
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated carbohydrates as chemical probes for molecular recognition studies. Current status and perspectives
Bruno
Linclau
 a,
Ana
Ardá
b,
Niels-Christian
Reichardt
c,
Matthieu
Sollogoub
d,
Luca
Unione
e,
Stéphane P.
Vincent
f and
Jesús
Jiménez-Barbero
*bgh
a,
Ana
Ardá
b,
Niels-Christian
Reichardt
c,
Matthieu
Sollogoub
d,
Luca
Unione
e,
Stéphane P.
Vincent
f and
Jesús
Jiménez-Barbero
*bgh
aSchool of Chemistry, University of Southampton, Highfield, Southampton SO171BJ, UK
bCIC bioGUNE, Basque Research and Technology Alliance (BRTA), 48160 Derio, Spain. E-mail: jjbarbero@cicbiogune.es
cCIC biomaGUNE, Basque Research and Technology Alliance (BRTA), Donostia-San Sebastian, Spain
dSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, UMR 8232, 4 place Jussieu, 75005 Paris, France
eDepartment of Chemical Biology and Drug Discovery, Utrecht Institute for Pharmaceutical Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands
fDepartment of Chemistry, Laboratory of Bio-organic Chemistry, University of Namur (UNamur), B-5000 Namur, Belgium
gIkerbasque, Basque Foundation for Science, Maria Diaz de Haro 3, 48013 Bilbao, Spain
hDepartment of Organic Chemistry II, Faculty of Science and Technology, UPV/EHU, 48940 Leioa, Spain
First published on 10th June 2020
Abstract
This review provides an extensive summary of the effects of carbohydrate fluorination with regard to changes in physical, chemical and biological properties with respect to regular saccharides. The specific structural, conformational, stability, reactivity and interaction features of fluorinated sugars are described, as well as their applications as probes and in chemical biology.
 Bruno Linclau | Bruno Linclau is Professor of Organic Chemistry at School of Chemistry, University of Southampton. He earned his Licentiate in Sciences (Chemistry) degree from the University of Ghent (Belgium), where he also obtained his PhD in 1996 with Prof. Maurits Vandewalle. He carried out postdoctoral research with Professor Dennis P. Curran at the University of Pittsburgh, Pittsburgh PA (USA) with a fellowship from the Belgian American Educational Foundation. He joined the faculty at Southampton University in 1999. |
 Ana Ardá | Ana Ardá studied Chemistry at the University of A Coruña, from where she received her doctorate in Chemistry in 2006. Between 2006 and 2008 she carried out a postdoctoral stay in at Utrecht University, with Prof. Hans Kamerling where she started to work with carbohydrates. In 2008 she moved to Centro de Investigaciones Biológicas (CIB-CSIC) in Madrid to join Prof. Jiménez-Barbero's group. In 2014 she moved to CIC bioGUNE in the Basque Country, and since December 2015 she is a Ramón y Cajal Fellow. Her research interests are focused on the study of glycan–protein interactions through a multidisciplinary approach, with a special focus on NMR techniques. |
 Niels-Christian Reichardt | Niels Reichardt is group leader at CIC biomaGUNE. He received his PhD in Chemistry from Konstanz University, Germany. His postgraduate studies on projects in synthetic carbohydrate chemistry were carried out at the Institute for Chemical Research of the CSIC in Seville. After a short postdoctoral stay with Professor Nicola Winnsinger at the University Luis Pasteur, Strasbourg, he moved to industry to work as a Research Scientist for Glycoform Ltd, Oxford, developing synthetic therapeutic glycoproteins until 2006. In 2006, he became group leader for glycotechnology at CIC biomaGUNE in San Sebastian. His current research interests include the synthesis of oligosaccharides and glycomimetics of biological interest, array-based functional glycomics and the development of quantitative glycan profiling methods for biotechnology and diagnostics. |
 Matthieu Sollogoub | Matthieu Sollogoub is Professor of Molecular Chemistry at Sorbonne University where he conducts his research team on various aspects of carbohydrate chemistry. He focuses mainly on cyclodextrin functionalization for catalysis and supramolecular assembly, he also explores carbohydrate mimickry. He obtained his PhD at the Ecole Normale Supérieure (ENS) in Paris in 1999 with Prof. P. Sinaÿ. He carried out postdoctoral research at the University of Southampton with Prof. T. Brown and joined the faculty of ENS and Sorbonne in 2001. In 2011, he received the Carbohydrate Research Award for Creativity in Glycoscience. |
 Luca Unione | Luca Unione studied Chemistry at the University of Napoli Federico II (Italy). He received the PhD in 2016 in Madrid (Spain), in the framework of the ITN-Marie Skłodowska-Curie, working in the field of structural characterization of carbohydrates, glycomimetics and their interactions with glycan binding proteins. He carried out a postdoctoral stay in the company Atlas molecular Pharma founded by the Center for Cooperative Research in Bioscience (CIC bioGUNE) in Biscay (Spain). The main aim of the company is to discover first-in-class, innovative therapeutics for the treatment of Rare and Ultra-Rare Diseases. In 2019 he moved to Utrecht University (The Netherlands) as postdoctoral fellow under the International Human Frontier Science Program (HFSP). His research interest is focused on the understanding of the role of glycans as regulators in biomedical processes, with a special focus on chemo-enzymatic synthesis of glycans and the study of glycan–protein interactions through a multidisciplinary approach. |
 Stéphane P. Vincent | Stéphane Vincent received his Master and PhD degrees in bioorganic chemistry from the Université Louis Pasteur (Strasbourg, France) with Professor Charles Mioskowski. After postdoctoral studies first at The Scripps Research Institute in the research group of Professor Chi-Huey Wong and then in Strasbourg with Prof. Jean-Marie Lehn, he took up a permanent position as a CNRS researcher in 2001 at the Ecole Normale Supérieure (Paris). In 2004, he began an appointment at Université de Namur (Belgium), as Assistant Professor and then, Full Professor in 2014. His research interests include the glycosciences, biocatalysis, and mechanistic enzymology. |
 Jesús Jiménez-Barbero | Jesús Jiménez-Barbero is Ikerbasque Research Professor and Scientific Director of CIC bioGUNE since 2014. He received his PhD in 1987 at Madrid. After postdoctoral stays at Zürich, Mill Hill, and Pittsburgh, he returned to Madrid (CSIC) and started to work on protein–carbohydrate interactions. In 2002 he was promoted to CSIC Research Professor at CIB, where he developed his scientific activity until he moved to Bilbao. His scientific interest is focused in the field of molecular recognition and Chemical Glycobiology, employing a multidisciplinary approach that combines synthesis, biochemistry, molecular biology, molecular modeling, and especially NMR, enjoying a wide network of scientific collaborations worldwide. |
1 Introduction
Carbohydrates are essential molecules for life. However, in contrast to the other important classes of biomolecules, their broad significance beyond as an energy source has been questioned until recently. This has been due to the difficulties associated with their study, caused both by their structural and chemical complexity as well as their non-template driven biosynthesis. Their structural roles in plants and in bacterial cell walls have been long known, however their key roles in immune regulation processes, cell–cell interactions (e.g., fertilization, inflammation)1,2 or in host–pathogen interactions3 have only become clear more recently. In addition, their relationship with different diseases, such as cancer,4–7 infection, and autoimmunity,8 is nowadays well-established and is a research area under intense development.The progress made in the last decades in Glycoscience,9 advancing our understanding on the functional role of the molecular recognition processes of sugars by lectins and antibodies, as well as on the chemical and structural details of the enzymatic machineries for their processing has brought about the emerging field of glycomimetics.10 These synthetic sugar analogues, potential drug candidates, are small molecules designed to interact with glycan-binding or glycan-processing proteins.11 They are structurally related to carbohydrates but endowed with adapted properties through chemical modifications.12 One of the most employed alterations consists in introducing fluorine, the less abundant halogen in metabolites in Nature, but most abundant in pharmaceuticals,13–15 where its strategic incorporation is part of the drug development process to optimise physicochemical, adsorption, and distribution properties.16,17
However, the first examples of the employment of fluorine-containing sugar analogues typically had no drug design ambitions, but were used as probes for studying glycan–protein interactions. In particular, early studies utilized N-fluoroacetylglucosamine derivatives as probes for binding to different proteins, and exploited 19F chemical shift perturbations or line broadening effects upon binding.18–20 Several authors proposed to systematically substitute every –OH of the pyranose rings by a fluorine atom as a probe for deducing hydrogen bonding networks in sugar–receptor complexes.21–26
These pioneering works paved the way for further developments, which continue evolving, in which fluorine is introduced in sugar molecules not only for studying glycan binding phenomena, but also for interrogating glycosylation reactions (enzymatic and non-enzymatic), to stabilize glycosides, or as a manner to develop glycosidase inhibitors. This review will focus on the use of fluorinated carbohydrate derivatives in the frontier between chemistry and biology, and includes a brief overview on how fluorination influences the properties of saccharides. The use of fluorinated iminosugars has been reviewed recently27 and is not included here. The application of 19F NMR in the structural and conformational determination of fluorinated sugar derivatives has also been reviewed and will not be covered here.28
2 The different types of sugar fluorination
Sugar fluorination usually refers to the replacement of a hydroxyl group by a fluorine atom, which formally is a ‘deoxyfluorination’ operation. Perhaps the most famous example is 2-deoxy-2-fluoroglucose 2.1 (Fig. 1), whose 18F analogue is one of the most used PET imaging agents. However, there are other sugar fluorination options: in gemcitabine 2.2, an anti-cancer agent, a secondary alcohol is replaced by a CF2-group, formally a ‘deoxydifluorination’ operation. Sugar fluorination, in which a C–H is replaced by C–F, is also possible, and mono-, di- and trifluorination processes have all been described. The most known examples include 6,6,6-trifluorofucose 2.3, and 3-fluorinated sialic acid 2.4. Note that in the latter, C3 now becomes stereogenic (see below). Finally, the substitution of the endo or exocyclic (anomeric) oxygen for CHF/CF2 has also been described,29 generating fluoro-C-glycosyl compounds (O1) or fluoro-carbasugars (O5). An example of the latter is 2.5. There are also other types, such as C–H for C–RF (RF is perfluoroalkyl) modifications leading to branched sugar derivatives,30 which will not be covered here.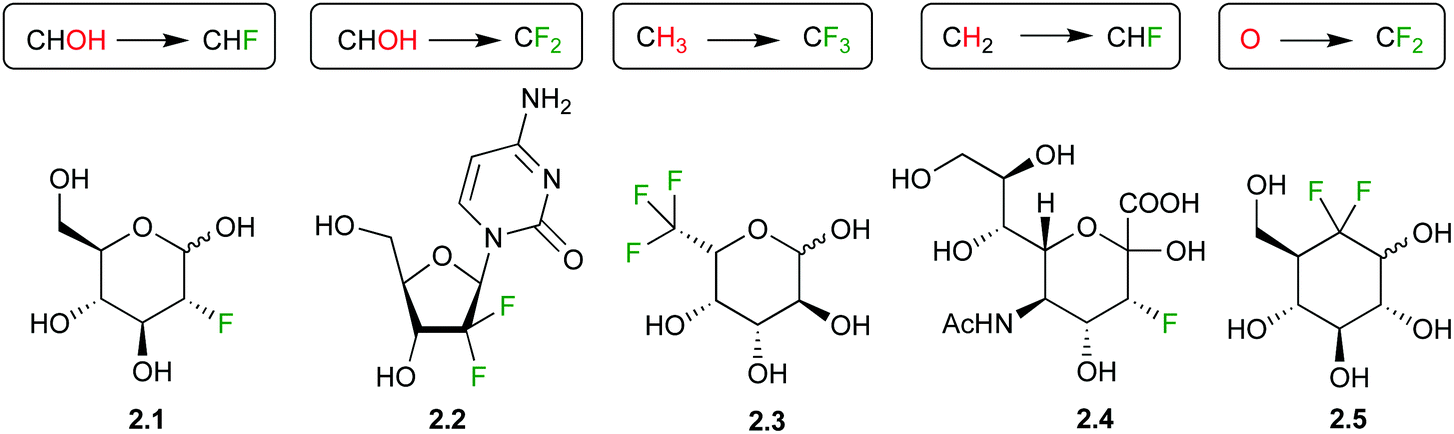 | ||
| Fig. 1 Examples of fluorinated carbohydrate derivatives featuring different fluorination types and motifs. | ||
As indicated above, a deoxyfluorination modification of a sugar is indicated by the prefix ‘deoxy’. Equally, if fluorination of a sugar is not accompanied by a deoxygenation reaction, then the position of fluorination is indicated. Hence, 2.6 is 6-fluorofucose (Fig. 2), although 6-deoxy-6-fluorogalactose is also correct. However, 2.1 should not be referred to as ‘2-fluoroglucose’, as this name corresponds to 2.7. Structure 2.7 is unstable and will decompose to the corresponding 2-keto derivative. However, fluorination at the 5-position as in 2.8 is possible. Although from a nomenclature point of view, 2.9 can thus be named ‘2-deoxy-2,2-difluoroglucose’, due to the loss of stereogenicity at C2, convention dictates that deoxysugar nomenclature is followed. Hence, 2.9 is referred to as 2-deoxy-2,2-difluoro-D-arabino-hexopyranose.
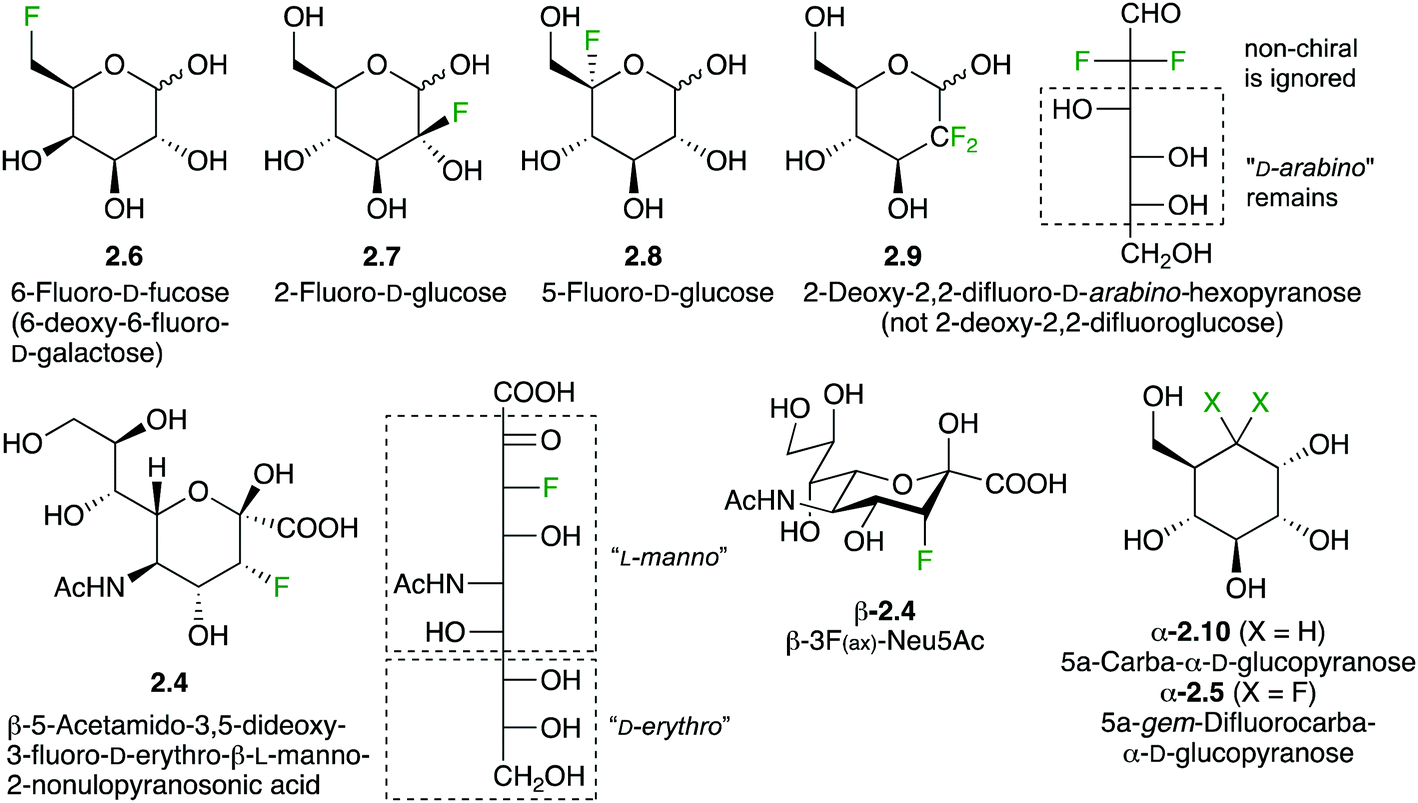 | ||
| Fig. 2 Typical nomenclature employed for fluorinated carbohydrate derivatives. | ||
Interestingly, 3-fluorinated sialic acid is often named after its C3-deoxyfluorinated nonulosonic acid derivative. Hence, 2.4 is named 5-acetamido-3,5-dideoxy-3-fluoro-D-erythro-L-manno-2-nonulopyranosonic acid. Using sialic acid as a naming base, the configuration at C3 needs to be specified, leading to (3R)-3F-Neu5Ac. However, a much more convenient way has been used for naming 3-fluorinated sialic acids by referring to the orientation of the fluorine substituent on the sialic acid chair conformation. Compound 2.4 is then named 3F(ax)-Neu5Ac. The difluorinated 2.10 is named according to carbasugar nomenclature as indicated.31
3 Influence of carbohydrate fluorination on physical properties
3.1 The fluorine atom and the C–F bond
Oxygen and fluorine are the two most electronegative atoms, but their difference in electronegativity results in a number of different properties. While they have a similar size, the higher ionization potential of the fluorine lone pair makes it a worse hydrogen bond acceptor with less polarizable lone pairs than oxygen. While the divalent oxygen allows for the presence of a covalent O–H bond, this is not the case for the monovalent fluorine, so a deoxyfluorination leads to the loss of hydrogen bond donating capacity at that position. Hence, this leads to a C–F group being much more hydrophobic than an alcohol group. A CH–OH is larger than a CH–F substituent, but similar in volume to a CF2-group.Due to its high electronegativity and absence of any other substituent (the alcohol oxygen also has an O–H bond), the C–F bond is highly polarized, which will affect the electron density of the carbohydrate structure. This is particularly pronounced when a C–H group is changed for C–F. Hence, adjacent functional groups are affected, which has consequences for their interactions with proteins.
3.2 The influence on carbohydrate structure and conformation
The conformation of the ligand is of great importance when considering protein–carbohydrate interactions.32 The presence of the fluorine may, in principle, affect the conformational properties of the fluorine-containing ligands at different levels, the monosaccharide rings, the pendant groups, the glycosidic linkages; as well as global physicochemical properties. | ||
| Fig. 3 Ring conformation of fluorinated carbohydrate derivatives. | ||
Only a few conformational studies have been performed for fluorocarbapyranoses. Nevertheless, conformational studies of α- and β-gem-difluorocarbaglucose, galactose and mannose derivatives using NMR, molecular modeling and, when possible, X-ray crystallography indicated that the presence of the CF2 moiety in the ring does not significantly affect the 4C1 chair conformation compared to the regular sugar.39 Furthermore, a study on 5a-gem-difluorocarbaidoses showed that it retained the conformational plasticity of its oxygenated counterpart.40 In the case of fluoro-C-glycosides, the situation is more diverse, as 4C1 but also other conformations (1C4 and 1S3) were observed in solution with galactose analogues.41
 | ||
| Fig. 4 Preferred conformations of the exocyclic fluoromethyl group. | ||
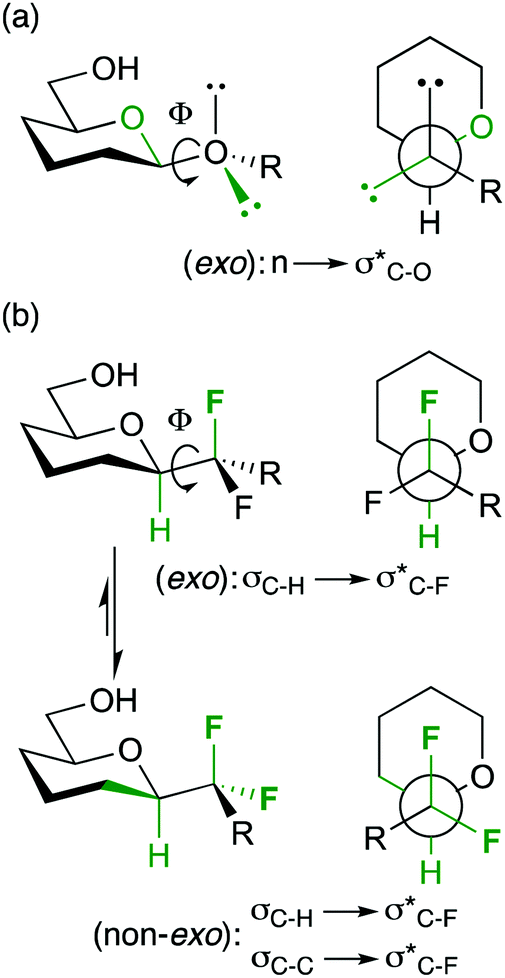 | ||
| Fig. 5 The anomeric and fluorine gauche effects determining anomeric conformation. | ||
Hence, the maximisation of σ*C–H/σ*C–F and σC–C/σ*C–F interactions might therefore stabilize “non-natural” conformations that would be high in energy for the parent O-glycoside, which has been demonstrated for a number of fluorinated C-glycosides including 3.941 (Fig. 6) and CHF and CF2C-glycoside analogues 3.12–3.14 of β-Gal-(1,1)-α-Man 3.10.50
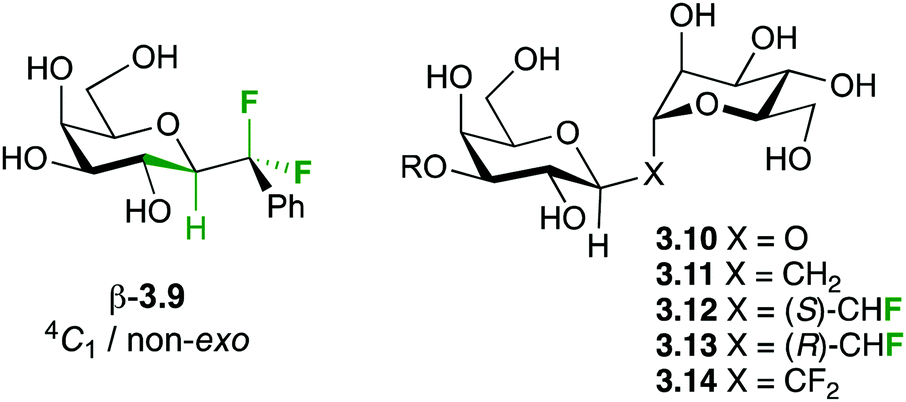 | ||
| Fig. 6 Substrates investigated for glycosidic bond conformation. | ||
In the exo-anomeric effect, hyperconjugation occurs between a lone pair orbital of the anomeric oxygen with the σ*C1–O5 bond, which is more favourable than with the σ*C1–C2 bond. This can be attributed to the distinct polarization of the C1–O5 bond, which results a larger σ* orbital centered on the less electronegative atom of the polarized bond. (3.15, Fig. 7a). This distinction disappears with carbasugars in which the C1–C2 and C1–C5a have the same polarity, therefore no preferential conformation is observed (3.16, Fig. 7b). In order to restore the conformational restriction caused by the exo-anomeric effect with carbasugars, a CF2-carbasugar as a “stereoelectronic” mimic for O5 was investigated. Indeed, it was demonstrated that the replacement of the endocyclic oxygen atom by a CF2 group in maltose induced a polarization of the C1–CF2 bond, which was shown to restore the exo-anomeric effect, and the preferential exo conformation (3.17, Fig. 7c).51
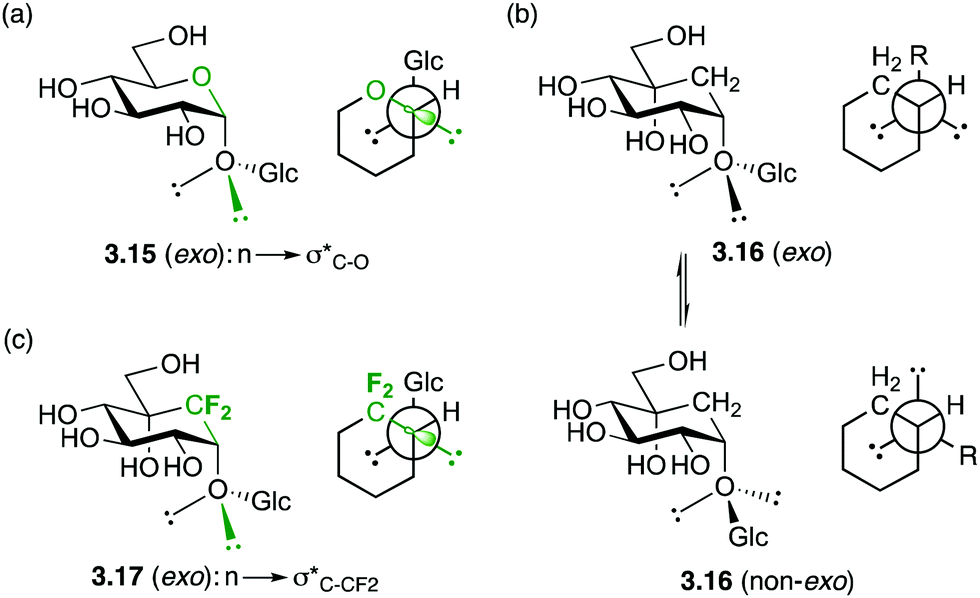 | ||
| Fig. 7 Schematic representation of the lone pair–σ* interactions responsible for the exo-anomeric effect and exo conformation in glycosides and gem-difluoro-carbaglycosides. | ||
| Deoxy-fluorination position | Glucose | Galactose | ||
|---|---|---|---|---|
| %α | %β | %α | %β | |
| — | 35 | 65 | 31 | 69 |
| 2 | 45 | 55 | 42 | 58 |
| 3 | 48 | 52 | 43 | 57 |
| 4 | 44 | 56 | 37 | 63 |
| 6 | 43 | 57 | 37 | 63 |
The solution-phase composition of galactose and its deoxyfluorinated analogues, in particular the equilibrium between the pyranose and furanose forms, has been extensively studied in the context of investigations of the enzyme galactose mutase (Table 2). In addition to the minor differences discussed above, galactose deoxyfluorination at the 2 or 3-position decreases the amount of furanose,53 an effect also observed for talose.54 However, galactose deoxyfluorination at the 6-position (to give 6-fluorofucose) results in an increase of furanose content,55 an effect that is further magnified upon trifluorination at this position.56 The same can be seen when the C6-hydroxymethyl group in altrose is replaced by a trifluoromethyl group.56 It is likely that fluorination at the 6-position reduces the nucleophilicity of the 5-OH group, favouring involvement of the 4-OH group in the hemiacetal ring formation.
| Mono-saccharide | Modification | α-Pyranose | β-Pyranose | α-Furanose | β-Furanose | Total furanose | Ref. |
|---|---|---|---|---|---|---|---|
| a Fucose. | |||||||
| Gal | — | 31.8 | 60.5 | 3.1 | 4.6 | 7.7 | 53 |
| 2F | 41.0 | 55.7 | 1.0 | 2.2 | 3.2 | 53 | |
| 3F | 40 | 58 | 0.7 | 1.6 | 2.3 | 53 | |
| 6F | 30 | 56 | 5 | 8 | 13 | 55 | |
| 6-Deoxya | 28 | 67 | 5 | 5 (3.7) | 56 and 57 | ||
| 6,6,6-Tri-Fa | 29 | 43 | 11 | 17 | 28 | 56 | |
| Alt | — | 30 | 41 | 18 | 11 | 29 | 56 |
| 6,6,6-Tri-F | 14 | 20 | 33 | 33 | 66 | ||
| Tal | — | 42.0 | 29.0 | 16.0 | 13.0 | 29 | 57 |
| 2F | 50 | 37 | 8.0 | 5.0 | 13 | 54 | |
The enzyme mutarotase catalyses the equilibrium between sugar anomers. Using two-dimensional exchange spectroscopy (2D-EXSY) at equilibrium, it was shown that mutarotase catalyses the rapid (time scale of a few seconds) exchange between the anomers of 4-deoxy-4-fluoro-D-glucose, but not that of glucose analogues with fluorination at the 2- and/or 3-position.58 This was explained by the electron withdrawing effect destabilizing the required free aldehyde group of the open form. Nevertheless, dissolution of a pure crystal of the β-anomer of 6-deoxy-6,6,6-trifluoroaltrose was reported to ‘soon’ give the equilibrium mixture.54
3.3 The stability of the glycosidic bond
The stronger inductive effect of fluorine compared to OH renders oxocarbenium formation more difficult for fluorinated carbohydrate derivatives compared to the native parents.59 This was realised very early on through hydrolysis studies of glycosyl phosphates and dinitrophenylglycosides.60 In general, hydrolysis rates decrease in the following order (Table 3): parent > 6-deoxyfluoro > 3-deoxyfluoro > 4-deoxyfluoro > 2-deoxyfluoro (with an inverse order for the deoxygenated analogues). Through a detailed study of dinitrophenylglycoside hydrolysis, it was established that the hydrolysis rates are largely dictated by the field effect on the oxocarbenium transition state, with significant electron deficient character on O5. Interestingly, dideoxydifluorination at the 3 or 4 and 6-positions leads to a hydrolysis rate equivalent to that of deoxyfluorination at 2-position, and dideoxymonofluorination actually leads to a higher rate constant than that of the parent derivative.| Compound | 105kobs (s−1) | |
|---|---|---|
| 25 °C | 45 °C | |
| a Extrapolated value. | ||
| Not substituted | 4.10 | |
| 2-Deoxy | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
|
| 2-Deoxy-2-fluoro | 0.068 | |
| 4-Deoxy | 111 | |
| 4-Deoxy-4-fluoro | 0.270 | |
| 2,6-Dideoxy-2,6-difluoro | 0.0075a | 0.19 |
| 3,6-Dideoxy-3,6-difluoro | 0.080a | 1.4 |
| 3,6-Dideoxy-6-fluoro | 6.1 | |
| 4,6-Dideoxy-4,6-difluoro | 0.032 | 0.79 |
| 4,6-Dideoxy-6-fluoro | 26 | |
The destabilizing effect on oxocarbenium transition state formation has been successfully exploited for ‘mechanism-based’ enzyme inhibition (first reports,61 reviews62). Deoxyfluorinated carbohydrates which display an excellent leaving group at the anomeric position can still undergo reaction with nucleophiles, including the catalytic nucleophile of retaining glycosidase enzymes, in contrast to the thus formed glycosyl-enzyme intermediate, which is formally a – less reactive – anomeric ester derivative. Hence, for glycosidases for which the hydrolysis of the glycosyl enzyme intermediate with the natural substrate is rate-determining, inactivation is possible by employing a fluorinated analogue, typically using fluorination adjacent to the anomeric center but also at the 5-position. In this regard, an interesting illustration of the effects of fluorination on anomeric reactivity is shown with the fluorinated maltoses 3.1863 and 3.1964 (Fig. 8), which were investigated as mechanism-based enzyme inhibitors for the maltosyl transferase GlgE1 from Streptomyces coelicor. While 3.18 was shown to form the covalent intermediate by reaction with the relevant nucleophilic Asp residue,63 the enzyme was able to subsequently effect hydrolysis. This could only be prevented with a E423A point mutation. However, with the trifluorinated maltose 3.19,64 the increased deactivation resulting from C2-difluorination rendered it too unreactive to react with the enzyme. Interestingly, this allowed crystallization of 3.19 in complex with GlgE1 (see below).
 | ||
| Fig. 8 Fluorinated maltose derivatives as mechanism-based inhibitors.63,64 | ||
Hence, disaccharides with a 2′-deoxyfluorination modification can still react with glycosidases, causing mechanism-based inhibition. Indeed, if the binding energy component provided by the natural aglycon (or the reducing sugar moiety) is large enough, then the rate of the initial reaction of the catalytic nucleophile with the fluorinated glycoside derivative can be increased. This fact has been exploited for the design of selective mechanism-based glycosidase inhibitors.65
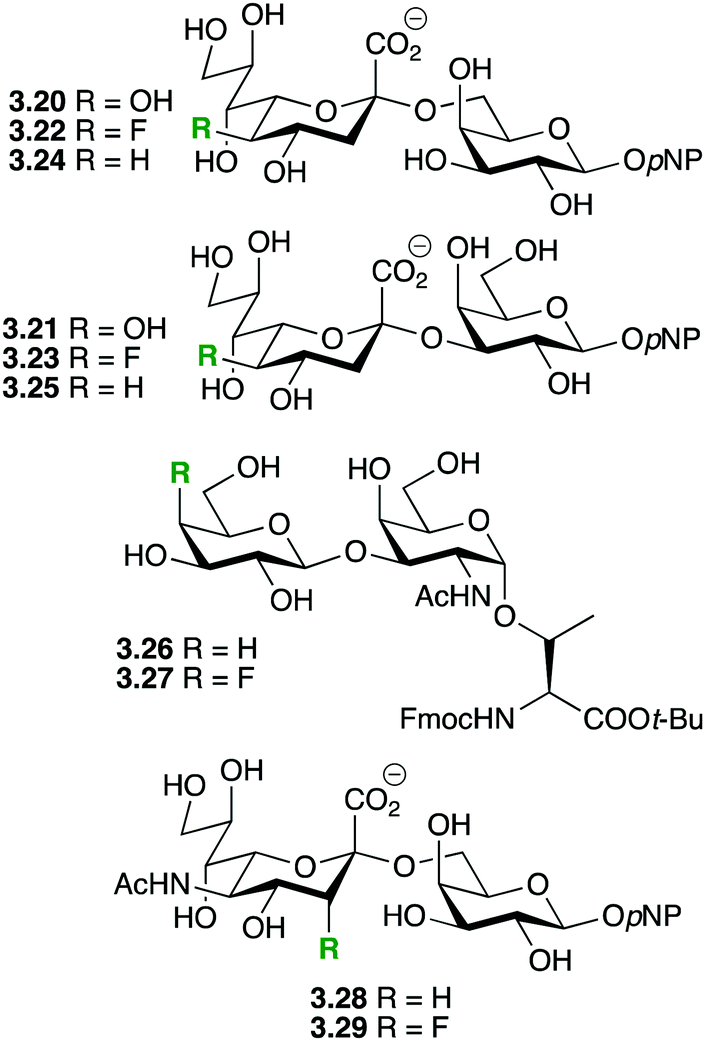
The same deactivating effect on oxonium formation would result in saccharides modified by deoxyfluorination at the non-reducing 2-position(s) being stabilized against both acid-catalysed and enzymatic hydrolysis. Recent examples of this strategy are shown in Fig. 9.
 | ||
| Fig. 9 Stabilisation of the glycosidic bond by fluorine introduction. | ||
Chen and coworkers showed that deoxyfluorination of the 5-OH of KDN in sialosides 3.20 and 3.21 leading to 3.22 and 3.23 did not significantly change the activity of the multifunctional Pasteurella multocida sialyltransferase (PmST1),66 which also possesses sialidase activity. In contrast, PmST1 displayed a significantly increased sialidase activity towards the 5-deoxygenated disaccharide 3.24, but not towards 3.25. It was concluded that the hydrogen bonding properties of the 5-OH group were not important for PmST1 activity. Hoffmann-Röder and coworkers investigated the enzymatic stability of the Thomson–Friedenreich antigen derivatives 3.26 and 3.27.67 A β-galactosidase catalyzed the hydrolysis of 3.16 with a half-life of approximately 3 h, while the fluorinated 3.27 remained intact over 7 h of incubation. Wong and coworkers showed that in contrast to 3.28, sialidases from C. perfingens and V. cholera were inactive towards 3.29.68a Also, 3.29 did not significantly inhibit the hydrolysis of 3.28.68
3.4 Lipophilicity
The lipophilicity of a compound is measured experimentally by determining its partition coefficient P between 1-octanol and water, and expressed as logP.69 It reflects a balance of the hydrophobic and hydrophilic features of the compound. This value is widely used in medicinal chemistry as a measure for membrane permeability, with optimum values between +1 and +3 (for orally available drugs). It also impacts affinity to proteins as it roughly relates to the extent of hydrophobic desolvation energy involved in the binding process. Hence, changing a compound's lipophilicity will affect affinity.
Carbohydrates are very hydrophilic, resulting in low logP values. However, there are very few experimental data available, in part reflecting the difficulty in measuring accurate sugar concentrations. The logP of Glc is reported to be −2.82 to −3.24.70 The replacement of hydroxyl groups by fluorine is expected to increase a compound's lipophilicity. Using a 19F NMR based method, the logP value has been experimentally quantified for a range of deoxyfluorinated carbohydrate derivatives (Fig. 10).35,46,71
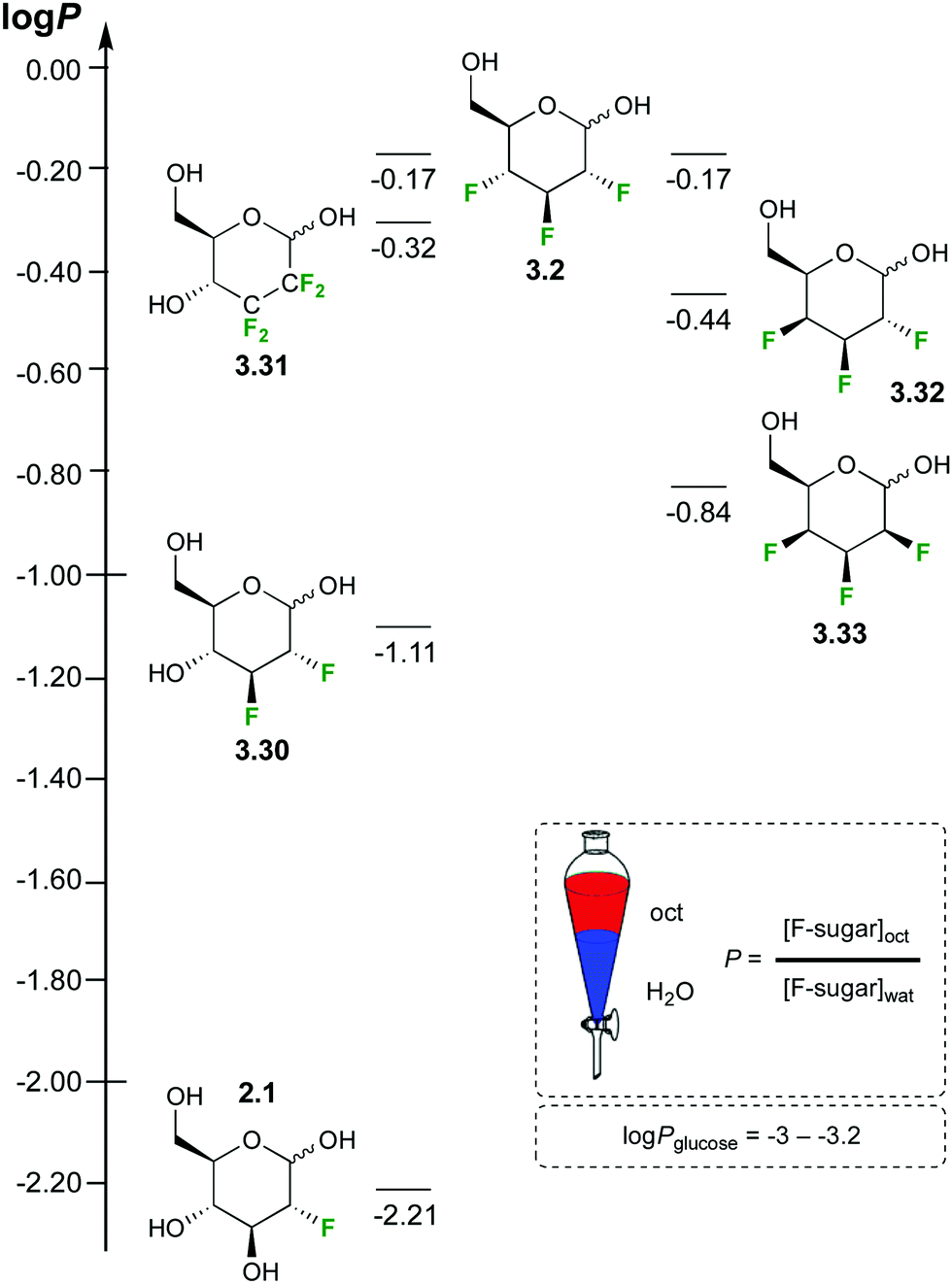 | ||
| Fig. 10 Lipophilicities of fluorinated sugar derivatives.35,46,71 | ||
For Glc derivatives, monodeoxyfluorination leads to a logP increase of almost an order of magnitude (−2.21 for 2.1), raising by another order of magnitude for a dideoxydifluorination (−1.11 for 3.30). Dideoxytetrafluorination leads to yet another order of magnitude increase (−0.32 for 3.31), as does trideoxytrifluorination in 3.2. The position of the fluorination (e.g. 6-deoxy-6-fluoroglucose, logP −2.36) and the carbohydrate stereochemistry (e.g. 2-deoxy-2-fluorogalactose, logP −2.37) also impact on the logP value (not shown). The lipophilicity of 2,3,4-trideoxy-2,3,4-trifluorinated monosaccharide derivatives can vary considerably, with the talose derivative 3.33 being much more polar compared to the Glc analogue 3.32.
4 Molecular interactions influenced by fluorination
4.1 CH–π interactions
Hydrogen bonding and CH–π interactions are the two key direct protein–sugar interactions, and both are influenced by fluorination. CH–π interactions are of paramount importance to stabilize the corresponding complexes72,73 and their importance is also substantiated by the relative abundance of aromatic amino acids in the binding pocket of lectins.74 CH–π interactions are usually described as weak polar interactions in which the delocalized electron density of sp2-hybridized aromatic moieties acts as an acceptor, while the hydrogen atom on the polarized C–H moiety acts as donor.75 In sugars, the CH donor groups are polarized by the geminal hydroxyl groups, making the CH-vector a suitable moiety for establishing polar interactions. Although oxygen and fluorine are the two most electronegative atoms, their intrinsic differences could influence their relative ability to participate in CH–π interactions with proteins, as demonstrated for hydrogen-bonded interactions. Moreover, in hydroxyl groups, the oxygen is already attached to a hydrogen atom, which reduces the electron withdrawing character of the C–O bond. Thus, the key question here is how fluorine influences the capacity of deoxyfluorosugars to participate in CH–π interactions.76 A simple NMR-based experimental approach has been used to evidence the preferred orientation of aromatic rings when interacting with saccharides in water solution.77A dynamic combinatorial approach for the study of carbohydrate/aromatic interactions has been presented.78 The experimental results from the analysis of a large data set of chemically diverse carbohydrate–aromatic complexes allowed the accurate determination of the structure–stability relationship that governs these weak interactions. The influence of the equatorial or axial orientation of the sugar polar groups on the strength of the carbohydrate–aromatic complex was also determined by using punctual OH → F substitutions. In particular, 2-deoxy-2-fluoro mannoside derivative showed enhanced stabilization properties with respect to the gluco-configurated analogue, suggesting that equatorial polar moieties might be involved in repulsive interactions with the aromatic units. Moreover, the introduction of a fluorine substituent at the anomeric center modulates the electron density at the endocyclic oxygen, thus fine-tuning the stability of the complexes and the orientation of the pyranose ring with respect to the aromatic moiety. These fundamental studies demonstrated the influence of fluorine substitution on CH/π interactions and provided the impetus to further investigate the role of ligand fluorination in carbohydrate–lectin interactions.
Additional evidences regarding the role of fluorine substitution in enhancing methyl–π interactions have been presented, along with an overall study that correlated the enthalpy contribution of these weak interactions as function of the hydrogen polarizability by none, one, or two fluorine substituents.79 The contribution from these studies lies in the elucidation of the polar character in methyl–π interactions in sugar/receptor recognition, thus complementing the advances in the study of the direct pyranose/aromatic stacking. The authors studied the interaction of wheat-germ agglutinin (WGA), a model lectin, with acetylated amino sugars by NMR and molecular modeling. DFT-based theoretical calculations suggested that the presence of fluorine atoms at the acetamide group should enhance the interaction between the fluorinated analogues and the aromatic residues of the protein, given the polarization of the C–H bond at the CHF2CONH– and CH2FCONH– functions by the electron-withdrawing fluorine atoms. On the contrary, the absence of any CH–π donor group in CF3CONH– derivatives should significantly reduce the binding energy due to the unfavorable contacts of the electron-rich fluorine atoms and the aromatic residues. To experimentally prove this hypothesis, the authors used an NMR-based strategy and demonstrated that the strength of the CH–π interaction significantly depends on the polarization degree of the CH–π hydrogen donor. The values for the binding constants (kD), derived by STD-NMR competition experiments, agreed with those derived by quantum mechanics calculations, revealing a difference of one order of magnitude between the monofluorinated and the trifluorinated analogues,79 as schematized in Fig. 11. The results from these studies offer the opportunity to exploit sugar fluorination as a strategy to enhance fundamental intermolecular interactions, with the consequent implications in drug design.
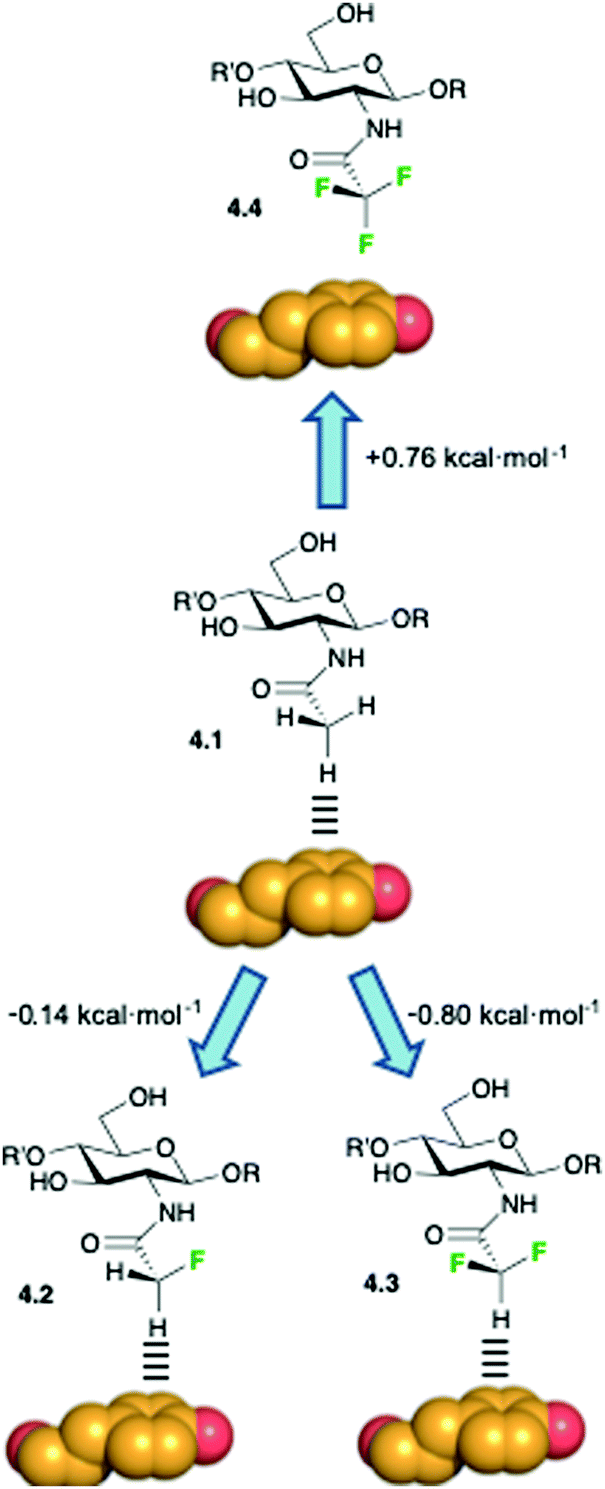 | ||
| Fig. 11 Modulation of the CH–π stacking interactions by diverse fluorination patterns. Free energy differences among the different fluorinated and not fluorinated derivatives in the WGA complexes were determined by STD-NMR competition experiments. | ||
4.2 Hydrogen bonding
One of the earliest applications of replacement of sugar hydroxyl groups by fluorine concerned investigations of the role of that hydroxyl group in the interaction with the protein. An unchanged binding affinity was interpreted as the OH group acting as a hydrogen acceptor or not participating in the interaction, whilst a much reduced affinity was interpreted to be caused by the loss in hydrogen bond donation from the ligand, providing information that can be used for ‘epitope mapping’.22–25 However, the high ionisation potential of organofluorine lone pairs makes fluorine only a weak hydrogen bond acceptor.80 In general, the stronger hydrogen-bond (HB) acceptor groups present in carbohydrates, as well as in water solvent, will be preferentially involved in intra- or intermolecular HB formation over C–F groups, whether in solution or in the solid phase. In crystal structures, where the resulting structure is a balance of many types of strong and weak interactions, it is not uncommon to see C–F bonds ‘ignored’ in hydrogen bonding networks, even for sugars with multiple C–F bonds.81This has been applied for the partial disruption of the dense hydrogen bonding network of polysaccharides such as cellulose, which is responsible for its mechanical and structural properties. Incorporation of two 3-deoxy-3-fluorinated glucose residues in a hexamer has led to more soluble analogues.82
In the absence of stronger hydrogen bond acceptors, intramolecular hydrogen bonding to fluorine in fluorinated carbohydrate derivatives has been demonstrated. In the solution phase, this is often apparent by the presence of a coupling constant between the alcohol hydrogen and the fluorine group (h1JOH–F, with the h1-prefix indicating hydrogen bond between the coupling partners).83 For example (Fig. 12), there is a 7.5 Hz coupling between the axial C2–OH and C4–F in the L-ribopyranose derivative 4.5,84 indicating a 6-membered ring intramolecular hydrogen bond. In 1,6-anhydro derivatives, this coupling is reduced due to the longer distance between the OH and the F groups (cf.4.6).85 Hydrogen bonding between vicinal fluorohydrins is weaker and h1J- are not always observed. An interesting case is 4.7,34b whose alcohol hydrogen displays a coupling constant to each fluorine atom, with a large 12.4 Hz 3JH–OH coupling indicative of the antiperiplanar orientation between the two coupling partners. Interestingly, fluorosugars have been used as probes to study intramolecular hydrogen bonding to fluorine.85,86
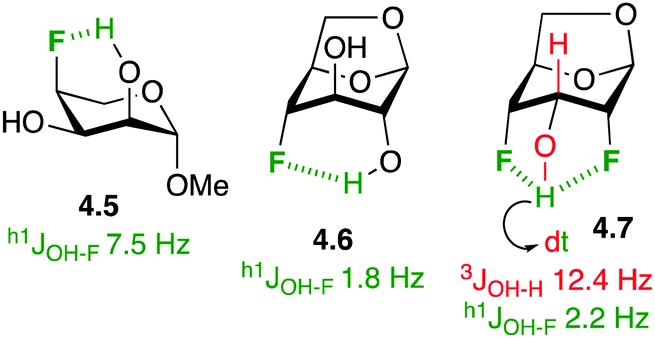 | ||
| Fig. 12 Intramolecular OH⋯F hydrogen bonding in some fluorinated carbohydrate derivatives. | ||
Crystal structures of protein-F–carbohydrate complexes often feature intermolecular hydrogen bonding contacts involving fluorine.87,88 The first example of a crystal structure involving a fluorinated trisaccharide 4.9 (Fig. 13)87 nicely showed how deoxyfluorination at the galactose 2-position lead to a very similar ligand conformation.
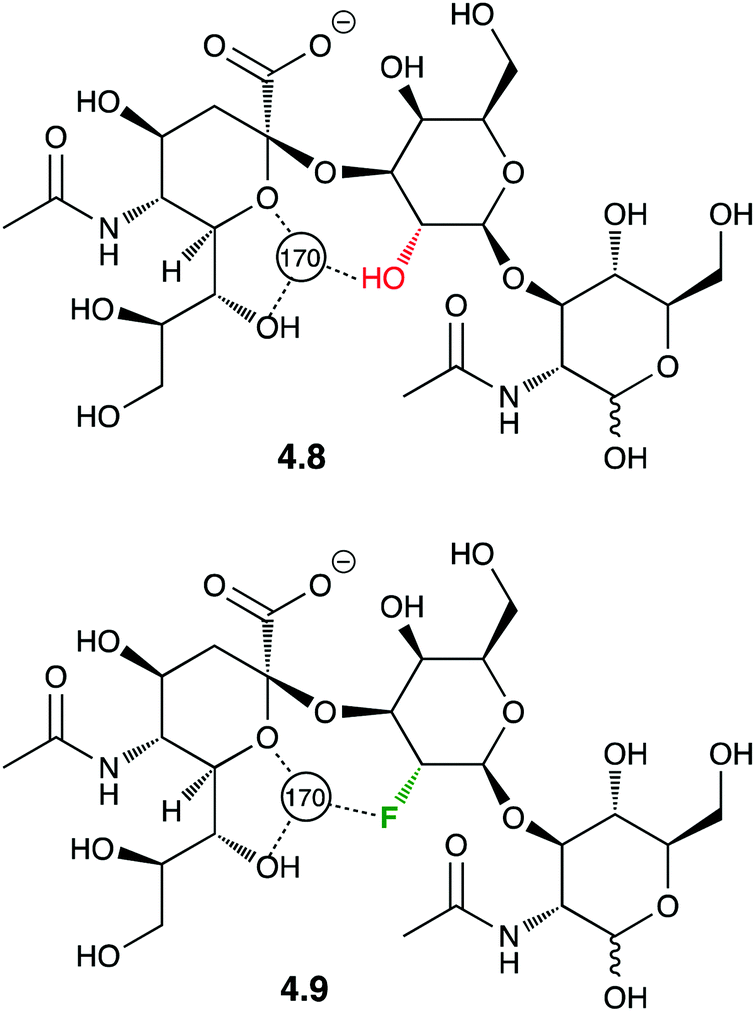 | ||
| Fig. 13 Crystal structure featuring intermolecular F⋯H2O hydrogen bond interactions. | ||
However, deoxyfluorination can also lead to incorporation of extra water molecules in the binding site. This has been illustrated by crystal structure analysis of the 6-deoxy-6-fluoro-D-galactose 2.6 complex with the arabinose binding protein (Fig. 14b), compared to that of the natural ligand (Fig. 14a).88 It was proposed that the repulsion between the fluorine atom and the carboxylate group caused a reorganisation, creating space for a water molecule which, in turn, enables hydrogen bond donation to the carboxylate group (which was lost upon 6-deoxyfluorination). In such cases, the energetics of binding are complicated, which has repercussions for ‘epitope mapping’ experiments.88
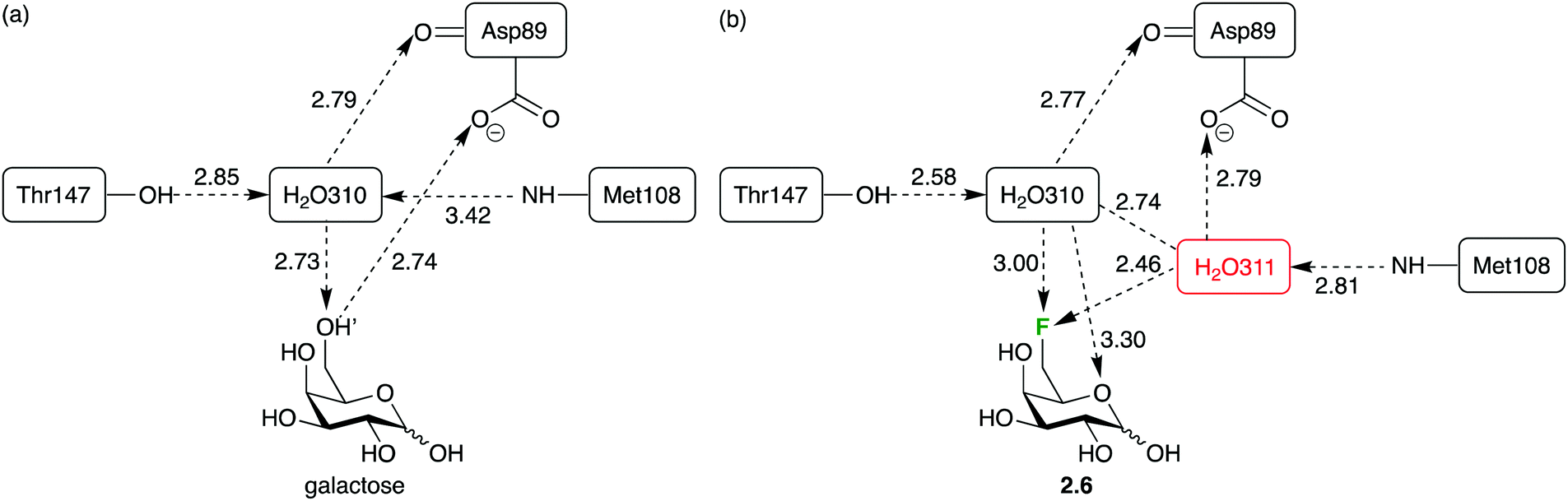 | ||
| Fig. 14 Sugar deoxyfluorination causing perturbation of glycan–lectin interactions: replacing a C–OH by a C–F bond allows incorporation of a new water molecule and formation of a new water-mediated hydrogen bond. | ||
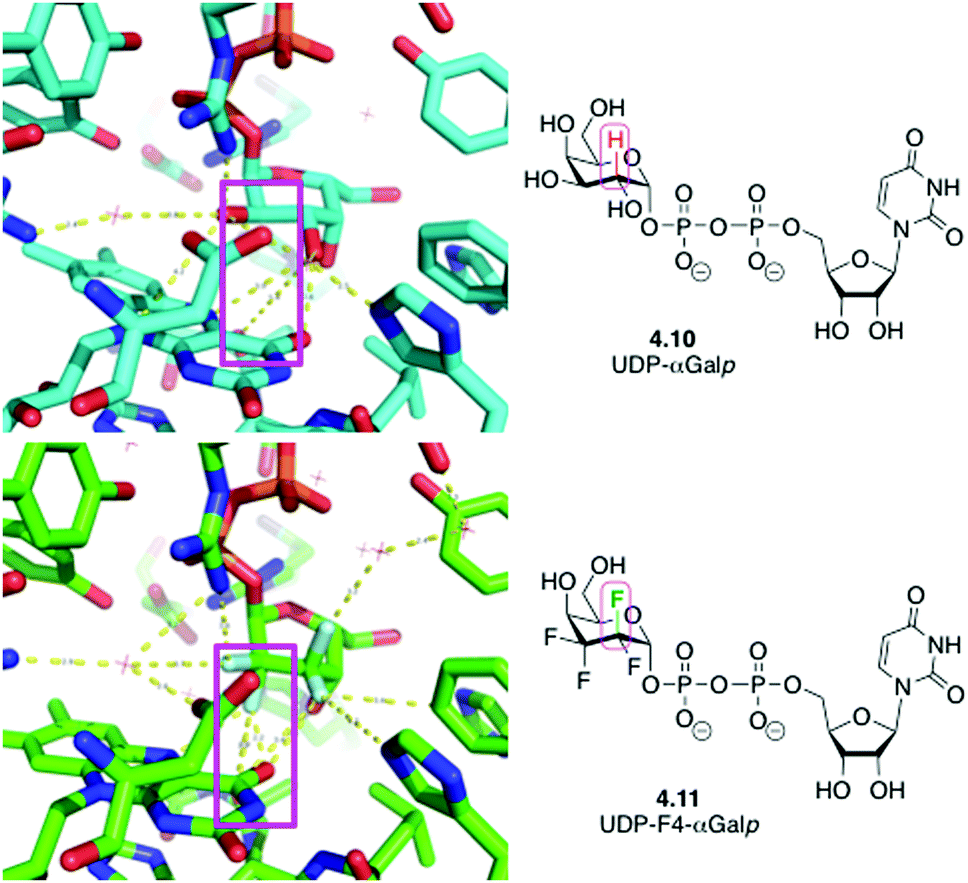
An interesting case has also been reported when replacing CHOH groups in UDP-galactopyranose 4.10 by CF2 moieties leading to 4.11 (Fig. 15).89a This substitution has also been shown to lead to additional intermolecular hydrogen bonding interactions involving the fluorine atom that replaced the hydrogen at C3 of 4.10,89a which may have contributed to the increased affinity of this analogue compared to the native ligand.89b
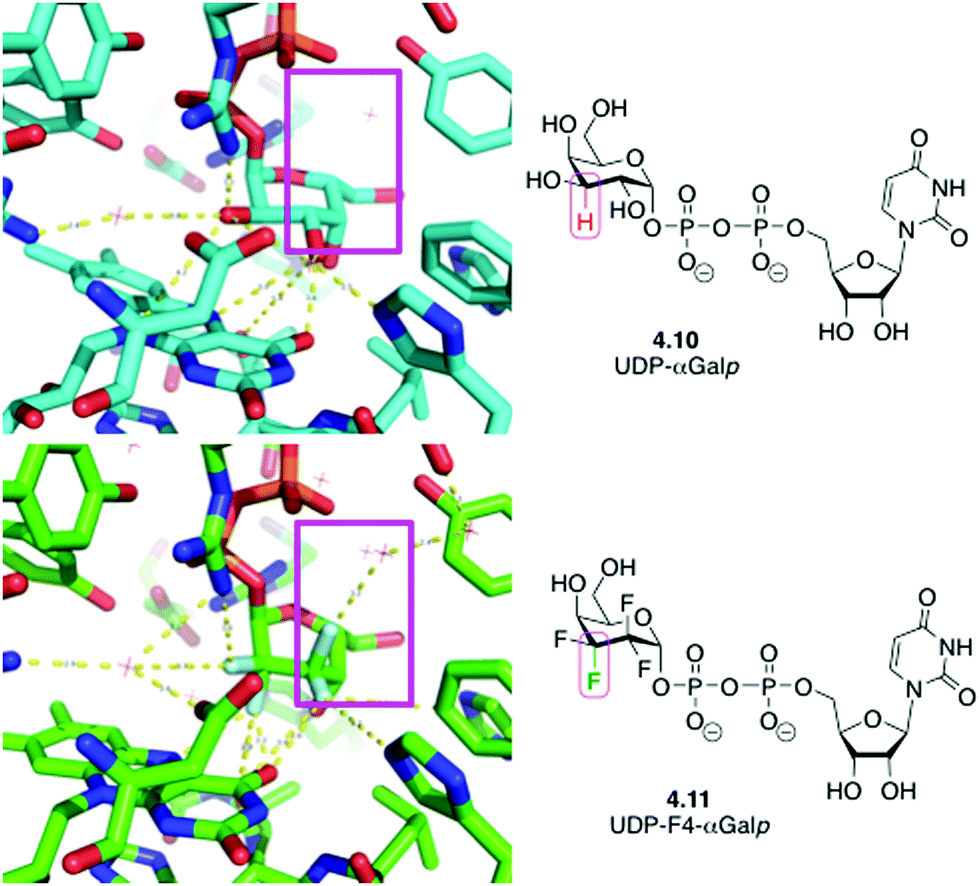 | ||
| Fig. 15 UDP-galactopyranose 4.10 and its tetrafluorinated analogue 4.11 in complex with UDP-galactopyranose mutase: replacing CHOH by CF2 allows formation of new water-mediated hydrogen bonds. | ||
A similar case can be observed for the fluorinated maltose 3.19, already mentioned in Fig. 9, in complex with GlgE1 (Fig. 16). The guanidinium group of Arg392 forms a bidentate hydrogen bonding interaction with the CF2-group in 3.19, while for 3.18 (as covalent complex with GlgE1) this residue forms a bidentate hydrogen bond with the fluorine and the anomeric oxygen.63,64
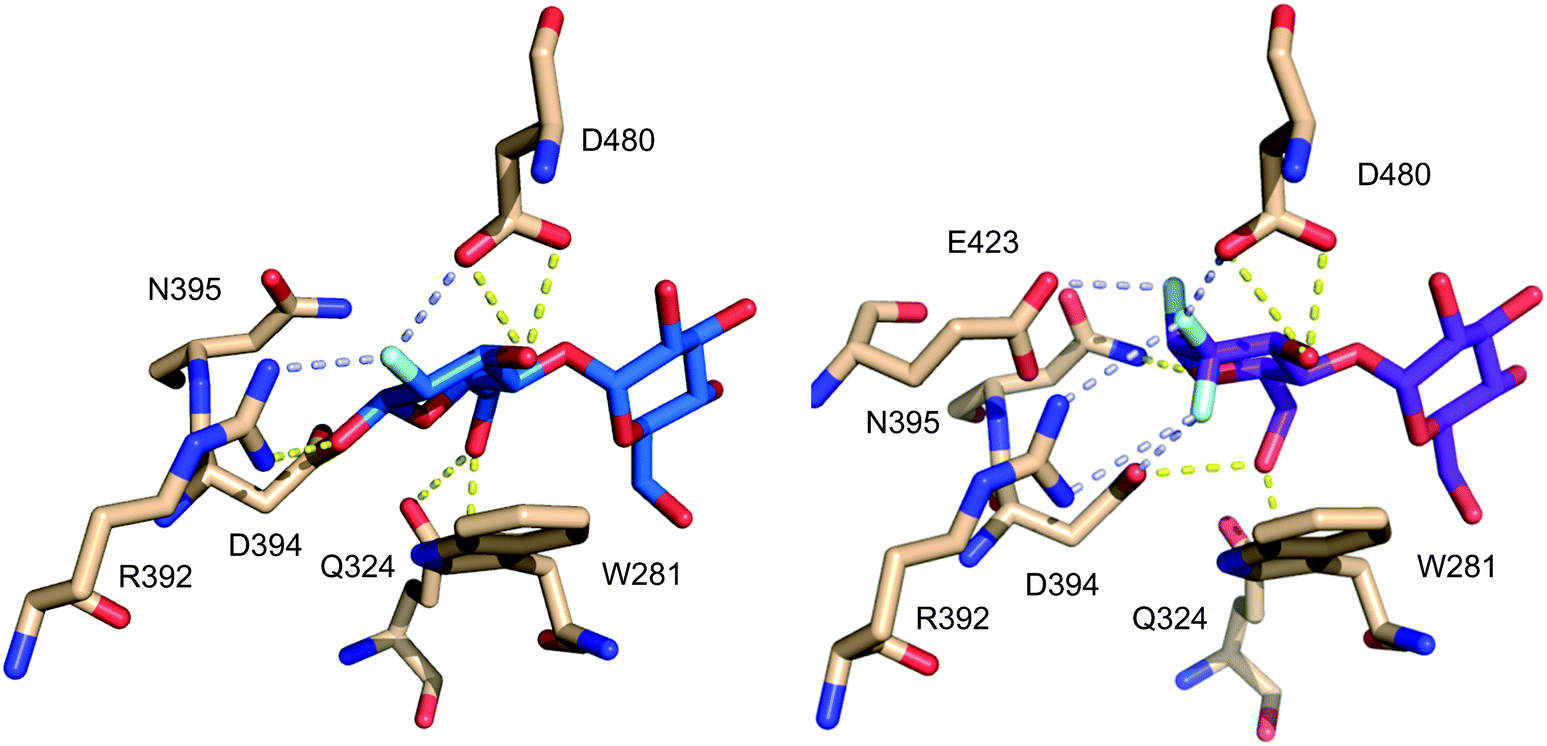 | ||
| Fig. 16 Comparison of CHF and CF2-motifs present in maltose fluorides 3.18 (left) and 3.19 (right) in complex with GlgE1 (PDB codes 4CN4 and 4U2Z).63,64 | ||
In a very original application, fluorine introduction has been successfully used to promote water binding in protein–carbohydrate complexes. Bridging water between the GlcNAc and threonine residues of Thr-containing glycopeptides was suspected to play an important role in binding to the anti-MUC1 antibodies (SM3), but never observed in crystal structures of 4.12 (Fig. 17) with SM3.90 It was hypothesised that GlcNAc residues featuring fluorinated amides may provide a more hydrophilic environment leading to stabilisation of water molecules. Calculations supported the presence of a hydrogen bond between the water and the fluorine. Indeed, crystal structures involving such fluorinated glycopeptides 4.13 and 4.14 did show bridging water molecules although the weak electron density of the fluorine atoms did suggest free rotation and hence did not support the presence of the F⋯HO hydrogen bond.
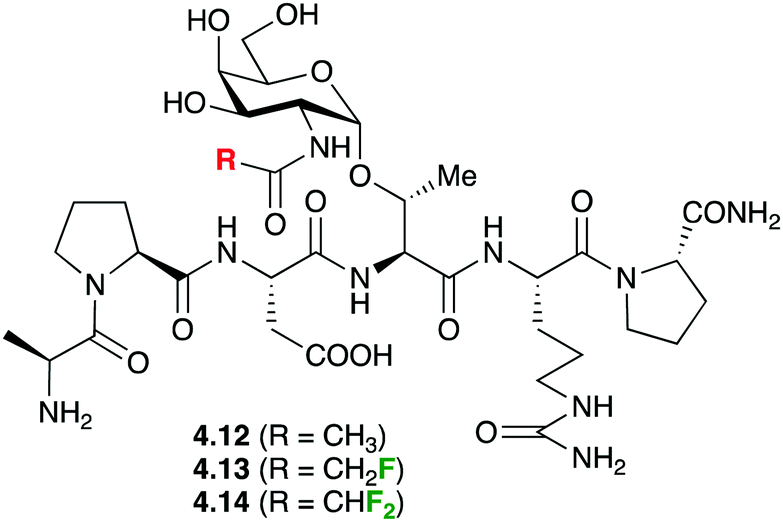 | ||
| Fig. 17 Fluorinated glycopeptides used in binding studies with MUC1 antibodies. | ||
Fluorine introduction will also influence hydrogen bonding capacities of the adjacent hydroxyl groups. So far, this has only been studied in non-carbohydrate model compounds, using alcohols and a standard acceptor (N-methyl pyrrolidinone).91 While, as expected,92 the electron withdrawing character of fluorine enhanced the hydrogen bond donating ability (also termed hydrogen bond acidity), the intramolecular hydrogen bonding to the fluorine atom actually leads to a reduced hydrogen bond acidity.93
4.3 C–F⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O and related interactions
O and related interactions
The occurrence of orthogonal C–F⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O arrangements was discovered during seminal studies involving a fluorine scan of thrombin inhibitors.94 The subsequent crystallographic database analysis pointed out the wider importance of this type of interaction, which was also identified for C–O(H)⋯CO.95 The shorter the C–F⋯CO contact, the more the C–F bond preferred to be positioned directly over the carbonyl carbon close to the carbonyl pseudotrigonal axis. Originally suggested to be a dipole interaction, other types such as n → π* and π–hole interactions have also been proposed,96 but will herein be referred to as a dipolar interaction. The C–F⋯CO interaction is weakly stabilizing and amounts to ca. 1 kJ mol−1.97 While C–H for C–F substitution in small bioactive molecules usually leads to affinity increases, it must be emphasized that other factors (for example, increase in lipophilicity) may also contribute as well.17,98
O arrangements was discovered during seminal studies involving a fluorine scan of thrombin inhibitors.94 The subsequent crystallographic database analysis pointed out the wider importance of this type of interaction, which was also identified for C–O(H)⋯CO.95 The shorter the C–F⋯CO contact, the more the C–F bond preferred to be positioned directly over the carbonyl carbon close to the carbonyl pseudotrigonal axis. Originally suggested to be a dipole interaction, other types such as n → π* and π–hole interactions have also been proposed,96 but will herein be referred to as a dipolar interaction. The C–F⋯CO interaction is weakly stabilizing and amounts to ca. 1 kJ mol−1.97 While C–H for C–F substitution in small bioactive molecules usually leads to affinity increases, it must be emphasized that other factors (for example, increase in lipophilicity) may also contribute as well.17,98
To the best of our knowledge, a direct comparison of C–O(H)⋯CO and C–F⋯CO interactions is not available yet. However, examples of the latter contacts in protein X-ray structures bound to fluorinated sugar derivatives have been described. For example (Fig. 18), the crystal structure of the bacterial heptosyltransferase WaaC in complex with a competitive inhibitor, ADP-2F-heptose 4.15, displays a clear C–F⋯CO interaction involving the Gly263/Thr262 linkage (d = 2.95 Å, F⋯CO 100.6°).99,100
 | ||
| Fig. 18 Dipolar interactions of the fluorine atom of ADP-2F-heptose 4.15 with heptosyltransferase WaaC (PDB 2H1H).99,100 See text for discussion. | ||
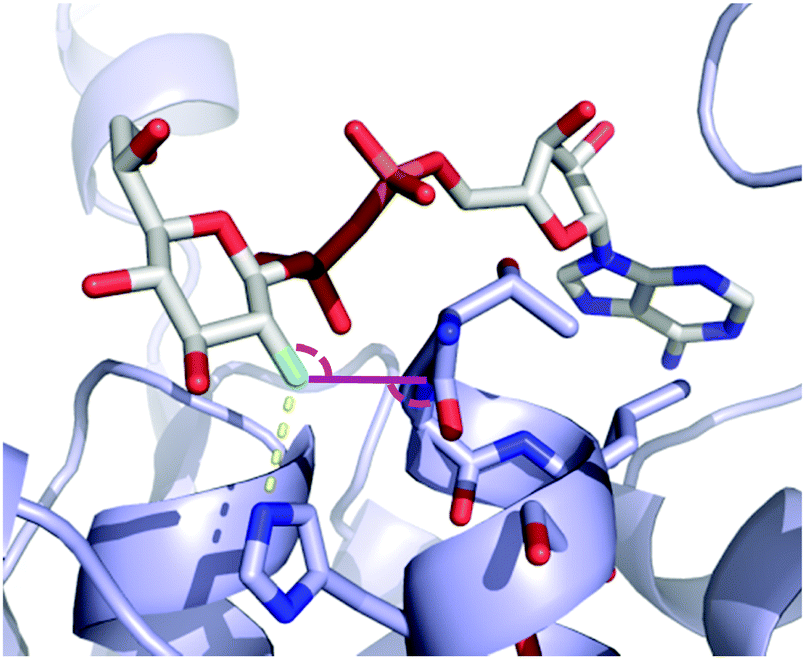
An interesting case arises for CHOH to CF2 replacement, which formally includes a C–H to C–F modification. This substitution was shown to allow for the establishment of an extra dipolar interaction. Fittingly, this feature has been depicted (Fig. 19) in the crystal structures of the mycobacterial galactose mutase UGM with Galp-UDP 4.10 and its tetrafluorinated derivative (4.11),89a which are very similarly positioned within the UGM binding site.89a The axial C–H group at the 2-position of galactose is replaced by a C–F bond, which is engaged in such an interaction with the carbonyl group of the FAD cofactor.
 | ||
| Fig. 19 CHOH for CF2 exchange allows introduction of an unexpected dipolar interaction.89a | ||
4.4 Polar hydrophobicity
The term ‘polar hydrophobicity’ was coined by DiMagno to capture the hydrophobic character of fluorine while possessing a large (C–F) dipole moment.37,101 In essence, while fluorine introduction can cause an increase in lipophilicity, it also allows for possible attractive dipole-mediated interactions, and the directional nature of the C–F dipole combined with the presence of multiple chiral centres make ‘polar hydrophobicity’ a useful concept. A compelling example is provided by the glycogen phosphorylase fluorinated inhibitors shown in Fig. 20.22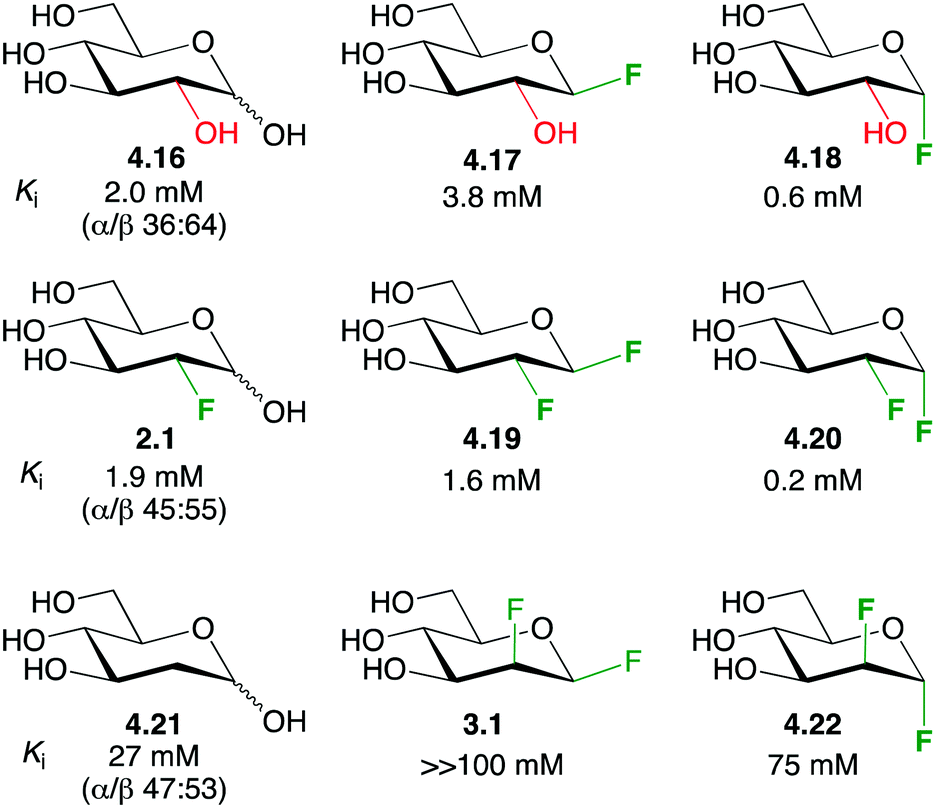 | ||
| Fig. 20 The enhanced affinity for glycogen phosphorylase of vicinal dideoxy-difluorinated carbohydrates.22 | ||
The inhibition constants of D-glucose 4.16 and 2-deoxy-2-fluoro-D-glucose 2.1 were similar. Compared to 4.16, the β-glucosyl fluoride 4.17 displayed a lower affinity, while the α-anomer 4.18 showed a higher affinity. Given that deoxyfluorination at the 2-position has little effect, similar low inhibition constants for 4.19 and 4.20 were expected. However, in each case, a significantly reduced value was found. Strikingly, the stereochemistry of the dideoxydifluorination proved to be important since the corresponding 1,2-dideoxy-1,2-difluoromannoses 3.1 and 4.22 exhibited a much decreased inhibition potency (more than two orders of magnitude), and merely deoxygenation (as in 4.21) led to lower binding. Therefore, according to the ‘polar hydrophobicity’ concept, the increased lipophilicity of the dideoxy-difluorinated glucoses may indeed increase affinity, but only when the C–F dipoles are correctly oriented (as in 4.19 and 4.20).
5 Fluorinated sugar derivatives as probes
5.1 Introduction
In this section, the focus is set on the use of 18F and 19F isotopes as bioanalytical probes for the study of kinetic transport phenomena, lectin–carbohydrate interactions or for in vivo imaging of tumors and drug distribution to name a few examples. The analytical methods employed to detect fluorinated carbohydrates cover 19F-NMR, 19F magnetic resonance imaging (MRI) and 18F positron emission tomography (PET). With 100% natural isotopic abundance, but low natural presence in biomolecules, its small size and large spectral resonance range of ca. 200 ppm, 19F is an attractive probe for studying metabolism and transport of biomolecules, including carbohydrates, by NMR spectroscopy.102 The fluorine atom has been used to substitute hydroxyl groups, protons or aldehydes giving rise to a C–F functional group that lies between the C–H and the C–OH in size, is chemically inert, retains the electronegativity of a hydroxyl group but loses its hydrogen donor capacity. This hydroxyl/fluorine exchange allows an evaluation of the contribution of individual hydrogen bonds in carbohydrate–protein binding events.21–25,1025.2 Membrane transport studies
Due to its excellent spectral range and the absence of background peaks in biological matrices, high resolution 19F-NMR is an ideal technique for studying kinetics and mechanism of carbohydrate metabolism and sugar–protein binding in vitro. This is the case for the glucose (Glu1) transporter mediated transport of D-glucose over the erythrocyte membrane, which has been studied by 19F-NMR and 19F-2D-EXSY-NMR by various groups over the years as a model for the development of glucose analogues for imaging and tumor targeting applications.34a,103,104 Both techniques take advantage of the shift to higher frequencies of fluorine resonances when moving from an extracellular to an intracellular environment.103 Where signal overlap between intra- and extracellular signals hinders peak integration, 19F-2D-EXSY-NMR can provide the required resolving power, with the intensity of cross peaks directly proportional to the flux.103,105Initial investigations with 2- and 3-deoxy-3-fluoro-D-glucose (2.1, 5.1, Fig. 21) showed uptake rates similar to glucose and a generally enhanced transport of the α-anomer,105 while the transport of the 4- and 6-deoxyfluoro analogues (5.2, 5.3) was only half of that of glucose.104 In contrast, higher fluorinated compounds like the hexafluoroglucose racemic analogue 5.4 were shuttled over the erythrocyte membrane with an order of magnitude higher transport efficiency compared to glucose.34a,101 However, a later synthesized trifluoroderivative of glucose 3.2 showed an overall decrease efflux efficiency to about 70% compared to 5.1, used as a reference.103
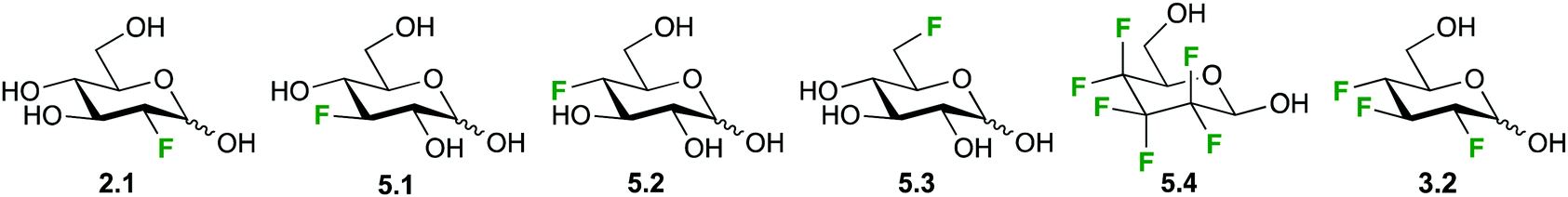 | ||
| Fig. 21 Selection of fluorinated monosaccharide structures employed in 19F-NMR spectroscopy for studying GLUT1-mediated glucose transport. | ||
Fluorinated fructose derivatives have been studied in GLUT2/GLUT5 mediated membrane transport, which is of relevance in breast cancer cell lines (2.1 is not transported by GLUT5). Clear dose-dependent inhibition of [14C]-D-fructose and -glucose by 6-deoxy-6-D-fluorofructose 5.5 (Fig. 22) was shown, with very low Ki values, indicating 5.5 binds to both transporters.106 Furthermore, by using [14C]-5.5, it could be demonstrated that actual transport into the cells is occurring. Similar results were found for the 3-deoxy-3-fluoroderivative 5.6 and for the fluorinated 2,5-anhydromannitol derivative 5.7.107
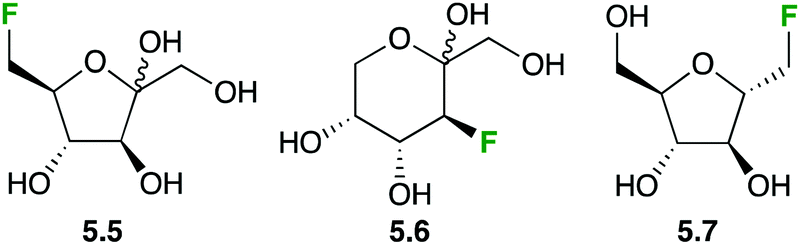 | ||
| Fig. 22 Fluorinated fructose analogues for the study of GLUT2 and GLUT5 transport. | ||
5.3 Lectin–carbohydrate interactions
19F-NMR spectroscopy has been extensively used for studying lectin–sugar interactions19–21 either employing 19F as a mere analytical probe or as an obvious method of choice for the analysis of fluoroglycomimetics designed for an enhanced interaction with the target protein. Allman et al. claimed that until 2009 most fluorinated carbohydrates showed lower affinity to lectins than their natural ligands.87b However, they completed the synthesis of a series of fluorinated sialylactosamine analogues and found similar or slightly higher binding affinity to the receptor TgMIC1 of the pathogen Toxoplasma gondii.87b The structural analysis of interaction of the fluorinated derivatives 5.9 and 4.9 (Fig. 23) with the TgMIC1 receptor showed a F-mediated hydrogen bond (see Section 4.2) that was also corroborated by 19F NMR. In fact, a shift to lower field of the 19F resonance was observed upon binding. Since monovalent carbohydrate–protein interactions usually do not lead to a measurable change in chemical shifts or line widths, the observed shift for fluorinated sialyl-lactosamine analogue 4.9 is remarkable.87b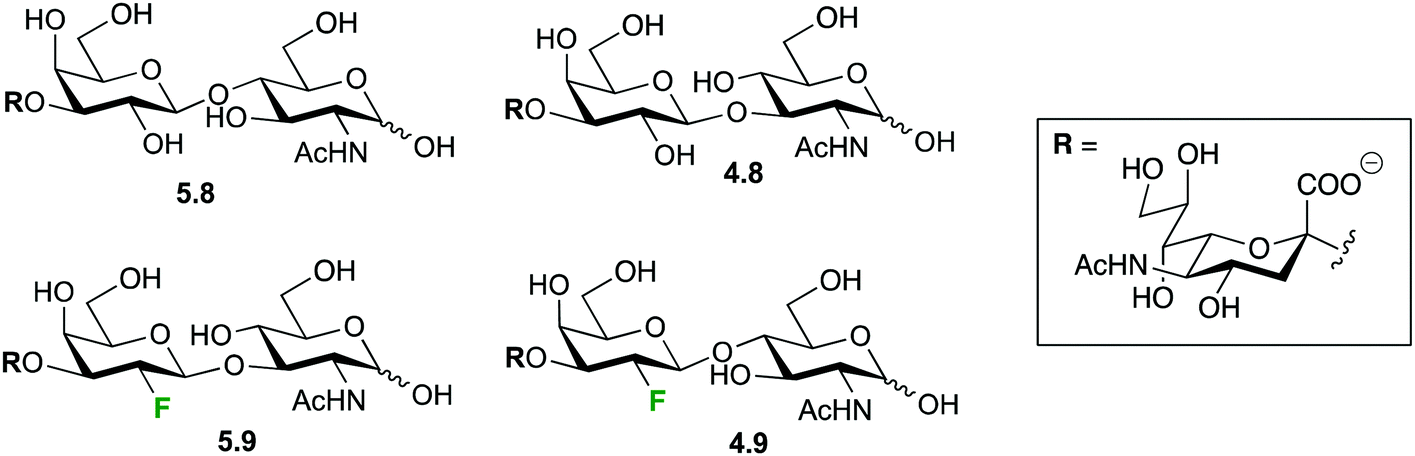 | ||
| Fig. 23 The sialylactosamine derivatives employed for studying interactions with the TgMIC1 receptor by 19F-NMR spectroscopy. | ||
Diercks et al. have developed an innovative 19F-STD NMR technique with greatly improved sensitivity for detecting binding events over the conventional 19F-NMR methods based changes of chemical shift.108 The interaction between Con A and 2-deoxy-2-fluoro-D-glucose 2.1 was used as model to develop a 19F-STD NMR methodology that avoids the quantification problems that existed with standard NMR methods19 and with previous versions of the 19F-STD NMR experiment,108 due to the requirement of J-coupling between the STD accepting hydrogen and the fluorine nucleus.
Alternatively, a T2-relaxation 19F-NMR-based method109 has been used to evaluate the interaction features and molecular insights of a small library of monofluorinated monosaccharides towards DC-SIGN, a calcium-dependent C-type lectin of biomedical interest, involved in viral infections. The method allowed the robust screening of a library of compounds, also permitted obtaining key information on the specific sugar–protein interactions, including the sugar hydroxyls involved in the coordination to the calcium atom. Moreover, a new binding mode of DC-SIGN to mannose through hydroxyl groups OH2 and OH3 was detected, along with the non ambiguous demonstration of the direct interaction of the lectin with Gal moieties, with implications for recognition of larger oligosaccharides, such as the histo blood type antigens.109
Also in the C-type lectin field, 19F R2-filtered NMR experiments have been employed within a fragment-based drug discovery program to determine novel Langerin ligands.110 In particular, the ManNAc analogue 5.10 (Fig. 24) was used as spy molecule in competitive binding assays versus a library of 2-deoxy-2-carboxamido-α-mannoside analogs 5.11, in which different molecular fragments were placed instead of the CF3 moiety. The analogues were initially chosen by virtual screening and the best hits were subsequently synthetized and submitted to the 19F R2-filtered NMR assay, which afforded the corresponding KI values in a fairly straightforward manner. Small, negatively charged substituents were found to substantially increase the affinities towards Langerin.110
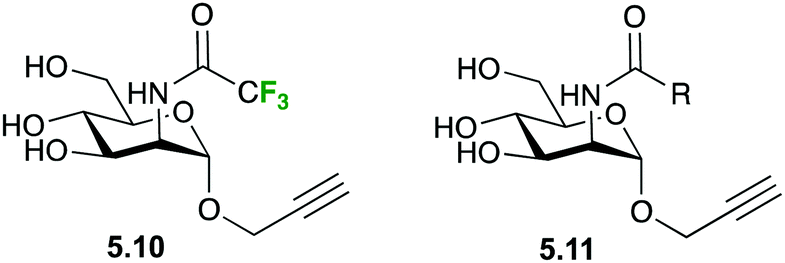 | ||
| Fig. 24 Spy molecule used in competitive binding assays of ManNAc analogues. | ||
19F-NMR experiments have also been used to analyse the recognition of fluorinated mannose-containing oligosaccharides by cyanovirin-N, a mannose binding anti-HIV lectin. In particular, the Manα(1 → 2)Man and Manα(1 → 2)Manα(1 → 2)-Man oligosaccharides showing a 2-deoxy-2-fluoro-α-D-mannopyranosyl unit at the non-reducing end (5.12 and 5.13, respectively, Fig. 25) were employed to evaluate the binding mode and to determine the kinetic and thermodynamic parameters of the interaction, which were further assessed through 1H–15N HSQC-based chemical shift perturbation analysis and ITC measurements. 2D 19F–19F exchange experiments also allowed detecting an additional binding mode for the trisaccharide, which could not be detected using regular 1H-based NMR experiments.111
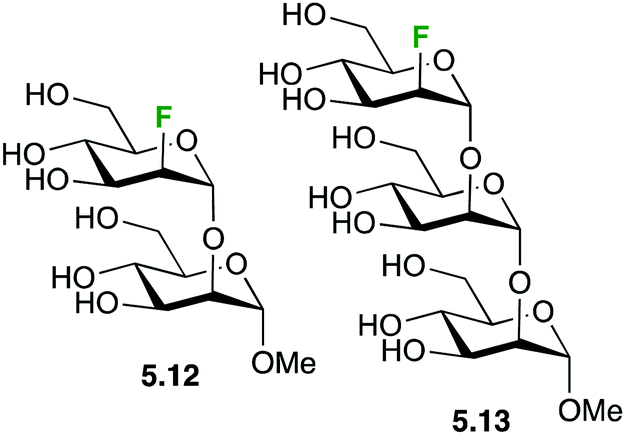 | ||
| Fig. 25 Fluorinated mannose-containing oligosaccharides. | ||
5.4 18F labeled sugars for PET imaging
 | ||
| Fig. 26 Labeling based on 2-deoxy-2-fluoro-D-glucose 2.1.113–118 | ||
The wide availability of 2.1 has prompted its use for the synthesis of related compounds, as (18F) labeled lactose,113 UDP-(18F)-2-deoxy-2-fluoro-D-glucose114 or tags to facilitate tracking of other biomolecules via PET.115,116 Wuest et al. has employed maleimidehexyloxime 5.7 as a reagent for tagging peptides and proteins after reaction with 2.1. As model, annexin 5, presenting a single cysteine, was labeled with 5.14 in 43–58% decay-corrected yield and its biodistribution and kinetics studied in a small animal xenograft tumour model.116 In a similar fashion, Boutureira et al. tagged proteins presenting accessible cysteine residues with (18F)2-deoxy-2-fluoro-D-thioglucose 5.15via disulfide or thioether linkages,117 while Maschauer et al. reported a slightly different approach for labeling two short peptides employing the [18F]-2-FGD-azide 5.16.118 Uptake and activity of the novel probe 5.17 and other derivatives was demonstrated in vitro on neurotensin receptor (NT-receptor) expressing cells and by autoradiography of rat brain slices, which showed an accumulation of the (18F) labeled probe in NT-receptor rich areas. Biodistribution studies in a nude xenograft mouse model with 5.17 showed rapid blood clearance, specific uptake by the kidneys with low uptake by other organs, showing sufficient signal to noise ratios for PET-scanning shortly after injection. Biodistribution of the glycosylated RGD peptide (18F)-2-deoxy-2-fluoro-D-glucose-RGD 5.18 showed a similar blood clearance but 3 times higher uptake by liver and kidney compared to the galactose analogue. With a good tumor/blood ratio of 2.4 and excellent metabolic stability 5.18 seems as a promising tool for future αvβ3-integrin imaging by PET.
Essentially the same approach for the labeling of proteins with 2-deoxy-2-(18F)fluoro-D-glucose via a triazole linkage was reported by Boutureira et al.119 Coupling of [18F]2-FDG azide 5.16 was achieved via copper catalyzed cycloaddition to the alkyne function of recombinant model protein with the non-canonical amino acid propargylglycin selectively incorporated via expanded genetic code methodology.120
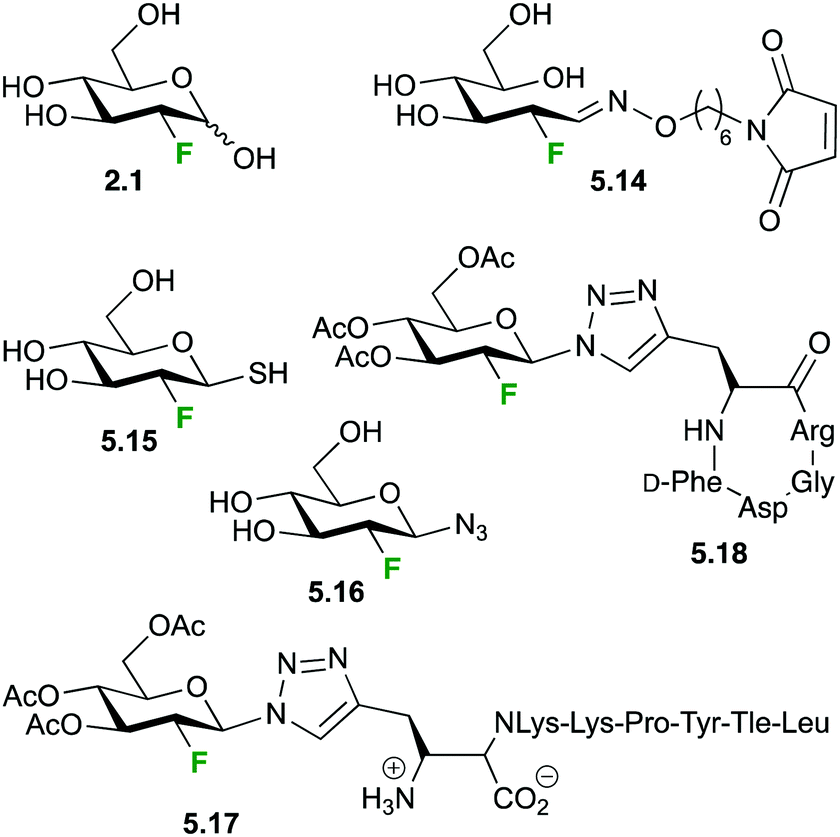
2-Deoxy-(18F)fluoro mannose 5.19 (Fig. 27) displays similar uptake profile as glucose and like 2-deoxy-2-fluoro-D-glucose it is not further metabolized after phosphorylation by hexokinase-1.121 Furumoto et al. synthesized 5.19,122 which showed a rapid cell uptake comparable to that of 2-deoxy-2-fluoro-D-glucose, with formation of a compound likely to be 2-deoxy-2-fluoro-D-mannose-6-phosphate. A biodistribution study in a tumor mouse model showed high uptake in tumor cells, relatively high uptake in brain and lower uptake in other tissues, which might provide an advantage for 2-deoxy-2-fluoro-D-mannose for imaging of brain tumours.122
 | ||
| Fig. 27 18F-Labeled mannose. | ||
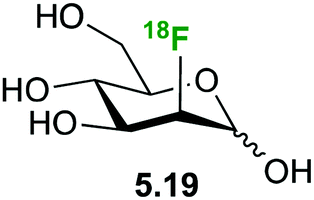
6-Deoxy-(18F)-fluorogalactose 2.6 (Fig. 28) has been synthesized as a PET tracer to study galactose metabolism in vivo,123 being incorporated 6 times faster into glycoconjugates than galactose itself.124,125 In contrast, 2-deoxy galactose is, in fact, a inhibitor of N-glycan synthesis with a similar potency as tunicamycin.124 However, it is taken up by the liver at a very high rate where it is phosphorylated and further converted to UDP-2-deoxy-2-(18F)fluoro-D-galactose 5.20, making it a viable tracer to study galactose metabolism.126
 | ||
| Fig. 28 18F-Labeled galactose derivative for the study of galactose metabolism by PET. | ||
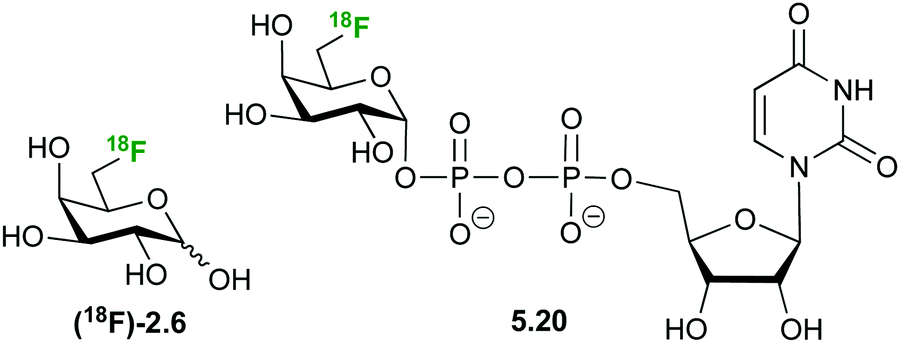
Based on the attractiveness of trehalose analogues as probes for bacterial infection imaging,127 Swarts demonstrated the selective uptake of 2-deoxy-2-(18F)fluorotrehalose 5.21 (Fig. 29) in Mycobacterium smegmatis, a non-pathogenic model organism for M. tuberculosis.112d
 | ||
| Fig. 29 Labeled trehalose as probe for bacterial infection.112d | ||
The importance of sialic acid as a terminal recognition element on larger oligosaccharides and the overexpression of sialylated structures on tumor cells have prompted interest in developing PET tracers to study sialic acid metabolism (Fig. 30). N-Acetyl-3-(18F)fluoro sialic acid (18F)-2.4 and N-acetyl-2-deoxy-2,3-(18F)difluorosialic acid 5.22 showed insufficient uptake to be of any use as PET tracer, probably due to the lack of membrane based sialic acid transporters and charge repulsion between the negatively charged membrane and the sialic acid derivatives.128,129
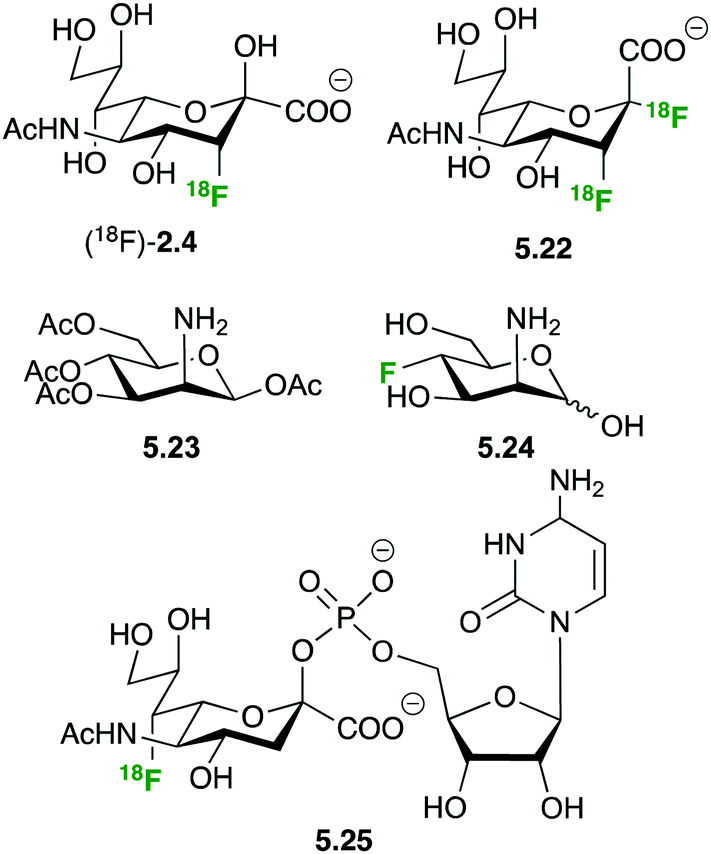 | ||
| Fig. 30 Fluorinated structure for studying sialic acid metabolism by PET. | ||
The peracetylated 2-deoxy-azido mannosamine 5.23 is a convenient sialic acid precursor widely employed for in vivo imaging of sialic acid integration into cell surface proteins by metabolic glycoengineering.130 Hartlieb et al. have developed synthetic routes towards non-radioactive 3-deoxy-fluoro-N-acetyl mannosamine 5.24, the precursor for CMP-7-deoxy-fluoro sialic acid 5.25.131
The synthesis of a 5-deoxy (18F)-fluoro ribose ((18F)FDR) 5.28 has also been achieved (Fig. 31)132,133 from S-adenosyl-L-methionine (SAM) 5.26. Subsequent hydrolysis of the nucleoside by a nucleosidase from Trypanosoma vivax (TvNH) produces the free radiolabeled (18F)-fluoro ribose 5.28 with decay correction in approximately 3 h.
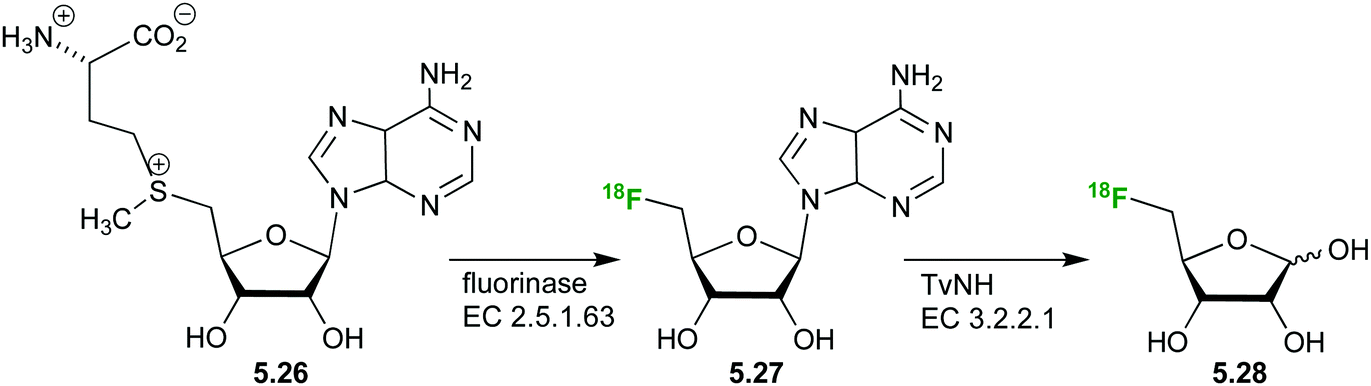 | ||
| Fig. 31 Radiolabeled ribose derivatives as tracers for studying ribose metabolism via PET (17–19), radiotracer 2-FDM, and precursor molecules. | ||
A biodistribution study has established (18F)FDR 5.27 as a suitable probe for PET imaging.133
5.5 19F MRI
Magnetic resonance imaging (MRI) is a radiation-free medical imaging modality that generates three-dimensional body images with anatomical detail for the detection of disease and monitoring treatment outcomes and excellent contrast for soft tissues.134,135 The relatively low sensitivity of MRI can be enhanced by the use of contrast agents.136 A huge gain in sensitivity is potentially also possible by employing fluorinated compounds as contrast agents.137 MRI however, has a 1000-fold lower sensitivity than PET and requires millimolar concentrations of fluorinated reporter molecules.136,137 Polyfluorinated sugars have been far less explored as contrast agents. Sufficient contrast can be obtained by employing labeled polysaccharides138 or functionalized nanoparticles137 as reporter molecules or also employing high concentrations of mono-fluorinated small molecules.139 Polymeric carbohydrates show suitable properties as scaffolds for the development of MRI contrast agents.138 Krawczyk et al. labeled with fluorine a series of natural polysaccharides and evaluated their properties as MRI contrast agents in cell and in vivo.138 Another strategy to increase fluorine density is the presentation of multiple fluorinated ligands on the surface of nanoparticles.137 Finally, 3-FDG has been used for in vivo brain imaging.139 Although 3-FDG is phosphorylated by hexokinase 300 times slower than its isomer 2-FDG, it was found to be a better substrate for the aldose reductase than D-glucose itself. Consequently, it may be employed as functional probe for the non-invasive study of glucose metabolism via the aldose reductase sorbitol (ARS) pathway.6 Applications
Section 5 has focused on the use of the fluorine-containing compounds as probes to monitor molecular interactions. We herein focus on their applications to monitor, modulate, and interfere with biological activities.6.1 Fluorine-containing sugars as inhibitors and chemical probes
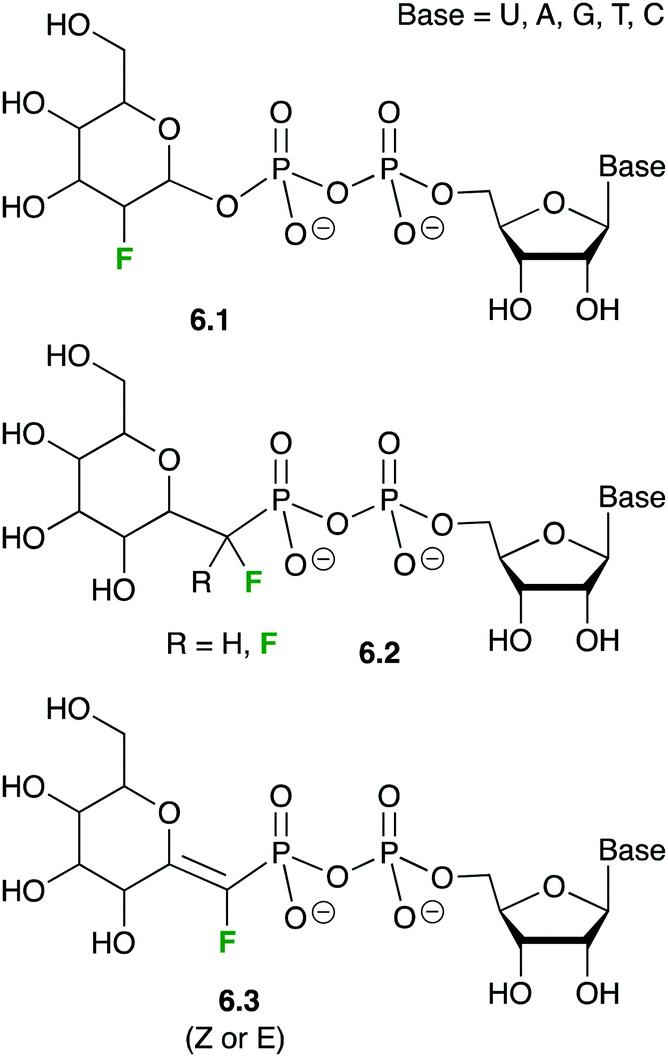 | ||
| Fig. 32 Fluorinated nucleotide-sugars as enzyme inhibitors or inactivators. | ||
In molecules 6.1, the electron-withdrawing character of the fluorine atom is believed to destabilize the transition state of the glycosylation reaction, which likely displays a substantial cationic character. Nucleotide-sugars 6.1 are thus donor substrates that are too slow for the enzymatic reaction to be observed, but that maintain the usually high affinity (low μM Km) of the natural substrates.141c,146 Thanks to this stability, XRD 3D-structures of GTs could be obtained in complex with nucleotide-fluorosugars 6.1, thus revealing the intimate contacts between the donor substrate and the enzyme.147
However, the very interesting inhibitory properties of molecules 6.1 could not be translated to in vivo applications or even in cell-based bioassays due to their inability to cross cell membranes. Such a limitation has recently been overcome thanks to a prodrug approach: indeed, it was shown that some precursors of 6.1 can be transported across the cell membrane. Once in the cytoplasm, they are enzymatically transformed into the fluorinated sugar-nucleotide, thus generating a GT inhibitor in situ. For instance, peracetylated 3-fluoro-sialic acid 6.4148 crosses the membrane of mammalian cells and is transformed into nucleotide-sugar 6.5 after intracellular enzymatic ester hydrolyses and CMP-transfer (Fig. 33), thus shutting down sialylation in cellulo.148 The same team demonstrated later on that the administration of 6.4 in mice dramatically decreased sialylated glycans in cells of a large set of tissues and resulted in deleterious effects on liver and kidney functions.149 The group of Adema then studied the effect of prodrug 6.4 on cancer cell lines, in vitro and in vivo, and showed reduced tumor growth and metastasis.150 A second generation of fluorinated pro-inhibitors 6.6 were then synthesized with a carbamate at C-5. The SialT inhibitory activities of the latter molecules 6.6 were found to be prolonged and enhanced thanks to a more efficient intracytoplasmic transformation into their nucleotide sugar active form.151 Interestingly, bacterial SialT can also be inhibited using the same prodrug approach, but this time the deprotected fluorinated pro-inhibitor 2.4 has to be used instead of 6.4. In pathogenic non-typeable Haemophilus influenzae, molecule 2.4 is taken up by the ATP-independent periplasmic transporter system. Its intracellular transformation into 6.5 triggers an inhibition of sialic acid incorporation into the bacterial lipo-oligosaccharide (LOS) resulting in enhanced serum-mediated killing.152
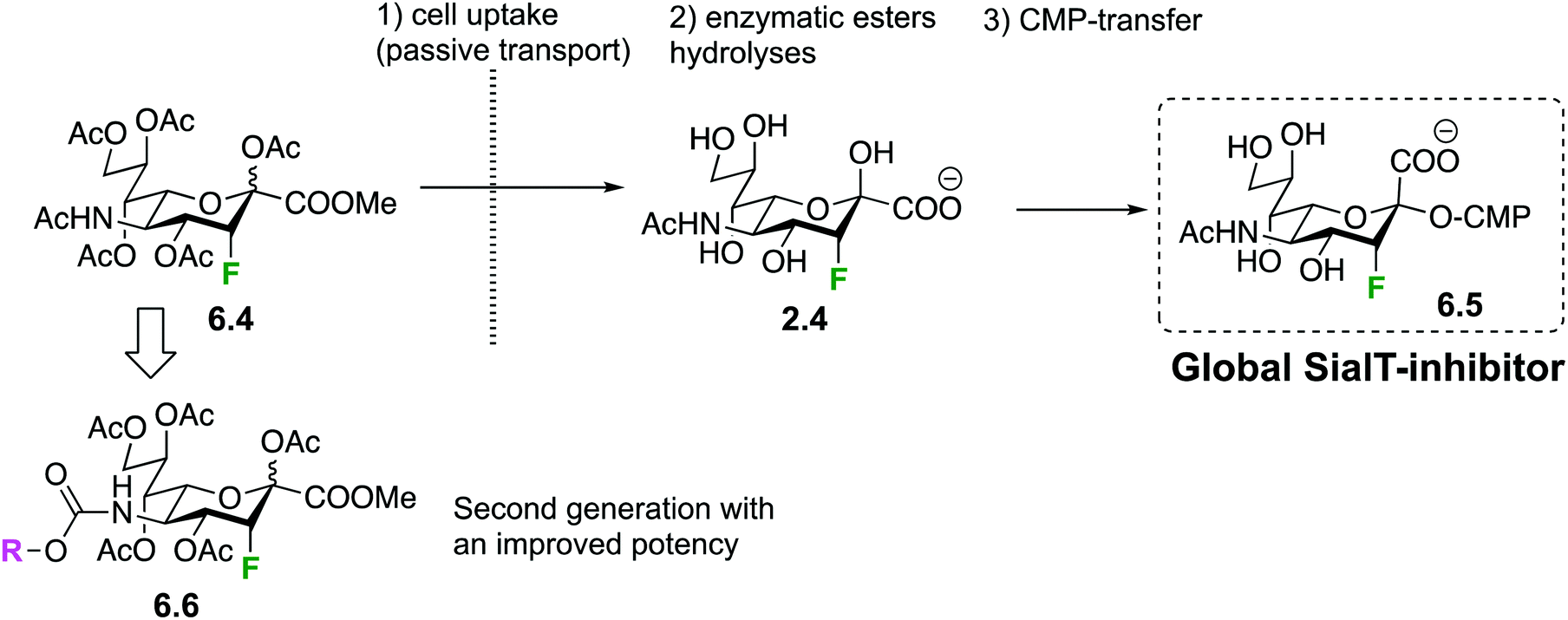 | ||
| Fig. 33 In cellulo generation of mammalian Sial-T global inhibitor 6.5. | ||
Given the biological relevance of fucosyltransferases (FucTs),140 the in cellulo generation of 2-deoxy-2-fluoro-fucose-GDP141c,d6.9 has been thoroughly studied by the same prodrug approach.148 The fluoro-fucoside 6.8 is an orally active inhibitor of protein and cellular fucosylation153 that was shown to inhibit several biological functions in transgenic sickle mice.154 Importantly, it was shown that the accumulation nucleotide-sugar 6.9 into the cells not only inhibit FucTs, but also shut down the biosynthesis of GDP-fucose, the natural FucT donor.148
A recent application of this fucosylation blockade has focused on the generation of non-fucosylated recombinant antibodies in CHO cells.155 Peracetylated fucostatin can be taken up in mammalian cells and enzymatically hydrolyzed to generate fucostatin 2.3156 (Fig. 34A) a 6,6,6-trifluorinated analogue of L-fucose. The latter is then transformed into GDP-fucostatin which blocks protein fucosylation by inhibiting the GDP-fucose de novo biosynthesis. Indeed, a cocrystal structure proved that GDP-fucostatin binds an allosteric site of GDP-mannose 4,6-dehydratase. This result can find important applications in the generation of non-fucosylated therapeutic antibodies. The latter have been shown to display an improved antibody-dependent cellular cytotoxicity, thus resulting in a better in vivo efficacy, which is a key parameter for instance in oncology clinical trials. The same strategy was later on exploited for the production of low-fucosylated monoclonal antibodies from murine hybridoma cells.157 In the galactose series, mono and difluorinated galactosides of general structure 6.10 (Fig. 34B) were shown to inhibit UDP-galactose biosynthesis in epithelial cells and fibroblasts, resulting in a metabolically induced galactosemic phenotype.158
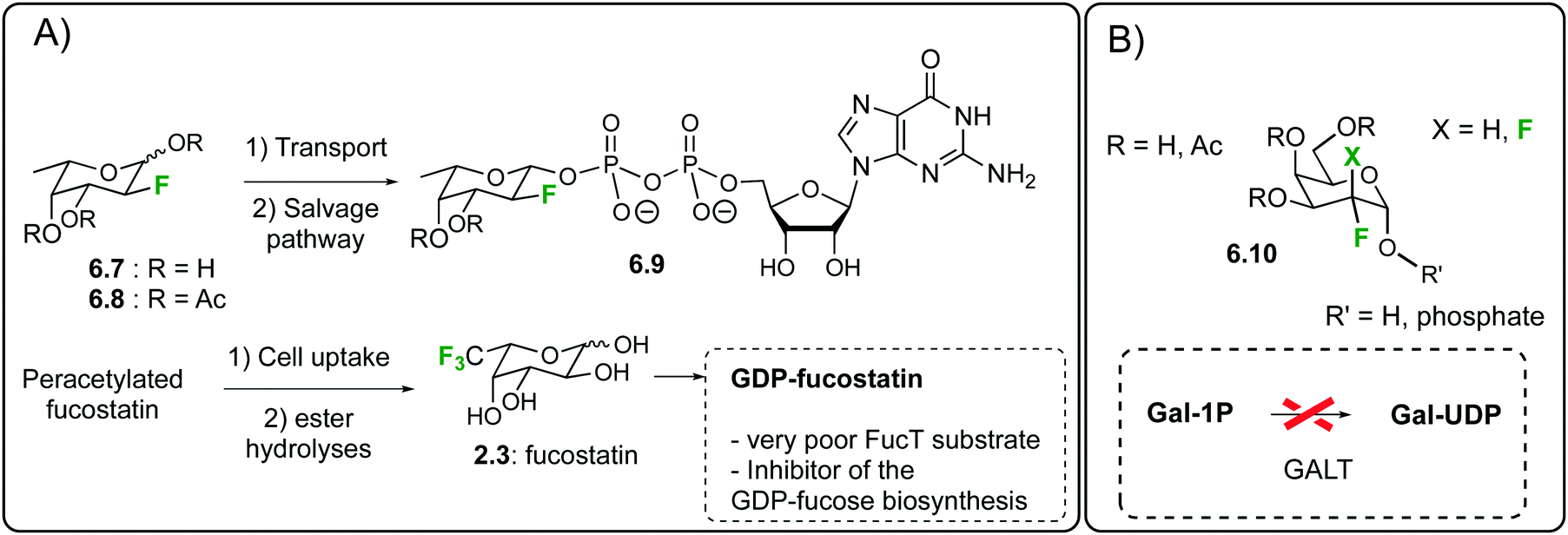 | ||
| Fig. 34 In cellulo generation of (A) fucosylation inhibitors 6.9 and GDP-fucostatin (B) galactose-1-phosphate uridyltransferase inhibitors. | ||
Very recently, a monofluorinated analogue of 2,4-diacetamido-2,4,6-trideoxygalactose, which is exclusively found in bacteria, has been shown to dramatically reduce Helicobacter pylori's ability to synthesize glycoproteins and led to diminished growth, motility, and biofilm formation.159 The proposed mode of action was here again the in situ generation of a glycosyltransferase inhibitor.
 | ||
| Fig. 35 2,3-Difluorosialosides as covalent sialidase inhibitors. | ||
 | ||
| Fig. 36 Highly fluorinated carbohydrate analogues. | ||
6.2 Synthetic vaccines and therapeutic antibodies
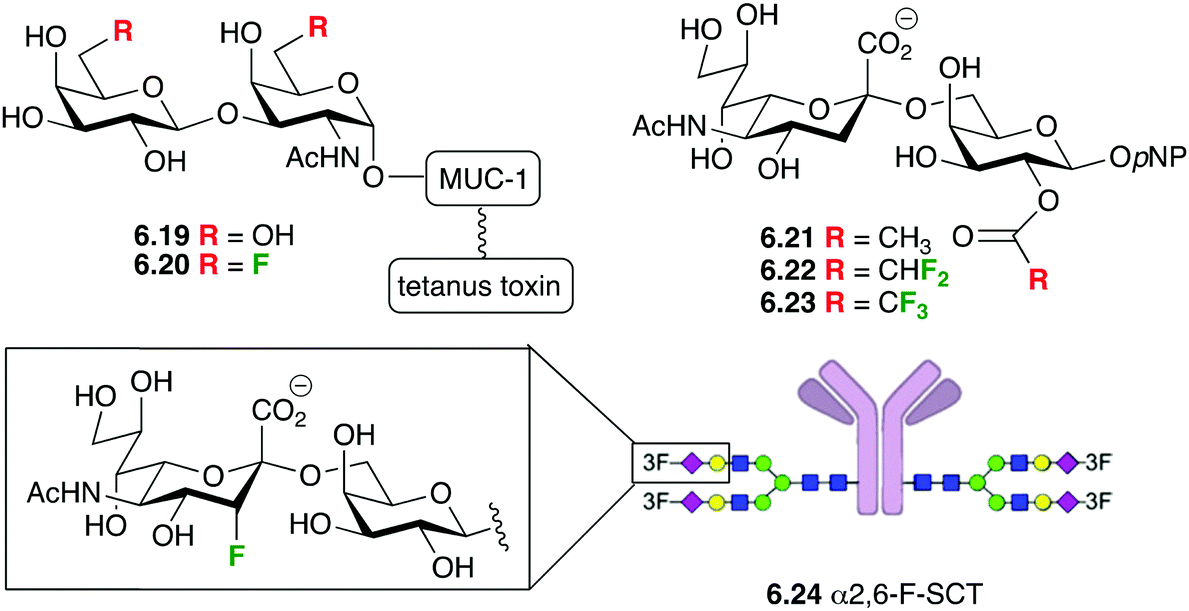
A particularly exciting application of fluorinated carbohydrates is their incorporation in synthetic antitumour vaccine and therapeutic antibodies.169 Synthetic vaccines with natural sugar antigens tend to be sensitive to enzymatic degradation in vivo, which can be overcome with fluorination (see Section 3.3). Maintaining the immunological efficiency requires that the fluorination does not negatively impact on the generation of the required type of antibodies, and that the thus generated antibodies bind with the tumour cells.The groups of Hoffmann-Röder and Kunz reported that fluorinated Thomsen–Friedenreich (TF) conjugates such as 6.19 and 6.20 (Fig. 37) generated a very strong immune response in mice, and their antibodies were shown to strongly bind to the epithelial tumour MCF-7 cells.170 Later on, Hoffmann-Röder showed that the antisera derived from the immunizations against a broader range of TF-MUC analogues fluorinated at the 6 and the 2-positions showed very little differences in the binding to the fluorinated and non-fluorinated antigens.170 These findings support the concept of using fluorinated analogues of tumour-associated carbohydrate antigen (TACA) for the design of synthetic vaccines. In addition, Yang et al. showed that fluorinated STn (another important TACA) antigens such as 6.22 and 6.23 significantly improved antigenicity compared to the nonfluorinated 6.21, and that the resulting antisera strongly reacted with STn positive tumor cells.171
 | ||
| Fig. 37 Fluorinated sugars as part of synthetic vaccines and antibodies. | ||
Interestingly, when 3-fluorinated sialic acids were incorporated in the monoclonal antibody rituximab (6.24), this glycoform showed similar binding and avidity to FcγRIIIa as the nonfluorinated rituximab, further showing the potential of fluorinated carbohydrates in the field of therapeutic antibodies.68b
7 Conclusions and perspectives
Fluorinated carbohydrate derivatives have been used in glycosciences, from glycochemistry to glycobiology, for over 30 years.28 The significant synthetic advances made in their synthesis, and in their derivatization (including glycosylation) have cemented their importance in glycobiology. This review provides an extensive summary of the effects of carbohydrate fluorination both with regard to changes in physical, chemical and biological properties.The influence of fluorination on carbohydrate conformation, with a focus on ring conformation, glycosidic bond and exocyclic methylene group conformation is described.51 This includes a comparison with C-glycosides and carbasugars.
The influence of fluorination on glycosidic bond stability and how this has been exploited is described.60 Finally, the novel direction of investigating carbohydrate lipophilicity is included.71
The influence of fluorination on intermolecular interactions is described in detail, both involving the C–F bond itself, as well as involving adjacent functional groups and C–H bonds.76 The recent advances in fluoro-containing glycomimetics synthesis,29 especially in terms of regio and stereo selective fluorine substitution, together with the development of novel strategies for biophysical characterization addressed the influence of fluorine in sugar/protein CH–π interactions and offer opportunities to exploit sugar fluorination to modulate intermolecular interactions.78 The already widespread use of fluorine in pharmaceuticals combined with strategic site-specific fluorine substitution may further boost for a rational drug design that combine the intrinsic advantages of drug fluorination with enhanced binding affinity to the target protein receptors.
With the exception of 2-FDG the development of fluorinated carbohydrate molecular imaging probes of PET112 and MRI137 has been less developed. Further investigation of other fluorinated monosaccharide isomers is likely to provide PET probes with higher organ selectivity. On the other hand the development of carbohydrate contrast agents for F-MRI has been hampered by the difficulty of introducing a sufficient number of chemically identical fluorine atoms in the probes and the associated problems in solubility. Recent efforts employing polysaccharide and oligosaccharide scaffolds might overcome the current technical difficulties and provide carbohydrate based contrast agents with additional targeting function.
From the enzymology viewpoint, fluorinated carbohydrates have long been recognized as valuable tools for probing enzymatic mechanisms at the molecular level, for a wide range of glycosyl-processing biocatalysts.141 During the last decade novel strategies have emerged to generate in cellulo potent fluorinated mechanism-based inhibitors, not only on whole cell assays, but also in vivo. The latter developments open new horizons in the development of drugs, for instance in the field of infectious diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. L. thanks the EPSRC (EP/P019943/1) for funding. L. U. acknowledges funding from the postdoctoral fellowship from the Human Frontier Science Program Organization (HFSP) Grant No. LT000747/2018-C. N. C. R. acknowledges funding from Agencia Estatal de Investigación (Spain) Grant No. CTQ2017-90039-R and RTC-2017-6126-1 and the Maria de Maeztu Units of Excellence Program. MDM-2017-0720. The group at Bilbao thank the Agencia Estatal de Investigacion of Spain (Grant RTI2018-094751-B-C21), the European Research Council (RECGLYCANMR, ERC AdG 788143), and the Severo Ochoa Excellence Accreditation SEV-2016-0644. SV acknowledges funding by FRS-FNRS (PDR grant number T.0102.18).References
- R. L. Schnaar, J. Allergy Clin. Immunol., 2015, 135, 609 CrossRef CAS PubMed.
- Y. van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593 CrossRef CAS PubMed.
- M. E. Taylor and K. Drickamer, FEBS J., 2019, 286, 1800 CrossRef CAS PubMed.
- M. J. Kailemia, D. Park and C. B. Lebrilla, Anal. Bioanal. Chem., 2016, 409, 395 CrossRef PubMed.
- E. Rodríguez, S. T. T. Schetters and Y. van Kooyk, Nat. Rev. Immunol., 2018, 18, 204 CrossRef PubMed.
- N. Taniguchi and Y. Kizuka, in Glycosylation and Cancer, ed. R. R. Drake and L. E. Ball, 2015, vol. 126, pp. 11–51 Search PubMed.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540 CrossRef CAS PubMed.
- E. Maverakis, K. Kim, M. Shimoda, M. E. Gershwin, F. Patel, R. Wilken, S. Raychaudhuri, L. R. Ruhaak and C. B. Lebrilla, J. Autoimmun., 2015, 57, 1 CrossRef CAS PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS PubMed.
- (a) R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef CAS PubMed; (b) R. Hevey, Biomimetics, 2019, 4 Search PubMed; (c) A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS PubMed.
- P. Valverde, A. Ardá, N.-C. Reichardt, J. Jiménez-Barbero and A. Gimeno, MedChemComm, 2019, 10, 1678 RSC.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- J. Wang, M. Sańchez Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- D. O’Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- R. A. Dwek, P. W. Kent and A. V. Xavier, Eur. J. Biochem., 1971, 23, 343 CrossRef CAS PubMed.
- F. Millett and M. A. Raftery, Biochemistry, 1972, 11, 1639 CrossRef CAS PubMed.
- P. Midoux, J.-P. Grivet and M. Monsigny, FEBS Lett., 1980, 120, 29 CrossRef CAS PubMed.
- C. P. J. Glaudemans and P. Kováč, Mol. Immunol., 1985, 22, 651 CrossRef CAS PubMed.
- I. P. Street, C. R. Armstrong and S. G. Withers, Biochemistry, 1986, 25, 6021 CrossRef CAS PubMed.
- C. P. J. Glaudemans, Chem. Rev., 1991, 91, 25 CrossRef CAS.
- P. Fernàndez, J. Jiménez-Barbero and M. Martín-Lomas, Carbohydr. Res., 1994, 254, 61 CrossRef.
- P. Fernàndez, F. J. Cañada, J. Jiménez-Barbero and M. Martín-Lomas, Carbohydr. Res., 1995, 271, 31 CrossRef.
- C. E. Miller, L. A. Mulard, E. A. Padlan and C. P. J. Glaudemans, Carbohydr. Res., 1998, 309, 219 CrossRef CAS PubMed.
- M. Bilska-Markowska, A. Szwajca and B. Marciniak, J. Fluorine Chem., 2019, 227, 109364 CrossRef CAS.
- (a) V. Wray, J. Chem. Soc., Perkin Trans. 2, 1976, 1598 RSC; (b) A. A. E. Penglis, Adv. Carbohydr. Chem. Biochem., 1981, 38, 195 CrossRef CAS; (c) M. Michalik, M. Hein and M. Frank, Carbohydr. Res., 2000, 327, 185 CrossRef CAS PubMed; (d) R. Csuk and B. I. Glanzer, Adv. Carbohydr. Chem. Biochem., 1989, 46, 73 CrossRef.
- E. Leclerc, X. Pannecoucke, M. Ethève-Quelquejeu and M. Sollogoub, Chem. Soc. Rev., 2013, 42, 4270 RSC.
- R. Plantier-Royon and C. Portella, Carbohydr. Res., 2000, 327, 119 CrossRef CAS PubMed.
- (a) T. Suami and S. Ogawa, Adv. Carbohydr. Chem. Biochem., 1990, 48, 21 CrossRef CAS PubMed; (b) A. Deleuze, C. Menozzi, M. Sollogoub and P. Sinaÿ, Angew. Chem., Int. Ed., 2004, 43, 6680 CrossRef CAS PubMed.
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618 CrossRef CAS PubMed.
- S. G. Withers, I. P. Street and S. J. Rettig, Can. J. Chem., 1986, 64, 232 CrossRef CAS.
- (a) S. Bresciani, T. Lebl, A. M. Z. Slawin and D. O’Hagan, Chem. Commun., 2010, 46, 5434 RSC; (b) L. Quiquempoix, Z. Wang, J. Graton, P. G. Latchem, M. Light, J.-Y. Le Questel and B. Linclau, J. Org. Chem., 2019, 84, 5899 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, J. St-Gelais, P. A. Johnson and D. Giguère, Nat. Commun., 2018, 9, 4721 CrossRef PubMed.
- (a) B. Linclau, S. Golten, M. Light, M. Sebban and H. Oulyadi, Carbohydr. Res., 2011, 346, 1129 CrossRef CAS PubMed; (b) B. Linclau, S. Golten and M. Light, J. Carbohydr. Chem., 2011, 30, 618 CrossRef CAS; (c) S. Golten, C. Q. Fontenelle, R. S. Timofte, L. Bailac, M. Light, M. Sebban, H. Oulyadi and B. Linclau, J. Org. Chem., 2016, 81, 4434 CrossRef CAS PubMed.
- H. W. Kim, P. Rossi, R. K. Shoemaker and S. G. DiMagno, J. Am. Chem. Soc., 1998, 120, 9082 CrossRef CAS.
- L. D. Hall and J. F. Manville, Can. J. Chem., 1969, 47, 19 CrossRef CAS.
- J. Sardinha, S. Guieu, A. Deleuze, M. C. Fernández-Alonso, A. P. Rauter, P. Sinaÿ, J. Marrot, J. Jiménez-Barbero and M. Sollogoub, Carbohydr. Res., 2007, 342, 1689 CrossRef CAS PubMed.
- L. Unione, B. Xu, D. Díaz, S. Martín-Santamaría, A. Poveda, J. Sardinha, A. P. Rauter, Y. Blériot, Y. Zhang, F. J. Cañada, M. Sollogoub and J. Jiménez-Barbero, Chem. – Eur. J., 2015, 21, 10513 CrossRef CAS PubMed.
- M. Kolympadi, M. Fontanella, C. Venturi, S. André, H.-J. Gabius, J. Jiménez-Barbero and P. Vogel, Chem. – Eur. J., 2009, 15, 2861 CrossRef CAS PubMed.
- (a) K. Bock and J. O. Duus, J. Carbohydr. Chem., 1994, 13, 513 CrossRef CAS; (b) K. N. Kirschner and R. J. Woods, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10541 CrossRef CAS PubMed.
- (a) L. D. Hall and L. Evelyn, Chem. Ind., 1968, 183 CAS; (b) L. Evelyn and L. D. Hall, Carbohydr. Res., 1976, 47, 285 CrossRef CAS.
- L. Phillips and V. Wray, J. Chem. Soc. B, 1971, 1618 RSC.
- M. Sharma, R. J. Bernacki, B. Paul and W. Korytnyk, Carbohydr. Res., 1990, 198, 205 CrossRef CAS PubMed.
- J. St-Gelais, M. Bouchard, V. Denavit and D. Giguère, J. Org. Chem., 2019, 84, 8509 CrossRef CAS PubMed.
- (a) J. F. Espinosa, F. J. Cañada, J. L. Asensio, M. Martín-Pastor, H. J. Dietrich, M. Martín-Lomas, R. R. Schmidt and J. J. Jimenez-Barbero, J. Am. Chem. Soc., 1996, 118, 10862 CrossRef CAS; (b) B. López-Méndez, C. Jia, Y. Zhang, P. Sinaÿ, L.-H. Zhang, J. Jiménez-Barbero and M. Sollogoub, Chem. – Asian J., 2008, 3, 51 CrossRef PubMed.
- A. García-Herrero, E. Montero, J. L. Muñoz, J. F. Espinosa, A. Vián, J. L. García, J. L. Asensio, F. J. Cañada and J. J. Jimenez-Barbero, J. Am. Chem. Soc., 2002, 124, 4804 CrossRef PubMed.
- (a) C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2016, 57, 92 CrossRef; (b) L. Hunter, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed.
- (a) R. W. Denton, K. A. Tony, J. J. Hernandez-Gay, F. J. Canada, J. Jimenez-Barbero and D. R. Mootoo, Carbohydr. Res., 2007, 342, 1624 CrossRef CAS PubMed; (b) J. Perez-Castells, J. J. Hernandez-Gay, R. W. Denton, K. A. Tony, D. R. Mootoo and J. Jimenez-Barbero, Org. Biomol. Chem., 2007, 5, 1087 RSC.
- B. Xu, L. Unione, J. Sardinha, S. Wu, M. Ethève-Quelquejeu, A. P. Rauter, Y. Blériot, Z. Zhang, S. Martín-Santamaría, D. Díaz, J. Jiménez-Barbero and M. Sollogoub, Angew. Chem., Int. Ed., 2014, 53, 9597 CrossRef CAS PubMed.
- L. Kerins, S. Byrne, A. Gabba and P. V. Murphy, J. Org. Chem., 2018, 83, 7714 CrossRef CAS PubMed.
- J. N. Barlow and J. S. Blanchard, Carbohydr. Res., 2000, 328, 473 CrossRef CAS PubMed.
- T. Haradahira, A. Kato, M. Maeda, Y. Torii, Y. I. Ichiya and K. Masuda, Appl. Radiat. Isot., 1992, 43, 627 CrossRef CAS.
- R. J. Abraham, E. J. Chambers and W. A. Thomas, Magn. Reson. Chem., 1994, 32, 248 CrossRef CAS.
- R. C. Bansal, B. Dean, S.-I. Hakomori and T. Toyokuni, J. Chem. Soc., Chem. Commun., 1991, 796 RSC.
- J. C. Errey, M. C. Mann, S. A. Fairhurst, L. Hill, M. R. McNeil, J. H. Naismith, J. M. Percy, C. Whitfield and R. A. Field, Org. Biomol. Chem., 2009, 7, 1009 RSC.
- D. Shishmarev, L. Quiquempoix, C. Q. Fontenelle, B. Linclau and P. W. Kuchel, Aust. J. Chem., 2020, 73, 117 CrossRef CAS.
- L. Lebedel, A. Ardá, A. Martin, J. Désiré, A. Mingot, M. Aufiero, N. A. Font, R. Gilmour, J. Jiménez-Barbero, Y. Blériot and S. Thibaudeau, Angew. Chem., Int. Ed., 2019, 58, 13758 CrossRef CAS PubMed.
- (a) S. G. Withers, D. J. MacLennan and I. P. Street, Carbohydr. Res., 1986, 154, 127 CrossRef CAS; (b) S. G. Withers, M. D. Percival and I. P. Street, Carbohydr. Res., 1989, 187, 43 CrossRef CAS; (c) M. N. Namchuk, J. D. McCarter, A. Becalski, T. Andrews and S. G. Withers, J. Am. Chem. Soc., 2000, 122, 1270 CrossRef CAS.
- (a) I. P. Street, J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9970 CrossRef CAS PubMed; (b) S. G. Withers, I. P. Street, P. Bird and D. H. Dolphin, J. Am. Chem. Soc., 1987, 109, 7530 CrossRef CAS.
- (a) B. P. Rempel and S. G. Withers, Glycobiology, 2008, 18, 570 CrossRef CAS PubMed; (b) L. I. Willems, J. Jiang, K.-Y. Li, M. D. Witte, W. W. Kallemeijn, T. J. N. Beenakker, S. P. Schröder, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chem. – Eur. J., 2014, 20, 10864 CrossRef CAS PubMed; (c) C. Tysoe and S. G. Withers, Curr. Top. Med. Chem., 2014, 14, 865 CrossRef CAS PubMed; (d) W. W. Kallemeijn, M. D. Witte, T. Wennekes and J. M. F. G. Aerts, Adv. Carbohydr. Chem. Biochem., 2014, 71, 297 CrossRef PubMed.
- K. Syson, C. E. M. Stevenson, A. M. Rashid, G. Saalbach, M. Tang, A. Tuukkanen, D. I. Svergun, S. G. Withers, D. M. Lawson and S. Bornemann, Biochemistry, 2014, 53, 2494 CrossRef CAS PubMed.
- S. Thanna, J. J. Lindenberger, V. V. Gaitonde, D. R. Ronning and S. J. Sucheck, Org. Biomol. Chem., 2015, 13, 7542 RSC.
- J. D. McCarter, W. Yeung, J. Chow, D. Dolphin and S. G. Withers, J. Am. Chem. Soc., 1997, 119, 5792 CrossRef CAS.
- H. Cao, Y. Li, K. Lau, S. Muthana, H. Yu, J. Cheng, H. A. Chokhawala, G. Sugiarto, L. Zhang and X. Chen, Org. Biomol. Chem., 2009, 7, 5137 RSC.
- M. Johannes, M. Reindl, B. Gerlitzki, E. Schmitt and A. Hoffmann-Röder, Beilstein J. Org. Chem., 2015, 11, 155 CrossRef PubMed.
- (a) X.-L. Sun, Y. Kanie, C.-T. Guo, O. Kanie, Y. Suzuki and C.-H. Wong, Eur. J. Org. Chem., 2000, 2643 CrossRef CAS; (b) H.-J. Lo, L. Krasnova, S. Dey, T. Cheng, H. Liu, T.-I. Tsai, K. B. Wu, C.-Y. Wu and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 6484 CrossRef CAS PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863 CrossRef CAS PubMed.
- (a) D. Bas, D. Dorison-Duval, S. Moreau, P. Bruneau and C. Chipot, J. Med. Chem., 2002, 45, 151 CrossRef CAS PubMed; (b) M. F. Mazzobre, M. V. Roman, A. F. Mourelle and H. R. Corti, Carbohydr. Res., 2005, 340, 1207 CrossRef CAS PubMed.
- B. Linclau, Z. Wang, G. Compain, V. Paumelle, C. Q. Fontenelle, N. Wells and A. Weymouth-Wilson, Angew. Chem., Int. Ed., 2016, 55, 674 CrossRef CAS PubMed.
- J. L. Asensio, A. Ardá, F. J. Cañada and J. Jimenez-Barbero, Acc. Chem. Res., 2013, 46, 946 CrossRef CAS PubMed.
- L. Montalvillo-Jiménez, A. G. Santana, F. Corzana, G. Jiménez-Osés, J. Jimenez-Barbero, A. M. Gómez and J. L. Asensio, J. Am. Chem. Soc., 2019, 141, 13372 CrossRef PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152 CrossRef CAS PubMed.
- M. I. Chávez, C. Andreu, P. Vidal, N. Aboitiz, F. Freire, P. Groves, J. L. Asensio, G. Asensio, M. Muraki, F. J. Cañada and J. Jimenez-Barbero, Chem. – Eur. J., 2005, 11, 7060 CrossRef PubMed.
- E. Jiménez-Moreno, G. Jiménez-Osés, A. M. Gómez, A. G. Santana, F. Corzana, A. Bastida, J. Jimenez-Barbero and J. L. Asensio, Chem. Sci., 2015, 6, 6076 RSC.
- M. C. Fernández-Alonso, F. J. Cañada, J. Jimenez-Barbero and G. Cuevas, J. Am. Chem. Soc., 2005, 127, 7379 CrossRef PubMed.
- A. G. Santana, E. Jiménez-Moreno, A. M. Gómez, F. Corzana, G. Jiménez-Osés, J. Jimenez-Barbero and J. L. Asensio, J. Am. Chem. Soc., 2013, 135, 3347 CrossRef CAS PubMed.
- (a) L. P. Calle, B. Echeverria, A. Franconetti, S. Serna, M. C. Fernández-Alonso, T. Diercks, F. J. Cañada, A. Ardá, N. C. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2015, 21, 11408 CrossRef CAS PubMed; (b) L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. J. Reichardt and J. Jiménez-Barbero, Chem. – Eur. J., 2017, 23, 3957 CrossRef CAS PubMed.
- (a) H. J. Schneider, Chem. Sci., 2012, 3, 1381 RSC; (b) P. A. Champagne, J. Desroches and J.-F. Paquin, Synthesis, 2015, 306 CAS.
- (a) J. D. Dunitz and R. Taylor, Chem. – Eur. J., 1997, 3, 89 CrossRef CAS; (b) G. Mehta and S. Sen, Eur. J. Org. Chem., 2010, 3387 CrossRef CAS; (c) K. Muller, Chimia, 2014, 68, 356 CrossRef CAS PubMed.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 13127 CrossRef CAS PubMed.
- K. Pervushin, A. Ono, C. Fernández, T. Szyperski, M. Kainosho and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14147 CrossRef CAS PubMed.
- K. Nakai, Y. Takagi and C. T. Tsuchiya, Carbohydr. Res., 1999, 316, 47 CrossRef CAS PubMed.
- B. Bernet and A. Vasella, Helv. Chim. Acta, 2007, 90, 1874 CrossRef CAS.
- (a) G. T. Giuffredi, V. Gouverneur and B. Bernet, Angew. Chem., Int. Ed., 2013, 52, 10524 CrossRef CAS PubMed; (b) L. Quiquempoix, E. Bogdan, N. J. Wells, J.-Y. Le Questel, J. Graton and B. Linclau, Molecules, 2017, 22, 518 CrossRef PubMed.
- (a) J. A. Garnett, Y. Liu, E. Leon, S. A. Allman, N. Friedrich, S. Saouros, S. Curry, D. Soldati-Favre, B. G. Davis, T. Feizi and S. Matthews, Protein Sci., 2009, 18, 1935 CrossRef CAS PubMed; (b) S. A. Allman, H. H. Jensen, B. Vijayakrishnan, J. A. Garnett, E. Leon, Y. Liu, D. C. Anthony, N. R. Sibson, T. Feizi, S. Matthews and B. G. Davis, ChemBioChem, 2009, 10, 2522 CrossRef CAS PubMed.
- P. S. Vermersch, J. J. G. Tesmer and F. A. Quiocho, J. Mol. Biol., 1992, 226, 923 CrossRef CAS PubMed.
- (a) K. E. van Straaten, J. R. A. Kuttiyatveetil, C. M. Sevrain, S. A. Villaume, J. Jiménez-Barbero, B. Linclau, S. P. Vincent and D. A. R. Sanders, J. Am. Chem. Soc., 2015, 137, 1230 CrossRef CAS PubMed; (b) I. N'Go, S. Golten, A. Ardá, J. Cañada, J. Jiménez-Barbero, B. Linclau and S. P. Vincent, Chem. – Eur. J., 2014, 20, 106 CrossRef PubMed.
- I. A. Bermejo, I. Usabiaga, I. Compañón, J. Castro-López, A. Insausti, J. A. Fernández, A. Avenoza, J. H. Busto, J. Jiménez-Barbero, J. J. Asensio, J. M. Peregrina, G. Jiménez-Osés, R. Hurtado-Guerrero, E. J. Cocinero and F. Corzana, J. Am. Chem. Soc., 2018, 140, 9952 CrossRef CAS PubMed.
- J. Graton, F. Besseau, A. M. Brossard, E. Charpentier, A. Deroche and J.-Y. Le Questel, J. Phys. Chem. A, 2013, 117, 13184 CrossRef CAS PubMed.
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004, p. 16 CrossRef; (b) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- (a) J. Graton, Z. Wang, A.-M. Brossard, D. Gonçalves Monteiro, J.-Y. Le Questel and B. Linclau, Angew. Chem., Int. Ed., 2012, 51, 6176 CrossRef CAS PubMed; (b) J. Graton, G. Compain, F. Besseau, E. Bogdan, J. M. Watts, L. Mtashobya, Z. Wang, A. Weymouth-Wilson, N. Galland, J.-Y. Le Questel and B. Linclau, Chem. – Eur. J., 2017, 23, 2811 CrossRef CAS PubMed.
- J. A. Olsen, D. W. Banner, P. Seiler, U. Obst Sander, A. d’Arcy, M. Stihle, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 2507 CrossRef CAS PubMed.
- (a) R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788 CrossRef CAS PubMed; (b) R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427 CrossRef CAS PubMed.
- K. J. Kamer, A. Choudhary and R. T. Raines, J. Org. Chem., 2013, 78, 2099 CrossRef CAS PubMed.
- (a) F. Hof, D. M. Scofield, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 5056 CrossRef CAS PubMed; (b) F. R. Fischer, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 8270 CrossRef CAS PubMed.
- L. Xing, C. Keefer and M. F. Brown, J. Fluorine Chem., 2017, 198, 47 CrossRef CAS.
- H. Dohi, R. Périon, M. Durka, M. Bosco, Y. Roué, F. Moreau, S. Grizot, A. Ducruix, S. Escaich and S. P. Vincent, Chem. – Eur. J., 2008, 14, 9530 CrossRef CAS PubMed.
- S. Grizot, M. Salem, V. Vongsouthi, L. Durand, F. Moreau, H. Dohi, S. Vincent, S. Escaich and A. Ducruix, J. Mol. Biol., 2006, 363, 383 CrossRef CAS PubMed.
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622 CrossRef CAS PubMed.
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132 CrossRef CAS.
- J. R. Potts, A. M. Hounslow and P. W. Kuchel, Biochem. J., 1990, 266, 925 CAS.
- D. Shishmarev, C. Q. Fontenelle, I. Kuprov, B. Linclau and P. W. Kuchel, Biophys. J., 2018, 115, 1906 CrossRef CAS PubMed.
- T. M. O'Connell, S. A. Gabel and R. E. London, Biochemistry, 1994, 33, 10985 CrossRef PubMed.
- B. J. Trayner, T. N. Grant, F. G. West and C. I. Cheeseman, Bioorg. Med. Chem., 2009, 17, 5488 CrossRef CAS PubMed.
- O.-M. Soueidan, B. J. Trayner, T. N. Grant, J. R. Henderson, F. Wuest, F. G. West and C. I. Cheeseman, Org. Biomol. Chem., 2015, 13, 6511 RSC.
- (a) T. Diercks, A. S. Infantino, L. Unione, J. Jiménez-Barbero, S. Oscarson and H. J. Gabius, Chem. – Eur. J., 2018, 24, 15761 CrossRef CAS PubMed; (b) T. Diercks, J. P. Ribeiro, F. J. Cañada, S. André, J. Jiménez-Barbero and H.-J. Gabius, Chem. – Eur. J., 2009, 15, 5666 CrossRef CAS PubMed; (c) J. P. Ribeiro, T. Diercks, J. Jiménez-Barbero, S. André, H. J. Gabius and F. J. Cañada, Biomolecules, 2015, 5, 3177 CrossRef CAS PubMed.
- (a) J. D. Martínez, P. Valverde, S. Delgado, C. Romanò, B. Linclau, N. C. Reichardt, S. Oscarson, A. Ardá, J. Jiménez-Barbero and F. J. Cañada, Molecules, 2019, 24, 2337 CrossRef PubMed; (b) P. Valverde, S. Delgado, J. D. Martínez, J. B. Vendeville, J. Malassis, B. Linclau, N. C. Reichardt, F. J. Cañada, J. Jiménez-Barbero and A. Arda, ACS Chem. Biol., 2019, 14, 1660 CrossRef CAS PubMed.
- E. C. Wamhoff, J. Hanske, L. Schnirch, J. Aretz, M. Grube, D. V. Silva and C. Rademacher, ACS Chem. Biol., 2016, 11, 2407 CrossRef CAS PubMed.
- E. Matei, S. André, A. Glinschert, A. S. Infantino, S. Oscarson, H. J. Gabius and A. M. Gronenborn, Chem. – Eur. J., 2013, 19, 5364 CrossRef CAS PubMed.
- (a) B. Beuthien-Baumann, K. Hamacher, F. Oberdorfer and J. Steinbach, Carbohydr. Res., 2000, 327, 107 CrossRef CAS PubMed; (b) G. R. Morais, R. A. Falconer and I. Santos, Eur. J. Org. Chem., 2013, 1401 CrossRef; (c) B. Barrios-Lopez and K. Bergstrom, Curr. Radiopharm., 2016, 9, 180 CrossRef PubMed; (d) S. Peña-Zalbidea, A. Y. T. Huang, H. W. Kavunja, B. Salinas, M. Desco, C. Drake, P. J. Woodruff, J. J. Vaquero and B. M. Swarts, Carbohydr. Res., 2019, 472, 16 CrossRef PubMed.
- G. Bormans and J. Verbruggen, J. Labelled Compd. Radiopharm., 2001, 44, 417 CrossRef CAS.
- O. Prante, K. Hamacher and H. H. Coenen, J. Labelled Compd. Radiopharm., 2007, 50, 55 CrossRef CAS.
- S. Maschauer, M. Pischetsrieder, T. Kuwert and O. Prante, J. Labelled Compd. Radiopharm., 2005, 48, 701 CrossRef CAS.
- F. Wuest, M. Berndt, R. Bergmann, J. van den Hoff and J. Pietzsch, Bioconjugate Chem., 2008, 19, 1202 CrossRef CAS PubMed.
- O. Boutureira, G. J. L. Bernardes, F. D'Hooge and B. G. Davis, Chem. Commun., 2011, 47, 10010 RSC.
- S. Maschauer, J. Einsiedel, R. Haubner, C. Hocke, M. Ocker, H. Hübner, T. Kuwert, P. Gmeiner and O. Prante, Angew. Chem., Int. Ed., 2010, 49, 976 CrossRef CAS PubMed.
- O. Boutureira, F. D'Hooge, M. Fernandez-Gonzalez, G. J. L. Bernardes, M. Sanchez-Navarro, J. R. Koeppe and B. G. Davis, Chem. Commun., 2010, 46, 8142 RSC.
- S. Smolskaya and Y. A. Andreev, Biomolecules, 2019, 9, 255 CrossRef CAS PubMed.
- H. Fukuda, T. Matsuzawa, Y. Abe, S. Endo, K. Yamada, K. Kubota, J. Hatazawa, T. Sato, M. Ito, T. Takahashi, R. Iwata and T. Ido, Eur. J. Nucl. Med., 1987, 7, 294 CrossRef PubMed.
- S. Furumoto, R. Shinbo, R. Iwata, Y. Ishikawa, K. Yanai, T. Yoshioka and H. Fukuda, J. Nucl. Med., 2013, 54, 1354 CrossRef CAS PubMed.
- (a) K. Ishiwata, et al. , Int. J. Radiat. Appl. Instrum., Part B, 1989, 16, 775 CrossRef CAS; (b) K. Ishiwata, et al. , Int. J. Radiat. Appl. Instrum., Part B, 1989, 16, 247 CrossRef CAS.
- N. Loch, C. C. Geilen, I. Spörndle, F. Oberdorfer, D. Keppler, R. Tauber and W. Reutter, FEBS Lett., 1991, 294, 217 CrossRef CAS PubMed.
- M. J. Morin, C. W. Porter, C. R. Petrie, W. Korytnyk and R. J. Bernacki, Biochem. Pharmacol., 1983, 32, 553–561 CrossRef CAS PubMed.
- M. Sørensen, K. S. Mikkelsen, K. Frisch, L. Bass, B. M. Bibby and S. Keiding, J. Nucl. Med., 2011, 52, 1566 CrossRef PubMed.
- K. M. Backus, H. L. Boshoff, C. S. Barry, O. Boutureira, M. K. Patel, F. D'Hooge, S. S. Lee, L. E. Via, K. Tahlan, C. E. Barry III and B. G. Davis, Nat. Chem. Biol., 2011, 7, 228 CrossRef CAS PubMed.
- I. Martinez-Duncker, R. Salinas-Marin and C. Martinez-Duncker, Int. J. Mol. Imaging, 2011, 28, 3497 Search PubMed.
- K. Ishiwata, M. Tomura, T. Ido, R. Iwata, K. Sato, J. Hatazawa, M. Kameyama and Y. Imahori, Int. J. Radiat. Appl. Instrum., Part B, 1990, 17, 363 CrossRef CAS.
- D. H. Dube and C. R. Bertozzi, Curr. Opin. Chem. Biol., 2003, 7, 616 CrossRef CAS PubMed.
- S. Hartlieb, A. Guenzel, R. Gerardy-Schahn, A. K. Muenster-Kuehnel, A. Kirschning and G. Draeger, Carbohydr. Res., 2008, 343, 2075 CrossRef CAS PubMed.
- M. Onega, J. Domarkas, H. Deng, L. F. Schweiger, T. A. D. Smith, A. E. Welch, C. Plisson, A. D. Gee and D. O'Hagan, Chem. Commun., 2010, 46, 139 RSC.
- S. Dall’Angelo, N. Bandaranayaka, A. D. Windhorst, D. J. Vugts, D. van der Born, M. Onega, L. F. Schweiger, M. Zanda and D. O’Hagan, Nucl. Med. Biol., 2013, 40, 464 CrossRef PubMed.
- R.-J. M. van Geuns, P. A. Wielopolski, H. G. de Bruin, B. J. Rensing, P. M. A. van Ooijen, M. Hulshoff, M. Oudkerk and P. J. de Feyter, Prog. Cardiovasc. Dis., 1999, 42, 149 CrossRef CAS PubMed.
- (a) I. L. Pykett, et al. , Radiology, 1982, 143, 157 CrossRef CAS PubMed; (b) H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 4485 CrossRef CAS PubMed.
- (a) J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, NMR Biomed., 2011, 24, 114 CrossRef CAS PubMed; (b) Y. Jian-xin, D. K. Vikram, C. Weina and P. M. Ralph, Curr. Med. Chem., 2005, 12, 819 CrossRef PubMed.
- O. Michelena, D. Padro, C. Carrillo-Carrión, P. del Pino, J. Blanco, B. Arnaiz, W. J. Parak and M. Carril, Chem. Commun., 2017, 53, 2447 RSC.
- T. Krawczyk, M. Minoshima, F. Sugihara and K. Kikuchi, Carbohydr. Res., 2016, 428, 72 CrossRef CAS PubMed.
- I. L. Kwee, T. Nakada and P. J. Card, J. Neurochem., 1987, 49, 428 CrossRef CAS PubMed.
- (a) Z. Tu, Y. N. Lin and C. H. Lin, Chem. Soc. Rev., 2013, 42, 4459 RSC; (b) L. Tedaldi and G. K. Wagner, Med. Chem. Commun., 2014, 5, 1106 RSC; (c) L. Wang, Y. Liu, L. Wu and X. L. Sun, Biochim. Biophys. Acta, 2016, 1864, 143 CrossRef CAS PubMed; (d) C. Breton, L. Š. Najdrová, C. Jeanneau, J. Koca and A. Imberty, Glycobiology, 2006, 16, 29R CrossRef CAS PubMed.
- (a) S.-H. Choi, M. W. Ruszczycky, H. Zhang and H.-W. Liu, Chem. Commun., 2011, 47, 10130 RSC; (b) P. T. Nyffeler, S. G. Duron, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192 CrossRef CAS PubMed; (c) M. Burkart, S. P. Vincent, A. Düffels, B. Murray, S. Ley and C.-H. Wong, Bioorg. Med. Chem., 2000, 8, 1937 CrossRef CAS PubMed; (d) B. W. Murray, V. Wittmann, M. D. Burkart, S.-C. Hung and C.-H. Wong, Biochemistry, 1997, 36, 823 CrossRef CAS PubMed; (e) H. Dohi, R. Périon, M. Durka, M. Bosco, Y. Roué, F. Moreau, S. Grizot, A. Ducruix, S. Escaich and S. P. Vincent, Chem. – Eur. J., 2008, 14, 9530 CrossRef CAS PubMed; (f) M. D. Burkart, S. P. Vincent and C.-H. Wong, Chem. Commun., 1999, 1525 RSC.
- (a) L. Dumitrescu, G. Eppe, A. Tikad, S. El Bkassiny, W. Pan, S. Gurcha, G. Besra, A. Arda, J. Jiménez-Barbero and S. P. Vincent, Chem. – Eur. J., 2014, 20, 15208 CrossRef CAS PubMed; (b) S. Habib, F. Larnaud, E. Pfund, T. M. Barragán, T. Lequeux, C. Ortiz Mellet, P. G. Goekjian and D. Gueyrard, Org. Biomol. Chem., 2014, 12, 690 RSC; (c) A. Caravano, H. Dohi, P. Sinaÿ and S. P. Vincent, Chem. – Eur. J., 2006, 11, 3114 CrossRef PubMed.
- (a) J. Delbrouck, L. Chêne and S. P. Vincent, Fluorosugars as inhibitors of bacterial enzymes, in Fluorine in Life Sciences: Pharmaceuticals, Medical Diagnostics, and Agrochemicals, ed. G. Haufe and F. Leroux, Academic Press, Elsevier, 2018, p. 241 Search PubMed; (b) Y. Jina, D. Bhattasali, E. Pellegrini, S. M. Forget, N. J. Baxtera, M. J. Cliffa, M. W. Bowler, D. L. Jakeman, G. M. Blackburna and J. P. Waltho, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12384 CrossRef PubMed; (c) S. M. Forget, D. Bhattasali, V. C. Hart, T. S. Cameron, R. T. Syvitskiad and D. L. Jakeman, Chem. Sci., 2012, 3, 1866 RSC; (d) C. Cocaud, R. B. Zheng, T. L. Lowary, T. Poisson, X. Pannecoucke, C. Nicolas and O. R. Martin, Carbohydr. Res., 2018, 461, 45 CrossRef CAS PubMed; (e) S. M. Forget, E. A. C. Bushnell, R. J. Boyd and D. L. Jakeman, Can. J. Chem., 2016, 94, 1 CrossRef; (f) F. Massicot, R. Plantier-Royon, J.-L. Vasse and J.-B. Behr, Carbohydr. Res., 2018, 464, 2 CrossRef CAS PubMed; (g) J.-S. Zhu, N. E. McCormick, S. C. Timmons and D. L. Jakeman, J. Org. Chem., 2016, 81, 8816 CrossRef CAS PubMed; (h) M. W. Loranger, S. M. Forget, N. E. McCormick, R. T. Syvitski and D. L. Jakeman, J. Org. Chem., 2013, 78, 9822 CrossRef CAS PubMed; (i) J.-S. Zhu, K. M. Stiers, E. Soleimani, B. R. Groves, L. J. Beamer and D. L. Jakeman, J. Org. Chem., 2019, 84, 9627 CrossRef CAS PubMed; (j) B. P. Rempel, M. B. Tropak, D. J. Mahuran and S. G. Withers, Angew. Chem., Int. Ed., 2011, 50, 10381 CrossRef CAS PubMed; (k) J.-S. Zhu, K. M. Stiers, S. M. Winter, A. D. Garcia, A. F. Versini, L. J. Beamer and D. L. Jakeman, ACS Omega, 2019, 4, 7029 CrossRef CAS PubMed.
- (a) G. Eppe, P. Peltier, R. Daniellou, C. Nugier-Chauvin, V. Ferrieres and S. P. Vincent, Bioorg. Med. Chem. Lett., 2009, 19, 814 CrossRef CAS PubMed; (b) P. A. Frantom, J. K. Coward and J. S. Blanchard, J. Am. Chem. Soc., 2010, 132, 6626 CrossRef CAS PubMed; (c) S. S. Lee, S. Y. Hong, J. C. Errey, A. Izumi, G. J. Davies and B. G. Davis, Nat. Chem. Biol., 2011, 7, 631 CrossRef CAS PubMed; (d) S.-I. Nishimura, M. Hato, S. Hyugaji, F. Feng and M. Amano, Angew. Chem., Int. Ed., 2012, 51, 3386 CrossRef CAS PubMed; (e) S. S. Lee, I. R. Greig, D. J. Vocadlo, J. D. McCarter, B. O. Patrick and S. G. Withers, J. Am. Chem. Soc., 2011, 133, 15826 CrossRef CAS PubMed; (f) R. Pongdee and H.-W. Liu, Bioorg. Chem., 2004, 32, 393 CrossRef CAS PubMed; (g) Q. Zhang and H.-w. Liu, J. Am. Chem. Soc., 2001, 123, 6756 CrossRef CAS PubMed; (h) A. Burton, P. Wyatt and G.-J. Boons, J. Chem. Soc., Perkin Trans. 1, 1997, 2375–2383 RSC; (i) P. Valverde, J.-B. Vendeville, K. Hollingsworth, A. Mattey, T. Keenan, H. Chidwick, H. Ledru, K. Huonnic, K. Huang, M. Light, N. Turner, J. Jimenez-Barbero, M. C. Galan, M. A. Fascione, S. Flitsch, W. B. Turnbull and B. Linclau, Chem. Commun., 2020 10.1039/D0CC02209H.
- (a) P. Peltier, M. Beláňová, P. Dianišková, R. Zhou, R. B. Zheng, J. A. Pearcey, M. Joe, P. Brennan, C. Nugier-Chauvin, V. Ferrières, T. L. Lowary, R. Daniellou and K. Mikušová, Chem. Biol., 2010, 17, 1356 CrossRef CAS PubMed; (b) C. D. Brown, M. S. Rusek and L. L. Kiessling, J. Am. Chem. Soc., 2012, 134, 6552 CrossRef CAS PubMed; (c) V. L. Schultz, X. Zhang, K. Linkens, J. Rimel, D. E. Green, P. L. DeAngelis and R. J. Linhardt, J. Org. Chem., 2017, 82, 2243 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521 CrossRef CAS PubMed.
- (a) K. Persson, H. D. Ly, M. Dieckelmann, W. W. Wakarchuk, S. G. Withers and N. C. Strynadka, Nat. Struct. Biol., 2001, 8, 166 CrossRef CAS PubMed; (b) S. Grizot, M. Salem, V. Vongsouthi, L. Durand, F. Moreau, H. Dohi, S. Vincent, S. Escaich and A. Ducruix, J. Mol. Biol., 2006, 363, 383 CrossRef CAS PubMed; (c) L. S. Ni, H. A. Chokhawala, H. Z. Cao, R. Henning, L. Ng, S. S. Huang, H. Yu, X. Chen and A. J. Fisher, Biochemistry, 2007, 46, 6288 CrossRef CAS PubMed; (d) R. P. Gibson, J. P. Turkenburg, S. J. Charnock, R. Lloyd and G. J. Davies, Chem. Biol., 2002, 9, 1337 CrossRef CAS PubMed; (e) W. Offen, C. Martinez-Fleites, M. Yang, E. Kiat-Lim, B. G. Davis, C. A. Tarling, C. M. Ford, D. J. Bowles and G. J. Davies, EMBO J., 2006, 25, 1396 CrossRef CAS PubMed; (f) C. P. C. Chiu, A. G. Watts, L. L. Lairson, M. Gilbert, D. Lim, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, Nat. Struct. Mol. Biol., 2004, 11, 163–170 CrossRef CAS PubMed.
- C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, A. Dell, S. M. Haslam and J. C. Paulson, Nat. Chem. Biol., 2012, 8, 661 CrossRef CAS PubMed.
- M. S. Macauley, B. M. Arlian, C. D. Rillahan, P.-C. Pang, N. Bortell, M. C. Marcondes, S. M. Haslam, A. Dell and J. C. Paulson, J. Biol. Chem., 2014, 289, 35149 CrossRef CAS PubMed.
- (a) C. Büll, T. J. Boltje, M. Wassink, A. M. A. de Graaf, F. L. van Delft, M. H. den Brok and G. J. Adema, Mol. Cancer Ther., 2013, 12, 1935 CrossRef PubMed; (b) C. Büll, T. J. Boltje, E. A. W. van Dinther, T. Peters, A. M. A. de Graaf, J. H. W. Leusen, M. Kreutz, C. G. Figdor, M. H. den Brok and G. J. Adema, ACS Nano, 2015, 9, 733 CrossRef PubMed; (c) C. Büll, T. J. Boltje, N. Balneger, S. M. Weischer, M. Wassink, J. J. van Gemst, V. R. Bloemendal, L. Boon, J. van der Vlag, T. Heise, M. H. den Brok and G. J. Adema, Cancer Res., 2018, 78, 3574 Search PubMed.
- T. Heise, J. F. A. Pijnenborg, C. Büll, N. van Hilten, E. D. Kers-Rebel, N. Balneger, H. Elferink, G. J. Adema and T. J. Boltje, J. Med. Chem., 2019, 62, 1014 CrossRef CAS PubMed.
- T. Heise, J. D. Langereis, E. Rossing, M. I. de Jonge, G. J. Adema, C. Büll and T. J. Boltje, Cell Chem. Biol., 2018, 25, 1 CrossRef PubMed.
- N. M. Okeley, S. C. Alley, M. E. Anderson, T. E. Boursalian, P. J. Burke, K. M. Emerton, S. C. Jeffrey, K. Klussman, C. L. Law, D. Susman, B. E. Toki, L. Westendorf, W. Zeng, X. Zhang, D. R. Benjamin and P. D. Senter, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5404 CrossRef CAS PubMed.
- J. D. Belcher, C. Chen, J. Nguyen, F. Abdulla, P. Nguyen, M. Nguyen, N. M. Okeley, D. R. Benjamin, P. D. Senter and G. M. Vercellotti, PLoS One, 2015, 10, e0117772 CrossRef PubMed.
- J. G. Allen, M. Mujacic, M. J. Frohn, A. J. Pickrell, P. Kodama, D. Bagal, T. San Miguel, E. A. Sickmier, S. Osgood, A. Swietlow, V. Li, J. B. Jordan, K. W. Kim, A. C. Rousseau, Y. J. Kim, S. Caille, M. Achmatowicz, O. Thiel, C. H. Fotsch, P. Reddy and J. D. McCarter, ACS Chem. Biol., 2016, 11, 2734 CrossRef CAS PubMed.
- M. M. Achmatowicz, J. G. Allen, M. M. Bio, M. D. Bartberger, C. J. Borths, J. T. Colyer, R. D. Crockett, T.-L. Hwang, J. N. Koek, S. A. Osgood, S. W. Roberts, A. Swietlow, O. R. Thiel and S. Caille, J. Org. Chem., 2016, 81, 4736 CrossRef CAS PubMed.
- N. C. McKenzie, N. E. Scott, A. John, J. M. White and E. D. Goddard-Borger, Carbohydr. Res., 2018, 465, 4 CrossRef CAS PubMed.
- V. Janes, S. Grabany, J. Delbrouck, S. Vincent, J. Gottschalk, L. Elling and F.-G. Hanisch, Cells, 2020, 9, 607 CrossRef PubMed.
- D. A. Williams, K. Pradhan, A. Paul, I. R. Olin, O. T. Tuck, K. D. Moulton, S. S. Kulkarni and D. H. Dube, Chem. Sci., 2020, 11, 1761–1774 RSC.
- (a) A. G. Watts, I. Damager, L. M. Amaya, A. Buschiazzo, P. Alzari, A. C. Frasch and S. G. Withers, J. Am. Chem. Soc., 2003, 125, 7532 CrossRef CAS PubMed; (b) S. Buchini, A. Buschiazzo and S. G. Withers, Angew. Chem., Int. Ed., 2008, 47, 2700 CrossRef CAS PubMed.
- C.-S. Tsaia, H.-Y. Yena, M.-I. Lina, T.-I. Tsaia, S.-Y. Wanga, W.-I. Huanga, T.-L. Hsua, Y.-S. E. Chenga, J.-M. Fanga and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2466 CrossRef PubMed.
- J. H. Kim, R. Resende, T. Wennekes, H. M. Chen, N. Bance, S. Buchini, A. G. Watts, P. Pilling, V. A. Streltsov, M. Petric, R. Liggins, S. Barrett, J. L. McKimm-Breschkin, M. Niikura and S. G. Withers, Science, 2013, 340, 71 CrossRef CAS PubMed.
- S. Buchini, F. X. Gallat, I. R. Greig, J. H. Kim, S. Wakatsuki, L. M. G. Chavas and S. G. Withers, Angew. Chem., Int. Ed., 2014, 53, 3382 CrossRef CAS PubMed.
- L. Dirr, I. M. El-Deeb, P. Guillon, C. J. Carroux, L. M. G. Chavas and M. von Itzstein, Angew. Chem., Int. Ed., 2015, 54, 2936 CrossRef CAS PubMed.
- Z. Gao, M. Niikura and S. G. Withers, Angew. Chem., Int. Ed., 2017, 56, 6112 CrossRef CAS PubMed.
- (a) R. S. Timofte and B. Linclau, Org. Lett., 2008, 10, 3673 CrossRef CAS PubMed; (b) J. A. Boydell, V. Vinader and B. Linclau, Angew. Chem., Int. Ed., 2004, 43, 5677 CrossRef PubMed; (c) B. Linclau, A. J. Boydell, R. S. Timofte, K. J. Brown, V. Vinader and A. C. Weymouth-Wilson, Org. Biomol. Chem., 2009, 7, 803 RSC; (d) M. J. Corr and D. O’Hagan, J. Fluorine Chem., 2013, 155, 72 CrossRef CAS; (e) V. Denavit, D. Lainé, C. Bouzriba, E. Shanina, P. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chem. – Eur. J., 2019, 25, 4478 CrossRef CAS PubMed; (f) L. Quiquempoix, Z. Wang, J. Graton, P. G. Latchem, M. Light, J.-Y. Le Questel and B. Linclau, J. Org. Chem., 2019, 84, 5899 CrossRef CAS PubMed.
- A. Ioannou, E. Cini, R. S. Timofte, S. L. Flitsch, N. J. Turner and B. Linclau, Chem. Commun., 2011, 47, 11228 RSC.
- B. P. Rempel and S. G. Withers, Aust. J. Chem., 2009, 62, 590 CrossRef CAS.
- (a) J. Wang, H. Li, G. Zou and L.-X. Wang, Org. Biomol. Chem., 2007, 5, 1529 RSC; (b) Q. Wang and Z. Guo, ACS Med. Chem. Lett., 2011, 2, 373 CrossRef CAS PubMed.
- (a) A. Hoffmann-Röder, S. Kaiser, S. Wagner, N. Gaidzik, D. Kowalczyk, U. Westerlind, B. Gerlitzki, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2010, 49, 8498 CrossRef PubMed; (b) S. Wagner, C. Mersch and A. Hoffmann-Röder, Chem. – Eur. J., 2010, 16, 7319 CrossRef CAS PubMed; (c) M. Johannes, T. Oberbillig and A. Hoffmann-Röder, Org. Biomol. Chem., 2011, 9, 5541 RSC; (d) A. Hoffmann-Röder and M. Johannes, Chem. Commun., 2011, 47, 9903 RSC; (e) T. Oberbillig, C. Mersch, S. Wagner and A. Hoffmann-Röder, Chem. Commun., 2012, 48, 1487 RSC; (f) M. Johannes, M. Reindl, B. Gerlitzki, E. Schmitt and A. Hoffmann-Röder, Beilstein J. Org. Chem., 2015, 11, 55 Search PubMed; (g) A. Baumann, S. Marchner, M. Daum and A. Hoffmann-Röder, Eur. J. Org. Chem., 2018, 3803 CrossRef CAS.
- F. Yang, X. J. Zheng, C. X. Huo, Y. Wang, Y. Zhang and X. S. Ye, ACS Chem. Biol., 2011, 6, 252–259 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |