
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Semi-biological approaches to solar-to-chemical conversion
Xin
Fang
 ,
Shafeer
Kalathil
and
Erwin
Reisner
*
,
Shafeer
Kalathil
and
Erwin
Reisner
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 15th June 2020
Abstract
This review presents a comprehensive summary of the recent development in semi-artificial photosynthesis, a biological-material hybrid approach to solar-to-chemical conversion that provides new concepts to shape a sustainable future fuelled by solar energy. We begin with a brief introduction to natural and artificial photosynthesis, followed by a discussion of the motivation and rationale behind semi-artificial photosynthesis. Then, we summarise how various enzymes can be combined with synthetic materials for light-driven water oxidation, H2 evolution, CO2 reduction, and chemical synthesis more broadly. In the following section, we discuss the strategies to incorporate microorganisms in photocatalytic and (photo)electrochemical systems to produce fuels and chemicals with renewable sources. Finally, we outline emerging analytical techniques to study the bio-material hybrid systems and propose unexplored research opportunities in the field of semi-artificial photosynthesis.
 Xin Fang | Xin Fang obtained his PhD degree from the University of Cambridge under the supervision of Prof. Erwin Reisner. His doctoral research focused on establishing efficient solar-to-chemical conversion avenues with biocatalysts in the form of enzymes and whole cells. His research broadly traverses the fields of biocatalysis, materials science, electrochemistry, and photocatalysis, with the aim to shift the paradigm of artificial photosynthesis by rationally integrating biocatalysts with synthetic components. Before that, he studied at Fudan University and Sichuan University for a Master and Bachelor's degree in polymer science, respectively. |
 Shafeer Kalathil | Shafeer Kalathil obtained his PhD in Chemical Engineering at Yeungnam University under the supervision of Prof. Moo Hwan Cho, where he worked on microbial fuel cells and microbial cell factories. He was a JSPS Fellow at the University of Tokyo (with Prof. Kazuhito Hashimoto) and a Postdoctoral Fellow at KAUST (with Prof. Pascal Saikaly). He joined the group of Prof. Erwin Reisner (University of Cambridge) as a Marie Curie Fellow to work on the microbial electrosynthesis of value-added products from waste-carbon. His current research focuses on the integration of light-harvesting materials with microbes to produce industrially relevant chemicals from carbon dioxide. |
 Erwin Reisner | Erwin Reisner received his PhD degree from the University of Vienna (with Prof. Bernhard K. Keppler), and postdoctoral training at the Massachusetts Institute of Technology (with Prof. Stephen J. Lippard) and the University of Oxford (with Prof. Fraser A. Armstrong) in biological inorganic chemistry, before moving to the University of Cambridge where he is currently the Professor of Energy and Sustainability. His laboratory explores the interface of chemical biology, synthetic chemistry, materials science and engineering relevant to the development of solar-driven processes for the sustainable synthesis of fuels and chemicals. |
1 Introduction
The consequences of anthropogenic carbon emissions call for innovative strategies to develop renewable energy technologies. Photovoltaics (PV) is the leading technology for solar energy conversion,1,2 but it shows disadvantages in intermittency and long-distance electricity transmission. Artificial photosynthesis is a process that converts solar energy into fuels and it thereby circumvents these drawbacks by storing solar energy in chemical bonds using synthetic light absorbers and catalysts.3,4 While synthetic catalysts still encounter challenges in solar-to-chemical conversion, nature provides evolutionarily-optimised biocatalysts. This review summarises how this biological machinery can be leveraged to catalyse light-driven reactions by an emerging technology termed “semi-artificial photosynthesis”, in which biocatalysts in the form of enzymes or microorganisms are incorporated with particles or structurally-crafted electrodes to produce fuels or chemicals.1.1 Natural photosynthesis
Photosynthesis occurs in photoautotrophs such as cyanobacteria, algae and higher plants, which harvest solar energy to produce biomass and O2 from CO2 and H2O. Photosynthesis is accomplished by two phases of reactions: the light reaction uses solar energy to generate the energy carrier adenosine triphosphate (ATP) and the reduced nicotinamide adenine dinucleotide phosphate (NADPH) as the reducing agent; the dark reaction then uses the ATP and NADPH to reduce atmospheric CO2 into carbohydrates through the Calvin cycle.5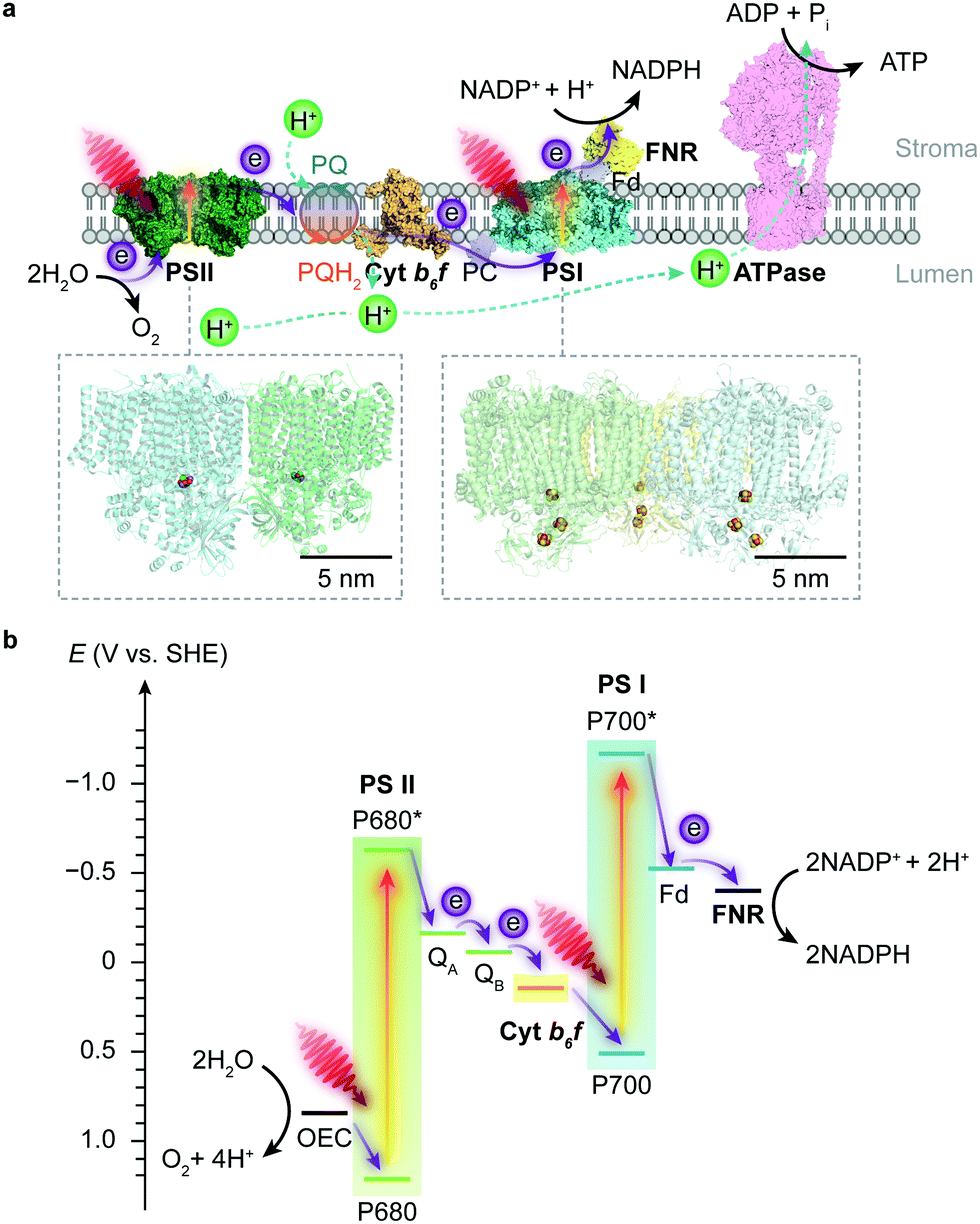 | ||
| Fig. 1 Schematic illustration of the light reaction in natural photosynthesis. (a) Electron and proton transfer pathways in the thylakoid membrane. At the start of the photosynthetic chain is PSII that oxidises H2O and releases O2 and protons upon light absorption. The electrons are then delivered to PSI via a plastoquinone (PQ) pool, cyt b6f and plastocyanin (PC). Electrons are photoexcited for a second time at PSI to reduce NADP+ to NADPH via a ferredoxin (Fd) and a ferredoxin–NADP+ reductase (FNR). The water oxidation and electron transport also induce proton translocation from the stroma to the lumen, which generates a proton gradient and chemiosmosis driving the synthesis of ATP by ATP synthase (ATPase). Protein data bank ID: PSII (4ub6), cyt b6f (4h44), PC (1bxu), PSI (5oy0), Fd-FNR (2yvj), ATPase (6fkf). (b) Energy level diagram of the Z-scheme electron transfer in the thylakoid membrane. The redox carriers are placed at their midpoint redox potentials at pH 7.0. | ||
Photorespiration can consume 25% of carbon photosynthetically fixed by C3 plants and this carbon loss is more severe at higher temperature. Some tropical plants, namely C4 plants, overcome the disadvantageous photorespiration by deploying an additional C4 pathway to increase the local concentration of CO2. In these plants, phosphoenolpyruvate is first carboxylated into a four-carbon compound oxaloacetate, which undergoes further transformations and decarboxylation to release CO2. The released CO2 is then scavenged by Rubisco and drawn into the Calvin cycle with minimal photorespiration.6
Some cyanobacteria can also fix atmospheric N2 using solar energy when nitrogen-containing substrates (NH3, NO3−, etc.) are not available.19,20 However, nitrogenase (N2ase), the enzyme responsible for N2 fixation, is sensitive to oxygen.21 Therefore, cyanobacteria have developed strategies to separate the oxygenic photosynthesis and N2 fixation spatially (in different cells) or temporally (during the night) to protect the N2ase from irreversible damage.20
1.2 Artificial photosynthesis
Artificial photosynthesis does not reproduce the exact reactions occurring in photoautotrophs, but exploits light absorbers and catalysts to produce fuels and chemicals with earth-abundant feedstock chemicals such as H2O and CO2. A prototype reaction is light-driven water splitting, which produces H2 as a suitable energy carrier to overcome the intermittency of solar irradiation.22,23 Solar water splitting has been under intense investigation over several decades, but it still faces challenges arising from solar light harvesting, catalyst stability, interfacial charge transfer and the water oxidation reaction.24 The four-electron water oxidation to oxygen involves multiple bond rearrangements and concerted proton release, and thus causes both a thermodynamic barrier and kinetic sluggishness that confront the existing catalytic chemistry.25Thermodynamics determines the suitability of light absorbers from an energetic perspective (Fig. 2), as illustrated by the water splitting reaction, where the energy band positions should straddle the electrochemical potentials of the half reactions, i.e., hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) (Fig. 3a). The band gap of the light absorbers should not be too wide to enable the visible light (400–800 nm, band gap: 1.53–3.10 eV) utilisation.26 The thermodynamic redox potentials of light absorbers should also be properly aligned with respect to their energy band positions to prevent themselves from photoinduced corrosion.27
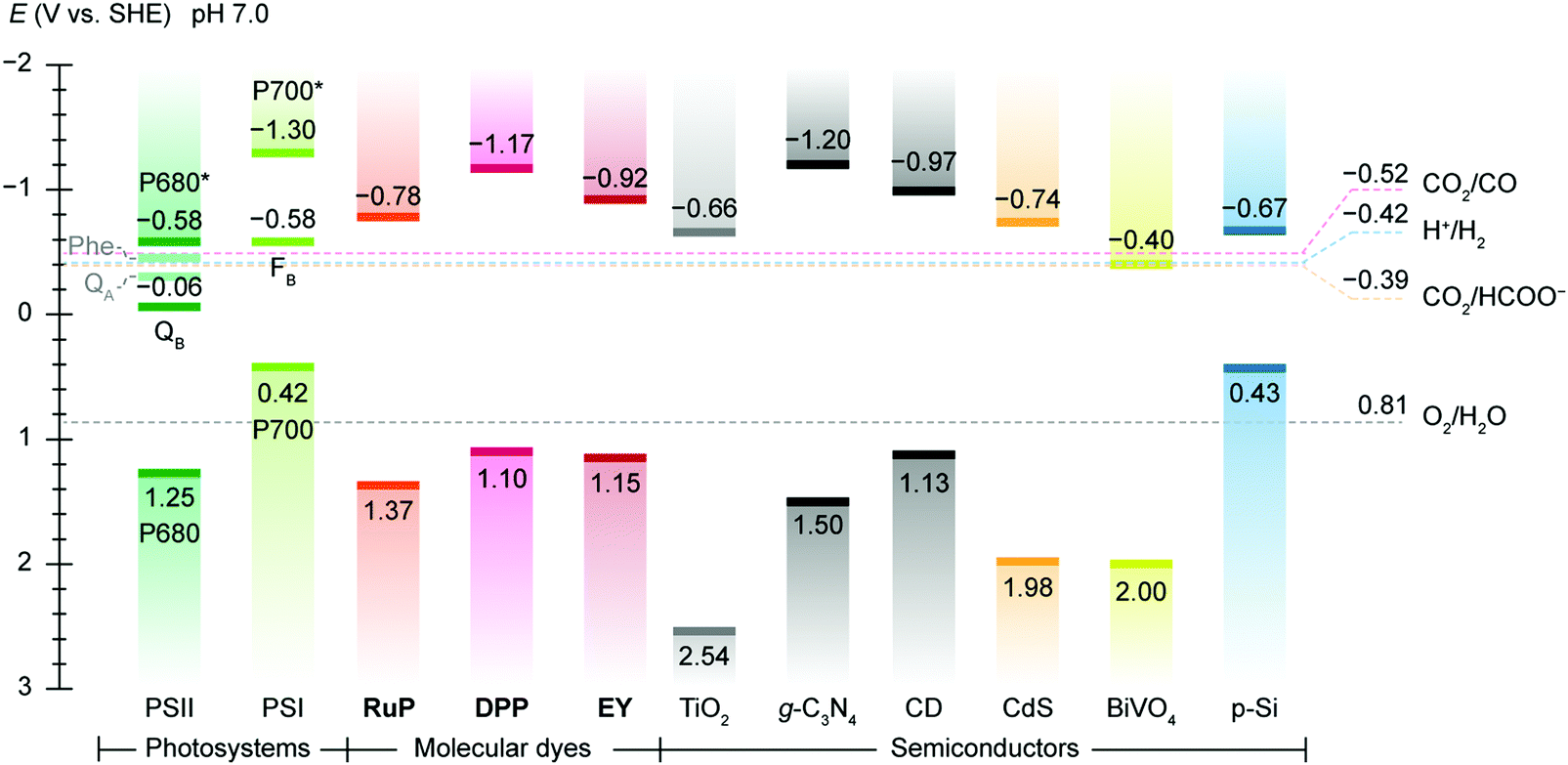 | ||
| Fig. 2 Energy band edges of light absorbers used in semi-artificial photosynthesis. The band edge locations of photosystems are represented by redox potentials of their reaction centre chlorophylls, i.e. P680 (PSII) and P700 (PSI) and terminal electron acceptors, i.e., QB (PSII) and a [Fe–S] cluster FB (PSI).119 For molecular dyes, the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) are used as conceptual equivalents of conduction band edge and valence band edge in semiconductors. Data source: DPP,147RuP,147Eosin Y (EY),292 TiO2,293g-C3N4,294 carbon dot (CD),283,295 CdS,296 BiVO4,157 p-Si.162 The redox potentials CO2/CO (catalysed by CODH),186 H+/H2 (catalysed by H2ase),133 CO2/HCOO− (catalysed by FDH),183 and O2/H2O are displayed as references. The energy band edges and redox potentials are corrected with respect to SHE at pH 7.0 via the Nernstian relationship (25 °C). | ||
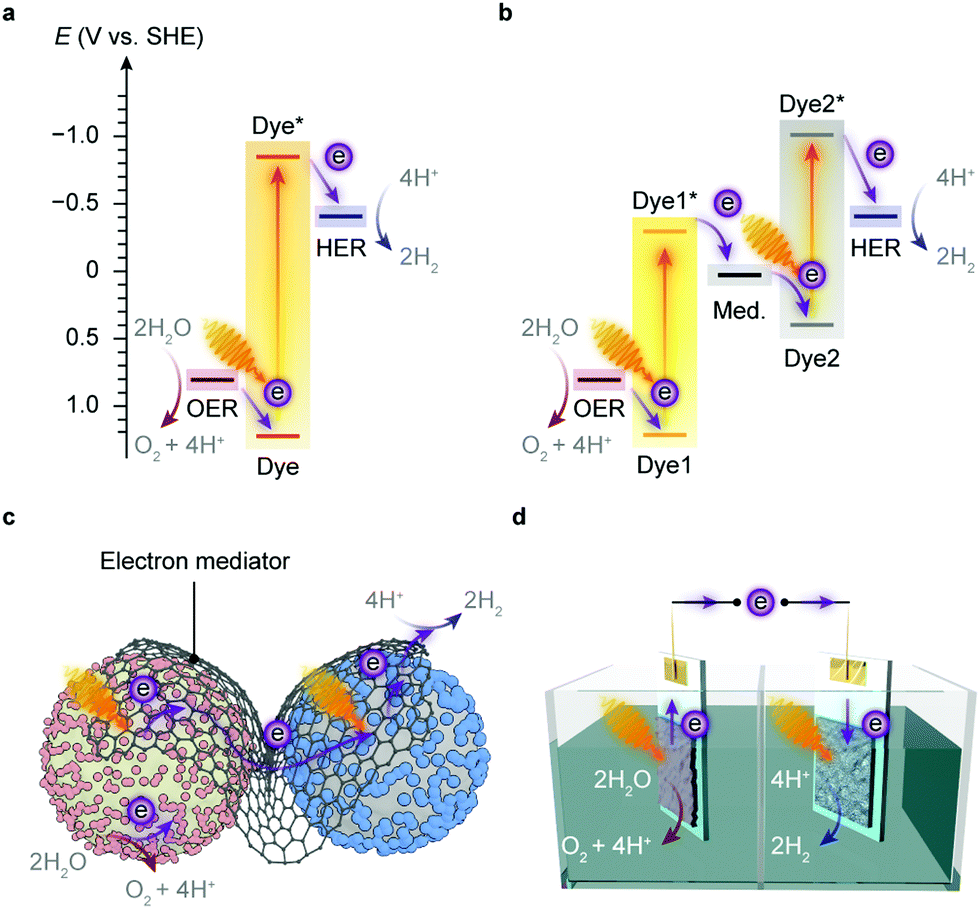 | ||
| Fig. 3 Artificial approaches to solar-to-chemical conversion. (a and b) Energy level diagrams of light-driven water splitting at pH 7.0: single-step excitation (a) and two-step excitation (b). Dye and Dye*: the light absorber in the ground and excited state, respectively. Med.: redox mediator. (c) Representation of a two-step photocatalytic water-splitting system with semiconductor particles and HER and OER co-catalysts. The HER and OER photocatalysts are interfaced with a solid-state electron mediator. (d) Representation of a PEC tandem water-splitting system. | ||
Several semiconductors whose electronic structures suffice in theory for overall water splitting with visible light have been under investigation, such as C3N4, CdS and Y2Ti2O5S2.22 An alternative strategy is to align the band position of two light absorbers and catalyse HER and OER with the aid of electron mediators (Fig. 3b),22,28 which resembles the photosynthetic electron transfer (“Z-scheme”) in the thylakoid membrane (Fig. 1b). This two-step excitation system can expand the light absorption by harvesting lower-energy photons,22,29 and can bring together materials that can only drive either HER or OER alone.30–32 Such tandem systems enable larger driving forces for water splitting, and permit higher theoretical solar-to-hydrogen conversion efficiencies than the single light-absorber systems.33 Yet two light absorbers also multiply chances of charge recombination and pose challenges to balance the electron transfer kinetics of the half reactions.3,22
The implementation of artificial photosynthesis is often envisioned in two forms: first, light absorbers are loaded with electrocatalysts and then suspended in a photoreactor (Fig. 3c).34 Second, solar fuel synthesis is performed in a photoelectrochemical (PEC) cell, where catalysts are immobilised on photoelectrodes and photoinduced electrons flow across the external circuit (Fig. 3d).35 From a techno-economical viewpoint, the photocatalytic system may be more advantageous, as PEC systems may be required to achieve up to ∼25% solar-to-hydrogen (STH) conversion efficiency to rival with petrol in energy prices, primarily due to higher investment in installations, whereas a STH efficiency of 5–10% would possibly suffice to render photocatalytic reactors cost-competitive.4,34 However, the highest STH conversion efficiency of particle-based systems is around 1%,28 whereas that of electrode-based systems can typically achieve more than 10%.36,37 PEC systems also have several merits that are appealing for fundamental research and practical applications: (1) immobilisation of photocatalysts on electrodes enables in-depth studies of half-reactions individually without sacrificial reagents and allows for in situ probing of photoredox chemistry through spectroscopic methodologies;38–40 (2) from an application's perspective, the electrode-based configuration enables separation of H2 and O2 in a two-compartment configuration,41 and can be transformed into a continuous fuel-production system with a flowing electrolyte solution.42 On the other side, drawbacks emerge: (1) the preparation of photoelectrodes requires conductivity and high film-forming capability of semiconductors; (2) the operation of a PEC cell can generate pH gradients and cause mass transfer limitations that account for substantial potential loss and subject electrodes to increasingly corrosive environments;43,44 (3) the internal resistance often necessitates external bias voltages to drive the electron flow from a photoanode to a photocathode.
Sustainable transformation of CO2 to chemicals and fuels would provide a means to close the loop of the anthropogenic carbon cycle and offer a viable solution for carbon capture and utilisation.45 The linear CO2 molecule is chemically stable, and thus activating the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond for the ensuing endothermic reactions may incur a substantial energy penalty, which is reflected in the negative potential (−1.9 V vs. SHE, pH 7.0) for the one-electron reduction to CO2˙−.46–48 Although such a high energy barrier can be overcome by large electrochemical potentials or high temperature, it results in poor energetic efficiencies for subsequent fuel synthesis. The activation of CO2 is followed by a stepwise proton and/or electron transfer, which gives rise to furcate reaction pathways towards miscellaneous products.45,49 In addition, due to the low solubility of CO2 in water (0.033 mol L−1 at 25 °C, 100 kPa),46 the reduction of CO2 usually competes with H2 evolution from H2O, which further reduces the selectivity and the conversion efficiency.48 Current photocatalytic CO2 reduction systems can produce CO and formate, but still struggle to produce multicarbon chemicals such as ethylene and ethanol selectively, which challenges the economic exploitation of this approach.50
O bond for the ensuing endothermic reactions may incur a substantial energy penalty, which is reflected in the negative potential (−1.9 V vs. SHE, pH 7.0) for the one-electron reduction to CO2˙−.46–48 Although such a high energy barrier can be overcome by large electrochemical potentials or high temperature, it results in poor energetic efficiencies for subsequent fuel synthesis. The activation of CO2 is followed by a stepwise proton and/or electron transfer, which gives rise to furcate reaction pathways towards miscellaneous products.45,49 In addition, due to the low solubility of CO2 in water (0.033 mol L−1 at 25 °C, 100 kPa),46 the reduction of CO2 usually competes with H2 evolution from H2O, which further reduces the selectivity and the conversion efficiency.48 Current photocatalytic CO2 reduction systems can produce CO and formate, but still struggle to produce multicarbon chemicals such as ethylene and ethanol selectively, which challenges the economic exploitation of this approach.50
Artificial photosynthesis has recently been extended to drive reactions such as N2 reduction. Today, industrial N2 reduction relies on the energy-demanding Haber–Bosch process that requires H2, high temperature (300–500 °C) and high pressure (150–200 atm).51,52 The Haber–Bosch process also entails considerable CO2 emission during the production of the inlet H2 from natural gas (1.87 kg CO2 per 1 kg NH3).51 The intense energy consumption and environmental stress arising from the Haber–Bosch process have spurred explorations in sustainable routes for N2-to-NH3 transformation. Photocatalytic N2 reduction uses solar light to replace fossil fuels as the energy source and water instead of natural gas as the hydrogen source and operates at room temperature and atmospheric pressure.53–56 The prospect of this approach largely depends on the photocatalysts that generate photoelectrons to reduce the stable and inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. However, the existing photocatalytic N2 reduction systems still display low solar-to-chemical conversion efficiencies (typically below 0.02%),51,55,57 and current studies encounter technical difficulties in reliable NH3 quantification and elimination of probable experimental artefacts.58
N bond. However, the existing photocatalytic N2 reduction systems still display low solar-to-chemical conversion efficiencies (typically below 0.02%),51,55,57 and current studies encounter technical difficulties in reliable NH3 quantification and elimination of probable experimental artefacts.58
2 Semi-artificial photosynthesis
Artificial photosynthesis establishes solar-to-fuel pathways to address the global energy challenge, but the bottlenecks are key-step reactions during the fuel-forming process, such as water oxidation and selective CO2 activation. Biology is highly capable of tackling these synthetic challenges through the naturally-refined biocatalytic machinery. Enzymes comprise only a handful of earth-abundant metal atoms as catalytic centres and cofactors, which are embedded in polypeptide chains (Fig. 4a).46 Enzymes perfectly orchestrate the electron and proton transfer, reactant delivery, bond rearrangement and product removal at the active site, thereby reducing the energy threshold and accelerating the reaction kinetics. Enzymes widely employ steric hindrance and electrostatic or hydrogen bond interactions to stabilise selected intermediates and transition states, and therefore provide an efficient reaction path towards a single product.59 The catalytic prowess of enzymes has inspired synthetic endeavours to mimic their active sites and ligand environments, and to exploit the biological strategies to stabilise intermediates and control selectivity.60–62 Although synthetic mimics are making progress, they have not yet reproduced the performance of their natural models in terms of catalytic rate, selectivity and electrochemical overpotentials under benign aqueous conditions.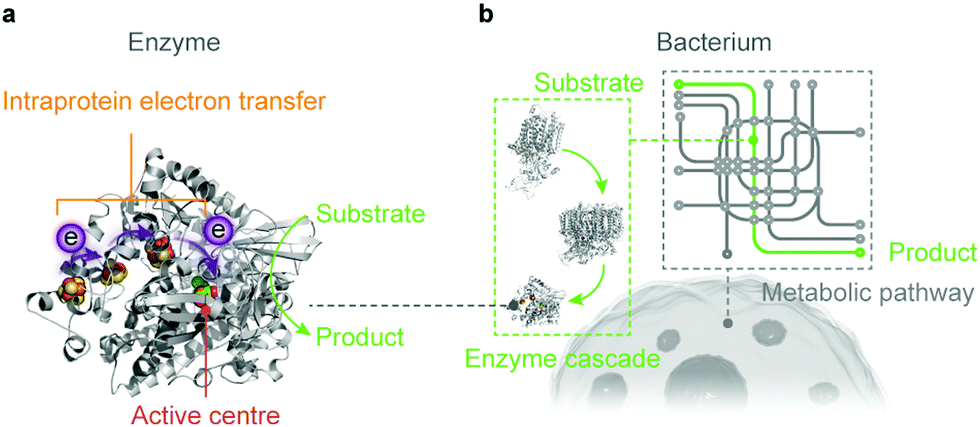 | ||
| Fig. 4 Biocatalysts in the form of an enzyme (a) and a bacterium (b) used in semi-artificial photosynthesis. | ||
Microorganisms can produce complex multicarbon compounds from simple feedstocks through their intracellular pathways (Fig. 4b). They can also maintain metabolic homeostasis at varying environmental and nutritional conditions. The enzymatic machinery embedded in its well-controlled and confined native environment benefits from the innate regulation mechanisms and can be repaired and regenerated when necessary. On the other side, microorganisms rank their physiological needs to survive ahead of synthetic efficiency to produce target chemicals. Carbon and electron fluxes in the metabolism are partially directed towards biomass synthesis for cell growth and maintenance, which reduces the pathway efficiency in chemical synthesis. Moreover, rerouting biochemical pathways towards desired products encounters resistance from the intracellular regulation, which renders such alteration problematic.63,64
Artificial photosynthesis, however, permits more flexibility in system design and modification. Taking light harvesting as an example: most chlorophylls in nature have a minimum absorption to green light (λ = 500–600 nm),13 which partially accounts for the low photosynthetic efficiency, whereas in artificial photosynthesis, broadband absorption can be readily achieved with a variety of semiconductors and molecular dyes.11 Furthermore, artificial systems allow for coupling fuel-forming reduction reactions with useful oxidative chemical transformations. Several studies have already demonstrated that electrons could be extracted from biomass, organic compounds and even plastics to produce H2 with versatile light-absorbing systems.65–69 Artificial photosynthesis systems reduce the dissipation of energy and electrons along the pathway, enabling high solar-to-fuel efficiencies routinely surpassing their natural counterparts.70–73 Artificial photosynthesis is also empowered by an array of analytical techniques such as electrochemistry, spectroscopy and operando methodologies,40,45,74,75 which are still less commonly employed for quantitative studies in biological research. The well-defined features of synthetic materials permit systematic investigations to understand reaction mechanisms and establish structure–function relationships for system design and optimisation.76
Semi-artificial photosynthesis provides a hybrid approach to solar-to-chemical conversion, by integrating the biocatalytic machinery (enzymes and microbes) with synthetic materials (dyes, electrocatalysts, semiconductors, electrodes, mediators).77 The photosynthetic biohybrid systems seek to outsource tasks to the components that can perform them best and thus combine strengths of both while bypassing limitations of each. In such hybrid systems, enzymes and microbes function often as catalysts to drive endergonic and complex chemical reactions, whereas synthetic materials are commonly scaffolds to immobilise biocatalysts and functional components that carry out light absorption, charge transfer, chemical transformations and product separation. Several synthetic materials such as polymers and metal organic frameworks can also provide additional protection to fragile enzymes for more sustained operation against environmental stressors.78–80 According to the form of biocatalyst, semi-artificial photosynthesis falls into two categories: the enzymatic and the microbial system. Each of these systems can be further distinguished as either a colloidal suspension or a PEC cell (Fig. 5).
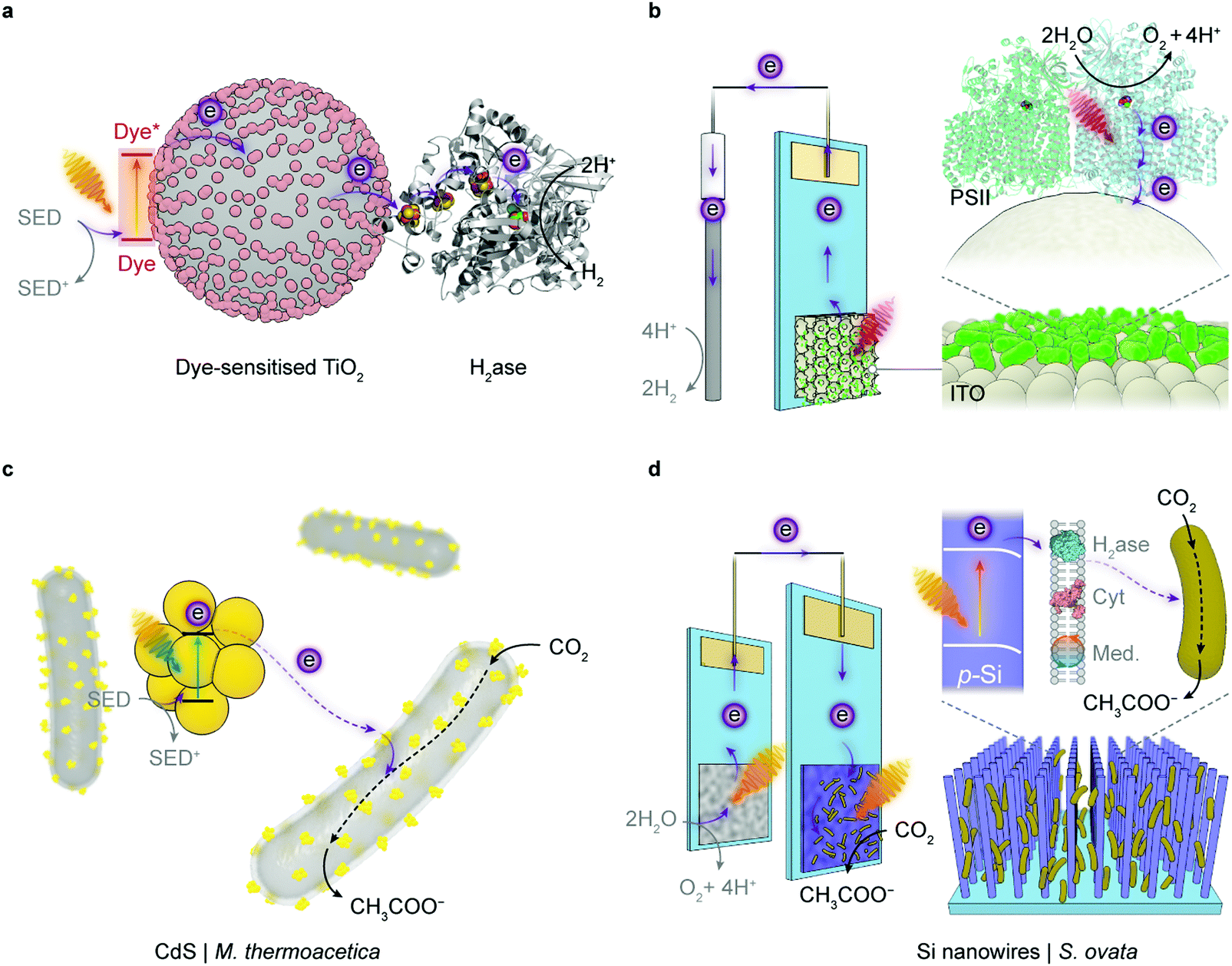 | ||
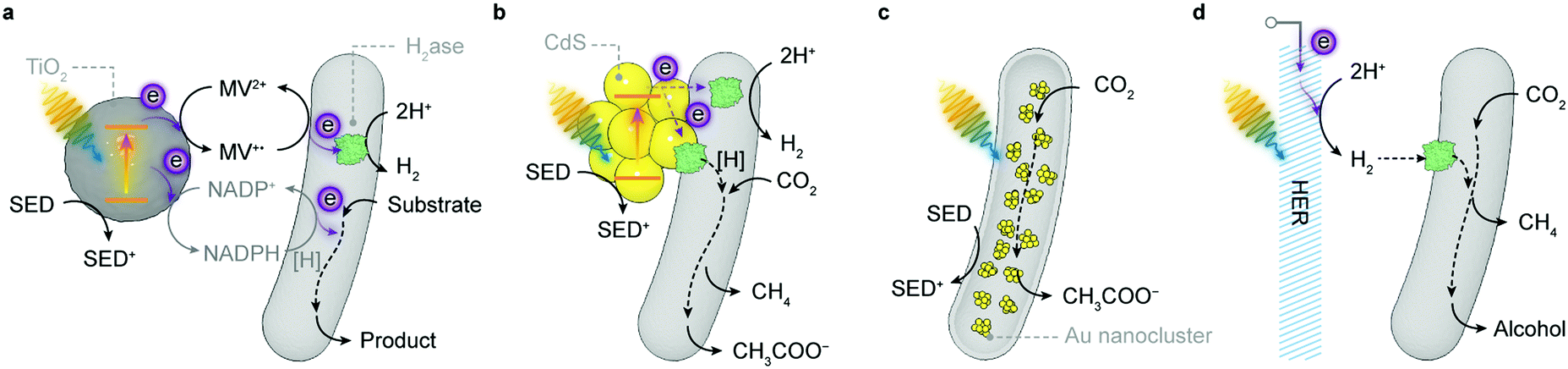
| Fig. 5 Representative semi-artificial photosynthesis systems. (a) A colloidal system with dye-sensitised TiO2 nanoparticles and H2ase.145 Under irradiation, the dye is excited and donates electrons to the conduction band of TiO2. The photoelectrons are then delivered to the catalytic centre of H2ase for proton reduction via intraprotein electron relays. The oxidised dye is regenerated by extracting electrons from a sacrificial electron donor (SED). (b) An electrode system with a three-dimensional indium tin oxide (ITO) electrode and PSII.102,120 PSII is wired by the porous ITO scaffold and photocatalyses water oxidation, aided by an applied potential. (c) A colloidal system with light absorbing CdS nanoparticles deposited on the CO2-reducing bacterium M. thermoacetica.226 The excited CdS transfers photoelectrons to the bacterium, which generates reducing equivalents to produce acetate from CO2via the Wood–Ljungdahl pathway. The holes in CdS are quenched by the SED. (d) A hybrid tandem system with TiO2 nanowires as the photoanode and p-Si nanowires integrated with acetogenic bacteria S. ovata as the photocathode.248 When irradiated, the TiO2 electrode oxidises water while p-Si nanowires generate photoelectrons that are delivered to the interfacing S. ovata via extracellular electron transfer pathways. S. ovata uses these electrons to reduce CO2 into acetate through the Wood–Ljungdahl pathway. | ||
3 Enzymatic hybrid systems
An enzymatic hybrid system employs separated electroactive or photoactive enzymes as catalysts or light absorbers, to function in concert with synthetic components in driving reactions selectively at high rates and yields (Fig. 5a and b). Amongst a myriad of enzymes available in nature, only a handful of them are of interest in synthetic reactions starting from the simplest feedstocks (e.g., H2O, CO2 and N2). Translation of their catalytic ability in vivo into advantages in vitro is confronted with several challenges. Whereas synthetic catalysts are either integral parts of electrodes or discrete entities (particles or molecules), these redox enzymes are proteins with molecular weights of 104–106 g mol−1 and areal footprints of 50–400 nm2. Their active sites (catalytic centres) are embedded within the protein matrices, which prevent indiscriminate reactions with substrate analogues. These enzymes have evolved intraprotein electron relays such as haems, quinones, and [Fe–S] clusters that can carry electrons between active sites and redox partners.81 The electron relays define directional electron transfer pathways within the protein and allow electrochemistry to activate, monitor and modulate the enzymatic redox chemistry.82 The current knowledge of the structure, functionality and catalytic mechanism within the enzymes discussed in this section has already been reviewed previously and is therefore not discussed here.59,83–86To approach maximal catalytic rate, enzymes are expected to accelerate the chemical reactions towards diffusion controlled kinetics.87 As such, enzymes should engage with electrodes in an “electroactive” orientation to enable rapid electron transfer kinetics between the distal relay centre and the electrode surface. In this regard, the enzyme–electrode interface must be rationally engineered with respect to topology, porosity and surface chemistry (hydrophilicity, surface charge, functional moieties, etc.). From a kinetic perspective, redox enzymes reduce the activation energy and thus, minimise the electrochemical overpotential needed to drive a reaction.59,88 Therefore, enzymes can often catalyse redox reactions very close to their thermodynamic potentials and reduce the energy penalty arising from large overpotentials. The low overpotentials can simplify system design by judiciously pairing with light absorbers and electron mediators according to their energy levels, and carry out reactions that were otherwise not possible with conventional electrocatalysts.
3.1 Water oxidation
Water is the “ideal” terminal electron source for the downstream fuel-forming reactions in artificial photosynthesis, but only few molecular catalysts with earth-abundant elements can handle this multielectron/proton process in an efficient fashion, which is further complicated by the combination with light absorbers. PSII, however, assigns the tasks of light harvesting and water oxidation to antenna proteins and the oxygen evolution centre (OEC), respectively, and orchestrates photochemistry in concert with catalysis in a stepwise manner via efficient intraprotein electron relays.89 However, the intrinsic fragility of PSII under light irradiation limits the longevity of its catalytic activity (τ1/2 < 30 min) and thereby strangles the continuous downstream synthesis with isolated PSII.90Antenna complexes in PSII harvest solar light and funnel the light energy to P680, where charge separation occurs within a few picoseconds.91 This is followed by an electron transfer towards a pheophytin molecule (PheD1) and later to plastoquinone A (QA) (Fig. 6a). The resulting highly oxidising PD1+ in P680 can extract an electron from a redox-active tyrosine YZ (TyrZ) and then initiate water oxidation at the Mn4CaO5 cluster.91 On the stroma side, QA is tightly bound to the D2 protein and acts as a single-electron acceptor, whereas QB in the D1 protein can accept two electrons and be fully protonated.92 The charge transfer from QA to QB is aided by a non-haem iron midway between them.93 The formed QBH2 departs from the reaction centre and is replenished by the plastoquinone pool in the thylakoid membrane.
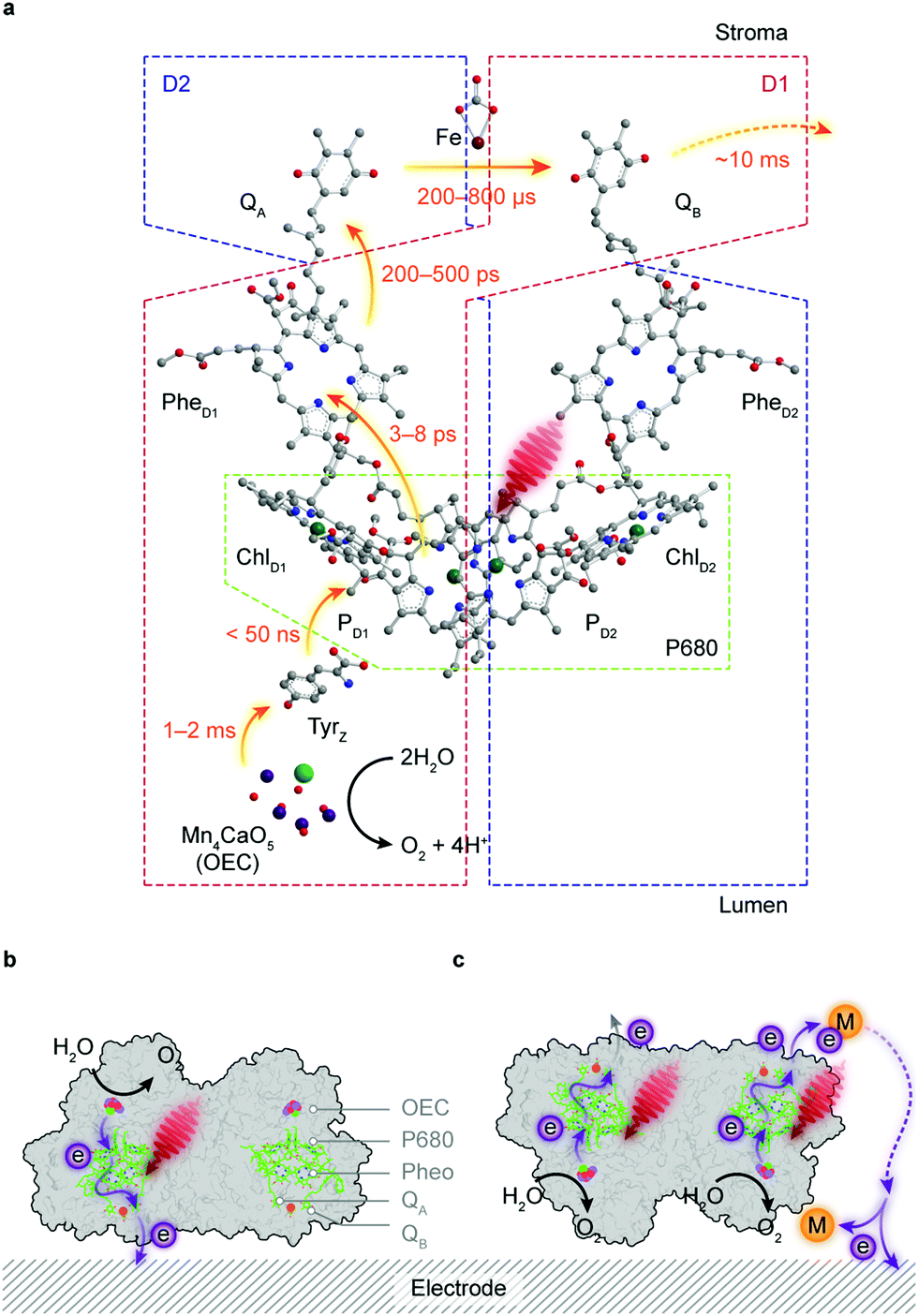 | ||
| Fig. 6 Electron transfer pathways in PSII-photoelectrochemistry. (a) Schematic representation of the electron transfer pathways (indicated by the orange arrow) in the reaction centre complex of PSII. (b and c) Electron transfer at the PSII-electrode interface. Direct electron transfer can only take place provided that the stromal side of PSII intimately interfaces with the electrode (the electroactive orientation, b). PSII cannot donate electrons to the electrode in unsuitable orientations unless there are diffusional redox couples that can mediate electrons between QB and the electrode (c). | ||
PSII is a biological model OER photocatalyst that inspires synthetic endeavours to mimic its core components,61,94–96 and stimulates mechanistic investigations to understand its functionality.97–99 Wiring PSII to an electrode allows the resulting biohybrid electrode to supply photoelectrons from water, and permits protein film-photoelectrochemistry (PF-PEC) to benchmark enzymatic activity in vitro, dissect photoinduced electron transfer pathways, and repurpose biogenic electrons to drive endergonic reactions.90,100
Attempts to interface PSII with an electrode surface started several decades ago,101 when isolated PSII was deposited on a Pt electrode and generated a photocurrent of a few microamperes. However, the minuscule amount of PSII loaded on the electrode renders in-depth studies and proof-of-concept demonstrations unfeasible. Underlying PSII-PEC is the electrode that provides a physical scaffold to immobilise enzymes and an artificial electron acceptor for the biogenic electrons. PSII's photochemistry and the ensuing current output can be greatly influenced by the material, morphology and physical property of the electrode scaffold, which dictates the strength of protein binding, the capacity of protein loading, the accessibility to intraprotein electron relays, the depth of light penetration, and the transport of reactants and products therein.102
The directionality of the intraprotein electron flow makes the interfacial electron transfer highly dependent on the protein orientation. QB, the terminal electron acceptor in PSII, can only undertake outward electron transfer if the stromal side of PSII is in close proximity to an electrode surface (Fig. 6b). The electron transfer via intrinsic plastoquinones, namely, direct electron transfer (DET) can be registered as an anodic photocurrent by chronoamperometry under irradiation, and verified in control experiments by the removal of the Mn4CaO5 cluster or addition of 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) that inhibits the QB site and interrupts the electron flow from QA.103,104 DET often results in a relatively low photocurrent because PSII loading may be low and a significant portion of the enzymes may be in an unfavourable orientation or remote from the electrode surface (Fig. 6c). This problem can be mitigated by using diffusional mediators such as 2,6-dichloro-1,4-benzoquinone (DCBQ, Em = 0.32 V vs. SHE, pH 7.0) to shuttle electrons from the QB site to the electrode (Fig. 6c).103 This mediated electron transfer (MET), in biological and artificial systems, comes at an expense in both energetics and kinetics, as extra energy is needed to drive the redox turnover of electron mediators and the diffusion of mediators is likely to govern the overall rate of electron transfer.
From both fundamental and practical viewpoints, an electrode with a large number of proteins being wired in an electroactive orientation is desired to produce a high photocurrent that can afford reliable analysis and proof-of-concept demonstrations (Fig. 7a). The making of such electrodes enables new possibilities to create solar-to-fuel pathways unattainable in biology. The chronology of the development of PSII electrode design will be outlined below.100
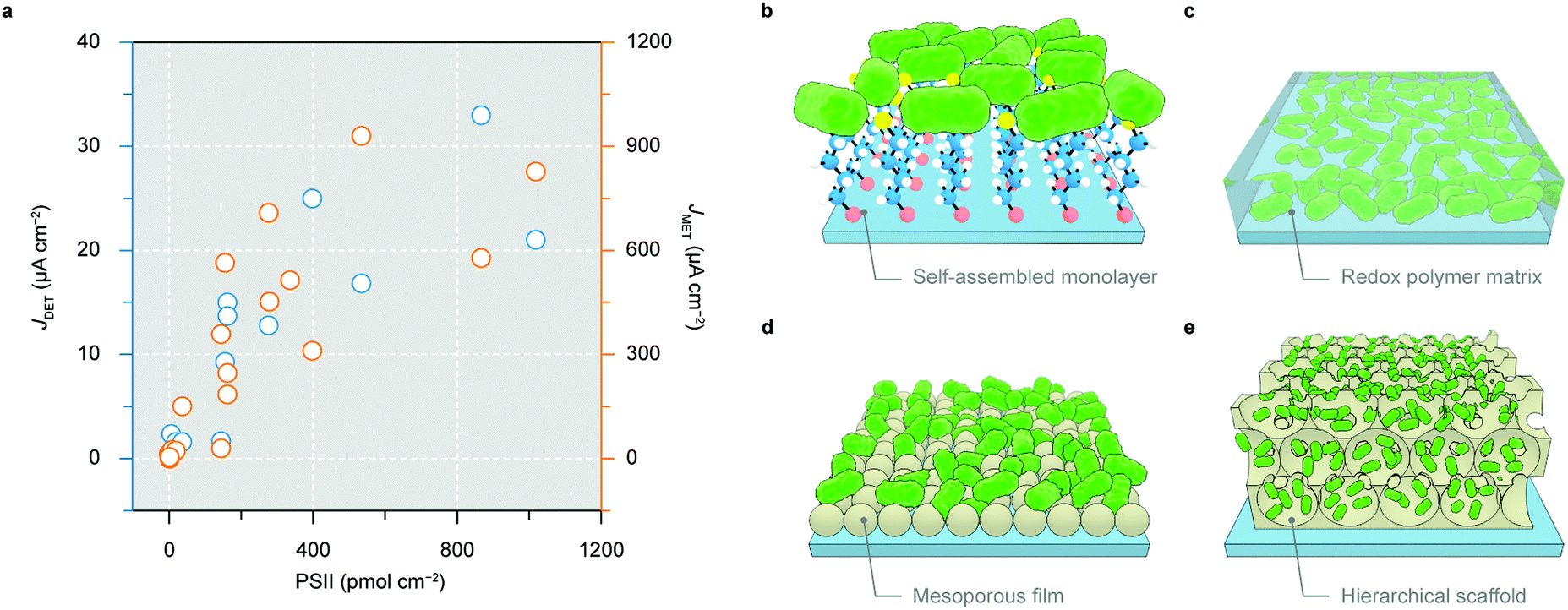 | ||
| Fig. 7 Electrode architectures to wire PSII for semi-artificial photosynthesis. (a) Correlation between protein loading in electrodes and the resulting photocurrent by DET (blue dots, the left axis) and MET (orange dots, the right axis). (b–e) PSII wired to a flat electrode via a self-assembled monolayer (b), and a redox-polymer matrix (c); PSII immobilised in a mesoporous film with nanoparticles (d), and a three-dimensional hierarchical scaffold (e). | ||
In the nascent stage of PSII-PEC, PSII with a polyhistidine tag (His-tag) at its stromal side was immobilised on flat Au electrodes by a self-assembled monolayer (Fig. 7b).105–108 The interaction between electrode-anchored nickel-nitrilotriacetic acid and His-tags permitted site-selective binding of PSII on Au electrodes. However, the flatness of the electrode surface limited the protein loading typically below 10 pmol cm−2 and thus resulted in submicroampere photocurrents.108 Electrodes using a redox polymer matrix as a non-diffusional mediator to electrically wire a high amount of proteins enable redox centres on the polymer backbone to access intraprotein electron relays, regardless of their orientations and distances (Fig. 7c).109 A polymer with the Os3+/Os2+ redox couple (E1/2 = 0.39 V vs. SHE) was employed as both an immobilisation matrix and an electron mediator to PSII, which gave rise to a photocurrent density up to 45 μA cm−2.110
The second generation of electrodes are mesoporous films made with metal oxides such as Fe2O3, TiO2 and ITO.104,111–115 A mesoporous ITO (meso-ITO) film with a thickness of 2–10 μm and a pore size up to 100 nm can be made with a high degree of tunability (Fig. 7d).104,111,116,117 The meso-ITO afforded a PSII loading of 19 pmol cm−2, which resulted in a DET photocurrent of 1.6 μA cm−2 and a mediated photocurrent of 22 μA cm−2.104 The surface chemistry of ITO can be modified to covalently bind PSII in favour of an electroactive orientation.111 The meso-ITO film can also be deposited onto the disk of a rotating ring disk electrode to detect the O2 production on the ring and study the oxygenic photoreactivity in PSII.118
The state-of-the-art third generation electrodes for PSII-PEC feature a hierarchical inverse opal structure with both macroporosity and mesoporosity (Fig. 7e).102,119–127 This electrode architecture has interconnected macropores that provide a large surface area accessible by protein diffusion. The skeleton is made of a mesoporous structure consisting of (semi)conducting and hydrophilic nanoparticles (e.g., ITO or TiO2) where proteins are adsorbed. The inverse opal electrodes can be fabricated via a co-assembly method using polystyrene beads as the structural template and nanoparticles as the electrode material. Such method makes the electrode structure easily variable to fit biocatalysts with different dimensions.128,129 The hierarchical structure also benefits both mass transport and light transmission, and permits high PSII loading in the range of 30–1000 pmol cm−2 (depending on the film thickness).119–122
A 40 μm thick inverse opal-ITO (IO-ITO) electrode with PSII attained a high photocurrent of 17 μA cm−2 for DET and 930 μA cm−2 in the presence of a DCBQ mediator.120 Os-based polymers can further be incorporated in the IO-ITO scaffold to improve the electrical wiring between PSII and the electrode to eliminate the need for diffusional additives.121 Compared to flat electrodes,110 the IO-ITO electrode with the polymer matrix achieved a higher photocurrent of 381 μA cm−2 due to higher PSII (336 pmol cm−2) loading.121 The high photocurrent allowed for reliable O2 quantification to calculate the Faraday efficiency, which was not possible with flat electrodes.
PSII immobilisation on porous ITO electrodes also allowed PF-PEC to dissect unexpected electron transfer pathways at the enzyme–electrode interface.100 DET from QA to the ITO electrode was observed when inhibiting PSII with DCBQ, showing the possibility to release electrons upstream of QB.104,130 Furthermore, analysing cathodic current contributions at low potentials allowed identifying a competing pathway stemming from photoinduced O2 reduction, presenting a short-circuit to the natural water oxidation process.118,130
Although PSII is the only enzyme that operates in nature for water oxidation to O2, it is not the only enzyme that can perform this reaction in vitro. Laccase, a blue multicopper oxidase that couples the oxidation of organic substrates with the reduction of O2 to H2O, has been shown to catalyse the reverse water oxidation reaction when immobilised on an electrode (E > 1.2 V vs. SHE, pH 7.4).131 In an attempt to use this enzyme for light-driven water oxidation, laccases were adsorbed on a semiconducting In2S3 electrode (band gap: 2.0 eV) as a visible-light absorber.132
3.2 Hydrogen production
H2ases are metalloenzymes that can catalyse the interconversion between H2 and H+/e− with a TOF of >1000 s−1.84,133 The HER activity of H2ases per active site is therefore comparable with that of the benchmark Pt catalyst.134,135 There are three phylogenic classes of H2ases termed according to the metallic centres at their active sites: [NiFe]–H2ase, [FeFe]–H2ase and [Fe]–H2ase, but only [NiFe]– and [FeFe]–H2ases incorporate [Fe–S] clusters as electron relay centres and can catalyse the reversible proton reduction to H2.136 These [Fe–S] clusters are spaced 10–14 Å apart, which permits sequential electron tunnelling towards the active sites at a rate (107 s−1) faster than catalysis.137,138These intraprotein electron relays can couple the catalytic turnover at the active site with the electron exchange through the electrode, and thus enable protein film electrochemistry (PFE) to probe the H2–H+ interconversion by deciphering resulting voltammograms and chronoamperograms.135,139 PFE reveals that [FeFe]–H2ases are active for both proton reduction and H2 oxidation yet extremely sensitive to O2, whereas the more O2-tolerant [NiFe]–H2ases usually show strong catalytic bias towards H2 oxidation and the reverse H+ reduction is often susceptible to H2 inhibition.136
[NiFeSe]–H2ases are a subclass of [NiFe]–H2ases with a selenocysteine residue coordinated to the nickel at the active site instead of a cysteine.138 [NiFeSe]–H2ases are kinetically more biased than other [NiFe]–H2ases towards H2 evolution without substantial product inhibition (Fig. 8i and j). The presence of selenium provides additional protection to the nickel centre from oxidative attacks, and hence confers better oxygen tolerance.138,140,141
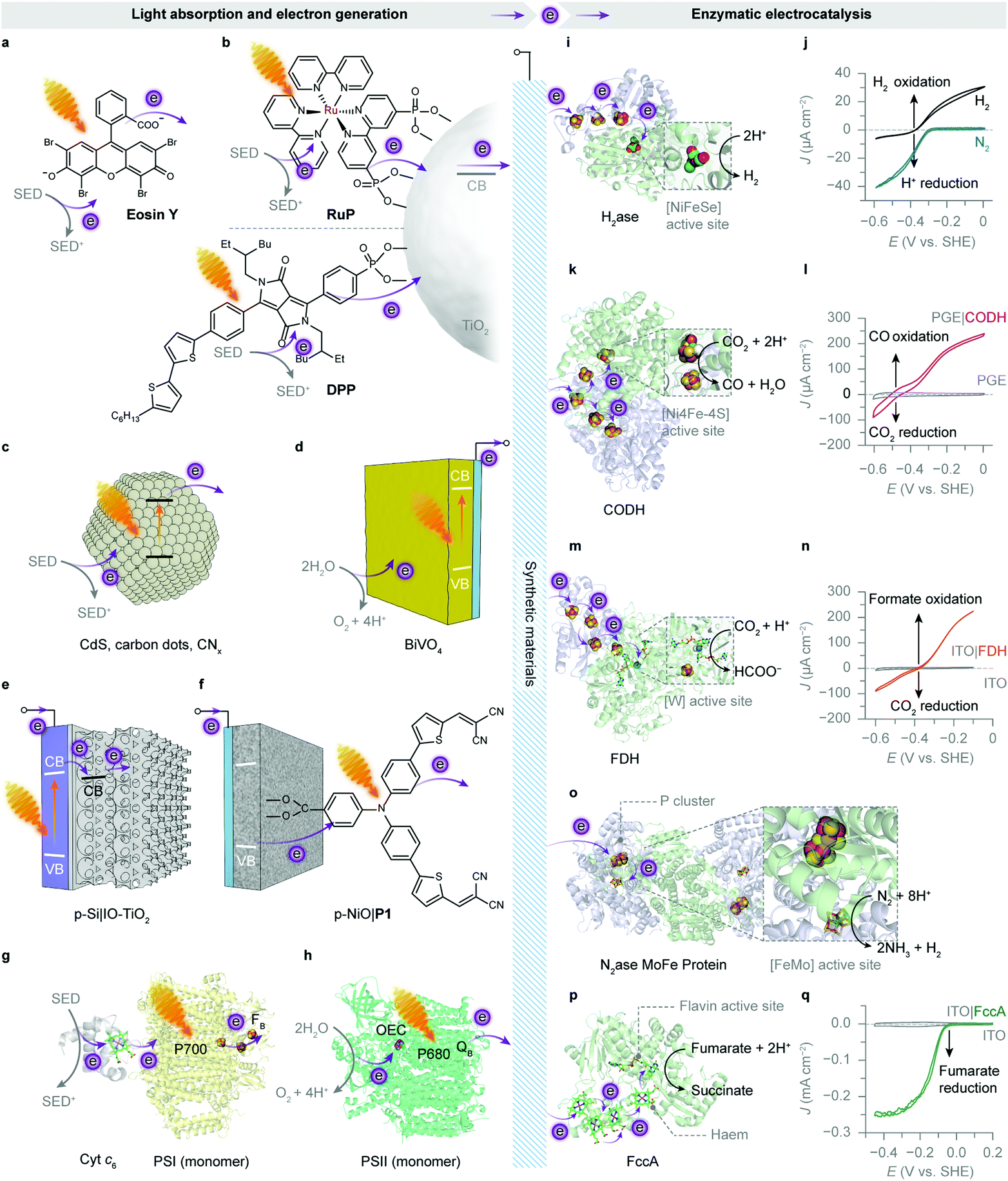 | ||
| Fig. 8 Enzymatic hybrid systems for semi-artificial photosynthesis. (a–h) Light harvesting and low-potential electron generation by abiotic and biotic light absorbers. (i–q) Enzymatic electrocatalysis. (a) An excited xanthene dye Eosin Y (λmax = 539 nm297). (b) Photoexcited electrons from the RuP or diketopyrrolopyrrole (DPP) dye are injected into the conduction band (CB) of TiO2 and further directed to biocatalysts. (c) Irradiation of semiconducting particles, such as polymeric carbon nitride (CNx), carbon dots and CdS, results in photoinduced electron transfer to biocatalysts. (d) A monoclinic BiVO4 electrode absorbs light and generates photoelectrons while using water as the electron donor. (e) An IO-TiO2 scaffold on a p-Si electrode as the photocathode for biocatalysts. Under irradiation, the excited p-Si electrode injects electrons into the CB of IO-TiO2, which are further delivered to the interfacing biocatalysts. (f) A P1 dye-sensitised p-type NiO electrode as the photocathode for biocatalysts. Light absorption by the P1 dye provokes electron transfer from the valence band (VB) of NiO, followed by an electron injection into the biocatalysts adsorbed on the NiO scaffold. (g) The reaction centre chlorophylls (P700, λmax = 700 nm) in PSI are excited by visible light irradiation. The photogenerated electrons are transferred to the terminal [Fe–S] cluster FBvia intraprotein electron relays. The luminal side of PSI is tethered with a cyt c6. Shown here is the core complex of a PSI monomer (protein data bank ID: 5oy0). (h) An excited PSII monomer (protein data bank ID: 4ub6) transfers photoinduced electrons to the terminal electron acceptor (QB) at the stromal side. The generated holes oxidise water to O2 at the OEC. (i) The protein structure of a [NiFeSe]–H2ase (protein data bank ID: 1cc1). Exogenous electrons are delivered via [Fe–S] clusters to the [NiFeSe] active site for proton reduction. (j) Protein film voltammogram of H2ase adsorbed on a TiO2 electrode in N2 or H2, at pH 6.0. Reproduced from ref. 162 with permission from Wiley-VCH, copyright 2016.162 (k) The protein structure of a CODH (protein data bank ID: 1jqk). Electrons are transferred via [Fe–S] clusters to the [NiFeS] active sites to drive the CO2 reduction into CO. (l) Protein film voltammetry scans of CODH adsorbed on a pyrolytic graphite edge (PGE) electrode (control: a bare PGE electrode, grey trace) in CO2/CO (50%/50%) at pH 6.0. Reproduced from ref. 159 with permission from the American Chemical Society, copyright 2013.159 (m) The protein structure of a FDH (protein data bank ID: 1h0h). Electrons are transferred through the intraprotein [Fe–S] relays to the [W] active site for the reduction of CO2 to formate. (n) Protein film voltammogram of reversible reduction of CO2 to formate by an FDH-loaded mesoporous ITO electrode (control: a bare ITO electrode, grey trace) in CO2/NaHCO3 containing formate, at pH 6.5. Reproduced from ref. 193 with permission from Wiley-VCH, copyright 2019.193 (o) The protein structure of a [MoFe] protein from N2ase (protein data bank ID: 4wes). Exogenous electrons access the [MoFe] active site via a P cluster ([8Fe7S]) to transform N2 into NH3. (p) The protein structure of fumarate reductase (FccA, protein data bank ID: 1d4c). Haem cofactors function as intraprotein electron relays to direct electrons towards the flavin active site for fumarate reduction to succinate. (q) Protein film voltammogram of FccA adsorbed on an IO-ITO electrode (control: a bare IO-ITO electrode, grey trace) in 1 mM fumarate at pH 7.0. Reproduced from ref. 117 with permission from the Royal Society of Chemistry, copyright 2016.117 | ||
[NiFeSe]–H2ase is therefore a common model biocatalyst and has been rationally coupled with various light absorbers for photocatalytic HER (Fig. 8). Early attempts employed dyes to generate photoelectrons, which were carried to H2ases by diffusional electron mediators such as methyl viologen (MV), in the presence of a sacrificial electron donor (SED).142,143 A more recent study shows that an organic dye Eosin Y could directly transfer photoelectrons to [NiFeSe]–H2ases without any electron mediators (Fig. 8a).144 The homogeneous system produced 0.5 μmol h−1 H2 for up to 15 h, corresponding to a TOFH2ase of 13.9 s−1. Notably, this Eosin Y–H2ase system also demonstrated some tolerance towards oxygen: more than 80% of photoactivity was retained with 5% O2 and the system remained photoactive even at aerobic conditions (21% O2) for 3 h. The system performance was limited by the interfacial electron transfer between dye molecules and H2ases.
To this end, semiconductors were used as light absorbers with immobilised H2ases. Photogenerated electrons can thereby stream directly from the conduction band towards the enzyme's distal relay centre (Fig. 8b). Co-adsorbing [NiFeSe]–H2ases on RuP–TiO2 particles through the putative interaction between the TiO2 surface and side-chain carboxylates near the distal [Fe–S] cluster yielded 3.56 μmol H2 in the first hour of irradiation and a benchmark TOFH2ase of 50 s−1.145,146 Replacing RuP with a metal-free diketopyrrolopyrrole (DPP) dye to sensitise TiO2 also attained similar HER activity (Fig. 8b).147 Likewise, [FeFe]–H2ases were immobilised on CdTe nanoparticles or CdS nanorods that had their surfaces modified with 3-mercaptopropionic acid.148,149 The negatively-charged surface induced [FeFe]–H2ases to interface with the semiconductors at their positive patch close to the distal [Fe–S] cluster. Transient absorption spectroscopy elucidated that the electron transfer occurred between the conduction band of CdS and the distal [Fe–S] cluster in [FeFe]–H2ase, regardless of the enzyme activity, at a rate (∼100 ns) comparable with that of the charge recombination in CdS.150 The rate of electron transfer is several orders of magnitude slower than that in synthetic systems (10−3–1 ns),151 probably due to the longer tunnelling distance (>10 Å) between the semiconductor surface and the distal [Fe–S] cluster. Nevertheless, the interfacial electron transfer is fast enough to outpace the incident photon flux, rendering the light absorption by CdS performance-limiting.150
The H2 evolution rate of the above systems declined over a few hours, presumably due to dye degradation or an unstable material–enzyme interface. Thus, robust carbon-based light absorbers such as carbon nitride (CNx) and carbon dots were utilised to improve the long-term stability (Fig. 8c).152–154 The CNx–H2ase hybrids remained photoactive for 48 h, totalling 2.5 μmol of H2 production and a turnover number (TON) of >50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.152 But the weak interaction between CNx and H2ases rendered the TOFH2ase much lower (1.5 s−1, in the first 4 h) than that of RuP–TiO2–H2ase hybrids. This problem can be partially resolved by blending CNx with TiO2 that has a high affinity for H2ases.153 The ternary hybrids exhibited better photocatalytic activity with a higher TOFH2ase of 7.8 s−1 within the first 4 h of irradiation and a longer H2 evolution span of 72 h. The photostability of H2ases was even better than their synthetic analogue, a molecular DuBois-type Ni bis(diphosphine) catalyst, that decomposed within 3 h of irradiation.152 [NiFeSe]–H2ases adsorbed on amine-capped carbon dots attained similar activity: a TOFH2ase of 1.1 s−1, H2 production of 2.6 μmol and a total TON of 52000 in 48 h.154 A comparison with carboxylic acid-capped carbon dots echoes with the previous finding that [NiFeSe]–H2ases prefer to electrostatically interact with positive surfaces, and underlines the importance of semiconductor surface chemistry that controls the electronic communication with enzymes.148,149,155
000.152 But the weak interaction between CNx and H2ases rendered the TOFH2ase much lower (1.5 s−1, in the first 4 h) than that of RuP–TiO2–H2ase hybrids. This problem can be partially resolved by blending CNx with TiO2 that has a high affinity for H2ases.153 The ternary hybrids exhibited better photocatalytic activity with a higher TOFH2ase of 7.8 s−1 within the first 4 h of irradiation and a longer H2 evolution span of 72 h. The photostability of H2ases was even better than their synthetic analogue, a molecular DuBois-type Ni bis(diphosphine) catalyst, that decomposed within 3 h of irradiation.152 [NiFeSe]–H2ases adsorbed on amine-capped carbon dots attained similar activity: a TOFH2ase of 1.1 s−1, H2 production of 2.6 μmol and a total TON of 52000 in 48 h.154 A comparison with carboxylic acid-capped carbon dots echoes with the previous finding that [NiFeSe]–H2ases prefer to electrostatically interact with positive surfaces, and underlines the importance of semiconductor surface chemistry that controls the electronic communication with enzymes.148,149,155
The aforementioned systems employ SEDs such as triethanolamine (TEOA) or ethylenediaminetetraacetic acid (EDTA) to expediently bypass the kinetic difficulty of water oxidation. To enable water to supply electrons for reductive reactions, synthetic chemistry and biology utilise different approaches. Artificial photosynthesis can carry out water oxidation separately by a photoanode in a PEC system. BiVO4 has a band structure well-suited for solar water oxidation, but it is insufficient to reduce protons (Fig. 2 and 8d).156,157 A photocathode hosting HER catalysts is thus required to further energise the electrons that are withdrawn from water by the BiVO4 photoanode. The electrochemical window of ITO (−0.6 to +2 V vs. SHE, pH 7.0) limits its application at negative potentials needed to drive reducing reactions.158 In view of this, ITO can be substituted with semiconductive TiO2 to fabricate porous electrodes that are applicable under cathodic conditions.159–161
To this end, [NiFeSe]–H2ases were interfaced with p-type silicon that was stabilised by a mesoporous TiO2 layer.162,163 The TiO2 layer is necessary to protect the underlying p-Si from unwanted surface passivation,163,164 but also to assist in binding [NiFeSe]–H2ases. An ensuing problem is that enzymes having a large footprint show a low loading capacity on the mesoporous TiO2 layer.163,165 As such, a hierarchical IO-TiO2 structure was built atop the p-Si to increase the H2ase loading (up to 120 pmol cm−2) (Fig. 8e).166 A 4 nm TiO2 interlayer was deposited on the surface of p-Si by atomic layer deposition to protect the electrode from the formation of an insulating SiO2 layer, followed by the construction of the IO-TiO2 scaffold. The p-Si|IO-TiO2|H2ase electrode gave a photocurrent onset potential of 0.0 V vs. SHE, pH 6.0, which is more positive than that of a water oxidising BiVO4–TiCo photoanode (−0.1 V vs. SHE, pH 6.0). Thus the BiVO4–TiCo||p-Si|IO-TiO2|H2ase tandem PEC system afforded overall solar water splitting without an external voltage, resulting in a stoichiometric production of H2 (0.47 μmol, ηF = 98%) and O2 (0.20 μmol, ηF = 84%) after 5 h of irradiation.166 [FeFe]–H2ases have also been interfaced with black Si electrodes with submicron porosity, but they are prone to desorb from the electrode surface due to weak interfacial interactions.167
Instead of using a p-type semiconductor, the H2ase-loaded IO-TiO2 scaffold was also interfaced with an encapsulated lead halide perovskite solar cell that generated a photovoltage of 0.9 V. In a tandem configuration with a BiVO4–TiCo photoanode, the resulting system produced H2 coupled to O2 evolution with a STH efficiency of 1.1%.168
To streamline the electron transfer in biological H2 production, photosystems and H2ases could be directly wired via molecular wires, redox polymers, protein subunits or external circuits to eliminate the diffusion-governing steps.18,120,166,169–171 For example, PSI has been connected with [FeFe]–H2ases by 1,6-hexanedithiol to allow photogenerated electrons to tunnel between the two proteins (Fig. 8g).18,169,172
Alternatively, PSII and H2ases were immobilised on IO-ITO electrodes and compartmentalised in a PEC cell (Fig. 8h).120 The onset potential of an IO-ITO|H2ase cathode (−0.4 V vs. SHE, pH 6.5) remained close to the thermodynamic value of proton reduction, whereas electrons from P680 were attenuated during the intraprotein electron transfer in PSII (Fig. 2). The energy is further offset by the use of electron mediators such as DCBQ that aid in the electron transfer from PSII to the electrodes. As such, a minimum bias voltage of 0.6 V was required to drive the photoelectrons from PSII towards H2ase. At an applied voltage of 0.9 V, this biohybrid PEC cell (IO-ITO|PSII||IO-ITO|H2ase) generated 0.52 μmol O2 (ηF = 104%) and 0.96 μmol H2 (ηF = 98%).120 Note that the system performance with respect to photocurrent, onset potential and longevity was limited by the IO-ITO|PSII photoanode. To reduce the bias voltage in the future, one could possibly extract electrons from the upstream electron relays such as QA or pheophytin (Fig. 2).104,130
An easier way is to substitute for an electron mediator with a more negative redox potential than DCBQ whilst also introducing a second light absorber to reduce the bias voltage. This has been achieved by the use of 3,5-di-tert-butyl-1,2-benzoquinone (DTBoQ, Em = 0.29 V vs. SHE) as the electron mediator, which shifted the photocurrent onset by 0.1 V earlier than DCBQ. When paired with a p-Si|IO-TiO2|H2ase photocathode, the required voltage was reduced to 0.24 V. At 0.4 V, the tandem PEC cell produced 0.125 μmol H2 (ηF = 91%) within 3 h.166
At the anodic side, a second light absorber can be judiciously selected to imitate the function of PSI or to complement the light absorption of PSII. A working example is DPP that has a negative excited-state redox potential (−1.15 V vs. SHE) and strong light absorption between 400 and 550 nm (λmax = 496 nm).147DPP and PSII were loaded in an IO-TiO2 scaffold as the light absorbers and electrically connected by an Os-based redox polymer (POs) hydrogel.119 Thus, a vectorial electron transfer from water to the TiO2 electrode was established to prime electrons with sufficient reducing power for HER. Moreover, DPP conferred supplementary green-light absorption on this PSII-containing photoanode, thereby enabling a panchromatic sensitivity to visible light. By directly wiring the IO-TiO2–DPP|POs-PSII photoanode with an IO-ITO|H2ase cathode, autonomous solar water splitting can be carried out using wired enzymes without a bias voltage.119 After 1 h of irradiation at zero bias, 0.015 μmol H2 was detected with a Faraday efficiency of 76%. H2ases at the cathode were not driven to their full capacity in catalysing proton reduction, due to the photodegradation of PSII in vitro. An initial applied bias STH conversion efficiency of 0.14% was obtained at a voltage of 0.3 V during the first hour.119 PSI, the native second light absorber in the Z-scheme, has also been rewired with PSII and H2ase by redox polymers in an attempt to reconstruct a biological H2 production pathway in vitro.173
3.3 Carbon dioxide reduction
From a viewpoint of carbon products, the biomass produced by the Calvin cycle is not ideal to substitute for petrochemicals as feedstocks for the chemical industry.174 However, biology has evolved more than one pathway for CO2 fixation,175 and the reductive acetyl-coenzyme A (acetyl-coA) pathway (Wood–Ljungdahl pathway) is an energy-conserving CO2-fixing pathway, through which inorganic carbon is assimilated into cellular metabolism in the form of formate and CO by carbon monoxide dehydrogenase (CODH) and formate dehydrogenase (FDH).176–178 Both [NiFe]-CODH and metal-dependent FDH can catalyse the interconversion between CO2 and their two-electron reduced carbon products, CO and formate, respectively. They control the binding of CO2 and the transfer of electrons and protons to partition intermediates on the reduction pathway towards CO or its hydrated form, formate.46[NiFe]-CODH is an O2-sensitive metalloenzyme with a [NiFe]-active site from anaerobes such as Moorella thermoacetica and Carboxydothermus hydrogenoformans (Fig. 8k). It efficiently catalyses CO oxidation with a TOF up to 40000 s−1 and CO2 reduction with a TOF up to 45 s−1, at a nearly thermodynamic potential (−0.5 V vs. SHE, pH 6.7) (Fig. 8l).46,179,180 CO2 is bound to the [NiFe]-cluster and undergoes a bifunctional attack by the electrophilic iron centre and the nucleophilic nickel centre, which enables a two-electron pathway for CO2 activation.181,182 Such two-electron reduction is also applied by [Mo]- or [W]-FDH, a metalloenzyme reversibly interconverting CO2 and formate with minimal overpotentials (−0.39 V vs. SHE, pH 7.0) (Fig. 8m and n).183,184 It has been determined that [W]-FDH isolated from Syntrophobacter fumaroxidans exhibited a TOF up to 3400 s−1 for formate oxidation and 280 s−1 for CO2 reduction (with MV as the redox partner).183 Both [W]- and [Mo]-FDH can achieve ∼100% Faraday efficiency for electrocatalytic CO2 reduction with formate as the sole product at modest overpotentials.183,184 These enzymes provide an efficient two-electron CO2 reduction pathway that bypasses the thermodynamically-uphill formation of CO2˙−.185
In view of the close thermodynamic redox potentials for CO2/CO (−0.52 V vs. SHE, pH 7.0), proton reduction (−0.42 V vs. SHE, pH 7.0) and CO2/HCOO− (−0.39 V vs. SHE, pH 7.0) (Fig. 2), similar strategies can be employed to drive CO2 reduction with solar energy. [NiFe]-CODH was adsorbed on RuP–TiO2 nanoparticles for light-driven CO2 reduction to CO in the presence of an SED (Fig. 8b). During 4 h of irradiation, the RuP–TiO2–CODH hybrids produced ∼5 μmol CO, corresponding to an average TOFCODH of 0.15 s−1.186 The relatively low turnover rate compared with H2ase hybrids was partly due to a smaller driving force for CO2 reduction than proton reduction, as the conduction band edge of TiO2 (−0.52 V vs. SHE, pH 6.0) nears the thermodynamic potential for CO2 reduction to CO. Factors such as interaction with TiO2 and enzyme orientation also affect performance. The enzyme activity began to decrease after 3 h, and the low activity might be partly due to inefficient dye regeneration.186,187 CODH has also been integrated in a PEC system comprising a P1 dye-sensitised p-type NiO cathode (Fig. 8f).188 The excited P1 dye receives an electron from the valence band of the NiO electrode, followed by electron injection into an adjacent CODH, via intraprotein electron relay centres, down to its [NiFe]-active site, where CO2 was reduced to CO (Fig. 8f).
Light-driven CO2-to-formate conversion has been previously realised by FDH-based photoredox systems, where electrons from SEDs are energised by photosensitisers and delivered to FDH via electron mediators,78,189–191 or by an FDH-based PEC system, where photoelectrons from the semiconductor photocathode p-InP were mediated to FDH by the MV/MV2+ redox couple.192 The diffusional mediator can be eliminated by immobilising FDH on RuP–TiO2 nanoparticles (Fig. 8b).193 FDH showed a TOF of 11 s−1 in the first 6 h of photocatalysis and yielded 2.6 μmol formate.193 Quartz crystal microbalance and infrared spectroscopy revealed that TiO2 binds FDH strongly through both electrostatic interaction and chemisorption. FDH can also be wired with an electrode to work in tandem with a photoanode.122,194,195 A recent study coupled an FDH-functionalised IO-TiO2 cathode with an IO-TiO2–DPP|POs-PSII tandem photoanode to drive CO2-to-formate reaction with the aid of a small bias voltage.122 After 1 h of irradiation with a bias of 0.3 V, 0.046 μmol of formate (ηF = 70%) was detected as the only product in the cathodic chamber of the PEC cell. The photocurrent decayed by half after 8 min, arising from the photodamage of PSII in the anode. Self-driven CO2 reduction was recently achieved by a PV-PEC tandem system with a perovskite solar cell, a BiVO4–FeOOH photoanode and a TiN nanoshell|FDH cathode.194 The resulting PV-PEC tandem cell yielded formate at a rate of 1.06 μmol h−1 with a Faraday efficiency of 83% in 8 h, corresponding to a solar-to-fuel efficiency of 0.08%. The formate production could last for three days, and the decrease in activity was ascribed to the degradation of the perovskite solar cell outside the PEC cell. FDH can also be loaded on a TiO2-deposited CuFeO2–CuO photocathode to couple with a BiVO4–FeOOH photoanode for autonomous light-driven CO2 reduction.196 This bias-free PEC cell allowed CO2-to-formate conversion at a rate of 0.098 μmol h−1 with a Faraday efficiency of 33.5% in 8 h.
The product of CO2 reduction can be extended from formate to methanol via an enzyme cascade.197–199 A prototypical PEC system employed CoPi-modified α-Fe2O3 and BiFeO3 as the photoanode and photocathode, respectively, and contained a mixture of enzymes, i.e., FDH, formaldehyde dehydrogenase and alcohol dehydrogenase, in the cathodic chamber.198 Electrons extracted from water are energised by photoelectrodes and vectored through enzyme cascades to reduce CO2 to methanol via NADH and diffusional electron mediators. Instead of being directly wired on the photocathode, enzymes in this system were dispersed in the catholyte, and depended on NADH, the reducing agent, to carry out redox reactions. As such, despite the well-aligned band positions, an external voltage (0.8 V) was needed to efficiently regenerate NADH and produce methanol (1.31 mM in 6 h).198 An enzyme cascade was also immobilised in a silica matrix to work with an “artificial thylakoid” in a colloidal system.199 The “artificial thylakoid” constituted a microporous protamine–TiO2 hollow sphere (microcapsule) with CdS nanoparticles deposited on its luminal surface. The TiO2 microcapsule received electrons from the excited CdS and exported electrons to regenerate NADH that furnished the enzyme cascade with reducing equivalents. In this way, photocatalytic oxidation and enzymatic CO2 reduction were decoupled so that the enzymes were protected from photoinduced reactive oxygen species (ROS). This biohybrid system has demonstrated a high rate and yield in renewing NADH and thus generated 85 μM methanol from CO2 in 2 h.199
3.4 Chemical synthesis
With enzymes as biocatalysts, photogenerated electrons from light absorbers can realise a broad spectrum of reactions of synthetic interest, such as N2 reduction, reduction of CC bonds, formation of C–C bonds and hydroxylation of C–H bonds etc. Several recent studies are briefly highlighted here.
Biology converts N2 into metabolically tractable NH3 using N2ases under physiological conditions. N2ase is a two-component system comprising a [MoFe] protein and an electron-transfer [Fe] protein. The two proteins associate and dissociate during catalysis, sequentially delivering electrons and energy to the [MoFe] active site, where two NH3 and one H2 are generated from one N2 and eight protons and electrons (Fig. 8o).86 To leverage N2ases for light-driven N2 fixation, the isolated [MoFe] proteins were adsorbed on a CdS nanorod to form a biohybrid complex, where light energy replaces ATP hydrolysis to drive the enzymatic turnover in N2.200 The photoexcited CdS nanorods generated electrons with low reducing potential (−0.74 V vs. SHE, pH 7.0), which were then forwarded to the interfacing [MoFe] proteins for N2 reduction. The CdS–[FeMo] protein hybrid attained an NH3 production rate of 315 nmol mgN2ase−1 min−1 and an average TOFN2ase of 1.25 s−1. The NH3 production lasted up to 5 h with a TON of 1.1 × 104 molNH3 molN2ase−1.200
The reduction of CC bonds is exemplified by the conversion of fumarate to succinate, which can be catalysed by the flavoenzyme fumarate reductase (FccA) (Fig. 8p and q).201 The light-driven fumarate reduction can be either performed in a colloidal system or within a PEC cell, using RuP-sensitised TiO2 nanoparticles, and carbon dots or a W–BiVO4–CoPi electrode, respectively (Fig. 8b–d).154,202 The FccA adsorbed on RuP–TiO2 exhibited a TON of 5800 over 4 h of irradiation, corresponding to a TOFFccA of 0.4 s−1,202 while that on amine capped carbon dots achieved a similar TOFFccA of 0.47 s−1 (averaged in the first 2 h of irradiation) with improved photostability up to 24 h.154 Photoinduced enzyme degradation was likely to account for the slowdown of productivity. In a PEC system with a W–BiVO4–CoPi photoanode, the turnover rate of the immobilised FccA dropped down to ∼0.01 s−1.202
Another example is the use of old yellow enzymes (NADPH dehydrogenases) for stereoselective CC reduction. Light absorbers including Au–TiO2 or N-doped carbon dots were employed to produce photoelectrons that drive the regeneration of reducing equivalents such as NAD(P)H and its synthetic substitute, for trans-hydrogenation of conjugated CC bonds.203,204 With molecular xanthene dyes, photoelectrons could be directly transferred to the prosthetic flavin moiety in the old yellow enzyme, eliminating the need of using costly nicotinamide cofactors.205 Such light-driven enzymatic catalysis yielded enantioselective products (enantiomeric excess > 90%) with conversion yields up to 80–90%.204,205
Photochemical C–C bond formation has recently been realised in a CdS–2-oxoglutarate:ferredoxin oxidoreductase (OGOR) hybrid system.206 The OGOR catalyses the amalgamation of CO2 and succinate into 2-oxoglutarate and is part of the reductive tricarboxylic acid cycle responsible for CO2 fixation in many autotrophic microorganisms. The irradiated CdS generates reducing equivalents to drive the catalytic turnover at OGOR which involves large substrates, significant conformational changes during catalysis, and eventual formation of C–C bonds.206
Functionalisation of C–H bonds provides a straightforward and atom-economical access towards a plethora of organic products,207 where enzymes such as cyt P450 show their synthetic advantages. The hallmark reaction of cyt P450 is the hydroxylation of C–H bonds, during which the delivery of electrons from the NADH reductase is synchronised with the activation of oxygen at the haem centre. The disadvantage of activity dependence on NADH cofactors and redox partners can be overcome by a semi-biological solution. A Ru(II)-diimine dye was covalently attached to a mutant haem domain of a cyt P450, which enabled excited dyes to inject electrons into the haem centre to catalyse the hydroxylation of lauric acid at an initial rate of 2.1 s−1 with a TON of more than 900 in 2 h, without the aid of NADH and reductase.208
4 Microbial hybrid systems
Microbial cells include an entire system of biochemical pathways and can produce metabolites of synthetic interest with high specificity at physiological conditions. Microorganisms utilise numerous enzyme cascades to maintain the intracellular metabolism. Different metabolic pathways are spatially organised to divert metabolites or enzymes that can react promiscuously, to maintain selectivity, concentrate reactants to drive unfavourable reactions, and protect enzymes or unstable intermediates from harmful cytoplasmic contents.209 Moreover, these biosynthetic pathways are under dynamic regulation to keep cellular functionality in tune with physiological needs at different conditions. These features allow microbes to synthesise complex products from the simplest and stable feedstocks (e.g., H2O, CO2, N2, etc.) and render microbial catalysis resilient to environmental variations. Appealing to synthetic chemistry is their ability to produce multicarbon products from CO2via inherent carbon assimilation pathways such as the Wood–Ljungdahl pathway and the Calvin cycle.175,178Such synthetic complexity is challenging to achieve with individual enzymes and a fixed reaction stoichiometry. Microbial catalysis can be further empowered by synthetic biology that employs genetic tools to access and engineer the intracellular metabolism, and create new pathways beyond the native metabolic pattern.210 Compared with enzymatic counterparts, the microbial hybrid systems are less explored, but have provoked increasing interest in solar-to-chemical conversion, due to the possibility to synthesise complex chemicals, achieve better stability and prospects for scalability.211,212 In the following discussions, we will summarise the recent achievements that take advantage of the microbial metabolism for chemical synthesis in solar energy conversion with photocatalysis and (photo)electrochemistry (Table 1).
| Microbe | Light absorber | Microbial reaction | Ref. |
|---|---|---|---|
| E. coli | TiO2 | H+ → H2 | 221 |
| E. coli | CdS | H+ → H2 | 222 |
| R. capsulata | Bi2O3 | H+ → H2 | 218 |
| R. capsulata | dye-TiO2 | H+ → H2 | 219 |
| R. eutropha | PV | CO2 → biomass, isopropanol | 240 |
| M. barkeri | CdS | CO2 → methane | 225 |
| M. barkeri | TiO2/InP | CO2 → methane | 239 |
| M. thermoacetica | CdS | CO2 → acetate | 226 |
| M. thermoacetica | Au | CO2 → acetate | 231 |
| M. thermoacetica | PDI/PFP | CO2 → acetate | 234 |
| S. ovata | Si nanowires | CO2 → acetate | 248 |
| S. ovata | Si nanowires | CO2 → acetate | 250 |
| Clostridium sp. | PV | CO2 → butanol, hexanol | 212 |
| R. palustris | CdS | CO2 → PHB, carotenoids | 232 |
| S. cerevisiae | InP | Glucose → shikimic acid | 238 |
| G. sulfurreducens | BiVO4 | Fumarate → succinate | 262 |
| X. autotrophicus | PV | N2 → NH3 | 243 |
Whereas enzymes catalyse biochemical reactions with specific substrates, products and stoichiometry, microorganisms are more versatile due to a set of diverse metabolic pathways involving a multitude of enzymes. Moreover, the microbial hybrid systems are being developed to establish an efficient conversion strategy of solar energy to desired multicarbon chemicals, a catalytic prospect particularly suitable for microorganisms.213 Thus, we summarise progress for microbial hybrid systems from a different perspective: instead of organising this section by individual reactions enabled by such systems, we put the emphasis on key questions imperative for research in this field: how microorganism and synthetic materials are integrated into hybrid structures, how non-photosynthetic microbes are empowered with light-absorbing capabilities, and how fluxes of reducing equivalents are guided into intracellular metabolic pathways.
4.1 Microbial photocatalysis in colloidal systems
The discussion begins with the microbial hybrids operating in colloidal systems (Fig. 5c). We consider the HER as a model reaction in the context of solar-to-fuel conversion. Photosynthetic H2 production can transiently take place in nature by microalgae and cyanobacteria under anaerobiosis, during which electron flux on the photosynthetic chain is shifted away from its normal path towards H2ase.17,214 However, such process is O2-sensitive and competes with NADPH-dependent biological processes. Although the O2-sensitivity can be partially alleviated by protective encapsulation,215,216 the rate and yield of H2 evolution are intrinsically limited by the light reaction, which saturates at fairly low light intensities (10–20% of solar light).214 Whereas these drawbacks can be partially overcome by streamlining the solar-to-H2 pathway with isolated H2ases and light absorbers, laborious purification and fragility of the enzymes diminish the practicality of this approach.An alternative are microbial hybrids where non-photosynthetic live cells are employed as biocatalysts, and the task of light harvesting is outsourced to synthetic light absorbers. Several early studies illustrate this strategy: H2ase- (and N2ase-) containing bacteria such as Clostridium butyricum, Rhodopseudomonas capsulata, Rhodospirillum rubrum were coupled with semiconductor nanoparticles, specifically, TiO2 and Bi2O3via MV as the electron mediator (Fig. 9a). In the presence of SEDs, photogenerated electrons were delivered into cells where H2 evolved.217–219 The low growth rate of these bacteria limits the availability of biocatalysts. As such, Escherichia coli (E. coli), a well-established bacterial chassis for synthetic biology,220 was engineered to heterologously express genes encoding [FeFe]–H2ases from Clostridium acetobutylicum.221 The recombinant E. coli was mixed with TiO2 nanoparticles and MV, and such hybrid system produced 216 μmol H2 during 15 h of irradiation.
 | ||
| Fig. 9 Photosynthetic microbial hybrid systems. (a) Irradiated semiconducting nanoparticles (e.g., TiO2) transfer photoexcited electrons to microbes to produce H2 or other products via the MV2+/MV+ or NADP+/NADPH redox couples in the presence of a SED. (b) CdS nanoparticles are deposited on the surface of microbes and deliver photoelectrons directly to the intracellular pathways for solar-to-chemical conversion and oxidise the SED under illumination. (c) Au nanoclusters are absorbed by microbes as intracellular light absorbers. Photoinduced electrons are delivered by cytoplasmic redox mediators to the Wood–Ljungdahl pathway that reduces CO2 into acetate. (d) Microbes are introduced in the cathodic chamber of an electrolyser and leverage the produced H2 as the reducing equivalents to transform CO2 into CH4 or multicarbon alcohols. | ||
To enable visible light absorption, CdS was employed, and instead of inducing heterologous expression of [FeFe]–H2ases, E. coli can produce H2 in anaerobic conditions with endogenously synthesised [NiFe]–H2ases. Moreover, CdS nanoparticles were precipitated on the bacterial surface to directly supply photoelectrons without diffusional mediators (Fig. 9b).222 It has been found that photogenerated electrons interacted with the intracellular metabolism and guided more reducing equivalents along the pathway for H2 production. To extend its aerobic uses, the CdS–E. coli hybrid was further encapsulated with biomimetic silica to protect the bacterium from oxygen.216 Under aerobic conditions, the hybrid system continuously produced H2 within 96 h of irradiation.
The catalytic ability of microorganisms is better exemplified in CO2 reduction, where they can produce complex organic products not readily accessible through synthetic methods.211 One example is CO2 reduction to CH4, which involves eight electron transfers that can facilely diverge to form a wide range of products.223 In the realm of biology, this reaction is handled by methanogenic archaea as part of their energy-conserving metabolism.224 In light of this, Methanosarcina barkeri (M. barkeri), an anaerobic methanogen, was employed as the catalytic machinery for CO2-to-CH4 conversion.225 Like the aforementioned CdS–E. coli hybrids,216,222 CdS nanoparticles were deposited on the surface of the M. barkeri as the light absorber. Photoelectrons produced by the excited CdS were delivered to the intracellular pathways via membrane-bound cytochromes or H2ases and were used by the bacteria for methanogenesis (Fig. 9b). The CdS–M. barkeri hybrid produced 13.7 μmol CH4 after three-day irradiation with a quantum efficiency of 0.34%.225 Isotopic labelling experiments confirmed that the CH4 was derived from CO2 reduction. The CH4 evolution halted after three days, likely due to the depletion of the SED cysteine.
CdS nanoparticles can also be interfaced with Moorella thermoacetica (M. thermoacetica), an acetogenic and electrotrophic bacterium. M. thermoacetica could directly use photogenerated electrons from the excited CdS to reduce CO2 into acetate via the intracellular Wood–Ljungdahl pathway, while holes produced by CdS were quenched by cysteine (Fig. 9b).226 A peak quantum yield of 2.44% was recorded at low-intensity simulated sunlight, which is comparable to the year-long averages (∼0.2–1.6%) for plants and algae.227
The electron transfer pathways between CdS and M. thermoacetica was further elucidated by transient absorption spectroscopy: photoelectrons were transported via membrane-bound electron acceptors such as cytochromes, ferredoxin, flavoproteins and menaquinones at the initial stage (3 h) of photosynthesis; at longer duration (24 h), a membrane-bound H2ase-mediated pathway dominated, which correlated with increased rates of acetate generation and thus a higher quantum efficiency.228 A latest proteomic study revealed that the photoexcited CdS induced changes to the metabolic pattern of M. thermoacetica: a host of enzymes associated with the tricarboxylic acid cycle and glycolysis were up-regulated. Both pathways together with the Wood–Ljungdahl pathway constitute the energy-conservation scheme of the CdS–M. thermoacetica hybrid system.229
To make the hybrid system more sustainable, the added cysteine, the SED to CdS, could be regenerated photocatalytically by TiO2 nanoparticles loaded with Mn(II) phthalocyanine co-catalysts, which ultimately extracted electrons from water and delivered them to the CdS–M. thermoacetica hybrids via the cysteine/cystine redox couple.230 The light absorbers can be replaced with those that are more biocompatible and accessible, for example, Au nanoclusters.231 Au nanoclusters could be ingested by M. thermoacetica and directly deliver photoexcited electrons with longer lifetime to the Wood–Ljungdahl pathway via cytoplasmic redox mediators (Fig. 9c). The intracellular interplay between photochemistry and metabolism bypassed kinetic and energetic drawbacks stemming from transmembrane diffusion. The Au nanoclusters also could reduce ROS formed during irradiation, thus improving the cellular viability in this hybrid system and increasing acetate production with a higher quantum yield of ∼2.9%.231
A similar protocol is applicable to various microorganisms with different physiological functionality. For example, CdS nanoparticles were precipitated on the photosynthetic bacterium Rhodopseudomonas palustris for CO2 fixation.232 Under visible light, the excited CdS generated additional reducing equivalents for the innate Calvin cycle, which promoted the production of biomass and valuable multicarbon compounds such as carotenoids and poly-β-hydroxybutyrate (PHB), as well as the photosynthetic efficiency. When CdS was hybridised with Thiobacillus denitrificans (T. denitrificans), an autotrophic denitrifier, the resulting CdS–T. denitrificans hybrid could use the photoelectrons for NO3− reduction to N2O.233
In light of its cytotoxicity, CdS has been substituted with organic semiconductors, i.e., perylene diimide derivative (PDI) and poly(fluorene-co-phenylene) (PFP) as the photosensitiser.234 PDI and PFP could be immobilised on the surface of M. thermoacetica through electrostatic and hydrophobic interactions, which enabled efficient electron transport across the cellular membrane. Such microbial hybrid system registered a quantum efficiency of 1.6%.
Light-driven fumarate reduction to succinate has been achieved by an enzymatic hybrid system with flavoenzymes (FccA),154,202 but such reaction can also be accomplished by a dye-sensitised whole cell containing flavocytochromes.235 This hybrid system comprised of photosensitisers (Eosin Y or proflavine), Shewanella oneidensis MR-1 (S. oneidensis), an electron mediator (MV) and a SED. Another model reaction is C–H bond functionalisation. Cyt P450 was employed to catalyse the hydroxylation of lauric acid with a covalently attached Ru(II) light absorber that was meant to deliver photoelectrons to the haem domain without the need of redox partners.208 The same strategy also applies in a microbial hybrid system: the cyt P450 was expressed in an engineered E. coli bacterial chassis and Eosin Y was absorbed into the cytoplasm and directly bound to the haem domain of the enzyme.236 Under visible light irradiation and with an SED, photoexcited Eosin Y can continuously supplement electrons to sustain the catalytic turnover of the cyt P450 more than 18 h (average TOFP450: 2.5 × 10−4 s−1).236
A comparison with the previous P450–Ru(II) dye hybrids illustrates the disparity that epitomises general differences between enzymatic and microbial systems. First, the P450–Ru(II) dye hybrids need post-translational modification to link the Ru(II) dye molecule to the specific cysteine residue, and such modification is specific to a particular type of cyt P450 and may affect the activity when applied to different cyt P450 variants.208 In contrast, the whole cell system has demonstrated success with both bacterial and human cyt P450 in producing high value chemicals such as drugs and steroids.236 Lastly, the enzyme hybrids showed an initial TOFP450 (2.1 s−1) several orders of magnitude higher than the whole cell system, but the activity of the former began to languish within 2 h (average TOFP450: 1.25 × 10−3 s−1), while productivity of the latter can be compensated by the prolonged longevity (>18 h) and the increased quantity of biocatalysts.208,236
Besides bacteria, eukaryotic cells such as yeasts are also favoured as a platform cell factory for biosynthesis of fuels and chemicals, as their well-studied biology primes genetic and analytical tools to unravel the mechanisms governing electron transport and metabolic flux in biohybrid systems.64 The intracellular redox reactions are dependent on cofactors, namely NADH or NADPH, as hydride sources and their generation is strongly intertwined with the central carbon metabolism.237 The expense of NAD(P)H precludes a stoichiometric supply to sustain the biosynthesis, and an effective cofactor regeneration was achieved photocatalytically by hybridising the yeast strain Saccharomyces cerevisiae (S. cerevisiae) with semiconducting InP nanoparticles (Fig. 9a).238S. cerevisiae was genetically engineered to suppress the cytosolic synthesis of NADPH and to enhance the carbon flux towards shikimic acid production. The NADPH was regenerated by the inward electron transport by irradiated InP nanoparticles bound on the cell surface. A maximum light-to-chemical conversion efficiency of 1.58% was attained within 12 h of irradiation.238
4.2 Electrolysis coupled to homogeneous microbial synthesis
Another biohybrid system integrated planktonic M. barkeri with a PEC cell system consisting of a water oxidising TiO2 photoanode and a p-InP–Pt photocathode, to couple artificial H2 evolution with biological methanogenesis. The unassisted water splitting under irradiation generated H2 in the cathodic chamber, which was consumed by the methanogens therein as reducing equivalents to reduce CO2 to CH4 (Fig. 9d). This hybrid system produced 68.8 nmol CH4 over three days, corresponding to a Faraday efficiency of 74%.239Similar strategies can be extended to other strains of bacteria to obtain CO2-reducing products that are more feasible for storage and transport.240,241 For example, in a water electrolyser inoculated with genetically-engineered Ralstonia eutropha, a water oxidising CoPi anode and a Co–P alloy cathode worked together at an applied voltage of 2.0 V to furnish the bacteria with H2.241 The bacteria transformed CO2 and H2 following a desired route into an array of fuels and chemical products such as poly(3-hydroxybutyrate), isopropanol, isobutanol and 3-methyl-1-butanol (Fig. 9d). The energy input can be readily outsourced to a photovoltaic device with a routinely attained efficiency of 18%,2 and the resulting system could anticipate a solar-to-chemical conversion efficiency of >7%.241,242 Such a modular platform has furthermore been leveraged beyond CO2 reduction, towards more complex ambient NH3 synthesis from N2 and H2O with the H2-uptake diazotrophic microorganism Xanthobacter autotrophicus.243 In the above hybrid systems, H2 acted as the vector of reducing power to connect microbial metabolism with artificial electrolysis. However, the low solubility of H2 (0.79 mM, 25 °C) and inefficient H2 delivery from the electrode limit the rate of throughput. A recent study shows that this bottleneck can be alleviated by introducing perfluorocarbon nanoemulsions as biocompatible H2 carriers to increase the H2 availability around cells, accelerate the H2 transfer and thereby improve the productivity of CO2 reduction.244
4.3 Photoelectrosynthesis with immobilised microorganisms
Instead of suspending microbes in solution, they can also be immobilised on electrodes to catalyse redox reactions driven by light or electricity (Fig. 10a). Such systems are referred to as microbial (photo)electrosynthesis. Underlying this process is the electron exchange ability of microorganisms with the interfacing electrode (Fig. 10b–d).245 This can either occur directly between the electrode and microorganism or be mediated by H2 and diffusional redox couples. The direct electron transfer for microbial electrosynthesis has the advantage of bypassing the handicap of low solubility of H2 and the diffusional limitations of electron shuttles. Besides, it avoids the potential loss from intermittent H2 generation and the potential toxicity of some mediators (e.g., MV).246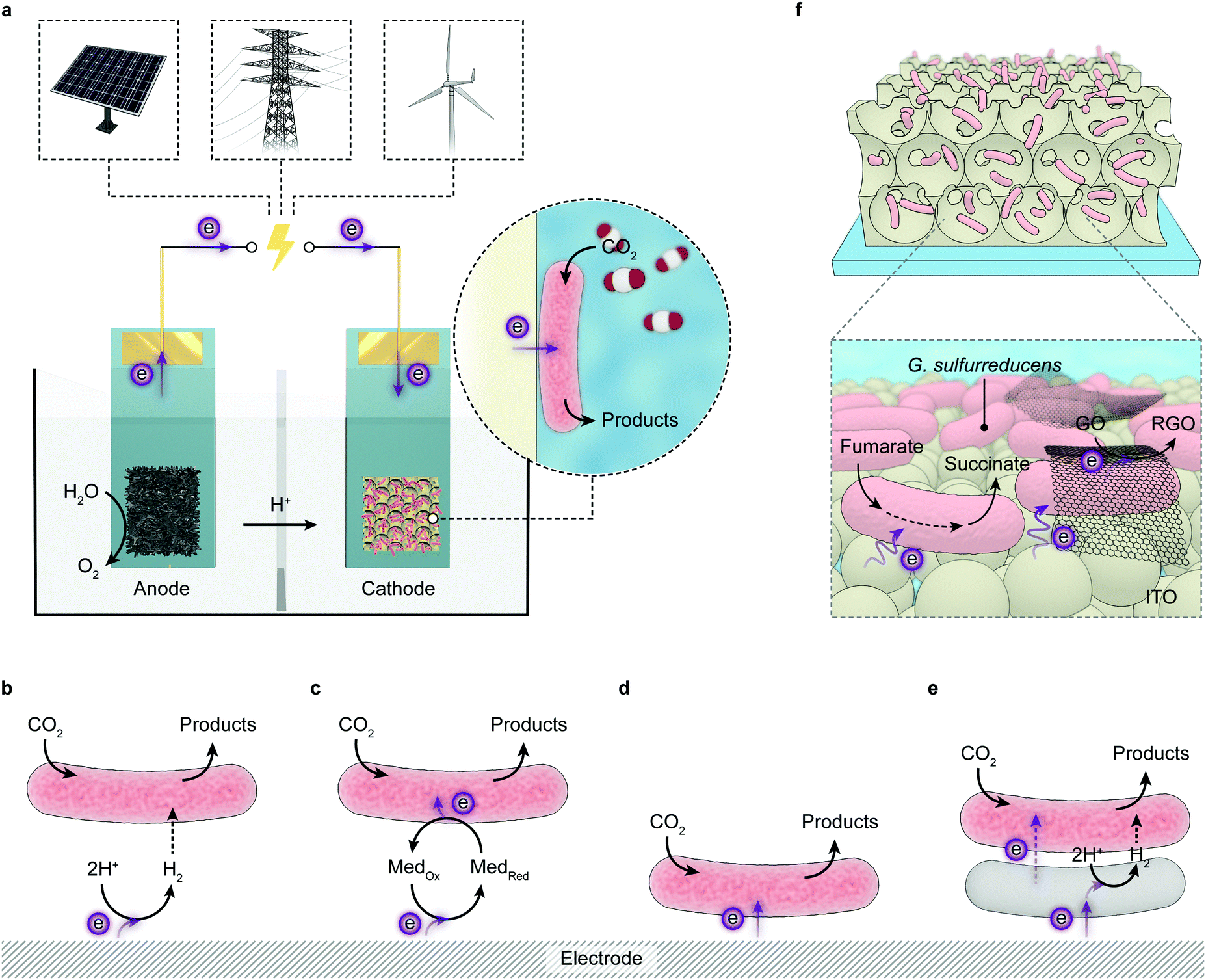 | ||
| Fig. 10 Microbial electrosynthesis. (a) Schematic representation of a microbial electrosynthetic cell powered with electricity that can be derived from renewable sources (solar light, wind, etc.). At the anode chamber, an electrode donor (e.g., H2O) is oxidised, providing electrons. The cathode chamber contains a microbial hybrid electrode where microorganisms receive electrons from the interfacing electrode scaffold and drive the reduction reaction (e.g. CO2 reduction). (b–e) Mechanisms for electron transfer from electrodes to microorganisms. Electrons from the electrode can be delivered to the microorganisms by H2 (b), redox mediators (Med), (c), direct electron transfer (d) or via interspecies electron/H2 transfer (e). (f) An IO-ITO|G. sulfurreducens hybrid electrode, in which G. sulfurreducens can directly receive electrons from the cathode and reduce fumarate or graphene oxide (GO). Reproduced from ref. 262 with permission from the National Academy of Sciences of the United States of America (NAS), copyright 2020. | ||
From a practical perspective, the advantages over a planktonic system largely resemble the benefits of heterogeneous catalysis over homogeneous systems such as easy catalyst separation and recycling. One can readily and flexibly apply the microbial electrode for photosynthesis, either by pairing with a photoelectrode in a PEC cell or connecting with a solar cell for PV-driven electrolysis. Of more interest to fundamental studies is that the hybrid electrode system permits electrochemical toolsets to access the intracellular redox chemistry via the current exchange at the biointerface.
Oxygenic photosynthetic microorganisms such as cyanobacteria have been immobilised on IO-ITO electrodes with tailored porosity to perform semi-artificial photosynthesis.128,129 The cyanobacterium encloses the entire photosynthetic apparatus within the cell, rendering themselves more robust than isolated PSII to carry out light-driven water oxidation. The cyanobacterial biofilm produced much lower photocurrent densities than those of PSII, but with increased longevity (>5 days).128 Compared with the IO-ITO|PSII electrodes, the IO-ITO|biofilm systems exhibited different photocurrent profiles, which is derived from different electron transfer mechanisms at the interface. The cyanobacterium likely relies on intracellular redox mediators that are released under irradiation to carry electrons towards the electrode surfaces.247
There are only few reports to directly interface non-photosynthetic microorganisms with photoelectrodes.248,249 The difficulty is in part due to the large footprint of cells, which requires appropriate electrode geometry to achieve a high loading density while maintaining light penetration. A notable example is an array of Si nanowires that served both as a light-absorbing semiconductor and a physical scaffold for the acetogenic bacterium, Sporomusa ovata (S. ovata) (Fig. 5d).248 The bacteria were integrated with the Si nanowires by taking photoexcited electrons from the conduction band to sustain their metabolism. The resulting biohybrid electrode was assembled with a photoanode made of water oxidising TiO2 nanowires for autonomous photoreduction of CO2 to acetate. The PEC cell produced a stable photocurrent of 0.3 mA cm−2 for more than 120 h, yielding an acetate titre of 20 mM with a Faraday efficiency of 86%. The overall solar-to-chemical conversion efficiency registered 0.38% during 200 h operation. Acetate could further be activated into acetyl-coA, a common biochemical intermediate, to access a variety of biosynthetic fine chemicals. The downstream synthesis could be performed by genetically-engineered E. coli that transformed acetate into n-butanol, polyhydroxybutyrate polymer, and isoprenoid compounds.248 Recently, the acetate production was accelerated as a result of a larger bacterial population and a better bacterium-nanowire interface, which was realised by improving the local pH environment inside the electrode.250 As such, the solar-to-chemical conversion efficiency was increased to 3.6% over seven days when coupling to a photovoltaic device.
Few light-absorbing semiconductors have been optimised to fit the dimension of microorganisms. Integration of microbes into the electrode scaffold also poses challenges on the chemical stability, biocompatibility, conductivity and surface chemistry of the semiconducting materials, not least the impact on light transmission. A simpler strategy is to immobilise microorganisms on electrodes with tailored morphologies while outsourcing light harvesting to photoanodes or external PV devices. This approach decouples light absorption from microbial metabolism, and thereby suppresses unwanted parasitic reactions and eliminates possible oxidative stresses stemming from photoinduced ROS. The simplified design also grants more flexibility in the making of electrode architectures to enhance the loading capacity and engineer the electrode surface to improve the interaction. Therefore, more efforts converge on interfacing microbial cells with tailored electrodes and improving the interfacial electron transfer and mass transport. Such exploitation can capitalise on decades of research in microbial fuel cells,251 and be guided by recent understandings of the interaction between synthetic materials and microorganisms.252,253 As these hybrid electrodes can readily operate with light absorbing electrodes or devices, to broaden the scope of discussion, this section will continue with a survey of recent progress in microbial electrosynthesis, where sessile bacteria exchange electrons with electrodes and carry out synthetic reactions.
An early study of microbial electrosynthesis employed graphite electrodes to supply Geobacter sulfurreducens (G. sulfurreducens) with electrons for fumarate reduction.254 This is reminiscent of a previously described electrode–bacterium hybrid system, where H2 acted as the electron vector (Fig. 10b). Yet this scenario has been ruled out because the electrode barely produced H2 at the applied potential (−0.3 V vs. SHE, pH 6.8). Instead, G. sulfurreducens was able to directly receive electrons from the electrode (Fig. 10d).255 The same reaction could also be accomplished by S. oneidensis cultured on a graphite electrode or in a three-dimensional reduced graphene oxide (RGO) scaffold.256,257
An envisioned application for microbial electrosynthesis is bioremediation, namely, decontaminating the wastewater possibly powered by solar energy.258 For example, under applied negative potentials, G. sulfurreducens could precipitate soluble U(VI) ions on electrodes, which can be easily collected.259 Bacteria such as Geobacter lovleyi and Anaeromyxobacter dehalogenans with the ability to respire with chlorinated compounds can also be exploited to dechlorinate contaminants (e.g., tetrachloroethene, 2-chlorophenol) in this manner.260,261
More recently, an IO-ITO electrode scaffold was employed to culture a large population of G. sulfurreducens (Fig. 10f).262 The resulting biohybrid electrode catalyses the reduction of soluble fumarate and heterogenous graphene oxide (GO), with electrons from an external power source or an irradiated dye-sensitised (RuP–TiO2) or water oxidising BiVO4 photoanode.262 The microbe-modified IO-ITO electrode enabled high current densities and also served as a platform underpinning a variety of analytical methods to decipher the electrical interplay at the biointerface.263
However, Geobacter and Shewanella strains lack CO2-fixation pathways. Autotrophic microorganisms that can source energy from inorganic chemical reactions (chemolithotrophs) or light (photoautotrophs) for carbon assimilation are more promising in this regard, as they can adapt to use an electrode as the electron donor to generate reducing power.264 Acetogens such as S. ovata, Clostridium ljungdahlii, and M. thermoacetica have been shown able to directly receive electrons from electrodes and reduce CO2 into acetate with Faraday efficiencies of >80%. Moreover, the electron uptake and CO2 fixation can also be decoupled using two bacteria strains (Acetobacterium woodii and Desulfobacterium corrodens), which can separately receive electrons from the electrode and reduce CO2, respectively, and communicate by means of interspecies H2 transfer (Fig. 10e).265
As microbial electrosynthesis systems are anticipated to operate together with PV devices, the ultimate solar-to-chemical efficiency will to a large extent depend on the biochemical pathway that dictates the energetics and stoichiometry of the substrate–product conversion. Whereas a sulphur-oxidising phototroph Prosthecochloris aestaurii can sustain its photosynthesis with electrons supplied from a cathode,266 its application for photoelectrosynthesis is largely unfeasible because the Calvin cycle is rather inefficient for solar fuel production and genetic tools to expand their synthetic diversity are not yet developed for some less studied microorganisms. Compared with methanogens of which CH4 is the only attainable product, acetogens enable a wide repertoire of chemicals such as ethanol, isopropanol, n-butanol and 2,3-butanediol, which can be further extended by metabolic engineering.267,268
The alliance of microbial metabolism with photocatalysis and (photo)electrochemistry is formed by virtue of the translocation of reducing power from abiotic materials to biotic entities. This can be achieved by interfacing microorganisms with light absorbers/electrodes, where electrons are either directly transferred to microorganisms or shuttled inward via in situ generated electron carriers (e.g. H2). Alternatively, electrons can be delivered to planktonic cells by diffusive redox mediators. Sessile microorganisms on electrodes can manage a high Faraday efficiency up to 90%. Yet the slow electron uptake (typically < 1 mA cm−2), stemming from the kinetic disparity between the electrochemical and biochemical processes, which is less amenable to improvement by interface engineering, diminishes the economic feasibility of this approach.269 Thus a technically favoured strategy may be to decouple the extracellular electrochemistry and intracellular biochemistry in a modular fashion, as exemplified by a recent microbial electrosynthesis setup that combined PV, electrolysis and fermentation modules.212 CO2 and H2O was electrochemically reduced into H2 and CO in a PV-powered electrolyser at a current density of 50 mA cm−2. The abiotically generated syngas was then fed into a microbial fermenter where a mixed culture of Clostridium autoethanogenum and Clostridium kluyveri worked together to produce butanol and hexanol at an energetic efficiency of 78% and an overall Faraday efficiency of ∼100%.212 As such, the highest attainable solar-to-chemical efficiency of the integrated system could be projected at 15%, assuming that the existing PV modules deliver a routine energy conversion efficiency of 20%.212
5 Future perspectives
Semi-artificial photosynthesis opens a wide spectrum for new solar-to-chemical conversion designs by rationally integrating biocatalysts in the form of proteins and whole cells with synthetic components, which traverses the fields of enzymology, microbiology, materials science, electrochemistry, and photocatalysis. The ultimate goal is to catalyse the transformation of H2O, CO2, and N2 into fuels, chemicals and fertilisers via a sustainable path and synthesise value-added fine chemicals with high yield and selectivity with renewable energy sources. Here we briefly propose several promising opportunities for the future development.At the biotic side, more enzymes or bacteria relevant to fuel/chemical production can be added into the inventory. The methodologies established with model biocatalysts on bespoke electrodes and semiconductor particles can be extrapolated to new biocatalysts to create new reaction pathways. For example, RuP–TiO2 particles were used in early work to adsorb H2ases for photocatalytic HER.145 The use of the same light absorber was later extended to CODH, FDH and FccA.186,193,202 CdS nanorods developed for CdS–H2ase complexes have been employed to drive N2-to-NH3 conversion with N2ases.149,200 Likewise, CdS nanoparticles have been in situ deposited on the surface of E. coli and M. thermoacetica.222,226 It is reasonable to envision that a similar strategy can be applied to other acetogens such as S. ovata and methanogens like M. barkeri.
Biocatalysts are widely-appreciated for their high product selectivity, yet such catalytic prowess is confined to naturally-occurring reactions. One way to expand the reaction scope is orchestrating various catalysts, including biotic and synthetic, to drive cascade reactions towards more complex and valuable products.270 For example, an enzyme cascade has been introduced into a tandem PEC cell to reduce CO2 into methanol.198 More opportunities are emerging in this respect to judiciously construct reaction pathways and sensibly direct electron and carbon flux towards desired products.
The second approach arises when advancing genetic techniques meet with enzymology and microbiology. With the derived methodologies, enzymes can be engineered with improved promiscuity and stability. PSII has been introduced with a polyhistidine tag near the stromal side in order to enhance the linkage with Ni-terminated moieties on an electrode surface.108 Such modifications and single site mutations more broadly are available for other enzymes to improve the interaction with light-absorbing materials. Pathways in microorganisms can be altered to maximise the chemical production or heterologously introduced to unlock unnatural reactions. Metabolic engineering can allow heterotrophic E. coli to synthesise sugar from CO2,271 and cyanobacteria to drive the reduction of CC bonds,272 or produce valuable aromatic compounds directly from CO2.273 Engineered microbes with additional pathways can work with light absorbers to yield synthetically useful products beyond their natural metabolites.238
Moreover, the genetic toolkit can alter the existing or enable new physiological functionalities in model microorganisms. For example, current production of electrogenic microorganisms can be enhanced by increasing metabolic fluxes, promoting electron shuttle secretion, encouraging biofilm formation, etc.274 Heterologous electron conduits have been transplanted into E. coli to confer extracellular electron transfer capability.274,275S. oneidensis has been engineered to metabolise glucose as carbon and energy source.276 Beyond this, these techniques also provide powerful means to generate insights into the poorly understood questions with regard to microbial metabolism strategies and electron uptake pathways, which, in turn, will facilitate the rational design of next-generation biohybrid systems.
In parallel, progress at the artificial side can be translated into strengths to stimulate the advancement of semi-artificial photosynthesis systems. Developments in materials science and synthetic chemistry will provide more opportunities in tuning a material's composition, morphology and property at various levels to improve the interaction with biocatalysts or adapt to different reaction conditions. A recent report demonstrated the application of a metal–organic framework to confine FDH within its precisely defined volume and porosity to stabilise enzymatic activity at non-physiological conditions.277 A metal–organic framework was also employed to encapsulate anaerobic bacteria (M. thermoacetica) to protect them against oxidative stress arising from irradiation.215 Such a strategy is feasibly applicable to other enzymes and bacteria. Another emerging possibility is to foster interaction with biocatalysts at a sub-entity level, instead of forming heterogeneous biointerfaces. This has been realised by fusing dye molecules with cyt P450 to replace native redox partners,208 or anchoring Pt nanoparticles with PSI for light-driven H2 evolution.278 At a subcellular scale, Au nanoclusters or molecular dyes have been exploited to intervene pathways within cells with light irradiation as bioorthogonal opportunities for intracellular modulation.231,236,279 Furthermore, the intracellular metabolism can be coupled with extracellular redox transformations to manufacture functional materials such as polymers and inorganic particles.280–282 These materials may provide additional benefits to promote extracellular electron transfer or cytoprotection.281
Advanced photochemistry provides another thrust in future development. Many photosynthetic biohybrid systems are limited by light absorbers that display non-ideal absorption and driving force to perform reactions, or suffer from degradation faster than the inactivation of biocatalysts. Potential solutions may arise from the iterative cycle of design, synthesis and characterisation in the field of artificial photosynthesis, which yields a variety of photocatalysts with fine-tuned properties and morphologies. Progress in this field can instantly contribute to the construction and optimisation of biohybrid systems. For example, carbon nitride and carbon dots emerged as an attractive visible-light absorber due to their well-fit band structure, low cost, and robustness.66,283 Carbon nitride–H2ase and carbon dot–H2ase complexes have been shown effective in photocatalytic H2 production.152–154 As such, more applications can be anticipated by replacing H2ase with other enzymes, or substituting the toxic CdS to interface with acetogenic bacteria. Studies into these newly-derived systems will gain more knowledge of the biotic–abiotic interplay and generate guidelines for the assembly of better systems.
Finally, semi-artificial photosynthesis, in a broader context, also enables a suite of powerful analytical techniques to delve into fundamental questions relevant to physiological functionality, biocatalytic redox chemistry and biotic–abiotic interaction (Fig. 11).284
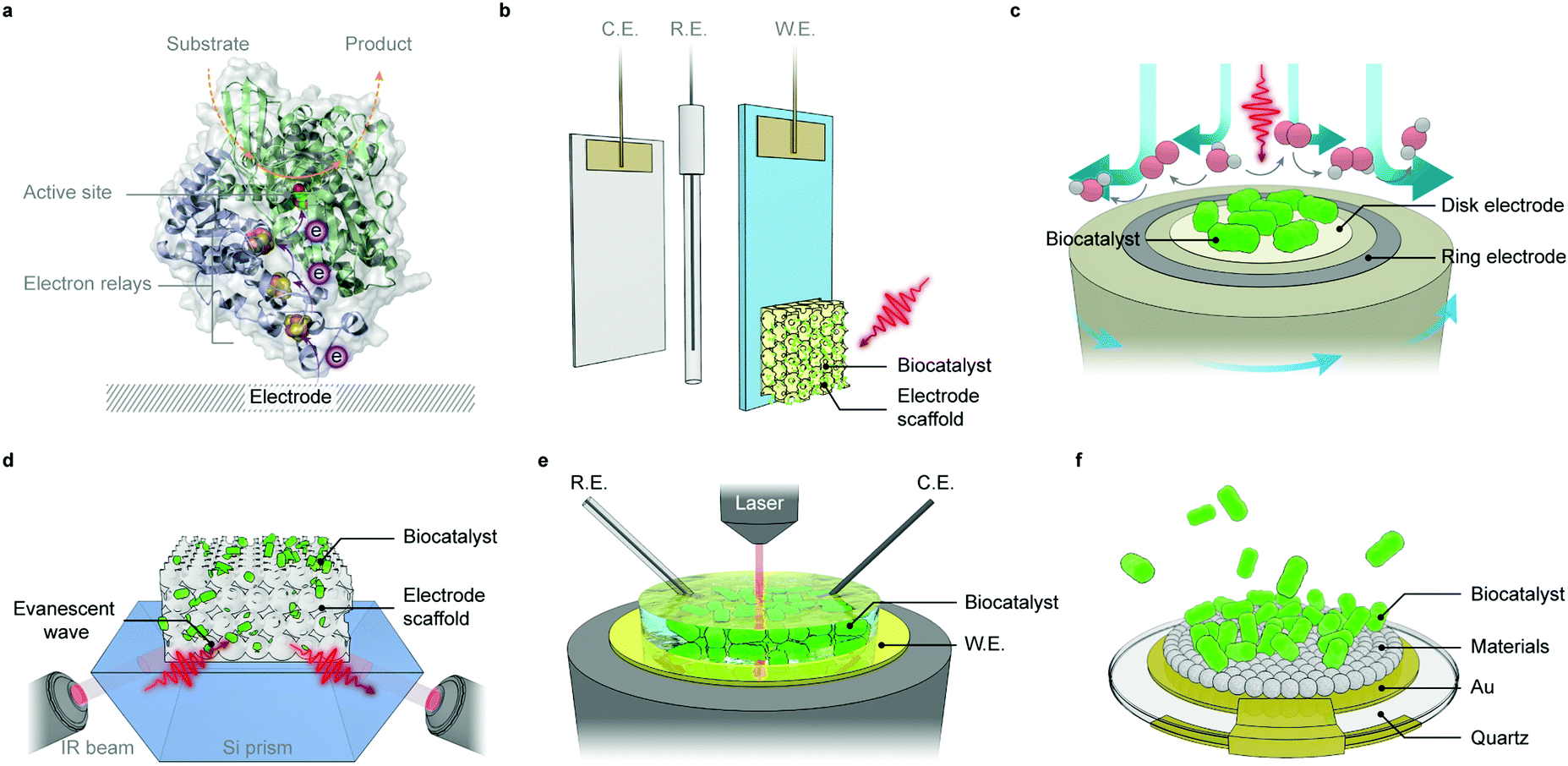 | ||
| Fig. 11 Analytical techniques employed in semi-artificial photosynthesis.284 (a) An enzyme–electrode interface. Electrons from the electrode are delivered to the active site via intraprotein electron relays. (b) A three-electrode setup comprising a working electrode (W.E.), counter electrode (C.E.) and reference electrode (R.E.) for protein film (photo)electrochemistry and microbial biofilm (photo)electrochemistry. (c) A rotating ring disk electrode with a biocatalyst-adsorbed disk electrode and a Pt ring electrode. The rotation of the apparatus generates a convective flow that hydrodynamically carries reaction products from the disk to the ring electrode for product analysis. (d) An ATR-IR setup with an electrode scaffold deposited on an optical waveguide crystal (e.g., a Si prism). The internal reflection generates an evanescent wave that penetrates into the electrode scaffold by typically ∼500 nm. (e) In situ spectroelectrochemistry that combines vibrational/electronic spectroscopy with biofilm electrochemistry. Spectroscopic signatures with mechanistic significance can be captured during the course of redox variation with accurate electrochemical control. (f) A typical piezoelectric chip (electrode materials deposited on a Au substrate) for quartz crystal microbalance analysis. Through the mass-frequency relationship, the mass variation stemming from the biocatalyst binding or desorption can be monitored, enabling quantitative insights into bio-electrode interaction and biocatalyst loading capacity. | ||
Wiring enzymes or cells to an electrode allows electrochemical methods to probe their redox chemistry and physiological functionality under turnover and non-turnover conditions, through efficient interfacial electron transfer (Fig. 11a).59,82,285,286 The simplicity of the electrochemical apparatus (typically in a three-electrode electrochemical cell) renders it readily compatible with an extra light source to investigate the light-driven redox reactions of photoactive enzymes (Fig. 11b).90 Besides stationary electrodes, rotating electrodes, namely rotating disk electrodes or rotating ring disk electrodes, provide an effective way to eliminate the limitations in mass transport that often obscures catalytic features.287 The rotating ring disk electrode, with an additional ring working electrode surrounding the central disk, enables quantification of reaction products formed at the disk electrode (Fig. 11c).118
Complementary to electrochemical methods are spectroscopic techniques, which allow for in situ detection of electronic and vibrational changes with relevance to enzymatic or cellular functionalities under electrochemical control and provide structural or mechanistic insights that were otherwise intractable with electrochemistry alone. For instance, ATR-IR was previously deployed to track molecular catalysts and reaction intermediates during catalytic cycles,288 while it also assisted to probe the enzyme–electrode interaction in H2ase–TiO2, FDH–TiO2 and PSII–ITO systems (Fig. 11d).102,166,193 Their versatility renders these tools readily transferable to biohybrid assemblies, and offers a high degree of flexibility to work together with (photo)electrochemistry in operando studies (Fig. 11e).
Time-resolved pump–probe spectroscopy, namely transient absorption spectroscopy, is universally applied in artificial photosynthesis systems to look into kinetic aspects of charge separation and transfer, which take place on a timescale (10−12–10−3 s) intractable by other methods.289 With such temporal resolution, electron transfer at a semiconductor–enzyme interface and the ensuing catalytic turnover can be deconvoluted, enabling unique strength in unravelling kinetic intricacies in photosynthetic biohybrid systems. This technique has been exploited to track the charge transfer in CdS nanorod–H2ase hybrids and revealed that photogenerated electrons transferred from CdS to the distal relay centre in H2ase at a rate comparable to that of their relaxation process.150 In a more complex microbial hybrid system (CdS-sensitised M. thermoacetica), transient absorption spectroscopic analysis provided kinetic insights into the electrical interplay between photoexcited semiconductors and acetogenic bacteria, and contributed to the elucidation of pathways connecting extracellular photochemistry and intracellular metabolism.228
Beyond electrochemistry and spectroscopy, quartz crystal microbalance (with dissipation) enables a sensitive method to measure the mass change on a piezoelectric quartz chip (Fig. 11f).76 This allows quantification of enzyme loadings in an electrode scaffold, which is not easily accessible by electrochemical or spectroscopic means and can be further extrapolated to evaluate the nature of enzyme interaction with electrode materials.166,193 More tools are continuously being added into the toolbox. Recently, electron paramagnetic resonance spectroscopy has been combined with protein film electrochemistry to study metalloenzymes (Cu–Zn superoxide dismutase) adsorbed on a mesoporous ITO electrode under precise potential control.290 Nanoscale secondary ion mass spectrometry was applied to study the metabolic activity of G. sulfurreducens biofilms.291 These emerging analytic techniques can provide fresh insights from different perspectives to produce a more comprehensive picture of biohybrid systems.
Semi-artificial photosynthesis synergises functional components from different disciplines. Advances in each field will yield new opportunities to further the development and implementation of biohybrid systems. Conclusions drawn from these studies will deepen the current understanding of the biology-materials interplay and form concrete steps to expedite the evolution of semi-artificial photosynthesis towards maturity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a CSC-Cambridge PhD scholarship (to X. F.), a Marie Sklodowska-Curie fellowship (EMES, 744317 to S. K.), and an ERC Consolidator Grant “MatEnSAP” (682833 to E. R.). The authors thank Dr Annika Eisenschmidt, Ms Esther Edwardes Moore, Prof. Julea Butt and Prof. Michael de Volder for the helpful feedback.References
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley-Blackwell, 2nd edn, 2014 Search PubMed.
- G. G. B. Tcherkez, G. D. Farquhar and T. J. Andrews, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7246–7251 CrossRef CAS PubMed.
- T. J. Erb and J. Zarzycki, Curr. Opin. Biotechnol., 2018, 49, 100–107 CrossRef CAS PubMed.
- M. Hagemann and H. Bauwe, Curr. Opin. Chem. Biol., 2016, 35, 109–116 CrossRef CAS PubMed.
- X.-G. Zhu, S. P. Long and D. R. Ort, Annu. Rev. Plant Biol., 2010, 61, 235–261 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- X.-G. Zhu, S. P. Long and D. R. Ort, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS PubMed.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- D. J. Lea-Smith, P. Bombelli, R. Vasudevan and C. J. Howe, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 247–255 CrossRef CAS PubMed.
- S. Rumpel, J. F. Siebel, C. Farès, J. Duan, E. Reijerse, T. Happe, W. Lubitz and M. Winkler, Energy Environ. Sci., 2014, 7, 3296–3301 RSC.
- S. I. Allakhverdiev, V. Thavasi, V. D. Kreslavski, S. K. Zharmukhamedov, V. V. Klimov, S. Ramakrishna, D. A. Los, M. Mimuro, H. Nishihara and R. Carpentier, J. Photochem. Photobiol., C, 2010, 11, 101–113 CrossRef CAS.
- J. Benemann, Nat. Biotechnol., 1996, 14, 1101–1103 CrossRef CAS PubMed.
- C. E. Lubner, A. M. Applegate, P. Knörzer, A. Ganago, D. A. Bryant, T. Happe and J. H. Golbeck, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20988–20991 CrossRef CAS PubMed.
- H. Bothe, O. Schmitz, M. G. Yates and W. E. Newton, Microbiol. Mol. Biol. Rev., 2010, 74, 529–551 CrossRef CAS PubMed.
- A. M. Muro-Pastor and W. R. Hess, Trends Microbiol., 2012, 20, 548–557 CrossRef CAS PubMed.
- P. Fay, Microbiol. Rev., 1992, 56, 340–373 CrossRef CAS.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- J. K. Hurst, Science, 2010, 328, 315 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- R. Kobayashi, T. Takashima, S. Tanigawa, S. Takeuchi, B. Ohtani and H. Irie, Phys. Chem. Chem. Phys., 2016, 18, 27754–27760 RSC.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- S. Keene, R. Bala Chandran and S. Ardo, Energy Environ. Sci., 2019, 12, 261–272 RSC.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
- E. Pastor, F. Le Formal, M. T. Mayer, S. D. Tilley, L. Francàs, C. A. Mesa, M. Grätzel and J. R. Durrant, Nat. Commun., 2017, 8, 14280 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- A. Vilanova, T. Lopes, C. Spenke, M. Wullenkord and A. Mendes, Energy Storage Mater., 2018, 13, 175–188 CrossRef.
- M. R. Singh, K. Papadantonakis, C. Xiang and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2760–2767 RSC.
- J. Jin, K. Walczak, M. R. Singh, C. Karp, N. S. Lewis and C. Xiang, Energy Environ. Sci., 2014, 7, 3371–3380 RSC.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- A. Banerjee, B. D. Yuhas, E. A. Margulies, Y. Zhang, Y. Shim, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 2030–2034 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- C. Wombwell and E. Reisner, Chem. – Eur. J., 2015, 21, 8096–8104 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pécaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- J. W. Lee, D. Na, J. M. Park, J. Lee, S. Choi and S. Y. Lee, Nat. Chem. Biol., 2012, 8, 536–546 CrossRef CAS PubMed.
- J. Nielsen and Jay D. Keasling, Cell, 2016, 164, 1185–1197 CrossRef CAS PubMed.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W.-h. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- N. Heidary, K. H. Ly and N. Kornienko, Nano Lett., 2019, 19, 4817–4826 CrossRef CAS PubMed.
- N. Kornienko, N. Heidary, G. Cibin and E. Reisner, Chem. Sci., 2018, 9, 5322–5333 RSC.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS PubMed.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- H. Li, D. Buesen, S. Dementin, C. Léger, V. Fourmond and N. Plumeré, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS PubMed.
- A. Heller, Acc. Chem. Res., 1990, 23, 128–134 CrossRef CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- B. Mondal, J. Song, F. Neese and S. Ye, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- W. J. Albery and J. R. Knowles, Biochemistry, 1976, 15, 5631–5640 CrossRef CAS PubMed.
- A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 5250–5254 CrossRef CAS PubMed.
- W. Lubitz, M. Chrysina and N. Cox, Photosynth. Res., 2019, 142, 105–125 CrossRef CAS PubMed.
- M. Kato, J. Z. Zhang, N. Paul and E. Reisner, Chem. Soc. Rev., 2014, 43, 6485–6497 RSC.
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 26–43 CrossRef CAS PubMed.
- S. De Causmaecker, J. S. Douglass, A. Fantuzzi, W. Nitschke and A. W. Rutherford, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19458–19463 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233–2237 CrossRef CAS PubMed.
- S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250–3256 CrossRef CAS PubMed.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- X. Wei, X. Su, P. Cao, X. Liu, W. Chang, M. Li, X. Zhang and Z. Liu, Nature, 2016, 534, 69–74 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- A. Agostiano, A. Ceglie and M. D. Monica, Bioelectrochem. Bioenerg., 1984, 12, 499–507 CrossRef CAS.
- X. Fang, K. P. Sokol, N. Heidary, T. A. Kandiel, J. Z. Zhang and E. Reisner, Nano Lett., 2019, 19, 1844–1850 CrossRef CAS PubMed.
- K. Brinkert, F. Le Formal, X. Li, J. Durrant, A. W. Rutherford and A. Fantuzzi, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1497–1505 CrossRef CAS PubMed.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2012, 134, 8332–8335 CrossRef CAS PubMed.
- J. Maly, C. Di Meo, M. De Francesco, A. Masci, J. Masojidek, M. Sugiura, A. Volpe and R. Pilloton, Bioelectrochemistry, 2004, 63, 271–275 CrossRef CAS PubMed.
- J. Maly, J. Krejci, M. Ilie, L. Jakubka, J. Masojídek, R. Pilloton, K. Sameh, P. Steffan, Z. Stryhal and M. Sugiura, Anal. Bioanal. Chem., 2005, 381, 1558–1567 CrossRef CAS PubMed.
- A. Badura, B. Esper, K. Ataka, C. Grunwald, C. Wöll, J. Kuhlmann, J. Heberle and M. Rögner, Photochem. Photobiol., 2006, 82, 1385–1390 CrossRef CAS PubMed.
- N. Terasaki, M. Iwai, N. Yamamoto, T. Hiraga, S. Yamada and Y. Inoue, Thin Solid Films, 2008, 516, 2553–2557 CrossRef CAS.
- A. Ruff, Curr. Opin. Electrochem., 2017, 5, 66–73 CrossRef CAS.
- A. Badura, D. Guschin, B. Esper, T. Kothe, S. Neugebauer, W. Schuhmann and M. Rögner, Electroanalysis, 2008, 20, 1043–1047 CrossRef CAS.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2013, 135, 10610–10613 CrossRef CAS PubMed.
- K. K. Rao, D. O. Hall, N. Vlachopoulos, M. Grätzel, M. C. W. Evans and M. Seibert, J. Photochem. Photobiol., B, 1990, 5, 379–389 CrossRef CAS.
- W. Wang, Z. Wang, Q. Zhu, G. Han, C. Ding, J. Chen, J.-R. Shen and C. Li, Chem. Commun., 2015, 51, 16952–16955 RSC.
- J. Li, X. Feng, J. Fei, P. Cai, J. Huang and J. Li, J. Mater. Chem. A, 2016, 4, 12197–12204 RSC.
- J. Li, X. Feng, Y. Jia, Y. Yang, P. Cai, J. Huang and J. Li, J. Mater. Chem. A, 2017, 5, 19826–19835 RSC.
- B. Reuillard, K. H. Ly, P. Hildebrandt, L. J. C. Jeuken, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2017, 139, 3324–3327 CrossRef CAS PubMed.
- C.-Y. Lee, B. Reuillard, K. P. Sokol, T. Laftsoglou, C. W. J. Lockwood, S. F. Rowe, E. T. Hwang, J. C. Fontecilla-Camps, L. J. C. Jeuken, J. N. Butt and E. Reisner, Chem. Commun., 2016, 52, 7390–7393 RSC.
- N. Kornienko, J. Z. Zhang, K. P. Sokol, S. Lamaison, A. Fantuzzi, R. van Grondelle, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2018, 140, 17923–17931 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 3, 944–951 CrossRef CAS.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- K. P. Sokol, D. Mersch, V. Hartmann, J. Z. Zhang, M. M. Nowaczyk, M. Rögner, A. Ruff, W. Schuhmann, N. Plumeré and E. Reisner, Energy Environ. Sci., 2016, 9, 3698–3709 RSC.
- K. P. Sokol, W. E. Robinson, A. R. Oliveira, J. Warnan, M. M. Nowaczyk, A. Ruff, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2018, 140, 16418–16422 CrossRef CAS PubMed.
- K. R. Stieger, S. C. Feifel, H. Lokstein, M. Hejazi, A. Zouni and F. Lisdat, J. Mater. Chem. A, 2016, 4, 17009–17017 RSC.
- D. Ciornii, M. Riedel, K. R. Stieger, S. C. Feifel, M. Hejazi, H. Lokstein, A. Zouni and F. Lisdat, J. Am. Chem. Soc., 2017, 139, 16478–16481 CrossRef CAS PubMed.
- M. Riedel, W. J. Parak, A. Ruff, W. Schuhmann and F. Lisdat, ACS Catal., 2018, 8, 5212–5220 CrossRef CAS.
- M. Riedel, J. Wersig, A. Ruff, W. Schuhmann, A. Zouni and F. Lisdat, Angew. Chem., Int. Ed., 2019, 58, 801–805 CrossRef CAS PubMed.
- D. Ciornii, A. Kölsch, A. Zouni and F. Lisdat, Nanoscale, 2019, 11, 15862–15870 RSC.
- J. Z. Zhang, P. Bombelli, K. P. Sokol, A. Fantuzzi, A. W. Rutherford, C. J. Howe and E. Reisner, J. Am. Chem. Soc., 2018, 140, 6–9 CrossRef CAS PubMed.
- T. Wenzel, D. Härtter, P. Bombelli, C. J. Howe and U. Steiner, Nat. Commun., 2018, 9, 1299 CrossRef PubMed.
- J. Z. Zhang, K. P. Sokol, N. Paul, E. Romero, R. van Grondelle and E. Reisner, Nat. Chem. Biol., 2016, 12, 1046–1052 CrossRef CAS PubMed.
- M. Pita, D. M. Mate, D. Gonzalez-Perez, S. Shleev, V. M. Fernandez, M. Alcalde and A. L. De Lacey, J. Am. Chem. Soc., 2014, 136, 5892–5895 CrossRef CAS PubMed.
- C. Tapia, S. Shleev, J. Conesa, A. L. De Lacey and M. Pita, ACS Catal., 2017, 7, 4881–4889 CrossRef CAS.
- S. V. Hexter, F. Grey, T. Happe, V. Climent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS PubMed.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- C. Wombwell, C. A. Caputo and E. Reisner, Acc. Chem. Res., 2015, 48, 2858–2865 CrossRef CAS PubMed.
- F. A. Armstrong, N. A. Belsey, J. A. Cracknell, G. Goldet, A. Parkin, E. Reisner, K. A. Vincent and A. F. Wait, Chem. Soc. Rev., 2009, 38, 36–51 RSC.
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS PubMed.
- M. C. Marques, C. Tapia, O. Gutiérrez-Sanz, A. R. Ramos, K. L. Keller, J. D. Wall, A. L. De Lacey, P. M. Matias and I. A. C. Pereira, Nat. Chem. Biol., 2017, 13, 544–550 CrossRef CAS PubMed.
- I. Okura, Coord. Chem. Rev., 1985, 68, 53–99 CrossRef CAS.
- I. Okura, N. Kaji, S. Aono, T. Kita and A. Yamada, Inorg. Chem., 1985, 24, 451–453 CrossRef CAS.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- J. Warnan, J. Willkomm, J. N. Ng, R. Godin, S. Prantl, J. R. Durrant and E. Reisner, Chem. Sci., 2017, 8, 3070–3079 RSC.
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef CAS PubMed.
- K. Wu, H. Zhu, Z. Liu, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690–5694 RSC.
- G. A. M. Hutton, B. Reuillard, B. C. M. Martindale, C. A. Caputo, C. W. J. Lockwood, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2016, 138, 16722–16730 CrossRef CAS PubMed.
- O. Rüdiger, J. M. Abad, E. C. Hatchikian, V. M. Fernandez and A. L. De Lacey, J. Am. Chem. Soc., 2005, 127, 16008–16009 CrossRef PubMed.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, PLoS One, 2014, 9, e107942 CrossRef PubMed.
- A. Bachmeier, V. C. C. Wang, T. W. Woolerton, S. Bell, J. C. Fontecilla-Camps, M. Can, S. W. Ragsdale, Y. S. Chaudhary and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 15026–15032 CrossRef CAS PubMed.
- T. E. Rosser and E. Reisner, ACS Catal., 2017, 7, 3131–3141 CrossRef CAS.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- C.-Y. Lee, H. S. Park, J. C. Fontecilla-Camps and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 5971–5974 CrossRef CAS PubMed.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef CAS PubMed.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- D. H. Nam, J. Z. Zhang, V. Andrei, N. Kornienko, N. Heidary, A. Wagner, K. Nakanishi, K. P. Sokol, B. Slater, I. Zebger, S. Hofmann, J. C. Fontecilla-Camps, C. B. Park and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 10595–10599 CrossRef CAS PubMed.
- Y. Zhao, N. C. Anderson, M. W. Ratzloff, D. W. Mulder, K. Zhu, J. A. Turner, N. R. Neale, P. W. King and H. M. Branz, ACS Appl. Mater. Interfaces, 2016, 8, 14481–14487 CrossRef CAS PubMed.
- E. Edwardes Moore, V. Andrei, S. Zacarias, I. A. C. Pereira and E. Reisner, ACS Energy Lett., 2020, 5, 232–237 CrossRef CAS PubMed.
- C. E. Lubner, R. Grimme, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 404–414 CrossRef CAS PubMed.
- H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed.
- C. Tapia, R. D. Milton, G. Pankratova, S. D. Minteer, H.-E. Åkerlund, D. Leech, A. L. De Lacey, M. Pita and L. Gorton, ChemElectroChem, 2017, 4, 90–95 CrossRef CAS.
- C. E. Lubner, P. Knörzer, P. J. N. Silva, K. A. Vincent, T. Happe, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 10264–10266 CrossRef CAS PubMed.
- F. Zhao, P. Wang, A. Ruff, V. Hartmann, S. Zacarias, I. A. C. Pereira, M. M. Nowaczyk, M. Rögner, F. Conzuelo and W. Schuhmann, Energy Environ. Sci., 2019, 12, 3133–3143 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- G. Fuchs, Annu. Rev. Microbiol., 2011, 65, 631–658 CrossRef CAS PubMed.
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1873–1898 CrossRef CAS PubMed.
- K. Schuchmann and V. Müller, Nat. Rev. Microbiol., 2014, 12, 809–821 CrossRef CAS PubMed.
- C. A. R. Cotton, C. Edlich-Muth and A. Bar-Even, Curr. Opin. Biotechnol., 2018, 49, 49–56 CrossRef CAS PubMed.
- S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 165–195 CrossRef CAS PubMed.
- A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328–10329 CrossRef CAS PubMed.
- J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560–8564 CrossRef CAS PubMed.
- M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 54, 8337–8339 CrossRef CAS PubMed.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- A. Bassegoda, C. Madden, D. W. Wakerley, E. Reisner and J. Hirst, J. Am. Chem. Soc., 2014, 136, 15473–15476 CrossRef CAS PubMed.
- W. E. Robinson, A. Bassegoda, E. Reisner and J. Hirst, J. Am. Chem. Soc., 2017, 139, 9927–9936 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393–2399 RSC.
- A. Bachmeier, S. Hall, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 13518–13521 CrossRef CAS PubMed.
- S. Ikeyama and Y. Amao, Sustainable Energy Fuels, 2017, 1, 1730–1733 RSC.
- Y. Amao, Sustainable Energy Fuels, 2018, 2, 1928–1950 RSC.
- Y. Amao, Chem. Lett., 2017, 46, 780–788 CrossRef CAS.
- B. A. Parkinson and P. F. Weaver, Nature, 1984, 309, 148–149 CrossRef CAS.
- M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. C. Pereira and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS PubMed.
- S. K. Kuk, Y. Ham, K. Gopinath, P. Boonmongkolras, Y. Lee, Y. W. Lee, S. Kondaveeti, C. Ahn, B. Shin, J.-K. Lee, S. Jeon and C. B. Park, Adv. Energy Mater., 2019, 9, 1900029 CrossRef.
- S. Y. Lee, S. Y. Lim, D. Seo, J.-Y. Lee and T. D. Chung, Adv. Energy Mater., 2016, 6, 1502207 CrossRef.
- S. K. Kuk, J. Jang, J. Kim, Y. Lee, Y. S. Kim, B. Koo, Y. W. Lee, J. W. Ko, B. Shin, J.-K. Lee and C. B. Park, ChemSusChem, 2020 DOI:10.1002/cssc.202000459.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, Small, 2016, 12, 4753–4762 CrossRef CAS PubMed.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed.
- S. Zhang, J. Shi, Y. Sun, Y. Wu, Y. Zhang, Z. Cai, Y. Chen, C. You, P. Han and Z. Jiang, ACS Catal., 2019, 9, 3913–3925 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- P. Taylor, S. L. Pealing, G. A. Reid, S. K. Chapman and M. D. Walkinshaw, Nat. Struct. Biol., 1999, 6, 1108–1112 CrossRef CAS PubMed.
- A. Bachmeier, B. J. Murphy and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 12876–12879 CrossRef CAS PubMed.
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 CrossRef PubMed.
- J. Kim, S. H. Lee, F. Tieves, D. S. Choi, F. Hollmann, C. E. Paul and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 13825–13828 CrossRef CAS PubMed.
- S. H. Lee, D. S. Choi, M. Pesic, Y. W. Lee, C. E. Paul, F. Hollmann and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 8681–8685 CrossRef CAS PubMed.
- H. Hamby, B. Li, K. E. Shinopoulos, H. R. Keller, S. J. Elliott and G. Dukovic, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 135–140 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- N.-H. Tran, D. Nguyen, S. Dwaraknath, S. Mahadevan, G. Chavez, A. Nguyen, T. Dao, S. Mullen, T.-A. Nguyen and L. E. Cheruzel, J. Am. Chem. Soc., 2013, 135, 14484–14487 CrossRef CAS PubMed.
- C. M. Agapakis, P. M. Boyle and P. A. Silver, Nat. Chem. Biol., 2012, 8, 527–535 CrossRef CAS PubMed.
- M. J. Smanski, H. Zhou, J. Claesen, B. Shen, M. A. Fischbach and C. A. Voigt, Nat. Rev. Microbiol., 2016, 14, 135–149 CrossRef CAS PubMed.
- K. K. Sakimoto, N. Kornienko and P. Yang, Acc. Chem. Res., 2017, 50, 476–481 CrossRef CAS PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- S. Cestellos-Blanco, H. Zhang, J. M. Kim, Y.-X. Shen and P. Yang, Nat. Catal., 2020, 3, 245–255 CrossRef CAS.
- M. L. Ghirardi, A. Dubini, J. Yu and P.-C. Maness, Chem. Soc. Rev., 2009, 38, 52–61 RSC.
- Z. Ji, H. Zhang, H. Liu, O. M. Yaghi and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10582–10587 CrossRef CAS PubMed.
- W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu and J. Zhao, Sci. Adv., 2018, 4, eaap9253 CrossRef PubMed.
- A. A. Krasnovsky and V. V. Nikandrov, FEBS Lett., 1987, 219, 93–96 CrossRef.
- P. Maruthamuthu, S. Muthu, K. Gurunathan, M. Ashokkumar and M. V. C. Sastri, Int. J. Hydrogen Energy, 1992, 17, 863–866 CrossRef CAS.
- K. Gurunathan, J. Mol. Catal. A: Chem., 2000, 156, 59–67 CrossRef CAS.
- S. Pontrelli, T.-Y. Chiu, E. I. Lan, F. Y. H. Chen, P. Chang and J. C. Liao, Metab. Eng., 2018, 50, 16–46 CrossRef CAS PubMed.
- Y. Honda, H. Hagiwara, S. Ida and T. Ishihara, Angew. Chem., Int. Ed., 2016, 55, 8045–8048 CrossRef CAS PubMed.
- B. Wang, C. Zeng, K. H. Chu, D. Wu, H. Y. Yip, L. Ye and P. K. Wong, Adv. Energy Mater., 2017, 7, 1700611 CrossRef.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- R. K. Thauer, A.-K. Kaster, H. Seedorf, W. Buckel and R. Hedderich, Nat. Rev. Microbiol., 2008, 6, 579–591 CrossRef CAS PubMed.
- J. Ye, J. Yu, Y. Zhang, M. Chen, X. Liu, S. Zhou and Z. He, Appl. Catal., B, 2019, 257, 117916 CrossRef CAS.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed.
- A. W. D. Larkum, Curr. Opin. Biotechnol., 2010, 21, 271–276 CrossRef CAS PubMed.
- N. Kornienko, K. K. Sakimoto, D. M. Herlihy, S. C. Nguyen, A. P. Alivisatos, C. B. Harris, A. Schwartzberg and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11750–11755 CrossRef CAS PubMed.
- R. Zhang, Y. He, J. Yi, L. Zhang, C. Shen, S. Liu, L. Liu, B. Liu and L. Qiao, Chem, 2020, 6, 234–249 Search PubMed.
- K. K. Sakimoto, S. J. Zhang and P. Yang, Nano Lett., 2016, 16, 5883–5887 CrossRef CAS PubMed.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- B. Wang, Z. Jiang, J. C. Yu, J. Wang and P. K. Wong, Nanoscale, 2019, 11, 9296–9301 RSC.
- M. Chen, X.-F. Zhou, Y.-Q. Yu, X. Liu, R. J.-X. Zeng, S.-G. Zhou and Z. He, Environ. Int., 2019, 127, 353–360 CrossRef CAS PubMed.
- P. Gai, W. Yu, H. Zhao, R. Qi, F. Li, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 7224–7229 CrossRef CAS PubMed.
- S. F. Rowe, G. Le Gall, E. V. Ainsworth, J. A. Davies, C. W. J. Lockwood, L. Shi, A. Elliston, I. N. Roberts, K. W. Waldron, D. J. Richardson, T. A. Clarke, L. J. C. Jeuken, E. Reisner and J. N. Butt, ACS Catal., 2017, 7, 7558–7566 CrossRef CAS.
- J. H. Park, S. H. Lee, G. S. Cha, D. S. Choi, D. H. Nam, J. H. Lee, J.-K. Lee, C.-H. Yun, K. J. Jeong and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 969–973 CrossRef CAS PubMed.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- J. Guo, M. Suástegui, K. K. Sakimoto, V. M. Moody, G. Xiao, D. G. Nocera and N. S. Joshi, Science, 2018, 362, 813–816 CrossRef CAS PubMed.
- E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C. Y. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461–11466 CrossRef CAS PubMed.
- J. P. Torella, C. J. Gagliardi, J. S. Chen, D. K. Bediako, B. Colón, J. C. Way, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2337–2342 CrossRef CAS PubMed.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- C. Liu, K. K. Sakimoto, B. C. Colón, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6450–6455 CrossRef CAS PubMed.
- R. M. Rodrigues, X. Guan, J. A. Iñiguez, D. A. Estabrook, J. O. Chapman, S. Huang, E. M. Sletten and C. Liu, Nat. Catal., 2019, 2, 407–414 CrossRef CAS.
- K. Rabaey and R. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed.
- D. R. Lovley, Environ. Microbiol. Rep., 2011, 3, 27–35 CrossRef CAS PubMed.
- A. J. McCormick, P. Bombelli, R. W. Bradley, R. Thorne, T. Wenzel and C. J. Howe, Energy Environ. Sci., 2015, 8, 1092–1109 RSC.
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Y. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS PubMed.
- F. Qian, H. Wang, Y. Ling, G. Wang, M. P. Thelen and Y. Li, Nano Lett., 2014, 14, 3688–3693 CrossRef CAS PubMed.
- Y. Su, S. Cestellos-Blanco, J. M. Kim, Y.-x. Shen, Q. Kong, D. Lu, C. Liu, H. Zhang, Y. Cao and P. Yang, Joule, 2020, 4, 800–811 CrossRef CAS.
- S. Li, C. Cheng and A. Thomas, Adv. Mater., 2017, 29, 1602547 CrossRef.
- K. K. Sakimoto, N. Kornienko, S. Cestellos-Blanco, J. Lim, C. Liu and P. Yang, J. Am. Chem. Soc., 2018, 140, 1978–1985 CrossRef CAS PubMed.
- V. Flexer and L. Jourdin, Acc. Chem. Res., 2020, 53, 311–321 CrossRef CAS PubMed.
- K. B. Gregory, D. R. Bond and D. R. Lovley, Environ. Microbiol., 2004, 6, 596–604 CrossRef CAS PubMed.
- S. M. Strycharz, R. H. Glaven, M. V. Coppi, S. M. Gannon, L. A. Perpetua, A. Liu, K. P. Nevin and D. R. Lovley, Bioelectrochemistry, 2011, 80, 142–150 CrossRef CAS.
- D. E. Ross, J. M. Flynn, D. B. Baron, J. A. Gralnick and D. R. Bond, PLoS One, 2011, 6, e16649 CrossRef CAS.
- Y.-C. Yong, Y.-Y. Yu, X. Zhang and H. Song, Angew. Chem., Int. Ed., 2014, 53, 4480–4483 CrossRef CAS.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef CAS PubMed.
- K. B. Gregory and D. R. Lovley, Environ. Sci. Technol., 2005, 39, 8943–8947 CrossRef CAS.
- S. M. Strycharz, T. L. Woodard, J. P. Johnson, K. P. Nevin, R. A. Sanford, F. E. Löffler and D. R. Lovley, Appl. Environ. Microbiol., 2008, 74, 5943–5947 CrossRef CAS.
- S. M. Strycharz, S. M. Gannon, A. R. Boles, A. E. Franks, K. P. Nevin and D. R. Lovley, Environ. Microbiol. Rep., 2010, 2, 289–294 CrossRef CAS PubMed.
- X. Fang, S. Kalathil, G. Divitini, Q. Wang and E. Reisner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5074–5080 CrossRef CAS PubMed.
- N. Heidary, N. Kornienko, S. Kalathil, X. Fang, K. H. Ly, H. F. Greer and E. Reisner, J. Am. Chem. Soc., 2020, 142, 5194–5203 CrossRef CAS PubMed.
- N. J. Claassens, D. Z. Sousa, V. A. P. M. dos Santos, W. M. de Vos and J. van der Oost, Nat. Rev. Microbiol., 2016, 14, 692–706 CrossRef CAS PubMed.
- J. S. Deutzmann and A. M. Spormann, ISME J., 2016, 11, 704–714 CrossRef PubMed.
- P. T. Ha, S. R. Lindemann, L. Shi, A. C. Dohnalkova, J. K. Fredrickson, M. T. Madigan and H. Beyenal, Nat. Commun., 2017, 8, 13924 CrossRef CAS.
- B. Schiel-Bengelsdorf and P. Dürre, FEBS Lett., 2012, 586, 2191–2198 CrossRef CAS PubMed.
- C. M. Humphreys and N. P. Minton, Curr. Opin. Biotechnol., 2018, 50, 174–181 CrossRef CAS PubMed.
- N. J. Claassens, C. A. R. Cotton, D. Kopljar and A. Bar-Even, Nat. Catal., 2019, 2, 437–447 CrossRef CAS.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- N. Antonovsky, S. Gleizer, E. Noor, Y. Zohar, E. Herz, U. Barenholz, L. Zelcbuch, S. Amram, A. Wides, N. Tepper, D. Davidi, Y. Bar-On, T. Bareia, D. G. Wernick, I. Shani, S. Malitsky, G. Jona, A. Bar-Even and R. Milo, Cell, 2016, 166, 115–125 CrossRef CAS.
- K. Köninger, Á. Gómez Baraibar, C. Mügge, C. E. Paul, F. Hollmann, M. M. Nowaczyk and R. Kourist, Angew. Chem., Int. Ed., 2016, 55, 5582–5585 CrossRef PubMed.
- J. Ni, H.-Y. Liu, F. Tao, Y.-T. Wu and P. Xu, Angew. Chem., Int. Ed., 2018, 57, 15990–15994 CrossRef CAS PubMed.
- M. A. TerAvest and C. M. Ajo-Franklin, Biotechnol. Bioeng., 2016, 113, 687–697 CrossRef CAS PubMed.
- M. A. TerAvest, T. J. Zajdel and C. M. Ajo-Franklin, ChemElectroChem, 2014, 1, 1874–1879 CrossRef CAS.
- D. Choi, S. B. Lee, S. Kim, B. Min, I.-G. Choi and I. S. Chang, Bioresour. Technol., 2014, 154, 59–66 CrossRef CAS PubMed.
- Y. Chen, P. Li, H. Noh, C.-W. Kung, C. T. Buru, X. Wang, X. Zhang and O. K. Farha, Angew. Chem., Int. Ed., 2019, 58, 7682–7686 CrossRef CAS PubMed.
- I. J. Iwuchukwu, M. Vaughn, N. Myers, H. O'Neill, P. Frymier and B. D. Bruce, Nat. Nanotechnol., 2009, 5, 73–79 CrossRef PubMed.
- Y. V. Lee and B. Tian, Nano Lett., 2019, 19, 2189–2197 CrossRef CAS PubMed.
- G. Fan, C. M. Dundas, A. J. Graham, N. A. Lynd and B. K. Keitz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4559–4564 CrossRef CAS PubMed.
- T. Song, H. Zhang, H. Liu, D. Zhang, H. Wang, Y. Yang, H. Yuan and J. Xie, Bioresour. Technol., 2017, 243, 573–582 CrossRef CAS PubMed.
- C. M. Dundas, A. J. Graham, D. K. Romanovicz and B. K. Keitz, ACS Synth. Biol., 2018, 7, 2726–2736 CrossRef CAS.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- N. Kornienko, K. H. Ly, W. E. Robinson, N. Heidary, J. Z. Zhang and E. Reisner, Acc. Chem. Res., 2019, 52, 1439–1448 CAS.
- M. del Barrio, M. Sensi, C. Orain, C. Baffert, S. Dementin, V. Fourmond and C. Léger, Acc. Chem. Res., 2018, 51, 769–777 CrossRef CAS PubMed.
- E. Marsili, J. B. Rollefson, D. B. Baron, R. M. Hozalski and D. R. Bond, Appl. Environ. Microbiol., 2008, 74, 7329–7337 CrossRef CAS PubMed.
- T. Reda and J. Hirst, J. Phys. Chem. B, 2006, 110, 1394–1404 CrossRef CAS PubMed.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef CAS PubMed.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- K. Abdiaziz, E. Salvadori, K. P. Sokol, E. Reisner and M. M. Roessler, Chem. Commun., 2019, 55, 8840–8843 RSC.
- G. L. Chadwick, F. Jiménez Otero, J. A. Gralnick, D. R. Bond and V. J. Orphan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20716–20724 CrossRef CAS PubMed.
- Z. S. Wang, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 22449–22455 CrossRef CAS PubMed.
- R. Beranek, Adv. Phys. Chem., 2011, 786759 Search PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402–9406 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
| This journal is © The Royal Society of Chemistry 2020 |