
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The accuracy challenge of the DFT-based molecular assignment of 13C MAS NMR characterization of surface intermediates in zeolite catalysis†‡
Alexander A.
Kolganov
a,
Anton A.
Gabrienko
 ab,
Ivan Yu.
Chernyshov
c,
Alexander G.
Stepanov
ab and
Evgeny A.
Pidko
*cd
ab,
Ivan Yu.
Chernyshov
c,
Alexander G.
Stepanov
ab and
Evgeny A.
Pidko
*cd
aBoreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences, Prospekt Akademika Lavrentieva 5, Novosibirsk 630090, Russia
bFaculty of Natural Sciences, Department of Physical Chemistry, Novosibirsk State University, Pirogova Str. 2, Novosibirsk 630090, Russia
cTheoMAT Group, ChemBio Cluster, ITMO University, Lomonosova Street 9, Saint Petersburg, 191002, Russia
dInorganic Systems Engineering group, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, Delft 2629 HZ, The Netherlands. E-mail: E.A.Pidko@tudelft.nl
First published on 30th September 2020
Abstract
The influence of the model and method choice on the DFT predicted 13C NMR chemical shifts of zeolite surface methoxide species has been systematically analyzed. Twelve 13C NMR chemical shift calculation protocols on full periodic and hybrid periodic–cluster DFT calculations with varied structural relaxation procedures are examined. The primary assessment of the accuracy of the computational protocols has been carried out for the Si–O(CH3)–Al surface methoxide species in ZSM-5 zeolite with well-defined experimental NMR parameters (chemical shift, δ(13C) value) as a reference. Different configurations of these surface intermediates and their location inside the ZSM-5 pores are considered explicitly. The predicted δ value deviates by up to ±0.8 ppm from the experimental value of 59 ppm due to the varied confinement of the methoxide species at different zeolite sites (model accuracy). The choice of the exchange–correlation functional (method accuracy) introduces ±1.5 ppm uncertainty in the computed chemical shifts. The accuracy of the predicted 13C NMR chemical shifts for the computational assignment of spectral characteristics of zeolite intermediates has been further analyzed by considering the potential intermediate species formed upon methane activation by Cu/ZSM-5 zeolite. The presence of Cu species in the vicinity of surface methoxide increases the prediction uncertainty to ±2.5 ppm. The full geometry relaxation of the local environment of an active site at an appropriate level of theory is critical to ensure a good agreement between the experimental and computed NMR data. Chemical shifts (δ) calculated via full geometry relaxation of a cluster model of a relevant portion of the zeolite lattice site are in the best agreement with the experimental values. Our analysis indicates that the full geometry optimization of a cluster model at the PBE0-D3/6-311G(d,p) level of theory followed by GIAO/PBE0-D3/aug-cc-pVDZ calculations is the most suitable approach for the calculation of 13C chemical shifts of zeolite surface intermediates.
1. Introduction
DFT calculations for the prediction of 13C NMR chemical shifts for different surface intermediates could provide crucial support to mechanistic NMR studies in zeolite catalysis. DFT calculations have been widely employed to compute the chemical shifts of various organic molecules,1 solids,2 and organometallic compounds.3 NMR chemical shift calculation has found its own niche in zeolite catalysis, namely for the calculation of 27Al NMR to support the assignment of the experimental spectra.4 This technique is being actively used in combination with 27Al 3Q MQMAS NMR spectroscopy for the investigation of Al atom distribution throughout the zeolite framework.4,5 However, this has not yet been extensively applied for the mechanistic studies of hydrocarbon conversion on zeolites, raising, therefore, concerns regarding the accuracy and spectral resolution of the DFT-based assignments. There are a number of issues that could limit the accuracy of the calculated chemical shifts. First, the choice of the method of calculations, in particular the choice of exchange–correlation functional and the basis set for the electronic structure calculations, is important.6,7 Secondly, the selection of a model to represent the structure and local zeolite environment of the examined species is also important.8 The screening of both the calculation method limitations and chosen model effect on the accuracy of calculations could determine the most reliable approach for the prediction of 13C NMR chemical shifts based on DFT analysis of the species adsorbed on zeolites.High resolution solid-state 13C MAS NMR is a very efficient tool for studying the adsorption and transformations of hydrocarbon species in zeolites.9–13 In particular, this technique can provide information on the pathways of methane chemical activation and conversion in the pores of metal-containing zeolite catalysts.14–24 The detection and correct identification of the methane activation products, surface bound intermediates, based on observed 13C NMR chemical shifts could give valuable insights into the reaction mechanism. The combination of the experimental techniques and computational chemistry methods is a powerful approach for the accurate description of the surface products.
Recent studies have successfully employed the 13C MAS NMR technique to study methane transformation on copper-containing zeolites.25–29 The reported spectroscopic data have shown the presence of few different signals with similar 13C chemical shifts in the range of 50–65 ppm.30,31 The unambiguous assignment of such signals to specific molecular configurations based solely on the experimental results is very challenging. It is thus not surprising that different assignments have been proposed for the same signals by different research groups.25–29 In particular, the origin of the signal at 62–63 ppm is highly disputed.26,28,29,32–34 Initially, this signal was assigned to a [Cu(μ-OCH3)Cu] structure.26 Other studies suggested that this signal should be attributed to methanol adsorbed to a dual Cu+ ([Cu(μ-CH3OH)Cu])27 or a mono-Cu+ site.28
Herein, we present a study on the advantages and limitations of common DFT strategies for rationalizing the 13C NMR spectra of zeolite adsorbed reaction intermediates. ZSM-5 zeolite is one of the most widely studied and applied zeolite materials in catalysis and has been selected as the main object of this computational investigation. Both model and methodological inaccuracies have been examined by calculating 13C NMR chemical shifts of the Si–O(CH3)–Al methoxides, chosen as the reference with a reliably determined chemical shift of 59 ppm, placed at the different locations within the zeolite framework. The determination of the prediction ability of the examined calculation protocols for 13C NMR chemical shifts of various proposed intermediates of the methane activation on copper-containing zeolites has been carried out.
2. Computational details
Models
Synthetic MFI-type zeolites (ZSM-5) can exist in the form of either monoclinic (P21/n) or orthorhombic (Pnma, Pn21a, or P212121) phase. A reversible phase transformation is possible under various conditions.35–37 For simplicity, the orthorhombic MFI unit cell with lattice parameters of a = 20.241 Å, b = 20.015 Å and c = 13.439 Å as optimized using DFT with an all-silica MFI periodic model was used for the periodic DFT calculations.38 To compensate for the positive charge of the extra-framework species, one or two framework Si atoms in MFI unit cell were substituted by Al. Si–O(CH3)–Al methoxides were placed along the 12 different locations in the zeolite framework (Fig. S1, ESI†). In the case of structures containing BAS, H+ was bonded to the Si(T1)–(O−)–Al(T2) oxygen atom. Cu+ cations in all models were coordinated to the two oxygen atoms connected to the Al atom at the T1 position (Al(T1)–O–Si(T2) and Al(T1)–O–Si(T5)). All models containing the [Cu–O–Cu]2+ core were placed in the α site of the zeolite framework with aluminium atoms placed in the T1 and T7 positions.Each Cu atom in the [Cu–O–Cu]2+ motif was coordinated to the two framework oxygen atoms: Al(T1)–O–Si(T2), Al(T1)–O–Si(T5) for the first one and Al(T7)–O–Si(T8), Al(T7)–O–Si(T11) for the second one. The resulting Si/Al ratios in the lattice of the periodic zeolite models were 95 (1 Al models) and 47 (2 Al models). After the optimization of the fully periodic zeolite structures, cluster models representing the local zeolite environment at the zeolite site were directly constructed from the periodic models.
“Two-ring” cluster models were constructed for the zeolite frameworks where methoxide located near the channel intersection (Fig. 1a). “Three-ring” models were constructed in the cases where methoxide located in the middle zeolite “ring” between the other two zeolite “rings” (Fig. 1b). For the models where methoxide is located in the cage between the two intersecting zeolite channels, the two-channel clusters were constructed (Fig. 1c). Dangling O atoms were substituted by H atoms at the ∼1.6 Å distance.39 To avoid the presence of Al–H bonds in the cluster models, the zeolite framework was extended by one or two Si atoms near the aluminum atoms.
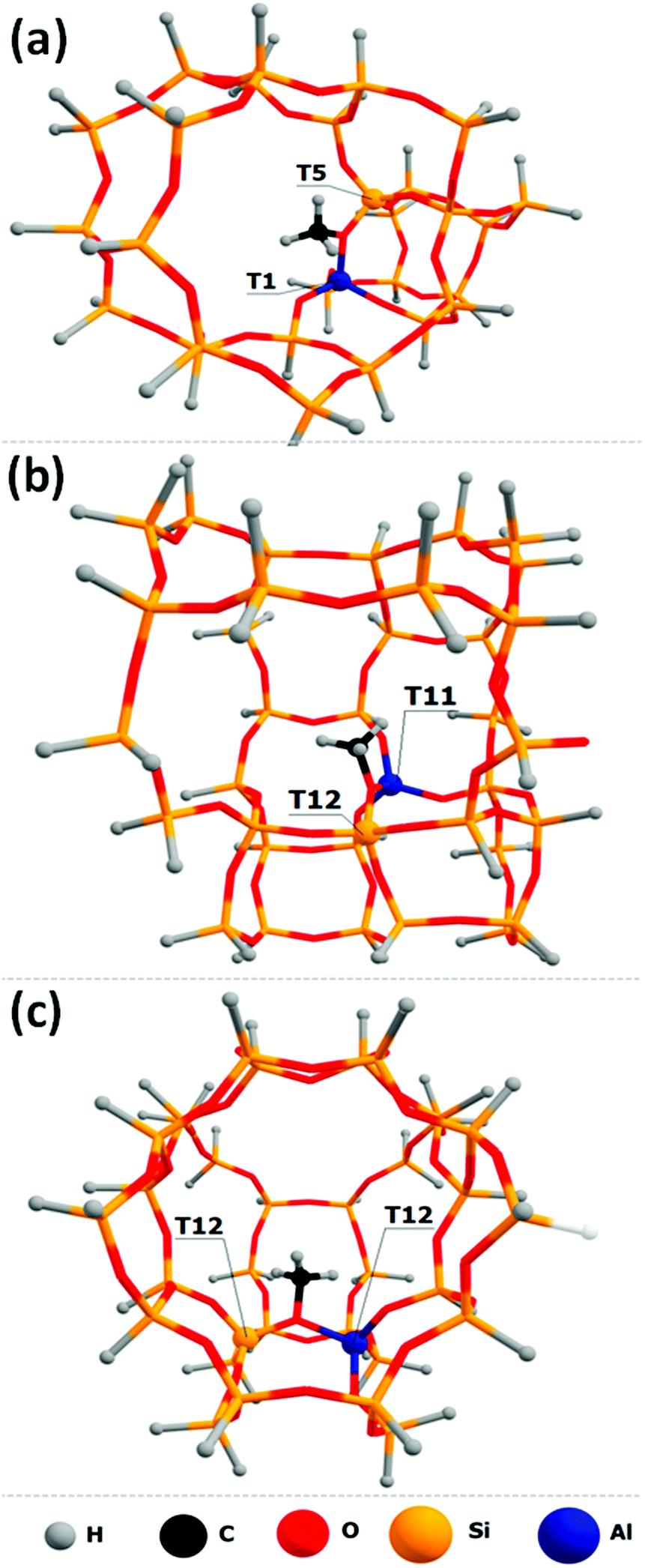 | ||
| Fig. 1 Cluster models of T1–T5 (a), T11–T12 (b) and T12–T12 (c) Si–O(CH3)–Al methoxides. | ||
Methods
Periodic DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP 5.3.5).40 The exchange–correlation (XC) energy term was described using the generalized gradient approximation PBE functional.41 To take into account the van der Waals interactions, semi-empirical D3 dispersion correction by Grimme42 with Becke–Johnson damping43 was applied. A plane-wave basis set with a cutoff energy of 450 eV in combination with the projected augmented wave (PAW) method was used.44 Brillouin zone-sampling was restricted to the Γ point.The spin-unrestricted calculations of 13C NMR chemical shifts were carried out using the cluster models directly constructed from the zeolite framework. All finite model cluster calculations were carried out using the Gaussian 16 program (Revision B.01).45
Two different computational strategies were implemented for the chemical shift calculations of the cluster models directly constructed from optimized periodic models, namely, (i) single-point electronic structure (SP) and chemical shift calculation directly on the geometry from periodic DFT or (ii) full optimization (OPT) of the cluster. The terminal framework O atoms in the clusters were replaced by H atoms at the respective positions.39 Six DFT functionals were used for the cluster calculations: the generalized gradient approximation PBE functional was chosen in order to be consistent with the method used in periodic DFT calculations, hybrid exchange–correlation functionals PBE046 and HSE06,47–50 long-range corrected wB97xD functional51 and hybrid mPW1PW9152,53 and B3PW9154 functionals. D3BJ42,43 dispersion corrections were implemented to PBE, PBE0 and B3PW91 functionals. In total, 12 different calculation protocols have been examined. 6-311G(d,p)55,56 basis was used for the cluster geometry optimization. During the cluster geometry optimization all dangling H atoms were fixed in their positions at a Si–H distance of ∼1.6 Å.39 The gauge-independent atomic orbital (GIAO)57 method along with aug-cc-pVDZ basis set was implemented for the NMR chemical shift calculation of the zeolite cluster 13C atoms.58 The choice of the functionals and basis set is based on the analysis of the 13C chemical shift prediction ability of the various techniques carried out in the study.613C chemical shifts were calculated using the following formula: δ = σref − σ + δref, where σref and σ are isotropic chemical shielding of the reference and calculated species, respectively. Tetramethylsilane (TMS) was chosen as the reference (δref = 0). σref values were calculated using the same calculation protocols. All σref results are shown in Table S1 (ESI†).
3. Results and discussion
Zeolite-bound Si–O(CH3)–Al methoxide species are characterized using a 13C chemical shift at 56–59 ppm, depending on the zeolite framework type.16,22,26,32–34,59,60 Methoxide groups formed within the MFI framework give a signal at about 59 ppm on the surface of H-ZSM-5.34 Therefore, such species can be used as the benchmark to investigate various possible uncertainties of the multiple DFT calculation protocols. In this study, we explicitly examine how the local environment affects the predicted chemical shifts by placing the Al atom into different T-sites throughout the framework of ZSM-5 zeolite (MFI framework type). There are twelve distinct T-sites in the MFI framework, and ten of them are examined here to analyze the influence of the model, i.e., the position of the Al-site and therefore the location of Si–O(CH3)–Al species, on the calculated chemical shift. Note, that there is more than one possible position for the methyl group for each T-site occupied by Al, i.e., the methyl group can be attached to two Si–O–Al–O–Si bridges related to the same Al-site. Therefore, the studied models were denoted further as T(Al)-site–T(Si)-sites”. T1–T5, T7–T11, T9–T10, and T12–T12 methoxides are shown in Fig. 2 as examples. All examined optimized structures of the methoxides are presented in Fig. S1 (ESI†). The relative stabilities of these intermediates computed using periodic PBE calculations and chemical shift values are reported in Tables S2 and S3 (ESI†), respectively.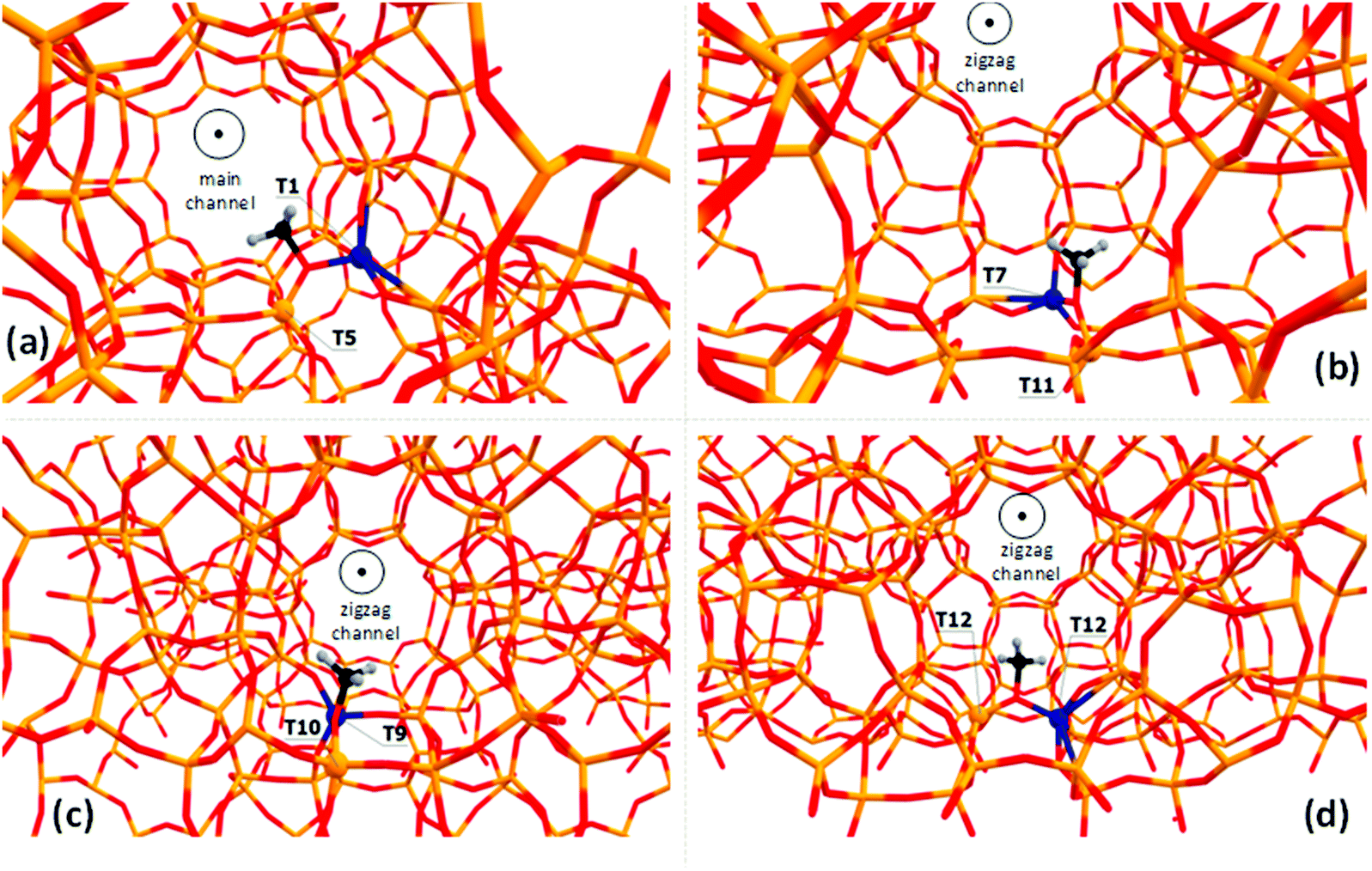 | ||
| Fig. 2 The locations of the T1–T5 (a), T7–T11 (b), T9–T10 (c), T12–T12 and (d) methoxides examined in this study. All models can be found in the ESI† (Fig. S1). | ||
Firstly, the relative stability of the methoxide intermediates at different zeolite sites was examined and the results are summarized in Fig. 3. The T9–T6 configuration provides the highest stability for the surface methoxide, with stabilities of the alternative structures being within approximately 30 kJ mol−1. An exceptionally low stability is predicted for the T7–T7 configuration (ΔE = 82 kJ mol−1). However, this structure was still taken into account and its chemical shifts were calculated to see the correlation between the relative energy and the chemical shift.
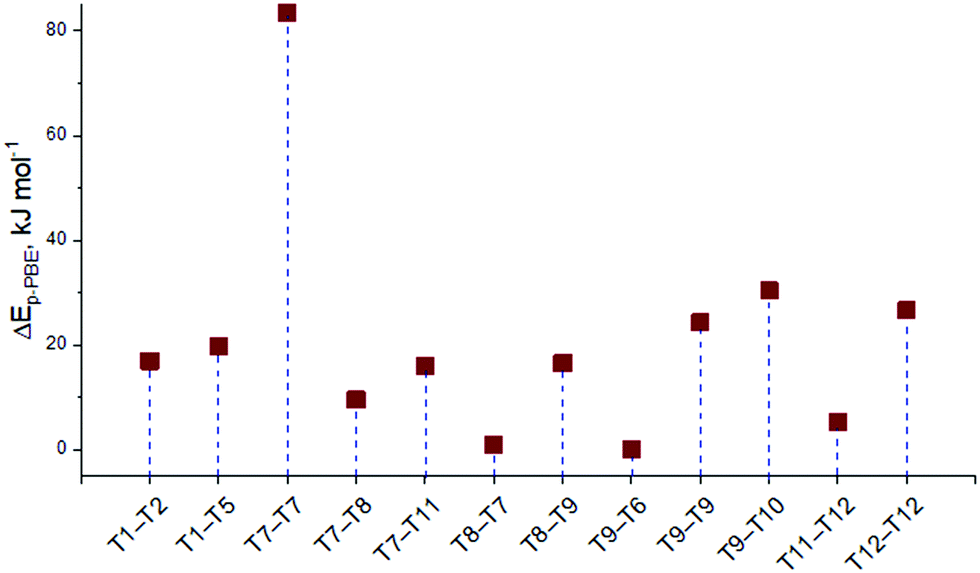 | ||
| Fig. 3 The relative stabilities of the surface methoxides placed at the different sites of the ZSM-5 zeolite. The energy of the most stable methoxide T9–T6 is set as zero point. | ||
The calculation of the 13C NMR chemical shifts of methoxide intermediates by means of a linear response as implemented in VASP 5.3.5 using the full periodic zeolite models (see Section S2, ESI†) gave unsatisfactory results. The respective computed δ(13C) values were 91.6 ppm (T1–T2 methoxide), 89.6 ppm (T7–T8 methoxide), 91.1 ppm (T9–T9 methoxide) and 89.9 ppm (T9–T9 methoxide), which are all much higher than the experimental value of 59 ppm. An improved accuracy of the predicted chemical shifts for the periodic system could in principle be achieved using a higher level of theory and more stringent computational parameters. The associated increase in the demand for computational resources rendered such a direct periodic modelling approach impractical for this case. Therefore, in this work a cluster modelling approach was used to accurately calculate the NMR spectral parameters of intrazeolite intermediates.
In order to calculate the methoxide chemical shift values, two main approaches were implemented. The single point (denoted as SP) approach was implemented by using the clusters directly constructed from the periodic model without further geometry relaxation. The main point of the SP calculations is to preserve the geometry features derived from periodic calculations which are on the higher “model” level. Another protocol is the cluster geometry optimization (denoted as OPT). This approach enables carrying out structural relaxation on a higher level of theory (hybrid GGA) compared to the periodic calculations (GGA), which, in turn, yields more accurate local geometry in the context of the current computational method. However, during the relaxation of the cluster model, essential structural features due to the extended zeolite structure could be lost resulting in artefacts that could not be found in the periodic models.8
To demonstrate possible inaccuracies arising from the choice of calculation method, 6 different functionals were implemented. Since the same basis set (aug-cc-pVDZ) was used for all calculations, the protocols were named “FUNCTIONAL(SP)” or “FUNCTIONAL(OPT)” for the single point and optimized models, respectively.
The results of chemical shift calculations for the surface methoxide species are summarized in Fig. 4. One can see that for all methods the 13C NMR chemical shift of the most unstable T7–T7 methoxide is predicted to be much higher than those of other configurations. For the rest of the species, there is no correlation observed between the relative stability and the value of the chemical shift. On comparing the results obtained from PBE-D3(SP) and PBE0-D3(SP) calculation protocols (Fig. 4), methoxide 13C NMR chemical shifts calculated using the PBE-D3 method are slightly higher than those obtained using the PBE0-D3 method. The mean chemical shift values are equal, 62.1 ± 0.8 ppm and 61.5 ± 0.7 ppm for the PBE-D3 and PBE0-D3 methods, respectively. Note, these two values are remarkably different from the experimental values of 58–59 ppm.
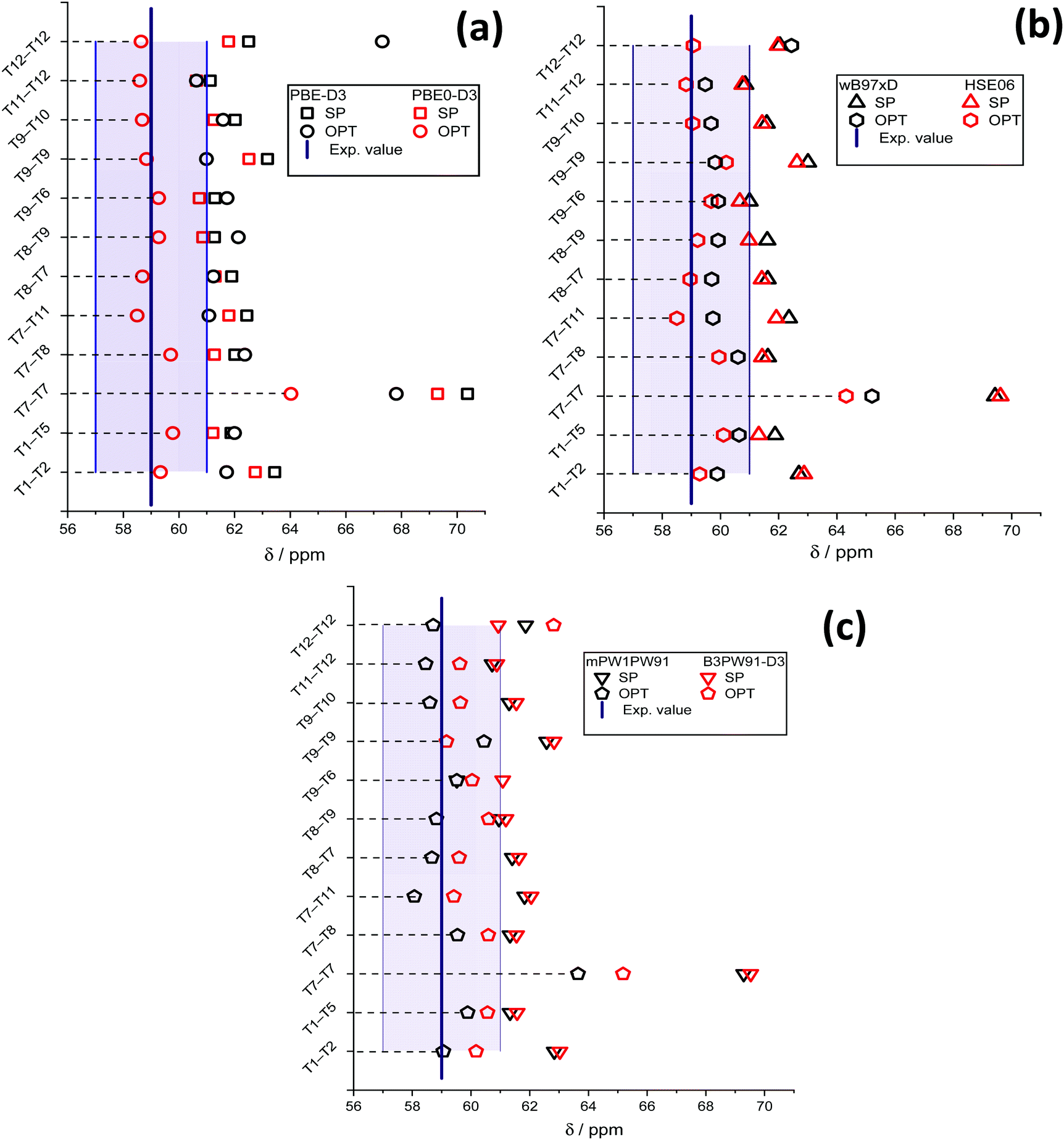 | ||
| Fig. 4 13C NMR chemical shifts of different methoxides and the experimental values calculated using PBE-D3 and PBE0-D3 (a), wB97xD and HSE06 (b), and mPW1PW91 and B3PW91-D3 (c) functionals. The filled area indicates the half-width of the experimentally observed signal31 at 59 ppm. | ||
For PBE-D3(OPT), the deviation of the chemical shift values increases. The notable increase of the T12–T12 methoxide chemical shift is related to a significant change of the methoxide orientation during the PBE-D3/6-311G(d,p) geometry optimization (Fig. S4, ESI†).
The values obtained using the PBE0-D3(OPT) protocol are much closer to the experimental ones with the mean value of 59.0 ± 0.5 ppm. Similar trends are observed for the data obtained using the wB97xD, HSE06 (Fig. 4b), mPW1PW91 and B3PW91-D3 (Fig. 4c) functionals. The results obtained from SP procedures with different exchange–correlation functionals are very close to each other. The XC-functional related uncertainty for the methoxide species derived from the method choice is ±0.3 ppm.
After cluster geometry optimization, all chemical shift values are decreased, with the HSE06(OPT) and mPW1PW91(OPT) results being closer to the experimental value. The configuration of the methoxide species inside the zeolite pores has a profound impact on the predicted chemical shift. In particular, we note a strong increase of the predicted chemical shift value for the T12–T12 methoxide when wB97xD and B3PW91-D3 functionals are used. The mean values of the predicted chemical shifts for different computational methods and model configurations are summarized in Table 1.
| Calculation protocol | Mean δ(13C)/ppm |
|---|---|
| PBE-D3(SP) | 62.1 ± 0.8 |
| PBE-D3(OPT) | 61.5 ± 0.6 |
| PBE0-D3(SP) | 61.5 ± 0.7 |
| PBE0-D3(OPT) | 59.0 ± 0.5 |
| wB97xD(SP) | 61.8 ± 0.7 |
| wB97xD(OPT) | 59.9 ± 0.4 |
| HSE06(SP) | 61.7 ± 0.7 |
| HSE06(OPT) | 59.4 ± 0.6 |
| mPW1PW91(SP) | 61.4 ± 0.9 |
| mPW1PW91(OPT) | 59.1 ± 0.7 |
| B3PW91-D3(SP) | 61.6 ± 0.7 |
| B3PW91-D3(OPT) | 59.9 ± 0.5 |
The observed differences between the experimental chemical shifts and the values predicted for different methoxide configurations are attributed to the fact that the experimental data can be viewed as a statistical average over all different possible configurations inside the zeolite pores, whereas the calculations provide the deterministic prediction for one specific configuration within the ensemble. In this respect, the computed values using the hybrid GGA functionals with OPT procedure appear to fall very well within the experimental ranges of 13C NMR chemical shifts for Si–O(CH3)–Al in ZSM-5 zeolite (Fig. 5).31 On the other hand, the GGA-level PBE-D3(OPT) procedure gives far less accurate results.
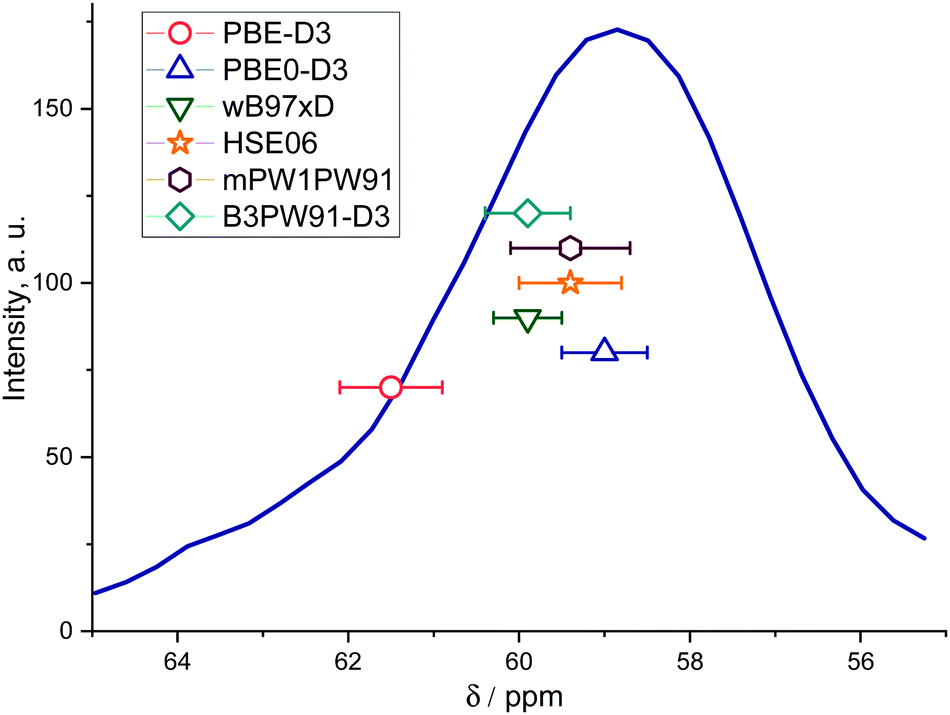 | ||
| Fig. 5 Mean calculated 13C chemical shifts of the Si–O(CH3)–Al methoxides placed in different locations throughout the ZSM-5 zeolite framework and the chemical shift value deviations superimposed on the experimental 13C CP MAS NMR spectrum. | ||
Fig. 6 presents the XC-related uncertainty of the mean 13C chemical shift values for each calculated methoxide (Fig. 6). All mean values and deviations are reported in Table S4 (ESI†). It is clearly seen that the obtained chemical shifts are not significantly influenced by the chosen calculation DFT functional in the case of SP calculations. The value deviations are in the range of 0.2–0.5 ppm. Concerning mean 13C chemical shifts calculated using cluster OPT procedures, it is clearly seen that XC-related uncertainties are larger compared to single point calculations. The results obtained demonstrate that the deviation of the chemical shifts calculated using the cluster optimization model is in the range of 0.8–1.5 ppm except in the case of T12–T12 methoxide (±3.7 ppm) because of a conformational change due to the cluster relaxation.
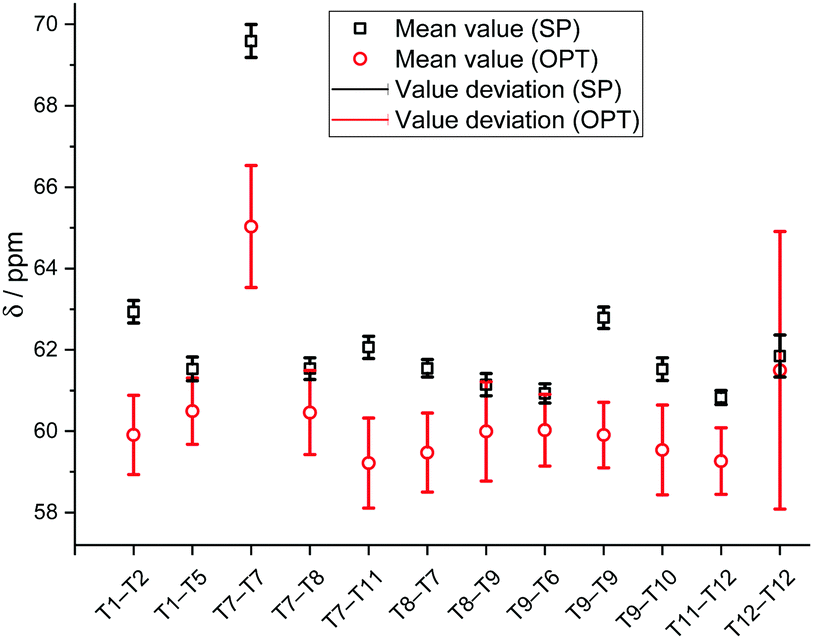 | ||
| Fig. 6 Mean calculated 13C chemical shifts of the Si–O(CH3)–Al methoxides calculated using various calculation techniques and XC functional-related uncertainties. | ||
The role of structural relaxation is very important for the 13C NMR chemical shift calculations.61 It was previously observed that the level of theory employed for the geometry optimization could give rise to deviations in the calculated 13C chemical shift values for transition metal complexes by up to 15 ppm.7 In our case, it follows from the reported results that cluster geometry optimization on a higher level of theory (h-GGA) compared to periodic calculations (GGA) is the key to obtaining accurate 13C NMR chemical shift values. However, cluster geometry optimization could lead to a local conformation change that can significantly change the predicted chemical shifts. The current analysis identifies the PBE0-D3(OPT) technique as providing the best results in terms of the agreement between theory and experiment (59 ppm experimental value vs. 59 ± 0.5 ppm calculated one).
Next, we analyzed the performance of different DFT methods for the prediction of 13C NMR chemical shifts of various proposed intermediates of the methane activation on copper-containing zeolites. Different possible intermediates were considered, namely, the surface methoxide Si–O(CH3)–Al (in the T1–T2 location), methanol adsorbed on BAS (Brønsted acid site), dimethyl ether (DME) adsorbed on BAS, methanol adsorbed on Cu+ cation, dimethyl ether adsorbed on Cu+, methanol adsorbed between two copper atoms [Cu(μ-CH3OH)Cu]2+, [Cu(μ-OCH3)Cu]+ species, and Si–O(CH3)–Al methoxide located in the vicinity of the [Cu(μ-OH)Cu]+ site.
All structures containing [Cu–O–Cu]2+ sites are believed to be possible products of methane activation using the Cu/ZSM-5 catalyst (Fig. S3, ESI†). The calculated chemical shifts are presented in Fig. 7.
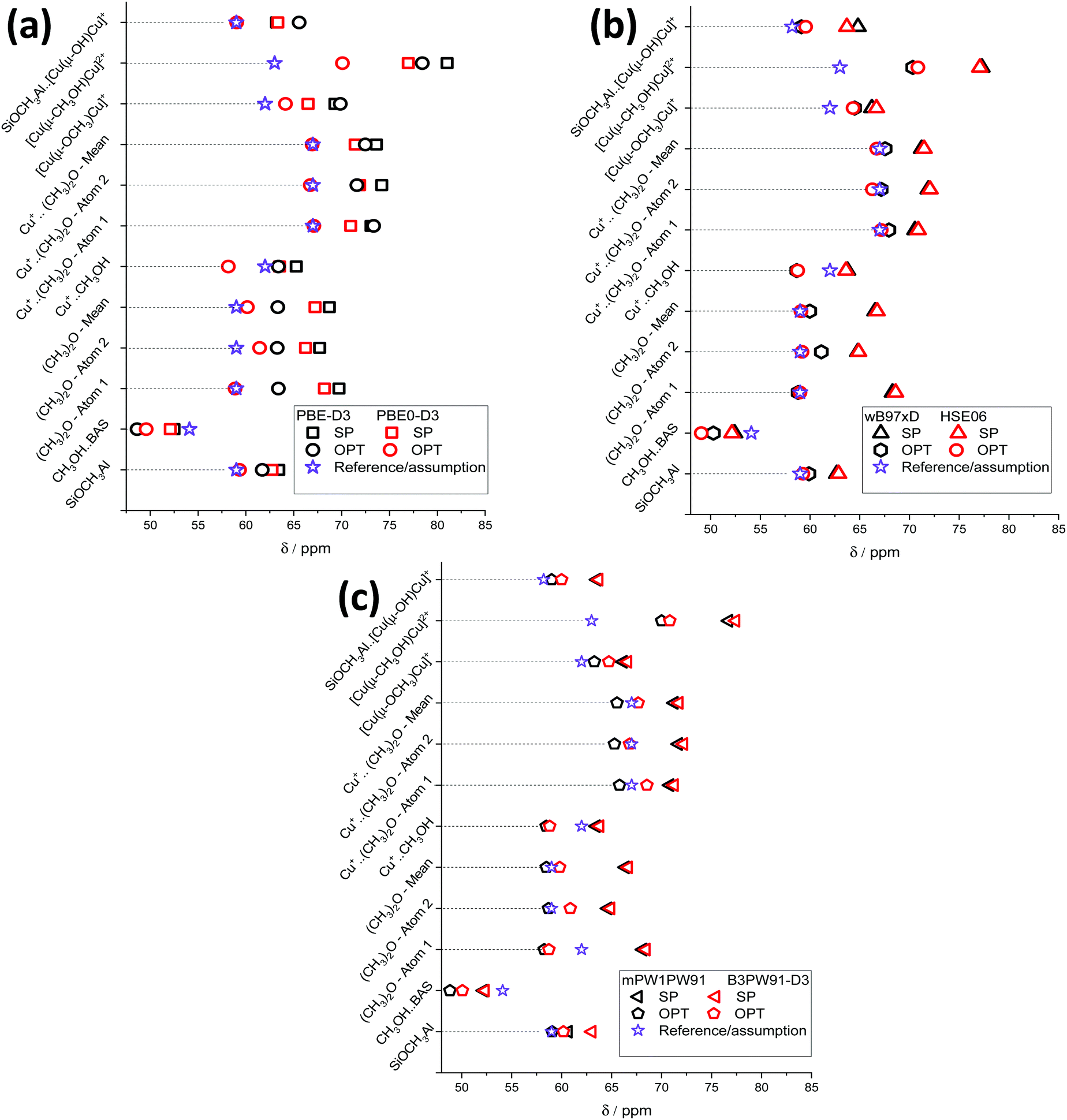 | ||
| Fig. 7 13C NMR chemical shifts of different methane activation surface products and their reference values or assignments calculated using PBE-D3 and PBE0-D3 (a), wB97xD and HSE06 (b), and mPW1PW91 and B3PW91-D3 (c) functionals. | ||
The general trends observed for the model case of the surface methoxides (Fig. 4) are well reproduced for the various hydrocarbon species on Cu/ZSM-5 (Fig. 7). The chemical shifts obtained using the SP scheme depend only slightly on the choice of the XC functional. The exception is SP-PBE, where uniformly higher δ values are predicted. Cluster geometry relaxation leads to a significant decrease in the resulting 13C NMR chemical shifts. SP calculations predict chemical shifts of 52–53 ppm for methanol adsorbed on BAS, which is in good agreement with the experimental values of 53–54 ppm.26,28,29,32–34 After the cluster relaxation in OPT scheme, predicted shifts decrease the values to 49–51 ppm, which are close to the chemical shift of “free” methanol.62
For BAS adsorbed DME, SP calculations predict a difference of about 4 ppm between the chemical shifts of the two carbon atoms caused by the differences in O–C interatomic distances in the optimized periodic zeolite models. The mean chemical shift values for each C atom are 68–69 ppm and 64–65 ppm. After the cluster geometry optimization, a conformational change of dimethyl ether takes place (Fig. S5, ESI†). The chemical shift values of two different atoms obtained using PBE-D3, HSE06 and mPW1PW91 techniques are equalized to 58–59 ppm after geometry optimization. However, calculations using PBE0-D3, wB97xD and B3PW91-D3 functionals resulted in slight differences between δ values for C-1 (58–59 ppm) and C-2 (60–61 ppm) atoms.
For methanol adsorbed on the Cu+ cation, the SP calculations give δ values of 63–64 ppm which are closer to the experimental assignment of the signal at 62 ppm27,28 than the chemical shifts of 58–59 ppm obtained using the OPT procedure. Interestingly, in the case of DME adsorbed on a Cu+ cation, no significant difference between the chemical shifts of C-1 and C-2 is observed in spite of the pronounced conformational change during the cluster geometry optimization (Fig. S6, ESI†). The chemical shifts predicted using SP calculations (71–74 ppm) are much higher compared to the experimental assignment of 67.0 ppm.27,28 However, the values obtained using the OPT procedure (65–67 ppm) are in good agreement with the experimental values.
For the bridging methoxide [Cu(μ-OCH3)Cu]+ intermediate, the SP and OPT procedures yield a δ value in the ranges of 66–69 ppm and 63–64 ppm, respectively. The latter are close to the experimental assignment of the signal at 62 ppm made by Narsimhan et al.26 Methanol adsorbed on dual Cu+ site ([Cu(μ-CH3OH)Cu]2+) is characterized by the calculated chemical shifts in the range of 70–81 ppm, which is much larger than the previous assignment of the signal at 63 ppm to such sites made on the basis of DFT calculations with a simplistic 10 T-atom cluster model.27
The calculated 13C chemical shift values of surface-bound methoxide Si–O(CH3)–Al neighboring the [Cu(μ-OH)Cu]+ site are very close to the values obtained for the Cu-free models with the exception of the results obtained with OPT-PBE-D3 that yields a slightly higher δ value of 66 ppm.
The influence of the XC-related uncertainty can be demonstrated by examining the mean 13C chemical shift values and their deviations for each calculated species (Fig. 8). All mean values are reported in Table S6 (ESI†). For the di-Cu-containing structures and DME on single Cu+ site, the XC-related uncertainties are in the range of ±1.5 and ±2.4 ppm (SP), ±2.2 and ±3.3 ppm (OPT). This is probably due to the higher sensitivity of the results for the models containing two heavy atoms to the choice of XC functional.
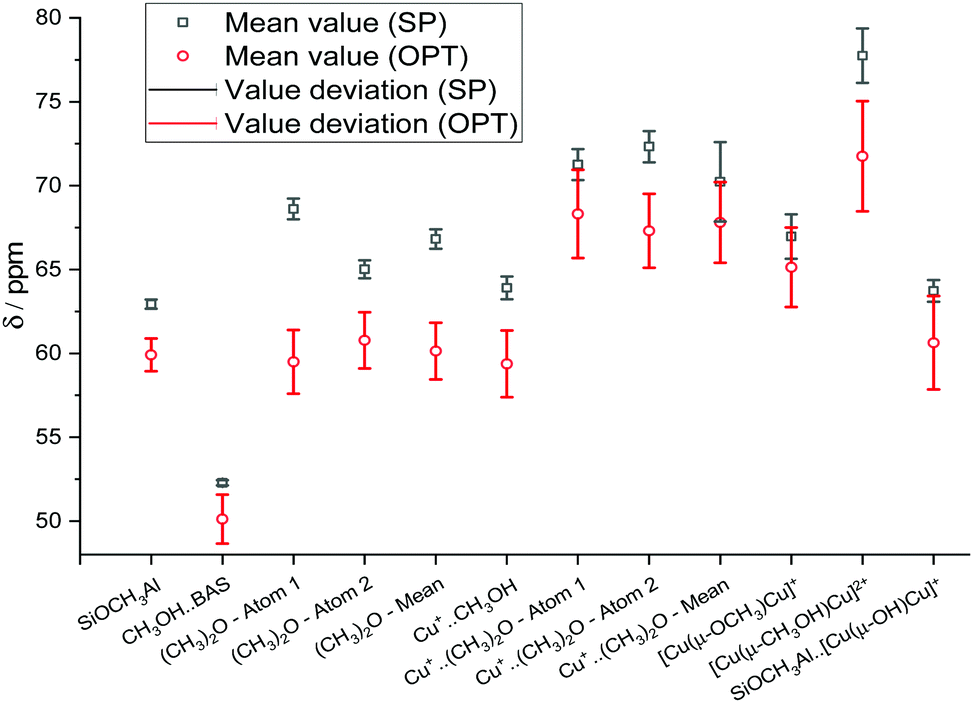 | ||
| Fig. 8 Mean calculated 13C chemical shifts calculated using various calculation techniques and XC functional-related uncertainties. | ||
Indeed, the XC-related uncertainty in the predicted chemical shifts is quite small for DME/BAS, despite the significant conformational change happening during the geometry optimization. Our analysis suggests that the OPT procedure is the most reliable computational protocol in providing computed chemical shifts in line with the experimental data.
The presented results show that the accurate calculation of the 13C NMR chemical shifts of intrazeolite intermediates requires the use of the h-GGA level of theory. This is in line with the previous computational studies on adsorption and reactivity of zeolite-based catalysts. Although general trends in the energetics of hydrocarbon activation by H-forms63,64 and TM-modified zeolites65,66 can be predicted well using the GGA and meta-GGA methods, the quantitative analysis of such systems requires more accurate h-GGA calculations.63,67
From a set of XC functionals considered here, similar performance is shown for almost all methods with the exception of the GGA PBE-D3 functional. The optimal performance in predicting chemical shifts for C1 intermediates in zeolite pores is observed for the PBE0-D3(OPT) protocol.
4. Conclusion
Model and XC functional-related uncertainties of twelve 13C NMR chemical shift calculation protocols were analyzed. The influence of the model inaccuracy and local zeolite confinement was demonstrated using the 13C NMR chemical shift DFT calculations of Si–O(CH3)–Al methoxide benchmark species located in 12 different positions throughout the ZSM-5 zeolite framework. The analysis was also extended to the possible methane activation products on the surface Cu/ZSM-5 zeolite.It is established that the effect of local confinement on the calculated chemical shifts influences chemical shifts values up to ±0.9 ppm. The XC-related uncertainty is ±0.3 ppm in the case of single point calculations. However, the cluster geometry optimization increases the value range up to ±1.5 ppm. This effect significantly increases (up to ±3.3 ppm) for the case of structures containing copper atoms. The results obtained indicate that cluster geometry optimization on the hybrid GGA level of theory is the crucial step to obtain local geometry that provides the basis for accurate prediction of the chemical shift values of Si–O(CH3)–Al species. Out of the examined calculation protocols, the pPBE-D3BJ/450eV//PBE0-D3BJ/6-311G(d,p)//GIAO-PBE0-D3BJ/aug-cc-pVDZ technique is the most appropriate one for the calculation of 13C NMR chemical shifts of the hydrocarbon species formed on the zeolite surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Foundation for Basic Research (grant no. 20-33-90093). This work was also supported, in part, by the Russian Academy of Sciences within the framework of budget project no. AAAA-A17-117041710084-2 for Boreskov Institute of Catalysis. NWO is acknowledged for providing access to SurfSara supercomputer resources.References
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS.
- A. H. Greif, P. Hrobarik, J. Autschbach and M. Kaupp, Phys. Chem. Chem. Phys., 2016, 18, 30462–30474 RSC.
- E. Dib, T. Mineva, E. Veron, V. Sarou-Kanian, F. Fayon and B. Alonso, J. Phys. Chem. Lett., 2018, 9, 19–24 CrossRef CAS.
- K. Chen, S. Horstmeier, V. T. Nguyen, B. Wang, S. P. Crossley, T. Pham, Z. Gan, I. Hung and J. L. White, J. Am. Chem. Soc., 2020, 142, 7514–7523 CrossRef CAS.
- E. Toomsalu and P. Burk, J. Mol. Model., 2015, 21, 244 CrossRef.
- J. Vicha, J. Novotny, M. Straka, M. Repisky, K. Ruud, S. Komorovsky and R. Marek, Phys. Chem. Chem. Phys., 2015, 17, 24944–24955 RSC.
- E. A. Pidko, ACS Catal., 2017, 7, 4230–4234 CrossRef CAS.
- J. F. Haw, B. R. Richardson, I. S. Oshio, N. D. Lazo and J. A. Speed, J. Am. Chem. Soc., 1989, 111, 2052–2058 CrossRef CAS.
- M. V. Luzgin, A. G. Stepanov, A. Sassi and J. Sommer, Chem. – Eur. J., 2000, 6, 2368–2376 CrossRef CAS.
- A. G. Stepanov, M. V. Luzgin, S. S. Arzumanov, H. Ernst and D. Freude, J. Catal., 2002, 211, 165–172 CAS.
- E. G. Derouane, H. He, S. B. Derouane-Abd Hamid, D. Lambert and I. Ivanova, J. Mol. Catal. A: Chem., 2000, 158, 5–17 CrossRef CAS.
- J. Xu, A. M. Zheng, X. M. Wang, G. D. Qi, J. H. Su, J. F. Du, Z. H. Gan, J. F. Wu, W. Wang and F. Deng, Chem. Sci., 2012, 3, 2932–2940 RSC.
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev, I. G. Danilova, I. P. Prosvirin, V. V. Kriventsov, V. I. Zaikovskii, D. Freude and A. G. Stepanov, ACS Catal., 2017, 7, 1818–1830 CrossRef CAS.
- M. V. Luzgin, D. Freude, J. Haase and A. G. Stepanov, J. Phys. Chem. C, 2015, 119, 14255–14261 CAS.
- A. A. Gabrienko, S. S. Arzumanov, I. B. Moroz, I. P. Prosvirin, A. V. Toktarev, W. Wang and A. G. Stepanov, J. Phys. Chem. C, 2014, 118, 8034–8043 CrossRef CAS.
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev, D. Freude, J. Haase and A. G. Stepanov, J. Phys. Chem. C, 2011, 115, 13877–13886 CrossRef CAS.
- S. S. Arzumanov, A. A. Gabrienko, D. Freude and A. G. Stepanov, Catal. Sci. Technol., 2016, 6, 6381–6388 RSC.
- Y. G. Kolyagin, I. I. Ivanova and Y. A. Pirogov, Solid State Nucl. Magn. Reson., 2009, 35, 104–112 CrossRef CAS.
- Y. G. Kolyagin, I. I. Ivanova, V. V. Ordomsky, A. Gedeon and Y. A. Pirogov, J. Phys. Chem. C, 2008, 112, 20065–20069 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, S. S. Arzumanov, L. V. Pirutko, A. G. Stepanov and G. I. Panov, J. Catal., 2013, 300, 47–54 CrossRef CAS.
- M. V. Luzgin, V. A. Rogov, S. S. Arzumanov, A. V. Toktarev, A. G. Stepanov and V. N. Parmon, Catal. Today, 2009, 144, 265–272 CrossRef CAS.
- J. F. Wu, W. D. Wang, J. Xu, F. Deng and W. Wang, Chem. – Eur. J., 2010, 16, 14016–14025 CrossRef CAS.
- X. M. Wang, J. Xu, G. D. Qi, B. J. Li, C. Wang and F. Deng, J. Phys. Chem. C, 2013, 117, 4018–4023 CrossRef CAS.
- A. A. Gabrienko, S. A. Yashnik, A. A. Kolganov, A. M. Sheveleva, S. S. Arzumanov, M. V. Fedin, F. Tuna and A. G. Stepanov, Inorg. Chem., 2020, 59, 2037–2050 CrossRef CAS.
- K. Narsimhan, V. K. Michaelis, G. Mathies, W. R. Gunther, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2015, 137, 1825–1832 CrossRef CAS.
- A. A. Kolganov, A. A. Gabrienko, S. A. Yashnik, E. A. Pidko and A. G. Stepanov, J. Phys. Chem. C, 2020, 124, 6242–6252 CrossRef CAS.
- V. L. Sushkevich, R. Verel and J. A. V. Bokhoven, Angew. Chem., Int. Ed., 2019, 132, 920–928 CrossRef.
- M. Dyballa, K. Thorshaug, D. K. Pappas, E. Borfecchia, K. Kvande, S. Bordiga, G. Berlier, A. Lazzarini, U. Olsbye, P. Beato, S. Svelle and B. Arstad, ChemCatChem, 2019, 11, 5022–5026 CrossRef CAS.
- A. A. Gabrienko, S. S. Arzumanov, M. V. Luzgin, A. G. Stepanov and V. N. Parmon, J. Phys. Chem. C, 2015, 119, 24910–24918 CrossRef CAS.
- A. A. Gabrienko, S. S. Arzumanov, I. B. Moroz, A. V. Toktarev, W. Wang and A. G. Stepanov, J. Phys. Chem. C, 2013, 117, 7690–7702 CrossRef CAS.
- W. Wang, M. Seiler and M. Hunger, J. Phys. Chem. B, 2001, 105, 12553–12558 CrossRef CAS.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS.
- V. Bosacek, J. Phys. Chem., 1993, 97, 10732–10737 CrossRef CAS.
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Nature, 1978, 272, 437–438 CrossRef CAS.
- B. F. Mentzen, J. M. Letoffe and P. Claudy, Thermochim. Acta, 1996, 288, 1–7 CrossRef CAS.
- B. F. Mentzen and F. Lefebvre, Mater. Res. Bull., 1997, 32, 813–821 CrossRef CAS.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- A. A. Arvidsson, V. P. Zhdanov, P. A. Carlsson, H. Gronbeck and A. Hellman, Catal. Sci. Technol., 2017, 7, 1470–1477 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. F. Izmaylov, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2006, 125, 104103 CrossRef.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. Mclean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- T. Blasco, M. Boronat, P. Concepcion, A. Corma, D. Law and J. A. Vidal-Moya, Angew. Chem., Int. Ed., 2007, 46, 3938–3941 CrossRef CAS.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260–15267 CrossRef CAS.
- S. E. Ashbrook, M. Cutajar, C. J. Pickard, R. I. Walton and S. Wimperis, Phys. Chem. Chem. Phys., 2008, 10, 5754–5764 RSC.
- J.-F. Wu, X.-D. Gao, L.-M. Wu, W. D. Wang, S.-M. Yu and S. Bai, ACS Catal., 2019, 9, 8677–8681 CrossRef CAS.
- D. Jain, Q. Zhang, J. Hightower, V. Gustin, A. Asthagiri and U. S. Ozkan, ChemCatChem, 2019, 11, 5945–5950 CrossRef CAS.
- P. N. Plessow and F. Studt, J. Phys. Chem. Lett., 2020, 11, 4305–4310 CrossRef CAS.
- M. H. Mahyuddin, A. Staykov, A. G. Saputro, M. K. Agusta, H. K. Dipojono and K. Yoshizawa, J. Phys. Chem. C, 2020, 124, 18112–18125 CrossRef CAS.
- M. H. Mahyuddin, Y. Shiota and K. Yoshizawa, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC.
- T. Anggara, C. Paolucci and W. F. Schneider, J. Phys. Chem. C, 2016, 120, 27934–27943 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculated chemical shift values and methoxide model structures. See DOI: 10.1039/d0cp04439c |
| ‡ We have made the complete dataset for this work openly available at https://doi.org/10.4121/13026671.v1. |
| This journal is © the Owner Societies 2020 |