
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe phenoxyl group-modulated interplay of cation–π and σ-type interactions in the alkali metal series†
Giacomo
Prampolini
 *a,
Marco
d'Ischia
b and
Alessandro
Ferretti
*a
*a,
Marco
d'Ischia
b and
Alessandro
Ferretti
*a
aIstituto di Chimica dei Composti OrganoMetallici (ICCOM-CNR), Area della Ricerca, via G. Moruzzi 1, I-56124 Pisa, Italy. E-mail: giacomo.prampolini@pi.iccom.cnr.it; alessandro.ferretti@pi.iccom.cnr.it
bDipartimento di Scienze Chimiche, Universitá di Napoli Federico II, I-80126 Napoli, Italy
First published on 30th October 2020
Abstract
The interaction potential energy surfaces (IPESs) of four alkaline metal cations (Na+, K+, Rb+ and Cs+) complexed with phenol and catechol were explored by accurate ab initio calculations to investigate the interplay of different noncovalent interactions and their behavior along the alkali metal series and upon –OH substitution. Selected one-dimensional interaction energy curves revealed two different minimum energy configurations for all phenol– and catechol–metal complexes, characterized either by cation–π or σ-type interactions. For each investigated complex several two-dimensional IPES maps were also computed, exploiting the computational advantages of the MP2mod approach. The size of the alkali cation was found to play a similar role in modulating both kinds of complexes, as the interaction strength always decreases along the metal series, from Na+ to Cs+. Conversely, the number of hydroxyl substituents markedly affected cation–π complexes vs. σ-type ones. As a most relevant finding, in catechol–metal complexes the strength of cation–π interactions is around half that of the σ-type ones. It is argued that the combined effect of cation dimensions and hydroxyl substitution in catechol–Na+ complexes makes σ-type configurations remarkably more stable and easily accessible than cation–π ones. Besides shedding new light on the origin of biological phenomena connected with underwater adhesion, the quantum mechanical interaction energy database provided herein may offer a useful reference for tuning accurate force fields, suitable for molecular dynamics simulations, where environmental effects might be also taken into account.
1 Introduction
Since the seminal paper of Feynman1 concerning the pure electrostatic nature of intermolecular forces, noncovalent interactions (NCI)2,3 have attracted massive attention in many different fields, because of the crucial role they play in medicine, biological processes, energy storage, supramolecular chemistry and advanced materials, to cite a few. Notwithstanding their origin is electrostatic, it is a common practice to classify the different kinds of NCIs, depending on their strength, involved species, behavior in water, etc., thus leading to the definition, for instance, of hydrophobic interactions,4 hydrogen bonds,5 π–π interactions,6 halogen bonds,7 and anion–π8 or cation–π interactions.9 Limiting the discussion to the latter kind, since their early definition by Dougherty and co-workers,9–11 the importance of cation–π interactions is nowadays well established and their role is relevant to many research topics and applications, ranging from biology12–14 to materials science.15–24 In most of the aforementioned cases, however, cation–π interactions may compete with other NCIs, and the resulting biological functions or material properties are often the result of a delicate balance, settled among all contributions.8 Very recent examples of such a complex interplay can be for instance found in the ion-driven assembly of biological catecholamines,14 or in the competition between cation–π interactions and hydrogen bonding in mussel-inspired adhesion.22In this framework, computational approaches and modeling can prove themselves to be very useful, due to their intrinsic capability to unravel the different contributions to the total energy of the system. On the one hand, given the large dimensions of both systems of biological interest and advanced materials, computational feasibility requires to resort to techniques rooted in classical physics and statistics, as molecular dynamics or MonteCarlo techniques,25,26 whose reliability is based in turn on the force-field (FF) employed to describe the investigated targets. Unfortunately, as far as cation–π interactions are concerned, the reliability of standard FFs has been often questioned, although FF's refinements with more complex models have recently been suggested.27–29 On the other hand, notwithstanding their application is generally limited by the system size, quantum mechanical methods are able to accurately distinguish among the different kinds of NCIs, and help to unveil the separate role of each contribution, at least for model systems of reduced complexity. Indeed, the long-lasting effort in accurately computing the cation–π interaction energy in benzene prototype complexes with alkali metal ions (M+),30–36 accompanied by its experimental validation through gas-phase collision induced dissociation (CID) measures,32,37–41 has allowed for a deeper insight into cation–π interaction patterns, revealing key features that are able to clarify the behavior of more complex systems.
From a biological point of view, benzene–M+ complexes can in fact be considered as the simplest model for the phenylalanine–M+ pair, where cation–π interactions are expected to be the primary source of attraction. Yet, if other biological functions have to be considered, model systems more complex then benzene–M+ should be taken into account. Phenol–M+ and catechol–M+ complexes would be in this sense the perfect candidates for mimicking the behavior of e.g. tyrosine or dihydroxy–phenylalanine (DOPA) in the presence of ions, but the picture remarkably changes with respect to benzene complexes. As pointed out in the extensive work of Vaden and Lisy,42–44 the hydroxyl substitution significantly influences the interaction patterns of the phenol–M+ complexes with respect to the ones found for benzene. On the same foot, in a subsequent study, Wheeler's group reported not negligible effects triggered by substitution on π–π interactions involving two aromatic rings.45 In both cases, the lone pair of the oxygen atoms within the hydroxyl substituents can directly interact with the other species (either the alkali ion42 or the neighboring π cloud45), giving rise to another kind of NCI that can in principle compete and modulate the principal source of interactions. More specifically, concerning the phenol–M+ complexes, several studies28,33,42,43,46–48 agree in indicating that the competition between cation–π and these cation-lone pair or σ-type42 interactions is pivotal in governing the behavior of complexes between aromatic moieties with hydroxyl substituents and alkali metal cations. According to Vaden and Lisy,42 the cation–π interactions are dominant in the popular en face or “top” conformations (i.e. when the ion is found along the ring Ĉ6 axis), where benzene and phenol show a similar behavior,42,46–48 whereas σ-type interactions, settled between the ion and the oxygen lone pair, become decisive in alternative geometries,42,46–48 where the ion interacts more closely with the hydroxyl group. When considering subsequent OH substitutions, along with its p- and m-isomers hydroquinone and resorcinol, catechol occupies a central position, but only few studies concerning its interaction with ions have been reported up to our knowledge in the literature, notwithstanding its relevance in cutting edge topics related to melanin research, including metal binding, antioxidant defense, adhesion and polymer buildup.19–24,49,50 In fact, except a few studies concerning the interactions of indole-containing moieties with specific cations,47,51–53 a systematic and reliable comparison of the competition between cation–π and σ-type interactions in catechol–M+ complexes is still missing.
Very recently,36 we have benchmarked the performances of a number of quantum mechanical methods, based both on wave-function (WF) approaches and density functional theory (DFT), in delivering accurate estimates of the cation–π interaction energy of benzene–M+, phenol–M+ and catechol–M+ complexes (M = Na+, K+, Rb+ and Cs+). On average, the best results were obtained with the WF based MP2mod approach,36 which delivered, at a lower computational cost among all tested methods, the best average standard deviation of about 5 kJ mol−1 with respect to highly accurate Coupled Cluster (CC) calculations, purposely carried out with single, double, and perturbatively included connected triple excitations (CCSD(T)), on several geometries of all investigated cation–π complexes. The better accuracy of MP2mod predictions with respect to both DFT benchmarked functionals and standard MP2 techniques54 relies on specifically modified basis sets, where the exponents of the polarization functions on each atom of the aromatic moiety are tuned to reproduce the aromatic homo-dimer interaction energies of a reference CCSD(T) training set,55–58 whereas for metal cations a standard def2-TZVPPD36,59 is employed. Moreover, MP2mod computational feasibility allowed us to compute several two-dimensional cross-sections of the cation-aromatic species interaction energy surface (IPES), revealing interesting features occurring both along the phenol–catechol and the alkali metal series. Such a database consisted of hundreds of benzene–M+, phenol–M+ and catechol–M+ interaction energies, computed at the MP2mod level on geometries obtained by displacing the metal cation on top of the carbon skeleton, i.e. on a plane parallel to the aromatic ring, to maximize the interaction of the cation with the π cloud. Besides the expected “en face” configuration, new stable arrangements appeared when the cation is displaced toward the oxygen atoms in the substituted species. In some cases, and in particular for catechol and the smaller cations, these attractive regions appeared even deeper than the standard “en face” ones, which phenol and catechol share with benzene.
In the present work, the insurgence upon hydroxyl substitution of alternative configurations of a stable complex between aromatic species and alkali cations (Na+, K+, Rb+ and Cs+) will be further investigated, extending the MP2mod sampling to other regions of the configurational space. In fact, the previous results strongly suggest that possible σ-type interactions between the alkali metal and the hydroxyl substituents in phenol and, most likely, in catechol, might lead to rather stable dimer arrangements, which might compete with standard cation–π ones, eventually affecting the intricate interplay among NCI in larger systems. Indeed, the major aim of this paper is to reliably explore the large portions of the IPESs of all investigated pairs, thus allowing a sound and unbiased comparison among all involved species, which could help in the rationalization of both the consequences of hydroxyl substitution and the role of cation dimensions on complex stability. The paper is organized as follows. In the next section all the computational details are reported, together with a description of the IPES regions that are going to be sampled at the MP2mod level. The discussion of the results is organized into three parts: first, mono-dimensional interaction energy curves are presented for all investigated complexes, to ascertain the position and the stability of possible minima in the proximity of the hydroxyl substituents; next, the interplay between intermolecular forces and the rotation of the hydroxyl groups around the C–O bond is analyzed in terms of two-dimensional energy maps; in the last subsection, the cation-aromatic moiety IPES in samples along the whole configurational space, and the interplay between cation–π and σ-type interactions are discussed in detail. Finally, our main conclusions are collected in the last section.
2 Computational details
All calculations were performed with the Gaussian16 suite of programs,60 using the specific MP2mod approach,36,55–58 briefly described in the following, or CCSD(T) techniques.The MP2mod calculations are carried out at the MP2 level but with modified basis sets, always taking into account the basis set superposition error (BSSE), by the standard Counterpoise (CP) correction.61 Such a scheme was recently validated by us,36 specifically for the compounds herein investigated. As described in ref. 36, the specific basis set employed for the cation–π complexes in MP2mod calculations consists of: (i) the modified 6-31G**(0.32,0.20)58 for all atoms in benzene, (ii) the modified 6-31G**(0.27, 0.34, 0.36)57 for all atoms in phenol and catechol, (iii) the large def2-TZVPPD59 basis set, considering explicitly all core electrons, for the two smaller Na+ and K+ metal cations, and (iv) the def2-TZVPPD59 basis set with a consistent effective core potential (ECP)62 for Rb+ and Cs+, as suggested by Armentrout and co-workers.37,39,41 Further details can be found in ref. 36 and in the original papers.55–58,63
The binding energy ΔE of each considered noncovalent complex, composed by an aromatic molecule A and a metal ion M+, is computed within the super-molecule approach, as
| ΔE = EA⋯M+ − EA − EM+ | (1) |
One-dimensional curves are extracted from the whole interaction potential energy surface (IPES) by computing ΔE(R) at different dimer arrangements, built by translating M+ with respect to the aromatic ring center of either benzene, phenol or catechol along a displacement vector R. In the top panels of Fig. 1, the selected vectors are shown as an example for catechol: Rtop, along the (z) axis perpendicular to the aromatic plane (xy), Rside, along the (x) axis lying within the ring plane and bisecting the aromatic C–C bond between the carbons bearing the oxygen atoms, and RO, the vector connecting the ring center with one oxygen atom. Furthermore, the conformational flexibility of the substituted aromatic moieties is also taken into account, by considering two different conformers for both phenol and catechol, built by rotating one hydroxyl group through the δ dihedral, placed either at 0° or at 180° (see panel b) in Fig. 1. As detailed in Fig. S1 in the ESI,† similar dimer arrangements were built for all considered A⋯M+ pairs.
 | ||
| Fig. 1 Set up of the dimer geometries employed in the one- and two-dimensional IPES's scans. (a) Cation–π interacting arrangements: M+ cation is displaced along Rtop (blue arrow), which coincides with the benzene ring Ĉ6 axis. (b) σ-Type interacting arrangements: M+ cation is displaced within the xy plane containing the aromatic moiety, either along Rside (blue arrow, bisecting the 1,2 aromatic C–C bond of the ring) or RO (orange arrow). The δ dihedral, ruling the rotation of the hydroxyl group, shown in the δ = 180° conformation, is also indicated with a black arrow. (c) Spherical coordinates considered in the two-dimensional scans. | ||
The interplay between noncovalent interactions and the conformational flexibility of the hydroxyl substituents is further investigated by computing at the MP2mod level two-dimensional scans ΔE(ρ,δ), ΔE(θ,δ), and ΔE(ϕ,δ), where the position of the metal ion M+ is varied along the spherical coordinates (ρ, θ and ϕ) shown in panel (c) of Fig. 1, while the full δ rotation is explored. Moreover, in order to explore the IPES of each considered A⋯M+ pair more in detail, two-dimensional landscapes, ΔE(ρ,ϕ), ΔE(ρ,θ), and ΔE(ϕ, θ) are also computed at the MP2mod level on a large number of dimer arrangements, built by moving the metal ion around the aromatic moiety as shown in panel (c) of Fig. 1.
Finally, since MP2mod was originally devised and validated specifically for noncovalent interactions involving aromatic species, to further assess MP2mod's accuracy, supplementary CCSD(T) reference calculations were carried out at selected geometries, where the ion is closer to the OH substituents. All CCSD(T) calculations were performed with the def2-TZVPPD basis set, using ECP's for the larger Rb+ and Cs+ ions, consistently with the reference data employed in ref. 36.
3 Results and discussion
3.1 1D curves
In our recent work36 concerning the cation–π interactions established between benzene, phenol, and catechol with alkali ions (Na+, K+, Rb+, and Cs+), it was found that the presence of one or more oxygen atoms induces significant differences among the three aromatic species. As our previous investigation was limited to cation–π interaction energy landscapes, only the complex arrangements obtained by displacing the metal ion in a plane parallel to the aromatic ring were considered, and it was noticed36 that, in particular for catechol and smaller ions, new stable minima appeared when the ion was shifted toward the hydroxyl substituents. To further explore the interaction between the cation and the hydroxyl substituents, several new geometrical arrangements are here explored, where the contribution of σ-type forces is expected to be maximized.The left panel of Fig. 2 shows several interaction energy profiles ΔE(R), obtained by displacing, with respect to benzene, phenol or catechol, the Na+ cation along the Rside and RO vectors, shown in Fig. 1. Similar results, obtained for all other ions, are shown in Fig. S2 and S3 in the ESI.† For all considered aromatic moieties, the Rside approaching direction clearly leads to more stable minima, where the ion can get closer to the oxygen lone pair (see also Table A and B in the ESI†) and, at the same time, to the aromatic electron π cloud. Yet, a striking effect is observed along the aromatic series, and the stability of the catechol⋯Na+ complex (170 kJ mol−1) is twice the one found for phenol (80 kJ mol−1), and around six times that of benzene (26 kJ mol−1). A somewhat smaller, but still significant effect was also found for all other ions, as shown in Fig. S2 and S3 (ESI†). It should be noticed that, since even benzene shows a fairly stable minimum along the Rside direction, the σ-type interaction cannot be ascribed solely to the electrostatic attraction between the cation and the oxygen lone pair (ΔEOx), but also to a second stabilizing term, accounting for the interaction of the alkali metal with the aromatic electron density (ΔEAr). In the case of alkali metal ions, ΔEAr is dominated by a favorable induction energy between the charged species and the polarizable π electron cloud. Interestingly, a less pronounced (<2 kJ mol−1) yet not negligible interaction energy has been recently reported64 between benzene and alkali metal atoms, suggesting a third source of interaction, routed in the dispersion forces established between the metal and aromatic polarizable electron clouds. By comparing the interaction energy trends along the alkali metal atoms series reported in ref. 64 with the ones reported in this work and in ref. 36 for alkali cations, it is evident that the increasing cation size has an opposite effect with respect to neutral atoms, where the interaction strength increases from Na to Cs, whereas it decreases when ions are considered.
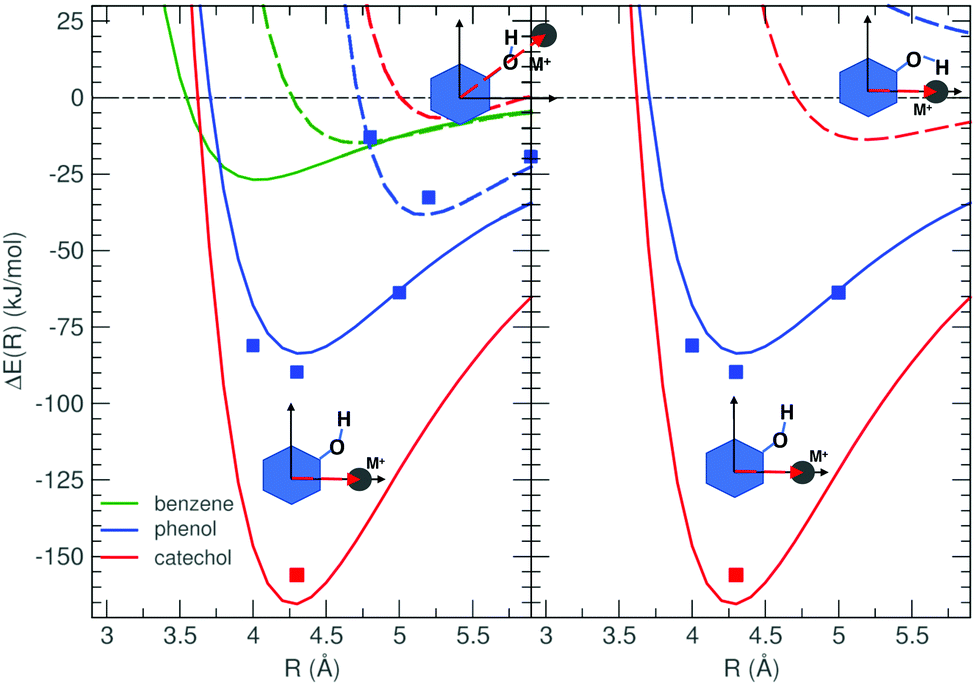 | ||
| Fig. 2 MP2mod (lines) interaction energy profiles (ΔE(R)) between Na+ and benzene (green), phenol (blue) and catechol (red). CCSD(T)/def2-TZVPPD (squares) data are also reported as the reference. Left panel: The metal ion is displaced along either the Rside (solid lines) or RO (dashed lines) vectors, defined in panel (b) of Fig. 1 (see also Fig. S1 in the ESI†). Right panel: The metal ion is displaced along Rside, while the aromatic moiety, either phenol or catechol, is placed in the δ = 180° (solid lines) or δ = 0° (dashed lines) conformation. | ||
As far as phenol and catechol are concerned, two different molecular conformations are also considered, differing in the δ dihedral (see panel b of Fig. 1), which rules the rotation of one hydroxyl group. From the relaxed torsional energy profile, computed at the MP2mod level on both isolated phenolic species and displayed in Fig. S6 in the ESI,† the two equivalent minima of phenol (at δ = 0° and δ = 180°) are found to be separated by a barrier of ∼10 kJ mol−1 at 90°, whereas, in catechol, two non-degenerate minima were found, with a significantly higher (∼27 kJ mol−1) and a non-symmetric barrier between them. The latter features can be ascribed to the formation/breaking, upon δ rotation, of an internal hydrogen bond (HB) between the two catechol's hydroxyl groups, which can be established in the absolute minimum conformation at 0°, and is lost in the local minimum at 180°, causing the increase (∼19 kJ mol−1) of the aromatic moiety's internal energy. To preliminary evaluate the effect of the hydroxyl flexibility on σ-type interactions, in the right panel of Fig. 2, the Na+ ion is displaced, along Rside, with respect to phenol and catechol, each considered in their two minima, namely with δ at 0° (dashed lines) or 180° (solid lines). When δ = 0°, it appears that strong σ-type interactions cannot take place for neither one of the phenolic species, as the rotated hydrogen atom prevents Na+ to bound to the aromatic molecule. In phenol, the curve does not present any stable minimum, as the steric repulsion with the hydrogen atom overcomes the attraction between the ion and the lone pair of one single oxygen atom, whereas in catechol, the combined presence of two oxygen atoms causes a weak σ-type interaction to take place, yet at a rather long distance (>5 Å). Although less impressive, similar trends were registered for all considered alkali ions, as shown in the ESI,† in Fig. S2 and S3. The results discussed so far indicate that rather strong σ-type interactions can be settled between alkali metal ions and both considered phenolic species, given that the cation is approaching along the Rside vector and the aromatic moiety rotates one hydroxyl group in its δ = 180° conformation. In fact, in the phenol complexes, the σ-type interaction can take place without increasing the internal energy of the organic moiety, as the 0° and 180° conformers are degenerate. In catechol, despite the 180° conformer is 20 kJ mol−1 higher in energy with respect to the 0° one, the large gain in terms of intermolecular energy (>125 kJ mol−1 for Na+) largely compensates the intramolecular term.
Once the stability of M+⋯A dimers in σ-type arrangements has been assessed, it is now interesting to compare such complexes with those held together by cation–π interactions, investigated in previous work.36 ΔE profiles of cation–π and σ-type interacting complexes, formed between phenol and Na+ or K+ (left panel) and between catechol and the same ions, are displayed in Fig. 3, whereas those involving larger Rb+ and Cs+ ions are reported in Fig. S4 and S5 (ESI†). The comparison highlights the different behaviors of the interaction energy in the two aromatic species for the various cations. Our previous results36 indicate that the effect of hydroxyl substituents on cation–π interactions is mainly located in the vicinity of the C–O bonds, while the well depth of the en face configuration remains essentially unaltered, as both phenol and catechol cation–π interaction curves (dashed lines) show essentially the same shape as the benzene⋯M+ profiles for the corresponding M+ ions. As far as the different considered cations are concerned, the depth of the cation–π minimum decreases along the alkali metal series, while the equilibrium M+⋯A distance is shifted to larger values. Turning to the σ-type curves (solid lines) computed in the present work, notwithstanding approaching along the Rtop directions allows the metal ion to settle closer to the ring center with respect to the Rside direction, in phenol the computed profiles show shapes and depths similar to the cation–π, for all considered cations. This does not hold in the case of catechol, where the σ-type curves always exhibit a more stable minimum, particularly deep for the Na+ cation (>160 kJ mol−1), but still significant for the larger ones (∼100, 90 and 80 kJ mol−1 for K+, Rb+ and Cs+, respectively, as shown in Fig. S5, ESI†). The difference between cation–π and σ-type minima decreases on going from Na+ to Cs+, being about 60 kJ mol−1 for the former and less than 10 kJ mol−1 for the latter.
 | ||
| Fig. 3 MP2mod (lines) interaction energy profiles ΔE(R), computed in dimer arrangements obtained by displacing the ion either along Rtop (dashed lines) or Rside (solid lines), as shown in the insets. CCSD(T)/def2-TZVPPD (squares) data are also reported as reference. Left: Phenol–Na+ (red) and phenol–K+ (green). Right: Catechol–Na+ (red) and catechol–K+ (green). | ||
As the MP2mod was originally conceived to reproduce π–π interactions,55–58 the accuracy of the method in handling other types of interactions calls for further validations. For this reason, in ref. 36, the accuracy of MP2mod results in computing the cation–π interaction energy was successfully validated against high level CCSD(T) calculations, carried out with the large def2-TZVPPD basis set, resulting in an average standard deviation of ∼5 kJ mol−1. Here the interaction energy in several σ-type interacting geometries has been computed again at the CCSD(T)/def2-TZVPPD level, for selected M+⋯A pairs, and compared with the MP2mod counterparts. Table 1 reports the interaction energy computed at CCSD(T) and MP2mod levels for several selected arrangements, where either cation–π or σ-type interactions are maximized. From these data, also displayed in Fig. 2, 3 and Fig. S2–S5 (in the ESI†), it appears that the standard deviation found for σ-type complexes with respect to the higher level method (5.3 kJ mol−1) is very similar with to the one achieved for cation–π interactions (6.0 kJ mol−1), and almost coincident to the global one reported in ref. 36. To avoid the computational burden associated with the CCSD(T) calculations, only the minimum geometries for catechol⋯Na+ and catechol⋯K+ complexes were subjected to the same test, and the results are reported in Table 2. The MP2mod energies again agree with the CCSD(T) reference values within few kJ mol−1, and, more important, the CCSD(T) ratio between cation–π and σ-type ΔE's (0.63 and 0.64 for Na+ and K+, respectively) is almost quantitatively reproduced by MP2mod (0.63 and 0.67), implying that this method can be used with confidence to investigate with balanced and reliable estimates the whole set of accessible dimer configurations of M+⋯A complexes. Exploiting the computational convenience and reliability of MP2mod calculations,36 we can therefore afford a systematic investigation of two-dimensional interaction energy landscapes, consisting of hundreds of ab initio calculations, obtained in few hours on a standard multi-core machine. We separate the discussion into two parts, one related to the interplay between the internal flexibility of the aromatic moiety (consisting in the OH rotation) and the cation displacement in σ-type arrangements, and a second part where the pathways connecting the cation–π and σ-type minima are extensively explored.
| Ion | Interaction | R (Å) | ΔECCSD(T) | ΔEMP2mod |
|---|---|---|---|---|
| Na+ | Cation–π | 1.90 | −41.7 | −44.1 |
| Na+ | Cation–π | 2.45 | −97.7 | −104.6 |
| Na+ | Cation–π | 3.20 | −67.9 | −73.9 |
| Na+ | σ-Type | 4.00 | −81.0 | −67.9 |
| Na+ | σ-Type | 4.30 | −89.7 | −83.6 |
| Na+ | σ-Type | 5.00 | −63.8 | −62.9 |
| K+ | Cation–π | 2.40 | −33.6 | −40.8 |
| K+ | Cation–π | 2.90 | −71.0 | −76.0 |
| K+ | Cation–π | 4.20 | −34.7 | −37.0 |
| K+ | σ-Type | 4.40 | −48.7 | −50.1 |
| K+ | σ-Type | 4.70 | −62.4 | −61.0 |
| K+ | σ-Type | 5.30 | −50.9 | −51.2 |
| Rb+ | Cation–π | 2.60 | −30.7 | −36.2 |
| Rb+ | Cation–π | 3.05 | −64.0 | −67.9 |
| Rb+ | Cation–π | 3.80 | −46.4 | −48.5 |
| Rb+ | σ-Type | 4.50 | −32.1 | −35.8 |
| Rb+ | σ-Type | 4.90 | −57.2 | −58.2 |
| Rb+ | σ-Type | 5.60 | −42.5 | −44.2 |
| Cs+ | Cation–π | 2.80 | −34.7 | −36.2 |
| Cs+ | Cation–π | 3.20 | −60.4 | −67.9 |
| Cs+ | Cation–π | 4.20 | −37.0 | −48.5 |
| Cs+ | σ-Type | 4.70 | −29.6 | −34.4 |
| Cs+ | σ-Type | 5.10 | −49.0 | −58.3 |
| Cs+ | σ-Type | 5.80 | −38.0 | −39.7 |
| All | Cation–π | Std. dev. (kJ mol−1): 6.0 | ||
| All | σ-Type | Std. dev. (kJ mol−1): 5.3 | ||
| Ion | Interaction | R (Å) | ΔECCSD(T) | ΔEMP2mod |
|---|---|---|---|---|
| Na+ | Cation–π | 2.50 | −96.4 | −104.3 |
| Na+ | σ-Type | 4.30 | −156.0 | −165.6 |
| K+ | Cation–π | 2.80 | −70.3 | −78.4 |
| K+ | σ-Type | 4.60 | −109.6 | −116.5 |
3.2 Interplay between torsional internal energy and σ-type interactions
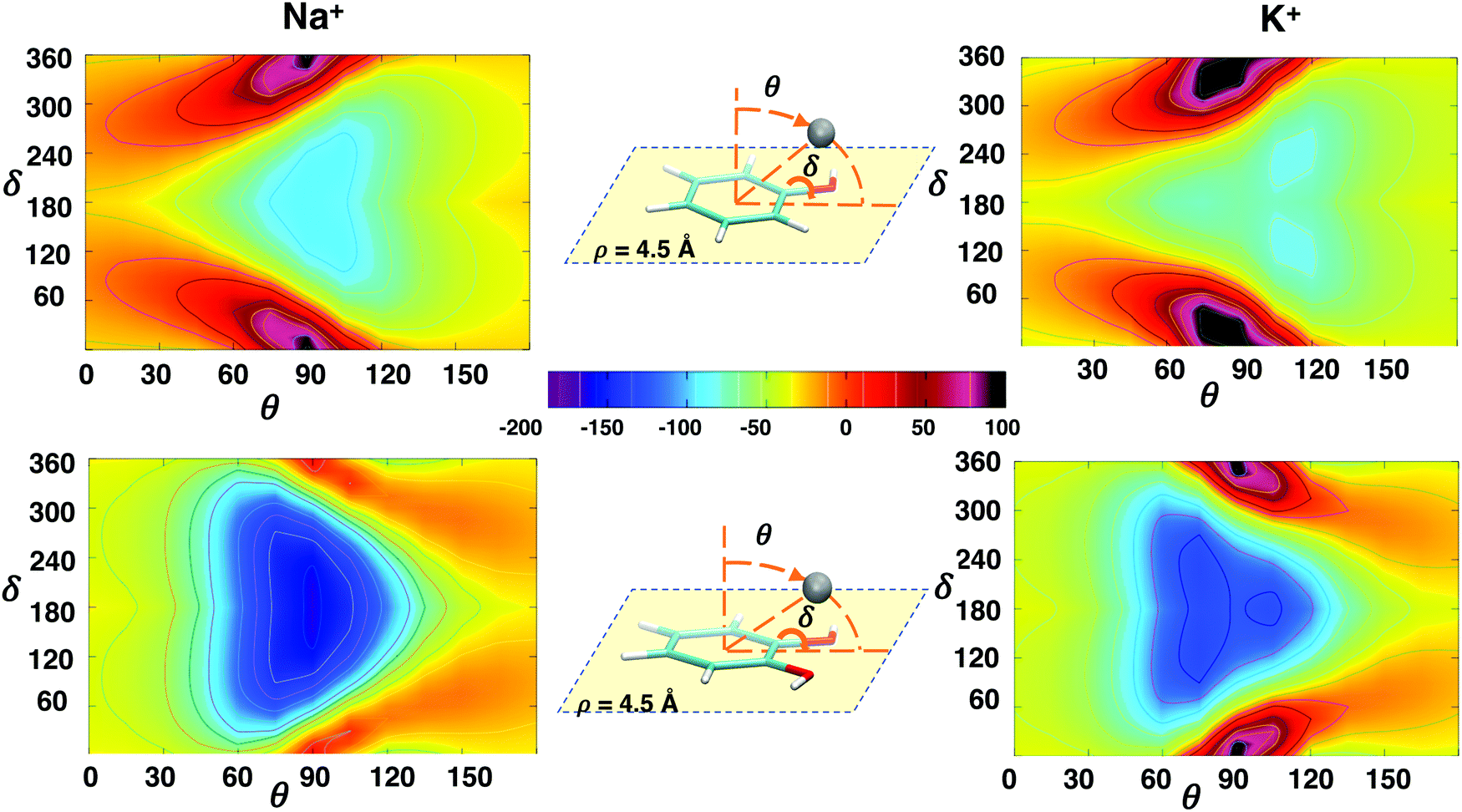
The interaction energy landscapes involving the δ rotation of the hydroxyl group around the C–O bond and each of the three spherical coordinates (ρ, θ and ϕ, shown in panel c of Fig. 1) describing the position of the metallic ion are displayed in Fig. 4–6 for Na+ and K+ ions, whereas those concerning with Rb+ and Cs+ are shown in Fig. S7–S9 in the ESI.†Fig. 4 shows the changes in the interaction energy upon δ rotation and by varying the distance ρ of the cation from the center of the ring along the Rside direction (i.e. lying within the aromatic plane, at ϕ = 0° and θ = 90°), for the phenol–Na+/phenol–K+ (top) and the catechol–Na+/catechol–K+ pairs (bottom). All the ΔE(ρ, δ) maps show the same qualitative behavior: in all cases, a clearly defined minimum is found, in a significantly attractive well centered at δ = 180° ± 50°, with ρ distances in between 4 Å and 5 Å. On the same foot, at small ρ's and with δ approaching 0°, a steep high energy region appears clearly, due to the cation–hydrogen repulsion, which is more evident as the ion dimensions increase (see also Rb+ and Cs+ in the ESI†), whereas a much smoother climb back is registered when the ion is moved to larger distances. Nonetheless, significant quantitative differences appear between the two aromatic species. First, the minimum depth is always larger in catechol, due to the stronger σ-type interaction, which can also be seen as a chelating interaction. As observed in the one dimensional curves, this effect is particularly remarkable for the catechol–Na+ pair, whose interaction is characterized by a very stable, wide and well defined minimum (∼−170 kJ mol−1). Next, it might be also worth noticing how the combined effect of the double –OH substitution causes the minimum energy region to extend to large ρ displacements, so it is likely that the cation feels a significant attractive force even when far from catechol, if approaching within the plane containing the aromatic ring (i.e. θ = 90°), along the Rside direction. It yet remains to be established if other approaching directions, maintaining θ = 90° at different ϕ angles, are possible, and if σ-type patterns of similar strength can be settled. | ||
| Fig. 4 2D interaction energy plots ΔE(ρ,δ) computed at MP2mod level between phenol (top) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled PES region, where the metal ion (gray sphere) is moved within the plane containing the aromatic ring, is shown in the insets together with the investigated dihedral. In the color palette, the interaction energy is reported in kJ mol−1. | ||
 | ||
| Fig. 5 2D interaction energy plots ΔE(ϕ,δ) computed at the MP2mod level between phenol (top) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled PES region, where the metal ion (gray sphere) is moved within the plane containing the aromatic ring, at a distance ρ = 4.5 Å, is shown in the insets together with the investigated dihedral. In the color palette, the interaction energy is reported in kJ mol−1. | ||
 | ||
| Fig. 6 2D interaction energy plots ΔE(θ,δ) computed at the MP2mod level between phenol (top) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled PES region, where the metal ion (gray sphere) is moved keeping a fixed distance ρ = 4.5 Å, is shown in the insets together with the investigated dihedral. In the color palette, interaction energy is reported in kJ mol−1. | ||
Fig. 5 shows the ΔE(ϕ,δ) landscapes, for phenol (top) and catechol (bottom), considering the Na+ and K+ cations, while the maps obtained for Rb+ and Cs+ are reported in Fig. S8 in the ESI.† In all cases, as shown in the inner panels, the cation is displaced counter-clockwise, within the aromatic plane (θ = 90°), at a fixed ρ distance of 4.5 Å, compatible with most of the minima found during the previous ρ, δ scan. The qualitative picture is again very similar among all considered pairs, and two minimum energy regions appear: one is centered at ϕ = 0° (i.e. along the Rside direction), δ being in the [120°–240°] range, whereas another is found around ϕ = 60°, when δ is rotated in the [−60° to +60°] range. Yet, significant differences are clearly visible both between the two aromatic species and among the investigated ions. Indeed, as far as phenol is concerned, the two minima present similar depths (−88 kJ mol−1 and −91 kJ mol−1 for Na+ and −67 kJ mol−1 and −61 kJ mol−1 for K+), and are both accessible by ϕ rotation, when the ion approaches the Rside vector facing the oxygen lone pair. In catechol, conversely, the minimum around ϕ = 0° is remarkably more stable (−158 kJ mol−1 and −109 kJ mol−1, for Na+ and K+, respectively) than the other one (−98 kJ mol−1 and −72 kJ mol−1), notwithstanding being surrounded by rather high barriers (>100 kJ mol−1), due to the presence of the second substituent, that compromise its accessibility by ϕ rotation. In contrast, when δ = 0° and the intra-molecular HB is settled, the minimum region at ϕ ∼ 60° is similar to those found in phenol, and, although less deep, it becomes accessible when the cation approaches the oxygen atom from the opposite side with respect to the hydrogen. All the above discussed features are more evident with Na+, whereas they lose contrast when the ion dimensions are increased. As can be observed by looking at Fig. S8 (ESI†), the minima found for the catechol–Cs+ pair are barely visible (−5 kJ mol−1 and −18 kJ mol−1 at ϕ = 0° and ϕ = 60°, respectively), suggesting that the σ-type interaction for larger ions is not so effective at the selected distance (ρ = 4.5Å). In fact, as shown in Fig. S4 and S5 (ESI†), the increasing dimensions of the considered metal ion not only shift the minimum at larger distances, but also result in significantly less deep wells.
Finally, it is interesting to achieve a deeper insight into the interplay between δ rotation and the polar angle θ, which shifts the ion from a position on top of the aromatic ring (θ = 0°), where the cation–π interaction was shown to be maximized,36 to the aromatic plane (θ = 90°), where the ion can more closely interact with the hydroxyl substituents. To this end, Fig. 6 shows the ΔE(θ,δ) landscapes, obtained by computing at MP2mod level the interaction energy between the investigated aromatic species and Na+ and K+, as a function of the θ and δ coordinates when ϕ is fixed at 0° and the distance ρ is again 4.5 Å. At difference with the previous cases, the interaction energy maps obtained for phenol and catechol are clearly distinguishable even from a first visual inspection. Notwithstanding that in all cases ΔE at θ = 0°/180° does not show any dependence on δ, significant differences appear between the investigated aromatic species when the ion is displaced toward θ = 90°. In fact, when the metal cation is displaced toward the phenol plane it experiences a rather smooth gradient, which goes from a slight attraction (e.g. ∼−20 kJ mol−1, for Na+) to a more defined minimum (−91 kJ mol−1), which slowly loses intensity when the ion is rotated beneath the aromatic ring (θ = 180°). In the vicinity of the plane, the δ rotation becomes effective in modulating the interaction patterns, and significant barriers appear when δ is placed in the 0°–30° range. A very different picture appears for catechol: the second hydroxyl substitution increases the strength of the σ-type interaction, in turn increasing the gradient on going from the position centered on top of the π cloud (θ = 0°) to the one within the aromatic plane, in between the two oxygen atoms, widening and further stabilizing the minimum region around θ = 90°, and eventually reducing the repulsive effect of the δ rotation. Such differences between phenol and catechol are augmented as the ion dimensions increase. On going from Na+ to Cs+ (see also Fig. S9, ESI†), the minimum region elongates toward θ larger than 90° for phenol, whilst smaller than 90° for catechol. In all cases the δ rotation becomes more effective on the interaction energy patterns with the increasing dimensions of the ion, and the single minimum found for Na+ gradually splits into three well separated minima, among which the most stable are found at θ ∼ 120° for the complexes involving phenol, and at θ ∼ 60° for catechol.
Based on the above findings, it can be concluded that the δ rotation can modulate the interaction energy between the considered alkali ions and the phenolic systems, affecting the pathways that lead to the stable σ-type minima and contributing to differentiate between phenol and catechol behavior. Yet, especially for the smaller ions, the σ-type interaction strength (in the −170 to 100 kJ mol−1 range) and the gradients leading the ion to the stable minima seem to be sufficient to overwhelm the rotational barrier (around 10 kJ mol−1 and 30 kJ mol−1 for phenol and catechol, respectively, see Fig. S6, ESI†), thus allowing the ions to settle in the resulting σ-type minima. For this reason, in the following discussion, where the A–M+ IPES is further explored along two spherical coordinates, the δ dihedral was fixed at 180°, unless otherwise stated.
3.3 Competition of non-covalent interactions
In this section we want to discuss in more detail the interplay between cation–π and σ-type interactions, which might differently contribute to determine the different minimum energy configurations of the resulting complexes. To this end, for all investigated phenol–M+ and catechol–M+ pairs, two dimensional maps were obtained by scanning the IPES at the MP2mod level along three pairs of spherical coordinates, namely (ϕ, ρ), (ρ, θ) and (ϕ, θ). Furthermore, to better understand both, the effect of hydroxyl substitution, and the nature of the σ-type interaction, the same landscapes were also purposely computed for the benzene–M+ complexes.The first IPES's cross-section explored by the MP2mod sampling is the one spanned by the radial distance ρ and the azimuthal angle ϕ, i.e. when the ion is displaced, within the aromatic plane (θ0σ = 90°), at different positions with respect to the ring center. The results shown in Fig. 7 and 8 confirm the preliminary findings obtained in the ΔE(ϕ,δ) scan: the approaching direction more effective to establish σ-type interactions is the one along the Rside vector (ϕ0σ = 0°), where the most stable σ-type minimum was found for all investigated phenol–M+ and catechol–M+ complexes, at ρ0σ distances varying between 4.2 Å (catechol–Na+) and 5.2 Å (phenol–Cs+), as reported in Table 3. It might be worth noting that also all the benzene–M+ pairs present a minimum at ϕ0σ = 0°, which is degenerate for symmetry with the other five versors ( = ±60°, ±120° and 180°) that bisect each aromatic C–C bonds. If the total σ-type interaction energy (ΔEσ) in the vicinity of the [ϕ0σ, ρ0σ] minimum can be expressed as a sum of the previously discussed ΔEOx and ΔEAr contributions, for each benzene–M+ σ-type complex ΔEOx clearly vanishes, due to the absence of hydroxyl substituents, and ΔEAr = ΔEbenzσ, which accounts for the interaction between the cation and the aromatic ring within the plane θ0σ = 90°. Based on the rather scarce sensitivity of the en face cation–π configurations on OH substitution,36 it could be also hypothesized that ΔEAr is not much affected by the presence of hydroxyl substituents, and the ΔEAr value obtained for the benzene complexes could be extended in first approximation to all the investigated aromatic species. Following this idea, along with the minimum total σ-type-interaction energy (ΔEσ), Table 3 also reports the ΔEOx contribution, estimated, for all investigated pairs, as
= ±60°, ±120° and 180°) that bisect each aromatic C–C bonds. If the total σ-type interaction energy (ΔEσ) in the vicinity of the [ϕ0σ, ρ0σ] minimum can be expressed as a sum of the previously discussed ΔEOx and ΔEAr contributions, for each benzene–M+ σ-type complex ΔEOx clearly vanishes, due to the absence of hydroxyl substituents, and ΔEAr = ΔEbenzσ, which accounts for the interaction between the cation and the aromatic ring within the plane θ0σ = 90°. Based on the rather scarce sensitivity of the en face cation–π configurations on OH substitution,36 it could be also hypothesized that ΔEAr is not much affected by the presence of hydroxyl substituents, and the ΔEAr value obtained for the benzene complexes could be extended in first approximation to all the investigated aromatic species. Following this idea, along with the minimum total σ-type-interaction energy (ΔEσ), Table 3 also reports the ΔEOx contribution, estimated, for all investigated pairs, as
| ΔEOx = ΔEσ − ΔEAr ≃ ΔEσ − ΔEbenzσ | (2) |
 | ||
| Fig. 7 2D interaction energy plots ΔE(ϕ,ρ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
 | ||
| Fig. 8 2D interaction energy plots ΔE(ϕ,ρ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Rb+ (left) or Cs+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
| M+ | ΔEσ(ρ0σ) | ΔEAr@ρ0σ | ΔEOx@ρ0σ | |||
|---|---|---|---|---|---|---|
| Benzene–M+ | Phenol–M+ | Catechol–M+ | Benzene–M+ | Phenol–M+ | Catechol–M+ | |
| Na+ | −26.8 (4.0) | −96.2 (4.2) | −164.5 (4.2) | −25.8 (4.2) | −70.4 | −138.7 |
| K+ | −17.1 (4.6) | −67.3 (4.8) | −119.1 (4.8) | −15.6 (4.8) | −51.7 | −103.5 |
| Rb+ | −14.3 (4.8) | −58.1 (5.0) | −103.8 (5.0) | −13.1 (5.0) | −45.0 | −90.7 |
| Cs+ | −13.3 (5.0) | −51.6 (5.2) | −93.2 (5.2) | −12.0 (5.2) | −39.6 | −81.1 |
 | ||
| Fig. 9 2D interaction energy plots ΔE(ρ,θ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
 | ||
| Fig. 10 2D interaction energy plots ΔE(ρ,θ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Rb+ (left) or Cs+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
The last computed interaction energy maps are the ones concerning with the azimuthal and polar angle, ΔE(ϕ,θ), displayed in Fig. 11 and 12. For a more sound comparison among the ions, each scan was performed by fixing the ion radial distance ρ to  , i.e. in the vicinity of ρ0σ distance found for the considered A–M+ pair, being
, i.e. in the vicinity of ρ0σ distance found for the considered A–M+ pair, being  4.0 Å, 4.4 Å, 4.5 Å and 4.7 Å, for Na+, K+, Rb+ and Cs+, respectively. The insurgence of the σ-type minimum in the (0, 90) region upon substitution is again clearly visible in all figures, together with the decrease of the interaction strength with increasing ion dimensions. A closer look at the top panels, where the IPES's cross section for the benzene molecule is displayed, reveals a smooth attractive region of about −50 kJ mol−1 for Na+, which extends to θ ∼ 60°, notwithstanding the repulsion of the hydrogens as the ion approaches the aromatic plane, at θ = 90°. In phenol, the appearance of the σ-type minimum is accompanied, at large θ, by the insurgence of a high repulsive barrier near ϕ = 30°, which corresponds to the position of the oxygen atom. Yet, in all cases the σ-type minimum remains well accessible by ϕ rotation, if the ion is approaching from negative ϕ angles. Turning to catechol, on the one hand the combined effect of the two available lone pairs enforces the σ-type minimum, increasing both its depth and wideness, on the other hand the steric encumbrance of the two hydroxyl groups limits its accessibility through ϕ rotation, leaving to the ion only the previously discussed (ρ, θ) path as an effective channel to reach the phenolic unit in a σ-type complex.
4.0 Å, 4.4 Å, 4.5 Å and 4.7 Å, for Na+, K+, Rb+ and Cs+, respectively. The insurgence of the σ-type minimum in the (0, 90) region upon substitution is again clearly visible in all figures, together with the decrease of the interaction strength with increasing ion dimensions. A closer look at the top panels, where the IPES's cross section for the benzene molecule is displayed, reveals a smooth attractive region of about −50 kJ mol−1 for Na+, which extends to θ ∼ 60°, notwithstanding the repulsion of the hydrogens as the ion approaches the aromatic plane, at θ = 90°. In phenol, the appearance of the σ-type minimum is accompanied, at large θ, by the insurgence of a high repulsive barrier near ϕ = 30°, which corresponds to the position of the oxygen atom. Yet, in all cases the σ-type minimum remains well accessible by ϕ rotation, if the ion is approaching from negative ϕ angles. Turning to catechol, on the one hand the combined effect of the two available lone pairs enforces the σ-type minimum, increasing both its depth and wideness, on the other hand the steric encumbrance of the two hydroxyl groups limits its accessibility through ϕ rotation, leaving to the ion only the previously discussed (ρ, θ) path as an effective channel to reach the phenolic unit in a σ-type complex.
 | ||
| Fig. 11 2D interaction energy plots ΔE(ϕ,θ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Na+ (left) or K+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
 | ||
| Fig. 12 2D interaction energy plots ΔE(ϕ,θ) computed at the MP2mod level between benzene (top), phenol (middle) or catechol (bottom) and Rb+ (left) or Cs+ (right) cations. The sampled IPES region is shown on the left, together with the investigated spherical coordinates. In the color palette, the interaction energy is reported in kJ mol−1. | ||
4 Conclusions
The preliminary one-dimensional interaction energy scans, carried out at the MP2mod level along the selected paths of the three-dimensional IPES, for complexes formed by three aromatic molecules (namely, benzene, phenol and catechol) and alkali ions of increasing dimensions (Na+, K+, Rb+ and Cs+) have revealed additional stable minima for the phenolic species, in different configurations with respect to the cation-π en face arrangements. Their stability can be traced back to the presence of one or more hydroxyl substituents on the aromatic ring and to the consequent establishing of σ-type interactions between the ion and the oxygen atoms. In fact, contrary to the “en face” configurations, where the ion is located along the vector perpendicular to the aromatic plane, thus maximizing the cation–π interactions, these σ-type minima, found in both phenol and catechol, are located within the plane containing the aromatic ring, in the vicinity of the oxygen atoms, and show similar (phenol) or even more attractive (catechol) interaction energies than those found at the cation–π geometries. The existence of such stable σ-type configurations in the investigated complexes, as well as the ratio between the MP2mod interaction energies of cation–π and σ-type minima in both phenol–M+ and catechol–M+ dimers, is in an excellent agreement with higher level CCSD(T)/def2-TZVPPD, thus confirming MP2mod's reliability in computing both cation–π and σ-type interactions.This last validation has allowed us to confidently exploit the MP2mod computational affordability to extend, with hundreds of new points for each complex, the sampling of the investigated IPES to a four-dimensional configurational space, which includes the three spherical coordinates determining the position of the ion and the dihedral governing the rotation of one hydroxyl substituent. The collected energy database was eventually organized in two-dimensional interaction energy maps and their features were compared among both the aromatic species and along the alkali metal series. As far as the former comparison is concerned, the analysis of the IPES landscapes confirmed the remarkable effect of hydroxyl substitution. In agreement with early studies reported for phenol–M+ complexes,46–48 the presence of OH groups significantly increases the stability of σ-type complexes, whose interaction energy is comparable to the one recently reported for the cation–π, “en face” configurations.36 The stability of the σ-type configurations significantly increases in catechol, where the favorable interaction with the oxygen lone pair is found to increase linearly with the number of substituents, and the combined chelating interaction causes the insurgence of much deeper minima with respect to phenol. The net preference registered for the smaller ions to bind with catechol in σ-type configurations strongly suggests that the energy barriers (e.g. breaking of the internal hydrogen bond through hydroxyl δ rotation) which may prevent the accessibility of σ-type's geometries might be easily overcome in virtue of the total energy gain. Moreover, the computed landscapes evidenced favorable energy channels connecting the “en face” configurations to regions in which the ion may interact directly with the oxygen's lone pair, suggesting that the σ-type interaction could play an important role in influencing the delicate balance among NCI when catechol–ion complexes are embedded in a more complex environment. Turning to the effect of the cation dimensions, the comparison of the interaction energy landscapes along the alkali metal series reveals rather monotonic trends, where the strength of the interaction diminishes with the increasing charge/mass ratio of the ion. Yet, there is a significant difference between the other ions and Na+, whose smaller dimensions allow it to more closely approach the hydroxyl substituents, consequently increasing the stability of the resulting σ-type complexes.
Overall, these results support the reported role of cation–π interactions in K+-aided adhesion of polydopamine films and the lack of effects of Na+. Underwater adhesion and film deposition are the result of a complex balance of NCI, which are fine-tuned by cation properties, and inter-species (cohesion) vs. intra-species (adhesion) effects. Nonetheless, care should be taken in extending the conclusions of the present work straightforwardly to condensed phase properties, as the present modeling does not take into account the critical effects connected with oligomer-type stacking interactions of the metal/catechol complexes, the role of water in competing for binding of the cation to the phenolic OH groups, and the role of the metal counterion, which is emerging as a critical contributor. In fact, notwithstanding the deep insight on the relative gas-phase stability of different noncovalent complexes that reliable interaction energy landscapes can offer, it is worth recalling that the presence of other species might of course alter the aforementioned conclusions. For instance, Vaden and Lisy42–44 reported that in aqueous solvents phenol–M+ complexes are in generally more stable in the σ-configuration with respect to the cation–π one, but, while with Na+ the dominant configurations are those in which the phenol hydroxyl group is not involved in hydrogen bonds with the surrounding water molecules, with K+, the phenol OH group is involved in a hydrogen bond with the solvent. The present findings on phenol complexes are consistent with the results of ref. 42 and those on catechol seem to suggest that a similar, even enhanced difference between the Na+ and K+ behavior should be expected for complexes involving catechol. Yet, if a full comprehension of the intricate interplay among all NCI has to be achieved, the whole complex + solvent system should be considered, resorting to techniques such as molecular dynamics,28,65 which is beyond the aims of the present work. Nonetheless, the here reported interaction energy database can be of outermost importance as a reference for accurately tuning the FF parameters describing the ion interaction with the considered aromatic units. Indeed, MP2mod databases have already been exploited by some of us to parameterize accurate quantum-mechanically derived FFs, achieving more than encouraging results in the simulation of condensed phase properties.66–69 Work in progress is aimed at extending the study to the interaction of metal cations with other catechol-containing systems, like the melanin/polydopamine component 5,6-dihydroxy-indole, and to the involvement of counterions in establishing complementary sets of non-covalent interactions. The overall aim of these studies is to gain a quantitative perspective of cation–π interactions in the complex interplay of NCI underpinning natural phenomena such as mussel byssus adhesion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MdI acknowledges MIUR (PRIN PROJECT 2017YJMPZN) for partial support.References
- R. P. Feynman, Forces in Molecules, Phys. Rev., 1939, 56, 340–343 CrossRef CAS.
- S. Scheiner, Noncovalent Forces, Springer International Publishing, Switzerland, 2015 Search PubMed.
- P. Hobza and J. Řezáč, Introduction: Noncovalent Interactions, Chem. Rev., 2016, 116, 4911–4912 CrossRef CAS.
- D. Chandler, Interfaces and the driving force of hydrophobic assembly, Nature, 2005, 437, 640–647 CrossRef CAS.
- G. R. Desiraju, A Bond by Any Other Name, Angew. Chem., Int. Ed., 2011, 50, 52–59 CrossRef CAS.
- D. Fagnani, A. Sotuyo and R. Castellano, Comprehensive Supramolecular Chemistry II, Elsevier, 2017, pp. 121–148 Search PubMed.
- P. Metrangolo and G. Resnati, Halogen Bonding: Fundamentals and Applications; Structure and Bonding, Springer, Berlin Heidelberg, 2008 Search PubMed.
- X. Lucas, A. Bauzá, A. Frontera and D. Quiñonero, A thorough anion–π interaction study in biomolecules: On the importance of cooperativity effects, Chem. Sci., 2016, 7, 1038–1050 RSC.
- J. C. Ma and D. A. Dougherty, The cation–π interaction, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- R. Kumpf and D. Dougherty, A mechanism for ion selectivity in potassium channels: computational studies of cation–π interactions, Science, 1993, 261, 1708–1710 CrossRef CAS.
- S. Mecozzi, A. P. West and D. A. Dougherty, Cation–π interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative guide, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566–10571 CrossRef CAS.
- D. A. Dougherty, The Cation–π Interaction, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS.
- A. Sigel, H. Sigel and R. K. O. Sigel, The Alkali Metal Ions: Their Role for Life, Metal Ions in Life Sciences, Springer International Publishing, Cham, 2016, vol. 16 Search PubMed.
- S. Hong, Y. Wang, S. Y. Park and H. Lee, Progressive fuzzy cation–π assembly of biological catecholamines, Sci. Adv., 2018, 4, eaat7457 CrossRef.
- A. S. Mahadevi and G. N. Sastry, Cation–π Interaction: Its Role and Relevance in Chemistry, Biology, and Material Science, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS.
- E. R. Tiekink, Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists, Coord. Chem. Rev., 2017, 345, 209–228 CrossRef CAS.
- D. Li, Z. Liu, J. Song, H. Li, B. Zhang, P. Yin, Z. N. Zheng, J. E. Roberts, M. Tsige, C. L. Hill and T. Liu, Cation Translocation around Single Polyoxometalate-Organic Hybrid Cluster Regulated by Electrostatic and Cation–π Interactions, Angew. Chem., Int. Ed., 2017, 129, 3342–3346 CrossRef.
- J. Luo, K. Chen, P. Yin, T. Li, G. Wan, J. Zhang, S. Ye, X. Bi, Y. Pang, Y. Wei and T. Liu, Effect of Cation–π Interaction on Macroionic Self-Assembly, Angew. Chem., Int. Ed., 2018, 57, 4067–4072 CrossRef CAS.
- S. Kim, J. Huang, Y. Lee, S. Dutta, H. Young Yoo, Y. Mee Jung, Y. Jho, H. Zeng and D. S. Hwang, Complexation and coacervation of like-charged polyelectrolytes inspired by mussels, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E847–E853 CrossRef CAS.
- M. A. Gebbie, W. Wei, A. M. Schrader, T. R. Cristiani, H. A. Dobbs, M. Idso, B. F. Chmelka, J. Herbert Waite and J. N. Israelachvili, Tuning underwater adhesion with cation–π interactions, Nat. Chem., 2017, 9, 473–479 CrossRef CAS.
- H. Birkedal, Mimicking mussel mechanics, Nat. Chem., 2017, 9, 408–409 CrossRef CAS.
- S. Park, S. Kim, Y. Jho and D. S. Hwang, Cation–π Interactions and Their Contribution to Mussel Underwater Adhesion Studied Using a Surface Forces Apparatus: A Mini-Review, Langmuir, 2019, 35, 16002–16012 CrossRef CAS.
- L. Xie, L. Gong, J. Zhang, L. Han, L. Xiang, J. Chen, J. Liu, B. Yan and H. Zeng, A wet adhesion strategy via synergistic cation–π and hydrogen bonding interactions of antifouling zwitterions and mussel-inspired binding moieties, J. Mater. Chem. A, 2019, 7, 21944–21952 RSC.
- G. D. Degen, P. R. Stow, R. B. Lewis, R. C. Andresen Eguiluz, E. Valois, K. Kristiansen, A. Butler and J. N. Israelachvili, Impact of Molecular Architecture and Adsorption Density on Adhesion of Mussel-Inspired Surface Primers with Catechol–Cation Synergy, J. Am. Chem. Soc., 2019, 141, 18673–18681 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Clarendon, Oxford, 1987 Search PubMed.
- D. Frenkel and B. Smith, Understanding Molecular Simulations, Academic Press, San Diego, 1996 Search PubMed.
- Q.-S. Du, S.-Y. Long, J.-Z. Meng and R.-B. Huang, Empirical formulation and parameterization of cation–π interactions for protein modeling, J. Comput. Chem., 2012, 33, 153–162 CrossRef CAS.
- E. A. Orabi, R. L. Davis and G. Lamoureux, Drude polarizable force field for cation–π interactions of alkali and quaternary ammonium ions with aromatic amino acid side chains, J. Comput. Chem., 2020, 41, 472–481 CrossRef CAS.
- A. Turupcu, J. Tirado-Rives and W. L. Jorgensen, Explicit Representation of Cation−π Interactions in Force Fields with 1/ r 4 Nonbonded Terms, J. Chem. Theory Comput., 2020, 16(11), 7184–7194 CrossRef CAS.
- J. B. Nicholas, B. P. Hay and D. A. Dixon, Ab Initio Molecular Orbital Study of Cation–π Binding between the Alkali-Metal Cations and Benzene, J. Phys. Chem. A, 1999, 103, 1394–1400 CrossRef CAS.
- D. Feller, D. A. Dixon and J. B. Nicholas, Binding enthalpies for alkali cation-benzene complexes revisited, J. Phys. Chem. A, 2000, 104, 11414–11419 CrossRef CAS.
- J. C. Amicangelo and P. B. Armentrout, Absolute binding energies of alkali-metal cation complexes with benzene determined by threshold collision-induced dissociation experiments and ab initio theory, J. Phys. Chem. A, 2000, 104, 11420–11432 CrossRef CAS.
- M. S. Marshall, R. P. Steele, K. S. Thanthiriwatte and C. D. Sherrill, Potential energy curves for cation–π interactions: Off-axis configurations are also attractive, J. Phys. Chem. A, 2009, 113, 13628–13632 CrossRef CAS.
- K. Ansorg, M. Tafipolsky and B. Engels, Cation–π Interactions: Accurate Intermolecular Potential from Symmetry-Adapted Perturbation Theory, J. Phys. Chem. B, 2013, 117, 10093–10102 CrossRef CAS.
- Y. Zhang, S. Chen, F. Ying, P. Su and W. Wu, Valence Bond Based Energy Decomposition Analysis Scheme and Its Application to Cation–π Interactions, J. Phys. Chem. A, 2018, 122, 5886–5894 CrossRef CAS.
- A. Ferretti, M. D’Ischia and G. Prampolini, Benchmarking Cation–π Interactions: Assessment of Density Functional Theory and Möller-Plesset Second-Order Perturbation Theory Calculations with Optimized Basis Sets (MP2mod) for Complexes of Benzene, Phenol, and Catechol, J. Phys. Chem. A, 2020, 124, 3445–3459 CrossRef CAS.
- V. N. Bowman, A. L. Heaton and P. B. Armentrout, Metal Cation Dependence of Interactions with Amino Acids: Bond Energies of Rb+ to Gly, Ser, Thr, and Pro, J. Phys. Chem. B, 2010, 114, 4107–4114 CrossRef CAS.
- P. B. Armentrout, Y. Chen and M. T. Rodgers, Metal Cation Dependence of Interactions with Amino Acids: Bond Energies of Cs+ to Gly, Pro, Ser, Thr, and Cys, J. Phys. Chem. A, 2012, 116, 3989–3999 CrossRef CAS.
- P. B. Armentrout, B. Yang and M. T. Rodgers, Metal Cation Dependence of Interactions with Amino Acids: Bond Energies of Rb+ and Cs+ to Met, Phe, Tyr, and Trp, J. Phys. Chem. B, 2013, 117, 3771–3781 CrossRef CAS.
- M. T. Rodgers and P. B. Armentrout, Cationic Noncovalent Interactions: Energetics and Periodic Trends, Chem. Rev., 2016, 116, 5642–5687 CrossRef CAS.
- A. A. Clark, B. Yang, M. T. Rodgers and P. B. Armentrout, Experimental and Computational Study of the Group 1 Metal Cation Chelates with Lysine: Bond Dissociation Energies, Structures, and Structural Trends, J. Phys. Chem. B, 2019, 123, 1983–1997 CrossRef CAS.
- T. D. Vaden and J. M. Lisy, Characterization of hydrated Na + (phenol) and K + (phenol) complexes using infrared spectroscopy, J. Chem. Phys., 2004, 120, 721–730 CrossRef CAS.
- T. D. Vaden and J. M. Lisy, Competition between cation–π interactions and intermolecular hydrogen bonds in alkali metal ion-phenol clusters. I. Phenol dimer, J. Chem. Phys., 2005, 123, 074302 CrossRef.
- T. D. Vaden and J. M. Lisy, Competition between cation–π interactions and intermolecular hydrogen bonds in alkali metal ion-phenol clusters. II. Phenol trimer, J. Chem. Phys., 2006, 124, 214315 CrossRef.
- S. E. Wheeler, Understanding substituent effects in noncovalent interactions involving aromatic rings, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS.
- V. Ryzhov and R. C. Dunbar, Interactions of phenol and indole with metal ions in the gas phase: Models for Tyr and Trp side-chain binding, J. Am. Chem. Soc., 1999, 121, 2259–2268 CrossRef CAS.
- S. Hoyau, K. Norrman, T. B. McMahon and G. Ohanessian, A Quantitative Basis for a Scale of Na+ Affinities of Organic and Small Biological Molecules in the Gas Phase, J. Am. Chem. Soc., 1999, 121, 8864–8875 CrossRef CAS.
- R. C. Dunbar, Metal Cation Binding to Phenol: DFT Comparison of the Competing Sites, J. Phys. Chem. A, 2002, 106, 7328–7337 CrossRef CAS.
- S. Memoli, A. Napolitano and M. D'Ischia, Giovanna Misuraca; Palumbo, A.; Prota, G. Diffusible melanin-related metabolites are potent inhibitors of lipid peroxidation, Biochim. Biophys. Acta, 1997, 1346, 61–68 CrossRef.
- Z. Xu, Mechanics of metal-catecholate complexes: The roles of coordination state and metal types, Sci. Rep., 2013, 3, 2914 CrossRef.
- R. C. Dunbar, Binding of Na+, Mg+, and Al+ to the π faces of naphthalene and indole: Ab initio mapping study, J. Phys. Chem. A, 1998, 102, 8946–8952 CrossRef CAS.
- L. J. Juszczak and A. S. Eisenberg, The Color of Cation–π Interactions: Subtleties of Amine-Tryptophan Interaction Energetics Allow for Radical-like Visible Absorbance and Fluorescence, J. Am. Chem. Soc., 2017, 139, 8302–8311 CrossRef CAS.
- B. Ivanova and M. Spiteller, Cation–π-complex of Ag(I) ion with 1H-indole-5-carboxylic acid–Structural analysis and energetics of the M–L bonds, Inorg. Chim. Acta, 2018, 471, 219–222 CrossRef CAS.
- K. E. Riley, J. A. Platts, J. Řezáč, P. Hobza and J. G. Hill, Assessment of the Performance of MP2 and MP2 Variants for the Treatment of Noncovalent Interactions, J. Phys. Chem. A, 2012, 116, 4159–4169 CrossRef CAS.
- V. Barone, I. Cacelli, O. Crescenzi, M. D'Ischia, A. Ferretti, G. Prampolini and G. Villani, Unraveling the interplay of different contributions to the stability of the quinhydrone dimer, RSC Adv., 2014, 4, 876–885 RSC.
- G. Prampolini, I. Cacelli and A. Ferretti, Intermolecular interactions in eumelanins: a computational bottom-up approach. I. small building blocks, RSC Adv., 2015, 5, 38513–38526 RSC.
- V. Barone, I. Cacelli, A. Ferretti and G. Prampolini, Noncovalent Interactions in the Catechol Dimer, Biomimetics, 2017, 2, 18 CrossRef.
- M. Jacobs, L. Greff Da Silveira, G. Prampolini, P. R. Livotto and I. Cacelli, Interaction Energy Landscapes of Aromatic Heterocycles through a Reliable yet Affordable Computational Approach, J. Chem. Theory Comput., 2018, 14, 543–556 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- S. Boys and F. Bernardi, The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. Leininger, A. Nicklass, W. Küchle, H. Stoll, M. Dolg and A. Bergner, The accuracy of the pseudopotential approximation: Non-frozen-core effects for spectroscopic constants of alkali fluorides XF (X = K, Rb, Cs), Chem. Phys. Lett., 1996, 255, 274–280 CrossRef CAS.
- I. Cacelli, F. Lipparini, L. G. da Silveira, M. Jacobs, P. R. Livotto and G. Prampolini, Accurate interaction energies by spin component scaled Moller-Plesset second order perturbation theory calculations with optimized basis sets (SCS-MP2(mod)): Development and application to aromatic heterocycles, J. Chem. Phys., 2019, 150, 234113 CrossRef.
- P. Wójcik, T. Korona and M. Tomza, Interactions of benzene, naphthalene, and azulene with alkali-metal and alkaline-earth-metal atoms for ultracold studies, J. Chem. Phys., 2019, 150, 234106 CrossRef.
- M. Albertí and N. F. Lago, Competitive solvation of K+ by C6H6 and H2O in the K+-(C6H6)n-(H2O)m (n = 1–4; m = 1–6) aggregates, Eur. Phys. J. D, 2013, 67, 73 CrossRef.
- I. Cacelli, G. Cinacchi, G. Prampolini and A. Tani, Computer Simulation of Solid and Liquid Benzene with an Atomistic Interaction Potential Derived from Ab Initio Calculations, J. Am. Chem. Soc., 2004, 126, 14278–14286 CrossRef CAS.
- I. Cacelli, C. F. Lami and G. Prampolini, Force-field Modeling through Quantum Mechanical Calculations: Molecular Dynamics Simulations of a Nematogenic Molecule in its Condensed Phases, J. Comput. Chem., 2009, 30, 366–378 CrossRef CAS.
- I. Cacelli, A. Cimoli, P. R. Livotto and G. Prampolini, An Automated Approach for the Parameterization of Accurate Intermolecular Force-Fields: Pyridine as a Case Study, J. Comput. Chem., 2012, 33, 1055 CrossRef CAS.
- L. Greff da Silveira, M. Jacobs, G. Prampolini, P. R. Livotto and I. Cacelli, Development and Validation of Quantum Mechanically Derived Force-Fields: Thermodynamic, Structural, and Vibrational Properties of Aromatic Heterocycles, J. Chem. Theory Comput., 2018, 14, 4884–4900 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp03707a |
| This journal is © the Owner Societies 2020 |