
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shining light on the electronic structure and relaxation dynamics of the isolated oxyluciferin anion†
Anand M.
Patel
 ,
Alice
Henley
,
Michael A.
Parkes
,
Mariana
Assmann
,
Graham A.
Worth
,
James C.
Anderson
and
Helen H.
Fielding
*
,
Alice
Henley
,
Michael A.
Parkes
,
Mariana
Assmann
,
Graham A.
Worth
,
James C.
Anderson
and
Helen H.
Fielding
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.h.fielding@ucl.ac.uk
First published on 12th August 2020
Abstract
Firefly bioluminescence is exploited widely in imaging in the biochemical and biomedical sciences; however, our fundamental understanding of the electronic structure and relaxation processes of the oxyluciferin that emits the light is still rudimentary. Here, we employ photoelectron spectroscopy and quantum chemistry calculations to investigate the electronic structure and relaxation of a series of model oxyluciferin anions. We find that changing the deprotonation site has a dramatic influence on the relaxation pathway following photoexcitation of higher lying electronically excited states. The keto form of the oxyluciferin anion is found to undergo internal conversion to the fluorescent S1 state, whereas we find evidence to suggest that the enol and enolate forms undergo internal conversion to a dipole bound state, possibly via the fluorescent S1 state. Partially resolved vibrational structure points towards the involvement of out-of-plane torsional motions in internal conversion to the dipole bound state, emphasising the combined electronic and structural role that the microenvironment plays in controlling the electronic relaxation pathway in the enzyme.
1 Introduction
Bioluminescence is the production and emission of light by a living organism. It is one of the most spectacular processes in nature and is observed widely in terrestrial and marine organisms.1,2 The light is produced by catalytic oxidation of a small molecule (luciferin) by an enzyme (luciferase). The most studied bioluminescence reactions are those of jellyfish3 and fireflies,4 of which the brightest is the bioluminescence of the North American Photinus pyralis firefly (quantum yield, Φ = 0.41).5 In fireflies, luciferase (Luc) catalyses adenylation and oxidation of D-luciferin (D-LH2) by O2 in the presence of adenosine triphosphate (ATP) and Mg2+ co-factors, to form the deprotonated keto-oxyluciferin anion (phenolate-keto) in its first electronically excited singlet state, S1 (Fig. 1). Subsequent relaxation to the ground electronic state, S0, results in the emission of yellow-green light (λmax = 558 nm).5 Despite the fact that all bioluminescent beetle species use the same small molecule and the same reaction to produce light, the colour of the emission varies from green to red depending on the species,6,7 or in the case of the Pyrophorus plagiophthalamus click beetle, even between individuals.6,8 Wavelength shifts are also observed when changing Luc in vitro and have been attributed to different amino acid residues.9 Thus, it is clear that the microenvironment of the luciferin plays a key role in defining its electronic properties.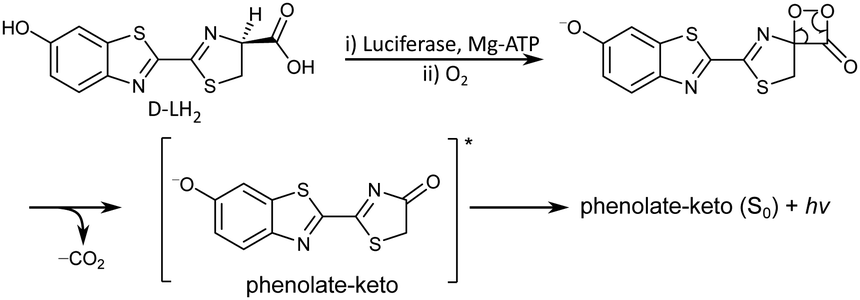 | ||
| Fig. 1 Reaction mechanism of firefly bioluminescence. | ||
Bioluminescence imaging has revolutionised the biochemical and biomedical sciences.4,10–13 Since it relies on the expression of Luc to catalyse the bioluminescence of a luciferin substrate, it does not require an external light source, unlike fluorescence imaging, so there are no background photons. Consequently, bioluminescence imaging has a higher signal/noise ratio than fluorescence imaging,14 which is particularly attractive for bioanalytical methods that require enhanced sensitivity, such as visualising tumour growth, gene expression, protein–protein interactions, cell proliferation, migration, differentiation and total cell distribution in small animals.15 Advances in bioluminescence imaging require brighter and multi-coloured near-infrared emitting luciferins for improved in vivo tissue penetration16 and new multiparametric techniques.17 To date, modifications to luciferins have successfully red-shifted the light emission; however, this has come at the cost of reducing the bioluminescence quantum yield.18 One of the research aims of our groups is to improve the fundamental understanding of bioluminescence for the rational design of new bioluminescence tools.19–22 The focus of the work described in this paper is to improve our understanding of the electronic structure of the light-emitting oxyluciferin (OL).
The solution chemistry of firefly OL is very complex. In physiologically relevant pH conditions, OL exists as a triple equilibrium of six chemical forms as a result of keto–enol tautomerism of the 4-thiazolone moiety and deprotonation of the two hydroxyl groups (Fig. 2).23–25 Using a chemical oriented multivariate data analysis procedure, Rebarz et al. were able to unravel the absorption spectra of the individual components, their pH dependent profiles and distributions, and accurate values for the phenol-phenolate, enol-enolate and keto–enol equilibrium constants.26 Ghose et al. used a similar procedure to determine the steady-state and time-resolved emission profiles of the individual components and accurate values for the excited-state phenol-phenolate, enol-enolate and keto–enol equilibrium constants,27 confirming the importance of excited-state keto–enol tautomerism reported in earlier work.28 Recently, Gosset et al. used femtosecond transient absorption spectroscopy and nanosecond fluorescence decay measurements to investigate the picosecond dynamics of the excited-state proton transfer reactions and excited state keto–enol conversion in aqueous buffer as a function of pH.29 A global and target state analysis of steady-state and time-resolved emission spectra of firefly OL complexed with Luc from the Japanese firefly Luciola cruciata revealed that the light-emission process of luciferin/Luc in vitro is complex and pH-dependent.30 The fluorescence spectra of the neutral forms of OL have been shown to be too blue-shifted to be responsible for the light emission process,23–26 which is consistent with the anionic form of luciferin being required to trigger electron transfer from the benzothiazole building block to lower the activation energy for collapse of the strained peroxide intermediate.31–34 However, the chemical identity of the firefly bioluminescence emitter remains uncertain.
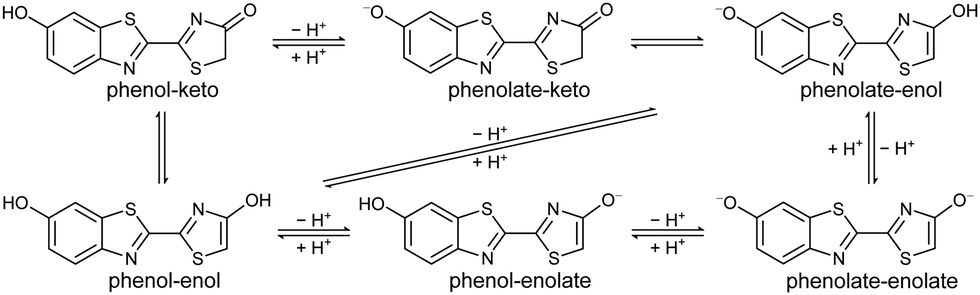 | ||
| Fig. 2 Chemical equilibria of firefly OL. | ||
Studying the intrinsic electronic structure and dynamics of the different chemical forms of firefly OL in the gas phase, free from perturbations from solvent or enzyme environments, is the perfect starting point for a bottom-up approach to unravelling the role of the microenvironment in controlling the bioluminescence wavelengths and quantum yields. To the best of our knowledge, there are only three reported experimental studies of firefly luciferin and OL in the gas phase. Action absorption spectra of firefly luciferin and OL have been recorded by monitoring the formation of charged and neutral fragments as a function of photon energy.35,36 The study of the OL anion showed that solvation by a single water molecule produced a blueshift in the absorption spectrum, suggesting that water molecules in the enzyme binding site could contribute to the wavelength shift observed in pH-sensitive Lucs.35 We reported a combined photoelectron spectroscopy and computational study of the deprotonated luciferin and its red-shifted analogue, infraluciferin.37 This study showed that the microenvironment of the luciferin controlled its conformation in the enzyme. Competing light-induced internal conversion, electron emission and decarboxylation processes were also observed. Here, we present the first combined photoelectron spectroscopy and computational study of the singly deprotonated anionic chemical forms of the OL emitter. Photoelectron spectra of a series of model OL analogues with well-defined deprotonation sites (Fig. 3), allow us to unravel the contributions of the three chemical forms.
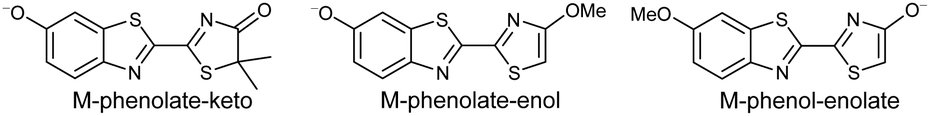 | ||
| Fig. 3 Model analogues of firefly OL studied in this work. | ||
2 Methods
2.1 Anion photoelectron spectroscopy
OL, 2-(6-hydroxybenzo[d]thiazol-2-yl)-5,5-dimethylthiazol-4(5H)-one (1), 2-(4-methoxythiazol-2-yl)benzo[d]thiazol-6-ol (2) and 2-(6-methoxybenzo[d]thiazol-2-yl)thiazol-4-ol (3) (Section S1, ESI†) were synthesised according to synthetic procedures in the literature.24,26,38 Anions (Fig. 2 and 3) were generated by addition of a drop of 28% ammonia solution to 1 mM solutions (5 mL) of OL, prepared in either dry MeCN or dry MeOH, and 1, 2 and 3, prepared in dry MeOH.Photoelectron spectra were recorded using our anion photoelectron imaging spectrometer which combines electrospray ionisation (ESI), a quadrupole mass filter, hexapole ion trap and velocity map imaging. The design of our spectrometer has been described in detail in an earlier publication39 and subsequent improvements to the operating procedures have been detailed in more recent publications.40,41 Briefly, negative ion ESI produced deprotonated anions of the chromophore of interest which were mass-selected using a quadrupole mass filter. These ions were then accumulated in a hexapole ion trap and thermalised using He gas before being focussed into the source region of the photoelectron imaging spectrometer where they interacted perpendicularly with a collimated ultraviolet (UV) laser beam with diameter around 3 mm. Wavelengths for photoionisation in the range 294–359 nm were generated by frequency-doubling the output of a dye laser, pumped by the second harmonic of a nanosecond Nd:YAG laser (532 nm). The power at the exit of the chamber was 1–5 mW. Photoelectrons generated from the interaction between the ions and the laser light were velocity-mapped onto a two-dimensional charge-coupled device camera coupled to a phosphor screen. Laser-only images were recorded and subtracted from the total signal. The resulting photoelectron images were inverted using the pBASEX method42 and the electron kinetic energy was calibrated using the photodetachment spectrum of I−.43 The kinetic energy resolution of the spectrometer is ΔE/E < 5%.
2.2 Quantum chemistry calculations
The minima of all OL− and model analogue anions, and their corresponding neutral radicals, were optimised using density functional theory44 (DFT) with the B3LYP45–48 hybrid functional and the 6-311++G(2df,2pd)49–51 basis set. Vibrational analysis was performed using the same level of theory to ensure true minima were reached and for use in photoelectron spectrum (PES) simulations (see below). These calculations were performed with the Gaussian0952 program. Vertical detachment energies (VDEs) were calculated using the equation-of-motion coupled-cluster method with single and double excitations for the calculation of ionisation potentials53 (EOM-IP-CCSD) as implemented in the Q-Chem program package,54 with the aug-cc-pVDZ55 basis set; in earlier work, this method was found to agree well with experimental VDEs of luciferin and infra-luciferin.37 Vertical excitation energies (VEEs) were obtained using the algebraic diagrammatic construction method to second order56,57 (ADC(2)) with the aug-cc-pVDZ basis set. These calculations were performed using the Turbomole computational chemistry software package with the resolution of the identity approximation.58 The excited states above the detachment threshold contain continuum states. To account for the interaction of resonance states with the continuum, a basis set with diffuse functions is necessary. The size of the basis set determines the number of continuum states that are calculated. Moreover, the continuum states obtained will change depending on the number of states calculated, but the lower lying continuum states converge with increasing number of excited states.59 For this reason, we used the aug-cc-pVDZ basis set and calculated up to 25–35 excited states of A′ and A′′ symmetry to ensure the convergence of the lower energy states.Stick photoelectron spectra of M-phenolate-keto were calculated using ezSpectrum (version 3.0).60 This required the equilibrium geometries, harmonic frequencies, and normal mode vectors of the (initial) ground electronic and (target) neutral radical states of M-phenolate-keto for calculating the overlaps between the initial and target vibrational wavefunctions using the parallel normal mode approximation. The vibrational temperature of the anions was assumed to be 300 K and the minimum intensity threshold was set to 0.001. The maximum numbers of vibrational quanta in the anion and neutral radical were limited to 2 and 4, respectively. The resulting stick spectra were convoluted with Gaussian instrumental profiles with full-width at half maxima (FWHM) equivalent to the instrumental resolution of the experimental spectrum, ΔE/E = 3.4% at 0.8 eV eKE.
3 Results
3.1 Photoelectron spectra
359 nm (3.45 eV) photoelectron spectra of M-phenolate-keto, M-phenolate-enol and M-phenol-enolate model analogues are presented in the top panel of Fig. 4; corresponding photoelectron spectra of OL− generated by ESI from MeOH and MeCN are presented in the bottom panel. The spectra are recorded as a function of electron kinetic energy, eKE, and are presented as a function of electron binding energy, eBE = hν − eKE. The corresponding photoelectron images are presented in Fig. S1 and S2 (ESI†).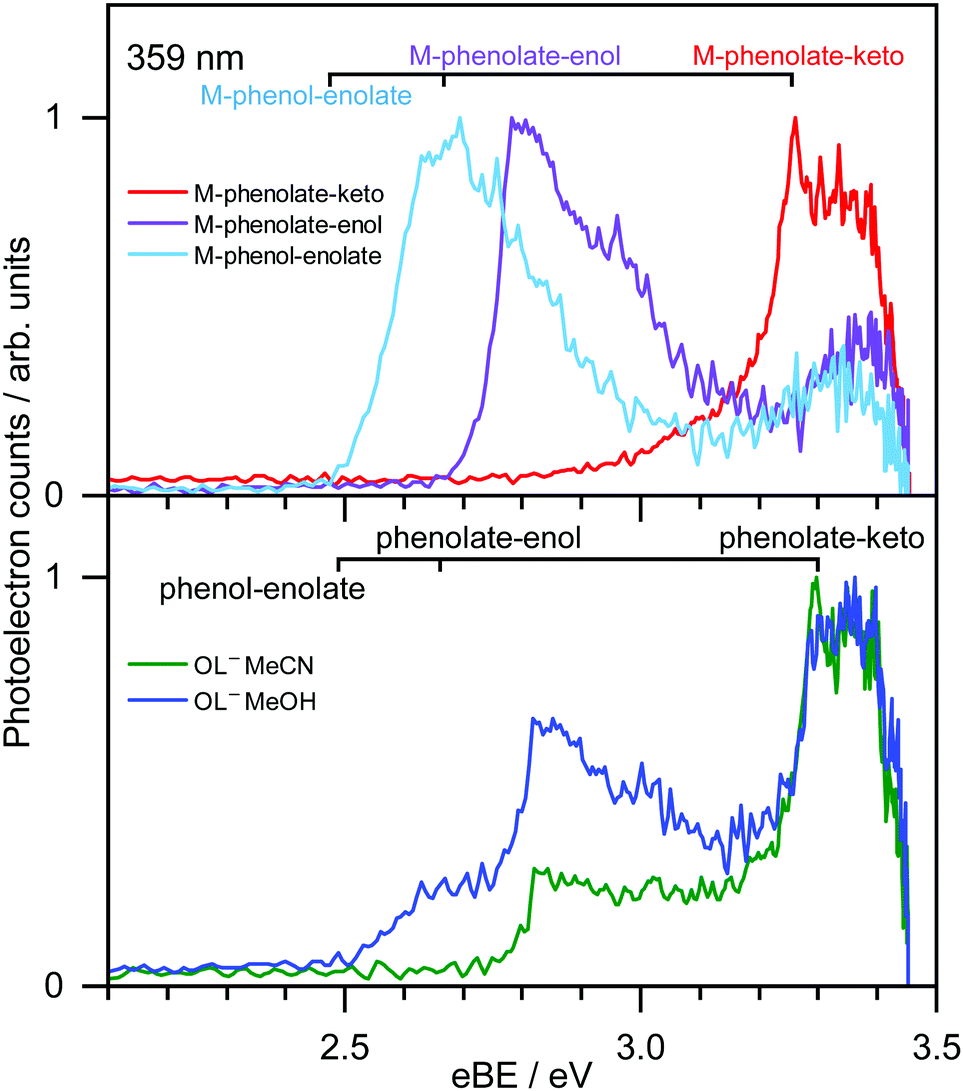 | ||
| Fig. 4 359 nm (3.45 eV) photoelectron spectra of M-phenolate-keto (red), M-phenolate-enol (purple) and M-phenol-enolate (light blue) generated by ESI of 1 mM MeOH solutions, plotted as a function of eBE (top). Photoelectron spectra of OL− generated by ESI from 1 mM dry MeOH (blue) and MeCN (green) solutions, plotted as a function of eBE (bottom). Combs mark EOM-IP-CCSD/aug-cc-pVDZ calculated VDEs of the trans conformers. | ||
The M-phenolate-keto photoelectron spectrum is characterised by a single, structured peak which rises to a maximum around 3.3 ± 0.1 eV. The M-phenolate-enol and M-phenol-enolate photoelectron spectra are red-shifted with respect to the M-phenolate-keto photoelectron spectrum and have broad, unresolved peaks with maxima around 2.8 ± 0.1 eV and 2.7 ± 0.1 eV, respectively. They both also have features at high eBE (low eKE). The baseline of the 359 nm photoelectron spectrum for M-phenolate-keto is non-zero from 1.3–2.8 eV. We attribute this to two-photon detachment, which is consistent with calculated detachment energies (Table 1). There is also a tail on the low eBE edge of the main detachment peak that we attribute to detachment from a fragment anion (Fig. S4, ESI†).
| Anion | ΔE | Geometry | D0 | D1 | D2 | Exp. | |||
|---|---|---|---|---|---|---|---|---|---|
| VDE | Hole | VDE | Hole | VDE | Hole | ||||
| trans-phenolate-keto | 0.00 |
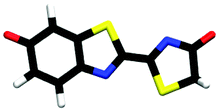
|
3.30 | πH | 5.07 | nO(ph) | 5.39 | πH−1 | 3.30 ± 0.14 |
| trans-phenolate-enol | 0.68 |
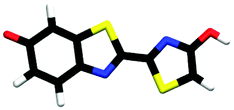
|
2.66 | πH | 4.38 | nO(ph) | 4.81 | πH−1 | 2.82 ± 0.11 |
| trans-phenol-enolate | 0.79 |
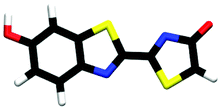
|
2.49 | πH | 4.18 | nO(en) | 5.07 | πH−1 | 2.63 ± 0.20 |
| cis-phenolate-keto | 0.23 |
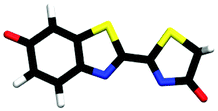
|
3.31 | πH | 5.12 | nO(ph) | 5.46 | πH−1 | |
| cis-phenolate-enol | 0.88 |
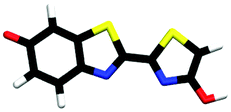
|
2.69 | πH | 4.42 | nO(ph) | 4.83 | πH−1 | |
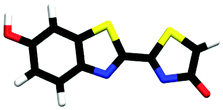
| cis-phenol-enolate | 1.05 |
|
2.50 | πH | 4.17 | nO(en) | 5.11 | πH−1 | |
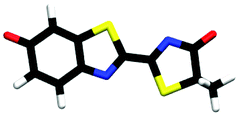
| trans-M-phenolate-keto | 0.00a |
|
3.26 | πH | 5.04 | nO(ph) | 5.35 | πH−1 | 3.26 ± 0.12 |
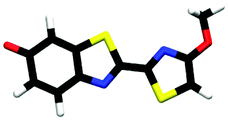
| trans-M-phenolate-enol | 0.00b |
|
2.65 | πH | 4.38 | nO(ph) | 4.80 | πH−1 | 2.78 ± 0.11 |
| trans-M-phenol-enolate | 0.25b |
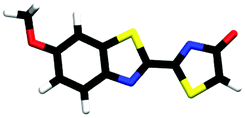
|
2.45 | πH | 4.15 | nO(en) | 4.99 | πH−1 | 2.69 ± 0.10 |
| cis-M-phenolate-keto | 0.23a |
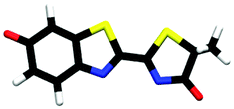
|
3.27 | πH | 5.09 | nO(ph) | 5.41 | πH−1 | |
| cis-M-phenolate-enol | 0.19b |
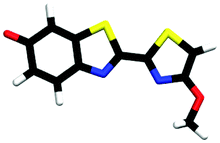
|
2.68 | πH | 4.41 | nO(ph) | 4.79 | πH−1 | |
| cis-M-phenol-enolate | 0.51b |
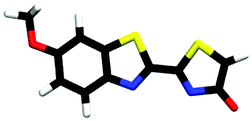
|
2.46 | πH | 4.14 | nO(en) | 5.04 | 0H−1 | |
The photoelectron spectrum of OL− generated by ESI from MeOH has three features that, from comparison with the spectra of the M-phenolate-keto, M-phenolate-enol and M-phenol-enolate analogues, can be attributed to contributions from all three deprotomers of the singly deprotonated ion. Similarly, it can be deduced that the photoelectron spectrum of OL− generated by ESI from MeCN can be attributed to contributions solely from phenolate-keto and phenolate-enol OL−. The spectra of M-phenolate-keto, M-phenolate-enol and M-phenol-enolate are red-shifted by around 0.05 eV with respect to those of the phenolate-keto, phenolate-enol and phenol-enolate forms of OL−; this can be understood in terms of the model anions being slightly destabilised by the weak electron-donating inductive effect of the methyl groups. By fitting shifted photoelectron spectra of the model analogues (Fig. S7, ESI†), the ratio of contributions to the spectra from the phenolate-keto, phenolate-enol and phenol-enolate forms of OL− is found to be approximately 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 following ESI from MeOH solution and the ratio of contributions to the spectra from phenolate-keto and phenolate-enol forms of OL− is approximately 4:1 following ESI from MeCN solution.
:4:1 following ESI from MeOH solution and the ratio of contributions to the spectra from phenolate-keto and phenolate-enol forms of OL− is approximately 4:1 following ESI from MeCN solution.
Photoelectron spectra of model analogues recorded in the range 359 nm (3.45 eV) to 294 nm (4.22 eV) are presented in Fig. 5, along with corresponding photoelectron spectra of OL− electrosprayed from MeOH and MeCN solutions. The overall profiles are different for the three analogues, so we discuss them separately. For M-phenolate-keto, the feature with a maximum around 3.3 ± 0.1 eV is independent of wavelength and thus attributed to direct detachment to the ground electronic state of the corresponding neutral radical, D0. The rising edge of the 359 nm spectrum is broader and this broadening decreases with increasing photon energy. As discussed above, we attribute this to photodetachment of a fragment anion. Apart from in the vicinity of this broadening, the photoelectron angular distributions (PADs) are characterised by slightly negative β2 values across the whole spectrum for all photon energies (Fig. S3, ESI†). There is evidence of some unresolved vibrational structure (3.3–4.0 eV), suggesting that the anion and corresponding neutral radical are reasonably rigid. As the photon energy is increased, a continuum of photoelectron kinetic energies is observed from eBE = D0 towards eBE ∼ hν. The photoelectron counts decrease over the highest 0.5 eV to zero when eBE = hν.
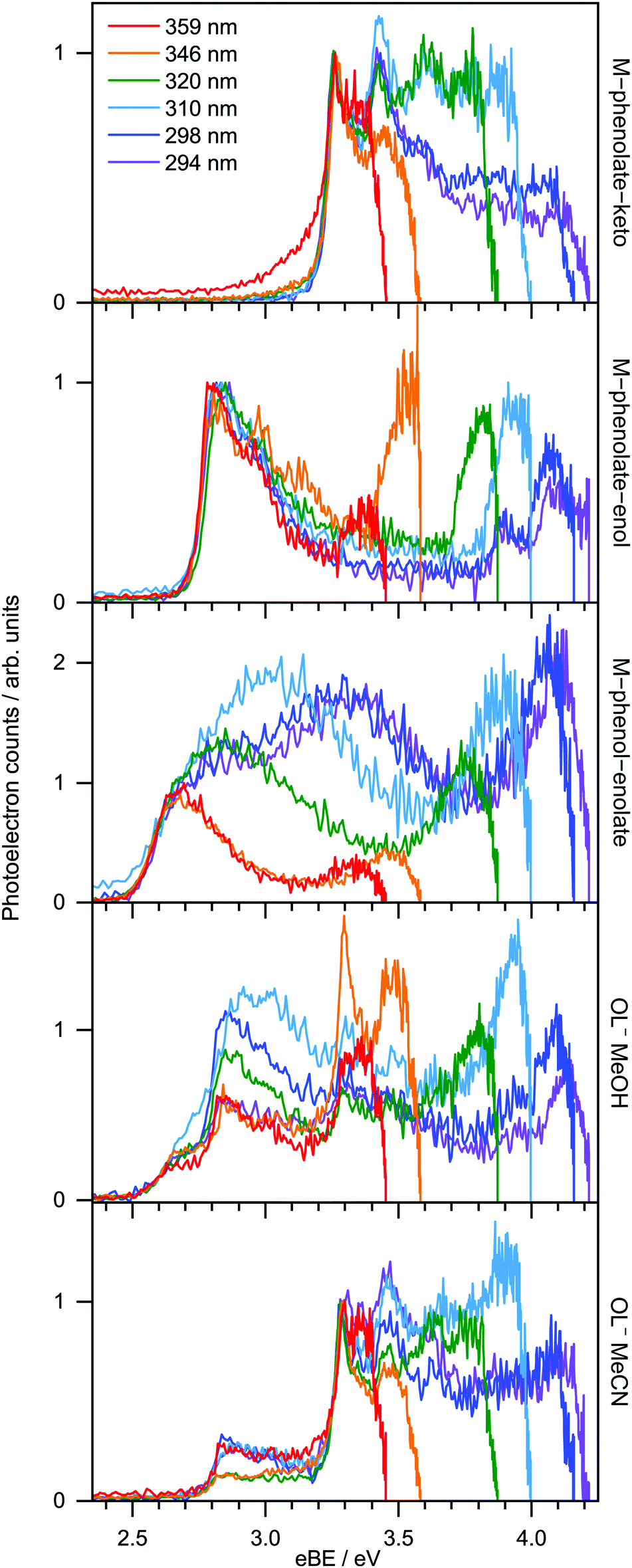 | ||
| Fig. 5 Photoelectron spectra recorded at 294 nm (4.22 eV), 298 nm (4.16 eV), 310 nm (4.00 eV), 320 nm (3.87 eV), 346 nm (3.58 eV) and 359 nm (3.45 eV) for M-phenolate-keto, M-phenolate-enol, M-phenol-enolate and OL− sprayed from MeOH and MeCN. The spectra of the model analogues are normalised to their rising edges and the OL− spectra are normalised to the peaks around 2.6 eV (MeOH) and 3.3 eV (MeCN). | ||
For M-phenolate-enol, the feature with a maximum around 2.8 ± 0.1 eV is independent of wavelength and attributed to direct detachment to the ground electronic state of the corresponding neutral radical, D0. This feature is completely unresolved in most of the spectra although there is clear evidence of oscillatory structure across the 346 nm spectrum. In the 310–294 nm photoelectron spectra, there is a feature around 3.9 eV that remains constant with increasing photon energy. This could be attributed to direct detachment to D1. There is an additional feature at high eBE that shifts linearly with photon energy. It does not have the characteristic exponential profile associated with thermionic emission (Fig. S8, ESI†),61–65 so must be attributed to autodetachment from a resonance, or resonances, lying in the detachment continuum. As with M-phenolate-keto, the PADs are characterised by slightly negative β2 values across the whole spectrum for all photon energies (Fig. S5, ESI†).
For M-phenol-enolate, the feature with a maximum around 2.7 ± 0.1 eV is independent of wavelength and attributed to direct detachment to the ground electronic state of the corresponding neutral radical, D0. This feature is completely unresolved in all the photoelectron spectra. There is a tail on the low eBE edge of this peak in the 310 nm spectrum that we believe is detachment from a fragment anion, similar to M-phenolate-keto. Similar to M-phenolate-enol, there is an additional feature at high eBE that shifts linearly with photon energy and does not have the characteristic exponential profile associated with thermionic emission (Fig. S9, ESI†), so it is also attributed to autodetachment from a resonance, or resonances, lying above the detachment threshold. Unlike the M-phenolate-keto and M-phenolate-enol spectra, there is significant broadening on the high eBE side of the peak associated with direct detachment to D0; this changes shape and shifts to higher eBE with increasing photon energy. In the 346–294 nm photoelectron spectra plotted as a function of eKE (Fig. S10, ESI†), it can be seen that this broadening is attributed to a feature at constant eKE ∼ 0.9 eV. As a result of the propensity for conserving vibrational energy during autodetachment, indirect detachment following photoexcitation of an excited state Sn with excess vibrational energy, Ev = hν − E(Sn), where E(Sn) is the adiabatic excitation energy (AEE) of Sn, will result in the emission of photoelectrons with eKE ∼ hν − E(D0) − Ev, where E(D0) is the ADE. This last expression can be rewritten as, eKE ∼ E(Sn) − E(D0); i.e. the photoelectrons are emitted with eKE corresponding to the Sn − D0 energy difference (Fig. 6). Thus, the additional feature at 0.9 eV eKE can be attributed to detachment from a resonance with an onset around 3.5 eV (∼355 nm). Broadening of photoelectron spectra, following photoexcitation of resonances above the detachment threshold, was observed in our study of deprotonated luciferin and infraluciferin ions and has been observed in photoelectron spectra of a range of other similar sized molecular anions recorded by various groups66–68 including our own.69 The PADs support our interpretation of a resonance: β2 ∼ 0 across the whole spectrum at all photon energies apart from the feature with eKE ∼ 0.9 eV where β2 < 0 (Fig. S6, ESI†).
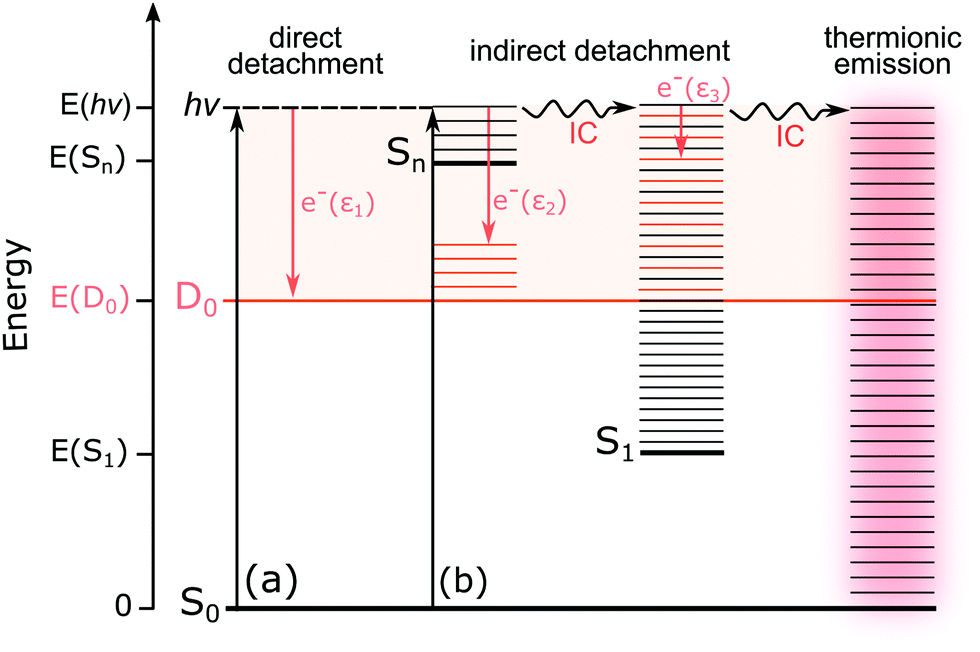 | ||
| Fig. 6 Schematic energy level diagram illustrating the possible excited-state pathways for the OL− anions. Thin horizontal black lines represent the vibrational levels of the electronic states of the anion and the pale orange shaded area represents the electron detachment continuum. Vertical orange arrows represent the eKE of direct and indirect electron detachment processes and the thin horizontal orange lines represent the vibrational energy left in the neutral radical following electron detachment (determined by the propensity for conservation of vibrational energy). The horizontal black arrows represent some of the possible internal conversion (IC) processes and thermionic emission (TE). (a) Direct photodetachment from S0 to the D0 continuum gives electrons with eKE, ε1 ∼ hv − VDE; in this particular example, VDE = ADE. (b) Indirect detachment following photoexcitation of Sn with excess vibrational energy, gives electrons with eKE, ε2 ∼ E(Sn) − E(D0). Indirect detachment following photoexcitation of Sn and subsequent IC to S1 gives electrons with eKE, ε3 ∼ 0. TE from S0 following photoexcitation of Sn and subsequent IC back to S0 gives electrons with an eKE Boltzmann distribution profile. | ||
3.2 Computational results
Table 1 lists the structures of the optimised OL− anions and the model analogues, together with the first three vertical detachment energies (VDEs) and the leading molecular orbitals from which the electrons are detached. For the phenolate-keto, phenolate-enol and phenol-enolate forms of the trans and cis conformers of OL−, the relative energies are around 0.0, 0.7 and 0.8 eV; this can be attributed to the degree of conjugation, reflected in the increasing length of the central C–C bond: 1.407, 1.424 and 1.434 Å.Potential energy scans of the OL− anions, and their corresponding neutral radicals, along the trans–cis coordinate (Fig. 7) show that for the phenolate-keto and phenolate-enol forms, the two minima correspond to planar trans and cis conformers. For the phenol-enolate, the two minima correspond to a planar trans conformer and a cis conformer in which the planes of the benzothiazole and thiazole moieties are twisted ∼13° with respect to one another. The same is true for the cis-M-phenol-enolate analogue. This can be explained by considering the resonance structures of the anions. For the phenolate-keto and phenolate-enol forms, the two resonance structures effectively delocalise the negative charge throughout the molecule; however, for the phenol-enolate form, the charge is localised on the thiazole moiety and the increased repulsion between the two sulphur atoms in the cis conformation forces the molecule to twist around the central C–C bond.
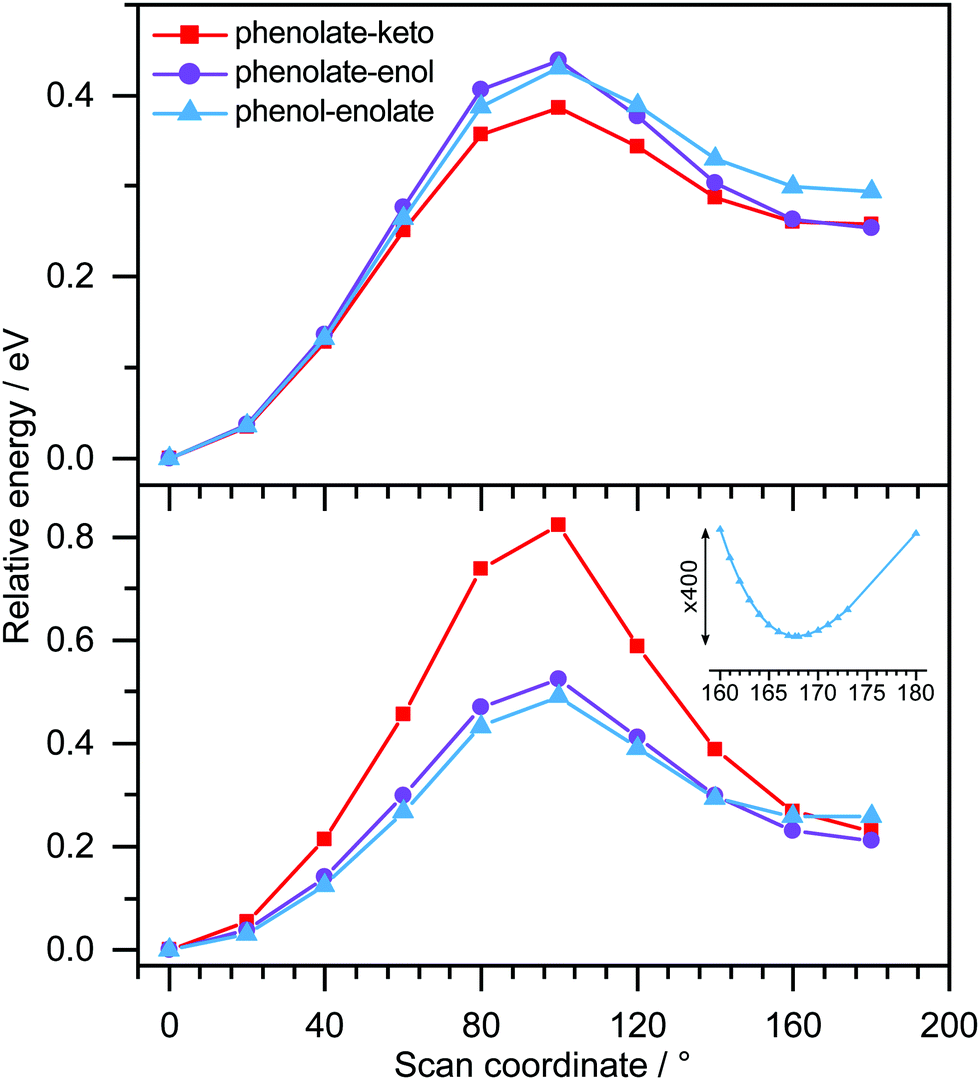 | ||
| Fig. 7 Relaxed potential energy scans of the OL− anions (bottom) and corresponding OL neutral radicals (top) as a function of twisting around the central C–C bond, starting from the trans-conformer at 0°, using the B3LYP/6-311++G(2df,2pd) method. The inset shows an expanded region of the potential energy scan for the phenol-enolate showing the minimum around 167°. | ||
As can be seen from Table 1, the D0 states correspond to electron holes in the highest occupied molecular orbitals (HOMOs), which have π character, πH. For phenolate-keto and phenolate-enol, the HOMOs are delocalised across the molecules, but for phenol-enolate, it is localised on the thiazole moiety. The D1 states correspond to electron holes in non-bonding orbitals on O atoms (nO). For phenolate-keto and phenolate-enol, the nO orbitals are localised on the phenolate O atoms, nO(ph), but for phenol-enolate, it is localised on the enolate O atom, nO(en). The D2 states correspond to electron holes in the HOMO−1 orbitals, which have π character, πH−1. For the phenolate-keto and phenolate-enol, these orbitals are localised on the benzothiazole rings, but for the phenol-enolate, it is delocalised across the molecules. The molecular orbitals of the model analogues are very similar to those of the corresponding OL− molecules. The HOMO, HOMO−1, nO(ph) and nO(en) orbitals for both the cis and trans forms of phenolate-keto, phenolate-enol and phenol-enolate OL− and corresponding model analogues are shown in Tables S1 and S2 (ESI†); it is worth noting that the orbitals are almost identical for the cis and trans forms.
The VEEs for the trans forms of phenolate-keto, phenolate-enol and phenol-enolate are listed in Table 2 and Tables S3–S5 (ESI†). We assume that the corresponding VEEs for the cis forms and model analogues will be similar since the VDEs and leading molecular orbitals from which detachment occurs are so similar. Our calculated VEEs for the S1 states of the trans-phenolate-keto, trans-phenolate-enol and trans-phenol-enolate OL− molecules are 2.20 eV (564 nm), 2.18 eV (569 nm) and 1.64 eV (756 nm), respectively. The values for the phenolate-keto and phenolate-enol OL− molecules are in good agreement with the maximum of an action absorption measurement of OL− sprayed from methanol solution (548 ± 10 nm).35 In the 3.45–4.22 eV photon energy range employed in our measurements, for trans-phenolate-keto and trans-phenolate-enol OL−, the electronically excited states with most significant oscillator strengths (f) are Feshbach resonances in the D0 continuum, lying at 3.68 eV (f = 0.18) and 3.87 eV (f = 0.12), respectively. For trans-phenol-enolate OL−, the only electronic transition with significant oscillator strength lying within this range is an excited shape resonance at 3.73 eV (f = 0.02), although there is a Feshbach resonance lying slightly higher at 4.34 eV (f = 0.25).
| State | Character | VEE | Oscillator strength | ||
|---|---|---|---|---|---|
| eV | nm | ||||
| Phenolate-keto | S1 | πH → π1* | 2.20 | 564 | 0.80 |
| S4 | πH−1 → π1* | 3.68 | 337 | 0.18 | |
| Phenolate-enol | S1 | πH → π1* | 2.18 | 569 | 0.65 |
| S11 | πH−1 → π1* | 3.87 | 320 | 0.12 | |
| Phenol-enolate | S1 | πH → π1* | 1.64 | 756 | 0.15 |
| S13 | πH → πdiff* | 3.73 | 332 | 0.02 | |
| S23 | πH−1 → π1* | 4.34 | 286 | 0.25 | |
4 Discussion
From the EOM-IP-CCSD calculated values of VDEs of OL− and the model anions (Table 1), we assign the peaks in the experimental photoelectron spectra around 3.3 eV, 2.8 eV and 2.6 eV (Fig. 4 and 5) to direct detachment to the ground electronic states of the neutral radicals, D0, of the phenolate-keto, phenolate-enol and phenol-enolate forms. The differences between the calculated values for the cis and trans forms is 0.01 eV, which is below the resolution of our measurements and means that we are unable to distinguish between the conformational isomers. The calculated VDEs are all within 0.15 eV of the experimental measurements, which is within the expected error of the method.70Similar to other studies of deprotonated anions formed by ESI, we find that the ratio of the contributions of the OL− deprotomers to the photoelectron spectra is dependent on the solvent used for the electrospray. The spectra recorded following ESI from MeCN are almost exclusively of phenolate-keto, the most stable species in vacuo, whereas the spectra recorded following ESI from MeOH have contributions from all three deprotomers. Interestingly, these observations contrast with conclusions drawn from the action absorption spectra of OL− sprayed from MeOH that were reported by Støckel et al.35 They observed a broad band (95 nm FWHM) centred around 548 ± 10 nm. Although they attributed this entirely to the action absorption spectrum of phenolate-keto, our calculations of the VEEs for the S1 state of phenolate-keto and phenolate-enol (564 nm and 569 nm, Tables S3 and S4, ESI†) suggest that it would be difficult to rule out a contribution from the phenolate-enol form to the action absorption spectrum. There is no evidence in the action absorption spectrum to suggest that the phenol-enolate form (VEE 756 nm, Table S5, ESI†) makes a significant contribution; this could be because the contribution is small (it only contributes 10% to the 359 nm photoelectron spectrum presented in Fig. 4) or that differences in the electrospray conditions71,72 result in an insignificant contribution from the phenol-enolate form.
In the 4.16 eV (298 nm) and 4.22 eV (294 nm) photoelectron spectra of M-phenolate-enol, a peak was observed around 3.9 eV which remains constant with increasing photon energy. This could be attributed to direct detachment to an electronically excited state of the neutral radical, D1. However, the calculated D1 VDE is 4.38 eV. This detachment threshold corresponds to detachment from a non-bonding orbital on the phenolate O atom and it is possible that the error in this calculation is larger than that for detachment from a π orbital (D0). Nonetheless, we do not see any evidence for detachment to the lower-energy D1 state (EOM-IP-CCSD VDE = 4.15 eV) in M-phenol-enolate (from a non-bonding orbital on the enolate O atom) and therefore we cannot assign the 3.9 eV peak in the M-phenolate-enol photoelectron spectrum to detachment to D1 with certainty.
The photoelectron spectra of M-phenol-enolate broaden on the high eBE side of the direct detachment peak and give rise to a feature at 0.9 eV eKE that is attributed to detachment from a resonance with an onset around 3.5 eV. Our ADC(2) calculations for trans-phenol-enolate show that the only transition with significant oscillator strength in this wavelength range is to a ππ* state at 3.73 eV, 1.24 eV above the S0–D0 VDE (Table 2). This ππ* state has excited shape resonance character with respect to D0 and is, therefore, strongly coupled to the D0 continuum. As a result, this ππ* state is expected to undergo rapid autodetachment to the D0 continuum.
We now turn our attention to the high eBE (low eKE) regions of the photoelectron spectra of the model analogues. In the M-phenolate-keto photoelectron spectra, a continuum of photoelectrons is observed from around eBE = D0 towards eBE ∼ hν, whereas in the M-phenolate-enol and M-phenol-enolate photelectron spectra, there are relatively narrow features (∼0.14 eV) at high eBE that are centred around 0.1 eV eKE (Fig. S8 and S9, ESI†).
The continuum of photoelectrons extending all the way to zero kinetic energy in the photoelectron spectra of M-phenolate-keto suggests that there is significant transfer of electronic energy into nuclear degrees of freedom. Our ADC(2) calculations for trans-phenolate-keto show that the only transition with significant oscillator strength in this wavelength range is to a ππ* state at 3.68 eV, 0.38 eV above the S0–D0 VDE (Table 2). This ππ* state has Feshbach resonance character with respect to D0 and is, therefore, only weakly coupled to the D0 continuum. As a result, it is expected to have a reasonably long autodetachment lifetime and internal conversion to lower lying electronically excited states will be able to compete with autodetachment. The S1 state has ππ* character and lies 1.1 eV below the D0 VDE; however, the vibrational levels of S1 that lie above the D0 detachment threshold have shape resonance character with respect to D0 and may, therefore, couple strongly to the detachment continuum. The continuum of low eKE photoelectrons is indicative of internal conversion from the photoexcited Feshbach resonance followed by internal conversion to S1 and subsequent vibrational autodetachment from S1. This would require the S1 state to be long-lived with respect to internal conversion to the ground-state in the gas phase, which would be consistent with solution phase measurements where the fluorescence lifetime of M-phenolate-keto has been found to be 1.9 ns (Φ = 0.26) in MeCN.24 Similar observations of low eKE photoelectrons attributed to autodetachment from lower lying electronically excited states populated by internal conversion from higher lying photoexcited states have been observed in photoelectron spectra of deprotonated luciferin and infraluciferin anions37 and the red Kaede protein.73 The fact that there is an efficient electronic relaxation pathway from a higher lying electronically excited state to the S1 state is significant because it suggests that isolated M-phenolate-keto could function as a UV activated fluorophore for bioimaging applications.
The low eKE peaks in the photoelectron spectra of M-phenolate-enol and M-phenol-enolate remain at constant eKE as photon energy increases across the range 3.45–4.22 eV, so they must arise from indirect electron detachment. They do not have the characteristic exponential profiles associated with thermionic emission from the ground electronic state. The fact that they are almost identical in all the photoelectron spectra recorded over this reasonably wide range of photon energies suggests that they must arise from detachment from an electronically excited state whose potential energy surface is parallel to that of the neutral, such as a weakly bound non-valence state.74–76 This is likely to be a dipole-bound state (DBS) due to the high dipole moments of the M-phenolate-enol and M-phenol-enolate neutral radicals (5.4 D and 6.0 D, respectively, for the trans forms) which are large enough to bind electrons.
For M-phenolate-enol, our ADC(2) calculations show that the only transition with significant oscillator strength in the 359–294 nm wavelength range is to a ππ* state at 3.87 eV, 1.21 eV above the S0–D0 VDE (Table 2). This ππ* state has Feshbach resonance character with respect to D0, similar to M-phenolate-keto. Consequently, internal conversion from this ππ* state to lower lying electronically excited states is expected to be able to compete efficiently with autodetachment. Therefore, we propose that the photoexcited Feshbach resonance undergoes internal conversion to a DBS, either directly or via S1 (Fig. 8). Subsequently, the DBS can autodetach to D0 and the reasonably narrow low eKE signal arises as a result of the similarity between the potential energy curves of the DBS and D0. Similar to M-phenolate-keto, if the S1 state of M-phenolate-enol is involved, its relatively long lifetime with respect to internal conversion back to the electronic ground-state would be consistent with solution phase measurements where the fluorescence lifetime has been found to be 4.88 ns (Φ = 0.47) in aqueous solution.27 Internal conversion from a high lying electronic state to a non-valence state has also been observed in a model photoactive yellow protein (PYP) chromophore.77
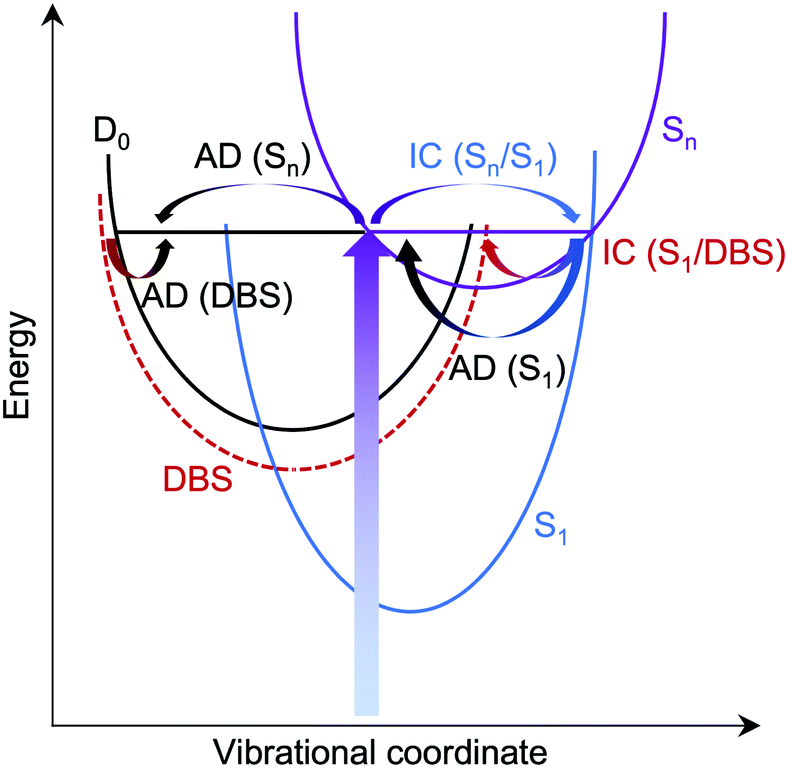 | ||
| Fig. 8 Schematic potential energy diagram illustrating possible autodetachment (AD) and internal conversion (IC) processes, following photoexcitation of a resonance in the D0 continuum, Sn. AD from S1 is responsible for the continuum of low eKE electrons observed in phenolate-keto photoelectron spectra, AD from Sn is responsible for the broadening observed on the high eBE edge of the peak associated with direct detachment in phenolate-enol photoelectron spectra and S1/DBS IC followed by AD from the DBS is responsible for the low eKE peaks in the phenolate-enol and phenolate-enolate photoelectron spectra. | ||
In addition to the peak centered around 0.1 eV, the 346 nm photoelectron spectrum of M-phenolate-enol has four sharp (∼0.01 eV) equally spaced peaks (∼0.024 eV) (Fig. 5 and Fig. S8, ESI†), characteristic of a vibrational progression.78 These peaks could be attributed to ν6 (167 cm−1), ν7 (184 cm−1), ν8 (192 cm−1) or ν9 (222 cm−1) progressions (Fig. S16, ESI†). Although the observation of a vibrational progression may, at first, seem inconsistent with the Δv = −1 propensity for pure vibrational autodetachment from a DBS,78 in M-phenolate-enol, vibrational autodetachment could take place from high-lying vibrational states of various vibrational modes of the DBS populated following internal conversion from the Feshbach resonance. Interestingly, an oscillatory structure is observed across the entire photoelectron spectrum (Fig. 5). This could be attributed to photoexcitation of low frequency vibrations in the Feshbach resonance that undergo competing autodetachment; however, time-resolved photoelectron spectroscopy measurements and quantum dynamics calculations would be required to confirm this.
For M-phenol-enolate, our observation of a low eKE feature attributed to autodetachment from a DBS, in addition to autodetachment from the photoexcited excited shape resonance described above, implies a bifurcation on the excited shape resonance potential energy surface with rapid internal conversion to the DBS competing with autodetachment.
Since the resonances excited in both M-phenolate-keto and M-phenolate-enol are Feshbach resonances, the molecular orbitals involved are very similar and the dipole moment of the M-phenolate-keto neutral radical is also large enough to bind an electron in a DBS (∼4.0 D), it is curious that the relaxation pathways are so different. A hint may lie in the observation of unresolved vibrational structure in the spectra of M-phenolate-keto. Our simulations of M-phenolate-keto photoelectron spectra (Fig. 9 and Fig. S14, ESI†) are dominated by in-plane vibrational modes (Fig. S15, ESI†). This can be explained by the relatively high barrier to rotation around the central C–C bond: ∼0.8 eV for phenolate-keto OL− compared to ∼0.5 eV for phenolate-enol OL− (Fig. 7). If out-of-plane torsions are also required for S1/DBS internal conversion, it is possible that this relaxation pathway is inhibited in M-phenolate-keto. This would also be consistent with the observation of out-of-plane torsional motions in the low eKE component of the 346 nm M-phenolate-enol photoelectron spectrum.
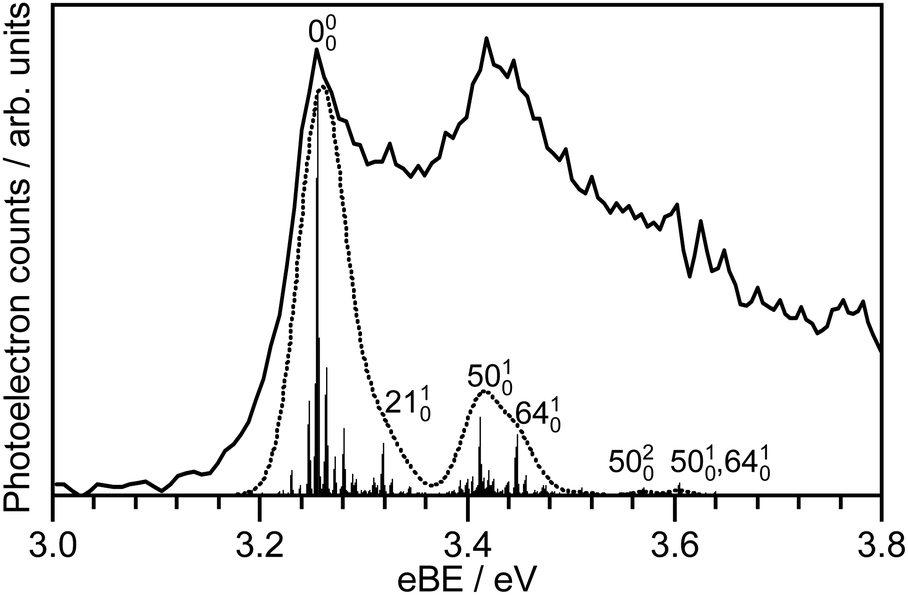 | ||
| Fig. 9 Calculated trans-M-phenolate-keto S0–D0 stick spectrum at 300 K convoluted with the instrument function (dashed line) and compared with the experimental 294 nm (4.22 eV) photoelectron spectrum (solid line). | ||
5 Conclusion
We have used photoelectron spectroscopy and quantum chemistry calculations to probe the electronic structure and relaxation dynamics of the higher lying electronically excited states of oxyluciferin bioluminescence emitters in the gas phase, free from perturbations with a solvent or enzyme environment, for the first time. We have shown that changing the deprotonation site has a dramatic influence on the relaxation dynamics following UV photoexcitation. We find that the phenolate-keto form undergoes internal conversion to the fluorescent S1 state, followed by vibrational autodetachment. In contrast, we find evidence to suggest that the phenolate-enol and phenol-enolate forms undergo internal conversion to a dipole bound state, possibly via the fluorescent S1 state, followed by vibrational autodetachment. Partially resolved vibrational structure in both the phenolate-keto and phenolate-enol forms leads us to propose that out-of-plane torsional motions around the central C–C bond are required for internal conversion to the DBS and that it is the restriction of these out-of-plane torsional motions in the phenolate-keto form that effectively turns off this relaxation pathway. This is significant because it highlights the value of photoelectron spectroscopy studies of isolated biochromophores in improving our understanding of the electronic and structural roles that complex biological environments play in controlling electronic relaxation pathways.The involvement of DBSs in the relaxation pathway from higher lying electronic states is also significant. Recent time-resolved photoelectron spectra revealing the involvement of DBSs in the electronic relaxation of analogues of the photoactive yellow protein chromophore77 have led to the suggestion that non-valence states may play an important role in the electronic relaxation mechanisms of other biochromophore anions in the gas phase.76 This work supports this idea and shows how the involvement of non-valence states can be identified from frequency-resolved photoelectron spectroscopy measurements following photoexcitation of higher lying electronic states. To determine the timescales of the relaxation process and the key vibrational modes will require time-resolved photoelectron spectroscopy measurements and quantum dynamics calculations, which we plan to do.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC through the award of DTA PhD studentships and grants EP/L005646/1 and EP/L005697/2. We are grateful to Dr Frank Otto for computational support and Dr Abil Aliev for NMR support.Notes and references
- S. H. Haddock, M. A. Moline and J. F. Case, Annu. Rev. Mar. Sci., 2010, 2, 443–493 CrossRef PubMed.
- E. N. Harvey, Bioluminescence, Academic Press, New York, 1952 Search PubMed.
- O. Shimomura, Bioluminescence: Chemical Principles and Methods, World Scientific, Singapore, 2006 Search PubMed.
- Z. M. Kaskova, A. S. Tsarkova and I. V. Yampolsky, Chem. Soc. Rev., 2016, 45, 6048–6077 RSC.
- Y. Ando, K. Niwa, N. Yamada, T. Enomoto, T. Irie, H. Kubota, Y. Ohmiya and H. Akiyama, Nat. Photonics, 2007, 2, 44 CrossRef.
- W. H. Biggley, J. E. Lloyd and H. H. Seliger, J. Gen. Physiol., 1967, 50, 1681–1692 CrossRef CAS PubMed.
- V. R. Viviani, Cell. Mol. Life Sci., 2002, 59, 1833–1850 CrossRef CAS PubMed.
- U. Stolz, S. Velez, K. V. Wood, M. Wood and J. L. Feder, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14955–14959 CrossRef CAS PubMed.
- V. R. Viviani, F. G. C. Arnoldi, A. J. S. Neto, T. L. Oehlmeyer, E. J. H. Bechara and Y. Ohmiya, Photochem. Photobiol. Sci., 2008, 7, 159–169 RSC.
- A. Kirkpatrick, T. Xu, S. Ripp, G. Sayler and D. Close, in Bioluminescence, ed. H. Suzuki, IntechOpen, London, 2019, ch. 1 Search PubMed.
- M. A. Paley and J. A. Prescher, MedChemComm, 2014, 5, 255–267 RSC.
- G. Morciano, A. C. Sarti, S. Marchi, S. Missiroli, S. Falzoni, L. Raffaghello, V. Pistoia, C. Giorgi, F. Di Virgilio and P. Pinton, Nat. Protoc., 2017, 12, 1542–1562 CrossRef CAS PubMed.
- L. Mezzanotte, M. van’t Root, H. Karatas, E. A. Goun and C. W. G. M. Löwik, Trends Biotechnol., 2017, 35, 640–652 CrossRef CAS PubMed.
- T. Troy, D. Jekic-McMullen, L. Sambucetti and B. Rice, Mol. Imaging, 2004, 3, 9–23 CrossRef CAS PubMed.
- C. M. Rathbun and J. A. Prescher, Biochemistry, 2017, 56, 5178–5184 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- B. R. Branchini, T. L. Southworth, D. M. Fontaine, D. Kohrt, C. M. Florentine and M. J. Grossel, Sci. Rep., 2018, 8, 5990 CrossRef PubMed.
- R. Podsiadły, A. Grzelakowska, J. Modrzejewska, P. Siarkiewicz, D. Słowiński, M. Szala and M. Świerczyńska, Dyes Pigm., 2019, 170, 107627 CrossRef.
- C. L. Stowe, T. A. Burley, H. Allan, M. Vinci, G. Kramer-Marek, D. M. Ciobota, G. N. Parkinson, T. L. Southworth, G. Agliardi, A. Hotblack, M. F. Lythgoe, B. R. Branchini, T. L. Kalber, J. C. Anderson and M. A. Pule, eLife, 2019, 8, e45801 CrossRef PubMed.
- J. C. Anderson, H. Grounds, A. P. Jathoul, J. A. H. Murray, S. J. Pacman and L. Tisi, RSC Adv., 2017, 7, 3975–3982 RSC.
- J. C. Anderson, C.-H. Chang, A. P. Jathoul and A. J. Syed, Tetrahedron, 2019, 75, 347–356 CrossRef CAS.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS PubMed.
- P. Naumov, Y. Ozawa, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 11590–11605 CrossRef CAS PubMed.
- T. Hirano, Y. Hasumi, K. Ohtsuka, S. Maki, H. Niwa, M. Yamaji and D. Hashizume, J. Am. Chem. Soc., 2009, 131, 2385–2396 CrossRef CAS PubMed.
- P. Naumov and M. Kochunnoonny, J. Am. Chem. Soc., 2010, 132, 11566–11579 CrossRef CAS PubMed.
- M. Rebarz, B.-M. Kukovec, O. V. Maltsev, C. Ruckebusch, L. Hintermann, P. Naumov and M. Sliwa, Chem. Sci., 2013, 4, 3803–3809 RSC.
- A. Ghose, M. Rebarz, O. V. Maltsev, L. Hintermann, C. Ruckebusch, E. Fron, J. Hofkens, Y. Mély, P. Naumov, M. Sliwa and P. Didier, J. Phys. Chem. B, 2015, 119, 2638–2649 CrossRef CAS PubMed.
- K. M. Solntsev, S. P. Laptenok and P. Naumov, J. Am. Chem. Soc., 2012, 134, 16452–16455 CrossRef CAS PubMed.
- P. Gosset, G. Taupier, O. Crégut, J. Brazard, Y. Mély, K.-D. Dorkenoo, J. Léonard and P. Didier, J. Phys. Chem. Lett., 2020, 11, 3653–3659 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. J. DeSa, P. Naumov and K. M. Solntsev, J. Am. Chem. Soc., 2016, 138, 16252–16258 CrossRef CAS PubMed.
- A. P. Schaap and S. D. Gagnon, J. Am. Chem. Soc., 1982, 104, 3504–3506 CrossRef CAS.
- M. Vacher, I. Fdez. Galván, B.-W. Ding, S. Schramm, R. Berraud-Pache, P. Naumov, N. Ferré, Y.-J. Liu, I. Navizet, D. Roca-Sanjuán, W. J. Baader and R. Lindh, Chem. Rev., 2018, 118, 6927–6974 CrossRef CAS PubMed.
- J. A. Koo, S. P. Schmidt and G. B. Schuster, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 30–33 CrossRef CAS PubMed.
- L. Yue, Y.-J. Liu and W.-H. Fang, J. Am. Chem. Soc., 2012, 134, 11632–11639 CrossRef CAS PubMed.
- K. Støchkel, C. N. Hansen, J. Houmøller, L. M. Nielsen, K. Anggara, M. Linares, P. Norman, F. Nogueira, O. V. Maltsev, L. Hintermann, S. B. Nielsen, P. Naumov and B. F. Milne, J. Am. Chem. Soc., 2013, 135, 6485–6493 CrossRef PubMed.
- K. Støchkel, B. F. Milne and S. B. Nielsen, J. Phys. Chem. A, 2011, 115, 2155–2159 CrossRef PubMed.
- J. L. Woodhouse, M. Assmann, M. A. Parkes, H. Grounds, S. J. Pacman, J. C. Anderson, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2017, 19, 22711–22720 RSC.
- O. V. Maltsev, N. K. Nath, P. Naumov and L. Hintermann, Angew. Chem., Int. Ed., 2014, 53, 847–850 CrossRef CAS PubMed.
- A. R. McKay, M. E. Sanz, C. R. S. Mooney, R. S. Minns, E. M. Gill and H. H. Fielding, Rev. Sci. Instrum., 2010, 81, 123101 CrossRef CAS PubMed.
- A. Henley, A. M. Patel, M. A. Parkes, J. C. Anderson and H. H. Fielding, J. Phys. Chem. A, 2018, 122, 8222–8228 CrossRef CAS PubMed.
- J. L. Woodhouse, A. Henley, M. A. Parkes and H. H. Fielding, J. Phys. Chem. A, 2019, 123, 2709–2718 CrossRef CAS PubMed.
- G. A. Garcia, L. Nahon and I. Powis, Rev. Sci. Instrum., 2004, 75, 4989–4996 CrossRef CAS.
- R. J. Peláez, C. Blondel, C. Delsart and C. Drag, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 125001 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- P. U. Manohar and A. I. Krylov, J. Chem. Phys., 2008, 129, 194105 CrossRef PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- R. A. Kendall, T. H. D. Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- J. Schirmer, Phys. Rev. A, 1982, 26, 2395–2416 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 28, 2299 CrossRef CAS.
- TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- E. Epifanovsky, I. Polyakov, B. Grigorenko, A. Nemukhin and A. I. Krylov, J. Chem. Theory Comput., 2009, 5, 1895–1906 CrossRef CAS PubMed.
- V. Mozhayskiy and A. Krylov, ezSpectrum, http://iopenshell.usc.edu/downloads/ezspectrum.
- C. E. Klots, J. Chem. Phys., 1993, 98, 1110–1115 CrossRef CAS.
- C. E. Klots, J. Chem. Phys., 1994, 100, 1035–1039 CrossRef CAS.
- R. Mabbs, E. Surber and A. Sanov, Analyst, 2003, 128, 765–772 RSC.
- J. B. Wills, F. Pagliarulo, B. Baguenard, F. Lépine and C. Bordas, Chem. Phys. Lett., 2004, 390, 145–150 CrossRef CAS.
- B. Climen, F. Pagliarulo, A. Ollagnier, B. Baguenard, B. Concina, M. A. Lebeault, F. Lépine and C. Bordas, Eur. Phys. J. D, 2007, 43, 85–89 CrossRef CAS.
- R. F. Gunion, M. K. Gilles, M. L. Polak and W. C. Lineberger, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 601–620 CrossRef CAS.
- Y. Toker, D. B. Rahbek, B. Klærke, A. V. Bochenkova and L. H. Andersen, Phys. Rev. Lett., 2012, 109, 128101 CrossRef CAS PubMed.
- C. W. West, J. N. Bull, A. S. Hudson, S. L. Cobb and J. R. R. Verlet, J. Phys. Chem. B, 2015, 119, 3982–3987 CrossRef CAS PubMed.
- A. Henley and H. H. Fielding, Int. Rev. Phys. Chem., 2019, 38, 1–34 Search PubMed.
- P. U. Manohar, J. F. Stanton and A. I. Krylov, J. Chem. Phys., 2009, 131, 114112 CrossRef PubMed.
- E. Janusson, A. V. Hesketh, K. L. Bamford, K. Hatlelid, R. Higgins and J. S. McIndoe, Int. J. Mass Spectrom., 2015, 388, 1–8 CrossRef CAS.
- H. Xia and A. B. Attygalle, Anal. Chem., 2016, 88, 6035–6043 CrossRef CAS PubMed.
- J. Tay, M. A. Parkes, K. Addison, Y. Chan, L. Zhang, H. C. Hailes, P. C. Bulman Page, S. R. Meech, L. Blancafort and H. H. Fielding, J. Phys. Chem. Lett., 2017, 8, 765–771 CrossRef CAS PubMed.
- K. D. Jordan and F. Wang, Annu. Rev. Phys. Chem., 2003, 54, 367–396 CrossRef CAS PubMed.
- J. Simons, J. Phys. Chem. A, 2008, 112, 6401–6511 CrossRef CAS PubMed.
- J. R. R. Verlet, C. S. Anstöter, J. N. Bull and J. P. Rogers, J. Phys. Chem. A, 2020, 124, 3507–3519 CrossRef CAS PubMed.
- J. N. Bull, C. S. Anstöter and J. R. R. Verlet, J. Phys. Chem. A, 2020, 124, 2140–2151 CrossRef CAS PubMed.
- H.-T. Liu, C.-G. Ning, D.-L. Huang, P. D. Dau and L.-S. Wang, Angew. Chem., Int. Ed., 2013, 52, 8976–8979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and 1H NMR spectra; photoelectron images; decomposition of OL− spectra; low eKE features; resonance; calculated VDEs, VEEs, vibrational modes, photoelectron spectra; coordinates of optimised geometries. See DOI: 10.1039/d0cp03276j |
| This journal is © the Owner Societies 2020 |