
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrafast spectroscopy on water-processable PCBM: rod–coil block copolymer nanoparticles†
Lucia
Ganzer
 a,
Stefania
Zappia
b,
Mattia
Russo
a,
Anna Maria
Ferretti
c,
Varun
Vohra
d,
Marianna
Diterlizzi
b,
Maria Rosa
Antognazza
e,
Silvia
Destri
*b and
Tersilla
Virgili
*a
a,
Stefania
Zappia
b,
Mattia
Russo
a,
Anna Maria
Ferretti
c,
Varun
Vohra
d,
Marianna
Diterlizzi
b,
Maria Rosa
Antognazza
e,
Silvia
Destri
*b and
Tersilla
Virgili
*a
aIFN-CNR, Dipartimento di Fisica, Politecnico di Milano, Milano I-20132, Italy. E-mail: tersilla.virgili@ifn.cnr.it
bIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), Consiglio Nazionale delle Ricerche (CNR), Milano I-20133, Italy. E-mail: silvia.destri@scitec.cnr.it
cIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), Consiglio Nazionale delle Ricerche (CNR), Milano I-20138, Italy
dDepartment of Engineering Science, the University of Electro-Communications (UEC), 1-5-1 Chofugaoka, Chofu, Tokyo 182-858, Japan
eCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, Milano 20133, Italy
First published on 11th November 2020
Abstract
Using ultrafast spectroscopy, we investigate the photophysics of water-processable nanoparticles composed of a block copolymer electron donor and a fullerene derivative electron acceptor. The block copolymers are based on a poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] rod, which is covalently linked with 2 or 100 hydrophilic coil units. In both samples the photogenerated excitons in the blend nanoparticles migrate in tens of ps to a donor/acceptor interface to be separated into free charges. However, transient absorption spectroscopy indicates that increasing the coil length from 2 to 100 units results in the formation of long living charge transfer states which reduce the charge generation efficiency. Our results shed light on the impact of rod–coil copolymer coil length on the blend nanoparticle morphology and provide essential information for the design of amphiphilic rod–coil block copolymers to increase the photovoltaic performances of water-processable organic solar cell active layers.
Introduction
Conjugated polymers can be employed in various innovative applications and consequently, the study of their optical and electronic properties has recently been gaining increasing interest.1,2 In particular, the low cost of their chemical synthesis and the facile tuning of their opto-electronic properties through simple chemistry could play an important role in the development of a new generation of electronic devices such as photovoltaic windows.3–5 In fact, conjugated polymers can be applied to the field of photovoltaics as active materials in organic solar cells (OSCs). OSC active layers are composed of two types of organic semiconducting materials: electron donors, which are generally conjugated polymers, and electron-acceptors such as [6,6]-phenyl-C61-butyric acid methyl ester (hereafter PCBM). The OSC efficiency can be increased through several strategies including broadening of the conjugated polymer absorption or increasing the exciton dissociation efficiency. The most common strategy to maximize the efficiency of OSC is to mix the donor and acceptor materials in an appropriate composition and solvent to form bi-continuous intermixed active layers with an interpenetrating network. Such active layer morphology produces a large donor/acceptor interface, thus providing a large number of exciton dissociation sites.6,7 There are two pathways for the polaron generation: the prompt formation from hot excitons generated near the interface on a time scale of <100 fs and the delayed formation via the exciton migration to the donor/acceptor interface on a time scale of ∼10 ps.8 In particular, extensive spectroscopic investigations have been carried out recently on the photophysics of the charge transfer state (CTS) formed at the molecular interface between polymers and fullerene derivatives.9–13 CTS consists of partially separated, Coulombically bound charge pairs, where the hole and electron are primarily localized on the highest occupied molecular orbital (HOMO) of the donor (D) and on the lowest unoccupied molecular orbital (LUMO) of the acceptor (A), respectively.14 Their wave function overlap results in the formation of hybrid ground and excited states, lying within the optical gap of the two materials. The ratio between full ionization and geminate recombination of CTS is the main factor in determining the photovoltaic performances.15–17As reported in several past studies, material design and deposition conditions produce OSC active layers with different nanocompositions and nanostructures which strongly affect the photovoltaic performances of the devices.9,10,18–22 For example, the lack of well-defined phase separation when blending amorphous polymers with PCBM can result in inefficient charge generation in amorphous polymer:PCBM thin films.9,11,20 Additionally, a higher polymer crystallinity will also generally lead to better charge transport properties.21,22 Therefore, a particular attention should be given to the active layer morphology, which strongly impacts the charge generation and transport efficiencies.
Although OSCs are being commercialized through several companies around the world nowadays, reaching their full potential would require printing efficient devices on large-areas through solution processes. The solvent that are generally employed for efficient OSC manufacturing are hazardous organic solvents like halogenated ones, which are strictly regulated by laws.23 Therefore, reducing or eliminating the use of these toxic solvents is mandatory before implementing large-scale production of OSCs. In fact, the high volume of aromatic and halogenated solvents used for the fabrication of organic photovoltaics on laboratory scale still remains one of the most important drawbacks in the industrial development of this technology. The preparation of polymer water-processable nanoparticles through the miniemulsion approach is a way to overcome this issue and considerably decrease the use of halogenated and aromatic solvents.
Note that several strategies are currently being investigated to manufacture OSCs using environment-friendly solvents or to minimize the volume of solvent employed.4,24 From a toxicity point-of-view, water is the ideal choice as OSC processing solvent. Consequently, the concept of water-dispersed conjugated polymer nanoparticle inks have gained considerable attention for the green processing of OSC active layers.12 The nanoparticles can be prepared through the miniemulsion25 or nanoprecipitation26 procedures, and present a good optical stability. The particle fabrication method drastically influences the size of the blend particles and their photophysics which, in turn, strongly affects their photovoltaic performances.27
Both nanoparticle fabrication methods rely on the use of chloroform or other organic solvents like toluene for the preliminary solubilization of the hydrophobic active blend. Nevertheless, the quantities of toxic solvent employed for this preliminary step are much smaller with respect to the traditional fabrication approaches. Furthermore, the final step of the miniemulsion process, which consists in the complete removal of the hazardous solvent through evaporation, provides a great opportunity to deal with the hazardous solvents. Unlike conventional deposition methods that disperse the solvent directly into the environment, controlled separation of the toxic solvent from the aqueous dispersion can be integrated into a proper circular industrial plant through condenser systems during the miniemulsion preparation itself to recover and recycle the hazardous solvents.
Here we investigate the photophysics of water-processable rod–coil block copolymer-based nanoparticles through ultrafast transient absorption (TA) spectroscopy. The polymers we study are composed of a poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2, 1-b;3,4-b′]dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (PCPDTBT) conjugated rod coupled with various 4-vinylpyridine (4VP) based flexible coils. These amphiphilic block copolymers produced with structurally different coils (chemical nature and coil length) enable the surfactant-free synthesis of stable aqueous dispersions of block copolymer-only nanoparticles and block copolymer:PCBM blend nanoparticles.
PCPDTBT was the first push–pull low bandgap copolymer and has thus been extensively studied. It exhibits a broad absorption spectrum that extends beyond 800 nm and respectable power conversion efficiencies when employed in OSCs.28–30 Through this photophysical study, we correlate the molecular structure of the block copolymer with the charge generation dynamics in blend nanoparticles, thus confirming the impact of coil length and structure on the organisation at the nanoscale level within the blend nanoparticles and on the charge separation efficiency. The pump–probe measurements reveal that different photo-induced species are formed in blend nanoparticles depending on whether they were fabricated with short or long coils. More precisely, we observe the formation of new photo-induced species that can be associated with a peculiar nanoscale arrangement of block copolymer and PCBM within the nanoparticles obtained when using block copolymers with long coils.
Experimental details
Synthesis and preparation of material
MilliQ grade ultrapure water with a resistivity of ∼18 MΩ cm at 25 °C is used for the formation of aqueous nanoparticle suspensions through the miniemulsion process. PCPDTBT and PCBM were purchased from Luminescence Technology Corp. and Sigma-Aldrich respectively, and employed as received without further purification.The block copolymer depicted in Fig. 1a were synthesized according to previous reports.31,32 The nanoparticle fabrication process is summarized below and a detailed description of the procedure can be found elsewhere.32
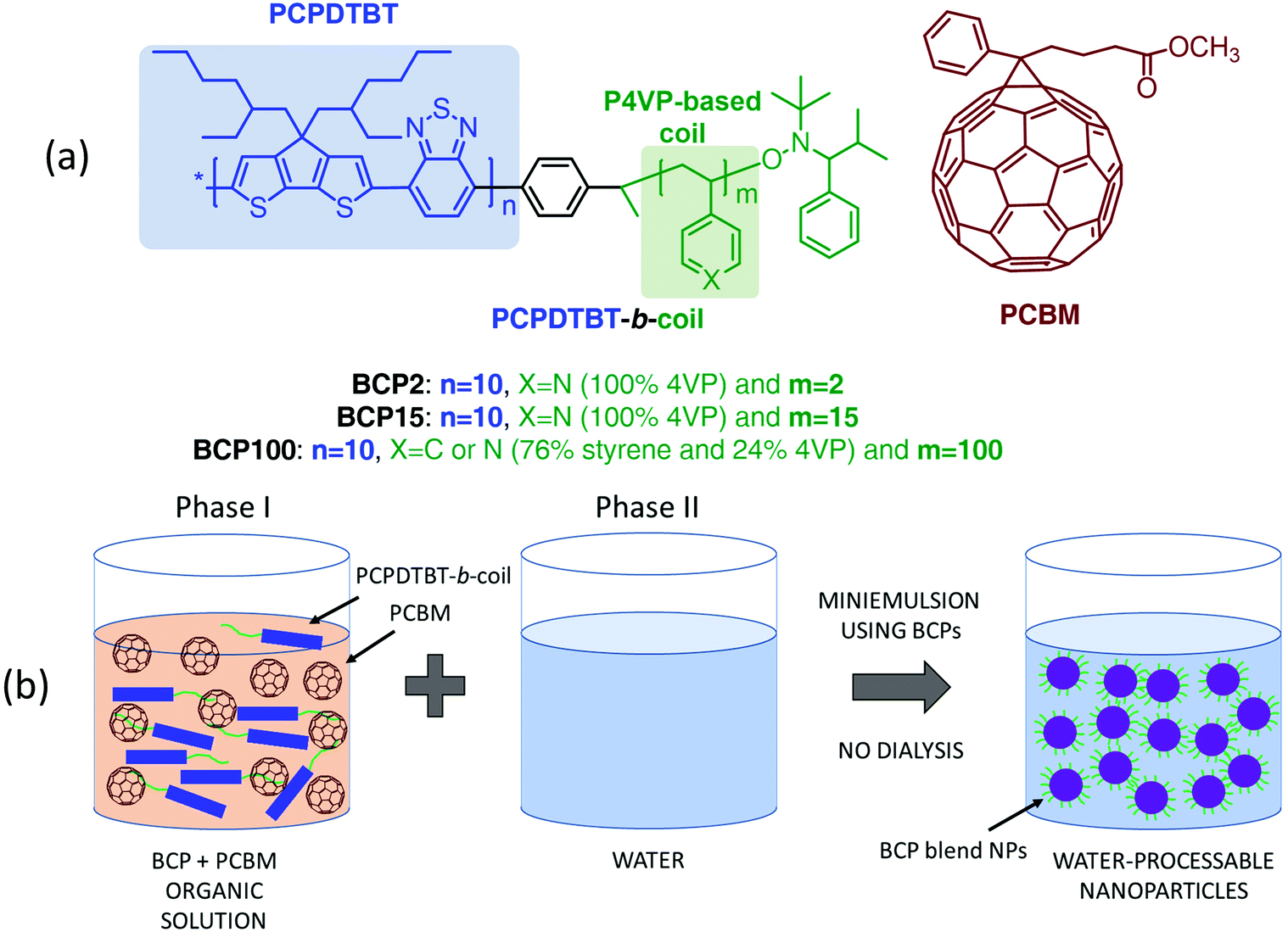 | ||
| Fig. 1 (a) Molecular structures of PCBM and block copolymers composed of PCPDTBT conjugated rods (highlighted in blue) and hydrophilic P4VP or poly4-vinylpirydine-co-styrene coils (in green). (b) Schematic representation of the preparation of water-suspended blend nanoparticles produced with the miniemulsion technique employing block copolymers as bifunctional materials (surfactant and electron donor functions). | ||
Block copolymers are first dissolved in an organic solvent (chloroform) with or without PCBM (phase I). Then, 60 μL of the organic solution are poured dropwise into 1 mL of ultrapure water at 40 °C (phase II) and stirred vigorously for an additional hour to form the macroemulsions. We employed magnetic stirring in all the procedures for the sample preparation. At the beginning, the speed was kept around 500 rpm during the injection of the organic solution containing the active blend in water. Then the speed was increased to around 1200 rpm for 30 minutes for the macroemulsion preparation, before the sonication of the sample. The sonication was performed with Ultrasonic Cleaner CP102 DIGIT (CEIA S.p.A.), for 10 minutes at room temperature. After that, the speed of the stirring was maintained around 200 rpm for 30 minutes at 60 °C allowing the slow evaporation of chloroform from the miniemulsion to achieve stable suspensions of the organic blends.
The nanoparticle films characterized throughout this study are prepared by drop casting the miniemulsion onto cleaned quartz substrates. On the other hand, the CTRL films are prepared by spin-coating PCPDTBT or PCPDTBT:PCBM solutions from chloroform.
Absorption and ultrafast TA spectroscopy
Linear absorption spectra were acquired with a JASCO V-750 spectrophotometer.A fraction of the output light of a Ti:sapphire laser (Libra, Coherent) characterized by 100 fs pulse duration, 1 kHz repetition rate and 800 nm central wavelength was used for the pump–probe experiments. Pump excitation wavelengths of 400 nm and 650 nm were used for the characterization of neat and blend samples, respectively. The 400 nm pulse is produced by second harmonic generation in a 1 mm-thick barium borate crystal, while 650 nm light comes from a non-collinear optical parametric amplifier. The broadband probe extending in the visible and NIR regions results from white light continuum generation in a 3 mm-thick sapphire plate pumped by 800 nm light. Using bandpass filters, we select either the visible or the NIR range. The delay between pump and probe pulses is controlled by a translation stage and the pump beam is modulated by a mechanical chopper with a 500 Hz frequency. The differential transmission (ΔT/T) of the probe is measured as a function of probe wavelength and pump–probe delay through a SP2150 Acton spectrometer from Princeton Instruments. The pump energy is adjusted to a fluence around 45 μJ cm−2. Measurements were performed with parallel polarisations between pump and probe beams.
TEM characterization
The TEM micrographs were collected at 200 kV using a ZEISS LIBRA200 FE equipped with a second generation in column omega energy filter. The characterized samples correspond to nanoparticles drop-casted on a copper grid coated with a silicon monoxide support film, which are then dried in air. The nanoparticle diameters were analyzed using the i-TEM Imaging Platform from Olympus and the mean diameters were calculated using dm = ∑dini/∑ni, where ni is the number of particles of dimension di.33Measurements on CW photo-induced absorption
Quasi-CW photo-induced absorption experiments have been performed in transmission geometry, in air and at room temperature. A 560 nm laser diode served as the pump beam, modulated by a mechanical chopper operating at frequency f = 670 Hz. A 8 mm diameter laser beam was obtained using a telescope, in order to guarantee optimal overlap with the probe beam. The probe was generated by a 100 W tungsten halogen lamp, focused on the sample by spherical mirrors in order to avoid chromatic aberrations. Transmitted light is dispersed by a monochromator. The changes in transmission under photoexcitation were detected using a calibrated Si photodiode and measured with a lock-in amplifier referenced to the modulation frequency. All spectra have been corrected for the photoluminescence and transmission spectrum.Results
The blend nanoparticles are formed through miniemulsion using PCPDTBT block copolymers with various 4VP-based coil structures mixed with PCBM in a 1 to 3 ratio (Fig. 1). Independently on the coil length, 4VP units interact with water thus conferring a hydrophilic character to the block copolymers which can fulfil the surfactant function necessary for the formation of nanoparticles using the miniemulsion process. The block copolymers also act as electron donor through the presence of the PCPDTBT rod. We previously demonstrated that PCPDTBT-b-P4VP block copolymers:PCBM nanoparticles can be successfully employed to produce OSC active layers with similar photovoltaic performances as those deposited directly from toxic organic solvents.31 Here, we particularly focus on the two following block copolymers (BCPs) and the resulting nanoparticles. The first one contains a short flexible segment with only 2 4VP repeating units and from now on we will refer to it as BCP2. The second one (hereafter BCP100) is characterised by a coil of a random copolymer containing a mean of 100 repeating units with a composition of 76% of styrene and 24% of 4VP.34 In other words, BCP2 and BCP100 contain 2 and 24 units of 4VP, respectively. In the following part of the manuscript, we will refer to nanoparticles prepared with and without PCBM as blend and neat nanoparticles, respectively. Note that both the neat and blend nanoparticles are prepared following the same procedure as described in Fig. 1(b) and detailed in the Experimental section.Fig. 2 displays transmission electron microscope (TEM) images of blend nanoparticles deposited from the suspensions. A mean diameter of ∼149 nm and ∼134 nm was measured for blend BCP2 and blend BCP100 nanoparticles, respectively. Despite their larger mean diameter, the blend BCP2 nanoparticles have a smaller size dispersion of ±45 nm with respect to that of the blend BCP100 nanoparticles with a value of ±68 nm. The minimum and maximum diameters of blend BCP2 nanoparticles are 74 nm and 368 nm, respectively. On the other hand, BCP100 nanoparticles have diameters between 56 nm and 310 nm. Blend BCP2 nanoparticles also exhibit a more regular shape compared to BCP100 ones.
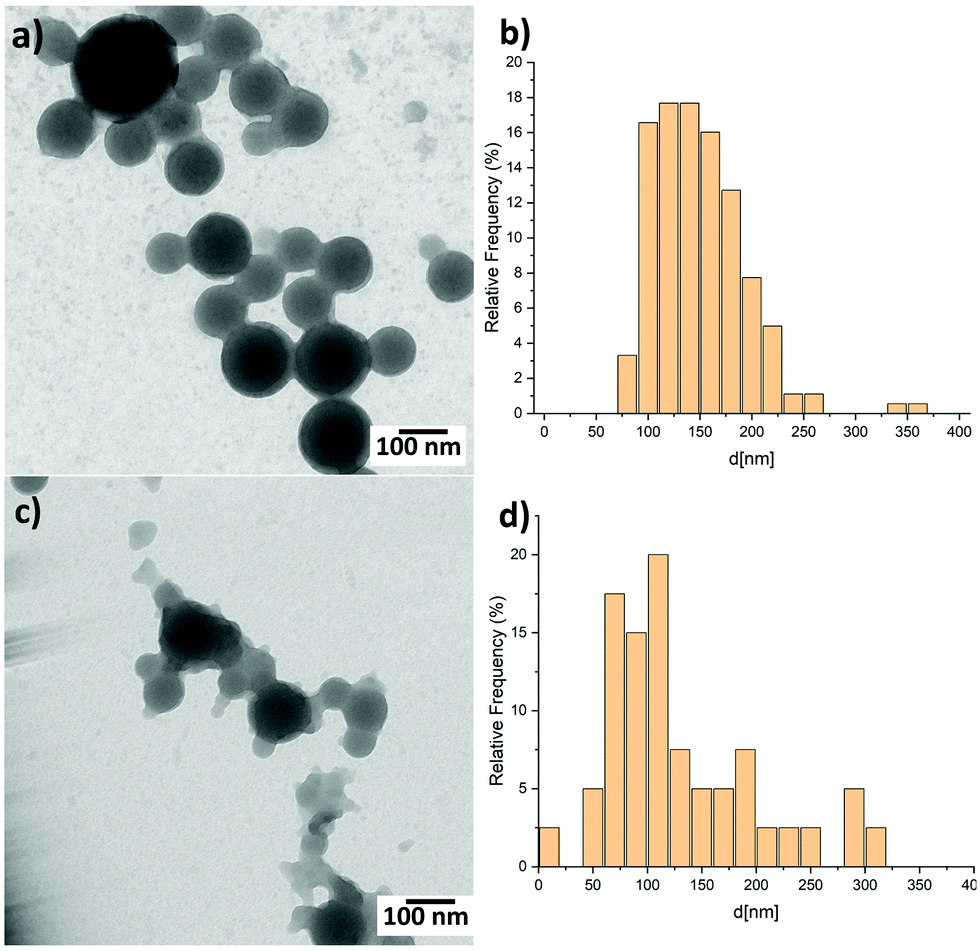 | ||
| Fig. 2 TEM images of (a) blend BCP2 nanoparticles and (c) blend BCP100 nanoparticles together with (b and d) the corresponding size distribution histograms. | ||
Unlike the blend BCP2 nanoparticles, the blend BCP100 nanoparticles show the presence of disordered polymers around the nanoparticles. Nevertheless, the blend nanoparticles exhibit large increases in diameters compared to the corresponding neat ones that have mean diameters of ∼36 nm and ∼70 nm, respectively.35 The increasing in nanoparticle dimensions upon addition of PCBM to the organic solution indicate that PCBM is successfully included into the blend nanoparticles, which will significantly influence the blend nanoparticles morphology. In fact, a high contrast between the inner and outer parts of the blend nanoparticles can be observed which suggests a high PCBM concentration in the inner parts of the nanoparticles.35 Note that the contrast is more evident in the blend BCP2 nanoparticles with respect to the blend BCP100 nanoparticles.
We employ thin films spin-coated from chloroform solutions as control (CTRL) reference films. Neat CTRL and blend CTRL correspond to the PCPDTBT only and the PCPDTBT:PCBM blend films, respectively. These are compared with neat and blend nanoparticle thin films prepared by drop-casting the aqueous suspensions. Fig. 3 shows all the absorption spectra, which have been first mathematically corrected for reducing the scattering contribution and then normalised at the peaks around 700 nm. The two peaks centred at 410 nm and 730 nm in the neat films (filled circles) can clearly be ascribed to the PCPDTBT rod.31 In comparison with the neat CTRL film, the neat nanoparticles samples display a broadening of both absorption peaks, which could be caused by PCPDTBT aggregate formation within the nanostructures.36
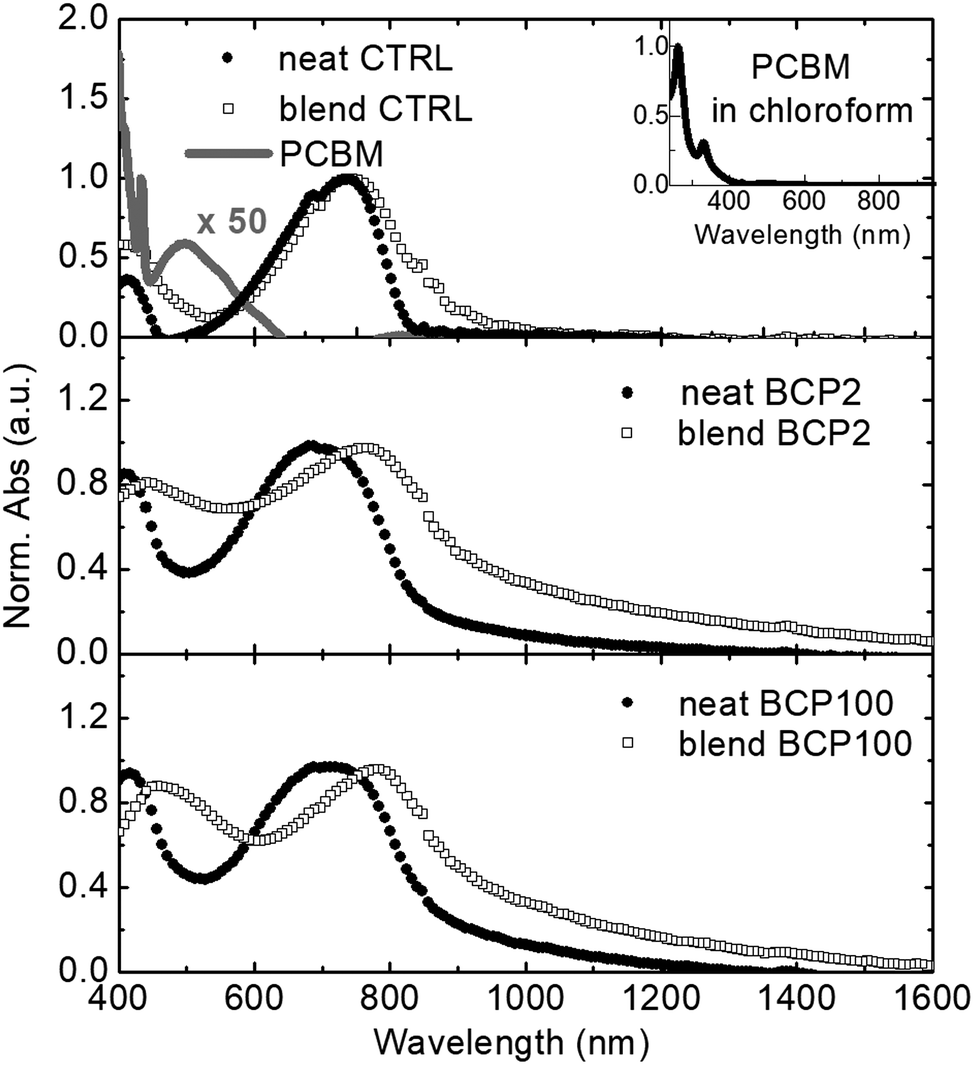 | ||
| Fig. 3 Normalised linear absorption spectra of neat and blend films (filled circles and empty squares, respectively). From top to bottom: control (CTRL), BCP2 and BCP100 films. Normalized and 50 times enhanced absorption spectra of PCBM in chloroform are presented in the inset and top panel, respectively. All the absorption spectra have been mathematically corrected for reducing the scattering contribution. | ||
As shown in the inset of Fig. 3, PCBM exhibits a strong absorption band in the ultraviolet region.31,37 To emphasize the contribution of PCBM in the visible spectral region, a 50 times enhanced PCBM absorption spectrum is presented in the top panel of Fig. 3. Despite the weak character of the PCBM absorption band around 500 nm, the large amount of PCBM present in the blend films results in a broadening and a red shift of the low energy absorption peak of PCPDTBT. This effect is stronger in the nanoparticle samples compared to the CTRL film. Shifts of around 20 nm and 40 nm can be observed for BCP2 and BCP100, respectively. Possible causes for the peak broadening and shift include difference in nanoparticle size and dispersity and related changes in light scattering contributions or different levels of molecular order within the nanoparticles.
We performed TA measurements on nanoparticles and CTRL films to elucidate the impact of thin film morphology on the formation of photo-induced species in PCPDTBT:PCBM blends. In general, positive differential transmissions are assigned to ground state photobleaching (GSB) or stimulated emissions (SEs) while negative features in the TA spectra are attributed to photoinduced absorption (PA) bands.38
Fig. 4 displays TA spectra at different pump–probe delays, in the visible and near infrared (NIR) regions for the CTRL, BCP2 and BCP100 neat nanoparticle films. We have used 400 nm light as pump excitation (details are reported in the Experimental section).
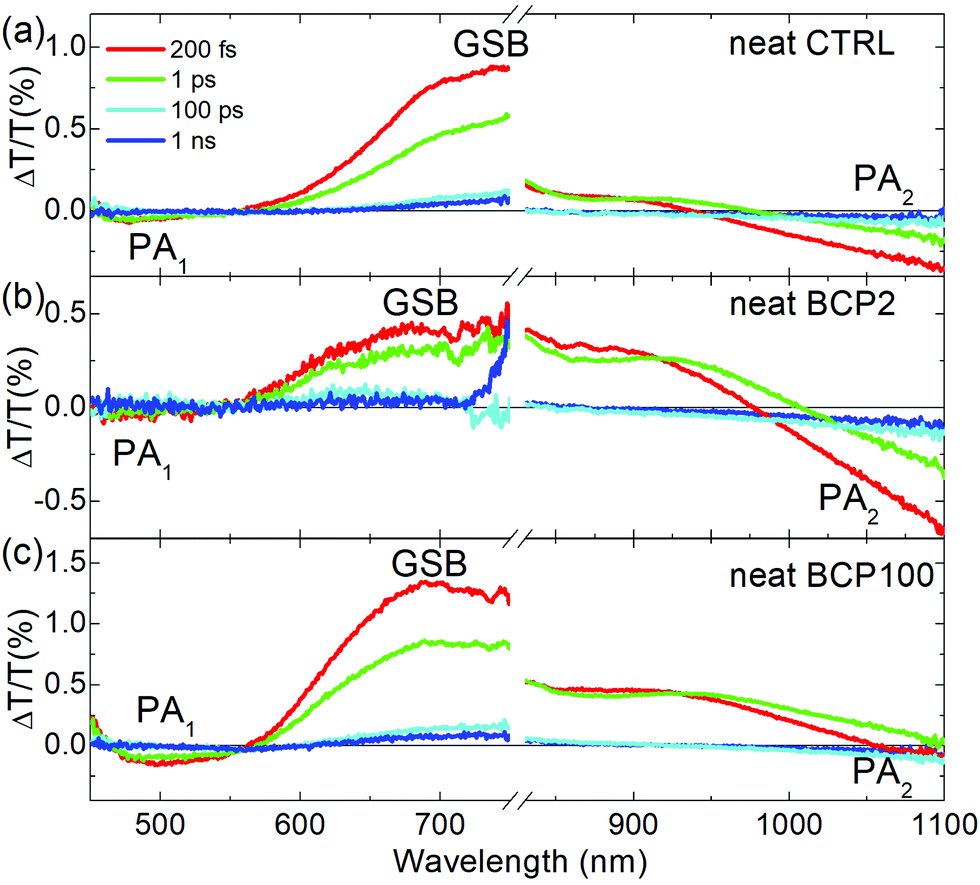 | ||
| Fig. 4 TA spectra of (a) neat CTRL, (b) neat BCP2 nanoparticles and (c) neat BCP100 nanoparticles films collected with a 400 nm excitation at different time delays between pump and probe beams. TA visible spectra of neat BCP2 nanoparticles were multiplied by 5. | ||
All samples exhibit two distinct bands in the visible range: a broad positive band centred at ∼680 nm corresponding to the GSB and a negative PA band at ∼500 nm, which we will refer to as PA1. In the neat films, GSB and PA1 have the same temporal decays (Fig. S1, ESI†), indicating that both bands are the signature of the same photoexcited population, the excitons. Fig. S1 (ESI†) shows the temporal dynamic of the PA1 photoinduced absorption band at around 500 nm in comparison with the bleaching signal at around 700 nm for the three neat solid state films. The fact that the decay traces are similar in the different spectral regions is a strong indication that they can be attributed to the same photoexcited population, the excitons. These findings are consistent with previous reports on the photophysical characterization of PCPDTBT.39
A second PA band (PA2) can be found in the NIR region. As shown in Fig. S2 (ESI†), the dynamics of this band are different from those of GSB and PA1, which indicates PA2 is attributed to a different photoinduced species. CW photo-induced absorption measurements (Fig. S3, ESI†) evidenced that the negative band in the NIR region (PA2) can be assigned to the formation of a charged population. Indeed, this technique reveals the presence of long-living species, usually either triplet states or charged states. From previous reports,39,40 we know that in our samples triplet states are really unlikely while this class of copolymers is characterised by formation of charge states; thus PA2 is associated to charged species. In the neat CTRL film, the PA2 band is wide and relatively intense which contrasts with the neat nanoparticle films, in which the PA2 band is weak and overlaps with the GSB. Moreover, the TA measurements presented in Fig. 4 and in Fig. S3 (ESI†) clearly indicate that the PA2 band intensity decreases with increasing coil length in the neat nanoparticle samples respect the GSB positive signal. We can thus conclude that increasing the coil length from 2 repeating units (BCP2) to 100 repeating units (BCP100) results in a decrease in charge formation efficiency in the neat nanoparticles.
To investigate the impact of PCBM inclusion into the various films, we also performed TA measurements on blend CTRL, BCP2 nanoparticle and BCP100 nanoparticle films (Fig. 5). In this case, the samples are excited at 650 nm to selectively excite PCPDTBT outside of the PCBM absorption range.
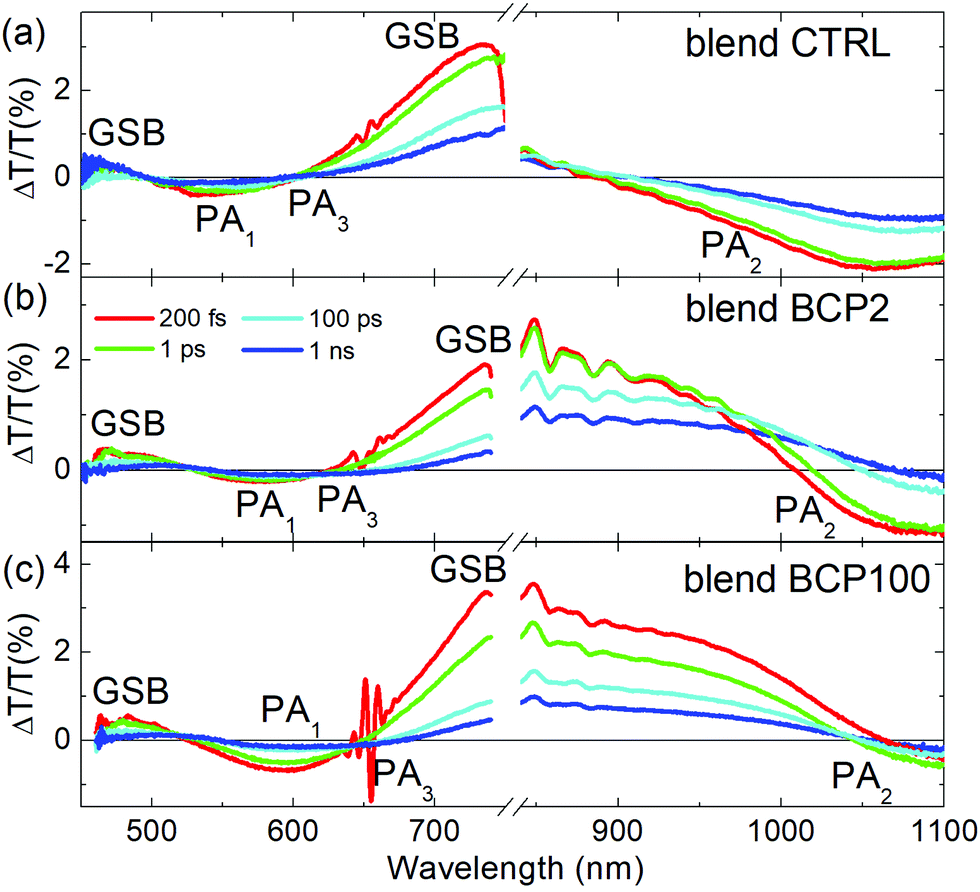 | ||
| Fig. 5 TA spectra of (a) blend CTRL, (b) blend BCP2 nanoparticles and (c) blend BCP100 nanoparticles films collected with a 650 nm excitation at different time delays between pump and probe beams. NIR TA spectra of blend BCP2 nanoparticles were divided by 2. | ||
In addition to the PCPDTBT GSB at around 680 nm, a second GSB is found at around 470 nm. Additionally, a clear red-shift of the PA1 bands is observed for all blend samples with respect to their corresponding neat films. Larger shifts are observed for nanoparticles with respect to CTRL films, which is in agreement with the observations from the linear absorption spectra (Fig. 3). The broadening of the negative photoinduced band at 550 nm is another clear difference found between neat and blend films. As the probe delay increases, we can observe the formation of a new negative band in the blend films (nanoparticles and CTRL), which we identify as PA3. We attribute the PA3 band appearance to the electron transfer from PCPDTBT moieties to PCBM.10,40
Blend CTRL films exhibit a much higher PA2 intensity relative to the GSB intensity compared to neat CTRL films. In other words, we can observe an increase of charge population at the expense of the exciton population, as expected from such intimately mixed PCPDTBT/PCBM blends.40,41 Unlike the CTRL blend films, the exciton spectra in the nanoparticle blends (GSB bands around 700–750 nm) exhibit a strong red shift towards the NIR region. Due to this red shift, the positive GSB signal corresponding to the presence of excitons overlaps with the negative PA2 associated with charge formation. Because of this strong overlap, it becomes very difficult to distinguish the individual dynamics of GSB and PA2.
A deeper analysis of the results obtained from the blend films in the visible region was performed by comparing TA spectra at an early probe delay of 200 fs and at a long 100 ps one (Fig. 6a–c). The spectra were normalized at the minimum of the PA1 bands. In this way it is possible to see clearly the appearance of the PA3 band at long delays (red lines in Fig. 6a–c), at around 620–650 nm, whose contribution is weaker in the CTRL. Unlike the other two blend samples, a positive band around 525 nm appears at long probe delays in the blend BCP100 nanoparticles. The temporal behaviours of the new photoinduced bands (highlighted by arrows in Fig. 6a–c) are presented in Fig. 6d and e. In the CTRL blend film the forming negative band (PA3) extendes from 625 nm to 725 nm with the peak at 665 nm. It overlaps with the ground state bleaching positive signal (GSB). To estimate the temporal behaviour of the PA3 band, we took as a reference of the bleaching decay (GSB) the dynamics at 725 nm and the signal at 665 nm as the sum of the two decays GSB + PA3. Calculating the difference between the signals at 665 nm and at 725 nm (Fig. S4 in the ESI†), we pointed out the PA3 temporal behaviour. The signals from CTRL, BCP2 and BCP100 blend nanoparticles have different growing time, defined as the time required to reach the maximum signal. The positive band formed around 525 nm (labelled C in Fig. 6e) and the negative one at 640 nm (labelled D in Fig. 6e) in the BCP100 blend nanoparticles reach their maximum at around 4 ps and at around 20 ps, respectively. On the other hand, the negative signal for the blend CTRL and blend BPC2 nanoparticles sample reach their maxima at around 10 and 40 ps (in Fig. 6d labelled A and B, respectively).
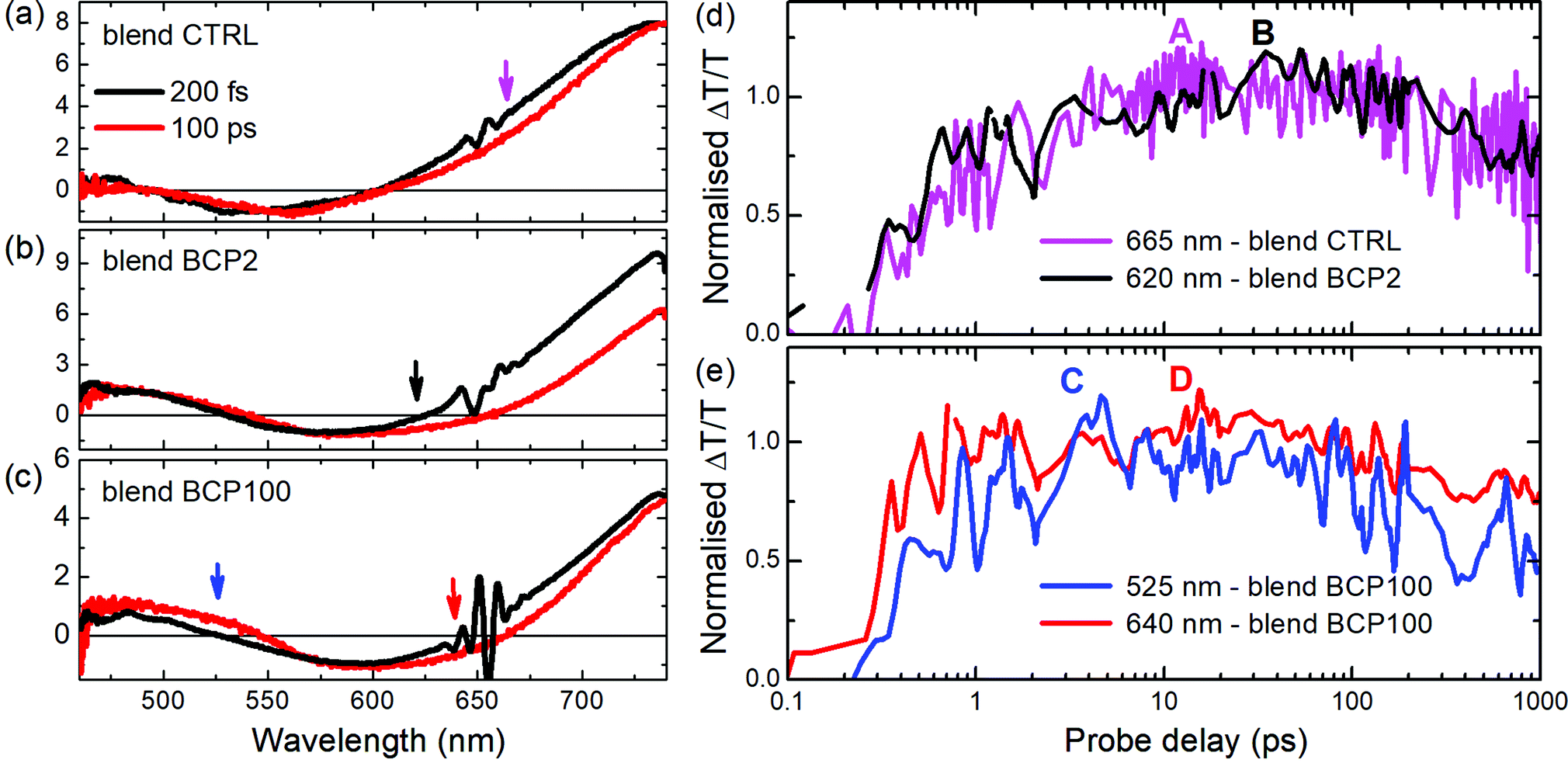 | ||
| Fig. 6 TA spectra in blend CTRL (a), BCP2 (b) and BCP100 (c) films at two different pump–probe delays; spectra have been normalized at the minimum of the photo-induced band around 600 nm in order to compare the temporal evolution of the bands. Inset: TA normalised spectra in blend CTRL. In (d and e) normalised temporal dynamics at different probe wavelengths in blend CTRL, BCP2 and BCP100 films. Arrows in (a–c) indicate the probe wavelengths shown in (d and e). Pump excitation: 650 nm. | ||
Discussion
As discussed above, in donor/acceptor (D/A) blend films, charges can be generated almost instantly (<100 fs) from hot excitons located near the D/A interface or on a ps time scale via exciton migration to the D/A interface. In our blend films, the appearance of the PA3 band around 620–680 nm indicates that photoinduced charges are populated in the electron acceptor (PCBM) within a timeframe of tens of ps. As the mechanism is not instantaneous, the proposed generation mechanism is that the charges are created by exciton separation at the D/A interface; for the nanoparticles those interfaces are placed inside the PCBM-rich nanoparticle core. Interestingly, the blend BCP100 nanoparticles exhibit also the formation of charge transfer states at around 525 nm with a growing time of around 4 ps (see Fig. 6e, blue line). These charge transfer states are formed at the interface between PCPDTBT and PCBM in the nanoparticles with the hole and electrons localized on PCPDTBT and PCBM, respectively.10,41,42 This intermediate state in which holes and electrons are still bound to each other is in direct competition with the formation of free charges.Our results thus highlight different charge transfer behaviours in the two blend nanoparticles. In the blend BCP100 nanoparticles, the photo-induced exciton can dissociate following two different mechanisms. The first mechanism involves the formation of charge transfer states at a PCPDTBT/PCBM interface in around 4 ps while in the second mechanism, the exciton is separated into free charges in around 20 ps.
The fact that the charge transfer state time formation is faster than the one for free charges mean that the BCP100 nanoparticle films contain more charge transfer state forming interfaces with respect to free charge generating ones. In contrast, only charge generating interfaces are found in the blend BCP2 nanoparticles and CTRL film where excitons dissociate into free charges in around 40 ps and 10 ps respectively. The distinct behaviours in the two nanoparticles can be associated with the different morphologies obtained when using BCP2 and BCP100. A detailed analysis of the morphological study can be found elsewhere for different nanoparticle preparation preocedures36 but relevant results are summarized below for nanoparticles prepared using rod–coil block copolymers.35 Carbon to sulphur (C/S) atomic ratio investigations in various nanoparticles reveal major morphological differences between BCP2- and BCP100-based nanoparticles. In the neat BCP2 nanoparticles, the C/S ratio increases when moving towards the nanoparticle edges which suggests the formation of a thin asymmetric carbon-rich shell.32 On the other hand, BCP100-based nanoparticles do not exhibit such well-defined core–shell structure and have a less defined shape. Moreover, in the BCP100 nanoparticles thin films, neighbouring nanoparticles are sometimes connected and thus the single nanostructure results less recognizable.32 According to the results presented in Fig. 2, blend nanoparticle diameters are approximately 2.8 and 1.9 folds larger than neat nanoparticle ones for BCP2 and BCP100, respectively. This suggests that interactions between the polymeric chain and PCBM are stronger in the case of short coil units. Thus, the quantity of PCBM included in the BCP2 blend is higher than in the BCP100 one causing an increasing in the size of the BCP2 nanoparticles with respect to the BCP100 ones.
The exciton's ability to dissociate into free charges at the D/A interface strongly depends on the order and crystallinity of the donor and acceptor domains.15 Charge generation efficiency increases with increasing domain order which is consistent with the morphological and photophysical results reported here. The less defined BCP100-based nanoparticles with a more disordered nanomorphology favour the formation of charge transfer states with respect to free charge generation that occurs at more ordered D/A interfaces. Unlike BCP2, the coil of BCP100 is not composed exclusively of 4VP units but also contains a large amount of styrene. The presence of styrene units could also have a major impact on the neat and blend nanoparticle photophysics. To ensure that the different charge generation mechanisms observed in BCP2- and BCP100-based blend nanoparticles are not simply related to the presence of styrene units, we performed TA measurements on blend BCP15 nanoparticles. Similarly to BCP2, BCP15 is a rod–coil BCP in which the coil is composed exclusively of 4VP units and in particular a mean of 15 repeating units of 4VP (Fig. 1a). TA spectra and temporal dynamics of BCP15 blend nanoparticles are very similar to those of BCP100 blend nanoparticles (Fig. S5, ESI†). These similar behaviours suggest that the key factor controlling the formation of long-lived interfacial charge transfer states is the coil length and not the presence of styrene units in BCP100. The coil length, which has a major impact on the nanoparticle nanomorphology, should thus be tuned to promote the formation of free charges to ensure efficient operation of nanoparticle-based OSCs.
Conclusions
In conclusion, using ultrafast spectroscopy we have studied the photophysics of water-processable nanoparticles formed through a modified miniemulsion process. PCPDTBT rods connected with either short (2 4VP units) or long (100 randomly polymerized styrene and 4VP units) coil segments simultaneously act as electron donor and surfactant. These amphiphilic rod–coil block copolymers were blended with PCBM, an electron acceptor, and the produced blend nanoparticles exhibit coil length dependent nanomorphologies. A clear correlation can be found between the formation of free charge generation D/A interfaces and the level of order in the nanoparticles. In less defined blend nanoparticles produced with long coils, long-lived charge transfer states are formed at the D/A interface which translates into a non-efficient exciton separation mechanism. Designing amphiphilic rod–coil block copolymer chains with relatively short coils ensures an efficient dissociation of excitons into free charges at the D/A interface.Our study thus emphasizes the importance of molecular design optimization to produce environmentally-safe yet efficient OSC active layers based on amphiphilic rod–coil block copolymers. Furthermore, we confirmed that the level of order in the thin active layers is a fundamental parameter to achieve high photovoltaic performances in OSCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Regione Lombardia project “I-Zeb”.References
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- T. Sano, S. Inaba and V. Vohra, ACS Appl. Energy Mater., 2019, 2, 2534–2540 CrossRef CAS.
- V. Vohra, Chem. Rec., 2019, 19, 1166–1178 CrossRef CAS.
- J. Cabanillas-Gonzalez, T. Virgili, G. Lanzani, S. Yeates, M. Ariu, J. Nelson and D. D. C. Bradley, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 14211 CrossRef.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS.
- S. K. Gupta, K. Dharmalingam, L. S. Pali, S. Rastogi, A. Singh and A. Garg, Nanomater. Energy, 2013, 2, 42–58 CrossRef CAS.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2010, 132, 6154–6164 CrossRef CAS.
- M. Scarongella, J. De Jonghe-Risse, E. Buchaca-Domingo, M. Causa, Z. Fei, M. Heeney, J. E. Moser, N. Stingelin and N. Banerji, J. Am. Chem. Soc., 2015, 137, 2908–2918 CrossRef CAS.
- T. Virgili, G. Grancini, E. Molotokaite, I. Suarez-Lopez, S. K. Rajendran, A. Liscio, V. Palermo, G. Lanzani, D. Polli and G. Cerullo, Nanoscale, 2012, 4, 2219–2226 RSC.
- T. J. Savenije, J. E. Kroeze, M. M. Wienk, J. M. Kroon and J. M. Warman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 1–11 CrossRef.
- B. Jana, A. Ghosh and A. Patra, J. Phys. Chem. Lett., 2017, 8, 4608–4620 CrossRef CAS.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS.
- D. Veldman, Ö. Ipek, S. C. J. Meskers, J. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kroon, S. S. Van Bavel, J. Loos and R. A. J. Janssen, J. Am. Chem. Soc., 2008, 130, 7721–7735 CrossRef CAS.
- C. R. McNeill, S. Westenhoff, C. Groves, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 19153–19160 CrossRef CAS.
- M. Scarongella, A. A. Paraecattil, E. Buchaca-Domingo, J. D. Douglas, S. Beaupré, T. McCarthy-Ward, M. Heeney, J. E. Moser, M. Leclerc, J. M. J. Fréchet, N. Stingelin and N. Banerji, J. Mater. Chem. A, 2014, 2, 6218–6230 RSC.
- D. Jarzab, F. Cordella, M. Lenes, F. B. Kooistra, P. W. M. Blom, J. C. Hummelen and M. A. Loi, J. Phys. Chem. B, 2009, 113, 16513–16517 CrossRef CAS.
- O. V. Mikhnenko, H. Azimi, M. Scharber, M. Morana, P. W. M. Blom and M. A. Loi, Energy Environ. Sci., 2012, 5, 6960–6965 RSC.
- M. C. Scharber, C. Lungenschmied, H. J. Egelhaaf, G. Matt, M. Bednorz, T. Fromherz, J. Gao, D. Jarzab and M. A. Loi, Energy Environ. Sci., 2011, 4, 5077–5083 RSC.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H. J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS.
- E. M. Ward, P. A. Schulte, K. Straif, N. B. Hopf, J. C. Caldwell, T. Carreón, D. M. De Marini, B. A. Fowler, B. D. Goldstein, K. Hemminki, C. J. Hines, K. Husgafvel Pursiainen, E. Kuempel, J. Lewtas, R. M. Lunn, E. Lynge, D. M. McElvenny, H. Muhle, T. Nakajima, L. W. Robertson, N. Rothman, A. M. Ruder, M. K. Schubauer-Berigan, J. Siemiatycki, D. Silverman, M. T. Smith, T. Sorahan, K. Steenland, R. G. Stevens, P. Vineis, S. H. Zahm, L. Zeise and V. J. Cogliano, Environ. Health Perspect., 2010, 118, 1355–1362 CrossRef CAS.
- S. Inaba, R. Arai, G. Mihai, O. Lazar, C. Moise, M. Enachescu, Y. Takeoka and V. Vohra, ACS Appl. Mater. Interfaces, 2019, 11, 10785–10793 CrossRef CAS.
- N. P. Holmes, M. Marks, P. Kumar, R. Kroon, M. G. Barr, N. Nicolaidis, K. Feron, A. Pivrikas, A. Fahy, A. D. de, Z. Mendaza, A. L. D. Kilcoyne, C. Müller, X. Zhou, M. R. Andersson, P. C. Dastoor and W. J. Belcher, Nano Energy, 2016, 19, 495–510 CrossRef CAS.
- S. Gärtner, A. J. Clulow, I. A. Howard, E. P. Gilbert, P. L. Burn, I. R. Gentle and A. Colsmann, ACS Appl. Mater. Interfaces, 2017, 9, 42986–42995 CrossRef.
- T. Virgili, C. Botta, M. M. Mróz, L. Parrenin, C. Brochon, E. Cloutet, E. Pavlopoulou, G. Hadziioannou and M. Geoghegan, Front. Chem., 2019, 7, 1–9 CrossRef.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- K. L. Jae, L. M. Wan, C. J. Brabec, J. Yuen, S. M. Ji, Y. K. Jin, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS.
- S. Zappia, G. Scavia, A. M. Ferretti, U. Giovanella, V. Vohra and S. Destri, Adv. Sustainable Syst., 2018, 2, 1700155 CrossRef.
- A. M. Ferretti, S. Zappia, G. Scavia, U. Giovanella, F. Villafiorita-Monteleone and S. Destri, Polymer, 2019, 174, 61–69 CrossRef CAS.
- A. Borodzinski and M. Bonarowska, Langmuir, 1997, 7463, 5613–5620 CrossRef.
- A. E. Di Mauro, M. Toscanini, D. Piovani, F. Samperi, M. L. Curri, M. Corricelli, L. De Caro, D. Siliqi, R. Comparelli, A. Agostiano, S. Destri and M. Striccoli, Eur. Polym. J., 2014, 60, 222–234 CrossRef CAS.
- S. Zappia, A. M. Ferretti, G. Scavia, U. Giovanella, V. Vohra, T. Virgili, L. Ganzer, M. Russo, E. Arias, I. Moggio and S. Destri, 2nd International Conference on Interface Properties in Organic and Hybrid Electronics, 2019, p. 93.
- Y. K. Choi, D. Lee, S. Y. Lee, T. J. Shin, J. Park and D. J. Ahn, Macromolecules, 2017, 50, 6935–6944 CrossRef CAS.
- J. Müllerová, M. Kaiser, V. Nádaždy, P. Šiffalovič and E. Majková, Sol. Energy, 2016, 134, 294–301 CrossRef.
- G. Cerullo, C. Manzoni, L. Lüer and D. Polli, Photochem. Photobiol. Sci., 2007, 6, 135–144 RSC.
- G. Grancini, N. Martino, M. R. Antognazza, M. Celebrano, H. J. Egelhaaf and G. Lanzani, J. Phys. Chem. C, 2012, 116, 9838–9844 CrossRef CAS.
- M. Tong, N. E. Coates, D. Moses, A. J. Heeger, S. Beaupré and M. Leclerc, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1–6 CrossRef.
- G. Grancini, M. Maiuri, D. Fazzi, A. Petrozza, H. J. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Nat. Mater., 2013, 12, 29–33 CrossRef CAS.
- G. Grancini, D. Polli, D. Fazzi, J. Cabanillas-gonzalez, G. Cerullo and G. Lanzani, J. Phys. Chem. Lett., 2011, 2, 1099–1105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the block copolymers, CW photo-induced spectroscopy data and the comparison of ultrafast decay times. See DOI: 10.1039/d0cp05478j |
| This journal is © the Owner Societies 2020 |