
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox potentials along the redox-active low-barrier H-bonds in electron transfer pathways†
Keisuke
Saito‡
 ab,
Manoj
Mandal‡
a and
Hiroshi
Ishikita
*ab
ab,
Manoj
Mandal‡
a and
Hiroshi
Ishikita
*ab
aResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan. E-mail: hiro@appchem.t.u-tokyo.ac.jp; Fax: +81-3-5452-5083; Tel: +81-3-5452-5056
bDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8654, Japan
First published on 15th September 2020
Abstract
Low-barrier H-bonds form when the pKa values of the H-bond donor and acceptor moieties are nearly equal. Here, we report redox potential (Em) values along two redox-active low-barrier H-bonds in the water-oxidizing enzyme photosystem II (PSII), using a quantum mechanical/molecular mechanical approach. The low-barrier H-bond between D1-Tyr161 (TyrZ) and D1-His190 is located in the middle of the electron transfer pathway. When the proton is at D1-His190, Em(TyrZ) is the lowest and can serve as an electron donor to the oxidized chlorophyll PD1˙+. Em(TyrZ) and Em(D1-His190) are equal, and the TyrZ⋯D1-His190 pair serves as an electron acceptor to Mn4CaO5 when the proton is at TyrZ. In the low-barrier H-bond between D1-His215 and plastoquinone QB, located at the terminus of the electron transfer pathway, the driving force of electron transfer and electronic coupling between QA and QB are maximized when the proton arrives at QB. It seems likely that local proton transfer along redox-active low-barrier H-bonds can alter the driving force or electronic coupling for electron transfer.
The driving force of water oxidation in photosystem II (PSII) is provided by light-induced charge separation in the reaction center. In PSII, the reaction center has a pair of chlorophylls (PD1/PD2), accessory chlorophylls (ChlD1/ChlD2), pheophytins (PheoD1/PheoD2), and plastoquinones (QA/QB) in the heterodimeric D1/D2 protein subunit pairs (Fig. 1a).1 Electronic excitation of ChlD1 leads to the formation of a charge-separated state [PD1PD2]˙+PheoD1˙−;2 electron transfer subsequently occurs via QA to QB. QB reduction is linked to QB protonation. After the second electron transfer, doubly protonated QBH2 is released from the binding site toward the bulk water region: the quinone pool. In [PD1PD2]˙+, the cationic state is more populated on PD1 than PD2 (PD1˙+
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PD2˙+ = 80:203–6). PD1˙+ has a significantly high redox potential (Em) for one-electron oxidation (>1100 mV6–9), which makes PD1˙+ abstract electrons from the substrate water molecules at the Mn4CaO5 cluster (700–800 mV10) via redox-active D1-Tyr161 (TyrZ; Fig. 1b).
:PD2˙+ = 80:203–6). PD1˙+ has a significantly high redox potential (Em) for one-electron oxidation (>1100 mV6–9), which makes PD1˙+ abstract electrons from the substrate water molecules at the Mn4CaO5 cluster (700–800 mV10) via redox-active D1-Tyr161 (TyrZ; Fig. 1b).
 | ||
| Fig. 1 (a) Electron transfer pathways in PSII (PDB code: 3ARC). Low-barrier H-bond pairs are labeled in red. Black arrows indicate electron transfer between cofactors. The dotted line indicates pseudo-C2 axes. (b) Low-barrier H-bond between TyrZ and D1-His190 near Mn4CaO5 cluster. (c) Typical potential energy profile of standard H-bonds. OA and OD stand for the acceptor and donor oxygen moieties, respectively. (d) Typical potential energy profile of low-barrier H-bonds (LBHBs). In low-barrier H-bonds, OA and OD cannot be distinguishable due to the same pKa values. | ||
TyrZ must form a hydrogen bond (H-bond) with D1-His190 to serve as a redox-active cofactor. Notably, TyrZ and D1-His190 form a significantly short (2.46 Å11) low-barrier H-bond.12,13 Low-barrier H-bonds can form only when the pKa values of the H-bond donor and acceptor moieties are nearly equal.14,15 The shape of the potential energy curve of a low-barrier H-bond is symmetric, while the curve of a standard H-bond is asymmetric because pKa(donor) > pKa(acceptor)16 (Fig. 1c and d). The pKa value of tyrosine (∼10) is higher than that of histidine (∼7) in water. In the PSII protein environment, a cluster of water molecules (whose positions are fixed by the components of the Mn4CaO5 cluster) donates a stable H-bond to the phenolic O site of TyrZ and decreases pKa(TyrZ) to the level of pKa(D1-His190), forming a low-barrier H-bond.12,13
In PSII, low-barrier H-bond formation is also observed at the terminus of the electron transfer pathway (QB and the H-bond partner D1-His215) when the second electron transfer from QA˙− to QBH˙ occurs.17 D1-His215 is the ligand of the non-heme Fe complex, and the Nδ site forms an H-bond with the carbonyl O site of QB. The presence of the cationic Fe facilitates the deprotonation of D1-His215 and proton uptake by QBH−. Specifically, decreasing the pKa value of ∼1418 for deprotonation of singly protonated histidine to the level of the pKa value of ∼1119 for deprotonation of doubly protonated plastoquinone. It seems likely that the low-barrier H-bond between D1-His215 and QB is not stable because it should not inhibit the release of the product QBH2 from the PSII binding site toward the quinone pool. A lingering question is why the redox-active low-barrier H-bond between TyrZ and D1-His190 is stable enough to function repeatedly in each S-state transition, while the bond between D1-His215 and QB functions only once before QBH2 is released.
Initially, the redox-active group was identified as D1-His190 based on observations of radical formation in the Ca2+-depleted PSII.20,21 Later, it was proposed that TyrZ, not D1-His190, was the origin of the radical state.22 Since then, TyrZ has been regarded as a redox-active group in the electron transfer pathway. Indeed, Em(TyrZ) is lower than Em(D1-His190), however the Em difference is not significantly large when the proton is at the TyrZ moiety.10Em values of the H-bond donor and acceptor moieties along low-barrier H-bonds have not been reported. Here, we report Em of the two redox-active low-barrier H-bonds, considering the entire electron and proton transfer pathways quantum-chemically in the presence of the PSII protein environment.
Results
Low-barrier H-bond switching from an electron donor to an acceptor
The potential-energy profile of the H-bond shows that TyrZ and D1-His190 form a low-barrier H-bond. The difference between pKa(TyrZ) and pKa(D1-His190) is nearly zero along the low-barrier H-bond. However, the difference between Em(TyrZ) (i.e., Tyr–O−/Tyr–O˙) and Em(D1-His190), (i.e., N–His–N−/N–His–N˙ or HN–His–N/HN–His–N˙+) depends on the H+ position (Fig. 2, right panel). Em(TyrZ) decreases and Em(D1-His190) increases as the proton moves from TyrZ to D1-His190. When H+ arrives at the lower Em moiety (D1-His190), Em(TyrZ) is the lowest and the driving force for electron transfer from TyrZ to oxidized chlorophyll PD1˙+ is the largest. The highest occupied molecular orbital (HOMO) is predominantly localized at the TyrZ moiety. Thus, the TyrZ moiety can serve as an electron donor to PD1˙+ most effectively when the proton is at the D1-His190 moiety. Note that the H+ atom position does not affect the Em values of other cofactors in the electron transfer pathway. | ||
| Fig. 2 Potential energy profile of the H-bond between TyrZ and D1-His190 (right vertical axis in kcal mol−1) and the cofactor Em values in the S1 to S2 transition (left vertical axis in mV). The Mn oxidation states are (Mn1, Mn2, Mn3, Mn4) = (III, IV, IV, III) in S1 and (III, IV, IV, IV) in S2, i.e., dangling Mn4 (Fig. 1b) is the oxidation site in the S1 to S2 transition. Left panels show the HOMO when (a) H+ is at the D1-His190 moiety (OTyrZ⋯H = 1.38 Å) with the HOMO population of TyrZ:D1-His190 = 96.1:0.4 and (b) at the TyrZ moiety (1.02 Å) with the HOMO population of TyrZ:D1-His190 = 51.4:44.7. Red arrows indicate electron transfer from TyrZ to PD1. Blue arrows indicate electron transfer (ET) from Mn4(III) to the TyrZ/D1-His190 pair. Cyan arrows indicate movement of the proton between the D1-His190 and TyrZ moiety. The gray dotted vertical line indicates the H+ position where Em(TyrZ) and Em(His190) are nearly equal and the TyrZ/His190 pair serves as an electron acceptor, finalizing the electron transfer process. | ||
In contrast, when H+ is at the TyrZ moiety, Em(TyrZ) and Em(D1-His190) are equal, and the highest occupied molecular orbital (HOMO) is delocalized over the two moieties (Fig. 2b, left panel). In this case, the driving force for election transfer from the oxygen-evolving complex (i.e., Mn4III/IV) to the TyrZ⋯D1-His190 pair is largest (Fig. 2b, right panel). Thus, the entire TyrZ⋯D1-His190 moiety cooperatively serves as an electron acceptor when electron transfer occurs from the oxygen-evolving complex.
Low-barrier H-bond maximizing electronic coupling and driving force
As an electron transfers to QBH˙, a low-barrier H-bond forms between D1-His215 and QBH˙/QBH−,17 facilitating QBH2 release from the binding site in PSII. We analyzed Em(N–His–N˙/N–His–N−) and Em(QBH˙/QBH−) along the low-barrier H-bond between D1-His215 and QB. Note that Em(QBH˙/QBH−) can be analyzed based on the (fully occupied) HOMO of QBH−, which is the same as the (singly occupied) HOMO (SOMO) of QBH˙.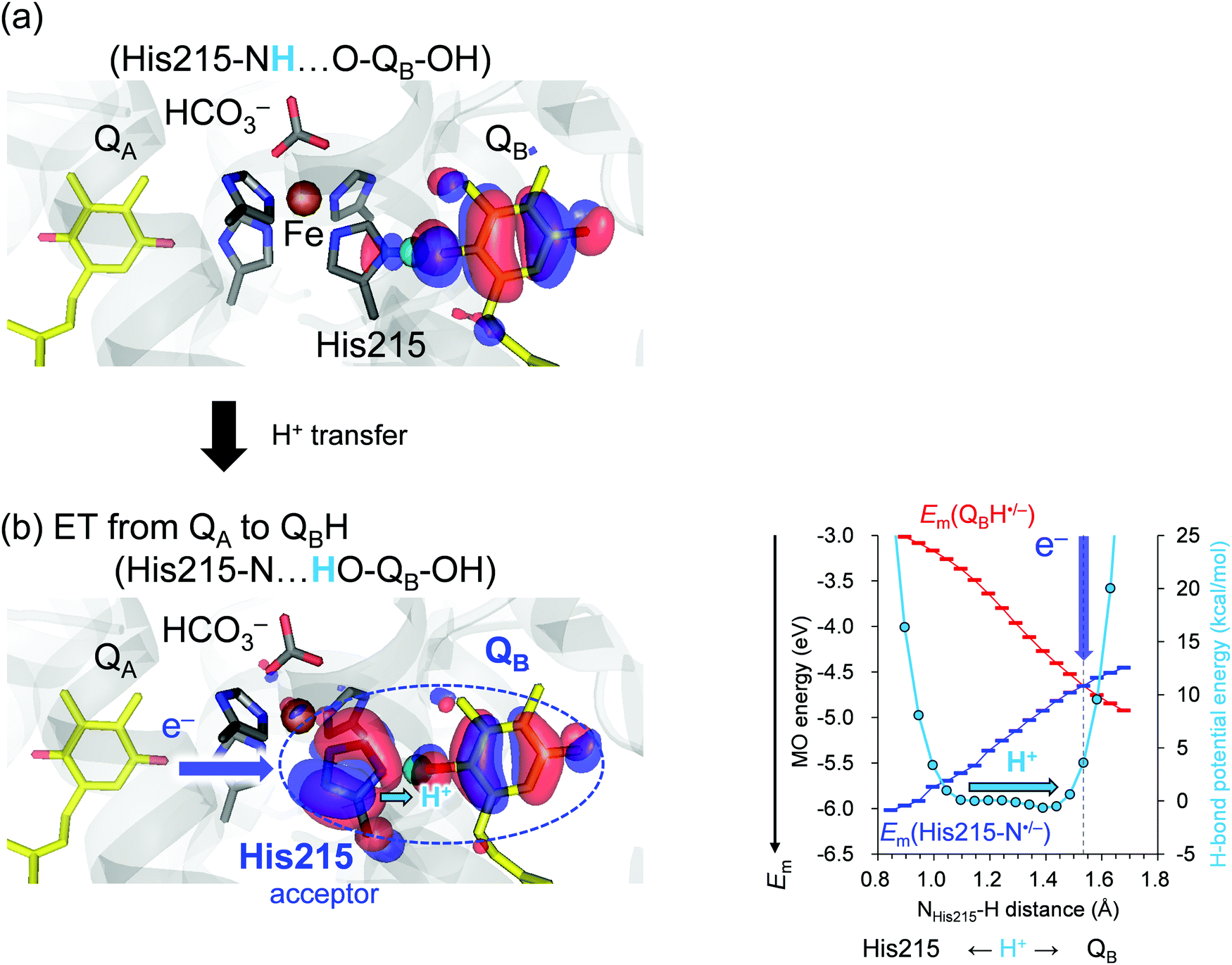 | ||
| Fig. 3 Potential energy profile of the H-bond between D1-His215 and QB (right vertical axis in kcal mol−1) and the QB MO energy [≈Em(QBH˙/QBH−)] in the S1 to S2 transition (left vertical axis in eV). Left panels show the HOMO when (a) H+ is at the D1-His215 moiety (NHis215⋯H = 1.15 Å) with the HOMO population of QB:D1-His215 = 97.4:1.9 and (b) at the QB moiety (1.54 Å) with the HOMO population of QB:D1-His215 = 37.6:41.8. Blue arrows indicate electron transfer (ET) from QA˙− to QBH˙. Cyan arrows indicate movement of the proton from the D1-His215 to the QB moiety. The gray dotted vertical line indicates the H+ position where Em(His215) and Em(QB) are nearly equal and the His215/QB pair serves as a terminal electron acceptor, finalizing the electron transfer process. | ||
Discussion
The redox-active low-barrier H-bond between TyrZ and D1-His190 plays a dual role in electron transfer by serving as both an electron donor and an electron acceptor. The movement of the proton by only 0.35 Å from the D1-His190 moiety along the low-barrier H-bond alters Em by ∼120 mV and switches its role from an electron donor to acceptor (Fig. 2). As the proton moves back to the TyrZ moiety in the low-barrier H-bond, Em(TyrZ) and Em(D1-His190) become equal within the barrier-less potential (OTyrZ⋯H = 1.02 Å; Fig. 2b, right panel). Thus, TyrZ⋯D1-His190 can serve as both an electron donor and acceptor in the middle of the electron transfer pathway during the S-cycle without being destabilized by proton movement. Until now, it has been thought that D1-His190 is not redox-active, but that TyrZ is22 (however see ref. 20 and 21). However, this result suggests that both TyrZ and D1-His190 are redox-active components in the electron transfer pathway, in particular when abstracting an electron from the oxygen-evolving complex (Fig. 2b).On the other hand, movement of the proton by only 0.35 Å along the low-barrier H-bond between D1-His215 and QBH˙/QBH− increases Em(QBH˙/QBH−) to the level of Em(D1-His215), leading to delocalization of the HOMO over the D1-His215 and QB moiety. The involvement of D1-His215 in the electron acceptor decreases the donor-to-acceptor distance from [QA]-to-[QB] to [QA]-to-[D1-His215] and increases the electronic coupling between the donor QA and the acceptor QBH˙/QBH−, because the electronic coupling increases as the electron donor-to-acceptor distance decreases.23,24 The role of the redox-active low-barrier H-bond in increasing electronic coupling is observed specifically for D1-His215⋯QBH˙/QBH−, since the axis of the low-barrier H-bond is consistent with the axis of the electron transfer pathway and delocalization of the HOMO over D1-His215 and QB decreases the substantial donor–acceptor distance (Fig. 3, left panel).
In contrast to the TyrZ⋯D1-His190 H-bond, the difference between Em(D1-His215) and Em(QB) is small but not zero within the barrier-less potential as the proton moves to the QB moiety (HD1-His215⋯H = 1.43 Å; Fig. 3b, right panel). Indeed, the HOMO is still predominantly localized at QB (Fig. S1, ESI†). Only after the proton exceeds the barrier-less potential by 0.1 Å is the HOMO evenly delocalized over D1-His215 and QB and the difference between Em(D1-His215) and Em(QB) reaches zero (HD1-His215⋯H = 1.53 Å, Fig. 3b, right panel). In reality, the low-barrier H-bond is unlikely to exist when HD1-His215⋯H = 1.53 Å because the potential energy profile was obtained assuming the presence of the H-bond (see Methods). Consistently, the donor–acceptor N⋯O distance (ND1-His215⋯OQB) is 2.47 Å when the proton is located at the D1-His215 moiety (ND1-His215–H⋯OQB), and it is lengthened to 2.55 Å when the proton arrives at the QB moiety (ND1-His215⋯H–OQB). Thus, QBH2 is released from the binding site (D1-His215) when H+ leaves the barrier-less potential (after cleavage of the low-barrier H-bond). The irreversibility of the reaction is characteristic of, and required for, QB serving as the terminal electron acceptor in the electron transfer pathway. Intriguingly, the corresponding change in the donor–acceptor distance in response to the H+ movement is absent in the TyrZ⋯D1-His190 H-bond (OTyrZ–H⋯ND1-His190 = 2.52 Å and OTyrZ⋯H–ND1-His190 = 2.50 Å). This is consistent with the role of the TyrZ⋯D1-His190 pair in the middle of the electron transfer pathway, which involves reversibly donating and accepting an electron during the entire S-cycle. As reported, environmental fluctuations may affect the potential energy profile,25 particularly for standard H-bonds. Proton transfer is energetically uphill in the standard H-bond (Fig. 1c). To overcome this energy barrier, fluctuation/rearrangement of the protein environment is a prerequisite for unstable protonation of the acceptor moiety. In contrast, proton transfer is barrier-less in the low-barrier H-bond, which does not require the corresponding fluctuation/rearrangement of the protein environment (Fig. 1d). Consistently, the B-factors of TyrZ and D1-His190 are specifically low11 because the H-bond network is fixed by the PSII protein electrostatic environment, namely the Mn4CaO5 cluster and the ionized ligand residues.12
For the redox-active low-barrier H-bonds to serve as a redox-active cofactor in the electron transfer pathway, the presence of the protein electrostatic environment is a prerequisite. First, TyrZ and D1-His190 cannot form a low-barrier H-bond in the absence of the PSII protein environment12 because of the difference in pKa (∼3) between tyrosine and histidine. Second, the protein electrostatic environment is required for the formation of the downhill electron transfer pathway that proceeds from the Mn4CaO5 cluster via TyrZ, as it increases Em(PD1) by ∼400 mV with respect to Em(Chla).6,26 Thus, the environment, in which the pKa values of the proton donor and acceptor moieties are equal and the Em values of the electron donor and acceptor moieties are in the tunable range by proton transfer, is ultimately provided by the common protein electrostatic environment.
In summary, the pKa values of the two moieties are equal, but the Em values depend on the H+ position in redox-active low-barrier H-bonds. The results also suggest that redox-active low-barrier H-bonds can differ in their characteristics, whether they serve as a redox-active cofactor in the middle of the electron transfer pathway (TyrZ⋯D1-His190) or in the terminus of the electron acceptor pathway (D1-His215⋯QB). These findings provide a key to understanding how nature optimizes electron and proton transfer in biological systems using abundantly available protons.
Methods
The atomic coordinates of PSII were obtained from the PSII crystal structure (PDB code, 3ARC).11 The atomic charges of the other cofactors in the MM region were taken from a previous study.27E m calculations for the electron transfer pathway via TyrZ⋯D1-His190
The HOMO energy level is largely correlated with Em for one-electron oxidation (e.g., ref. 28 and 29). For the Mn4CaO5 cluster, the HOMO in Sn corresponds to the molecular orbital, which predominantly contributes to the release of an electron in the Sn to Sn+1 transition. We included all redox-active cofactors (Mn4CaO5 cluster, TyrZ, PD1, and PD2) simultaneously in the QM region30,31 (see below), identified HOMOs of Mn4CaO5, TyrZ, PD1, and PD2 in S1 on the basis of the Mulliken population analysis32 (results provided in a previous study10), and obtained the Em values (note, Em(S1/S2) for the Mn4CaO5 cluster). The Em values of the redox sites are calculated using eqn (1):| Em = −0.15499EHOMO + 1205.65, | (1) |
QM/MM calculations
The unrestricted density functional theory method was employed with the B3LYP functional using the QSite program.33 B3LYP is a widely used functional to calculate redox potentials and HOMO and LUMO energy levels (e.g., ref. 34 and 35). Using B3LYP can also make a comparison with our previous studies for Mn4CaO5,10,36 TyrZ,12,13 PD1,6,37 and QB17 in PSII. In the QM region, all the atomic coordinates were fully relaxed. In the MM region, the positions of the H atoms were optimized using the OPLS2005 force field,38 while the positions of the heavy atoms were fixed.To investigate the H-bond between TyrZ and D1-His190, the LACVP* basis set was employed. The Mn4CaO5 cluster was considered to be in S1 with antiferromagnetically coupled Mn ions; the resulting Mn oxidation states (Mn1, Mn2, Mn3, Mn4) and the total spin S were S = 8/2 (↑ ↓ ↑↑) in S1. Note that the difference in S (for example, S = 0 in S139) did not affect the calculated Em values (≤3 mV).10 O1–O5 was considered to be unprotonated (O2−). The four water ligand molecules, W1–W4, were considered to be water molecules (H2O). The Mn4CaO5 geometry was obtained from our previous studies.40,41 The initial-guess wavefunctions were obtained using the ligand field theory42 implemented in the QSite program. The QM region was defined as the Mn4CaO5 cluster (including the ligand side-chains of D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, and CP43-Glu354, and backbone of D1-Ala344), ligand water molecules of W1–W4, O4–water chain (W539, W538, and W393), Cl-1 binding site (Cl-1, W442, W446, and side-chains of D1-Asn181 and D2-Lys317), second-sphere ligands (side-chains of D1-Asp61 and CP43-Arg357), the H-bond network of TyrZ (side-chains of D1-Tyr161, D1-His190, and D1-Asn298), diamond-shaped cluster of water molecules12 (W5, W6, and W7), and PD1/PD2. The MM region was defined as the entire PSII protein, as in a previous study.10 D1-His337 was considered to be protonated,43 whereas all other titratable groups were in the standard protonation states. See ref. 10 for the QM/MM-optimized atomic coordinates.
To investigate the H-bond between D1-His215 and QB, the LACVP**+ basis set was employed. The QM region was defined as [QB, D1-His252, D1-Ser264, bicarbonate, Fe, D1-His215, D1-His272, D2-His214, and D2-His268]. We assumed a high-spin state (S = 2) of Fe2+ and set the spin multiplicity of the system to S = 2 in the calculations for QBH− and QBH2. The MM region was defined as the D1 and D2 protein subunits, as in a previous study.17 D1-His252 was considered to be protonated before QBH2 formation,44 whereas all other titratable groups were in the standard protonation states. See ref. 17 for the QM/MM-optimized atomic coordinates.
To obtain the potential energy profiles of the O⋯H⋯N H-bond, the QM/MM optimized geometry was used as the initial geometry. The H atom under investigation was moved between O and N by 0.05 Å before the geometry was optimized by constraining the O–H and H–N distances, and the energy was calculated. This procedure was repeated until the H atom reached the O and N atoms. This approach, which is based on the single QM/MM-optimized geometry, provides the unique minimum-energy pathway, in particular for the H-bond between TyrZ and D1-His190, as demonstrated by analyzing proton-transfer pathways in the different protein conformations.12
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JST CREST (JPMJCR1656 to H. I.), JSPS KAKENHI (JP18H05155, JP18H01937, JP20H03217, and JP20H05090 to H. I., JP18H01186 to K. S., and JP16H06560 to K. S.), and the Interdisciplinary Computational Science Program in CCS, University of Tsukuba (K. S.).References
- J. R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 Search PubMed.
- H. Tamura, K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16373–16382 Search PubMed.
- S. E. J. Rigby, J. H. A. Nugent and P. J. O'Malley, Biochemistry, 1994, 33, 10043–10050 Search PubMed.
- B. A. Diner, E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas and D. A. Chisholm, Biochemistry, 2001, 40, 9265–9281 Search PubMed.
- T. Okubo, T. Tomo, M. Sugiura and T. Noguchi, Biochemistry, 2007, 46, 4390–4397 Search PubMed.
- K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J.-R. Shen and H. Ishikita, J. Am. Chem. Soc., 2011, 133, 14379–14388 Search PubMed.
- V. V. Klimov, S. I. Allakhverdiev, S. Demeter and A. A. Krasnovskii, Dokl. Akad. Nauk SSSR, 1979, 249, 227–230 Search PubMed.
- A. W. Rutherford, J. E. Mullet and A. R. Crofts, FEBS Lett., 1981, 123, 235–237 Search PubMed.
- F. Rappaport, M. Guergova-Kuras, P. J. Nixon, B. A. Diner and J. Lavergne, Biochemistry, 2002, 41, 8518–8527 Search PubMed.
- M. Mandal, K. Kawashima, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2020, 11, 249–255 Search PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 Search PubMed.
- K. Saito, J.-R. Shen, T. Ishida and H. Ishikita, Biochemistry, 2011, 50, 9836–9844 Search PubMed.
- K. Kawashima, K. Saito and H. Ishikita, Biochemistry, 2018, 57, 4997–5004 Search PubMed.
- A. Warshel, A. Papazyan and P. A. Kollman, Science, 1995, 269, 102–106 Search PubMed.
- C. N. Schutz and A. Warshel, Proteins, 2004, 55, 711–723 Search PubMed.
- C. L. Perrin and J. B. Nielson, Annu. Rev. Phys. Chem., 1997, 48, 511–544 Search PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 954–959 Search PubMed.
- T. C. Bruice and G. L. Schmir, J. Am. Chem. Soc., 1958, 80, 148–156 Search PubMed.
- R. Hasegawa, K. Saito, T. Takaoka and H. Ishikita, Photosynth. Res., 2017, 133, 297–304 Search PubMed.
- A. Boussac, J.-L. Zimmermann, A. W. Rutherford and J. Lavergne, Nature, 1990, 347, 303–306 Search PubMed.
- A. Boussac and A. W. Rutherford, Biochemistry, 1992, 31, 7441–7445 Search PubMed.
- B. J. Hallahan, J. H. A. Nugent, J. T. Warden and M. C. W. Evans, Biochemistry, 1992, 31, 4562–4573 Search PubMed.
- C. C. Moser, J. M. Keske, F. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796–802 Search PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 Search PubMed.
- T. Vasilevskaya, M. G. Khrenova, A. V. Nemukhin and W. Thiel, J. Comput. Chem., 2016, 37, 1801–1809 Search PubMed.
- H. Ishikita, W. Saenger, J. Biesiadka, B. Loll and E.-W. Knapp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9855–9860 Search PubMed.
- K. Kawashima and H. Ishikita, Chem. Sci., 2018, 9, 4083–4092 Search PubMed.
- T. Watanabe and M. Kobayashi, in Chlorophylls, ed. H. Scheer, CRC Press, Boca Raton, FL, 1991, pp. 287–303 Search PubMed.
- D. D. Méndez-Hernández, P. Tarakeshwar, D. Gust, T. A. Moore, A. L. Moore and V. Mujica, J. Mol. Model., 2013, 19, 2845–2848 Search PubMed.
- S. G. Abuabara, L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2005, 127, 18234–18242 Search PubMed.
- P. Joshi, V. Shewale, R. Pandey, V. Shanker, S. Hussain and S. P. Karna, J. Phys. Chem. C, 2011, 115, 22818–22826 Search PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 Search PubMed.
- QSite, version 5.8, Schrödinger, LLC, New York, NY, 2012 Search PubMed.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 Search PubMed.
- D. Coskun, S. V. Jerome and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 1121–1128 Search PubMed.
- K. Saito, M. Mandal and H. Ishikita, Biochemistry, 2020, 59, 3216–3224 Search PubMed.
- H. Tamura, K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16373–16382 Search PubMed.
- J. L. Banks, H. S. Beard, Y. X. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 Search PubMed.
- D. Koulougliotis, D. J. Hirsh and G. W. Brudvig, J. Am. Chem. Soc., 1992, 114, 8322–8323 Search PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 Search PubMed.
- K. Kawashima, T. Takaoka, H. Kimura, K. Saito and H. Ishikita, Nat. Commun., 2018, 9, 1247 Search PubMed.
- G. Vacek, J. K. Perry and J. M. Langlois, Chem. Phys. Lett., 1999, 310, 189–194 Search PubMed.
- S. Nakamura and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 9364–9375 Search PubMed.
- H. Ishikita and E.-W. Knapp, J. Am. Chem. Soc., 2005, 127, 14714–14720 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04265j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2020 |