
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-to-ultraviolet (<340 nm) photon upconversion by triplet–triplet annihilation in solvents†
Yoichi
Murakami
 *ab,
Ayumu
Motooka‡
a,
Riku
Enomoto
a,
Kazuki
Niimi
c,
Atsushi
Kaiho
c and
Noriko
Kiyoyanagi
c
*ab,
Ayumu
Motooka‡
a,
Riku
Enomoto
a,
Kazuki
Niimi
c,
Atsushi
Kaiho
c and
Noriko
Kiyoyanagi
c
aSchool of Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: murakami.y.af@m.titech.ac.jp
bPRESTO, JST, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cNippon Kayaku Co., Ltd., 3-31-12 Shimo, Kita-ku, Tokyo 115-8588, Japan
First published on 11th November 2020
Abstract
In this article, visible-to-ultraviolet photon upconversion (UV-UC) by triplet–triplet annihilation in the emission range shorter than 340 nm, which has not been explored well, is presented and the relevant physicochemical characteristics are elucidated. Investigations were carried out in several deaerated solvents using acridone and naphthalene derivatives as a sensitizer and emitter, respectively. Both upconversion quantum efficiency and sample photostability under continuous photoirradiation strongly depended on the solvent. The former dependence is governed by the solvent polarity, which affects the triplet energy level matching between the sensitizer and emitter because of the solvatochromism of the sensitizer. To elucidate the latter, first we investigated the photodegradation of samples without the emitter, which revealed that the sensitizer degradation rate is correlated with the difference between the frontier orbital energy levels of the sensitizer and solvent. Inclusion of the emitter effectively suppressed the degradation of the sensitizer, which is ascribed to fast quenching of the triplet sensitizer by the emitter and justifies the use of ketonic sensitizers for UV-UC in solvents. A theoretical model was developed to acquire insight into the observed temporal decays of the upconverted emission intensity under continuous photoirradiation. The theoretical curves generated by this model fitted the experimental decay curves well, which allowed the reaction rate between the emitter and solvent to be obtained. This rate was also correlated with the difference between the frontier orbital energy levels of the emitter and solvent. Finally, based on the acquired findings, general design guidelines for developing UV-UC samples were proposed.
Introduction
Photon upconversion (UC) is a technology to convert presently wasted sub-bandgap photons into those with higher energies (i.e., light of shorter wavelength), which are useful in many fields including photovoltaics and photocatalysis. To date, UC using triplet–triplet annihilation (TTA) between organic molecules has been widely explored because of its applicability to low-intensity and non-coherent light.1–5 Most of the previous studies focused on visible-to-visible UC.1–40 If TTA-UC technology can be reliably extended to the ultraviolet (UV) region (<400 nm), it will become suitable for a broader range of applications, such as for hydrogen generation by water splitting using anatase titanium dioxide (α-TiO2), which has a band gap of 3.2 eV (λgap ∼ 385 nm).41Since the pioneering studies by Castellano and co-workers42,43 and Merkel and Dinnocenzo,44 there have been multiple reports45–52 exploring UC of visible light to UV light (UV-UC). Here, the principle of TTA-UC is briefly described (Fig. 1a). First, a sensitizer molecule absorbs a low-energy photon (visible photon in this context) and transforms to the excited singlet (S1) state, which immediately converts to the triplet (T1) state with a certain quantum yield through intersystem crossing. If the energy of the T1 state of the emitter is similar to or lower than that of the sensitizer, the T1 energy of the sensitizer can be transferred to the emitter (triplet energy transfer; TET), creating a T1 emitter (Fig. 1b). When two T1 emitters interact and undergo TTA, an S1 emitter can be generated from which an upconverted photon (UV photon in this context) is emitted as delayed fluorescence.
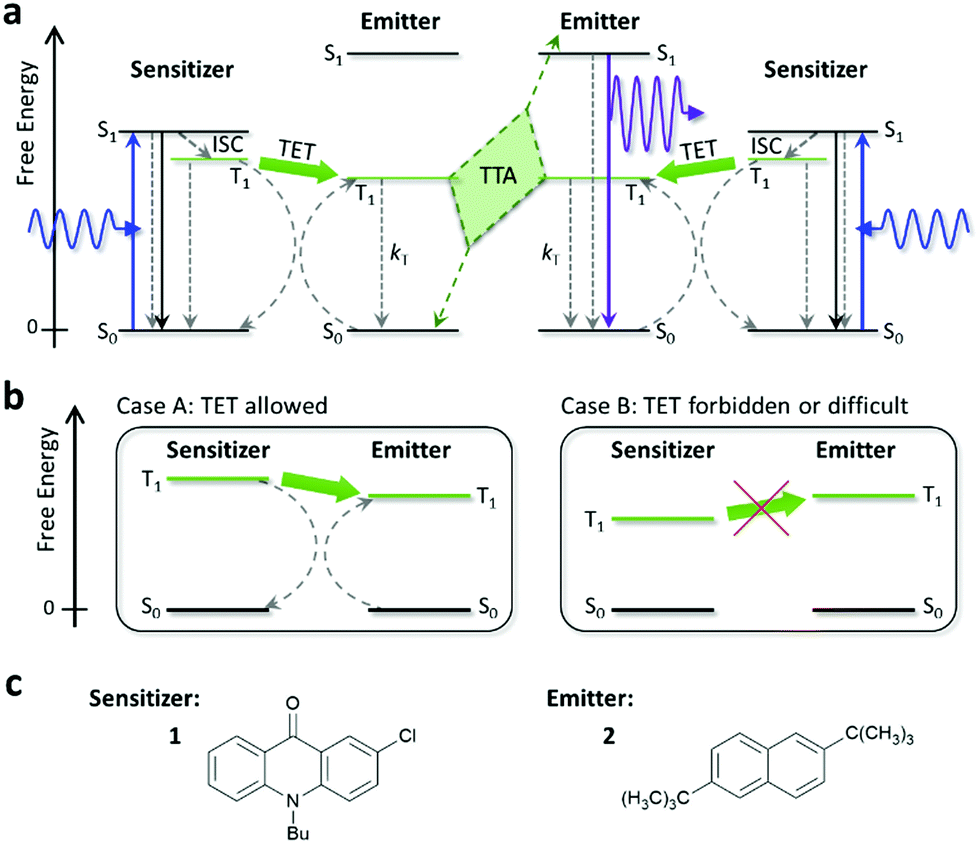 | ||
| Fig. 1 (a) Schematic diagram of the process of TTA-UC. ISC, TET, and TTA mean intersystem crossing, triplet energy transfer, and triplet–triplet annihilation, respectively. Solid and dashed arrows represent radiative and non-radiative processes, respectively. (b) Schematic depictions of two cases where TET is allowed (Case A) and forbidden or difficult (Case B). (c) Molecular structures of the sensitizer 1 and emitter 2 used in this study. | ||
Most previous UV-UC studies were carried out using pyrene or a derivative, whose UC emission maxima range between ca. 375 and 425 nm,42,45,50 or 2,5-diphenyloxazole (PPO), whose UC emission maxima range between 350 and 400 nm,43,44,46,48,49,51 as the emitter. For PPO, 2,3-butanedione (biacetyl) has often been used as the sensitizer.43,46,49 As far as we surveyed, except for our previously published open patent documents53 on which this study is based and a recent report using iridium complex sensitizers in water,54 both of which were UV-UC to wavelengths shorter than 340 nm, the shortest emission peak wavelength reported for UV-UC is 343–344 nm using terphenyl as the emitter.47,50 Therefore, UV-UC with emission maxima shorter than 340 nm has not been well explored thus far.
Shortening emission wavelengths further is meaningful for the following reasons. First, although λgap of α-TiO2 is ca. 385 nm, which was determined by tangentially extrapolating its absorbance or reflectance spectrum to the horizontal axis,55 a general characteristic of semiconductors is that their absorption coefficient is low near λgap.56 For example, sufficient absorption is attained only below ca. 350 nm in the case of α-TiO2 nanoparticles.55,57 Second, the quantum efficiency of water-splitting photocatalysts increases with the energy of incident photons.58 This present article investigates UV-UC with emission maxima shorter than 340 nm and elucidates the relevant physicochemical characteristics.
However, we have noticed that such UV-UC, whether the samples used in this article or other samples such as those made using biacetyl and/or PPO, is accompanied by non-trivial or sometimes remarkable photodegradation, although such characteristics were not explicitly presented and discussed previously. Only recently, Lee et al.50 showed fast photodegradation caused by continuous photoirradiation at 455 nm in deaerated tetrahydrofuran (THF) when PPO and terphenyl were used as emitters. They showed that, among the emitters tested, only pyrene exhibited satisfactory photostability in deaerated THF.50
Previously, we reported visible-to-visible UC in systems using an ionic liquid as the solvent.16,21–23,28 These samples, when properly sealed, exhibited excellent photostability and their lifetime exceeded several years (Section S1 of the ESI†). However, when the same ionic liquid was combined with the sensitizer and emitter used in the present study for UV-UC (Fig. 1c), such photostability was not observed (Fig. S1, ESI† and also below). We also found that the combination of biacetyl and PPO in deaerated dimethylformamide (DMF), which were used previously,46,49 showed poor stability under continuous photoirradiation (Fig. S2, ESI†).
Based on these observations, we consider that UV-UC at wavelengths shorter than ca. 370 nm tends to suffer from low photostability, presumably because the use of high-energy triplet states may induce photochemical reactions, such as hydrogen abstraction from the solvent. This is an unaddressed issue that should be investigated before UV-UC technology is used in applications. Therefore, it is important to obtain understanding of the governing factors and/or mechanism of such photodegradation in UV-UC.
In this study, based on our previous technological findings regarding UV-UC,53 we develop UV-UC samples that exhibit photoemission peaks in the 320–340 nm range. We find that both the UC quantum efficiency (ΦUC) and photostability of these samples depend on the solvent. To understand this phenomenon, we conduct a systematic investigation by performing both experiments and theoretical analysis. The aim of this article is to elucidate the factors governing such solvent dependence and obtain general guidelines for designing UV-UC systems with high UC efficiency and photostability.
Experimental
We used 10-butyl-2-chloro-9(10H)-acridinone (1) and 2,6-di-tert-butylnaphthalene (2) as the sensitizer and emitter, respectively (Fig. 1c). Both 1 and 2 (purity: >98%) were purchased from TCI; 1 was recrystallized before use and 2 was used as received. We chose 1, in which the photoexcitation is the n→π* transition, because the small overlap between the n and π* orbitals around its carbonyl group leads to a small S1–T1 energy gap and the n, π* state has a high quantum yield of S1-to-T1 intersystem crossing (ΦT,sen),59 both of which are desirable for sensitizers for TTA-UC. After testing several acridones, we found that 1 was preferable over the other candidates because of its visible absorption in the 400–425 nm range (Fig. S3, ESI†) and ability to undergo TET with naphthalenes. We chose 2 because of its relatively high fluorescence quantum yield and suitable fluorescence spectrum for the purpose of this study.Samples were prepared using the solvents listed in Table 1. Details of the solvents are given in Table S1 in the ESI.† We included D-limonene because it has been reported to prevent degradation of solutes in visible-to-visible UC by functioning as a strong antioxidant that quickly scavenges residual oxygen.60 Additionally, in the former half of this study, we included the ionic liquid [C4dmim][NTf2] as a reference solvent because it enables highly stable red-to-blue UC16,21–23 (Fig. S1, ESI†). Throughout this report, the concentrations of 1 and 2 were 2 × 10−4 and 2 × 10−3 M, respectively. A continuous-wave laser with an emission wavelength of 405 nm and spot diameter of 0.8 mm was used as the excitation source unless otherwise specified. The absorption spectra of 1 and 2 are depicted in Fig. S3 in the ESI† and their fluorescence spectra are shown in Fig. 2a.
| Solvent 〈ET(30)〉a | A 405nm | Φ F,sen | Φ T,sen | Φ F,emi | Φ UC/% (Exct. intensity/W cm−2) | k sen,degr /M s−1 | k emi,rxn /s−1 |
|---|---|---|---|---|---|---|---|
| (P′)b | |||||||
| a From ref. 62 unless otherwise specified. b From ref. 63. c N,N-Dimethylformamide. d Measured using Reichardt's dye but no absorption peak was found in the visible to near-infrared region. e Measured using Reichardt's dye. f Absorbance of 1 at 405 nm with an optical path length of 1 mm. g Determined by assuming the Ermolev's rule ΦT,sen = 1 − ΦF,sen. h Excitation intensity where the T1 state of 1 is generated at 1.9 × 10−3 M s−1. i Excitation intensity where the T1 state of 1 is formed at 1.77 × 10−3 M s−1. j Obtained by the procedure described in Section S13 of the ESI. k Obtained from the fit to the experimental decay curves of the UC emission intensity using our model; see Fig. 4c. | |||||||
| Hexane 〈30.9〉 | 0.067 | 0.006 | 0.994 | 0.33 | 4.5 (0.40)h | 7.35 × 10−7 | 5.73 × 10−3 |
| (0.1) | 8.2 (1.75) | — | |||||
| Ethyl acetate 〈38.0〉 | 0.20 | 0.274 | 0.726 | 0.39 | 1.9 (0.20)h | 2.35 × 10−6 | 1.92 × 104 |
| (4.4) | 4.9 (1.74) | — | |||||
| Toluene 〈33.9〉 | 0.23 | 0.191 | 0.809 | 0.57 | 2.2 (0.17)h | 1.56 × 10−5 | 4.47 × 10−1 |
| (2.4) | |||||||
| Acetonitrile 〈45.6〉 | 0.15 | 0.598 | 0.402 | 0.43 | 0.38 (0.50)h | 4.09 × 10−7 | 3.29 × 104 |
| (5.8) | |||||||
| DMFc 〈43.2〉 | 0.17 | 0.593 | 0.407 | 0.49 | 0.015 (0.44)h | 1.48 × 10−5 | — |
| (6.4) | |||||||
| D-Limonene 〈N/A〉d | 0.20 | 0.030 | 0.970 | 0.16 | ≅0 (0.15)h | 1.95 × 10−5 | — |
| (N/A) | |||||||
| [C4dmim][NTf2] 〈40.9〉e | 0.15 | 0.575 | 0.425 | 0.58 | 0.25 (0.44)i | 1.15 × 10−6 | — |
| (N/A) | |||||||
| Methanol 〈55.4〉 | 0.16 | 0.657 | 0.343 | 0.37 | ≅0 (0.52)h | — | — |
| (5.1) | |||||||
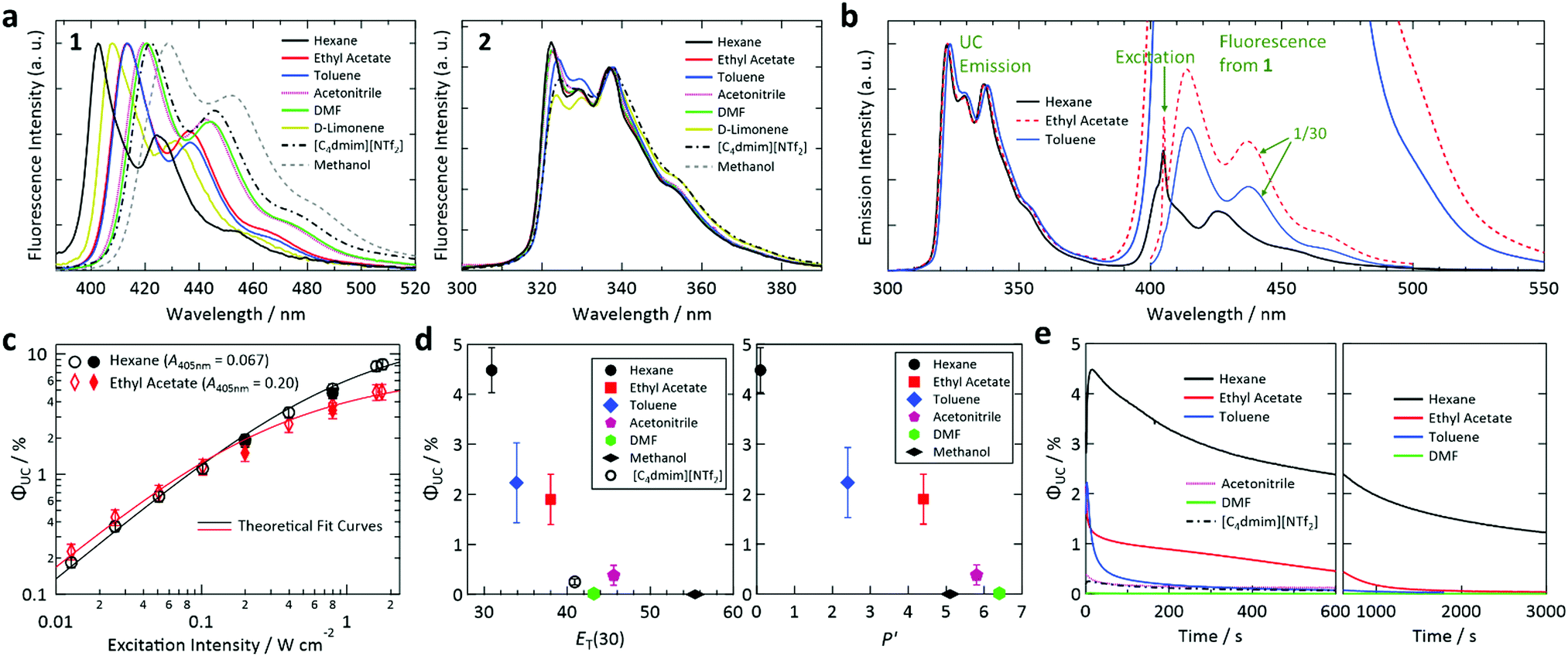 | ||
| Fig. 2 (a) Fluorescence spectra of 1 (left) and 2 (right) in the solvents used in this study, acquired using an absolute quantum yield spectrometer and degassed dilute solutions (concentrations of the order of 10−4–10−5 M). (b) Photoemission spectra of the samples prepared with hexane, ethyl acetate, and toluene acquired upon excitation at 405 nm. (c) Dependence of the upconversion (UC) quantum efficiency of the samples prepared using hexane and ethyl acetate on excitation intensity. Open symbols denote data acquired while increasing excitation intensity and filled symbols represent data acquired with decreasing excitation intensity. (d) UC quantum efficiencies plotted against ET(30) (left) and solvent polarity scale P′ (right). (e) Temporal profiles of UC quantum efficiency, which is proportional to UC emission intensity, measured for samples prepared using different solvents under continuous photoirradiation at 405 nm. In (b, d and e), the excitation light intensity was chosen such that the irradiation generated the T1 state of 1 at a rate of 1.9 × 10−3 M s−1 (1.77 × 10−3 M s−1 for [C4dmim][NTf2]). See the main text for the details. | ||
Except for the sample with [C4dmim][NTf2], all samples, which contained both 1 and 2 or only 1, underwent at least seven (typically eight or nine) freeze–pump–thaw (FPT) cycles using our FPT system to carefully remove dissolved air. Our FPT system consisted of a small glass jar (#33.010007.11A.710, EVAC) with a flange, an O-ring-sealed stainless-steel (SUS) flange coupling to it, and all-SUS vacuum line consisting of a flexible metal hose and Swagelok valves and tube fittings. The vacuum line was connected to an oil-free dry scroll pump (nXDS15i, Edwards) able to attain a vacuum of ca. 1 Pa. To efficiently remove dissolved gas, the volume of liquid in the jar was small (ca. 2 mL). The increase of solute concentration induced by the FPT cycles was negligible for all samples, as confirmed by their unchanged absorbance in UV-vis measurements. Typically, the emergence of bubbles in the liquid ended within three or four FPT cycles, and thus the aforementioned number of FPT cycles was believed to be sufficient. After the FPT cycles, the glass jar coupled with a closed SUS valve was detached from the vacuum line and transferred into a vacuum-type SUS glovebox containing freshly replaced nitrogen gas (purity: >99.998%). In the glovebox, the liquid sample was injected into a square glass capillary (inner dimensions: 1 × 1 mm, outer dimensions: 2 × 2 mm, length: 27 mm) with a closed end. The open top of the capillary was immediately closed with a low-melting-point solder as previously described.16,21–23 The seals were checked by placing the capillary under vacuum for a long period (hours or days); an effective seal was confirmed by the sample volume remaining constant. This sealing method works for at least several years (e.g., the sample in Fig. S1, ESI†). For the sample with [C4dmim][NTf2], oxygen and moisture were removed by stirring the sample, which had a small volume (<600 μL), in an open vial at 60 °C and 200 rpm for 2 h inside a nitrogen-filled glovebox equipped with a gas-purification system (OMNI-LAB, VAC; oxygen and moisture: <1 ppm) before it was sealed in a glass capillary inside the glovebox.
Time-resolved measurements of the UC emission intensity were carried out using nanosecond light pulses generated from an optical parametric oscillator (OPO; NT-242, Ekspla) at 410 nm and 20 Hz. The fluorescence quantum yields of 1 and 2 (ΦF,sen and ΦF,emi, respectively) and their fluorescence spectra were acquired by an absolute quantum yield spectrometer (Quantaurus-QY, Hamamatsu) using a quartz cell (1 × 1 cm); the solute concentration for these measurements (of the order of 10−5 to 10−4 M) was chosen so that the absorptance of each sample in the integrating sphere was between 0.35 and 0.55 at the excitation wavelength.
In reference experiments, photodegradation was controllably induced in a sample using a setup where the excitation laser beam was expanded to irradiate almost the entire volume of a sample liquid (ca. 2 mL) in a hermetically sealed glass vial from below (see Fig. S4 in the ESI† for details). In these experiments, the photoirradiation was continued until each molecule of 1 in the sample turned to the T1 state 85 times on average. The duration of photoirradiation was set by assuming that the initial absorbance of 1 at 405 nm did not change during the course of the irradiation. All photoemission spectra in this report were corrected by the wavelength-dependent sensitivities of the grating in a monochromator and CCD array detector as described in our previous reports.16,21–23
All quantum-chemical simulations were carried out using Gaussian 16® at the B3LYP/6-31G++(d,p) level.
Results and discussion
The fluorescence and absorption spectra of sensitizer 1 exhibited large solvatochromic shifts whereas those of emitter 2 showed small solvatochromic shifts (see Fig. 2a for the fluorescence spectra and Fig. S3 in the ESI† for the optical absorption spectra). This behavior is ascribed to the large (negligible) permanent dipole moment of 1 (2) (Fig. S5, ESI†). Fig. 2b shows photoemission spectra of samples prepared using hexane, ethyl acetate, and toluene upon excitation at 405 nm. The UC emission spectra were structured with the emission maximum at 322 nm and other peaks in the range of 320–340 nm. The photoemission spectra also contained peaks originating from fluorescence from the S1 state of 1 in the 400–500 nm range. The intensity of this fluorescence relative to that of the UC emission varied considerably between samples, which is partially attributed to the difference of ΦF,sen in these solvents (ΦF,sen = 0.006, 0.274, and 0.191 in hexane, ethyl acetate, and toluene, respectively; cf.Table 1). Re-absorption of UC fluorescence from 2 by 1 is weak because of the transmission window of 1 from 310–370 nm (Fig. S6, ESI†).The dependence of ΦUC of the samples with hexane and ethyl acetate on excitation intensity was determined (Fig. 2c). For ΦUC in this article, we customarily describe efficiency in percent and thus the maximum is 100%, which is twice the maximum UC quantum yield of 0.5. The emission intensity between 310 and 380 nm was used to calculate ΦUC; i.e., the emission between 380 and 405 nm was not used to exclude the tail of the fluorescence and thermally induced UC emission. The procedure used to determine ΦUC is described in Section S9 of the ESI.† As shown in Fig. 2c, the samples with hexane and ethyl acetate attained high ΦUC of 8.2% and 4.9%, respectively, at an excitation intensity of ca. 1.75 W cm−2. The data points in Fig. 2c were acquired while first increasing the excitation intensity and then while decreasing the excitation intensity to confirm the reproducibility of the ΦUC values. Although ΦUC measured while decreasing the excitation intensity were slightly lower than those obtained with increasing excitation intensity for both samples, the differences were smaller than the related error bars and thus ΦUC values were considered reproducible.
In Fig. 2c, theoretical curves31 were fitted to the data points. From the fitting, excitation threshold intensities (Ith) for the samples with hexane and ethyl acetate of ca. 1.3 and 0.43 W cm−2, respectively, were obtained. These high Ith values and the solvent dependence of Ith are explained as follows. First, Ith strongly depends on the absorbance of the sample at the excitation wavelength; Ith decreases as the absorbance increases and vice versa.61 The samples with hexane and ethyl acetate exhibited low absorbances of 0.067 and 0.20 (corresponding optical absorption coefficients of 0.154 and 0.461 mm−1), respectively, at the fixed excitation wavelength of 405 nm used in this study. Therefore, the high Ith here mainly arose from the low absorbance of 1 at the excitation wavelength and the solvent dependence mainly originated from the large solvatochromic shift of 1 (cf.Fig. 2a). Besides this factor, the variation of the triplet lifetime (τT) of 2 in different solvents (ca. 114 μs in hexane and 47 μs in ethyl acetate) should also contribute to the solvent dependence of Ith.
We found that ΦUC and photostability strongly depended on the solvent. To systematically compare ΦUC and the rates of photoinduced changes of the samples prepared using different solvents, in the following experiments we set the laser power irradiated onto the sample sealed in a glass capillary (see the Experimental section for details) such that the irradiation generated the T1 state of 1 at a rate of 1.9 × 10−3 M s−1 in the photoirradiation volume (a cylinder with a diameter of 0.8 mm and length of 1 mm). At this rate, each 1 molecule in the volume transitions to the T1 state 9.5 times per second. Note that the rate was 1.77 × 10−3 M s−1 in the sample with [C4dmim][NTf2]. The actual laser power irradiated onto each sample, which was in the range of 0.73–2.6 mW or 0.15–0.52 W cm−2, was determined using the absorbance at 405 nm (A405nm) and ΦT,sen listed in Table 1. The ΦT,sen values were estimated assuming Ermolev's rule of ΦT ≈ 1 − ΦF.59
The determined ΦUC values are plotted against the polarity scales ET(30)62 and P′63 in Fig. 2d. We were unable to determine the ET(30) value for D-limonene because it did not exhibit an absorption peak in the visible to near-infrared range in a solution of Reichardt's dye. No P′ values for D-limonene or [C4dmim][NTf2] were found in the literature. From these plots, we found that ΦUC is correlated with the solvent polarity and decreases as the polarity increases.
As mentioned above, 1 has a large dipole moment (Fig. S5, ESI†) and thus exhibits a large bathochromic shift as the solvent polarity increases, whereas 2 does not (Fig. 2a). Therefore, as the solvent polarity increases, the T1 level of 1 is considered to be lowered relative to that of 2, making TET thermodynamically unfavorable (i.e., Case B in Fig. 1b). The solvent dependence of ΦUC of our samples is mainly attributed to this mechanism, which is supported by our calculation of the dependence of the triplet energies of 1 and 2 on solvent permittivity (Fig. S7, ESI†). In addition, the solvent dependences of ΦT,sen and ΦF,emi (Table 1) should also affect ΦUC.
The stability of the samples under continuous photoirradiation strongly depended on the solvent (Fig. 2e). For example, UC emission in toluene decayed rapidly whereas that in hexane lasted much longer; the reason for this behavior is investigated below. It is noted that no UC emission was observed when D-limonene and methanol were used (Table 1). While the lack of UC emission in methanol can be explained by the above discussion regarding Fig. 2d, the reason for the absence of UC emission in D-limonene is unclear. It may be caused by the high reactivity of D-limonene, which has a reactive unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, with high-energy triplet states of 1 and 2.
C bond, with high-energy triplet states of 1 and 2.
Here, we note the following three points. First, although the use of solvents with different certified purities resulted in a minor but recognizable effect on the intensity of UC emission, this difference did not alter the qualitative profile of the temporal UC emission intensity change (Fig. S8, ESI†). Second, the temporal decays of the UC emission intensity observed in Fig. 2e were not considered to be governed by residual oxygen in the solvents, which was the case in previous visible-to-visible UC studies.64–68 This is partly because the use of D-limonene, which scavenged residual oxygen efficiently and helped to attain stable visible-to-visible UC,60 completely suppressed the UC emission in the present study. That the UC emission decays observed in Fig. 2e were not caused by residual oxygen was also supported by the thorough FPT treatment and tightly sealed samples used here. Third, the decay rate of the UC emission in hexane in the present study is much slower than that of a previously reported biacetyl/PPO/DMF system46,49 when compared using the similar triplet generation rate on 1 (Fig. S2, ESI†).
In the following investigations, we excluded the sample with methanol because it did not realize UC and the sample with [C4dmim][NT2] because its UC efficiency was low and the photochemical reaction with a molten salt is complex.
To understand the solvent-dependent photostability of our samples, first, we investigated samples containing only 1. When each sample in a glass capillary was excited at a triplet generation rate of 1.9 × 10−3 M s−1, the decay rate of the fluorescence intensity of 1 depended on the type of solvent (Fig. 3a and Fig. S9 in the ESI† for the fluorescence intensities and spectra, respectively). We confirmed that the photoirradiation induced a decrease of the absorbance of 1 (Fig. 3b and Fig. S10 in the ESI†), indicating that photoirradiation induced degradation of 1. To compare the degradation rates of 1 in different solvents, we determined the rate of sensitizer degradation ksen,degr [M s−1] from the decay curves in Fig. 3a by applying a double-exponential fit (see Section S13 of the ESI† for the procedure used to calculate ksen,degr).
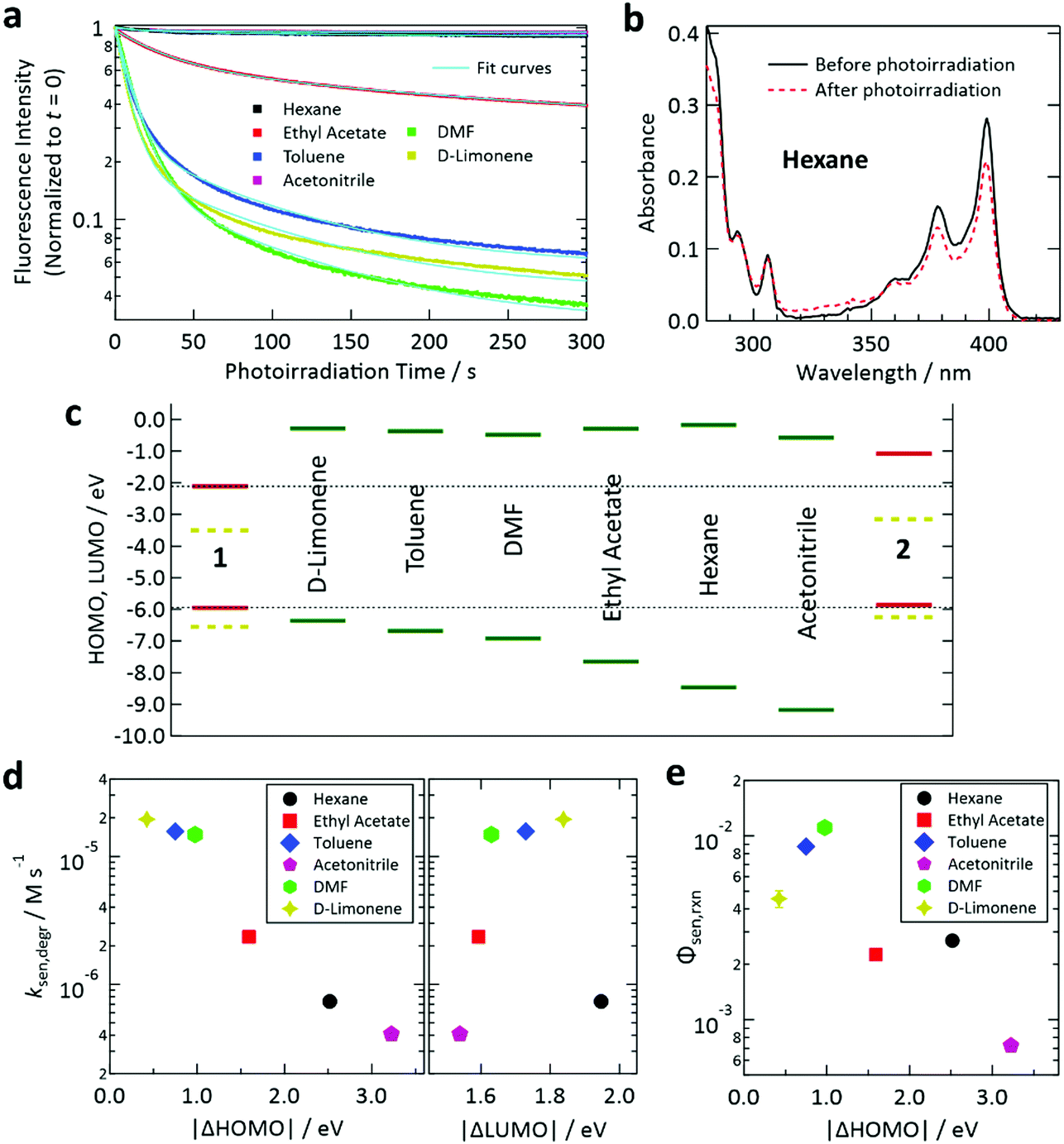 | ||
| Fig. 3 (a) Temporal decay profiles of the fluorescence intensity of deaerated samples containing only sensitizer 1 under continuous irradiation at 405 nm. The excitation light intensity for each case (cf.Table 1) was chosen so that the T1 state of 1 was generated at a rate of 1.9 × 10−3 M s−1. (b) Decrease of the absorbance of 1 in deaerated hexane (optical path length: 1 mm) induced by photoirradiation at 405 nm using the setup and conditions described in Section S5 of the ESI.† (c) Calculated HOMO and LUMO levels of 1, 2, and the solvents. The SOMO levels of the T1 states of 1 and 2 are shown as yellow dashed lines. (d) Degradation rates of fluorescence intensities determined from the results in panel (a) plotted against the difference between the HOMO levels of 1 and the solvent (left) and that between the LUMO levels of 1 and the solvent (right). (e) Reaction quantum yield between the T1 state of 1 and solvent for the samples containing only 1 estimated using the optical absorption changes in Fig. S10 in the ESI.† | ||
Here we use the frontier orbital theory to discuss the observed photoinduced degradation of 1 in the solvents. Generally, excited states of ketones such as the T1 state of 1 have n, π* electronic configuration where n and π* are singly occupied molecular orbitals (SOMOs) and can serve as electron-accepting and -donating orbitals, respectively.59 Generally, such SOMOs interact with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of an adjacent molecule and create new orbitals into which electrons from both molecules are partially or fully transferred; such a charge transfer generally allows energetic stabilization and may lead to formation of an excited-state complex.59 For the n, π* state of ketones, such intermolecular interaction with a ground-state molecule such as a solvent molecule may cause hydrogen abstraction from the latter because of the half-filled orbital on the oxygen atom of the ketone. Hydrogen abstraction by ketones has been widely studied.69–71 Two factors are proposed to govern this intermolecular reaction: (i) the energetic proximity of the frontier orbitals of the two interacting molecules and (ii) the constructive spatial overlap of these orbitals.59
To study factor (i), we calculated the HOMO and LUMO levels of 1, 2, and the solvents, as depicted in Fig. 3c. In this figure, SOMO levels of the T1 states of 1 and 2 are also shown. From the relation between ksen,degr and the energetic separations of the HOMOs and LUMOs between 1 and the solvents (denoted as Δ|HOMO| and Δ|LUMO|, respectively), we found a clear correlation of ksen,degr with Δ|HOMO|, whereas no obvious correlation was found between ksen,degr and Δ|LUMO| (Fig. 3d). The same tendency was also observed when the difference between the ionization energies of 1 and the solvents (which physically corresponds to Δ|HOMO|) and that between their electron affinities (which corresponds to Δ|LUMO|) were plotted (Fig. S11, ESI†). These results reveal that the electron transfer from the solvent to 1 is the rate-limiting step of this photodegradation, which can be interpreted as an electron transfer-initiated hydrogen abstraction process.71,72 We also estimated the quantum efficiency of the degradation of the T1 state of 1 in each solvent (Φsen,rxn) from the decease of the absorbance of 1 induced by the controlled photoirradiation (cf. Fig. S4, ESI†). The procedure followed to calculate Φsen,rxn is described in Section S15 of the ESI.† Although the scatter of the data points is larger than that in the case of ksen,degr, a similar correlation with Δ|HOMO| was also found for Φsen,rxn (Fig. 3e).
Next, we investigated photoinduced changes of samples containing both 1 and 2. For the sample with hexane, photodegradation of 1 was suppressed by the presence of 2 × 10−3 M of 2, as recognized from the invariance of the optical absorption spectrum of 1 during photoirradiation (Fig. 4a). This suppression is ascribed to prompt TET from 1 to 2 in hexane, which drastically shortens the lifetime of the T1 state of 1, meaning that 2 strongly suppresses the probability of 1 reacting with the solvent. A similar tendency was also found for the samples with other solvents (Fig. S10 and S12 in the ESI†). However, for the sample with DMF, the decrease in the absorbance of 1 was not well suppressed (Fig. S12, ESI†); this could be because of inefficient TET from 1 to 2 caused by the relatively high polarity of DMF (cf.Fig. 2d and 1b). The suppressed photodegradation of 1 in hexane induced by addition of 2 was also evidenced by the invariance of the fluorescence emission intensity of 1 even after 80 min of photoirradiation (Fig. 4b); the similar tendency was also seen for the samples with other solvents (see Fig. S9 and S13 in the ESI†).
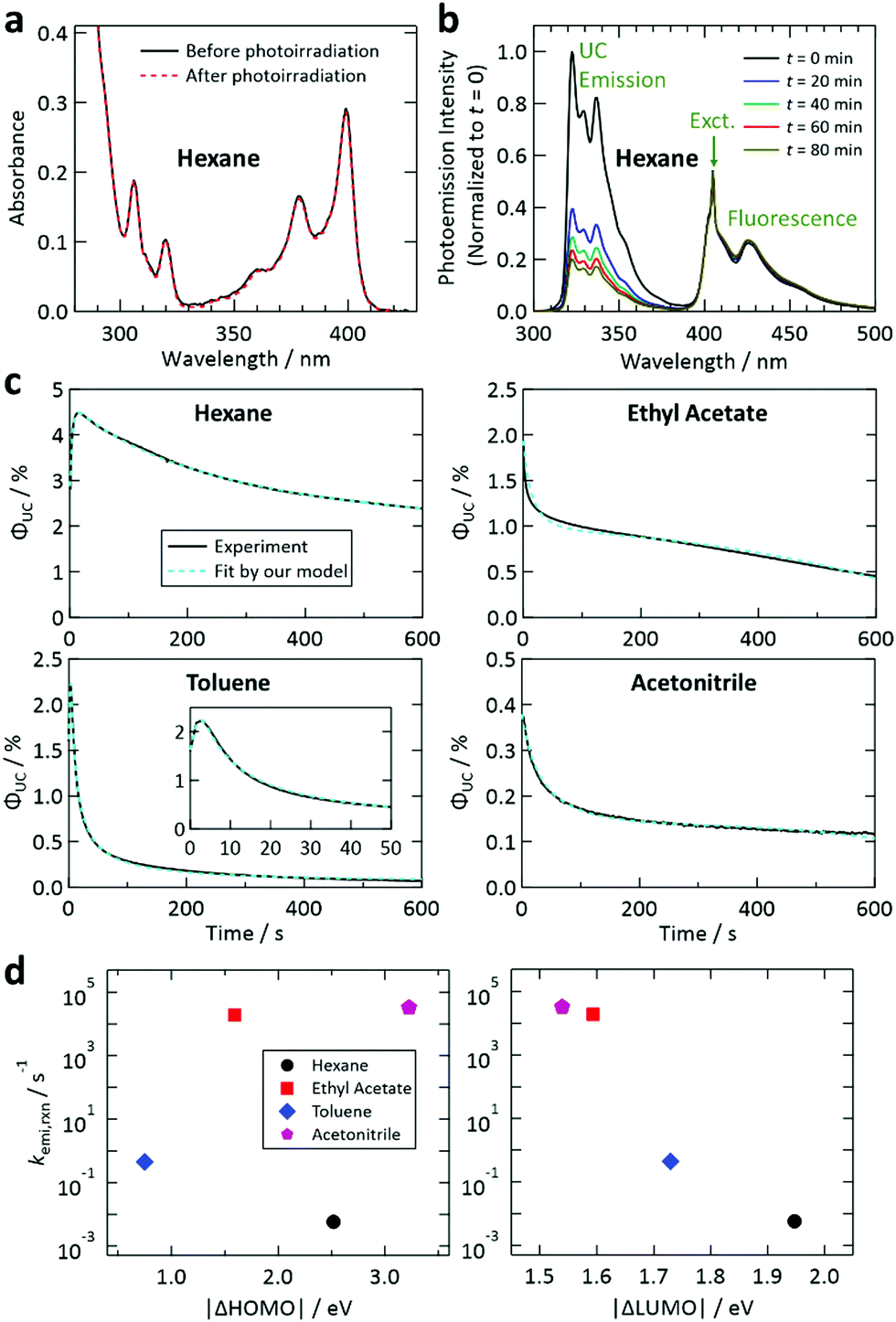 | ||
| Fig. 4 (a) Demonstration of the suppression of the decrease of the absorbance of 1 by addition of 2 in hexane using the same conditions as in Fig. 3b. (b) Demonstration of the suppression of the decrease of the fluorescence of 1 upon addition of 2 in hexane, which was measured using the same conditions as in Fig. 3a. See also Fig. S9 and S13 in the ESI.† (c) Fit of the temporal decay curves of the UC quantum efficiency shown in Fig. 2e (solid curves) by the theoretical model proposed in this study (dashed curves). (d) Reaction rates between the T1 state of 2 and the solvents, obtained from the fittings shown in (c), plotted against the difference between the HOMO levels of 2 and the solvent (left) and that between the LUMO levels of 2 and the solvent (right). | ||
Our results reveal that by adding an energy-accepting emitter at sufficient concentration (of the order of 10−3 M), preferable aspects of ketones as the sensitizer (cf. first paragraph of the Experimental section) can be harnessed for UV-UC while effectively suppressing the drawback of using triplet ketones; i.e., the relatively high reactivity of their T1 state. Considering the viscosities of the solvents employed in this study (ca. 0.3–0.6 mPa s at room temperature), the diffusion-controlled rate constant kdiff was estimated to be 1–2 × 1010 M−1 s−1 using the following equation16,59
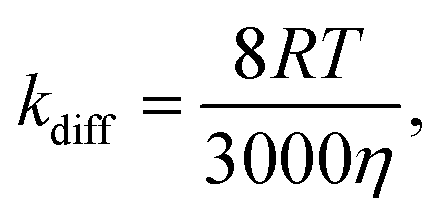 | (1) |
The rate of the reaction between the T1 state of 1 and ground state of 2 was considered to be negligible, even though their ground-state HOMO levels are close (Fig. 3c), for the following reasons. First, a bimolecular reaction rate is proportional to the product of the concentrations of the two species involved. In our samples, the concentration of 2 (2 × 10−3 M) was much lower than that of the solvents (7–20 M). Second, the interaction time between the T1 state of 1 and ground state of 2 should be very short because such an encounter immediately causes an exothermic TET,11 unlike the interaction between the T1 state of 1 and the solvent, which can last much longer.
We have reached the point to discuss the temporal decay curves of the UC emission intensity under continuous photoirradiation in Fig. 2e. To analyze these decay curves, we developed the theoretical model described below. First, we postulate that the triplet emitter (E*) becomes a new species (ε) by reacting with a surrounding solvent molecule (sol) at a rate of kemi,rxn [s−1]. This ε is assumed to quench both E* and the triplet sensitizer (S*) at the kdiff given by eqn (1). Therefore,
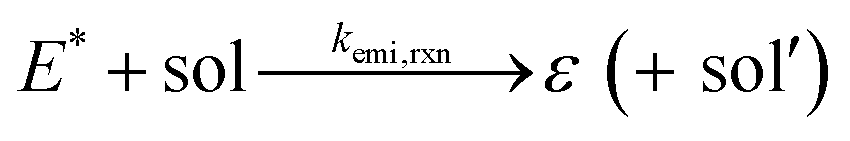 | (2) |
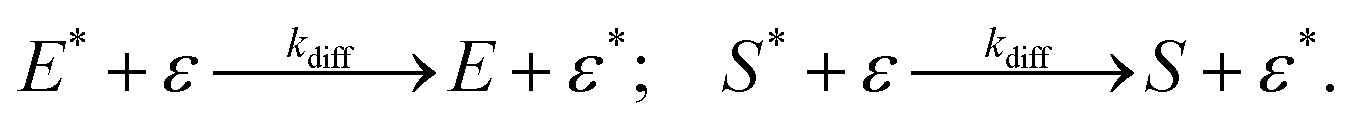 | (3) |
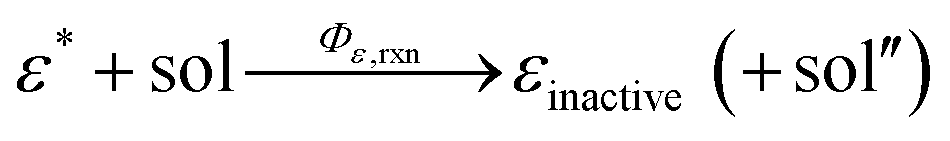 | (4) |
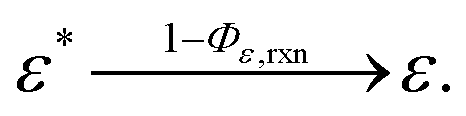 | (5) |
In addition, we consider initial impurity species in the solvent, Q, which quenches both E* and S*. Similar to the case of ε, we introduce the kinetic relations of
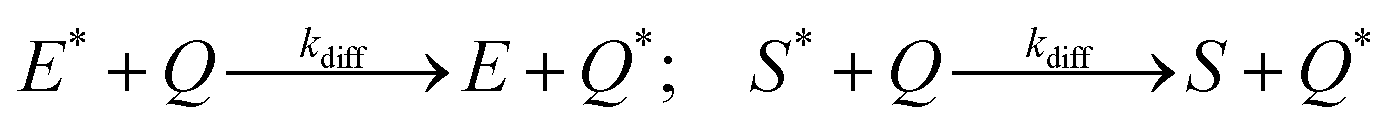 | (6) |
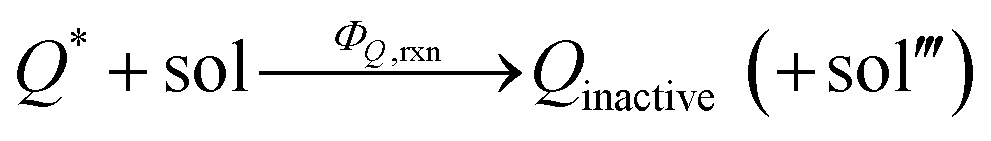 | (7) |
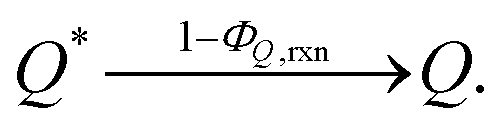 | (8) |
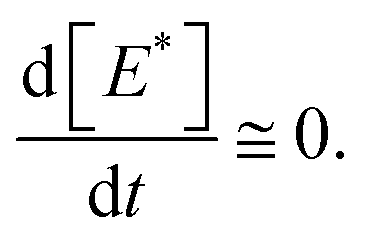 | (9) |
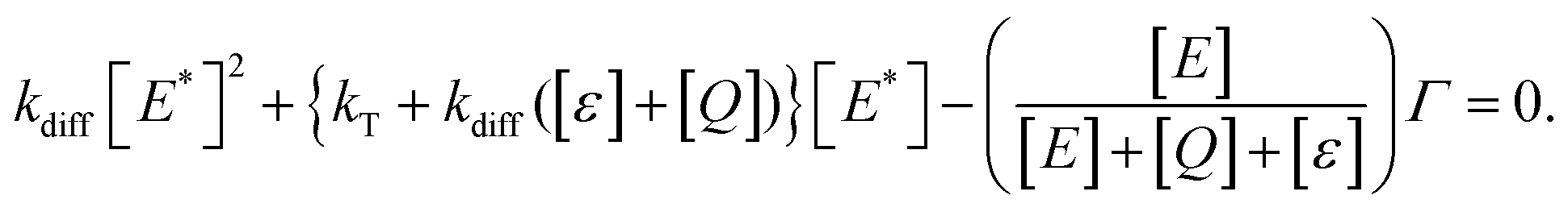 | (10) |
Fig. 4c shows the results of the fittings of this model to the experimental curves for the samples with hexane, ethyl acetate, toluene, and acetonitrile. In all cases, the agreement between the model and experimental curves was good, suggesting that the model has captured the physicochemical characteristics of the present system. All these fittings resulted in Q ≤ 5 × 10−4 M (i.e., molar fraction of 0.005% or lower), which does not contradict the certified purities of the solvents. It is noted that processes represented by eqn (4) and (7) are necessary to fit our theoretical model to the experimental curves.
In Fig. 4d, the values of kemi,rxn obtained from the fitting are plotted against |ΔHOMO| and |ΔLUMO|. It is noted that the magnitude of kemi,rxn, which represents the chemical instability of the triplet emitter, strongly depended on the solvent. In this figure, a correlation was found only for |ΔLUMO| and kemi,rxn. The same tendency was also observed when the difference between the ionization energies of 2 and the solvents and that between the electron affinities of 2 and the solvents were plotted (Fig. S15, ESI†). These results suggest that the process described by eqn (2) is limited by electron transfer from 2 to the solvent; i.e., electron transfer in the opposite direction to that in the reaction between 1 and the solvent discussed above. We did not carry out further detailed investigation of the reaction mechanism because it is beyond the scope of the present study. Nevertheless, the findings acquired from our experimental and theoretical investigations revealed that the photostability of this UV-UC system is controlled by the energetic difference between the relevant frontier orbital levels of the solute (1 or 2) and solvent, and that these photodegradation reactions are rate-limited by the electron transfer between molecules.
Conclusions
Using sensitizer 1 and emitter 2, UV-UC to a shorter wavelength than 340 nm (maximum intensity at 322 nm) was achieved in various solvents. Both ΦUC and the photostability of 1 under continuous photoirradiation depended on the solvent. The use of hexane yielded the highest ΦUC of 8.2%, which is close to that of 10.2% reported for UV-UC in the 350–400 nm range achieved using a nanocrystal sensitizer and PPO,51 and also the highest photostability among the tested solvents. We found that ΦUC was mainly governed by solvent polarity, which varied the relative T1 energy-level matching between 1 and 2 because of the solvatochromic shift imposed on 1. The solvent dependence of ΦT,sen and ΦF,emi should also affect ΦUC.When the samples were prepared without 2, ksen,degr was large in most of the tested solvents and found to be correlated with Δ|HOMO| between 1 and the solvent. This correlation indicated that the photodegradation of 1 was rate-limited by electron transfer from the solvent to 1 and likely to be an electron transfer-initiated hydrogen abstraction process. However, when the energy acceptor 2, which quenches the T1 state of 1, was added to the samples, the degradation of 1 was effectively suppressed. This finding justifies the use of a ketonic sensitizer for UV-UC as long as the emitter concentration is higher than the order of 10−3 M in non-viscous solvents.
We developed a theoretical model and the curves generated by this model fitted the experimentally acquired temporal decay curves of the UC emission intensity well. This fitting provided several insights into the characteristics of the present UV-UC system. For example, the initial rapid rise of the UC emission intensity for the sample with hexane (cf.Fig. 4c) was ascribed to the presence of a trace amount of impurities (Q ∼ 1.9 × 10−7 M). Furthermore, kemi,rxn obtained from the fitting was correlated with Δ|LUMO|, which revealed that the photodegradation of 2 was rate-limited by electron transfer to the solvent. These findings indicate that the energetic difference between the frontier orbitals of the solute and solvent is the primary factor determining the photostability. Besides this viewpoint, the frontier orbital theory also addresses the importance of spatial overlap between two frontier orbitals involved in a reaction. Decreasing such overlap by addition of bulky groups to solutes may enhance their photostability.
Overall, this experimental and theoretical study has provided several fundamental insights regarding UV-UC in solvents. As general design guidelines for sample development, one should optimize solvent polarity to maximize ΦUC and use a combination of solvent and solute whose frontier energy levels are as far apart as possible to enhance solute photostability. These guidelines have not previously been explicitly proposed for UV-UC or visible-to-visible UC. The physicochemical insights obtained from this study will help to establish stable and efficient UV-UC systems in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We cordially thank Prof. Susumu Kawauchi (Tokyo Institute of Technology) for valuable comments and discussion regarding the quantum-chemical simulations. We also thank Dr Natasha Lundin for correcting the English used in this report. Part of this work was supported by JSPS KAKENHI (Grant no. 17H03183, Y. M.).References
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- P. E. Keivanidis, S. Baluschev, T. Miteva, G. Nelles, U. Scherf, A. Yasuda and G. Wegner, Adv. Mater., 2003, 15, 2095–2098 CrossRef CAS.
- D. V. Kozlov and F. N. Castellano, Chem. Commun., 2004, 2860–2861 RSC.
- S. Baluschev, P. E. Keivanidis, G. Wegner, J. Jacob, A. C. Grimsdale, K. Müllen, T. Miteva, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2005, 86, 061904 CrossRef.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- S. Baluschev, V. Yakutkin, T. Miteva, G. Wegner, T. Roberts, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, New J. Phys., 2008, 10, 013007 CrossRef.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- T. Miteva, V. Yakutkin, G. Nelles and S. Baluschev, New J. Phys., 2008, 10, 103002 CrossRef.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171–1174 CrossRef CAS.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795–1799 CrossRef CAS.
- Y. Murakami, Chem. Phys. Lett., 2011, 516, 56–61 CrossRef CAS.
- A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139–143 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- C. E. McCusker and F. N. Castellano, Chem. Commun., 2013, 49, 3537–3539 RSC.
- X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334–2338 CrossRef CAS.
- Y. Murakami, H. Kikuchi and A. Kawai, J. Phys. Chem. B, 2013, 117, 2487–2494 CrossRef CAS.
- Y. Murakami, H. Kikuchi and A. Kawai, J. Phys. Chem. B, 2013, 117, 5180–5187 CrossRef CAS.
- Y. Murakami, T. Ito and A. Kawai, J. Phys. Chem. B, 2014, 118, 14442–14451 CrossRef CAS.
- S. H. C. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS.
- R. Vadrucci, C. Weder and Y. C. Simon, Mater. Horiz., 2015, 2, 120–124 RSC.
- X. Yu, X. Cao, X. Chen, N. Ayres and P. Zhang, Chem. Commun., 2015, 51, 588–591 RSC.
- S. Hisamitsu, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 11550–11554 CrossRef CAS.
- Y. Murakami, Y. Himuro, T. Ito, R. Morita, K. Niimi and N. Kiyoyanagi, J. Phys. Chem. B, 2016, 120, 748–755 CrossRef CAS.
- H. Kim, O. S. Kwon, S. Kim, W. Choi and J. H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 RSC.
- K. Börjesson, P. Rudquist, V. Gray and K. Moth-Poulsen, Nat. Commun., 2016, 7, 12689 CrossRef.
- K. Kamada, Y. Sakagami, T. Mizokuro, Y. Fujiwara, K. Kobayashi, K. Narushima, S. Hirata and M. Vacha, Mater. Horiz., 2017, 4, 83–87 RSC.
- J. Han, F. Zhang, J. You, Y. Hiroaki, S. Yamada, T. Morifuji, S. Wang and X. Li, Photochem. Photobiol. Sci., 2017, 16, 1384–1390 RSC.
- K. Narushima, S. Hirata and M. Vacha, Nanoscale, 2017, 9, 10653–10661 RSC.
- R. Vadrucci, A. Monguzzi, F. Saenz, B. D. Wilts, Y. C. Simon and C. Weder, Adv. Mater., 2017, 29, 1702992 CrossRef.
- Ł. Bujak, K. Narushima, D. K. Sharma, S. Hirata and M. Vacha, J. Phys. Chem. C, 2017, 121, 25479–25486 CrossRef.
- Y. Murakami, S. K. Das, Y. Himuro and S. Maeda, Phys. Chem. Chem. Phys., 2017, 19, 30603–30615 RSC.
- S. Hisamitsu, N. Yanai, H. Kouno, E. Magome, M. Matsuki, T. Yamada, A. Monguzzi and N. Kimizuka, Phys. Chem. Chem. Phys., 2018, 20, 3233–3240 RSC.
- A. Abulikemu, Y. Sakagami, C. Heck, K. Kamada, H. Sotome, H. Miyasaka, D. Kuzuhara and H. Yamada, ACS Appl. Mater. Interfaces, 2019, 11, 20812–20819 CrossRef CAS.
- Y. Che, X. Yuan, F. Cai, J. Zhao, X. Zhao, H. Xu and L. Liu, Dyes Pigm., 2019, 171, 107756 CrossRef CAS.
- A. Tokunaga, L. M. Uriarte, K. Mutoh, E. Fron, J. Hofkens, M. Sliwa and J. Abe, J. Am. Chem. Soc., 2019, 141, 17744–17753 CrossRef CAS.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 55, 8269–8285 CrossRef.
- W. Zhao and F. N. Castellano, J. Phys. Chem. A, 2006, 110, 11440–11445 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912–5917 CrossRef CAS.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef CAS.
- P. Duan, N. Yanai and N. Kimizuka, Chem. Commun., 2014, 50, 13111–13113 RSC.
- M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. J. Wangelin, Chem. – Eur. J., 2015, 21, 15496–15501 CrossRef CAS.
- N. Yanai, M. Kozue, S. Amemori, R. Kabe, C. Adachi and N. Kimizuka, J. Mater. Chem. C, 2016, 4, 6447–6451 RSC.
- V. Gray, P. Xia, Z. Huang, E. Moses, A. Fast, D. A. Fishman, V. I. Vullev, M. Abrahamsson, K. Moth-Poulsen and M. L. Tang, Chem. Sci., 2017, 8, 5488–5496 RSC.
- M. Barawi, F. Fresno, R. Pérez-Ruiz and V. A. de la Peña O’Shea, ACS Appl. Energy Mater., 2019, 2, 207–211 CrossRef CAS.
- H. L. Lee, M. S. Lee, H. Park, W. S. Han and J. H. Kim, Korean J. Chem. Eng., 2019, 36, 1791–1798 CrossRef CAS.
- S. He, X. Luo, X. Liu, Y. Li and K. Wu, J. Phys. Chem. Lett., 2019, 10, 5036–5040 CrossRef CAS.
- S. Hisamitsu, J. Miyano, K. Okumura, J. Ka-Ho Hui, N. Yanai and N. Kimizuka, ChemistryOpen, 2020, 9, 14–17 CrossRef CAS.
- Y. Murakami, T. Ito, R. Morita, K. Niimi and N. Kiyoyanagi, worldwide patent application WO2015115556A1, international publication date, Aug 6, 2015, https://patents.google.com/patent/WO2015115556A1/en; Jpn Pat., JP2014023668, JP6436356B2; US Pat., US20170084758A1, US10175557B2; CN Pat., CN201580006015.1A, CN105940083B.
- B. Pfund, D. M. Steffen, M. R. Schreier, M. S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468 CrossRef CAS.
- R. López and R. Gómez, J. Sol-Gel Sci. Technol., 2012, 61, 1–7 CrossRef.
- M. Fox, Optical Properties of Solids, Oxford University Press, Oxford, 2001 Search PubMed.
- D. Reyes-Coronado, G. Rodriguez-Gattorno, M. E. Espinosa-Pesqueira, C. Cab, R. de Coss and G. Oskam, Nanotechnology, 2008, 19, 145605 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, 2009 Search PubMed.
- J. Ma, S. Chen, C. Ye, M. Li, T. Liu, X. Wang and Y. Song, Phys. Chem. Chem. Phys., 2019, 21, 14516–14520 RSC.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299 CrossRef CAS.
- J. P. Cerón-Carrascoa, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, J. Phys. Org. Chem., 2014, 27, 512–518 CrossRef.
- L. Komsta, M. Waksmundzka-Hajnos and J. Sherma, Thin Layer Chromatography in Drug Analysis, CRC Press, Boca Raton, 2013 Search PubMed.
- F. Marsico, A. Turshatov, R. Peköz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057–11064 CrossRef CAS.
- C. Mongin, J. H. Golden and F. N. Castellano, ACS Appl. Mater. Interfaces, 2016, 8, 24038–24048 CrossRef CAS.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 RSC.
- Q. Liu, M. Xu, T. Yang, B. Tian, X. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9883–9888 CrossRef CAS.
- S. Wan, J. Lin, H. Su, J. Dai and W. Lu, Chem. Commun., 2018, 54, 3907–3910 RSC.
- C. Walling and M. J. Gibian, J. Am. Chem. Soc., 1965, 87, 3361–3364 CrossRef CAS.
- W. J. Leigh, E. C. Lathioor and M. J. St. Pierre, J. Am. Chem. Soc., 1996, 118, 12339–12348 CrossRef CAS.
- E. C. Lathioor and W. J. Leigh, Photochem. Photobiol., 2006, 82, 291–300 CrossRef CAS.
- S. Inbar, H. Linschitz and S. G. Cohen, J. Am. Chem. Soc., 1981, 103, 1048–1054 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Photostability of visible-to-visible UC in an ionic liquid, photostability of UV-UC using biacetyl and PPO in DMF, optical absorption spectra of the sensitizer and emitter used in this study, information about the solvents used in this study, experimental setup to controllably induce photodegradation, calculated dipole moments of the sensitizer and emitter, spectral overlap between emitter fluorescence and sensitizer absorption, calculated permittivity dependence of the sensitizer and emitter triplet energies, determination of ΦUC, effect of solvent purity on temporal decay profiles of ΦUC, temporal changes of fluorescence spectra of the sensitizer in the absence of the emitter during photoirradiation in different solvents, photoirradiation-induced changes of optical absorption spectra of samples containing only the sensitizer in different solvents, procedure to calculate ksen,degr, plots of ksen,degr against ionization energy and electron affinity, procedure to calculate Φsen,rxn, photoirradiation-induced changes of optical absorption spectra of samples containing both the sensitizer and emitter in different solvents, temporal changes of fluorescence spectra of the sensitizer in the presence of the emitter during photoirradiation in different solvents, effect of photoirradiation on the triplet lifetime of the emitter, calculation details of our theoretical model, and plots of kemi,rxn against ionization energy and electron affinity. See DOI: 10.1039/d0cp04923a |
| ‡ Present address: FANUC Corp., Oshino-mura, Yamanashi 401-0597, Japan. |
| This journal is © the Owner Societies 2020 |