
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging unconventional organic solvents for C–H bond and related functionalization reactions
Congjun
Yu
 a,
Jesús
Sanjosé-Orduna
b,
Frederic W.
Patureau
*a and
Mónica H.
Pérez-Temprano
*b
a,
Jesús
Sanjosé-Orduna
b,
Frederic W.
Patureau
*a and
Mónica H.
Pérez-Temprano
*b
aInstitute for Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: Frederic.Patureau@rwth-aachen.de
bInstitute of Chemical Research of Catalonia (ICIQ), Avinguda dels Països Catalans 16, 43007 Tarragona, Spain. E-mail: mperez@iciq.es
First published on 2nd March 2020
Abstract
Solvent engineering is an increasingly essential topic in the chemical sciences. In this context, some recently appeared unconventional solvents have shown their large potential in the field of C–H bond functionalization reactions. This review aims not only at recognizing and classifying a short selection of these emerging solvents, in particular halogenated ones, but also at providing a medium term perspective of the possibilities they will offer for synthetic method development.
 Congjun Yu | Congjun Yu was born in 1990 in Shandong, China. He got his BSc at Shanghai Normal University in 2013. After that, he studied at the Shanghai Institute of Organic Chemistry (SIOC), CAS, and obtained his MSc in 2016 under the supervision of Prof. Zhang. Since January 2017, he joined Prof. Patureau's group now at the Institute of Organic Chemistry of the RWTH Aachen University, where he is pursuing his PhD degree. He has been focusing on developing novel strategies to achieve selective cross-dehydrogenative coupling (CDC) reactions. |
 Jesús Sanjosé-Orduna | Jesús Sanjosé-Orduna obtained his BSc and MSc Degrees from the University of Barcelona in 2014 and 2015, respectively. He then joined the research group of Mónica H. Pérez-Temprano at the Institute of Chemical Research of Catalonia (ICIQ), where he is pursuing his PhD degree. During his thesis, he has investigated the intricacies of Cp*Co-catalyzed C–H functionalization reactions, not only to provide a comprehensive mechanistic picture of these transformations but also to improve their efficiency based on fundamental knowledge. |
 Frederic W. Patureau | Frederic W. Patureau was born in 1982 in France. He got his BSc (2003) and MSc (2005) at the University of Paris-Sud Orsay. He obtained his PhD degree in the group of Prof. Reek at the University of Amsterdam (UvA) in 2009. He was then a Humboldt postdoctoral fellow in the group of Prof. Glorius at the University of Münster (WWU Münster). He started his independent career as a Junior-Professor at the TU Kaiserslautern University (2011–2018). Since 2018, he is Professor at the Institute of Organic Chemistry of the RWTH Aachen University. His research is focused on investigating novel strategies for pragmatic cross-couplings and other metal catalyzed reactions. |
 Mónica H. Pérez-Temprano | Mónica H. Pérez-Temprano received her BSc (2005) and MSc (2006) from the University of Valladolid. She carried out her PhD thesis (2011) under the supervision of Prof. Espinet and Prof. Casares at the University of Valladolid. In 2012, she moved to the University of Michigan to work under the supervision of Prof. Sanford. In 2015, she began her independent career as Group Leader at the Institute of Chemical Research of Catalonia (ICIQ). Her research interests are focused on the rational design of efficient and sustainable approaches for the synthesis of organic molecules using fundamental organometallic chemistry. |
Key learning points(1) Much C–H bond functionalization reactivity can be gained by looking beyond conventional organic solvents.(2) Key transitions states can be decisively controlled by carefully designed solvents. (3) The field of unconventional solvent mediated C–H bond functionalization is still in its infancy. (4) So far, interestingly, the majority of these unconventional solvents are (per-)halogenated (but this could evolve in the future). (5) More unconventional solvent enabled C–H bond functionalization reactivity can be expected in the coming years, as well as new privileged organic solvents. |
Introduction
The organic solvents in which chemical processes occur are rarely a topic of discussion,1 and if so, often only on a very basic level. In the past few years, however, incredible reaction discoveries were enabled by new organic solvents, in particular halogenated ones such as HFIP, trifluoroethanol, trichloroethanol, tetrachloroethylene and others, which were not usually employed in organic synthetic methods before. Some of these new unconventional solvents often have an “extra-ordinary” (out of the ordinary) quality to them: the new reactions don’t or barely operate in their absence. What makes them so special? How will this change organic and organometallic synthesis, in particular for direct C–H bond functionalization coupling reactions? This tutorial review is focused on those new organic solvent effects on C–H bond functionalization reactivity. It should be noted that due to early reviews on one of these leading emerging solvents: Hexafluoroisopropanol (HFIP),2–4 only the selected (most recent) insights on that particular solvent will be covered. Conversely, with HFIP no longer being the only unconventional organic solvent on the shelf, a more inclusive overview on this rising topic was increasingly needed. With this tutorial review, we hope to support the key learning points mentioned above and thereby encourage further research efforts in these areas, in particular for applications in innovative (C–H bond functionalization based) synthetic method development. Moreover, by choice of the authors, ionic liquids, supercritical CO2, or water are not covered in this selective tutorial review. While some of the key learning points certainly apply to these media as well, and while each of the latter media could justify a review in its own right, the choice has been made to focus priory on the emerging, often not yet fully recognized, unconventional organic solvent enabled C–H bond functionalization reactivities.Defining “unconventional organic solvent”
For the purpose of this tutorial review focused on direct C–H bond functionalization, an unconventional (or not yet conventional) solvent may be defined as a seldom utilized organic solvent with reactivity supporting properties that go beyond solvents broadly considered as conventional. This expression is meant to support the discussion and key learning points.HFIP and TFE: super polar, super H-bond donors
Hexafluoroisopropanol (HFIP) is arguably the most popular oxo-halogenated unconventional solvent at the moment for C–H bond functionalization reactions, closely followed by trifluoroethanol (TFE).2–4 Therefore, of all the selected unconventional solvents covered in this review, HFIP and TFE already benefit from (somewhat) deeper mechanistic understanding. Their main features are a high polarity (for an organic solvent), combining both a hydrophobic as well as a hydrophilic side, and a mildly acidic OH group (pKa analogous to that of a phenol). This is combined with excellent single H-bond donor properties, a phenomenon enjoying growing consensus in the scientific community to explain its extra-ordinary reactivity enabling character.2–4 Interestingly, this is also associated with a very strong shielding effect at the oxygen atom, easily measured in 17O NMR spectroscopy, in comparison to the non-fluorinated analogues (Scheme 1).5 All these properties ensure highly specific solvation of transitions states, typically decreasing activation energies.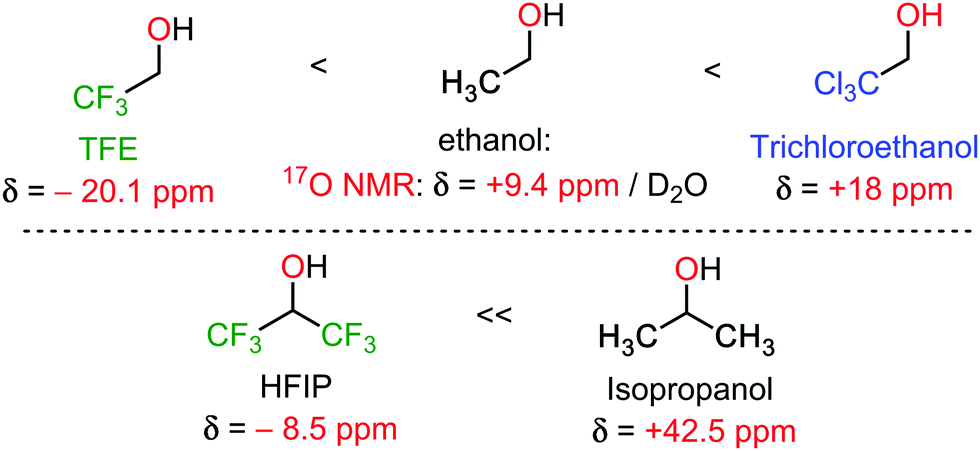 | ||
| Scheme 1 17O NMR of some conventional and unconventional solvents.5 | ||
As mentioned above, different reviews have covered the effect of perfluorinated alcohols on organic transformations. Therefore, this section will focus on a very selected number of recent examples in the context of C–H functionalization reactions, one of the cornerstones of modern synthetic chemistry.
In 2018, some of us revealed the beneficial effect of HFIP in Cp*Co(III)-catalysed C–H functionalization reactions. Initially, the authors observed that the addition of HFIP as additive accelerated dramatically the C–H activation of N-(2-pyrimidylindole) by [Cp*Co(MeCN)3](BF4)2, from 24 h to 1 h (Scheme 2a).6 Driven by this extraordinary boosting effect, they next investigated the global influence of HFIP in catalysis. They selected as benchmark transformations the reaction between N-(2-pyrimidylindole) and diphenylacetylene, as coupling partner. Using these starting materials, several literature precedents have described chemodivergent reactivity depending on the reaction conditions: hydroarylation vs. oxidative annulation (Scheme 2b).7,8 These reactivity patterns represented an ideal test case for investigating the role of HFIP since, a priori, it could play opposite effects. In both cases, the authors discovered that the addition of HFIP enables the use of milder conditions for obtaining the corresponding product in almost quantitative yields, but presumably due to different reasons. In the formation of the annulated product, it seems that its high polarity is crucial for solubilizing the salts present in the reaction mixture. On the other hand, in the hydroarylation reaction, its role as proton source probably facilitates the protodemetalation step of the catalytic cycle.
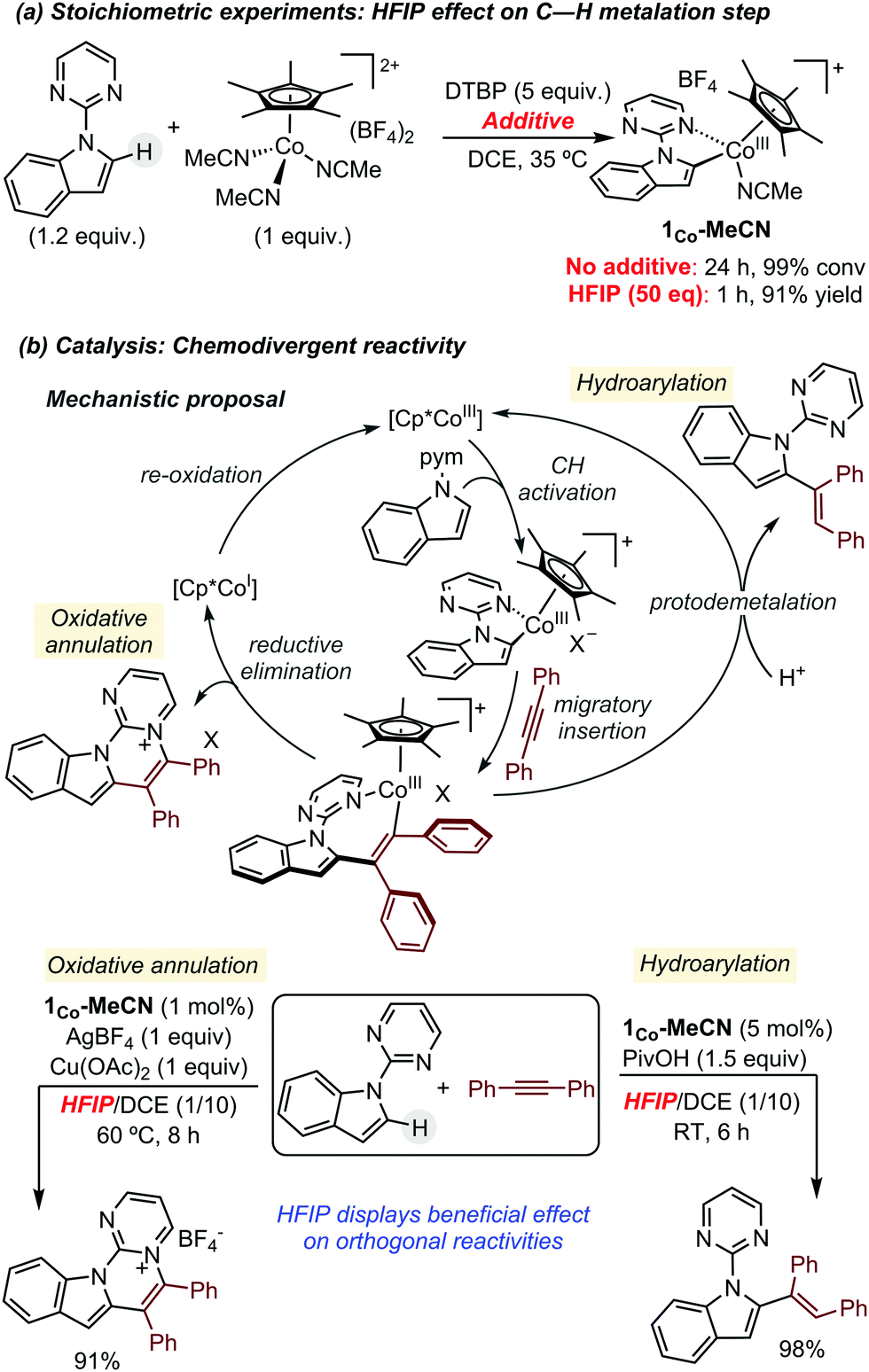 | ||
| Scheme 2 Beneficial effect of HFIP in Cp*Co-catalysed C–H functionalization reactions using alkynes as coupling partners. | ||
Over the past year, different literature precedents have shown solvent-controlled C–H functionalization processes when using HFIP and/or TFE. Miura and co-workers have demonstrated the selective C–H alkenylation and alkylation of 2-pyridones with acrylates, depending on the employed solvent (Scheme 3a).7 Using DMF, the authors observed the expected formation of the C6-alkenylated product. In sharp contrast, HFIP promoted the alkylation processes due to its capability to act as a proton source. Loh and co-workers have reported selective Cp*Co-catalysed C–H aminomethylation or hydroxymethylation of heteroarenes using 1,2-oxadetidine as coupling partner (Scheme 3b).8 The formation of the aminomethylated product was favoured when using PhCF3 as solvent, while 2,2,2-trifluoroethanol (TFE) was the best choice for developing the hydromethylation protocol. Interestingly, Rovis et al. have reported an unexpected regiodivergent Cp*Ir(III)-catalysed alkene diamination protocol (Scheme 3c).9 The authors observed the formation of five- or six- membered ring lactams by the employment of HFIP and TFE, respectively. They hypothesised that the higher acidity of HFIP compared to TFE is the responsible of the regioselectivity.
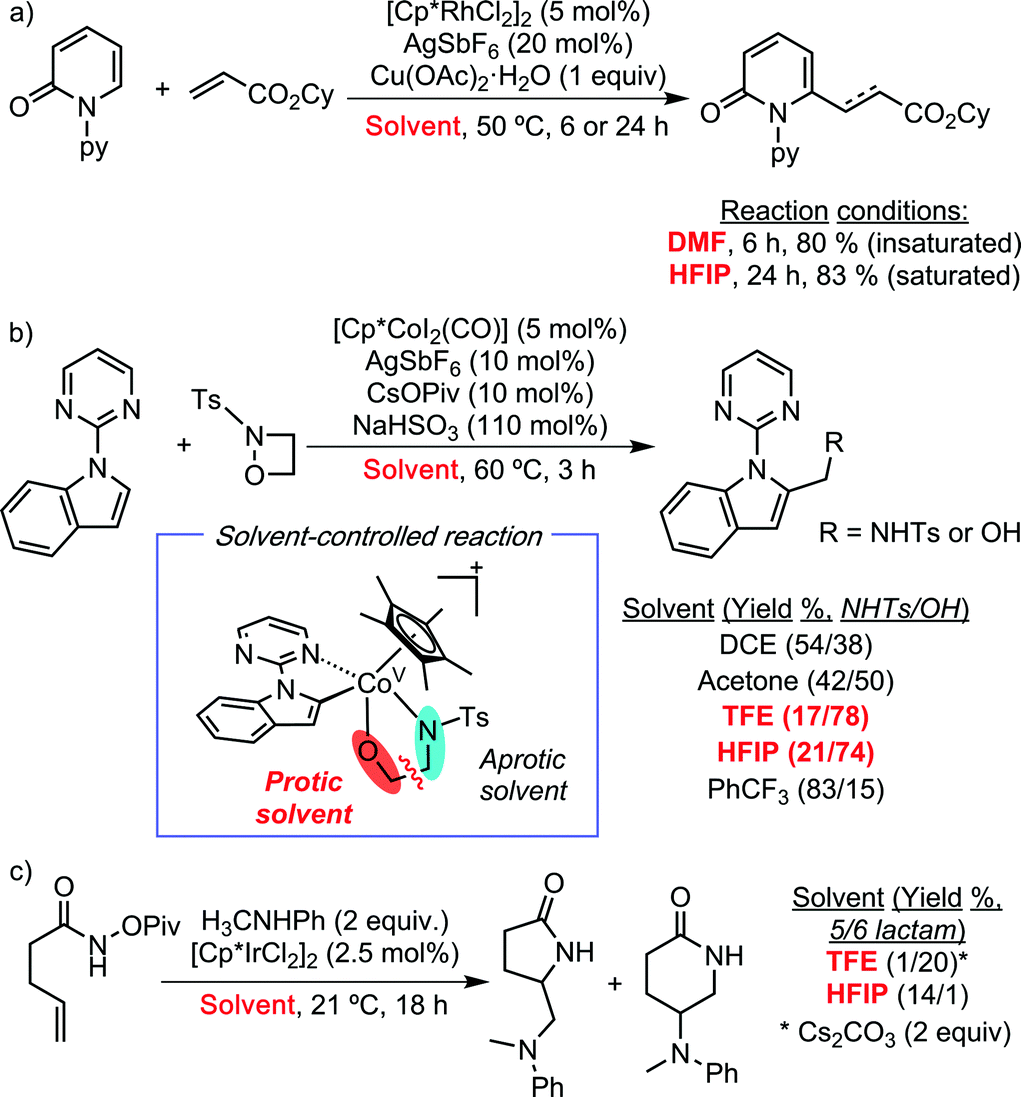 | ||
| Scheme 3 Chemoselective Cp*MIII-catalysed C–H functionalization reactions using TFE and/or HFIP. | ||
Apart from the mentioned examples, other research groups have also shown during the last year the extraordinary capability of TFE or HFIP to promote different C–H functionalization processes using (Cp*)-based Group 9 (Co, Rh or Ir) pre-catalysts. Chang and co-workers have developed a synthetic protocol for accessing γ-lactams via C–H amidation, through a Ir-carbonylnitrenoid intermediate, using a pre-catalyst supported by bidentate ligands (Scheme 4).10 In this particular case, 2,2,2-trifluoroethanol exhibited a higher performance than HFIP. This methodology featured a wide substrate scope, good group tolerance and scalability.
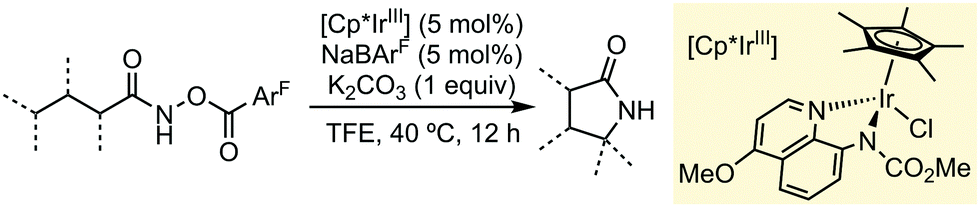 | ||
| Scheme 4 Synthesis of γ-lactams via Cp*IrIII-catalysed C–H functionalization reactions. | ||
These protic perfluorinated solvents have also been exploited to develop novel stereoselective transformations (Scheme 5). In this regard, Cramer et al. have disclosed the highly efficiency of TFE in the enantioselective synthesis of cyclopentenylamines, using a chiral CpxRhI(cod) pre-catalyst, in terms of yield and enantioinduction (Scheme 5a).11 The authors proposed that the strong ionizing ability of the perfluorinated alcohol favours the generation of the active rhodium(III) species involved in the C–H activation step. This is supported by the low conversions observed in less polar solvents such as dichloromethane or toluene. Following the same strategy, Cramer has recently developed the enantioselective synthesis of dihydroisoquinolones with chiral cyclopentadienyl cobalt complexes (Scheme 5b).12 In this case, the employment of HFIP as solvent improved the reactivity, when compared to TFE.
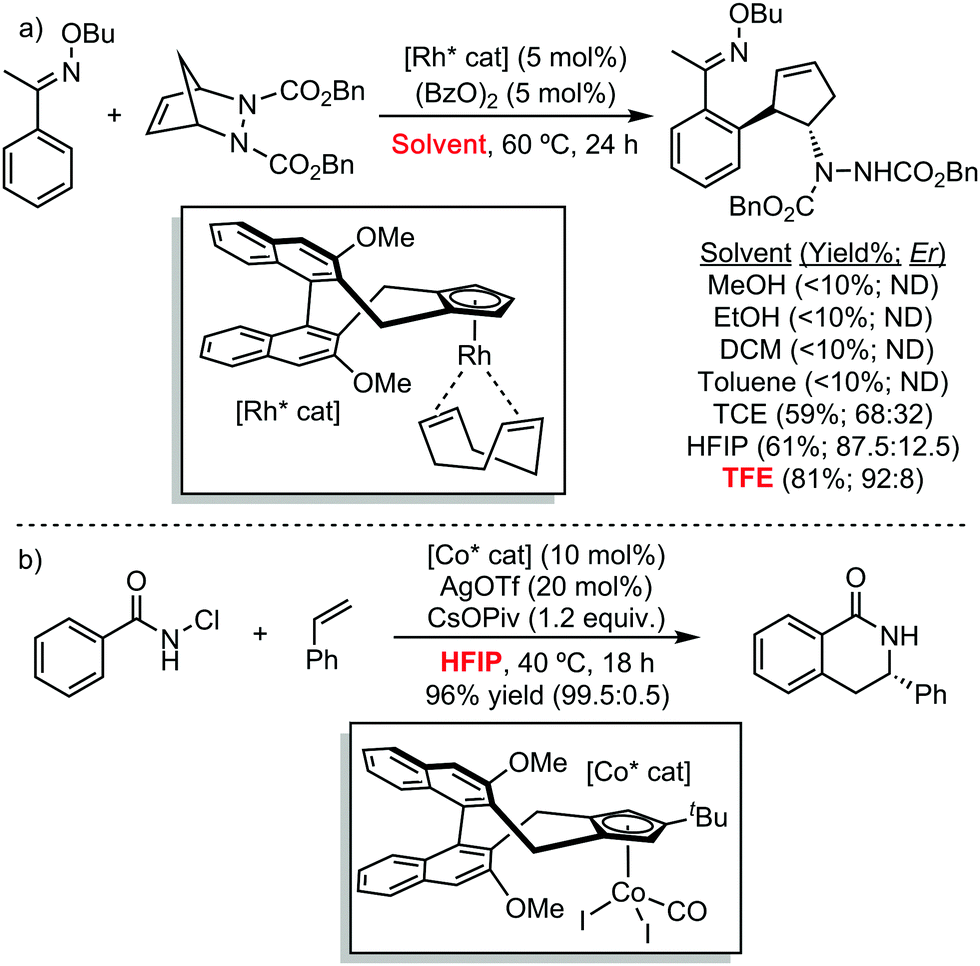 | ||
| Scheme 5 Stereoselective C–H functionalization reactions. | ||
Other research groups have taken advantage of HFIP to engineer different C–C and C–heteroatom bond-forming reactions using other transition metals pre-catalysts, or even in their absence. For example, Larrosa has described a Pd/Ag protocol for the α-arylation of benzo[b]thiophenes.13 In the context of early transition metals, Donohoe has exploited HFIP unique properties, such as its high polarity and hydrogen bonding capability, to promote the stereoselective synthesis of oxygen-containing heterocycles by titanium complexes, using allyl or benzyl alcohols as alkylating reagent (Scheme 6a).14 Preliminary mechanistic studies showed the formation of a titanium species, containing two hexafluoroisopropoxy units, which presumably are involved in the cyclization reaction. Ritter and co-workers have reported a Fe-catalysed aromatic C–H amination, using [MsO–NH3](OTf) as nitrogen source, for the synthesis of unprotected anilines (Scheme 6b).15 The versatility of this methodology is attributed to the use of HFIP. The formation of a HFIP–triflate hydrogen bond, supported experimentally, enhances the reactivity of [MsO–NH3](OTf) as oxidant. Moreover, the presence of HFIP increases the electrophilicity of putative cationic radical species, making the addition of the nitrogen source facile to more electron-poor arenes. The same group has developed a metal-free late-stage C–O formation protocol, using bis(methanesulfonyl)peroxide as terminal oxidant and hexafluoroisopropanol as solvent (Scheme 6c).16 This example is particularly remarkable since, a priori, O-centred radicals generated from the peroxide could participate in the hydrogen-atom abstraction from HFIP, hindering the desired reactivity. HFIP has also been applied as solvent in the development of photocatalytic C–H functionalization processes. In this context, Chen and He have shown its extraordinary effect, compared to CH2Cl2 or even TFE, in the remote Csp3–H heteroarylation of free aliphatic alcohols using hypervalent iodine as oxidant under irradiation (Scheme 7a).17
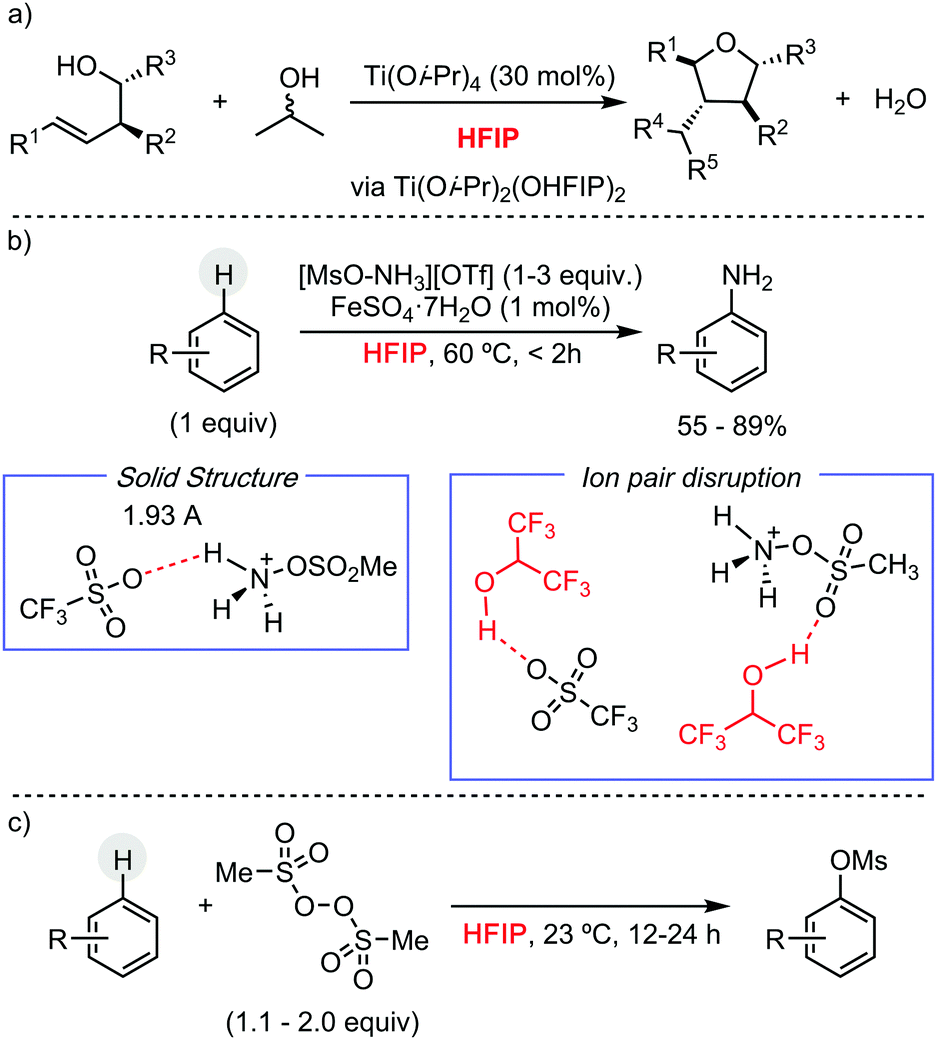 | ||
| Scheme 6 C–C and C–heteroatom bond-forming reactions promoted by HFIP. | ||
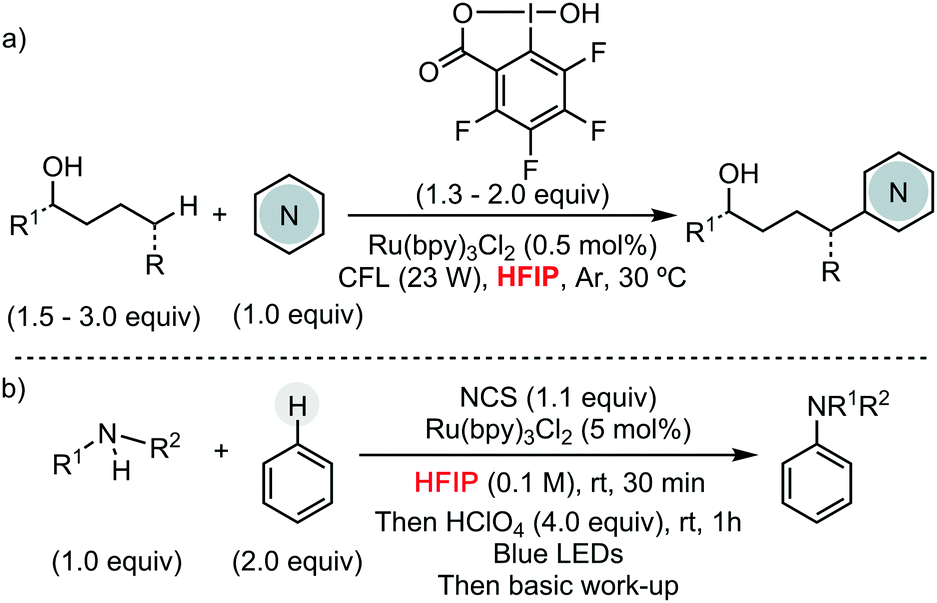 | ||
| Scheme 7 Photoredox C–H functionalizations. | ||
Leonori has described the regioselective amination of simple arenes using alkyl amines as coupling partners (Scheme 7b).18 The authors explored different reaction conditions, being HFIP selected as the solvent of choice when activating weakly electron-rich arenes. This solvent was also used in a parallel screening approach for the late-stage functionalization of complex molecules relevant for drug discovery. Recently, HFIP was also utilized in some other high profile synthetic methods, such as meta- and para-selective C–H bond functionalizations by Sunoj and Maiti (Scheme 8).19–22 Clearly, HFIP will remain a top unconventional organic solvent on the shelf of synthetic method developers in the years to come.
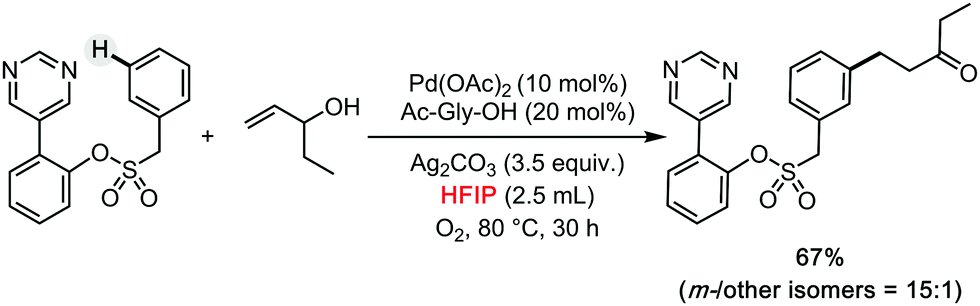 | ||
| Scheme 8 meta-Selective C–H bond functionalizations by Maiti.19–22 | ||
Trichloroethanol: the new HFIP? or a completely uncharted territory?
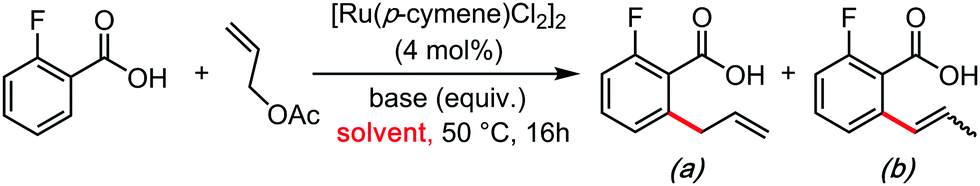
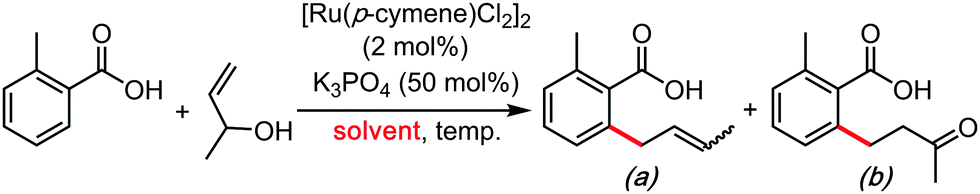
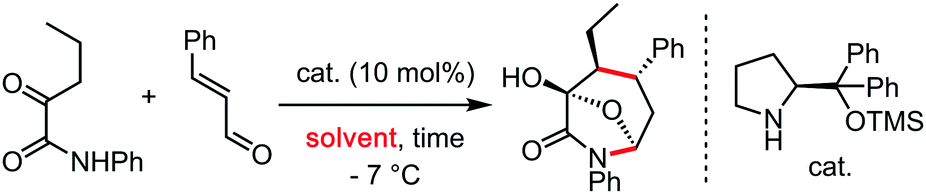
In the wake of the unconventional reactivity enabled by HFIP, Trichloroethanol has very recently appeared as a unique solvent for organometallic catalysis and C–H bond functionalization coupling reactions. This effect seems very different from the previous paragraph on HFIP and trifluoroethanol, as trichloroethanol seems to outperform the former solvents in those new synthetic methods. This could have something to do with intra and inter-molecular H-bonding network differences, completely altering the stabilization of the various key transitions states. These were mainly explored by the Gooßen group.23–25 They recently developed a series of carboxylate-directed C–H allylations with vinyl acetates (Table 1), allyl amines (Table 2), allyl alcohols or ethers (Table 3). In their condition optimizations, trichloroethanol was always found to be the best solvent for the reactions, especially compared with HFIP and TFE. Moreover, in a very different transformation, Bonne and Rodriguez26 reported an organocatalyzed enantioselective synthesis of aza-seven-membered rings in which trichloroethanol performed as an extra-ordinary solvent to achieve both high diastereoselectivity and enantioselectivity (Table 4).|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Yield (%) | |
| a | b | |||
| 1 | K2CO3 (0.5) | NMP | 18 | 3 |
| 2 | K2CO3 (0.5) | 1,4-Dioxane | 5 | — |
| 3 | K2CO3 (0.5) | toluene | Trace | — |
| 4 | K2CO3 (0.5) | EtOH | 28 | Trace |
| 5 | K2CO3 (0.5) | HFIP | 15 | 13 |
| 6 | K2CO3 (0.5) | AcOH | — | — |
| 7 | K2CO3 (0.5) | TFE | 60 | 5 |
| 8 | K2CO3 (0.5) | Trichloroethanol | 60 | Trace |
|
|
|||
|---|---|---|---|
| Entry | Additive (mol%) | Solvent | Yield (%) |
| 1 | — | Toluene | Trace |
| 2 | — | Dioxane | Trace |
| 3 | — | MeOH | 8 |
| 4 | — | tert-Amyl alcohol | Trace |
| 5 | — | HFIP | 47 |
| 6 | — | TFE | 30 |
| 7 | — | Trichloroethanol | 52 |
| 8 | CF3CO2H (30) | Trichloroethanol | 80 |
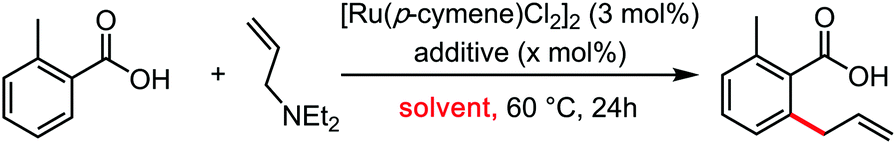
|
|
||||
|---|---|---|---|---|
| Entry | Temp. (°C) | Solvent | Yield (%) | |
| a (E/Z) | b | |||
| 1 | 60 | Toluene | 13 (2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
6 |
| 2 | 60 | CH3CN | 8 (2:1) |
3 |
| 3 | 60 | tert-Amyl alcohol | 16 (1.8:1) |
— |
| 4 | 60 | HFIP | 27 (2:1) |
— |
| 5 | 60 | TFE | 68 (1.7:1) |
— |
| 6 | 60 | Trichloroethanol | 80 (2:1) |
— |
| 7 | 50 | Trichloroethanol | 89 (2:1) |
— |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time | Yield (%) | dr | ee |
| 1 | Toluene | 15 h | 0 | — | — |
| 2 | DCM | 15 h | 0 | — | — |
| 3 | CH3CN | 15 h | 0 | — | — |
| 4 | EtOH | 15 h | 92 | 3:1 |
97 |
| 5 | Trichloroethanol | 24 h | 91 | 5:1 |
99 |
| 7 | Same (at −7 °C) | 3 days | 94 | 7:1 |
99 |
It can be reasonably assumed that the intra and inter-molecular H-bonding networks greatly differ from trifluoroethanol, to HFIP, to trichloroethanol, thus greatly affecting stabilizing interactions with key transitions states. This is again well illustrated in Scheme 1, wherein the shielding pattern of the oxygen atom greatly differs from CF3 to CCl3, probably betraying significant H-bonding differences. In other words, there seem to exist a great margin of transition state and reactivity tuning, simply by structural design of the oxo-halogenated solvent. It is therefore reasonable to expect that HFIP and trichloroethanol mediated reactivity will continue flourishing in the coming years. Moreover, the discovery of other unconventional oxo-halogenated solvents can be expected as well.
Tetrachloroethylene: π-acid effect, “Knochel Trick,” and enantioselective supramolecular catalysis
Just as Trichloroethanol in the previous section, tetrachloroethylene (C2Cl4) is also not a very common solvent in synthetic organic chemistry, not to mention in C–H bond functionalization reactions. It is nonetheless a cheap chemical commodity, used notably for dry-cleaning, for industrial degreasing, and as a synthetic precursor.27 However, as a perchlorinated solvent, its solubilizing capacity is sometimes only moderate. It is therefore often utilized in combination with other co-solvents. Herein, we shall discuss only the most recent examples, in particular those which are metal catalysed, and in which C2Cl4 plays an essential role. While the examples that we selected are relatively few, and mechanistically not sufficiently investigated, the decisive role of C2Cl4 in those transformation may have a profound impact on the field of synthetic method development.One the best leads to understand the effect of C2Cl4 is probably the “Knochel trick.” In a pioneering and inspiring 1998 work, Knochel reported a Ni-catalysed C(sp3)–C(sp3) bond forming cross coupling reaction.28 The overall reaction (Scheme 9) runs under basic/reductive organo-zinc conditions. The authors found that catalytic amounts of 3-trifluoromethylstyrene had a pronounced acceleration effect on the rate of the reaction. Analogously structured trifuoromethyl-ketones were also investigated, however none was found as efficient as the 3-trifluoromethylstyrene catalyst, considerably increasing the efficiency of the reaction. In a second contribution in 1999, Knochel clearly formulated the hypothesis along which the electron-poor olefin would facilitate the rate limiting reductive elimination step through transient π-acid coordination to the active metal centre.29
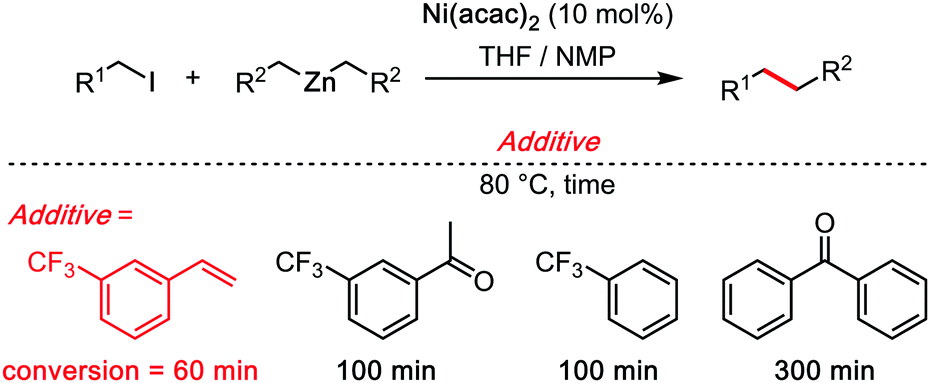 | ||
| Scheme 9 The “Knochel trick,” 1998. | ||
The “Knochel trick” clearly suggests that when faced with a rate limiting reductive elimination that will not yield, one can simply use a π-acidic olefin additive or co-solvent. It has been twenty years since Knochel formulated the concept, yet, in spite of numerous cases wherein reductive elimination controls and limits the rate and scope of the coupling reaction, many of which concern C–H bond functionalization reactions, the “Knochel trick” has been only seldom utilized.30 Of course, styrenes are rarely chemically inert additives. For instance, they may insert in all sorts of metal–carbon bonds, they may trigger undesired redox processes, they may even polymerize. What this concept arguably needs is a somewhat cheaper and more stable olefinic π-acid, perhaps, for example, tetrachloroethylene (C2Cl4). Crystallographic proof of the ability of C2Cl4 to really bind to transition metal catalysts is very scarce, although this may be solely due to limited general interest in such organometallic motives. In 2014, Figueroa and co-authors reported a beautiful crystal structure of a TCE–Pd0 complex (Scheme 10), thereby demonstrating the affinity of TCE for low oxidation state metal centres.31 Therein, the characteristic Pd–CCl2 distance: 2.042(3) Å, and the pronounced loss of planarity of the C2Cl4 ligand suggest a relatively strong η2 coordination mode. This crystallographic data therefore confirms the validity of applying “Knochel's trick” with C2Cl4 as additive/co-solvent.
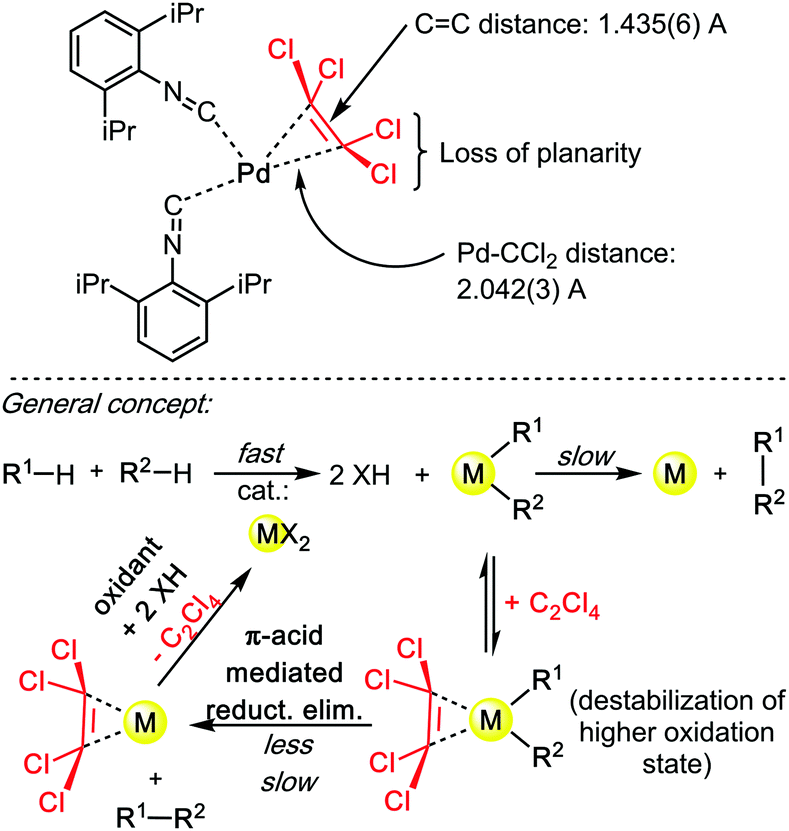 | ||
| Scheme 10 Figueroa's X-rayed TCE–Pd0 complex (2014), and general concept (beneath). | ||
Some of us have started doing so in 2013, in the Ru-catalysed dehydrogenative C1–N dimerization of carbazoles,32 a system that was revisited with a mechanistic investigation, in collaboration with the Maseras group, in 2018 (Scheme 11).33 Therein, C2Cl4 was found to be a superior co-solvent (chlorobenzene is utilized as an additional co-solvent in order to improve solubility).
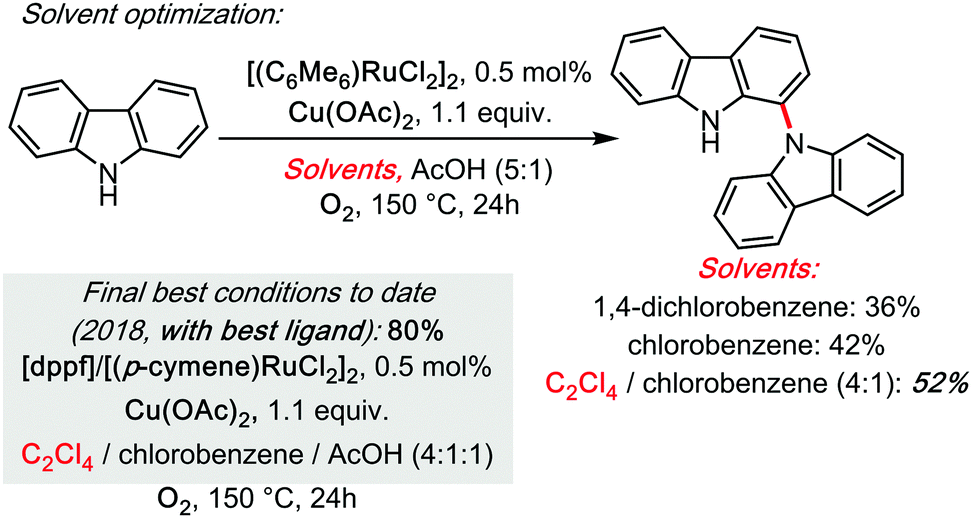 | ||
| Scheme 11 Ru catalysed cross dehydrogenative carbazolation of carbazoles (homo-coupling) under TCE, 2013–2018. | ||
Moreover, the advanced DFT calculations of Maseras (as well as our kinetic investigations) leave no doubt as to the rate limiting character of the C1–N (cooperative polynuclear) reductive elimination step. It is therefore not surprising that C2Cl4 would be found a superior co-solvent for this process, by analogy to Knochel's system. Some of us were moreover able to transpose this reactivity to a true hetero-coupling: the Ruthenium catalysed cross dehydrogenative ortho N-carbazolation of diarylamines, in 2014.34 This remains at the moment the only direct access to this motif in a single step (Scheme 12).
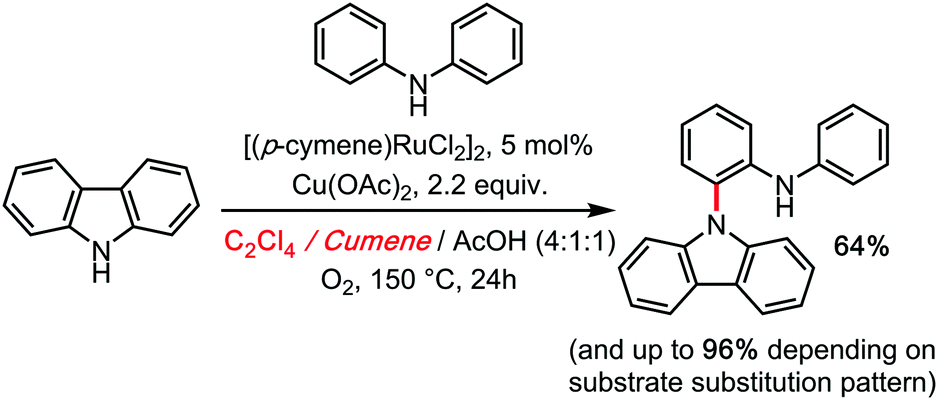 | ||
| Scheme 12 Ru catalyzed cross dehydrogenative carbazolation of diarylamines (hetero-coupling) under TCE, 2014. | ||
Interestingly, it should be mentioned that C2Cl4 was also utilized as a privileged solvent in metal free (enantioselective) coupling reactions. Recently for example, Papai and Takemoto reported an asymmetric hetero-Michael addition of α,β-unsaturated carboxylic acids under organo-catalytic conditions (N–H bond functionalization, Scheme 13).35 Some of the best results in terms of conversion and especially enantiomeric excess were obtained in C2Cl4 as a solvent. The highly halogenated character of the solvent possibly imposes a good compromise between dissolving capacity and maximized H-bonding strength/efficiency between substrates and supramolecular chiral catalyst – thus a very different mode of action than that of for example HFIP. In general, we suspect also other beneficial effects of the C2Cl4 solvent, such as solvating/stabilizing π-complexes with electron-rich substrates.
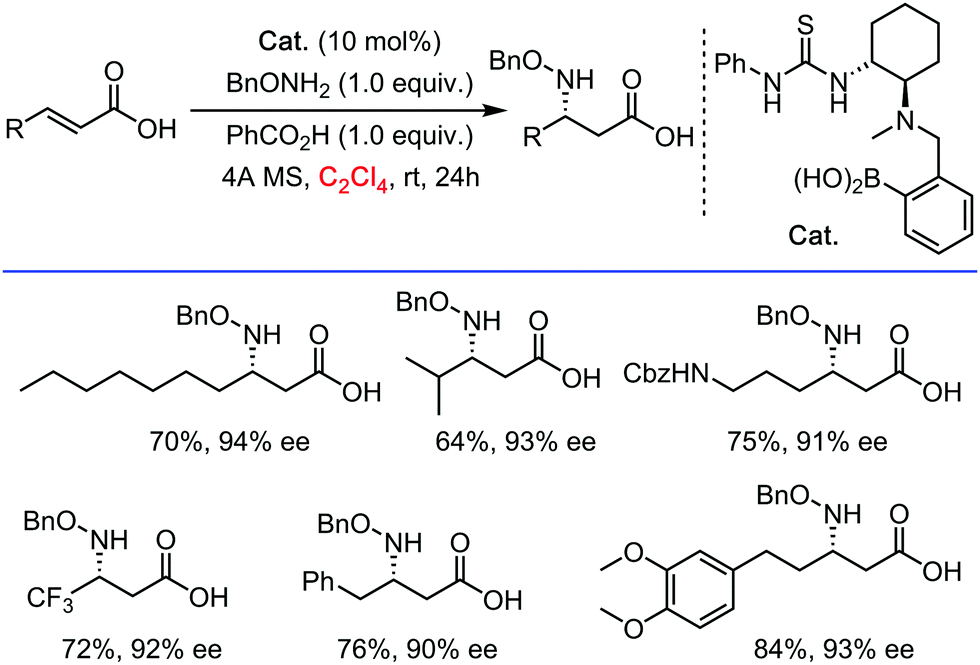 | ||
| Scheme 13 Asymmetric hetero-Michael addition of α,β-unsaturated carboxylic acids under organo-catalytic conditions, 2018. | ||
Overall, it can be said that the π-acid induced reductive elimination strategy is a promising concept. We anticipate that method developers with reductive elimination issues will increasingly incorporate the “Knochel trick” in their optimization, in particular with cheap co-solvents such as C2Cl4 or analogous. This is an occurrence which we expect to increase in the field of C–H bond functionalization, as the elementary steps before reductive elimination (i.e. C–H bond activation) become increasingly facile with improved catalyst and ligands designs.
Beyond halides: the case of cumene: oxidant activator, radical reservoir, and “Mukaiyama Trick” for C–H bond functionalizations and cross dehydrogenative couplings
As a last example, it is also possible to consider halide-free unconventional solvents with reactivity enabling properties that go beyond those of conventional organic solvents in C–H bond functionalization/cross dehydrogenative coupling reactions. Until recently, the use of cumene as a solvent has remained anecdotic. Nevertheless, this cheap commodity solvent, also the main component in Hock's phenol synthesis process,36 has allowed significant progress in some redox active chemical transformations. Its main feature and difference from classical aromatic solvents is its pronounced radical character. Indeed, its tertiary position accommodates a rather persistent carbon centred radical. The consequence of this specific feature is well illustrated when comparing cumene to some classical solvents in the direct oxidation of diarylmethane to benzophenones with O2, under additive and catalyst free conditions (Scheme 14). Recently, Ren and Wang found such a reaction to be feasible in cyclohexane, nevertheless requiring 5 bars of dioxygen gas in order to achieve high yields in this additive free process (Method A).37 Moreover, interestingly, they found that methanol, acetonitrile, DMF, and even toluene do not accommodate this reaction. Cumene however, has been demonstrated by some of us to accommodate this very same ultra-simple reaction at only 1 bar of dioxygen gas, under otherwise similar reaction conditions (Method B).38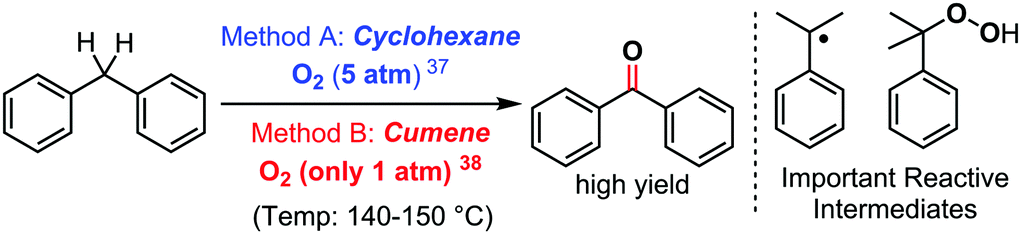 | ||
| Scheme 14 Oxidation of diarylmethanes with O2 under additive free conditions: the importance of the solvent, 2017–2019. | ||
This effect is what Maes39 recently described as the “Mukaiyama trick.”40 Its principle, which Mukaiyama did not exemplify on cumene but rather on other components, relies on the ability of cumene to readily activate dioxygen towards the cumyl radical and other activated species such as the cumyl hydroperoxide. The so called “Mukaiyama trick” is the concept of intercepting this radical oxidation process in order to trigger the oxidation of another species, such as the diarylamethane substrate (Scheme 15), which would otherwise require harsher and/or catalytic reaction conditions. Thus, the cumene solvent is an organic activator of dioxygen, analogously to, for example, Cupper salts/catalysts. This of course may come to the cost of potential by-products (for example hydroxylated or dehydrogenated cumyl species), although not necessarily if the cumyl radical only serves as radical chain propagator. This is what one could call the “Radical Reservoir effect.”
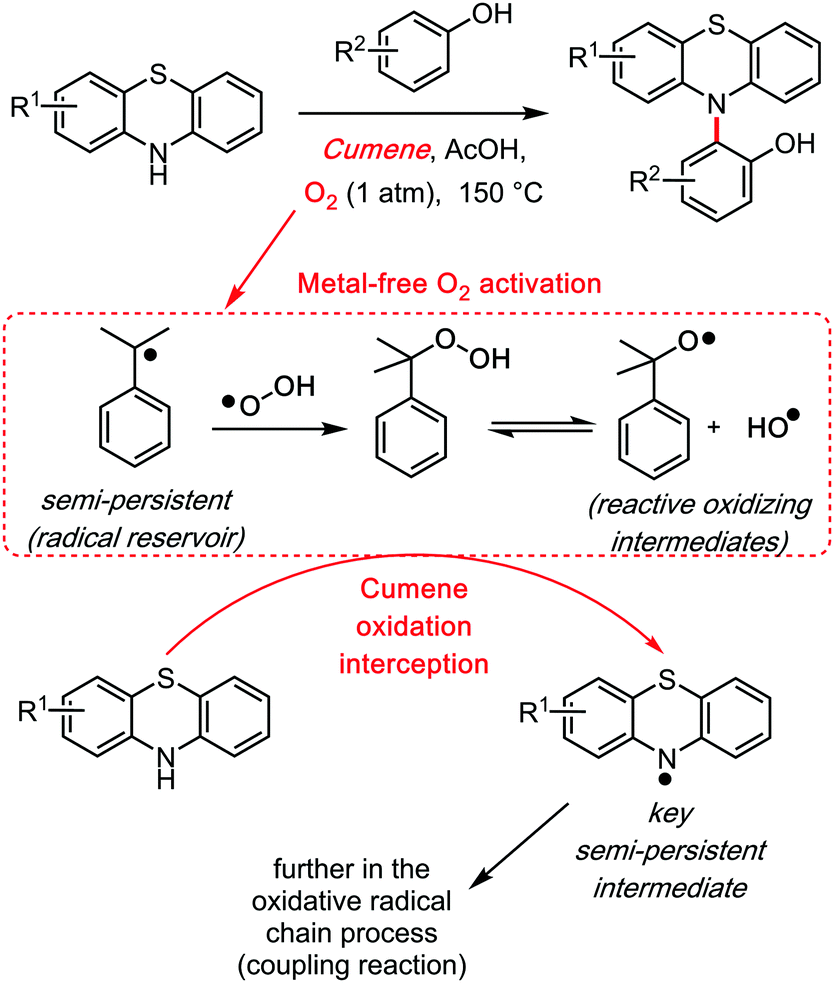 | ||
| Scheme 15 The cumene/O2 mediated oxidative phenothiazination of phenols, 2015. | ||
The first clear radical involvement of the cumene solvent, in a cross dehydrogenative reaction in which it is not also the target substrate, may be the additive free phenol phenothiazine coupling reaction (2015, Scheme 15).41 In that reaction, the phenothiazine substrate intercepts the oxidized cumyl intermediates to form a persistent, EPR characterizable42 N-centred radical species, which then triggers the C–N bond formation with phenols. Thus, the cumene solvent allowed the first O2 mediated, additive and catalyst free, intermolecular cross dehydrogenative amination reaction.
Cumene has therefore a reactivity enabling character, especially under mildly oxidative conditions. A year later, in 2016, Zhou and Cai reported another cross dehydrogenative amination reaction with cumene as solvent, together with a copper salt (Scheme 16).43
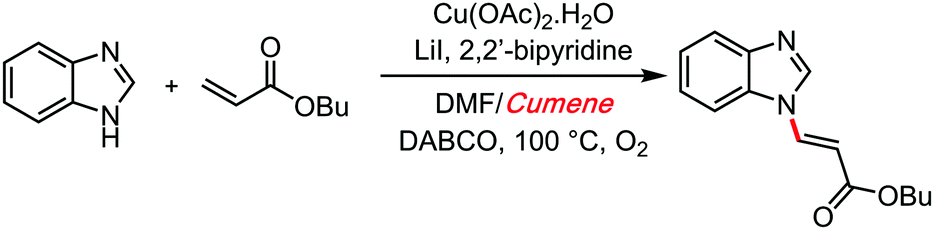 | ||
| Scheme 16 Zhou and Cai's reaction with cumene, 2016. | ||
In 2017, Bao reported a Palladium nanoparticle catalysed cross dehydrogenative coupling of aldehydes with aryl pyrazoles, wherein the pyrazole unit serves as coordinative directing group (Scheme 17).44 Interestingly, the oxidizing driving force of that reaction is an organic peroxide, with cumene as a solvent. Unfortunately, this interesting contribution is not extensive on the role of the solvent. However, a radical chain propagator role may be suggested on the basis of comparison with other systems discussed in the present review (i.e.Scheme 14).
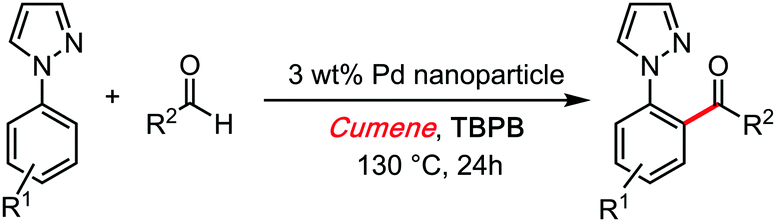 | ||
| Scheme 17 Bao's reaction with cumene, 2017. | ||
Also in 2017, a particularly interesting case of Rh(II) catalysed carbene insertion coupling reaction reported by Lu and Wang,45 demonstrated a clear superiority of cumene over any other tested solvent (Table 5). This might suggest the involvement of radical chains in that reaction, possibly stimulated by the cumene solvent's radical reservoir effect.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temp. (°C) | Time (h) | Solvent | Yield (%) |
| 1 | Rh2(Oct)4 | 80 | 24 | DCE | 60 |
| 2 | Rh2(Oct)4 | 80 | 24 | CHCl3 | 38 |
| 3 | Rh2(Oct)4 | 80 | 24 | MeCN | 24 |
| 4 | Rh2(Oct)4 | 80 | 24 | 1,4-Dioxane | 41 |
| 5 | Rh2(Oct)4 | 80 | 24 | DMF | NR |
| 6 | Rh2(Oct)4 | 80 | 24 | Cyclohexane | 28 |
| 7 | Rh2(Oct)4 | 80 | 24 | Benzene | 56 |
| 8 | Rh2(Oct)4 | 80 | 24 | Toluene | 58 |
| 9 | Rh2(Oct)4 | 80 | 24 | Xylene | 65 |
| 10 | Rh2(Oct)4 | 80 | 24 | Chlorobenzene | 35 |
| 11 | Rh2(Oct)4 | 80 | 24 | Nitrobenzene | 25 |
| 12 | Rh2(Oct)4 | 80 | 24 | Cumene | 87 |
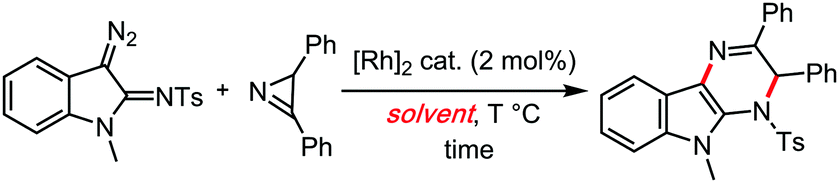
In 2018, some of us also reported a Copper catalysed dehydrogenative aminomethylation of phenols, with a peroxide as the oxidant and cumene as the solvent.46 There too, the radical reservoir role of the cumene solvent might explain its clear superiority compared to all other tested solvents (Scheme 18). It was notably verified that the C–H benzylic position of the solvent is important. Indeed, cumene (and toluene), were found to be considerably superior solvents in this reaction compared to benzene, or tBu-benzene. It should be noted that replacing cumene with cumene-D12 did not have a measurable impact on the initial rate of the reaction, making it difficult to obtain any experimental kinetic evidence for the radical role of cumene. TEMPO, a classical radical scavenger, does suppress the reaction. Moreover, the fact that tBu-benzene is a significantly less competent solvent than cumene leaves little doubt as to its probable radical role in this cross dehydrogenative coupling reaction.
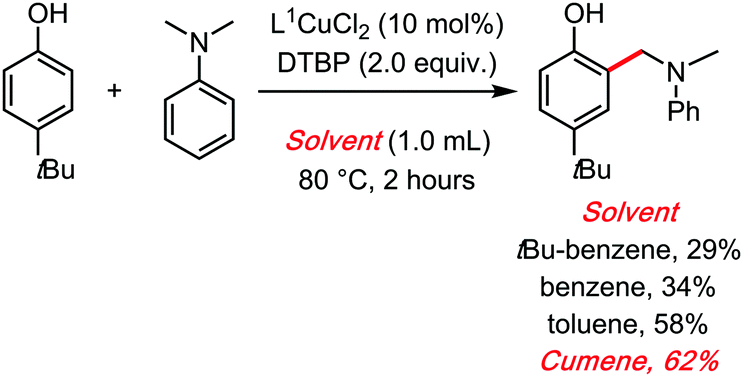 | ||
| Scheme 18 Cu catalysed Aminomethylation of phenols in cumene, 2018. | ||
In summary, cumene seems to be a particularly well suited solvent for oxidative transformations, or reactions involving radical chain processes, or both. Its C-centred radical persistency seems to be a key feature of this reactivity enabling character. cumene being a very cheap solvent, its use in novel oxidative and/or radical synthetic method development is expected to radically increase in the coming years, in particular in aerobic and/or peroxide containing systems.
Conclusions
In conclusion, we have presented a short selection of emerging solvents in the field of synthetic method development, in particular for C–H bond functionalization reactions.47 Our non-exhaustive short-list includes: HFIP, trifluoroethanol, trichloroethanol, C2Cl4, and one emerging non-halogenated unconventional solvent: cumene. For several of these solvents, an understanding of some of the mode(s) of action is starting to emerge. Cumene is a radical oxidant activator, a semi persistent radical reservoir, and a “Mukaiyama Trick” reducer. Cumene is therefore an excellent choice in systems in which oxidation or redox processes are the limiting factor. Tetrachloroethylene is a cheap and stable π-acid, a reductive elimination enabler (“Knochel Trick”), among other properties. It is an excellent co-solvent in systems in which a reductive elimination step would be rate determining. HFIP is a very polar and excellent H-bond donor. It is well suited to lower the energies of certain polar transition states, or to increase chemo and/or enantio selectivity by tuning the activation energy differences of various mechanistic pathways. Its range of application is therefore considerable, and is likely to increase in the coming years. Trichloroethanol, a structural similar and cheaper solvent, seems to possess completely different properties. Thus, the discipline of emerging unconventional organic solvent enabled C–H functionalization reactivity has started to flourish in the past few months. We expect this field to considerably expand in the coming years, in terms of number and versatility of applications, in terms of number and design of unconventional solvents on the shelf, and especially in terms of understanding of their modes of actions, which remains largely insufficient at this point. These are exciting times ahead!Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. W. P. acknowledges ERC project 716136: 2O2ACTIVATION, and DFG project PA 2395/2-1 for financial support on this and related topics. M. H. P.-T. thanks CERCA Programme/Generalitat de Catalunya and the Spanish Ministry of Economy, Industry and Competitiveness (MINECO: CTQ2016-79942-P, AIE/FEDER, EU) for the financial support. COST action CA15106 (CHAOS – C–H Activation in Organic Synthesis) is also acknowledged.Notes and references
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, 2006, p. 145 Search PubMed.
- J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394 RSC.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- S. K. Sinha, T. Bhattacharya and D. Maiti, React. Chem. Eng., 2019, 4, 244 RSC.
- A. Bernhardt, H. Kelm and F. W. Patureau, ChemCatChem, 2018, 10, 1547 CrossRef.
- J. Sanjosé-Orduna, J. M. S. Toro and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2018, 57, 11369 CrossRef.
- S. Hazra, K. Hirano and M. Miura, Asian J. Org. Chem., 2019, 8, 1097 CrossRef.
- S. Li, P. Shi, R.-H. Liu, X.-H. Hu and T.-P. Loh, Org. Lett., 2019, 21, 1602 CrossRef.
- J. H. Conway Jr and T. Rovis, J. Am. Chem. Soc., 2018, 140, 135 CrossRef.
- S. Huh, S. Y. Hong and S. Chang, Org. Lett., 2019, 21, 2808 CrossRef.
- S.-G. Wang and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 2514 CrossRef.
- K. Ozols, Y.-S. Jang and N. Cramer, J. Am. Chem. Soc., 2019, 141, 5675 CrossRef.
- C. Colletto, A. Panigrahi, J. Fernández-Casado and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 9638 CrossRef.
- Y. Zhu, I. Colomer, A. L. Thompson and T. J. Donohoe, J. Am. Chem. Soc., 2019, 141, 6489 CrossRef.
- E. M. D'Amato, J. Börgel and T. Ritter, Chem. Sci., 2019, 10, 2424 RSC.
- J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026 CrossRef.
- G.-X. Li, X. Hu, G. He and G. Chen, Chem. Sci., 2019, 10, 688 RSC.
- A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Dougles and D. Leonori, Nat. Chem., 2019, 11, 426 CrossRef.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef PubMed.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751 CrossRef.
- A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147 RSC.
- S. Bag, R. Jayarajan, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 3182 CrossRef.
- A. S. Trita, A. Biafora, M. P. Drapeau, P. Weber and L. J. Gooßen, Angew. Chem., Int. Ed., 2018, 57, 14580 CrossRef.
- X.-Q. Hu, Z. Hu, G. Zhang, N. Sivendran and L. J. Gooßen, Org. Lett., 2018, 20, 4337 CrossRef.
- X.-Q. Hu, Z. Hu, A. S. Trita, G. Zhang and L. J. Gooßen, Chem. Sci., 2018, 9, 5289 RSC.
- S. Goudedranche, D. Pierrot, T. Constantieux, D. Bonne and J. Rodriguez, Chem. Commun., 2014, 50, 15605 RSC.
- E.-L. Dreher, T. R. Torkelson and K. K. Beutel, Chlorethanes and Chloroethylenes, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, 2011 Search PubMed.
- R. Giovannini, T. Stüdemann, G. Dussin and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 2387 CrossRef CAS.
- R. Giovannini, T. Stüdemann, A. Devasagayaraj, G. Dussin and P. Knochel, J. Org. Chem., 1999, 64, 3544 CrossRef CAS.
- X. Hu, Chem. Sci., 2011, 2, 1867 RSC.
- A. E. Carpenter, A. J. McNeece, B. R. Barnett, A. L. Estrada, C. C. Mokhtarzadeh, C. E. Moore, A. L. Rheingold, C. L. Perrin and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 15481 CrossRef CAS.
- M.-L. Louillat and F. W. Patureau, Org. Lett., 2013, 15, 164 CrossRef CAS.
- A. W. Jones, C. K. Rank, Y. Becker, C. Malchau, I. Funes-Ardoiz, F. Maseras and F. W. Patureau, Chem. – Eur. J., 2018, 24, 15178 CrossRef CAS.
- M.-L. Louillat, A. Biafora, F. Legros and F. W. Patureau, Angew. Chem., Int. Ed., 2014, 53, 3505 CrossRef CAS.
- N. Hayama, R. Kuramoto, T. Földes, K. Nishibayashi, Y. Kobayashi, I. Pápai and Y. Takemoto, J. Am. Chem. Soc., 2018, 140, 12216 CrossRef CAS.
- M. Weber, M. Weber and M. Kleine-Boymann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2004 Search PubMed.
- X. Tian, X. Cheng, X. Yang, Y.-L. Ren, K. Yao, H. Wang and J. Wang, Org. Chem. Front., 2019, 6, 952 RSC.
- A. Malekafzali, K. Malinovska and F. W. Patureau, New J. Chem., 2017, 41, 6981 RSC.
- H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946 CrossRef CAS.
- K. Narasaka, A. Morikawa, K. Saigo and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1977, 50, 2773 CrossRef CAS.
- M.-L. Louillat-Habermeyer, R. Jin and F. W. Patureau, Angew. Chem., Int. Ed., 2015, 54, 4102 CrossRef CAS.
- M. Goswami, A. Konkel, M. Rahimi, M.-L. Louillat-Habermeyer, H. Kelm, R. Jin, B. de Bruin and F. W. Patureau, Chem. – Eur. J., 2018, 24, 11936 CrossRef CAS PubMed.
- T.-T. Lai, D. Xie, C.-H. Zhou and G.-X. Cai, J. Org. Chem., 2016, 81, 8806 CrossRef CAS.
- P. Yang and Y.-S. Bao, RSC Adv., 2017, 7, 53878 RSC.
- H. Ding, Z. Wang, S. Bai, P. Lu and Y. Wang, Org. Lett., 2017, 19, 6514 CrossRef CAS.
- C. Yu and F. W. Patureau, Angew. Chem., Int. Ed., 2018, 57, 11807 CrossRef CAS.
- During the finalizing phase of this review, this related and interesting review appeared: J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164 RSC.
| This journal is © The Royal Society of Chemistry 2020 |