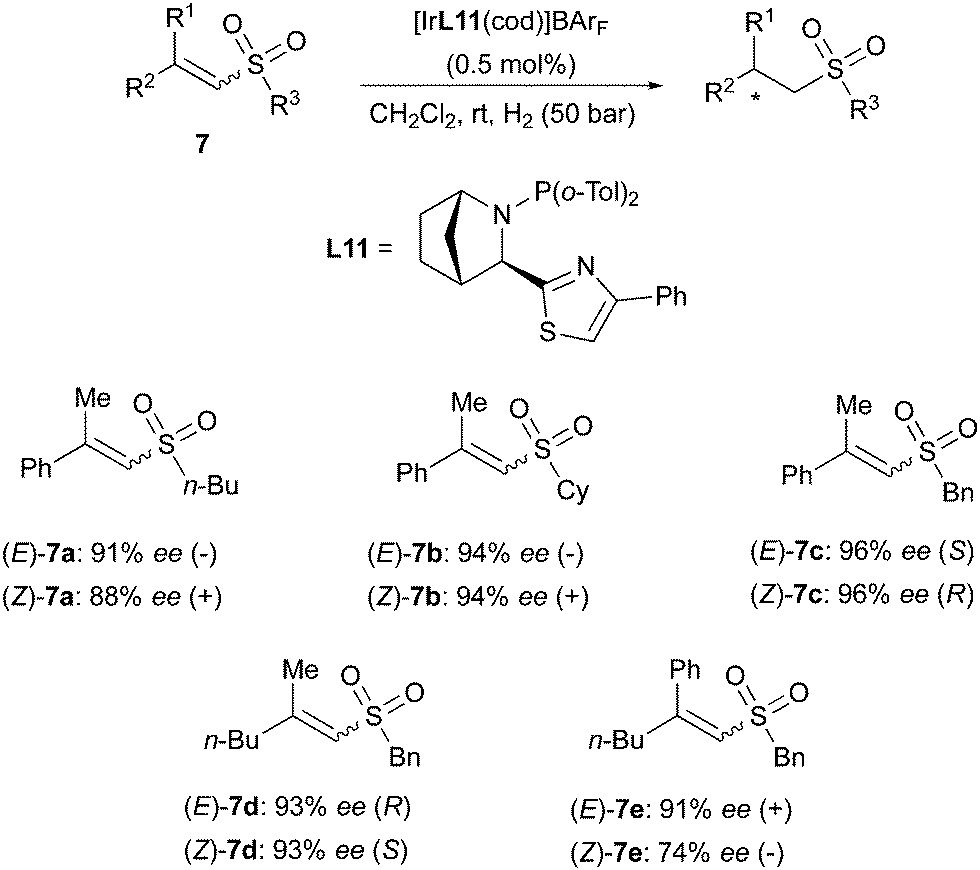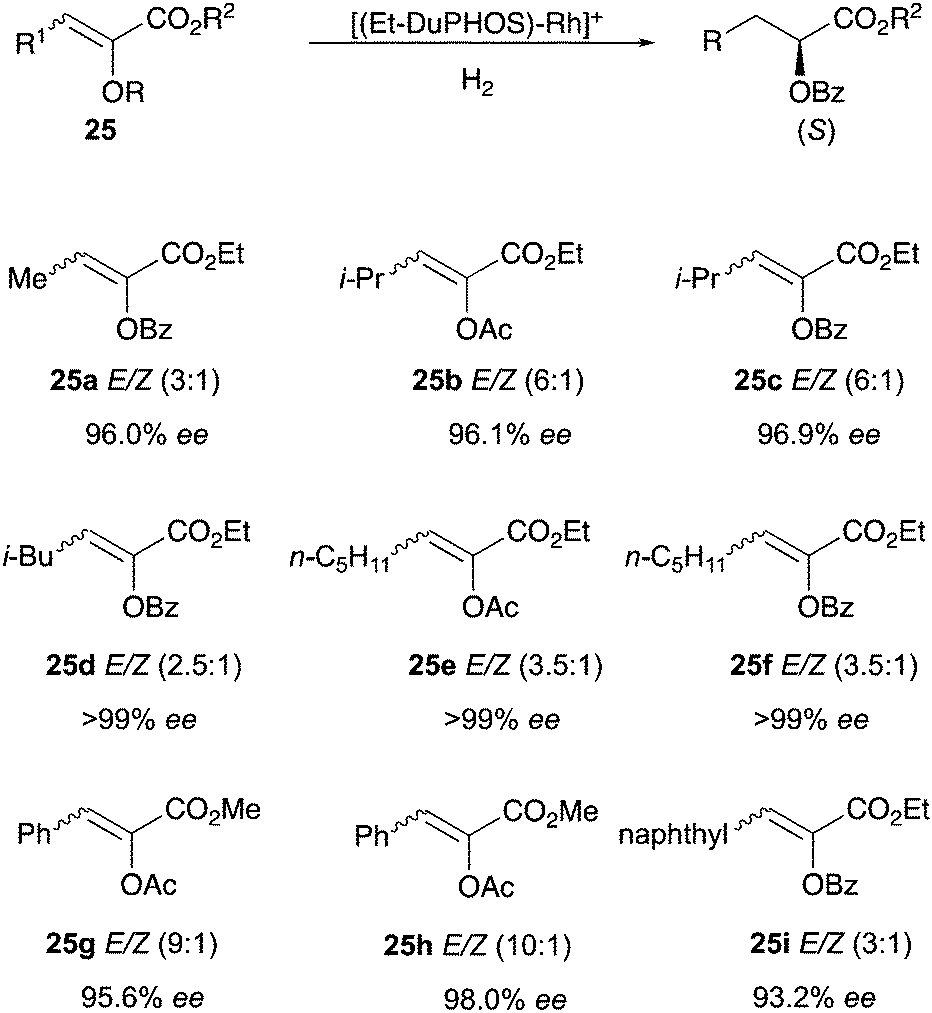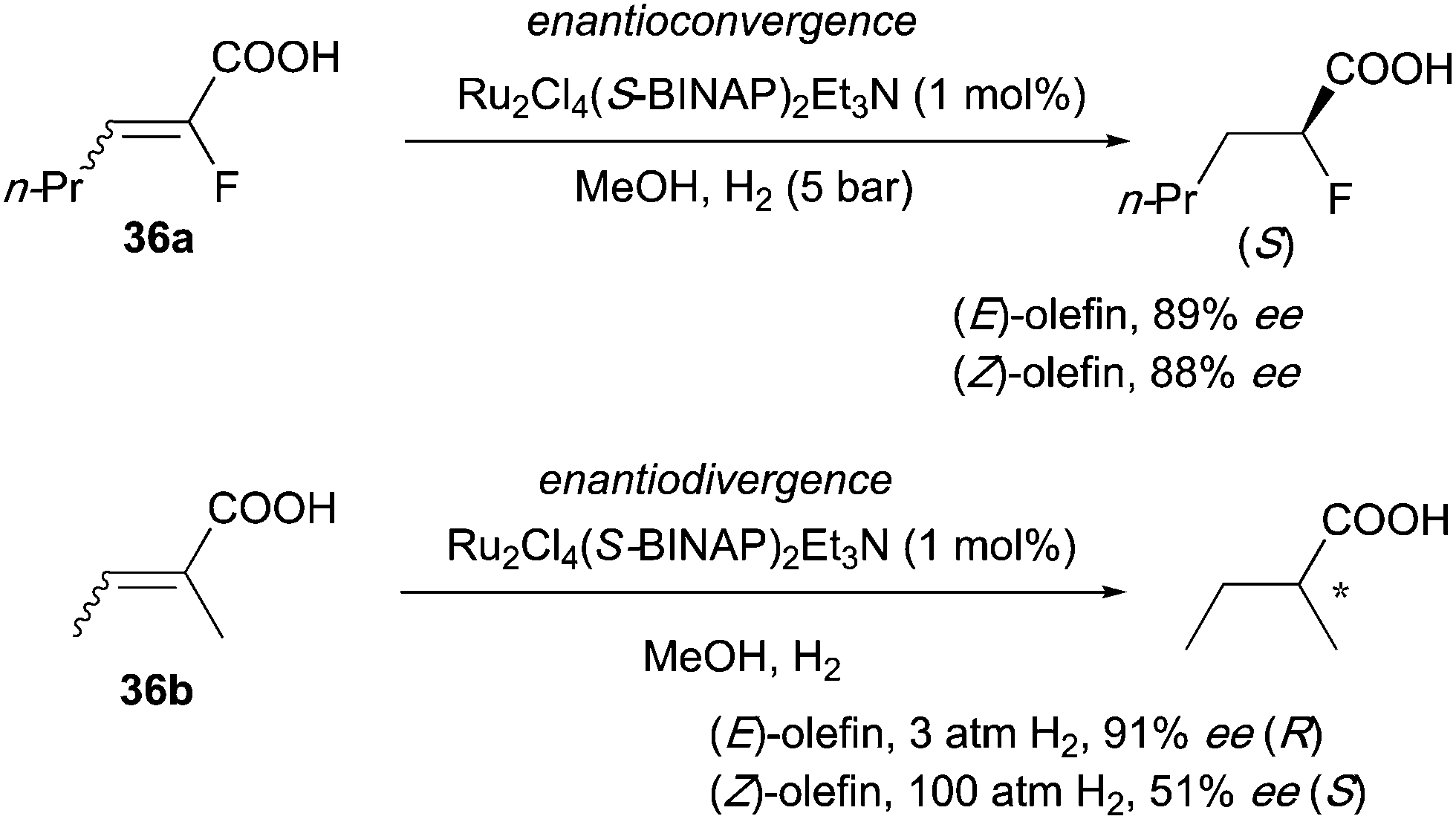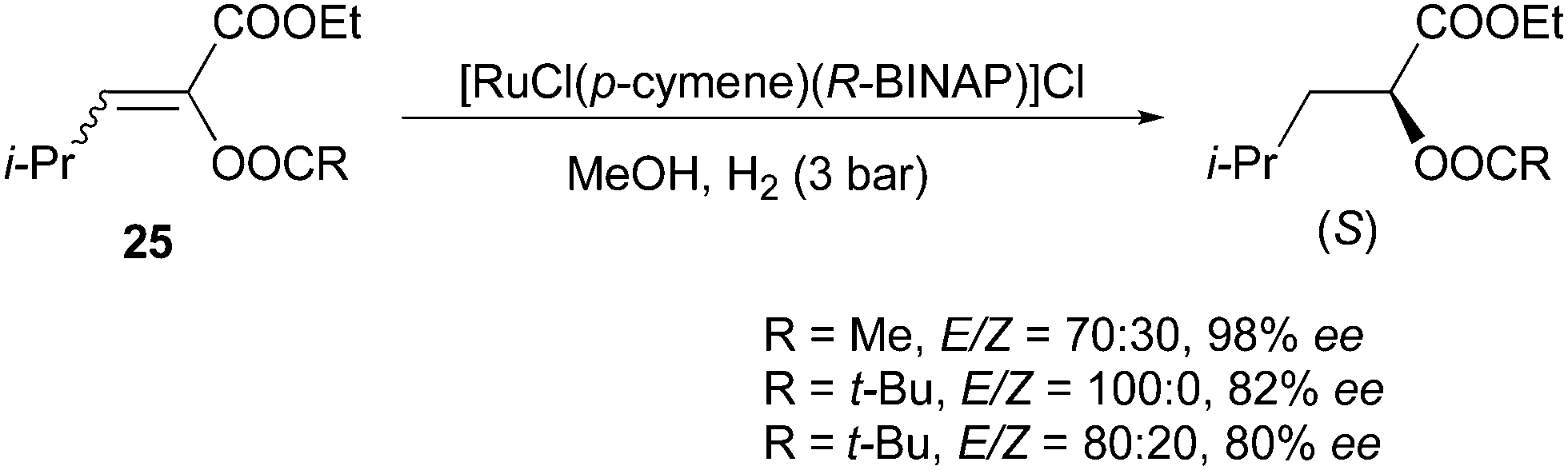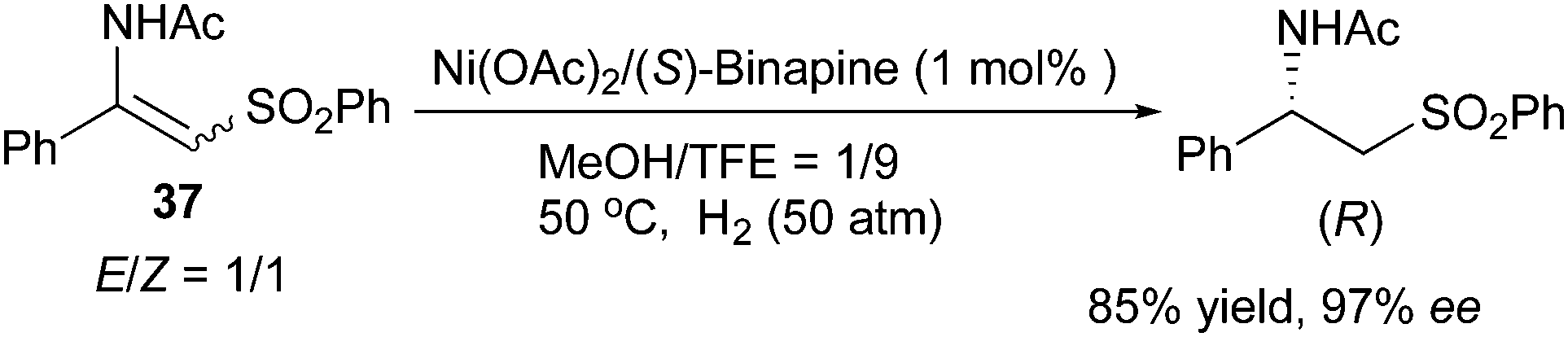DOI:
10.1039/C9CS00138G
(Review Article)
Chem. Soc. Rev., 2020,
49, 2504-2522
Enantioconvergent and enantiodivergent catalytic hydrogenation of isomeric olefins
Received
4th July 2019
First published on 23rd March 2020
Abstract
The asymmetric catalytic hydrogenation of olefins is one of the most widely studied and utilised transformations in asymmetric synthesis. This straightforward and atom-economical strategy can provide excellent enantioselectivity for a broad variety of substrates and is widely relevant for both industrial applications and academic research. In many instances the hydrogenation is stereospecific in the regard that the E–Z-geometry of the olefin governs the stereochemistry of the hydrogenation, producing an enantiodivergent outcome. Interestingly, the possibility to hydrogenate E- and Z-isomer mixtures to a single stereoisomer in an enantioconvergent manner has been reported. This avoids the need for synthesis of geometrically pure alkene starting materials and therefore constitutes a significant practical advantage. This review article aims to provide an overview of the different stereochemical outcomes in the hydrogenation of olefins. Although the field is well developed and selectivity models have been proposed for a number of catalytic systems, an organized collection of enantioconvergent results, as opposed to the more common enantiodivergent case, might promote new investigation into these phenomena.

Luca Massaro
| Luca Massaro was born in 1993 in Rome, Italy. He received his MSc degree in Organic and Biomolecular Chemistry from Sapienza University of Rome in 2018. Soon after, he joined Prof. Pher G. Andersson's group at Stockholm University as PhD student. His research focuses on catalytic asymmetric hydrogenations. |

Jia Zheng
| Jia Zheng received his BS and ME degrees from Jinan University (JNU) in 2009 and 2012. He then studied organic chemistry with Prof. Huanfeng Jiang at South China University of Technology (SCUT), where he obtained his PhD in 2016. He's been doing postdoctoral research in the field of asymmetric hydrogenation with Prof. Pher G. Andersson at the Department of Organic Chemistry at Stockholm University Since 2017. |

Cristiana Margarita
| Cristiana Margarita graduated in Organic Chemistry from Sapienza University of Rome in 2014. In 2019 she received her PhD from Stockholm University with a thesis on enantio- and regioselective catalytic hydrogenations, under the supervision of Prof. Pher G. Andersson. |

Pher G. Andersson
| Pher G. Andersson was born in 1963 in Växjö, Sweden. He was educated at Uppsala University where he received his BSc in 1988 and his PhD in 1991. After postdoctoral research at Scripps Research Institute with Prof. K. B. Sharpless, he returned to Uppsala where he became Docent in 1994 and full Professor in 1999. Since 2010 he also holds the position of Honorary Professor at the University of KwaZulu-Natal, South Africa. As of January 2013, he is Professor of Organic Chemistry at Stockholm University. His main research interests involve organometallic chemistry, stereoselective synthesis, and asymmetric catalysis. |
1. Introduction
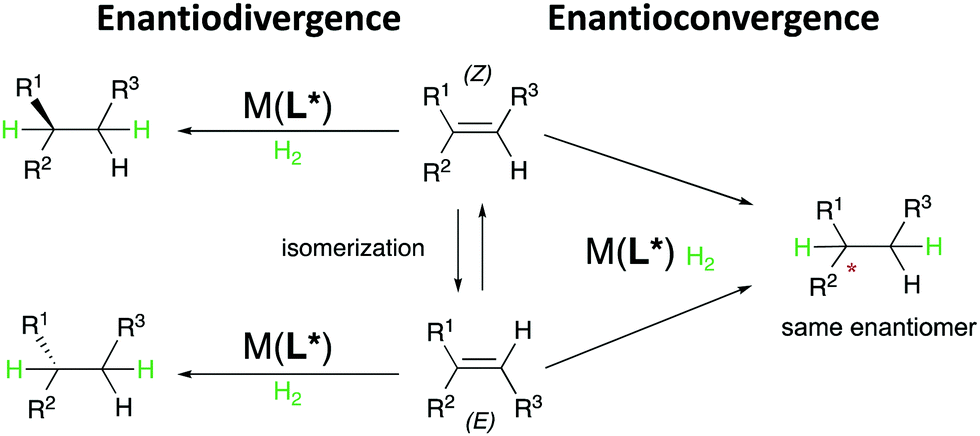
The transition-metal catalyzed asymmetric hydrogenation of prochiral olefins constitutes one of the most practical and useful methods in modern stereoselective synthesis. The advantageous features of this transformation comprise perfect atom-economy, nearly quantitative yields, high enantioselectivity, low catalyst loadings, and mild reaction conditions, which together define it as a powerful tool for the generation of stereogenic centers in target molecules and synthetic intermediates. Accordingly, catalytic asymmetric hydrogenation has found wide application both in academic research and industrial processes since the pioneering work of Knowles and Noyori, which was awarded the Nobel Prize in 2001. Since then, the development of novel, efficient catalytic systems for asymmetric hydrogenation has flourished, granting a great expansion of the reaction scope, development of new catalysts, and optimized chiral ligands.1–4 As a result, a large number of olefinic substrates (having diverse structural and electronic properties) can now be hydrogenated in a highly enantioselective manner. When analyzing the stereochemical outcome of the hydrogenation reaction, an influence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C geometry on the selectivity and reactivity is commonly observed. For example, the Ir-catalyzed asymmetric hydrogenation of non-functionalized (Z)-alkenes often results in lower enantiomeric excesses than the corresponding (E)-configured alkenes. In addition, the most generally encountered outcome when hydrogenating E/Z isomeric olefins is that they result in opposite enantiomers (enantiodivergent). On the other hand, the less frequent, but more useful enantioconvergent hydrogenation of E/Z isomers has also been observed, for instance in the case of Rh-P,P-catalyzed hydrogenations of certain classes of functionalized olefins. In this case, both the E and Z isomers are transformed into the same enantiomer of the product. The implications of the two different outcomes also impact the strategy in which the hydrogenation is employed in organic synthesis. On one side, the enantiodivergent outcome can grant the possibility of using the same enantiomer of catalyst to hydrogenate either the (E)- or (Z)-olefin individually to produce the desired enantiomer of the product, (provided that high enantioselectivity is reached in both cases). On the other hand, the enantioconvergent hydrogenation can allow for the direct asymmetric hydrogenation of mixtures of E- and Z-olefins, avoiding the need for preparation of geometrically pure alkenes or for their time-consuming separation.
C geometry on the selectivity and reactivity is commonly observed. For example, the Ir-catalyzed asymmetric hydrogenation of non-functionalized (Z)-alkenes often results in lower enantiomeric excesses than the corresponding (E)-configured alkenes. In addition, the most generally encountered outcome when hydrogenating E/Z isomeric olefins is that they result in opposite enantiomers (enantiodivergent). On the other hand, the less frequent, but more useful enantioconvergent hydrogenation of E/Z isomers has also been observed, for instance in the case of Rh-P,P-catalyzed hydrogenations of certain classes of functionalized olefins. In this case, both the E and Z isomers are transformed into the same enantiomer of the product. The implications of the two different outcomes also impact the strategy in which the hydrogenation is employed in organic synthesis. On one side, the enantiodivergent outcome can grant the possibility of using the same enantiomer of catalyst to hydrogenate either the (E)- or (Z)-olefin individually to produce the desired enantiomer of the product, (provided that high enantioselectivity is reached in both cases). On the other hand, the enantioconvergent hydrogenation can allow for the direct asymmetric hydrogenation of mixtures of E- and Z-olefins, avoiding the need for preparation of geometrically pure alkenes or for their time-consuming separation.
The occurrence of these two possible outcomes for asymmetric hydrogenation is presented in the two main sections of this review, further divided according to the type of transition metal used in the catalysis. Out of the enormous number of reports concerning asymmetric hydrogenation of olefins, we have selected studies in which pure E and Z olefins were directly compared in the reaction or used as E/Z mixtures.
2. Enantiodivergent hydrogenations
Due to their prevalence in the literature, the main section of the review is dedicated to enantiodivergent results in the hydrogenation of (E)- or (Z)-configured olefins. An overview of reports in which the different geometry of the olefin produces opposite enantiomers is discussed for each transition-metal subsection.
2.1 Iridium
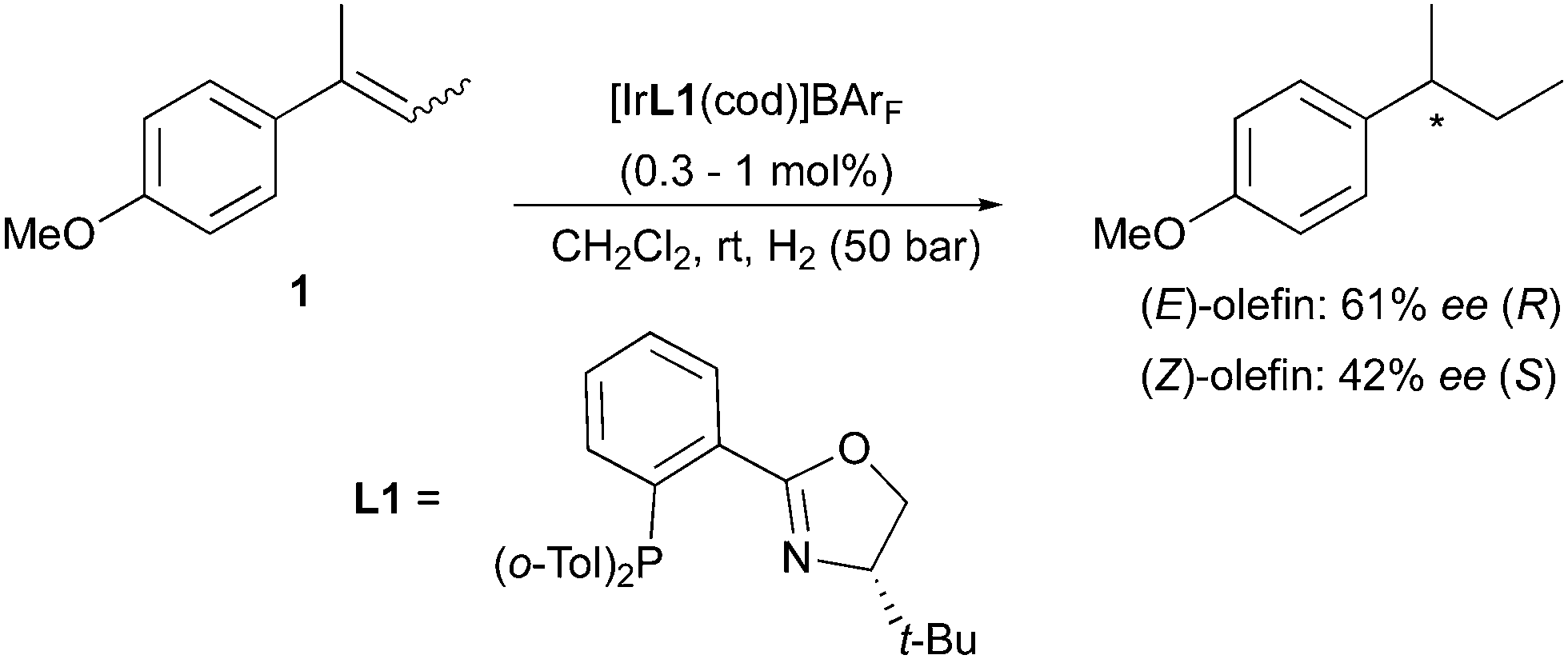
Numerous examples can be found in the literature on the use of chiral iridium catalysts (with N,P or NHC ligands) resulting in the enantiodivergent hydrogenation of trisubstituted olefins with different geometry. N,P-ligated Ir-complexes have proved to be highly efficient catalysts for the asymmetric hydrogenation of alkenes lacking coordinative groups such as amides. Alkyl-substituted alkenes can be challenging substrates for this transformation, providing lower ee since the similar properties of the substituents cause a more arduous enantiofacial selection by the catalysts. In this case the process relies on steric effects, discriminating between the smallest substituent, normally a hydrogen atom, and a more hindered group on the less substituted terminus of the olefin (the non-prochiral site). This type of enantioselection results in (E) and (Z)-alkenes undergoing hydrogenation to give opposite enantiomeric products. Competing alternative pathways, such as double bond migrations, can also occur in the hydrogenation of (Z)-olefins, producing more complicated reaction outcomes and lower levels of enantioselectivity. However, with the development of Ir-based catalysts not requiring coordinative groups on the prochiral substrates, asymmetric hydrogenation could be extended to a wide variety of unfunctionalized olefins. Initial studies focused on the preparation of efficient chiral Ir-complexes for such transformations, and employed a common set of aryl–alkyl-substituted olefins to test the new catalysts performance. Out of these selected substrates, 2-(4-methoxyphenyl)-2-butene was often evaluated in both the (E)- and (Z)-isomeric forms. This allowed the assessment of the stereochemical behavior exhibited by several different classes of Ir-catalysts towards the asymmetric hydrogenation of geometrical isomers of non-functionalized alkenes. In the earliest report on the use of Ir–PHOX complexes for the asymmetric hydrogenation of olefins, the Pfaltz group developed a series of highly efficient chiral catalysts that provided excellent enantioselectivities (up to 98% ee) for several aryl-substituted alkenes. Despite high conversions, moderate ee were initially obtained in the case of the (E)- and (Z)-2-(4-methoxyphenyl)-2-butene isomers (61% and 42% respectively). However, it was already noticed that the two substrate isomers furnished opposite enantiomers of the hydrogenated product when using the same chiral catalyst 1 (Scheme 1).5,6
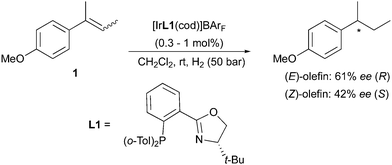 |
| | Scheme 1 | |
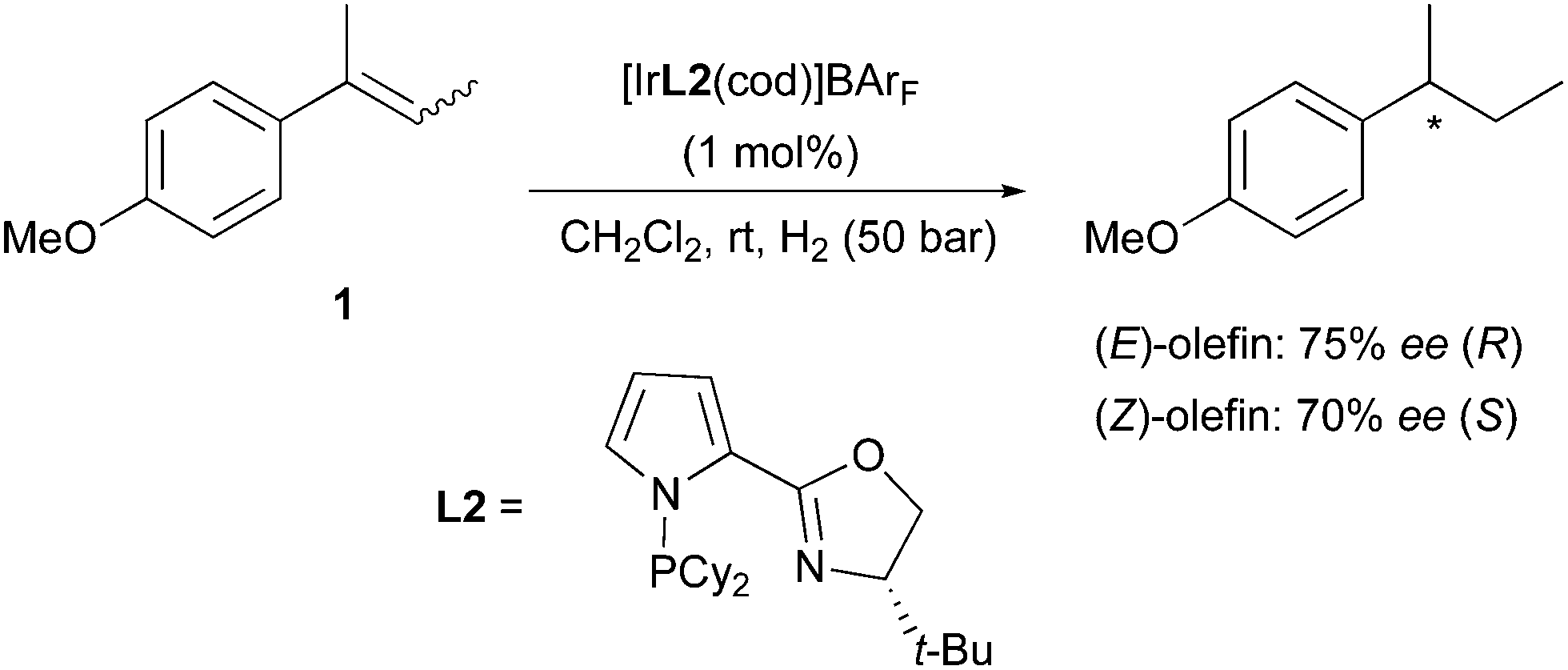
A few years later, a new set of N,P-ligands containing a pyrrole structure (PyrPHOX) afforded an improvement of selectivity in the hydrogenation of both (E)- and (Z)-2-(4-methoxyphenyl)-2-butene. The stereochemical outcome of the reactions matched previous observations, as opposite absolute configurations were obtained after the hydrogenation (Scheme 2).7
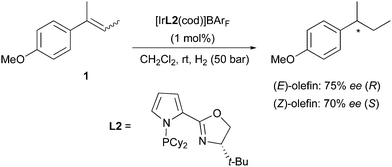 |
| | Scheme 2 | |
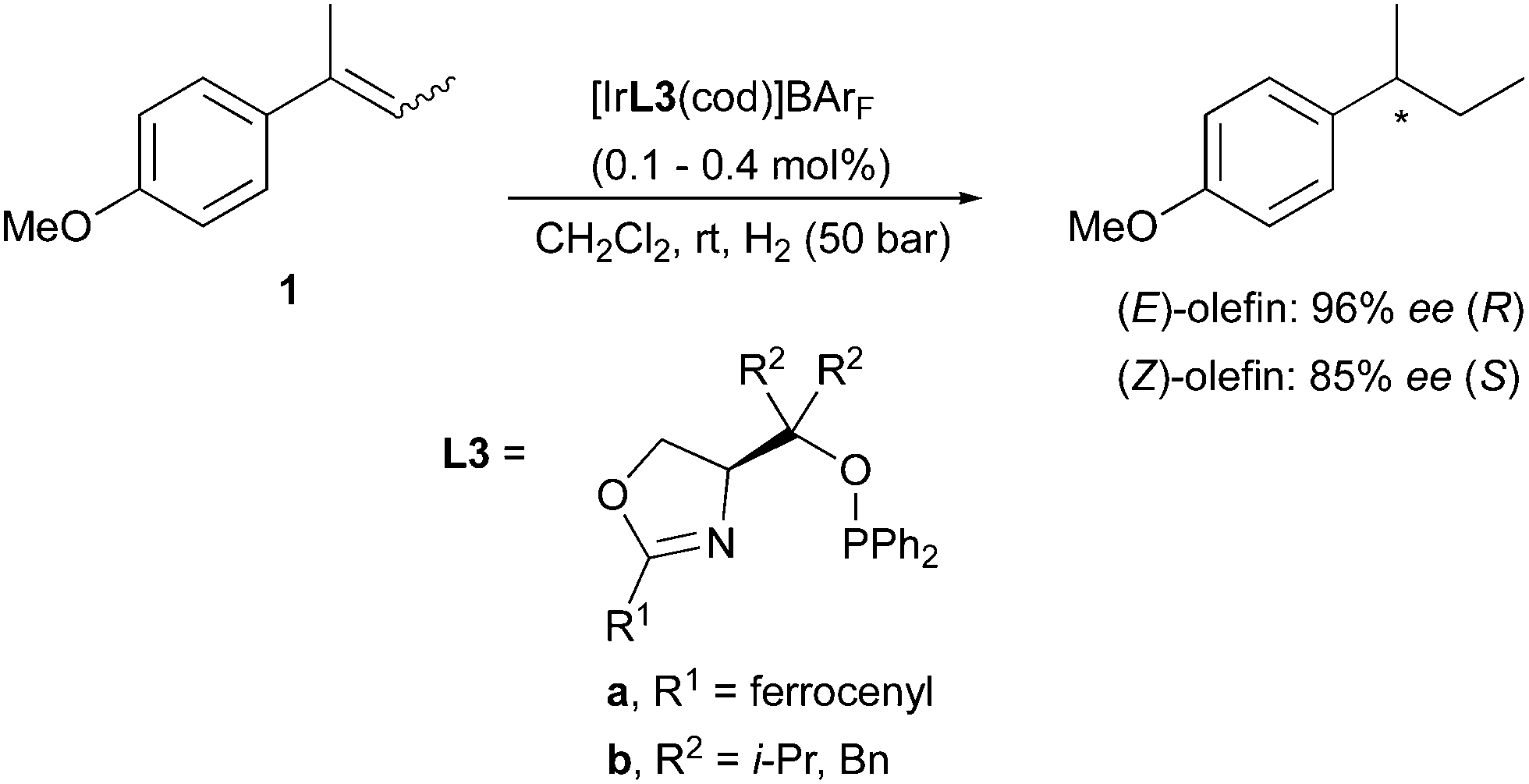
This trend for enantiodivergent hydrogenations of these substrates was also observed in several reports as ligand development continued. A class of successful phosphinite-oxazoline Ir-catalysts allowed high enantioselectivity in this reaction, with the better result obtained for the (E)-isomer (Scheme 3).8
 |
| | Scheme 3 | |
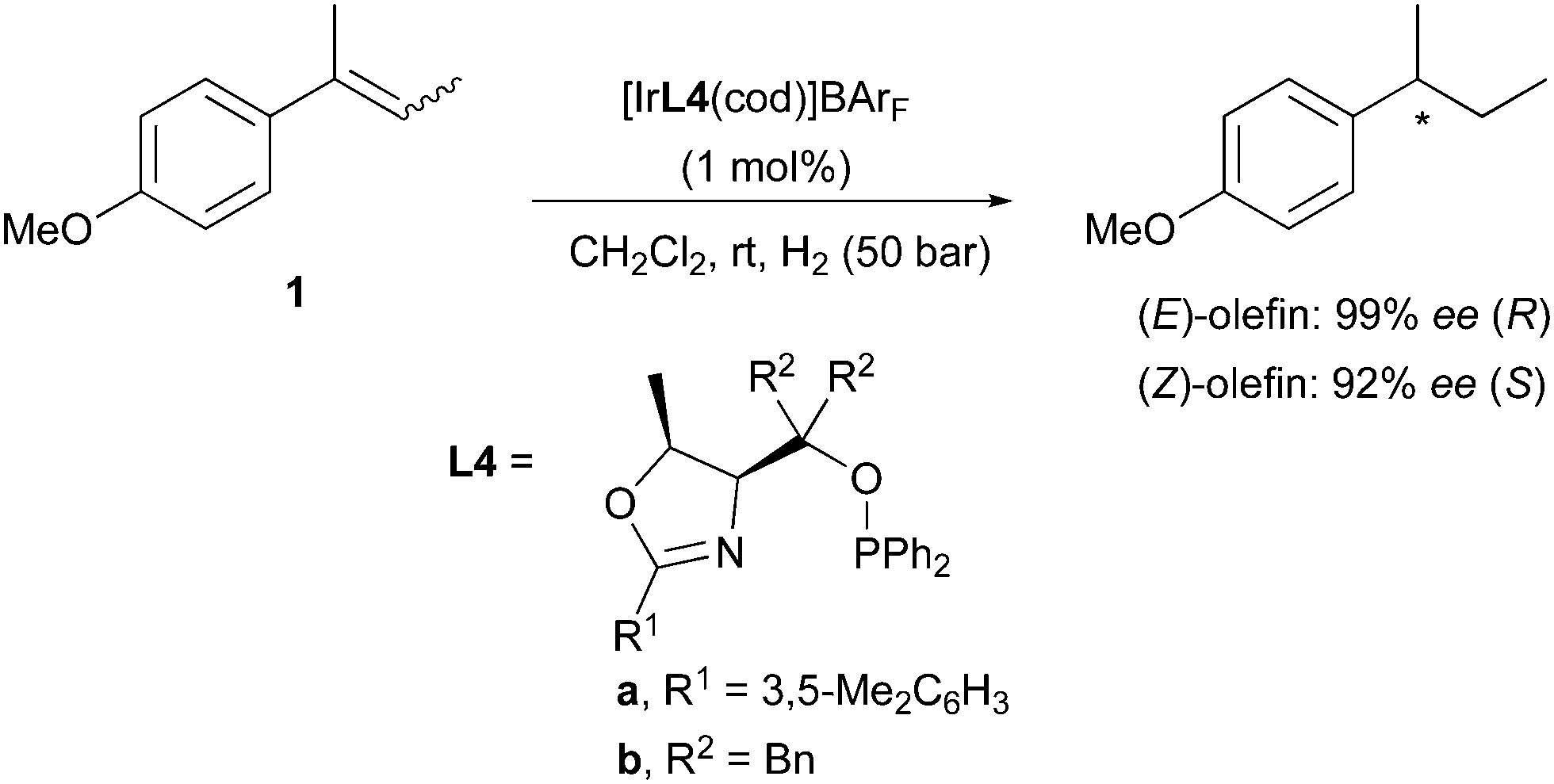
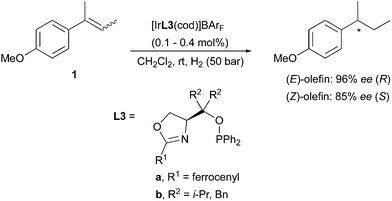
A variation on this type of ligand structure resulted in a group of catalysts that gave the best enantioselectivities achieved for these olefins. Threonine-derived phosphinite oxazoline L4 was found to hydrogenate (E)- and (Z)-2-(4-methoxyphenyl)-2-butene in a stereodivergent manner in 99% and 92% ee, respectively (Scheme 4).9
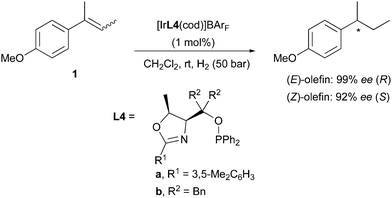 |
| | Scheme 4 | |
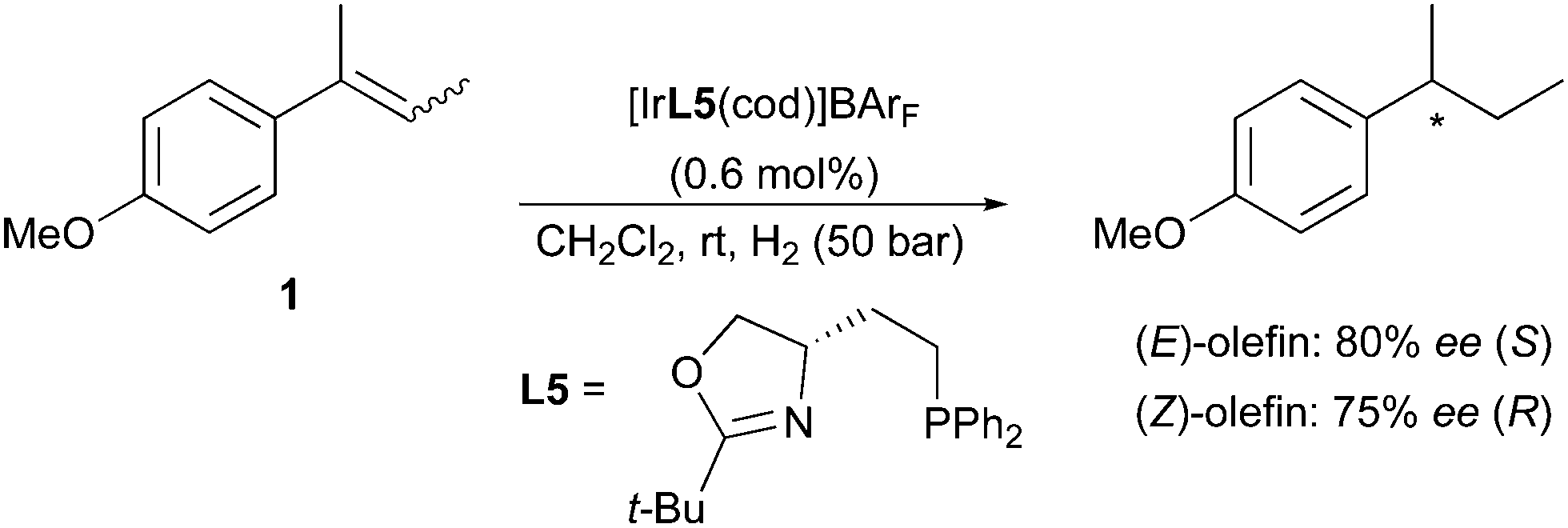
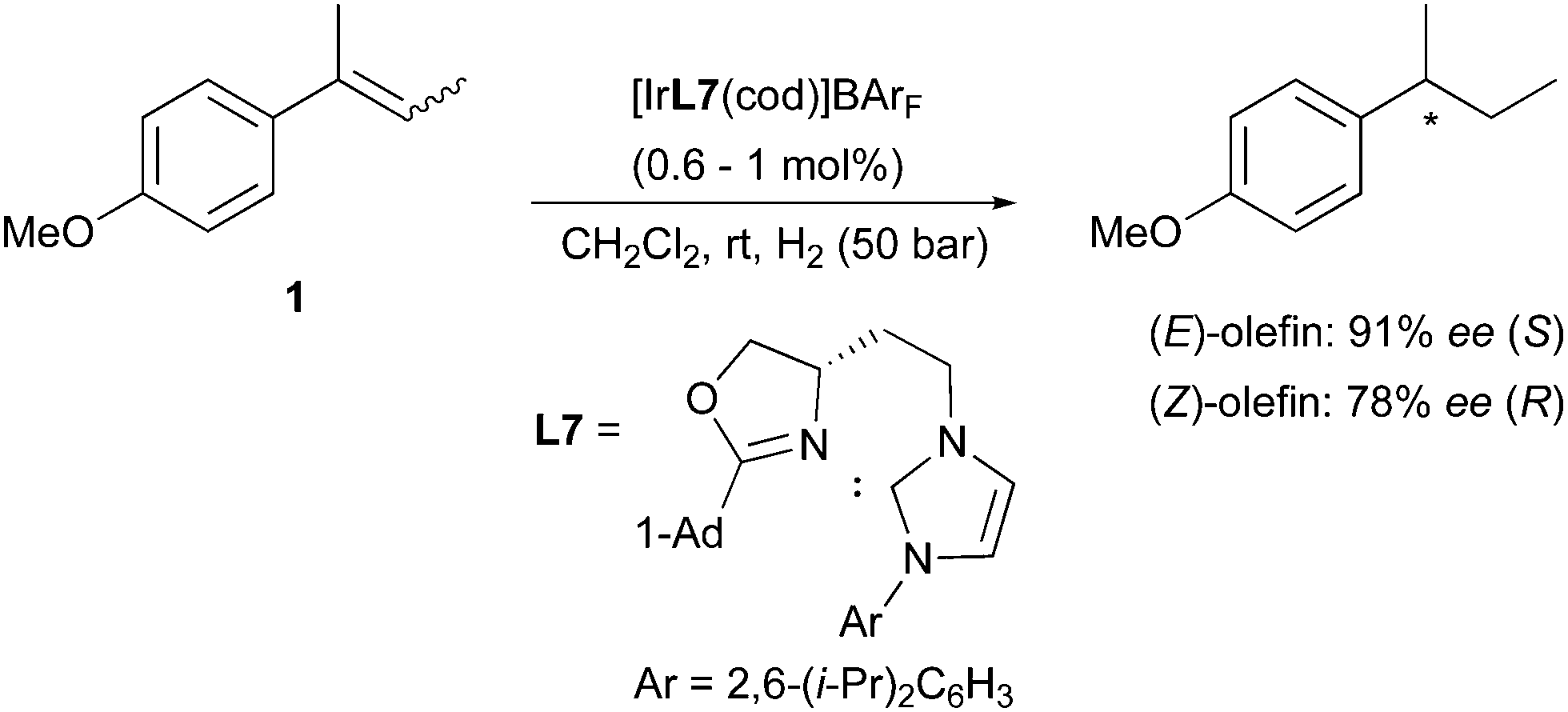
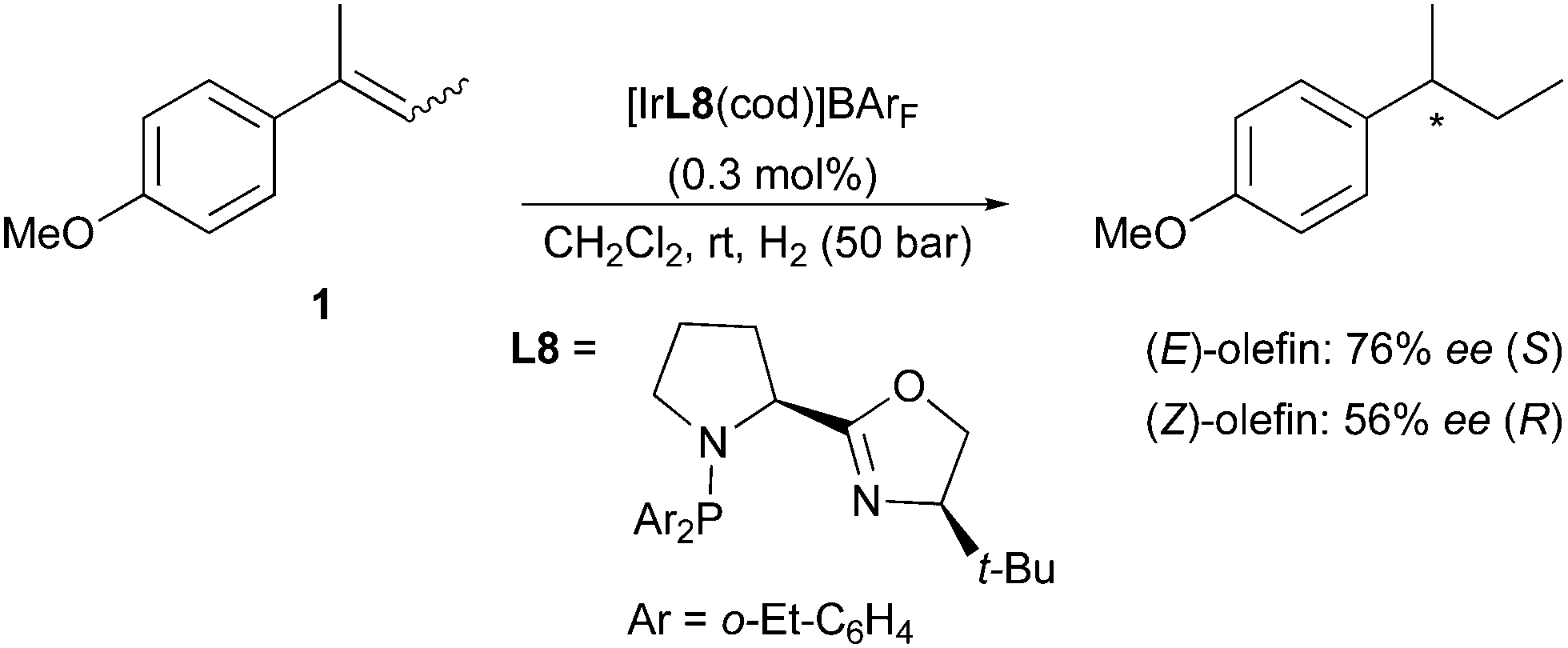
Employing the less-substituted oxazoline chiral ligand L5 did not alter the stereochemical outcome of this reaction. Again, the same enantiodivergence was also reported by the Burgess group, even if the smaller ligand employed could not provide the ee achieved by its phosphinite analogues (Scheme 5).10 When the oxazoline ring on the PHOX ligand class was exchanged for an imidazoline (PHIM), the resulting Ir-catalysts showed good selectivity in the asymmetric hydrogenation of minimally functionalized olefins. Although the results for the isomeric standard olefins were inferior to those afforded by the phosphinite-containing catalysts, it still confirmed the consistent generation of opposite enantiomeric products (Scheme 6).11
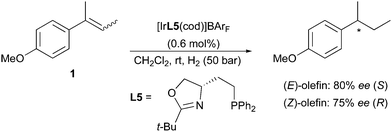 |
| | Scheme 5 | |
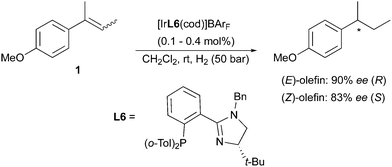 |
| | Scheme 6 | |
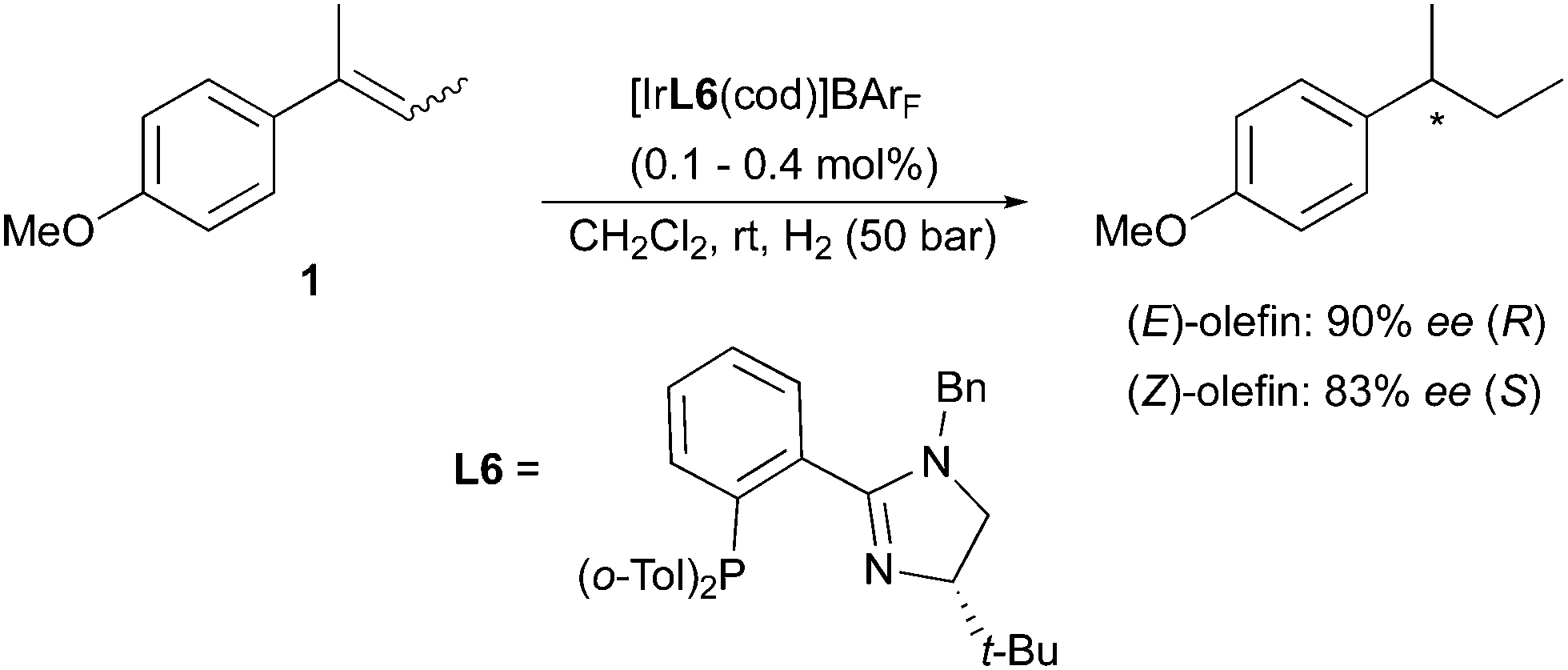
(E)- and (Z)-2-(4-methoxyphenyl)-2-butene were also successfully hydrogenated by means of Ir-catalysts containing chiral N-heterocyclic-carbene (NHC) ligands developed in the Burgess group. The NHC replaces the phosphine binding site compared to the structure of the various Crabtree-type N,P-ligands designed for Ir-catalyzed hydrogenation. The electronic properties of the N,P and NHC ligands can differ to a certain extent, for example NHC-containing Ir-complexes were found to be much less acidic than their phosphine counterparts under hydrogenation conditions.12 However, the mechanism of stereoselection relies on steric factors in either case, and this results in similarly enantiodivergent hydrogenations of unfunctionalized olefins (Scheme 7).13,14
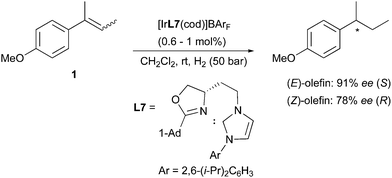 |
| | Scheme 7 | |
A series of proline-derived phosphine-oxazoline ligands were also tested in the hydrogenation of simple olefins, although not reaching the levels of enantioselectivity obtained with other Ir-catalysts. When comparing the results from the asymmetric hydrogenation of (E)- and (Z)-2-(4-methoxyphenyl)-2-butene, this system again confirmed the previously observed stereodivergence (Scheme 8).15
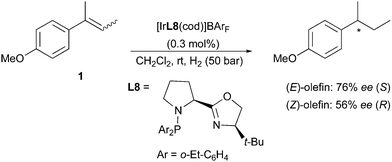 |
| | Scheme 8 | |
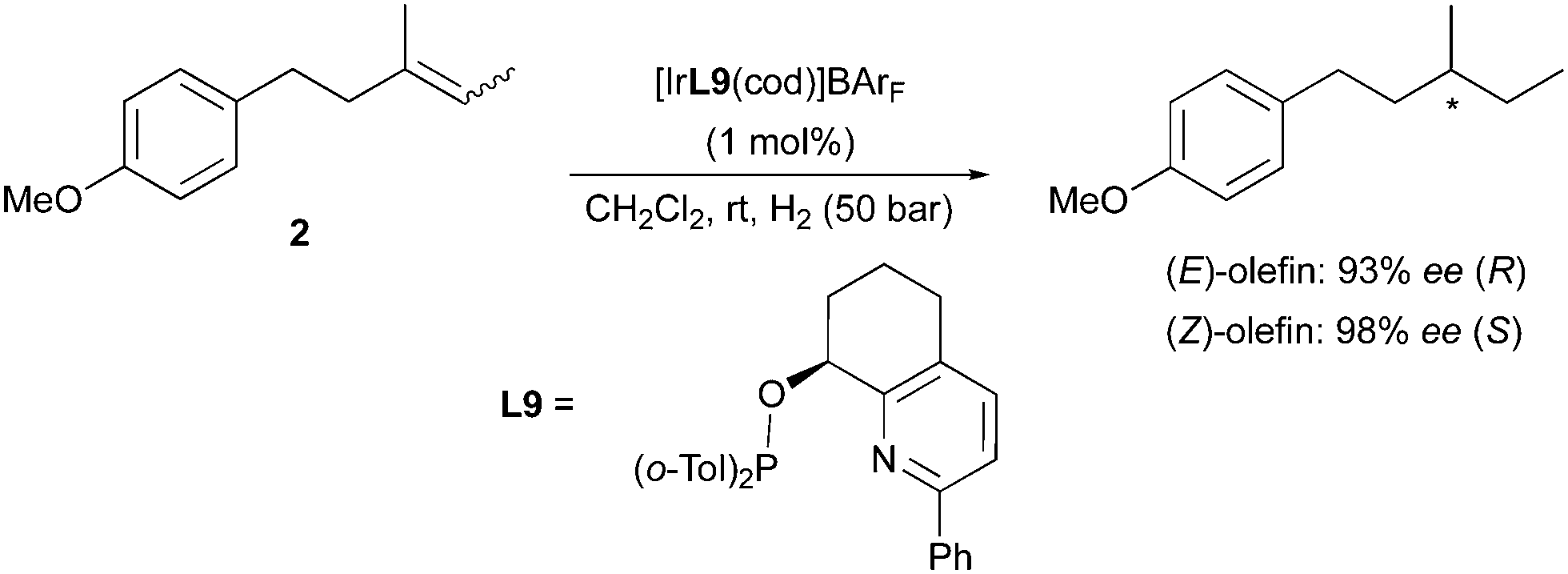
A higher homologue of this substrate was used by the Pfaltz group as model for the study on the particularly challenging asymmetric hydrogenation of purely alkyl-substituted olefins. In this case the aryl group was moved to a remote position with regards to the prochiral double bond, and excellent enantiomeric excesses were afforded via the employment of highly selective pyridine-based catalysts for both geometrical isomers 2 of the olefin (Scheme 9).16 This case also showed consistency with the observed enantiodivergent outcome in Ir-catalyzed hydrogenations. Surprisingly, for this substrate the (Z)-isomer delivered a higher ee.
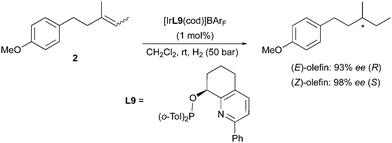 |
| | Scheme 9 | |
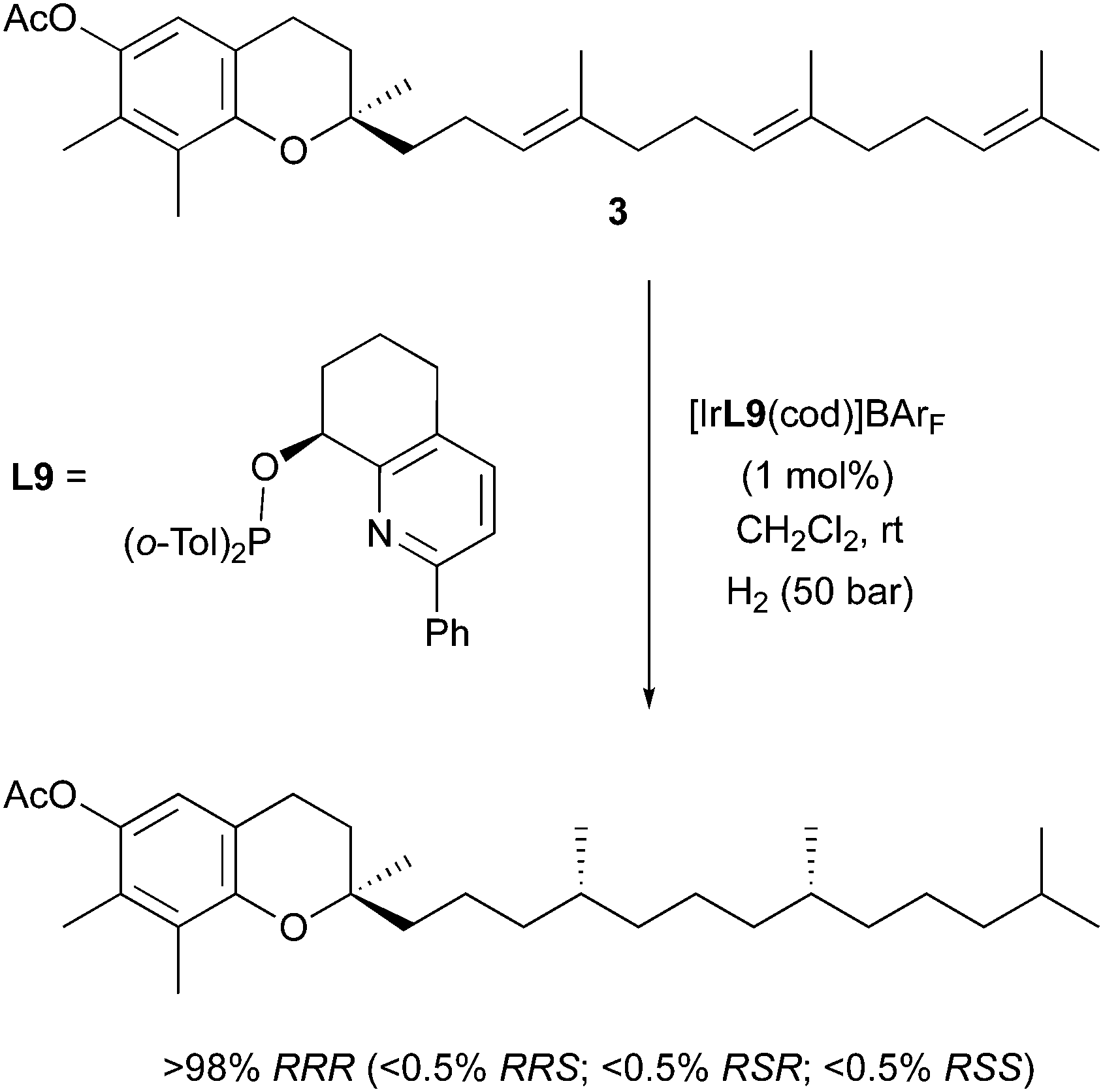
In the same study, this stereodivergence in the hydrogenation of alkyl-substituted olefins was also observed when these pyridine-based catalysts were applied to the preparation of a natural product. γ-Tocotrienylacetate was subjected to the asymmetric hydrogenation procedure, installing two stereogenic centers in one step, to selectively prepare stereoisomers of tocopherol (Scheme 10). Since the molecule contains more than one prochiral double bond in this case, individual control of their geometry allows the control of the relative stereochemistry of the preferred diastereomeric product. In the reported application, two (E)-configured olefins gave rise to stereocenters of (RR) configuration, and the major diastereoisomer of tocopherol was obtained in excellent selectivity (>98% RRR).
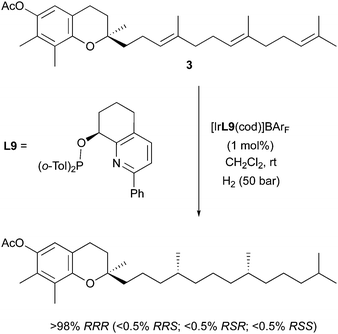 |
| | Scheme 10 | |
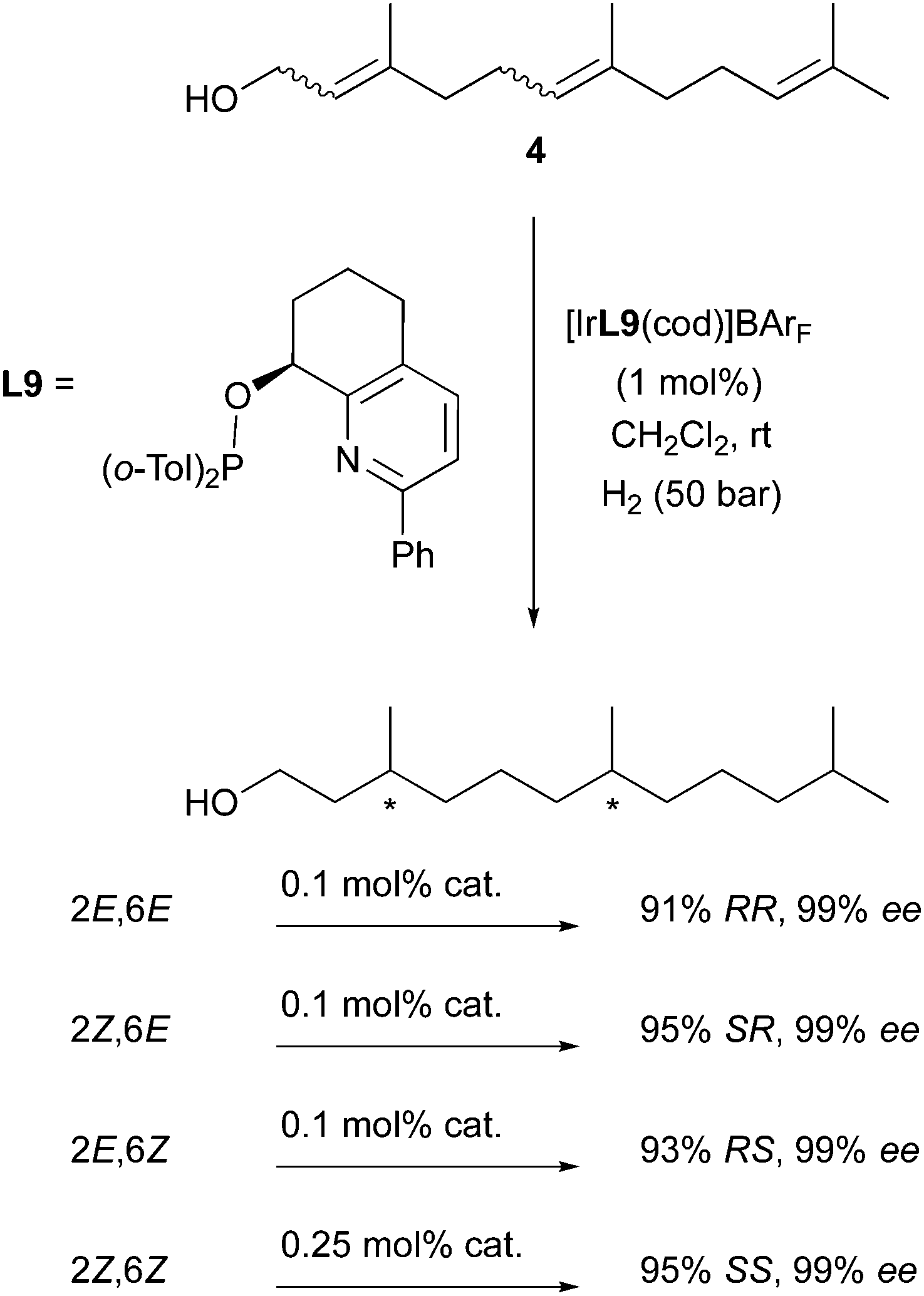
Following this successful strategy, the Pfaltz group also studied the stereodivergent hydrogenation of other polyenes such as farnesol derivatives. With the employment of a single Ir-catalyst (containing the highly efficient pyridyl-ligand L9) and by systematic variation of the olefins geometry, it was elegantly demonstrated that the enantiodivergence allowed access to each of the hexahydrofarnesol stereoisomers with excellent levels of selectivity (Scheme 11).17
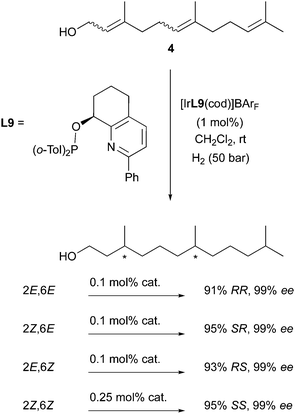 |
| | Scheme 11 | |
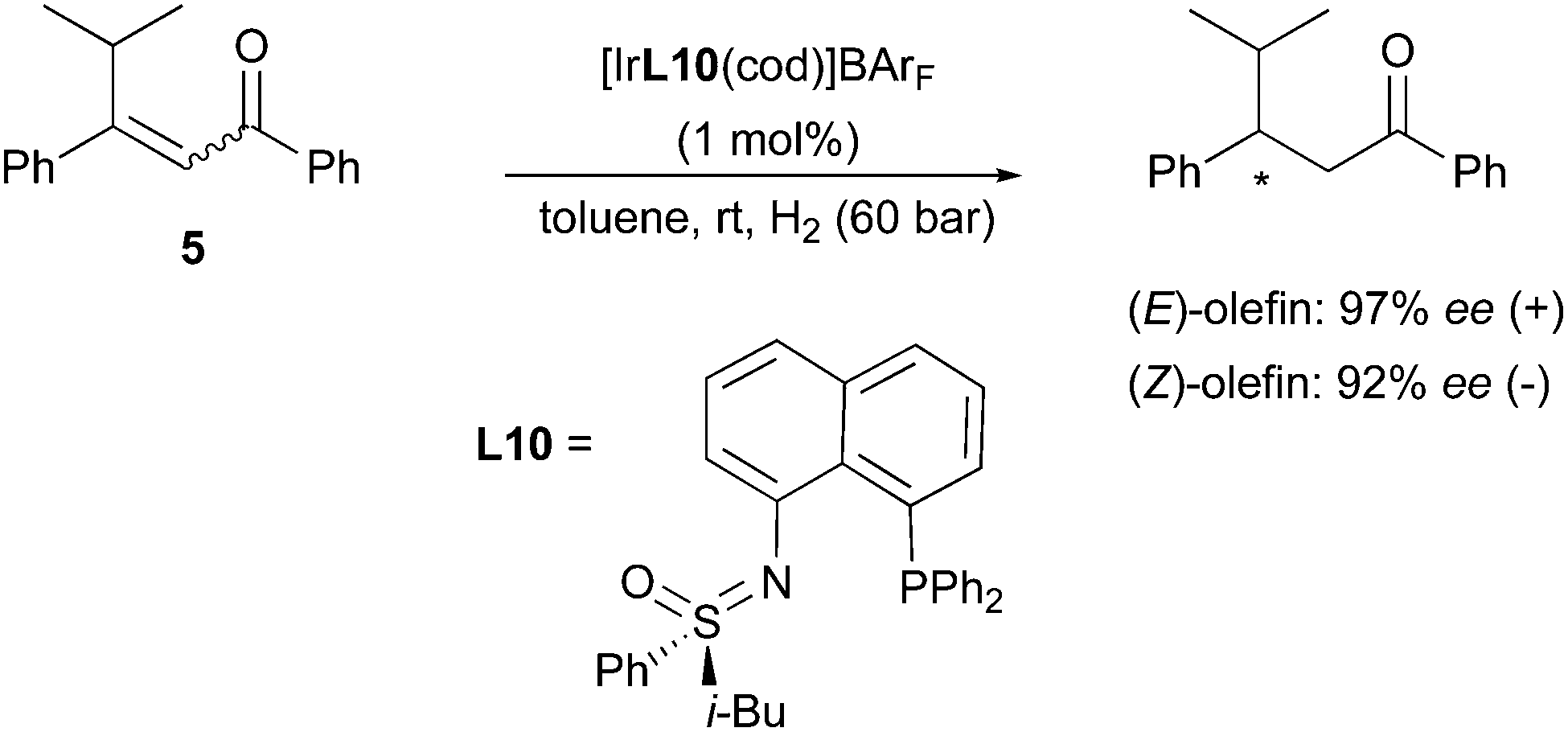
As shown in these contributions, the development of the highly efficient Ir-N,P-catalysts for the asymmetric hydrogenation of unfunctionalized and minimally functionalized olefins resulted in a great expansion of the substrate scope where Rh and Ru-complexes could not perform efficiently. The ongoing investigation of the potential of the Ir-catalyzed asymmetric hydrogenation was further extended to many classes of challenging or particularly interesting functionalized alkenes, whose highly enantioselective reduction was still an issue. In some of the studies concerning the hydrogenation of functionalized prochiral olefins, the same stereodivergent outcome encountered with alkyl-substituted substrates was observed. For example, in 2008 Bolm reported on the asymmetric hydrogenation of the CC in enones, a group of substrates for which the method can be hampered by undesired carbonyl reduction. The Ir-catalyst employed for this hydrogenation contained a sulfoximine-derived N,P-ligand, which proved very efficient towards β-aryl-substituted enones. A pair of geometrical isomers, (E)- and (Z)-5, was also evaluated with this system, and resulted in an enantiodivergent outcome reaching 97% and 92% ee, respectively (Scheme 12).18
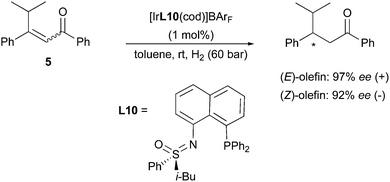 |
| | Scheme 12 | |
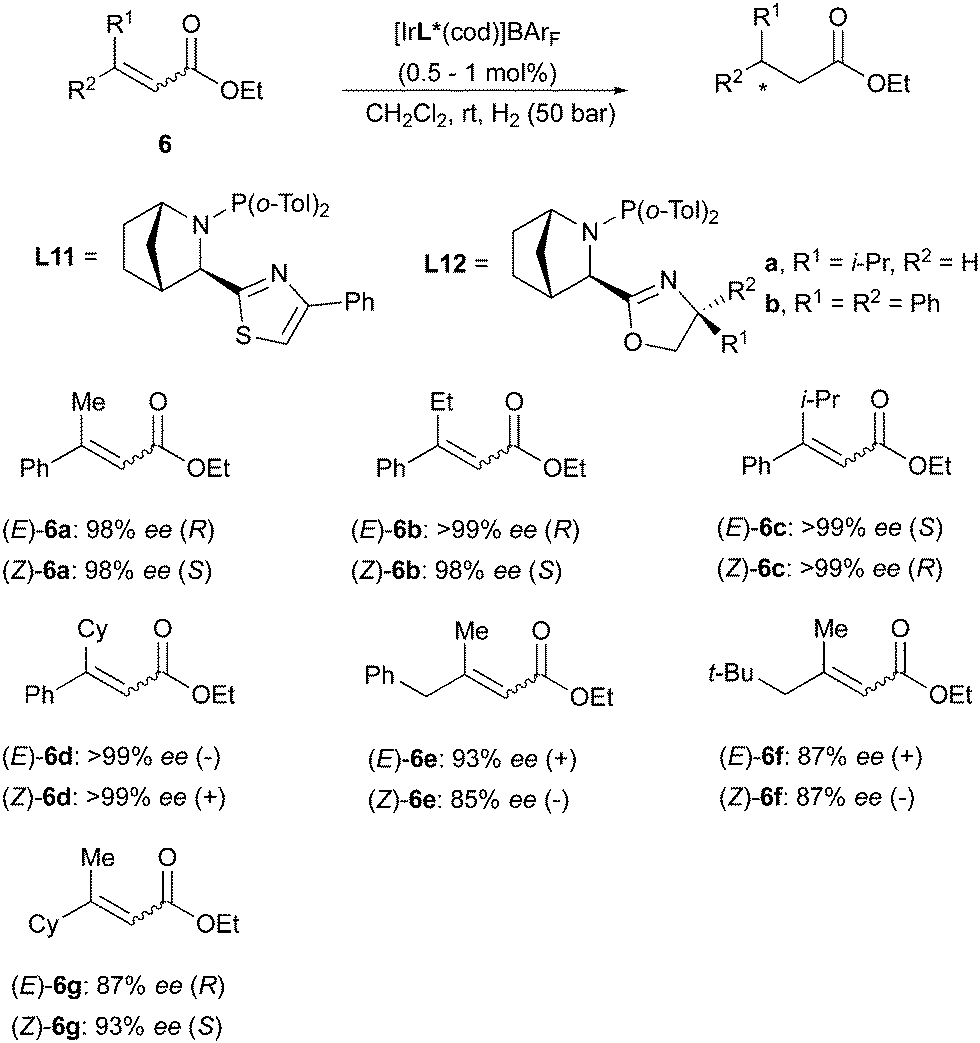
A similar divergent outcome was observed in the Ir-catalyzed asymmetric hydrogenation of unsaturated esters reported by the Andersson group in 2012. When using the bicyclic N,P ligands L11 and L12 for hydrogenation of a range of (E)- or (Z)-substrates, opposite enantiomers of the corresponding products were formed in high enantioselectivity (Scheme 13).19 The absolute configurations observed experimentally were found to be in agreement with those predicted by a stereoselectivity model developed by the group.20 This quadrant model is based on the steric hindrance generated by the chiral ligand around the iridium center, dictating the preferential mode of coordination of the olefinic substrates. According to the model, the favored coordination mode of (E)- and (Z)-olefins should generate hydrogenated products of opposite configuration, as confirmed by experimental results.20 In a later work, the Andersson group has reported the asymmetric hydrogenation of a broad variety of unsaturated sulfones. In this case as well, (E)- and (Z)-alkenes were reduced to chiral products of opposite absolute configuration by means of the same bicyclic N,P-ligand. However, the hydrogenation of the (Z)-configured sulfones turned out to be more problematic and lower conversions were generally observed, despite maintaining high stereoselectivity (Scheme 14).21
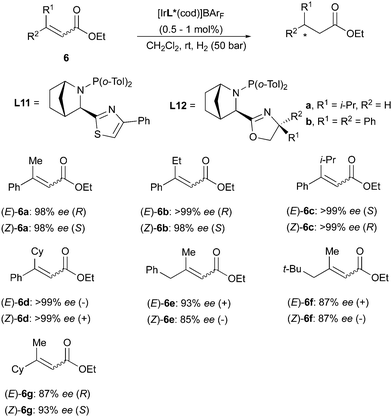 |
| | Scheme 13 | |
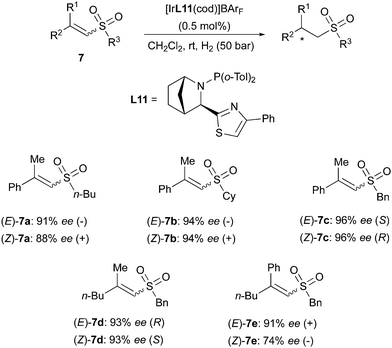 |
| | Scheme 14 | |
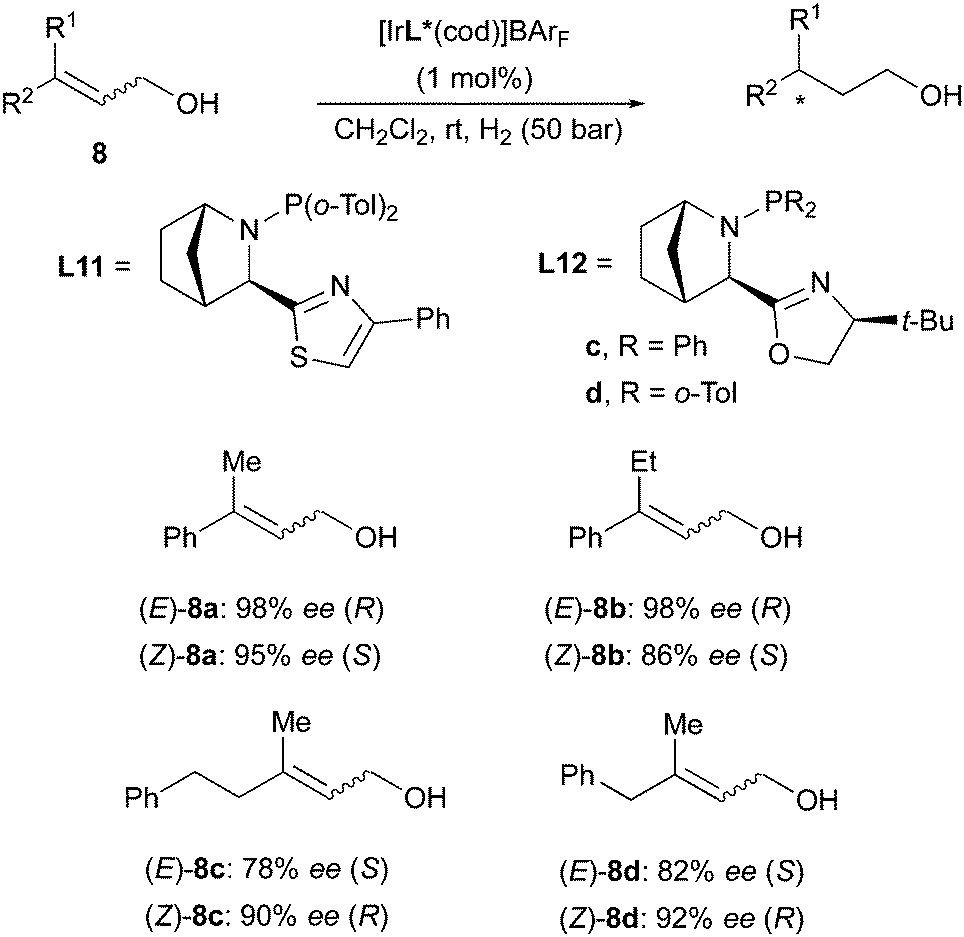
Another example of enantiodivergence was reported in the Ir-catalyzed hydrogenation of allylic alcohols. Employing the same class of Ir-catalysts used for esters and sulfone derivatives, the Andersson group reported the hydrogenation of several aryl- and alkyl-substituted allylic alcohols in both (E)- and (Z)-configuration. The different geometrical isomers of the olefins were found to consistently give opposite enantiomers of the products (Scheme 15). The stereochemical outcome was again consistent with the quadrant model prediction.22
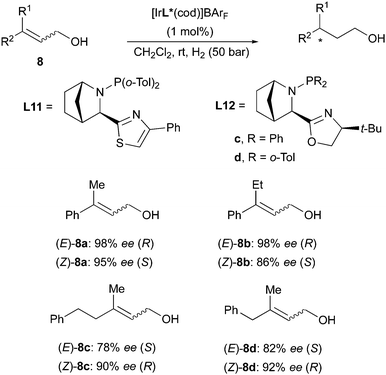 |
| | Scheme 15 | |
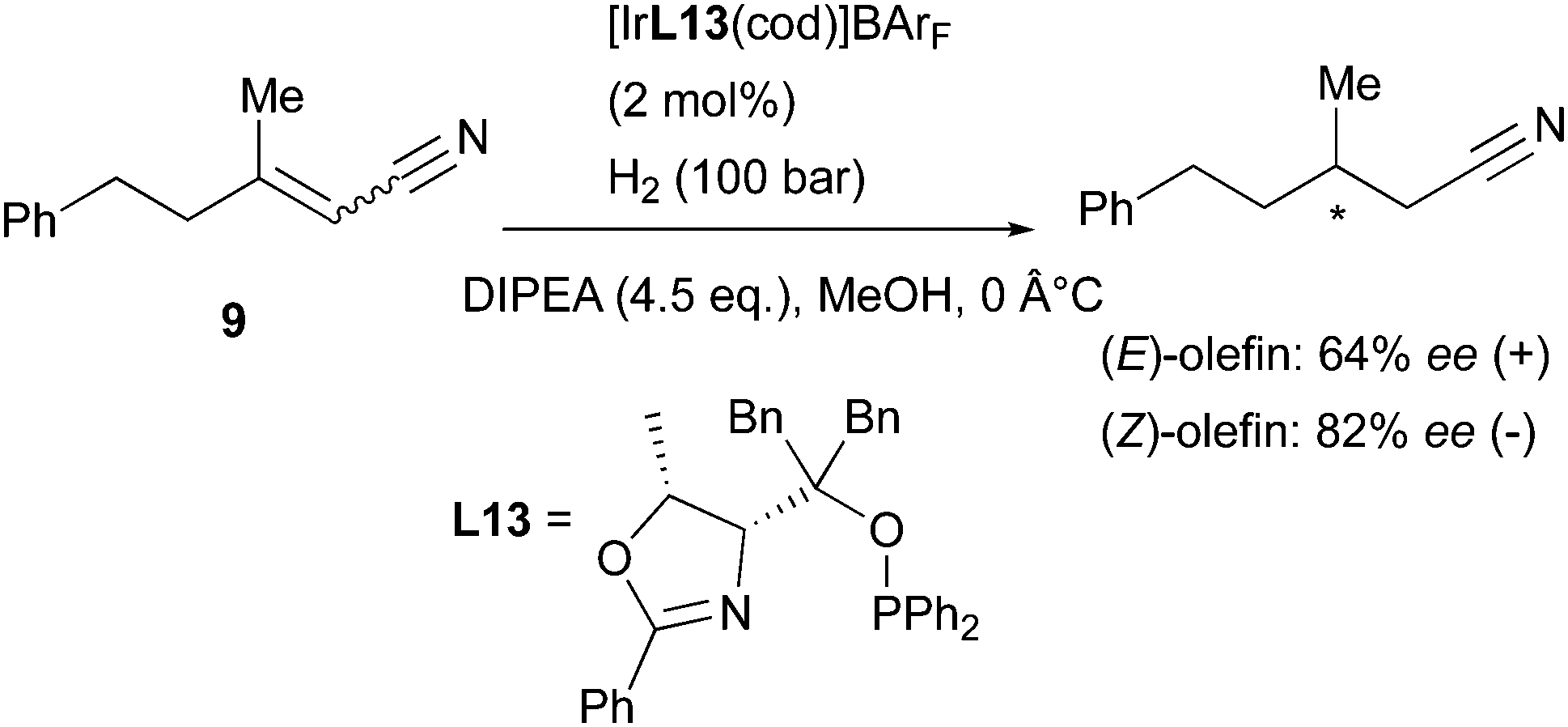
The Pfaltz group has studied the asymmetric hydrogenation of an interesting class of functionalized olefins: α,β-unsaturated nitriles. This functional group has been reported to directly coordinate to the Ir-catalyst, and the reduction was found to be successful only when operated under alkaline conditions. The reaction is likely to occur via a different mechanism compared to the Ir-N,P-catalyzed hydrogenation of other types of alkenes. However, when a pair of (E)- and (Z)-isomers were tested, a stereodivergent behavior was found also for unsaturated nitriles (Scheme 16). The hydrogenated product was obtained in much better selectivity for the (Z)-isomer in this case, but a clear preference toward formation of opposite enantiomers was observed.23
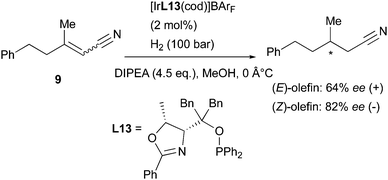 |
| | Scheme 16 | |
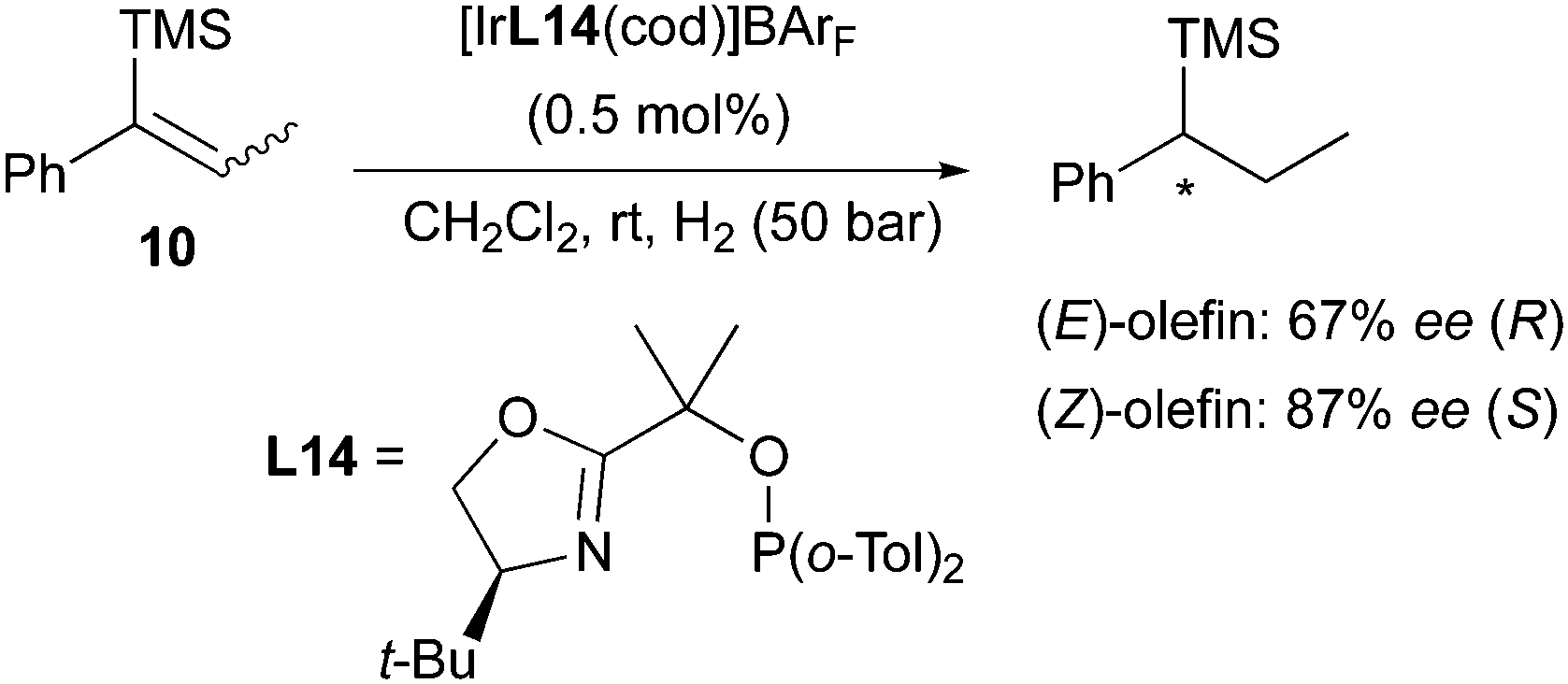
In a more recent work, the same enantiodivergent outcome was found when hydrogenating two isomeric vinylsilanes with a PHOX catalyst. In this case the (Z)-olefin was reduced in higher selectivity than the (E)-counterpart, and again producing opposite enantiomeric products (Scheme 17).24
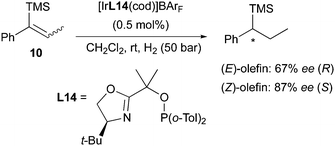 |
| | Scheme 17 | |
2.2 Rhodium
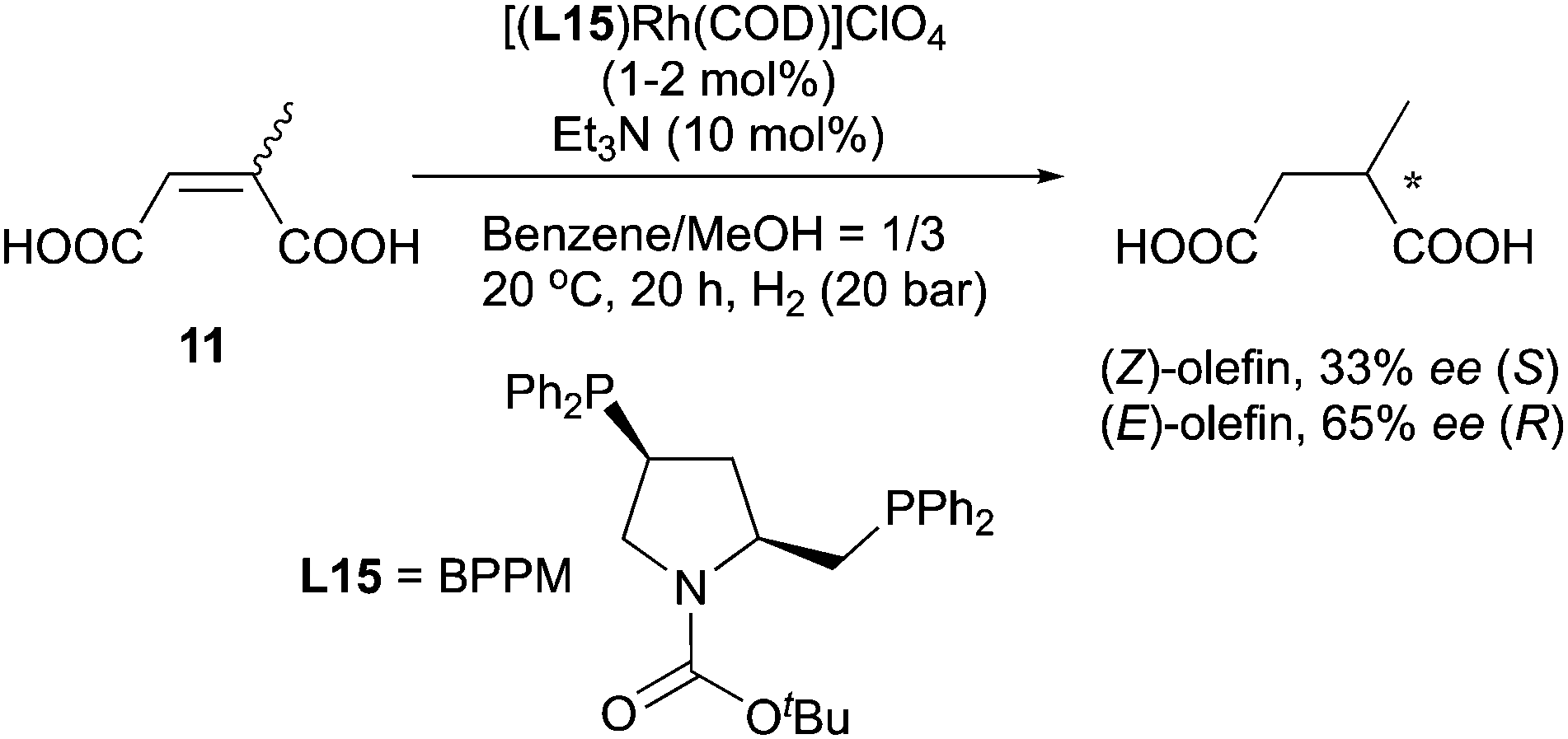
As early as 1980 the enantiodivergence phenomenon in Rh-catalyzed hydrogenation was reported by Ojima's group.25 By using [(BPPM)Rh(COD)]ClO4 as catalyst, an E/Z pair of N-(acylamino)cinnamic acids was found to give opposite enantiomeric products upon reduction (Scheme 18). It was suggested that the chiral recognition taking place in the olefin complexation step led to the observed enantiodivergence.
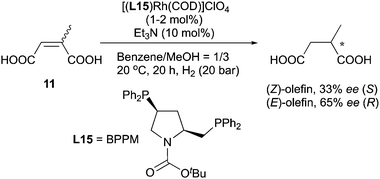 |
| | Scheme 18 | |
Later the same year, the same phenomenon was observed when Noyori and coworkers applied their newly developed BINAP ligand in the asymmetric hydrogenation of α-(acylamino)acrylic acids.26,27 When using (S)-BINAP as ligand, (R)-N-benzoylphenylalanine was obtained from the (Z)-substrate with excellent ee while the (E)-substrate was converted into the (S)-configuration product with a slightly lower ee (Scheme 19).
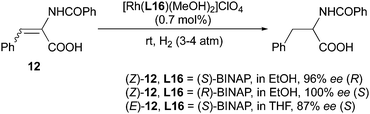 |
| | Scheme 19 | |
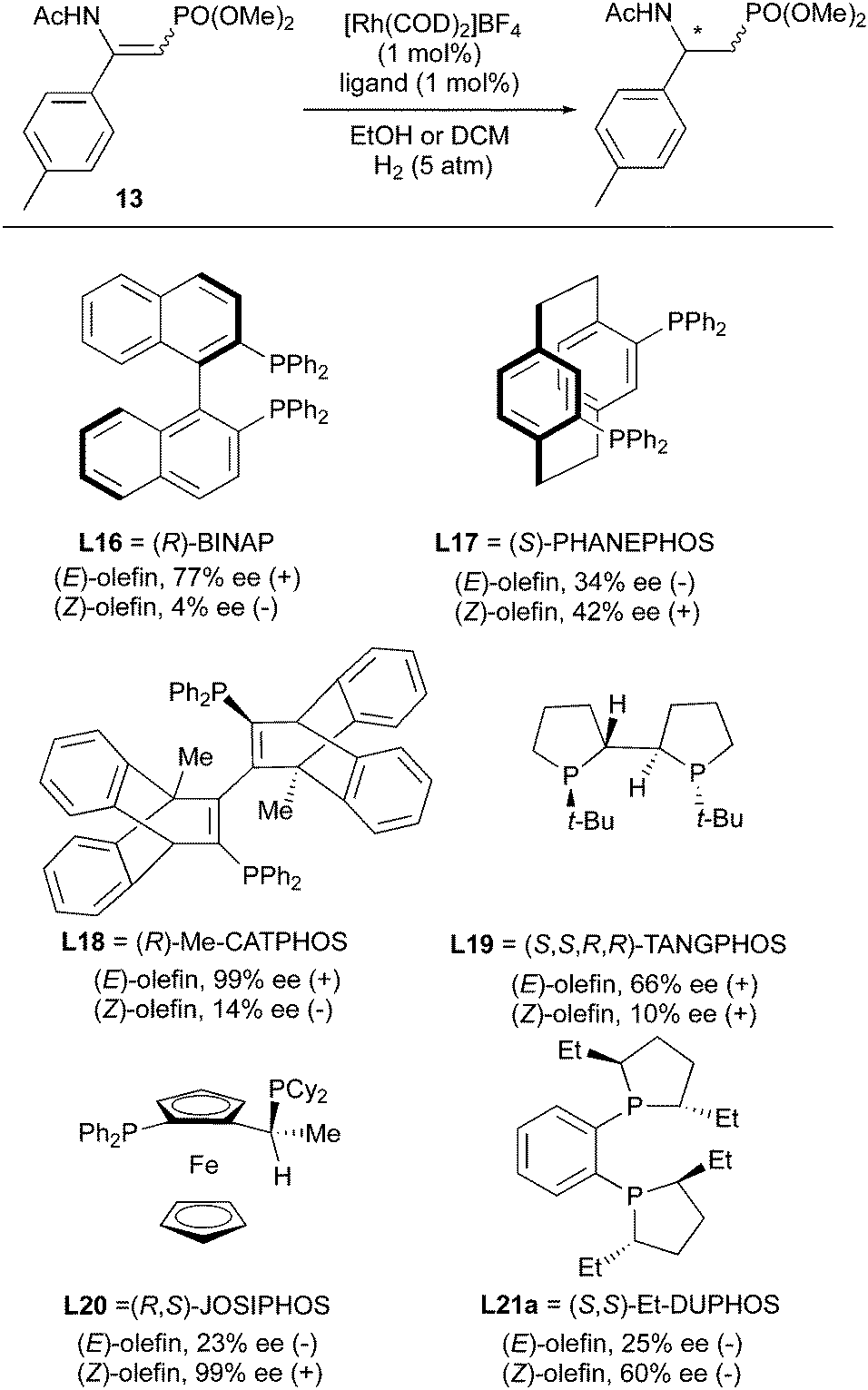
Additional ligands were evaluated in the studies of asymmetric hydrogenation of β-enamidophosphonates (Scheme 20).28,29 Doherty and Smyth developed the Me-CATPHOS ligand which was utilized to hydrogenate both (E)- and (Z)-β-enamidophosphonate with 99% and 17% ee, respectively. Other ligands like BINAP, JOSIPHOS, Et-DUPHOS, TANGPHOS, PHANEPHOS showed a “match and mismatch” effect as well, and gave higher ee for one isomer than the other. Enantiodivergence occurred in all the cases except for TANGPHOS, which gave 66% ee (+) and 10% ee (+). However, 10% is too low a value to provide a useful level of enantioconvergence.
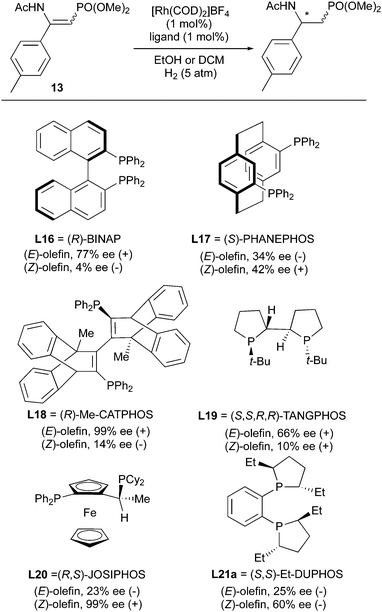 |
| | Scheme 20 | |
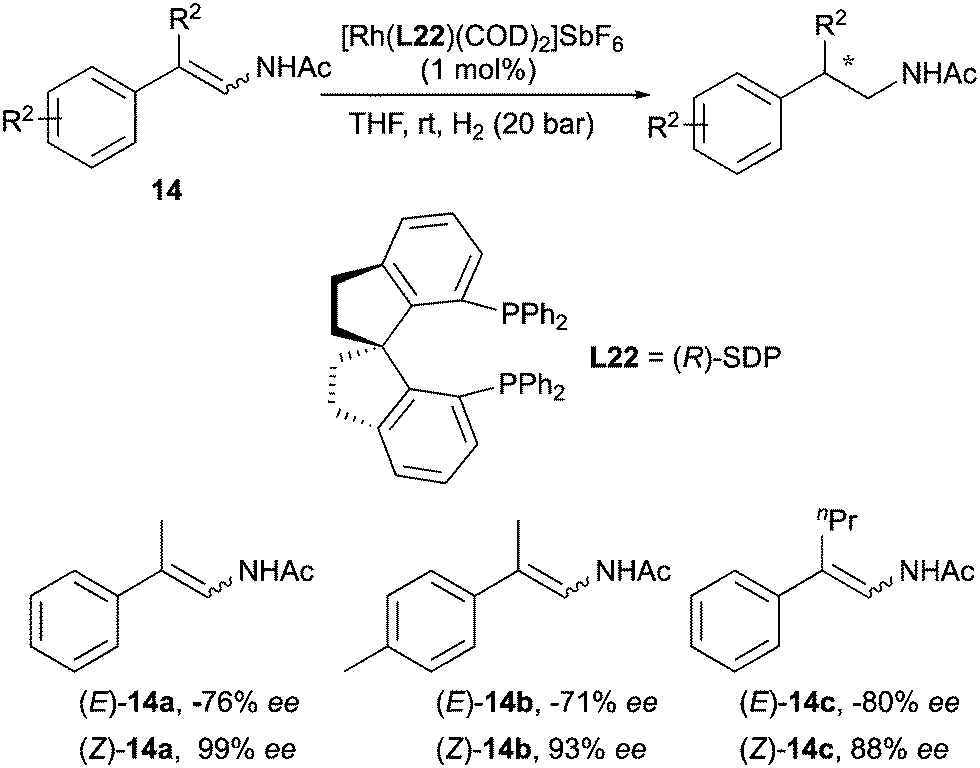
The Zhang group developed an asymmetric hydrogenation of simple (Z)-β-branched enamides by employing a bisphosphine ligand (R)-SDP (Scheme 21).30 When (E)-enamides were examined the desired products were obtained with comparatively lower enantioselectivities and opposite configuration.
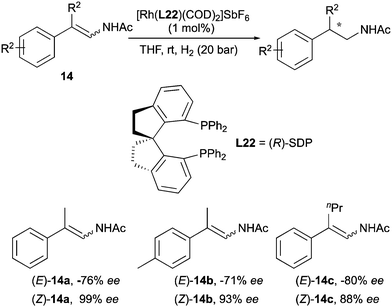 |
| | Scheme 21 | |
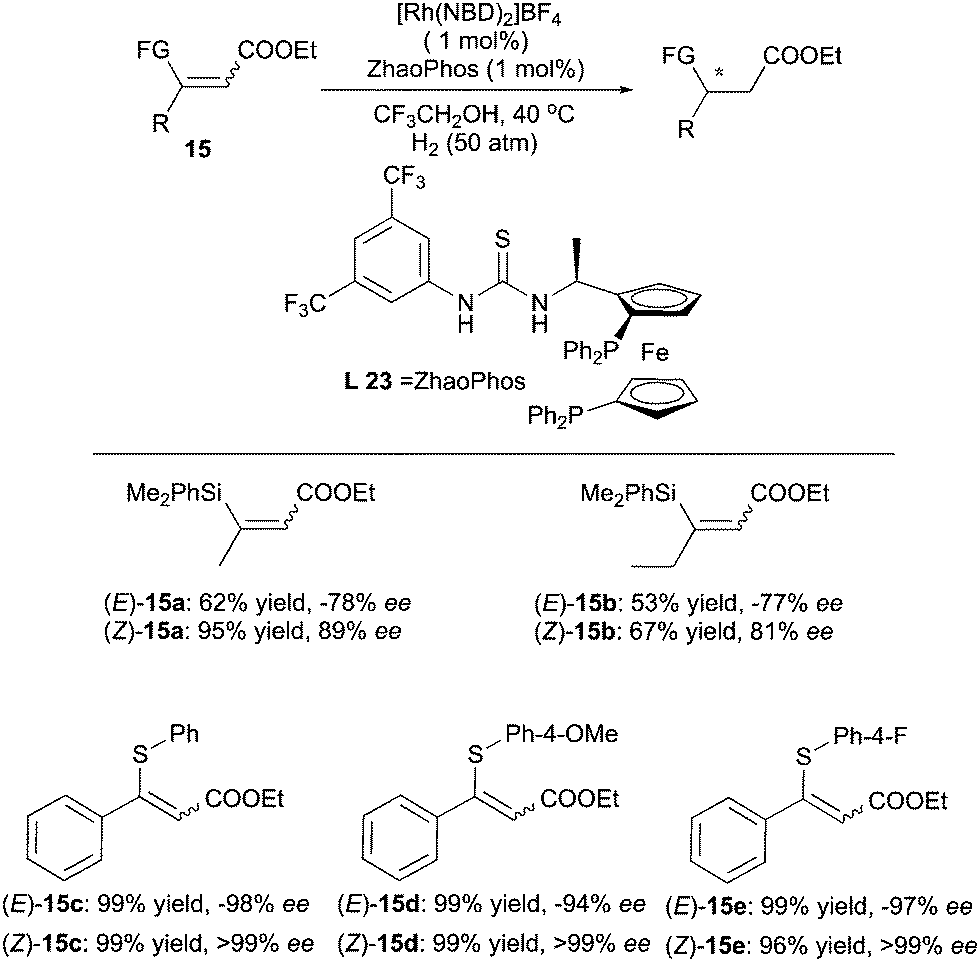
The ZhaoPhos ligand developed by the Zhang's group was also found to give enantiodivergent products in the hydrogenations of functionalized α,β-unsaturated esters (Scheme 22). When β-silyl-substituted esters were employed, the use of ZhaoPhos led to opposite configuration of the products in the hydrogenation of the (E)- and (Z)-olefins.31 The ee values deriving from (Z)-olefins were slightly higher than those obtained for (E)-olefins. Enantiodivergence was again observed when the silyl group was replaced by a thio group.32 In this case, both (E) and (Z)-substrates were hydrogenated to give products of opposite configuration with excellent ee.
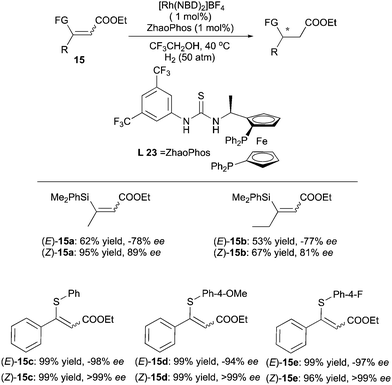 |
| | Scheme 22 | |
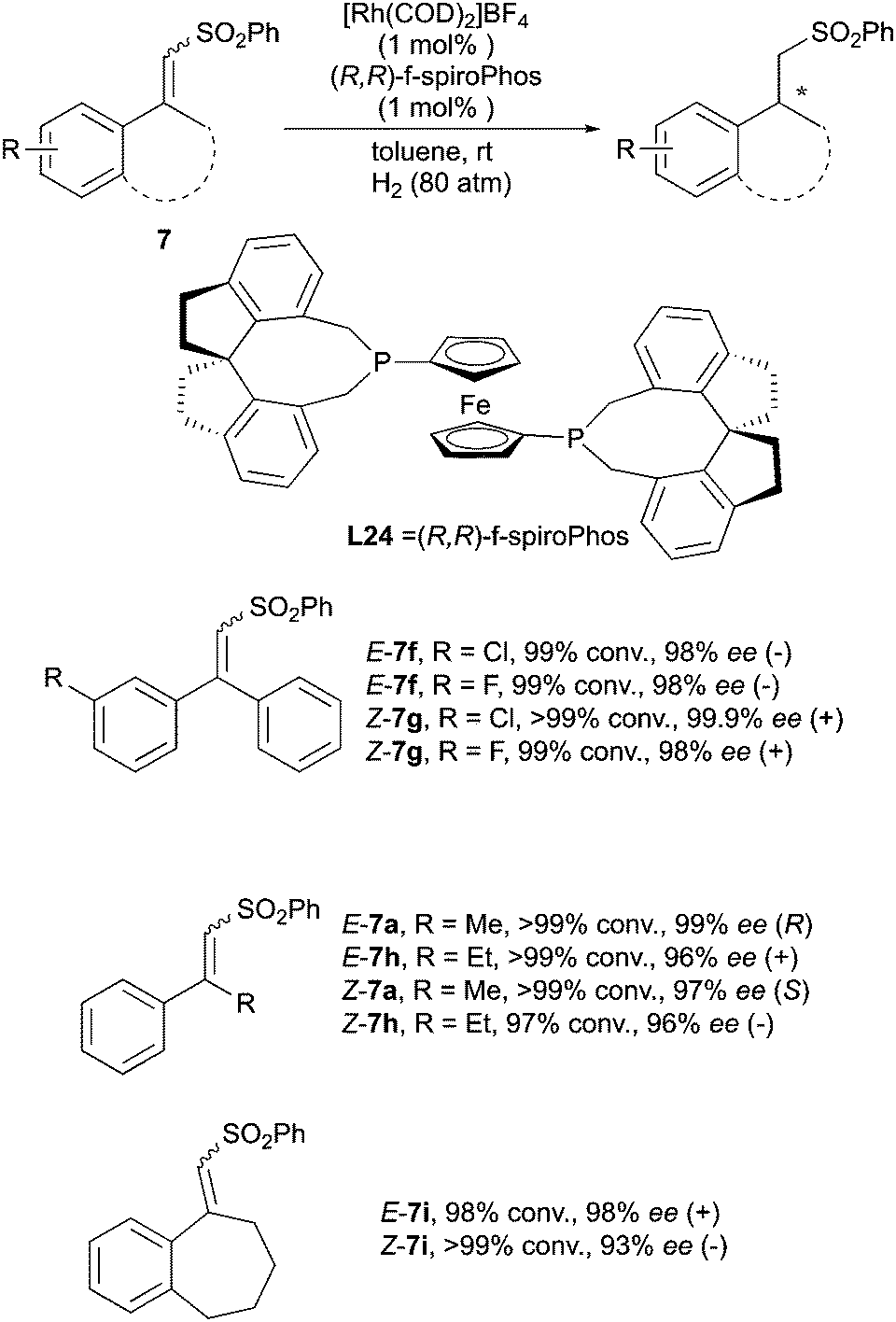
Recently the Hou group reported the asymmetric hydrogenation of unsaturated sulfones with excellent enantioselectivity by using a Rh–(R,R)-f-spiroPhos complex (Scheme 23). The Rh catalysis showed clear enantiodivergence in hydrogenating this class of compounds.33
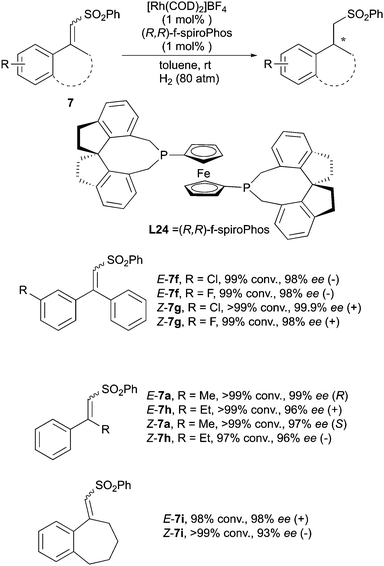 |
| | Scheme 23 | |
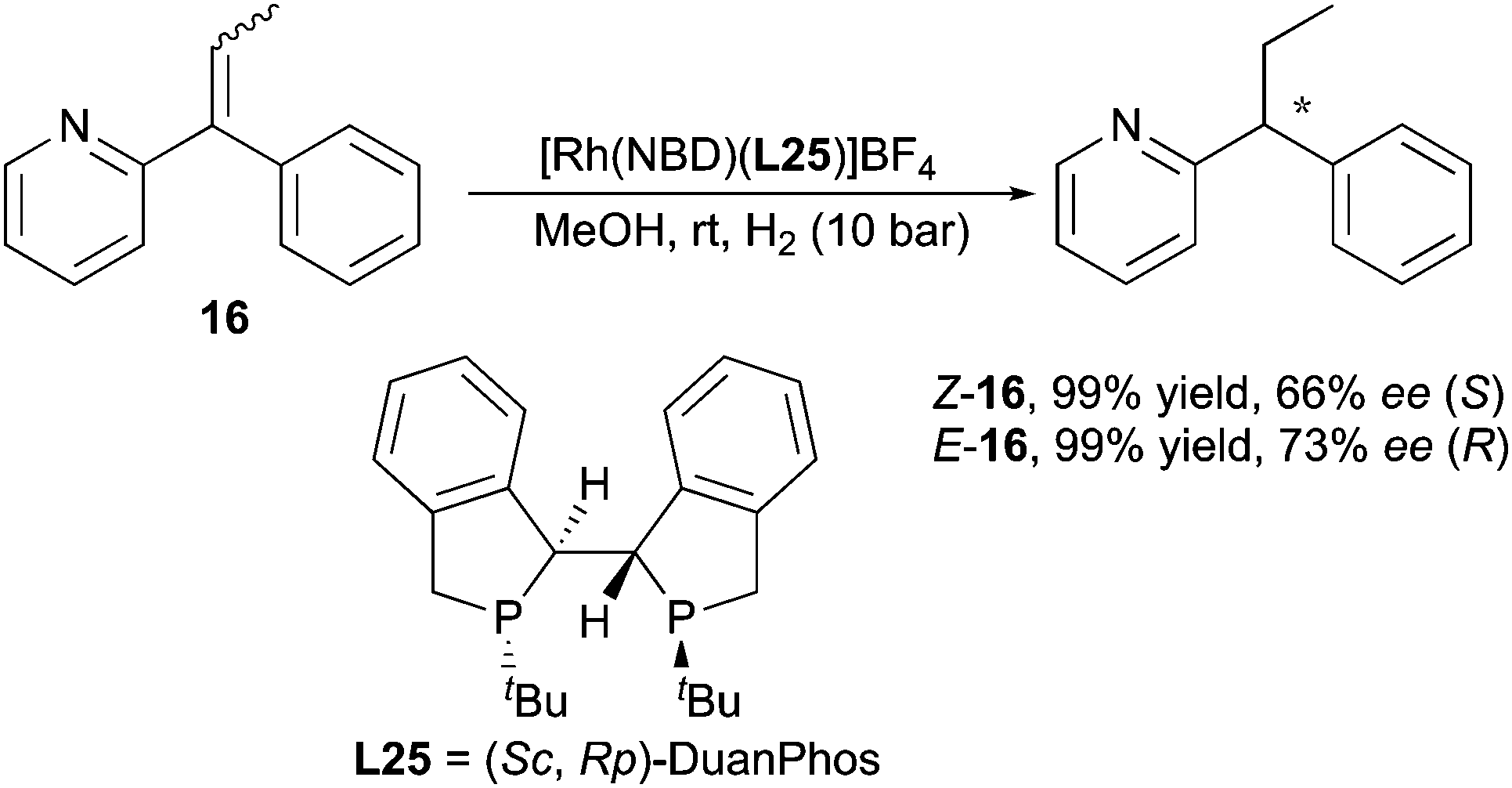
In the hydrogenation of pyridine-containing 1,1-diarylalkenes reported by Zhang, it was claimed that chelation played a critical role, since the replacement of the 2-pyridine group with other aryl groups gave no conversion. However, this chelation of the substrate did not influence the enantiodivergent outcome when geometrically pure pyridine-containing olefins were evaluated. 66% and 73% ee were obtained from (Z)- and (E)-olefins, yielding products with opposite optical rotation (Scheme 24).34
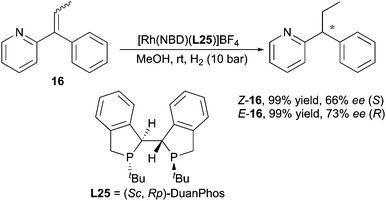 |
| | Scheme 24 | |
2.3 Ruthenium
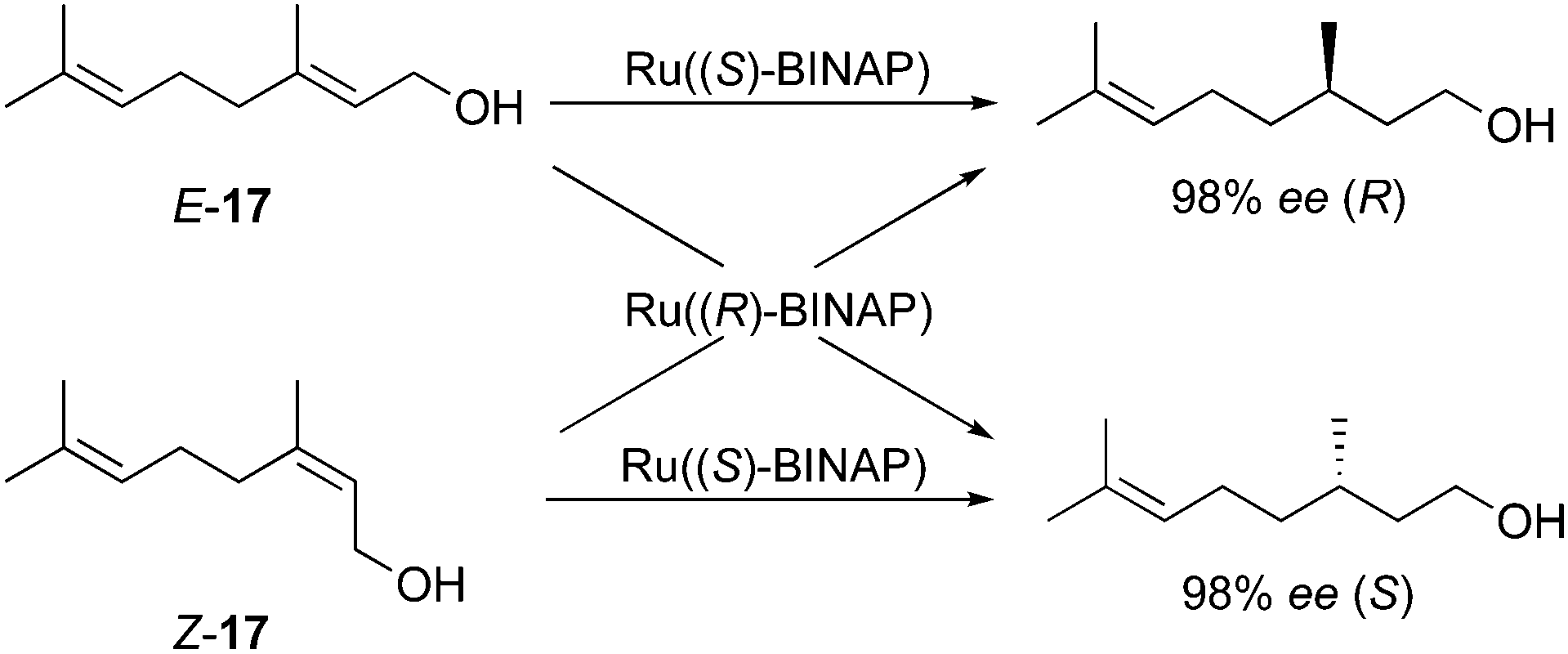
This shorter section covers the renowned ruthenium–BINAP catalyzed hydrogenation of allylic alcohols. In 1987, Noyori applied the BINAP ligand in a ruthenium catalytic system for the hydrogenation of allylic alcohols providing one of the first examples describing the enantiodivergence and its significance in synthesis.35 The hydrogenation of stereochemically pure geraniol (E-17) and nerol (Z-17) proceeded smoothly to give opposite enantiomeric alcohols with excellent enantioselectivities, leaving the unfunctionalized alkene moieties untouched (Scheme 25).
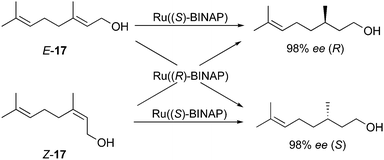 |
| | Scheme 25 | |
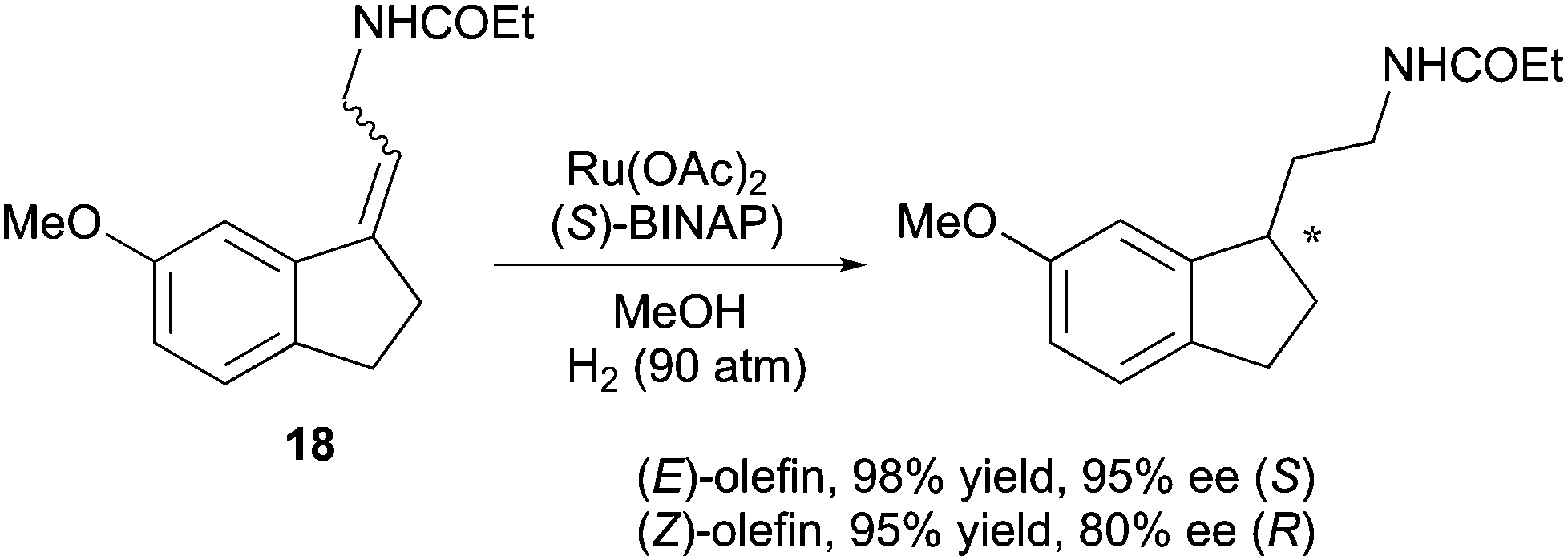
By using the Ru/BINAP system, Uchikawa and co-workers obtained enantiodivergence when hydrogenating an exocyclic double bond on an indane scaffold. The same divergent reactivity was detected for both geometrical isomers, while the (E)-configured substrate provided a higher ee than the (Z)-substrate (Scheme 26).36
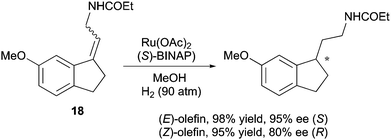 |
| | Scheme 26 | |
3. Enantioconvergent hydrogenations
This second main section covers the less frequent cases of enantioconvergent hydrogenations of E/Z-isomeric olefins, which has been successfully demonstrated by a limited number of catalytic systems applied to specific substrates. The subsections are again divided by the metal employed in the reactions.
3.1 Iridium
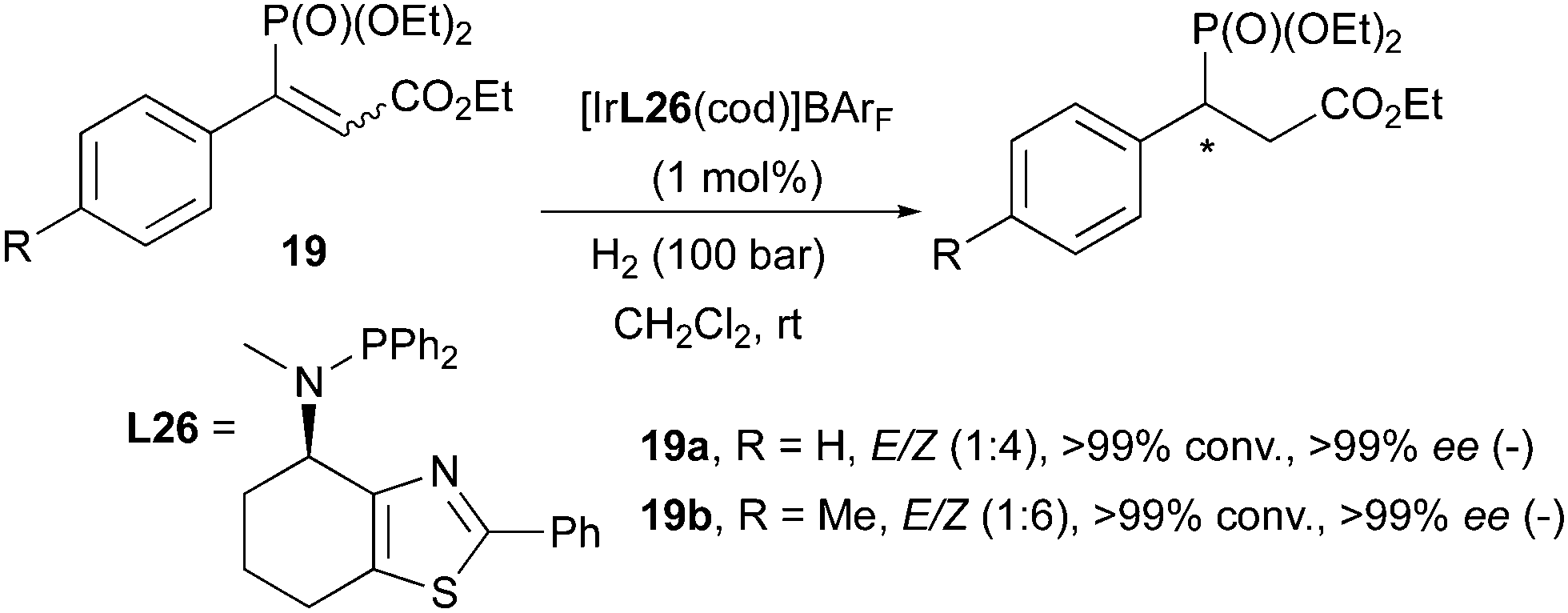
There are some reports in which iridium-catalyzed enantioconvergent hydrogenation of olefins has been observed. In these examples, Ir-N,P catalysts were used in the hydrogenation of variously functionalized olefins (nitroalkenes, maleic acid diesters, vinylphosphonates) and generated a different stereochemical outcome compared to the majority of the cases.
In 2009 the Andersson group reported the Ir-catalyzed enantioselective hydrogenation of prochiral organophosphorus compounds, such as 1-arylvinylphosphonates and carboxyethylvinylphosphonates. Among the latter class of substrates, an interesting phenomenon was observed: (E)- and (Z)-configured olefins containing the phosphonate substituent led to the same enantiomer upon hydrogenation, despite differences in reactivity (the reaction was found to be much slower for the (E)-isomer). In virtue of this stereoconvergent behavior, it was possible to directly employ E/Z mixtures of the substrates in the hydrogenation and full conversions with excellent enantioselectivities were achieved (>99% ee, Scheme 27). Enantioconvergence in asymmetric hydrogenations had not been observed for Ir-catalysts before, and it was suggested that substrate chelation might be a directing factor in this case.37
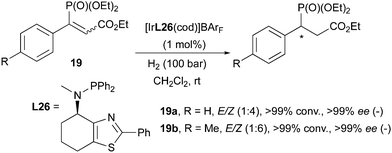 |
| | Scheme 27 | |
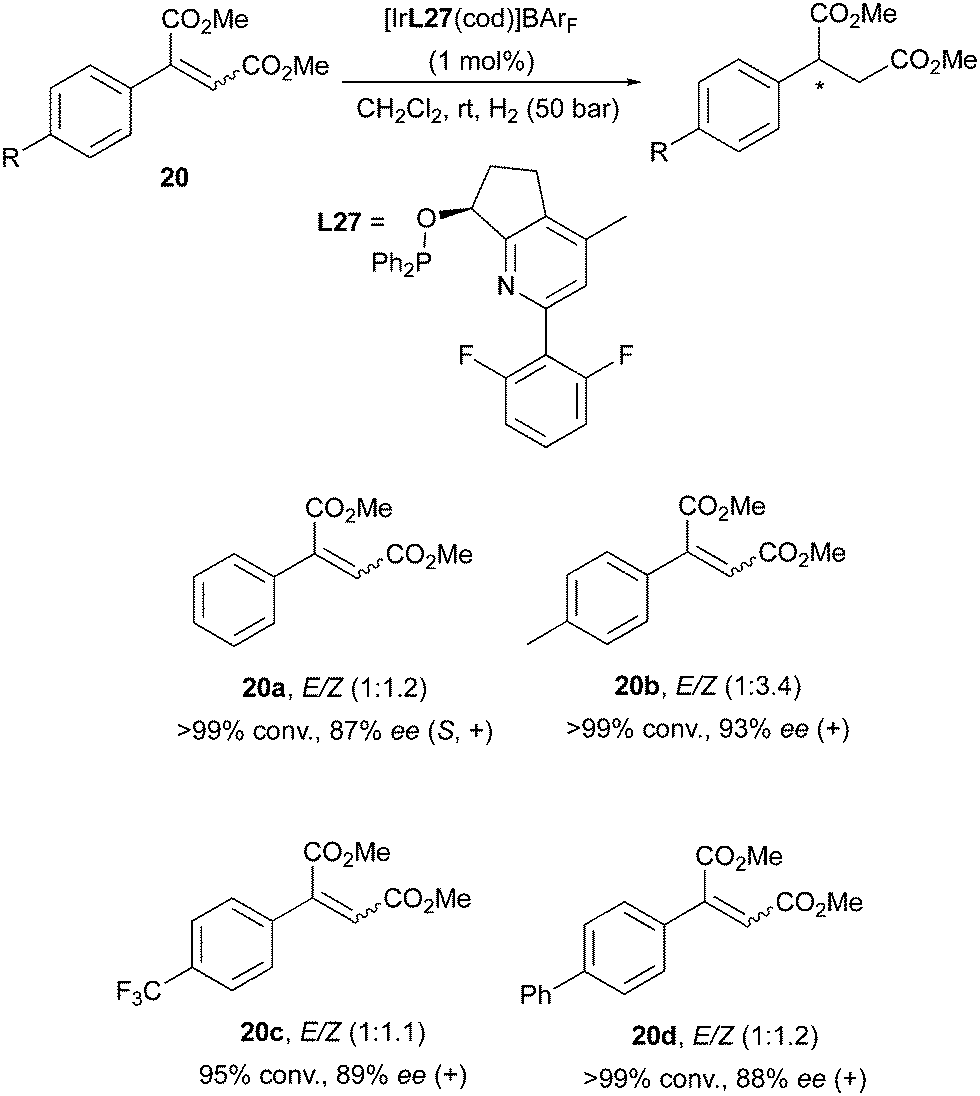
While this surprising outcome is a rare event for Ir-catalysis, it has not remained an isolated case. A stereoconvergent Ir-catalyzed hydrogenation was also reported by the Pfaltz group in the investigation of maleic acid diesters as substrates. It was found that the fluorinated, pyridine-based N,P-ligand could hydrogenate E/Z mixtures of the diester substrate, leading to high enantioselectivities (Scheme 28).38
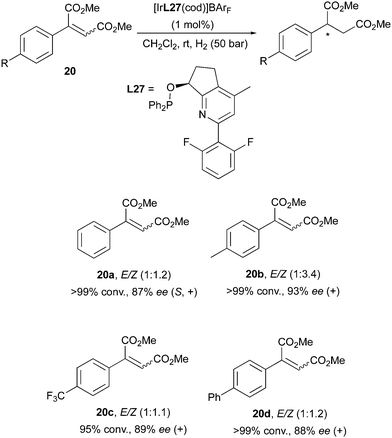 |
| | Scheme 28 | |
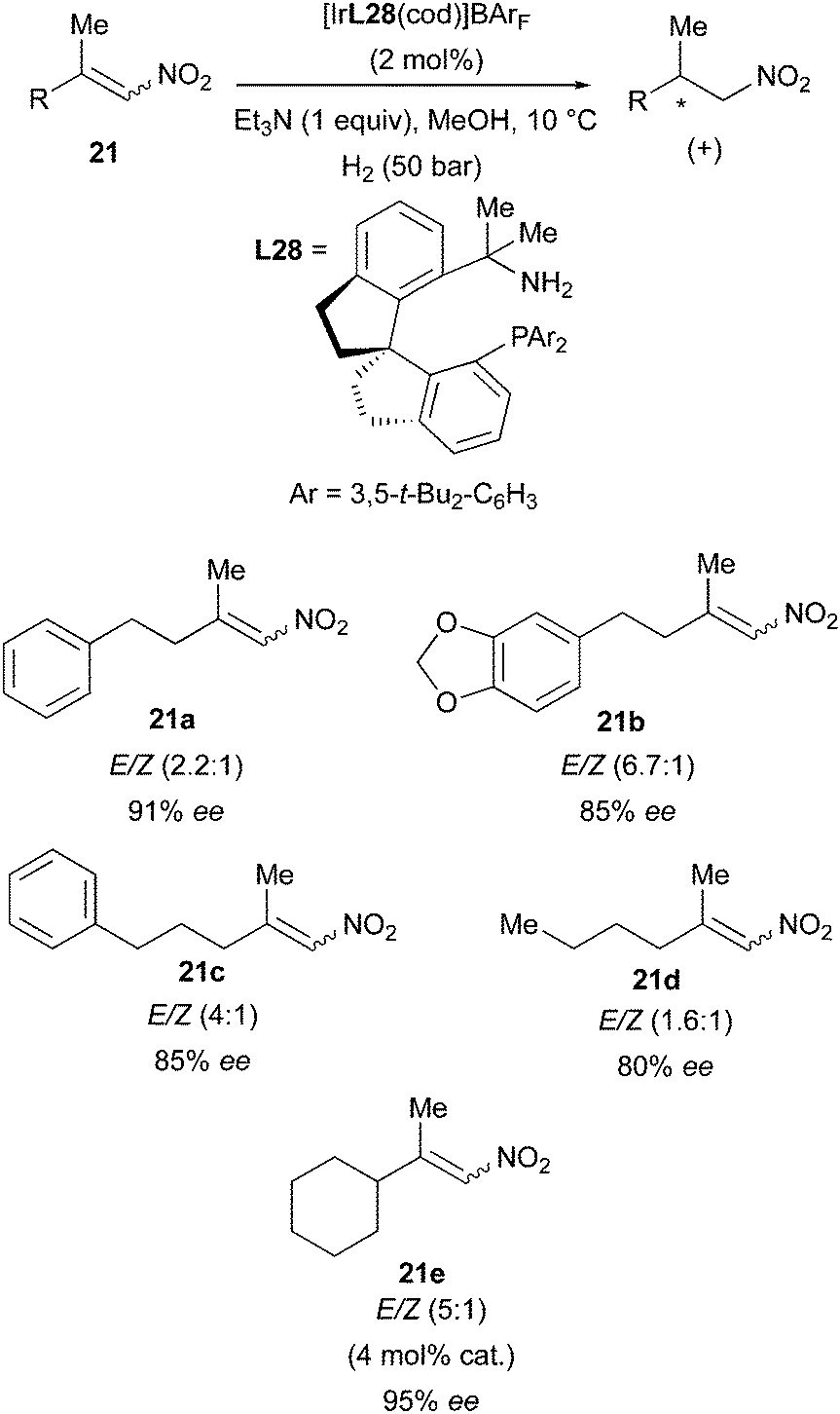
Another example in which Ir-catalyzed hydrogenation produced an enantioconvergent outcome was reported by the Zhou group in 2016, when they employed SpiroBAP ligands (N,P) in the successful asymmetric hydrogenation of β,β-dialkyl-nitroalkenes. Several E/Z mixtures of these substrates could be handled by the Ir-catalysts, which afforded the saturated products in up to 95% ee (Scheme 29).39
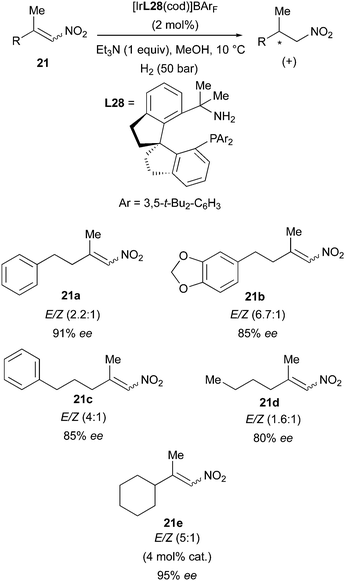 |
| | Scheme 29 | |
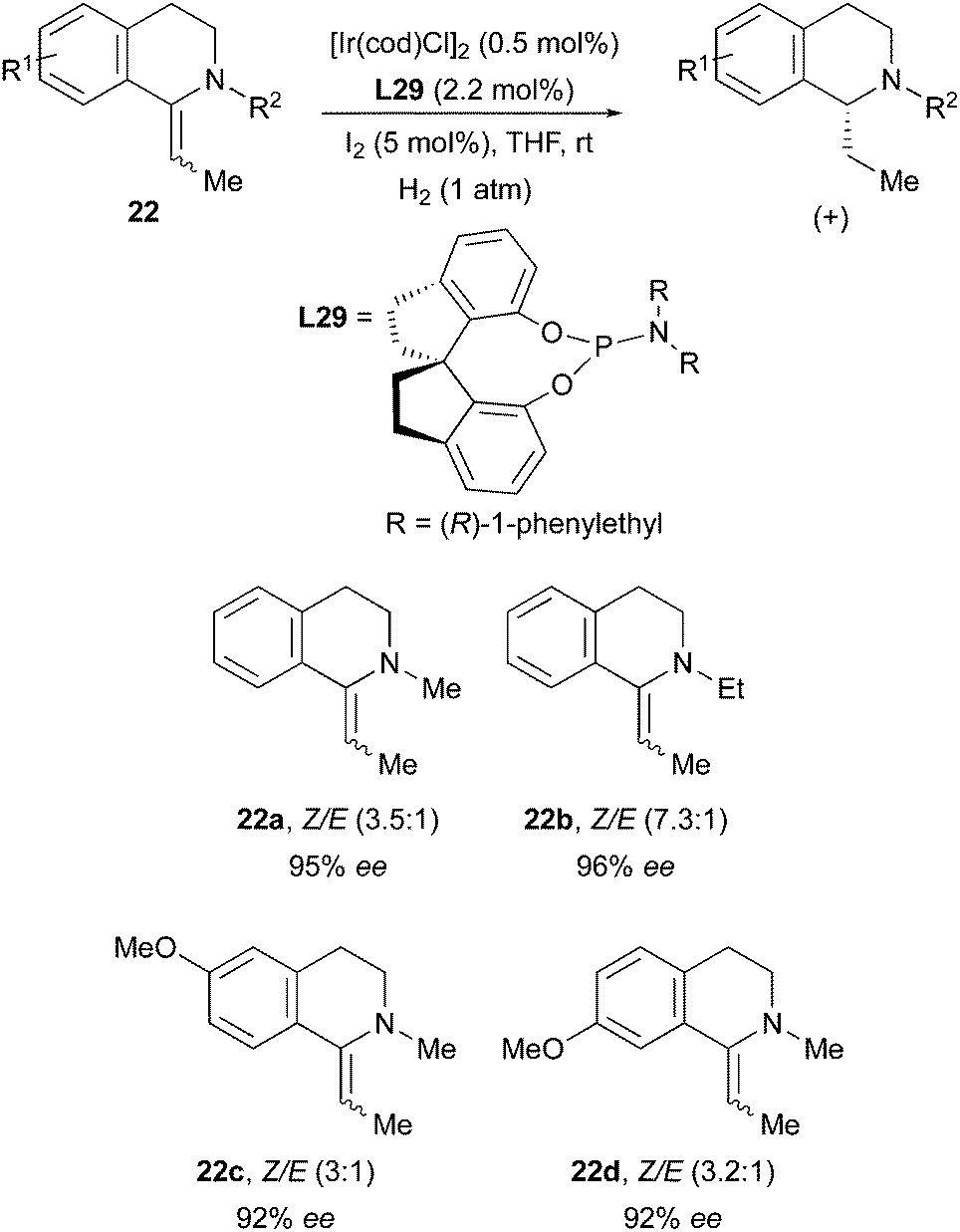
The Zhou group had previously observed another enantioconvergent Ir-catalyzed hydrogenation, when employing a monodentate phosphoroamidite ligand on tetrahydroisoquinolines. Also in this case E/Z mixtures of the exocyclic olefins could be reduced in high enantioselectivity (Scheme 30).40
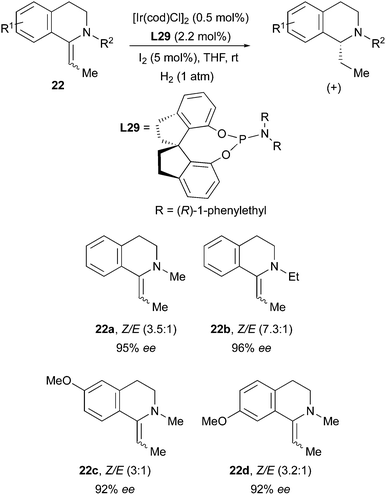 |
| | Scheme 30 | |
3.2 Rhodium
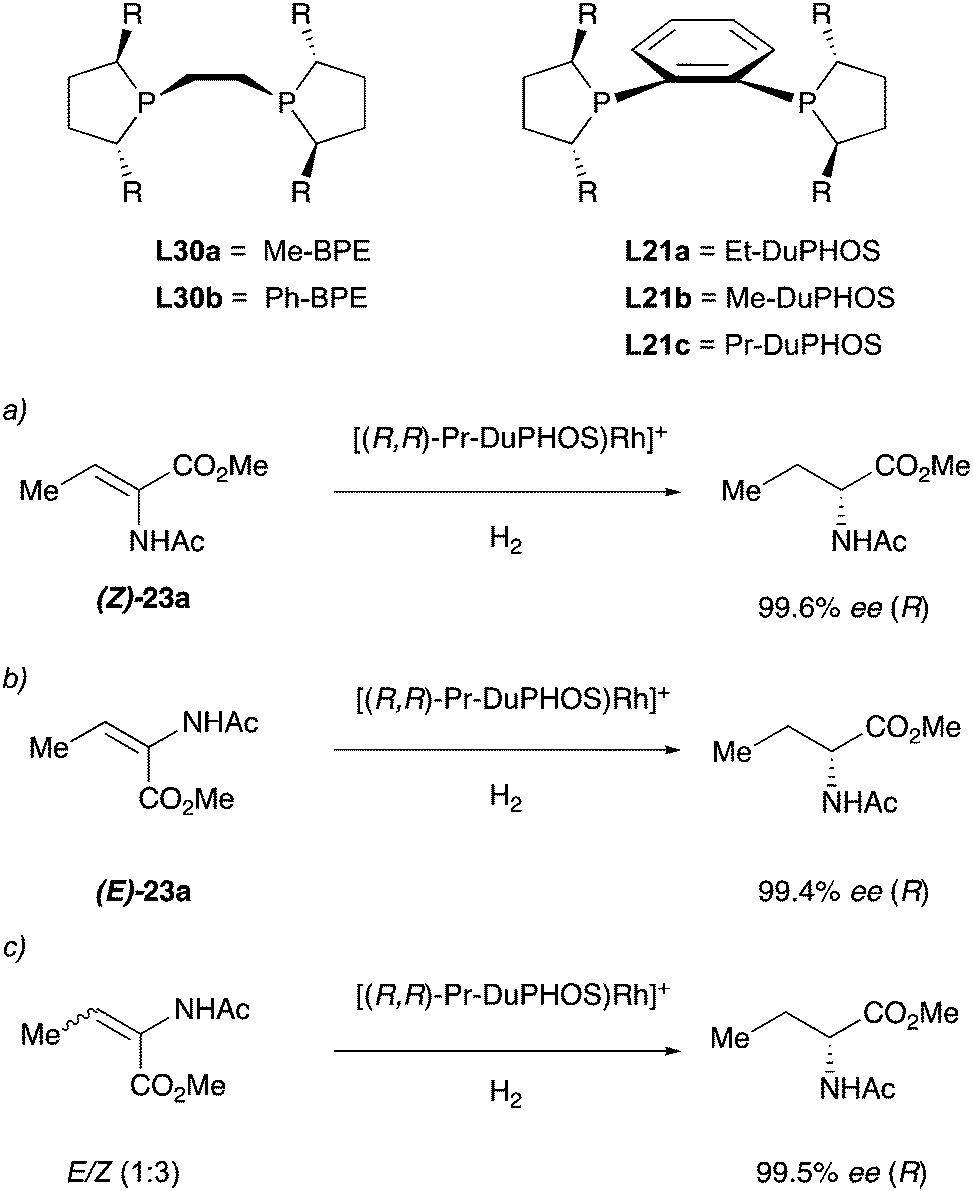
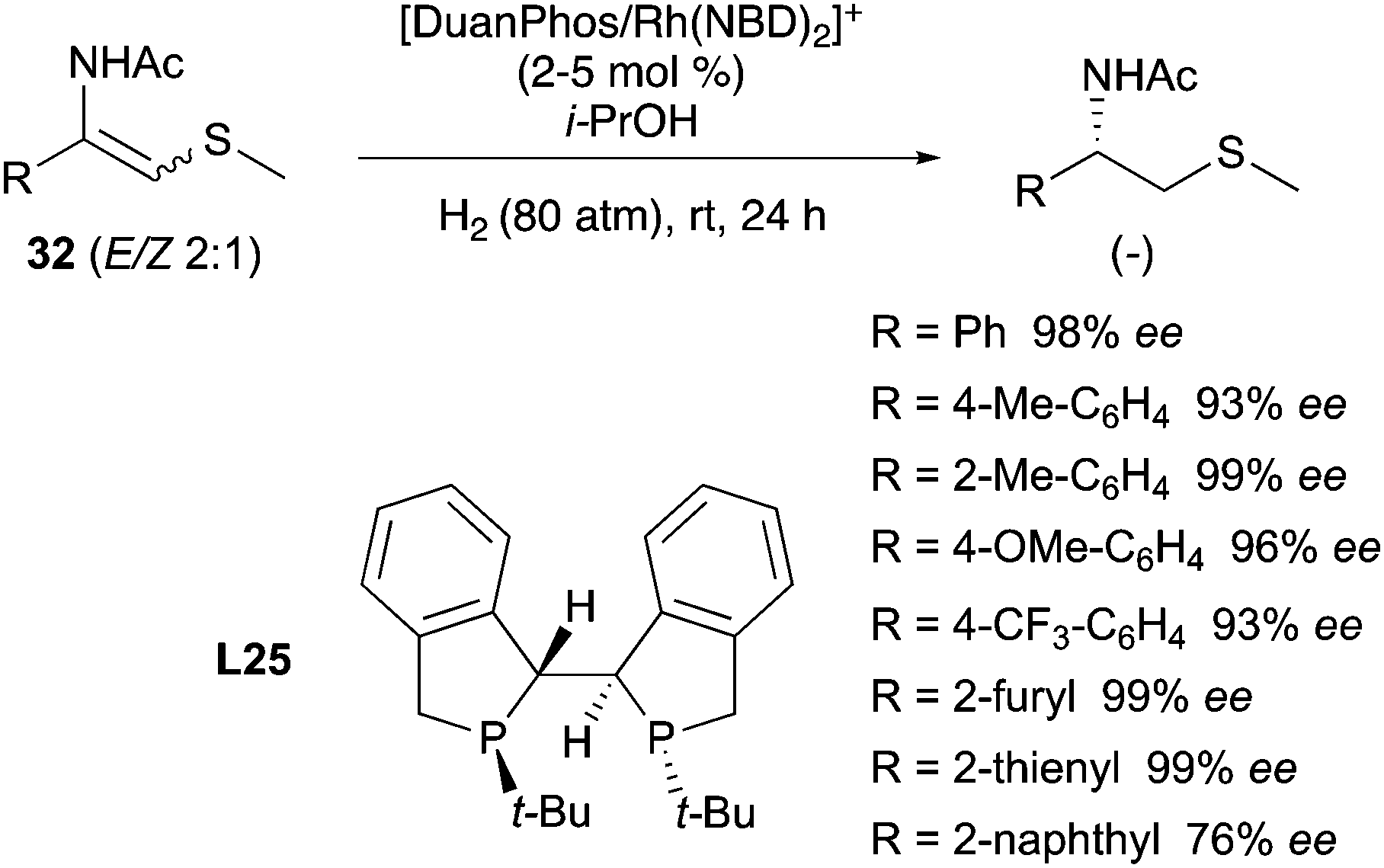
In 1993 the Burk group developed a new class of ligands with a C2-symmetric bis(phospholane) backbone for the asymmetric hydrogenation of enamides (Scheme 31). The Rh catalysts obtained with these ligands were tested in the hydrogenation of different enamides bearing the acyl and cbz protecting groups. Notably, the authors found that using the DuPHOS–Rh catalysts, both (Z)- and (E)-isomeric enamides could be hydrogenated in high enantiomeric excess to products having the same absolute configuration.41 For example, hydrogenation of methyl (Z)-α-acetamidobutenoate 23a with (R,R)-PrDuPHOS–Rh afforded methyl (R)-2-acetamidobutyrate in more than 99% ee. Hydrogenation of the corresponding (E)-23a isomer with the same catalyst again afforded the (R)-product in 99% ee (Scheme 31, entries a and b). When the reaction was performed on the mixture of the two isomeric olefins it again resulted in 99% ee (Scheme 31, entry c). This is, to the best of our knowledge, the first example of enantioconvergence for (E)- and (Z)-olefins in asymmetric hydrogenation.42
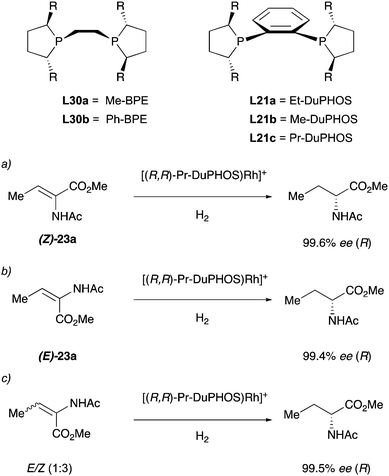 |
| | Scheme 31 | |
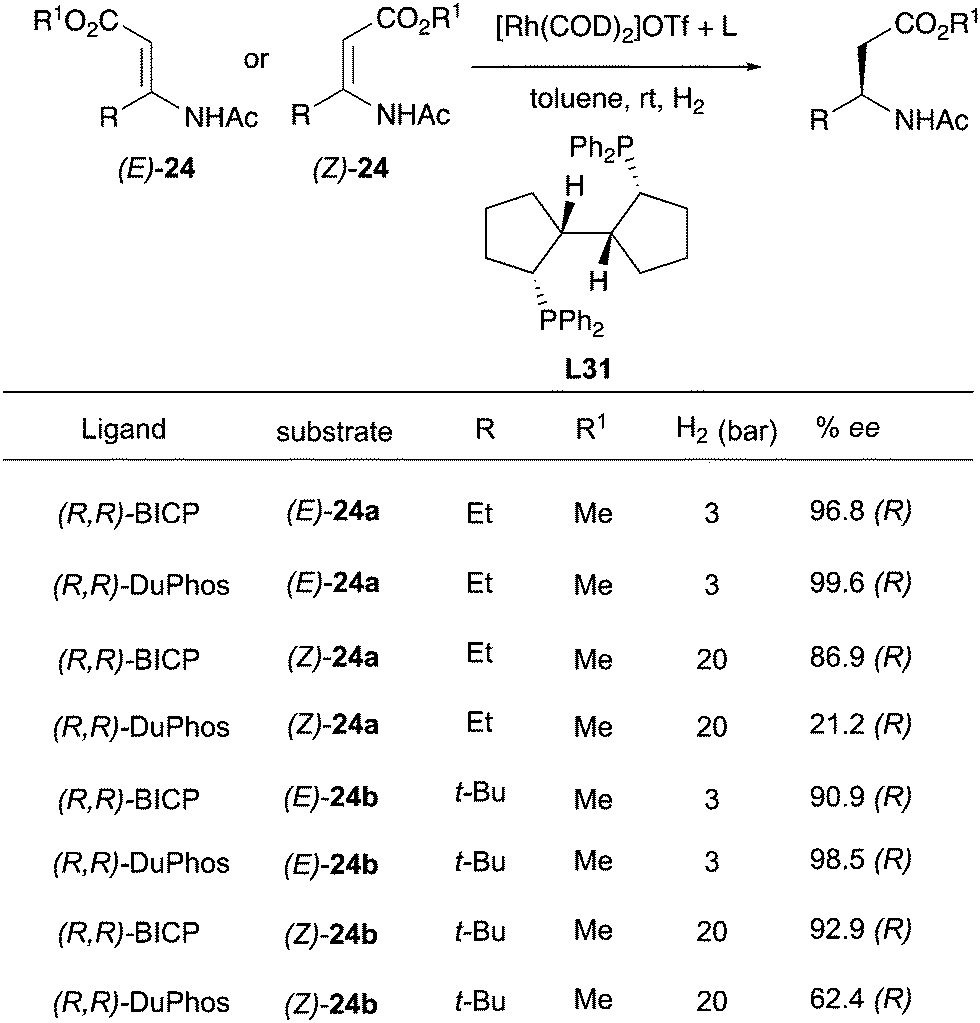
In the following years, the Zhang group developed an asymmetric hydrogenation of β-(acylamino)-acrylates using the DuPHOS ligand and a new ligand L31, showing a convergent enantioselective outcome even if the less reactive (Z)-isomer provided lower ee (Scheme 32).43
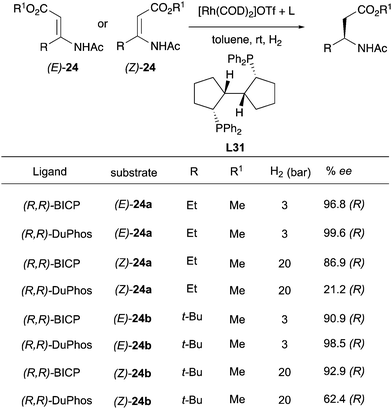 |
| | Scheme 32 | |
In 1998 enol ester substrates were hydrogenated using a cationic Et–DuPHOS–Rh catalyst showing high levels of enantioselectivity (93–99% ee) (Scheme 33). Substrates bearing β-substituents could be employed as E/Z isomeric mixtures with no negative effect on the selectivity. Also in this case, deuterium experiments indicated that no significant E/Z isomerization of the substrates occurred during the course of the reactions.44
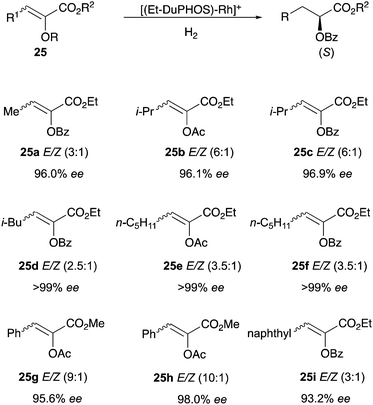 |
| | Scheme 33 | |
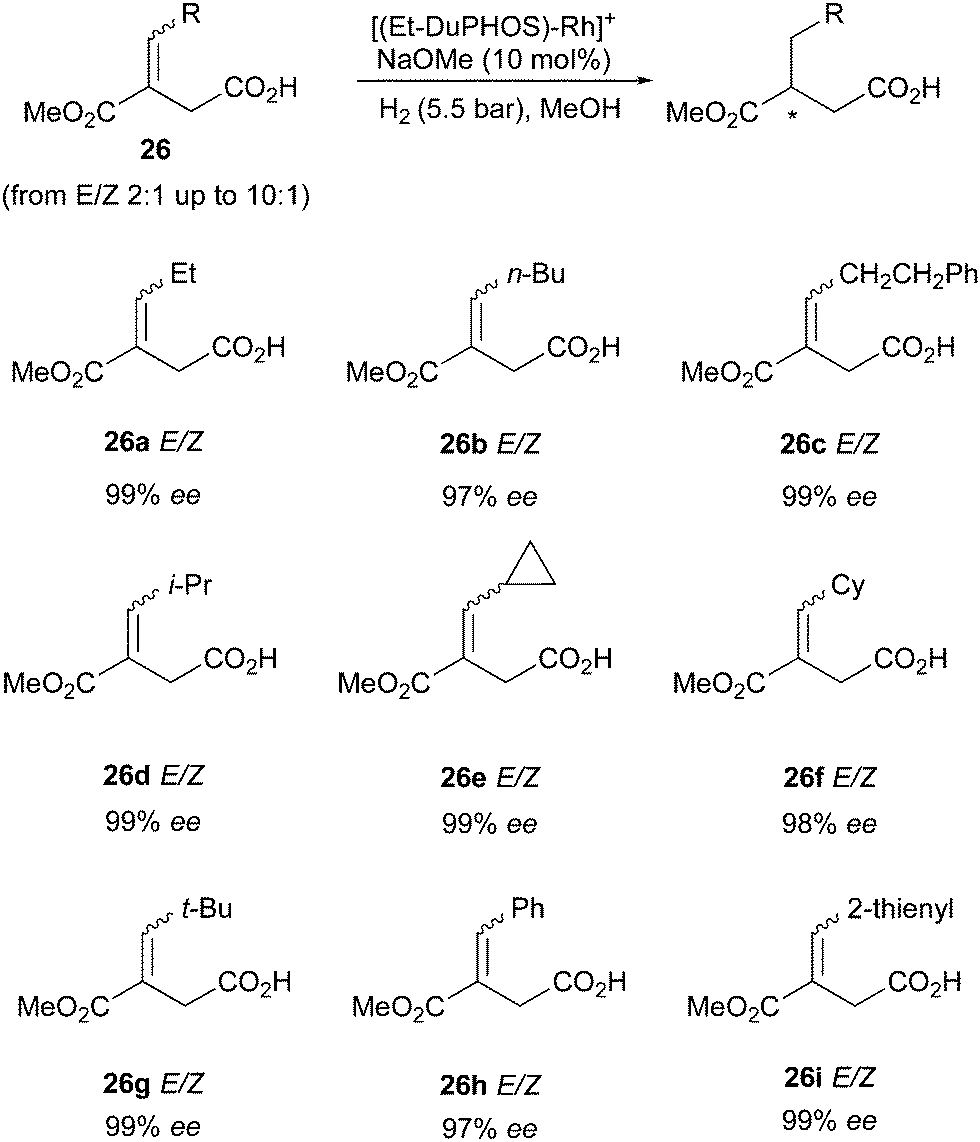
Another class of compounds that DuPHOS ligands were able to hydrogenate efficiently as mixtures of (E)- and (Z)-isomers were itaconates (Scheme 34). The synthesis of compound 26 is not trivial in terms of controlling the E/Z-diastereoselectivity, but since the mixture of the two isomers could be hydrogenated in higher than 97% ee, this problem was alleviated.45
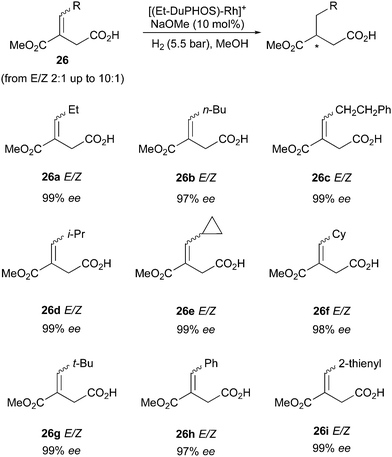 |
| | Scheme 34 | |
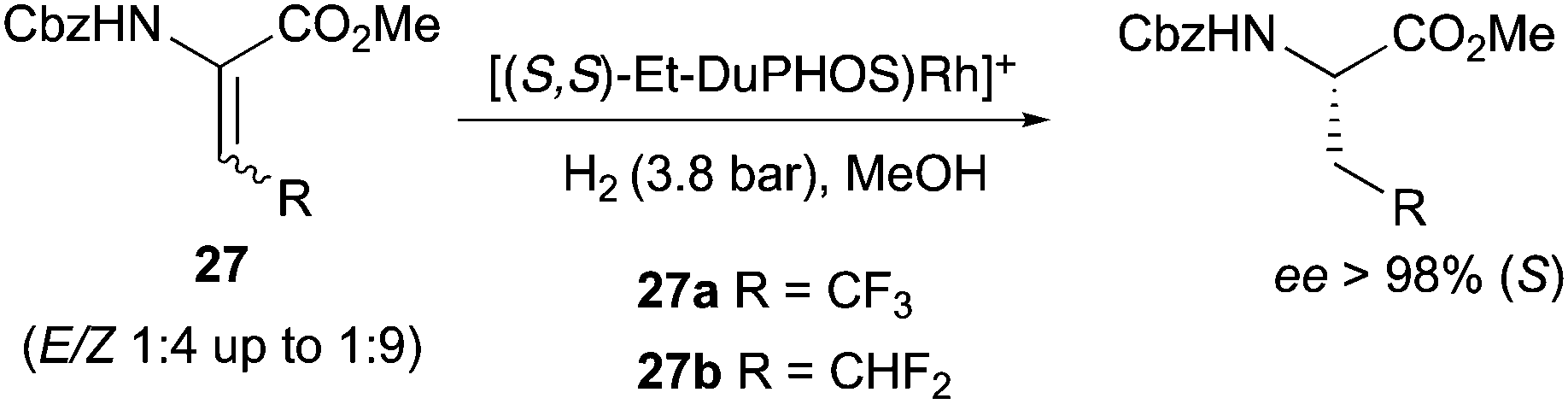
DuPhos ligands were also employed in the synthesis of fluoro-containing amino acids, allowing the highly enantioselective hydrogenation of (E) and (Z) isomer mixtures. The enantioselectivity was proved to be independent from the E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio since both mixtures 1:4 and 1:9 produced the same ee (Scheme 35).46
:Z ratio since both mixtures 1:4 and 1:9 produced the same ee (Scheme 35).46
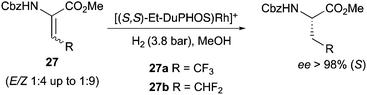 |
| | Scheme 35 | |
Another interesting example was observed by the Schmidt group when hydrogenating α-acyloxyacrylates with Rh and Ru catalysts (Scheme 36). The pure E- and Z-isomers and mixtures of variable ratios of these substrates resulted in the same enantiomer under the hydrogenation conditions, with very similar selectivity.47
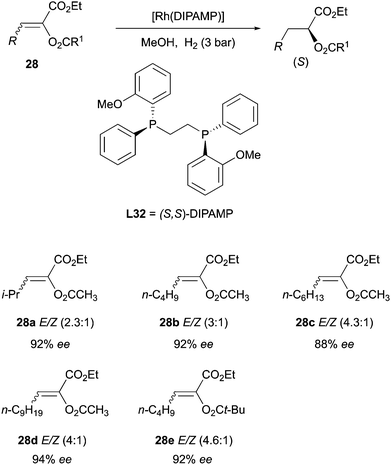 |
| | Scheme 36 | |
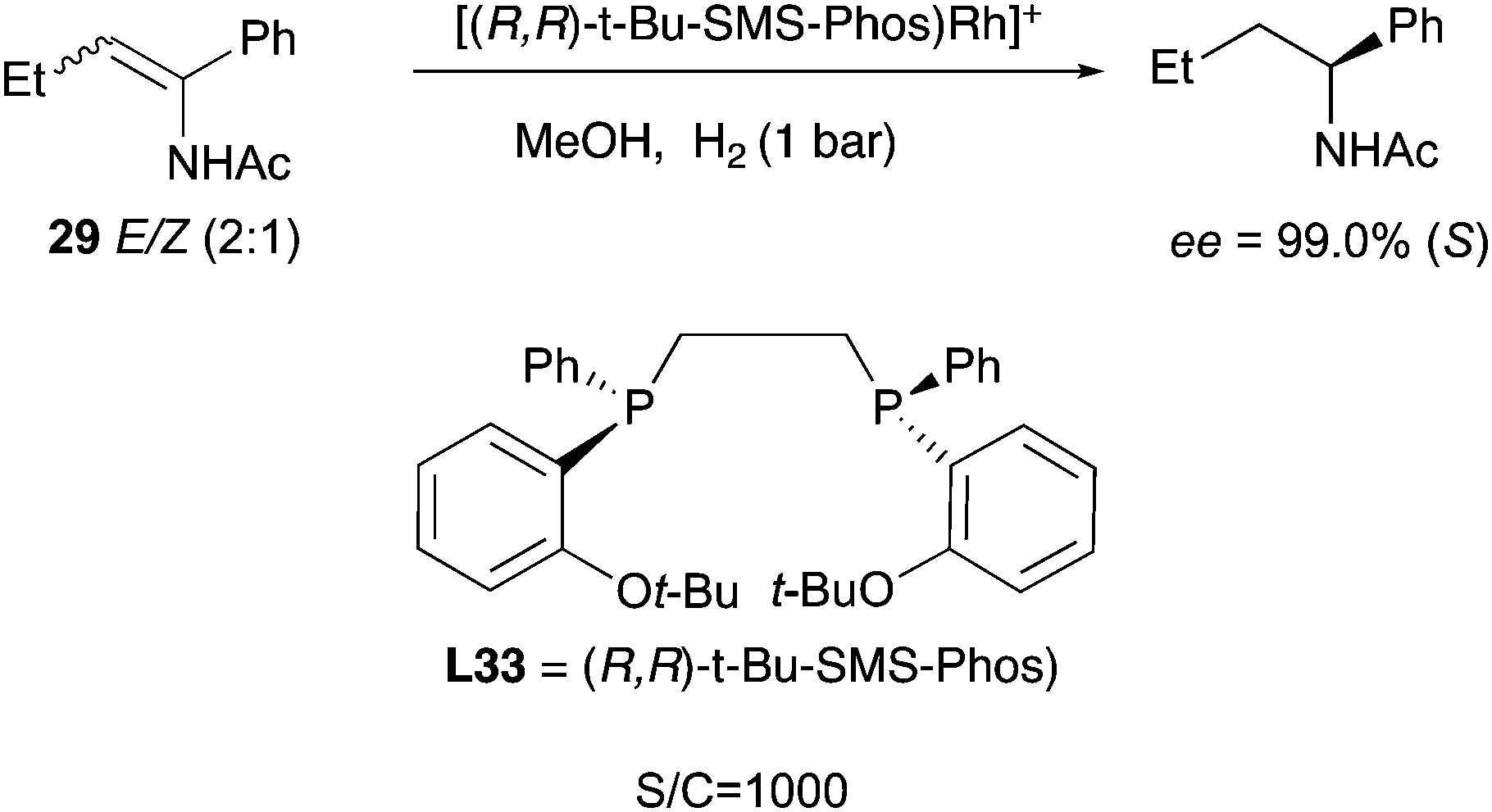
The continuous development and improvement of the diphosphine ligands led to the design of ligand L33, which was able to efficiently hydrogenate a wide number of enamides under mild conditions, outperforming many other catalysts. The geometrical isomers of 29 were hydrogenated to almost perfect convergence and in this case the catalyst loading could be decreased to 0.1 mol% (Scheme 37).48
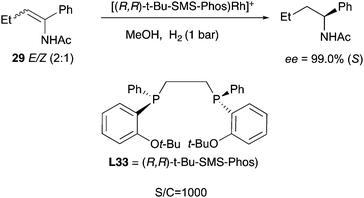 |
| | Scheme 37 | |
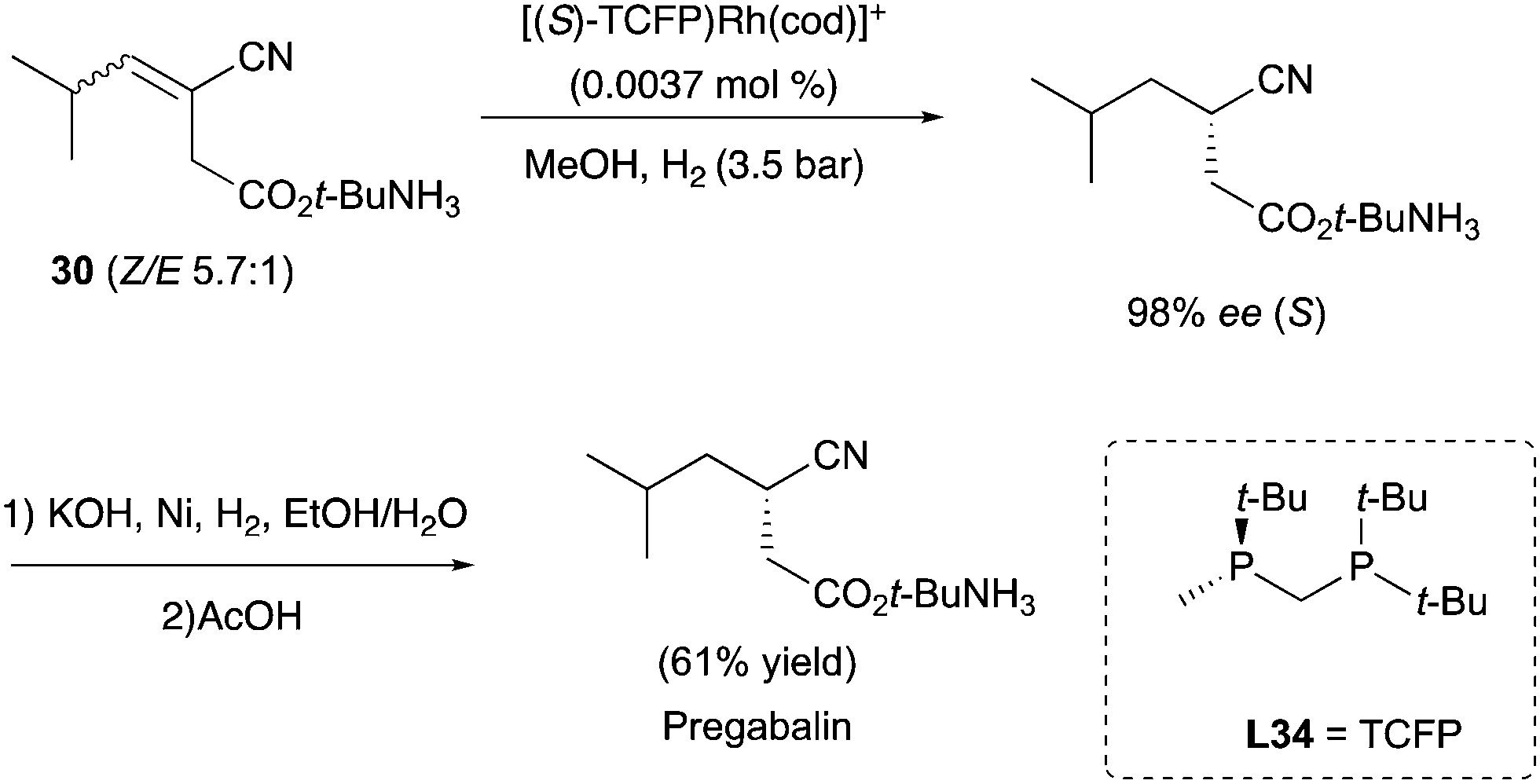
The enantioconvergent hydrogenation was also exploited to target important precursors for drugs. For example, the diphosphine ligand L34 was nicely employed in the asymmetric hydrogenation of E/Z mixture of the cyanoester substrates, targeting the chiral precursor of Pregabalin (Scheme 38).49–51
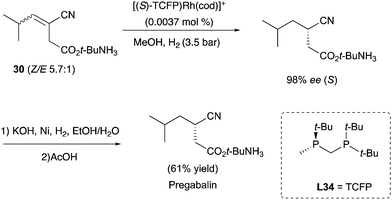 |
| | Scheme 38 | |
The Challenger group developed a multikilogram synthesis of Imagabalin hydrochloride using ligand L34 (Scheme 39).52
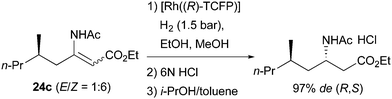 |
| | Scheme 39 | |
Another scalable process exploiting the usefulness of the enantioconvergence was developed by Grayson in search of a convenient route to (S)-β-phenylalanine. Different ligands were screened, the best level of selectivity was obtained with a diphosphine ligand based on camphor (Scheme 40).53
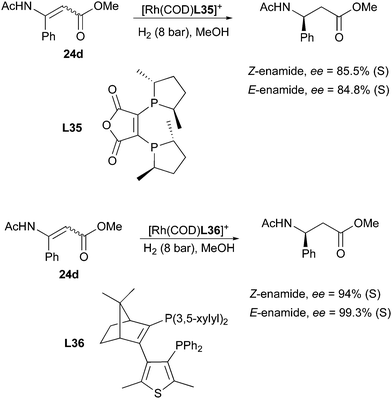 |
| | Scheme 40 | |
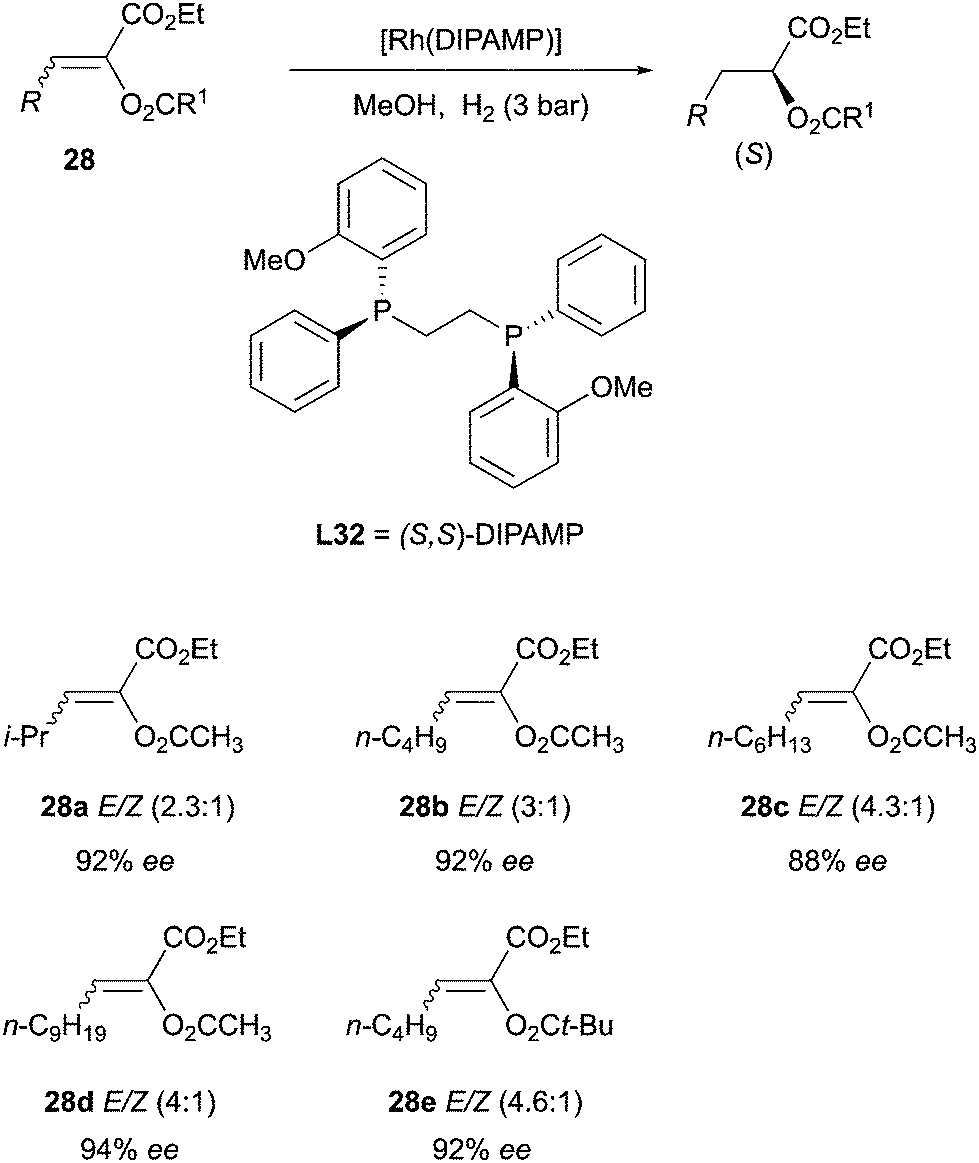
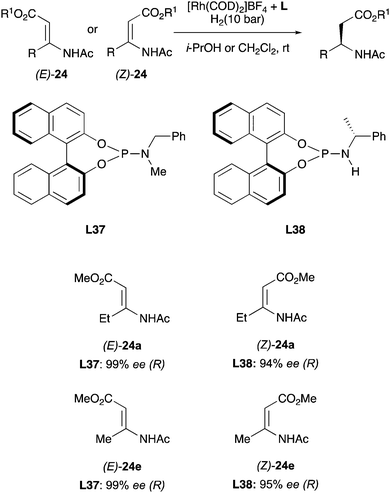
In the early 2000s major breakthroughs in the development of monodentate phosphorus ligands, applied to asymmetric hydrogenations were achieved by different research groups.54 In particular, Feringa and co-workers developed a new series of phosphoramidites which were successfully employed in the hydrogenation of β-(acylamino)-acrylates (Scheme 41).55
 |
| | Scheme 41 | |
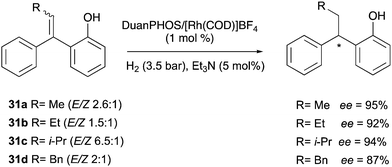
In 2011 Wang reported the hydrogenation of trisubstituted olefinic substrates in various E/Z mixtures. It was also demonstrated that the 2′-hydroxyl groups could be readily removed in high yield without loss of ee from the products (Scheme 42).56
 |
| | Scheme 42 | |
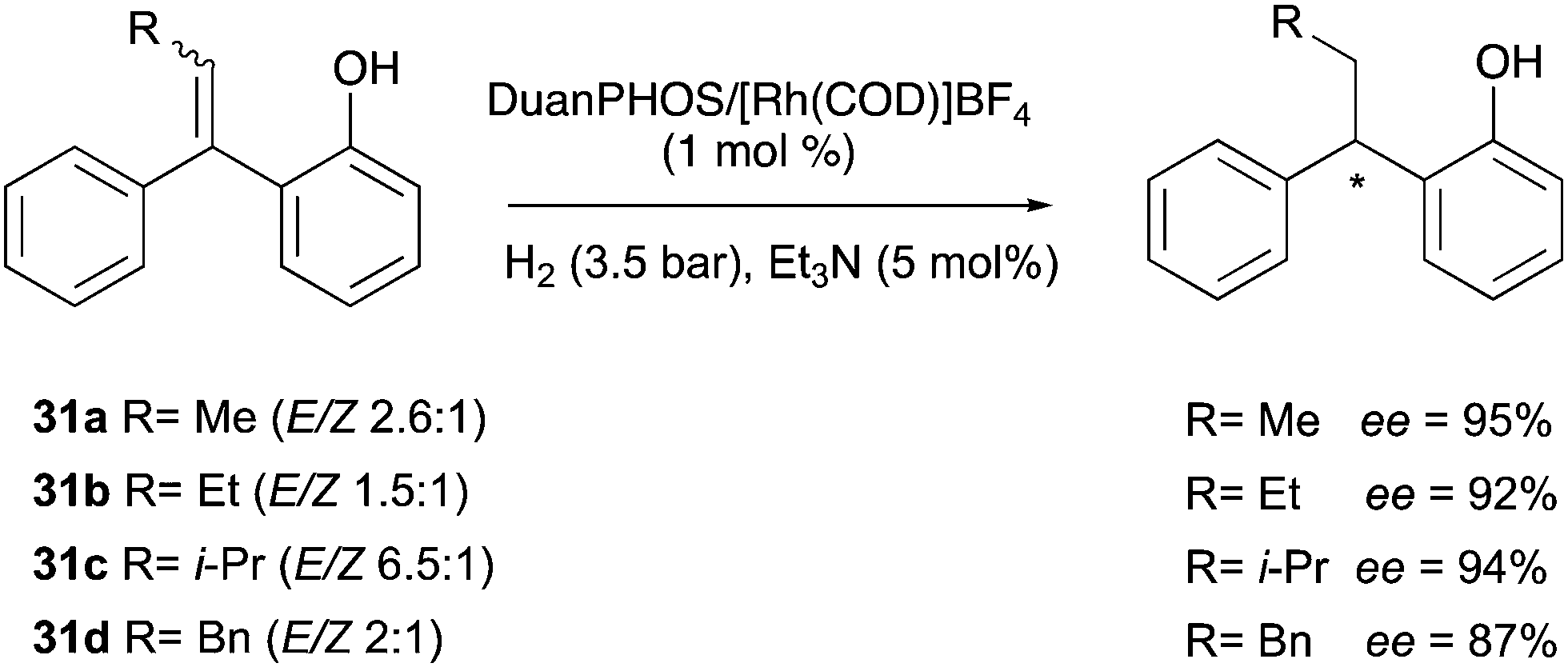
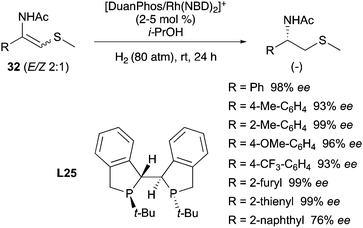
In 2017, a different class of substrates were found to behave similarly. The Zhang group was able to hydrogenate variously substituted β-acetylaminovinylsulphides with excellent ee, enabling the possibility of synthesizing useful drugs from mixtures of olefins. Out of eight ligands only the DuanPhos showed high reactivity and selectivity in presence of 2:1 mixtures of (E) and (Z) isomers, with isopropanol as the best solvent (Scheme 43).57
 |
| | Scheme 43 | |
3.3 Ruthenium
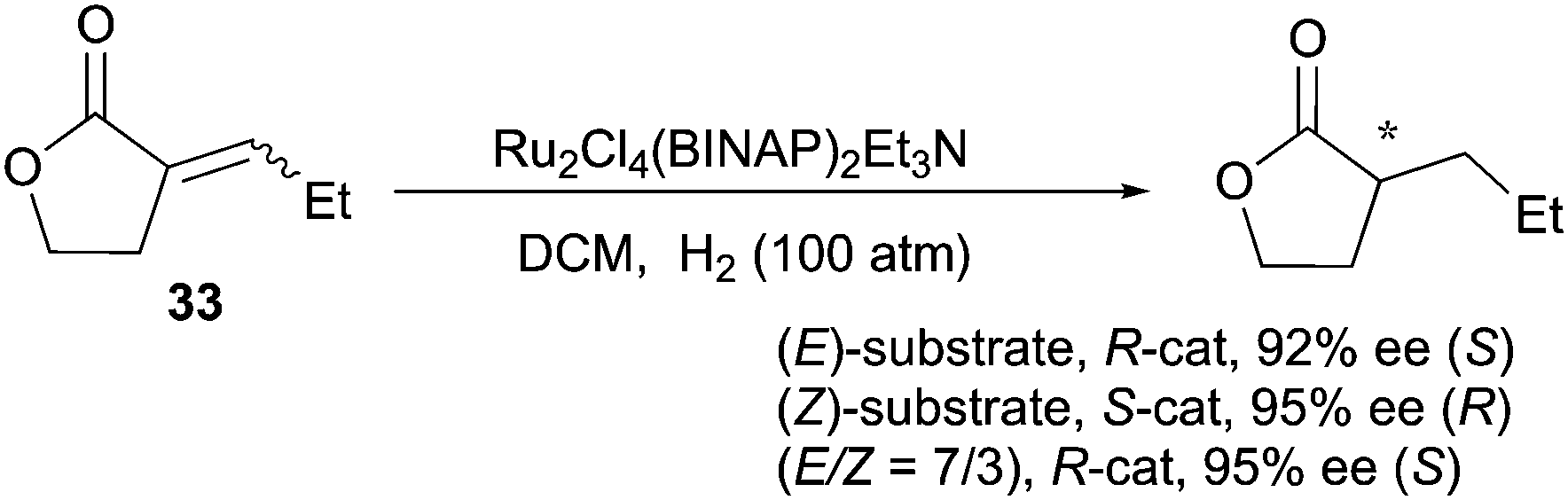
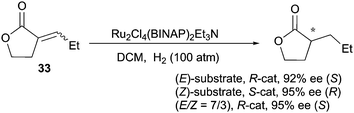
Examples of enantioconvergence observed with the Ru–BINAP catalytic system mainly concern prochiral CC bonds in proximity to functional groups. For example, when (E)- and (Z)-unsaturated lactones were evaluated in the Ru-catalyzed asymmetric hydrogenation, they afforded the same enantiomeric outcome (92% and 95% ee) (Scheme 44).58 Moreover, isomeric mixtures (E/Z = 7:3) were hydrogenated with same enantioselectivity (95% ee), which indicates a clear enantioconvergent transformation. This behavior was suggested to be due to a chelation of the carbonyl moiety, but isomerization of the (Z)- to the (E)-olefin could not be ruled out.
 |
| | Scheme 44 | |
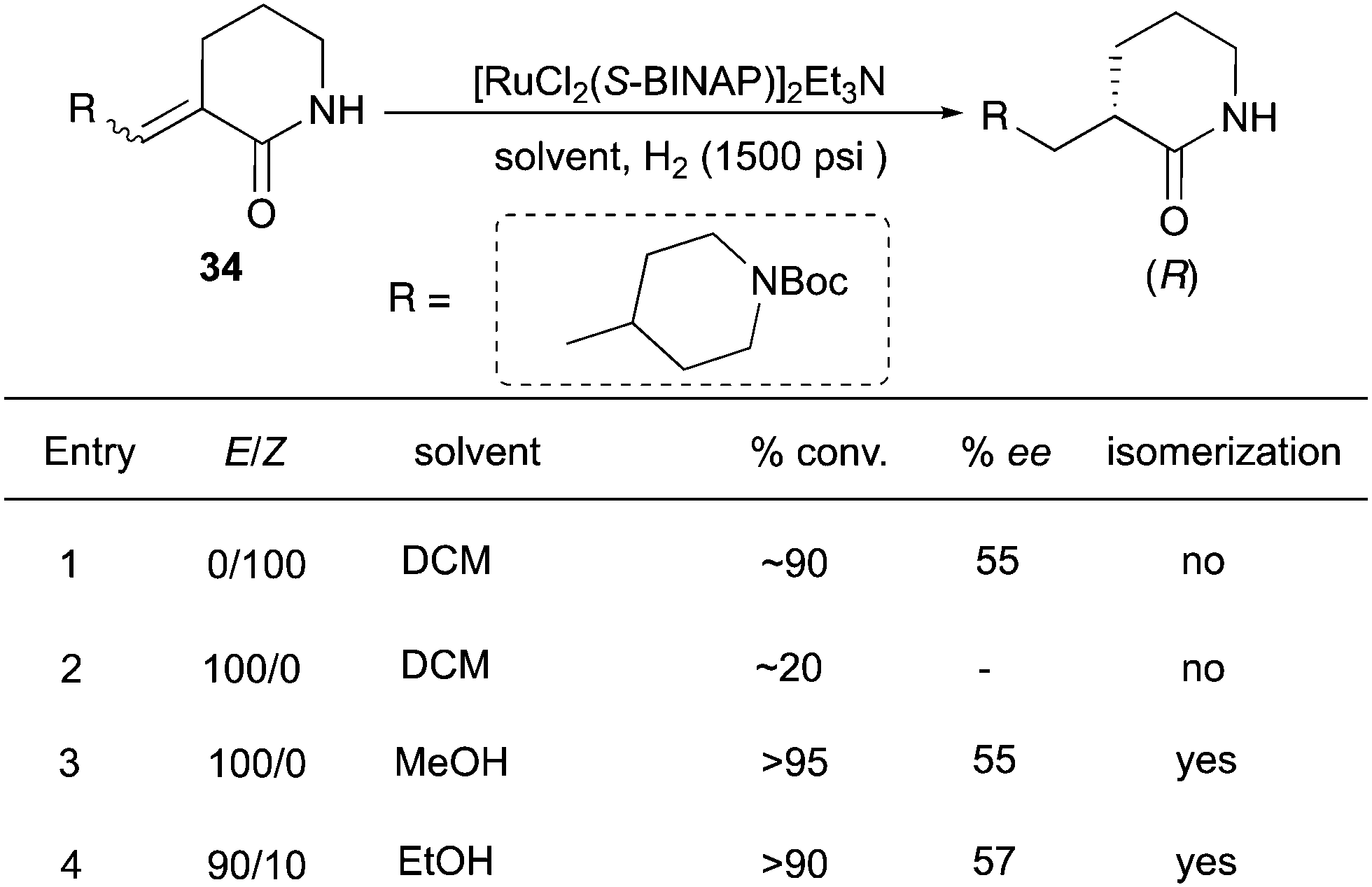
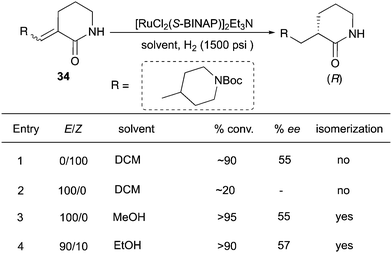
Similarly, asymmetric hydrogenations of unsaturated lactams (E, Z and mixtures) gave products with the same configuration, although in moderate ee (Scheme 45).59 In this case, the solvent played a crucial role. The reaction of (Z)-substrates proceeded much faster in DCM than for (E)-substrates, giving 90% conversion with 55% ee (entries 1 and 2). Interestingly, when the same reaction was conducted in MeOH, a fast isomerization took place to produce the (Z)-olefin, which was sequentially hydrogenated (entry 3). In ethanol the (E) to (Z) isomerization occurred to some extent, but the reaction still led to good conversion of the E/Z mixture with similar enantioselectivity.
 |
| | Scheme 45 | |
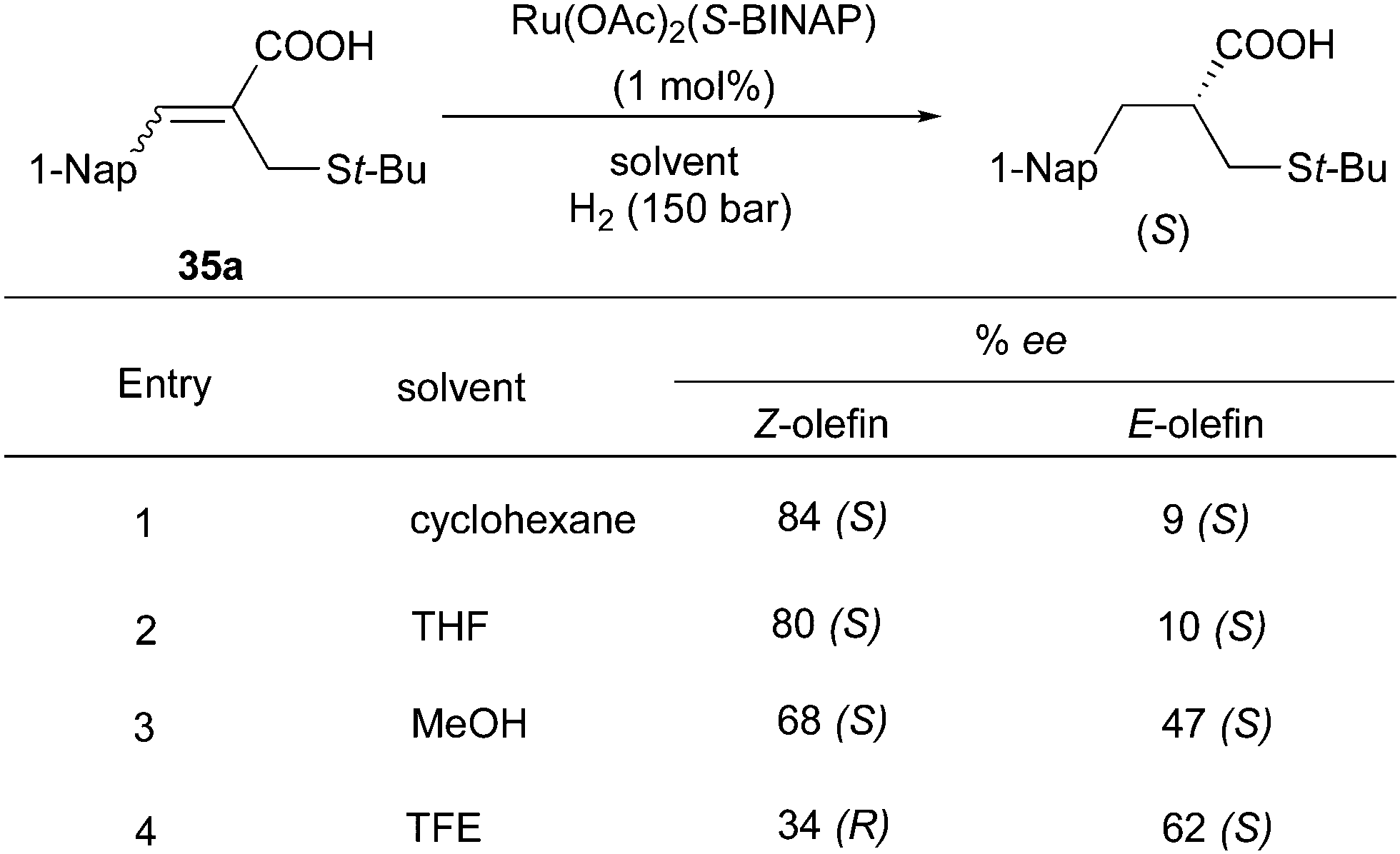
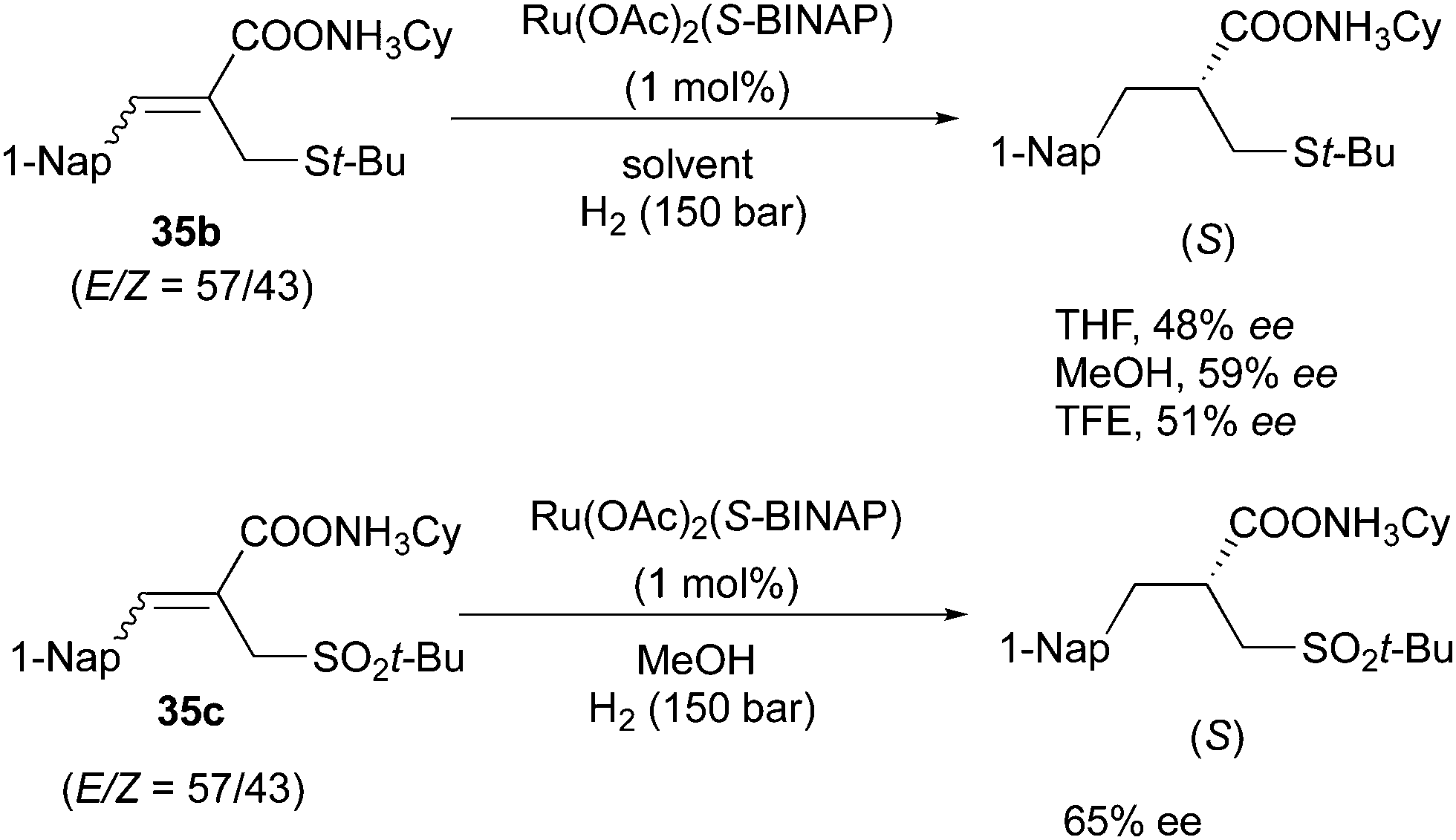
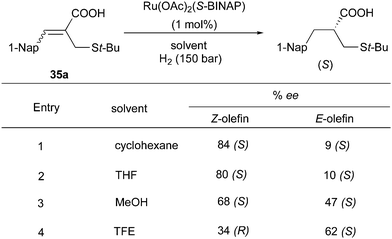
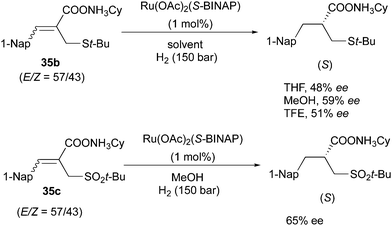
The Ru/BINAP system also showed enantioconvergence in the asymmetric hydrogenation of some acyclic compounds containing a carbonyl group. In the hydrogenation of (Z)-unsaturated carboxylic acids, the solvent effects are significant.60,61 In the non-polar solvent cyclohexane, 84% ee was obtained for the (S)-product deriving from the hydrogenation of the (Z)-olefin, while the highly polar solvent TFE led to a reversed π-face selectivity generating the product with opposite configuration. By contrast, for the (E)-substrate the (S)-product was formed with low enantioselectivity in cyclohexane, but moderate in TFE (Scheme 46). The isomeric mixtures of cyclohexylammonium salts were hydrogenated to provide the (S)-configuration product in moderate ee, which were not particularly affected by solvents (Scheme 47). The authors suggested that the enantioconvergence could stem from Ru chelation to the carboxylate group rather than to the sulfur atom or CC. Later, this strategy was applied as a key step into a 6 step total synthesis, converting the olefin mixture into the saturated product with 65% ee.
 |
| | Scheme 46 | |
 |
| | Scheme 47 | |
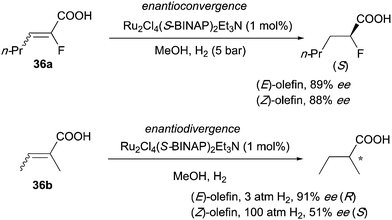
In the asymmetric hydrogenation of 2-fluoro-2-alkenoic acids, developed by the group of Saburi, both (E)- and (Z)-substrates gave similar enantioselectivities and afforded the product with the same configuration (Scheme 48).62 However, enantiodivergence was found when the fluoride was replaced with a methyl group, which could mean that the presence of the electronegative fluorine atom is directly influencing the chelation ability of the substrate.
 |
| | Scheme 48 | |
Schmidt and co-workers developed the synthesis of α-acyloxyacrylates, which were sequentially hydrogenated by Rh and Ru catalysts (Scheme 49).47 Similarly, Ru gave the same enantioconvergence as Rh in the asymmetric hydrogenation of this class of compounds. For a mixture of acetate-substituted olefins with a 70:30 E/Z ratio, 98% ee was obtained. In addition, no apparent effect was found when varying the E/Z ratio from 100:0 to 80:20 of the pivalate substituted olefin.
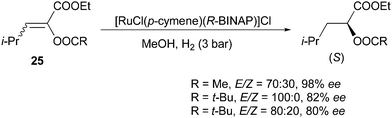 |
| | Scheme 49 | |
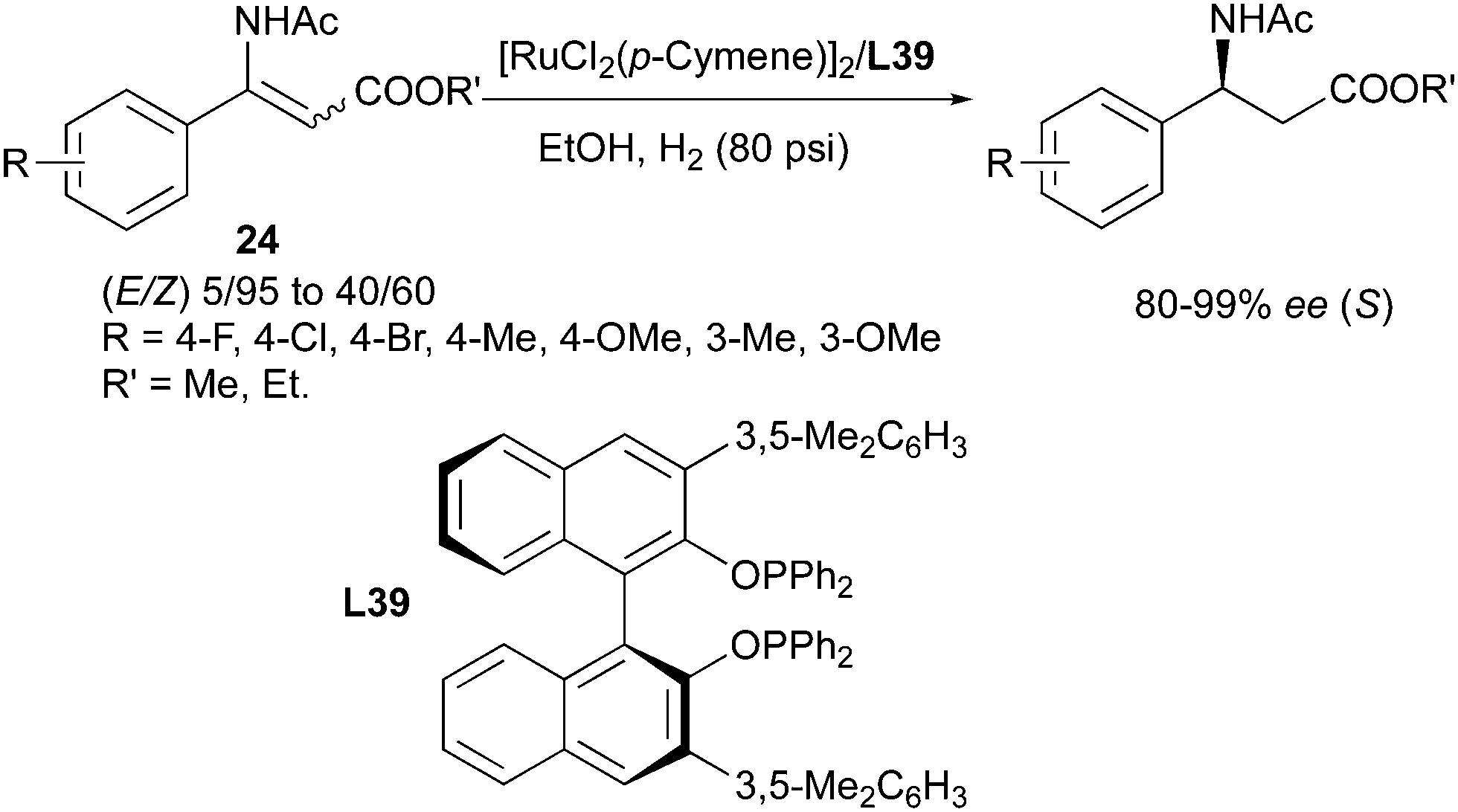
Beside the BINAP ligand, the o-BINAPO ligand developed by Zhang's group also provided efficient enantioconvergent transformations (Scheme 50).63 In the hydrogenation of isomeric mixtures of α-(acylamino)acrylates, the modified o-BINAPO ligand L39 led to high enantioselectivity, up to 99% ee. In the successive substrate evaluation, a variety of E/Z mixtures with ratios from 5:95 to 40:60 were smoothly converted into the (S)-configured product, with high enantioselectivities.
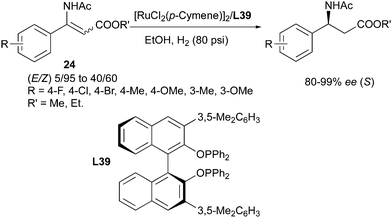 |
| | Scheme 50 | |
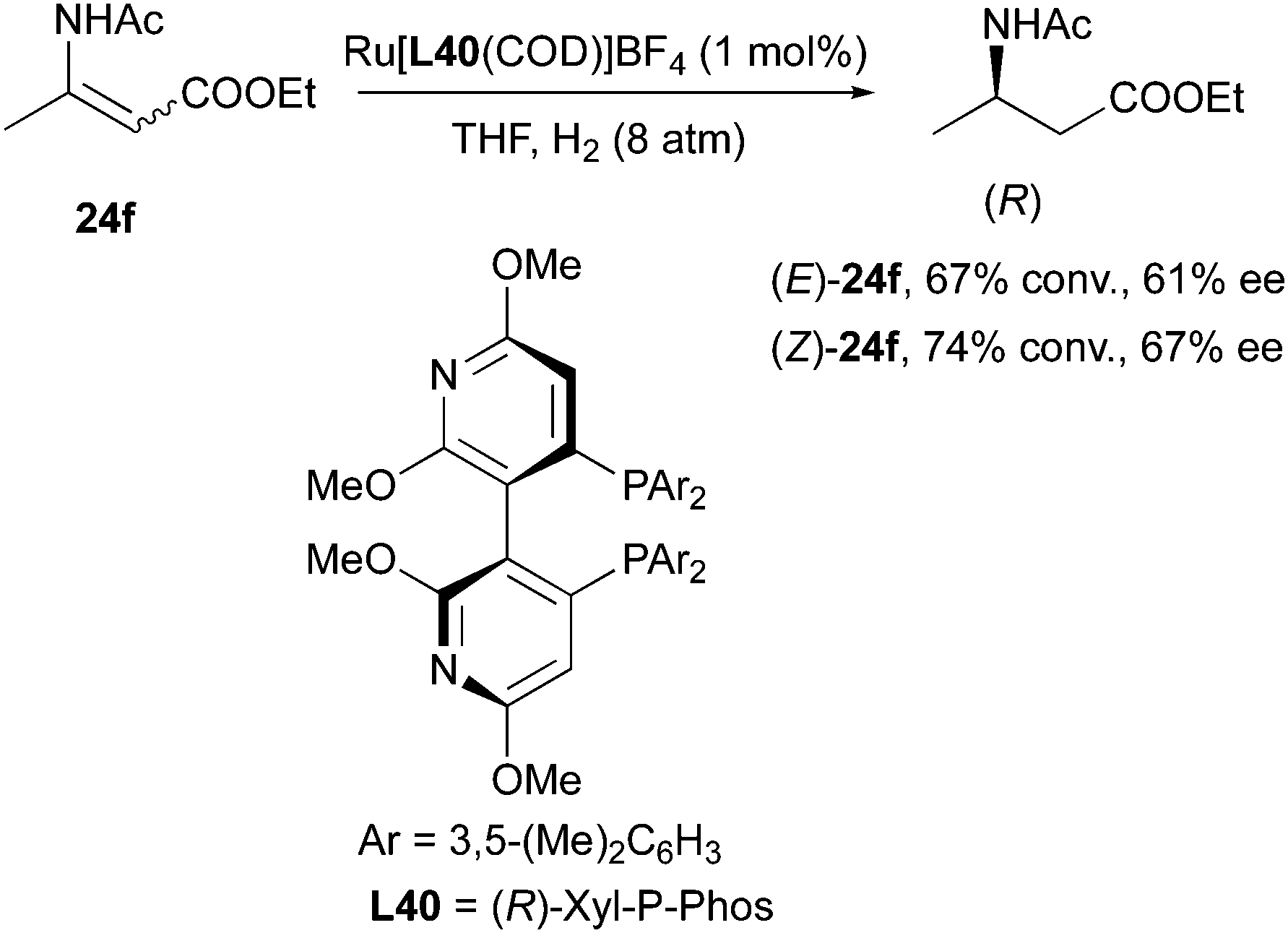
Similarly, in the study of asymmetric hydrogenation of alkyl-substituted-(acylamino)acrylates, Yeung and Chan observed enantioconvergent hydrogenation when using Ru[(R)-Xyl-P-Phos(COD)]BF4 as catalyst. In this case both (E)- and (Z)-ethyl 3-acetamidobut-2-enoates were converted into (R)-product in THF, albeit with moderate conversion and ee (Scheme 51).64
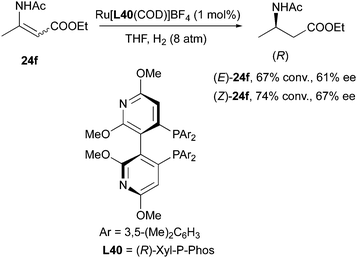 |
| | Scheme 51 | |
3.4 Other metals
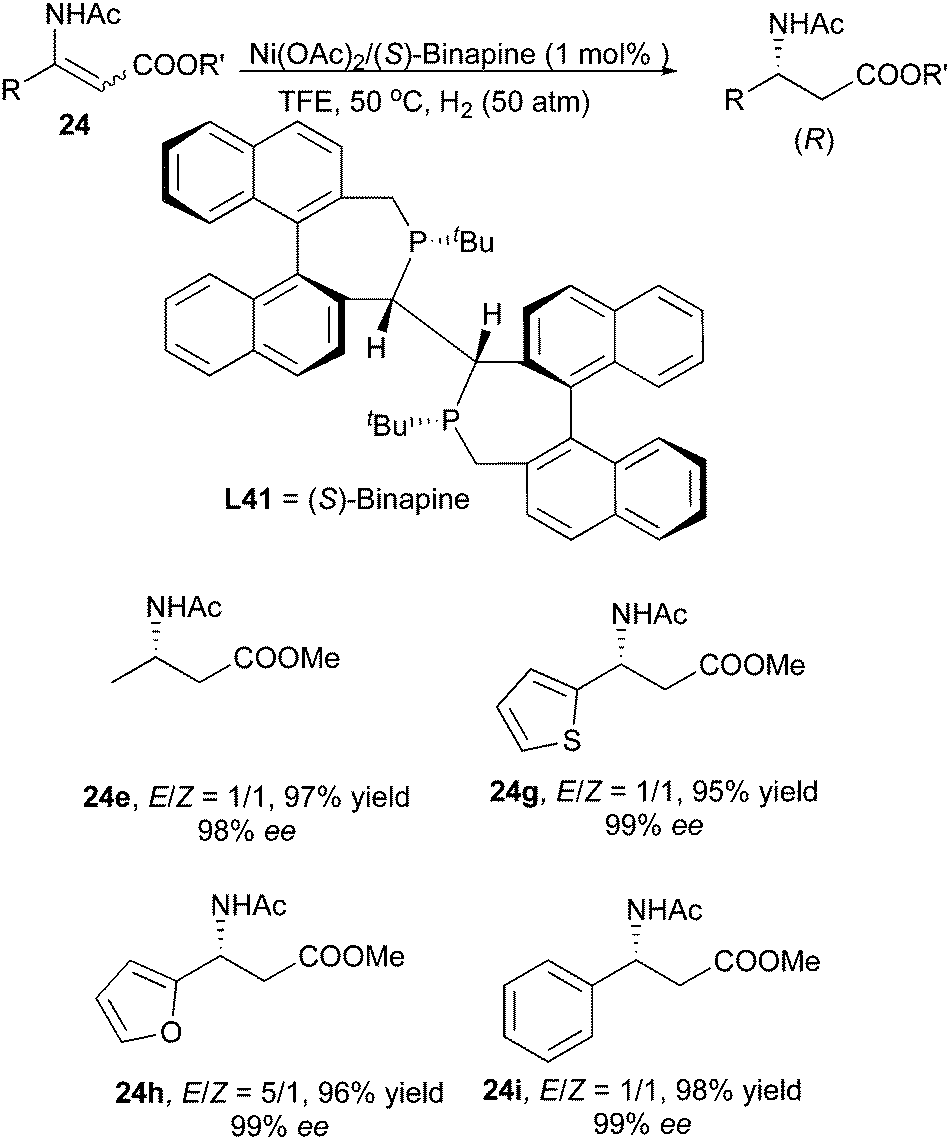
Recently, Lv and Zhang developed a Ni/Binapine system for the asymmetric hydrogenation of β-(acylamino)acrylates. Similarly to the Ru/o-BINAPO system, enantioconvergence was observed when hydrogenating the same compounds with the new nickel system. Both aromatic and aliphatic substrates with E/Z ratios of 1:1 to 5:1 could be hydrogenated to afford β-amino acid derivatives in up to 99% ee (Scheme 52).65 Later, this system was applied to the hydrogenation of E/Z mixtures of β-acetylamino vinylsulfones, which showed enantioconvergence in the hydrogenation giving 97% ee (Scheme 53).66
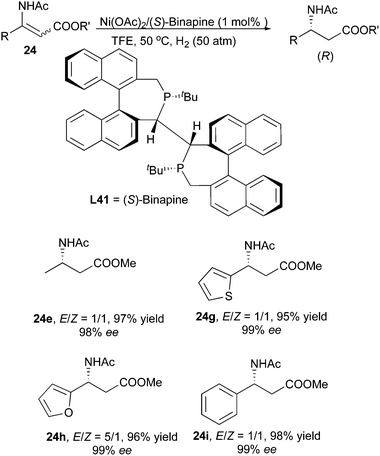 |
| | Scheme 52 | |
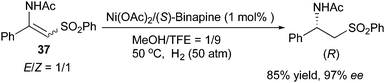 |
| | Scheme 53 | |
4. Mechanistic investigations
In the following section we briefly categorize the proposed mechanisms for the hydrogenations, with a focus on the stereoselective outcome attempting to rationalize the factors determining divergence and convergence in the examples in which both (E) and (Z) isomers were analyzed.
4.1 Enantiodivergence
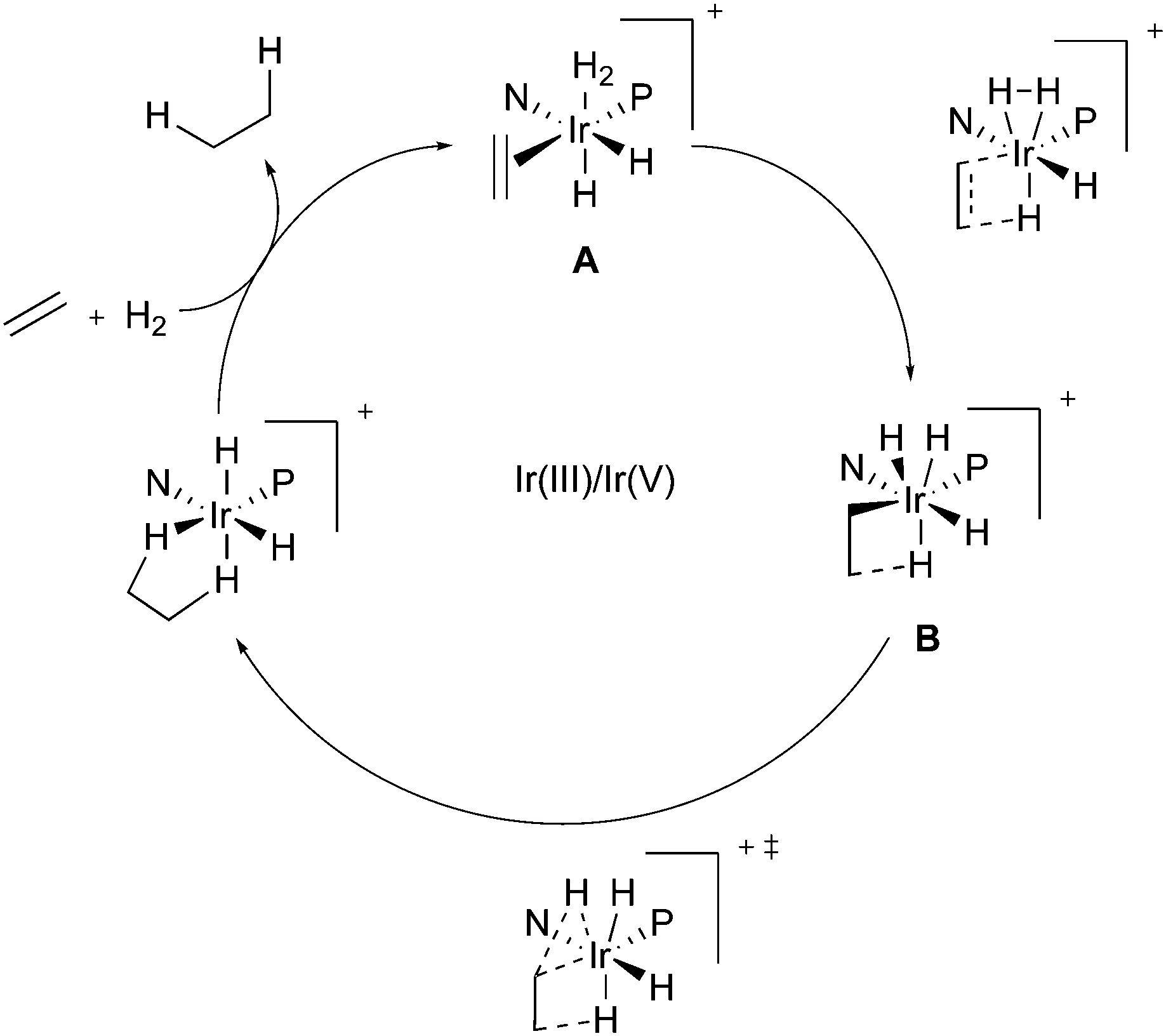
In the context of Ir-N,P-catalysts, in 2003, Brandt and co-workers proposed a mechanism involving an Ir(III)/Ir(V) catalytic cycle.67 The migratory insertion step, which was identified as rate-determining due to the significant calculated energy barrier, was suggested to occur simultaneously to and to be also facilitated by the oxidative addition of the coordinated hydrogen molecule to form the Ir(V) species (Fig. 1).
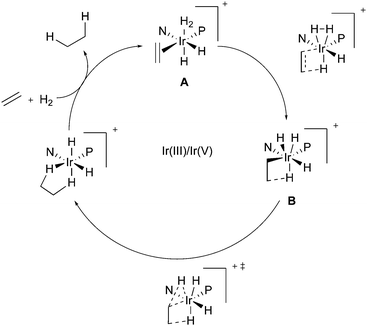 |
| | Fig. 1 Ir(III)/Ir(V) catalytic pathway for the iridium catalyzed hydrogenation. | |
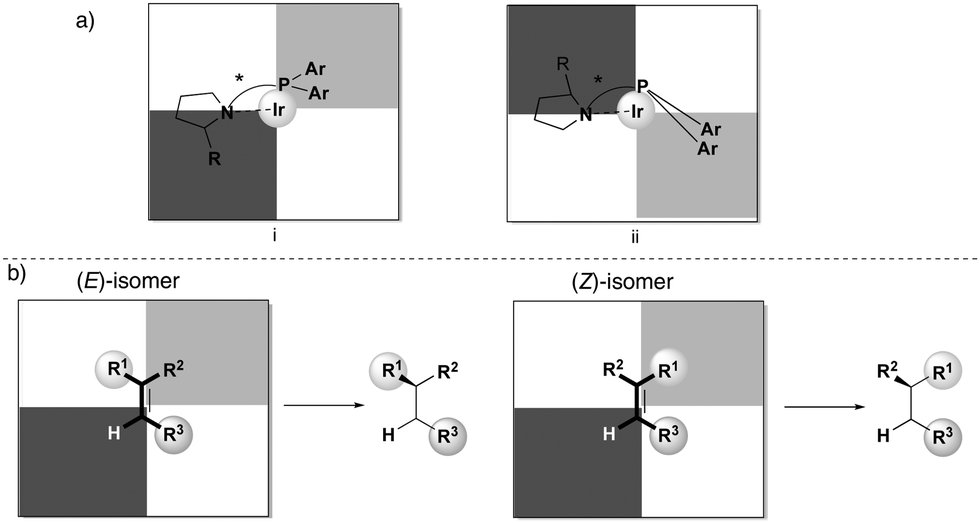
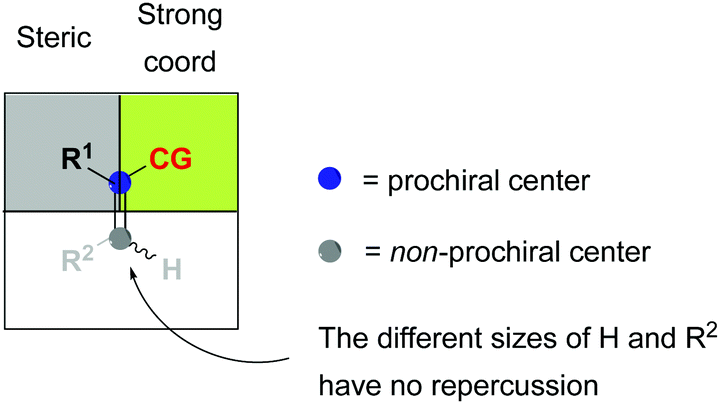
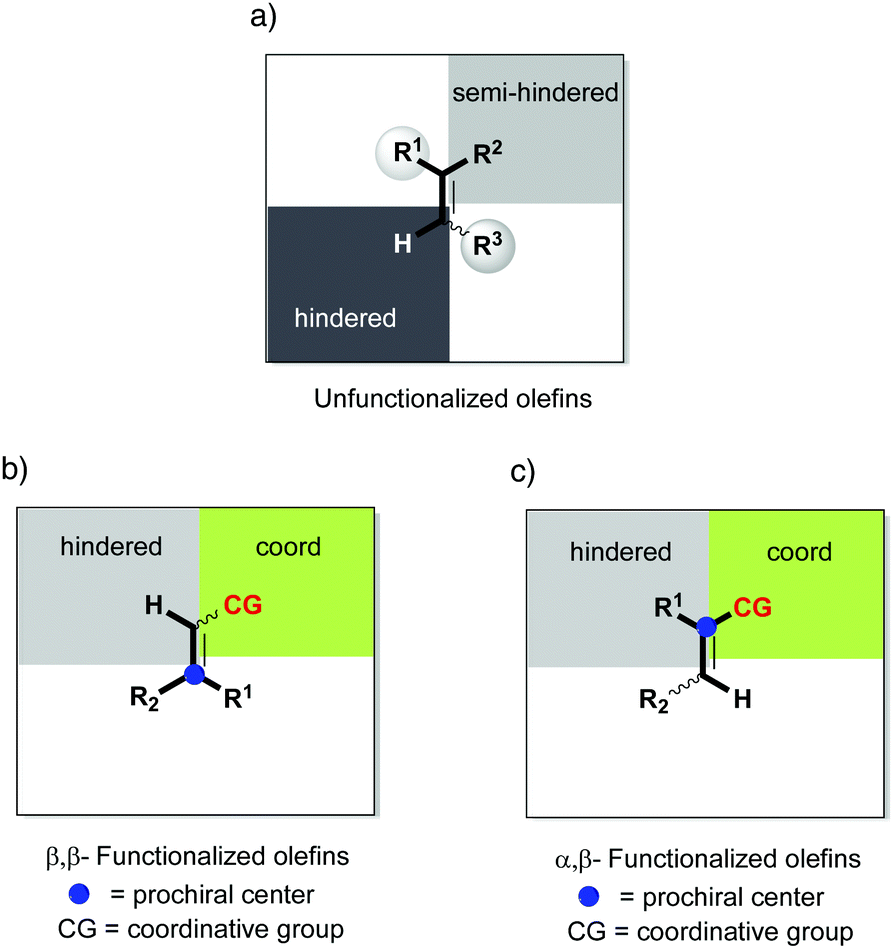
In order to rationalize the stereoselectivity of the hydrogenation, the steric environment around the incoming olefin, which is coordinated trans to phosphorus, must be taken into account. Andersson and co-workers presented a quadrant model describing this, depicted from the perspective of the coordinated alkene (Fig. 2). The gray quadrants represent the most hindered areas, which dictate the preferential coordination of one face of the alkene in order to minimize steric interactions (depending on the ligand structure and absolute configuration). Since the preferential coordination depends on the position of the smallest substituent (H) on the substrate, olefins are not discriminated on the prochiral carbon atom, and this results in the formation of opposite enantiomers upon hydrogenation of an (E)- or (Z)-olefin. Moreover, DFT calculations indicate that this model is reliable for a wide number of unfunctionalized olefins, as also verified experimentally.20
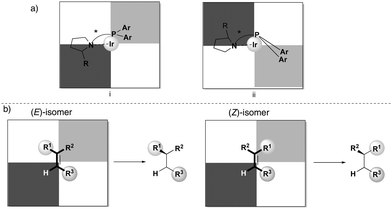 |
| | Fig. 2 (a) Quadrant selectivity model. (b) Hydrogenation of isomeric olefins. | |
The hydrogenation is also enantiodivergent when a chelating group is present close to the non-prochiral carbon, such as β,β-disubstituted functionalized olefins. The Burgess group proposed a model involving the chelation of the carbonyl group to the Ir metal center and in following studies several pathways involving a chelation were suggested to explain the enantioselective outcome obtained from α,β-unsaturated molecules.68–70 In 2016, Bolm and co-workers investigated the mechanism of the hydrogenation of α,β-unsaturated ketones using an Iridium sulfoximine catalyst by means of DFT studies. Substrate chelation was proposed in this case as well and, in contrast with previous studies, the coordination of the olefin was proposed to be trans to the ligand nitrogen atom. Interestingly, also in this case (Z)- and (E)-olefins produced opposite enantiomers (Fig. 3).71
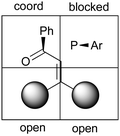 |
| | Fig. 3 Quadrant selectivity model based on coordination. | |
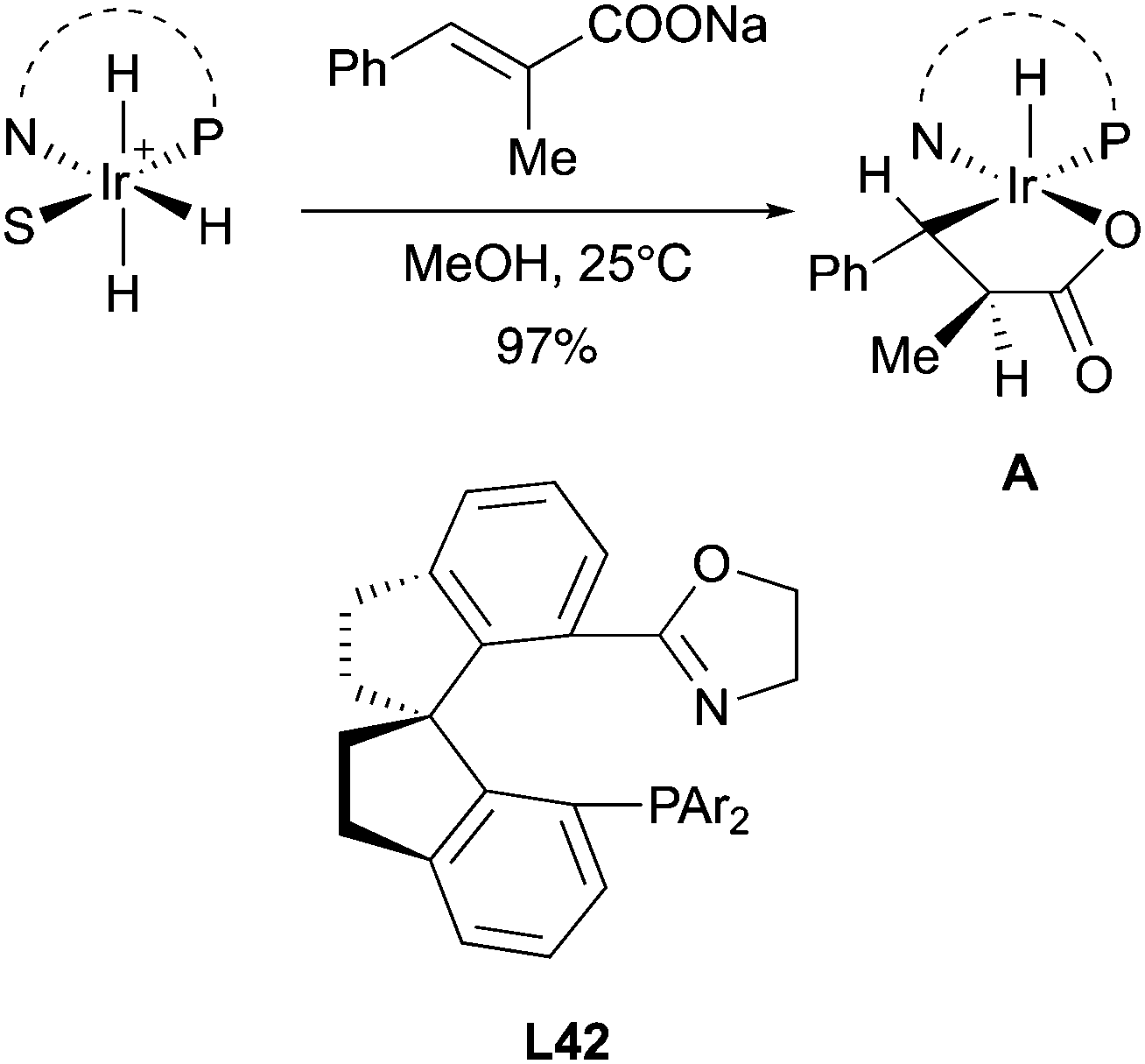
In 2017, the Zhou group studied the mechanism of the asymmetric hydrogenation of unsaturated carboxylic acid catalyzed by Ir–SIPHOX catalysts L42. Notably, it was possible to isolate the Ir(III) migratory insertion intermediate A, which could not undergo reductive elimination in the absence of hydrogen gas providing another strong evidence to support the involvement of an Ir(III)/Ir(V) catalytic cycle (Scheme 54).72
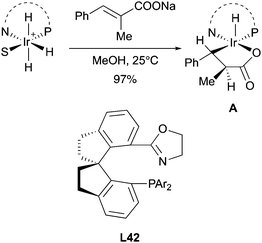 |
| | Scheme 54 | |
Overall, different mechanistic pathways are proposed to compete, especially in presence of chelating groups, which can affect which of the enantiotopic faces of the double bond approaches the metal center. Nevertheless, one necessary requirement for divergent hydrogenation seems to be related to the non-prochiral carbon dictating the preferred face of coordination of the olefin. On the other hand, a chelating group can influence the coordination of the olefin and, moreover, direct the first migratory insertion that is crucial for the stereochemical outcome. However, if the coordinating functional group is bonded to the non-prochiral carbon, steric effects can still be considered to prevail, hence the geometry of the double bond still being strongly determining. Based on the above discussion, we proposed a general model for enantiodivergent hydrogenations, including unfunctionalized olefins and β,β-disubstituted functionalized olefins (Fig. 4).
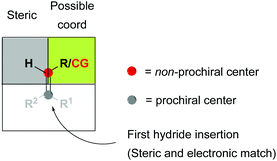 |
| | Fig. 4 General model for enantiodivergent hydrogenations. CG = coordinative group. | |
4.2 Enantioconvergence
4.2.1 Chelation effect.
Enantioconvergence mainly occurs in the hydrogenations of functionalized olefins, and is proposed to be due to either chelation or isomerization. In contrast to β,β-disubstituted functionalized olefins, α,β-disubstituted functionalized olefins with chelating group tend to undergo enantioconvergent hydrogenation. Mechanistic studies of the Rh-catalyzed asymmetric hydrogenation began with the discovery of the efficient (P,P)-Rh catalysts. The most studied mechanism regarded the asymmetric hydrogenation of α- or β-dehydroamino acids.73 Several mechanistic pathways have been proposed depending on the characteristics of the substrates and catalysts. One of the first was made by Halpern and co-workers and considered the coordination of substrate happening prior to the oxidative addition of the hydrogen molecule. The calculations showed that the oxidative addition was the rate-determining step followed by a rapid and irreversible migratory insertion.74–76
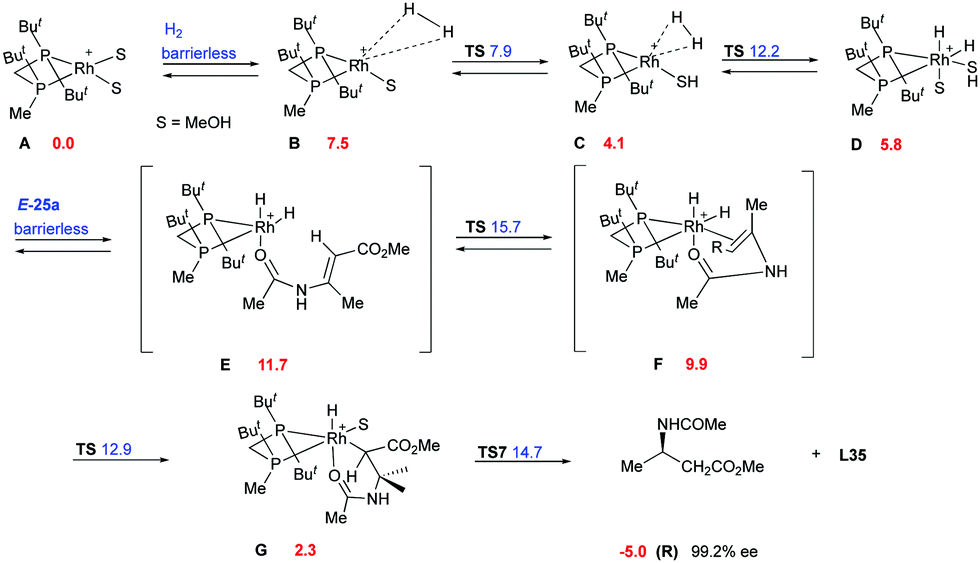
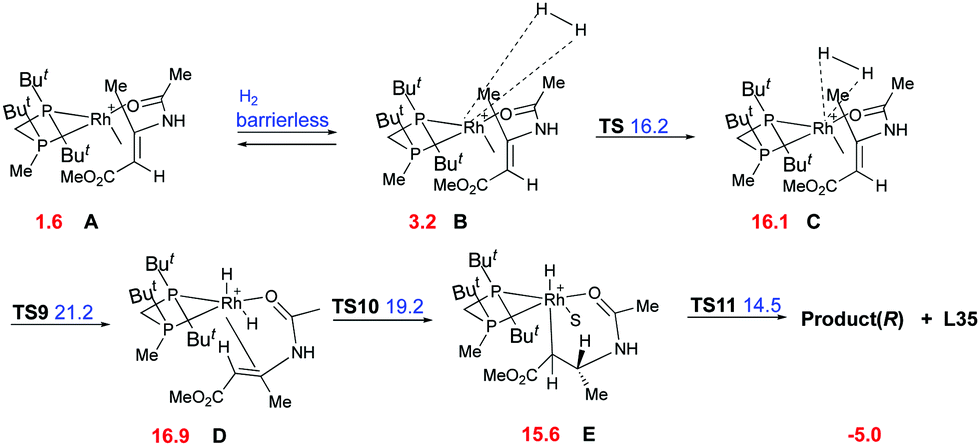
Imamoto and co-workers did an extensive experimental and computational study on the hydrogenation of β-dehydroamino acids aiming to justify the lower reactivity and selectivity observed for the (Z)-isomers.77 Two main pathways were proposed, the dihydride (Fig. 5) and the unsaturated (Fig. 6), which would lead to the same enantiomer but with different grade of selectivity. In these two pathways the coordination step is the main difference and it depends on the chelation ability of the substrate.
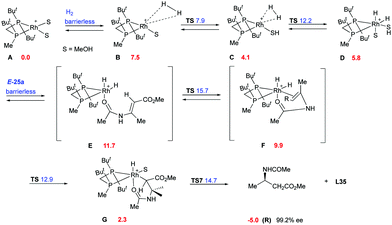 |
| | Fig. 5 Dihydride reaction pathway. | |
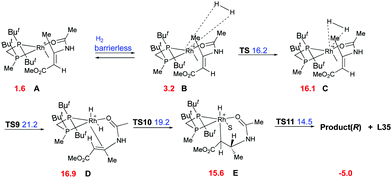 |
| | Fig. 6 Unsaturated pathway. | |
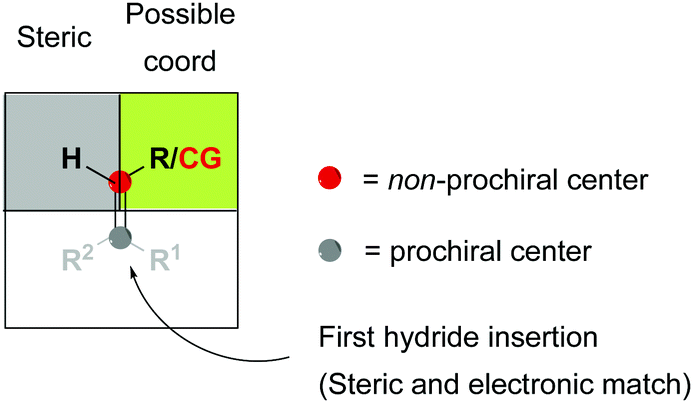
The binding ability of the (Z)-isomer was revealed to be stronger than for the respective (E)-isomer, favoring the less selective unsaturated pathway. In most asymmetric hydrogenations with Rh-catalysts, the presence of a strong chelating functional group is necessary to ensure the desired reactivity. If this group resides on the prochiral carbon, the R groups in the β position do not determine the enantioselective outcome, even though they affect how the olefin fits into the catalyst pocket. This means that the coordination of the functional group will also control the selectivity between the enantiotopic faces of the double bond regardless of the geometry of the olefins (Fig. 7). Consequently, a number of convergent hydrogenations of isomeric mixtures were developed with this particular type of substrate and Rh catalysts.
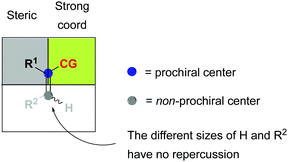 |
| | Fig. 7 General model for convergent hydrogenations. CG = coordinative group. | |
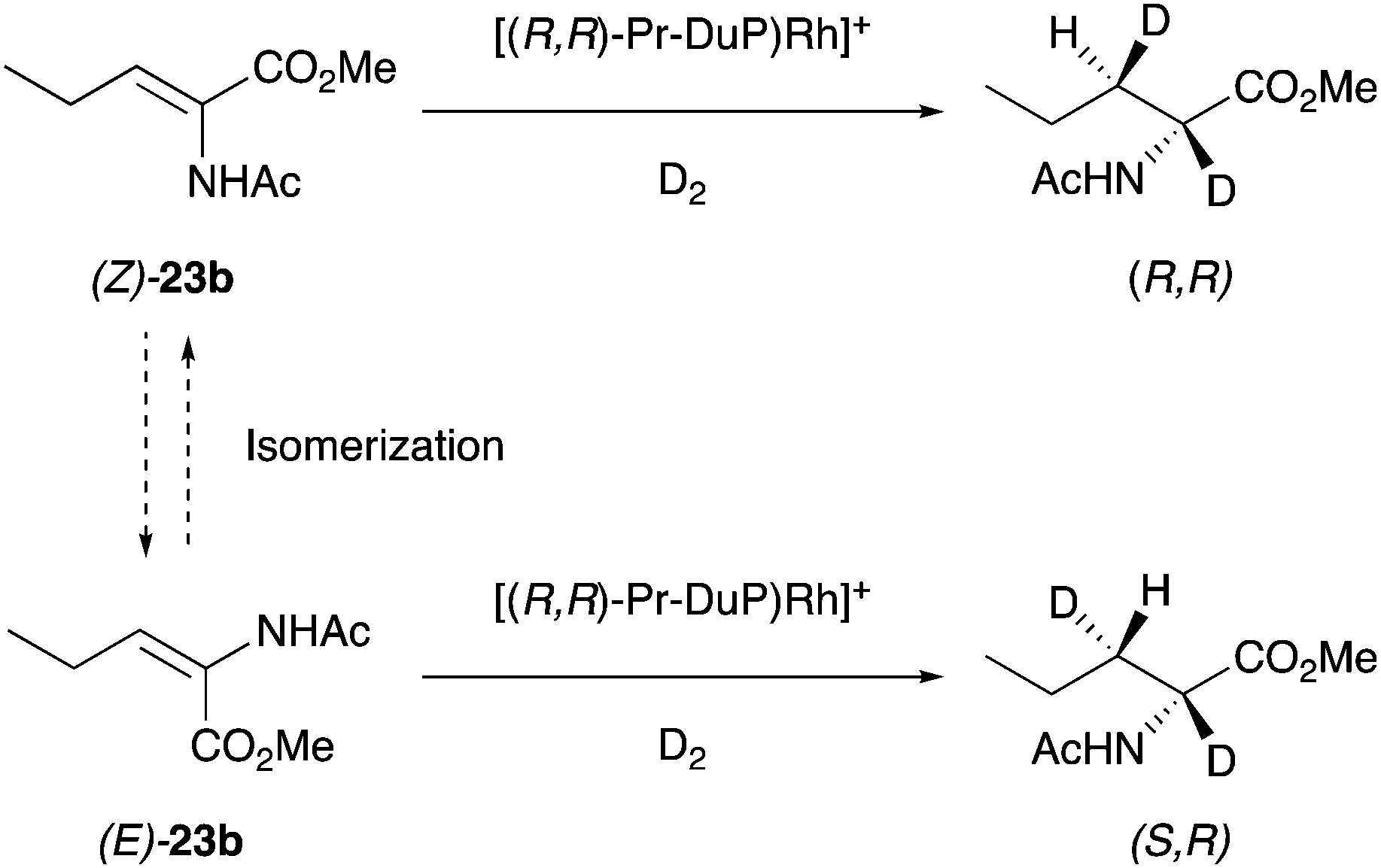
4.2.2 Isomerization.
In Rh-catalyzed enantioconvergent hydrogenations, Burk first proposed a mechanism involving isomerization of the olefins during the hydrogenation process and attempted to confirm this by deuterium experiments.41 Assuming a stereospecific cis-addition of D2 to the enamide carbon–carbon double bond, deuteration of (E)- and (Z)-enamides should produce diastereomeric products (Scheme 55). However, an isomerization of the starting material could lead to a convergent reaction that produces predominantly one diastereomer. In the hypothetical case of fast isomerization from E to Z, and that Z is hydrogenated faster than E, the same diastereomer would be produced from both diastereomers of the starting enamide. However, the results of the experiments excluded the presence of any form of isomerization since deuteration of (Z)-23b produced only one diastereomer (R,R). Likewise, deuteration of (E)-23b afforded specifically the other diastereomer (S,R). These results instead suggest that the stereochemical outcome is determined by chelation. In spite of this, Burk provided an important possible pathway to explain how enantioconvergent hydrogenation could take place.
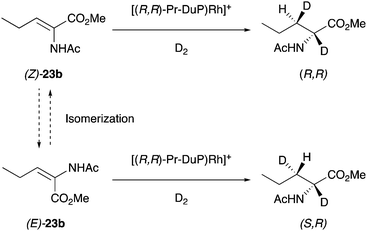 |
| | Scheme 55 | |
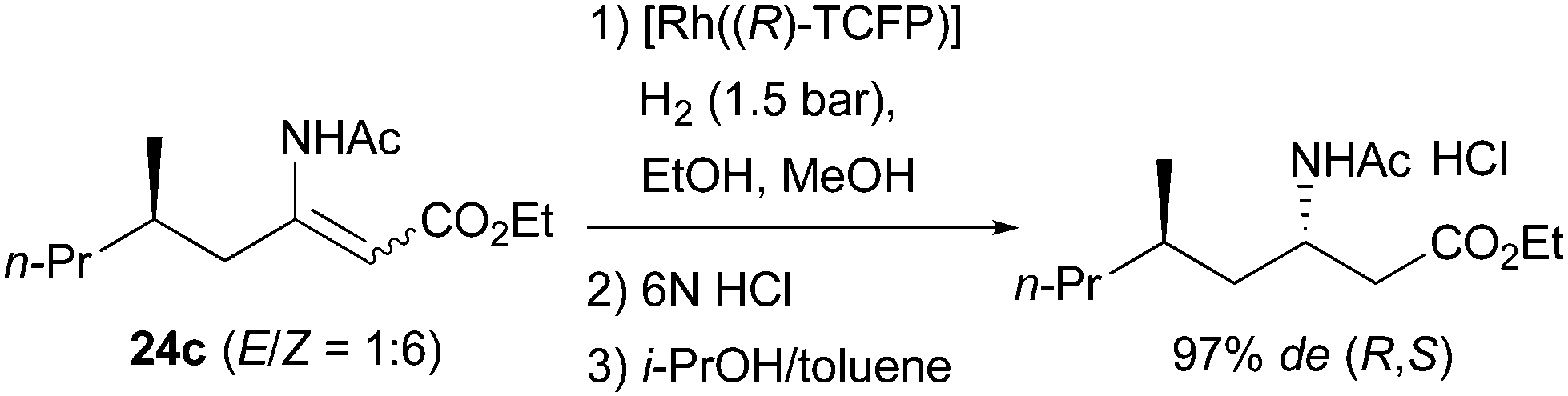
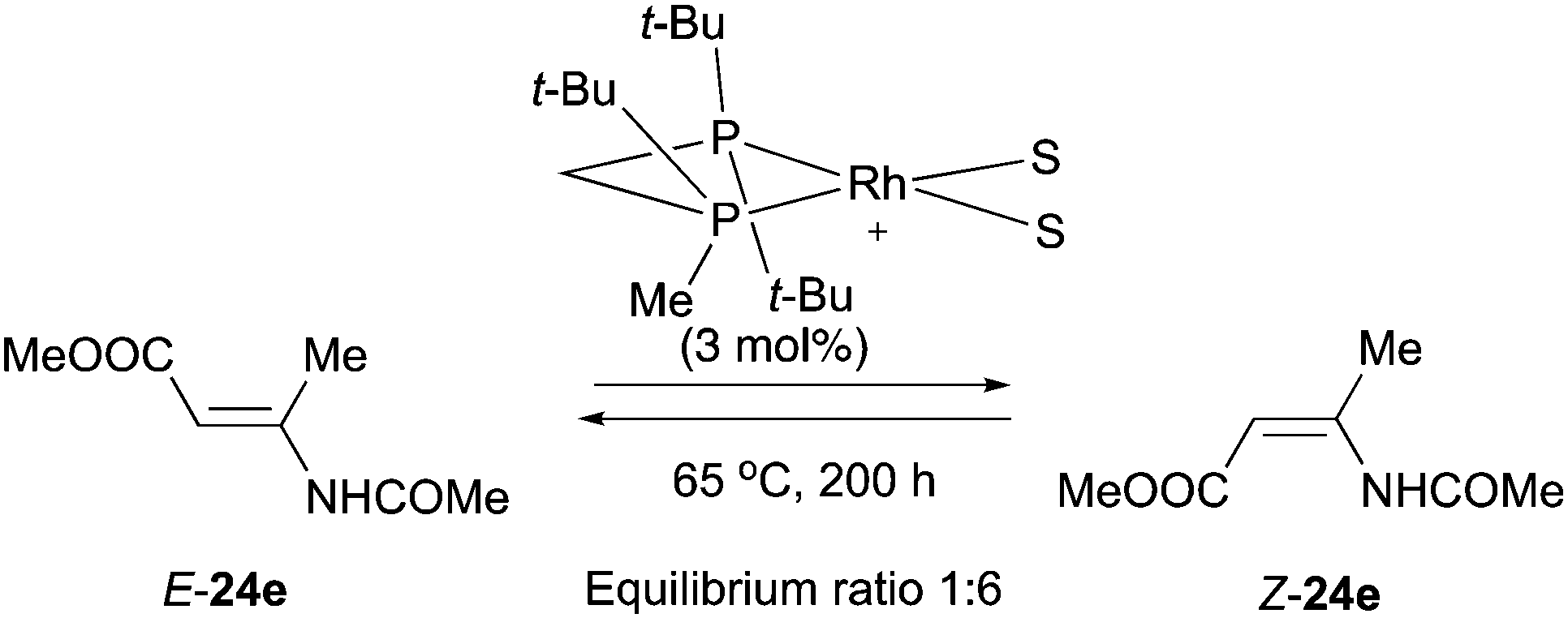
A related example was reported by Imamoto and Gridnev who were able to detect the catalytic isomerization of compound 24e in the presence of a Rh-complex. However, the isomerization was attested to be irrelevant for the catalytic cycle due to the high activation barrier at room temperature (Scheme 56).77
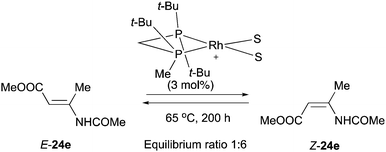 |
| | Scheme 56 | |
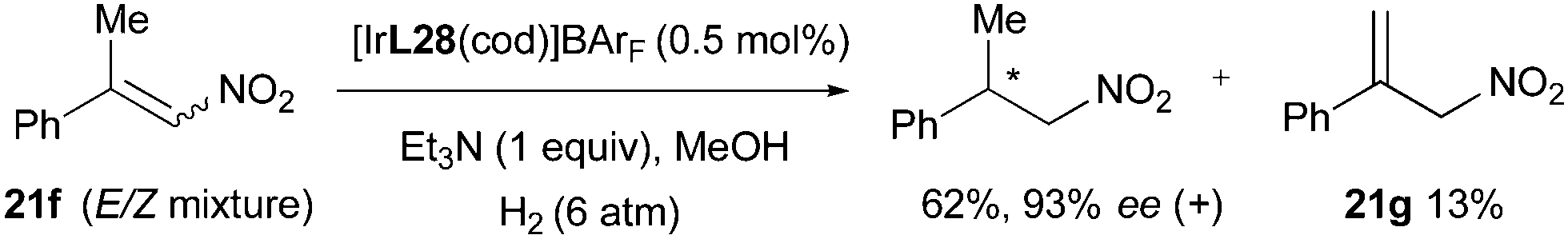
Finally, in the iridium-catalyzed hydrogenation of β,β-disubstituted nitroalkenes the reaction was found to proceed through an isomerization that produced terminal olefin intermediates and led to an enantioconvergent outcome. The formation of terminal alkene 21g was observed during the hydrogenation of an E/Z isomeric mixture 21f and the equilibrium between isomers could also be supported by deuterium experiments (Scheme 57).
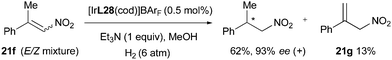 |
| | Scheme 57 | |
5. Conclusions
In the majority of the reported asymmetric hydrogenations of trisubstituted olefins, the geometry of the double bond in the substrate will control the stereochemical outcome of the reaction, leading to enantiodivergence (Fig. 8). For the iridium-catalyzed hydrogenation of unfunctionalized olefins, the enantiodivergence results from the mechanism where the olefin fits into the catalyst pocket by placing the smallest substituent (H) into the most sterically hindered quadrant (Fig. 9a). The same phenomenon was observed in Rh- and Ru-catalyzed hydrogenations of functionalized olefins, where the chelating groups were connected to the non-prochiral carbon (Fig. 9b). In both of these cases, the catalyst discriminates between the Re and Si faces of the olefin by the substituents on the non-prochiral terminus of the olefin and results in enantiodivergent reactions.
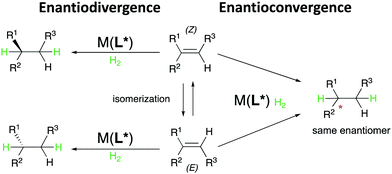 |
| | Fig. 8 Enantiodivergent and enantioconvergent pathways. | |
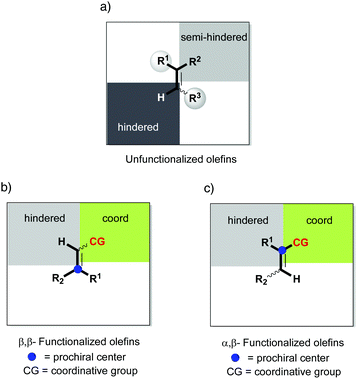 |
| | Fig. 9 Proposed selectivity models. | |
Considering the enantioconvergent hydrogenations that have been observed, the stereochemical outcome has been explained by either chelating effects or isomerization processes. Whereas the chelating effects have been supported by deuterium experiments and theoretical calculations, the cases that have been proposed to involve isomerization processes are still theoretical hypotheses that await further evidence. For the iridium-catalyzed hydrogenations, no unfunctionalized olefin has been reported to result in an enantioconvergent reaction. In a few cases Ir showed the ability to achieve enantioconvergent outcomes when hydrogenating functionalized olefins, possibly due to either chelation or isomerization processes. In contrast, Rh and Ru catalysts showed a more consistent performance in terms of enantioconvergence when the chelating groups were situated at the prochiral carbon (Fig. 9c).
Despite the above-mentioned progress, the development of versatile catalytic systems for enantioconvergent hydrogenations is still in high demand. Generally, the enantioconvergent reactions that involve chelation are often rather specific and substrate-dependent. Therefore, future challenges will involve development of enantioconvergent hydrogenations that take place via isomerization of the olefin. This is a demanding task since; (1) the catalysts must distinguish between isomeric olefins, resulting in clear preference for the matched over the mismatched isomer and (2) in the mismatched case, after delivering the first hydride, a reverse beta-hydride elimination should allow isomerization of the mismatched olefin into the matched isomer. In order to tackle these difficulties, both in depth investigations of mechanism and computational studies and new catalytic systems are required.
This field provides plenty of challenges and exciting opportunities for chemists to develop highly efficient hydrogenations that are not dependent on the geometry of the double bond. This would dramatically simplify olefin preparation and reduce costs in industry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Swedish Research Council (VR), Stiftelsen Olle Engkvist Byggmästare, Knut and Alice Wallenberg Foundation (KAW 2016.0072 and KAW 2018:0066) supported this work. We are also thankful to Mark Nolan and Adriano Zerbini for proofreading the manuscript.
Notes and references
- C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS.
- C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346–1356 CrossRef CAS PubMed.
- X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS PubMed.
- J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed.
- A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 CrossRef CAS PubMed.
- D. G. Blackmond, A. Lightfoot, A. Pfaltz, T. Rosner, P. Schnider and N. Zimmermann, Chirality, 2000, 12, 442–449 CrossRef CAS PubMed.
- P. G. Cozzi, N. Zimmermann, R. Hilgraf, S. Schaffner and A. Pfaltz, Adv. Synth. Catal., 2001, 343, 450–454 CrossRef CAS.
- J. Blankenstein and A. Pfaltz, Angew. Chem., Int. Ed., 2001, 40, 4445–4447 CrossRef CAS PubMed.
- F. Menges and A. Pfaltz, Adv. Synth. Catal., 2002, 344, 40–44 CrossRef CAS.
- D.-R. Hou, J. Reibenspies, T. J. Colacot and K. Burgess, Chem. – Eur. J., 2001, 7, 5391–5400 CrossRef CAS.
- F. Menges, M. Neuburger and A. Pfaltz, Org. Lett., 2002, 4, 4713–4716 CrossRef CAS PubMed.
- Y. Zhu, Y. Fan and K. Burgess, J. Am. Chem. Soc., 2010, 132, 6249–6253 CrossRef CAS PubMed.
- M. T. Powell, D.-R. Hou, M. C. Perry, X. Cui and K. Burgess, J. Am. Chem. Soc., 2001, 123, 8878–8879 CrossRef CAS PubMed.
- M. C. Perry, X. Cui, M. T. Powell, D.-R. Hou, J. H. Reibenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113–123 CrossRef CAS PubMed.
- G. Xu and S. R. Gilbertson, Tetrahedron Lett., 2003, 44, 953–955 CrossRef CAS.
- S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher and A. Pfaltz, Science, 2006, 311, 642–644 CrossRef CAS PubMed.
- A. Wang, B. Wüstenberg and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 2298–2300 CrossRef CAS PubMed.
- S.-M. Lu and C. Bolm, Chem. – Eur. J., 2008, 14, 7513–7516 CrossRef CAS PubMed.
- J.-Q. Li, X. Quan and P. G. Andersson, Chem. – Eur. J., 2012, 18, 10609–10616 CrossRef CAS PubMed.
- T. L. Church, T. Rasmussen and P. G. Andersson, Organometallics, 2010, 29, 6769–6781 CrossRef CAS.
- B. K. Peters, T. Zhou, J. Rujirawanich, A. Cadu, T. Singh, W. Rabten, S. Kerdphon and P. G. Andersson, J. Am. Chem. Soc., 2014, 136, 16557–16562 CrossRef CAS PubMed.
- J.-Q. Li, J. Liu, S. Krajangsri, N. Chumnanvej, T. Singh and P. G. Andersson, ACS Catal., 2016, 6, 8342–8349 CrossRef CAS.
- M.-A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2014, 53, 8668–8671 CrossRef PubMed.
- A. Wang, M. Bernasconi and A. Pfaltz, Adv. Synth. Catal., 2017, 359, 2523–2529 CrossRef CAS.
- I. Ojima, T. Kogure and N. Yoda, J. Org. Chem., 1980, 45, 4728–4739 CrossRef CAS.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- A. Miyashita, H. Takaya, T. Souchi and R. Noyori, Tetrahedron, 1984, 40, 1245–1253 CrossRef CAS.
- S. Doherty, C. H. Smyth, A. Harriman, R. W. Harrington and W. Clegg, Organometallics, 2009, 28, 888–895 CrossRef CAS.
- S. Doherty, J. G. Knight, A. L. Bell, S. El-Menabawey, C. M. Vogels, A. Decken and S. A. Westcott, Tetrahedron: Asymmetry, 2009, 20, 1437–1444 CrossRef CAS.
- J. Zhang, C. Liu, X. Wang, J. Chen, Z. Zhang and W. Zhang, Chem. Commun., 2018, 54, 6024–6027 RSC.
- Z. Zhang, Z. Han, G. Gu, X.-Q. Dong and X. Zhang, Adv. Synth. Catal., 2017, 359, 2585–2589 CrossRef CAS.
- G. Liu, Z. Han, X.-Q. Dong and X. Zhang, Org. Lett., 2018, 20, 5636–5639 CrossRef CAS PubMed.
- Q. Yan, G. Xiao, Y. Wang, G. Zi, Z. Zhang and G. Hou, J. Am. Chem. Soc., 2019, 141, 1749–1756 CrossRef CAS PubMed.
- H. Yang, E. Wang, P. Yang, H. Lv and X. Zhang, Org. Lett., 2017, 19, 5062–5065 CrossRef CAS PubMed.
- H. Takaya, T. Ohta, N. Sayo, H. Kumobayashi, S. Akutagawa, S. Inoue, I. Kasahara and R. Noyori, J. Am. Chem. Soc., 1987, 109, 1596–1597 CrossRef CAS.
- K. Fukatsu, O. Uchikawa, M. Kawada, T. Yamano, M. Yamashita, K. Kato, K. Hirai, S. Hinuma, M. Miyamoto and S. Ohkawa, J. Med. Chem., 2002, 45, 4212–4221 CrossRef CAS PubMed.
- P. Cheruku, A. Paptchikhine, T. L. Church and P. G. Andersson, J. Am. Chem. Soc., 2009, 131, 8285–8289 CrossRef CAS PubMed.
- M. Bernasconi, M.-A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2014, 53, 5385–5388 CrossRef CAS PubMed.
- Y.-B. Yu, L. Cheng, Y.-P. Li, Y. Fu, S.-F. Zhu and Q.-L. Zhou, Chem. Commun., 2016, 52, 4812–4815 RSC.
- P.-C. Yan, J.-H. Xie, G.-H. Hou, L.-X. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2009, 351, 3243–3250 CrossRef CAS.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125–10138 CrossRef CAS.
- In the following years the Burk group reported the asymmetric hydrogenation of arylalkylenamides using different DuPHOS ligands (J. Am. Chem. Soc., 1996, 118, 5142–5143). Although the authors presented the catalytic system as tolerant towards mixtures of (E) and (Z) isomers, the reported NMR chemical shifts of the starting materials, assigned for (E)/(Z) mixtures, actually match the two rotamers of the single (Z) isomer. This was found out by comparing the starting material NMR data of both isolated (E) and (Z) isomers reported by other independent research groups (J. Chem. Soc., Perkin Trans. 1, 1985, 831–836; Org. Lett., 2010, 12, 2214–2217). Other papers on asymmetric hydrogenation of enamides referred to Burk's work over the years, reporting the same incoherencies (J. Org. Chem., 1998, 63, 9590–9593; Tetrahedron: Asymm., 1998, 9, 1863–1866; Org. Lett., 1999, 1, 1679–1681; J. Org. Chem., 2000, 65, 5871–5874; Angew. Chem., Int. Ed., 2002, 41, 847–849). Due to the mismatched data, the aforementioned works have not been included in the enantioconvergent section of this review.
- G. Zhu, Z. Chen and X. Zhang, J. Org. Chem., 1999, 64, 6907–6910 CrossRef CAS PubMed.
- M. J. Burk, C. S. Kalberg and A. Pizzano, J. Am. Chem. Soc., 1998, 120, 4345–4353 CrossRef CAS.
- M. J. Burk, F. Bienewald, M. Harris and A. Zanotti-Gerosa, Angew. Chem., Int. Ed., 1998, 37, 1931–1933 CrossRef CAS.
- Z. Hu and W. Han, Tetrahedron Lett., 2008, 49, 901–902 CrossRef CAS.
- U. Schmidt, J. Langner, B. Kirschbaum and C. Braun, Synthesis, 1994, 1138–1140 CrossRef CAS.
- B. Mohar and M. Stephan, Adv. Synth. Catal., 2013, 355, 594–600 CAS.
- H.-P. Wu and G. Hoge, Org. Lett., 2004, 6, 3645–3647 CrossRef CAS PubMed.
- M. J. Burk, P. D. de Koning, T. M. Grote, M. S. Hoekstra, G. Hoge, R. A. Jennings, W. S. Kissel, T. V. Le, I. C. Lennon, T. A. Mulhern, J. A. Ramsden and R. A. Wade, J. Org. Chem., 2003, 68, 5731–5734 CrossRef CAS PubMed.
- G. Hoge, J. Am. Chem. Soc., 2003, 125, 10219–10227 CrossRef CAS PubMed.
- M. Birch, S. Challenger, J.-P. Crochard, D. Fradet, H. Jackman, A. Luan, E. Madigan, N. McDowall, K. Meldrum, C. M. Gordon, M. Widegren and S. Yeo, Org. Process Res. Dev., 2011, 15, 1172–1177 CrossRef CAS.
- J. I. Grayson, J. Roos and S. Osswald, Org. Process Res. Dev., 2011, 15, 1201–1206 CrossRef CAS.
- I. V. Komarov and A. Börner, Angew. Chem., Int. Ed., 2001, 40, 1197–1200 CrossRef CAS PubMed.
- D. Peña, A. J. Minnaard, J. G. de Vries and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 14552–14553 CrossRef PubMed.
- X. Wang, A. Guram, S. Caille, J. Hu, J. P. Preston, M. Ronk and S. Walker, Org. Lett., 2011, 13, 1881–1883 CrossRef CAS PubMed.
- W. Gao, H. Lv and X. Zhang, Org. Lett., 2017, 19, 2877–2880 CrossRef CAS PubMed.
- T. Ohta, T. Miyake, N. Seido, H. Kumobayashi, S. Akutagawa and H. Takaya, Tetrahedron Lett., 1992, 33, 635–638 CrossRef CAS.
- J. Y. L. Chung, D. Zhao, D. L. Hughes, J. M. McNamara, E. J. J. Grabowski and P. J. Reider, Tetrahedron Lett., 1995, 36, 7379–7382 CrossRef CAS.
- H. Jendralla, Tetrahedron: Asymmetry, 1994, 5, 1183–1186 CrossRef CAS.
- G. Beck, H. Jendralla and B. Kammermeier, Tetrahedron, 1994, 50, 4691–4698 CrossRef CAS.
- M. Saburi, L. Shao, T. Sakurai and Y. Uchida, Tetrahedron Lett., 1992, 33, 7877–7880 CrossRef CAS.
- Y.-G. Zhou, W. Tang, W.-B. Wang, W. Li and X. Zhang, J. Am. Chem. Soc., 2002, 124, 4952–4953 CrossRef CAS PubMed.
- J. Wu, X. Chen, R. Guo, C.-h. Yeung and A. S. C. Chan, J. Org. Chem., 2003, 68, 2490–2493 CrossRef CAS PubMed.
- X. Li, C. You, S. Li, H. Lv and X. Zhang, Org. Lett., 2017, 19, 5130–5133 CrossRef CAS PubMed.
- J. Long, W. Gao, Y. Guan, H. Lv and X. Zhang, Org. Lett., 2018, 20, 5914–5917 CrossRef CAS PubMed.
- P. Brandt, C. Hedberg and P. G. Andersson, Chem. – Eur. J., 2003, 9, 339–347 CrossRef CAS PubMed.
- J. Zhou, J. W. Ogle, Y. Fan, V. Banphavichit, Y. Zhu and K. Burgess, Chem. – Eur. J., 2007, 13, 7162–7170 CrossRef CAS PubMed.
- F. Maurer and U. Kazmaier, J. Org. Chem., 2013, 78, 3425–3428 CrossRef CAS PubMed.
- Y. Liu, I. D. Gridnev and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1901–1905 CrossRef CAS PubMed.
- J. Engel, S. Mersmann, P.-O. Norrby and C. Bolm, ChemCatChem, 2016, 8, 3099–3106 CrossRef CAS.
- M.-L. Li, S. Yang, X.-C. Su, H.-L. Wu, L.-L. Yang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 541–547 CrossRef CAS PubMed.
- I. D. Gridnev and T. Imamoto, Chem. Commun., 2009, 7447–7464, 10.1039/B912440C.
- J. Halpern, D. P. Riley, A. S. C. Chan and J. J. Pluth, J. Am. Chem. Soc., 1977, 99, 8055–8057 CrossRef CAS.
- A. S. C. Chan, J. J. Pluth and J. Halpern, J. Am. Chem. Soc., 1980, 102, 5952–5954 CrossRef CAS.
- J. Halpern, Science, 1982, 217, 401 CrossRef CAS PubMed.
- I. D. Gridnev, Y. Liu and T. Imamoto, ACS Catal., 2014, 4, 203–219 CrossRef CAS.
Footnote |
| † These two authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jia
Zheng†
,
Jia
Zheng†