
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electronic structure of the aqueous permanganate ion: aqueous-phase energetics and molecular bonding studied using liquid jet photoelectron spectroscopy†
Karen D.
Mudryk
 ab,
Robert
Seidel
cd,
Bernd
Winter
e and
Iain
Wilkinson
*a
ab,
Robert
Seidel
cd,
Bernd
Winter
e and
Iain
Wilkinson
*a
aLocally-Sensitive & Time-Resolved Spectroscopy, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, D-14109 Berlin, Germany. E-mail: iain.wilkinson@helmholtz-berlin.de
bFachbereich Physik, Freie Universität Berlin, Arnimallee 14, D-14195 Berlin, Germany
cOperando Interfacial Photochemistry, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Strasse 15, D-12489 Berlin, Germany
dFachbereich Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, D-12489 Berlin, Germany
eFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany
First published on 8th September 2020
Abstract
Permanganate aqueous solutions, MnO4−(aq.), were studied using liquid-micro-jet-based soft X-ray non-resonant and resonant photoelectron spectroscopy to determine valence and core-level binding energies. To identify possible differences in the energetics between the aqueous bulk and the solution–gas interface, non-resonant spectra were recorded at two different probing depths. Similar experiments were performed with different counter ions, Na+ and K+, with the two solutions yielding indistinguishable anion electron binding energies. Our resonant photoelectron spectroscopy measurements, performed near the Mn LII,III- and O K-edges, selectively probed valence charge distributions between the Mn metal center, O ligands, and first solvation shell in the aqueous bulk. Associated resonantly-enhanced solute ionisation signals revealed hybridisation of the solute constituents’ atomic orbitals, including the inner valence Mn 3p and O 2s. We identified intermolecular coulombic decay relaxation processes following resonant X-ray excitation of the solute that highlight valence MnO4−(aq.)–H2O(l) electronic couplings. Furthermore, our results allowed us to infer oxidative reorganisation energies of MnO4˙(aq.) and adiabatic valence ionisation energies of MnO4−(aq.), revealing the Gibbs free energy of oxidation and permitting estimation of the vertical electron affinity of MnO4˙(aq.). Finally, the Gibbs free energy of hydration of isolated MnO4− was determined. Our results and analysis allowed a near-complete binding-energy-scaled MnO4−(aq.) molecular orbital and a valence energy level diagram to be produced for the MnO4−(aq.)/MnO4˙(aq.) system. Cumulatively, our mapping of the aqueous-phase electronic structure of MnO4− is expected to contribute to a deeper understanding of the exceptional redox properties of this widely applied aqueous transition-metal complex ion.
Introduction
The aqueous-phase permanganate ion, MnO4−(aq.), is well-known for its properties as a strong oxidizing agent over a broad range of pH, acting as a redox precursor of several manganese species with different oxidation states, namely the MnO42−(aq.), MnO43−(aq.), MnO2(s), Mn(H2O)63+(aq.), and Mn(H2O)62+(aq.) complexes.1 Thus, MnO4−(aq.) is considered a versatile oxidizer suitable for green chemistry,2 oxidation of micro-pollutants,3 detection of organic compounds,4 water disinfection,5–7 and synthesis of energy materials.8 This transition-metal complex has also been applied in energy conversion and storage systems, such as MnO4−(aq.)/MnO42−(aq.) redox flow batteries9 and microbial fuel cells.10The aforementioned applications rely on the same fundamental process, that is, an electron-transfer reaction between a reduced solute or an electrode (acting as the electron donor) and MnO4−(aq.) (acting as the electron acceptor). During this type of reaction, the electronic and nuclear geometric configuration of both the donor and acceptor, as well as the dielectric and microscopic electronic polarization of the surrounding solvent molecules, are generally changed.11 The energetics associated with these changes can be expressed as a reorganization energy which can be broken down into vibrational, λi, and solvational, λ0, components. It has previously been demonstrated that (total) oxidative reorganisation energies, λox, may be directly extracted from photoelectron spectroscopy (PES) measurements,12–14 as reported here. While dielectric properties of the solvent are required to calculate λ0, detailed knowledge of the electronic structure of the reactants in the reaction medium is a prerequisite to calculate λi.12–16 Experimentally, such electronic structure information may be garnered using PES techniques.17–19
Solute- and solvent-induced electronic polarization effects affect the reaction energetics and pathways that facilitate a redox process,11 with charge distributions within the solute and in the solution more generally determining the kinetics and mechanisms involved in the aforementioned applications. In an aqueous solution, generally, the electronic structure of the solute and the solvent are correlated and influenced by each other.20,21 Geometric structural changes may also arise due to solute–solvent interactions.22–24 Furthermore, in transition-metal complexes, the redox potential varies depending on the electronic charge distributions/molecular bonding interactions between the metal centre and its ligands.17,25,26 Hence, aqueous-phase electronic structure studies are expected to provide an abundance of insights into the redox behaviour of MnO4−(aq.) and its role in determining the efficiency of associated electron-transfer processes.
Liquid microjet-based soft X-ray PES (XPS) allows valence- and core-level ionization energies of aqueous solutes and the associated solvent to be determined on an absolute energy scale.17,27 The technique also allows aqueous–gas interfaces or (predominantly) bulk-liquid environments to be probed through tuning of ionising photon energies. Thereby the probing depth of the (photo)electrons in the solution is varied, enabling preferential surface- versus predominantly bulk-sensitive probing.27–29 Further electronic structure insights may be garnered by performing resonantly-enhanced photoelectron spectroscopy (RPES) experiments.17,30,31 Through excitation at core-to-virtual-valence resonances, signals from solutes with relatively low ionization cross-sections or local concentrations can be selectively enhanced and detected.32 Furthermore, the energetics of resonant (valence) photoelectron signals generated following core-to-virtual-valence excitation and solute intramolecular relaxation allow the atomic parentages of specific molecular bonding interactions to be interrogated.17 Additionally, such experiments potentially access non-local relaxation phenomena, such as Intermolecular Coulombic Decay (ICD), and intermolecular electronic interactions between solute and solvent molecules.33 Here we utilize these salient features of aqueous-phase PES to elucidate the electronic structure of the aqueous permanganate ion. We aim to characterize both the solute's complex intra-molecular electronic structure as well as identify signatures of electronic interactions between the permanganate ions and hydrating water molecules.
To our knowledge, all previous PES studies of the permanganate ion have focused on gaseous MnO4−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 and solid KMnO435–38 samples. By extending such studies to the aqueous phase, we thus provide experimental insight into the effect of hydration on the electronic structure and redox properties of the widely applied aqueous permanganate ion. Specifically, we report soft X-ray liquid microjet non-resonant and resonant PES experiments from MnO4− aqueous solutions. We present absolute binding energies (BEs) of the valence orbitals and of the O 1s and Mn 2p3/2,1/2 core levels (with respect to the vacuum level). We also explore the effect of interfacial versus liquid-bulk hydration and counter-ion (Na+versus K+) on the measured valence BEs at relatively low permanganate ion concentrations (∼0.2 M). In addition, we use Mn LII,III- and O K-pre-edge RPES measurements to confirm and expand on direct PES determinations of the MnO4− BEs and elucidate ionisation energies that are otherwise difficult to extract. The RPES results are also used to identify the atomic parentages and confirm hybridizations of a number of the valence molecular orbitals.39–41 Furthermore, intermolecular solute and solvent electronic coupling phenomena are identified through detection of resonant ICD processes following soft X-ray photoexcitation. Our cumulative results are used to construct an aqueous-phase molecular orbital diagram for the MnO4−(aq.) anion, as well as an electronic state energy level diagram for the MnO4−(aq.) anion/MnO4˙(aq.) radical system. We additionally infer (valence ionisation) reorganisation energies of oxidation, λox, and adiabatic ionisation energies (AIEs, equivalent to the Gibbs free energy of oxidation in the case of the lowest energy ionising transition) of the MnO4−(aq.) ion. Furthermore, we estimate the vertical electron affinity (VEA) of the MnO4˙(aq.) radical, and the Gibbs free energy of hydration, ΔGhyd, of isolated MnO4−. Combinatorially, our direct PES and RPES results allow us to produce a comprehensive compilation of the electronic energy level structure of the permanganate ion in bulk aqueous solution and at the aqueous-solution–gas interface.
34 and solid KMnO435–38 samples. By extending such studies to the aqueous phase, we thus provide experimental insight into the effect of hydration on the electronic structure and redox properties of the widely applied aqueous permanganate ion. Specifically, we report soft X-ray liquid microjet non-resonant and resonant PES experiments from MnO4− aqueous solutions. We present absolute binding energies (BEs) of the valence orbitals and of the O 1s and Mn 2p3/2,1/2 core levels (with respect to the vacuum level). We also explore the effect of interfacial versus liquid-bulk hydration and counter-ion (Na+versus K+) on the measured valence BEs at relatively low permanganate ion concentrations (∼0.2 M). In addition, we use Mn LII,III- and O K-pre-edge RPES measurements to confirm and expand on direct PES determinations of the MnO4− BEs and elucidate ionisation energies that are otherwise difficult to extract. The RPES results are also used to identify the atomic parentages and confirm hybridizations of a number of the valence molecular orbitals.39–41 Furthermore, intermolecular solute and solvent electronic coupling phenomena are identified through detection of resonant ICD processes following soft X-ray photoexcitation. Our cumulative results are used to construct an aqueous-phase molecular orbital diagram for the MnO4−(aq.) anion, as well as an electronic state energy level diagram for the MnO4−(aq.) anion/MnO4˙(aq.) radical system. We additionally infer (valence ionisation) reorganisation energies of oxidation, λox, and adiabatic ionisation energies (AIEs, equivalent to the Gibbs free energy of oxidation in the case of the lowest energy ionising transition) of the MnO4−(aq.) ion. Furthermore, we estimate the vertical electron affinity (VEA) of the MnO4˙(aq.) radical, and the Gibbs free energy of hydration, ΔGhyd, of isolated MnO4−. Combinatorially, our direct PES and RPES results allow us to produce a comprehensive compilation of the electronic energy level structure of the permanganate ion in bulk aqueous solution and at the aqueous-solution–gas interface.
Experimental
Measurements
Experiments were primarily performed at the U49/2 PGM-1 beamline42 with the SOL3PES end-station43 at the BESSY II synchrotron facility (Helmholtz-Zentrum Berlin für Materialien und Energie). Select experiments were repeated at the P04 beamline44 with the EASI end-station45 at the PETRA III synchrotron facility (Deutsches Elektronen-Synchrotron). MnO4−(aq.) solutions were prepared at room temperature, either by diluting aliquots of a commercial NaMnO4(aq.) solution (Aldrich, 40 wt%) or by dissolving KMnO4 crystals (Roth, ≥99% and Fluka, ≥99%) in millipure water (18.2 MΩ cm resistivity at 25 °C). Following preparation, all sample reservoirs containing MnO4−(aq.) solutions were shielded from ambient light to prevent photochemical reactions.46 After ultrasonic degassing, the samples were introduced into the liquid-jet-synchrotron-beam interaction vacuum chamber of the end-stations as horizontal, free-flowing liquid microjets.30 The solutions were injected at 0.7–1.1 ml min−1 flow rates through 25 μm or 35 μm orifice diameter quartz capillary nozzles using high-performance liquid chromatography (HPLC) pumps. All sample delivery materials were chemically inert towards MnO4−(aq.). The jet was electrically grounded to the interaction chamber by means of a gold wire immersed in the solution close to the injection point. A cooling system connected to the liquid jet nozzle holder allowed the temperature of the solution to be regulated prior to injection. Generally, both the temperature and sample flow rate were optimized to maximize jet stability during measurements. However, specific sample temperatures (in the 280 ± 3 K range) were adopted in situations where PES peak spectral widths are compared or used to derive additional parameters. The pressures in the interaction chambers were maintained between 5 × 10−4 and 1 × 10−3 mbar using a combination of turbo molecular pumping (∼2000 or ∼2700 L s−1 pumping speed for water vapour in the SOL3PES and EASI instruments, respectively) and two (SOL3PES) or three (EASI) liquid nitrogen-filled cold traps (∼4.5 × 104 L s−1 pumping speed for water vapour per trap in both instruments).43,45The U49/2 PGM-1 beamline provided linearly, horizontally-polarized light and a focal spot size of approximately 100 × 80 μm2 (horizontal × vertical).42 The P04 beamline provided circularly-polarized light and a reduced vertical focal spot size of approximately 180 × 33 μm2,44 resulting in significantly diminished gas-phase contributions to the recorded spectra. The beamlines were protected from the liquid samples introduced into the end-station interaction chambers using windowless differential pumping chamber assemblies.43,45 The photon flux at U49/2 PGM-142 (measured after the last focusing mirror in the beamline) is of the order of 1013 ph s−1 in the 85–500 eV photon energy range or 1012 ph s−1 in the 500–1500 eV photon energy range, with a slightly higher photon flux available in the latter energy range at the sample position at P04.44 All spectra were recorded at maximum photon flux, with corresponding peak intensities of ∼103 W cm−2 under our experimental conditions. The associated absorbed X-ray doses (D) on the samples were of the order of 10−3 Gy. As expected, spectra recorded at 635 eV photon energy and full and reduced (50% and 10%) flux showed no indication of sample radiation damage.
The photoelectrons emitted from the liquid jet and the surrounding gas-phase molecules were detected using differentially-pumped hemispherical electron analysers (Scienta Omicron R4000 HiPP-2 in the SOL3PES setup,43 or HiPP-3 in the EASI instrument45). The electron collection axis of the analyser was set orthogonal or at magic angle to the light propagation and light polarization axes in all experiments. The liquid jet propagated orthogonally to both the light propagation and analyser electron collection axes. The analyser parameters were set to maximize electron collection by adopting an analyser entrance slit aperture larger than the limiting aperture located at the exit of the analyser pre-lens and the entrance of the hemisphere.43,47 The liquid jet position was optimized by maximizing the count rate of the lowest energy ionising transition (1b1−1 peak) of liquid water (in experiments performed at photon energies lower than 540 eV) or of the liquid water Auger electron signal (in experiments performed at photon energies higher than 540 eV), thereby minimizing the gas-phase contributions to the spectra. The energy resolution of the U49/2 PGM-1 beamline and SOL3PES end-station experimental setup was determined by recording gas-phase Ar XPS spectra under identical beamline and analyser conditions to those adopted in the liquid jet experiments. Voigt profile fits to Ar 3p or 2p XPS spectra allowed us to determine instrumental resolutions of ∼53 meV, ∼310 meV, and ∼400 meV at 150 eV, 530 eV, and 638 eV, respectively. In the P04 beamline and EASI endstation experiments, total energy resolutions of ∼410 meV, ∼460 meV, and ∼480 meV were estimated at 635 eV, 1135 eV, and 1251 eV photon energies, respectively.
Non-resonant, surface-sensitive valence photoelectron spectra were recorded at photon energies of 150 eV. This corresponds to a ∼130 eV photoelectron kinetic energy (KE) and probing of all signal components at similar depths of ∼1 nm.28,29,48 Surface-and-bulk-sensitive valence photoelectron measurements were performed at a 635 eV photon energy resulting in ∼600 eV KEs. Corresponding electron effective attenuation lengths (EALs) are estimated to be ∼4 nm, with the associated signals predominantly attributed to the aqueous bulk.28,29,48 We approximately reproduced such electron kinetic energies and EALs in additional non-resonant surface- and bulk-sensitive core-level photoelectron spectra by appropriately tuning the ionizing photon energies. Predominantly bulk solution probing depths of ∼4 nm were assured in our valence RPES experiments by the high photon energies, and therefore electron emission energies, associated with the core-to-virtual valence energy gaps (see below).
In order to compare solute and solvent signals, 0.05 M NaCl(aq.) solutions (Aldrich, ≥99% NaCl crystals dissolved in millipure water) were used to produce (nearly pure) water reference samples with the required conductivity to mitigate liquid jet streaming potentials17,49 and X-ray-pulse-induced charging effects.50
Mn LII,III-pre-edge (Mn 2p to virtual-valence excitation) RPES measurements were performed by scanning the photon energy between 641.8–661.9 eV (with 0.15 eV photon energy steps) and collecting electrons associated with the outer-valence-band BE range, 5–42 eV. Similarly, O K-pre-edge (O 1s to virtual-valence excitation) RPES measurements were performed by scanning over the 526.4–537.4 eV photon energy range (with 0.1 eV photon energy steps) and collecting electrons within the 6–40 eV outer-valence BE range. We integrated the spectral intensities over the different BE ranges and plotted the results as a function of excitation photon energy to produce partial electron yield (PEY) proxies of X-ray absorption spectroscopy (XAS) spectra. From here onwards we refer to these data as PEY-XAS spectra. Additional photoelectron spectra were recorded over extended BE ranges at select off-resonant and resonant photon energies to isolate resonant-signal-enhancements with high signal-to-noise ratios and span the complete MnO4−(aq.) (inner- and outer) valence region.
Data treatment
Shirley baselines51 were fit and subtracted from each photoelectron spectrum. Multiple baselines were generally sequentially subtracted, with each baseline covering a specific BE range. This was particularly so in the spectra extending over the entire valence band, where three baseline fits were implemented to include the outer (∼5–20 eV and ∼20–40 eV) and inner (∼40–90 eV) valence regions. Although an imperfect solution for aqueous-phase spectra, the Shirley background may be judiciously applied over relatively narrow energy ranges. Generally, consistent results were obtained when applying alternative background treatment procedures, for example subtracting cubic baselines from each of the above mentioned spectral ranges. Non-resonant and resonant signals were fit using sets of Voigt functions. For valence signals for which the Lorentzian component of the Voigt fits and life-time broadening effects were confirmed to be negligible, more robust Gaussian fits were adopted and the latter fit results are reported instead. We adopt the common procedure of calibrating the valence photoelectron BE scales using the peak centres extracted from fits to the liquid water lowest energy ionisation peak, 1b1(l)−1, with an assigned BE of 11.31 ± 0.04 eV.‡49 For the core-level BE data, we utilise the similarly commonly adopted procedure of calibrating the photoelectron BE scale using the peak centres extracted from fits to liquid water 1a1−1 (O 1s−1) ionisation peaks with an assigned BE of 538.1 ± 0.1 eV.52 In the case of the 1t2 (Mn 2p) spectra and solute feature BE calibrations, 1a1 (O 1s) spectra were recorded under identical conditions to enable energetic referencing. For the RPES data, the centre of the water 1b1(l) peak in the lower photon energy off-resonant spectra was used to calibrate the BE scale of the related resonant spectra and RPES maps. Furthermore, associated resonant and off-resonant spectra were BE smoothed (5-point adjacent-averaging) with the resulting off-resonant signals being subtracted from the resonant data in order to isolate resonant enhancements. (Given that the resonant and off-resonant spectra were recorded using a 100 meV BE step size, a 5-point adjacent-average smoothing resulted in a 500 meV effective smoothing/averaging range. Due to the generally ≥1 eV spectral widths of aqueous-phase photoelectron spectroscopy features, such smoothing was found to have a negligible effect on the extracted BEs but notably enhanced our ability to highlight multi-component solute features.) We note that, for resonant spectra recorded near the Mn LII,III- pre-edges – and the associated off-resonant data – care was taken to ensure that the spectral region close to ∼20 eV BE remained positive after the background correction procedure. This resulted in more consistent results overall, particularly between data treatments adopting different baseline subtraction methods. MnO4−(aq.) BEs and peak widths at FWHM were determined from average values extracted from fits to different data sets, with the associated errors corresponding to standard deviations. Fitting errors and the uncertainty in the water 1b1(l) BE value used to calibrate the BE scale were considered individually for each data set to determine the errors. A cubic baseline was fit and subtracted from the Mn L-pre-edge PEY-XAS data in order to precisely extract associated resonant photon energies. In both the O K- and Mn L-pre-edge PEY-XAS data sets, the peaks associated with different resonances were fit using a set of Gaussian functions. In the Mn LII,III-pre-edge experiments, the photon energy was calibrated using Voigt profile fits to O 1s XPS spectra recorded with the beamline fundamental and weak second harmonic. The associated electron KE differences precisely defined the implemented photon energies. The O 1s – 4a1 resonance in liquid water at 535.0 eV53 was used to calibrate the photon energy in the O 1s RPES measurements.
Results and discussion
Molecular structure of MnO4−(aq.)
To assign the PES features described throughout this Section, we first consider the nuclear geometric symmetry of the MnO4− anion in aqueous solution. Existing theoretical calculations highlight the retention of an average tetrahedral anion symmetry in moving from the gas-54 to the (bulk) aqueous-phase.§ Accordingly, a Td point group is utilised along with existing (isolated-molecule) molecular orbital diagrams54–56 to assign and label the MnO4−(aq.) PES signals. We assume here (and validate below) that the same symmetry assignment holds (on average) for aqueous-phase MnO4− both in the aqueous bulk and at the solution–gas interface. We note that the average Mn–O bond lengths (162 pm and 163 pm, respectively)57 and the breathing vibrational mode frequencies (ν1 ∼ 104 meV) and anharmonicity constants of permanganate58 were found to be near identical in KMnO4 aqueous solutions and in the crystalline solid. Collectively, these results imply that the permanganate ion retains a similar, tetrahedral geometry in vacuum, as a component of a solid, and – most importantly in this case – in bulk aqueous solution.Based on the above and our PES-based determination of the aqueous MnO4−(aq.) ionisation energetics described below, a valence and core-level molecular orbital diagram is presented for the aqueous sodium permanganate system in Fig. 1. The associated orbital energies are scaled in accordance with the average BEs extracted from our cumulative data and reported in the following sections. Hence, Fig. 1 provides a simplified schematic of the overall system energetics. We note, however, that such a single-configurational depiction of the permanganate anion electronic structure is of qualitative use only.54
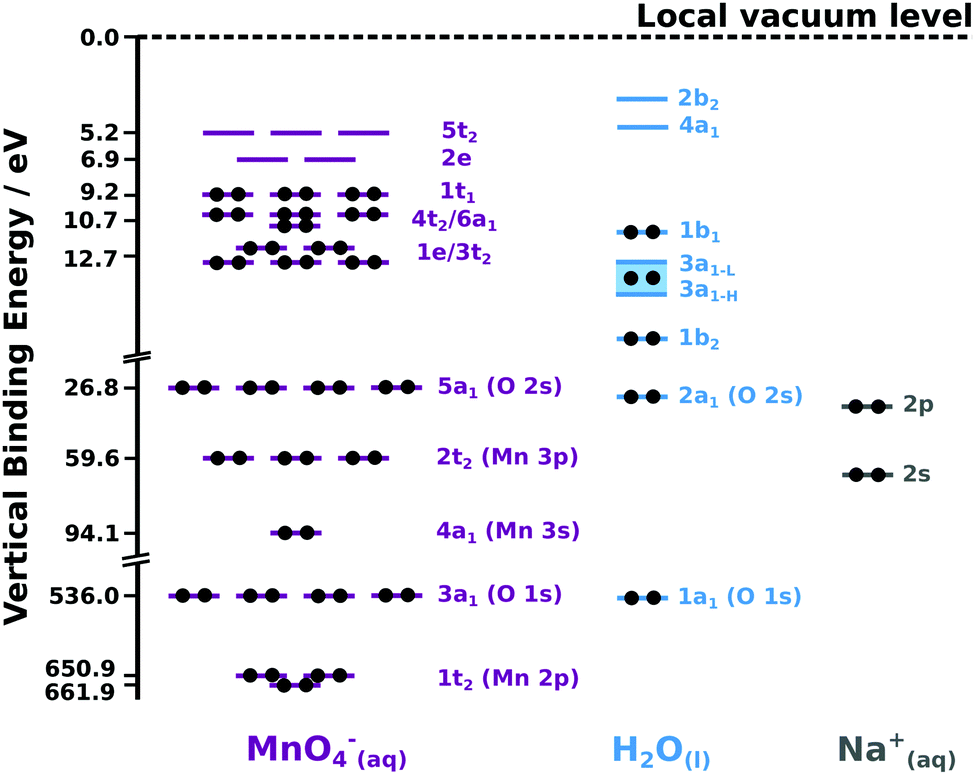 | ||
| Fig. 1 Experimentally determined, binding-energy-scaled molecular orbital diagram of the aqueous NaMnO4 solution depicting the predominant electronic ground state configurations of the associated solution components. orbital occupations are denoted by black dots. (left) MnO4−(aq) orbitals represented by purple bars. the 1t2 (Mn 2p) and 3a1 (O 1s) core; 4a1 (Mn 3s), 2t2 (Mn 3p), and 5a1 (O 2s) inner valence; 3t2, 1e, 4t2, 6a1, and 1t1 outer valence; and 2e and 5t2 unoccupied or virtual valence orbital BEs are exclusively labelled on the ordinate. The MnO4−(aq) diagram component is adapted from the isolated-molecule diagrams reported in ref. 40 and 54. (right) associated molecular orbital diagrams for the aqueous solvent (light blue bars and fills) and Na+(aq) counter ion (grey bars). These diagrams are based on fits to the data reported here and in ref. 49 and 60. | ||
Permanganate anion molecular orbitals are shown in purple in Fig. 1, with the corresponding aqueous solvent and sodium cation orbitals shown in light blue and grey, respectively. (A splitting of the H2O(l) 3a1 molecular orbital due to intermolecular anti-bonding and bonding contributions, respectively 3a1-L and 3a1-H,59 is indicated by a light-blue fill). The positions of the MnO4−(aq.) virtual valence orbitals (2e and 5t2) were determined through fits to Na/KMnO4(aq.) UV-visible absorption spectra and are consistent with our RPES and PEY-XAS results (see ESI,† Fig. S1A). We depict the predominant permanganate electronic ground state orbital occupancies40 using black circles in Fig. 1. The ionisation features discussed in the following sections are labelled in accordance with the Fig. 1 molecular orbital labels.
The predominant electronic ground state occupancies of the chemically active outer-valence and two lowest energy virtual-valence molecular orbitals of MnO4− are qualitatively described by a (3t2)6(1e)4(6a1)2(4t2)6(1t1)6(2e)0(5t2)0 configuration.40,56 These molecular orbitals are linear combinations of the frontier Mn 3d, 4s, and 4p and O 2p atomic orbitals.40 The 3t2 and 1e features are primarily attributed to Mn 3d and O 2p metal–ligand bonding interactions. The 6a1 feature is principally ascribed to Mn 4s and 3p and O 2pσ bonding interactions. The 4t2 and 1t1 orbitals are essentially O-localized, Mn–O non-bonding orbitals, with respective degrees of O–O ligand bonding and anti-bonding character. The virtual valence 2e and 5t2 orbitals are the Mn–O anti-bonding analogues of the 3t2 and 1e bonding features. The experimentally-determined valence BEs of the aqueous solvent (light blue lines) and Na+ counter ion (light grey lines) orbitals are also shown in Fig. 1, to facilitate direct comparison to the anion energetics.
The experimentally-determined BEs of the majority of the solute and aqueous solvent core-levels are additionally shown in Fig. 1. The O-centred permanganate anion 3a1 solute and water 1a1 solvent orbitals have similar BEs, both corresponding to removal of O 1s core/inner shell electrons. The Mn-centred and spin–orbit-split 1t2 orbitals of the anion are also shown, corresponding to removal of Mn 2p core/inner-shell electrons.
Direct (non-resonant) photoelectron spectra
This section presents a subset of the experimental spectra used to nearly fully map the electronic structure of MnO4−(aq.) solutions and to produce – and then move beyond – the simplified schematic shown in Fig. 1. This includes identification of electronic structure fingerprints arising from the solute as well as those arising from electronic interactions between the aqueous anionic solute and solvent molecules.Fig. 2 shows 0.2 M NaMnO4(aq.) solution XPS spectra (purple curves) measured at 635 eV (panels A and B), 1135 eV (panel C), and 1251 eV (panel D) photon energies; resulting in aqueous bulk spectral sensitivities. Reference spectra from 0.05 M NaCl(aq.) solutions, measured under similar conditions, are also shown to highlight the aqueous solvent signals (light blue curves). Multi-component Voigt and/or Gaussian profile fits to the data were performed to model the cumulative spectral profiles (see the ESI,† Fig. S2). Furthermore, as an independent and additional analysis, the baseline subtracted and intensity normalised solvent data was subtracted from the corresponding NaMnO4(aq.) solution data to produce difference spectra (red curves). For the spectra shown in Panel A, 3-point adjacent-averaging was applied to the solution and solvent spectra before subtraction. (Here, the solution and solvent spectra are presented with a 150 meV BE step size. Hence, a 3-point adjacent-average smoothing resulted in a 450 meV effective smoothing/averaging range, similar to the RPES difference spectra.) The results of Gaussian or Voigt profile fits to the difference spectra peaks are additionally shown in the various panels of Fig. 2 (purple and grey fits highlight anion and cation peaks, respectively). The relatively weak inner and outer valence features in panels A and B were found – with the exception of peak 6 – to be essentially unaffected by life-time broadening effects, with the Voigt and Gaussian profile fits yielding near-identical Gaussian peak widths. There, Gaussian fits were found to be more robust than their Voigt profile equivalents and were accordingly adopted. Cumulative, multi-peak Voigt fits to the BE-calibrated, baseline-corrected data and Gaussian and Voigt fit combinations to the solution–solvent difference spectra were found to yield similar BE results for all peaks. All MnO4−(aq.) solute features have been assigned, with the remaining signals in Fig. 2 associated with the water solvent and Na+(aq.) solute counter-ion contributions. The latter peaks have been assigned in accordance with ref. 60 and 61, respectively.
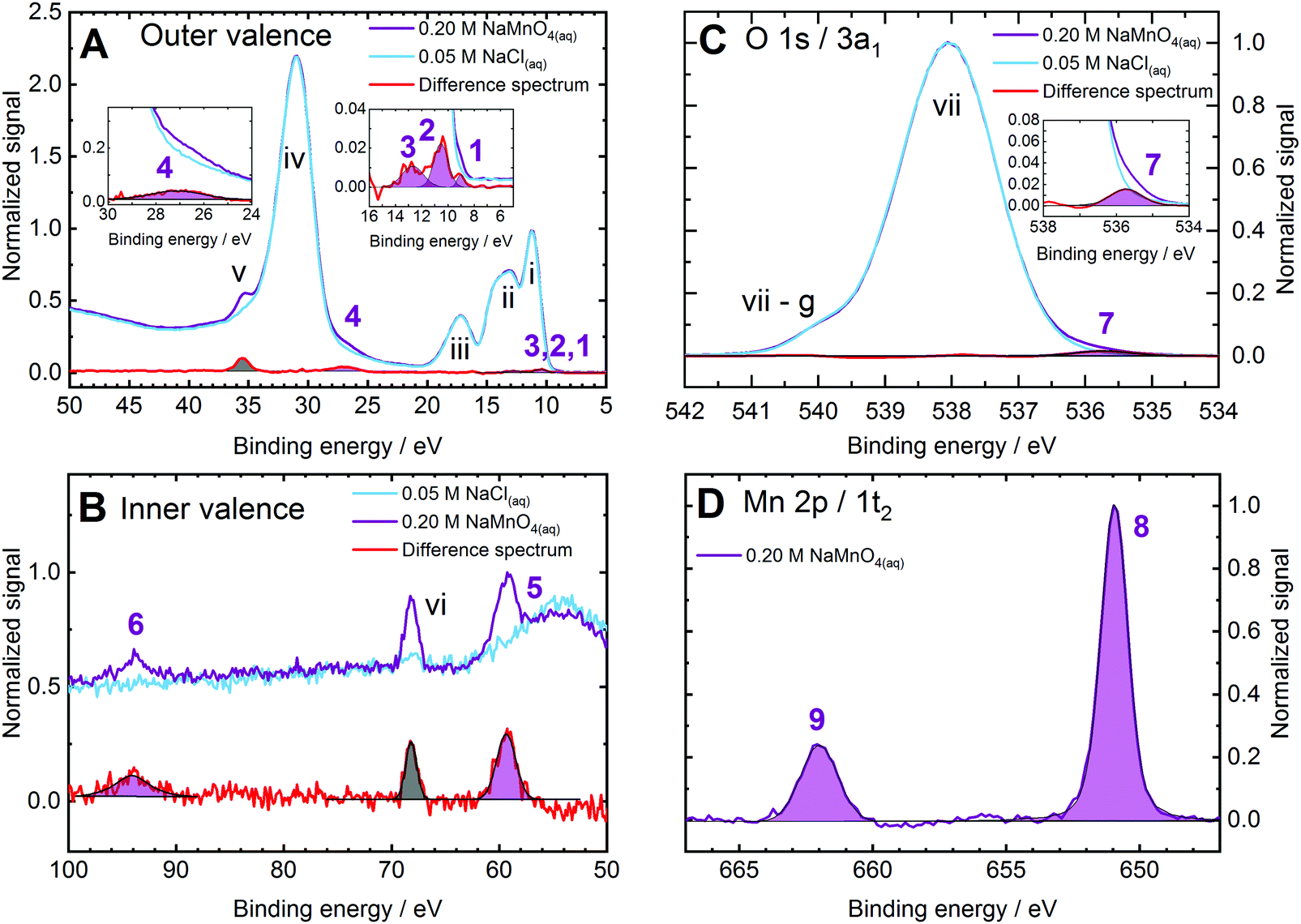 | ||
| Fig. 2 Bulk-sensitive X-ray photoelectron spectra of 0.2 M aqueous NaMnO4 (purple curves) and 0.05 M sodium chloride (blue curves, representative of the aqueous solvent) solutions recorded at the P04 beamline, PETRA III (panels a, c, and d) and at the U49/2 PGM-1 beamline, BESSY II (panel b). Solution–solvent difference spectra (red curves) are plotted in panels (a)–(c) to highlight solute features. Gaussian (panel a and peaks 5 and vi in panel b) or Voigt (peak 6 in panel b and all features in panels c and d) profile fits to the solute anion and cation signals are illustrated by purple and grey fills, respectively. (a) Valence band and solution–solvent difference spectra recorded at a 635 eV photon energy. The right inlay shows the MnO4−(aq.) lowest energy ionizing transition features (peaks 1 (1t1−1), 2 (4t2−1/6a1−1), and 3 (1e−1/3t2−1)) while the left inlay shows an oxygen-centred inner valence ionization feature (peak 4 (5a1−1)). (b) As for panel a with a focus on a higher binding energy range and highlighting additional MnO4−(aq.) inner valence features (peak 5 (2t2−1) and 6 (4a1−1)). (c) Oxygen 1s core ionization and difference spectra recorded at a photon energy of 1135 ev, with the MnO4−(aq.) oxygen-ligand-associated feature (peak 7 (3a1−1)) highlighted in the panel inlay. (d) Manganese 2p, MnO4−(aq.) core ionization spectrum recorded at a photon energy of 1251 eV and highlighting the spin-orbit split Mn(vii) solute metal centre features (peaks 8 and 9 (1t2−1)). the MnO4−(aq.) BEs extracted from this figure and similar data sets are summarized in Table 1. The solvent and counter-ion Na+ ionization features are labelled with lower-case roman numerals and the associated binding energies are summarized in Table 2. | ||
In the valence spectral regions shown in Fig. 2A and B, four distinct MnO4−(aq.) signals are readily extracted at 9.22 ± 0.08 eV (peak 1, panel A, right inset), 26.82 ± 0.04 eV (peak 4, panel A, left inset), 59.6 ± 0.1 eV (peak 5, panel B), and 94.1 ± 0.1 eV (peak 6, panel B) BEs. Within the adopted molecular orbital framework, peak 1 is attributed to a (1t1)−1 ionization process and corresponds to the removal of an electron from the highest occupied molecular orbital (HOMO) and first vertical ionization energy (VIE) of MnO4−(aq.). The MnO4−(aq.) peak 4 signal is assigned to a (5a1)−1 process, with peak 5 assigned as (2t2)−1 ionization, and peak 6 assigned as a (4a1)−1 process. The latter three features are thought to be predominantly associated with ionisation of the ligand O 2s, Mn 3p, and Mn 3s atomic orbitals, respectively.41,62 However, as will be shown in the following Resonant photoemission spectra section, these inner-valence orbital features are found to be hybridized.
Upon performing cumulative Gaussian fits to the solution–solvent difference spectra shown in the right inset of Fig. 2, panel A, it was possible to extract two more MnO4−(aq.) valence BEs: 10.67 ± 0.04 eV and 12.84 ± 0.09 eV (peaks 2 and 3). Based on Fig. 1 and associated ref. 40 and 54 these features are tentatively assigned to overlapping (4t2)−1/(6a1)−1 and (1e)−1/(3t2)−1 ionizing transitions, respectively. The assignments of these peaks to multiple final states is supported by their relatively large feature widths (see Table 1 for details); as well as the identification of narrower but spectrally overlapping resonantly enhanced ionisation features that will be discussed in the following Resonant photoemission spectra section.
| Label | Peak origin | BE/eV | FWHMG/eV |
|---|---|---|---|
| MnO4−(aq.) – XPS | |||
| 1 | (1t1)−1 | 9.22 ± 0.08 | 0.8 ± 0.1 |
| 2 | (4t2/6a1)−1 | 10.67 ± 0.04 | 1.2 ± 0.1 |
| 3 | (1e/3t2)−1 | 12.84 ± 0.09 | 1.9 ± 0.2 |
| 4 | (5a1)−1 | 26.82 ± 0.04 | 2.1 ± 0.6 |
| 5 | (2t2)−1 | 59.6 ± 0.1 | 2.3 ± 0.2 |
| 6 | (4a1)−1 | 94.1 ± 0.1 | 2 ± 4 (2.70 ± 0.07)* |
| 7 | (3a1)−1 | 536.0 ± 0.1 | 0.9 ± 0.3 |
| 8 | (1t2)−1 | 650.9 ± 0.1 | 0.93 ± 0.06 |
| 9 | (1t2)−1 | 661.9 ± 0.1 | 1.0 ± 0.9 |
| MnO4−(aq.) – RPES participator Auger | |||
| 2-a | (4t2)−1 | 9.85 ± 0.08 | 0.7 ± 0.2 |
| 3-a | (1e or 3t2)−1 | 12.66 ± 0.04 | 0.83 ± 0.04 |
| 5 | (2t2)−1 | 59.8 ± 0.4 | 2.6 ± 0.6 |
While peak 2 is reproducibly extracted from bulk- (and surface-sensitive) difference spectra data, the higher BE peak 3 feature directly underlies the gas-phase water 1b1 (HOMO) ionization vibronic peaks (that are generally and variably perturbed by liquid sample electrokinetic/ionization-induced charging). Accordingly, the ∼12.8 eV BE feature (peak 3) could only be isolated when tight vertical X-ray focusing conditions were implemented (as afforded at the P04 beamline) that minimized gas-phase spectral contributions. When larger vertical X-ray focal spot sizes and/or lower ionizing photon energies were implemented, this peak was superimposed on large gas-phase signals and could not be extracted (see ESI,† Fig. S3, for example). Hence, we additionally rely on our RPES experiments, reported in the subsequent section, to confirm this BE.
MnO4−(aq.) solution O 1s (3a1) and Mn 2p (1t2), as well as aqueous solvent reference O 1s (1a1), core-level XPS spectra were additionally recorded. Exemplary data, a solution–solvent O 1s difference spectrum, and associated Voigt fits to the data are shown in Fig. 2 panels C and D. Here the solute oxygen-ligand feature – peak 7 at 536.0 ± 0.1 eV, (3a1)−1 – is uniquely and reproducibly extracted and found to be shifted to lower BEs with respect to the O 1s, (1a1)−1, feature of the aqueous solvent by 2.1 eV. (Note that peak vii-g is associated with the gas-phase water O 1s contribution which is differentially shifted between the sample solution and aqueous reference spectra due to changes in electrokinetic and ionization-induced charging effects.) The solute-only Mn 2p3/2 and Mn 2p1/2 features – peaks 8 and 9 at 650.9 ± 0.1 eV and 661.9 ± 0.1 eV, respectively, (1t2)−1 – are found to be shifted to higher BEs by ∼12 eV with respect to metallic Mn.37,63,64 The large positive Mn 2p chemical shifts reflect the formal MnVII+ oxidation state of the MnO4−(aq.) metal centre and its covalent bonding to the O ligands. While the MnO4−(aq.) and H2O(l) oxygen atoms share the same formal OII− oxidation state, the respective negative chemical shift of the solute peak indicates a higher MnO4−(aq.) O-ligand charge density. This charge excess may be attributed to intramolecular (ligand–ligand) and potential intermolecular (ligand–water) O–O covalent bonding interactions.
BEs and peak FWHMs of all MnO4−(aq.) associated signals were extracted from fits to multiple bulk-sensitive data sets, similar to that shown in Fig. 2. The averaged results and their standard deviations, are summarised in the top part of Table 1. Gaussian peak FWHMs are specifically reported for liquid jet temperatures of 280 ± 3 K. The BE values listed on the ordinate of Fig. 1 correspond to the values summarised in Table 1. Corresponding averaged results from the BE peak fits to the water and Na+(aq.) spectral contributions to Fig. 2 and similar data sets are also illustrated in Fig. 1. These solvent and cation peak fit results are additionally summarized in Table 2.
| Label | Peak origin | BE/eV | FWHM/eV |
|---|---|---|---|
| H2O(l) – XPS | |||
| i | (1b1)−1 | 11.31 ± 0.04 | 1.42 ± 0.03 |
| ii-a | (3a1-L)−1 | 13.02 ± 0.05 | 1.6 ± 0.1 |
| ii-b | (3a1-H)−1 | 14.46 ± 0.06 | 1.6 ± 0.1 |
| iii | (1b2)−1 | 17.42 ± 0.04 | 2.32 ± 0.06 |
| iv | (2a1)−1 | 31.04 ± 0.04 | 2.97 ± 0.02 |
| vii | (1a1)−1 | 538.1 ± 0.1 | 1.65 ± 0.03 |
| Na+(aq.) – XPS | |||
| v | 2p | 35.65 ± 0.04 | 1.25 ± 0.08 |
| vi | 2s | 68.46 ± 0.04 | 1.6 ± 0.1 |
Exceptionally, one additional feature is extracted in the surface-sensitive 3a1−1 (O 1s) data that is not discernible in the bulk-sensitive experiments (see ESI,† Fig. S3B and S5B). We note either a slight increase in the width of peak vii in the solution spectra occurs with respect to the solvent reference spectra or that an additional ionisation feature emerges at a BE of 538.3 ± 0.2 eV (unfortunately, these two spectral forms cannot be differentiated due to the overlap of peak vii with its gas-phase counterpart, vii-g, on the high BE side of the 3a1−1/O 1s peak). Notably, the BE of this feature – marked with a † – can only be extracted from the difference spectra, with cumulative Voigt profile fits to the solution data yielding a single solute contribution to the 3a1−1 (O 1s) spectrum. This broadening of the solvent feature or additional solute peak may be attributable to a broader range of hydration environments being sampled at the aqueous–gas interface and/or a reduction of the solute molecular symmetry at the interface. We favour the former explanation due to the indiscernible effects of a potential reduction of molecular symmetry on the other surface-sensitive PES features. However, a firm assignment requires more in-depth experimental analysis (for example concentration-dependent, surface- and bulk-sensitive measurements) which is beyond the scope of the work presented here.
As a further cross-check of our measurements and results, we have additionally probed the effects of the counter-ion on the extracted BEs (see ESI,† Fig. S5). No BE shifts were observed in changing the counter-ion of the solution from Na+ to K+ at fixed 0.2 M concentration (note that such comparisons are only valid at such low concentrations due to the aqueous solubility limit of KMnO465). The consistency of the MnO4− anion BEs with counter-ion was preserved in both aqueous bulk- and interfacially-sensitive experiments, see Fig. S6, S10 and Tables S2, S5 in the ESI† for details. Accordingly, we attribute the solute signals reported here to separated and hydrated MnO4− anions and Na+/K+ cations (in spite of previous reports regarding partial aqueous dissociation of permanganate salts66).
As expected, the gas-phase permanganate anion is significantly less stable with respect to ionisation than the aqueous- and solid-phase species. The (1t1)−1/HOMO−1 VIE of isolated MnO4− was determined to be 4.91 ± 0.03 eV (relative to vacuum) by Gutsev et al.,34 with an associated AIE of 4.80 ± 0.1 eV also being extracted. The ∼4.3 eV disparity between the gas- and aqueous-phase VIEs reflects both the anion stabilisation afforded by the aqueous solvent – i.e. the Gibbs free energy of hydration, ΔG0hyd – and the (oxidative) solvent reorganisation energy – λox. Similar energetic offsets are observed between the gas- and aqueous-phase (4t2)−1/(HOMO−1)−1 VIEs (∼4.5 eV). However, the inference that the second aqueous phase ionisation feature is associated with two unresolved final states – i.e. a (4t2/6a1)−1 process – prevents a direct comparison of the second PES peaks across phases. We return to this point in the subsequent Absolute energetics and thermodynamics of MnO4−(aq.) solutions section.
Resonant photoemission spectra
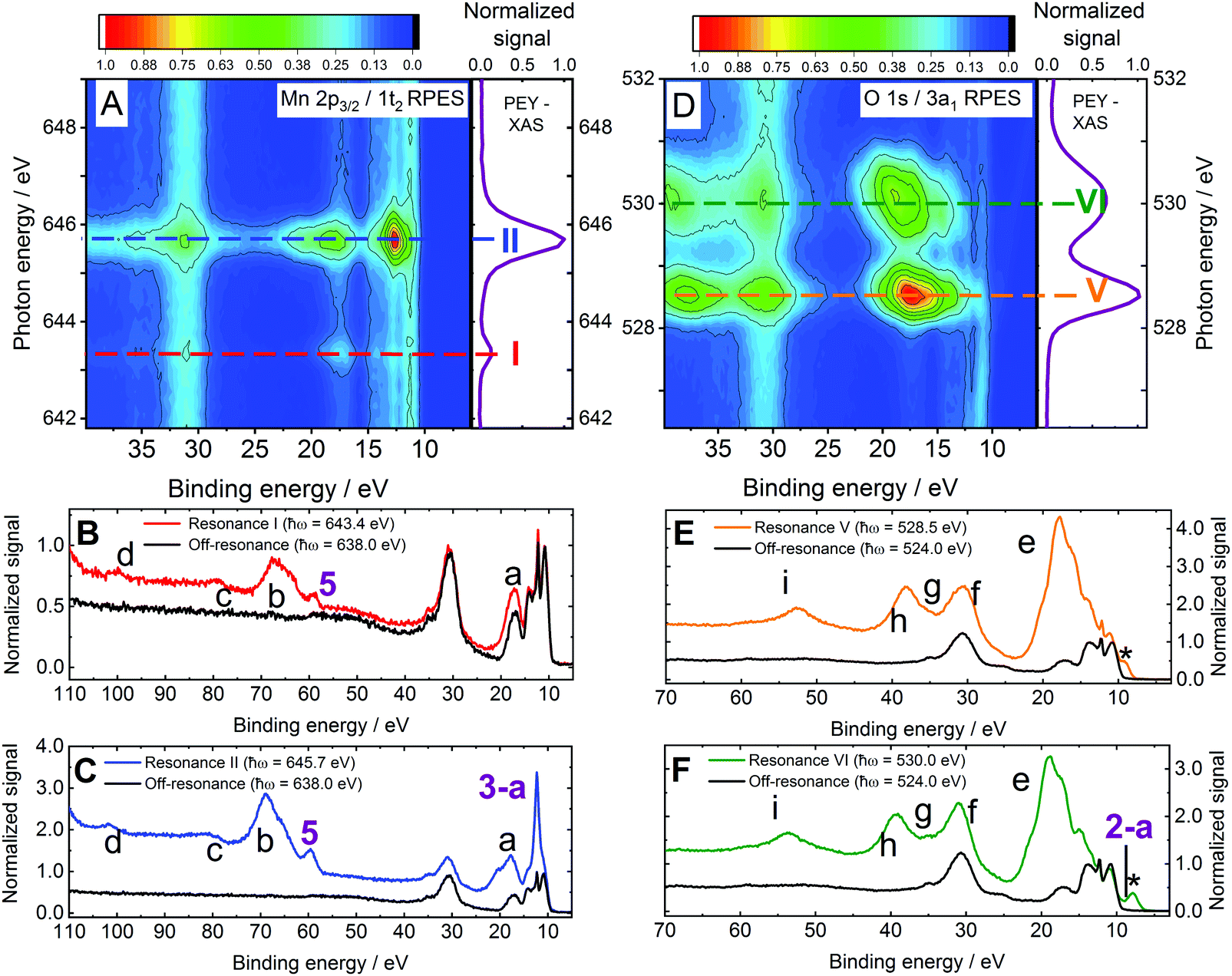 | ||
| Fig. 3 RPES spectra from 0.2 M NaMnO4(aq.) solutions recorded at the U49/2 PGM-1 beamline, BESSY II. (A) 1t2/Mn 2p3/2 to virtual valence RPES map (left) and PEY-XAS spectrum (right). (B) 1t2/Mn 2p3/2 → 2e resonant spectrum recorded at Resonance I. (C) 1t2/Mn 2p3/2 → 5t2 resonant spectrum recorded at Resonance II. (D) 3a1/O 1s to virtual valence RPES map (left) and PEY-XAS spectrum (right). (E) 3a1/O 1s → 2e resonant spectrum recorded at Resonance V. (F) 3a1/O 1s → 5t2 resonant spectrum recorded at Resonance VI. The photon energies associated with Resonances I, II, V, and VI are highlighted by dashed lines in panels a and d. Off-resonant spectra are shown as reference signals (i.e. without resonant-enhancement, black curves) in panels b, c, e, and f. Participator auger signals are labelled with numbers (see the bottom panel of Table 1) while spectator auger and potential ICD signals are labelled with letters (see Table 4). The asterisks highlight an artefact signal associated with ionisation of H2O(l) 1a1/O 1s electrons by the second harmonic of the beam line. Although the experiment strictly detects both the kinetic energies of the direct photoelectrons and Auger electrons, with a binding energy scale generally only having legitimate meaning for the former, we plot the abscissas on a binding energy scale here to enable ready comparison – particularly of participator auger features – with the direct XPS data presented in the previous section. | ||
| Resonance | Transition | Photon energy/eV |
|---|---|---|
| I | 1t2 (Mn 2p3/2) → 2e | 643.4 ± 0.1 |
| II | 1t2 (Mn 2p3/2) → 5t2 | 645.7 ± 0.1 |
| III | 1t2 (Mn 2p1/2) → 2e | 654.2 ± 0.2 |
| IV | 1t2 (Mn 2p1/2) → 5t2 | 656.0 ± 0.2 |
| V | 3a1 (O 1s) → 2e | 528.5 ± 0.1 |
| VI | 3a1 (O 1s) → 5t2 | 530.0 ± 0.1 |
In order to probe Auger contributions from both inner and outer valence orbitals, 0.2 M NaMnO4(aq.) solution PES spectra were recorded with improved signal-to-noise ratios at each of the aforementioned resonant photon energies over an extended BE range. Fig. 3B and C show extended valence-band RPES spectra recorded at the Mn Resonances I (red curve) and II (blue curve), respectively. In each panel, a spectrum recorded at a lower, off-resonant photon energy (638.0 ± 0.1 eV) is additionally plotted as a non-resonant reference (black curves; note the absence of resonant enhancement signals therein). Corresponding spectra associated with Mn Resonances III and IV are shown in the ESI† (see Fig. S7, panels B and C). Fig. 3E and F show analogous full valence-band spectra recorded at O Resonances V (orange curve) and VI (green curve), respectively. The associated off-resonant reference spectrum shown in both panels (black curves) was recorded at a 524.0 ± 0.1 eV photon energy. Resonantly enhanced signals are labelled in panels B, C, E, and F of Fig. 3, with numbers that identify solute participator Auger signals (see Fig. 4, panels B1/C1) and letters that highlight solute spectator Auger signals and potential resonant non-local decay processes (see Fig. 4, panels B2/C2 and B3/C3, respectively). The asterisks in panels E and F highlight artefact signals associated with removal of H2O(l) 1a1 (O 1s) electrons through absorption of the unfiltered second harmonic of the U49/2 PGM-1 beamline. Notably, we similarly recorded RPES data from 0.15 M KMnO4−(aq.) solutions in the vicinity of the Mn LII/III- and O K-edges. Similar results were extracted with the two solutions, in all cases (compare Fig. 3 with ESI,† Fig. S6).
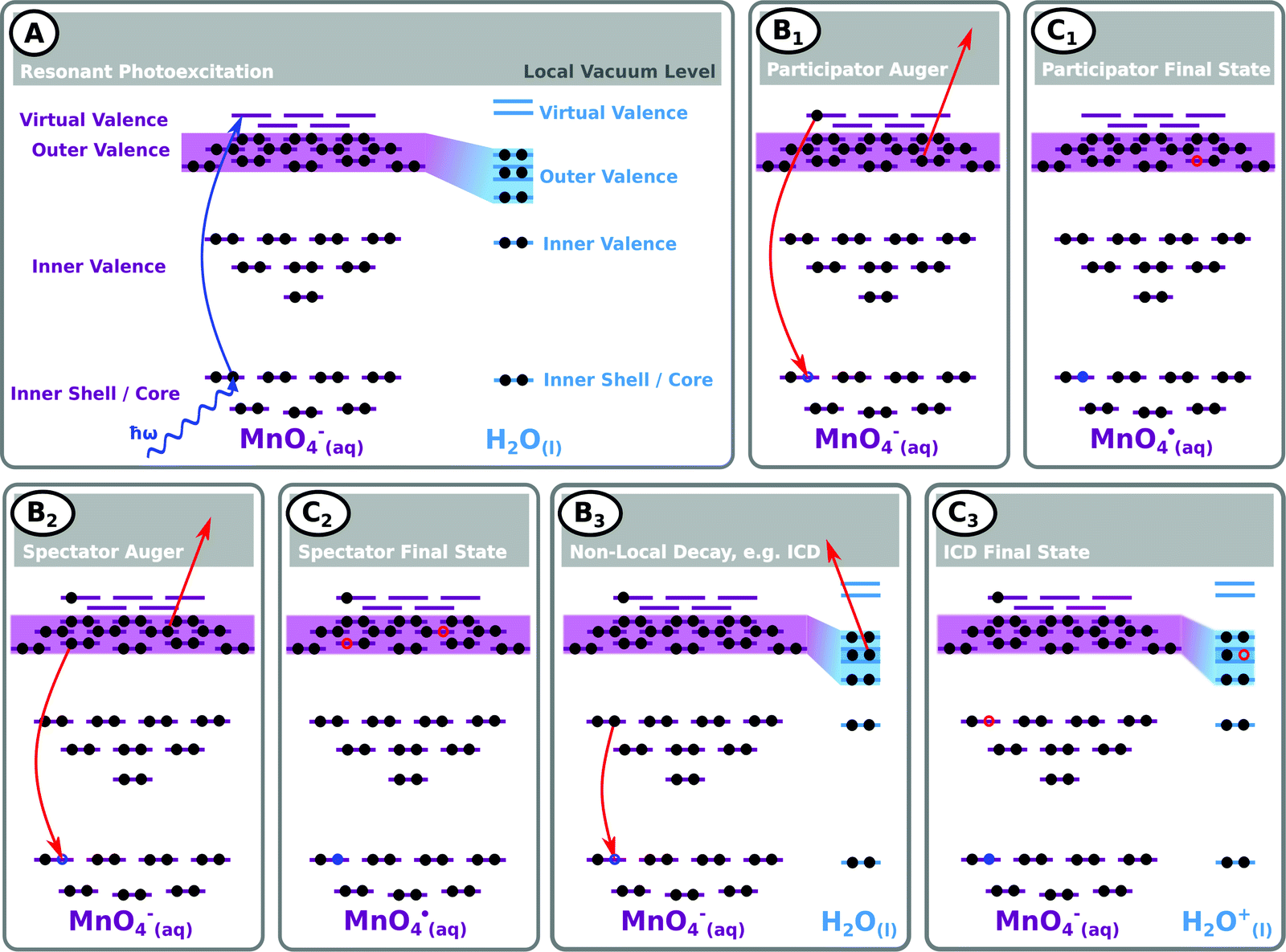 | ||
| Fig. 4 Schematic of exemplary RPES processes occurring in aqueous MnO4− solutions. (A) Depiction of the initial resonant excitation along with an overview of the photo-excited virtual valence, chemically active and delocalised outer valence, (more) localised but environmentally-sensitive inner valence, and inner shell/core orbitals. (B1) Highlights solute participator resonant auger processes that result in (C1) singly-ionised, single-hole aqueous permanganate radical final states and permit the extraction of MnO4− solute ionisation signals and BEs that are otherwise obscured by the dominant solvent background signal. (B2) Highlights a solute spectator resonant auger process that generates (C2) electronically excited states of the aqueous permanganate radical and allows molecular orbital hybridisation and intramolecular delocalisation phenomena to be assessed. (B3) An example of a potential non-local core-hole relaxation process, in this case resonant intermolecular coulombic decay (ICD). (C3) ICD charge-separated, electronically excited MnO4−(aq.) anion and highly unstable H2O+(l) cation final state. See the RPES photoemission spectra section in the main body of the text for further details. | ||
A schematic of possible processes contributing to the resonant enhancement signals associated with Fig. 3 (and the complementary ESI† data) is shown in Fig. 4. Fig. 4A illustrates the first step of the RPES process, the core-to-virtual-valence transition that generates a highly-excited and unstable, core-hole state localised at one of the atomic constituents of the MnO4−(aq.) solute. The short-lived (few- to sub-femtosecond) nature of the initially photo-excited core-hole state may lead to considerable broadening of the RPES signals (with ∼0.2 eV, ∼1.0 eV, and ∼0.4 eV broadenings expected near the O K, Mn LII, and Mn LIII edges, respectively69). In an aqueous solution, the core-hole may be refilled in a number of different ways; for example, by electrons in higher-lying valence solute, and potentially solvent,70 molecular orbitals. Due to the elementally localised and fleeting nature of the core-hole, the refilling is expected to selectively occur via molecular orbitals with similar atomic parentages (Mn for Resonances I–IV and O for Resonances V–VI). However, if appreciable electron delocalisation – for example, intra- or intermolecular charge transfer – occurs within the ultrashort core-hole lifetime, this refilling step may also be coupled to electron ejection from additional elements/molecular orbitals. Equivalently considering such intra- or intermolecular processes in the energy domain, electron ejection from additional elements/molecular orbitals can occur if the photo-excited species are significantly involved in bonding interactions/are hybridised with the solute valence manifold.
Fig. 4B1 illustrates an exemplary solute participator Auger decay process – where the solute core-hole is refilled by the electron that was promoted to the virtual valence orbital and a solute outer valence electron is simultaneously ejected. Notably, the process of refilling the core hole with the aforementioned solute outer valence electron and simultaneous ejection of the initially excited virtual-valence electron yields a similar electron kinetic energy and final state and is indistinguishable. Panel C1 shows the resulting final, singly oxidised solute (MnO4˙) state and highlights that such participator processes produce PES peaks with energy positions/nominal BEs similar to those generated through direct photoemission. Accordingly, such resonant enhancement features are identifiable due to their nominal BEs being near independent of the ionising photon energy. These participator Auger signals may also be significantly enhanced with respect to peaks in non-resonant/direct photoemission experiments due to both the increase in absorption cross-section at the excited resonances and potential constructive interference effects between the direct and participator Auger electron photoemission channels.
Fig. 4B2 shows an exemplary alternative intramolecular decay process, a spectator resonant Auger channel – where the initially excited electron remains in a solute virtual-valence orbital, the core-hole is refilled by an outer-valence electron of the solute, and another solute outer valence electron is simultaneously ejected. Panel C2 shows the resulting electronically excited, singly oxidised (MnO4˙) final state. In this case, the peak position/nominal BE of the emitted Auger electrons will vary with photon energy. However, their KEs will remain approximately constant at different resonances due to their dependence on the approximately constant molecular orbital energy gaps associated with the electrons involved in the de-excitation process. Panel C2 also implies that the energetics of the emitted spectator Auger electrons will be perturbed from the peak positions expected within a single-reference electronic structure framework. This is primarily due to the electron correlation differences between the electronically excited (valence) hole-pair spectator final states compared to the single-hole analogues produced through direct or participator resonant Auger ionisation.
With the solute embedded in an aqueous solvent, a number of non-local relaxation mechanisms may also occur, with the solvent molecules actively participating in the electronic deactivation processes. An example is shown in Fig. 4B3, which shows a resonant ICD process involving a core-hole refill by a solute inner valence electron, with simultaneous ejection of an outer valence electron from a solvent molecule in the solute's first hydration shell. The final state of such an ICD process is shown in panel C3, which highlights production of an electronically excited solute anion (MnO4−), with unchanged molecular charge, along with a highly unstable neighbouring H2O+(l) cation.71 Such processes can be identified by their characteristic autoionization features72,73 which are expected to produce PES features with near photon energy independent electron KEs.
In all of the RPES processes described above, the relaxation mechanisms are driven by electron correlation effects in the initially-localised intermediate photo-excited states. In this regard, selective resonant enhancement of solute ionisation signals, the associated system energetics, and attribution of those signals to processes involving specific molecular orbitals, provides a framework to experimentally determine atomic parentages of RPES-active molecular orbitals (or more generally, electronic states).19,67,74 Conversely, a knowledge of the atomic parentage of specific molecular orbitals (states) can aid in the assignment of RPES features. Furthermore, should dynamic charge transfer processes occur within a core-hole lifetime – for example, from a metal centre to a ligand or from a ligand to a hydrating solvent molecule – signatures of such ultrafast events (or intramolecular or intermolecular hybridisation from an energy-domain perspective) are expected to be reflected in the RPES spectra.68
With the aim of confirming and disentangling overlapping solute BEs, assessing parentages of different solute valence molecular orbitals, and interrogating bonding interactions between the MnO4− solute and solvent molecules, the extended NaMnO4(aq.) solution spectra shown in Fig. 3 were analysed in detail. As a starting point, the baseline-corrected off-resonant spectra were subtracted from the corresponding baseline-corrected resonant spectra (see the Data treatment sub-section for details). This isolated the enhancement signals at Resonances I, II, V, and VI (see Fig. 3). The Resonance III and IV data shown in ESI,† Fig. S7 was similarly treated. Fig. 5 shows the resonant-off-resonant difference spectra results for Mn (2p3/2) Resonances I and II on a KE scale, with the peaks highlighting electronic relaxation processes involving molecular orbitals with significant Mn-character. Similar results are shown for the Mn (2p1/2) Resonances III and IV in Fig. S8 in the ESI.† Corresponding data for the O (1s) Resonances V and VI are shown in Fig. 6A, with the encompassed peaks highlighting resonant enhancement features involving molecular orbitals with significant O-ligand character. Cumulative Voigt or Gaussian profile fits were applied to the data shown in Fig. 5, 6 and Fig. S8 (ESI†) to determine the energetics of the resonant enhancement peaks (see ESI,† Fig. S9). The results of these fits are summarized in Table S4 in the ESI.† Corresponding KMnO4(aq.) solution Resonance I, II, V, and VI results are shown in ESI,† Fig. S10, with associated fit results summarised in Table S5 (ESI†). Based on these extracted RPES KEs/nominal BEs and the calibrated photon energy resonances, all RPES peaks have been assigned. Assignments were made by considering the experimental BEs determined from the direct PES experiments (see Table 1), excitation energy gaps from our MnO4−(aq.) UV-visible (see Fig. S1 and ref. 56) and PEY-XAS spectra (Fig. 3 and Fig. S1, S7, ESI†), and the MnO4− electronic structure calculation literature.40,54,56 Participator Auger peak feature assignments and averaged nominal BEs are reported in the bottom panel of Table 1, with the spectator Auger and ICD feature assignments and associated averaged electron KEs presented in Table 4. The rational for the RPES peak assignments is discussed in the following.
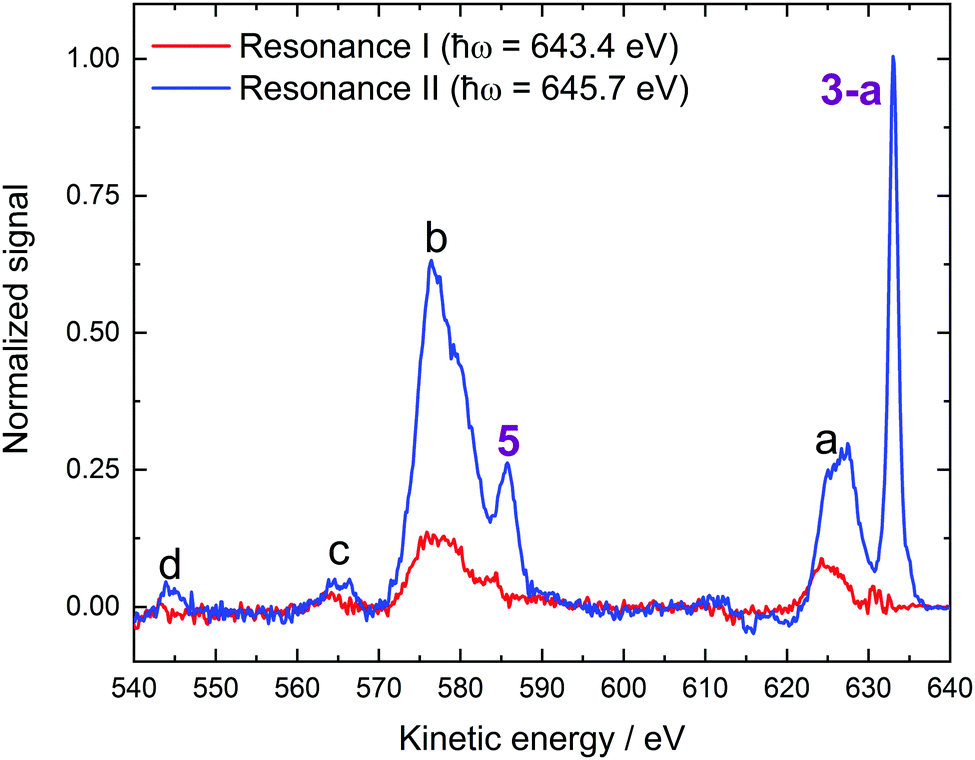 | ||
| Fig. 5 0.2 M NaMnO4(aq.) Resonance I (Mn 2p3/2 → 2e) and II (Mn 2p3/2 → 5t2) RPES difference spectra. Participator Auger signals are labelled with numbers while spectator auger signals are labelled with letters. The origins of the associated peaks are discussed in the main body of the text. | ||
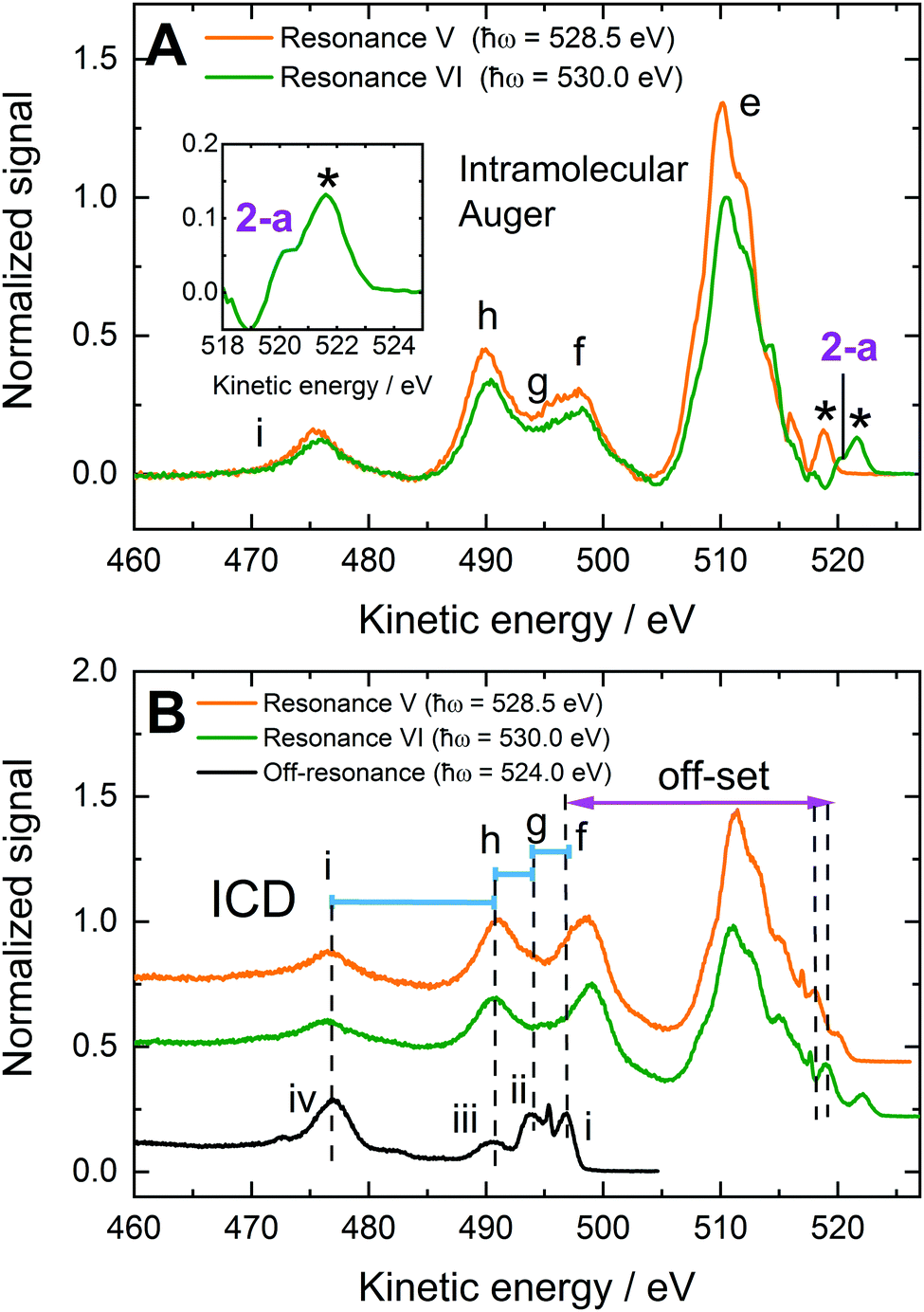 | ||
| Fig. 6 0.2 M NaMnO4(aq.) solution Resonance V and VI RPES data. (A) Resonance V (O 1s → 2e) and VI (O 1s → 5t2) RPES difference spectra. Participator Auger signals are labelled with numbers while spectator auger and potential ICD signals are labelled with letters. The asterisks highlight an experimental artefact signal associated with ionization of the H2O(l) O 1s molecular orbital with the residual, unfiltered second harmonic of the beam line. (B) As measured Resonance V and VI and spectrally offset off-resonant data highlighting the common energy gaps between peaks f, g, h, and i and the water valence orbitals. Similar energetic separations of the resonantly enhanced RPES features and the water peaks in the offset non-resonant spectra imply that resonant ICD processes occur following ligand-core-to-virtual-valence excitations of MnO4−(aq.). The spectral offset applied to the off-resonant data corresponds to the sum of the resonant-non-resonant photon energy difference, the 5a1 binding energy, and electron correlation effects. See the main body of the text for further details. | ||
| Signal | Assignment 1 | Assignment 2 | KE/eV |
|---|---|---|---|
| a | LIII-VV | — | 627.1 ± 0.4 |
| b | LIII-2t2,V | — | 576 ± 1 |
| c | LIII-2t2,5a1 | — | 565.7 ± 0.8 |
| d | LIII-4a1,V | — | 544.6 ± 0.1 |
| e | K-VV | — | 516.3 ± 0.1 |
| K-VV | — | 514.4 ± 0.2 | |
| K-VV | — | 512.2 ± 0.1 | |
| K-VV | — | 510.2 ± 0.2 | |
| K-VV | — | 508.0 ± 0.2 | |
| f | K-5a1,1b1(w) | K-5a1,V | 498.3 ± 0.5 |
| g | K-5a1,3a1(w) | K-5a1,V | 495 ± 1 |
| h | K-5a1,1b2(w) | — | 490.1 ± 0.2 |
| i | K-5a1,2a1(w) | — | 475.7 ± 0.2 |
Our Voigt profile fits to the Resonance II data reveal a significant lifetime broadening contribution to the total width of peak 3-a (0.77 ± 0.03 eV FWHM Lorentzian component of a 1.32 ± 0.01 eV total FWHM peak width). This broadening likely reflects constructive interference between the direct 1e−1 or 3t2−1 ionization channels and the respective LIII-1e5t2 or LIII-3t25t2 participator Auger processes, which respectively result in the same final states. Indeed, average ∼0.4 eV spectral broadenings are expected to be imparted onto (coherently) resonantly enhanced LIII-pre-edge peak widths, as channel interference exclusively occurs during the ∼1.8 fs (average) Mn 2p3/2 core-hole lifetime.69 The greater lifetime broadening contribution observed here seemingly reflects a shorter than average Mn 2p3/2 core-hole lifetime in the polyatomic, condensed-phase MnO4−(aq.) anion. Notably, similar results were obtained in the crystalline phase RPES studies reported by Reinert et al.67
A second Mn-associated participator Auger feature, peak 5, is observed in the Resonance I and II data shown in Fig. 5. These peaks occur at 583.9 ± 0.1 eV and 585.6 ± 0.1 eV KEs corresponding to respective 59.5 ± 0.1 eV and 60.1 ± 0.1 eV BEs (BEaverage = 59.8 ± 0.4 eV). Considering the MnO4−(aq.) BEs determined in the direct PES experiments, peak 5 is readily assigned as a resonant enhancement of the 2t2 (Mn 3p) inner valence ionisation feature. The resonant Auger processes responsible for peak 5 in the Resonance I and II difference spectra shown in Fig. 5 are therefore labelled LIII-2t22e and LIII-2t25t2, respectively.
We now consider the spectator Auger decay channel contributions to Fig. 5 (and the equivalent KMnO4−(aq.) data shown in Fig. S10A and B, ESI†). Starting with feature a, a spectral profile that appears to be made up of two or more peaks is observed to contribute to both the Resonance I and Resonance II spectra. The KE of the main feature is reported in Table 4. Similar features occur following excitation to Resonances III and IV, as shown in Fig. S8 in the ESI.† Feature a is notably narrower in the Resonance I and II cases (2p3/2 core-hole) with respect to the Resonance III and IV cases (2p1/2 core-hole), as expected based on the shorter LII- versus LIII-edge core-hole lifetimes of the latter (∼1.8 fs versus ∼0.7 fs, on average, respectively69). Furthermore, additional lower KE relaxation channels seemingly become accessible following higher-energy excitation to Resonances III and IV. Upon consultation of Table 1, feature a is generally attributed to L-VV spectator Auger processes, involving different pairs of Mn–O outer valence (V) bonding permanganate orbitals (6a1, 1e, 3t2). The broadening and spectral extension of feature a in the Resonance III and IV cases is then attributed to the onset of LII–LIIIV Coster–Kronig channels,76 as described in the ESI.†
Similarly to feature a, feature b appears to encompass two or more RPES peaks. Again, the KE of the main feature is reported in Table 4 and similar, but broadened, features are observed in the Resonance III and IV data shown in ESI,† Fig. S8. We generally attribute the peak b signal components to solute spectator Auger processes involving the 2t2 (Mn 3p) inner valence and solute Mn–O bonding outer valence electrons (6a1, 1e, 3t2), i.e. LIII-2t2V. The appearance of such peaks are indicative of outer-inner valence electronic coupling, i.e. Mn 3p hybridisation. Additionally, noting that the 6a1 orbital is predicted to have significant Mn 3p character – unlike the 1e and 3t2 orbitals – peak b may be tentatively primarily attributed to a LIII-2t26a1 spectator Auger process.40 As with peak a, LII–LIII2t2 Coster–Kronig transitions lead to a relative broadening and spectral extension of feature b in the Resonance III and IV excitation cases (see the ESI† for details).
A relatively weak additional spectator Auger signal – labelled peak c – occurs at an average KE value of 565.7 ± 0.8 eV and is present in both the Resonance I and II spectra. We tentatively assign these peaks to a LIII-2t25a1 spectator Auger process. This feature in particular suggests that the inner valence 2t2 (Mn 3p) and 5a1 (O 2s) and resonantly populated 2e and 5t2 virtual valence orbitals are coupled and that the Mn and O inner valence atomic orbitals are hybridised within the valence manifold. The corresponding signals in the Resonance III and IV data are thought to be buried under the aforementioned Coster–Kronig features.
Finally, additional weak resonantly-enhanced signals are observed in the Resonance I and II spectra at KEs close to 544.2 ± 0.1 eV. We label these features d and assign them to a LIII-4a1V spectator Auger emission process. Considering the MnO4−(aq.) 4a1 (Mn 3s) BE from Table 1 and neglecting electron correlation effects, we estimate a BE of ∼12.5 eV for the orbital involved in the LIII-4a1V process. This BE value is consistent with those extracted from signal 3 and RPES signal 3-a, suggesting a LIII-4a11e and/or LIII-4a13t2 process occurs. Such processes highlight the Mn-based character of the 1e and/or 3t2 orbitals, associated electronic coupling to the 4a1 orbitals, and Mn 3s inner-outer valence hybridisation.
We first consider the solute participator Auger decay channels. Given the non-bonding, predominant O 2p character of the 1t1 and 4t2 orbitals,40,56 associated participator Auger signals are expected to emerge in the 9–11 eV BE range (see Table 1). However, the analysis of this spectral region is complicated by its overlap with beamline second harmonic artefact signals (peaks marked by ‘*’ in Fig. 3E, F and 6A). Particularly, the Resonance V participator Auger decay channel associated with the 1t1 molecular orbital would be superimposed on this artefact. Hence, such a feature cannot be resolved using the difference spectrum shown in Fig. 6A alone. However, the K-2e1t1 participator peak is discernible from the photon energy dependence of the RPES map shown in Fig. 3D. This map specifically highlights an enhanced BE feature at 9.2 ± 0.1 eV over a ∼1 eV photon energy region in the vicinity of Resonance V, in agreement with the BE extracted from the direct PES spectra.
More generally, participator Auger decay signals are identifiable from the high signal-to-noise ratio, background subtracted spectra, like those presented in Fig. 6A. In the Resonance VI data, the lower KE shoulder to the second harmonic artefact signal (marked ‘*’ in Fig. 6A) is labelled peak 2-a and resolved at a KE of 520.1 ± 0.1 eV, corresponding to a BE of 9.85 ± 0.08 eV. Consulting Table 1 and noting the aforementioned predominant oxygen electronic character of the 4t2 molecular orbital, we ascribe this feature to a K-4t25t2 process. The 9.85 ± 0.08 eV BE is consequently appended to a 4t2−1 ionisation channel, with the associated BE included in the bottom panel of Table 1 with a peak 2-a label. In this particular case, we note that the reported BE may be somewhat biased by over-subtraction of the off-resonant background data. Although our results are consistent across multiple data sets which display varying degrees of over-subtraction, the participator BE value reported must, therefore, be considered a lower limit of the true value. We further note that any similar participator Auger signals in the Resonance V data would be buried under the second harmonic artefact signal.
The 4t2−1 BE peak (9.85 ± 0.08 eV, 2-a) discerned in the O-sensitive Resonance VI RPES data sets displays a relatively narrow average width (0.7 ± 0.2 eV) in comparison to the peak 2 feature extracted from the direct PES results (1.2 ± 0.1 eV at FWHM). Therefore, with the consistency of the RPES feature width across multiple data sets in mind, we infer that peak 2 in the direct PES experiments indeed consists of two overlapping components. The broader associated spectral profile would then correspond to the weighted average of the 4t2−1 and 6a1−1 features, resulting in the extracted peak 2 BE of 10.67 ± 0.04 eV. We note that this inference is further supported by the solid-phase ultraviolet PES KMnO4 experiments reported by Reinert et al.,37 which resolved the solid-state analogues of the peak 2 components using high-energy-resolution, near-threshold ionisation.
Additional participator Auger features potentially occur in the 515–520 eV KE region, although they cannot be unequivocally assigned. A particularly weak feature is observed close to a 518 eV KE in the Resonance VI data, which may be linked to a LIII-5t26a1 participator channel and relate to direct PES peak 2 and RPES peak 2-a. However, the KE and width of this feature cannot be reliably extracted due to the low signal intensity and the over-subtraction of the off-resonant spectrum in this region. Another, more intense, potential participator feature occurs at a KE of 516.3 ± 0.1 eV in the Resonance V data, corresponding to a calibrated BE of 12.18 ± 0.05 eV. Considering our assignment of peak 3-a to a selective enhancement of either the 1e−1 or 3t2−1 ionization channels, the 516.3 ± 0.1 eV peak could potentially be assigned as a participator Auger peak and to a selective K-1e5t2 ionisation process, with peak 3-a correspondingly being attributed to a selective LIII-3t25t2 participator Auger process. Hence, a 1e BE value of 12.18 ± 0.05 eV and a 3t2 BE value of 12.66 ± 0.04 eV (peak 3-a) could be tentatively inferred. Following such an inference, peak 3 in the direct PES experiments (with an extracted BE of 12.84 ± 0.09 eV) would then be associated with a weighted average of the two RPES participator peaks (with peak 3-a making the dominant contribution to the cumulative peak detected in direct PES). As appealing as such an assignment may be, we caution, however, that the ∼518 eV and 516.3 ± 0.1 eV potential participator peaks mentioned in this paragraph may be additionally and (perhaps more convincingly) alternatively attributed to spectator Auger processes, as discussed below. As such, the individual 1e and 3t2 BEs cannot be unambiguously determined, neither in our direct PES nor RPES experiments.
Focusing on the 9.0–10.5 eV participator BE region, a notable absence of any 1t1 or 4t2 orbital participator signals in the Mn-sensitive spectra (which are unperturbed by artefact signals) is highlighted. This confirms the predominant O-character of these molecular orbitals. Considering the higher BE 6a1, 1e, and 3t2 participator signals in the O K-pre-edge data, only features potentially associated with the 1e molecular orbital could be identified. Additional signatures of the 6a1, 1e, and 3t2 ionisation channels are either too weak, or are obscured by the beamline second harmonic artefact signal or K-VV spectator Auger (and potentially non-local intermolecular) decay signatures. Therefore, further insights into the O character and BEs of the 6a1, 1e, and 3t2 molecular orbitals can only be garnered through analysis of the spectator Auger signals, which we discuss in the following.
Considering the spectator Auger signals in Fig. 3E, F and 6A (and the KMnO4(aq.) data shown in Fig. S6 and S10, ESI†), a structured feature spanning KEs of 505–519 eV is found to dominate the Resonance V and VI spectra. This structure, labelled peak e, constitutes a manifold/convolution of four or more components with ∼2.1 eV sub-structure separations. We note that a very similar feature was observed in the solid-state KMnO4 RPES data reported by Reinert et al.,67 demonstrating that these peaks are associated with the MnO4− anion. The energy range spanned by feature e indicates its components correspond to K-VV spectator Auger processes. Possible processes associated with the 514.4 ± 0.2 eV, 512.2 ± 0.1 eV, 510.2 ± 0.2 eV, and 508.0 ± 0.2 eV KE peaks include the K-1t13t2, K-1e1e, K-6a13t2, and K-3t23t2 spectator Auger decay channels. Alternative and related assignments for the 516.3 ± 0.1 eV feature in the Resonance V data and the ∼518 eV KE peak in the Resonance VI data – discussed in the context of potential participator Auger channels above – would then be respective K-4t24t2 and K-1t11t1 processes. Indeed, when the common ∼2.1 eV sub-peak separations in the feature e progression are considered, we favour the assignment of the 516.3 ± 0.1 eV and ∼518 eV features to the aforementioned resonant spectator Auger processes (although the above-discussed participator channels may additionally underlie these peaks). We note that the resolution of up to six distinct spectral features spanning the peak e energy range and their assignment to K-VV spectator Auger processes suggests the entire valence band of MnO4−(aq.) is characterised by significant O character.
The signals spanning the KE range from 470–505 eV appear to be made up of three unresolved components and a lower KE feature that we label features f, g, h and feature i, respectively. These features have remarkably similar profiles in the Resonance V and VI RPES spectra recorded from all MnO4−(aq.) solutions. They are also significantly broadened with respect to the sub-components of signal e; from which multiple features separated by just ∼2 eV could be isolated. The electron KEs associated with signals f, g, h and i were determined to be 498.3 ± 0.5 eV, 495 ± 1 eV, 490.1 ± 0.2 eV, and 475.7 ± 0.2 eV, respectively, via a multi peak fit analysis (see ESI,† Fig. S9, S10 and Tables S4, S5). These energetics indicate involvement of the MnO4−(aq.) nominally non-bonding inner valence 5a1 (O 2s) electrons, with the features potentially being assignable to K-5a1V spectator Auger processes. The appearance of such peaks are again indicative of outer-inner valence orbital electronic coupling in MnO4−(aq.), i.e. in this case, O 2s valence hybridisation. Considering the 5a1 BE from the direct PES experiments (see Table 1), the following peak f, g, h and i BEs are respectively estimated (neglecting electron correlation): 10.9 ± 0.5 eV, 14 ± 1 eV, 19.1 ± 0.2 eV, and 33.5 ± 0.2 eV. Possible intramolecular solute spectator Auger assignments for peaks f and g would be K-5a1(4t2/6a1) and K-5a1(1e/3t2), respectively. However, peak h is more challenging to assign, with no solute BEs lying within ±6 eV of the expected ∼19 eV nominal BE of a 5a1 spectator Auger partner electron. Peak i also has no obvious spectator Auger signal assignment, with the closest match to its energetics being a K-5a15a1 process that would be shifted by more than 6 eV to higher KE in comparison to single-reference electronic structure expectations. With these assignment challenges in mind, we are driven to consider alternative, non-local core-hole relaxation mechanisms for the assignment of peaks h and i, and potentially f and g.
In Fig. 6B, we KE offset the off-resonant spectrum recorded at 524.0 ± 0.1 eV by the difference in the photon energies used to record the off-resonant and Resonance V and VI data (+4.5 ± 0.1 eV and +6.0 ± 0.1 eV, respectively) and the 5a1 BE (−26.82 ± 0.04 eV). This leads to overall off-resonant KE shifts of −22.3 ± 0.1 eV and −20.8 ± 0.1 eV with respect to the Resonance V and VI data, respectively. As a result, we see an excellent overlap – keeping in mind the additional differential peak shifting associated with electron correlation effects – of the water valence feature peak centres with the spectrally broadened f, g, h, and i features, which we attribute to resonant ICD.
We note that such ICD-induced mirroring of the liquid water photoelectron spectrum in RPES data has previously been observed following core-to-virtual valence resonant excitation of OH−(aq.).33 In that case, participator-like ICD processes – involving core-hole refill by the initially photoexcited electrons and simultaneous ejection of valence electrons from the solvent – were observed; as opposed to spectator-like (K-5a1V(w)) ICD processes ascribed to peaks f, g, h, and i in Fig. 6. Consequently, the question of whether such participator-ICD processes also contribute to the MnO4−(aq.) O K pre-edge data shown in Fig. 6 arises. Using similar arguments to those presented above, we can estimate the KE positions for such participator-like ICD electrons in Fig. 6. For water 1b1, 3a1, 1b2, and 2a1 electron ejection, respective KEs of 517.2 ± 0.1 eV, 514.8 ± 0.1 eV, 511.1 ± 0.1 eV, and 497.5 ± 0.1 eV and 518.7 ± 0.1 eV, 516.3 ± 0.1 eV, 512.6 ± 0.1 eV, and 499.0 ± 0.1 eV are expected at Resonances V and VI, respectively. Indeed, these KEs span the range of feature e in Fig. 6. However, the sharpness of the ionisation features in these spectral regions and the occurrence of a similar spectral profile in crystalline KMnO4 RPES data,67 imply that intramolecular spectator Auger decay processes dominate in the feature e region.
We finally emphasise that the resonant ICD features – being signatures of electronic coupling between the MnO4−(aq.) solute ligands and directly hydrating solvent molecules – were uniquely accessible in our RPES experiments, and only following detailed and careful analysis of the participator and spectator Auger channels. The relative intensity of resonant enhancement features f, g, h, and i, and particularly the occurrence of peaks h and i in what would otherwise be a sparsely populated region of the RPES spectrum, highlights the great potential of liquid-phase RPES to interrogate intermolecular bonding phenomena in the aqueous-phase, even at relatively low solute concentrations (0.2 M versus a ∼56 M water solvent concentration).
Absolute energetics and thermodynamics of MnO4−(aq.) solutions
Having extensively surveyed the electronic structure of MnO4−(aq.) in the previous sections, we utilise a selection of our results to extract aqueous-phase energetic parameters that underpin redox and more general chemical phenomena involving MnO4−(aq.). Of particular interest are the aqueous-phase adiabatic ionisation energies, AIE(aq.), characteristic minimum energy thresholds for liberating electrons to vacuum. With chemical reactions – including redox processes – primarily governed by the absolute energetics of the most readily liberated electrons, we focus our initial analysis on the lowest energy vertical ionising transition of MnO4−(aq.) (1t1−1), the associated production of the (1)2T1 state of MnO4˙ radical, and the MnO4−/MnO4˙ + e− redox half reaction. We begin by defining the free energetics of the associated chemical cycle.Fig. 7A highlights the free energetic relationships between the VIE and AIE of MnO4−(aq.), the VEA of MnO4˙(aq.), and the associated reorganisation free energies of oxidation (λox) and reduction (λred). The diagram depicts the following relation that can be utilised to extract the lowest AIE of MnO4−(aq.), equivalent to its Gibbs free energy of oxidation, ΔGox:
| AIE = VIE − λox = ΔGox | (1) |
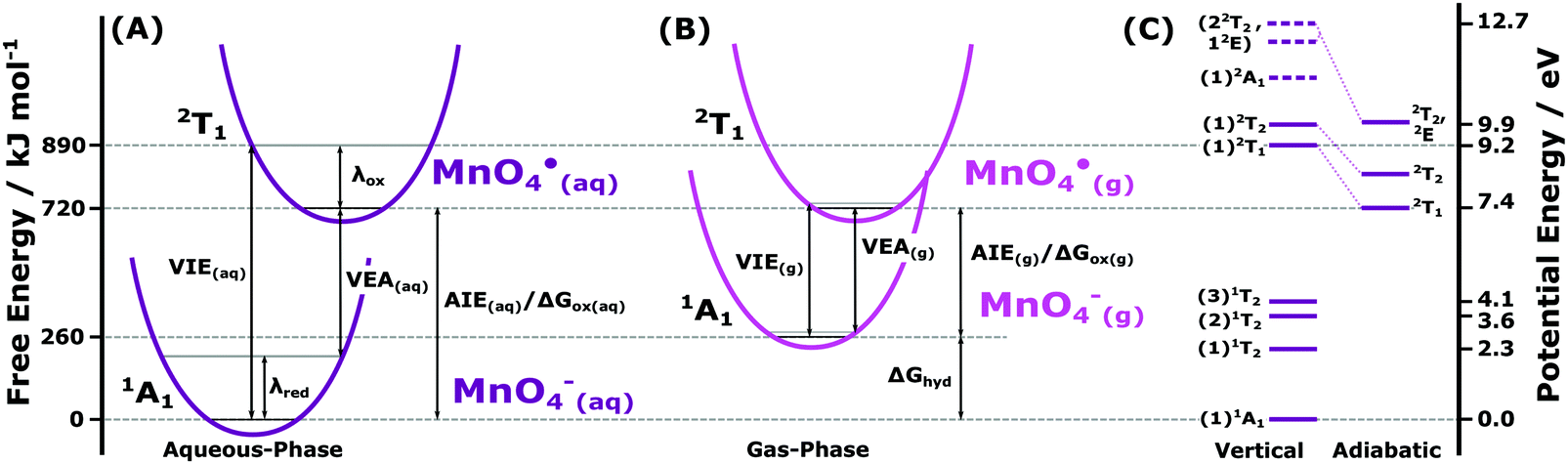 | ||
| Fig. 7 (A) Schematic quadratic potential energy curves of the MnO4−(aq.) anion and MnO4˙(aq.) radical, highlighting the parameters extracted from the lowest energy MnO4−(aq.) ionising transition. (B) Corresponding schematic of the MnO4−(g) anion and MnO4˙(g) radical potential energy curves, produced according to ref. 34 and energetically aligned so that the ground rovibronic state of the MnO4˙ radical occurs at the same energy in the aqueous- and gas-phase. the associated alignment energy corresponds to the vacuum level or +7.4 eV on the potential energy scale (relative to the MnO4−(aq.) anion ground state). (C) Electronic energy level structure of MnO4−(aq.) and MnO4˙(aq.) extracted from the spectroscopic results reported herein and potential energy referenced to the MnO4−(aq.) electronic ground state. On the left of diagram (C), vertical excitation energies and associated state energy levels of the optically active anion excited states of MnO4−(aq.) (see Fig. S1A in the ESI†) and valence electronically excited states of the MnO4˙(aq.) radical are shown. The solid lines are associated with the precisely determined MnO4−(aq.) and MnO4˙(aq.) state energies, with dashed lines denoting the equivocally inferred (1)2A1 (6a1−1), (1)2E (1e−1), and (2)2T2 (3t2−1) MnO4˙(aq.) state energetics at the equilibrium geometry of the MnO4−(aq.) ground state. For the two lowest VIEs, corresponding adiabatic MnO4˙(aq.) radical state energy levels, as extracted using the direct PES (2T1) and (2T2) RPES peak BEs/widths and linear response theory, are shown to the right. VIE: vertical ionisation energy, VEA: vertical electron affinity, λox/red: total reorganisation energy of the oxidised/reduced state, AIE: adiabatic ionisation energy, ΔGox: Gibbs free energy of oxidation, ΔGhyd: Gibbs free energy of hydration. | ||
It has previously been demonstrated that, within the linear response approximation, liquid-phase photoelectron feature widths can be related to λox using the equation below:12–14,77,78
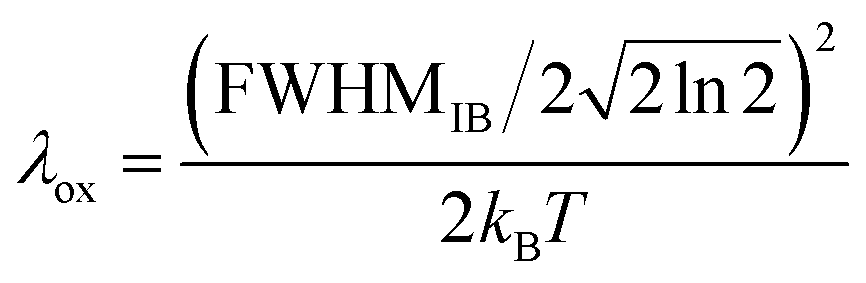 | (2) |
Notably, approximating the FWHMIB term in eqn (2) with a photoelectron feature width (for example, the average values reported in Table 1) will over and under estimate the values of λox and AIE, respectively. The overall ionisation peak width is only partially determined by the inhomogeneous broadening term, being further widened by experimental resolution limitations and potential lifetime broadening effects. Only by carefully accounting for the latter broadening mechanisms, can we expect to extract accurate FWHMIB terms and, hence, values of λox and AIE(aq.).
The Gaussian fits to our direct PES spectra recorded at sample temperatures of 277–278 K yield an average 0.8 ± 0.1 eV 1t1−1 (peak 1) feature width that we term FWHMG. As previously mentioned, our direct PES data analysis confirmed that lifetime broadening effects are negligible in the associated valence spectral region. The solution phase FWHMIB component for the 1t1−1 peak ionisation feature can therefore be determined as follows:
 | (3) |
Inserting our λox estimate and the lowest VIE of MnO4−(aq.) (9.22 ± 0.08 eV, see Table 1) into expression (1) yields an MnO4−(aq.) AIE and ΔGox value of 7.4 ± 0.2 eV or 720 ± 20 kJ mol−1. Importantly, we can use this (and other) AIE(aq.) result(s) to determine additional thermodynamic quantities of the MnO4−/MnO4˙ + e− system and/or connect the energetics of different chemical species to those of MnO4−(aq.), examples of which we present below.
The extracted AIE(aq.) value permits us to estimate the VEA(aq.) of MnO4˙ using the following expression, again within the linear response approximation:14,78
| VEA = 2AIE − VIE | (4) |
Based on the MnO4−(aq.) 1t1−1 ionising transition analysis above and the 4t2 BE and peak width extracted in our RPES analysis, we can similarly estimate the λox and AIE values for the first electronically excited state of the MnO4˙(aq.) radical. In this case our results correspond to a 283 K jet temperature (see Table 1) but are still comparable with our 277–278 K MnO4˙(aq.) ground state results (such a temperature variation leads to a less than 40 meV difference in λox and AIE values, well below our reported experimental uncertainties). The results of such an analysis, as well as the aforementioned findings reported in this section, are summarised in Table 5. Here, the energetics are presented along with the electronic state symmetries of the produced MnO4˙(aq.) radicals (assuming a retention of Td symmetry upon ionisation), with the assigned dominant molecular orbital symmetry of the removed electron given in parenthesis.
| Radical state | VIE(aq.)/eV | FWHMIB/eV | λ ox/eV | AIE(aq.)/eV | ΔGhyd/eV |
|---|---|---|---|---|---|
| (1)2T1 (1t1−1) | 9.22 ± 0.08 | 0.7 ± 0.1 | 1.8 ± 0.2 | 7.4 ± 0.2 | 2.6 ± 0.3 |
| (1)2T2 (4t2−1) | 9.85 ± 0.08 | 0.6 ± 0.2 | 1.5 ± 0.5 | 8.4 ± 0.5 | 2.3 ± 0.5 |
As noted in the introduction, a molecular orbital or single-configuration electronic structure formalism provides only a qualitative description of the electronic structure of the permanganate anion, with a multi-reference electronic structure treatment demonstrably being needed to quantitatively describe electronically excited states of MnO4−.54 Accordingly, we present our valence band spectroscopic results within a more general framework in Fig. 7C, as an electronic state energy level diagram of MnO4−(aq.) and MnO4˙(aq.). The diagram is energetically referenced to the vibronic ground state of MnO4−(aq.) and based on the energetics summarised in Table 5. Otherwise, vertical excitation energies for the electronically excited anion states were determined in our UV-visible absorption spectroscopy measurements (see ESI,† Fig. S1). The VIE values extracted directly from the non-resonant PES/RPES results and the AIE values determined from the associated BEs and peak widths are labelled ‘Vertical’ and ‘Adiabatic’ in Fig. 7C, respectively.
The AIE(aq.) values (equivalent to ΔGox) and associated state energetics extracted here are usefully pinned to a universal referencing standard (the vacuum level). They are expected to permit direct comparison between the electronic energetics of MnO4−(aq.) and redox reaction partner species (including other aqueous manganese oxides). Accordingly, complementary liquid-jet-PES experiments will enable (full, rather than just half) reaction free energies to be experimentally determined and chemical process propensities to be predicted. Similarly, the ionisation and state energetics (VIE(aq.), AIE(aq.)/ΔGox, VEA(aq.), and ΔGhyd values) reported here are expected to provide a stringent test for emerging multi-configurational electronic structure calculation methods that aspire to explicitly treat and elucidate the effects of hydration on complex solutes.80–83 We hope that the results presented in this section in particular will stimulate such theoretical investigations.
Conclusions
We have presented a comprehensive analysis of the electronic structure of aqueous-phase MnO4−(aq.) and MnO4˙(aq.) based on liquid jet direct PES and RPES experiments. Absolute electronic BE/VIE(aq.) values were determined for the majority of the valence manifold, as well as select core levels of MnO4−(aq.). This allowed us to produce an associated experimentally-determined-BE-scaled, near-complete molecular orbital diagram for this system. At 0.2 M concentrations, similar electronic energetics were identified (relative to the vacuum level) in the aqueous bulk and at the solution–gas interface, as well as upon switching the MnO4−(aq.) counter-ion from Na+(aq.) to K+(aq.). The aqueous and solid phase BEs of MnO4− were found to be similar, albeit with the aqueous phase BEs shifted to lower values by up to a few-hundred meV compared to their solid-state counterparts. Our aqueous phase results also highlight an increase of the lowest-energy ionising transition BEs by more than 4 eV upon hydration, i.e. in moving from the gas to the aqueous phase.Our RPES experiments reinforced our direct PES results and allowed us to isolate additional valence BEs that were otherwise obscured by other solute and/or solvent-associated signals. Within a molecular orbital framework, the RPES data additionally revealed specific atomic parentages of the MnO4−(aq.) valence molecular orbitals as well as hybridisation of the constituent outer and inner valence atomic orbitals. Furthermore, MnO4−(aq.)–H2O(l) ICD processes were identified, highlighting electronic coupling – and perhaps covalent bonding interactions – between the valence manifolds of the solute and solvent. Finally, careful analysis of our cumulative results allowed us to infer oxidative reorganisation energies, and adiabatic valence MnO4−(aq.) ionisation energies of the two lowest energy ionising transitions. An associated Gibbs free energy of oxidation of MnO4−(aq.) of 7.4 ± 0.2 eV (720 ± 20 kJ mol−1) was revealed. We additionally extracted a vertical electron affinity estimate of 5.7 ± 0.2 eV for the MnO4˙(aq.) radical. Finally, the Gibbs free energy of hydration of isolated MnO4− was most accurately determined to be 2.6 ± 0.2 eV or 260 ± 20 kJ mol−1.
Our absolute energy scale measurements are furthermore expected to permit direct comparison between the electronic energetics of MnO4−(aq.), other aqueous manganese oxide species, and/or redox reactive partners (be they bulk aqueous-phase reactants or interfacial species/materials). Hence, this work feeds into a larger effort to experimentally characterise not just half-reactions, but complete chemical transformations in aqueous solution and on an absolute energy scale. Furthermore, the results presented here constitute experimental benchmarks for state-of-the-art electronic structure calculations and associated spectral simulation methods. Calculations focused on the explicit treatment of hydration and its associated effects on electronically complex solutes80–83 as well as those focused on simulating X-ray spectra of increasingly complex systems84–86 all stand to benefit. Such studies, although technically challenging, are pre-requisite for the development of a deeper understanding of the energetics, dynamics, and reactivity of this unusual and ubiquitously applied aqueous transition-metal complex.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Helmholtz-Zentrum Berlin für Materialien und Energie and the Deutsches Elektronen-Synchrotron for the allocation of synchrotron radiation beamtime, the Chemistry Lab User Support teams at BESSY II and PETRA III, Mr Garlef Wartner for support during the BESSY II measurements, and Mr Sebastian Malerz and Dr Florian Trinter for support during the PETRA III measurements. We also acknowledge Prof. Stephan Thürmer for insightful discussions regarding aqueous-phase PES background correction methodologies. We also gratefully acknowledge Prof. Jógvan Magnus Haugaard Olsen for his support in sharing the optimised MnO4−(aq.) nuclear geometry files associated with ref. 80. RS thanks the German Research Foundation for granting an Emmy-Noether stipend (SE 2253/3-1).Notes and references
- C. C. Liang, in Encyclopedia of Electrochemistry of the elements, ed. A. J. Bard, Marcel Dekker, Inc., 1973, vol. I Search PubMed.
- N. Singh and D. G. Lee, Org. Process Res. Dev., 2001, 5, 599–603 CrossRef CAS.
- M. S. Roderick, J. L. Adcock, J. M. Terry, Z. M. Smith, S. Parry, S. M. Linton, M. T. Thornton, C. J. Barrow and P. S. Francis, J. Phys. Chem. C, 2013, 117, 10286–10293 CrossRef CAS.
- J. L. Adcock, N. W. Barnett, C. J. Barrow and P. S. Francis, Anal. Chim. Acta, 2014, 807, 9–28 CrossRef CAS.
- Y. Chang, Y. Bai and J. Qu, Chemosphere, 2016, 163, 73–80 CrossRef CAS.
- B. Liu, F. Qu, W. Chen, H. Liang, T. Wang, X. Cheng, H. Yu, G. Li and B. Van der Bruggen, Water Res., 2017, 125, 72–80 CrossRef CAS.
- P. Xie, J. Ma, J. Fang, Y. Guan, S. Yue, X. Li and L. Chen, Environ. Sci. Technol., 2013, 47, 14051–14061 CrossRef CAS.
- A. J. Gibson, K. G. Latham, R. C. Burns and S. W. Donne, Electrochim. Acta, 2017, 236, 198–211 CrossRef CAS.
- A. N. Colli, P. Peljo and H. H. Girault, Chem. Commun., 2016, 52, 14039–14042 RSC.
- T. R. Eliato, G. Pazuki and N. Majidian, Energy Sources, Part A, 2016, 38, 644–651 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- J. Moens, R. Seidel, P. Geerlings, M. Faubel, B. Winter and J. Blumberger, J. Phys. Chem. B, 2010, 114, 9173–9182 CrossRef CAS.
- R. Seidel, S. Thürmer, J. Moens, P. Geerlings, J. Blumberger and B. Winter, J. Phys. Chem. B, 2011, 115, 11671–11677 CrossRef CAS.
- D. Ghosh, A. Roy, R. Seidel, B. Winter, S. Bradforth and A. I. Krylov, J. Phys. Chem. B, 2012, 116, 7269–7280 CrossRef CAS.
- C. Adriaanse, M. Sulpizi, J. VandeVondele and M. Sprik, J. Am. Chem. Soc., 2009, 131, 6046–6047 CrossRef CAS.
- R. N. Tazhigulov and K. B. Bravaya, J. Phys. Chem. Lett., 2016, 7, 2490–2495 CrossRef CAS.
- R. Seidel, B. Winter and S. E. Bradforth, Annu. Rev. Phys. Chem., 2016, 67, 283–305 CrossRef CAS.
- D. Yepes, R. Seidel, B. Winter, J. Blumberger and P. Jaque, J. Phys. Chem. B, 2014, 118, 6850–6863 CrossRef CAS.
- R. Seidel, K. Atak, S. Thürmer, E. F. Aziz and B. Winter, J. Phys. Chem. B, 2015, 119, 10607–10615 CrossRef CAS.
- Y. Jiao, B. W. Adams, A. O. Dohn, K. B. Møller, H. Jónsson and C. Rose-Petruck, Phys. Chem. Chem. Phys., 2017, 19, 27266–27274 RSC.
- A. J. Orr-Ewing, Chem. Soc. Rev., 2017, 46, 7597–7614 RSC.
- B. Mennucci, J. M. Martínez and J. Tomasi, J. Phys. Chem. A, 2001, 105, 7287–7296 CrossRef CAS.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS.
- J. C. Hindman, J. Chem. Phys., 1962, 36, 1000–1016 CrossRef CAS.
- M. Kubin, M. Guo, T. Kroll, H. Löchel, E. Källman, M. L. Baker, R. Mitzner, S. Gul, J. Kern, A. Föhlisch, A. Erko, U. Bergmann, V. Yachandra, J. Yano, M. Lundberg and P. Wernet, Chem. Sci., 2018, 9, 6813–6829 RSC.
- G. Aullón and S. Alvarez, Theor. Chem. Acc., 2009, 123, 67–73 Search PubMed.
- R. Seidel, S. Thürmer and B. Winter, J. Phys. Chem. Lett., 2011, 2, 633–641 Search PubMed.
- N. Ottosson, M. Faubel, S. E. Bradforth, P. Jungwirth and B. Winter, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 60–70 CrossRef CAS.
- Y.-I. Suzuki, K. Nishizawa, N. Kurahashi and T. Suzuki, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 010302 CrossRef.
- B. Winter, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 139–150 CrossRef CAS.
- T. Miteva, N. V. Kryzhevoi, N. Sisourat, C. Nicolas, W. Pokapanich, T. Saisopa, P. Songsiriritthigul, Y. Rattanachai, A. Dreuw, J. Wenzel, J. Palaudoux, G. Öhrwall, R. Püttner, L. S. Cederbaum, J.-P. Rueff and D. Céolin, J. Phys. Chem. Lett., 2018, 9, 4457–4462 CrossRef CAS.
- H. Ali, R. Seidel, M. N. Pohl and B. Winter, Chem. Sci., 2018, 9, 4511–4523 RSC.
- E. F. Aziz, N. Ottosson, M. Faubel, I. V. Hertel and B. Winter, Nature, 2008, 455, 89–91 CrossRef CAS.
- G. Gutsev, B. K. Rao, P. Jena, X.-B. Wang and L.-S. Wang, Chem. Phys. Lett., 1998, 312, 598–605 CrossRef.
- M. Oku, J. Electron Spectrosc. Relat. Phenom., 1995, 74, 135–148 CrossRef CAS.
- M. Oku, T. Shishido and S. Kohiki, Surf. Sci. Spectra, 2004, 11, 59–65 CrossRef CAS.
- F. Reinert, S. Kumar, P. Steiner, R. Claessen and S. Hüfner, Z. Phys. B: Condens. Matter, 1994, 94, 431–438 CrossRef CAS.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, J. A. Bradley, S. R. Daly, S. A. Kozimor, W. W. Lukens, R. L. Martin, D. Nordlund, G. T. Seidler, D. K. Shuh, D. Sokaras, T. Tyliszczak, G. L. Wagner, T.-C. Weng and P. Yang, J. Am. Chem. Soc., 2013, 135, 1864–1871 CrossRef CAS.
- T. Ziegler, A Chronicle About the Development of Electronic Structure Theories for Transition Metal Complexes, 2011 Search PubMed.
- L. Jose, M. Seth and T. Ziegler, J. Phys. Chem. A, 2012, 116, 1864–1876 CrossRef CAS.
- K. H. Johnson and F. C. Smith, Phys. Rev. B: Solid State, 1972, 5, 831–843 CrossRef.
- Helmholtz-Zentrum Berlin für Materialien und Energie, J. Large-scale Res. Facil., 2016, 2, A72 CrossRef.
- R. Seidel, M. N. Pohl, H. Ali, B. Winter and E. F. Aziz, Rev. Sci. Instrum., 2017, 88, 073107 CrossRef.
- J. Viefhaus, F. Scholz, S. Deinert, L. Glaser, M. Ilchen, J. Seltmann, P. Walter and F. Siewert, Nucl. Instrum. Methods Phys. Res., Sect. A, 2013, 710, 151–154 CrossRef CAS.
- http://www.fhi-berlin.mpg.de/mp/winter/Main/ExperimentalMethods, accessed 02.07, 2020.
- G. Zimmerman, J. Chem. Phys., 1955, 23, 825–832 CrossRef CAS.
- S. K. Eriksson, M. Hahlin, J. M. Kahk, I. J. Villar-Garcia, M. J. Webb, H. Grennberg, R. Yakimova, H. Rensmo, K. Edström, A. Hagfeldt, H. Siegbahn, M. O. M. Edwards, P. G. Karlsson, K. Backlund, J. Åhlund and D. J. Payne, Rev. Sci. Instrum., 2014, 85, 075119 CrossRef.
- S. Thürmer, R. Seidel, M. Faubel, W. Eberhardt, J. C. Hemminger, S. E. Bradforth and B. Winter, Phys. Rev. Lett., 2013, 111, 173005 CrossRef.
- N. Kurahashi, S. Karashima, Y. Tang, T. Horio, B. Abulimiti, Y.-I. Suzuki, Y. Ogi, M. Oura and T. Suzuki, J. Chem. Phys., 2014, 140, 174506 CrossRef.
- S. Hellmann, C. Sohrt, M. Beye, T. Rohwer, F. Sorgenfrei, M. Marczynski-Bühlow, M. Kalläne, H. Redlin, F. Hennies, M. Bauer, A. Föhlisch, L. Kipp, W. Wurth and K. Rossnagel, New J. Phys., 2012, 14, 013062 CrossRef.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- B. Winter, E. F. Aziz, U. Hergenhahn, M. Faubel and I. V. Hertel, J. Chem. Phys., 2007, 126, 124504 CrossRef.
- L. Å. Näslund, J. Lüning, Y. Ufuktepe, H. Ogasawara, P. Wernet, U. Bergmann, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. B, 2005, 109, 13835–13839 CrossRef.
- N. M. S. Almeida, R. G. McKinlay and M. J. Paterson, Chem. Phys., 2015, 446, 86–91 CrossRef CAS.
- T. Ziegler, in Molecular Electronic Structures of Transition Metal Complexes II, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–38 DOI:10.1007/430_2011_47.
- P. Sharma, D. G. Truhlar and L. Gagliardi, J. Chem. Phys., 2018, 148, 124305 CrossRef.
- T. K. Sham and B. S. Brunschwig, J. Am. Chem. Soc., 1981, 103, 1590–1591 CrossRef CAS.
- W. Kiefer and H. J. Bernstein, Mol. Phys., 1972, 23, 835–851 CrossRef CAS.
- M. N. Pohl, E. Muchová, R. Seidel, H. Ali, Š. Sršeň, I. Wilkinson, B. Winter and P. Slavíček, Chem. Sci., 2019, 10, 848–865 RSC.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Faubel and I. V. Hertel, J. Phys. Chem. A, 2004, 108, 2625–2632 CrossRef CAS.
- B. Winter, R. Weber, I. V. Hertel, M. Faubel, P. Jungwirth, E. C. Brown and S. E. Bradforth, J. Am. Chem. Soc., 2005, 127, 7203–7214 CrossRef CAS.
- A. P. Mortola, H. Basch and J. W. Moskowitz, Int. J. Quantum Chem., 1973, 7, 725–737 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- J. K. A. C. Thompson, D. T. Atwood, E. M. Gullikson, M. R. Howells, J. B. Kortright, Y. Liu, A. L. Robinson, J. H. Underwood, K. Kim, I. Lindau, P. Pianetta, H. Winick, G. P. Williams and J. H. Scofield, X-Ray Data Booklet, Lawrence Berkeley National Laboratory, University of California, Berkeley, California 94720, 3rd edn, 2009 Search PubMed.
- W. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 92nd edn, 2011 Search PubMed.
- A. Pflueger, Ann. Phys., 1903, 12, 430 CrossRef.
- F. Reinert, P. Steiner, R. Zimmermann, R. Claessen and S. Hüfner, Z. Phys. B: Condens. Matter, 1995, 99, 229–234 CrossRef.
- M. A. Brown, M. Faubel and B. Winter, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2009, 105, 174–212 RSC.
- J. L. Campbell and T. Papp, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- I. Unger, R. Seidel, S. Thürmer, M. N. Pohl, E. F. Aziz, L. S. Cederbaum, E. Muchová, P. Slavíček, B. Winter and N. V. Kryzhevoi, Nat. Chem., 2017, 9, 708–714 CrossRef CAS.
- Z.-H. Loh, G. Doumy, C. Arnold, L. Kjellsson, S. H. Southworth, A. Al Haddad, Y. Kumagai, M.-F. Tu, P. J. Ho, A. M. March, R. D. Schaller, M. S. Bin Mohd Yusof, T. Debnath, M. Simon, R. Welsch, L. Inhester, K. Khalili, K. Nanda, A. I. Krylov, S. Moeller, G. Coslovich, J. Koralek, M. P. Minitti, W. F. Schlotter, J.-E. Rubensson, R. Santra and L. Young, Science, 2020, 367, 179–182 CAS.
- S. Thürmer, I. Unger, P. Slavíček and B. Winter, J. Phys. Chem. C, 2013, 117, 22268–22275 CrossRef.
- B. Winter, U. Hergenhahn, M. Faubel, O. Björneholm and I. Hertel, J. Chem. Phys., 2007, 127, 094501 CrossRef.
- S. Thürmer, R. Seidel, W. Eberhardt, S. E. Bradforth and B. Winter, J. Am. Chem. Soc., 2011, 133, 12528–12535 CrossRef.
- M. Oku, J. Electron Spectrosc. Relat. Phenom., 1995, 74, 135–148 CrossRef CAS.
- D. Coster and R. D. L. Kronig, Physica, 1935, 2, 13–24 CrossRef CAS.
- P. Winget, E. J. Weber, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2000, 2, 1231–1239 RSC.
- Y. Tateyama, J. Blumberger, M. Sprik and I. Tavernelli, J. Chem. Phys., 2005, 122, 234505 CrossRef.
- M. Bühl, J. Phys. Chem. A, 2002, 106, 10505–10509 CrossRef.
- J. M. Olsen and E. Hedegård, Phys. Chem. Chem. Phys., 2017, 19, 15870–15875 RSC.
- C. Bistafa, Y. Kitamura, M. Nagaoka and S. Canuto, J. Phys. Chem. B, 2018, 122, 9202–9209 CrossRef CAS.
- D. Asthagiri, L. R. Pratt, M. E. Paulaitis and S. B. Rempe, J. Am. Chem. Soc., 2004, 126, 1285–1289 CrossRef CAS.
- M. Uudsemaa and T. Tamm, Chem. Phys. Lett., 2004, 400, 54–58 CrossRef CAS.
- S. Sreekantan Nair Lalithambika, R. Golnak, B. Winter and K. Atak, Inorg. Chem., 2019, 58, 4731–4740 CrossRef CAS.
- S. S. N. Lalithambika, K. Atak, R. Seidel, A. Neubauer, T. Brandenburg, J. Xiao, B. Winter and E. F. Aziz, Sci. Rep., 2017, 7, 40811 CrossRef CAS.
- E. Pluhařová, C. Schroeder, R. Seidel, S. E. Bradforth, B. Winter, M. Faubel, P. Slavíček and P. Jungwirth, J. Phys. Chem. Lett., 2013, 4, 3766–3769 CrossRef.
- J. M. H. Olsen and E. D. Hedegård, Phys. Chem. Chem. Phys., 2017, 19, 15870–15875 RSC.
- W. F. Egelhoff, Surf. Sci. Rep., 1987, 6, 253–415 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04033a |
| ‡ We choose to reference our data to this more recent value, rather than the originally determined 11.16 ± 0.04 eV.60 Note that current experiments are not able to directly access absolute solution-phase binding energies, hence, requiring the adoption of an energetic reference; see discussion in ref. 59 for details. |
| § Established by comparison of the isolated molecule geometries in ref. 54 with the optimised aqueous-phase nuclear geometries associated with ref. 87 which were kindly shared with us during the drafting of this manuscript [JMH Olsen, Personal Communication, 09.04.2019]. |
| ¶ Here, the work function has been assumed to be representative of a KMnO4 crystal-face-independent value.88 |
| This journal is © the Owner Societies 2020 |