
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDOPC versus DOPE as a helper lipid for gene-therapies: molecular dynamics simulations with DLin-MC3-DMA†
Inna
Ermilova
 * and
Jan
Swenson
* and
Jan
Swenson
Department of Physics, Chalmers University of Technology, Gothenburg, Sweden. E-mail: ina.ermilova@gmail.com; inna.ermilova@chalmers.se; jan.swenson@chalmers.se; Tel: +46-728487773
First published on 9th December 2020
Abstract
Ionizable lipids are important compounds of modern therapeutic lipid nano-particles (LNPs). One of the most promising ionizable lipids (or amine lipids) is DLin-MC3-DMA. Depending on their pharmaceutical application these LNPs can also contain various helper lipids, such as phospho- and pegylated lipids, cholesterol and nucleic acids as a cargo. Due to their complex compositions the structures of these therapeutics have not been refined properly. Therefore, the role of each lipid in the pharmacological properties of LNPs has not been determined. In this work an atomistic model for the neutral form of DLin-MC3-DMA was derived and all-atom molecular dynamics (MD) simulations were carried out in order to investigate the effect of the phospholipid headgroup on the possible properties of the shell-membranes of LNPs. Bilayers containing either DOPC or DOPE lipids at two different ratios of DLin-MC3-DMA (5 mol% and 15 mol%) were constructed and simulated at neutral pH 7.4. The results from the analysis of MD trajectories revealed that DOPE lipid headgroups associated strongly with lipid tails and carbonyl oxygens of DLin-MC3-DMA, while for DOPC lipid headgroups no significant associations were observed. Furthermore, the strong associations between DOPE and DLin-MC3-DMA result in the positioning of DLin-MC3-DMA at the surface of the membrane. Such an interplay between the lipids slows down the lateral diffusion of all simulated bilayers, where a more dramatic decrease of the diffusion rate is observed in membranes with DOPE. This can explain the low water penetration of lipid bilayers with phosphatidylethanolamines and, probably, can relate to the bad transfection properties of LNPs with DOPE and DLin-MC3-DMA.
1 Introduction
Modern gene-therapies are widely using lipid-nanoparticles (LNPs) as vehicles for the delivery of nucleic acids. These LNPs can be employed for curing a large range of diseases, such as cancer,1,2 amyloidosis,3 Alzheimer's disease,4 Ebola,5 modern coronavirus COVID-196,7 and many others. Depending on their applications, these LNPs can contain phospholipids, cholesterol, pegylated lipids, ionizable lipids and a cargo (a nucleic acid).8,9 Such a variety of components leads to complex structures of large sizes. These complex structures are very challenging to elucidate by experiments, such as X-ray and neutron diffraction, and cryogenic transmission electron microscopy (cryo-TEM). The obstacle here is the polydispersity of therapeutics, which can be seen in the variety of sizes of LNPs10 in the same mixture and in the diversity of textures within a single particle.11 Typically, the experimental studies have to be combined with structural modelling,12,13 despite the computational limits, in order to refine the texture of the LNPs on an atomistic level and reveal the exact mechanisms behind the pharmacological properties of therapeutics.There were attempts to come closer to understanding these mechanisms by creating LNPs where one component was selected to be a variable. Comparative experimental studies were performed using formulations with changes of either a phospholipid (so-called “helper lipid”14,15) or an ionizable lipid.
In one of such studies Kulkarni et al.16 have investigated LNPs for plasmid DNA delivery using various phospholipids (DOPE, DOPC, SOPC, and DSPC) as well as ionizable lipids, cholesterol and pegylated lipids. The ratio between the compounds was the same. The in vitro studies showed that LNPs containing DOPE as a helper lipid were more potent in a sense of transfection properties when the ionizable lipid was DLin-KC2-DMA. In the same study when the ionizable lipid was DLin-MC3-DMA the best helper lipids were DOPC and SOPC16 and DOPE exhibited the worst transfection properties.
For the siRNA encapsulation together with DLin-KC2-DMA the best phospholipid was DSPC, according to Kulkarni et al.17 However, there were no experiments in their work showing the efficacy of LNPs in the sense of transfection.
Jayaraman et al.18 have performed a more detailed study where they compared 56 ionizable lipids as compounds of siRNA LNPs: DLin-MC3-DMA exhibited the best gene-silencing activity in formulations containing DSPC, cholesterol and pegylated lipids.
In 2018 the first siRNA based drug, produced by Alnylam for curing amyloidosis, was approved.19–21 This became a game-changer in the pharmaceutical industry in the race for RNA-based vaccines.21 This drug contains DLin-MC3-DMA. The most potent ionizable lipid became very attractive for other pharmaceutical companies which are working on developing mRNA vaccines.22,23
Nevertheless, even for the approved new drug, the precise mechanisms of interactions between DLin-MC3-DMA and various phospholipids and the structure of the therapeutic were not disclosed. The lack of this information made medicines for gene-therapies attractive for computational studies.
A small number of theoretical studies have been done with DLin-KC2-DMA, but no computational study has been performed with DLin-MC3-DMA, probably, due to the absence of numerical models. Ramenzapour et al.24 combined MD simulations with cryo-TEM and SAXS for investigating membranes, formed by POPC, cholesterol and the ionizable lipid, at acidic and neutral pH. According to their study, whole molecules of DLin-KC2-DMA preferred to reside in the center of the lipid bilayer at pH 7.4 while at higher pH the ionizable lipids were positioned with headgroups towards the membrane surface and their tails were spread between the tails of POPC.24
Leung et al.25 went towards the lower resolution in computer simulations by carrying out coarse-grained (CG) simulations of whole LNPs and combining them with 31P NMR and cryo-TEM techniques. Their vehicles contained DLin-KC2-DMA, siRNA duplexes, DSPC, cholesterol and pegylated lipid. The images obtained from snapshots from CG simulations were quite similar to the cryo-TEM images. However, the mechanisms behind phase formation and intermolecular interactions were not disclosed due to the low resolution of the CG simulations.
In this theoretical work, we are beginning to employ the multiscale simulation approach26–28 by starting from simple systems and atomistic MD simulations with the derivation of the numerical model for one of the most potent ionizable lipids, which will help us to derive CG models in future. The primary attention is focused on understanding the mechanisms behind interactions between different phospholipids and DLin-MC3-DMA. Four model membranes containing various amounts of DLin-MC3-DMA (Fig. 1(a)) and DOPC and DOPE lipids (Fig. 1(c) and (d)) at neutral pH serve as possible pieces of LNP shells. The reason for the selection of the specific phospholipids was to compare the behavior of DLin-MC3-DMA in membranes built of lipids containing identical tails, but different headgroups. This can unveil possible reasons of efficacies of LNPs. For instance, from MD trajectories one can calculate mass density profiles for whole systems and for different parts of the molecules. This determines the partitioning of molecules in lipid bilayers, including the water permeabilities of membranes. Computed lateral diffusion coefficients give an idea of the molecular velocities in planes parallel to membrane surfaces. Radial distribution functions (RDFs) demonstrate associations between pairs of atoms, indicate the possible formation of hydrogen bonds and, perhaps, reveal the mechanisms of interactions between the lipids.
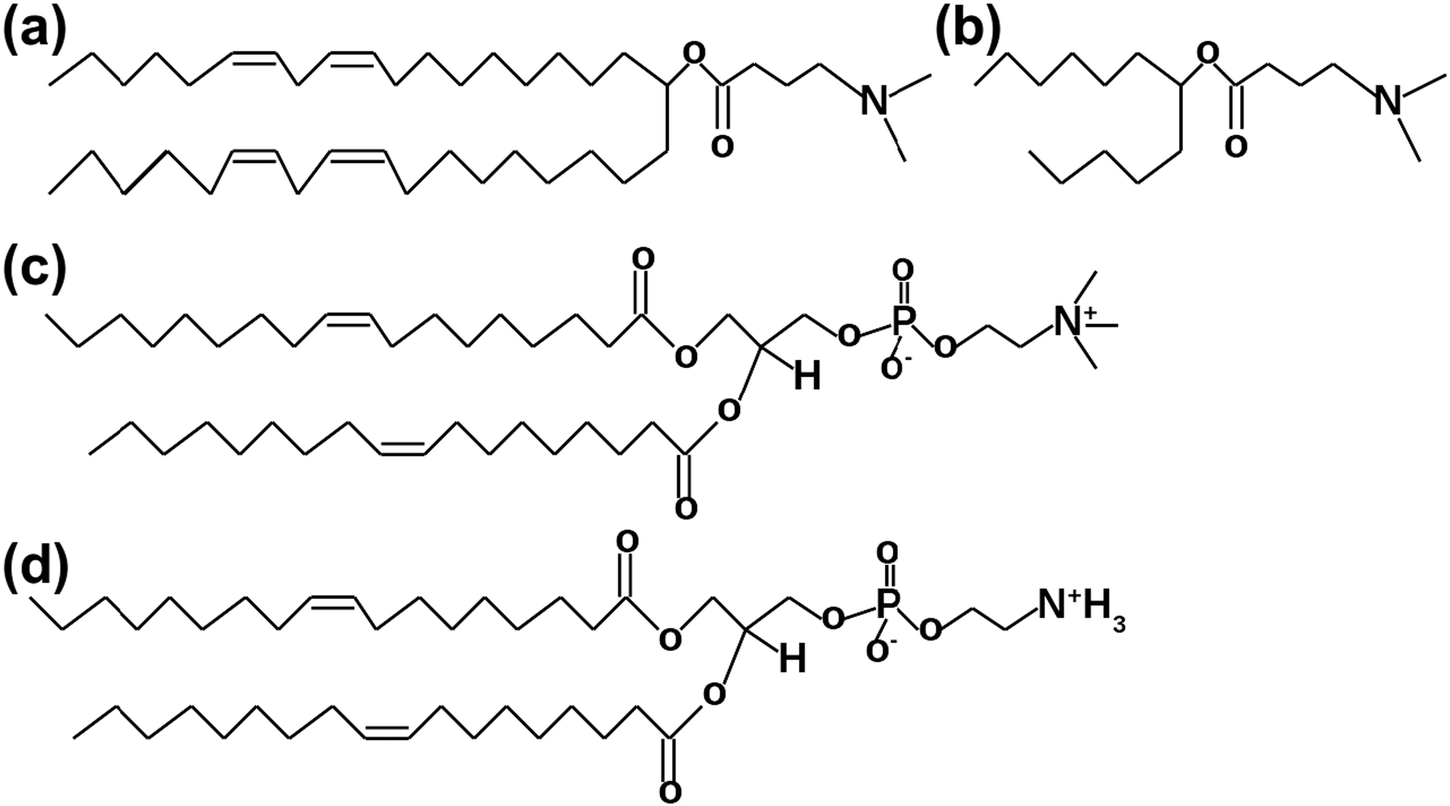 | ||
Fig. 1 Lipids which were used in MD simulations. (a) DLin-MC3-DMA. (b) The part of DLin-MC3-DMA which was used for the FF parametrization. (c) DOPC,18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Δ9 – cis) PC. (d) DOPE, 18:1(Δ9 – cis) PE. :1 (Δ9 – cis) PC. (d) DOPE, 18:1(Δ9 – cis) PE. | ||
Moreover, free energy calculations with well-tempered metadynamics29–31 simulations are employed for the investigation of water penetration through four studied model membranes. A thermodynamic characteristic of this process, such as free energy (ΔG°), is known to indicate if the process can proceed spontaneously (ΔG°< 0). A comparison between the binding free energy values will reveal which membrane is more permeable. For such studies well-tempered metadynamics simulations are considered as the most promising approach32 due to its ability to sample only “interesting” parts of free energy landscape. Therefore, the information about the binding free energies of water to membranes can shed light on the efficacy of LNPs containing in their outer shells DLin-MC3-DMA with either DOPC or DOPE lipids.
2 Models and simulation setup
Parametrization of the model for DLin-MC3-DMA
The model for DLin-MC3-DMA was developed by applying the same philosophy as was done in the case of polyunsaturated phospholipids.34 Since the pH around an LNP is considered as neutral, a neutral model for the ionizable lipid was derived and the total molecular charge was set to zero. Since we will not use the ionized form of this compound, we will call it the “amine” lipid until the end of this paper.The strategy for the derivation of parameters was the same as the one used in previous versions of the SLipids force field (FF)34–36 for polyunsaturated phospholipids. The general form of the SLipids FF can be written as:
| EFF = Ebonded + Enon-bonded, | (1) |
| Ebonded = Eangles + Edihedrals + Ebonds + EUrey–Bradley | (2) |
| Enon-bonded = ELennard-Jones + ECoulomb. | (3) |
In this work the accent was on calculations of partial atomic charges and parameters for dihedrals for the headgroup of DLin-MC3-DMA, while the partial charges for the lipid tails were slightly adjusted from the ones which were derived in the earlier version of the SLipids FF.34 Dihedrals for the DLin-MC3-DMA tails were taken from the SLipids FF for polyunsaturated lipids.34
Since quantum chemical calculations have high costs in terms of computational time and computer memory,37,38 the smaller model compound for DLin-MC3-DMA was utilized (Fig. 1(b)). 50 random conformations were generated for this molecule. Then the computations of partial atomic charges were carried out using the B3LYP39–41 level of theory with the basis set cc-pVTZ42 in Gaussian09 software43 together with the restrained electrostatic potential approach (RESP)44 in the R.E.D. software.45 The lipid headgroups were placed in a polarizable continuum with a dielectric constant of 78.4 in the IEFPCM model46,47 in order to mimic the solvent effects and their induced polarization on the charge distribution. The final set of partial atomic charges can be found in Fig. S1 of the ESI.†
After computations of partial atomic charges a set of new parameters for dihedrals was derived using the same philosophy as in the previous version of the SLipids FF.34
Fig. S2 in the ESI† illustrates the dihedrals which were chosen for the parametrization. For the parametrization of the dihedrals the molecule from Fig. 1(b) was used as well. As in the SLipids FF for polyunsaturated lipids, the original molecule was optimized using the second order Møller–Plesset perturbation theory48 with the cc-pVDZ basis set.42 Then conformations were created for every value of the dihedral by rotating the optimized molecule around the dihedral of interest with a step of 10 degrees in the interval from −180 to 180 degrees, keeping other degrees of freedom frozen. For computing the relative quantum mechanical energies of different conformations the hybrid method for interaction energies (HM-IE)49 was employed which was expressed by relation (4):
| ECCSD(T)/BBS = ECCSD(T)/SBS + ECCSD(T)/BBS − ECCSD(T)(SBS) ≈ ECCSD(T)/SBS + EMP2/BBS − EMP2/SBS | (4) |
After completing the high order quantum chemical calculations the resulting values were used for fitting the dihedral potential with equation (5):
| Edihedral = (ECCSD(T)/BBS − EMD) − (ECCSD(T)/BBS − EMD)min | (5) |
MD simulation set-ups
Compositions of simulated systems are presented in Table 1. Firstly, two lipid bilayers were created, where one was containing only DOPC and the other one was built by only DOPE lipids. Every bilayer was built out of 100 single boxes containing two phospholipids: one was in the outright position and the other one was inverted (but not mirrored). The resulting bilayers were utilized for creating membranes with various contents of DLin-MC3-DMA. In these lipid bilayers in selected places (at a distance of ∼10 Å) phospholipids were deleted and optimized molecules of DLin-MC3-DMA were inserted instead of them.| Lipid | Number of PCs/Pes | Number of DLin-MC3-DMA | Number of waters (TIP3p54) |
|---|---|---|---|
| DOPC | 190 | 10 | 8000 |
| DOPC | 170 | 30 | 8000 |
| DOPE | 190 | 10 | 8000 |
| DOPE | 170 | 30 | 8000 |
| DOPC (pure) | 200 | 0 | 8000 |
| DOPE (pure) | 200 | 0 | 8000 |
Then every created system was equilibrated for 300 ns in the NPT ensemble and each production run was 300 ns. A pressure of 1 atm during simulations was supported by a semi-isotropic pressure coupling scheme using the Berendsen barostat.55 The temperature was held at 298 K by the velocity rescaling thermostat.56 The algorithm employed for the integration of the Newtonian equation of motion was leap-frog57 with a time step of 2 fs and a Verlet cut-off scheme58 with the van der Waals type cut-off and a cut-off radius of 1.2 nm. Bonds were constrained using the LINCS algorithm59,60 with 12 iterations. MD simulations and analysis of the resulting trajectories were done using built-in routines in gromacs-4.6.7 software.61 Systems containing pure DOPC and DOPE lipid bilayers were simulated for the sake of comparison. The generalized snapshots from starting configurations for systems with 5 and 15% of DLin-MC3-DMA can be observed in Fig. S4 in the ESI.† Placing phospholipids instead of the amine lipids will give an idea about starting configurations for systems containing only DOPC/DOPE lipids and water.
Well-tempered metadynamics simulation set-ups
4 systems, containing DLin-MC3-DMA, were utilized for well-tempered metadynamics29,62 simulations from previously equilibrated classical MD simulations. For every lipid bilayer 5 different starting configurations were created where the selected water molecule was located in various parts of the system. Later those configurations were used for performing 5 parallel well-tempered metadynamics simulations per lipid bilayer.63The collective variable (CV) was chosen as the Z-component of the distance between the centers of mass of the water molecule and a lipid bilayer (see Fig. 2). The Gaussian functions were of height 1.2 kJ mol−1. Their width was 0.05 nm (the parameter σ) which were deposited every 500 steps (the value of the parameter PACE). Due to the small size of the water molecule, the bias factor, γ, was set to 10.0.
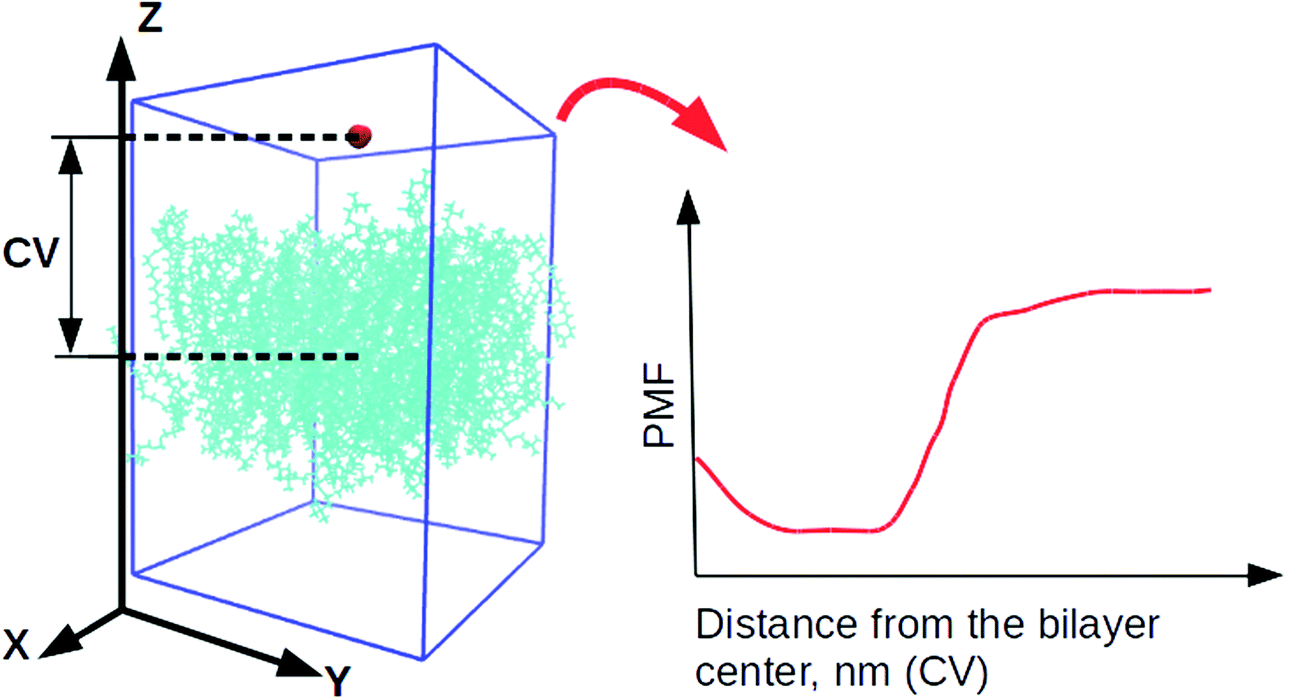 | ||
| Fig. 2 Definition of the collective variable for well-tempered metadynamics simulations. A small red molecule is a water molecule, cyan molecules are lipids. All other water molecules were omitted for clarity. “CV” is the collective variable, and “PMF” is the potential of mean force. | ||
All simulations were performed in the NVT ensemble at a temperature of 298 K using the velocity rescaling thermostat.56 The integrator for Newtonian equations of motions was leap-frog57 with a time step of 2 fs and a cut-off scheme Verlet58 where a cutoff radius was 1.2 nm. The LINCS algorithm was employed for constraining bonds with 12 iterations.59,60 The software used as an MD engine was gromacs-4.6.7 with plumed-2.1.262 for the free energy calculation. Every simulation was carried out for 150 ns.
3 Results and discussion
Fig. 3 shows final snapshots of the simulated systems. In (a) and (b) no significant differences can be observed in the positioning of DLin-MC3-DMA in DOPC and DOPE membranes respectively: the amine lipids are situated close to the surfaces of the bilayers. Fig. 3(c) and (d) present systems with lower contents of phospholipids. In this case DLin-MC3-DMA exhibits a preference to be located at the surface of the membrane with DOPE while in the case of DOPC the amine lipid is partitioned in the center of the bilayer to a large extent.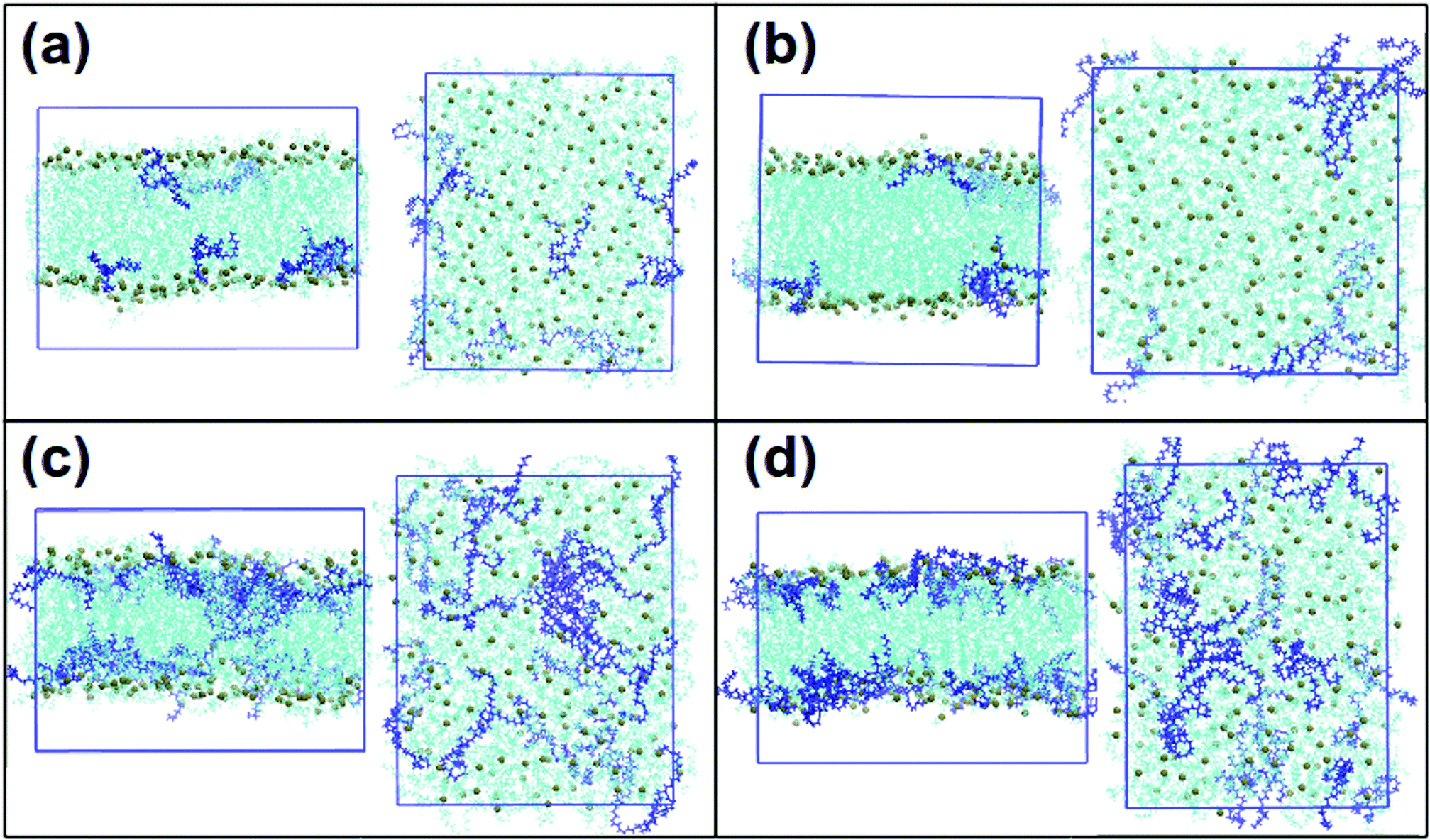 | ||
| Fig. 3 Snapshots of the final frames of the systems: frontal views and views from the top of the box. (a) DOPC and 5% of DLin-MC3-DMA, (b) DOPE and 5% of DLin-MC3-DMA, (c) DOPC and 15% of DLin-MC3-DMA and (d) DOPE and 15% of DLin-MC3-DMA. Phospholipids are visualized by cyan color with phosphorus shown as spheres of dark lime color. DLin-MC3-DMA is visualized by the dark blue color. The molecular graphics software is VMD.33 | ||
Fig. 3 shows also that the amine lipid tends to cluster in all simulated systems.
The analysis of snapshots is important, but it does not give the information about the statistics over the whole simulation time. That is why more detailed analysis of trajectories was carried out.
3.1 Lateral diffusion, average area per lipid, and mass density profiles
The lateral diffusion coefficient is one of the key characteristics of a lipid bilayer.64,65 It can determine how fast lipids move in two dimensions, and give an idea about the viscosity of membranes64 and their fluidity.66–68 The membrane fluidity and its fusogenicity have an impact on a drug delivery process.69
Table 2 shows lateral diffusion coefficients for the investigated systems, which were computed using a correction for the artificial center of mass motion of each monolayer as it was done earlier by Jämbeck et al.35 The experimental lateral diffusion coefficient at a temperature of 298 K was available only for the pure DOPC lipid bilayer, which was about 0.932 × 10−7 cm2 s−171 and comparable with our computed value. Nevertheless, regardless of such a good agreement with experiment it is important to note that the computed values are sensitive to the box shape and can have an error of up to 50% due to the artifacts from periodic boundary conditions according to B. A. Camley et al.72 and R. M. Venable et al.73 Taking into account this fact we can try to compare the results for simulations with the amine lipid.
| System | Lateral diffusion of PC/PE | Lateral diffusion of DLin-MC3-DMA |
|---|---|---|
| DOPC + 5%DLin-MC3-DMA | 1.15 ± 0.40 | 1.01 ± 0.45 |
| DOPC + 15%DLin-MC3-DMA | 0.94 ± 0.40 | 0.65 ± 0.40 |
| DOPE + 5%DLin-MC3-DMA | 1.00 ± 0.45 | 1.15 ± 0.30 |
| DOPE + 15%DLin-MC3-DMA | 0.66 ± 0.45 | 0.55 ± 0.20 |
| DOPC (pure) | 0.98 ± 0.42 | — |
| DOPE (pure) | 1.20 ± 0.42 | — |
In systems containing DOPC the diffusion of phosphatidylcholine as well as the diffusion of the amine lipid slows down when the amount of DLin-MC3-DMA increases. An even stronger effect is observed in systems with DOPE, where the addition of DLin-MC3-DMA decreases the diffusion of both lipids more dramatically than in the case of membranes with DOPC. This can be considered as a reason for why LNPs containing DLin-MC3-DMA and DOPE do not exhibit good transfection in plasmid DNA delivery, because in the process of the DNA delivery16 the lateral diffusion and the membrane fusion are important factors.
However, the lateral diffusion is a consequence of intra- and intermolecular interactions.74,75 Therefore, the differences in diffusion indicate that there may be strong associations between DOPE headgroups and DLin-MC3-DMA. For checking up this assumption one can calculate other properties of simulated membranes.
One such property is the area per lipid.76,77 In every system there were 100 lipids per leaflet. According to this number of lipids the average area per lipid was calculated for each membrane by dividing the area of the simulation box in a plane perpendicular to the z-axis by the number of lipids in a leaflet (see eqn (6)):
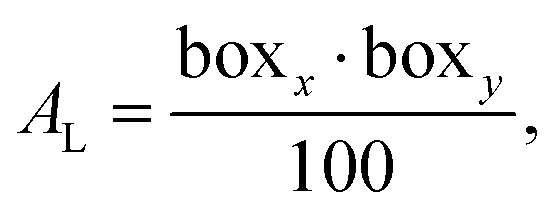 | (6) |
| System | Area per lipid | The membrane thickness |
|---|---|---|
| DOPC + 5%DLin-MC3-DMA | 70.98 ± 0.3 | 33.9 ± 0.1 |
| DOPC + 15%DLin-MC3-DMA | 73.01 ± 0.3 | 33.1 ± 0.1 |
| DOPE + 5%DLin-MC3-DMA | 63.52 ± 0.3 | 37.0 ± 0.1 |
| DOPE + 15%DLin-MC3-DMA | 68.46 ± 0.4 | 33.7 ± 0.1 |
| DOPC (pure) | 69.00 ± 1.2 | 35.6 ± 0.1 |
| DOPE (pure) | 63.35 ± 1.0 | 38.2 ± 0.1 |
Another property is the mass density of the lipid bilayer, measured in the direction perpendicular to the plane of the membrane surface. Mass density profiles are similar to electron density profiles, which can be obtained using X-ray scattering. The highest value of the total mass density is associated with lipid headgroups and the lowest value shows where the bilayer center is situated.78
Fig. 4 demonstrates mass density profiles for the simulated systems as well as for lipid bilayers containing pure phospholipids. Both (a) and (b) parts of the figure exhibit the same trend in shifting the peak of the mass density towards the bilayer center with increasing concentration of DLin-MC3-DMA. This can be interpreted as thinning of the bilayer since the highest value of mass density is associated with the position of the lipid headgroup (see Table 3 for the computed membrane thickness values). Nevertheless, in order to get an exact picture of where the various parts of the lipid molecules are situated one shall compute contributions to the mass density profiles from those parts.
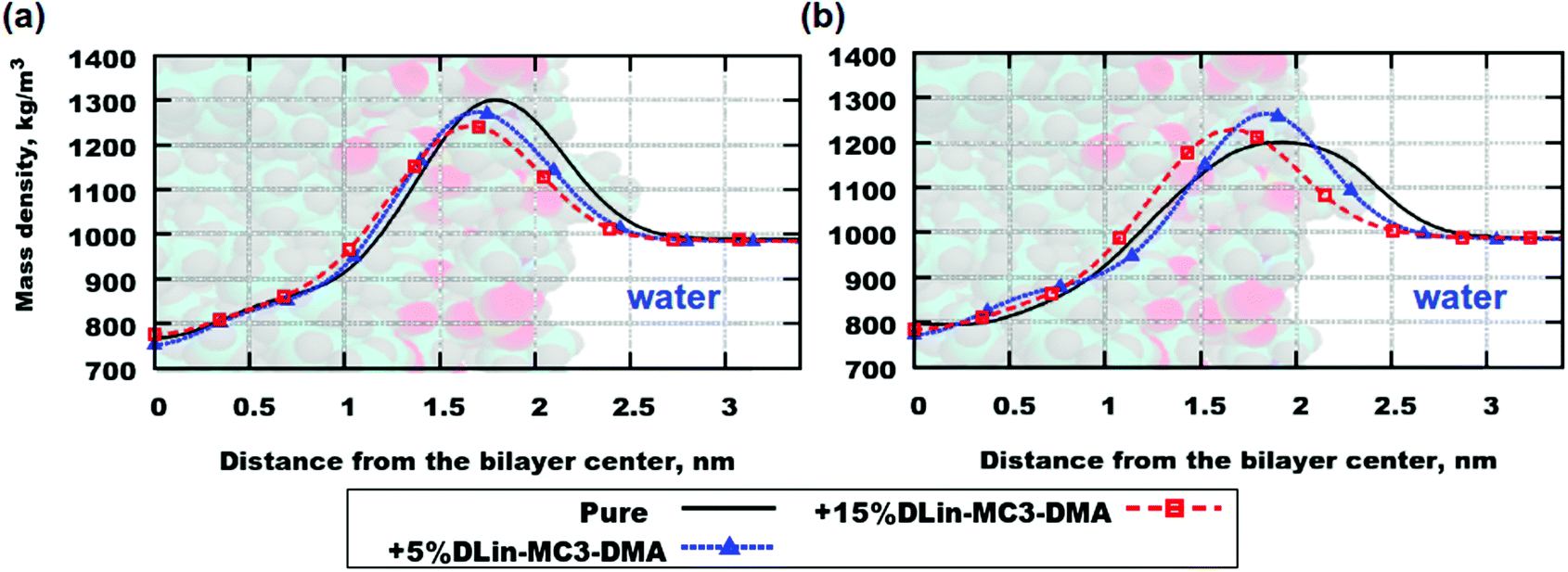 | ||
| Fig. 4 Mass density profiles for the following systems: (a) DOPC and (b) DOPE. Labels: “Pure” stands for lipid bilayers containing only phospholipids, +5% DLin-MC3-DMA means that it was 5% of the amine lipid and +15% DLin-MC3-DMA means that it was 15% of the amine lipid. | ||
Fig. 5 displays contributions to mass density profiles from headgroups and tails for both lipids. Already at the lowest concentration of the amine lipid (5 mol%) differences in profiles for the two phospholipids can be observed (Fig. 5(a) and (b)): the position of the peak for the DLin-MC3-DMA tail is in the DOPE headgroup region while in the case of DOPC this peak appears in the region for the phospholipid tails. The headgroup of the amine lipid “prefers” to be situated between phospholipid headgroups in both DOPC and DOPE membranes.
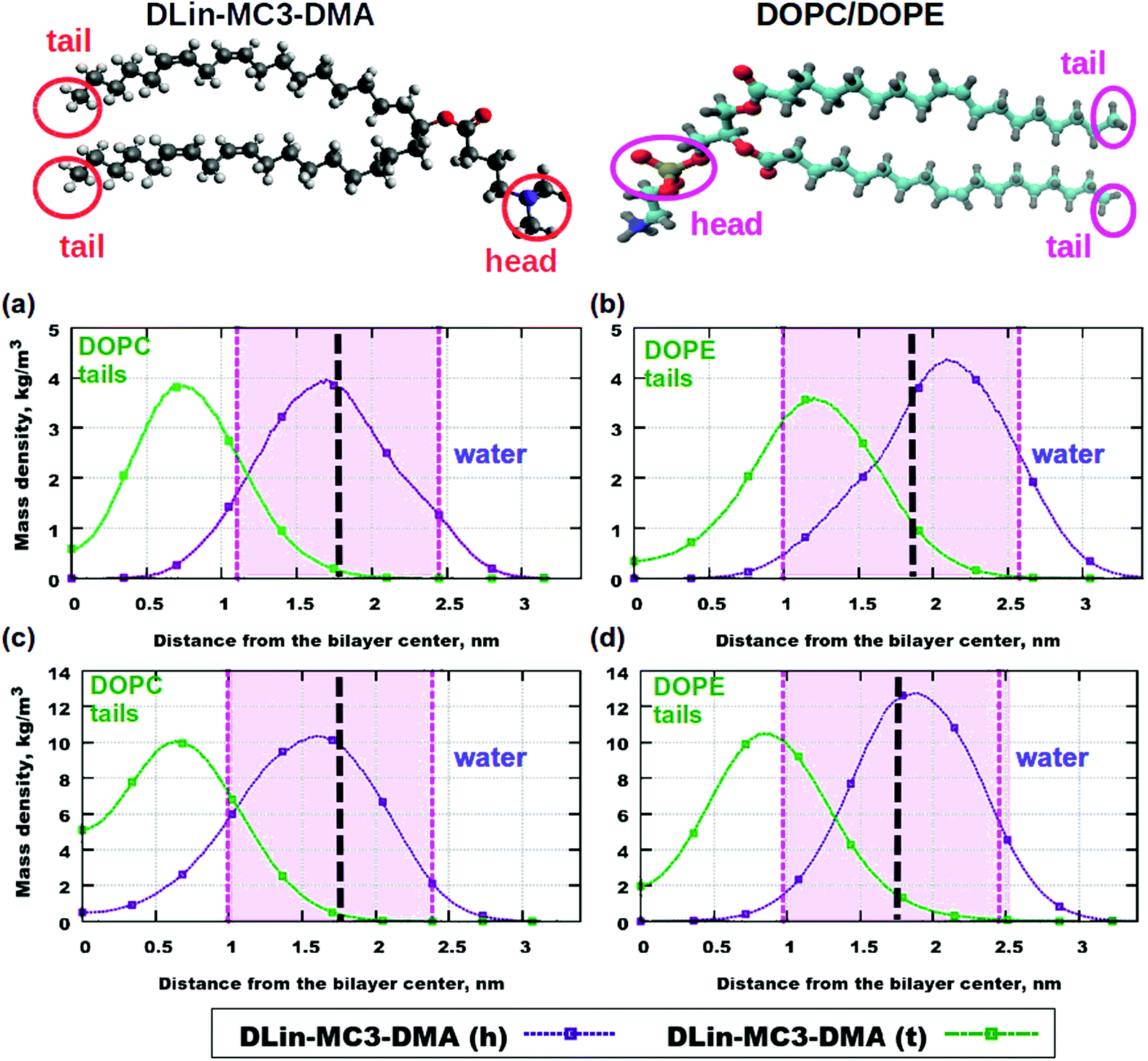 | ||
| Fig. 5 Contributions to the mass density profiles: (a) DOPC and 5% of DLin-MC3-DMA, (b) DOPE and 5% of DLin-MC3-DMA, (c) DOPC and 15% of DLin-MC3-DMA and (d) DOPE and 15% of DLin-MC3-DMA. Pink areas show the locations of the PO4 headgroups of the phospholipids. DLin-MC3-DMA (h) and DLin-MC3-DMA (t) denote the headgroup and the tail of the lipid respectively. On the image representing the structure of DLin-MC3-DMA the parts which were used in calculations of partial molecular mass densities are denoted by red circles. From the head group only the nitrogen and two carbons were taken for calculations in order to get a “comparable” profile, while from tails carbons with hydrogens were taken for computations. From the structure of phospholipids on the picture the PO4-groups were considered as the representatives of the lipid headgroups, while tails were denoted with CH3-groups in the ends of tails. Parts of DLin-MC3-DMA were colored in a different way: light gray color – hydrogen, dark gray – carbon, blue – nitrogen, and red – oxygen. Black dashed lines denote the points of maximum for the mass densities of PO4-groups. Lipids on top are visualized using Avogadro70 (DLin-MC3-DMA) and VMD33 (DOPC/DOPE) software. | ||
When the amount of the amine lipid is 15 mol% (Fig. 5(c) and (d)) the peak for the DLin-MC3-DMA tail is shifted to just outside the area for the DOPE headgroup. In the DOPC membrane the position of this peak does not change, compared to lipid bilayers with 5 mol% of the amine lipid. Moreover, a higher amount of the DLin-MC3-DMA tails resides in the center of the DOPC membrane than in the lipid bilayer with DOPE. The headgroup of the amine lipid “prefers” to be located in the region of the phospholipid headgroups, but in the membrane with DOPC a small amount was detected even in the center of the lipid bilayer. Such a location of DLin-MC3-DMA is slightly similar to the one which was observed by Ramenzapour et al.24 in simulations of DLin-KC2-DMA in a membrane containing POPC and cholesterol at a neutral pH.
However, lipids are not the only molecules present in the simulated systems. Due to their motion the motion of water can be affected. The water can penetrate the membranes as well. Fig. S6 in the ESI† shows mass density profiles for water in the simulated membranes. In pure phospholipid bilayers there were no significant amounts of water detected in the center of the bilayers, but the addition of the amine lipid has changed the penetration of the membranes. For instance, the amount of 5 mol% has made the DOPE lipid membrane permeable (Fig. S6(b), ESI†), while in the case of DOPC the penetration was more negligible (Fig. S6(a), ESI†). At a higher concentration of DLin-MC3-DMA (15 mol%) the situation was the other way around and the water penetration of the bilayer containing DOPC is higher than that at 5 mol% of the amine lipid, while for the system containing DOPE the mass density of water in the bilayer center is almost equal to zero.
These findings suggest that there are two competing effects which determine the water penetration in the investigated membranes. The effects dominate in systems with DOPE and DLin-MC3-DMA. At the lowest concentration of DLin-MC3-DMA the peak of the mass density for the CH3-groups of the lipid tails is observed inside the PO4-group region (Fig. 5(b)). This location of small amounts of hydrophobic parts can be the reason for a proton-transfer disruption, because in the pure phospholipid bilayers the phosphate groups have “the proton-collecting antenna effect” according to Brändén et al.79 and Yamashita et al.80 Furthermore, water molecules around phosphatidylcholine headgroups do not have the same preferential orientations as they have around phosphatidylethanolamine headgroups.81 Therefore the presence of extraneous CH3-groups can cause pore formation in DOPE membranes more than in lipid bilayers with DOPC.
At 15 mol% in lipid bilayers with DOPC the water penetration gets slightly higher with the increase in the concentration of the amine lipid. This is correlated with the fact that a small amount of headgroups of DLin-MC3-DMA reside in the center of the membrane composed of DOPC (Fig. 5(c)). These headgroups can play the role of “transporters” of water molecules through the membrane. In the lipid bilayer with DOPE containing the same amount of DLin-MC3-DMA the decrease in water penetration can be explained by the slight change in the location of the lipid tails of DLin-MC3-DMA. The maximum of the mass density profile for the CH3-group of the amine lipid shifted towards the carbonyl groups of DOPE which can give rise to a hydrophobic layer in that region and prevent the water from entering the membrane.
This information about the contributions to the mass densities from different parts of the molecules allows us to only speculate that the slow lateral diffusion at higher concentrations of the amine lipids can be related to strong interactions and, perhaps, associations between the headgroups of phospholipids and parts of DLin-MC3-DMA. Particularly, in membranes with DOPE lipid tails of the amine lipid seem to associate with phospholipid headgroups. To elucidate this, we have calculated RDFs for correlations between different groups of the two components.
3.2 Radial distribution functions
RDFs are important characteristics of intra- and intermolecular associations which can occur in various systems. They can also explain the locations of molecules and be related to forces causing molecular motions. For the simulated bilayers intermolecular RDFs were of high interest.From the computed mass density profiles and diffusion coefficients we got indications for the fact that there could be forces occurring between the lipid headgroups which could “help” the amine lipid to stay closer to the DOPE membrane's surface or help DLin-MC3-DMA to penetrate the bilayer with DOPC. In order to validate this hypothesis RDFs between pairs of atoms in lipid headgroups were calculated. Fig. 6 shows such correlation functions. In (a) one can see a high peak at a distance less than 2 Å for hydrogen atoms in the amine-group of DOPE and the oxygen atom in the carbonyl group of DLin-MC3-DMA. This peak indicates possible formation of a hydrogen bond between these lipid groups. For the system containing DOPC (in the same part of the figure) such RDFs were calculated for the methyl hydrogens of the choline group. In this case hydrogen bonding does not occur between chosen pairs of atoms in DOPC and DLin-MC3-DMA. However, in the case of DOPC, hydrogens from the glycerol group seem to associate more with the carbonyl oxygen from DLin-MC3-DMA (Fig. 6(b)) and for DOPE such association takes place only at 15 mol% of amine lipid in the system.
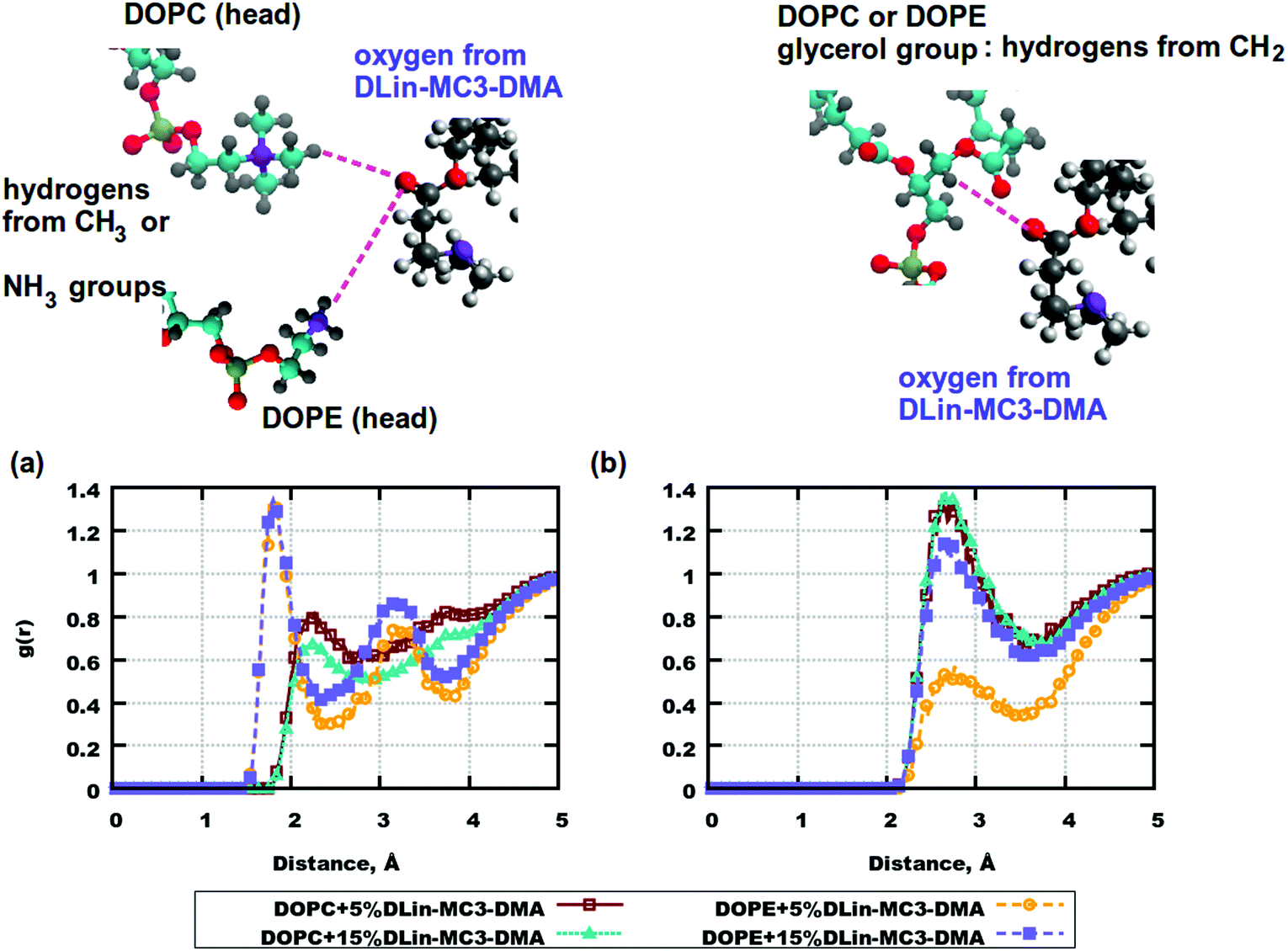 | ||
| Fig. 6 RDFs between pairs of atoms in phospholipid and DLin-MC3-DMA headgroups. (a) RDFs between oxygen from DLin-MC3-DMA and hydrogens from the CH3-group of DOPC/hydrogens from the amine group of DOPE. (b) RDFs between oxygen from DLin-MC3-DMA and hydrogens from the CH2-group of the glycerol group in DOPC/DOPE. Parts of the phospholipids were colored in the following way: cyan color – carbon, blue color – nitrogen, yellow color – phosphorus, red color – oxygen, and gray color – hydrogen. Parts of DLin-MC3-DMA were colored in a different way: light gray color – hydrogen, dark gray – carbon, blue – nitrogen, and red – oxygen. | ||
Another considerable group of RDFs are RDFs between carbonyl oxygens in phospholipid tails and hydrogens in the headgroup of the amine lipid. Fig. S7(a) in the ESI† demonstrates strong associations between hydrogens from the CH-group of DLin-MC3-DMA and the carbonyl oxygens in the phospholipids. However, this association is rather weak for the system containing DOPE and 5% of the amine lipid.
Fig. S7(b–d) (ESI†) presents significant peaks for the pair of atoms labeled as “b”, “c” and “d”, but taking into account the chemical structure of DLin-MC3-DMA it can be concluded that these peaks are likely consequence of the associations illustrated in Fig. S7(a) (ESI†) or even Fig. 6(a).
From computations for lipid headgroups follows that interactions between them are not strong enough to be considered as possible hydrogen bonding. This leaves a question about possible strong associations involving lipid tails. RDFs between lipid tails were calculated for all combinations of pairs of atoms, but no significant peaks were observed. However, the lipid tails of DLin-MC3-DMA were found to be associated with the headgroups of DOPE. Fig. 7 shows RDFs between hydrogens from amine and methyl groups of phospholipids and carbons from the tails of the amine lipid. High peaks can be observed for carbons on positions (b) and (c) at a distance of about 2.1 Å. In the case of DOPC, RDFs between hydrogens in the methyl groups and carbons on the DLin-MC3-DMA tails show peaks as well, but the distance is shifted to about 2.5 Å and the peak value is significantly lower.
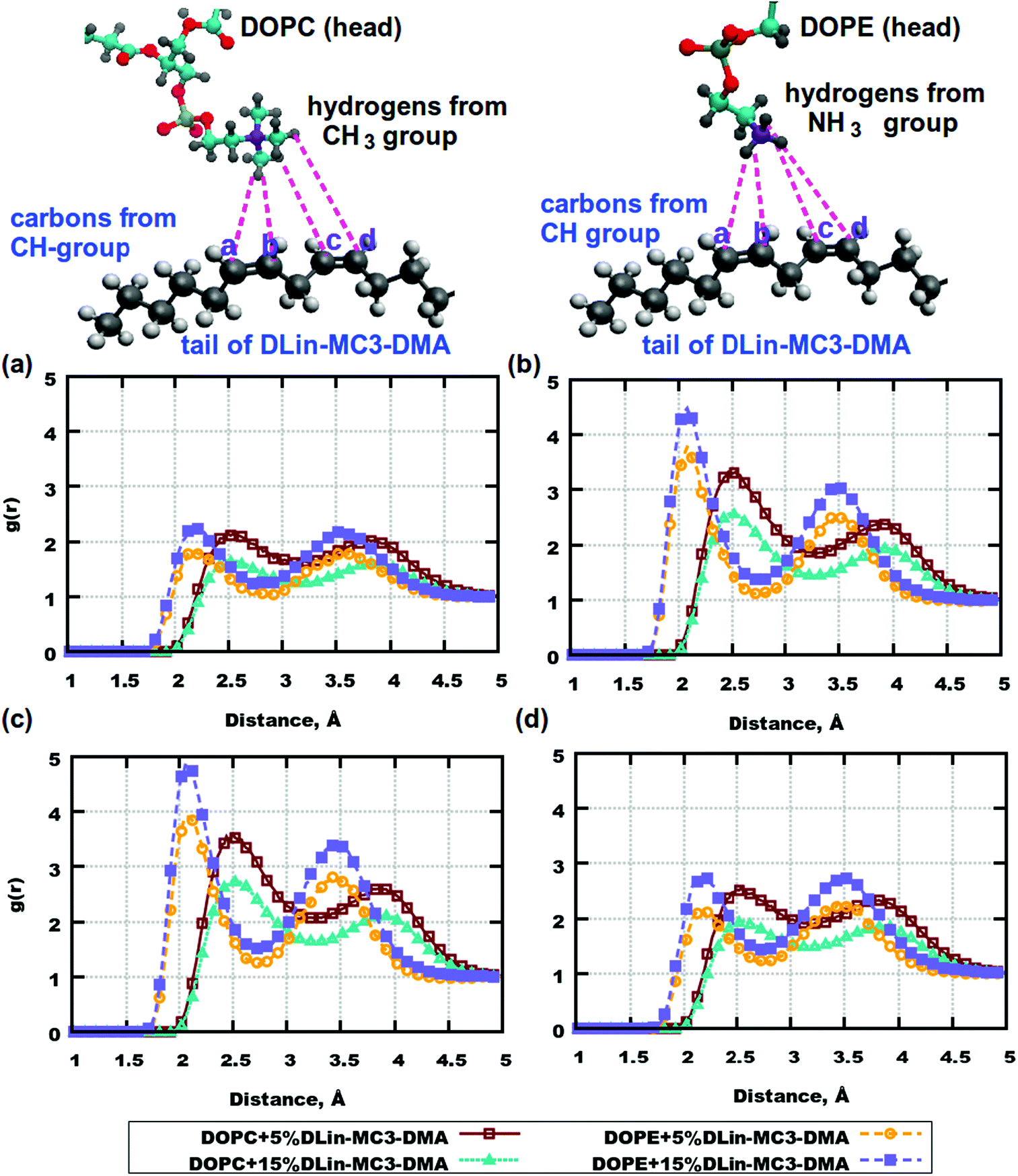 | ||
| Fig. 7 RDFs between pairs of atoms in phospholipid headgroups and in the tail of DLin-MC3-DMA. (a) RDFs between carbons labeled as “a” in DLin-MC3-DMA and hydrogens from the CH3-group of DOPC/hydrogens from the amine group of DOPE. (b) RDFs between carbons labeled as “b” in DLin-MC3-DMA and hydrogens from the CH3-group of DOPC/hydrogens from the amine group of DOPE. (c) RDFs between carbons labeled as “c” in DLin-MC3-DMA and hydrogens from the CH3-group of DOPC/hydrogens from the amine group of DOPE. (d) RDFs between carbons labeled as “d” in DLin-MC3-DMA and hydrogens from the CH3-group of DOPC/hydrogens from the amine group of DOPE. Parts of the phospholipids were colored in the following way: cyan color – carbon, blue color – nitrogen, yellow color – phosphorus, red color – oxygen, and gray color – hydrogen. Parts of DLin-MC3-DMA were colored in a different way: light gray color – hydrogen, dark gray – carbon, blue – nitrogen, and red – oxygen. | ||
The DOPC headgroup associates rather with tails of DLin-MC3-DMA, through hydrogens in the CH2-group. Fig. S8 in the ESI† demonstrates strong associations between hydrogens from the CH2-group in DOPC and carbons in positions (b) and (c) of DLin-MC3-DMA. In the case of DOPE the peak appears at a distance of 3 Å, which can occur due to an association with the NH3-group (Fig. 7).
RDFs between phospholipids and DLin-MC3-DMA give an idea about associations between these compounds and unveil the mechanisms of the proton transfer disruption in membranes with DOPE and the amine lipid. However, there is still a missing variable which could help to clarify the structures of membranes. This variable is the association between the amine lipids themselves. Fig. S9 in the ESI† demonstrates the strongest associations between the headgroups of the amine lipids which occur at distances of 2.4–3.4 Å. Comparing the values of RDFs between the amine lipids with RDFs between DLin-MC3-DMA and phospholipids it can be concluded that the amine lipids have a stronger tendency of aggregating with themselves than associating with DOPC or DOPE. The concentration of DLin-MC3-DMA affects its aggregation as well: RDFs have lower values at 15 mol% of the amine lipid than at 5 mol% (Fig. S9 in the ESI†). The reason for higher values of RDFs at the lower concentration of DLin-MC3-DMA is likely that the function was normalized to the average density of the lipid which is lower at 5 mol% than at 15 mol%, and therefore a given correlation becomes less weighted at a higher concentration of the amine lipid.
Finalizing this discussion we can conclude that there is valuable information about interactions occurring in LNPs by DLin-MC3-DMA and phospholipids. At 5 mol% of the amine lipid there is a slight structural difference between membranes with DOPC and DOPE, while at 15 mol% of DLin-MC3-DMA one can see clear discrepancies in lipid bilayers formed by each phospholipid together with the amine lipid. In membranes with DOPE the structure of domains appears as a hydrophobic net of DLin-MC3-DMA on the surface, which is formed by strong head-to-head aggregations between the amine lipids, weaker head-to-head associations between DOPE and DLin-MC3-DMA and relatively strong associations between the tails of the amine lipid and the DOPE headgroup. In the case of lipid bilayers with DOPC one can probably observe a “mixture” of different domains, where the amine lipids can be partitioned even in the center of the membrane.
3.3 Well-tempered metadynamics simulations
The resulting averaged and symmetrized potential of mean force (PMF) profiles are presented in Fig. 8. During the integration of files the minimum of each PMF was set to zero, which is seen on profiles as the area for the location of the water layer. The curve representing PMF for the lipid bilayer containing DOPE and 5% of DLin-MC3-DMA has the lowest free energy value in the bilayer center among all, which can imply that water will more likely penetrate this membrane than other ones. The highest values of PMF are observed for the membranes containing DOPC with 5% of DLin-MC3-DMA and DOPE with 15% of DLin-MC3-DMA, which could mean that these two lipid bilayers are least permeable.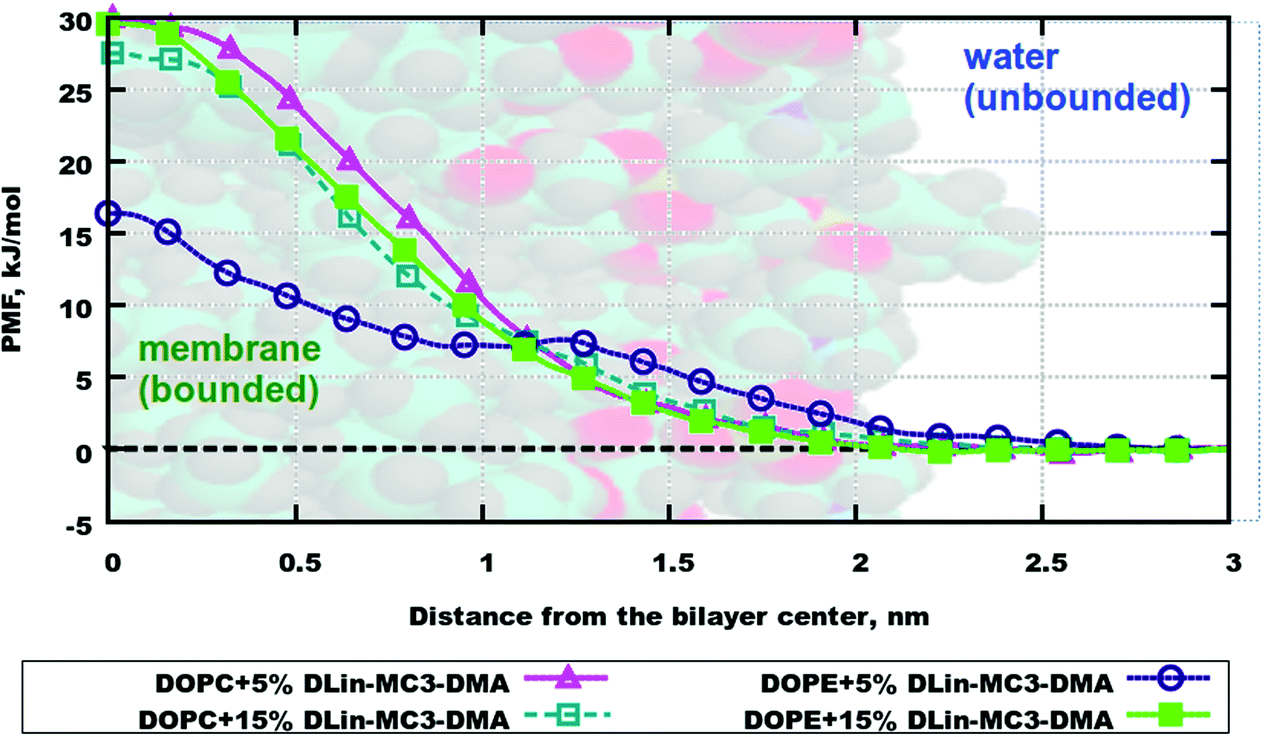 | ||
| Fig. 8 Averaged potential of mean force (PMF) profiles for 4 lipid bilayers. Values of PMF in the bilayer center demonstrate which membrane water would penetrate best (the smaller value means a better penetration). | ||
However, regardless of the fact that PMF can visualize the thermodynamics of a process very well, the actual value of the free energy can give more idea about the possibility for the process to proceed spontaneously. In the case of simulated systems the binding free energy value is the thermodynamic characteristics, which is showing if the process of water penetration through a membrane can go spontaneously. It can be computed according to the following eqn (7):
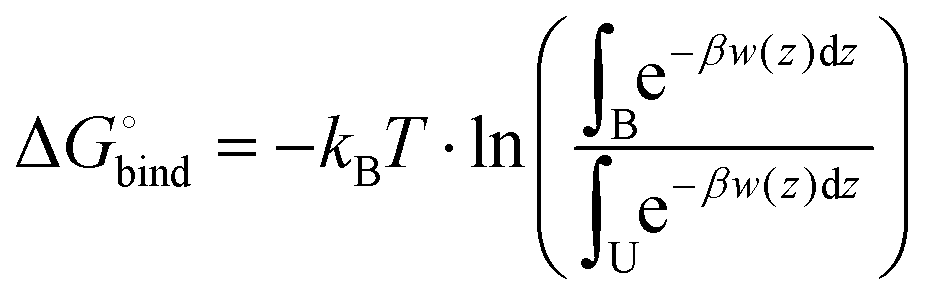 | (7) |
Table 4 shows binding free energies for a water molecule in all simulated systems. All integrals were calculated according to mass density profiles determined from the analysis of classical MD simulations (“bound” and “unbound” states). From the energetic point of view the lipid bilayer containing DOPE and 5% of DLin-MC3-DMA appears to be the most permeable, while all other membranes seem to be the least permeable from the similarity of PMF profiles and values of binding free energies. These findings are coherent with observations from classical MD simulations where the water penetration through membranes was detected from mass density profiles (Fig. S10–S17 presenting the information about the evolution of CVs and convergence are shown in the ESI.†)
| System | Binding free energy, kJ mol−1 |
|---|---|
| DOPC + 5%DLin-MC3-DMA | 2.41 ± 0.12 |
| DOPC + 15%DLin-MC3-DMA | 2.13 ± 0.12 |
| DOPE + 5%DLin-MC3-DMA | 0.14 ± 0.10 |
| DOPE + 15%DLin-MC3-DMA | 2.44 ± 0.12 |
Nevertheless, for proper investigations of the water penetration through a membrane the free energy is not the only factor which shall be taken into consideration: characteristics such as the diffusion of water molecules inside the bilayers and the membrane thicknesses82,83 are of importance. Although the latter ones can be determined, computation of diffusion for a single water molecule is rather a complex process. The reason is the differences in diffusion of water in various locations in systems (in water, by the membrane surface, between lipid tails or in the center of the membrane) and the compositions of the lipid bilayers.83 Therefore, we provide only thermodynamic characteristics of the water penetration process in this work.
4 Conclusions
In this work a neutral model for the amine lipid was derived and the atomistic MD simulations were carried out employing the new model.Regardless of the fact that our membranes were very simple they still give the information about certain domains formed in the shell of complex LNPs.11,84 Taking into consideration the structural information obtained from computed properties one can conclude that the amine lipid forms aggregates in DOPC lipid bilayers at a concentration of 15 mol% of DLin-MC3-DMA, while at the same concentration in DOPE the structures create hydrophobic nets on the surface of the membrane: between the phosphate and carbonyl groups of DOPE. These nets are in the case of our simulations seen as causes of a slow lateral diffusion at higher concentrations of the amine lipid and the pore formation in the DOPE membrane with the lowest content of DLin-MC3-DMA, probably, due to the proton transfer disruption around the phospholipid headgroups.79–81 However, regarding the proton transfer disruption we can only speculate out of scientific findings made by those who investigated similar problems using experimental or computational methods. It can be concluded that such a behavior of DLin-MC3-DMA in membranes with DOPE can be the reason why LNPs composed of these lipids have bad transfection properties.17
Nevertheless, considering only the fusogenic properties of LNPs might not be the best approach, because these vehicles interact with cell membranes which might have “incompatible” fusogenic properties. According to Sayers et al.85 one shall investigate the properties of cell membranes when working on the delivery of nucleic acids. Moreover employing more than one helper lipid in LNPs can also improve their properties. Epaxal®86,87 for treating hepatitis A and Inflexal V®88,89 for treating an influenza were developed and approved in the end of the twentieth century. Both drugs are known to be based on a mixture of DOPC and DOPE.90 MD simulations of lipid bilayers containing more than one helper lipid together with the amine lipid are considered for the future work together with more complex systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Swedish Research Council (Vetenskapsrådet), grant no. 2017-06716. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC). In the National Supercomputer Center (NSC) Tetralith cluster was employed for calculations through projects SNIC2019/3-280, SNIC2019/3-553 and SNIC2019/7-36. In High Performance Computing Center North (HPC2N) Kebnekaise cluster was used for simulations with the project numbers SNIC2019/5-74 and SNIC2020/5-45 and the storage was given in terms of projects SNIC2020/10-22 and SNIC2020/6-53. In Chalmers Centre for Computational Science and Engineering (C3SE) Hebbe and Vera clusters were utilized for calculations in projects SNIC/2018/3-490, SNIC2019/3-53, and C3SE/2020-1-15 with the storage given from projects SNIC2020/6-12 and C3SE605/17-3. We are grateful to Henrik Grönbeck from Chalmers University of Technology for giving the access to projects C3SE/2020-1-15 and C3SE605/17-3. Thanks to Jennifer Gilbert and Tommy Nylander from Lund University for the inspiration to work on this project.References
- C. Li, T. Li, L. Huang, M. Yang and G. Zhu, Chem. – Asian J., 2019, 14, 1570–1576 CrossRef CAS.
- N. Veiga, Y. Diesendruck and D. Peer, Adv. Drug Delivery Rev., 2020 DOI:10.1016/j.addr.2020.04.002.
- M. D. Benson, N. R. Dasgupta and B. P. Monia, Neurodegen. Dis. Manag., 2019, 9, 25–30 CrossRef.
- M. M. Wen, Front. Mol. Neurosci., 2016, 9, 129 Search PubMed.
- M. Meyer, E. Huang, O. Yuzhakov, P. Ramanathan, G. Ciaramella and A. Bukreyev, J. Infect. Dis., 2018, 217, 451–455 CrossRef CAS.
- C. Liu, Q. Zhou, Y. Li, L. V. Garner, S. P. Watkins, L. J. Carter, J. Smoot, A. C. Gregg, A. D. Daniels and S. Jervey, et al. , ACS Cent. Sci., 2020, 6, 315–331 CrossRef CAS.
- W.-H. Chen, U. Strych, P. J. Hotez and M. E. Bottazzi, Curr. Trop. Med. Rep, 2020, 1–4 Search PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS.
- S. Patel, R. C. Ryals, K. K. Weller, M. E. Pennesi and G. Sahay, J. Controlled Release, 2019, 303, 91–100 CrossRef CAS.
- J. Zhang, R. M. Haas and A. M. Leone, Anal. Chem., 2012, 84, 6088–6096 CrossRef CAS.
- V. N. Anghel, D. Bolmatov and J. Katsaras, Phys. Rev. E, 2018, 97, 062405 CrossRef CAS.
- A. Soper and K. Edler, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1652–1660 CrossRef CAS.
- P. Sillrén, J. Swenson, J. Mattsson, D. Bowron and A. Matic, J. Chem. Phys., 2013, 138, 214501 CrossRef.
- M. Scheideler, I. Vidakovic and R. Prassl, Chem. Phys. Lipids, 2020, 226, 104837 CrossRef CAS.
- P. P. Guimaraes, R. Zhang, R. Spektor, M. Tan, A. Chung, M. M. Billingsley, R. El-Mayta, R. S. Riley, L. Wang and J. M. Wilson, et al. , J. Controlled Release, 2019, 316, 404–417 CrossRef CAS.
- J. A. Kulkarni, J. L. Myhre, S. Chen, Y. Y. C. Tam, A. Danescu, J. M. Richman and P. R. Cullis, Nanomedicine, 2017, 13, 1377–1387 CrossRef CAS.
- J. A. Kulkarni, D. Witzigmann, J. Leung, Y. Y. C. Tam and P. R. Cullis, Nanoscale, 2019, 11, 21733–21739 RSC.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda and J. K. Narayanannair, et al. , Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS.
- X. Zhang, V. Goel and G. J. Robbie, J. Clin. Pharm., 2020, 60(5), 573–585, DOI:10.1002/jcph.1553.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS.
- K. Garber, Alnylam launches era of RNAi drugs, 2018.
- M. Y. Arteta, T. Kjellman, S. Bartesaghi, S. Wallin, X. Wu, A. J. Kvist, A. Dabkowska, N. Székely, A. Radulescu and J. Bergenholtz, et al. , Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3351–E3360 CrossRef CAS.
- E. Beltrán-Gracia, A. López-Camacho, I. Higuera-Ciapara, J. B. Velázquez-Fernández and A. A. Vallejo-Cardona, Cancer Nanotechnol., 2019, 10, 11 CrossRef.
- M. Ramezanpour, M. Schmidt, I. Bodnariuc, J. Kulkarni, S. Leung, P. Cullis, J. Thewalt and D. Tieleman, Nanoscale, 2019, 11, 14141–14146 RSC.
- A. K. Leung, I. M. Hafez, S. Baoukina, N. M. Belliveau, I. V. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, J. Phys. Chem. C, 2012, 116, 18440–18450 CrossRef CAS.
- G. S. Ayton, S. Izvekov, W. G. Noid and G. A. Voth, Curr. Top. Membr., 2008, 60, 181–225 CAS.
- M. L. Berkowitz, Biomembrane Simulations: Computational Studies of Biological Membranes, CRC Press, 2019 Search PubMed.
- S. A. Pandit and H. L. Scott, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 136–148 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef.
- M. Bonomi, A. Barducci and M. Parrinello, J. Comput. Chem., 2009, 30, 1615–1621 CrossRef CAS.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 1–13 Search PubMed.
- M. Invernizzi and M. Parrinello, J. Phys. Chem. Lett., 2020, 11, 2731–2736 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, et al. , J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- I. Ermilova and A. P. Lyubartsev, J. Phys. Chem. B, 2016, 120, 12826–12842 CrossRef CAS.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Phys. Chem. B, 2012, 116, 3164–3179 CrossRef.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Chem. Theory Comput., 2013, 9, 774–784 CrossRef.
- L. Goerigk, A. Karton, J. M. Martin and L. Radom, Phys. Chem. Chem. Phys., 2013, 15, 7028–7031 RSC.
- A. R. Ford, T. Janowski and P. Pulay, J. Comput. Chem., 2007, 28, 1215–1220 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- P. J. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,. J. B. Foresman Farkas, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian-09 Revision D.01, 2009 Search PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollman, J. Am. Chem. Soc., 2002, 115, 9620–9631 CrossRef.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- J. Tomasi, B. Mennucci and E. Cances, THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- C. S. Pomelli, J. Tomasi and V. Barone, Theor. Chem. Acc., 2001, 105, 446–451 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- J. B. Klauda, S. L. Garrison, J. Jiang, G. Arora and S. I. Sandler, J. Phys. Chem. A, 2004, 108, 107–112 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS.
- J. B. Klauda, V. Monje, T. Kim and W. Im, J. Phys. Chem. B, 2012, 116, 9424–9431 CrossRef CAS.
- J. B. Klauda, J. Chem. Phys., 2018, 149, 220901 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- H. J. C. Berendsen, D. van der Spoel and R. Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. F. M. Sciacca, F. Lolicato, G. D. Mauro, D. Milardi, L. D'Urso, C. Satriano, A. Ramamoorthy and C. La Rosa, Biophys. J., 2016, 111, 140–151 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- M. Deighan, M. Bonomi and J. Pfaendtner, J. Chem. Theory Comput., 2012, 8, 2189–2192 CrossRef CAS.
- P. Cullis, FEBS Lett., 1976, 70, 223–228 CrossRef CAS.
- E. Falck, M. Patra, M. Karttunen, M. T. Hyvönen and I. Vattulainen, Biophys. J., 2004, 87, 1076–1091 CrossRef CAS.
- A. Oshima, H. Nakashima and K. Sumitomo, Langmuir, 2019, 35, 11725–11734 CrossRef CAS.
- P. Fahey, D. Koppel, L. Barak, D. Wolf, E. Elson and W. Webb, Science, 1977, 195, 305–306 CrossRef CAS.
- C. M. Gee, A. C. Watkinson, J. A. Nicolazzo and B. C. Finnin, J. Pharm. Sci., 2014, 103, 909–919 CrossRef CAS.
- W. Qiang, Y. Sun and D. P. Weliky, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15314–15319 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- A. Filippov, G. Orädd and G. Lindblom, Langmuir, 2003, 19, 6397–6400 CrossRef CAS.
- B. A. Camley, M. G. Lerner, R. W. Pastor and F. L. Brown, J. Chem. Phys., 2015, 143, 12B604_1 CrossRef.
- R. M. Venable, H. I. Ingólfsson, M. G. Lerner, B. S. Perrin Jr, B. A. Camley, S. J. Marrink, F. L. Brown and R. W. Pastor, J. Phys. Chem. B, 2017, 121, 3443–3457 CrossRef CAS.
- A. Blume, Curr. Opin. Colloid Interface Sci., 1996, 1, 64–77 CrossRef CAS.
- S. Hollan, Haematologia, 1996, 27, 109–127 CAS.
- J. Nagle, Biophys. J., 1993, 64, 1476–1481 CrossRef CAS.
- D. M. Engelman, Nature, 1969, 223, 1279–1280 CrossRef CAS.
- J. B. Klauda, N. Kučerka, B. R. Brooks, R. W. Pastor and J. F. Nagle, Biophys. J., 2006, 90, 2796–2807 CrossRef CAS.
- M. Brändén, T. Sandén, P. Brzezinski and J. Widengren, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19766–19770 CrossRef.
- T. Yamashita and G. A. Voth, J. Phys. Chem. B, 2010, 114, 592–603 CrossRef CAS.
- A. Chernyshev and S. Cukierman, Biophys. J., 2006, 91, 580–587 CrossRef CAS.
- R. M. Venable, A. KraÌĹmer and R. W. Pastor, Chem. Rev., 2019, 119, 5954–5997 CrossRef CAS.
- S.-J. Marrink and H. J. Berendsen, J. Phys. Chem., 1994, 98, 4155–4168 CrossRef CAS.
- F. A. Heberle, R. S. Petruzielo, J. Pan, P. Drazba, N. Kučerka, R. F. Standaert, G. W. Feigenson and J. Katsaras, J. Am. Chem. Soc., 2013, 135, 6853–6859 CrossRef CAS.
- E. Sayers, S. Peel, A. Schantz, R. England, M. Beano, S. Bates, A. Desai, S. Puri, M. Ashford and A. Jones, Mol. Ther., 2019, 27, 1950–1952 CrossRef CAS.
- B. R. Holzer and M. Egger, Curr. Opin. Infect. Dis., 1995, 8, 186–190 CrossRef.
- P. A. Bovier, Expert Rev. Vaccines, 2008, 7, 1141–1150 CrossRef CAS.
- R. Mischler and I. C. Metcalfe, Vaccine, 2002, 20, B17–B23 CrossRef CAS.
- C. Herzog, I. C. Metcalfe and U. B. Schaad, Vaccine, 2002, 20, B24–B28 CrossRef.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, tables showing density profiles, radial distribution functions, and average values of torsional angles. See DOI: 10.1039/d0cp05111j |
| This journal is © the Owner Societies 2020 |