
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the location of the unpaired electron in spin–orbit changing collisions of NO with Ar†
Cornelia G.
Heid
a,
Imogen P.
Bentham
a,
Victoria
Walpole‡
a,
Razvan
Gheorghe
a,
Pablo G.
Jambrina
 b,
F. Javier
Aoiz
c and
Mark
Brouard
*a
b,
F. Javier
Aoiz
c and
Mark
Brouard
*a
aDepartment of Chemistry, University of Oxford, The Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: mark.brouard@chem.ox.ac.uk
bDepartamento de Química Física, Universidad de Salamanca, 37008, Salamanca, Spain
cDepartamento de Química Física, Facultad de Química, Universidad Complutense, 28040 Madrid, Spain
First published on 9th September 2020
Abstract
Understanding the molecular forces that drive a reaction or scattering process lies at the heart of molecular dynamics. Here, we present a combined experimental and theoretical study of the spin–orbit changing scattering dynamics of oriented NO molecules with Ar atoms. Using our crossed molecular beam apparatus, we have recorded velocity-map ion images and extracted differential and integral cross sections of the scattering process in the side-on geometry. We observe an overall preference for collisions close to the N atom in the spin–orbit changing manifold, which is a direct consequence of the location of the unpaired electron on the potential energy surface. In addition, a prominent forward scattered feature is observed for intermediate, even rotational transitions when the atom approaches the molecule from the O-end. The appearance of this peak originates from an attractive well on the A′ potential energy surface, which efficiently directs high impact parameter trajectories towards the region of high unpaired electron density near the N-end of the molecule. The ability to orient molecules prior to collision, both experimentally and theoretically, allows us to sample different regions of the potential energy surface(s) and unveil the associated collision pathways.
I. Introduction
To capture and elucidate collisional and reactive processes between molecules accurately and to a high level of detail is one of the biggest goals in chemical dynamics. In its pursuit, a joint effort between experiment and theory is in most cases indispensable. Calculations are needed, for instance, to assign transitions in high-resolution spectra,1,2 to verify product distributions of reactions,3–5 or to rationalize resonances observed in scattering experiments.6–11 Experiments, in turn, are required to test calculated potential energy surfaces (PESs)12–18 and to validate theoretical predictions19–21 and approximations.22–24Increasingly refined methods to control molecules in collisions and reactions have provided particularly sensitive probes for testing the accuracy of PESs. Recent work by Vogels et al., has challenged the accuracy of the PESs involved in the scattering of NO with para-H2.25 In their experiments, in which the velocity of the NO molecules was controlled by a Stark decelerator and the H2 molecules were slowed down via an ‘anti-seeding’ technique with Ne, they were able to resolve a Feshbach resonance around 14 cm−1. Comparison with the position of the resonance calculated on two very similar high-level coupled-cluster PESs,26,27 allowed to establish the more accurate of the two potentials, highlighting the sensitivity of their experimental approach. Likewise, measurements of the reaction rate of metastable He with H2 and its isotopologues16,18,28 showed that shape (or orbiting) resonances observed below 10 K could be used to benchmark quantum dynamics calculations to within 7 × 10−3 cm−1.18 Observations of resonances in other high-resolution experiments of molecular interactions, including in vibrational excitation studies,8,11 have demonstrated similar sensitivities to the corresponding calculated energy levels and PESs.5–7,9,14,29
In addition to probing the local energy landscape via dynamical resonances, initial alignment23,30–38 or orientation39–44 of selected reactant quantum states can be used to gauge the geometric preferences and uncover the associated reaction pathways of molecular encounters.37,38 In this case, specific portions of the PES can be explored selectively by confining the relative geometry of the interacting species. Spatial alignment (alignment parallel or perpendicular to a reference axis) through polarized laser light has revealed pronounced preferences for specific initial configurations,30,33–38 and even enabled the three-dimensional visualization of distinct microscopic reaction pathways.23
Molecular orientation, in which a specific end or side of a molecule can be directed towards a collision partner, is most widely achieved through hexapole state selection coupled with adiabatic passage into a static electric40–42,45,46 or magnetic43,44,47 field. The good agreement between experimental and calculated steric preferences obtained in studies of inelastic collisions of state selected NO(X) with Ar and He atoms40–42,48–51 have confirmed the accuracy of the NO(X) + He/Ar ground electronic PESs,52–54 while (unoriented) scattering experiments of electronically excited NO(A) with Ne17 pointed to shortcomings in the calculated PESs.55,56
Electric field orientation requires the molecule to be oriented to posses a permanent dipole moment and, if only the dominant first-order Stark effect is taken into account, to be open-shell.57 The open-shell nature of the ground 2Π state of these molecules leads to two spin–orbit manifolds, given by the sum of the projections of the electronic orbital (Λ) and spin (Σ) angular momenta onto the internuclear axis. For the particular case of the extensively studied 2Π diatomic NO, the values of Λ and Σ are ±1 and 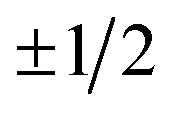 , respectively; the spin–orbit quantum number, Ω, can thus take on values of
, respectively; the spin–orbit quantum number, Ω, can thus take on values of 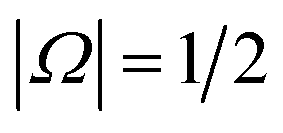 and
and 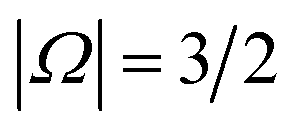 . The former corresponds to the spin–orbit ground state, and the latter corresponds to the spin–orbit excited state, which lies 123 cm−1 above the
. The former corresponds to the spin–orbit ground state, and the latter corresponds to the spin–orbit excited state, which lies 123 cm−1 above the 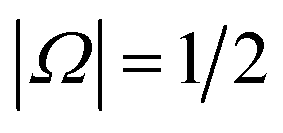 state.58 Depending on the collision energy, rotational excitations are possible to either of the two manifolds.
state.58 Depending on the collision energy, rotational excitations are possible to either of the two manifolds.
Integral and differential scattering cross sections have been measured for a range of (oriented and unoriented) open-shell molecules and collision partners, in particular OH46,59–63 and NO.64–67 The OH + Rg (Rg = rare gas) systems have been systematically studied by Scharfenberg et al.,62 who showed that the contribution of the spin–orbit excited states to the total scattering intensity significantly decreased with increasing polarizability and mass of the atom. This observation was in part correlated with the larger anisotropy and deeper attractive wells in the potentials for the heavier rare gases, suggesting that the more repulsive character of the PESs for the lighter atoms is conducive to spin–orbit excitations, which require an additional ∼140 cm−1 (relative to the spin–orbit conserving manifold) to be accessed.68 Consistent with this picture of a more impulsive, lower impact parameter collision are the more backward scattered differential cross sections measured for spin–orbit excited NO after collisions with Ar, Kr, and Xe.66,69–73
In the present work, we focus on transitions from the rotational and spin–orbit ground state to the spin–orbit excited manifold, 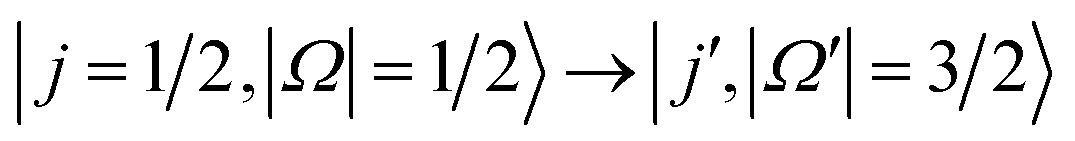 , in the scattering of oriented NO molecules with Ar, using a combination of experiment and theory to unravel the dynamics. In a first step, we compare the experimental data obtained in the side-on orientation with full QM calculations to establish very good agreement between the experimental and computational results. In the second step, we then simulate the experiments at the limit of infinite field orientation, again using a QM treatment. The calculated differential and integral cross sections provide new insights into the molecular forces that promote spin–orbit changing excitations. By controlling the initial relative orientation of the collision partners, we are able to probe selected parts of the potential energy surface; the resulting trends in steric preference, as a function of Δj = j′ − j (where j and j′ designate the initial and final rotational states, respectively), will be shown to be particularly sensitive to the location of the unpaired electron within the NO molecule.
, in the scattering of oriented NO molecules with Ar, using a combination of experiment and theory to unravel the dynamics. In a first step, we compare the experimental data obtained in the side-on orientation with full QM calculations to establish very good agreement between the experimental and computational results. In the second step, we then simulate the experiments at the limit of infinite field orientation, again using a QM treatment. The calculated differential and integral cross sections provide new insights into the molecular forces that promote spin–orbit changing excitations. By controlling the initial relative orientation of the collision partners, we are able to probe selected parts of the potential energy surface; the resulting trends in steric preference, as a function of Δj = j′ − j (where j and j′ designate the initial and final rotational states, respectively), will be shown to be particularly sensitive to the location of the unpaired electron within the NO molecule.
II. Methods
A. Experimental methods
The experimental setup is identical to the one described previously for detection of the spin–orbit conserving (ΔΩ = 0) rotational transitions.50,51 The NO molecules were expanded in a skimmed molecular beam containing about 15% NO in Ar. Using a hexapole, the molecules were state selected in their low-field seeking f Λ-doublet state64 and focused into the center of the scattering chamber where they were crossed with a neat argon beam at right angles. Prior to collision with the Ar atoms, the NO were adiabatically oriented in a static electric field located in the interaction region.In the electric field, the initially pure f Λ-doublet state evolves into a superposition state with contributions from both Λ-doublets:40,48,74
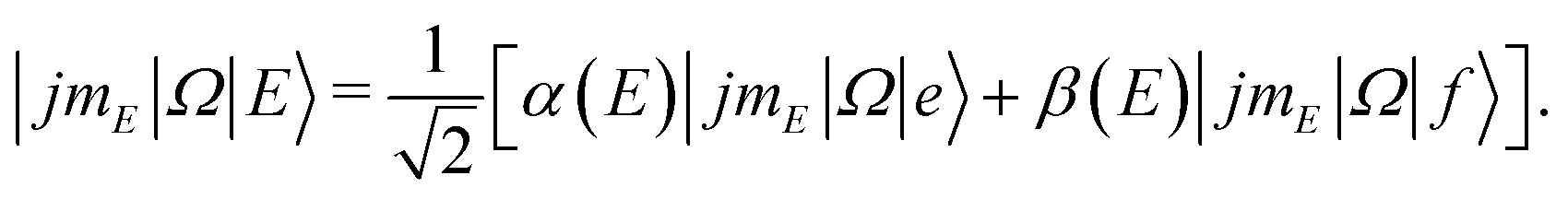 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between initial f and e states. (At infinite field, |α| = |β| = 1.)
:1 ratio between initial f and e states. (At infinite field, |α| = |β| = 1.)
The field was oriented perpendicular to the relative velocity vector (k = vAr − vNO) such that the Ar preferentially impacted on the side of the diatomic (see Fig. 1(a) and (b)). By switching the direction of the electric field every 200 laser shots, images for both side-on orientations were collected alternatingly. The spin–orbit rotationally excited (ΔΩ = 1) NO molecules were ionized by a (1 + 1′) Resonance Enhanced Multiphoton Ionization (REMPI) process75 with the resonant laser pulse (≈226 nm) coming from a dye laser, pumped by an excimer laser and tuned to a specific NO(A) ← NO(X) transition on the mixed Q21 + R11 branch. The ionization pulse was provided by the 308 nm fundamental of the XeCl excimer laser. The nascent NO ions were subsequently velocity mapped76 onto a dual MCP/phosphor screen detector and imaged77 with an intensified charge-coupled device camera connected to a data acquisition computer. The laser beams and the NO molecular beam were operated at 10 Hz, while the Ar beam was operated at 5 Hz to allow for background subtraction on a shot-to-shot basis.
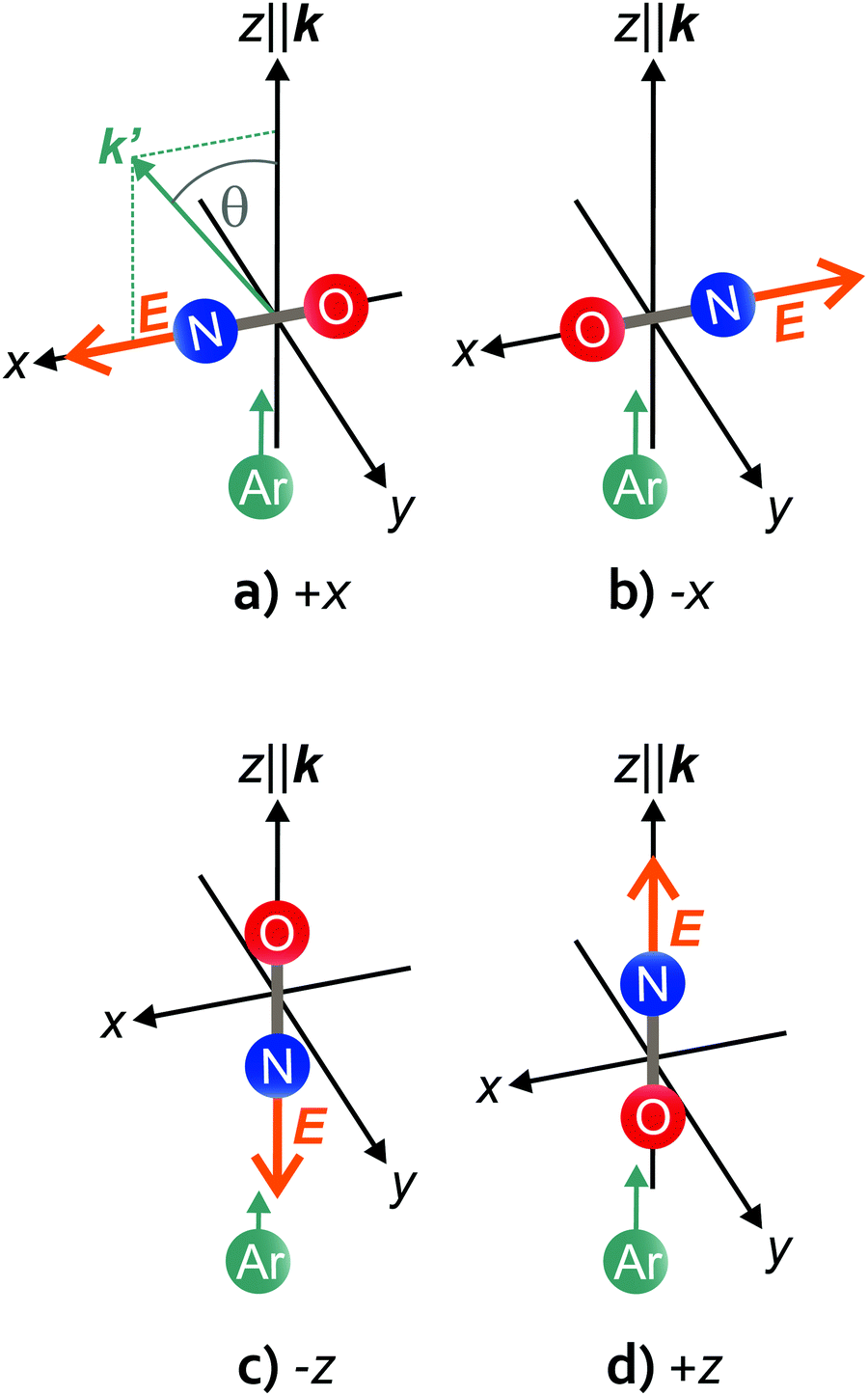 | ||
| Fig. 1 Schematic representations of the two side-on (top) and the two end-on (bottom) configurations in the scattering frame, in which the z-axis is defined parallel to the initial relative velocity vector, k = vAr − vNO, and k and k′ (= vAr′ − vNO′) define the +xz plane. The scattering angle, θ, is indicated in (a). The +x, −x, −z, and +z orientations, depicted in (a)–(d), are labelled according to the axis along which the electric field vector, E, is directed in each case. Note that the +x and the −z orientations correspond to repulsive collisions of the approaching Ar atom towards the N-side/N-end, whereas the −x and +z orientations correspond to repulsive collisions towards the O-side/O-end of the NO molecule. | ||
The DCSs for the two orientations of each of the measured spin–orbit excited transitions were extracted by fitting a linear combination of modified spherical harmonics basis functions to the sum and difference of the images for the two sides. From the fitted images, the relevant polarization moments were extracted and then used to calculate the DCSs. The method is described in detail in our earlier work50,51 (see in particular Section 4 in ref. 51).
We define the orientation in the two side-on configurations according to the electric field vector in the scattering frame. In the scattering frame, the initial relative velocity vector, k, is parallel to the z-axis and the +xz plane is defined by k and k′, the outgoing relative velocity vector (and the y-axis is chosen such that the frame is right-handed). If the electric field points along the +x-axis, the orientation is “+x”; if the field points along the −x-axis, the orientation is “−x”. The two side-on, as well as the two end-on geometries are depicted schematically in Fig. 1. Note that in the side-on configuration, the +x orientation corresponds to repulsive collisions off the N-side (a), and the −x orientation to repulsive collisions off the O-side of the molecule (b). A closer look at Fig. 1(a) and (b) shows that each of the two side-on geometries actually contains both configurations; if k′ was lying in the −x hemisphere, the +x orientation in panel (a) would become −x and vice versa for the −x orientation in panel (b). Therefore, the azimuthal angle of k′ (in the detector frame) has to be taken into account when analyzing the side-on data. In the end-on configuration, the distribution of k′ around k is symmetric, and N-end/O-end collisions are defined as “−z” and “+z”, irrespective of the azimuthal angle of k′ (Fig. 1(c) and (d)).
A consequence of the asymmetry of the side-on scattering process with respect to k is that the experimental velocity-map ion images contain the scattering distributions of both orientations to either side of k50,51 (with k pointing roughly diagonal from the top left to the bottom right corner of the image, dividing it into two parts – this is indicated in the top left image in Fig. 2). Furthermore, due to the variation in detection probability associated with the experimental geometry, the signal intensity is more intense on the lower left side and weaker towards the top right side of the images. The lower left side is where the laboratory velocity is close to zero (“slow” side) and ions in that region have a higher detection probability than ions closer to the top right side that move faster in the laboratory frame (“fast” side). Because most of the intensity in each configuration is located in the lower left portion of the image, we label the images according to the orientation on that side. For the analysis, however, both sides are taken into account.
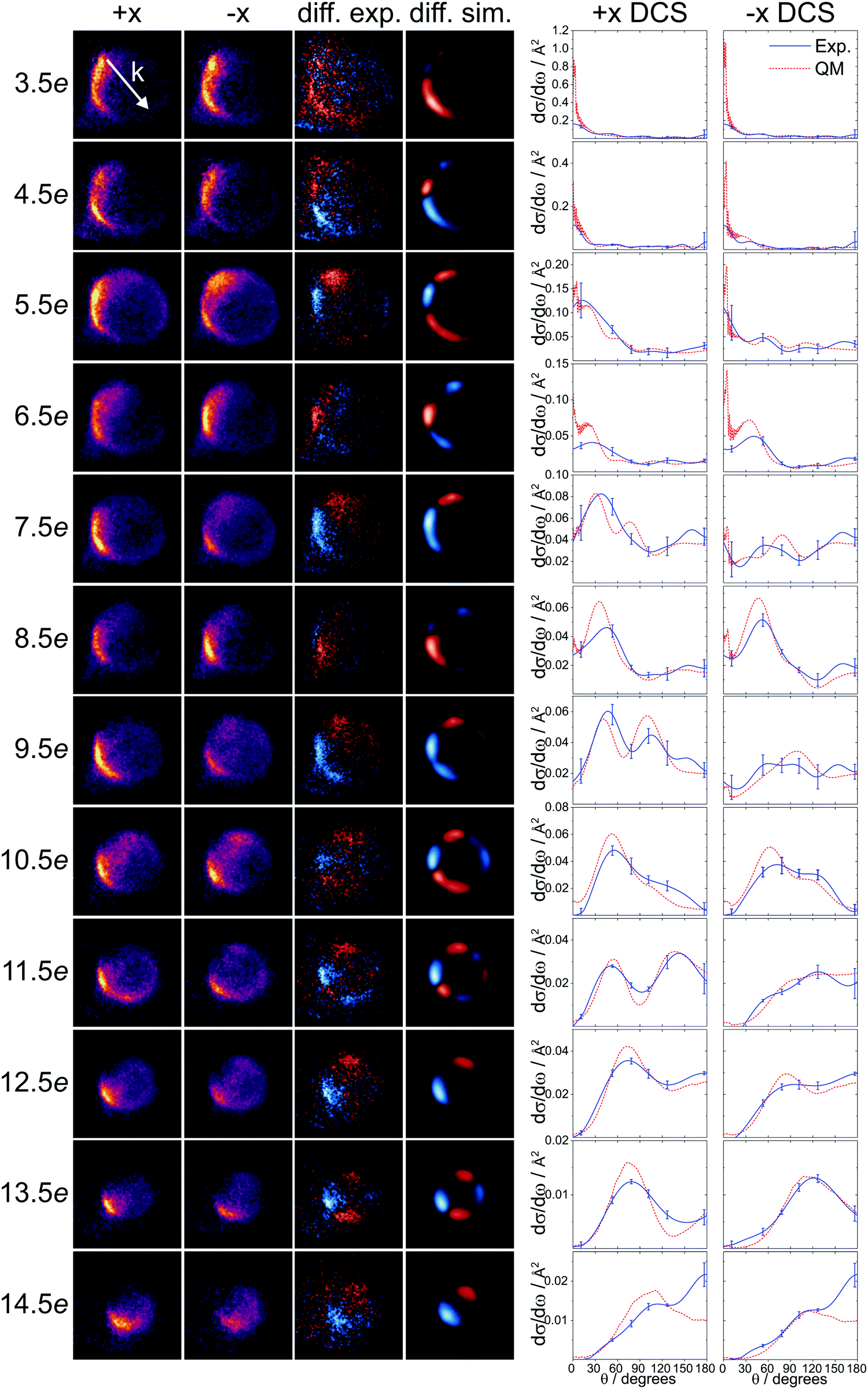 | ||
| Fig. 2 Experimental velocity-map ion images and DCSs compared with QM results (ΔΩ = 1). Experimental +x and −x ion images (columns 1 and 2), experimental and QM simulated difference images (columns 3 and 4), and differential cross sections for the +x and −x orientations (columns 5 and 6) are shown for j′ = 3.5e − 14.5e (top to bottom). The experimental DCSs (blue lines), with error bars corresponding to one standard deviation, are compared to the QM calculated DCSs (red dashed lines). The QM DCSs are averaged over the spread of the experimental collision energy. The relative velocity vector, k = vAr − vNO, is indicated by the white arrow in the top left image. | ||
B. Theoretical and computational methods
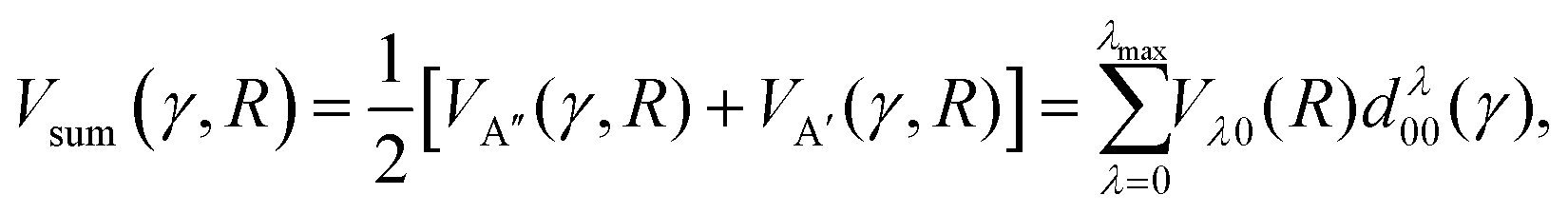 | (2) |
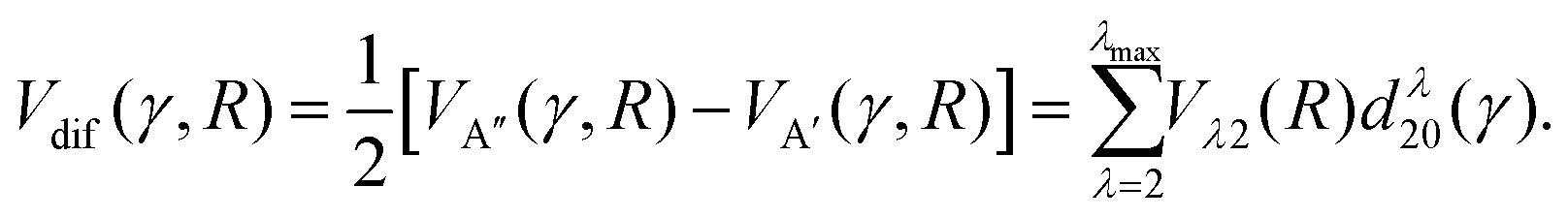 | (3) |
As shown in our previous work,50,51 the orientation dependent differential cross section (DCS) can be expressed in terms of the molecular bond-axis dependent polarization moments, or r-PDDCSs. The r-PDDCSs themselves are a function of the relevant scattering amplitudes, as defined in the ESI.† For a specific transition from an initial state 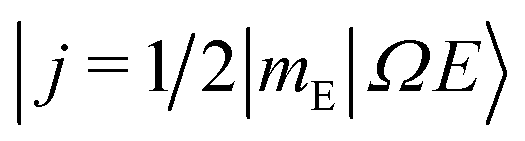 to a final state |j′Ω′E′〉, the DCS corresponds to:
to a final state |j′Ω′E′〉, the DCS corresponds to:
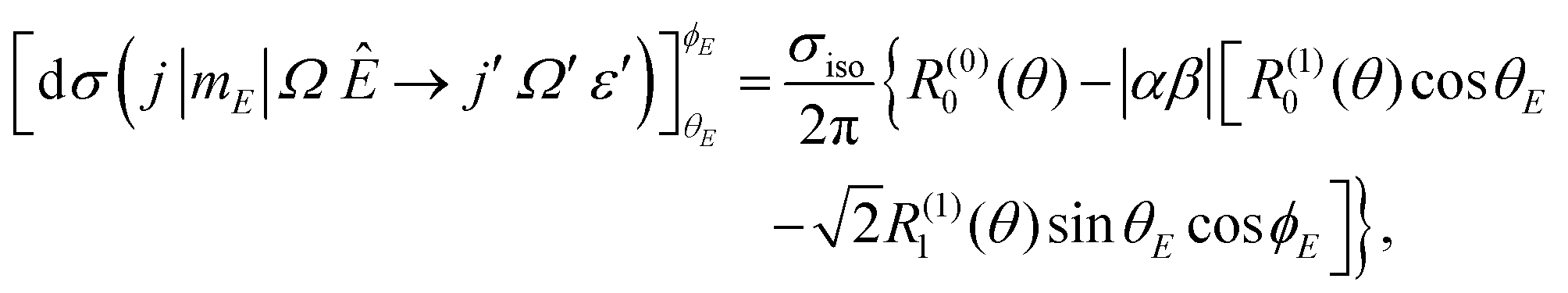 | (4) |
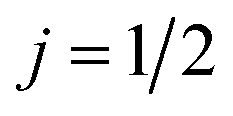 , in which case only the first-order orientation moments exist, R(1)0(θ) and R(1)1(θ) quantify orientation along the z- and x-axes, respectively, and R(0)0(θ) represents the isotropic moment. A thorough discussion of the formalism for oriented DCSs is given in the ESI.†
, in which case only the first-order orientation moments exist, R(1)0(θ) and R(1)1(θ) quantify orientation along the z- and x-axes, respectively, and R(0)0(θ) represents the isotropic moment. A thorough discussion of the formalism for oriented DCSs is given in the ESI.†
In previous work82,83 it was demonstrated that it is possible to define a quantum generalized deflection function (GDF), a quantum analog to the classical joint probability distribution of the scattering angle θ and the total angular momentum J, which can be written as:
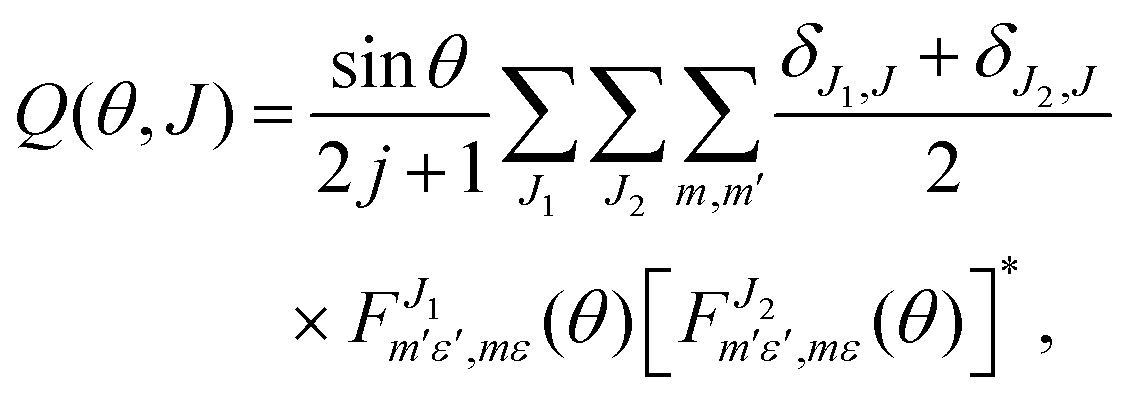 | (5) |
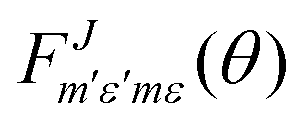 is defined as:
is defined as: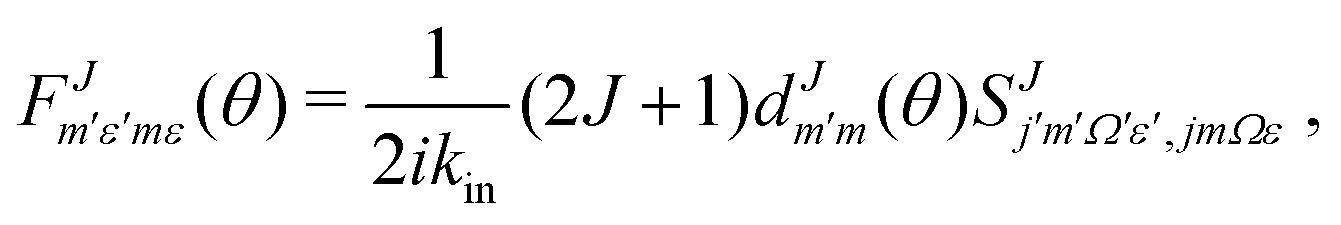 | (6) |
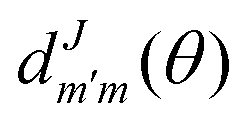 the reduced rotation matrix element, and
the reduced rotation matrix element, and 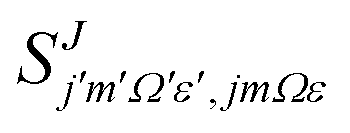 the scattering matrix element for the specific transition.
the scattering matrix element for the specific transition.
In contrast to its classical counterpart, the QM GDF also accounts for coherences between J-partial waves, which enables the observation of interference patterns and the disentanglement of the J-partial waves that contribute to constructive and destructive interference. The QM GDF can therefore provide valuable insights into the scattering mechanism.82
As shown in the ESI† (see also ref. 83), for electric field orientation of the 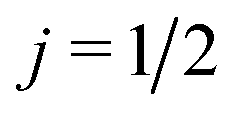 initial state, the orientation dependent QM GDF can be expressed as:
initial state, the orientation dependent QM GDF can be expressed as:
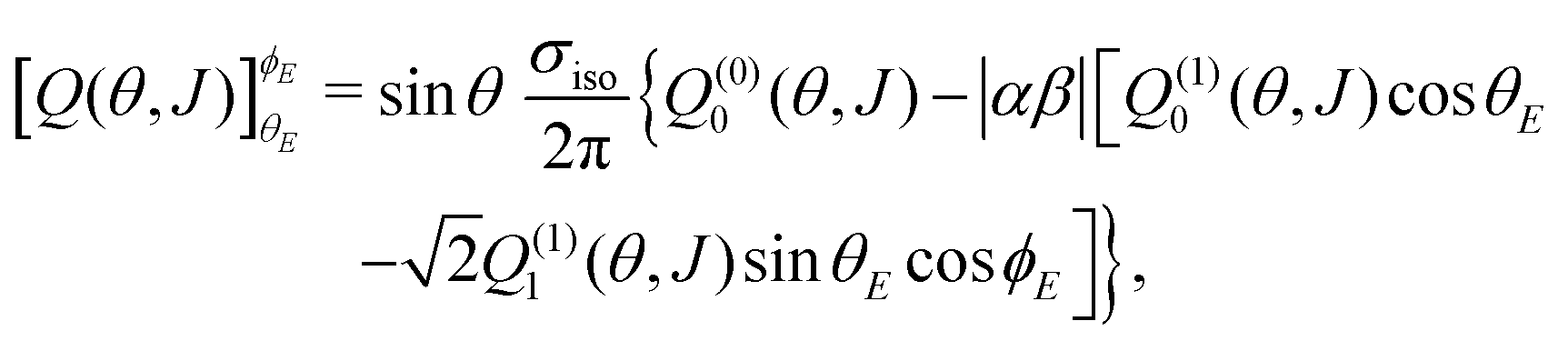 | (7) |
Integration of eqn (7) over θ yields the J-partial cross section,
 | (8) |
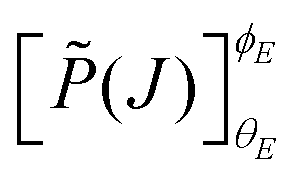 denoting the orientation dependent collision probability, or opacity function.
denoting the orientation dependent collision probability, or opacity function.
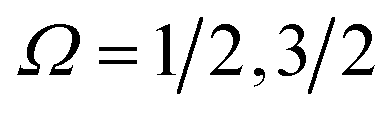 ), and both Λ-doublet levels were included in the scattering calculation. 190 partial waves (J = l + j, …, |l − j|) were used to ensure convergence. The scattering amplitudes for the individual Λ-doublet resolved transitions, obtained in the Hibridon calculation, were combined to determine the r-PDDCSs (see ESI†), which were then used to calculate the bond-axis oriented differential cross sections according to eqn (4).
), and both Λ-doublet levels were included in the scattering calculation. 190 partial waves (J = l + j, …, |l − j|) were used to ensure convergence. The scattering amplitudes for the individual Λ-doublet resolved transitions, obtained in the Hibridon calculation, were combined to determine the r-PDDCSs (see ESI†), which were then used to calculate the bond-axis oriented differential cross sections according to eqn (4).
To allow comparison with the experiment, the experimental velocity distribution was accounted for by averaging the DCSs calculated at seven different collision energies in the range between 590 and 710 cm−1, each weighted to a Gaussian distribution function with a mean collision energy of 651 cm−1 and a FWHM of 35 cm−1. For the theoretical analysis, the DCSs for the two side-on (±x) and the two end-on (±z) orientations were also calculated in the limit of infinite field (and at a fixed Ecoll = 651 cm−1), with calculations run separately on the A′ and A′′ PESs, as well as the sum and the full PESs. For the calculations on the A′, A′′, and sum PESs, which effectively describe a closed-shell system, only the physically meaningful spin–orbit conserving transitions were considered. On the full potential, the infinite field DCSs were computed for both spin–orbit manifolds.
III Results
A. Velocity-map ion images and differential cross sections
The experimental velocity-map ion images for the spin–orbit excited transitions following inelastic collisions of NO with Ar are presented in Fig. 2. The images for the +x and the −x orientations are shown in the first and second column, and the difference images, obtained by subtracting the −x image from the +x image, are shown in the third column. These are compared to the simulated difference images, using the QM polarization moments, in the fourth column. The intensity scale is identical in the +x and −x images for each rotational state; the intensities are therefore directly comparable.Overall, there is good agreement between the experimental and QM simulated data, indicating the high accuracy of the PESs used and a solid understanding of the experimental parameters. As the rotational excitation of the NO increases (from 3.5e to 14.5e, top to bottom), the outgoing kinetic energy decreases, resulting in smaller Newton spheres. It can also be seen that, in general, the distributions become more and more backward scattered as a function of increasing rotational excitation, reflecting the requirement for more impulsive encounters (and consequently more backward scattered trajectories) to access higher lying rotational levels.
Similar to our previous results for the spin–orbit conserving manifold50,51 and end-on oriented collisions,42,72 an alternation in overall scattering intensity between the +x and −x configurations is observed as a function of Δj. We have rationalized this alternation in steric preference by quantum interference, in which the sign of the phase shift between interfering scattering amplitudes changes as a function of Δj.50,51 In the current data, the alternation is most clearly seen in the difference images, where blue on the slow side of the image corresponds to a preference for the +x orientation (positive intensity), and red indicates a preference for the −x orientation (negative intensity). On the fast side of the images, the colors (and intensities) are inverted, as on this side, the +x orientation is subtracted from the −x orientation. For odd Δj transitions, the +x orientation is preferred, especially in the forward scattered direction. For even Δj transitions up to Δj = 10, the −x orientation, although to a smaller degree, dominates. For transitions with Δj ≥ 12, the +x orientation is preferred, irrespective of whether Δj is odd or even.
The differential cross sections extracted from the experimental images and the corresponding QM calculated DCSs for the +x and −x orientations are shown on the right-hand side of Fig. 2. The match between experiment and calculation is good; for j′ = 11.5e − 13.5e, the agreement is excellent, while for some of the other states, especially the intermediate ones around j′ = 6.5e − 9.5e, the agreement is somewhat poorer. We attribute the reduced quality of the fitted data for these particular states to the fact that the main features overlap with the region where the laboratory velocity is close to zero and the detection efficiency is highest.49,72 As a consequence, any small inaccuracies in the instrument function will lead to noticeable deviations from the true DCS.
Compared to the spin–orbit conserving DCSs, the DCSs in the current data set show intensity over the entire range of scattering angles, are generally more backward scattered, and have features that are broader and less sharp. The dominance of the +x orientation for odd Δj transitions is observed clearly, particularly in the forward scattered direction (θ ≤ 90°) for the transitions between j′ = 5.5e − 11.5e. For even and higher Δj transitions, on the other hand, the DCSs for the two side-on orientations are more similar, both in shape and magnitude.
The ion images and DCSs measured for the spin–orbit excited state in the end-on orientation have exhibited similar structures, and followed the same odd–even alternation, as the current experimental data, although the difference in intensity between the +z and −z geometries were somewhat more pronounced than between the two side-on orientations.72
B. Integral steric asymmetries (ISA)
The integral steric asymmetry quantifies the extent to which one orientation is preferred over the other. For the x-axis configuration, the steric asymmetry, Sx, is calculated from the integrated differential cross sections (eqn (4)) for the +x and −x orientations, σ±x: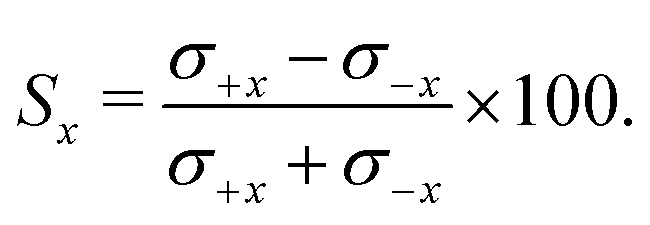 | (9) |
The ISA for z-axis orientation, Sz, is defined analogously as:
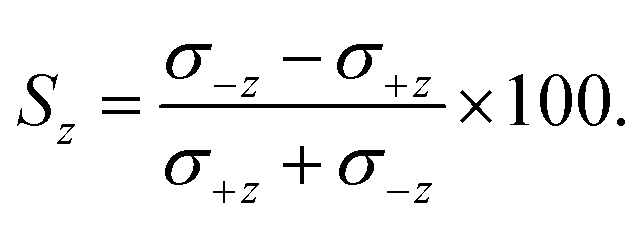 | (10) |
As will be discussed below, all collisions in the spin–orbit changing manifold are nearside, such that the +x/−z orientation corresponds to repulsive scattering off the N-side/N-end, while the −x/+z configuration corresponds to repulsive scattering off the O-side/O-end. A positive ISA therefore indicates a preference for repulsive scattering off the N-side/N-end, and a negative ISA indicates a preference for repulsive scattering off the O-side/O-end.
Fig. 3 shows the experimental ISA (blue) along with the collision energy averaged QM ISA (red) for the x-axis orientation from the current experiment (top panel) and, for comparison, the z-axis orientation from previous work in our group.85
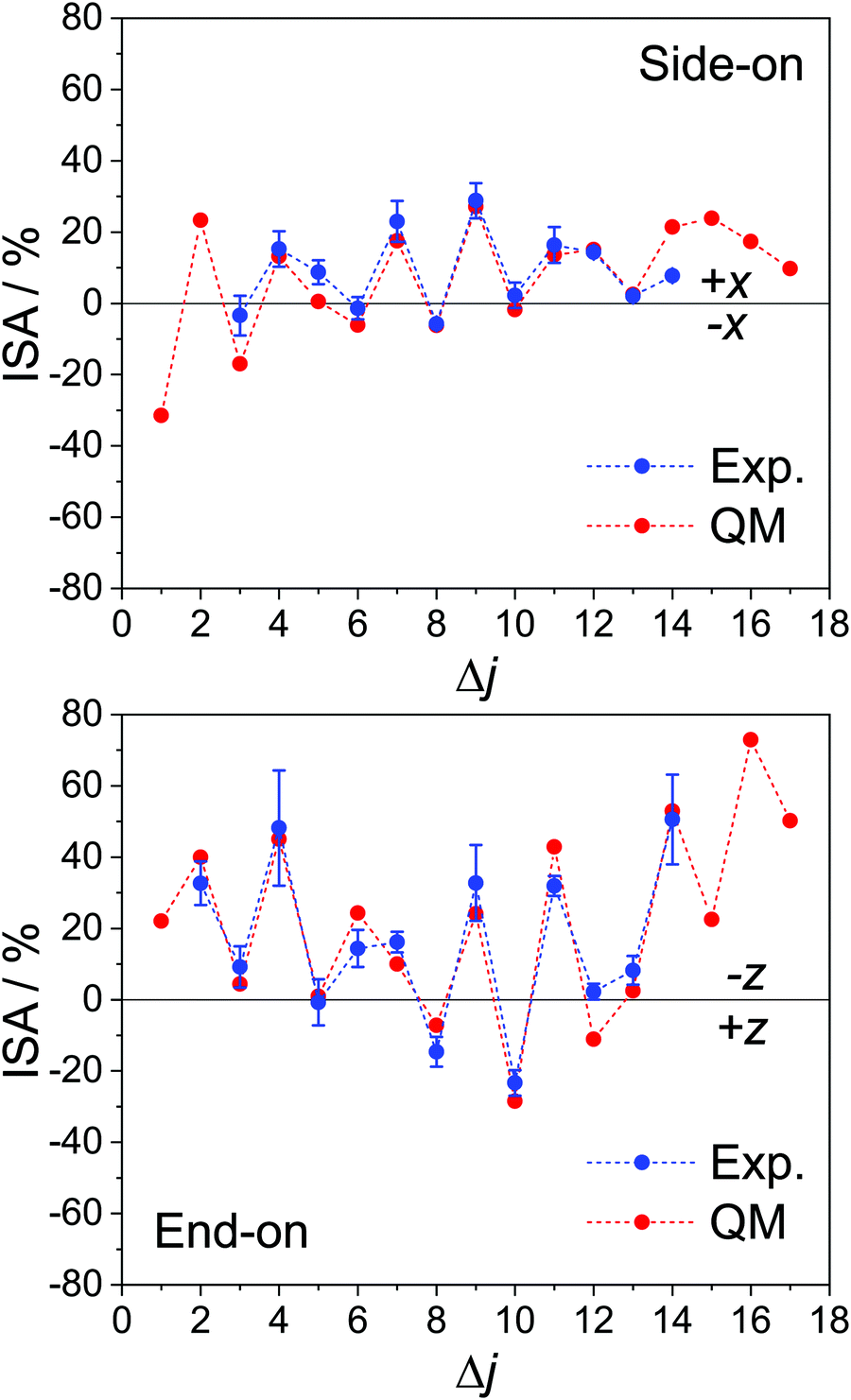 | ||
| Fig. 3 Experimental and QM calculated integral steric asymmetries for the spin–orbit excited manifold (ΔΩ = 1). The experimentally measured ISA is shown in blue, with error bars representing one standard deviation; the corresponding QM data, which have been averaged over the experimental collision energy distribution, are shown in red. The top panel is for the x-axis configuration, probed in the current experiment, and the bottom panel is for the z-axis orientation (with the experimental data taken from ref. 85). Both sets of data are for the final e Λ-doublet states. | ||
As with the DCSs, there is good agreement between the experimental and QM calculated ISAs, and the alternation in preference between odd and even Δj is evident. However, in both the side-on and the end-on configurations, the N-side/N-end preference is significantly larger in magnitude than the preference for the O-side/O-end. In fact, for Δj = 6, 8, 10 in the x-axis configuration, the ISA is close to zero. The observed trend points to an overall preference for collisions off the N-side/N-end of the molecule and contrasts with the steric asymmetries we have measured for the corresponding spin–orbit conserving transitions, for which the ISAs for adjacent Δj transitions are similar in magnitude42,51 (see Fig. S1 in the ESI†). Note that for Δj = 5 in the side-on orientation, the ISA is also approximately zero, even though the main feature in the forward scattered direction is larger in the +x DCS than in the −x DCS (Fig. 2). The small ISA is partly due to the fact that upon integration, the intensity is weighted by sinθ (to account for the solid angle), and partly because the −x DCS is consistently larger in the backward scattered region (θ ≥ 90°).
C. Calculated DCSs and QM deflection functions at infinite field
Experimentally, the extent to which the NO molecules can be oriented is limited in part by the electric field strength; in order to eliminate any effects due to incomplete initial orientation of the molecules, we have calculated the DCSs at infinite field, at which the mixing of the initial e and f Λ-doublets is complete (i.e., both states contribute equally to the initial superposition state, as |α| = |β| = 1). Note that even at infinite field, the initial orientation of the NO molecules is still defined by a relatively broad cosine probability distribution, in which the most probable orientation is the one in which the NO axis (N → O) is antiparallel to the electric field vector, but other orientations, weighted according to the cosine distribution, contribute as well (see, for example, Fig. 3 in ref. 42). Fig. 4 presents the infinite field DCSs for the +x (red), −x (blue dashed), +z (purple dashed) and −z (green) orientations, along with the maximized DCSs (gray shaded area), for Δj = 4e − 13e. The maximized DCS corresponds to the orientation (as defined by θE/ϕE) that maximizes eqn (4) at a given scattering angle. Note that at θ = 0°/180°, the DCSs for the −x and +x configurations are identical, since at those angles the orientation is along the ±z-axis.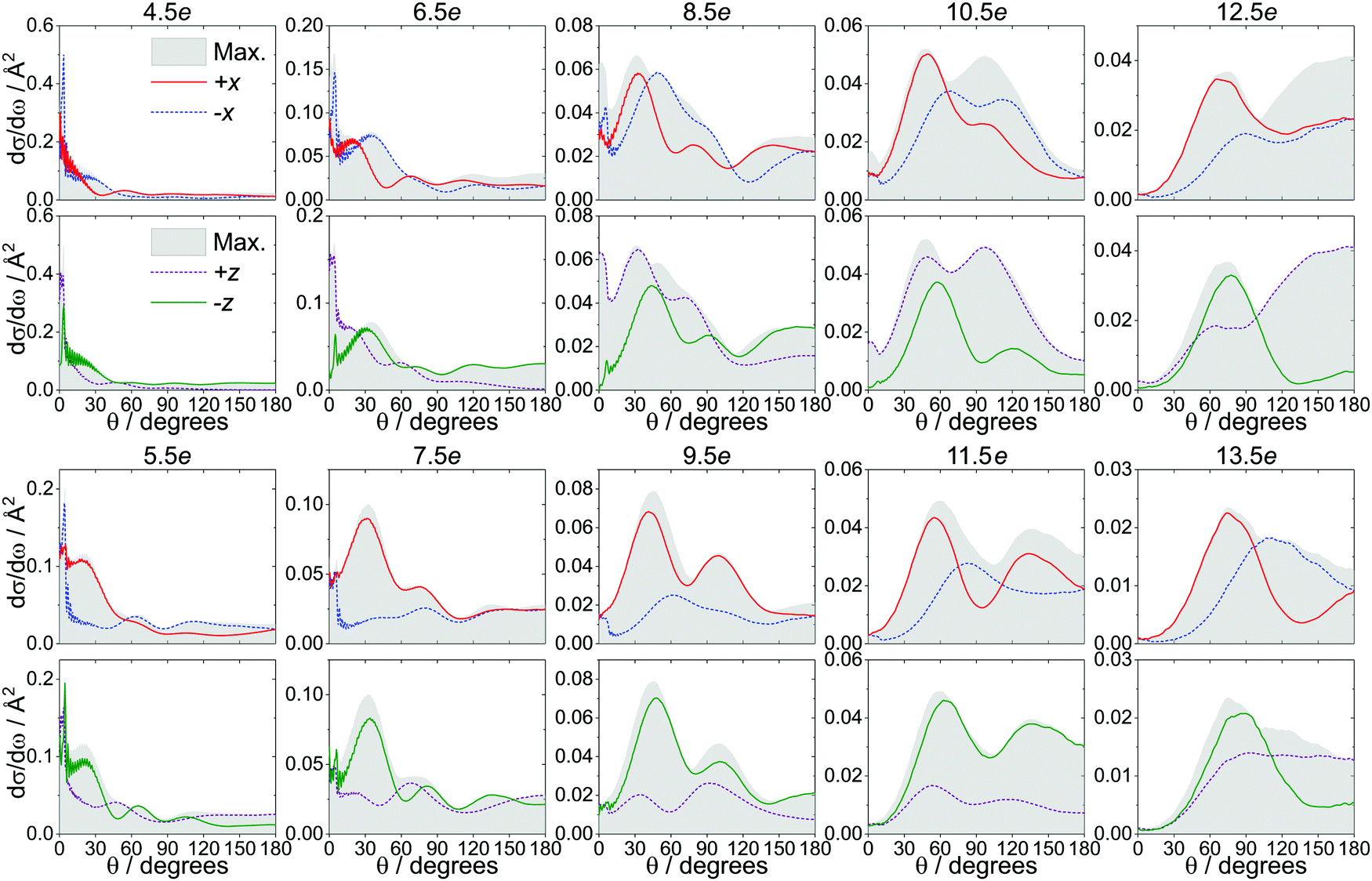 | ||
| Fig. 4 Comparison of the spin–orbit changing, infinite field DCSs for the side-on (red: +x, blue: −x) and end-on (purple: +z, green: −z) configurations with the maximized DCSs (gray shaded areas). The even transitions between Δj = 4–12 are shown in the two upper rows, and the odd transitions between Δj= 5–13 are shown in the two lower rows. | ||
For the odd Δj transitions, the trends are similar to the ones for the spin–orbit conserving manifold,50,51 with the +x and the −z orientations (collisions off the N-side/N-end) dominating in the side-on and the end-on configuration, respectively. In addition, the side-on DCSs are close to the maximized DCSs in the forward scattered region, while the end-on DCSs more closely match the maximized DCSs in the backward scattered region.50,51
However, the even Δj transitions, shown in the top two rows of Fig. 4, follow a different pattern. Most strikingly observed for Δj = 6, 8, 10 is a distinct peak in the very forward scattered direction that is maximized in the O-end (+z) and minimized in the N-end (−z) orientation, with the two side-on orientations representing intermediate cases. At larger values of θ, the magnitude of the +x DCS for Δj ≤ 8 is comparable to that of the −x DCS (which is expected to dominate for the even Δj) and becomes more dominant for Δj ≥ 10, where it closely matches the maximized DCS in the forward scattered region. This significantly diminishes the steric preference for the −x orientation for these even Δj states, in agreement with the increased importance of N-side/N-end collisions observed in the ISA (Fig. 3).
The J-partial cross sections, shown in Fig. S3 in the ESI,† corroborate the trends observed in the DCSs in Fig. 4; for the odd Δj transitions, the preference for +x and −z is clear, while for the even Δj transitions, the two side-on and the two end-on orientations are more similar. Differences are in general more evident at higher impact parameters, which are correlated with smaller scattering angles.
Fig. 5 shows the QM generalized deflection functions (divided by sinθ) for the final 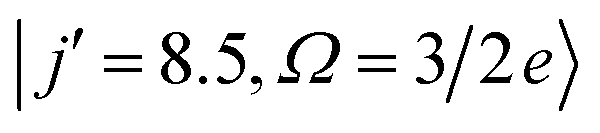 and
and 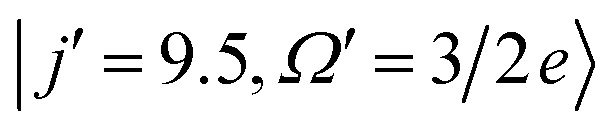 states. As described in Section II B 1. and in ref. 82 and 83, the QM deflection function represents the joint quasi-probability of the scattering angle and total angular momentum. It exposes the range of total angular momenta that contribute to each of the maxima in the DCS. By summing the contributions of all Js (integration or sum along the vertical axis), the DCS is recovered, and similarly, by integration over the scattering angle, the J-partial cross section is obtained.
states. As described in Section II B 1. and in ref. 82 and 83, the QM deflection function represents the joint quasi-probability of the scattering angle and total angular momentum. It exposes the range of total angular momenta that contribute to each of the maxima in the DCS. By summing the contributions of all Js (integration or sum along the vertical axis), the DCS is recovered, and similarly, by integration over the scattering angle, the J-partial cross section is obtained.
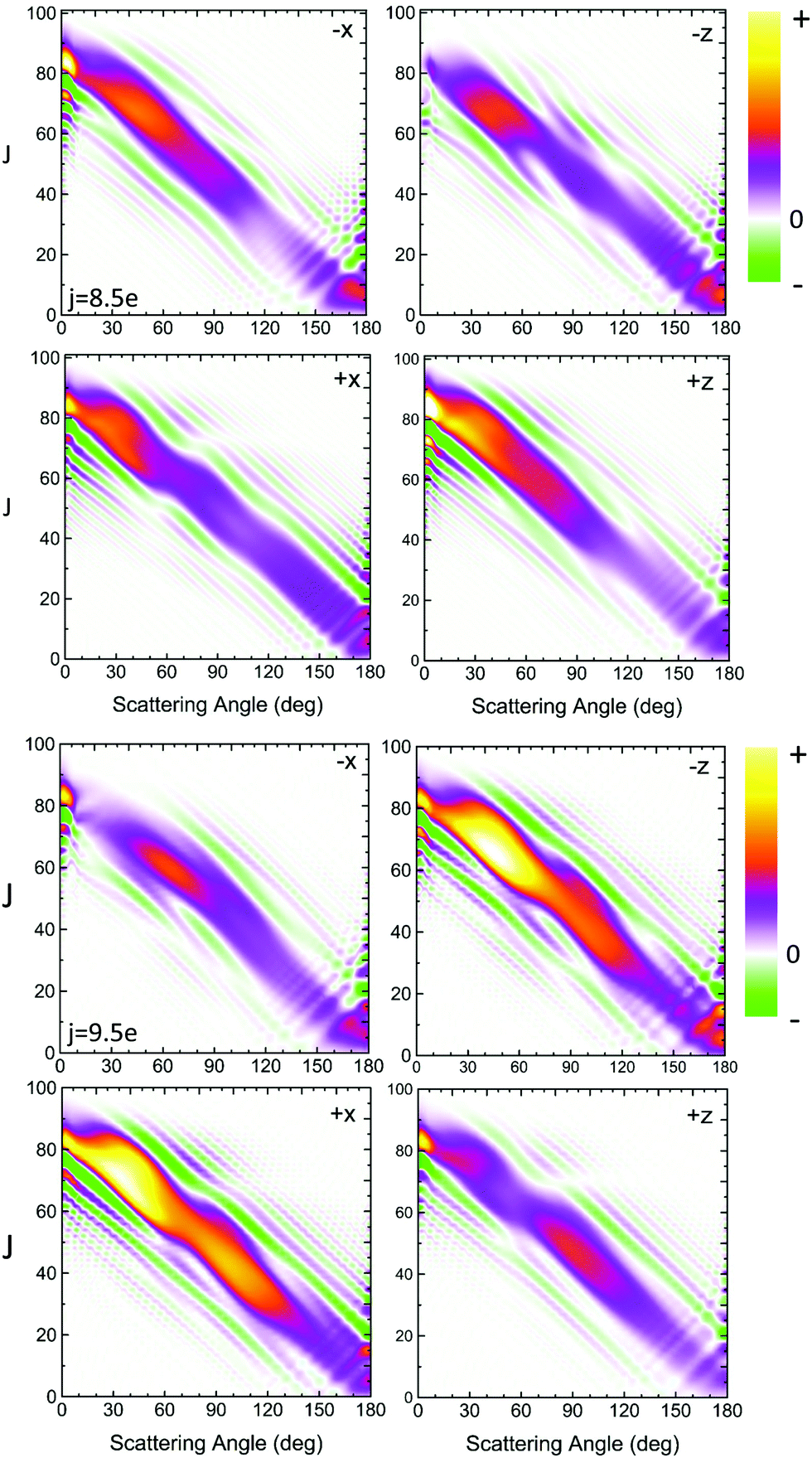 | ||
| Fig. 5 QM generalized deflection functions for the j′ = 8.5e (top) and j′ = 9.5e (bottom) final rotational states in the ΔΩ = 1 manifold. In both states, the +x and −z (N-side/N-end) and the −x and +z (O-side/O-end) orientations show similar features, with +x/−z clearly dominating in the odd Δj = 9 state. The forward peak (θ∼0°) is correlated with high impact parameters (J>80) and appears especially pronounced in the +z configuration for j′ = 8.5e. The generalized deflection functions (eqn (7)) have been divided by sinθ to highlight forward and backward scattering. | ||
The QM J–θ correlations in Fig. 5 are indicative of direct collisions: high J lead to low scattering angles, and vice versa. Although the GDFs extend over the entire range of scattering angles, the J–θ mutual dependence is diagonal and continuous, which rules out the presence of resonances or long-lived complexes. The negative slope indicates that all collisions are nearside. Moreover, the interval of partial wave coherences is small, and hence interference between different groups of partial waves is largely irrelevant except at the extreme forward angles.
In spite of these common features, the specific structure of the GDFs for the different orientations, and the even and odd Δj transitions, differ substantially, suggesting distinct scattering mechanisms. In both states shown in Fig. 5, the N-side (+x, bottom left) and N-end (−z, top right) and the O-side (−x, top left) and O-end (+z, bottom right) deflection functions exhibit similar features. For Δj = 8, the O-side/O-end orientations are slightly dominant over the N-side/N-end orientations, whereas for Δj = 9, the N-side/N-end orientations are significantly more intense than the O-side/O-end orientations. This again mirrors the overall dominance of N-side/N-end collisions over O-side/O-end collisions in the spin–orbit excited manifold, as observed in Fig. 3.
The prominent forward peak close to θ∼0° is strongly correlated with a small range of high impact parameters (J∼80–90). In the 8.5e final state, the feature is particularly intense in the +z orientation, but is only weak in the +x and essentially absent in the −z orientation. In the 9.5e state, the peak has similar intensity in all four configurations.
VI Discussion
A. N-side/N-end preference
The observed trends in the calculated DCSs and QM deflection functions for the even and odd Δj transitions indicate that the scattering dynamics into the spin–orbit excited manifold are fundamentally different to the dynamics into the spin–orbit conserving manifold. In order to unravel these differences, we turn to the potential energy surfaces that govern the scattering process in each case. As already mentioned, collisions of the NO molecule with a rare gas atom occur on the coupled A′ and A′′ potentials. In a quasi-classical approach, Zhang and Stolte recently used an expression which explicitly takes into account the position of the unpaired electron with respect to the triatomic plane:86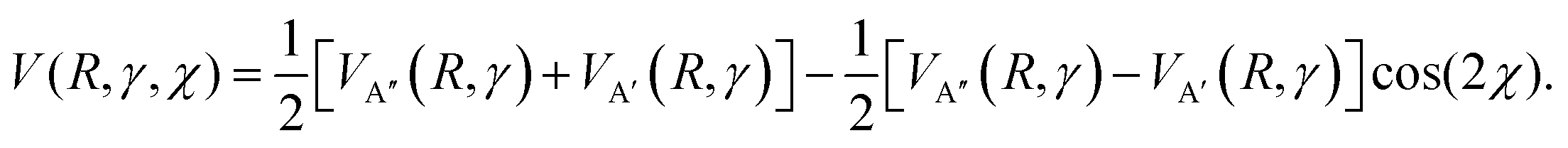 | (11) |
The first and second term (excluding the cos(2χ) dependence) are the sum and difference potentials introduced in eqn (2) and (3), and χ is the angle of the unpaired electron with respect to the triatomic plane.
For spin–orbit conserving transitions, it is favorable if the interaction between the unpaired electron and the approaching Ar atom is minimal. In order to effect a spin–orbit excitation, on the other hand, the Ar atom necessarily needs to interact with the unpaired electron. In the two limiting cases of the unpaired electron lying within (χ = 0°, preferred for spin–orbit changing transitions) or perpendicular (χ = 90°, preferred for spin–orbit conserving transitions) to the plane defined by the three atoms, eqn (11) gives:
| V(R,γ,χ = 0°) = VA′(R,γ) | (12) |
| V(R,γ,χ = 90°) = VA′′(R,γ) | (13) |
Consequently, we expect the spin–orbit changing manifold to be predominantly governed by the A′ potential, and the spin–orbit conserving transitions by the A′′ potential.
The A′, A′′, and sum PESs are plotted in Fig. 6. The red lines represent the repulsive core, while the blue lines indicate the attractive parts of the potential. As seen in the figure, the A′ PES is wider on the N-side and features a concave repulsive region on the side of the molecule. The extended width on the N-side indicates the location of the unpaired electron and collisions towards this end of the molecule are expected to be more likely to lead to spin–orbit excitation than collisions towards the O-side/O-end of the molecule. The A′′ and sum PESs, in contrast, are convex. The widest part of the A′′ PES is close to the center of the molecular axis and interaction with the unpaired electron, which in this case lies out of plane, is less efficient.
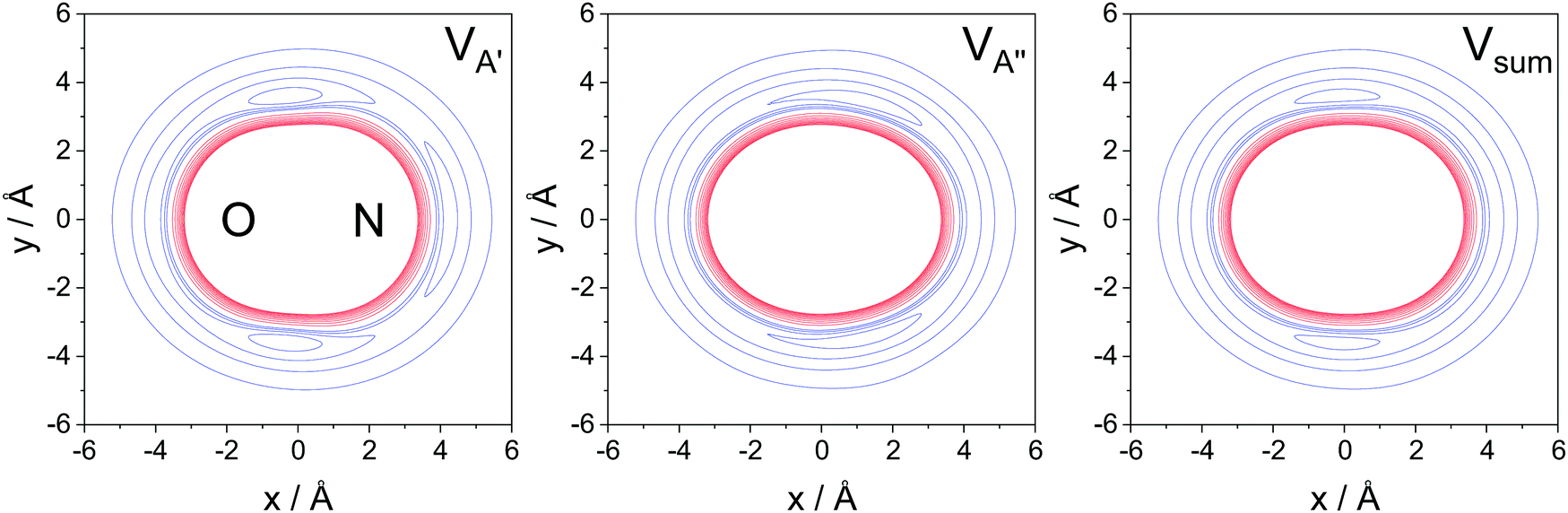 | ||
| Fig. 6 NO + Ar potential energy surfaces for the A′, A′′, and sum potentials (left to right). Repulsive parts of the PES are represented in red with an energy spacing of 100 cm−1; attractive parts are represented in blue with a spacing of 25 cm−1. The N-end and O-end of the molecule are indicated in the left panel. | ||
These considerations imply two things: firstly, the A′ potential, as indicated in eqn (12), plays a crucial role for spin–orbit changing transitions (and less so for spin–orbit conserving transitions). Secondly, the overall shift of the ISA towards an N-side/N-end preference is a direct consequence of the location of the unpaired electron. For odd Δj transitions, in which N-side/N-end collisions are also favored based on quantum interference, the trends are similar to the ones in the spin–orbit conserving manifold. For the even Δj transitions, in which quantum interference is constructive for O-side/O-end collisions, the required interaction with the unpaired electron renders trajectories towards the N-side/N-end more important than in the spin–orbit conserving manifold. This results in comparable contributions from N-side/N-end and O-side/O-end collisions, and consequently a relatively small steric asymmetry, for even Δj transitions in the spin–orbit excited manifold.
B. The prominent forward scattered peak
In addition to the overall N-side/N-end preference in the spin–orbit changing manifold, a second intriguing feature in our calculations is the prominent forward peak observed for even Δj transitions, particularly the states between Δj = 6–10, in the O-end orientation.Fig. 7 examines the contributions of the A′ (first column) and A′′ (second column) potentials to the total DCSs for the spin–orbit conserving (ΔΩ = 0, third column) and changing (ΔΩ = 1, fourth column) transitions for j′ = 8.5e. This state was chosen because of the prominence of its forward scattered peak, but the results for other intermediate Δj transitions are overall very similar. The DCSs for the side-on and the end-on orientations are shown at the top and bottom of Fig. 7, respectively. The DCSs obtained on the A′ potential exhibit a strong forward scattered peak in all orientations except the N-end (−z) configuration, whereas on the A′′ potential, only a small sharp peak within a broader feature in the forward direction is observed. The spin–orbit conserving transitions calculated on the full PES resemble the DCSs calculated on the A′′ PES, confirming that these transitions occur to a large extent on the A′′ potential. The spin–orbit changing transitions appear to originate from a combination of the A′ and A′′ potentials, and the strong forward peak is retained.
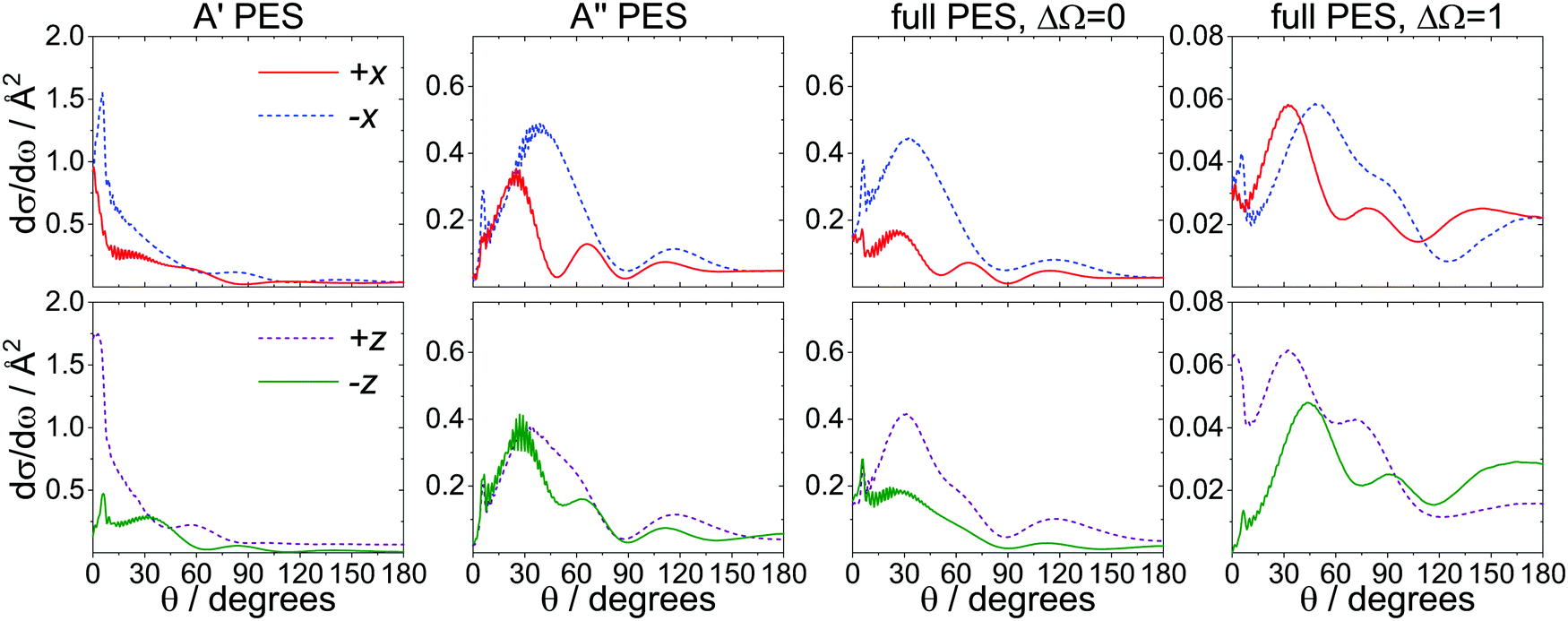 | ||
| Fig. 7 Infinite field QM DCSs for Δj = 8, calculated on the A′, A′′, and full potentials, in the ±x (top) and the ±z (bottom) orientations. The calculations run on the A′ and A′′ PESs (first and second columns) correspond to spin–orbit conserving transitions. The third and fourth columns show the DCSs calculated on the full potential for the spin–orbit conserving and changing manifolds, respectively. | ||
We have already established the importance of the A′ potential for the spin–orbit changing manifold, and Fig. 7 shows that the strong forward peak is a further signature of the A′ potential. The large impact parameters associated with the forward peak (see Fig. 5) suggest that Ar atoms approaching from the O-end (towards the N-end) interact particularly efficiently with the electron once they reach the region of the extended width on the N-side of the potential.
Based on the preceding analysis, it is tempting to attribute the forward scattered peak observed in the spin–orbit changing manifold exclusively to the concave shape of the repulsive core in the A′ PES. However, calculations run on truncated potentials, in which the attractive parts have been removed (see ESI†), reveal that this conclusion is incorrect. As shown in Fig. S5 in the ESI,† the prominent forward peak is completely absent in the DCSs calculated on the truncated PESs (similar results are obtained using a hard shell QM model87 – see ESI†). This is strong evidence that the attractive parts of the potential are essential for the appearance of the prominent forward peak in the spin–orbit changing manifold.
The potential energy surfaces shown in Fig. 6 feature an attractive well on the side of the molecule. The well on the side of the A′ potential runs roughly parallel to and is deepest around the concave indentation of the repulsive wall. It is conceivable that high impact parameter trajectories approaching from the O-end of the molecule are efficiently funnelled through this attractive well and brought into close contact with the extended part of high electron density, so as to promote spin–orbit excitation. The high impact parameter required for these kinds of trajectories results in minimal deflection of the Ar atom, as manifested in the sharp forward feature.
While the strong forward peak is most prominent for intermediate, even Δj transitions, it is also present in the odd Δj ≤ 11 transitions (see Fig. 4 and Fig. 5). However, due to the overall preference for N-side/N-end collisions for the odd states, the forward peak is overshadowed by the main feature in the forward direction. The peak is perhaps most evident in the −x geometry for Δj = 7, 9, where it is next to a minimum in the DCS. The prominence of the forward scattered feature in the even Δj transitions is thus a combination of the characteristic topology of the A′ PES and the quantum interference effects which determine the overall preference for a specific orientation at a given Δj.
In order for the prominent forward scattered feature to appear, the unpaired electron must be lying in (or at least very close to) the triatomic plane defined by the NO molecule and the Ar atom. Given the shape of the A′ potential, this is the most efficient way for large impact parameter collisions to interact with the electron, and thus promote a spin–orbit excitation. For lower impact parameters (and larger scattering angles), the atom will be able to interact with the electron within a larger range of χ angles, and the dynamics are determined by contributions from both the A′ and A′′ potentials.
V Conclusions
We have presented a combined experimental and theoretical study of the NO(X) spin–orbit changing transitions,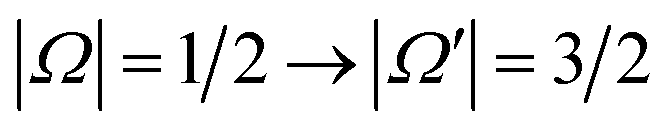 , following oriented collisions with argon atoms. The experimentally measured differential and integral cross sections for the side-on geometry have been shown to be in good agreement with quantum mechanical calculations. As in previous studies,42,50,51 quantum interference effects, manifest as oscillations in the steric preference for N-side or O-side collisions, have been observed.
, following oriented collisions with argon atoms. The experimentally measured differential and integral cross sections for the side-on geometry have been shown to be in good agreement with quantum mechanical calculations. As in previous studies,42,50,51 quantum interference effects, manifest as oscillations in the steric preference for N-side or O-side collisions, have been observed.
The theoretical analysis and discussion has focused on the overall shift of the ISA in the spin–orbit changing manifold towards an N-side/N-end preference and a prominent forward scattered peak, observed most intensely in the calculations for intermediate, even Δj transitions in the O-end orientation. We have rationalized the shift of the ISA towards positive values with the higher unpaired electron density, and consequently more facile spin–orbit excitation, closer to the N-side/N-end of the molecule, which leads to a competition between N-side/N-end and (otherwise preferred) O-side/O-end collisions for even Δj transitions. The ability to manipulate the initial relative orientation of the NO and the argon collision partners thus provides a direct probe of the unpaired electron's location and explains the importance of the A′ potential for scattering into the spin–orbit excited manifold. Our conclusions are consistent with the notion that spin–orbit excited transitions are primarily governed by the difference potential.78,79,81 The differences between the A′ and A′′ potentials are most significant on the N-side, where the A′ potential is substantially wider than the A′′ potential, and this is where spin–orbit changing collisions most likely occur.
QM generalized deflection functions and infinite field calculations of the DCSs carried out on the A′, A′′, sum, and full potentials revealed that the prominent forward feature corresponds to high impact parameter collisions and originates from the specific topology of the A′ PES, an interplay between the repulsive region of high electron density near the N-side of the molecule and the attractive well nested into the concave region of the potential. We have argued that the attractive well efficiently funnels high impact parameter trajectories from the O-end, bringing the Ar atom into close contact with the unpaired electron on the N-side of the molecule, and thus facilitating a change in spin–orbit quantum number.
Our study demonstrates how the ability to determine differential and integral cross sections for specific orientations of a diatomic molecule can yield very detailed insights into the scattering dynamics and the forces that govern them. Although the good agreement with the experimental measurements are further proof of the accuracy of the PESs used for the calculations, the angular resolution in our experiments is currently not high enough to resolve the sharp forward scattered feature predicted in the QM calculations (though there might be a hint of it in some of the images). However, in principle, it should be possible to combine our method of electric field orientation with a Stark decelerated crossed molecular beam setup;25,88–90 this would afford the angular resolution required to measure very sharp features in the DCSs. If such an experiment can be carried out successfully, it will provide a sensitive tool to test the accuracy of calculated potential energy surfaces and shed light on the associated scattering dynamics, potentially also in processes more complicated than simple diatom-atom scattering. In the specific case of our current work, the experimental confirmation of the prominent forward scattered feature in the DCSs for even Δj transitions would offer an ultimate test for the accuracy of the NO + Ar potentials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. J. A. and P. G. J. thank Prof. Enrique Verdasco for his help with the calculations. Funding by the UK EPSRC (to M. B. via Programme Grant EP/L005913/1 and EP/T021675/1) and the Spanish Ministry of Science and Innovation (grant MINECO/FEDER-PGC2018-096444-B-I00) is gratefully acknowledged. P. G. J. acknowledges funding by the Fundación Salamanca City of Culture and Knowledge (programme for attracting scientific talent to Salamanca).References
- P. Jankowski, A. R. W. McKellar and K. Szalewicz, Science, 2012, 336, 1147–1150 CrossRef CAS.
- P. Jankowski, L. A. Surin, A. Potapov, S. Schlemmer, A. R. W. McKellar and K. Szalewicz, J. Chem. Phys., 2013, 138, 084307 CrossRef.
- G. Czakó and J. M. Bowman, J. Am. Chem. Soc., 2009, 131, 17534–17535 CrossRef.
- C. Xiao, X. Xu, S. Liu, T. Wang, W. Dong, T. Yang, Z. Sun, D. Dai, X. Xu, D. H. Zhang and X. Yang, Science, 2011, 333, 440–442 CrossRef CAS.
- R. Otto, J. Ma, A. W. Ray, J. S. Daluz, J. Li, H. Guo and R. E. Continetti, Science, 2014, 343, 396–399 CrossRef CAS.
- M. Qiu, Z. Ren, L. Che, D. Dai, S. A. Harich, X. Wang, X. Yang, C. Xu, D. Xie, M. Gustafsson, R. T. Skodje, Z. Sun and D. H. Zhang, Science, 2006, 311, 1440–1443 CrossRef CAS.
- W. Dong, C. Xiao, T. Wang, D. Dai, X. Yang and D. H. Zhang, Science, 2010, 327, 1501–1502 CrossRef CAS.
- T. Wang, J. Chen, T. Yang, C. Xiao, Z. Sun, L. Huang, D. Dai, X. Yang and D. H. Zhang, Science, 2013, 342, 1499–1502 CrossRef CAS.
- J. B. Kim, M. L. Weichman, T. F. Sjolander, D. M. Neumark, J. Kłos, M. H. Alexander and D. E. Manolopoulos, Science, 2015, 349, 510–513 CrossRef CAS.
- M. Hapka, G. Chałasiński, J. Kłos and P. S. Żuchowski, J. Chem. Phys., 2013, 139, 014307 CrossRef.
- T. Yang, J. Chen, L. Huang, T. Wang, C. Xiao, Z. Sun, D. Dai, X. Yang and D. H. Zhang, Science, 2015, 347, 60–63 CrossRef CAS.
- K. Stark and H. Werner, J. Chem. Phys., 1996, 104, 6515–6530 CrossRef CAS.
- D. Skouteris, D. E. Manolopoulos, W. Bian, H.-J. Werner, L.-H. Lai and K. Liu, Science, 1999, 286, 1713–1716 CrossRef CAS.
- S. Chefdeville, Y. Kalugina, S. Y. T. van de Meerakker, C. Naulin, F. Lique and M. Costes, Science, 2013, 341, 1094–1096 CrossRef CAS.
- C. Naulin and M. Costes, Int. Rev. Phys. Chem., 2014, 33, 427–446 Search PubMed.
- E. Lavert-Ofir, Y. Shagam, A. B. Henson, S. Gersten, J. Kłos, P. S. Żuchowski, J. Narevicius and E. Narevicius, Nat. Chem., 2014, 6, 332–335 CrossRef CAS.
- T. F. M. Luxford, T. R. Sharples, K. G. McKendrick and M. L. Costen, J. Chem. Phys., 2016, 145, 174304 CrossRef.
- A. Klein, Y. Shagam, W. Skomorowski, P. S. Żuchowski, M. Pawlak, L. M. C. Janssen, N. Moiseyev, S. Y. T. van de Meerakker, A. van der Avoird, C. P. Koch and E. Narevicius, Nat. Phys., 2017, 13, 35–38 Search PubMed.
- C. L. Russell and D. E. Manolopoulos, Chem. Phys. Lett., 1996, 256, 465–473 CrossRef CAS.
- F. Lique, G. Li, H.-J. Werner and M. H. Alexander, J. Chem. Phys., 2011, 134, 231101 CrossRef.
- M. Tizniti, S. D. Le Picard, F. Lique, C. Berteloite, A. Canosa, M. H. Alexander and I. R. Sims, Nat. Chem., 2014, 6, 141–145 CrossRef CAS.
- S. M. Remmert, S. T. Banks, J. N. Harvey, A. J. Orr-Ewing and D. C. Clary, J. Chem. Phys., 2011, 134, 204311 CrossRef.
- F. Wang, K. Liu and T. P. Rakitzis, Nat. Chem., 2012, 4, 636 CrossRef CAS.
- H. Guo and K. Liu, Chem. Sci., 2016, 7, 3992–4003 RSC.
- S. N. Vogels, T. Karman, J. Kłos, M. Besemer, J. Onvlee, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, Nat. Chem., 2018, 10, 435–440 CrossRef CAS.
- T. de Jongh, T. Karman, S. N. Vogels, M. Besemer, J. Onvlee, A. G. Suits, J. O. F. Thompson, G. C. Groenenboom, A. van der Avoird and S. Y. T. van de Meerakker, J. Chem. Phys., 2017, 147, 013918 CrossRef.
- J. Kłos, Q. Ma, M. H. Alexander and P. J. Dagdigian, J. Chem. Phys., 2017, 146, 114301 CrossRef.
- Y. Shagam, A. Klein, W. Skomorowski, R. Yun, V. Averbukh, C. P. Koch and E. Narevicius, Nat. Chem., 2015, 7, 921–926 CrossRef CAS.
- A. Bergeat, J. Onvlee, C. Naulin, A. van der Avoird and M. Costes, Nat. Chem., 2015, 7, 349–353 CrossRef CAS.
- F. Wang, J. Lin and K. Liu, Science, 2011, 331, 900–903 CrossRef CAS.
- F. Wang and K. Liu, J. Chem. Phys., 2016, 145, 144305 CrossRef.
- F. Wang and K. Liu, J. Chem. Phys., 2016, 145, 144306 CrossRef.
- H. Pan, F. Wang, G. Czakó and K. Liu, Nat. Chem., 2017, 9, 1175–1180 CrossRef CAS.
- W. E. Perreault, N. Mukherjee and R. N. Zare, Science, 2017, 358, 356–359 CrossRef CAS.
- W. E. Perreault, N. Mukherjee and R. N. Zare, Nat. Chem., 2018, 10, 561 CrossRef CAS.
- T. R. Sharples, J. G. Leng, T. F. M. Luxford, K. G. McKendrick, P. G. Jambrina, F. J. Aoiz, D. W. Chandler and M. L. Costen, Nat. Chem., 2018, 10, 1148–1153 CrossRef CAS.
- J. F. E. Croft, N. Balakrishnan, M. Huang and H. Guo, Phys. Rev. Lett., 2018, 121, 113401 CrossRef CAS.
- P. G. Jambrina, J. F. E. Croft, H. Guo, M. Brouard, N. Balakrishnan and F. J. Aoiz, Phys. Rev. Lett., 2019, 123, 043401 CrossRef CAS.
- B. Friedrich, D. R. Herschbach, J.-M. Rost, H.-G. Rubahn, M. Renger and M. Verbeek, J. Chem. Soc., Faraday Trans., 1993, 89, 1539–1549 RSC.
- J. van Leuken, J. Bulthuis, S. Stolte and J. Snijders, Chem. Phys. Lett., 1996, 260, 595–603 CrossRef CAS.
- A. Gijsbertsen, H. Linnartz, C. A. Taatjes and S. Stolte, J. Am. Chem. Soc., 2006, 128, 8777–8789 CrossRef CAS.
- B. Nichols, H. Chadwick, S. D. S. Gordon, C. J. Eyles, B. Hornung, M. Brouard, M. H. Alexander, F. J. Aoiz, A. Gijsbertsen and S. Stolte, Chem. Sci., 2015, 6, 2202–2210 RSC.
- J. Zou, S. D. S. Gordon, S. Tanteri and A. Osterwalder, J. Chem. Phys., 2018, 148, 164310 CrossRef.
- S. D. S. Gordon, J. J. Omiste, J. Zou, S. Tanteri, P. Brumer and A. Osterwalder, Nat. Chem., 2018, 10, 1190–1195 CrossRef CAS.
- E. M. Jones and P. R. Brooks, J. Chem. Phys., 1970, 53, 55–58 CrossRef CAS.
- M. C. van Beek, G. Berden, H. L. Bethlem and J. J. ter Meulen, Phys. Rev. Lett., 2001, 86, 4001–4004 CrossRef CAS.
- A. Boca and B. Friedrich, J. Chem. Phys., 2000, 112, 3609–3619 CrossRef CAS.
- M. de Lange, M. Drabbels, P. Griffiths, J. Bulthuis, S. Stolte and J. Snijders, Chem. Phys. Lett., 1999, 313, 491–498 CrossRef CAS.
- M. Brouard, H. Chadwick, S. D. S. Gordon, B. Hornung, B. Nichols, F. J. Aoiz and S. Stolte, J. Chem. Phys., 2016, 144, 224301 CrossRef CAS.
- C. G. Heid, V. Walpole, M. Brouard, F. J. Aoiz and P. G. Jambrina, Nat. Chem., 2019, 11, 662–668 CrossRef CAS.
- V. Walpole, C. G. Heid, P. G. Jambrina, F. J. Aoiz and M. Brouard, J. Phys. Chem. A, 2019, 123, 8787–8806 CrossRef CAS.
- M. H. Alexander, J. Chem. Phys., 1999, 111, 7426–7434 CrossRef CAS.
- M. H. Alexander, J. Chem. Phys., 1999, 111, 7435–7439 CrossRef CAS.
- J. Kłos, G. Chałasiński, M. T. Berry, R. Bukowski and S. M. Cybulski, J. Chem. Phys., 2000, 112, 2195–2203 CrossRef.
- P. Pajón-Suárez, G. Rojas-Lorenzo, J. Rubayo-Soneira and R. Hernández-Lamoneda, Chem. Phys. Lett., 2006, 421, 389–394 CrossRef.
- H. Cybulski and B. Fernández, J. Phys. Chem. A, 2012, 116, 7319–7328 CrossRef CAS.
- Tutorials in Molecular Reaction Dynamics, ed. M. Brouard and C. Vallance, Royal Society of Chemistry, Cambridge, 2012 Search PubMed.
- C. Amiot, J. Mol. Spectrosc., 1982, 94, 150–172 CrossRef CAS.
- K. Schreel, J. Schleipen, A. Eppink and J. J. t. Meulen, J. Chem. Phys., 1993, 99, 8713–8722 CrossRef CAS.
- M. C. van Beek, J. J. ter Meulen and M. H. Alexander, J. Chem. Phys., 2000, 113, 637–646 CrossRef CAS.
- G. Paterson, S. Marinakis, J. Kłos, M. L. Costen and K. G. McKendrick, Phys. Chem. Chem. Phys., 2009, 11, 8804–8812 RSC.
- L. Scharfenberg, K. B. Gubbels, M. Kirste, G. C. Groenenboom, A. van der Avoird, G. Meijer and S. Y. T. van de Meerakker, Eur. Phys. J. D, 2011, 65, 189–198 CrossRef CAS.
- G. Sarma, S. Marinakis, J. J. ter Meulen, D. H. Parker and K. G. McKendrick, Nat. Chem., 2012, 4, 985–989 CrossRef CAS.
- J. J. van Leuken, F. H. W. van Amerom, J. Bulthuis, J. G. Snijders and S. Stolte, J. Phys. Chem., 1995, 99, 15573–15579 CrossRef CAS.
- H. Kohguchi, T. Suzuki and M. H. Alexander, Science, 2001, 294, 832–834 CrossRef CAS.
- A. Gijsbertsen, H. Linnartz, G. Rus, A. E. Wiskerke, S. Stolte, D. W. Chandler and J. Kłos, J. Chem. Phys., 2005, 123, 224305 CrossRef CAS.
- X.-D. Wang, P. A. Robertson, F. J. J. Cascarini, M. S. Quinn, J. W. McManus and A. J. Orr-Ewing, J. Phys. Chem. A, 2019, 123, 7758–7767 CrossRef CAS.
- J. Brown, C. Kerr, F. Wayne, K. Evenson and H. Radford, J. Mol. Spectrosc., 1981, 86, 544–554 CrossRef CAS.
- C. J. Eyles, M. Brouard, H. Chadwick, B. Hornung, B. Nichols, C.-H. Yang, J. Kłos, F. J. Aoiz, A. Gijsbertsen, A. E. Wiskerke and S. Stolte, Phys. Chem. Chem. Phys., 2012, 14, 5403–5419 RSC.
- C. J. Eyles, M. Brouard, H. Chadwick, F. J. Aoiz, J. Kłos, A. Gijsbertsen, X. Zhang and S. Stolte, Phys. Chem. Chem. Phys., 2012, 14, 5420–5439 RSC.
- M. Brouard, H. Chadwick, S. D. S. Gordon, B. Hornung, B. Nichols, J. Kłos, F. J. Aoiz and S. Stolte, J. Chem. Phys., 2014, 141, 164306 CrossRef CAS.
- M. Brouard, S. D. S. Gordon, B. Nichols, V. Walpole, F. J. Aoiz and S. Stolte, Phys. Chem. Chem. Phys., 2019, 21, 14173–14185 RSC.
- M. Brouard, H. Chadwick, S. D. S. Gordon, C. G. Heid, B. Hornung, B. Nichols, J. Kłos, P. G. Jambrina and F. J. Aoiz, Chin. J. Chem. Phys., 2020, 33, 217–233 CrossRef CAS.
- M. H. Alexander and S. Stolte, J. Chem. Phys., 2000, 112, 8017–8026 CrossRef CAS.
- R. Uberna, R. D. Hinchliffe and J. I. Cline, J. Chem. Phys., 1996, 105, 9847–9858 CrossRef CAS.
- A. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- M. H. Alexander, Chem. Phys., 1985, 92, 337–344 CrossRef CAS.
- M. H. Alexander, J. Chem. Phys., 1993, 99, 7725–7738 CrossRef CAS.
- R. N. Zare, Angular Momentum, Wiley-Interscience, New York, 1987 Search PubMed.
- M. H. Alexander, J. Chem. Phys., 1982, 76, 5974–5988 CrossRef CAS.
- P. G. Jambrina, M. Menéndez and F. J. Aoiz, Chem. Sci., 2018, 9, 4837–4850 RSC.
- P. G. Jambrina, M. Menéndez, A. Zanchet, E. García and F. J. Aoiz, Phys. Chem. Chem. Phys., 2019, 21, 14012–14022 RSC.
- HIBRIDON is a package of programs for the time-independent quantum treatment of inelastic collisions and photodissociation written by M. H. Alexander, D. E. Manolopoulos, H. Werner and B. Follmeg, with contributions by P. F. Vohralik, D. Lemoine, G. Corey, R. Gordon, B. Johnson, T. Orlikowski, A. Berning, A. D. Esposti, C. Rist, P. Dagdigian, B. Pouilly, G. van der Sanden, M. Yang, F. de Weerd, S. Gregurick and J. Kłos.
- M. Brouard, S. D. S. Gordon, A. Hackett Boyle, C. G. Heid, B. Nichols, V. Walpole, F. J. Aoiz and S. Stolte, J. Chem. Phys., 2017, 146, 014302 CrossRef CAS.
- X. Zhang and S. Stolte, Chem. Phys., 2018, 514, 4–19 CrossRef CAS.
- M. Brouard, B. Hornung and F. J. Aoiz, Phys. Rev. Lett., 2013, 111, 183202 CrossRef CAS.
- J. Onvlee, S. N. Vogels, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, New J. Phys., 2015, 17, 055019 CrossRef.
- A. von Zastrow, J. Onvlee, D. H. Parker and S. Y. T. van de Meerakker, EPJ Techn. Instrum., 2015, 2, 11 CrossRef.
- S. N. Vogels, Z. Gao and S. Y. T. van de Meerakker, EPJ Techn. Instrum., 2015, 2, 12 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04228e |
| ‡ Current address: Max Planck Institute for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen, Germany. |
| This journal is © the Owner Societies 2020 |