
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1D materials from ionic self-assembly in mixtures containing chromonic liquid crystal mesogens†
Carlos
Rodríguez-Abreu
 *ab,
Yury V.
Kolen'ko
c,
Kirill
Kovnir
de,
Margarita
Sanchez-Dominguez
f,
Rekha Goswami
Shrestha
g,
Partha
Bairi
h,
Katsuhiko
Ariga
gi and
Lok Kumar
Shrestha
*g
*ab,
Yury V.
Kolen'ko
c,
Kirill
Kovnir
de,
Margarita
Sanchez-Dominguez
f,
Rekha Goswami
Shrestha
g,
Partha
Bairi
h,
Katsuhiko
Ariga
gi and
Lok Kumar
Shrestha
*g
aInstituto de Química Avanzada de Cataluña, Consejo Superior de Investigaciones Científicas (IQAC-CSIC), Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: carlos.rodriguez@iqac.csic.es
bCIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Jordi Girona 18-26, 08034 Barcelona, Spain
cInternational Iberian Nanotechnology Laboratory (INL), Av. Mestre José Veiga, Braga, 4715-330, Portugal
dDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA
eAmes Laboratory, U.S. Department of Energy, Ames, Iowa 50011, USA
fCentro de Investigación en Materiales Avanzados (CIMAV, S.C.), Unidad Monterrey, Apodaca, Nuevo León 66628, Mexico
gInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Ibaraki, Japan. E-mail: SHRESTHA.Lokkumar@nims.go.jp
hDepartment of Physics, Jadavpur University, Kolkata 700032, India
iGraduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8561, Japan
First published on 3rd October 2020
Abstract
Ionic self-assembly is a simple yet powerful method to obtain robust nanostructures. Herewith, we use mixtures of oppositely-charged porphyrins that can act as mesogens to form chromonic liquid crystals in water, i.e., molecular stacks with orientational (nematic) or positional (hexagonal) order. Electrostatic locking coupled with π–π interactions between aromatic groups within the stacks, together with inter-stack hydrogen bonding induce formation of all-organic crystalline nanofibers with high aspect ratio (a few tenths of nanometers in width but several tenths of micrometers in length) and that display branching. The nanofibers prepared from metal-free porphyrin units feature interesting optical properties, including an absorption spectrum that is different from the simple sum of the individual spectra of the components, which is attributed to a striking aggregation-induced chromism. When in contact with some polar organic solvents the materials become fluorescent, as a result of disaggregation. In a proof-of-concept, the obtained self-assembled one-dimensional (1D) materials were carbonized (yield ca. 60%) to produce nitrogen-doped carbon nanofibers that can be used as active electrode materials for energy storage applications.
1. Introduction
Molecular self-assembly is an interesting and versatile method to produce nanostructures. Being spontaneous, it is intrinsically minimalistic in terms of energy input, and it does not require sophisticated equipment, which makes it amenable for scaling up.1 Self-assembly frequently takes place in aqueous media, which favors sustainability and biocompatibility.Molecular self-assembly is driven by weak interactions such as hydrogen-bonding, hydrophobic effects, electrostatics, van der Waals and π–π interactions, and certain building blocks allow for multiple interactions. Porphyrin are examples of such blocks and are associated to chromophores found in Nature that play an important role in biological functions, such as photosynthesis, molecular recognition and catalysis.2,3 These molecules can also hold different metals in the macrocycle via complexation with the pyrrole nitrogens; some of these porphyrin–metal complexes are found in nature and are biologically relevant (e.g. heme porphyrin). Porphyrin–metal complexes can also generate composite nanostructures.4
Semiconducting properties derived from aromaticity of porphyrins are of particular interest, and impart significant electron mobility, especially in stacked structures. Because of the aforementioned optoelectronic properties, self-assembled porphyrin nanomaterials have potential applications in photocatalysis, dye-sensitised solar cells, photodynamic therapy, hydrogen production, sensors, carbon dioxide reduction, and electrocatalysts for fuel cells, among others.5–9
Chromonic liquid crystals are a class of self-assembled structures formed in a solvent (usually aqueous) by planar molecular building blocks that tend to arrange into columns similar to thermotropic discotic liquid crystals. Chromonic assembly is driven by π–π interactions between the aromatic moieties. Porphyrins, being polyaromatic and planar with peripheral polar groups, have the structural characteristics required to form chromonic liquid crystals. In fact, thermotropic and lyotropic mesophases of the nematic and hexagonal type have been reported for some nonionic and ionic porphyrins.10,11 Chromonic liquid crystals can also be formed by a variety of molecules, notably dyes such as cyanines, xanthenes,12 azo compounds and perylenes. Among them, the dye sunset yellow is one of the most studied chromonic mesogens.13
Chromonic assemblies are soft materials and therefore fragile under certain conditions. To make them more robust, it is necessary to fix the structure. One strategy is their coassembly with inorganic species via sol–gel to form mesostructured oxides.14–17 Another possibility would be to use additional intermolecular interactions for fixation. In this regard, ionic self-assembly is a useful technique for the synthesis of nanomaterials, which consists in the coupling of structurally different ionic building blocks (charged tectons) by electrostatic interactions.18–20
There are several reports on ionic self-assembly of porphyrins, that result in the formation of nanorods,21,22 nanofibers,4 nanotubes,23 nanosheets,24 branched structures25 and other morphologies.26 However, to our knowledge, carboxy substituted porphyrins have not been used as anionic tectons in the assemblies. They are of interest since carboxy groups are present in natural porphyrins. Moreover, the carboxy group can improve selectivity in biotargeting and also facilitate binding and functionalization.
Nanostructured carbon is, in many cases, produced in several steps. A typical procedure is based on the use of a hard template such as mesoporous silica (in turn produced via surfactant templating) that is impregnated with a carbon precursor to be finally carbonized.16 It would then be interesting to produce directly carbon nanofibers from a self-assembled carbon precursor without the need of a hard template intermediate. In this regard, chromonics have an inherent ability to form 1D structures, that are useful in a plethora of applications,27–29 but literature on the direct carbonization of chromonic ionic assemblies is scarce. Moreover, the fabrication of 1D materials via self-assembly may have advantages in terms of e.g. costs and potential of scaling up as compared to other methods such as electrospinning or the use of porous alumina templates.30
Herein we present a report on the ionic self-assembly of chromonic mesogens in water, in which π–π stacking interactions also play a role. Our objective was to demonstrate that chromonic mesogens can serve as building blocks for the preparation of organic and carbon 1D materials with novel properties; the possibility of designing chromonic tectons with different architectures opens perspectives for the control of the materials’ dimensions. For that, we first explore and describe the mesomorphic chromonic behavior of a carboxy-substituted porphyrin, and then the mixtures of this anionic porphyrin with a cationic one (both free-base or metal free) are studied using a variety of characterization techniques. The formation of carbon fibers from the ionic assemblies is also presented to demonstrate the use of the fibers as hard templates and their potential applications in electrochemical supercapacitance are explored. This work builds on previous publications on porphyrin assembly2,26 to provide new insights, with the support of multiple instrumentation, on the formation of lyotropic liquid crystals in these systems and the fixation of such as liquid crystalline structures into 1D materials via ionic interactions. Moreover, to our knowledge this constitutes one of the few examples of the use of porphyrin-based 1D materials produced via wet chemistry as templates for the synthesis of carbon nanofibers.
2. Experimental
2.1 Materials
Mesogens tetrakis(4-carboxyphenyl)porphyrin (TCPP) and tetrakis(1-methylpyridinium-4-yl)porphyrin p-toluenesulfonate (TMPyP, Scheme 1b) were obtained from TCI Europe and were used without further purification. TCPP-Na (Scheme 1a) was obtained by neutralization with sodium hydroxide (TCPP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH molar ratio = 1:4). Millipore water (resistivity = 18.2 MΩ cm) was used in all experiments.
:NaOH molar ratio = 1:4). Millipore water (resistivity = 18.2 MΩ cm) was used in all experiments.
 | ||
| Scheme 1 Schematic molecular structures of (a) TCPPNa and (b) TMPyP. | ||
2.2 Preparation of ionic assemblies and carbon fibers
Aqueous solutions of TCPP-Na and TMPyP were prepared separately using Millipore water and then mixed at TCPP-Na:TMPyP molar ratio = 1:1. The mixture was left under stirring overnight in the dark. Then, the mixture was centrifuged at 4000 rpm and the precipitate was washed several times with Millipore water, until the supernatant was almost colorless. To obtain carbon fibers from the resultant 1D assemblies, the product was heat-treated at 1000 °C in a tubular furnace (KOYO, Tokyo, Japan) under a constant flux of nitrogen (120 cc min−1) with temperature ramp of 5 °C min−1 and hold time 1 h.
2.3 Characterizations
Small-wide angle X-ray scattering (SAXS/WAXS) patterns of liquid crystals were collected at 20 °C with an Anton Paar SAXSess mc2 instrument. Samples were placed in flame-sealed glass capillaries (wall thickness = 0.01 mm, diameter = 0.9 mm) for the measurement.SAXS measurements on dispersions were performed in the beamline NCD11 at ALBA Synchrotron Barcelona (Spain). The energy of the incident beam was 12.6 keV (λ = 0.995 Å). Samples were placed in wax-sealed quartz capillaries (Hilgenberg, Germany) with an outside diameter of 1.5 mm and wall thickness of 0.01 mm. The scattered X-rays were detected on a Pilatus 1 M detector (Dectris). Data on each sample were collected over 60 s.
Materials were also characterized by powder X-ray diffraction (XRD, Rigaku RINT2000), Raman spectroscopy (Jobin-Yvon T64000), scanning electron microscopy (SEM, Hitachi S-4800, operated at 10 kV), and transmission electron microscopy (TEM, JEOL Model JEM-2100F operated at 200 kV). A theta probe spectrometer (Thermo Electron Co. Germany) was used for the X-ray photoelectron spectroscopy (XPS) measurements. The instrument is equipped with a monochromatic Al-Kα radiation source (photon energy 15 keV, energy resolution ≤0.47 eV, space resolution ≤15 μm). C 1s, O 1s, and N 1s core-level XPS spectra were recorded in 0.05 eV steps. A built-in electron flood gun prevented sample charging. The Avantage™ software was used to deconvolute the XPS spectra. Data were fitted with line width and height as free parameters. Thermogravimetric analysis (TGA) was performed for the evaluation of thermal stability of carbon samples. The TGA measurements were performed on the Hitachi HT-Seiko 6300 instrument while heating samples from 30 to 1000 °C under argon atmosphere (temperature ramp 10 °C min−1). Nitrogen adsorption/desorption isotherm was recorded at 77.35 K on Quantachrome Autosorb-iQ2, and specific surface area, pore size distribution and pore volumes were estimated. Surface characterization of the samples was carried out in an atomic force microscope from Asylum Research, model MFP3D-SA, employing the Tapping mode technique (AC mode) in air. Samples were scanned in areas of 20 × 20 μm, 5 × 5 μm, 2.5 × 2.5 μm, 2 × 2 μm, 1.5 × 1.5 μm and 1 × 1 μm, in a speed interval from 0.20 to 0.50 Hz. A rectangular cantilever (AC240TS-R3) was used, with a nominal first resonance frequency of 70 kHz, a nominal spring constant of 2 N m−1 and a nominal radius of curvature of 7 nm.
2.4 Electrochemical measurements
Cyclic voltammetry (CV) and galvanostatic charge–discharge (CD) measurements were carried out on a three-electrode system in an aqueous electrolyte (1 M H2SO4) at 25 °C. Glassy carbon electrode (GCE) was modified with the prepared carbon fiber and used as the working electrode. Typically, carbon fiber was dispersed in solvent mixture (water:ethanol = 4:1; 1 mg mL−1) and sonicated for 60 min. The suspension (5 μL) was dropped onto the clean and dry GCE surface and dried at 60 °C for 3 h. Ionomer Nafion solution (5 μL: 5% in ethanol) was added on top of the carbon fiber as a binder and dried at 60 °C for 12 h before electrochemical measurements. A platinum wire and Ag/AgCl were used a counter and reference electrode, respectively. The CV and CD profiles were recorded at 25 °C on a CH instruments (CHI 850D Work station).
3. Results
3.1 Chromonic behavior of TCPPNa and TMPyP
Bottom-up fabrication of nanomaterials relies on the self-assembly of molecular tectons. Accordingly, the behavior of TCPPNa and TMPyP as mesogens was investigated by looking at the occurrence of chromonic liquid crystals in water. First, contact experiments (so called Lawrence tests) were carried out. TCPPNa was found to form lyotropic liquid crystals in contact with water, with nematic and hexagonal textures (Fig. 1a), whereas TMPyP only formed hydrated crystals with a spherulite texture (Fig. 1b). Anionic chromonics (e.g. anionic cyanine dyes) are generally more soluble than their cationic counterparts, and memorphism is determined by the characteristics of the peripheral charged groups and counterions.14 The degree of hydration of the counterion plays an important role in the solubility of these molecules. | ||
| Fig. 1 Polarized optical microscopy images of TCPPNa (a) and TMPyP (b) in contact with water. In the images, water penetration and diffusion occur from the black to the bright regions. N: nematic phase. M: hexagonal phase. | ||
The structure of the liquid crystals formed by TCPPNa was analyzed by SAXS/WAXS. At 30 wt% TCPPNa in water (Fig. 2a), the SAXS/WAXS pattern shows an inflexion point at ca. 2 nm−1 and a broad peak at ca. 5 nm−1, corresponding to correlation lengths between aggregates inside the nematic phase (which was identified by polarized optical microscopy); the absence of sharp peaks is typical in this phase, since it displays orientational but not positional order. There is also a peak at 16 nm−1, equivalent to a Bragg distance of 0.4 nm, which is ascribed to the stacking distance between TCPPNa molecules. This relatively short distance would suggest π–π interactions and cofacial arrangement along the column axis.
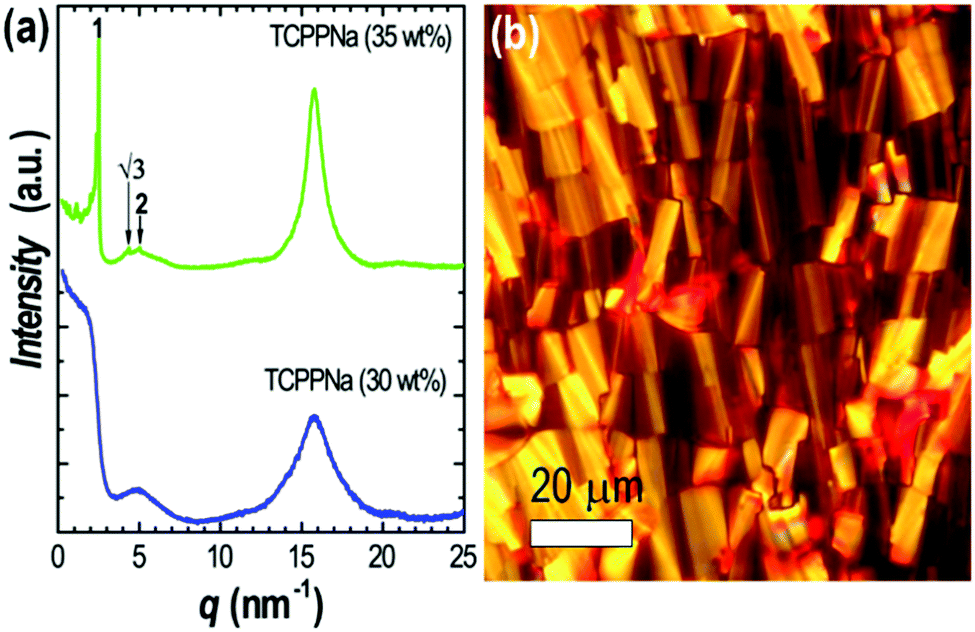 | ||
| Fig. 2 (a) SAXS/WAXS patterns of TCPPNa at 30 and 35 wt% in water, and (b) polarized optical microscope image of liquid crystal at 35 wt% in water. | ||
Upon increasing TCPPNa concentration to 35 wt% (Fig. 2a), a sharp and strong peak appears at ca. 2.5 nm−1; two weaker peaks are also observable at ca. √3 and 2 position ratios relative to the strongest peak (marked by arrows), indicating the presence of a hexagonal (columnar) phase, which agrees with the fan-like optical texture of the sample (Fig. 2b). Again, the stacking distance peak (at ca. 16 nm−1) is noticeable at this concentration.
The hexagonal phase can be viewed as formed by aligned molecular columns. The diameter of such columns (D) can be approximately estimated assuming cylindrical shape by D = 2d(2ϕ/(π√3))0.5, where ϕ is the volume fraction of TCPPNa, and d = 2π/q is the Bragg spacing, calculated from the scattering vector q of the first, strongest peak. If a density of ca. 1.5 nm is assigned to TCPPNa, then D = 1.6 nm, close to the size of a single TCPPNa molecule.31 This suggests that the hexagonal phase is formed by aligned, one-molecule-width porphyrin columns. SAXS measurements at higher temperatures indicated that the hexagonal phase with 35 wt% TPCPPNa is stable at least up to 80 °C, showing no appreciable change in the unit cell parameter.
3.2 Formation of fibers from porphyrin assemblies in water
The cationic and anionic porphyrins used herewith as tectons are separately fully soluble in water before mixing at the concentrations studied. These tectons already form aggregates in water from very low concentrations (e.g. dimers), as can be confirmed by UV-spectroscopy and widely reported in the literature.32–36 Mixing of the two oppositely charged porphyrins leads to the formation of an ionically assembled solid, since the net charge is zero or negligible, and therefore, the solubility of the precursor porphyrins is drastically reduced. Such solid forms, however, a dispersion in water with transient stability. The aspect of the two separated TCPP and TMPyP solutions and their mixture (ionic assembly) is shown in Fig. 3a. There is a change from reddish to green color upon assembly. Microscopic observation revealed that the obtained green dispersion is formed by birefringent fibers (Fig. 3b and c). The fibrous morphology of the assembly can also be seen in the AFM image (Fig. S1, ESI†). The change in optical properties is reflected in the UV-vis absorption spectra (Fig. 3d), i.e. the spectrum of the ionically assembled fibers in water is different from the sum of the individual spectra of the cationic and anionic precursor solutions, suggesting complexation.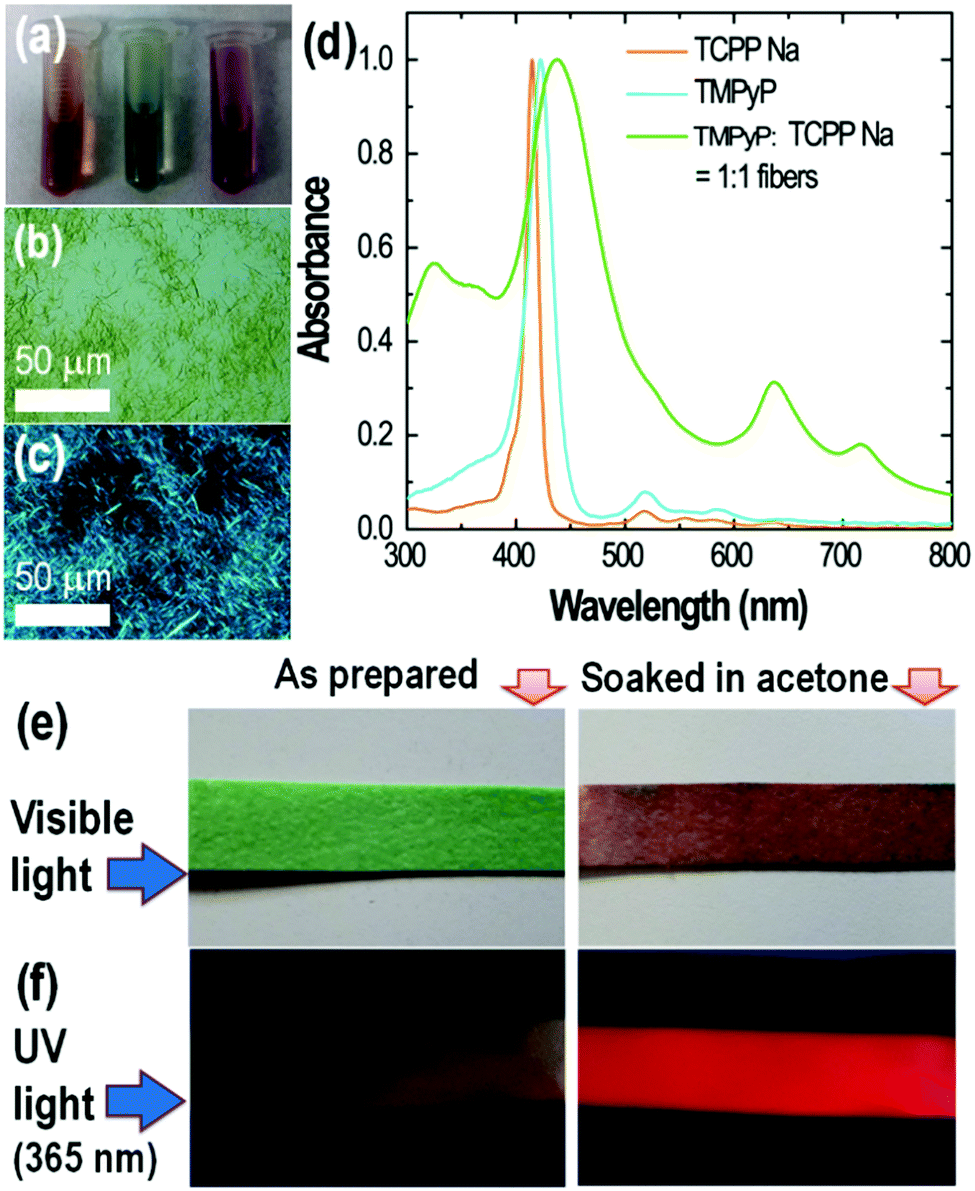 | ||
| Fig. 3 (a) Samples of TMPyP (left), TMPyP–TCPPNa equimolar mixture (middle) and TCPPNa (right) in water. The porphyrin concentration is 10−4 M. Microscopic images of TMPyP–TCPPNa equimolar mixture in water showing fibers under non-polarized (b) and polarized (c) lights. (d) Normalized UV-vis spectra of TMPyP, TCPPNa and TMPyP–TCPPNa equimolar mixture (i.e., fibers) in water. Solvatochromic behavior of porphyrin self-assembled fibers in acetone under visible light (e) and UV light irradiation (f). | ||
The Soret bands of TCPPNa and TMPyP are at 415 and 423 nm, respectively. Upon mixing and assembly, there is broadening and splitting of the band into a blue-shifted one at 330 nm and a red shifted one at 438 nm. This has been reported as a signature of molecular coupling in ionic self-assemblies with strong electronic interaction.8 Also, new Q-bands at 635 and 715 nm appear, i.e. different from the Q bands of TCPPNa and TMPyP. The features of the absorption spectrum of the fibers could be attributed to the presence of H and J-aggregates,23,37 with possible contribution of resonance light scattering additional to absorption at long wavelengths.24
The formed fibers were stable when redispersed in acidic (pH = 2) and alkaline (pH = 12) media, and also at high ionic strength (e.g., 20 g L−1 or 0.34 M NaCl), but were disrupted by certain organic polar solvents. Accordingly, if a filter paper with embedded fibers (i.e., by immersion in the aqueous fiber dispersion following by drying) is soaked in acetone, the fibers are disrupted into their building porphyrin blocks and the color changes from green to dark red-brownish (Fig. 3e and f). Ionic assembly results in quenching of the fluorescence relative to that of TCPPNa and TMPyP tectons in water.38–40 Therefore, the filter paper with embedded TMPyP–TCPPNa fibers is not fluorescent, but turns luminescent under UV-light after soaking in acetone (Fig. 3f), indicating that the fibers are disrupted and the photoluminescence of TCPPNa and TMPyP is somehow recovered.38,41 Some applications of this solvatochromic effect and disaggregation-induced fluorescence could be foreseen in sensing of volatile organic compounds and other substances.42,43
In order to get more insight into the structure of the TMPyP–TCPPNa fibers in aqueous dispersion, SAXS measurements were carried out (Fig. S2, ESI†). At low angles, the data can be fitted to a power law I(q) ∝ q−f where f is the fractal dimension. For the samples of TMPyP–TCPPNa fibers, f = 2.6, which implies a mass fractal object. A similar value (2.2) was found for carbon nanotubes,44 and it was interpreted as scattering from branched rope clusters, which is somehow in agreement with the microscopy observations described below.
We next analyzed the surface morphology of the TMPyP–TCPPNa fibers by scanning electron microscopy (SEM). As can be seen in Fig. 4a–d, self-assembly leads to the formation of nanofibers with multiple branches (shelf-like), as previously reported for certain porphyrin systems.25 The nanofibers, that can reach several tenths of micrometers in length, are constituted by bundles of molecular belts or tapes of around 30 nm in width. The fibers are also formed at much lower concentrations (10−5–10−4 M) of the precursor porphyrin solutions (Fig. S3, ESI†). Self-assembled TMPyP–TCPPNa fibers were also observed by transmission electron microscopy (TEM, Fig. 4e and Fig. S4a–d, ESI†). HRTEM images (Fig. 4f and Fig. S4e, f, ESI†) show aligned stripes inside the fibers (lattice fringes), with a width of ca. 1 nm, similar to the size of one porphyrin molecule. This indicates that the fibers are crystalline, as confirmed by X-ray diffraction (XRD, Fig. S5, ESI†) and spot type selected area electron diffraction (inset in Fig. 4f). The results suggest a hierarchical organization mechanism (see Scheme 2). We speculate that the porphyrin molecules first stack face-to-face via π–π interactions to form columns with monomolecular cross section. Then, these columns bundle to form the fibers, probably by hydrogen bonding although electrostatics can also play a role.2,45,46 This mechanism would be analogous to the assembly in the hexagonal chromonic liquid crystals described above. However, additional studies are needed to properly assess the contributions of each type of interactions in the self-assembly process.
 | ||
| Fig. 4 (a–d) SEM images of TMPyP–TCPPNa fibers, as well as (e) TEM and (f) HR-TEM images of the fibers. Inset in (f) shows selected area electron diffraction pattern. The concentration of the precursor porphyrin solutions was 0.05 M. | ||
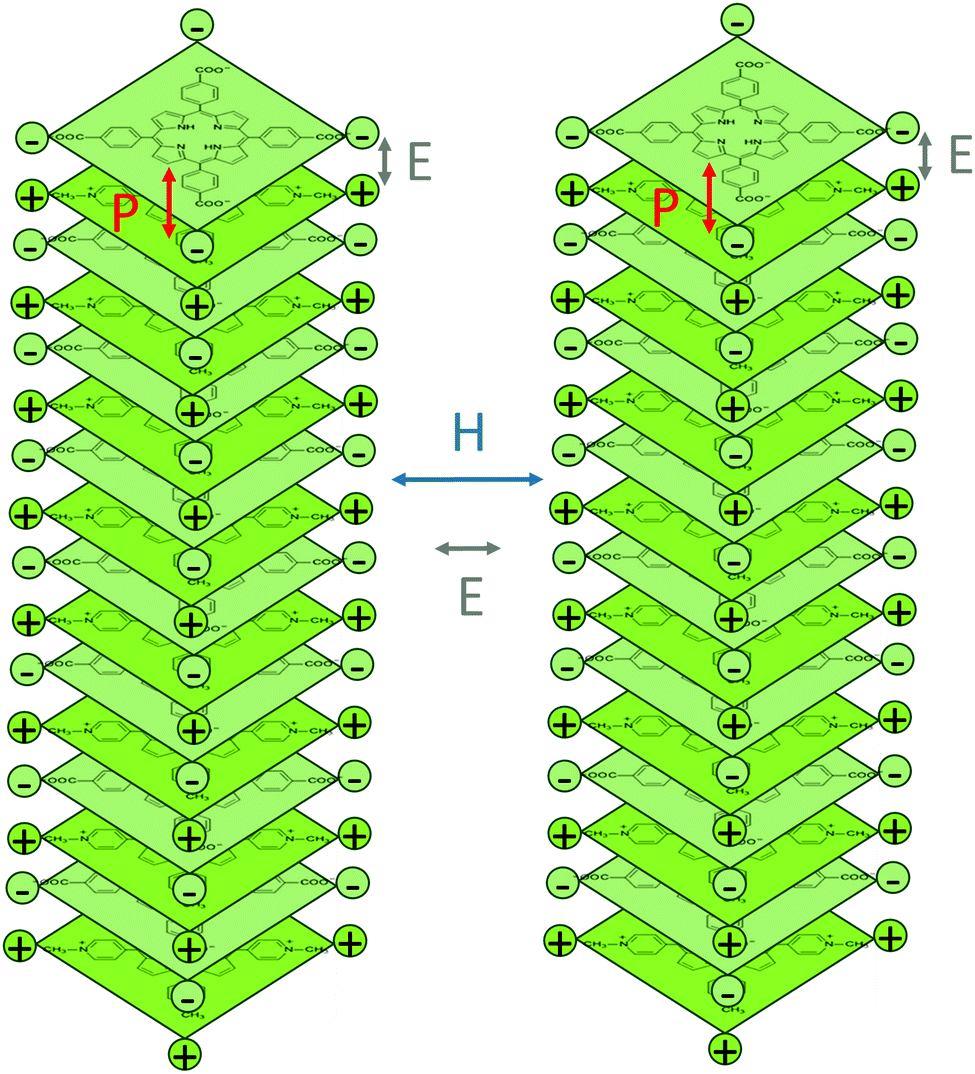 | ||
| Scheme 2 Possible configuration of porphyrin molecules in the columnar aggregates and interactions likely involved in the hierarchical self-assembly. P: π–π stacking; E: electrostatics; H: hydrogen bonding. | ||
Mazur et al. have carried out comprehensive crystallographic analyses of porphyrin self-assembled 1D structures.25,45,47 Indexing of the most of the diffraction peaks in the powder XRD pattern shown in Fig. S5 (ESI†) was possible in different unit cells. First it was attempted in the cell similar to one reported by Mazur et al. such indexing resulted in an orthorhombic primitive cell with unit cell parameters a = 30.60(5) Å, b = 22.32(2) Å, and c = 7.963(5) Å. An indexing with higher figure-of-merit was obtained in the cell with half of the volume as compared to one reported by Mazur: an orthorhombic primitive cell with a = 18.00(1) Å, b = 17.72(3) Å, and c = 8.830(7) Å. Alternatively, primitive tetragonal cell with volume similar to that of Mazur's cell volume can describe all the peaks: a = 17.69(1) Å, c = 17.92(2) Å. At this stage, powder XRD data is not sufficient to get a definitive answer regarding true symmetry and unit cell dimensions of the studied compounds, and a single crystal diffraction experiment is required.
XPS survey spectra, core level C 1s, O 1s, and N 1s with deconvoluted peaks (Fig. S6, ESI†) confirm that the surface of the assembled 1D fibers mainly contain C, O, and N elements. The N 1s signals observed at 401, 399 and 397 eV can be assigned to pyridinium, pyrrole and imine nitrogens, respectively. The ratios of the areas corresponding to the deconvoluted peaks are close to 1:1:1, suggesting that the TMPyP:TCPPNa molar ratio in the fibers is close to one, similar to native solution.
3.3 Formation of carbon fibers from porphyrin 1D assemblies
To explore the formation of carbon materials from the as-prepared TMPyP–TCPPNa fibers, thermogravimetric analysis (TGA) was performed (Fig. S7, ESI†), and it was found that the fibers have a high carbon yield (ca. 60% at 1000 °C). Following this result, the porphyrin nanofibers were drop casted from their aqueous dispersions on silicon wafers, and the films obtained after drying were carbonized at 1000 °C with a heating rate of 10 °C min−1 under N2 atmosphere. Notably, the carbonized materials preserve the fiber-like structure: they appear under SEM (Fig. 5a–d) as a mat of highly entangled fibers that are sintered. Fiber-like morphology with amorphous graphitic carbon structure can also be seen in TEM (Fig. 5e) and HRTEM images (Fig. 5f). Pyrolysis of the dry TMPyP–TCPPNa nanofibers powder at 1000 °C under the N2 atmosphere also yielded carbon fibers (Fig. S8 and S9, ESI†). | ||
| Fig. 5 (a–d) SEM images of the carbon fibers obtained by the carbonization of pristine TMPyP–TCPPNa fibers, together with the (e) TEM and (f) HR-TEM images of the carbon fiber. Inset in panel (f) shows selected area electron diffraction pattern. The concentration of the precursor porphyrin solutions was 0.05 M. | ||
XRD of the carbon fibers (Fig. 6a) showed two very broad peaks at 2θ of 25° and 44° indicating a graphite-like structure similar to the one observed in some multiwall carbon nanotubes or other graphitic carbons.48 The Raman scattering spectrum of the resultant carbon fibers displays two main peaks at 1331 and 1573 cm−1, corresponding to D (disordered) and G (graphitic) bands of carbon materials (Fig. 6b).49,50 Intensity ratio of G and D band, IG/IDca. 1.03 confirms the graphitic structure, which contribute to high conductivity of the carbon fibers. A broad band at ca. 2900 cm−1 (2D band, also known as G′) is interpreted as an overtone of D, and in the present case is weaker and broader than in highly ordered graphitic samples.51
 | ||
| Fig. 6 (a) XRD pattern, (b) Raman scattering spectrum, (c) XPS survey and XPS core level C 1s (d) and N 1s (e) spectra of the carbon fibers. Deconvoluted peaks are also included in (d) and (e). | ||
XPS survey spectrum shows C, O, and N as the major elements in the carbon fibers demonstrating that the high-temperature heat-treatment of the TMPyP–TCPPNa fibers results in the formation of heteroatoms (N and O) doped carbon fibers, which is desired in several applications such as in high-performance supercapacitance, electrocatalysis and photocatalysts. As it can be seen in Fig. 6d, XPS C 1s spectrum could be deconvoluted into four peaks centered at 284.2, 285.9, 285.8, and 287.7 eV, respectively corresponding to the C–C (sp3), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2), C–N (sp3), and O–CO bonding states of carbon.52 Similarly, XPS core level N 1s peaks could be deconvoluted into four peaks corresponding to pyrrolic (399.2 eV, weak), pyridinic (398.2 eV), graphitic (401.1 eV) and oxidized nitrogen (402.2 eV).53 XPS core level O 1s spectrum with deconvoluted peaks are presented in Fig. S10 (ESI†). From the XPS spectra the N/C ratio in the carbon fibers was estimated to be 0.065.
C (sp2), C–N (sp3), and O–CO bonding states of carbon.52 Similarly, XPS core level N 1s peaks could be deconvoluted into four peaks corresponding to pyrrolic (399.2 eV, weak), pyridinic (398.2 eV), graphitic (401.1 eV) and oxidized nitrogen (402.2 eV).53 XPS core level O 1s spectrum with deconvoluted peaks are presented in Fig. S10 (ESI†). From the XPS spectra the N/C ratio in the carbon fibers was estimated to be 0.065.
N2 adsorption/desorption isotherm of the carbon fiber (Fig. S11a, ESI†) displays mixed type-I/type-IV behavior typical of microporous materials with partial mesopores framework structure.53 The pore size distributions estimated by Barrett–Joyner–Halenda (BJH) method (Fig. S11b, ESI†) and density functional theory (DFT) method (Fig. S11c, ESI†) show peaks at 3.09 and 1.38 nm. The specific surface area and total pore volume were respectively ca. 194.2 m2 g−1 and 0.313 cm3 g−1. Usually, graphitization upon heat treatment is detrimental to surface area because structural defects are reduced.54
We further investigate supercapacitive properties of the derived C fibers. The electrode showed quasi-rectangular CV profiles over a wider range of scan rates from 5 to 300 mV s−1 (Fig. 7a) indicating the electrical double-layer capacitive behavior. The specific capacitance was ca. 205.6 F g−1 at 5 mV s−1. The carbon fiber electrode showed relatively good capacitance retention at fast sweep of 300 mV s−1 sustaining about 45.4% initial capacitance (Fig. 7b). Galvanostatic charge–discharge measurements performed at different current densities from 1 to 20 A g−1 (Fig. 7c) also confirm the supercapacitive behavior giving specific capacitance of 249.2 F g−1 at 1 A g−1 (Fig. 7d), which is higher than that of the commercial activated carbons and comparable to that of the high surface area nanoporous activated carbons derived from different biomass.42,46 The electrochemical (CV and CD) measurements demonstrate that the heteroatoms-doped carbon fiber obtained from the direct carbonization of the TMPyP–TCPPNa fibers could be a promising materials for energy storage applications. However, it is necessary a further improvement of surface textural properties including surface area and porosity by the chemical activation method with dehydrating agents such as potassium hydroxide or zinc chloride.
 | ||
| Fig. 7 (a) CV profiles at different scan rates (5–300 mV s−1), (b) the specific capacitance calculated from CV curves vs. scan rate, (c) galvanostatic CD profiles at different current densities (1–20 A g−1), and (d) the specific capacitance calculated from CD curves vs. current density. | ||
4. Discussion
The pyrolysis of polyaromatic compounds, especially those containing a complexed metal, for producing nanostructured carbons has been developed in the last decade, encouraged by their potential application as catalysts.55,56 Such efforts included the use of discotic mesophases as precursors anchored to hard templates such as porous alumina.57–59 Chromonic liquid crystals of indanthrone disulfonate coated on a solid substrate were also studied by Hurt as carbon precursors.60 Different from those approaches, the 1D materials introduced herein are produced by a hard-template-free process, which requires a robust self-assembled precursor that can retain morphology at high temperatures and upon carbonization with reasonable carbon yield. The collected experimental evidence suggests that the robustness comes from the strength of coupled interactions. On one hand, π–π interactions that produce very long stacks and liquid crystals as a result of inter-stack structuration. On the other hand, the electrostatic interactions between peripheral charged groups in the porphyrin molecules. Besides structural integrity, this coupled interactions give rise to singular optical properties that can be manipulated to certain extent, for instance by disrupting the ionic assembly with addition of a good solvent.The 1D materials studied here are formed from equimolar mixtures of oppositely charged porphyrin tectons; this equimolarity appears to be preserved within the 1D materials, as suggested by XPS. SAXS measurements in the aqueous assemblies confirms that anionic porphyrin can stack face to face to form one-molecule-width columns. But electrostatics and hydrogen bonding induce these columns to bundle so that thicker but still crystalline fibers are formed. The ionic self-assembly strategy presented herein have also been tested for a variety of porphyrin tectons both metallized and non-metallized (free-base), the main variation being the core metal (Cu, Ni, Ag, and Zn)4 and the peripheral groups, which mainly consist of ammonium as cations and sulfonate as anions. These peripheral groups determine the pH-dependent protonation states. In this respect, Mazur et al.25 obtained shelf-like porphyrin assemblies from tetra(4-aminophenyl)porphyrin (TAPP), and tetra(4-sulfonatophenyl)porphyrin (TSPP) under acidic pH for porphyrin ion charges of 2+ and 2−. They attributed the morphology to crystal splitting during initial oriented attachment growth followed by Ostwald ripening. In the present case, shelf-like structures are also produced although the charges are 4+ and 4− at the pH of the experiments. The morphology of the porphyrin assemblies is strongly dependent on precipitation conditions (e.g., solvent).26 For example, the ion pair of TMPyP and TSPP45 (quite close to the one studied herein) produced straight, non-branched fibers, a morphology quite different to the one reported herein. The reason can be probably found in the use of additional solvents during precipitation. An aromatic donor–acceptor interaction was suggested for that pair, which might be also the case for the present TMPyP–TCPPNa pair.
As for the optical properties, the appearance of new and broad absorption bands at long wavelengths that cause the striking changes in optical properties upon assembly have been marked as a signature of coherent coupling of the transition dipoles, similar to the case of TSPP–Sn(IV) meso-tetra(4-pyridyl)porphyrin dichloride,23 which also showed almost complete fluorescence quenching upon assembly. Notably, a ion pair TMPyP:TSPP similar to the one studied here does not show any new absorption bands upon ionic assembly, pointing to weaker electronic interaction and absence of delocalized states for photoexcitation.2
Although the original fiber-like morphology of the porphyrin precursors was preserved after carbonization, we did not observe ordered graphene layers as those obtained by Müllen et al. from discotic molecules.58 This could be attributed to both the method of carbonization and the chemical structure of the precursors. The obtained 1D carbons are nitrogen-doped by an endogenous source and this impacts the observed supercapacitor properties, which could be improved if the specific surface area is increased by introducing additional microporosity. Besides the simplicity of preparation, another advantage of using the present TMPyP–TCPPNa fibers as carbon precursors is that their aqueous dispersions can be formulated as inks that can be printed or patterned on substrates directly and subjected to carbonization.
5. Conclusions
One-dimensional (1D) nanomaterials were successfully prepared by simple mixing of oppositely charged aromatic building blocks. This robust fabrication strategy is based on self-assembly of porphyrins into molecular columns. The assembly is driven by π–π interactions among blocks, thus enabling chromonic liquid crystal phases. Further, the coupled ionic interactions between peripheral ionic groups lock the 1D structure and trigger hierarchical bundling into fibrous structures. This singular kind of organization produce interesting optical properties such as aggregation- and solvent-induced chromism and luminescence, promising for sensing applications The as-formed fiber-like materials are crystalline and robust enough to withstand carbonization and preserve their mat-like starting 1D morphology yielding carbon nanofibers with promising supercapacitive behavior. Further laboratory efforts are currently on the way for the improvement of properties and development of proofs-of-concept for sensing and supercapacitor applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
C. R. acknowledges the financial support from Generalitat de Catalunya (2017SGR1778 grant), and from the Spanish Ministry of Science, Innovation and Universities (CTQ2017-84998-P project). C. R. is also grateful to the Agencia Estatal de Investigación and to the European Regional Development Fund (Fondo Europeo de Desarrollo Regional, FEDER). We thank ALBA staff for their help and support in the Synchrotron SAXS experiments performed at BL11-NCD beamline at ALBA (Spain). This work is partially supported by supported by JSPS KAKENHI Grant Number JP 16H06518 (Coordination Asymmetry), CREST JST Grant Number JPMJCR1665, and Grants-in-Aid for Scientific Research (C) Grant Number 20K05590.References
- M. Lazzari, C. Rodríguez-Abreu, J. Rivas and M. Arturo López-Quintela, Self-assembly: A minimalist route to the fabrication of nanomaterials, J. Nanosci. Nanotechnol., 2006, 6, 892–905 CrossRef CAS.
- U. Mazur and K. W. Hipps, A Systematic Approach toward Designing Functional Ionic Porphyrin Crystalline Materials, J. Phys. Chem. C, 2018, 122, 22803–22820 CrossRef CAS.
- Y. Zhong, J. Wang and Y. Tian, Binary ionic porphyrin self-Assembly: Structures, and electronic and light-harvesting properties, MRS Bull., 2019, 44, 178–182 CrossRef.
- Z. Wang, K. J. Ho, C. J. Medforth and J. A. Shelnutt, Porphyrin nanofiber bundles from phase-transfer ionic self-assembly and their photocatalytic self-metallization, Adv. Mater., 2006, 18, 2557–2560 CrossRef CAS.
- S. V. Bhosale and D. Duc La, Nanoscale porphyrin superstructures: Properties, self-assembly and photocatalytic applications, SPR Nanosci., 2019, 5, 57–85 CAS.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Porphyrinoids for Chemical Sensor Applications, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS.
- J. Wang, Y. Zhong, X. Wang, W. Yang, F. Bai, B. Zhang, L. Alarid, K. Bian and H. Fan, PH-Dependent Assembly of Porphyrin-Silica Nanocomposites and Their Application in Targeted Photodynamic Therapy, Nano Lett., 2017, 17, 6916–6921 CrossRef CAS.
- J. Wang, Y. Zhong, L. Wang, N. Zhang, R. Cao, K. Bian, L. Alarid, R. E. Haddad, F. Bai and H. Fan, Morphology-Controlled Synthesis and Metalation of Porphyrin Nanoparticles with Enhanced Photocatalytic Performance, Nano Lett., 2016, 16, 6523–6528 CrossRef CAS.
- B. Baptayev, S. H. Lee, D. H. Kim and M. P. Balanay, Controlling aggregation using self-assembled axially coordinated pyridine-based ligands on porphyrin analogues for dye-sensitized solar cells, Chem. Phys. Lett., 2019, 730, 407–410 CrossRef CAS.
- N. B. Mckeown and J. Painter, Lyotropic and Thermotropic Mesophase Formation of Novel Tetra-[oligo(ethyleneoxy)]-substituted Phthalocyanines, J. Mater. Chem., 1994, 4, 1153–1156 RSC.
- N. Usol’tseva, Lyotropic behaviour of sheet-like chemical compounds: Amphotropy of phthalocyanine and porphyrin derivatives, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 288, 201–210 CrossRef.
- J. R. Magana, M. Homs, C. Solans, M. Obiols-Rabasa, L. M. Salonen and C. Rodríguez-Abreu, Self-Assembly and Formation of Chromonic Liquid Crystals from the Dyes Quinaldine Red Acetate and Pyronin y, J. Phys. Chem. B, 2016, 120, 250–258 CrossRef CAS.
- D. J. Edwards, J. W. Jones, O. Lozman, A. P. Ormerod, M. Sintyureva and G. J. T. Tiddy, Chromonic liquid crystal formation by edicol sunset yellow, J. Phys. Chem. B, 2008, 112, 14628–14636 CrossRef CAS.
- C. Rodríguez-Abreu, C. A. Torres and G. J. T. Tiddy, Chromonic liquid crystalline phases of pinacyanol acetate: Characterization and use as templates for the preparation of mesoporous silica nanofibers, Langmuir, 2011, 27, 3067–3073 CrossRef.
- C. Rodríguez-Abreu, C. Aubery-Torres, C. Solans, A. López-Quintela and G. J. T. Tiddy, Characterization of perylene diimide dye self-assemblies and their use as templates for the synthesis of hybrid and supermicroporous nanotubules, ACS Appl. Mater. Interfaces, 2011, 3, 4133–4141 CrossRef.
- J. R. Magana, Y. V. Kolen’Ko, F. L. Deepak, C. Solans, R. G. Shrestha, J. P. Hill, K. Ariga, L. K. Shrestha and C. Rodriguez-Abreu, From Chromonic Self-Assembly to Hollow Carbon Nanofibers: Efficient Materials in Supercapacitor and Vapor-Sensing Applications, ACS Appl. Mater. Interfaces, 2016, 8, 31231–31238 CrossRef CAS.
- J. R. Magana, C. Solans, L. M. Salonen, E. Carbó-Argibay, J. Gallo, G. J. T. Tiddy and C. Rodríguez-Abreu, Chromonic self-assemblies in a series of dialkyl-thiacarbocyanine dyes and generalization of a facile route for the synthesis of fluorescent nanostructured silica fibers, J. Taiwan Inst. Chem. Eng., 2018, 92, 134–142 CrossRef CAS.
- C. F. J. Faul and M. Antonietti, Ionic self-assembly: Facile synthesis of supramolecular materials, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- Y. Guan, S. H. Yu, M. Antonietti, C. Böttcher and C. F. J. Faul, Synthesis of supramolecular polymers by ionic self-assembly of oppositely charged dyes, Chem. – Eur. J., 2005, 11, 1305–1311 CrossRef CAS.
- C. F. J. Faul, Ionic self-assembly for functional hierarchical nanostructured materials, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS.
- A. D. Schwab, D. E. Smith, C. S. Rich, E. R. Young, W. F. Smith and J. C. De Paula, Porphyrin nanorods, J. Phys. Chem. B, 2003, 107, 11339–11345 CrossRef CAS.
- J. L. Jacobsen, P. E. Berget, M. C. Varela, T. Vu, N. E. Schore, K. E. Martin, J. A. Shelnutt, L. M. Santos and C. J. Medforth, Synthesis and nanostructures of 5,10,15,20-tetrakis(4-piperidyl)porphyrin, Tetrahedron, 2013, 69, 10507–10515 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, Porphyrin nanotubes by ionic self-assembly, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS.
- Y. Tian, C. M. Beavers, T. Busani, K. E. Martin, J. L. Jacobsen, B. Q. Mercado, B. S. Swartzentruber, F. van Swol, C. J. Medforth and J. A. Shelnutt, Binary ionic porphyrin nanosheets: electronic and light-harvesting properties regulated by crystal structure, Nanoscale, 2012, 4, 1695–1700 RSC.
- J. R. Eskelsen, K. J. Phillips, K. W. Hipps and U. Mazur, Hyperbranched crystalline nanostructure produced from ionic π-conjugated molecules, Chem. Commun., 2015, 51, 2663–2666 RSC.
- C. J. Medforth, Z. Wang, K. E. Martin, Y. Song, J. L. Jacobsen and J. A. Shelnutt, Self-assembled porphyrin nanostructures, Chem. Commun., 2009, 7261–7277 RSC.
- Z. Li, Z. Li, Z. Shi and X. Fang, Facet-Dependent, Fast Response, and Broadband Photodetector Based on Highly Stable All-Inorganic CsCu2I3 Single Crystal with 1D Electronic Structure, Adv. Funct. Mater., 2020, 30, 1–10 Search PubMed.
- W. Yang, K. Hu, F. Teng, J. Weng, Y. Zhang and X. Fang, High-Performance Silicon-Compatible Large-Area UV-to-Visible Broadband Photodetector Based on Integrated Lattice-Matched Type II Se/n-Si Heterojunctions, Nano Lett., 2018, 18, 4697–4703 CrossRef CAS.
- X. Xu, J. Chen, S. Cai, Z. Long, Y. Zhang, L. Su, S. He, C. Tang, P. Liu, H. Peng and X. Fang, A Real-Time Wearable UV-Radiation Monitor based on a High-Performance p-CuZnS/n-TiO2 Photodetector, Adv. Mater., 2018, 30, 1–8 Search PubMed.
- H. Hou, G. Shao, W. Yang and W. Y. Wong, One-dimensional mesoporous inorganic nanostructures and their applications in energy, sensor, catalysis and adsorption, Prog. Mater. Sci., 2020, 113, 100671 CrossRef CAS.
- N. Tio, S. Cherian and C. C. Wamser, Adsorption and Photoactivity of Tetra(4-carboxyphenyl)porphyrin (TCPP) on nanoparticulate TiO2, J. Phys. Chem. B, 2000, 50, 3624–3629 Search PubMed.
- M. Y. Choi, J. A. Pollard, M. A. Webb and J. L. McHale, Counterion-dependent excitonic spectra of tetra(p-carboxyphenyl)porphyrin aggregates in acidic aqueous solution, J. Am. Chem. Soc., 2003, 125, 810–820 CrossRef CAS.
- R. F. Pasternack, P. R. Huber, P. Boyd, G. Engasser, L. Francesconi, E. Gibbs, P. Fasella, G. Cerio Venturo and L. deC. Hinds, On the aggregation of meso-substituted water-soluble porphyrins, J. Am. Chem. Soc., 1972, 94, 4511–4517 CrossRef CAS.
- R. Lauceri, S. Gurrieri, E. Bellacchio, A. Contino, L. Monsu'scolaro, A. Romeo, A. Toscano and R. Purrello, J-Type Aggregates of the Anionic Meso-Tetrakis(4-Sulfonatophenyl)Porphine Induced by ‘Hindered’ Cationic Porphyrins, Supramol. Chem., 2000, 12, 193–202 CrossRef CAS.
- A. Iosif and U.-H. Grummt, Aggregation of Tetrakis(4-methylpyridy1)porphyrin and Tetrakis(4-sulphonatopheny1)porphyrin in Water, J. Prakt. Chem., 1997, 339, 420–425 CrossRef CAS.
- H. Ojadi, S. Emmanuel and L. Russell, Properties of Porphyrin Dimers, Formed by Pairing Cationic and Anionic Porphyrins, J. Am. Chem. Soc., 1985, 107, 7783–7784 CrossRef.
- C. Frigerio, J. P. G. Santos, P. Quaresma, S. L. H. Rebelo, A. Gomes, P. Eaton, E. Pereira, P. A. Carvalho, J. A. Shelnutt, L. Jiang, H. Wang and C. J. Medforth, Ionic self-assembly reactions of a porphyrin octacation, Tetrahedron, 2016, 72, 6988–6995 CrossRef CAS.
- F. J. Vergeldt, R. B. M. Koehorst, A. Van Hoek and T. J. Schaafsma, Intramolecular Interactions in the Ground and Excited State of Tetrakis(N-methylpyridyl) porphyrins, J. Phys. Chem., 1995, 4397–4405 CrossRef CAS.
- Z. Ou, H. Yao and K. Kimura, Organic Nanoparticles of Porphyrin without Self-aggregation, Chem. Lett., 2006, 35, 782–783 CrossRef CAS.
- A. S. D. Sandanayaka, Y. Araki, T. Wada and T. Hasobe, Structural and Photophysical Properties of Self-Assembled Porphyrin Nanoassemblies Organized by Ethylene Glycol Derivatives, J. Phys. Chem. C, 2008, 19209–19216 CrossRef CAS.
- M. Samperi, D. Limón, D. B. Amabilino and L. Pérez-García, Enhancing Singlet Oxygen Generation by Self-Assembly of a Porphyrin Entrapped in Supramolecular Fibers, Cell Rep. Phys. Sci., 2020, 1, 100030 CrossRef.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Solvatochromic covalent organic frameworks, Nat. Commun., 2018, 9, 3802 CrossRef.
- S. Ishihara, J. Labuta, W. Van Rossom, D. Ishikawa, K. Minami, J. P. Hill and K. Ariga, Porphyrin-based sensor nanoarchitectonics in diverse physical detection modes, Phys. Chem. Chem. Phys., 2014, 16, 9713–9746 RSC.
- D. W. Schaefer, J. Zhao, J. M. Brown, D. P. Anderson and D. W. Tomlin, Morphology of dispersed carbon single-walled nanotubes, Chem. Phys. Lett., 2003, 375, 369–375 CrossRef CAS.
- M. Adinehnia, B. Borders, M. Ruf, B. Chilukuri, K. W. Hipps and U. Mazur, Comprehensive structure-function correlation of photoactive ionic π-conjugated supermolecular assemblies: An experimental and computational study, J. Mater. Chem. C, 2016, 4, 10223–10239 RSC.
- C. M. Drain, A. Varotto and I. Radivojevic, Self-organized porphyrinic materials, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS.
- J. R. Eskelsen, Y. Qi, S. Schneider-Pollack, S. Schmitt, K. W. Hipps and U. Mazur, Correlating elastic properties and molecular organization of an ionic organic nanostructure, Nanoscale, 2014, 6, 316–327 RSC.
- T. Zhao, C. Hou, H. Zhang, R. Zhu, S. She, J. Wang, T. Li, Z. Liu and B. Wei, Electromagnetic wave absorbing properties of amorphous carbon nanotubes, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- C. Sengottaiyan, R. Jayavel, R. G. Shrestha, T. Subramani, S. Maji, J. H. Kim, J. P. Hill, K. Ariga and L. K. Shrestha, Indium Oxide/Carbon Nanotube/Reduced Graphene Oxide Ternary Nanocomposite with Enhanced Electrochemical Supercapacitance, Bull. Chem. Soc. Jpn., 2019, 92, 521–528 CrossRef CAS.
- L. K. Shrestha, L. Adhikari, R. G. Shrestha, M. P. Adhikari, R. Adhikari, J. P. Hill, R. R. Pradhananga and K. Ariga, Nanoporous carbon materials with enhanced supercapacitance performance and non-aromatic chemical sensing with C1/C2 alcohol discrimination, Sci. Technol. Adv. Mater., 2016, 17, 483–492 CrossRef CAS.
- D. B. Vidano, R. P. Fishbach, T. M. Willis and L. J. Loehr, Observation of Raman band shifting with excitation wavelength for carbons and graphites, Solid State Commun., 1981, 39, 341–344 CrossRef.
- R. Shibuya, T. Kondo and J. Nakamura, Active sites in nitrogen-doped carbon materials for oxygen reduction reaction, Carbon-Based Met. Catal. Des. Appl., 2018, 1–2, 227–249 Search PubMed.
- J. Wang, Y. Xu, B. Ding, Z. Chang, X. Zhang, Y. Yamauchi and K. C. W. Wu, Confined Self-Assembly in Two-Dimensional Interlayer Space: Monolayered Mesoporous Carbon Nanosheets with In-Plane Orderly Arranged Mesopores and a Highly Graphitized Framework, Angew. Chem., Int. Ed., 2018, 57, 2894–2898 CrossRef CAS.
- W. Lu and D. D. L. Chung, Preparation of conductive carbons with high surface area, Carbon, 2001, 39, 39–44 CrossRef CAS.
- G. Faubert, G. Lalande, R. Côté, D. Guay, J. P. Dodelet, L. T. Weng, P. Bertrand and G. Dénès, Heat-treated iron and cobalt tetraphenylporphyrins adsorbed on carbon black: Physical characterization and catalytic properties of these materials for the reduction of oxygen in polymer electrolyte fuel cells, Electrochim. Acta, 1996, 41, 1689–1701 CrossRef CAS.
- I. Herrmann, U. I. Kramm, S. Fiechter, V. Brüser, H. Kersten and P. Bogdanoff, Comparative study of the carbonisation of CoTMPP by low temperature plasma and heat treatment, Plasma Processes Polym., 2010, 7, 515–526 CrossRef CAS.
- K. Jian, H. S. Shim, A. Schwartzman, G. P. Crawford and R. H. Hurt, Orthogonal carbon nanofibers by template-mediated assembly of discotic mesophase pitch, Adv. Mater., 2003, 15, 164–167 CrossRef CAS.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Müllen, Carbonization of disclike molecules in porous alumina membranes: Toward carbon nanotubes with controlled graphene-layer orientation, Angew. Chem., Int. Ed., 2005, 44, 2120–2123 CrossRef CAS.
- L. Zhi, T. Gorelik, J. Wu, U. Kolb and K. Müllen, Nanotubes fabricated from Ni-naphthalocyanine by a template method, J. Am. Chem. Soc., 2005, 127, 12792–12793 CrossRef CAS.
- K. Jian, H. Xianyu, J. Eakin, Y. Gao, G. P. Crawford and R. H. Hurt, Orientationally ordered and patterned discotic films and carbon films from liquid crystal precursors, Carbon, 2005, 43, 407–415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic force microscopy (AFM) image of fibers, SEM, TEM and HR-TEM images of the fibers before and after pyrolysis, additional XPS data, XRD patterns, TG/DTA data and N2 sorption data (PDF). See DOI: 10.1039/d0cp04348f |
| This journal is © the Owner Societies 2020 |