
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanolubrication in deep eutectic solvents
James E.
Hallett
 ,
Hannah J.
Hayler
and
Susan
Perkin
*
,
Hannah J.
Hayler
and
Susan
Perkin
*
Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: susan.perkin@chem.ox.ac.uk
First published on 26th August 2020
Abstract
We report surface force balance measurements of the normal surface force and friction between two mica surfaces separated by a nanofilm of the deep eutectic solvent ethaline. Ethaline, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of choline chloride and ethylene glycol, was studied under dry conditions, under ambient conditions and with added water, revealing surface structural layers and quantised frictional response highly sensitive to water content, including regions of super-lubric behaviour under dry conditions and with added water. We also report exceptionally long-ranged electrostatic repulsion far in excess of that predicted by Debye–Hückel theory for a system with such high electrolyte content, consistent with previously reported observations of “underscreening” in ionic liquid and concentrated aqueous electrolyte systems [Smith et al., J. Phys. Chem. Lett., 2016, 7(12), 2157].
:2 mixture of choline chloride and ethylene glycol, was studied under dry conditions, under ambient conditions and with added water, revealing surface structural layers and quantised frictional response highly sensitive to water content, including regions of super-lubric behaviour under dry conditions and with added water. We also report exceptionally long-ranged electrostatic repulsion far in excess of that predicted by Debye–Hückel theory for a system with such high electrolyte content, consistent with previously reported observations of “underscreening” in ionic liquid and concentrated aqueous electrolyte systems [Smith et al., J. Phys. Chem. Lett., 2016, 7(12), 2157].
1 Introduction
Deep eutectic solvents (DESs) are a class of liquid of growing importance, and are often described as a sub-category or new class of ionic liquids (ILs).1 Like ILs, they are a liquid mixture of binary ion pairs with no neutral solvent. Unlike ILs, which are primarily composed of discrete anions and cations of one particular species, DESs contain a complex ionic component and can contain a variety of ionic species.2 The term deep eutectic solvent originates from the low melting temperature TM obtained at the eutectic composition.3 DESs are obtained by mixing a hydrogen bond-capable salt (typically a quaternary ammonium salt) with a metal salt or hydrogen bond donor species (HBD). In many cases these two components are crystalline solids at room temperature, whilst the mixture formed is liquid under ambient conditions and with relatively low TM. Furthermore, a wide range of biodegradable, inexpensive components such as amino acids and sugars can be used to form DESs,4,5 and it is this flexibility that makes them such an exciting prospect as a green solvent.2 Indeed, deep eutectic solvents have been proposed as environmentally friendly alternatives for processes including electrodeposition,6 metal oxide processing,7 organic synthesis,8 and the purification and manufacture of biodiesel.9,10Perhaps the most commonly studied DESs are derived from mixing choline chloride with urea, ethylene glycol or glycerol (so-called reline, ethaline and glyceline) and it is these systems that have been used to make general observations of the unique supramolecular structure of DES systems. It is generally accepted that DESs form due to charge delocalization between the salt anion and hydrogen bond donor,4 but the structural consequences of this unique formation mechanism are still an area of active investigation. Recent work used a combination of neutron diffraction and atomistic modelling to reveal the liquid structure of reline. Hammond and co-workers11 measured neutron diffraction data from isotopically substituted reline samples, combined with empirical potential structure refinement (EPSR) to reveal the 3D structure of the solvent, which, in addition to the predicted hydrogen bond network within the network, also showed significant spatial ordering between all the DES components. Subsequent work unveiled the role of water in the 3D structure of the solvent.12 Remarkably, even at 42 wt% water, the 3D spatial order of the DES was retained, as the water was sequestered into nanostructured domains around the cholinium cations. At higher concentrations the structure was disrupted, and was best described as an aqueous solution of the DES components. Recently the structure of DESs has been directly probed via atomic force microscopy (AFM). The nanostructure of reline, ethaline and glyceline at both graphite13 and platinum14 substrates has been investigated, and was found to be strongly influenced by surface potential, substrate material and water content. For platinum, at positive potentials, the innermost displaceable layer was of comparable thickness to choline (∼0.45 nm) and was thought to be in contact with an impenetrable chloride Stern layer. At negative potentials, the innermost displaceable layer for reline, ethaline and glyceline was too thin to correspond to any DES component other than the HBD, and was thought to be in contact with an impenetrable choline Stern layer. For graphite, the final displaceable layer corresponded to the HBD at all potentials, except for −1 V at which point it displayed the same behaviour as the platinum electrode. The difference was attributed to the atomically smooth graphite surface more strongly aligning counterions than the platinum surface. They also noted that the magnitude of the final layer push-through force increased with increasing positive potential. However it should be noted that, as AFM does not provide an absolute measure of separation, the hypothesised near-surface structure was based on geometric arguments of molecular size rather than a definitive measure of the liquid layer thickness. In addition to dry DESs, the influence of added water was also studied for the platinum electrode.14 It was shown that added water also increased the push-through force and number of layers at open circuit potential, up to a maximum degree of structuring at 40 wt% water for reline, 30 wt% water for glyceline and 50 wt% water for ethaline. However, application of a surface potential disrupted this structure, which was attributed to a rearrangement of the surface-ion composition, resulting in a surface structure closer to that of a salt solution at a charged interface. AFM measurements have also been performed using alkyl ammonium bromide – glycerol based ionic liquids on mica substrates.15 Mica substrates are well known to have a negative surface charge through dissolution of potassium cations,16 and the DES structure showed layering with a HBD-enriched near-surface layer consistent with the layering observed for an applied negative potential.
A promising application of deep eutectic solvents is that of a lubricant. In many industrial applications spent oil-based lubricants are environmentally damaging and rarely recycled,17 so the prospect of a biodegradeable alternative is an enticing one.18 Work to date has prioritised bulk measurements of the tribological properties of choline chloride-based DESs, particularly of sliding steel–steel contacts and comparable friction coefficients to oil-based lubricants has been reported.19 More recent work reported better lubrication performance from choline chloride-based DES, and found that adding graphene further reduced the coefficient of friction.20 Abbott et al.21 reported superior lubrication performance from DESs and from ionic liquids than from base oil for steel-steel contacts. Intriguingly they also reported outstanding corrosion resistance from DESs compared to base oil, when the lubricant had become contaminated with aqueous salt solution, and noted a potential application for DESs as marine lubricants. They propose that the formation of an interfacial barrier by the ionic component is the origin of the enhanced lubrication, and the enhancement is optimal at the eutectic composition,21,22 and it is the behaviour of this interfacial layer that we interrogate in this study.
The surface force balance is an ideal instrument to perform measurements of the structure and frictional properties of confined films, including aqueous systems,23 alkanes24 and ionic liquids.25 Of particular interest are ionic liquids, which can form layered structures at charged and uncharged interfaces.26–28 The coefficient of friction of the film has been shown to vary with the number of confined molecular layers, leading to so-called “quantised friction”.29–31 Intriguingly the structure of ionic liquid films can also be manipulated by changing the surface charge,32 which as a consequence also acts to change the frictional properties, leading to “switchable superlubricity”.33 The formation of a robust, low friction surface film has lead to ionic liquids being proposed as a novel lubricant system.34–36
In this work, we apply the surface force balance technique to the deep eutectic solvent ethaline, a 1:2 molar mixture of choline chloride and ethylene glycol. We report surface layering consistent with previous AFM measurements,13–15 but with greater force resolution enabling the detection of weaker surface layers than previously reported, and anomalous long-ranged forces consistent with ‘underscreening’. We measure the frictional response of the surface layers and report quantised friction in the superlubric regime. We also show that both the surface structure and the frictional response are highly sensitive to water content, either through adsorption of atmospheric humidity or through added water. Finally, we propose molecular ordering and mechanisms to rationalise the reported data.
2 Experimental setup
2.1 Surface force balance
Experiments were performed using a surface force balance (SFB). White light interferometry is used to determine the film thickness D, and normal force, FN, between semi-transparent reflective surfaces separated by a thin liquid film. In this work, the surfaces used were back-silvered mica sheets of equal thickness (typically 2–4 μm). These mica sheets were glued (silver-side down) on to cylindrical lenses (radius of curvature ∼ 10 mm) and mounted in the SFB in a crossed-cylinder geometry (Fig. 1). A droplet of deep eutectic solvent was injected between the two mica surfaces, forming a capillary bridge. White light interferometry was used to determine the thickness of the layer between the two silver mirrors, comprising the two equal-thickness mica sheets and the confined liquid film, with a precision of 0.1 nm. One of the lenses was mounted on a piezoelectric tube, capable of normal and lateral motion. The lenses were mounted across orthogonal leaf springs of known spring constants (shear spring kL = 388 or 441 ± 4 N m−1 and normal spring kN = 141 ± 3 or kN = 2670 ± 84 N m−1), such that any induced deflection of the springs by the piezoelectric tube could be used to extract the normal interaction force FN or the lateral force FL.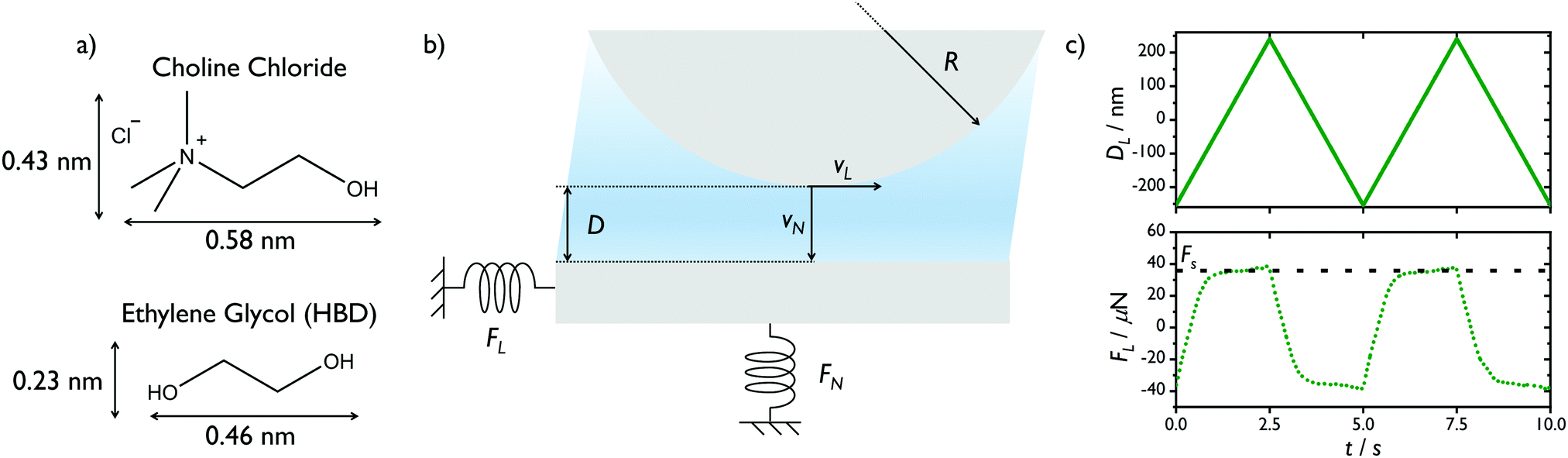 | ||
| Fig. 1 (a) Molecular structure and approximate dimensions13 of DES components choline chloride and ethylene glycol. The chloride ion diameter is approximately 0.36 nm.37 (b) Schematic of the surface force balance, showing the film thickness D, lens radius R, directions of normal and lateral velocity vN and vL and their corresponding force FN and FL. (c) Top panel: Applied lateral displacement DL during two shear cycles at 203 nm s−1. Bottom panel: Example of measured lateral friction FL during shear cycles. The horizontal line indicates the regime of continuous sliding which occurs at FS. | ||
For further information, the surface force balance technique is discussed in detail elsewhere.24,38
2.2 Materials
Mica (ruby muscovite) was of optical grade (S&J Trading). Mica facets of constant thickness (identified by their single interference colour) were attached to mica substrate and coated with an approximately 40 nm layer silver by evaporating silver shot (99.9999%, Alfa Aesar) using an HHV Auto306 (HVV Ltd) thermal evaporator. EPON 1004 (Shell) was used to attach mica facets to fused silica cylindrical lenses.Anhydrous ethylene glycol (Acros, 99.8%) and choline chloride (Sigma Aldrich, ≥99%) were used as received. Ethaline was prepared by mixing choline chloride and ethylene glycol in a 1:2 molar ratio. This mixture was subsequently maintained under a nitrogen atmosphere and stirred overnight at 60 °C until a homogeneous liquid was obtained. Sample mixtures were subsequently stored under nitrogen until use. Fresh samples were always prepared for use on the following day. Water content of an exemplary ethaline mixture prepared in this way was verified via coulometric Karl Fischer titration (Mettler Toledo) in an argon-filled glovebox, yielding a water content of 19.7 ppm.
All mica cleaving, sample preparation and SFB assembly were carried out in a laminar flow hood (Bassaire P3HF) to maintain dust free conditions, equivalent to ISO 5 class cleanroom environment.
2.3 Experimental procedure
Mica-coated lenses were prepared and mounted in the SFB chamber. Prior to sample injection, the thickness of the mica substrates was verified via a contact calibration in air. After retracting the lenses the sample liquid was then injected between the SFB lenses, and the chamber was sealed and purged with nitrogen. An open dish of fresh phosphorous pentoxide powder was also placed in the chamber as a desiccant to maintain dry conditions throughout the measurement. In order to study the role of water in the surface structure and lubrication properties of the confined deep eutectic solvent, water content was controlled by adding water to samples prepared as outlined above, to obtain samples of known water content before injecting the liquid between the lenses.In a typical measurement mica surfaces are brought together by moving one lens towards the other at fixed rate. At long distance the measured separation decreases linearly. At shorter distances surface forces become measureable and the separation as a function of time deviates from the linear baseline established at long distances. This deviation is then used to extract the normal force. We note that recent work38 has shown that viscous drainage of a confined fluid can result in a velocity-dependent hydrodynamic force that can be confused with any long-ranged static surface forces. We confirm that hydrodynamics are not modifying the measured surface force by performing measurements in the regime where the long-range forces have no dependence on approach velocity (typically ∼ 0.5 nm s−1).
We also perform measurements where the mica surfaces are sheared at fixed rate with a triangle wave function, while simultaneously decreasing their separation (Fig. 1c, top panel). The friction measurements reported here were performed at shearing velocity vs = 305 nm s−1, but were reproducible in the range 101–404 nm s−1. During these shearing experiments deflection of the shear springs are measured via an air-gap capacitance probe, allowing the lateral force FL to be determined as a function of time. As the mica layers confine the liquid to a few molecular layers, a measureable stick-slip friction force is measured and we define the kinetic friction force FS as the lateral force in the regime of continuous sliding (indicated by the dotted line in Fig. 1c, bottom panel). Shear velocities are deliberately much faster than the normal approach velocity, such that a complete shear cycle can be completed at a near constant normal force. We also note that the normal force profiles obtained with and without shear are essentially identical, confirming that shear is not inducing any change in the structure of the surface layers.
3 Results and discussion
3.1 Normal force measurements of ethaline
In this section we proceed to describe the interfacial structure of ethaline under dry, ambient and hydrated conditions. We present measurements of the normal force, and use molecular size arguments to suggest the arrangement of the confined layers in each system. | ||
| Fig. 2 Force profiles for (a) fresh dry and (b) 24 hour equilibrated ethaline. Triangles indicated retractions from adhesive minima. In both instances the inset shows a log-linear representation of the same data, and the dotted line gives an exponential fit to the long-range force. Radius of curvature 12 mm, mica thickness 3280 nm. Inset cartoons show possible structure, where green cations are choline (with grey hydroxyl groups), yellow anions are chloride, blue ovals are ethylene glycol and the dashed box indicates one detected layer. In (a) the mica is only weakly charged, yielding the so-called “checkerboard” layering. In (b) the magnitude of the charge is greater, resulting in the formation of a choline Stern layer and repelling chloride from the surface layers, resulting in a local excess of ethylene glycol in the form of a HBD layer (smaller dashed box). | ||
At separations smaller than 4 nm, a series of repulsive maxima and attractive minima emerge, which increase in magnitude as the surfaces approach contact. These maxima and minima are measured separately due to the spring-deflection method for detecting force. When dFN/dD ≥ kN, there is a spring instability and the surfaces “jump in” to a smaller separation. Similarly individual attractive minima are measured on retraction of the surfaces, which then “jump out” to long distances, the range of which can be used to determine the attractive minima strength.44 The oscillatory near-surface equilibrium interaction profile is commonly termed a “structural force”, and is the result of sequential layers of molecules being “squeezed out” from between the mica surfaces. The periodicity and decaying amplitude of the structural force can be interpreted to yield information on the molecular arrangement in the liquid film and has been widely studied in a range of solvents, including alkanes45 and ionic liquids.34
Different approaches have been used to define the layer thickness in confined liquid systems. One approach is to measure the jump in positions from consecutive layers and fit a trendline to define the layer thickness. Recently it was shown38,46 that this approach fails to take into account compression of the mica substrates, which is convolved with the layer compression and becomes increasingly pronounced at large normal force. It was also shown that fitting a layer thickness from minima rather than the maxima does not have this issue, as there is no mica compression for negative normal force. An alternative approach is to define the layer thickness as the jump-in distance (supposing an unchanged mica compression during the jump), which yields the same layer thickness as the minima fit. Gooneie et al.47 extended this approach by estimating the mica contribution to the layer compression to determine the degree of molecular compression within the film between jumps in. For a DES this compression could be due to a change in molecular order, as for a normal ionic liquid, or through a change in composition in the gap e.g. exclusion or concentration of the HBD. Therefore, we choose to define the step thickness as the jump-in distance, because the assumption that every layer is the same thickness or compresses by the same amount (required to fit a layer thickness as outlined above) might not be true for the multi-component system of a deep eutectic solvent. For instances where the layer thickness appears to be constant, we obtain good agreement between step sizes obtained from the jump-in distance and a fit of the minima.
From the structural force we are able to discern steps of thickness 0.45 ± 0.03 nm. At least three steps are present and are evidenced in the approach curve and in points which indicate the jump out force and distance on retraction. We note that the absolute magnitude of the squeeze-out force showed some variation between experiments of order 50%, which we attribute to small differences in DES composition or degree of mica ionisation, however the layer separations are highly reproducible. Based on dimensional arguments, we expect these steps to correspond to choline ions oriented approximately parallel to the mica surfaces, or ethylene glycol–chloride ion complexes. The orientation of the complexes is less clear due to possible variations in conformation of the ethylene glycol around the chloride. Chloride ions are too small to individually account for the measured step thickness, while surface force measurements of pure ethylene glycol yield steps of thicknesses ≤0.4 nm.13,48 For ionic liquids the measured interfacial structure at charged interfaces has been attributed to alternating layers of anions and cations.49,50 A measured step thus corresponds to the simultaneous squeeze-out of an anionic layer and a cationic layer, to maintain electroneutrality in the gap and to prevent adjacent layers being of like-charge. This argument rests on step thicknesses measured with SFB and AFM that are too large to solely correspond to one charged species, but can accommodate one layer of each species.34 For the measurements reported here the steps are too thin to be due to simultaneous squeeze-out of two oppositely charged layers. Instead, it appears that each layer is of mixed charge, such that only a single molecular layer needs to be squeezed out to maintain electroneutrality in the SFB gap (as opposed to the simultaneous squeeze-out of a cationic layer and an anionic layer). This hypothesised structure is illustrated in Fig. 2a, bottom right. This behaviour has been reported before in so-called mixed-layer “checkerboard” arrangements of ionic liquid cations and anions at interfaces.51–53 This hypothesis is supported by recent simulations of the DES reline. Mamme et al.54 used molecular dynamics to simulate the structure of reline at neutral and charged interfaces. They found that at negative and positive potentials a Stern layer was present at the interface of choline cations or chloride anions respectively, but beyond this innermost layer all of the other layers were of mixed composition, albeit with some variation in charge density. Intriguingly they also noted that the hydrogen bond donor urea was present in all layers and also had a surface excess for both negative and positive surface potentials. Mamme et al.55 also investigated water distribution in reline, which was shown to have a surface excess with applied potential. This supports the mixed-layer hypothesis reported here, although some charge oscillation was reported. From our measurements it is not possible to determine the ion ratios of the squeezed out layers, but we infer that they are electroneutral based on geometrical arguments. However, some charge oscillation in the equilibrium structure could be accommodated during an SFB measurement, whereby electroneutrality in the gap could be conserved by inter-layer exchange of charged species with each other or with smaller, uncharged HBDs (which are present in all layers) during layer squeeze-out. This is supported by recent simulations of shape-asymmetric ionic liquids by Merlet et al.56 Mixed composition layers have previously been reported for ionic liquids at uncharged interfaces,57 but Merlet et al. showed highly mixed first- and second-layers even with an applied surface potential, and reported “spontaneous exchange of ions between different layers of the electrolyte close to the electrode surface”. They attributed this behaviour to voltage-dependent in-plane ordering of the ionic species.
For ethaline that has been allowed to equilibrate with the mica substrates overnight in a dry nitrogen atmosphere (2b) we make two key observations. Firstly, the long range repulsion is still present and of comparable range to the fresh, dry sample. Secondly, we note that the surface layering has changed, and now in addition to the 0.45 nm steps there is also a 0.23 ± 0.03 nm step adjacent to the innermost surface layer. The only component in the DES with a spatial dimension small enough to correspond to this step is the HBD ethylene glycol, which is of comparable thickness. We therefore propose that the 0.23 nm step corresponds to ethylene glycol lying parallel to the mica surface.
We also note that for dry ethaline the final (immovable) layer thickness is ∼0.8 ± 0.05 nm after the final jump-in, while for equilibrated ethaline (for the same normal force) it is consistently slightly larger (∼0.9 ± 0.05 nm). Although absolute separation in any surface force balance measurement is susceptible to errors induced by misalignment of the optical path, or from flattening of the mica surfaces with increasing normal force,31,46 we note that this supports the idea that the interfacial structure is somehow changing as the system equilibrates.
We now speculate on the origin of the structural transition observed in the equilibrated sample. Mica substrates are well known to develop a negative surface charge through dissolution of potassium cations,16 and the DES structure for the equilibrated sample (Fig. 2b) displays layering with a HBD-enriched near-surface layer consistent with the layering observed with AFM14 for an applied negative potential. However, initially this is not the case and no HBD layer is observed. We therefore propose two mechanisms for the formation of the HBD layer in the equilibrated sample. Firstly, the equilibration time for the surface potential of mica is approximately 1 hour in ultrapure water.23 It is likely that the equilibration process is slower in the concentrated electrolyte environment of a deep eutectic solvent, so it may take several hours of equilibrating at large separations for the mica to become sufficiently charged for the surface-choline-HBD structure to form. As the magnitude of the surface charge increases more choline molecules will move to the surface to counter the negative surface charge, and are likely to orient at least somewhat perpendicularly to the negatively charged mica surface, with the choline charged group oriented towards the mica and the hydroxyl group towards the bulk liquid. In doing so they will exclude ethylene glycol which was previously at the surface, forming a HBD-rich layer adjacent to the choline Stern layer. This is illustrated in Fig. 2b, bottom right. One might expect the ethylene glycol layer to lie parallel to the mica surface to maximise hydrogen bonding between their hydroxyl groups and the exposed hydroxyl groups of the interfacial (perpendicularly oriented) choline, which will be of higher density than the parallel-oriented choline of the next molecular layer. Secondly, although the SFB chamber is maintained under nitrogen atmosphere with a phosphorous pentoxide dessicant, it is likely that some atmospheric humidity is eventually adsorbed by the hygroscopic DES sample. It is hard to verify if and by how much the water content is changing over the course of a measurement – the volume of liquid held between the mica surfaces (typically ∼ 20 μl) is much too little for Karl Fischer titration. Nevertheless, water is known to change the surface structure of ionic liquids measured with the SFB29,58–60 and so may also play a role in this process as well, either through enhancing the solvation of the surface potassium ions as outlined above, or by changing the inter-molecular interactions such as via enhanced hydrogen bonding. Hammond et al.12 note that small amounts of added water enhance the DES structure via strengthening the hydrogen bond network, rather than modifying it to a different structure, and we set about making a comparable measurement here.
In order to test the effect of small quantities of water in the DES, we take a dried sample and allow it to equilibrate with the ambient environment for several hours. Karl Fischer titration was performed on an exemplary sample, yielding a water content of 3800 ppm, comparable to values reported for undried DESs.14 We then perform measurements in the sealed SFB chamber, recording force profiles at different times after exposing the surfaces to the air-equilibrated liquid.
We immediately note that for this “ambient” sample, the surface layering is notably stronger with identical 0.45 nm ± 0.05 step size to the dry case (Fig. 3, 1 hour). This supports the hypothesis that small amounts of water enhance the hydrogen bond network in the liquid, which one would expect to increase the squeeze out force. However, the stronger molecular layering in the humid sample could also be due to enhancement of the mica surface charge via increased dissociation of the potassium ions by absorbed water. A similar dramatic enhancement of the squeeze out force by ambient water has also been reported in ionic liquids.59,60 We find that this structure is robust over several hours and many repeat measurements. We also allow the liquid film to equilibrate overnight at large separation and observe an additional structural transition (Fig. 3, 24 hours). It appears that the inner two steps are squeezed out simultaneously at a force between the previous two values, and the thickness of this intermediate step is 0.72 ± 0.05 nm. We also find more pronounced steps of thickness ∼ 0.7 nm at longer distances. We note that the longest single molecular component in ethaline is choline, of length ∼ 0.58 nm, therefore the layer thickness must no longer correspond to a single molecular layer. We propose that for sufficiently long equilibration times and sufficient adsorbed water, the mica surfaces are able to become charged enough to separate the counter-ions and co-ions into distinct layers, comparable to the oscillatory charge density structure of a conventional room temperature ionic liquid at a charged mica surface (shown schematically in Fig. 3). For alternating layers of anions and cations, a measured step must be of a cationic layer and an anionic layer to conserve electroneutrality. It is easy to see (Fig. 1a) that a layer of choline molecules oriented parallel to the mica surface, and a mixed layer of ethylene glycol (also oriented parallel to the mica surface) and chloride ions would have a thickness in the range 0.66–0.79 nm, so this is consistent with our hypothesis.
 | ||
| Fig. 3 Force profiles for ambient ethaline at two times: shortly after injecting the liquid (1 hour, light green) and after equilibration overnight (24 hours, dark green). The 1 hour profile is representative of runs performed over the first ∼10 hours of measurement. The 24 hours profile was also repeatable over ∼10 hours, which implies some sort of threshold condition was achieved (e.g. sufficient surface charge) between the force profiles rather than a continuous equilibration process. Cartoons are shown to the right illustrating the possible molecular structures in the circled positions in the force profiles. Dotted boxes in the cartoons indicate the squeezed-out layer detected immediately to the left of the circled points. Radius of curvature 8 mm, mica thickness 3153 nm. | ||
We also note that the layers appear to be highly compressible before finally reaching an instability and squeezing out a layer of molecules. This is consistent with the liquid-phase behaviour of a dilute monolayer, which can undergo compression before squeeze-out, such as through reorganisation of the layer, or by excluding other molecular species – in this instance likely through the exclusion of the uncharged species (ethylene glycol or water).
We therefore make three statements: firstly, that the humid sample is inherently more structured than the dry sample, which at least in part appears to be through internal hydrogen bonding throughout the liquid. Secondly, as for comparable measurements in ionic liquid systems,59 trace water enhances the surface charge of the mica by increasing the dissociation of potassium ions from the mica surface. This increase in surface charge is sufficient to rearrange the surface structure of the DESs from a mixed “checkerboard” structure to discrete layers of cations and anions that must be squeezed out simultaneously to maintain electro-neutrality in the gap. Thirdly, uncharged species (water and ethylene glycol) can be squeezed out continuously at the forces applied between steps.
The normal force profiles obtained for ethaline with 30 wt% and 50 wt% water are shown in Fig. 4. In both instances we note three significant differences when compared to dry and humid ethaline. At separations greater than 5 nm, a weak, long range repulsive force is measured, the range of which decreases with increasing water content. As in the previous section, we can fit this exponential decay to obtain λs. For 30 wt% water we obtain a λs of 6 ± 1 nm, and for 50 wt% water we obtain a λs of 4 ± 1 nm (insets, Fig. 4a and b respectively). For conventional Debye–Hückel screening the opposite trend would be expected, so these measurements also appear to display underscreening-like behaviour. We expect underscreening to become significant where (a/λD) > 1,43 a condition these state points fulfil so attributing our observation of decreasing screening length with increasing water content to underscreening is appropriate.
 | ||
| Fig. 4 Force profiles (dark green) for ethaline with (a) 30 wt% and (b) 50 wt% water continuous retractions (light greens) from different layers are shown to indicate adhesive minima. In both instances the inset shows a log-linear representation of a comparable measurement at the same concentration, and the dotted line gives an exponential fit to the long-range force. Radius of curvature (a) 11 mm (inset 12 mm) and (b) 8 mm (inset 10 mm). Mica thickness (a) 2388 nm (inset 5368 nm) (b) 2381 nm (inset 8032 nm). Inset cartoon in (b) shows possible structure of both hydrated films, following colouring convention of Fig. 2. Added water increases the magnitude of the mica surface charge and modifies the interactions between the DES components, resulting in alternating charged layers, such that a squeezed-out layer (shown in the dashed box) includes an anionic and a cationic layer to maintain electroneutrality in the film. Step thicknesses imply a similar structure for 30 wt% and 50 wt% water, but the greater persistence of layers for 30 wt% suggests that for 50 wt% the extra water molecules act to disrupt the DES structure further from the mica surface. | ||
At separations smaller than 5 nm a structural force emerges for both 30 wt% and 50 wt% water. We observe a similar structure with at least 3 molecular layers present and of comparable thickness for both concentrations. We measure outer step thicknesses of 0.74 ± 0.08 nm (0.82 ± 0.03 nm from jump outs) and 0.80 ± 0.08 nm (0.82 ± 0.02 nm from jump outs) for 30 wt% and 50 wt% water respectively. As for Fig. 3, the measured step thicknesses for both concentrations are too large to solely be due to a single molecular species, and instead appear to correspond to a pair of oppositely charged molecular layers. This is illustrated in Fig. 4b, bottom right. However, for the innermost measured layer we obtain a smaller step thickness of 0.50 ± 0.10 nm and 0.55 ± 0.10 nm for 30 wt% and 50 wt% water respectively. We also note that the squeeze-out force for the innermost layer is rather larger than for pure ethaline, such that some compression of the mica is necessary (resulting in an underestimation of the film thickness at closest approach, particularly for the thicker mica used for 50 wt% water) before the surfaces jump together. The adhesive minima are also notably stronger than for dry ethaline, with innermost layer adhesions of 16–20 mN m−1 for both 30 wt% and 50 wt% water. Previous reports of the adhesion of mica in pure water give a value of 15 ± 3 mN m−1,23 in good agreement with the values reported here. The observation of mica adhesion comparable to values reported for pure water, combined with the final step thickness of 0.55 ± 0.10 nm, is consistent with the picture of a Stern layer of perpendicularly-oriented choline ions being excluded from the film as e.g. the surface charge is regulated at such close confinement, leaving only a surface film of water ions in the gap. The enhancement of interfacial structure in ethaline by added water has been reported previously, but we also note some differences between our data and previous AFM measurements.14 At OCP two or three pronounced steps of thickness ∼ 1 nm were measured, but at negative applied potentials they reported that the layers in the interfacial structure measured at OCP “almost disappear when a potential is applied and are replaced with a short-range exponential decay”.
How can we consolidate these results? We note that the confined layers display a compression of around 0.1–0.3 nm before they are squeezed out. AFM approach speeds are typically at least an order of magnitude quicker than an equivalent SFB measurement,15 so are less sensitive to any slow rearrangements in the confined film and so could also result in a larger layer thickness. The interfacial structure of an electrolyte is intimately linked to the roughness, crystallinity and electrical (i.e., conductor or dielectric) properties of the surface.49,57 Furthermore, the local electrolytic environment experienced by an AFM tip can more rapidly change when a potential is applied (through flux of counter- and co-ions to and from the bulk14), such that an AFM measurement is not necessarily representative of the native interfacial structure. On the other hand a surface force balance measurement has a contact area several orders of magnitude larger, and so is both more representative of the native interfacial structure and less prone to short-time diffusive processes altering the apparent structure. We also note that differences in the interfacial structure of ionic liquids, measured with AFM and SFB, have been reported within the same manuscript.59
3.2 Frictional response
We now present measurements of the shear response of confined dry and wet ethaline films which we measure as a function of varying load.:1 and 2:1 ionic liquids.28,60,61 For dry ethaline the coefficient of friction of the confined molecular layers is superlubric (μ < 0.01) at all measured loads. One can also obtain a value for the contact pressure σc during the friction measurement using the Johnson–Kendall–Roberts (JKR) model for contact mechanics, which provides an estimate of the contact area for modest (<4 mN m−1) normal force.24 We note that this force is not uniform over the contact region and becomes less appropriate at larger normal force as the surfaces become more deformed,46 therefore we only report the contact pressure for the first (outermost) solid-like layer in the shear force for each sample. For the dry case we find σc = 23 ± 5 kPa. Although the contact pressure is comparable to the fresh sample (σc = 25 ± 5 kPa), after equilibration overnight the frictional response changes considerably. We noted earlier (Fig. 2b) that equilibration leads to the formation of a HBD-rich layer adjacent to a tightly-bound Stern layer. In the outermost measured molecular layers the coefficient of friction is still superlubric, but is remarkably different for the HBD and innermost layer. For the HBD layer the coefficient of friction increases markedly to 0.03 ± 0.01, and for the innermost layer it increases further to 0.12 ± 0.02. These values are comparable to those obtained previously for ionic liquids,28,60,61 but represents a change of more than a factor of 20 for the innermost layer compared to the dry case. These values compare favourably to bulk measurements of the friction coefficient of ethaline reported previously, which vary between 0.0220 and 0.621 depending on method, material and surface roughness. We propose that the improved performance reported here is due to the increased flatness of mica when compared to e.g. steel-steel contacts. While a rough surface has a reduced real contact area, the roughness also disrupts any long range ordered surface structure. By using atomically flat mica as a substrate, the surface layering can propagate over larger lengthscales, allowing continuous inter-layer sliding over a wide area. The data shown is representative of multiple repeats and we obtain a qualitatively similar quantised friction response during separate experiments, but the calculated friction coefficients can vary by up to factor of 2, which we attribute to small differences in composition (both ethylene glycol: choline chloride ratio and adsorbed water), crystallograpic misalignment of the mica substrates and variations in the precise contact geometry. A similar increase of friction coefficient with equilibration (but without structural transformation in the normal force) has been reported for dodecane45 and ionic liquid29 equilibrated against humid air. For dodecane the increase in friction coefficient with water content was attributed to the formation of insoluble crystalline salt bridges, which is unlikely to be the case here. On the other hand, the increase in friction coefficient for the ionic liquid was attributed to localisation of water molecules at the charged IL headgroups, and subsequent solidification of the film, which acts to increase the force required for adjacent layers to slide past each other. This hypothesis is more compatible with the DES system reported here. As water is accommodated in the nanostructure at the interface, it appears that either the active shear plane is changing, or that it is altered in such a way that sliding is resisted. Ashworth et al.62 reported many competing hydrogen-bond interactions in DESs – forming a so-called “alphabet soup”. Trace amounts of water can further enhance the hydrogen bond network in the DES,12 and so an increase in friction coefficient could be the result of enhanced inter-layer hydrogen bonding, which acts to resist sliding.
 | ||
| Fig. 5 Film thickness D and shear force FS as a function of normal force FN for (a) fresh dry and (b) 24 hour equilibrated ethaline. Coefficients of friction are shown for white-dotted quasi-linear regions. Radius of curvature 8 mm, mica thickness 3280 nm. | ||
 | ||
| Fig. 6 Film thickness D and shear force FS as a function of normal force FN for ethaline solutions with (a) 30 wt% and (b) 50 wt% water. Coefficients of friction are shown for white-dotted quasi-linear regions. Insets show measurements with a stiffer normal spring, which show the reproducibility of the frictional response for repeat measurements and at much greater normal force. Radius of curvature (a) 10 mm and (b) 8 mm. Mica thickness (a) 4261 nm and (b) 2381 nm. Insets – Radius of curvature (a) 12 mm and (b) 10 mm. Mica thickness (a) 2388 nm and (b) 8032 nm. | ||
4 Conclusions
In this paper, we have presented a detailed study of the normal and shear forces acting across nanoscale films of the deep eutectic solvent ethaline, confined between charge regulated atomically flat substrates. The liquid film produces a force profile characteristic of a structural force, with clear steps corresponding to the squeeze out of molecular layers on approach and adhesive minima on retraction. Intriguingly the layer thicknesses and strengths are strongly altered by both adsorbed and added water, significantly increasing the squeeze-out force.We also detect long-ranged repulsions consistent with ‘underscreening’, previously only measured in ionic liquid solutions and concentrated aqueous electrolytes. The lengthscale of the repulsion is maximal for dry ethaline, and decreases with added water, unlike for conventional Debye–Hückel screening.
The frictional response of the confined film was also measured as a function of normal force, yielding a quantised frictional response with a strong dependence on water content. Remarkably we report a re-entrant superlubric regime with water content, where dry ethaline and 50 wt% water display superlubric properties even at high compression, whereas humid ethaline and 30 wt% water have a quantised frictional response with non-superlubric friction coefficients comparable to conventional ILs. Friction coefficients compare favourably to previously reported values, and the lubrication provided even up to 50 wt% water supports the previous assertion that DESs could find use as marine lubricants.
This work highlights the interplay of interfacial charge and nanoscale structure, and the role of water in modifying the influence of both. At the nanoscale liquid structure is not homogeneous, and the amphiphilicity of molecules at the local level can have dramatic consequences for structure and dynamic properties, as recorded here for the lubrication of shearing surfaces at the nanoscale. Future work will explore other DESs, as well as the role of additives in their structural and frictional properties in confinement.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Charles Monroe and Andrew Wang for assistance with Karl Fischer titration measurements. Funding is gratefully acknowledged from The European Research Council (under Starting Grant No. 676861, LIQUISWITCH).Notes and references
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010–2011 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS.
- R. Li, Q. Chu and J. Liang, RSC Adv., 2015, 5, 44933–44942 RSC.
- H. Mou, J. Wang, D. Zhang, D. Yu, W. Chen, D. Wang and T. Mu, J. Mater. Chem. A, 2019, 7, 5719–5725 RSC.
- A. Rajawat, S. Khandelwal and M. Kumar, RSC Adv., 2014, 4, 5105–5112 RSC.
- A. E. Daz-Álvarez, J. Francos, B. Lastra-Barreira, P. Crochet and V. Cadierno, Chem. Commun., 2011, 47, 6208–6227 RSC.
- A. P. Abbott, P. M. Cullis, M. J. Gibson, R. C. Harris and E. Raven, Green Chem., 2007, 9, 868–872 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS.
- Z. Chen, B. McLean, M. Ludwig, R. Stefanovic, G. G. Warr, G. B. Webber, A. J. Page and R. Atkin, J. Phys. Chem. C, 2016, 120, 2225–2233 CrossRef CAS.
- O. S. Hammond, H. Li, C. Westermann, A. Y. Al-Murshedi, F. Endres, A. P. Abbott, G. G. Warr, K. J. Edler and R. Atkin, Nanoscale Horiz., 2019, 4, 158–168 RSC.
- Z. Chen, M. Ludwig, G. G. Warr and R. Atkin, J. Colloid Interface Sci., 2017, 494, 373–379 CrossRef CAS.
- R. Pashley and J. Israelachvili, Colloids Surf., 1981, 2, 169–187 CrossRef CAS.
- W. J. Bartz, Tribol. Int., 1998, 31, 35–47 CrossRef CAS.
- A. Syahir, N. Zulkifli, H. Masjuki, M. Kalam, A. Alabdulkarem, M. Gulzar, L. Khuong and M. Harith, J. Cleaner Prod., 2017, 168, 997–1016 CrossRef CAS.
- S. D. Lawes, S. V. Hainsworth, P. Blake, K. S. Ryder and A. P. Abbott, Tribol. Lett., 2010, 37, 103–110 CrossRef CAS.
- I. Garcia, S. Guerra, J. de Damborenea and A. Conde, Lubricants, 2019, 7, 37 CrossRef.
- A. P. Abbott, E. I. Ahmed, R. C. Harris and K. S. Ryder, Green Chem., 2014, 16, 4156–4161 RSC.
- E. I. Ahmed, A. P. Abbott and K. S. Ryder, AIP Conf. Proc., 2017, 1888, 020006 CrossRef.
- U. Raviv, P. Laurat and J. Klein, J. Chem. Phys., 2002, 116, 5167–5172 CrossRef CAS.
- J. Klein and E. Kumacheva, J. Chem. Phys., 1998, 108, 6996–7009 CrossRef CAS.
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572–6574 RSC.
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243–1247 RSC.
- F. Endres, N. Borisenko, S. Z. El Abedin, R. Hayes and R. Atkin, Faraday Discuss., 2012, 154, 221–233 RSC.
- O. Werzer, E. D. Cranston, G. G. Warr, R. Atkin and M. W. Rutland, Phys. Chem. Chem. Phys., 2012, 14, 5147–5152 RSC.
- A. M. Smith, M. A. Parkes and S. Perkin, J. Phys. Chem. Lett., 2014, 5, 4032–4037 CrossRef CAS.
- A. M. Smith, K. R. Lovelock, N. N. Gosvami, T. Welton and S. Perkin, Phys. Chem. Chem. Phys., 2013, 15, 15317–15320 RSC.
- R. Lhermerout and S. Perkin, Phys. Chem. Chem. Phys., 2020, 22, 455–466 RSC.
- J. Sweeney, F. Hausen, R. Hayes, G. B. Webber, F. Endres, M. W. Rutland, R. Bennewitz and R. Atkin, Phys. Rev. Lett., 2012, 109, 155502 CrossRef.
- H. Li, R. J. Wood, M. W. Rutland and R. Atkin, Chem. Commun., 2014, 50, 4368–4370 RSC.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- O. Y. Fajardo, F. Bresme, A. A. Kornyshev and M. Urbakh, J. Phys. Chem. Lett., 2015, 6, 3998–4004 CrossRef CAS.
- R. Lhermerout, C. Diederichs and S. Perkin, Lubricants, 2018, 6, 9 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Lhermerout and S. Perkin, Phys. Rev. Fluids, 2018, 3, 014201 CrossRef.
- A. Pandey, R. Rai, M. Pal and S. Pandey, Phys. Chem. Chem. Phys., 2014, 16, 1559–1568 RSC.
- A. R. Harifi-Mood and R. Buchner, J. Mol. Liq., 2017, 225, 689–695 CrossRef CAS.
- A. M. Smith, A. A. Lee and S. Perkin, J. Phys. Chem. Lett., 2016, 7, 2157–2163 CrossRef CAS.
- A. A. Lee, C. S. Perez-Martinez, A. M. Smith and S. Perkin, Faraday Discuss., 2017, 199, 239–259 RSC.
- A. A. Lee, C. Perez-Martinez, A. M. Smith and S. Perkin, Phys. Rev. Lett., 2017, 119, 026002 CrossRef.
- J. Israelachvili, Y. Min, M. Akbulut, A. Alig, G. Carver, W. Greene, K. Kristiansen, E. Meyer, N. Pesika and K. Rosenberg, et al. , Rep. Prog. Phys., 2010, 73, 036601 CrossRef.
- A. M. Smith, J. E. Hallett and S. Perkin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 25418–25423 CrossRef CAS.
- R. Lhermerout and S. Perkin, 2020, arXiv preprint arXiv:2001.01090.
- A. Gooneie, T. E. Balmer and M. Heuberger, Phys. Rev. Res., 2020, 2, 022026 CrossRef CAS.
- H. K. Christenson and R. G. Horn, J. Colloid Interface Sci., 1985, 103, 50–55 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS.
- C. Merlet, C. Péan, B. Rotenberg, P. A. Madden, P. Simon and M. Salanne, J. Phys. Chem. Lett., 2013, 4, 264–268 CrossRef CAS.
- T. Cremer, M. Stark, A. Deyko, H.-P. Steinrück and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS.
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. Lett., 2014, 5, 179–184 CrossRef CAS.
- K. Tomita, M. Mizukami, S. Nakano, N. Ohta, N. Yagi and K. Kurihara, Phys. Chem. Chem. Phys., 2018, 20, 13714–13721 RSC.
- M. H. Mamme, S. L. Moors, H. Terryn, J. Deconinck, J. Ustarroz and F. De Proft, J. Phys. Chem. Lett., 2018, 9, 6296–6304 CrossRef CAS.
- M. H. Mamme, S. L. Moors, E. A. Mernissi Cherigui, H. Terryn, J. Deconinck, J. Ustarroz and F. De Proft, Nanoscale Adv., 2019, 1, 2847–2856 RSC.
- C. Merlet, D. T. Limmer, M. Salanne, R. Van Roij, P. A. Madden, D. Chandler and B. Rotenberg, J. Phys. Chem. C, 2014, 118, 18291–18298 CrossRef CAS.
- H. Zhou, M. Rouha, G. Feng, S. S. Lee, H. Docherty, P. Fenter, P. T. Cummings, P. F. Fulvio, S. Dai, J. McDonough, V. Presser and Y. Gogotsi, ACS Nano, 2012, 6, 9818–9827 CrossRef CAS.
- A. M. Smith, K. R. Lovelock and S. Perkin, Faraday Discuss., 2013, 167, 279–292 RSC.
- H.-W. Cheng, J.-N. Dienemann, P. Stock, C. Merola, Y.-J. Chen and M. Valtiner, Sci. Rep., 2016, 6, 30058 CrossRef CAS.
- C. S. Perez-Martinez and S. Perkin, Langmuir, 2019, 35, 15444–15450 CrossRef CAS.
- A. M. Smith, K. R. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, J. Phys. Chem. Lett., 2013, 4, 378–382 CrossRef CAS.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- Faraday Discuss., 2013, 167, 347–370, (General Discussion) Search PubMed.
| This journal is © the Owner Societies 2020 |