained with the RYB colour code) the light emissive and absorption properties of which derive from the contribution of each single molecular component. The possibility to simultaneously control not only the type of chromophores employed, but also their relative molecular ratio, three-dimensional positioning, and interchromophoric interactions offers an incredibly powerful tool to meticulously and elegantly craft the absorptive and emissive properties of the assembly, conceivably enabling unlimited browsing through the colour spectrum, both in emission and in absorption.
1.2 Engineering approaches to functional multichromophoric architectures
Multichromophoric organic nanostructures have emerged as a new class of efficient light-emitting or light-absorbing architectures for light-harvesting, photosensitisation, photocatalysis, optoelectronic and sensor devices operating under low-energy-consumption regimes. This tutorial review has the scope to report a selection of relevant examples depicting bottom-up synthetic routes for the construction of these sophisticated multichromophoric nanostructures focusing on their application as artificial light-harvesting antennas (LHAs), and luminescent and chromogenic materials/devices. Specifically, we discuss the three main strategies developed to produce these nanostructures (Fig. 4): (i) the supramolecular mixing of dyes; (ii) the sequential interconnection of dyes through covalent bonding; and (iii) the templated assembly of molecular dyes. | ||
| Fig. 4 Bottom-up synthetic strategies for the creation of coloured nanoarchitectures. The RGB model is employed for definig the photophysical (absorption/emission) properties of the chromophores. | ||
2. Supramolecular mixing
Supramolecular architectures are good candidates for engineering multichromophoric structures as they can be formed from easy-to-prepare and modular components through non-covalent assembly. The co-assembly can be achieved by exploiting supramolecular interactions such as H-bonding, π–π stacking, host–guest inclusions, van der Waals forces, and metal coordination.In this context, supramolecular mixing provides a simple access to topologically complex systems, where non-covalent bonds bring molecules in close spatial proximity allowing for tight electronic communication. In this section (Fig. 5), we focus on the supramolecular structures prepared by serendipitous mixing of chromophores (e.g., organogels, nanocrystals, etc.) as well as pre-programmed supramolecular interactions (e.g., polymers, MOFs, etc.).
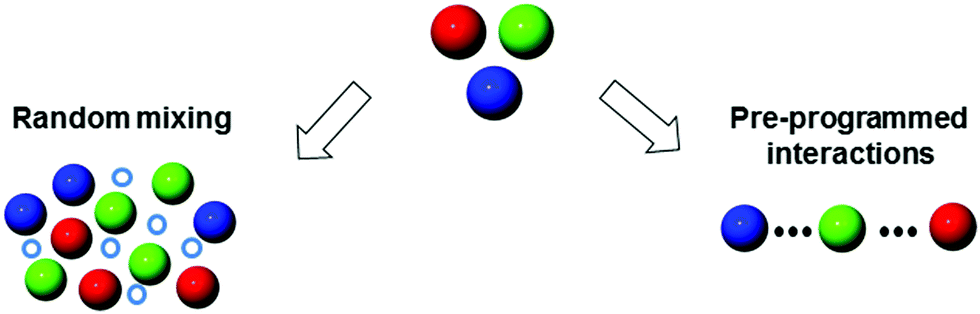 | ||
| Fig. 5 From serendipitous (organogel, crystal, etc.) to programmed (polymers, MOFs, etc.) supramolecularly-mixed coloured nanostructures. | ||
2.1 Organogels
Supramolecular mixing achieved through gelation can provide functional materials with tuneable optoelectronic properties, having potential applications in the construction of electronic devices as well as LHA.11 Chromophore-based organogels formed by non-covalent interactions are widely used for light harvesting due to their ability to facilitate the energy transfer processes and hence to increase the quantum efficiency of LHA. A significant number of LH organogels formed by simple mixing of chromophore-based donor and acceptor couples have been reported; here we discuss the most representative examples to demonstrate the concept of the approach. Ajayaghosh et al. used organogelator oligo(phenylenevinylene) (OPV) 1 as an energy donor to promote energy transfer to Rhodamine B 2 within the mixed organogel 1·2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol. ratio; Fig. 6a).12 The energy transfer between the two molecules was illustrated by the amplification of the acceptor emission at 620 nm with the concomitant quenching of OPV emission between 500 and 650 nm (λexc = 470 nm), as shown in Fig. 6b. Its efficiency was determined to be 90% from the quenching of the OPV fluorescence profile. Later, the same group reported that an even higher energy transfer was achieved when less than 2 mol% of a new acceptor 3 was encapsulated in the same OPV-based organogel, resulting in a 95% quenching of the OPV emission (Fig. 6c).13 Fluorescence microscopy images of gel fibres before and after encapsulation of the acceptor depict the occurrence of the energy transfer as the initial yellow emission of the gel turned to red in the presence of 3 upon excitation of the OPV units (Fig. 6d).
:1 mol. ratio; Fig. 6a).12 The energy transfer between the two molecules was illustrated by the amplification of the acceptor emission at 620 nm with the concomitant quenching of OPV emission between 500 and 650 nm (λexc = 470 nm), as shown in Fig. 6b. Its efficiency was determined to be 90% from the quenching of the OPV fluorescence profile. Later, the same group reported that an even higher energy transfer was achieved when less than 2 mol% of a new acceptor 3 was encapsulated in the same OPV-based organogel, resulting in a 95% quenching of the OPV emission (Fig. 6c).13 Fluorescence microscopy images of gel fibres before and after encapsulation of the acceptor depict the occurrence of the energy transfer as the initial yellow emission of the gel turned to red in the presence of 3 upon excitation of the OPV units (Fig. 6d).
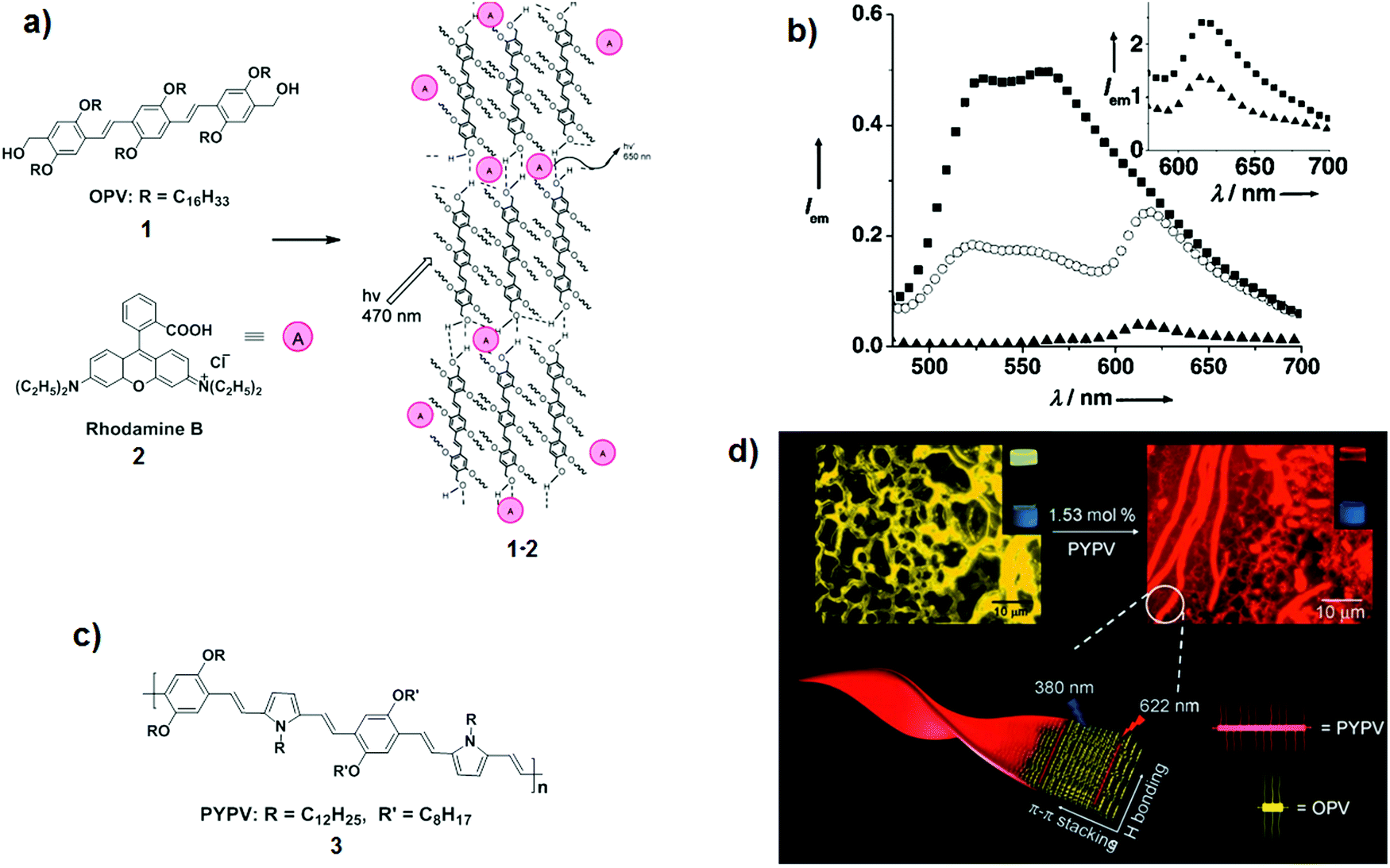 | ||
| Fig. 6 (a) Structures of energy donor OPV 1 and energy acceptor Rhodamine B 2 used to form organogel 1·2 and probable assembly pattern of Rhodamine B dispersed in the OPV gel; (b) fluorescence spectra in cyclohexane/CHCl3 (16:1) of 1 (black square), 1·2 (white circle), and 2 (black triangle) at λexc = 470 nm;12 (c) energy acceptor 3; (d) fluorescence microscopy images of OPV cyclohexane gel before and after encapsulation of 3 upon illumination at 365 nm.13 Adapted with permission from ref. 12 and 13. Copyrights 2003 and 2007, John Wiley & Sons, Inc. | ||
Similarly, in 2011, Del Guerzo and co-workers reported the preparation of LH gel nanofibers by co-assembly of a blue-light emitting donor 4 and green and red emitting acceptors 5 and 6 (Fig. 7a and b).14 Anthracene derivative 4 was exploited as a light-harvesting organogelator capable of hosting and sensitising the acceptors. Tetracene-based acceptors 5 and 6 were engineered to display green (λem = 502 nm, Φem = 53%) and orange-red (λem = 555 nm, Φem = 69%) emission signals, respectively, and to be structurally similar to anthracene-based donor 4. It was shown that, by changing the ratio of acceptors (from 0 to 0.020 equiv.), in co-assembled gel 4·5·6 the excitation energy migrates from donor moiety 4 to acceptors 5 and 6, modulating the colour of the emission. For instance, the addition of 0.012 equiv. of 5 and 6 to 4 gave a white-light emitting gel 4·5·6 under UV light (Fig. 7c and d), suggesting the occurrence of an energy transfer from molecule 4 to 5 and 6.
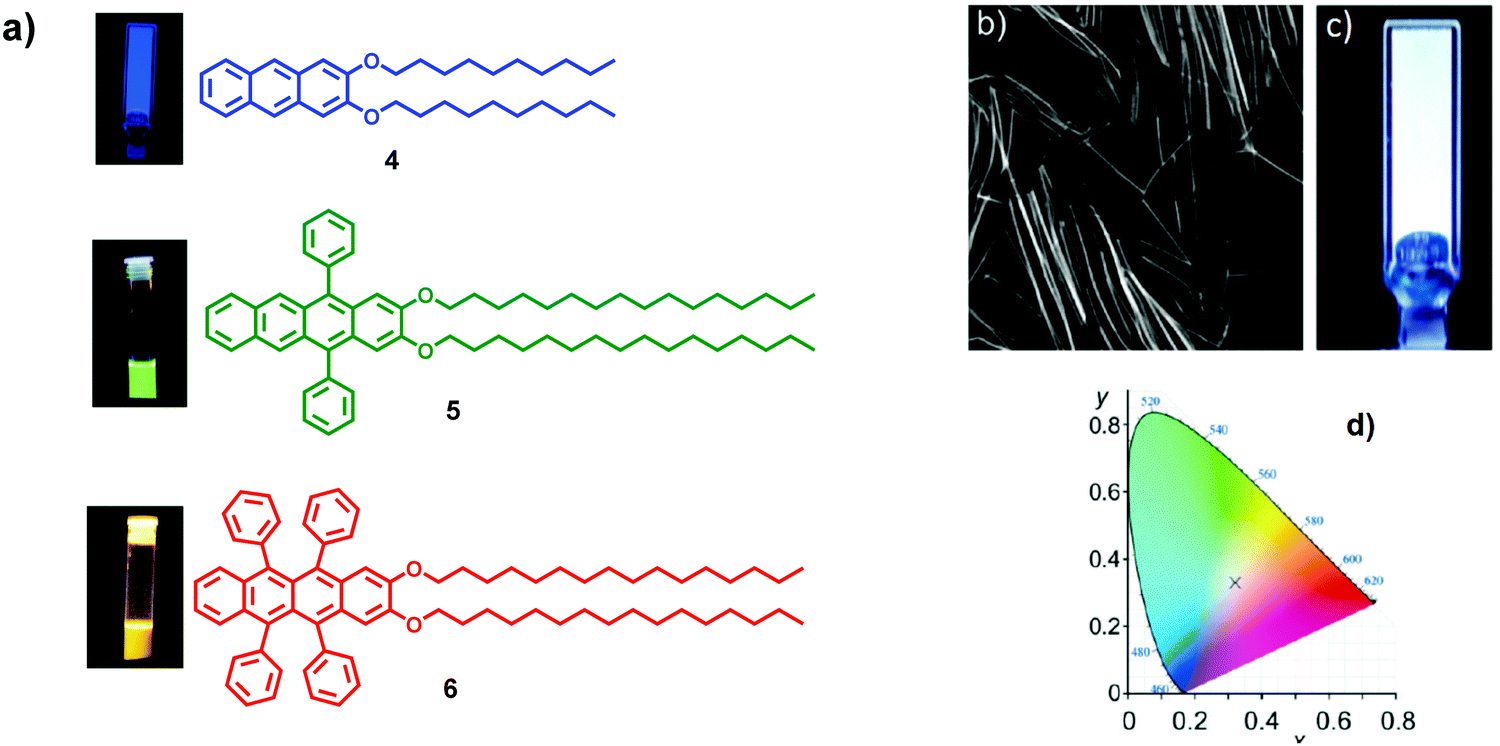 | ||
| Fig. 7 (a) Molecular structures of blue 4, green 5 and red 6 emitters and images of the gel of 4 in DMSO and the solutions of 5 and 6 in THF under UV light, λexc = 365 nm; (b) fluorescence intensity confocal microscopy image (30 × 30 μm, 80 × 80 nm/pixel, λexc = 385 nm, λem > 405 nm) of the W-gel 4·5·6; (c) image of W-gel 4·5·6 (2.0 mM of 4 in DMSO with 0.012 equiv. of 5 and 6) under UV light, λexc = 365 nm; (d) CIE 1931 chromaticity coordinates of W-gel 4·5·6 in DMSO: (x) 0.319, (y) 0.332. Adapted with permission from ref. 14. Copyright 2011, American Chemical Society. | ||
2.2 Organic nanocrystals
Another engineering approach is based on the co-assembly of organic nanocrystals. Yang and co-workers described difluoro-boron β-diketonate derivatives 7–9 mimicking chlorosomes.15Uniform nanocrystals 7·8 and 7·9 were obtained by co-assembling energy donor 7 with different amounts of either acceptor 8 or 9 (0.0001–0.1 mol%) in water, and in the presence of surfactant sodium dodecylbenzene sulfonate (Fig. 8a–c). Upon excitation at the maximum absorption wavelength of 7 (λexc = 480 nm), quenching of the donor's emission at 567 nm was observed when increasing the acceptor's doping ratio (Fig. 8d and e). Simultaneously, new bands appeared at 616 nm or 636 nm, ascribable to the emission of molecules 8 and 9, respectively. By means of steady-state and time-resolved fluorescence spectroscopy, the energy transfer efficiencies were estimated to be 95% and 90% for systems 7·8 and 7·9, respectively, at a doping ratio of 0.1 mol%. This indicates that the excitation energy absorbed by approximately 1000 donors was efficiently funnelled to a single acceptor. Moreover, the energy transfer results in the variation of the emission colour of 7·9, from yellow to orange and red upon increasing the doping ratio of acceptor 9 (Fig. 8f). At a doping concentration of 0.01 mol%, 28- and 29-fold enhancement of the acceptor emission for 7·8 and 7·9 nanocrystals, respectively, was observed.
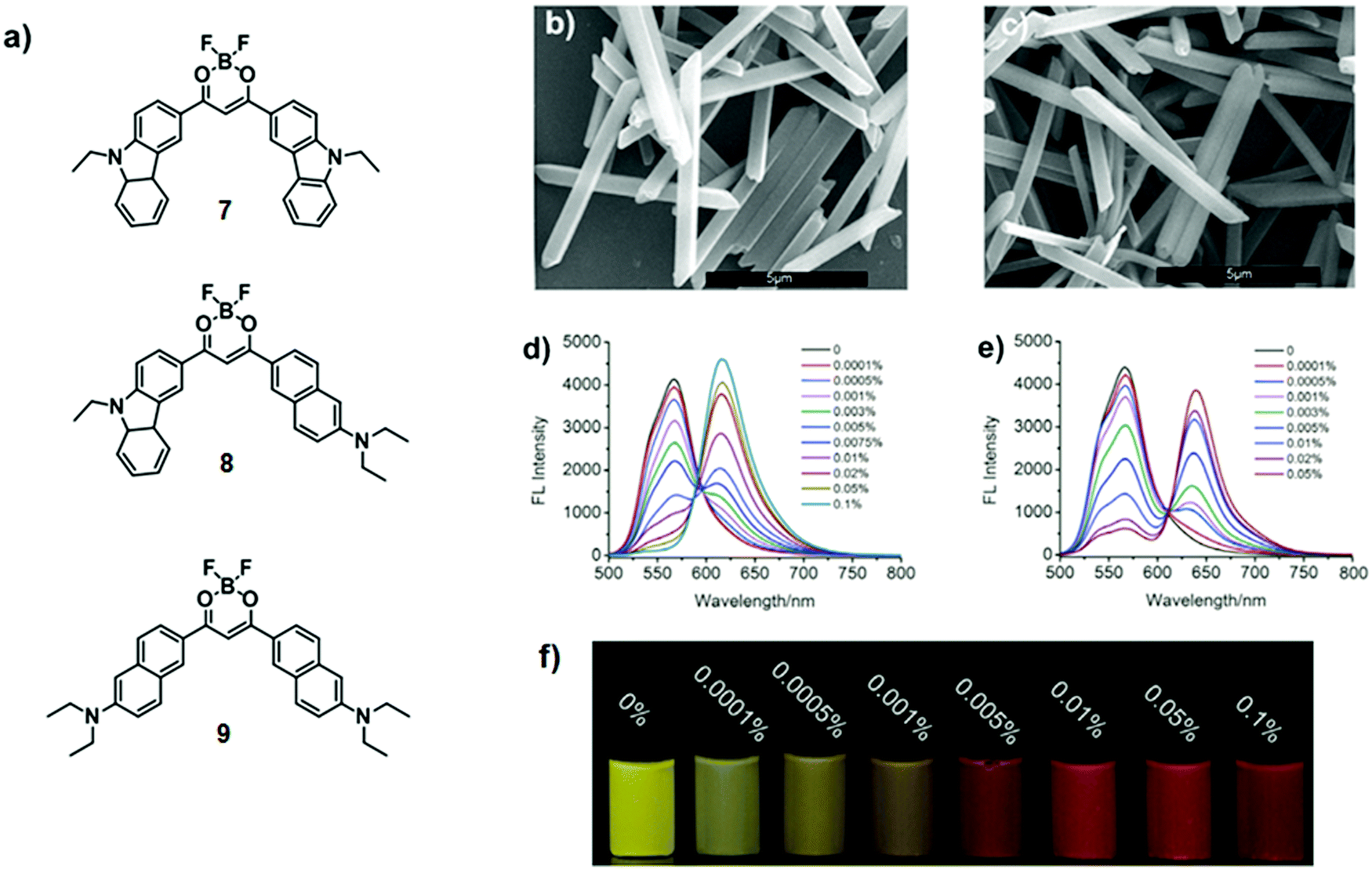 | ||
| Fig. 8 (a) Chemical structures of difluoroboron β-diketonate derivatives 7–9; (b and c) SEM-images of 7·8 and 7·9 nanocrystals doped with 0.1 mol% 8 and 9, respectively; (d and e) fluorescence spectra of aqueous dispersions of 7·8 and 7·9 nanocrystals doped with different amounts of 8 (d) and 9 (e), λexc = 480 nm; (f) fluorescence images of aqueous dispersions of nanocrystals 7·9 doped with different amounts of 9. Adapted with permission from ref. 15. Copyright 2016, John Wiley & Sons, Inc. | ||
Comparably, Hu and co-workers reported a simple method to construct materials with tuneable emissive colours using organic co-crystals.16 Octafluoronaphthalene (OFN; 10) – pyrene (11) 1,2,4,5-tetracyanobenzene (TCNB; 12) tri-component co-crystals 10·11·12 were obtained by introducing different amounts of TCNB acceptor into a pyrene–OFN 10·11 donor host via a competitive intermolecular interaction strategy. Blue emitting pyrene–OFN crystals arise from arene-perfluoroarene (AP) interactions between pyrene 11 and OFN 10, whilst pyrene–TCNB are obtained by charge transfer interactions between pyrene 11 and TCNB 12 (Fig. 9a–c). As depicted in Fig. 9d, absorption spectra of co-crystals 10·11·12 display negligible changes in a low doping ratio of 12 (0.5% and 1%), whereas at 3% of TCNB quenching of the pyrene–OFN-centred fluorescence at 403 nm was observed concomitant with an amplification of the pyrene–TCNB emission at 535 and 575 nm (λexc = 365 nm, Fig. 9e). Notably, the doped pyrene–OFN co-crystals exhibited tuneable emission colours, including white-light emission at a 1% doping ratio of TCNB 12.
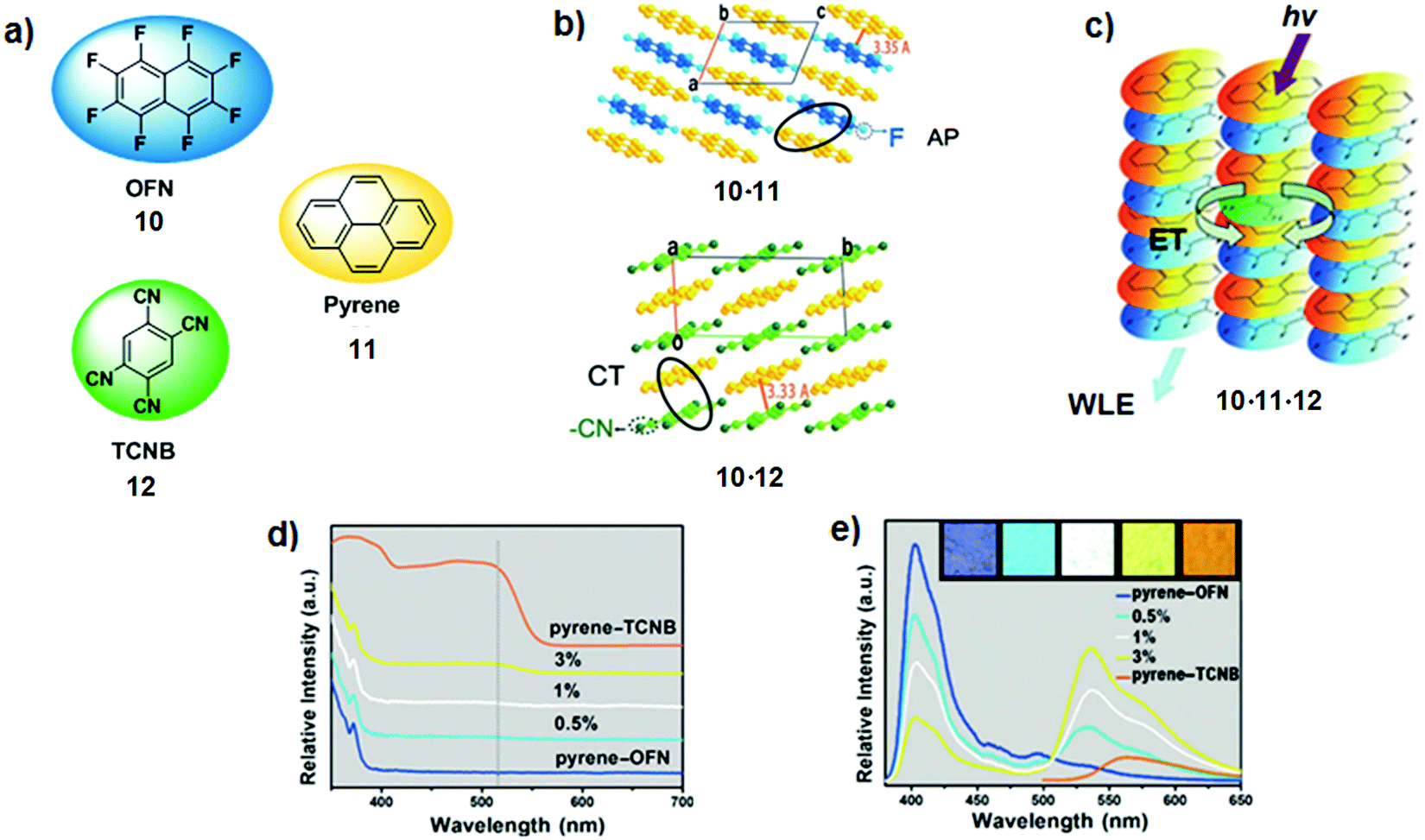 | ||
| Fig. 9 (a) Chemical structures of OFN, pyrene and TCNB 10–12; (b) molecular packing modes of pyrene–OFN 10·11 and pyrene–TCNB 11·12; (c) schematic illustration of organic cocrystal 10·11·12; (d) UV-Vis absorption spectra of pyrene–OFN, pyrene–TCNB and TCNB-doped pyrene–OFN (dashed line shows the absorption bands of pyrene–TCNB in doped pyrene–OFN cocrystals); (e) fluorescence spectra of pyrene–OFN with different TCNB doping ratios, λexc = 365 nm. The inset shows the photographs excited by a UV lamp. Adapted with permission from ref. 16. Copyright 2017, John Wiley & Sons, Inc. | ||
2.3 Supramolecular polymers
Other attractive architectures in the field are supramolecular polymers. They are peculiar polymers, in which monomers are held together via highly directional and reversible non-covalent interactions. Therefore, intermolecular interactions are responsible for the degree of polymerisation, chain lifetime, and conformational flexibility of the supramolecular assemblies. In this regard, in a seminal report, Schenning and co-workers described the formation of three-component supramolecular polymers through self-complementary hydrogen bonding interactions between ureido-pyrimidinone (UPy) units.17 Blue-emitting oligofluorene 13 (Φem = 50%), green-emitting oligo(phenylene–vinylene) 14 (Φem = 45%), and red-emitting perylenebisimide 15 (Φem = 55%) were functionalised with the UPy group at both extremities, giving RGB emissive bis-UPy chromophores (Fig. 10a and b). A large dimerisation constant Kdim (ca. 106 M−1 in CHCl3) between UPy moieties ensures the existence of bis-UPy chromophores 13, 14, and 15 as hydrogen-bonded polymers at given concentrations (10−6 to 10−5 M). Fluorescence titration experiments (Fig. 10c) of donor 13 with either acceptor 14 or 15 highlighted the quenching of the donor emission when the samples were excited at the maximum absorption wavelength of 13 (λexc = 364 nm). Concomitantly, the emission of acceptors 14 and 15 increased, suggesting that UPy-based hydrogen bonding interactions favour an efficient energy transfer. Simultaneous incorporation of luminophores 14 and 15 into the chains of 13 gave the supramolecular copolymer 13·14·15 containing all three RGB species. At a certain ratio (13/14/15 = 59:33:8), white-light emission was achieved through energy transfer (Fig. 10c and d).17 In contrast, white-light emission was not attained in mixtures of bare oligo(fluorene), oligo(phenylene–vinylene), and perylene bis-imide chromophores that lacked UPy units.
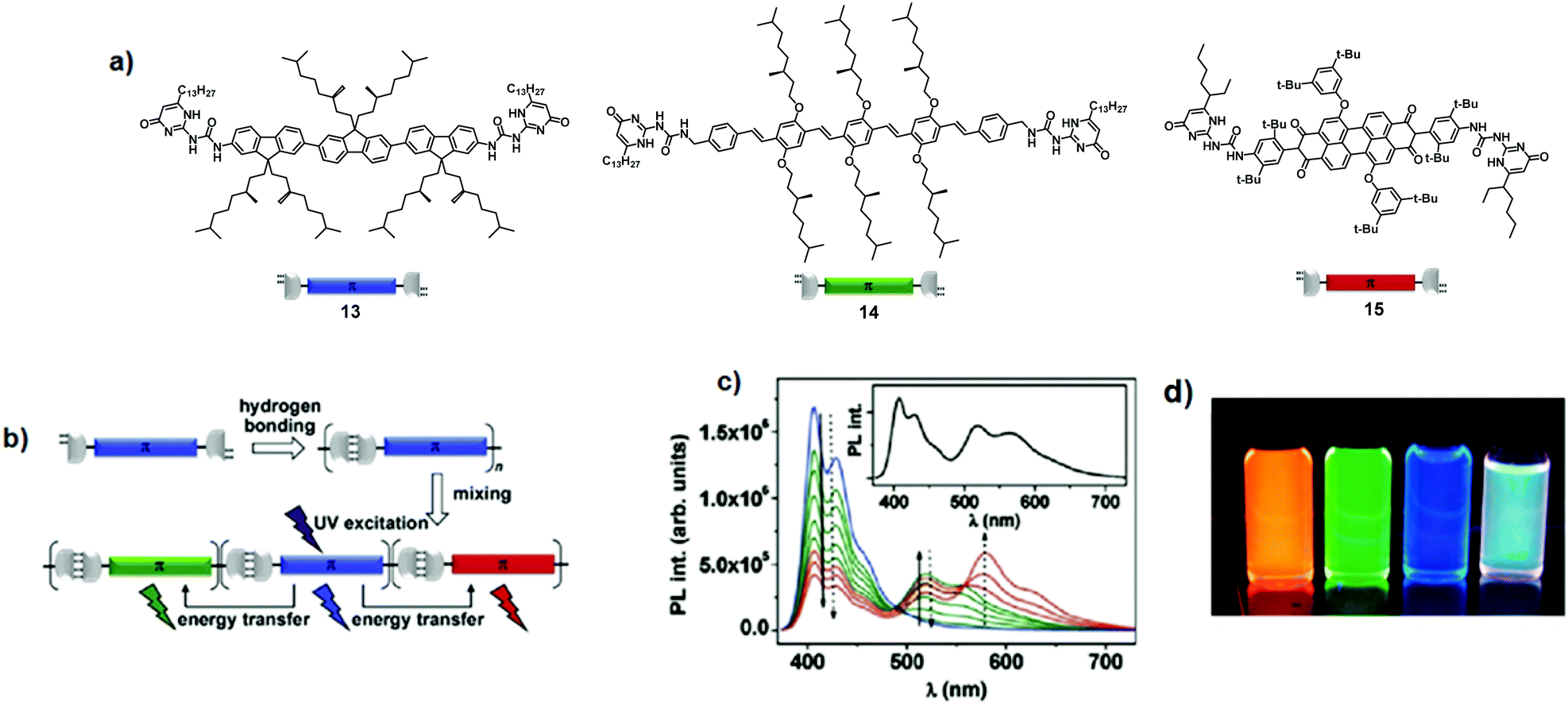 | ||
| Fig. 10 (a) Chemical structures of chromophores 13, 14 and 15; (b) supramolecular polymerisation; (c) photoluminescence spectra of titration experiments in CHCl3 (blue: pure 13, green: successive addition of 14, red: further addition of 15). The solid arrows indicate spectral changes upon addition of 14 to 13, the dotted arrows upon addition of 15 to a mixture of 13 and 14. The inset shows the emission spectrum corresponding to a ratio of 59:33:8. [13] = 1.6 × 10−6 M, λexc = 364 nm; (d) from left to right: solutions of pure chromophores 15, 14, 13 and a white-light emitting mixture 13·14·15 in CHCl3 (λexc = 365 nm). Adapted with permission from ref. 17. Copyright 2009, American Chemical Society. | ||
In a recent report, Lee et al. demonstrated the formation of supramolecular coordination polymers composed of multiporphyrin dendrimers 16M and multipyridyl porphyrins 17M (Fig. 11a and b).18 The polymer 16M·17M was built through axial coordination interactions of pyridyl groups of the energy acceptor 17M to the peripheral Zn(II)-porphyrins of the energy donor 16M. The formation of 16M·17M considerably reduces intramolecular energy transfer within the multiporphyrin dendrimers 16M and favours the intermolecular energy transfer to pyridyl bearing porphyrin 17M. As shown in Fig. 11c, dendrimer 16b exhibits very weak fluorescence emission from Zn(II)-porphyrins at 580 and 630 nm due to efficient energy transfer to the nonfluorescent focal Cu(II)-porphyrin acceptor. However, upon the addition of acceptor 17a, strong emission peaks appeared at 655 and 724 nm, coincident with the emission profile of 17a. Simultaneously, the Zn(II)-porphyrin emissions at 570 and 630 nm were quenched.
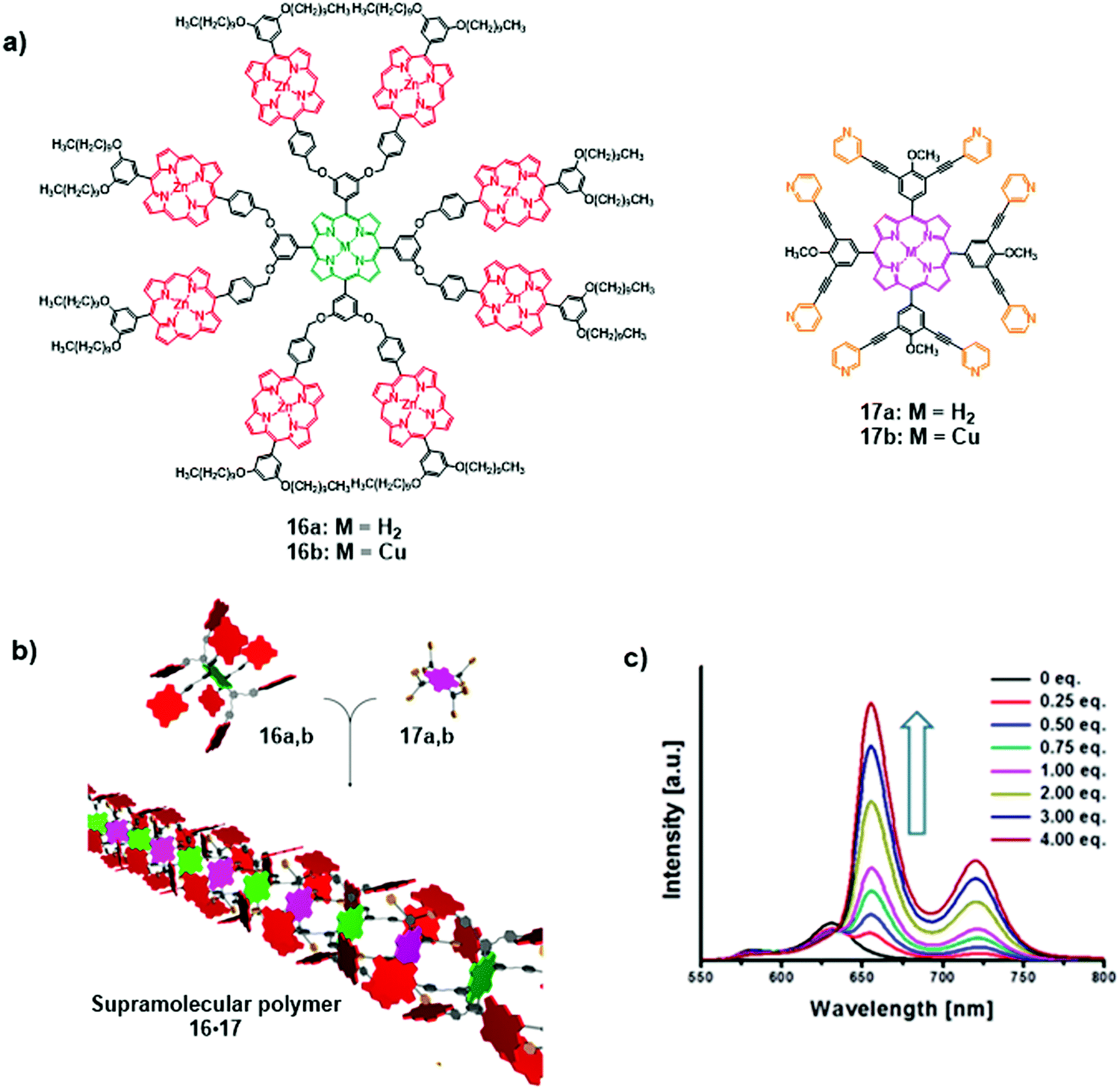 | ||
| Fig. 11 (a) Structures of multiporphyrin dendrimers 16a and b and multipyridyl porphyrins 17a and b; (b) supramolecular polymer 16·17; (c) fluorescence emission spectra of 16b upon the addition of 17a, λexc = 414 nm. Adapted with permission from ref. 18. Copyright 2015, American Chemical Society. | ||
In parallel, Wang et al. first described a FRET-capable supramolecular polymer formed through host–guest interactions.19 A boron-dipyrromethene (BODIPY)-bridged pillar[5]arene dimer 18, acting as an energy donor, and two BODIPY-based energy acceptors 19 and 20 were used to construct linear 18·19 and cross-linked 18·20 polymers (Fig. 12a). Both architectures displayed very strong absorption in a wide spectroscopic range from 300 to 700 nm. Moreover, upon the excitation of 18·19 (λexc = 470 nm) and 18·20 (λexc = 490 nm), the quenching of the emission of donor 18 at 522 nm was observed upon gradual addition of acceptors 19 and 20 (Fig. 12b and c). Concomitantly, the amplification of the emission bands at 595 nm or 669 nm, assigned to the emission of 19 or 20, respectively, was observed. Fig. 12 illustrates the changes in fluorescence emission before and after the addition of 19 or 20, highlighting the efficient energy transfer from donor 18 to acceptors 19 or 20. The FRET efficiencies were measured to be 51% for 18·19 and 63% for 18·20, suggesting a high stability of the formed host–guest assemblies.
 | ||
| Fig. 12 (a) Chemical structures of BODIPY-bridged pillar[5]arene dimer 18, BODIPY derivatives 19 and 20, and schematic illustration of linear 18·19 and cross-linked 18·20 polymers; (b) fluorescence emission spectra of 18·19 upon the addition of 19, λexc = 470 nm; (c) fluorescence emission spectra of 18·20 upon the addition of 20, λexc = 490 nm. The images display the changes in fluorescence emission colour. Adapted with permission from ref. 19. Copyright 2015, Royal Society of Chemistry. | ||
2.4 Metal–organic frameworks (MOFs)
The insertion of ligands displaying chromophoric properties into porous materials such as metal–organic frameworks (MOFs)20 is certainly an appealing approach to engineer functional systems in which the dyes are organised in a controlled fashion. By using chromophoric spacers between metal nodes, efficient LH hybrid materials could be obtained.21Lee et al. designed the pillared paddlewheel MOF Zn·21·22, comprising zinc clusters as nodes, porphyrintetraacid 21 as a paddlewheel linker and pyridine-functionalised BODIPY 22 as a pillar.22 The synthesis of the chromophoric MOF includes the initial assembly of tetraacid porphyrins with Zn(II) ions, followed by addition of an excess of BODIPY and heating of the resulting suspension at 80 °C for 20 h (Fig. 13a). Confocal laser scanning microscopy revealed the efficient energy transfer from the BODIPY units to the porphyrin struts by showing the luminescence only through the 650–710 nm filter (Fig. 13b).
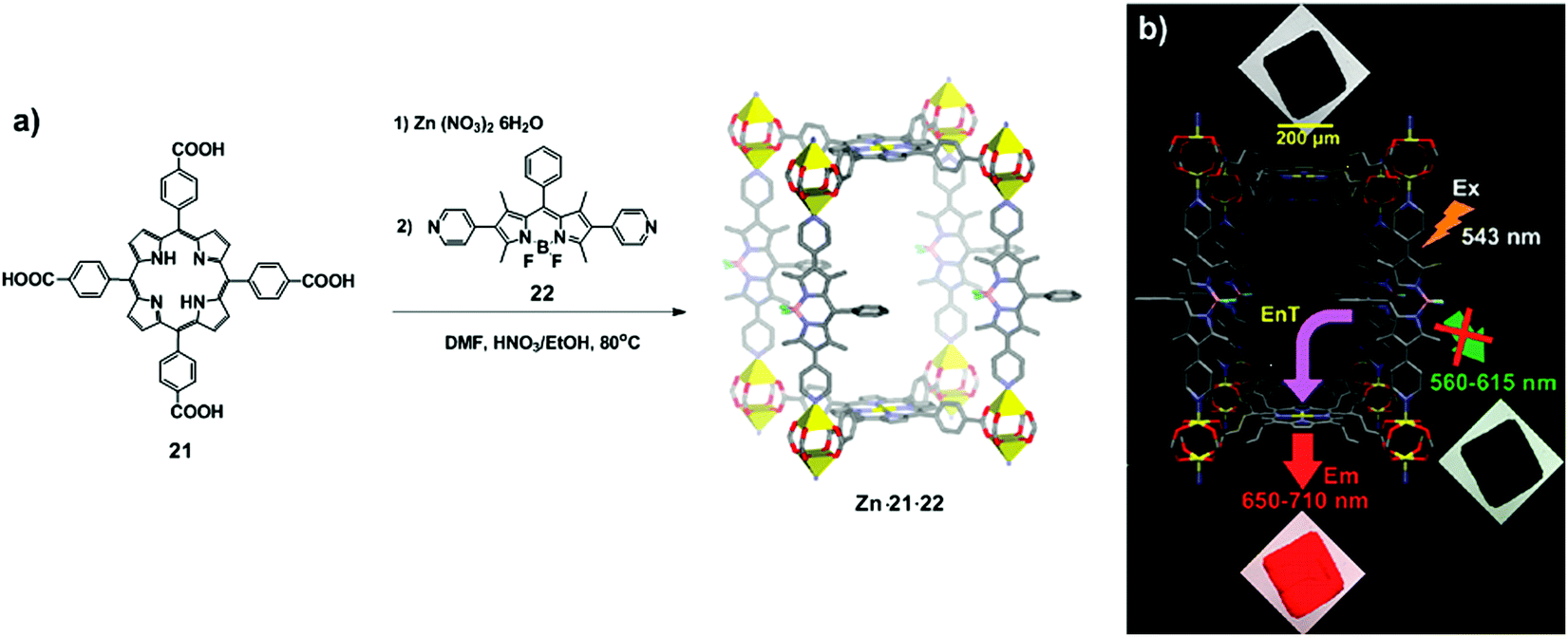 | ||
| Fig. 13 (a) Synthesis of Zn·21·22 by coordination of porphyrintetraacid 21 and BODIPY 22. Here, Zn·21·22 represented as a unit cell, containing Zn (yellow polyhedral), O (in red), N (in blue), B (in pink), F (in cyan), and C (in grey). (b) Confocal laser scanning microscopy (CLSM) images of crystals of Zn·21·22. For illustrative purposes, when the crystal is shown in black, there is no emission, while crystals in red or green denote the appropriate range of emission. Adapted with permission from ref. 22. Copyright 2011, American Chemical Society. | ||
3. Sequential covalent bonding
In contrast to the supramolecular mixing, the sequential route relies on the covalent assembly of “chromonomer” units into a multi-chromophoric architecture. Following this approach, various LH dyads and triads, macrocyclic arrays, dendritic porphyrins and multichromophores have been prepared. The synthesis of these coloured architectures usually involves multistep paths to interconnect the distinct chromophores. Herein, we report selected multichromophoric structures featuring different donor-to-acceptor ratios, interchromophoric distance and orientation; as well as tailored photophysical properties.3.1 Light-harvesting dyads and triads
Heyer and Ziessel have described LH dyads 23, 24 and triads 25, 26 bearing triphenylamine (TPA) as an electron donor and diketopyrrolopyrrole (DPP) and BODIPY dyes as electron acceptors.23 All fragments were covalently interconnected by a tolane spacer (thienyl–alkyne–phenyl for dyad 24a and triads 25, or phenyl–alkyne–phenyl for dyads 23, 24b and triads 26, Fig. 14). When the DPP moiety was selectively excited at 489 nm, the intensity of the donor emission at 568 nm was quenched while enhancing that of the acceptor at 659 nm. The excitation spectrum of the acceptor at λem = 669 nm was nearly identical to the absorption spectrum, suggesting an efficient energy transfer from DPP to the BODIPY moiety. Donor–acceptor dyads and triads were prepared in modular fashion via Pd-catalysed Sonogashira cross-coupling reactions (Fig. 15a). The dyads and triads exhibit very strong absorption throughout the whole range of visible light (300–700 nm). For instance, the UV-Vis absorption spectrum of dyad 24b displays three major absorption bands at 372, 489 and 645 nm, assigned to the absorption of DPP and BODIPY moieties (Fig. 15b). | ||
| Fig. 14 Molecular structures of dyads 23, 24 and triads 25, 26. Adapted with permission from ref. 23. Copyright 2015, American Chemical Society. | ||
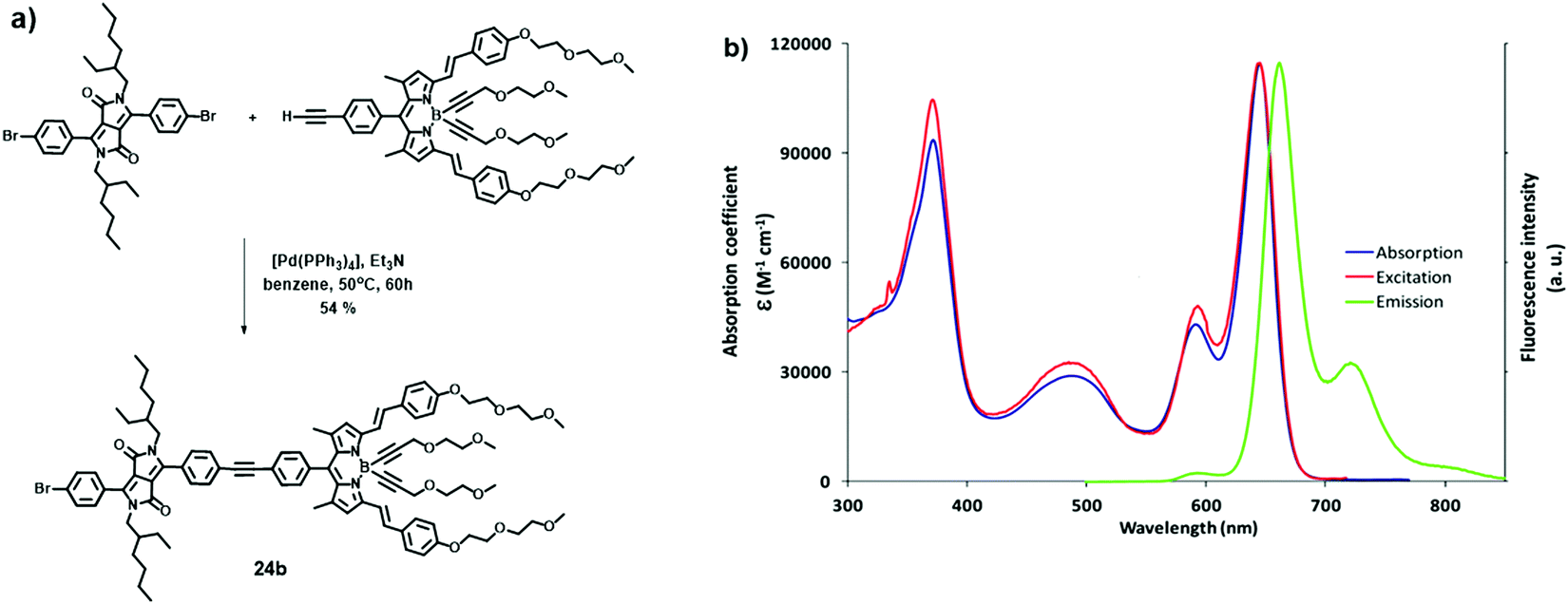 | ||
| Fig. 15 (a) Synthesis of dyad 24b; (b) UV-Vis absorption (blue line), fluorescence emission (green line, λexc = 489 nm) and excitation spectra (red line, λem = 669 nm) of dyad 24b at rt in THF. Adapted with permission from ref. 23. Copyright 2015, American Chemical Society. | ||
In a seminal paper, Würthner and co-workers reported the synthesis of a calix[4]arene-based multichromophoric architecture constructed of co-facially positioned perylene bisimide (PBI) dyes (Fig. 16a).24 UV-Vis absorption spectra of calix[4]arene-based LHA display the absorption of one orange PBI unit at 526 and 490 nm, whereas the absorption peaks at 579 and 701 nm originate from the violet PBIs and green PBIs, respectively (Fig. 16b). When the LH array was excited at the absorption maxima of the inner orange PBI (λexc = 490 nm), its emission was completely quenched. Simultaneously, the characteristic emission of outer green PBI moieties at 744 nm was observed, suggesting the presence of an efficient energy transfer from the inner orange PBI to the outer green PBI dyes. The excitation spectrum of calix[4]arene-based LHA at λem = 850 nm was nearly identical to the absorption spectrum, suggesting an efficient contribution of the yellow and orange PBIs to the green emission.
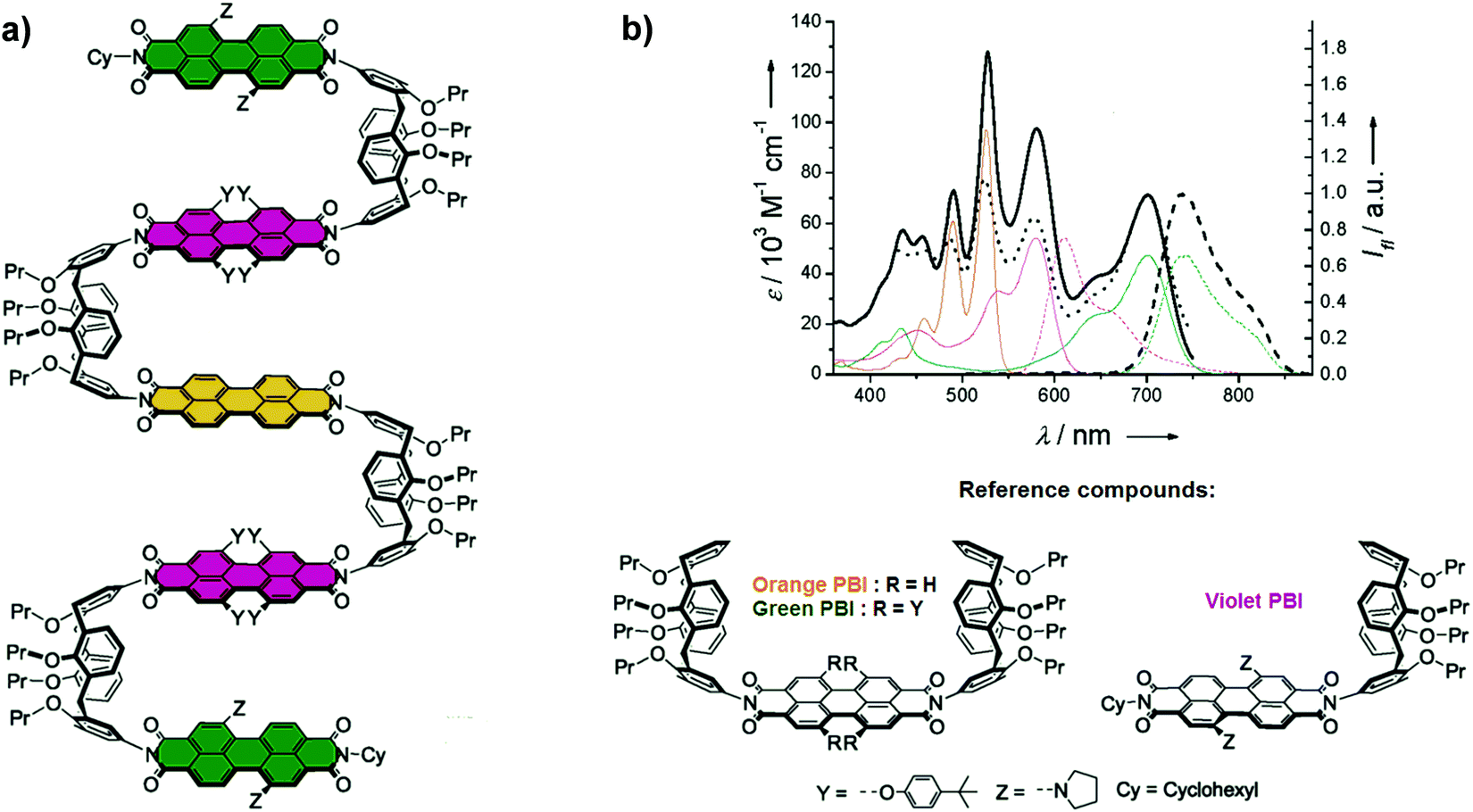 | ||
| Fig. 16 (a) Molecular structure of calix[4]arene-based LHA; (b) UV-Vis absorption (solid lines), fluorescence emission (dashed lines) spectra of the LHA (in black, λexc = 490 nm) along with those of the independent molecular units: orange PBI (in orange), green PBI (in green), violet PBI (in violet) at r.t. in CH2Cl2. Excitation spectrum of calix[4]arene-based LHA (black, dotted line, λem = 850 nm) at r.t. in CH2Cl2. Adapted with permission from ref. 24. Copyright 2006, American Chemical Society. | ||
3.2 Dendritic light-harvesting antennae
Chomophoric dendrimers are monodisperse tree-like macromolecules, incorporating chromophores in predefined sites of their structure. The dendritic approach gives structures in which the donor and acceptor molecules are organised in close proximity, favouring efficient energy transfer processes.25 Consequently, a plethora of dyes such as BODIPY, rylenes, porphyrins, and coordination-complex-based dyes have been used for the preparation of multichromophores depicting efficient LH properties.26 In a seminal work, Müllen and co-workers reported a multichromophoric rigid polyphenylene dendrimer bearing a terrylene diimide (TDI) core, four perylene monoimides (PMIs) and eight peripheral naphthalene monoimides (NMIs) separated by interchromophoric distances of ca. 2–3 nm (Fig. 17a). Dendrimer 27 absorbs in the whole visible spectrum and the normalisation of the absorption and emission spectra of the chromophores displays a nice overlap between the donor emissions and the acceptor absorptions (Fig. 17b and c). Fluorescence measurements were performed to characterise the unidirectional energy transfer from the periphery to the core by a FRET mechanism.27 The emission spectra clearly illustrate the highly efficient energy transfer from NMIs and PMIs to TDI by showing the quenching of the donor fluorescence at 430 and 560 nm and the amplification of the TDI fluorescence at 706 nm upon excitation of the donors (λexc = 360 and 490 nm). The FRET efficiency ΦET from PMIs to TDI within 27 was measured to be 99.5% by calculating the ratio between the fluorescence lifetime of the donor in the absence and presence of the acceptor. (τD and τDA estimated to be 4.3 and 0.02 ns respectively at λexc = 490 nm). The short lifetime of NMI in the triad (τD and τDA determined to be 0.33 and 0.04 ns respectively at λexc = 360 nm) suggested the presence of an efficient energy transfer from NDIs to TDI. | ||
| Fig. 17 (a) Structure of the rigid polyphenylenedendrimer 27 based on TDI (in red), PMIs (in green), NMIs (in blue); (b) UV-Vis absorption (black line) and emission fluorescence (coloured line) spectra upon excitation at 360 nm (blue), 490 nm (green), and 640 nm (red) in PhMe; (c) normalised absorption (solid) and fluorescence (solid + symbol) spectra of the distinct chromophores in PhMe. Adapted with permission from ref. 27. Copyright 2005, American Chemical Society. | ||
Several other porphyrin dendrimers have been successfully synthesised following this strategy. For instance, Okada and co-workers described a multiporphyrin architecture comprising eight tetraphenyl Zn porphyrins (TP-Por), four Zn porphyrins with two meso-phenyl and two meso-ethynyl groups (DE-Por), and a focal Zn porphyrin with four meso-ethynyl groups (TE-Por; Fig. 18a).28 As depicted in Fig. 18b, dendrimer 28 exhibits intense absorption across the 400–700 nm region with ε > 56000 M−1 cm−1. Notably, the direct linkage of ethynyl groups to the meso-positions of the porphyrin led to the red shift of the absorption bands, determining the following order of the energy levels of the lowest excited states: TP-Por > DE-Por > TE-Por. The excitation of TP-Por at 426 nm or DE-Por at 453 nm resulted in intense TE-Por-centred emissions at 707 nm and 780 nm (Fig. 18c and d). Steady-state and time-resolved fluorescence studies of dendrimer 28 showed the singlet excitation energy transfer from TP-Por* to DE-Por and TE-Por with total quantum efficiency of 96% as well as from DE-Por* to TE-Por in 98% quantum efficiency.
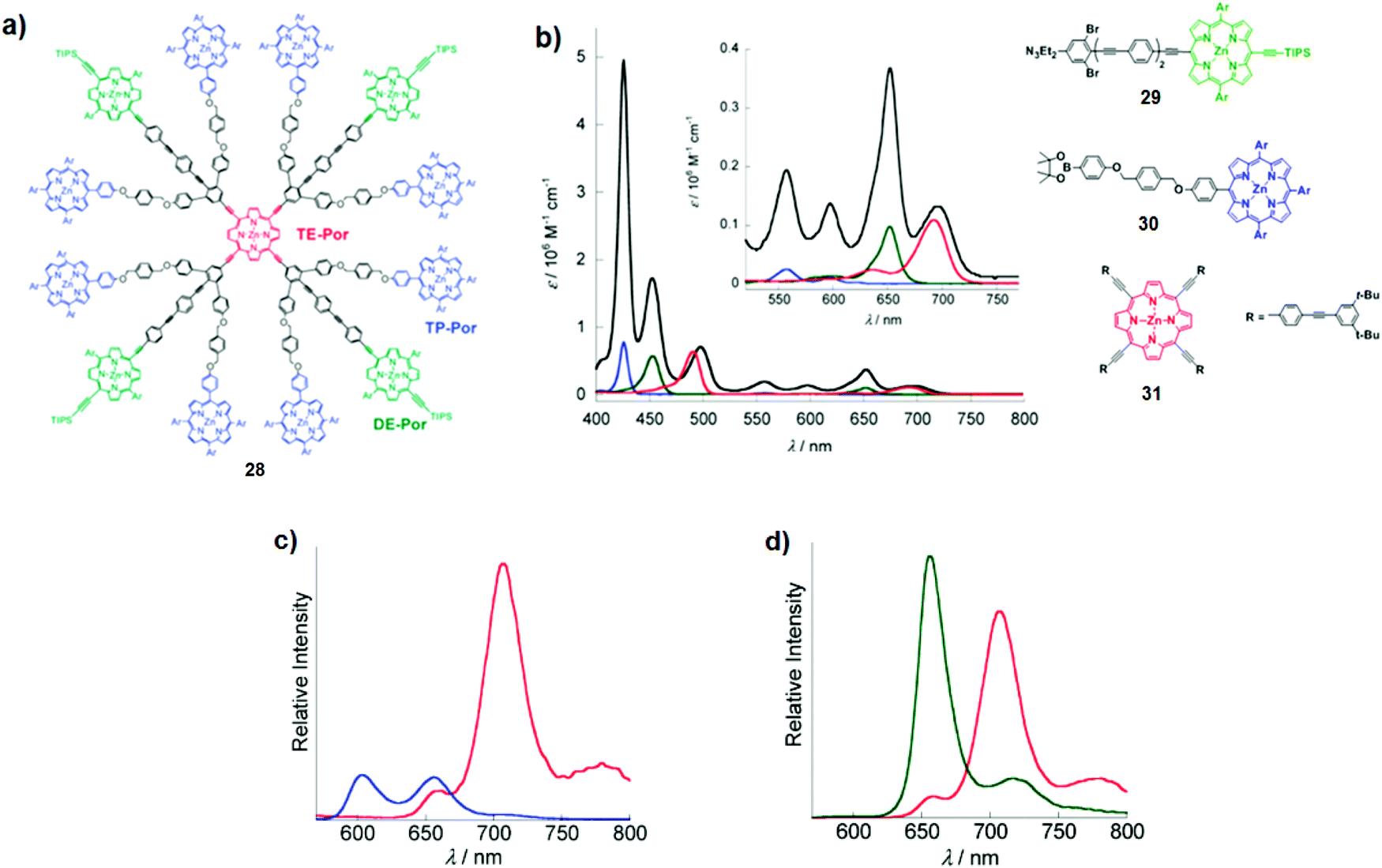 | ||
| Fig. 18 (a) Chemical structure of dendrimer 28; (b) UV-Vis absorption spectra of dendrimer 28 (black line) and model compounds: DE-Por29 (green line), TP-Por30 (blue line), and TE-Por31(red line) based monomers in THF. The inset shows expanded absorption spectra in the Q-band region; (c) steady-state fluorescence spectra of 28 (red line) and TP-Por derivative 30 (blue line) in THF, λexc = 426 nm; (d) steady-state fluorescence spectra of 28 (red line) and DE-Por derivative 31 (green line) in THF, λexc = 453 nm. Adapted with permission from ref. 28. Copyright 2011, American Chemical Society. | ||
3.3 Dynamic covalent engineering
Although the classical covalent approach offers large possibilities in the design of the molecular structures, the use of dynamic covalent chemistry gives access to large architectures that would otherwise be of difficult synthetic accessibility. In this field, Matile and co-workers took advantage of disulfide exchange for the assembly of π-stacks of differently core-substituted naphthalenediimdes (NDIs), affording complex multi-coloured surfaces synthesised with a high degree of precision at the molecular level (Fig. 19a).29 The approach was successfully applied to the construction of artificial photosystems with oriented redox gradients transporting holes and electrons to generate photocurrent. Later, the same group increased the complexity of their architectures and integrated additional chromophores through the sequential use of multiple orthogonal dynamic reactions such as the disulfide, acyl hydrazone and boronic ester exchanges. As an example, the dynamic incorporation of suitably ordered and oriented stacks of red riboflavin into assembly 33 generated a significant improvement of the photocurrent (Fig. 19b and c).30 | ||
| Fig. 19 (a) Self-organising surface-initiated polymerisation (SOIP) using NDI based initiator (A) and propagators (B, C and D) for the preparation of photosystem 32; (b) representation of the complex surface architecture of 33 with integration of riboflavin obtained through sequential orthogonal dynamic bonds; (c) photocurrent generated by 33 with the riboflavin channel (red solid) or without (black dashed). Adapted with permission from ref. 29 and 30. Copyright 2013 and 2015, Royal Society of Chemistry. | ||
Taking part in the growing interest for orthogonal dynamic covalent bonds, You and co-workers developed very recently an elegant and simple strategy for the construction of tuneable multicolour luminescent materials based on dynamic covalent switching systems and reaction networks (Fig. 20).31 The colour of the emitters was easily modulated through the adjustment of intramolecular ring/chain equilibrium by intermolecular dynamic covalent reactions (DCRs) with amines or the addition of base enabled the activation of switching (Fig. 20a). Furthermore, the diverse sensitivity of the switches toward pH and amine stimuli allowed the construction of communicating networks for multicolour switching (Fig. 20b). By changing the components and DCRs within the dynamic network the multistate optical switching, including white-light emission, was achieved.
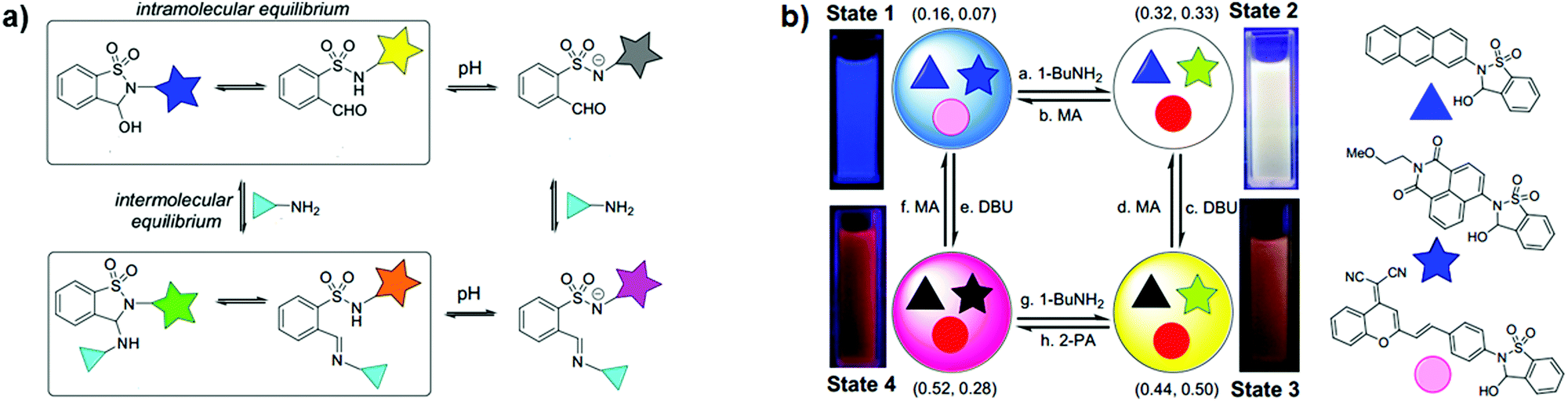 | ||
| Fig. 20 (a) DCRs within dynamic covalent switches, where the star represents the fluorophore moiety; (b) illustration of the multicolour emissive system with the interconversion between four states, related fluorescence images under a 365 nm UV lamp and CIE coordinates (MA – methanesulfonic acid; DBU – 1,8-diazabicyclo[5.4.0]undec-7-ene; 2-PA – pyridine-2-carboxaldehyde). Adapted with permission from ref. 31. Copyright 2019, American Chemical Society. | ||
4. Template route
In contrast to the architectures constructed with sequential approaches, templated multichromophoric assemblies are prepared exploiting programmed scaffolds that are decorated with chromophores only at a later stage of the synthesis (Fig. 21). Programming of the framework involves the introduction of appropriate receptor sites, which enable covalent or non-covalent assembly with dyes. This allows controlling crucial FRET parameters such as donor-to-acceptor ratio, orientation and distance between the dyes. In this respect, the use of a programmable template allows the appropriate positioning of the dyes, preventing the quenching of the fluorophore by excimer or exciplex formation, thereby favouring the FRET process. In this section, we describe: (i) the co-assembly between the pre-programmed template and dyes; and (ii) pre-formed chromophore-template conjugates. Our attention mainly focuses on the diversity of the templates (e.g., DNA, peptides, proteins, micelles, vesicles, organic polymers and inorganic materials), methodologies to assemble the chromophores, and the photophysical properties of the final architectures.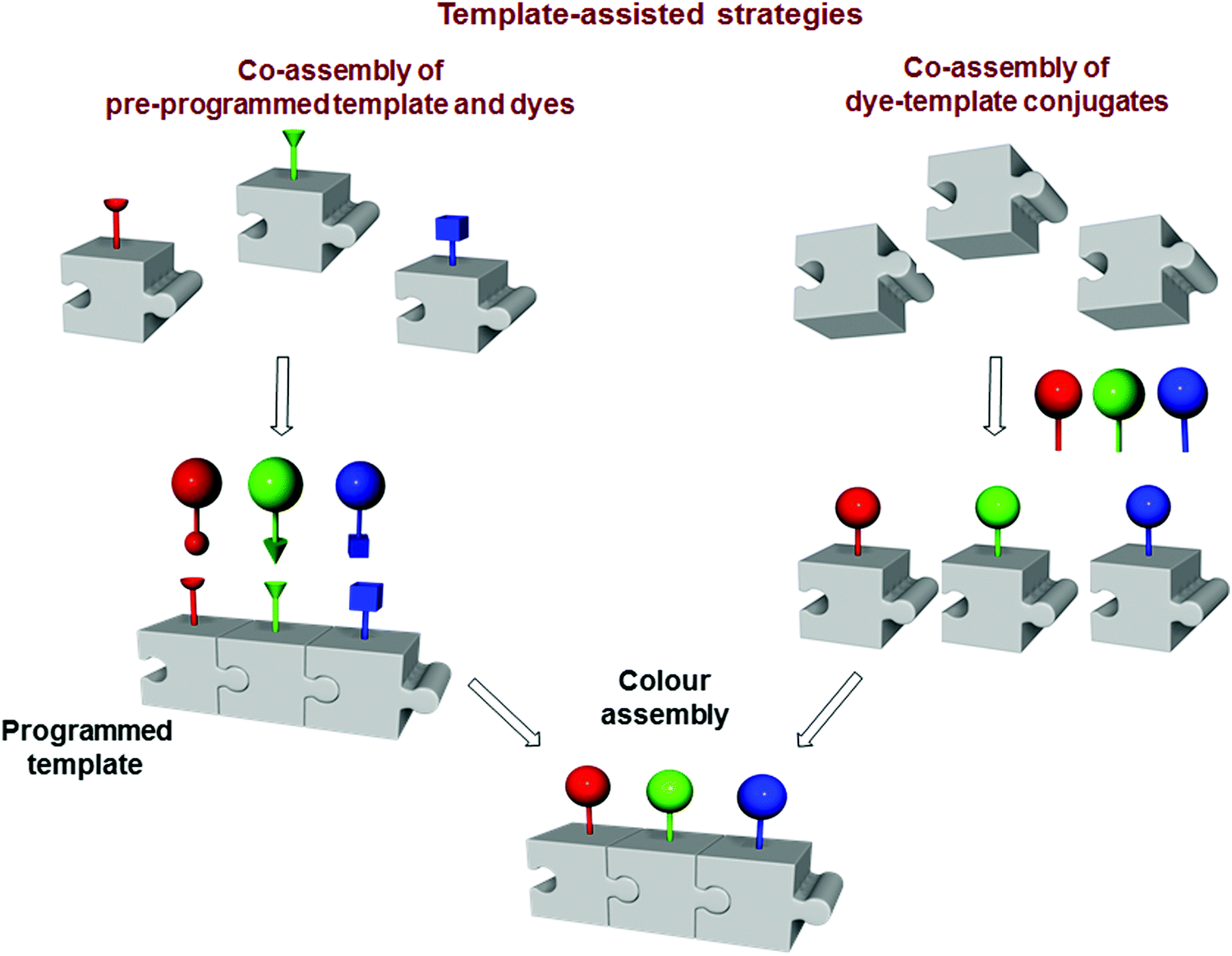 | ||
| Fig. 21 Schematic illustration of template-assisted strategies. | ||
4.1 Co-assembly between the pre-programmed template and dyes
:1 to 12:8, where the emission of Nr-dU at 675 nm became more intense by indirect donor excitation at 380 nm than by direct excitation at 558 nm (antenna effect > 1). The excitation spectra highlighted the important contribution of the Py-dU donor to the acceptor's emission (λem = 675 nm, Fig. 22e).
 | ||
| Fig. 22 (a) Structures of ethynylnile red (Nr-dU) and ethynylpyrene (Py-dU) containing 2′-deoxyuridine; (b) DNA-templated assemblies of Py-dU and Nr-dUvia hydrogen bonding; (c) UV-Vis absorption spectra of the assemblies (1.25 M in H2O + 2% DMSO); (d) assemblies containing Py-dU:Nr-dU from 20:0 to 0:20; (e) excitation spectra of assembled architectures at the emission of Nr-dU (λem = 675 nm). Adapted with permission from ref. 35. Copyright 2015, Royal Society of Chemistry. | ||
Polypeptides and short peptides have also been widely employed as pre-programmed scaffolds to spatially organise chromophores, as observed in natural LH complexes.36,37 Their intrinsic ability to self-assemble into highly organised nano-architectures via secondary, tertiary and quaternary structures makes them interesting candidates to tailor the distribution and orientation of dyes. In this respect, several examples involving the co-assembly between peptides and dyes (especially porphyrin derivatives) through π–π stacking, electrostatic, and metal coordination interactions have been reported. For instance, Moser and co-workers engineered LH systems based on four-helix proteins in which synthetic chlorin and bacteriochlorin derivatives were bonded to a histidine residue, which is a well-known Zn porphyrin-binding site.37
A covalent approach was also used to accommodate chromophores into α-peptides. Our group described the selective incorporation of yellow ethynylpyrene (Y-Py), red perylenediimide (R-PDI) and blue naphthalenediimide (B-NDI) into a pre-programmed α-peptide.38 This was achieved through the simultaneous use of three dynamic covalent bonds involving orthogonal reactions, namely disulfide, boronate and acyl hydrazone formation reactions (Fig. 23a and b). The coloured α-peptide absorbs throughout the entire visible spectrum with distinct peaks at 376, 398, 495, 527 and 612 nm, confirming its panchromatic absorption properties (Fig. 23c). However, investigation of the energy transfer using steady-state fluorescence spectroscopy revealed a low efficiency: 5% and 5.5% from Y-Py to B-NDI and from R-PDI to B-NDI, respectively. In a follow-up paper, the effect of various structural modifications on the energy transfer efficiency, such as the structure of chromophores as well as their spatial organisation on the α-peptides (e.g., interchromophoric distance, orientation, and order), was studied.39 Although an improvement of the energy-transfer efficiencies to 15 and 22% was achieved for the Y-Py → B-NDI and R-PDI → B-NDI processes, respectively, in-depth analysis of the conformational properties of the α-peptide-based assemblies performed by metadynamics simulations suggested that the antennas adopt peculiar conformations in which the chromophores collapse in π–π stacks. These arrangements quench the excited states and reduce the energy transfer efficiencies.
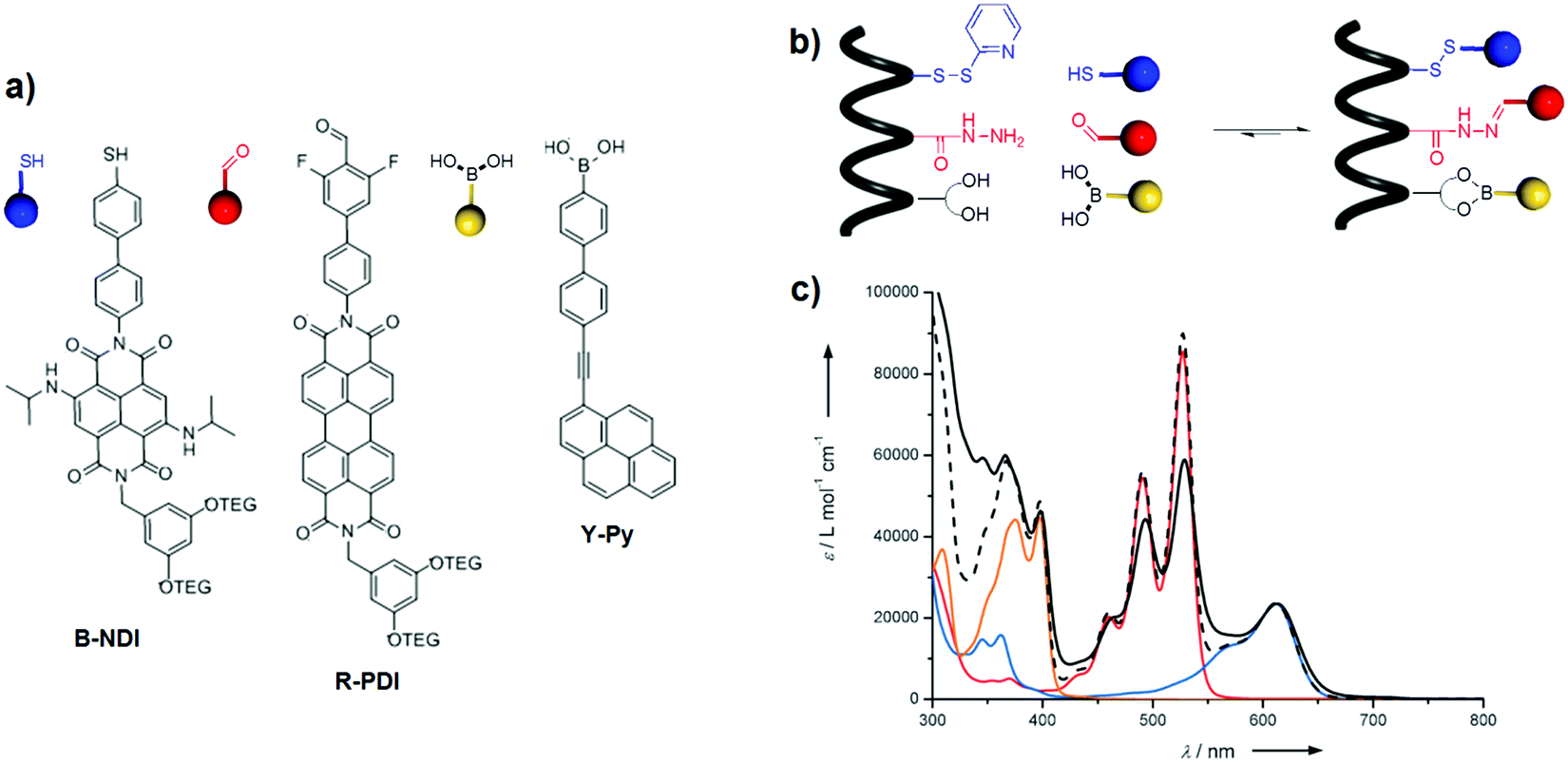 | ||
| Fig. 23 (a) Chemical structures of chromophores B-NDI, R-PDI, and Y-Py bearing a complementary sticky side to the receptor sites of pre-programmed α-peptide: thiol, aldehyde, boronic acid; (b) templated chromophoric assembly into pre-programmed peptide; and (c) absorption spectra of Y-Py (in yellow), R-PDI (in red), B-NDI (in blue) and arithmetic sum of the three spectra (dashed black line) in DMF. Absorption spectrum of the coloured peptide (solid black line) in DMF normalised on B-NDI with the arithmetic sum of absorption of the three dyes. Adapted with permission from ref. 38. Copyright 2015, John Wiley & Sons, Inc. | ||
 | ||
| Fig. 24 (a) Preparation of the surface-cross-linked micelle 36; (b) UV-Vis absorption spectra of DPA–SCM (CD = 23 μM) and (c) fluorescence emission spectra (λexc = 375 nm) of DPA–SCM (CD = 23 μM) upon addition of the acceptor EY ([EY] = 0, 0.08, 0.17, 0.33, 0.5, 0.67, 0.84, 1, 1.17, 1.34 μM) in THF. Adapted with permission from ref. 40. Copyright 2012, John Wiley & Sons, Inc. | ||
Vesicles, with a hollow lumen enclosed by an amphiphilic bilayer, were also considered as supramolecular templates. Chromophores can be loaded either into the isolated lumen, hydrophobic core layer or hydrophilic shell. In this way, this vesicular platform provides spatial separation of donor and acceptor molecules, which helps to avoid self-quenching between the chromophores. In this context, Guo and co-workers designed the vesicular antenna 37 formed from amphiphilic calix[5]arene (AmC5A) by encapsulation of 1-anilino-8-naphthalenesulfonate (1,8-ANS) into the calixarene cavity and entrapment of 4,7-bis(thien-2-yl)-2,1,3-benzothiadiazole (DBT) into the hydrophobic bilayer (Fig. 25a).41 Blue-light emitting 1,8-ANS and orange-light emitting DBT were employed as an energy donor and acceptor, respectively. Fluorescence titration experiments of AmC5A/1,8-ANS assembly with DBT exhibited the quenching of donor emission at 475 nm concomitantly to the amplification of that of the acceptor at 575 nm (λexc = 369 nm, Fig. 25b). At a donor/acceptor molar ratio of 10:1, the energy transfer efficiencies were calculated as 97% in phosphate-buffered saline (PBS) buffer (pH = 7.0). The efficient energy transfer from 1,8-ANS to DBT was proven also by decreasing the fluorescence lifetime of AmC5A/1,8-ANS from 5.1 ns to 1 ns upon the addition of DBT. The tuning of the emissive colour by adjusting the amount of DBT is observed in the chromaticity diagram (going from blue to orange). Notably, a pure white light emission with colour coordinates (0.31, 0.34) could be attained at a donor/acceptor ratio of 25:1 (Fig. 25c).
 | ||
| Fig. 25 (a) Preparation of the vesicular LHA 37; (b) fluorescence emission spectra of AmC5A/1,8-ANS in PBS buffer (10 × 10−3 M, pH = 7.0) with different concentrations of DBT, [1,8-ANS] = [AmC5A] = 20 × 10−6 M, λexc = 369 nm; (c) the CIE chromaticity diagram, showing the luminescent colour changes of AmC5A/1,8-ANS with different concentrations of DBT in PBS buffer. Adapted with permission from ref. 41. Copyright 2016, John Wiley & Sons, Inc. | ||
In an inspirational work, Würthner and co-workers reported the self-assembly of two amphiphilic PBIs into water-soluble vesicles loaded with a bispyrene based energy donor (Fig. 26a).42 Following the preferred conformation of the dimeric donor (stacked or unstacked), the energy transfer between the chromophores could be efficiently triggered using pH. At low pH, the donor molecules display monomeric purple-blue fluorescence, whereas under basic conditions an excimer emission could be observed, overlapping the absorption of the PBI membrane. Exceptionally, a white emission could be obtained when adjusting the pH at 9.0 (Fig. 26b).
 | ||
| Fig. 26 (a) Structure of amphiphilic PBIs and schematic representation of donor-loaded and photostabilised perylene vesicles with pH-tunable energy transfer; (b) photograph of loaded vesicles in aqueous solutions at different pH under UV lamp (366 nm) and CIE 1931 chromaticity diagram highlighting three points: fluorescence coordinates of the donor excimer, perylene membrane and white fluorescence coordinates. Adapted with permission from ref. 42. Copyright 2009, Springer Nature. | ||
:1:1) to white-light emission (39/40/41 = 1:1:0.2). In a successive study, the authors engineered antenna 44 by initial encapsulation of oxazole donor 43, followed by functionalisation of the channel entrances with stopcock BODIPY-based chromophore 42, the latter acting as an energy acceptor (Fig. 27c). As shown in Fig. 27d, upon selective excitation of the donor at 350 nm, the emission of 44 exhibits predominantly the features of the stopcock molecule at 610 nm, indicating the occurrence of a radial energy transfer from the inner to the outer surface of the crystal.
 | ||
| Fig. 27 (a) Schematic illustration of LH hybrid 38; (b) fluorescence spectra of LH hybrid 38 with loaded donor/acceptor ratio of 1:1:1 (solid black line), carbostyril 39 (blue dashed line), BODIPY 40 (green dashed line) and oxazine 41 (red dashed line), λexc = 375 nm. Inset shows the fluorescence image of 38 under 350/50 nm excitation and recording the emission with a long-pass filter of 400 nm; (c) functionalisation of zeolitic channel entrances with BODIPY-stopcock molecule 42; (d) normalised excitation (λem = 660 nm, dashed line) and emission (λexc = 350 nm, solid line) spectra of LH hybrid 44. Adapted with permission from ref. 44. Copyright 2017, MDPI AG (Basel, Switzerland). | ||
Multilayered clays are also widely exploited to spatially organise chromophores to facilitate LH processes. Among a wide variety of clays, layered magnesium silicate with attached aminopropyl groups (aminoclay) is the most applicable clay for the loading of dyes in its interlayer galleries. Since aminoclay (AC) is well soluble in water, and the charge repulsion between the protonated amine groups makes it easily exfoliable, supporting the incorporation of dyes. For instance, Rao et al. exploited aminoclay as a template for the non-covalent assembly of coronene (CS) 45 and perylene (PS) 46 tetracarboxylate salts, acting as an energy donor and acceptor, respectively.45 Green-emitting hybrid gel 45·46 was formed through electrostatic interactions between the negatively-charged carboxylate groups of 45 and 46 and the positively-charged aminoclay (Fig. 28a and b). When 5–7 mol% of 46 (relatively to 45) was loaded, upon excitation of 45 (λexc = 350 nm), amplified green acceptor emission at 475–600 nm reached a maximum. Further loading of 46 (> 10 mol%) resulted in a decrease of fluorescence emission with a concomitant bathochromic shift, attributed to the perylene stacking (Fig. 28c). This strategy was further applied for the preparation of hybrid 45·47 using sulforhodamine 47 as an energy acceptor, instead of perylene derivative 46.46 At 0.65 mol% loading of 47, pure white-light emission was attained (Fig. 28d).
 | ||
| Fig. 28 (a) Chemical structures of chromophores 45–47; (b and d) schematic illustrations of the co-assembled clay-chromophore hybrids 45·46 and 45·47, and the energy transfer processes; (c) emission spectra of hydrogel 45·46 (λexc = 350 nm) with different amounts of 46 and photographs of 45·46, made from both individual and mixed CS 45 and PS 46 dyes under visible (top) and UV (bottom) light. Adapted with permission from ref. 45 and 46. Copyright 2011 and 2013, John Wiley & Sons, Inc. | ||
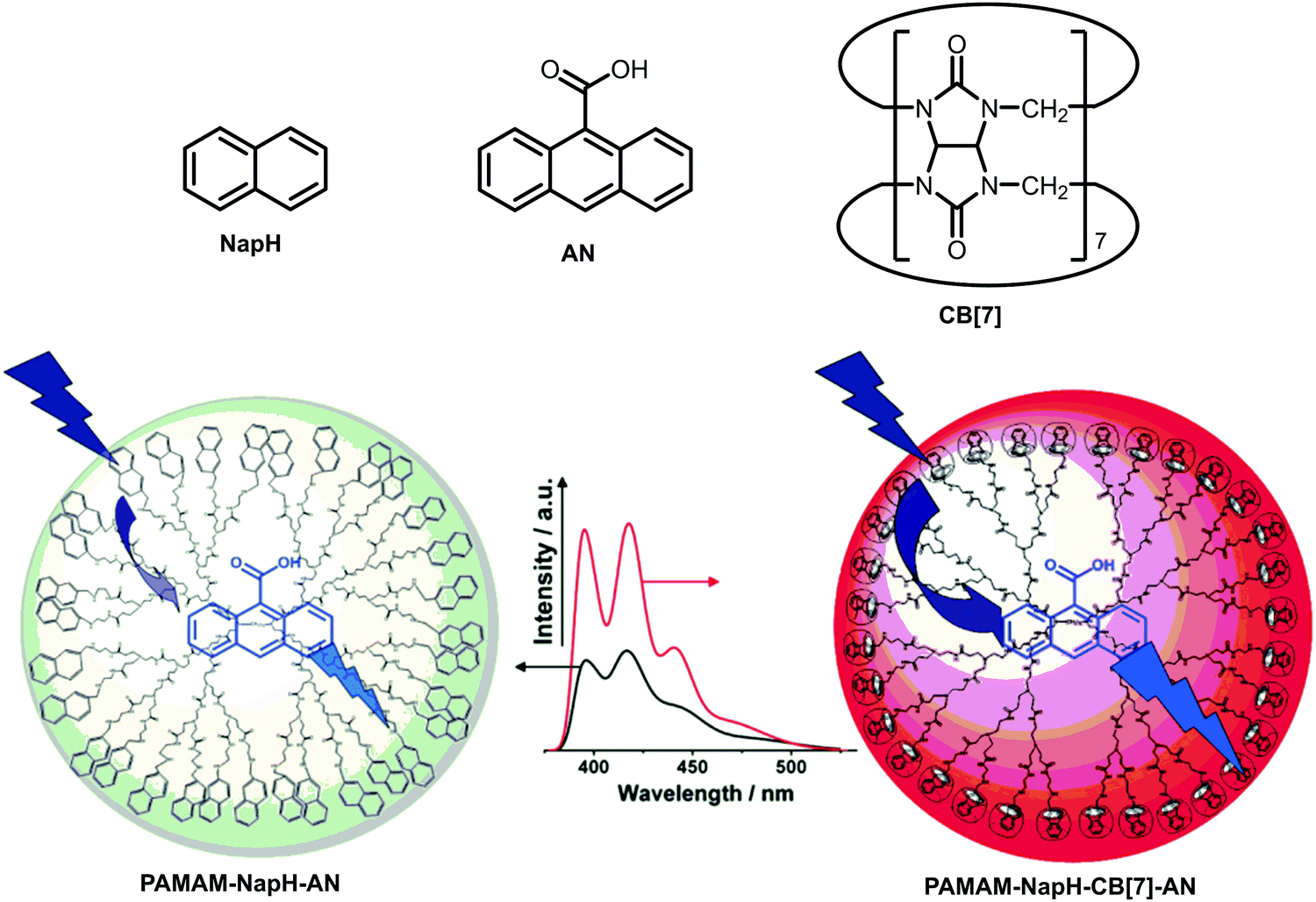 | ||
| Fig. 29 Schematic illustration of two dendritic systems: PAMAM decorated with NapH–AN complex (left) and PAMAM decorated with NapH–CB[7]–AN complex (right). Fluorescence spectra show the corrected emission of AN in PAMAM–NapH-CB[7] (red line) and PAMAM–NapH (black line) (λexc = 276 nm). Adapted with permission from ref. 47. Copyright 2009, American Chemical Society. | ||
4.2 Co-assembly between pre-formed chromophore-derived template conjugates
An alternative strategy to spatially organise chromophores into a multichromophoric system is to use templates bearing chromophores to organise the functional molecules into the final architecture. This approach includes the initial preparation of chromophore-derived proteins, and nucleic acids among the most exploited templates.:4:1), the self-assembled architecture absorbs throughout the entire visible spectrum and its excitation spectra, monitored at 650 nm, underlined the major contribution of the donors in the acceptor emission at 612 nm (Fig. 30c). The overall efficiency of the energy transfer in this system was estimated to be over 90%. Moreover, the amplification of the acceptor emission (at 612 nm) in the same ratio is clearly evidenced in the emission spectra (Fig. 30d).
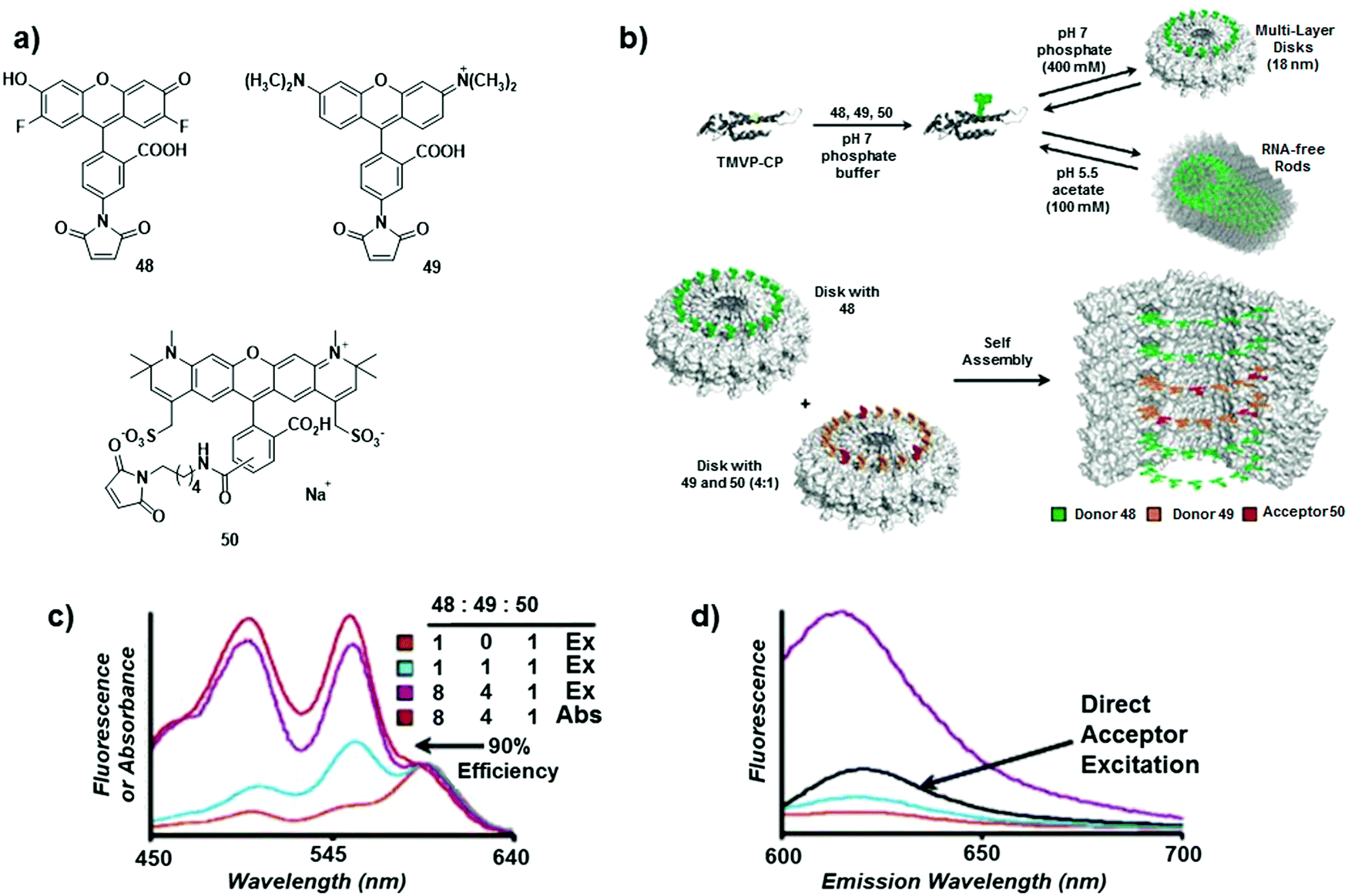 | ||
| Fig. 30 (a) Structures of chromophores 48, 49 and 50; (b) self-assembly of the chromophore–protein conjugates into rods or disks and spatial distribution of the chromophores in an antenna complex built with a molar ratio of 48/49/50 = 8:4:1; (c) excitation spectra of antenna complexes built with different D/A ratios. Absorption spectrum for the 8:4:1 system in red is normalised with the excitation spectra at 597 nm; (d) emission spectra of antenna complexes built with different D/A ratios, upon excitation of the donor (λexc = 495 nm) compared with emission spectra (in black) upon direct excitation of the acceptor unit (λexc = 588 nm). Adapted with permission from ref. 48. Copyright 2007, American Chemical Society. | ||
The construction of the efficient LH antennae upon the assembly of chromophore–peptide conjugates was also demonstrated by Harris et al.49 As in the previous example, various cysteine-containing β-polypeptides were covalently attached at different positions to synthetic bacteriochlorins, BC1, BC2 and BC3 absorbing at λmax = 713 nm, 729 nm and 758 nm, respectively. The chromophore-polypeptide conjugates, containing histidine residues, capable of coordinating acceptor Bacteriochlorophyll a (BChl a, λmax = 820 nm), were then mixed with α-polypeptide and BChl a to form various α,β-dyads (Fig. 31a and b). The energy transfer efficiencies were determined from 50 to 95% depending on the donor used and on the interchromophoric distance. The authors also reported the combination of three distinct α,β-dyads 51, 52 and 53 (in ratio 1:1:1) into complex 54, containing bacteriochlorins BC1, BC2 and BC3, respectively (Fig. 31a–c). This significantly increases the solar coverage and the overall energy transfer efficiency was estimated to be around 95%.
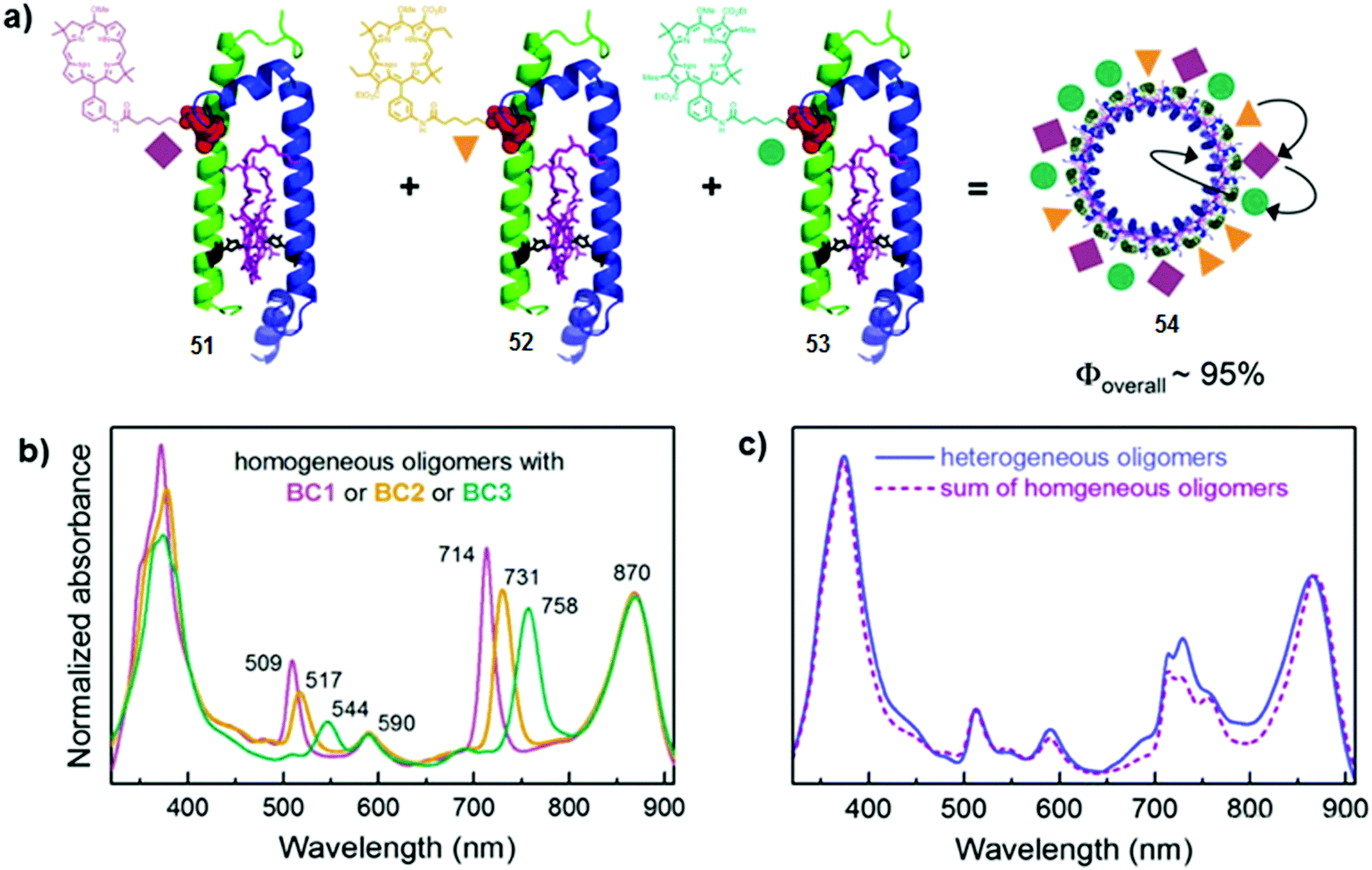 | ||
| Fig. 31 (a) Schematic representation of the self-assembly of α,β-dyads 51–53, containing α-polypeptide (blue helix), β-polypeptide (green helix) covalently attached to BC1, BC2 and BC3, and BChl a (purple) coordinated to a histidine residue (black), affording one possible arrangement of BC1 (mauve rhomb), BC2 (orange triangle) and BC3 (green circle) within LH1-type complex 54; (b) UV-Vis absorption spectra of α,β-dyads 51 (purple), 52 (orange) and 53 (teal); (c) UV-Vis absorption spectra of 54 and the calculated sum of the individual spectra of 51–53 (dashed purple line). Adapted with permission from ref. 49. Copyright 2014, Springer Nature. | ||
:6:1, the absorption spectrum consists of the linear combination of the individual profiles of the three chromophores and covers the entire visible spectrum (Fig. 32c). The FRET efficiency was calculated from the quenching of the primary donor emission and was estimated to be around 90% for T1 emission (Fig. 32d). The amplification of the acceptor emission was determined to be 85%, suggesting the presence of a strong antenna effect.
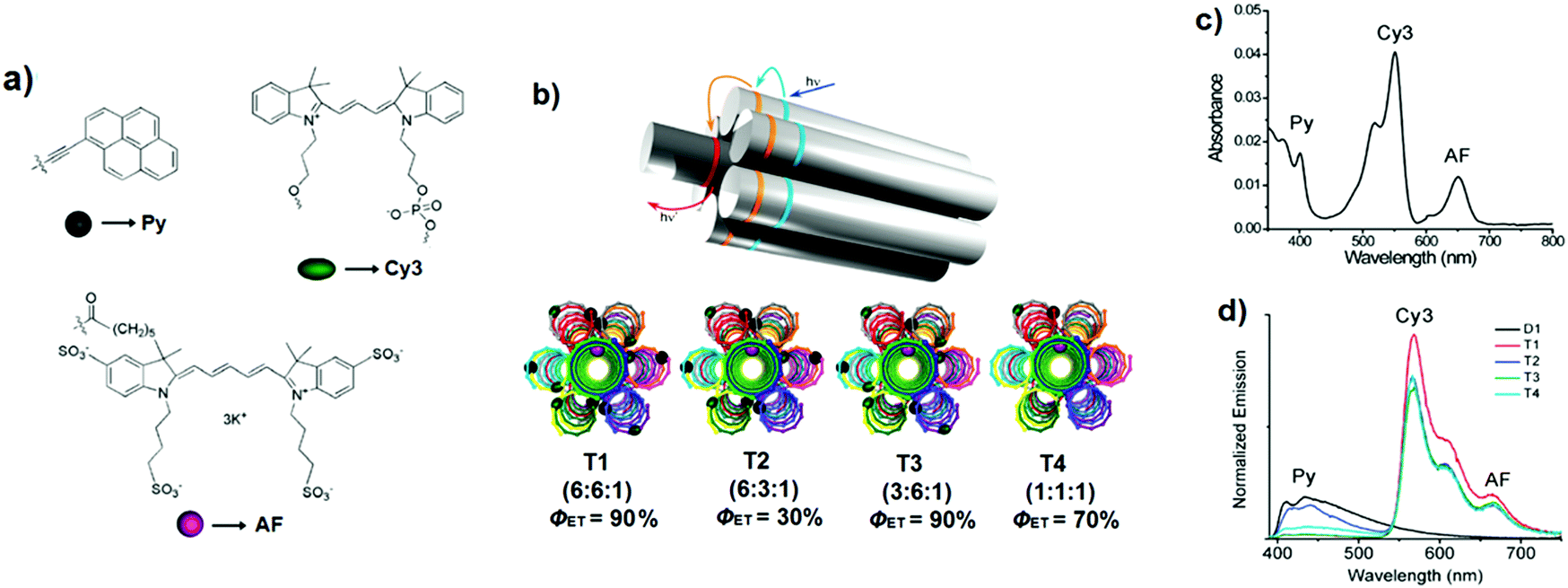 | ||
| Fig. 32 (a) Molecular structures of Py, Cy3 and AF; (b) schematic representation of self-assembled DNA nanoscaffolds called triads, containing different ratios (Py:Cy3:AF) of chromophores; (c) UV-Vis absorption spectrum of T1; (d) normalised emission spectra of D1 (6:0:0), T1–T4 (λexc = 380 nm). Adapted with permission from ref. 50. Copyright 2011, American Chemical Society. | ||
Following the same approach, Medintz and co-workers reported multi-dye FRET cascades on DNA-based architectures.51 The authors prepared 36 antenna constructs involving linear, bifurcated, Holliday junction, 8-arm star and branched structures spatially arranging up to five dyes (Fig. 33).
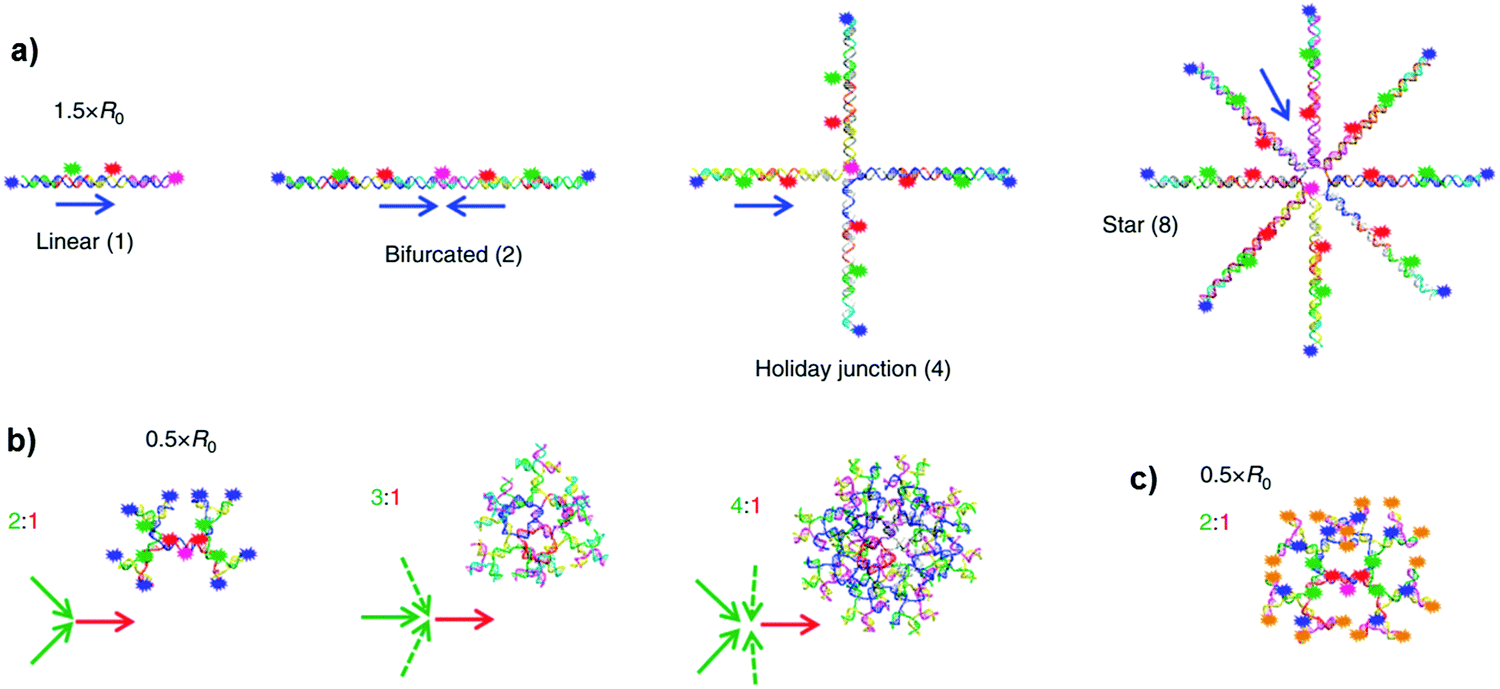 | ||
| Fig. 33 Schematic illustrations of: (a) [Cy3–Cy3.5–Cy5]n → Cy5 four-dye, three FRET step system with sequential donor–acceptor arrangements of Cy3 (blue), Cy3.5 (green), Cy5 (red) and Cy5.5 (pink) in linear (n = 1), bifurcated (n = 2), Holliday junction (n = 4) and star (n = 8) configurations. The blue arrows show the direction of the FRET pathway(s); (b) dendrimer FRET systems where each dye preceding the Cy5.5 has 2, 3 or 4 donors; (c) dendrimer five-dye FRET system with AF488 (orange), Cy3 (blue), Cy3.5 (green), Cy5 (red) and Cy5.5 (pink) dyes. Arrows demonstrate the general donor (green) to acceptor (red) architecture. Adapted with permission from ref. 51. Copyright 2014, Springer Nature. | ||
DNA-arranged networks were self-assembled through the hybridisation of shorter dye-containing DNA sequences, using a staggered or concatenated construction approach to achieve the closest dye spacing's in the double helices. Organic dyes such as cyanines (Cy3, Cy3.5, Cy5 and Cy5.5) and Alexa Fluor 488 (AF488) were attached to a DNA sequence through succinimidyl ester modifications to terminal or internal positions, or directly into the strand using phosphoramidite chemistry. By changing the number of dyes and their distances in different geometries, one-, three- and four-consecutive FRET pathways have been studied. It was found that, in contrast to the linear four-dye construct, the structures with each dye preceding the central-terminal Cy5.5 exhibited >500-fold increase in terminal exciton delivery efficiency within the assembly. This enhancement was attributed to the occurrence of multiple interacting FRET steps.
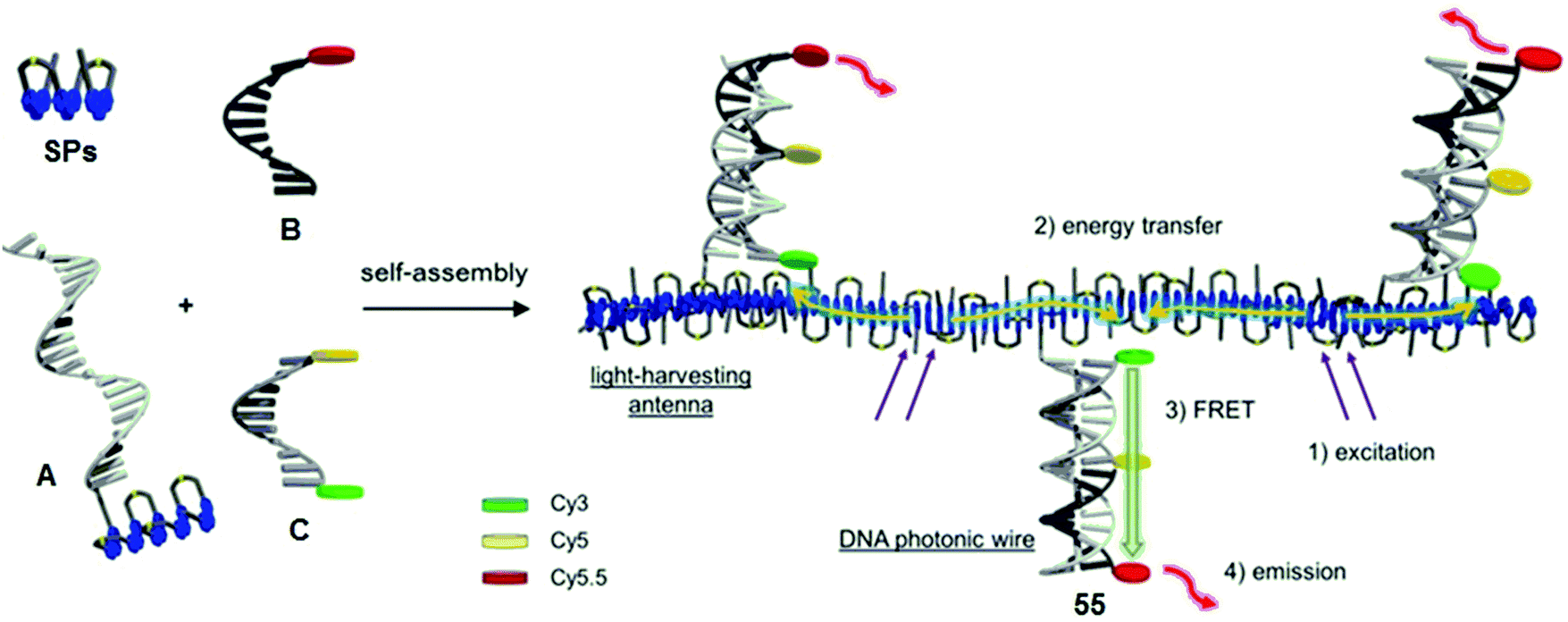 | ||
| Fig. 34 Schematic illustration of the supramolecular assembly of LHC 58. Excitation energy absorbed by phenanthrene units is transferred within the LH antenna to the acceptor dye (Cy5.5) through intermediate Cy3 and Cy5 donors via a FRET mechanism. Adapted with permission from ref. 53. Copyrights 2019, John Wiley & Sons, Inc. | ||
:0.75:1, white-light emission was achieved (Fig. 35c and d).
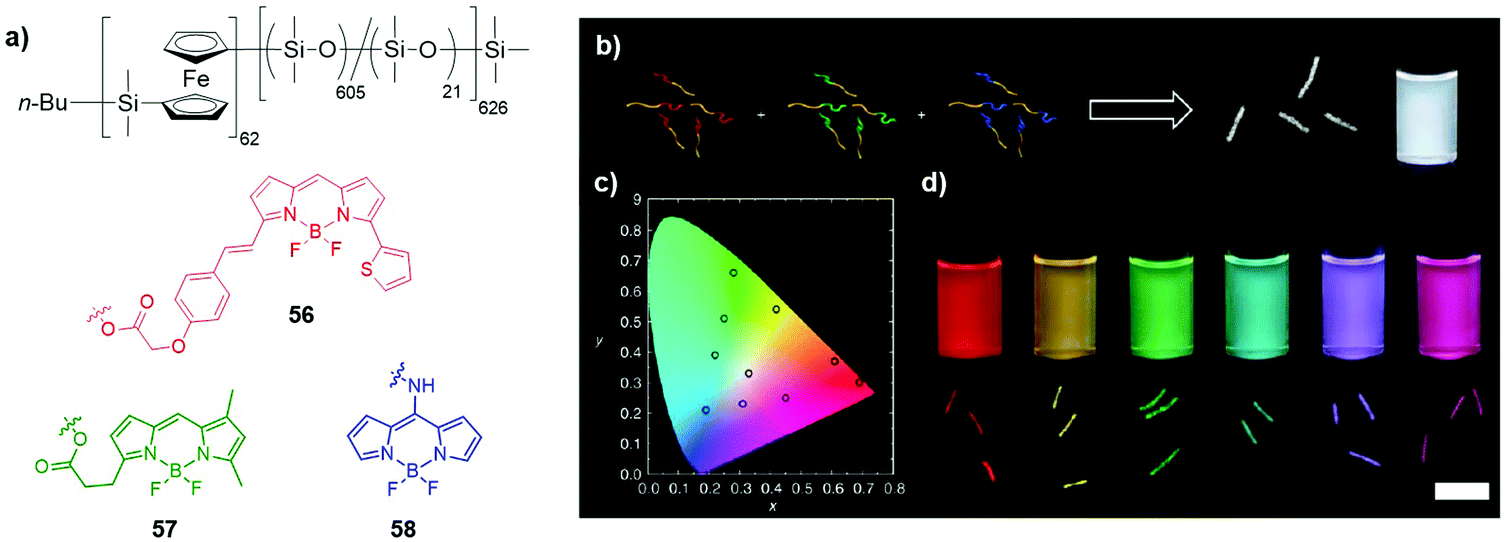 | ||
| Fig. 35 (a) Structures of BODIPY-functionalised block-copolymers 55–57; (b) schematic illustration of the self-assembly of 55–57 to form micelles of tuneable emission colour; white light-emitting micelles (molar ratio of 55/56/57 = 1.03:0.75:1) are given here as an example with the corresponding confocal fluorescence micrograph and raw photographic image of the micelle solution under UV irradiation at 365 nm; (c) CIE 1931 chromaticity diagram showing the colour coordinates of micelle solutions; (d) confocal fluorescence micrographs and raw photographic image of micelle solutions under UV irradiation at 365 nm; Adapted with permission from ref. 52. Copyright 2014, Springer Nature. | ||
In 2018, Kownacki et al. described an approach to produce efficient light-harvesting complexes by combining DNA nanoscaffolds with supramolecular phenanthrene polymers (SPs).52 Various LHCs were constructed, phenanthrene-containing A and cyanine-labelled B and C oligomers in a self-assembled fashion. Among them, complex 55 resulted in a photonic wire consisting of phenanthrene as the primary donor, Cy3 and Cy5 as intermediate donors and Cy5.5 as the acceptor (Fig. 34). The FRET efficiency was estimated to be 59%, suggesting a remarkable stepwise energy transfer process. The antenna effect was estimated to be from 1.4 up to 23, depending on the structure of the complex.
5. Conclusions
Among the different approaches to gain control over the colour of a nanostructure, the mixing of distinct chromophores absorbing at selected wavelengths is certainly a versatile method to engineer functional multichromophoric architectures. This approach is particularly effective for constructing light-harvesting antennas, tailored to capture a maximum of sunlight in a given environment. The photophysical processes occurring between dyes in a multichromophoric nanostructure play a crucial role in the functional properties of a multichromophoric antenna. For instance, efficient energy transfers are governed by the peculiar spatial arrangements of the dyes in the architecture, and the development of new protocols for organising given chromophores into architectures with tailored properties is pivotal if one wants to engineer a fully functional system. In this tutorial review we have rationalised the strategies that chemists use to engineer artificial multichromophoric nanostructures. In particular, we focused on the supramolecular mixing, covalent and non-covalent bottom-up synthesis, templated assembly, and impact that the resulting constructs have on the materials’ properties.The supramolecular mixing provides a simple, synthetically accessible way to access topologically complex architectures through non-covalent bonding interactions. Self-assembled light-harvesting systems, such as organic gels and supramolecular polymers with high donor/acceptor ratios, promote good energy-transfer efficiencies. In the supramolecular polymers approach, the non-covalent bonding of different monomers combines the advantages of small molecules with those of polymers and allows varying the morphology and composition as well as tuning the colour emission. On the other hand, supramolecular mixing through gelation provides an efficient method to accurately blend multiple components into highly organised nanostructures to facilitate energy migration in a controlled fashion. The efficiency of the energy transfer process in the gel is usually higher than that measured in solution, further demonstrating the importance of having an intimate contact between the donor–acceptor chromophoric couples. The relative positioning of the chromophores into these superstructures is however difficult to predict and control, and this often leads to a poor degree of spatial organisation and low material reproducibility.
The sequential interconnection of chromophores through covalent bonding to prepare dyads, triads, and dendritic structures allows the engineering of efficient light harvesting systems. For instance, significant energy transfer quantum yields could be obtained from dendritic structures due to their high degree of organisation and proximity between chromophores. However, their organic synthesis often involves complex multistep pathways, which are usually limited by the given chromophores. In this respect, the emergence of orthogonal dynamic covalent reactions has opened new horizons in the accessibility of covalent nanostructures of increased complexity.
To overcome the synthetic limitations of the covalent approach and the poor degree of molecular organisation of materials obtained from the supramolecular mixing of dyes, one can also consider templating scaffolds that, when properly programmed with reactive groups, can spatially arrange chromophores in a given organisation and concentration. Examples of nanostructured templates include nucleic acids, polymers, proteins, micelles, zeolites, and nanoparticles. Depending on the chemical structure of the template, one can use either covalent or non-covalent interactions to attach the dyes. This strategy has been applied either through the self-assembly of the dyes on the scaffold or through the co-assembly of the dye and the scaffold conjugates, producing light-harvesting systems working with energy transfer efficiencies close to unitary values. Emphasis is now on antenna systems that can not only efficiently transfer energy, but also on architectures tailoring the absorption of a given range of the UV-vis light, quantum maximisation of the light absorption, adaptation, amplification, self-healing, and replication.
With the perspective to engineer architectures featuring strong solar light absorption, self-healing properties and adaptive properties, applications towards functional devices transforming light into chemical energy seem to be the most exciting. The obvious promise is to integrate these systems in photocatalytic systems to trigger exploitable chemical processes. The approach would consist in coupling the antenna with suitable artificial reaction centres that, catalysing a given chemical transformation, can operate under solar exposure.54 The results available today in the field of chemical biology and molecular genetics further imply, perhaps more unexpectedly, much potential in the field of photo-biocatalysis. The idea here is to have programmed living organisms that, depending on the environment and on the available chromophores, would produce a tailored light-harvesting architecture coupled to a reaction centre triggering a given chemical transformation. If a high-energy content molecule is to be produced, i.e. NH3, H2, or CH4, this could open the way toward the engineering of photobioreactors for solar fuel production.
Although the integration of a tailorable organic mimicry of the natural light-harvesting system into an exploitable and functional photoreactor producing solar fuels directly from solar light seems quite far away, the development of quantitative synthetic routes allowing the formation of tailored multichromophoric architectures of increased complexity remains the most appealing perspective for the field to be successful.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors gratefully acknowledge the EU through the ERC Starting Grant “COLORLANDS” project, the H2020-MSCA-RISE “INFUSION” and the H2020-MSCA-ITN-ETN “PHOTOTRAIN” networks. D. B. also thanks the School of Chemistry at Cardiff University and the University of Vienna for generous financial support.References
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle, G. Govindjee and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 CrossRef CAS.
- K. Nassau, The Physics and Chemistry of Color: The Fifteen Causes of Color, John Wiley & Sons, Inc., 2nd edn, 2001 Search PubMed.
- M. V. Orna, The Chemical History of Color, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013 Search PubMed.
- Z. Heinrich, Color Chemistry: Synthesis, Properties and Applications of Organic Dyes and Pigments, John Wiley & Sons, Inc., 3rd, revised edn, 2003 Search PubMed.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- M. Klessinger, Chem. Unserer Zeit, 1978, 12, 1–11 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- F. Ishiwari, Y. Shoji and T. Fukushima, Chem. Sci., 2018, 9, 2028–2041 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332–335 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M.-T. Kao, A. G. L. Olive and A. Del Guerzo, J. Am. Chem. Soc., 2011, 133, 316–325 CrossRef CAS.
- P. Z. Chen, Y. X. Weng, L. Y. Niu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Angew. Chem., Int. Ed., 2016, 55, 2759–2763 CrossRef CAS.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclère, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS.
- H. Lee, Y.-H. Jeong, J.-H. Kim, I. Kim, E. Lee and W.-D. Jang, J. Am. Chem. Soc., 2015, 137, 12394–12399 CrossRef CAS.
- L.-B. Meng, D. Li, S. Xiong, X.-Y. Hu, L. Wang and G. Li, Chem. Commun., 2015, 51, 4643–4646 RSC.
- M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 8889–8935 CrossRef CAS.
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 3501–3510 RSC.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS.
- E. Heyer and R. Ziessel, J. Org. Chem., 2015, 80, 6737–6753 CrossRef CAS.
- C. Hippius, F. Schlosser, M. O. Vysotsky, V. Böhmer and F. Würthner, J. Am. Chem. Soc., 2006, 128, 3870–3871 CrossRef CAS.
- A. Adronov and J. M. J. Fréchet, Chem. Commun., 2000, 1701–1710 RSC.
- A. S. Abd-El-Aziz, A. A. Abdelghani, B. D. Wagner and R. Bissessur, Macromol. Rapid Commun., 2019, 40, 1800711 CrossRef.
- M. Cotlet, T. Vosch, S. Habuchi, T. Weil, K. Müllen, J. Hofkens and F. De Schryver, J. Am. Chem. Soc., 2005, 127, 9760–9768 CrossRef CAS.
- A. Uetomo, M. Kozaki, S. Suzuki, K. I. Yamanaka, O. Ito and K. Okada, J. Am. Chem. Soc., 2011, 133, 13276–13279 CrossRef CAS.
- M. Lista, E. Orentas, J. Areephong, P. Charbonnaz, A. Wilson, Y. Zhao, A. Bolag, G. Sforazzini, R. Turdean, H. Hayashi, Y. Domoto, A. Sobczuk, N. Sakai and S. Matile, Org. Biomol. Chem., 2013, 11, 1754 RSC.
- K.-D. Zhang, N. Sakai and S. Matile, Org. Biomol. Chem., 2015, 13, 8687–8694 RSC.
- H. Zou, Y. Hai, H. Ye and L. You, J. Am. Chem. Soc., 2019, 141, 16344–16353 CrossRef CAS.
- P. Ensslen and H.-A. Wagenknecht, Acc. Chem. Res., 2015, 48, 2724–2733 CrossRef CAS.
- Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS.
- F. D. Lewis, R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2018, 51, 1746–1754 CrossRef CAS.
- P. Ensslen, Y. Fritz and H.-A. Wagenknecht, Org. Biomol. Chem., 2015, 13, 487–492 RSC.
- Q. Zou, K. Liu, M. Abbas and X. Yan, Adv. Mater., 2016, 28, 1031–1043 CrossRef CAS.
- G. Kodali, J. A. Mancini, L. A. Solomon, T. V. Episova, N. Roach, C. J. Hobbs, P. Wagner, O. A. Mass, K. Aravindu, J. E. Barnsley, K. C. Gordon, D. L. Officer, P. L. Dutton and C. C. Moser, Chem. Sci., 2017, 8, 316–324 RSC.
- L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS.
- L. Rocard, D. Wragg, S. A. Jobbins, L. Luciani, J. Wouters, S. Leoni and D. Bonifazi, Chem. – Eur. J., 2018, 24, 16136–16148 CrossRef CAS.
- H. Q. Peng, Y. Z. Chen, Y. Zhao, Q. Z. Yang, L. Z. Wu, C. H. Tung, L. P. Zhang and Q. X. Tong, Angew. Chem., Int. Ed., 2012, 51, 2088–2092 CrossRef CAS.
- Z. Xu, S. Peng, Y. Y. Wang, J. K. Zhang, A. I. Lazar and D. S. Guo, Adv. Mater., 2016, 28, 7666–7671 CrossRef CAS.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS.
- K. V. Rao, A. Jain and S. J. George, J. Mater. Chem. C, 2014, 2, 3055–3064 RSC.
- L. Gartzia-Rivero, J. Bañuelos and I. López-Arbeloa, Materials, 2017, 10, 495 CrossRef.
- K. V. Rao, K. K. R. Datta, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2011, 50, 1179–1184 CrossRef CAS.
- K. V. Rao, K. K. R. Datta, M. Eswaramoorthy and S. J. George, Adv. Mater., 2013, 25, 1713–1718 CrossRef CAS.
- Y. Zeng, Y. Li, M. Li, G. Yang and Y. Li, J. Am. Chem. Soc., 2009, 131, 9100–9106 CrossRef CAS.
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS.
- M. A. Harris, J. Jiang, D. M. Niedzwiedzki, J. Jiao, M. Taniguchi, C. Kirmaier, P. A. Loach, D. F. Bocian, J. S. Lindsey, D. Holten and P. S. Parkes-Loach, Photosynth. Res., 2014, 121, 35–48 CrossRef CAS.
- P. K. Dutta, R. Varghese, J. Nangreave, S. Lin, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 11985–11993 CrossRef CAS.
- S. Buckhout-White, C. M. Spillmann, W. R. Algar, A. Khachatrian, J. S. Melinger, E. R. Goldman, M. G. Ancona and I. L. Medintz, Nat. Commun., 2014, 5, 5615 CrossRef CAS.
- M. Kownacki, S. M. Langenegger, S.-X. Liu and R. Häner, Angew. Chem., Int. Ed., 2019, 58, 751–755 CrossRef CAS.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372 CrossRef.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |